

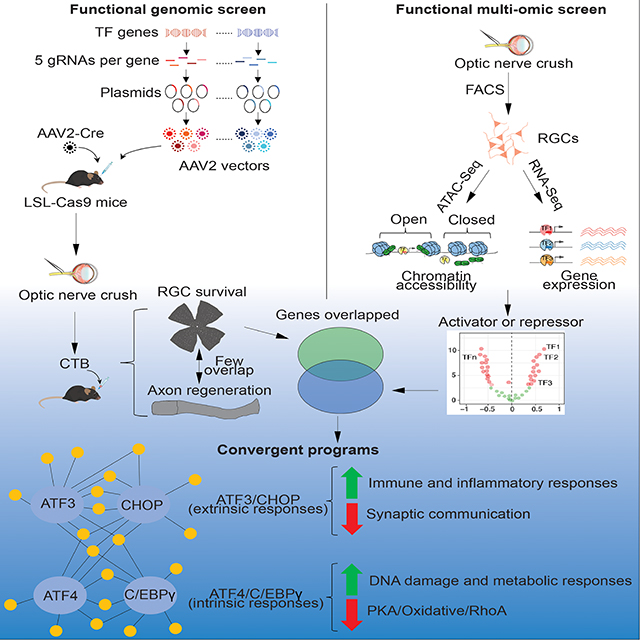
SUMMARY
Regulatory programs governing neuronal death and regenerative failure in neurodegenerative diseases remain poorly understood. In adult mice, optic nerve crush (ONC) injury by severing retinal ganglion cell (RGCs) axons, results in massive RGC death and regenerative failure. We performed an in vivo CRISPR/Cas9-based genome-wide screen of 1893 transcription factors (TFs) to seek repressors of RGC survival and axon regeneration following ONC. In parallel, we profiled the epigenetic and transcriptional landscapes of injured RGCs by ATAC-seq and RNA-seq to identify injury responsive TFs and their targets. These analyses converged on four TFs as critical survival regulators, of which ATF3/CHOP preferentially regulate pathways activated by cytokines and innate immunity and ATF4/C/EBPγ regulate pathways engaged by intrinsic neuronal stressors. Manipulation of these TFs protects RGCs in a glaucoma model. Our results reveal core transcription programs that transform an initial axonal insult into a degenerative process and suggest novel strategies for treating neurodegenerative diseases.
eTOC Blurb:
By independent in vivo genetic screen and multi-omic analysis of injured retinal ganglion cells, Feng et al identify four transcription factors, which act as complementary critical regulators of neuronal survival following injury. These genes regulate two distinct degenerative programs in response to extrinsic and intrinsic stressors, respectively.
Graphical Abstract
INTRODUCTION
Despite their vast clinical heterogeneity, most neurodegenerative diseases share certain pathological outcomes, the most devastating of which is neuronal death. Distal axon damage and dying back neuropathy have been observed in the early stage of many neurodegenerative diseases that lead to loss of neurons, including glaucoma, amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (Calkins et al., 2017; Edwards, 2019; Quigley, 2016; Quigley et al., 1983). However, the pathways that lead from axonal insults to neuronal death remain incompletely understood (Almasieh and Levin, 2017; Perlson et al., 2010).
To identify the mechanisms by which damage to axons in the adult central nervous system (CNS) result in neurodegeneration, we took advantage of the optic nerve crush (ONC) model. In this model, 80% of retinal ganglion cells (RGCs) are lost two weeks after injury and there is virtually no spontaneous axon regeneration (Aguayo et al., 1991; Williams et al., 2020). Here, axonal injury is clearly the primary insult and extensive neurodegeneration, and regenerative failure are pathological outcomes. Previous candidate gene approaches have revealed several pathways that affect RGC survival and/or axon regeneration (Williams et al., 2020), but a comprehensive view of the regulatory landscape governing this process is lacking.
Dissection of complex biological processes has often been advanced by unbiased large-scale genetic screening of many tissues including the CNS (Geschwind and Konopka, 2009; Kampmann, 2020; Parikshak et al., 2015). The clustered regularly interspaced short palindromic repeats (CRISPR) technology (Cong et al., 2013; Mali et al., 2013) provides a powerful means to conduct such screens in vivo. For analysis of ONC, intravitreal injection of AAV2 vectors efficiently transduces most RGCs with minimal effect on other cell types (Nawabi et al., 2015; Park et al., 2008). In addition, the massive and reproducible RGC loss and the short experimental duration of the ONC model allow for survival and regeneration to be analyzed efficiently. Since gene expression changes are key injury responses (Chandran et al., 2016; He and Jin, 2016; Hilton and Bradke, 2017; Moore et al., 2009; Varadarajan et al., 2022; Winter et al., 2022), we reasoned that an in vivo systematic screening of TFs would be a good starting point to identify the critical regulators of neuronal survival and/or axon regeneration.
An alternative approach to identifying TFs that control neuronal injury responses is epigenetic profiling. To control target gene expression, TFs need to bind to specific DNA sequence motifs, but the TF binding sites are not always in an accessible chromatin environment. Moreover, injury may significantly change the chromatin accessibility for TFs (Finelli et al., 2013; Gaub et al., 2010; Palmisano et al., 2019; Puttagunta et al., 2014). The assay of transposase-accessible chromatin using sequencing (ATAC-seq) is a high-throughput method that quantifies accessible chromatin genome-wide (Buenrostro et al., 2013; Buenrostro et al., 2015; Corces et al., 2017). Thus, integrative analysis of ATAC-seq and gene expression changes measured by RNA-sequencing (RNA-seq) in the same cell-type permits unbiased identification of injury-reactive TFs and their transcription targets.
In this study, we conducted multiple orthogonal, but complementary, functional genomic analyses, starting with a comprehensive, in vivo CRISPR screen of all known mammalian TFs. In parallel, we performed a RNA-seq coupled to an ATAC-seq analysis to define transcriptional networks and their drivers in intact and injured RGCs. Remarkably these different approaches converged on an overlapping set of TFs as critical regulators of neuronal injury responses, highlighting a core transcription program that mediates injury-induced neuronal degenerative outcomes. In a companion paper, Jacobi et al (accompany paper) utilized high-throughput single cell RNA-seq to analyze pathways downstream of several well-validated promoters of survival and regeneration (Park et al., 2008; Sun et al., 2011). Their results define molecular programs for survival, degeneration, and regeneration, providing complementary insights into these key aspects of neuronal injury responses.
RESULTS
Optimization of in vivo CRISPR screening
CRISPR has two major components: a guide RNA (sgRNA) and the CRISPR-associated endonuclease 9 (Cas9). By targeting a specific gene, the gRNA guides the non-specific Cas9 to desired DNA locations and introduces site-specific double strand breaks that generate insertion/deletion mutations through imprecise repair. Due to the high efficiency of generating sgRNAs, CRISPR has been utilized for genome-wide screens extensively in cultured cells (Shalem et al., 2014; Shalem et al., 2015) and more recently in in vivo models (Jin et al., 2020; Wertz et al., 2020). To perform an in vivo loss-of-function screen for TFs that promote degeneration and/or inhibit axon regeneration, we utilized the RIKEN and TFCat databases (Fulton et al., 2009; Kanamori et al., 2004) to generate a non-redundant list of 1893 transcription regulators in the mouse genome. We further optimized each of the individual steps to establish an efficient functional screening platform (Figure 1A).
Figure 1. In vivo CRISPR screen identifies negative transcriptional regulators of RGC survival and axon regeneration after ONC.
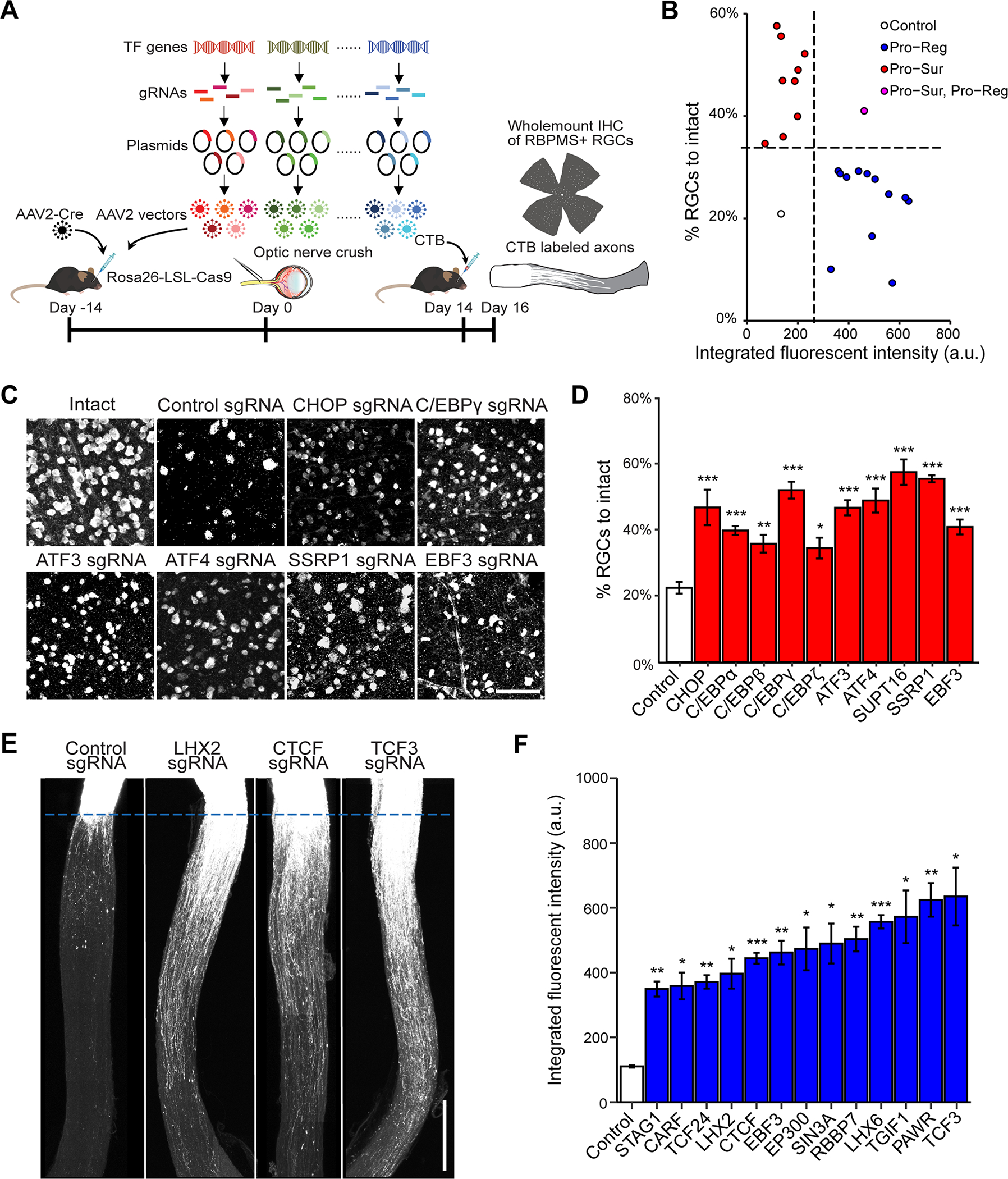
(A) Schematic illustration of the in vivo CRISPR screen in ONC model. AAV2 vectors encoding sgRNAs were injected intravitreally to Rosa26-LSL-Cas9 mice at 2 weeks before ONC. To ensure ablation efficiency, the mix of five different sgRNAs (from GeCKO mouse v2 CRISPR knockout library) were selected to target each gene candidate.
(B) The final lists of survival and regeneration hits were categorized into three groups: (1) deletion of the TF increasing axon regeneration without affecting RGC survival (Pro-Reg, 12 hits); (2) deletion of the TF increased RGC survival without affecting axon regeneration (Pro-Sur, 9 hits); (3) deletion of the TF promoted both RGC survival and axon regeneration (Pro-Sur, Pro-Reg, 1 hit). Each data point is the averaged results from RGC survival or axon regeneration analyses.
(C) Representative immunohistochemistry images of wholemount retinas showing improved survival after ONC injury by CRISPR ablation of individual TFs. Scale bar, 50 μm.
(D) Quantification of RGC survival with individual TF knockout. Data are shown as mean ± s.e.m. with n = 4–5 biological repeats. *p<0.05, **p<0.01, ***p<0.001, calculated by one-way ANOVA.
(E) Representative optic nerve images showing axon regeneration after ONC injury with individual TF knockout. Scale bar, 0.5 mm.
(F) Quantification of CTB labeled fluorescent intensity (from crush site) for all sgRNA hits that promote RGC axon regeneration. Data are shown as mean ± s.e.m. with n=5–6 biological repeats. *p<0.05, **p<0.01, ***p<0.001. Welch anova test, followed by Dunnett’s T3 adjustment.
Briefly, to maximize CRISPR knockout efficiency, we selected five sgRNAs targeting different regions of each TF gene from the well-characterized genome-scale CRISPR/Cas9 knockout (GeCKO) library (Shalem et al., 2014; Shalem et al., 2015). Then, we generated a library of 1893 pools of AAV-sgRNA vectors, with each pool containing 5 different sgRNA-bearing AAV vectors targeting the same TF genes (Swiech et al., 2015) (Figure 1A). To introduce sgRNAs and Cas9 to RGCs, we co-injected pools of AAVs expressing sgRNAs and Cre into the vitreous bodies of Rosa26-LoxP-STOP-LoxP-Cas9-GFP (LSL-Cas9) mice. At 2 weeks after viral injection, ONC was performed, followed 2 weeks later by quantitative procedures to assess the extent of RGC survival and axon regeneration (Figure S1A).
As a proof-of-principle test, we showed that introducing sgRNAs for Satb1, an established marker for ON-OFF direction-selective RGCs (Peng et al., 2017), resulted in efficient Satb1 removal, as indicated by immunohistochemistry (Figure S1B and S1C). Similarly, with its specific sgRNAs, CRISPR mediated targeting of Pten gene also led to efficient reduction of PTEN expression (Figure S1D–E) and produced the expected increases in RGC survival and axon regeneration (Figure S1F–S1I) comparable to those seen in conventional PTEN knockout mice (Park et al., 2008).
Identification of negative TF regulators of neuronal survival and/or axon regeneration
We then screened the effect of knockout of 1893 TFs, testing each gene in both eyes of one mouse. Genes whose knockout increased survival and/or regeneration (see Method) were retested in at least 3 additional eyes. Those producing consistent and significant increases in neuronal survival and/or axon regeneration were considered as true positive hits. This stringent screening protocol might miss TFs with modest effects (false negatives) but is expected to identify most, if not all, TFs with strong effects.
In total, from the 1893 TFs tested, we identified 10 genes as negative transcriptional regulators of neuronal survival (Figure 1B–D). Consistent with previous studies (Hu et al., 2012), CCCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP, also called Ddit3), was identified as an anti-survival factor, providing additional verification of our screen method (Figure 1C and 1D). Other survival hits included several other C/EBP family members (C/EBPα, C/EBPβ, C/EBPγ, and CEBPζ) and two ATF family members (ATF3, ATF4), as well as two FACT complex components (SSRP1 and SUPT16), and EBF3, a tumor suppressor. We observed a similar increased survival of RGCs in ATF3 knockout mice (Renthal et al., 2020), validating the phenotype observed with the CRISPR experiment (Figure S1J–M). Notably, the identified ATFs and C/EBPs are in the basic leucine zipper domain (bZIP) containing proteins family. While these evolutionarily conserved TFs have been implicated in regulating several biological processes, such as memory inhibition (Abel et al., 1998; Bartsch et al., 1995; Sidrauski et al., 2013), integrated stress response and cell death (Aime et al., 2020; Costa-Mattioli and Walter, 2020; Karuppagounder et al., 2016; Kovalchuke et al., 2019; Sun et al., 2013; Wortel et al., 2017), the predominance of ATF and C/EBP members in our survival hits were a surprise, as they were not previously identified in this capacity in the literature.
Although our focus here is on neuronal survival, we also assessed axon regeneration and identified 13 genes as negative regulators of this process (Figure 1B, E, F). This group included several genes implicated in epigenetic regulation (CTCF/STAG1, histone acetyltransferase EP300, RBBP7, and SIN3A), members of the BHLH family (TCF3, TCF24) and the homeobox family (LHX2, LHX6, TGIF1) genes, tumor suppressors PAWR/WT1, and EBF3 (Figure 1E and 1F). The only overlapping gene among the survival and regeneration lists is EBF3. This dissociation suggests that transcription programs for regulating neuronal survival and axon regeneration are largely separate; the companion paper based primarily on transcriptomics (Jacobi et al., accompanying paper) describes these distinct programs in detail.
Characterization of chromatin accessibility changes in RGCs following optic nerve crush
As a drastic insult, axotomy may trigger large-scale changes in gene expression via reorganization of chromatin. To assess genome-wide changes in chromatin accessibility following ONC, we performed ATAC-seq, which enables high-throughput quantification of open chromatin regions, as well as TF footprinting (Bentsen et al., 2020; Buenrostro et al., 2013). In tandem, we also performed bulk RNA sequencing, allowing us to directly assess whether identified chromatin alterations led to transcriptional changes of the TFs and their target genes. We optimized purification of the RGCs from intact or injured mice for the ATAC-seq and RNA-seq studies (Figure 2A). Based on published data (Tran et al., 2019), we reasoned that most critical transcriptional responses to injury would occur in the first few days after injury, when gene expression has already been altered, but RGC death has not started. Thus, we FACS-sorted RGCs at 0, 1 or 3 days after injury and subjected them to ATAC-seq and RNA-seq (Figure 2A).
Figure 2. Characterization of chromatin accessibility changes in retinal ganglion cells following optic nerve crush.
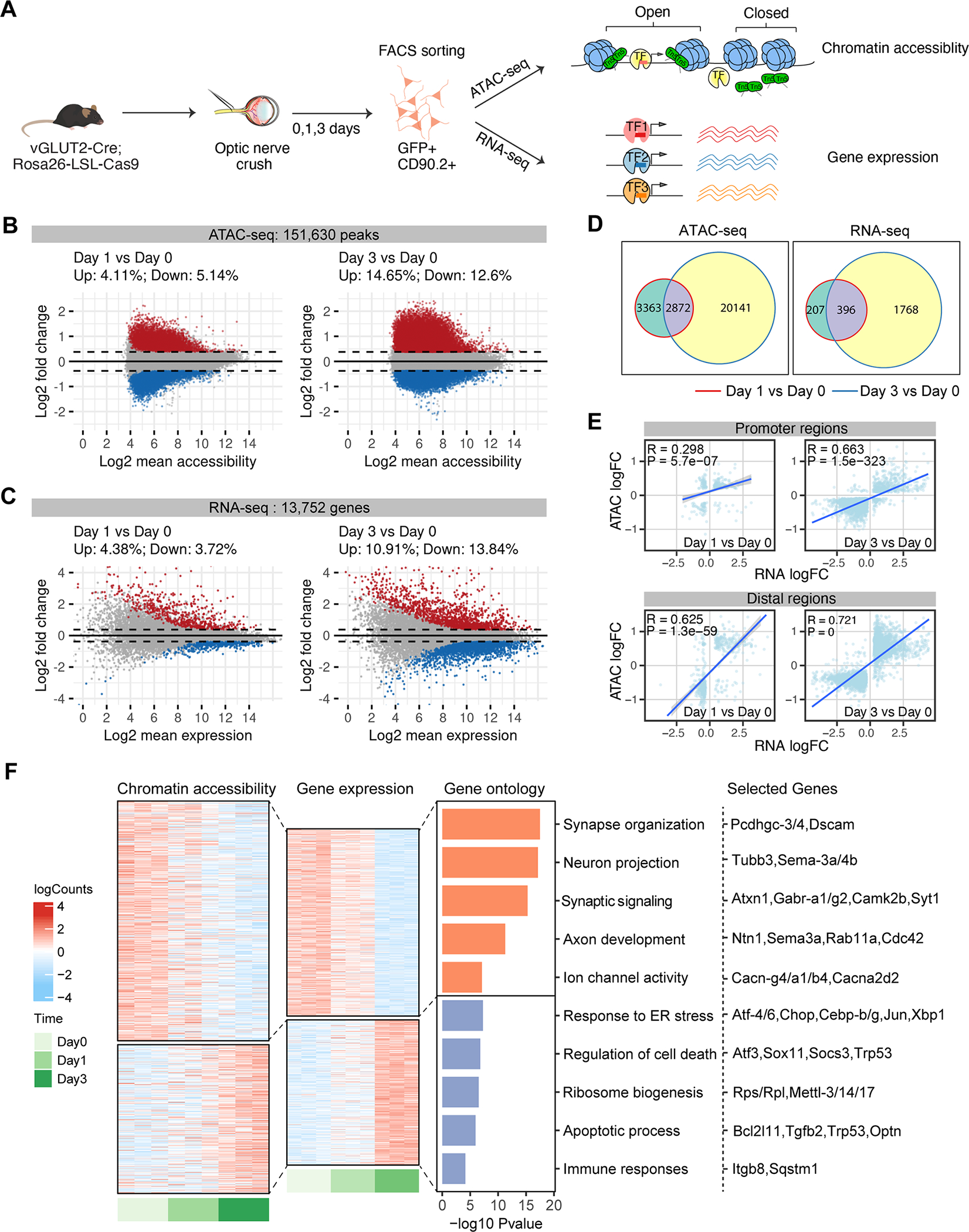
(A) Schematic diagram summarizing the overall experimental flow. vGLUT2-labeled RGCs were FACS-sorted at 0 (no crush) 1, 3 days following optic nerve crush. ATAC-seq and RNA-seq were performed on separate sets of injured RGCs, with n = 3–6 biological repeats in each time point.
(B-D) MA plots displaying differential accessible regions (DAR) (B) and differential expressed genes (DEGs) (C) in RGCs following injury. Each dot represents a peak region or a gene, and colored dots indicate DARs or DEGs (FDR p < 0.1, | log2 FC| > 0.3). Upregulated: red; Down-regulated: blue. (D) Overlap of DARs and DEGs at day 1 or day 3 following injury.
(E) Pearson correlations between injury-induced changes in gene expression and chromatin accessibility at the promoter and distal DARs. Using GENCODE annotations, we defined an ATAC-seq peak ± 2 kb of a gene’s transcription start site (TSS) as a promoter (proximal regulatory element), and non-promoter peaks ± 500 kb of TSS as distal regulatory regions of that gene. The differential accessibilities of these DARs (log2 FCs) were correlated and plotted against the differential expression of the linked genes. If several distal peaks are linked to the same gene, the average differential accessibility was used to correlate with differential expression.
(F) Chromatin accessibility and mRNA expression of linked peak-gene pairs. Heatmap colors indicate row-scaled chromatin accessibility (left) or mRNA expression levels (right). Bar plots represent negative log10 FDR-corrected p-values of top Gene Ontology (GO) terms associated with genes in each cluster. Representative genes in each term were displayed.
Following extensive quality control procedures (Figure S2A–G), we identified 151,630 reproducible accessible regions, or peaks, across the pooled ATAC-seq dataset. Importantly, we observed high correlations between biological replicates within peaks (Pearson’s correlation coefficient r > 0.95, Figure S2E). By analyzing chromatin accessibility changes in response to ONC, we identified 14,024 (~9.25%) differentially accessible regions (DARs) at day 1 post-injury and 41,346 (~27.25%) DARs at day 3 (Figure 2B, Table S1), with similar numbers of peaks bearing increased or reduced accessibility at both time points. Consistent with the ATAC-seq data, RNA-seq analysis on injured RGCs identified more changes at day 3 versus day 1: ~ 8.1% (1,115) differentially expressed genes (DEGs) on day 1, and ~ 24.75% DEGs (3,403) at day 3 (Figure 2C, Table S2). Only ~50% of DARs or DEGs at day 1 overlap with those identified at day 3 (Figure 2D). Thus, these results revealed injury-induced, time-dependent alterations in both genomic accessibility and gene expression patterns in RGCs.
DARs are enriched in more accessible chromatin regions that were previously annotated from mouse brain (Ernst and Kellis, 2012; Shen et al., 2012), including, but not confined to promoters and enhancers and depleted in heterochromatin and transcribed regions that are less accessible (Figure S2C), in agreement with previous findings from other regions and cells (de la Torre-Ubieta et al., 2018; Ernst and Kellis, 2012; Klemm et al., 2019; Shen et al., 2012). As the accessibility of both proximal promoter and distal regions such as enhancers is important for regulating gene expression (Roadmap Epigenomics et al., 2015; Spitz and Furlong, 2012), we further analyzed the relationships between gene expression and local chromatin accessibility patterns in our dataset (Figure 2E). The correlations across DEGs and DARs (both proximal and distal), increase at day 3 after injury compared to day 1, consistent with the expectation that changes in chromatin accessibility precede changes in gene expression. We also found that accessibility of distal DARs correlates more strongly with gene expression than promoter-proximal chromatin accessibility, a finding also observed in other cell types (Trevino et al., 2020).
We next identified significantly correlated peak-gene pairs (Table S3), defining two patterns of chromatin accessibility and gene expression in response to CNS injury (Figure 2F). The first cluster of peak-gene pairs includes those whose chromatin accessibility and gene expression are both down-regulated by injury. Functionally they are associated with synapse organization, synaptic transmission, cell adhesion and neuron projection (Figure 2F, Table S3). Among them are Pcdhgc3/4 and Dscam, which have been implicated in RGC survival (Chen et al., 2012; Keeley et al., 2012). The second is an injury up-regulated cluster, which is involved in the regulation of endoplasmic reticulum stress, ribosome biogenesis, cell death / apoptosis, immune processes, and RNA processing (Figure 2F, Table S3). Interestingly, in addition to several genes previously implicated in stress/injury response and neuronal death, such as Sox11, Socs3, Gadd45g (Fischer et al., 2004; Norsworthy et al., 2017; Sun et al., 2011) and glaucoma-associated gene Optn (Wiggs and Pasquale, 2017), this cluster also includes several death-promoting TF hits identified from our CRISPR screening, including Atf3, Atf4, CHOP, Cebpb, Cebpγ (Figure 1, Figure 2F). Thus, these findings suggest that CNS injury induces widespread chromatin accessibility changes driving the expression of genes that are less favorable for neuronal survival and axon regrowth.
Identification of TFs driving chromatin accessibility changes
To identify TFs whose expression was significantly correlated with changes in the accessibility of their binding motifs, we integrated ATAC-seq, RNA-seq and databases of TF-binding specificity. This approach is based on the rationale that the abundance of a TF and the accessibility of its binding motifs are associated with its gene regulatory activity (Corces et al., 2020; Trevino et al., 2020) (Figure 3A). By scanning DARs in the ATAC-seq data for TF motif occurrence, we identified a total of 1,310 significantly enriched TF binding motifs (Table S4). We than used ChromVAR (Schep et al., 2017) to compute changes in motif accessibility across injury conditions as deviation Z-scores, as an indicator of a TF’s activity at that position, which was further correlated with each TF’s mRNA expression from RNA-seq data (Table S4). Remarkably, among the top quartile of all TFs whose binding motifs showed the largest correlation between changes in their expression levels and accessibility of their binding motifs were four of the CRISPR screen hits ATF3, ATF4, C/EBPγ, CHOP/Ddit3 (Figure 3B, absolute Pearson correlations r > 0.5 and P < 0.05). mRNA and protein expression levels of all 4 TFs were consistently increased at day 3, as was the increased accessibility of their binding sites (Figure 3C and D).
Figure 3. Identification of transcription factors driving chromatin accessibility changes.
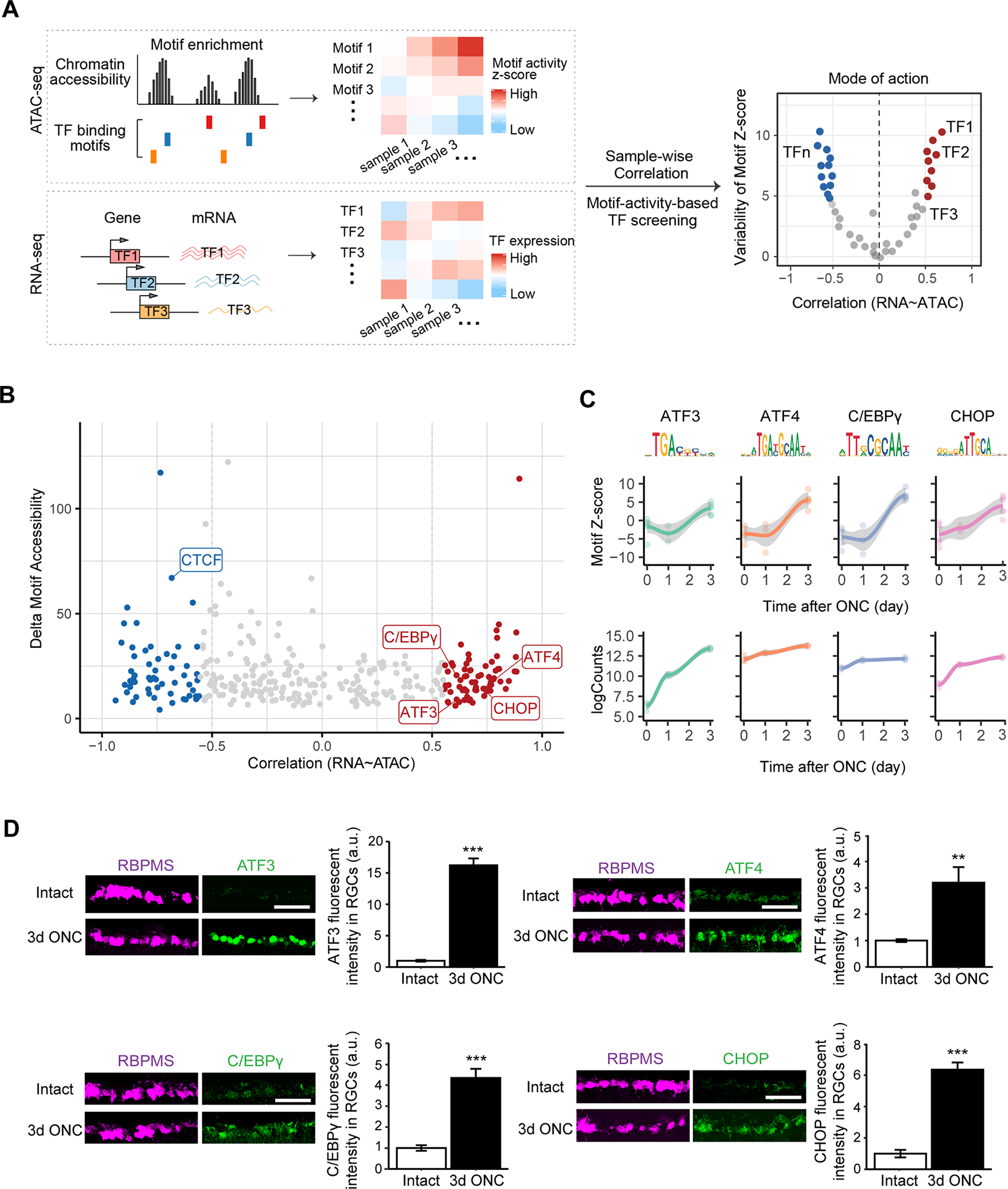
(A) A schematic diagram displaying an unbiased bioinformatic approach to identify TF regulators driving chromatin accessibility and gene expression changes in injured RGCs. In this approach, we first find TF binding motifs that are significantly enriched within differentially accessible regions (DAR). The degree of accessibility at enriched TF motifs was computed as deviation Z-scores, and were correlated with TF expression levels across samples to classify TF mode of actions.
(B) TF gene expression - motif accessibility correlations against maximum inter-sample difference in deviation Z-score. Each dot represents a TF, and colored dots indicate TFs whose gene expressions are significantly correlated or anti-correlated to motif deviations (Pearson’s correlation coefficient r > 0.5 and FDR < 0.1), and whose maximum cross-sample difference in deviation z-score is in the top 25% of all TFs. Th genes overlapped with CRISPR screen hits were highlighted.
(C) TF binding motif accessibility deviations and RNA-seq expression levels for ATF3, ATF4, C/EBPγ, CHOP/DDIT3.
(D) Representative immunohistochemical images showing injury-induced protein expression at 3d post-ONC of individual TFs. Data are shown as mean ± s.e.m. with n = 4. **p<0.01, ***p<0.001, calculated by two sample t test. Scale bar, 10 μm.
Characterization of functional TF targets by combinatorial analysis of DNA footprinting and RNA-seq of perturbed RGCs
For further computational and functional analysis we focused on a set of four genes – ATF3, ATF4, C/EBPγ and CHOP/Ddit3 – for several reasons. First, all four were hits in our CRISPR screen (Figure 1D). Second, these genes exhibited both increased chromatin accessibility and gene expression (Figures 2F and 3C). Third, ATAC-Seq data analysis implicated these genes as critical TFs that drive injury-induced responses in RGCs. (Figure 3B). In contrast, other hits from the CRISPR screen (SUPT16, SSRP1) are expressed in intact and injured RGCs with similar levels (Tran et al., 2019), suggesting a permissive role in injury responses. Finally, these four TF genes, which are members of an evolutionarily conserved family, may act synergistically via dimerization to regulate gene expression programs (Reinke et al., 2013). Together, these data support the hypothesis that this family of TFs act as critical transcriptional regulators of early injury responses in RGCs.
To characterize survival regulating pathways, we used two complementary methods to identify the downstream targets of ATF3/4, C/EBPγ and CHOP in injured RGCs (Figure 4A). First, we characterized TF binding to target genes via DNA footprinting. Since TF binding shields bound DNA elements from transposase-mediated digestion, protected DNA sequences are believed to be direct TF binding sites, or footprints (Figure S3). Using newly developed methods (Bentsen et al., 2020; Funk et al., 2020; Vierstra et al., 2020), which identify direct TF-DNA binding events (similar to ChIP-seq), we mapped genome-wide DNA footprints overlapping with each TF’s binding motifs, and quantified TF binding activities by leveraging ATAC-seq measurements of footprinted regions (Figure S3A). Notably, all four TFs become more active following injury, with increased binding depth and more open surrounding chromatin (Figure S3B–D), supporting their roles in driving injury-induced chromatin state changes. Mapping each TF’s footprints to individual genes revealed a highly significant correlation between footprint activities and gene expression changes induced by injury (Figure S3E).
Figure 4. Identification of direct target genes of the four survival TFs.

(A) A schematic diagram displaying integrative analysis of DNA-footprinting using ATAC-seq data and RNA-seq to identify each TF’s direct target genes. Chromatin-bound TF protects DNA elements from Tn5 transposase cleavage, creating single-nucleotide-resolution DNA “footprints” during ATAC-seq. Mapping these footprints would identify DNA regions directly bound by the TF, and the genes linked to TF footprinted regions. To identify genes that are directly regulated by each survival TF, RNA-seq was performed on RGCs with or without prior CRSIPR ablation of this TF at day 3 following injury. Genes that are footprinted by this TF and differentially expressed (up- or down-regulated, absolute logFC >0 and FDR < 0.1) upon ablation of this TF are considered as its direct target genes. N = 4–5 biological repeats in each condition, except for the group with ATF4 ablation (n = 2 biological repeats after outlier removal).
(B) A Chow -Rusky Venn diagram and an Upset plot showing the overlap of each TF’s direct targets. Similar to regular Venn diagram graphics, the Chow-Rusky Venn diagram is divided into 2n connected regions (n = 4 TF combinations). The regions forming loops indicate overlapped targets of colored TFs (e.g. 140 targets common to 4 TFs), while the other open connected regions indicate targets exclusive to certain sets of TFs (e.g. 820 targets unique to ATF3 that is red colored, but not to the other 3 TFs). In the Upset plot, the set size indicates the number of direct targets for each TF. The intersection size indicates the number of overlapped targets unique to dotted TF(s).
(C) A heatmap of correlation matrix showing similarity and dis-similarity in the expression levels of each TF’s direct target genes. Normalized RNA-seq counts of each TF’s direct target genes were correlated and clustered by the distances among samples from control RGCs (nontargeting sgRNA) or treated RGCs receiving individual CRISPR TF ablations. Colors in the heatmap indicate Pearson’s correlations between two samples. ATF3 and CHOP are more similar in the expression levels of their target genes, while ATF4 and C/EBPγ are more similar.
(D) Shared or unique target gene sets of each TF. Positive targets are defined as genes that are down-regulated (log2FC < 0, FDR < 0.1) when this TF is ablated. Negative targets are defined as genes that are up-regulated (log2FC > 0, FDR < 0.1) when this TF is ablated. Based on gene expression similarity in (C), each TF’s positive or negative targets were grouped together and clustered into three gene sets: a) genes that are uniquely controlled by ATF3 and CHOP, b) genes that are uniquely controlled by ATF4 and C/EBPγ, and c) genes that are shared by ATF3/CHOP and ATF4/C/EBPγ groups. Gene ontology on these three gene sets were performed and related GO terms with enrichment FDR value < 0.05 were presented.
The second method to characterize downstream, survival regulating pathways was to perform RNA-seq on injured RGCs from which ATF3, ATF4, C/EBPγ or CFIOP had been deleted (Figure 4A). We injected AAVs expressing gene-specific sgRNAs and mCherry reporter to the vitreous bodies of LSL-Cas9: Vlgut2-Cre mice, so that sgRNA-mediated knockouts of individual TFs occur selectively in RGCs (Zhang et al., 2019). At day 3 following optic nerve crush, transduced RGCs were FACS-purified for RNA-seq. Following quality control and outlier removal, we analyzed gene expression changes caused by each perturbation. We found that in comparison to control (non-targeting) gRNAs, knocking out ATF3, ATF4 or CFIOP resulted in ~1,800 differentially expressed genes (DEGs), whereas C/EBPγ depletion generated ~5,600 DEGs (Figure S4A, Table S5). Recognizing that genes identified by RNA-seq include both direct and indirect targets of specific TFs, we focused on the overlap between each TF’s DEGs from RNA-seq coupled to TF perturbations and its target genes predicted by footprint analysis (Table S6). We observed significant overlaps, with ~27–50% of the knockout-associated DEGs, identified as direct target genes of individual TFs (Figure S4B), supporting our hypothesis that RNA-seq identifies both direct and indirect transcriptional responses.
3. Identification of Two Degenerative Programs Dependent on ATF3/CHOP and C/EBPγ/ATF4 Respectively
Further analyses focused on the direct transcriptional targets of ATF3, ATF4, CHOP and C/EBPγ. As expected, direct targets include both down-regulated or up-regulated genes due to CRISPR knockout of each TF (Figure S4C), thus representing positively or negatively regulated genes of the TFs, respectively. By comparing the GO terms of these target genes of individual TFs, we found that the shared functions of these TFs are relevant to regulating responses to stimulus and cell death processes (Figure S4D), consistent with their roles in mediating RGC degeneration after injury (Figure S4D). Interestingly, several of these target genes, such as Srebf1 and Rora, which are shared targets of all 4 TFs, have been implicated in human glaucoma by a GWAS study (Gharahkhani et al., 2021).
To assess overlap among gene expression programs regulated by the four key TFs, we constructed a Venn diagram of their direct target genes. As shown in Figure 4B, ATF3 and C/EBPγ each regulate a unique set of genes, while most target genes of ATF4 or CHOP are shared by the other two TFs. Hierarchical clustering on the expression levels of the direct target genes of individual TFs revealed distinct gene expression patterns in injured RGCs after perturbation, one shared by knockout of ATF3 or CHOP, and the other by the knockout of ATF4 or CEBPγ (Figure 4C). We therefore group these directly targeted genes and their GO terms into different clusters: positive or negative targets shared by ATF3/CHOP, C/EBPγ/ATF4, or those shared by all four TFs (Figure 4D). Strikingly, ATF3/CHOP and ATF4/CEBPγ appear to regulate different positive and negative regulatory pathways (Figure 4D and 5A). The positive targets of ATF/CHOP mainly regulate neuronal responses to extrinsic factors, such as TNFR1/p38 MAPK signaling, TLR2 and other molecules in innate immunity (Figure 4D, 5A, S5A, S5D). Differential expression of TLR2 and Pkmyt1 under these conditions was verified by immunohistochemistry (Figure S5G–S5J). In contrast, ATF4/C/EBPγ’s positive targets are more relevant to cell intrinsic stress responses, such as cell cycle/DNA damage/checkpoint regulation, p53 and NAD signaling and autophagy. For example, Tnfrsf21 or death receptor 6 (DR6), a TNF receptor family member that triggers cell death (Benschop et al., 2009), is uniquely bound and activated by ATF3 and CHOP, but not C/EBPγ and ATF4 (Figure 5A–B, ATF3/CHOP positive; Figure S5).
Figure 5. Two distinct transcriptional programs regulated by ATF3/CHOP or C/EBPγ/ATF4 in injured RGCs.
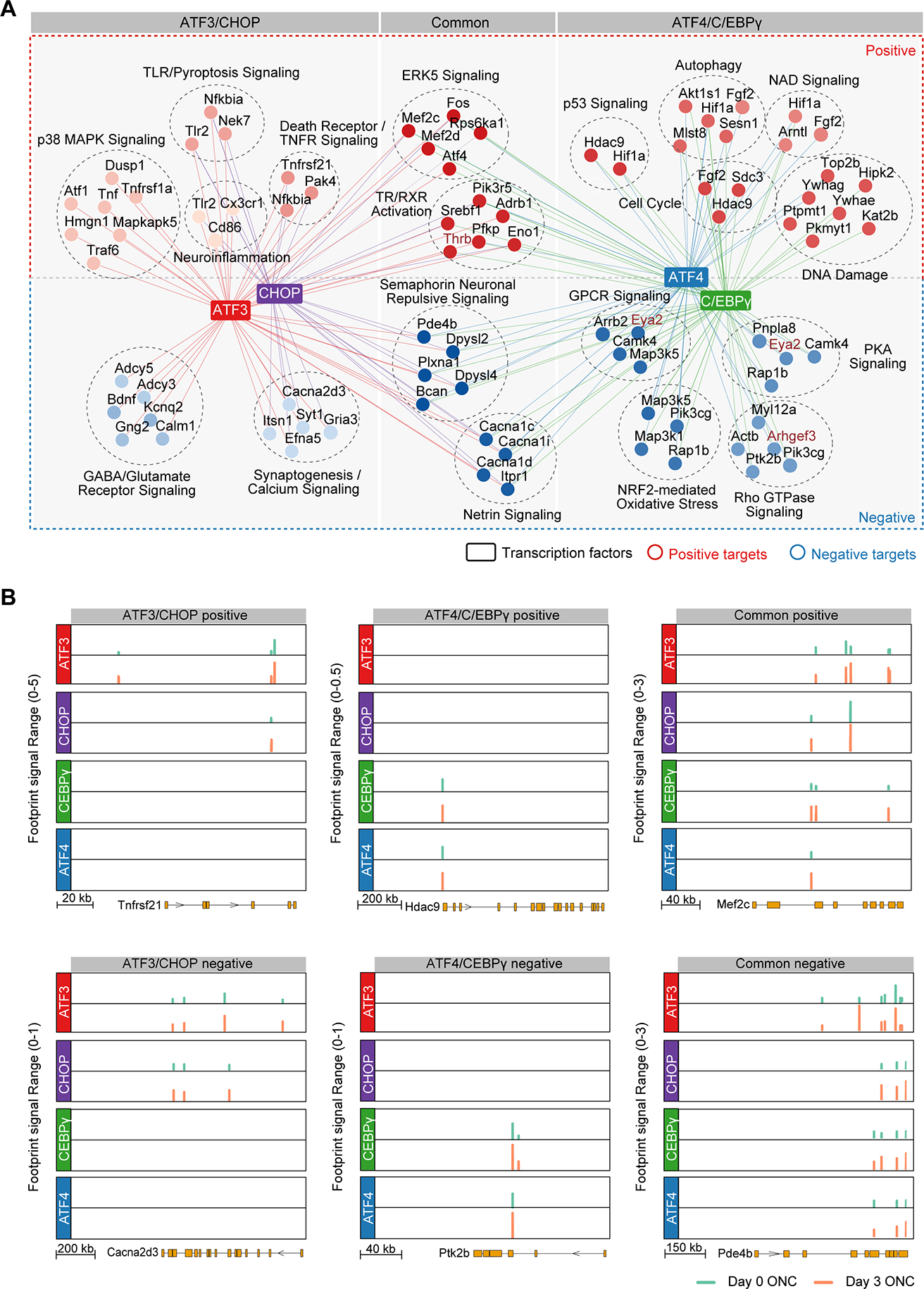
(A) A TF network plot showing the positive or negative targets and the associated biological pathways that are uniquely controlled by ATF3 and CHOP (ATF3/CHOP), uniquely controlled by ATF4 and C/EBPγ (ATF4/C/EBPγ), or shared by both ATF3/CHOP and ATF4/C/EBPγ (common). Representative genes in each pathway were shown. Each node indicates a gene. Node colors indicate the different GO terms, and edge colors indicate TFs.
(B-C) Genome browser views of each of 4 TF’s binding sites around representative target genes. Y-axis indicates Tn5-bias corrected footprint signals of each TF from control (day 0) or optic nerve crushed (day 3) RGCs. Shown representative genes, including both positive (B) and negative (C) targets, are selected from the three groups in (A).
In contrast, Hdac9, which regulates endogenous DNA repair (Wong et al., 2009), is a unique target of C/EBPγ and ATF4 (Figure 5A–B, C/EBPγ/ATF4 positive; Figure S5). Negative targets of ATF3/CHOP and ATF4/CEBPγ are also distinct. ATF3/CHOP negative targets are mainly associated with different aspects of synaptic function, suggesting that ATF3 and CHOP mediate direct injury-triggered disruption of RGCs’ physiology. In contrast, the negative targets of ATF4/C/EBPγ are highly relevant to protein kinase A signaling and Rho GTPase regulation, important processes related to intrinsic regulation of signaling and homeostasis. Thus, these results revealed two complementary programs in mediating injury-induced RGC degeneration.
Additive Effects of Co-knockout of TFs in Both Divisions Resulting in nearly Complete Protection of RGCs After Injury
If ATF3/CHOP and CEBPγ/ATF4 indeed regulate distinct degenerative pathways in injured RGCs, co-manipulating one TF from each of the two subgroups would be predicted to have additive effects and further improve RGC survival compared to individual CRISPR perturbations of each TF. To test this prediction, we deleted several pairs of the four TFs and assessed neuronal survival. Injury-induced alterations could regulate RGC survival by RGC- autonomous and RGC-nonautonomous mechanisms (Zhang et al., 2019). We revised our protocol by injecting gene-specific sgRNA-carrying AAVs to the vitreous bodies of the mice of LSL-Cas9: Vglut2-Cre. In this way, sgRNA-mediated knockouts of individual TFs occur selectively in RGCs but not in other retinal cell types (Zhang et al., 2019) (Figure S6A). As shown in Figure S6B and S6C, RGC-selective knockout of each of these TFs led to virtually identical survival outcomes in these mice as non-selective knockouts (Figure 1C and 1D), supporting the RGC-autonomous effects of these TFs.
To minimize the effect of diluting AAV2 vectors due to combining different pools of sgRNAs, we intravitreally injected two different sgRNA encoding AAV2 vectors together with AAV2-Cre into LSL-Cas9 (Figure 6A) and verified knockout efficiency by immunohistochemistry (Figure 6B–E, S6D–G). Co-ablation of one TF from each of the two subgroups led to higher RGC survival in three of four cases (ATF3/CEBPγ, ATF3/ATF4, and CEBPγ/CHOP) compared to single knockout (Figure 6F, 6G). In contrast, co-knockout of TFs within the same group, (ATF3/CHOP or ATF4/C/EBPγ) was not significantly more protective than single knockout (Figure 6F, 6G). All four of these genes are expressed in most RGC types (Figure S6H, I). Because 80–90% of RGCs are transduced by intravitreal injected AAVs (Jacobi et al., accompany paper), these results suggested that co-knockout of these TFs protect most RGC types from injury-induced degeneration. Thus, these observations provide functional validation of our bioinformatic predictions from integrative analysis of DNA-footprinting and RNA-seq, supporting the overlapping, yet complementary effects of ATF3/CHOP and ATF4/C/EBPγ on activating two parallel pro-death pathways following injury.
Figure 6. Functional interactions of the key TFs in injured RGCs.
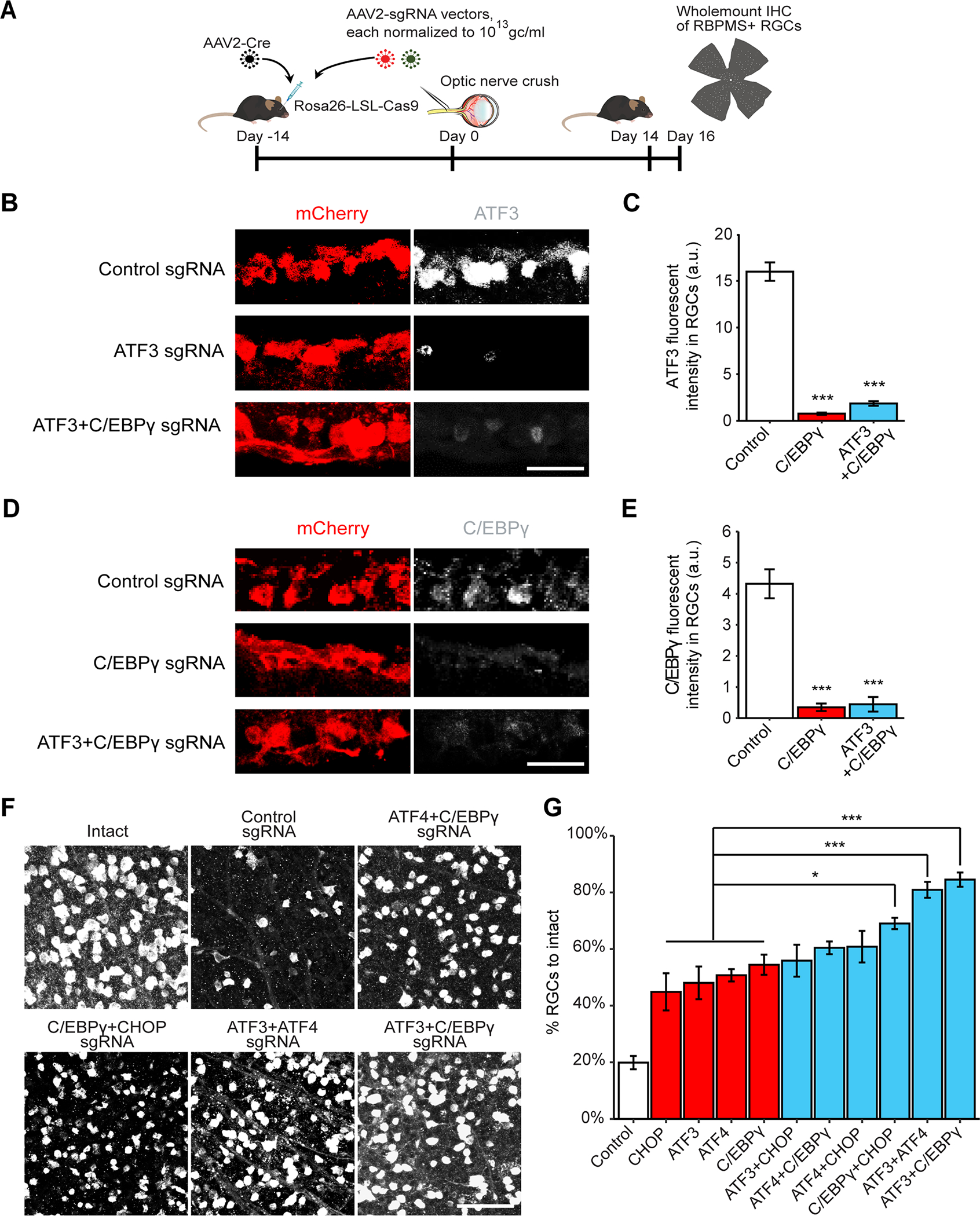
(A) Schematic illustration of CRISPR KO with sgRNAs targeting multiple genes. AAV2 vectors encoding sgRNAs were injected intravitreally to Rosa26-LSL-Cas9 mice at 2 weeks before ONC.
(B-C) Representative images (B) and quantification (C) of ATF3 immunohistochemical staining indicate the knockout efficiency of ATF3 sgRNA and ATF3+C/EBPγ sgRNA. Data are shown as mean ± s.e.m. with n = 4. ***p<0.001, calculated by one-way ANOVA. Scale bar, 10 μm.
(D-E) Representative images (D) and quantification (E) of C/EBPγ immunohistochemical staining indicate the knockout efficiency of C/EBPγ sgRNA and ATF3+C/EBPγ sgRNA. Data are shown as mean ± s.e.m. with n = 4. ***p<0.001, calculated by one-way ANOVA. Scale bar, 10 μm.
(F) Representative images of RGC survival with combinations of sgRNA hits injected to LSL-Cas9 mice. Scale bar, 50 μm.
(G) Quantification of RGC survival with combinations of sgRNA hits. Data are shown as mean ± s.e.m. with n = 4–5 biological repeats. *p<0.05, ***p<0.001, calculated by nonparametric Kruskal Wallis test, followed by Bonferroni adjustment.
Knockout of Identified TFs Protects RGCs in a Glaucoma Model
Identification of these transcriptional drivers of injury-induced neuronal degeneration and their downstream pathways provides new potential therapeutic targets. To directly test whether our findings extend to pathological conditions, we extended our knockout analysis to a glaucoma model. Glaucoma, a leading cause of irreversible blindness worldwide, is the result of progressive RGC loss (Quigley and Broman, 2006; Weinreb et al., 2014). Elevated intraocular pressure (IOP) is a major risk factor, and a proposed pathological mechanism is initial axonal damage and subsequent RGC death (Calkins et al., 2017; Guo et al., 2021; Nickells et al., 2012; Quigley et al., 1983; Sommer, 1989; Weinreb et al., 2016). Likewise, ocular hypertension in rodents leads to RGC death and has been widely used to model glaucoma (Cone et al., 2010; Sappington et al., 2010; Zhang et al., 2019). To induce consistent IOP elevation, we modified the conventional microbead/viscoelastic strategy and developed a novel viscobead occlusion experimental glaucoma model (Figure S7 and 7A–G). After injection into the anterior chamber, the viscobeads rapidly accumulated at the iridocorneal angle (Figure S7E–F, 7A–C) and induced consistent IOP elevation for at least 8 weeks in most mice (Figure 7D and S7F). Importantly, about 30% of RGC loss was consistently observed at 8 weeks after viscobeads injection, as assessed in retinas (Figure 7E–G) and optic nerves (Figure S7G–K).
Figure 7. CRISPR ablation of the identified TFs protects RGCs in a glaucoma model.
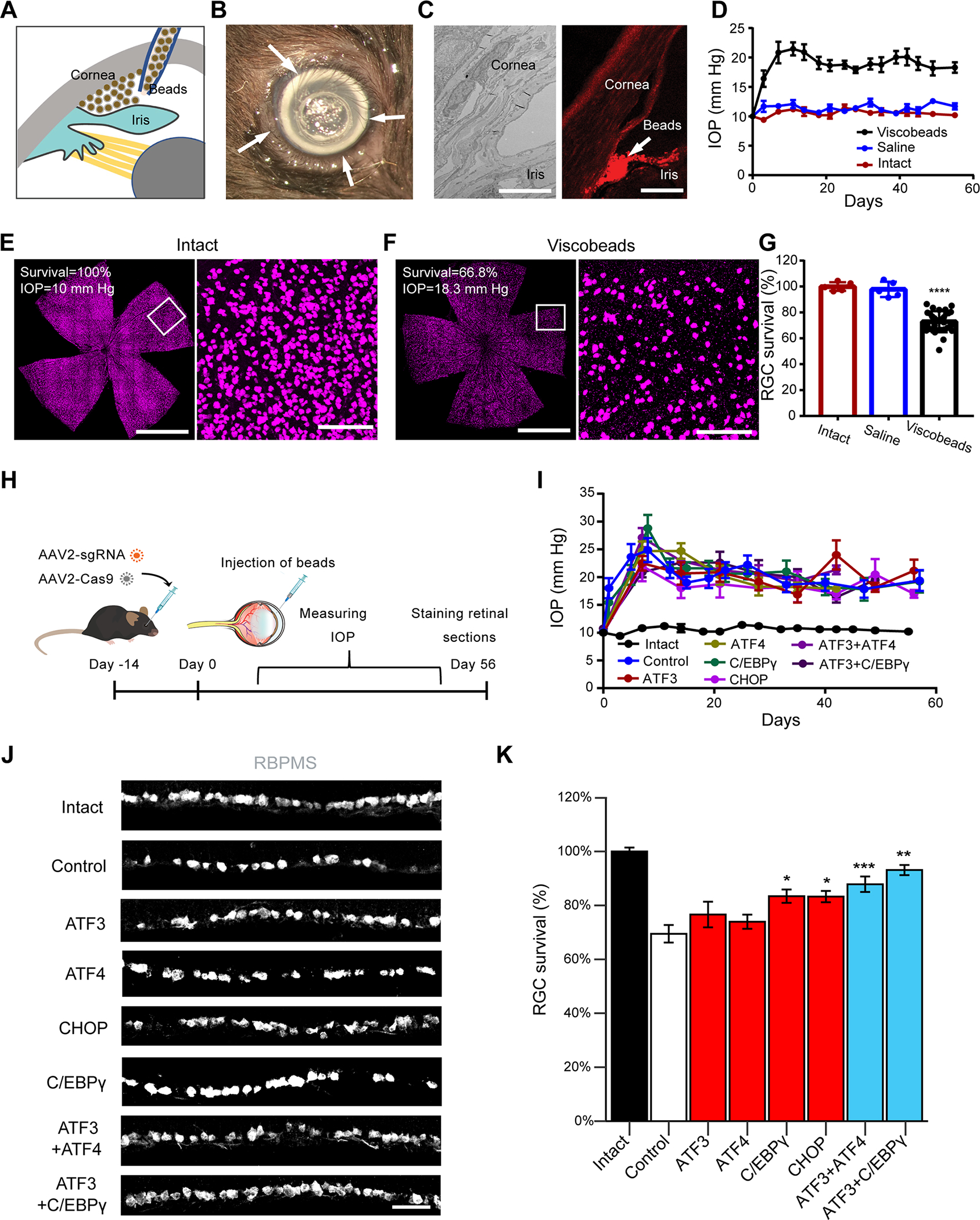
(A) Schematic illustration of the viscobeads occlusion experimental glaucoma model. Concentrated viscobeads were injected into the mouse anterior chamber to block aqueous drainage.
(B) A representative photograph of viscobeads accumulated at mouse iridocorneal angle 5 min after injection. Arrows show the white viscobeads were restricted at the iridocorneal angle.
(C) Viscobeads distribution in the mouse anterior chamber following intracameral injection. Left: A representative transmission electron microscopy (TEM) image of an intact mouse iridocorneal angle. Right: A representative fluorescence image showing rhodamine B labelled viscobeads accumulation at mouse iridocorneal angle. Scale bar, 10 μm (left) and 100 μm (right).
(D) Intraocular pressure (IOP) in the mice before and after the injection of viscobeads (n=10) or saline (n=5) and the naïve group (n=5). Data are shown as mean ± SD. (E, F) Representative wholemount retina confocal images from an intact mouse (E) and a mouse after 8 weeks from viscobeads injection (F). RBMPS antibody was used for the immunohistochemical staining of RGCs. Scale bar for the left panel for (F, G), 2 mm. Scale bar for the left panel for (F, G), 100 μm.
(G) Quantification of RGC survival 8 weeks after the viscobeads (n=31) or saline (n=5) injection and the naïve group (n=5). Data are shown as mean ± SD.
(H-K) Knockout of individual TFs protects RGCs in the glaucoma mice. (H) Schematic illustration. AAV2 vectors encoding sgRNAs were injected intravitreally at 2 weeks before beads injection. (I) IOP elevation in different group of mice. (J) Representative images of retinal sections showing RGC survival with sgRNA injections targeting indicated TFs. Scale bar, 20 μm. (K) Quantification of RGC survival after 8 weeks of viscobeads injection, including intact condition (n=7), control (non-targeting) sgRNA injection (n=10), ATF3 sgRNA injection (n=13), ATF4 sgRNA injection (n=9), C/EBPγ sgRNA injection (n=9), CHOP sgRNA injection (n=10), ATF3+ATF4 sgRNA injection (n=10), ATF3+C/EBPγ sgRNA injection (n=12). Data are shown as mean ± s.e.m.. **p<0.01, ***p<0.001, calculated by one-way ANOVA.
To ask whether the knockdown of ATF3, ATF4, C/EBPγ or CHOP, alone or in combination, would increase RGC survival, we injected the AAV2 vectors encoding the single-targeting or multiple-targeting sgRNAs together with AAV2-Cas9 (Wang et al., 2020) into the vitreous bodies of wildtype mice that had received bead injections two weeks previously (Figure 7H). None of the AAV injections altered IOP elevation (Figure 7I). However, in comparison to control sgRNA group, knockout of C/EBPγ and CHOP showed a statistically significant increase in RGC survival under the same level of elevated IOP (Figure 7J–K). Furthermore, we observed additive protective effects on RGCs and RGC axons for both ATF3+ATF4 and ATF3+C/EBPγ CRISPR groups, similar to our observations in the ONC model (Figure 6F–G, Figure S7L–M). These results support the translatability of these TFs and their target downstream pathways in multiple contexts. The TFs defined here may therefore represent synergistic molecular targets for neuroprotection after injury.
DISCUSSION
Through two orthogonal genome-wide strategies, in vivo CRISPR screening and combined assays of chromatin accessibility (ATAC-seq) and gene expression (RNA-seq), we identified several TFs that play key roles in injury-induced neuronal loss. We then focused on a set of four TFs – ATF3, ATF4, C/EBPγ and CHOP identified through these independent approaches. We showed that their activation and complementary action play key roles in the early engagement of different degeneration-regulatory pathways in injured RGCs. Manipulation of these TFs also led to significant neuroprotection in a newly modified glaucoma model.
Previously, several hypotheses have been proposed to explain axotomy-induced neuronal degeneration. For example, axotomy may deprive target-derived pro-survival signals, or initiate the production of “injury signals” that can be retrogradely transported to the cell bodies to elicit degenerative responses (Howell et al., 2013; Syc-Mazurek and Libby, 2019). In addition, injury may trigger rapid responses in surrounding cells, including connected neurons and glial cells along the nerve and in the retina, thus indirectly triggering neuronal responses (Baldwin et al., 2015; Liddelow and Barres, 2017; Williams et al., 2020). Therefore, it was thought that there would be many TFs and pathways regulating injury-elicited neuronal survival/death. Surprisingly, our large-scale CRISPR screening identified only a small number (10 out of 1893) of TFs that negatively regulate neuronal survival, which remarkably, are concentrated in the family of basic leucine zipper domain (bZIP), in particular, ATF and C/EBP members. Because of our stringent procedure, our screen missed some important survival regulators such as Jun (Syc-Mazurek et al., 2017). Importantly, four of these positive hits (ATF3/4, C/EBPγ and CHOP) were also identified as key injury regulators by independent studies of chromatin accessibility coupled to TF footprinting, which prompted us to focus on these candidate master regulatory genes. Our results showed that knockout of each of these four TFs individually significantly improves neuronal survival, while knockout of specific combinations had additive effects, which were remarkably consistent with our bioinformatic analyses of their distinct and overlapping regulatory targets. Furthermore, expression of these TFs is reduced in injured RGCs with pro-survival interventions, including Pten/Socs3 deletion with or without CNTF (Jacobi et al., accompanying paper). Encouraging protective effects of manipulating these TFs in an experimental glaucoma model points to these TFs as generalizable therapeutic targets to prevent neuronal loss. In terms of clinical relevance, a caveat is that we deleted genes before injury; future studies will assess whether manipulations after injury or following disease onset are also effective.
How might these TFs act in injured RGCs? Our results suggest that they are activated (both by expression and binding activity) at very early time points post-injury. Important mechanistic insights came from our analysis of functionally relevant direct target of these TFs, as activators or repressors. As expected, some targets shared by the four TFs are functionally relevant to cell death, such as ERK5 signaling, which has been previously implicated in mediating neurotrophin-mediated retrograde survival responses (Heerssen and Segal, 2002; Watson et al., 2001). Remarkably, our results revealed that ATF3/CHOP and C/EBPγ/ATF4 engage in two distinct neurodegeneration programs. ATF3/CHOP preferentially up-regulate genes related to neuronal responses to extrinsic factors, such as TNFR/IRF and neuroinflammation, but down-regulates genes related different aspects of synaptic transmission function. In contrast, genes induced by CEBPγ/ATF4 are more relevant to cellular responses to intrinsically generated stressors, such as the DNA damage response and the NAD/p53 pathway, perhaps relating to injury-triggered metabolic reprogramming and oxidative stress in injured RGCs. This might help mechanistically explain the results of a recent study showed that a sciatic nerve injury induces double-stranded breaks in axotomized sensory neurons (Cheng et al., 2021). Furthermore, CEBPγ/ATF4-suppressed genes are highly relevant to the intrinsic homeostatic processes, such as PKA and Rho signaling, which are crucial for neuronal survival regulation (Goldberg, 2004; Huang and Reichardt, 2001). Interestingly, several of the genes regulated by TFs have been implicated in human glaucoma by GWAS studies (Gharahkhani et al., 2021). Future studies will analyze the mechanistic basis of these TFs in regulating these diverse transcriptional programs.
In addition to RGC degeneration, some of these molecules and pathways have been suggested as important players in other types of neurodegeneration. For example, neuronal innate immune activation, a targeted process of ATF3/CHOP, has been observed in neurons expressing mutant Huntingtin (Lee et al., 2020). On the other hand, p53 is a key regulator of c9orf72 poly(PR) triggered neurodegeneration (Maor-Nof et al., 2021) as well as anterograde pathways of sensory axon degeneration (Simon et al., 2021). However, considering our finding that a seemingly simple axotomy could rapidly elicit multiple degenerative cascades in RGCs, it is conceivable that these different degeneration programs might be co-activated in other neurodegenerative diseases. This could contribute to unsatisfactory results from approaches that target single genes in neurodegenerative diseases. Thus, our results highlight the potential importance of manipulating multiple targets for effective neuroprotection.
Our analysis of the four TFs also suggests mechanisms that underlie important differences between responses of CNS and PNS neurons to injury, particularly prominent neuronal death in the CNS but not the PNS. For example, axotomized primary sensory neurons are known to have marked ATF3 up-regulation but do not die; instead, they mount regenerative responses (Cheng et al., 2020; Renthal et al., 2020). Thus, ATF3 is a critical pro-death gene in RGCs, but not in peripheral sensory neurons; indeed, ATF3 is pro-regenerative factor in both RGCs and PNS (Kole et al., 2020; Renthal et al., 2020; Seijffers et al., 2007). The mechanism by which ATF3 promotes different injury outcomes after different forms of injury remain mysterious. Furthermore, whereas ATF4/CEBPγ/CHOP are upregulated in injured RGCs, they exhibit minimal or no up-regulation in injured sensory neurons (Renthal et al., 2020). These differences may help explain why neuronal death is a prominent pathological event in CNS neurodegenerative diseases but occurs rarely in peripheral neuropathies.
A prerequisite of axon regeneration is neuronal survival. But it is less known how these processes are coordinated in injured neurons. Our results provide several insights. First, the CRISPR screen identified largely separate sets of TFs negatively regulating survival and regeneration. While most survival genes are ATF/CEBP members, most identified regeneration-inhibiting TFs are implicated in epigenetic control of gene expression. Furthermore, knockout of these epigenetic factors fails to promote neuronal survival, and their regeneration phenotypes are weaker than what is seen after knockout of PTEN and/or SOCS3 (Bei et al., 2016; Park et al., 2008; Smith et al., 2009; Sun et al., 2011). These results suggest that such epigenetic mechanisms may provide permissive control of axon regeneration, different from instructive regulation provided by PI3K/mTOR and JAK/STAT activation consequent of PTEN or SOCS3 inhibition (Smith et al., 2009; Sun et al., 2011; Jacobi et al., accompanying paper). Second, our results reveal both distinct and common effects of TFs on axon regeneration in CNS and PNS. For example, CTCF knockout inhibits axon regeneration in PNS (Palmisano et al., 2019), in stark contrast to its regeneration-promoting effects of its knockout in ONC, as shown here (Figure 1F). On the other hand, ATF3 is upregulated in both DRG and RGCs after injury, and it acts as a positive regulator of axon regeneration in both cases (Kole et al., 2020; Renthal et al., 2020). Indeed, ATF3 knockout completely abolished the regeneration phenotype triggered by PTEN deletion (Jacobi et al., accompanying paper). In light of the massive numbers of direct and indirect targets of ATF3 (this study, Renthal et al., 2020), we speculate that ATF3 may act as reprogramming factor, permitting the activation of axon regeneration program in injured neurons, even though such reprogramming process may render neurons more prone to cell death. Future studies are needed to develop combinatorial treatments that not only protect injured neurons from degeneration, but also promote their regenerative programs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhigang He (Zhigang.He@childrens.harvard.edu).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
RNA-sequencing and ATAC-sequencing data will be available from the Gene Expression Omnibus through series accession number GEO: GSE184547 and GSE190667. All other data referenced in this publication or code used to perform meta-analysis will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request
EXPERIMENTAL MODEL ANDRESOURCE AVAILABILITY
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All experimental procedures were performed in compliance with animal protocols approved by the IACUC at Boston Children’s Hospital and Harvard University. Mice aged at 4 weeks were used for optic nerve crush model. Male and female mice were used in this study at ratios dependent on litters available and with equal distributions across experiments conducted extemporaneously. B6J.129 Rosa26-floxed STOP-Cas9 (Rosa26-LSL-Cas9) (026175; Jackson Labs) and Vglut2-ires-Cre mouse strains (016963; Jackson Labs) were obtained from Jackson Laboratories. The ATF3f/f mouse strain was acquired from Dr. Clifford Woolf’s lab (Renthal et al., 2020).
METHOD DETAILS
Library Virus production
First, we utilized the RIKEN databases (Kanamori et al., 2004; Fulton et al., 2009) and generated a non-redundant list of 1893 transcription regulators across nearly the entire mouse genome. Second, to maximize knockout efficiency, we selected five sgRNAs targeting different regions of each TF from the well-characterized genome-scale CRISPR/Cas9 knockout (GeCKO) library (Shalem et al., 2014; Shalem et al., 2015). These sgRNAs were subcloned into pAV-U6-gRNA (Swiech et al., 2015) and the resultant expression vectors with sgRNAs for the same TF gene were mixed. As a result, we generated a library of 1893 pooled sgRNAs, each of which has 5 sgRNA-bearing plasmids for individual TF genes. Third, these plasmids were used to prepare AAV vectors for transducing RGCs in vivo. We chose serotype 2/2 (AAV2) because it transduces RGCs following intravitreal injection with high efficiency and reasonable selectivity (mostly RGCs and amacrine cells in the ganglion cell layer) (Nawabi et al., 2015; Norsworthy et al., 2017; Park et al., 2008). All AAV viral vectors were made by Boston Children’s Hospital Viral Core. AAV serotype 2 were used in our study as following: AAV2-Cre; AAV2-PTEN sgRNA; AAV2-sgRNAs for other screened TFs. The titers of all viral preparations were at least 5 × 1012 genome copies/mL for sgRNA injection targeting single gene. Viral titers for sgRNA combinations targeting multiple genes were adjusted to 1 × 1013 genome copies/mL for each target gene.
Surgical procedures
For all surgical procedures, mice were anaesthetized with ketamine and xylazine and received Buprenorphine as a postoperative analgesic.
Intravitreous AAV injection:
As previously described, intravitreal virus injection was performed two weeks before optic nerve crush injury to enable axon regeneration. Briefly, a pulled-glass micropipette was inserted near peripheral retina behind the ora serrata and deliberately angled to avoid damage to the lens. 2 ul of the combination of AAV2/2-CAG-Cre virus and AAV2-sgRNAs were mixed (1:3 mix) was injected for LSL-Cas9 mice (Platt et al., 2014). 2 ul AAV2/2 virus was injected for or LSL-Cas9; vGLUT2-Cre mice. For sgRNA combinations targeting multiple genes, the titer of each sgRNA encoding AAV was adjusted to 1 × 1013 genome copies/mL and mixed at a ratio of 1:1.
Optic nerve injury:
As previously described, optic nerve was exposed intraorbitally and crushed with fine forceps (Dumont #5 FST) for 5s, approximately 500um behind the optic disc. Afterwards, eye ointment was applied post-operatively to protect the cornea. Robust axon regeneration could be observed from 2 weeks post crush by Alexa-conjugated cholera toxin subunit B labeling.
Perfusions and tissue processing
For immunostaining, animals were given an overdose of anesthesia and transcardiacally perfused with ice cold PBS followed by 4% paraformaldehyde (PFA, sigma). After perfusion, optic nerves were dissected out and postfixed in 4% PFA overnight at 4°C. Tissues were cryoprotected by sinking in 30% sucrose in PBS for 48 hours. Samples were frozen in Optimal Cutting Temperature compound (Tissue Tek) using a dry ice and then sectioned at 12 mm for optic nerves.
Retinal wholemount staining and quantification of RGC survival
Dissected retinas were rinsed in PBS and then blocked in PBS with 1% Triton X-100 and 5% horse serum (wholemount buffer) overnight at 4 °C. Primary antibodies diluted in the wholemount buffer were then treated for 2–4 days at 4 °C, followed by three times of rinsing by PBS (10 min each time). Secondary antibodies (all with 1:500 dilution) were diluted in PBS and treated overnight at 4 °C. After five times of rinsing by PBS (10 min each time), retinas were mounted with Fluoromount-G (Southern Biotech, Cat. No. 0100–01).
Immunostaining and imaging analysis
Cryosections (12-μm thick) were permeabilized and blocked in blocking buffer (0.5% Triton X-100 and 5% horse serum in PBS) for 1 h at room temperature and overlaid with primary antibodies overnight at 4 Celsius degree (Table 1). For BrdU staining, cells or tissue sections were denatured with 2 N HCl for 30 minutes at 37 Celsius degree and then were neutralized with 0.1 M sodium borate buffer for 10 min before proceeding to normal blocking procedure. On the next day, the corresponding Alexa Fluor 488-, 594- or 647-conjugated secondary antibodies were applied afterwards (all secondary antibodies were purchased from Invitrogen). All stained sections were mounted with solutions with DAPI-containing mounting solution and sealed with glass coverslips. All immunofluorescence-labeled images were acquired using Zeiss 700 or Zeiss 710 confocal microscope. For each biological sample, 3–5 sections of each optic nerve were imaged were taken under 10× or 20× objectives for quantification. Positive cell numbers were then quantified manually using the Plugins/ Analyze /Cell Counter function in ImageJ software. For fluorescent intensity analysis, the images were first converted to 8-bit depth in ImageJ software and then, the mean intensity value was calculated by the build-in function: Analyze/Measure.
Tissue clearing, imaging and quantification of optic nerve regeneration
Mice injected with fluorophore tagged Cholera Toxin B (CTB) were perfused with 4% paraformaldehyde. Dissected optic nerves were then subjected to a modified procedure from previously published iDISCO tissue clearing method, which rendered the optic nerves transparency for direct fluorescent imaging (Renier et al., 2014). This procedure has been tested for better preservation of CTB florescence and the least change of optic nerve shape during tissue clearing. For dehydration, optic nerve samples were incubated in dark for 0.5h of 80% tetrahydrofuran (THF, Sigma-Aldrich 360589–500ML)/H2O and then switched to 100% THF for 1h. Then, samples were incubated in Dichloromethane (DCM, Sigma-Aldrich 270997–1L) for 20min (nerves should sink at the bottom). Samples were finally switched to dibenzyl ether (DBE, Sigma-Aldrich 33630–250ML) until complete transparency (at least 3h, but overnight is recommended). Transparent nerves can be stored in DBE without obvious fluorescence decay of CTB for at least 1 year. For imaging, processed nerves can be mounted in DBE and imaged under Zeiss 710 confocal microscope. Z-stack scanning and maximum projection of Z-stack images were used in order to capture all regenerated axons. For image analysis, fluorescent intensity profile along the nerve was generated by the build-in function of ImageJ: Analyze/Plot Profile. The crush site of maximum projected optic nerve image was identified by overlaying 5 single slice images at different imaging depth (z-direction) of the same nerve (Figure S1A). To calculate the integral of fluorescent intensity across the entire length of the nerve, a Matlab algorithm was developed by our lab to quantify the “area under curve” from the plot profile data generated by ImageJ.
Cell Preparation and Fluorescence-Activated Cell Sorting (FACS)
Retinas were dissected in AMES solution (Sigma A1420, equilibrated with 95% O2/5% CO2). Upon dissection, eyes and lenses were visually inspected for damage, blood, or inflammation, which were used as criteria for exclusion. Retinas were digested in papain and dissociated to single cell suspensions using manual trituration in ovomucoid solution. Cells were spun down at 450 g for eight minutes, resuspended in AMES+4% BSA to a concentration of 10 million cells per 100ml. 0.5ml of 0.2mg/ml anti-CD90.2-PE-Cy7 (Affymetrix eBioscience 25-0902-82) per 100ml of cells was incubated for 15 min, washed with an excess of media, spun down and resuspended again in AMES+4% BSA at a concentration of ~7 million cells per 1 ml. Just prior to FACS the live cell marker Calcein Blue (Invitrogen C1429) was added. Cellular debris, doublets, and dead cells (Calcein Blue negative) were excluded. For FACS experiments with vGLUT2-Cre; LSL-Cas9 mice, RGCs were collected based CD90.2 and GFP double positive expression. For purification of transduced RGCs with CRISPR perturbation (sgRNAs co-expressed with mcherry), cells were collected based on CD90.2, GFP and mcherry triple positive expression. Cells were collected into ~150ul of AMES+5% BSA.
Viscobead-induced experimental mouse glaucoma model
The elevation of intraocular pressure (IOP) was induced by injection of viscobeads to the anterior chamber of mouse eyes. The surgery procedures were modified by a well-established microbead occlusion model (Yang et al., 2012). Briefly, by using a standard double emulsion method, poly-d,l-lactic-co-glycolic acid (PLGA) / polystyrene (PS) core-shell microparticles (viscobeads) with 1–20 um size distributed were first fabricated at a concentration of 30% (v/v) in saline. The corneas of anesthetized mice were gently punctured near the center using a 33g needle (CAD4113, sigma). A bubble was injected through this incision site into the anterior chamber to prevent the possible leakage. Then, 1 uL viscobeads were injected into the anterior chamber. After 5 min when the viscobeads were accumulated at the iridocorneal angle, the mouse was applied antibiotic Vetropolycin ointment (Dechra Veterinary Products, Overland Park, KS) and placed on a heating pad for recovery.
Intraocular pressure measurement
The IOP measurements were performed using a TonoLab tonometer (Colonial Medical Supply, Espoo, Finland) according to product instructions. Mice were first anesthetized with a sustained isoflurane flow (3% isoflurane in 100% oxygen). Average IOP was generated automatically with five measurements after the elimination of the highest and lowest values.
RNA-Seq library preparation
RNA from 5,000 – 10,000 FACS-sorted RGCs were isolated with RNeasy Micro Kit (Qiagen), and RNA-seq libraries were prepared with SMART-Seq v4 Ultra Low Input RNA Kit (Clontech), following manufacturers’ protocols. The cDNA was fragmented to 300 base pairs (bp) using the Covaris M220 (Covaris), and then the manufacturer’s instructions were followed for end repair, adaptor ligation, and library amplification. The libraries were quantified by the Qubit dsDNA HS Assay Kit (Molecular Probes); Library size distribution and molar concentration of cDNA molecules in each library were determined by the Agilent High Sensitivity DNA Assay on an Agilent 2200 TapeStation system. Libraries were multiplexed into single pool and sequenced using a HiSeq4000 instrument (Illumina, San Diego, CA) to generate 69 bp pair-end reads. The average uniquely-mapped, non-duplicated reads are ~24 millions.
ATAC-Seq library preparation
ATAC-seq was performed as described with minor modifications (1). Briefly, 50,000 – 100,000 sorted RGCs were lysed in 100 μL ice-cold Resuspension Buffer (RSB, 10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, pH=7.4) with 0.1% NP40, 0.1% Tween- 20, and 0.01% Digitonin. Cell lysis were washed with 1 ml of cold ATAC-RSB containing 0.1% Tween-20 but NO NP40 or digitonin, and centrifuged at 4 °C for 10 minutes at 500 × g. Pelleted nuclei were resuspended gently in 50 μL transposition mix (25 ul 2× TD buffer, 2.5 ul transposase (100nM final), 16.5 ul PBS, 0.5 ul 1% digitonin, 0.5 ul 10% Tween-20, 5 ul H2O) and incubated for 30 minutes at 37 °C. DNA was cleaned using Zymo DNA Clean and Concentrator-5 Kit, and PCR amplified 5 cycles in a 50 μL reaction with Illumina Nextera adaptors using NEBNext High Fidelity 2× Master Mix. To determine the number of additional cycles to amplify the libraries, a side qPCR reaction was performed using 5 μL (10%) of the pre-amplified PCR. We used the Ct value of the qPCR at ¼ maximum fluorescence as the number of additional cycles. Final PCR products were cleaned using Zymo DNA Clean and Concentrator-5 Kit, and quantified by the KAPA Library Quantification kit prior to pooling and sequencing to an average depth of ~90M unique non-mitochondria reads per sample on the Illumina NovaSeq platform at 2×100 bp.
Processing of RNA-seq data
RNA-seq data was processed and analyzed using custom pipeline (available at https://github.com/icnn/RNAseq-PIPELINE) as described previously (Cheng et al., 2020; Norsworthy et al., 2017). Raw sequenced reads were mapped to the reference genome Mus musculus (mm10) refSeq (refFlat) using STAR with default parameter (Dobin et al., 2013). Data quality was assessed on base-quality calls, nucleotide composition of sequences, insert sizes, per cent of uniquely aligned reads and transcript coverage using custom scripts and Picard Tools (http://broadinstitute.github.io/picard). More than 85% of reads were mapped uniquely to reference genome. Total counts of read fragments aligned to candidate gene regions were derived using the HTSeq program (https://htseq.readthedocs.io) with mouse mm10 refSeq (refFlat table) as a reference and used as a basis for the quantification of gene expression. Only uniquely mapped reads were used for subsequent analyses. Sequencing depth was normalized between samples using TMM in edgeR (Robinson et al., 2010). Outlier samples were removed using WGCNA (Langfelder and Horvath, 2008), with absolute sample connectivity score more than 2.5 standard deviation away from the mean. Genes with no counts in over 50% of all samples were removed.
RNA-seq analysis – differential gene expression
Principle component analysis (PCA) of the normalized expression data (first five PCs) was correlated with potential technical covariates, including experimental batch, aligning and sequencing bias calculated from STAR and Picard respectively. Differential expression analysis was conducted with the Bioconductor package edgeR (Robinson et al., 2010), including covariates that are significantly correlated with expression PCs: ~ Genotype + AlignSeq.PC1 + AlignSeq.PC3 + AlignSeq.PC4. Statistical significance of differential expression was determined at FDR < 10% (q < 0.1).
Processing of ATAC-seq data
Alignment:
Raw sequencing fastq files were assessed for quality, adapter content and duplication rates with FastQC, trimmed using trim-galore (https://github.com/FelixKrueger/TrimGalore) and aligned with bowtie2 (Langmead and Salzberg, 2012): bowtie2 --very-sensitive -X 2000 -x [reference_genome] -1 [input.left] -2 [input.right] | samtools view -hb -S - | samtools sort -o [sample_label].bam. The reference genome was mouse GRCm38 vM11. Samtools (Danecek et al., 2021) was used to calculate the read statistics, and Picard Tools (http://broadinstitute.github.io/picard) was used to remove duplicate.
ATAC-seq data QC - Transcription start site enrichment, fragment size distribution, chromHMM enrichment:
Enrichment of open region signals at the transcription start site, an important quality control metric to evaluate ATAC-seq data, was measured by tssenrich (https://github.com/anthony-aylward/tssenrich). TSS positions were derived from RefSeq mm10, and the distance of each read within ± 2 kb centered on TSS were calculated. Read counts at each distance was summed up in a given library. A successful ATAC-seq library would form a characteristic shape with reads aggregated at the TSS, with an enrichment score > 7. Fragment size distribution is another quality control metric to visualize the nucleosome-sized periodicity resulting from chromatin digestion. This information was calculated by Picard Tools CollectInsertSizeMetrics. Enrichment of DA peaks within annotated genic regions of the genome or epigenetically annotated regions of the genome (Bogu et al., 2015) was calculated using the ratio between the (#bases in state AND overlap feature)/(#bases in genome) and the [(#bases overlap feature)/(#bases in genome) X (#bases in state)/(#bases in genome)] as we and others previously described (De La Torre Ubieta et al. 2018; Roadmap Epigenomics et al., 2015)
Peak calling and annotations:
MACS2 (Zhang et al., 2008) was used to call peaks on each sample with the following command: macs2 callpeak -g mm --treatment [sample BAM] --format BAMPE --call-summits --nolambda --keep-dup all --min-length 100 -q 0.05. Peaks that overlapped with ENCODE mm10 blacklisted regions were removed. Peaks from each sample were merged to a set of union peaks across all conditions using bedtools merge (Quinlan et al., 2010). DiffBind (Stark et al., 2011) was used to obtain a consensus peak set by calculating the overlap rate. We kept peaks overlapped in at least two of the fifteen samples, at which point the overlap rate starts to drop off geometrically, indicating a good agreement among the peaksets. With this threshold, we obtained a total of 151,630 consensus peaks, with an average merged peak width of 1282 bp. The consensus peaks were further annotated to the transcriptional start site of genes in a distance of ± 2 kb from the gene start using ChIPseeker. Gencode vM24 was used for annotations.
ATAC-seq analysis - differential accessibility:
We obtained the number of reads for consensus peaks (hereafter referred to as peaks) across samples using featureCounts from the Subread package (Liao et al., 2014), and the GC content using bedtools nuc. We then calculated normalization factors for each peak accounting for GC content, peak width, and total number of unique non-mitochondrial fragments sequenced using conditional quantile normalization from cqn package. We further performed principle component analysis (PCA) on cqn-normalized peak-count matrix using prcomp, and correlated technical and experimental variables to the first five PCs, including duplicate rate, fraction of reads in peaks (FRiP), TSS enrichment, batch, and injury conditions (Figure S2F). Batch effects significantly correlated with the peak-count PCs, which was then included as a covariate for differential accessibility analysis (Figure S2G). To obtain differential accessible peak-regions (DAR) comparing injured vs uninjured RGCs, we used a negative binomial regression with normalization based on the size factors from cqn (Hansen et al., 2012) and implemented in DEseq2 with default parameters (fitType=“parametric”, test=“Wald”) (Love et al., 2014). Normalized peak-count matrix with batch effects regressed out was used for subsequent analysis. MA-plot was used to visualize the general differential accessibility changes at 1- and 3-day following injury. Heatmaps and coverage plots of normalized reads within peaks were used to display DARs around specific genes, generated by ComplexHeatmap and Gviz respectively.
ATAC-seq analysis – linking proximal and distal regulatory elements to cognate genes:
Using GENCODE annotations, we defined an ATAC-seq peak ± 2 kb of a gene’s transcription start site (TSS) as a promoter (proximal regulatory element), and non-promoter peaks ± 500 kb of TSS as distal regulatory regions for that gene. To correlate differential accessible promoter/distal peaks to gene expressions, we computed the sample-wise Pearson’s correlations between normalized read counts from ATAC-seq and RNA-seq data. For genes with multiple distal regulatory links in the ± 500 kb window, the average accessibility of distal regulatory elements was used to correlate with gene expression. Next, to account for spurious associations, we generated a background null model by computing correlations between randomly selected peaks and randomly selected genes on different chromosomes. We calculated the mean and standard deviation of this null distribution of correlations, enabling us tocompute p-values for the test correlations. The correlative peak-gene pairs with FDR < 0.1 were further clustered by K-means and genes linked each cluster were annotated with gprofiler2 for general biological pathways in Gene Ontology (GO). GO terms were chosen based on their FDR-corrected p-values and relevance to the current study.
ATAC-seq analysis – transcription factor motif activity:
To find TF motifs within peaks, we used motif position weight matrices (PWMs) integrated from JASPAR2016, HOCOMOCCO v10, UniPROBE and SwissRegulon, which include a core set of 1,312 non-redundant motifs from human and mouse (Funk et al., 2020). This integrated PWMs were used to scan differential accessible regions for motif occurrence by motifmatchr, resulting in a binary peak-by-matches matrix. For each motif, we computed the odds ratio and the significance (P-value < 5×10−5) of enrichment comparing to background nucleotide frequencies across input peak regions using Fisher’s exact test. The degree of accessibility at enriched TF motifs across samples was computed by chromVAR (Schep et al., 2017), which quantitatively measures changes of ATAC-seq counts in peaks containing the TF motifs as deviation Z-scores. To correspond TF motifs to TF genes, ChromVAR deviation z-scores for each TF motif were correlated to the TF gene’s log2-transformed TPM values across samples. If the correlation between motif and gene expression is greater than 0.5 with an adjusted p-value less than 0.05 and a maximum cross-sample difference in deviation z-score that is in the top quartile, the TF is classified as a positive regulator of chromatin state; if the correlation is less than – 0.5 with an adjusted p-value less than 0.05 and a maximum deviation score in the top quartile, it is classified as a negative TF regulator of chromatin state. To visualize, we plotted the TF gene – motif correlations against the maximum cross-sample difference in deviation z-score for all TFs, with the top ‘TF hits’ meeting the criteria of positive or negative TF regulators of chromatin state described above.
ATAC-seq analysis – transcription factor footprinting:
Tn5 bias correction and footprinting:
The first step of fooptrinting is to correct Tn5 transposase cleavage bias in the ATAC-seq data. To do this, we first merged biological replicates from each condition by Picard Tools and downsampled ATAC-seq BAMs to a depth of 60 million reads using samtools. TOBIAS (Bentsen et al., 2020) ATACorrect module was applied to merged, downsampled reads within the consensus peaks to estimate the background bias of Tn5 transposase. Subtracting the background Tn5 insertion cuts from the uncorrected signals yields a corrected track, highlighting the effect of protein binding. The footprint score was calculated by TOBIAS ScoreBigWig, which measures both accessibility and depth of the local footprint, thus correlating with the presence of a TF at its target loci, and the chromatin accessibility of the regions where this TF binds.
Assigning TFs to footprints:
To match footprints to potential TF binding sites, and to estimate TF binding activity on its target loci, we applied TOBIAS BINDetect module to the corrected ATAC-seq signals within peaks, with the same set of TF motif PWMs used for ChromVAR as input. This method obtains the positions of TF binding sites, which are then mapped to the footprints for each condition. Each footprint site was assigned a log2FC (fold change) between two conditions, representing whether the binding site has larger/smaller TF footprint scores in comparison. To calculate statistics, a background distribution of footprint scores is built by randomly subsetting peak regions at ~200bp intervals, and these scores were used to calculate a distribution of background log2FCs for each comparison of two conditions. The global distribution of log2FC’s per TF was compared to the background distributions to calculate a differential TF binding score, which represents differential TF activity between two conditions. A P-value is calculated by subsampling 100 log2FCs from the background and calculating the significance of the observed change. By comparing the observed log2FC distribution to the background log2FC, the effects of any global differences due to sequencing depth, noise etc. are controlled. To visualize, we used soGGI to plot TF footprints, and bar graphs to show the global TF footprint activity changes comparing injured to uninjured RGCs.
Linking TF footprint sites to its targeted genes combining RNA-seq data:
Footprint sites which are bound by each of the four TF with increased footprint score after injury were considered as injury-responsive TF-footprints. Distribution of these footprints were further annotated with ChIPseeker (Yu et al., 2015), and the footprints located within ± 500 kb of a gene’s TSS were linked to that gene. We did not consider distal intergenic regions that are > ± 500 kb of TSS as linking them to a gene requires higher-order chromatin conformation data. Genes that are footprinted by the TF and differentially regulated in the RNA-seq upon CRISPR ablation of this TF are considered as direct target genes of each TF. The similarity and dis-similarity of each TF’s target genes were analyzed by calculating sample distances and then hierarchically clustered. Genes that are unique or common to each TF were annotated with gprofiler2 and Ingenuity Pathway Analysis (IPA) Software (Qiagen) for gene ontology analysis. GO terms were chosen based on their FDR-corrected p-values and relevance to the current study.
QUANTIFICATION AND STATISTICAL ANALYSIS
The normality and variance similarity were measured by Microsoft Excel and R programming Language before we applied any parametric tests. If criteria for parametric test were not met, we would then perform non-parametric tests (indicated in the figure legends). Two-tailed student’s t-test was used for the single comparison between two groups. The rest of the data were analyzed using one-way or two-way ANOVA depending on the appropriate design. Post hoc comparisons were carried out only when the primary measure showed statistical significance. P-value of multiple comparisons was adjusted by using Bonferroni’s correction. Error bars in all figures represent mean ± S.E.M. The mice with different litters, body weights and sexes were randomized and assigned to different treatment groups, and no other specific randomization was used for the animal studies.
Supplementary Material
ATAC-seq metadata, quality control metrics, and annotation of differentially accessible regions (DARs) comparing RGCs injured at day 1 or 3 versus uninjured condition (day 0). Related to Figure 2. (A) ATAC-seq metadata and quality control metrics including percent of duplicated reads after sequencing (DuplicateRate), non-duplicated, uniquely mapped reads (UniqMappedReads), total number of peaks from MACS2 (TotNarrowPeak), TSS enrichment, and fraction of reads in peaks (FRiP). Annotation of injured RGC DARs [absolute log fold changes (logFC) > 0.3 and false discover rate (FDR) < 0.1] comparing day 1 v.s. day 0 (B) or day 3 v.s. day 0 (C).
RNA-seq metadata, quality control metrics, and annotation of differentially expressed genes (DEGs) comparing injured RGCs at indicated time points versus uninjured ones (day 0). Related to Figure 2. (A) RNA-seq metadata and quality control metrics obtained from Picard Tools. Principle component analysis (PCA) was performed on the metrics and the first seven PCs accounting > 95% of variance was kept for further analysis. (B-C) Annotation of injured RGC DEGs (absolute logFC > 0.3 and FDR < 0.1) comparing day 1 v.s. day 0 (B) or day 3 v.s. day 0 (C).
Clustering and annotation of putative peak-gene links. Related to Figure 2. (A) Linked peak-gene pairs. Proximal or distal peaks that are differential accessible are linked to corresponding genes that are differentially expressed at day 1 or day 3 following optic nerve crush. The peak-gene pairs are grouped into two clusters: the 'up' clust er where the accessibility of linked peaks and the expression of corresponding genes are both up-regulated, and the 'down' cluster where both are down -regulated. (B) Gene Ontology of the up-regulated cluster. (C) Gene Ontology of the down-regulated cluster.
TF motif activity in injured RGCs, related to Figure 3. (A) ChromVAR-estimated motif matches in DARs. A list of non-redundant, human or mouse TF motifs (Methods) was used, named in the format of [species]-[database]-[corresponding TF]-[motif ID]. ChromVAR returns variability, which is the standard deviation of the motif z-scores computed across samples, and boostrap confidence intervals and p-value for the variability by resampling samples. These calculations are correlative to motif accessibility changes across samples. (B) Sample-wise correlations of motif activity to the expression levels of corresponding TF.
RNA-seq metadata, quality control metrics and annotation of differentially expressed genes comparing RGCs with or without CRISPR ablation of one of the 4 survival TFs (ATF3, ATF4, C/EBPγ CHOP). Related to Figure 4–5. (A). RNA-seq metadata and quality control metrics obtained from Picard Tools. (B -E) Annotation of DEGs in RGCs with ablation of ATF3, ATF4, C/EBPγ, CHOP, respectively.
Target genes regulated by ATF3/CHOP, or ATF4/C/EBPγ or by all 4 TFs predicted by DNA-footprinting and RNA-seq. Related to Figure 4–5. (A) Genes that are positively or negatively regulated by ATF3/CHOP, but not ATF4/C/EBPγ. (B) Genes that are positively or negatively regulated by ATF4/C/EBPγ, but not ATF3/CHOP. (C) Genest hat are positively or negatively regulated by all four TFs.
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-RBPMS | Raygene | Cat# A008712 |
| Chicken anti-GFP | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| Goat anti-RFP | Rockland | Cat# 200-101-379S |
| Mouse anti-mCherry (1C51) | Novus Biologicals | Cat# NBP1-96752SS; RRID: AB_11008969 |
| CD90.2 (Thy-1.2) Monoclonal Antibody (53-2.1), PE-Cyanine7 | Thermo Fisher Scientific | Cat# 25-0902-82; RRID: AB_469642 |
| Rabbit anti-ATF3 | LSBio | Cat# LS-C382181-50 |
| Rabbit anti-ATF4 | Cell Signaling Technology | Cat# 11815; RRID: AB_2616025 |
| Rabbit anti-C/EBPγ | ProteinTech | Cat# 12997-1-AP; RRID:AB_2877902 |
| Rabbit anti-CHOP | Cell Signaling Technology | Cat# 2895S |
| Rabbit anti-TLR2 | Abcam | Cat# ab213676 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 706-605-148; RRID: AB_2616025 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Thermo Fisher Scientific | Cat# A32758; RRID:AB_2340476 |
| Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 711-545-152; RRID:AB_2762833 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31570; RRID:AB_2536180 |
| Alexa Fluor 647 AffiniPure Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 711-605-152; RRID: AB_2492288 |
| Alexa Fluor 405 Donkey Anti-Rabbit IgG (H&L) | Abcam | Cat# ab175651 |
| DyLight™ 405 AffiniPure Donkey Anti-Guinea Pig IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 706-475-148 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Alexa-conjugated cholera toxin subunit B | Thermo Fisher Scientific | Cat# C34776 |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| B6J.129 Rosa26-floxed STOP-Cas9 mice | The Jackson Laboratory | Cat# JAX: 026175 |
| Vglut2-ires-Cre mice | The Jackson Laboratory | Cat# JAX: 016963 |
| Recombinant DNA | ||
| pAAV-U6-sgRNA-GFP | This study | N/A |
| pAAV-U6-sgRNA-mcherry | This study | N/A |
| pAAV-Cas9 | Obtained from Dr. Yang Hu | N/A |
| Software and algorithms | ||
| ImageJ | NIH | RRID: SCR_003070 |
| R 4.0.2 | R Foundation | N/A |
| MATLAB | Mathworks | N/A |
| Python (v3.7.3) | N/A | https://www.python.org/ |
| STAR (v2.7.5c) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| PicardTools (v2.25.0) | N/A | http://broadinstitute.github.io/picard/ |
| HTSeq | (Anders et al., 2015) | https://htseq.readthedocs.io/en/master/ |
| trim-galore (v0.6.0) | N/A | https://github.com/FelixKrueger/TrimGalore |
| bowtie2 (v2.4.2) | (Langmead and Salzberg, 2012) | https://github.com/BenLangmead/bowtie2 |
| samtools (v1.11) | (Danecek et al., 2021) | http://www.htslib.org/ |
| tssenrich (v1.3.0) | N/A | https://pypi.org/project/tssenrich/ |
| MACS2 (v2.2.7.1) | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| subread-featureCounts (v2.0.0) | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| bedtools (v2.30.0) | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| TOBIAS (v0.11.6) | (Bentsen et al., 2020) | https://github.com/loosolab/TOBIAS |
| DiffBind (v2.14.0) | (Ross-Innes et al., 2012) | http://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| edgeR (v3.30.3) | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| DEseq2 (v1.28.1) | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| cqn (v1.34.0) | (Hansen et al., 2012) | http://bioconductor.org/packages/release/bioc/html/cqn.html |
| WGCNA (v1.69) | (Langfelder and Horvath, 2008) | https://cran.r-project.org/web/packages/WGCNA/index.html |
| ChromVAR (v1.10.0) | (Schep et al., 2017) | https://bioconductor.org/packages/release/bioc/html/chromVAR.html |
| motifmatchr (v1.10.0) | N/A | https://bioconductor.org/packages/release/bioc/html/motifmatchr.html |
| ChIPseeker (v1.24.0) | (Yu et al., 2015) | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| TFBSTools (v1.26.0) | (Tan and Lenhard, 2016) | http://bioconductor.org/packages/release/bioc/html/TFBSTools.html |
| universalmotif (v1.6.4) | N/A | https://bioconductor.org/packages/release/bioc/html/universalmotif.html |
| GenomicRanges (v1.42.0) | (Lawrence et al., 2013) | http://bioconductor.org/packages/release/bioc/html/GenomicRanges.html |
| soGGi (v1.20.0) | N/A | https://bioconductor.org/packages/release/bioc/html/soGGi.html |
| Gviz (v1.32.0) | N/A | http://bioconductor.org/packages/release/bioc/html/Gviz.html |
| gprofiler2 (v0.2.1) | N/A | https://cran.r-project.org/web/packages/gprofiler2/index.html |
| Ingenuity Pathway Analysis (IPA) | N/A | https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis) |
| dplyr (v1.0.1) | N/A | https://cran.r-project.org/web/packages/dplyr/index.html |
| ggplot2 (v3.3.5) | N/A | https://cran.r-project.org/web/packages/ggplot2/index.html |
| ggpubr (v0.4.0) | N/A | https://cran.r-project.org/web/packages/ggpubr/index.html |
| cowplot (v1.1.1) | N/A | https://cran.r-project.org/web/packages/cowplot/index.html |
| RColorBrewer (v1.1-2) | N/A | https://cran.r-project.org/web/packages/RColorBrewer/index.html |
| ComplexHeatmap (v2.6.2) | (Gu et al., 2016) | https://www.bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Vennerable (v3.1.0) | N/A | https://github.com/js229/Vennerable |
| corrplot (v0.84) | N/A | https://cran.r-project.org/web/packages/corrplot/index.html |
| matrixStats (v0.61.0) | N/A | https://cran.rstudio.com/web/packages/matrixStats/index.html |
| Hmiscs (v4.4-2) | N/A | https://cran.r-project.org/web/packages/Hmisc/index.html |
| circlize (v0.4.13) | (Gu et al., 2014) | https://cran.r-project.org/web/packages/circlize/index.html |
| Deposited data | ||
| Raw and analyzed data: RNA-seq | This paper | GSE184547; GSE190667 |
| Raw and analyzed data: ATAC-seq | This paper | GSE184547 |
| mm10; | N/A | ftp://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz |
| Gencode vM24 | (Frankish et al., 2021) | https://www.gencodegenes.org/mouse/release_M24.html |
| Chromatin state maps of mouse brain | (Bogu et al., 2015) | https://github.com/gireeshkbogu/chromatin_states_chromHMM_mm9 |
| Blacklisted regions for mouse | (Amemiya et al., 2019) | https://github.com/Boyle-Lab/Blacklist/tree/master/lists |
Highlights:
In vivo CRISPR screen identifies repressors of CNS neuronal survival and regeneration Parallel multi-omic analysis characterizes pro-survival transcriptional networks These two comprehensive approaches converge on complementary survival programs Manipulation of these pro-survival programs is protective in a glaucoma model
ACKNOWLEDGEMENTS
This study was supported by grants K99 EY032181 to F.T., EY030204-01 to J.R.S and Z.H., R01EY021526 and R01EY026939 to Z.H., Wings for Life Spinal Cord Research Foundation to Y.C. and A.J., and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to C.J. W., D.H.G. and Z.H., Gilbert Family Foundation (to Z.H.) and the Medical Technology and Enterprise Consortium and DoD (W81XWH-16-1-0775 (to Z.H.). IDDRC and viral cores were supported by the grants from the NIH (HD018655 and P30EY012196).
Footnotes
DECLARATION OF INTERESTS
C.J.W. is a founder of Nocion Therapeutics and QurAlis. J.R.S. is a consultant for Biogen. Z.H. is an advisor of SpineX, Life Biosciences and Myro Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abel T, Martin KC, Bartsch D, and Kandel ER (1998). Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science 279, 338–341. [DOI] [PubMed] [Google Scholar]
- Aguayo AJ, Rasminsky M, Bray GM, Carbonetto S, McKerracher L, Villegas-Perez MP, Vidal-Sanz M, and Carter DA (1991). Degenerative and regenerative responses of injured neurons in the central nervous system of adult mammals. Philos Trans R Soc Lond B Biol Sci 331, 337–343. [DOI] [PubMed] [Google Scholar]
- Aime P, Karuppagounder SS, Rao A, Chen Y, Burke RE, Ratan RR, and Greene LA (2020). The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol Dis 136, 104725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasieh M, and Levin LA (2017). Neuroprotection in Glaucoma: Animal Models and Clinical Trials. Annu Rev Vis Sci 3, 91–120. [DOI] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin KT, Carbajal KS, Segal BM, and Giger RJ (2015). Neuroinflammation triggered by beta-glucan/dectin-1 signaling enables CNS axon regeneration. Proc Natl Acad Sci U S A 112, 2581–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, Bailey CH, and Kandel ER (1995). Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell 83, 979–992. [DOI] [PubMed] [Google Scholar]
- Bei F, Lee HHC, Liu X, Gunner G, Jin H, Ma L, Wang C, Hou L, Hensch TK, Frank E, et al. (2016). Restoration of Visual Function by Enhancing Conduction in Regenerated Axons. Cell 164, 219–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benschop R, Wei T, and Na S (2009). Tumor necrosis factor receptor superfamily member 21: TNFR-related death receptor-6, DR6. Adv Exp Med Biol 647, 186–194. [DOI] [PubMed] [Google Scholar]
- Bentsen M, Goymann P, Schultheis H, Klee K, Petrova A, Wiegandt R, Fust A, Preussner J, Kuenne C, Braun T, et al. (2020). ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun 11, 4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogu GK, Vizan P, Stanton LW, Beato M, Di Croce L, and Marti-Renom MA (2015). Chromatin and RNA Maps Reveal Regulatory Long Noncoding RNAs in Mouse. Mol Cell Biol 36, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–-21 29 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkins DJ, Pekny M, Cooper ML, Benowitz L, Lasker I.I.o.A., and Glaucomatous Neurodegeneration P (2017). The challenge of regenerative therapies for the optic nerve in glaucoma. Exp Eye Res 157, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran V, Coppola G, Nawabi H, Omura T, Versano R, Huebner EA, Zhang A, Costigan M, Yekkirala A, Barrett L, et al. (2016). A Systems-Level Analysis of the Peripheral Nerve Intrinsic Axonal Growth Program. Neuron 89, 956–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Alvarez FJ, Lefebvre JL, Friedman B, Nwakeze C, Geiman E, Smith C, Thu CA, Tapia JC, Tasic B, et al. (2012). Functional significance of isoform diversification in the protocadherin gamma gene cluster. Neuron 75, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Yin Y, Zhang A, Bernstein AM, Kawaguchi R, Gao K, Potter K, Gilbert H-Y, Ao Y, Ou J, et al. (2020). Transcription factor network analysis identifies REST/NRSF as an intrinsic regulator of CNS regeneration. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YC, Snavely A, Barrett LB, Zhang X, Herman C, Frost DJ, Riva P, Tochitsky I, Kawaguchi R, Singh B, et al. (2021). Topoisomerase I inhibition and peripheral nerve injury induce DNA breaks and ATF3-associated axon regeneration in sensory neurons. Cell Rep 36, 109666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone FE, Gelman SE, Son JL, Pease ME, and Quigley HA (2010). Differential susceptibility to experimental glaucoma among 3 mouse strains using bead and viscoelastic injection. Exp Eye Res 91, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Shcherbina A, Kundu S, Gloudemans MJ, Fresard L, Granja JM, Louie BH, Eulalio T, Shams S, Bagdatli ST, et al. (2020). Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat Genet 52, 1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304 e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA (2019). A Unifying Hypothesis for Alzheimer’s Disease: From Plaques to Neurodegeneration. Trends Neurosci 42, 310–322. [DOI] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2012). ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finelli MJ, Wong JK, and Zou H (2013). Epigenetic regulation of sensory axon regeneration after spinal cord injury. J Neurosci 33, 19664–19676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Petkova V, Thanos S, and Benowitz LI (2004). Switching mature retinal ganglion cells to a robust growth state in vivo: gene expression and synergy with RhoA inactivation. J Neurosci 24, 8726–8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Jungreis I, Lagarde J, Loveland JE, Mudge JM, Sisu C, Wright JC, Armstrong J, Barnes I, et al. (2021). Gencode 2021. Nucleic Acids Res 49, D916–D923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton DL, Sundararajan S, Badis G, Hughes TR, Wasserman WW, Roach JC, and Sladek R (2009). TFCat: the curated catalog of mouse andhumantranscriptionfactors. Genome Biol 10, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CC, Casella AM, Jung S, Richards MA, Rodriguez A, Shannon P, Donovan-Maiye R, Heavner B, Chard K, Xiao Y, et al. (2020). Atlas of Transcription Factor Binding Sites from ENCODE DNase Hypersensitivity Data across 27 Tissue Types. Cell Rep 32, 108029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub P, Tedeschi A, Puttagunta R, Nguyen T, Schmandke A, and Di Giovanni S (2010). HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ 17, 1392–1408. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, and Konopka G (2009). Neuroscience in the era of functional genomics and systems biology. Nature 461, 908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharahkhani P, Jorgenson E, Hysi P, Khawaja AP, Pendergrass S, Han X, Ong JS, Hewitt AW, Segre AV, Rouhana JM, et al. (2021). Genome-wide meta-analysis identifies 127 open-angle glaucoma loci with consistent effect across ancestries. Nat Commun 12, 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL (2004). Intrinsic neuronal regulation of axon and dendrite growth. Curr Opin Neurobiol 14, 551–557. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- Guo X, Zhou J, Starr C, Mohns EJ, Li Y, Chen EP, Yoon Y, Kellner CP, Tanaka K, Wang H, et al. (2021). Preservation of vision after CaMKII-mediated protection of retinal ganglion cells. Cell 184, 4299–4314 e4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Statistical Genomics: Methods and Protocols, Vol 1418 (Springer; New York: ). [Google Scholar]
- Hansen KD, Irizarry RA, and Wu Z (2012). Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, and Jin Y (2016). Intrinsic Control of Axon Regeneration. Neuron 90, 437–451. [DOI] [PubMed] [Google Scholar]
- Heerssen HM, and Segal RA (2002). Location, location, location: a spatial view of neurotrophin signal transduction. Trends Neurosci 25, 160–165. [DOI] [PubMed] [Google Scholar]
- Hilton BJ, and Bradke F (2017). Can injured adult CNS axons regenerate by recapitulating development? Development 144, 3417–3429. [DOI] [PubMed] [Google Scholar]
- Howell GR, Soto I, Libby RT, and John SW (2013). Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp Neurol 246, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Park KK, Yang L, Wei X, Yang Q, Cho KS, Thielen P, Lee AH, Cartoni R, Glimcher LH, et al. (2012). Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 73, 445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, and Reichardt LF (2001). Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24, 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Simmons SK, Guo A, Shetty AS, Ko M, Nguyen L, Jokhi V, Robinson E, Oyler P, Curry N, et al. (2020). In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M (2020). CRISPR-based functional genomics for neurological disease. Nat Rev Neurol 16, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M, Konno H, Osato N, Kawai J, Hayashizaki Y, and Suzuki H (2004). A genome-wide and nonredundant mouse transcription factor database. Biochem Biophys Res Commun 322, 787–793. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, Alim I, Khim SJ, Bourassa MW, Sleiman SF, John R, Thinnes CC, Yeh TL, Demetriades M, Neitemeier S, et al. (2016). Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med 8, 328ra329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley PW, Sliff BJ, Lee SC, Fuerst PG, Burgess RW, Eglen SJ, and Reese BE (2012). Neuronal clustering and fasciculation phenotype in Dscam- and Bax-deficient mouse retinas. J Comp Neurol 520, 1349–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- Kole C, Brommer B, Nakaya N, Sengupta M, Bonet-Ponce L, Zhao T, Wang C, Li W, He Z, and Tomarev S (2020). Activating Transcription Factor 3 (ATF3) Protects Retinal Ganglion Cells and Promotes Functional Preservation After Optic Nerve Crush. Invest Ophthalmol Vis Sci 61, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuke L, Mosharov EV, Levy OA, and Greene LA (2019). Stress-induced phosphoubiquitin formation causes parkin degradation. Sci Rep 9, 11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput Biol 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, et al. (2020). Cell Type-Specific Transcriptomics Reveals that Mutant Huntingtin Leads to Mitochondrial RNA Release and Neuronal Innate Immune Activation. Neuron 107, 891–908 e898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, and Barres BA (2017). Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, and Church GM (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, Blum JA, Castruita PA, Linares GR, Ruan K, et al. (2021). p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell 184, 689–708 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, and Goldberg JL (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawabi H, Belin S, Cartoni R, Williams PR, Wang C, Latremoliere A, Wang X, Zhu J, Taub DG, Fu X, et al. (2015). Doublecortin-Like Kinases Promote Neuronal Survival and Induce Growth Cone Reformation via Distinct Mechanisms. Neuron 88, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickells RW, Howell GR, Soto I, and John SW (2012). Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu Rev Neurosci 35, 153–179. [DOI] [PubMed] [Google Scholar]
- Norsworthy MW, Bei F, Kawaguchi R, Wang Q, Tran NM, Li Y, Brommer B, Zhang Y, Wang C, Sanes JR, et al. (2017). Sox11 Expression Promotes Regeneration of Some Retinal Ganglion Cell Types but Kills Others. Neuron 94, 1112–1120 e1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmisano I, Danzi MC, Hutson TH, Zhou L, McLachlan E, Serger E, Shkura K, Srivastava PK, Hervera A, Neill NO, et al. (2019). Epigenomic signatures underpin the axonal regenerative ability of dorsal root ganglia sensory neurons. Nat Neurosci 22, 1913–1924. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, and Geschwind DH (2015). Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, et al. (2008). Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322, 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YR, Tran NM, Krishnaswamy A, Kostadinov D, Martersteck EM, and Sanes JR (2017). Satb1 Regulates Contactin 5 to Pattern Dendrites of a Mammalian Retinal Ganglion Cell. Neuron 95, 869–883 e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Maday S, Fu MM, Moughamian AJ, and Holzbaur EL (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci 33, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttagunta R, Tedeschi A, Soria MG, Hervera A, Lindner R, Rathore KI, Gaub P, Joshi Y, Nguyen T, Schmandke A, et al. (2014). PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat Commun 5, 3527. [DOI] [PubMed] [Google Scholar]
- Quigley HA (2016). Understanding Glaucomatous Optic Neuropathy: The Synergy Between Clinical Observation and Investigation. Annu Rev Vis Sci 2, 235–254. [DOI] [PubMed] [Google Scholar]
- Quigley HA, and Broman AT (2006). The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Hohman RM, Addicks EM, Massof RW, and Green WR (1983). Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol 95, 673–691. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke AW, Baek J, Ashenberg O, and Keating AE (2013). Networks of bZIP protein-protein interactions diversified over a billion years of evolution. Science 340, 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renier N, Wu Z, Simon DJ, Yang J, Ariel P, and Tessier-Lavigne M (2014). iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910. [DOI] [PubMed] [Google Scholar]
- Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, Geschwind DH, and Woolf CJ (2020). Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes after Axonal Injury. Neuron 108, 128–144 e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington RM, Carlson BJ, Crish SD, and Calkins DJ (2010). The microbead occlusion model: a paradigm for induced ocular hypertension in rats and mice. Invest Ophthalmol Vis Sci 51, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, and Woolf CJ (2007). ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci 27, 7911–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. (2013). Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Belsky DM, Bowen ME, Ohn CYJ, O’Rourke MK, Shen R, Kim G, Pitts J, Attardi LD, Tessier-Lavigne M (2021). An anterograde pathway for sensory axon degeneration gated by a cytoplasmic action of the transcriptional regulator P53. Dec Cell 56, 976–984.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, and He Z (2009). SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron 64, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A (1989). Intraocular pressure and glaucoma. Am J Ophthalmol 107, 186–188. [DOI] [PubMed] [Google Scholar]
- Spitz F, and Furlong EE (2012). Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, et al. (2011). Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 480, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Liu J, Crary JF, Malagelada C, Sulzer D, Greene LA, and Levy OA (2013). ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J Neurosci 33, 2398–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, Sur M, and Zhang F (2015). In vivo interrogation of gene function in the mammalianbrain using CRISPR-Cas9. Nat Biotechnol 33, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syc-Mazurek SB, Fernandes KA, Wilson MP, Shrager P, and Libby RT (2017). Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol Neurodegener 12, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syc-Mazurek SB, and Libby RT (2019). Axon injury signaling and compartmentalized injury response in glaucoma. Prog Retin Eye Res 73, 100769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G, and Lenhard B (2016). TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics 32, 1555–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran NM, Shekhar K, Whitney IE, Jacobi A, Benhar I, Hong G, Yan W, Adiconis X, Arnold ME, Lee JM, et al. (2019). Single-Cell Profiles of Retinal Ganglion Cells Differing in Resilience to Injury Reveal Neuroprotective Genes. Neuron 104, 1039–1055 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon SJ, Huber N, Pritchard JK, Chang HY, Greenleaf WJ, and Pasca SP (2020). Chromatin accessibility dynamics in a model of human forebrain development. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadarajan SG, Hunyara JL, Hamilton NR, Kolodkin AL, and Huberman AD (2022). Central nervous system regeneration. Cell 185, 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Haugen E, et al. (2020). Global reference mapping of human transcription factor footprints. Nature 583, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, and Segal RA (2001). Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 4, 981–988. [DOI] [PubMed] [Google Scholar]
- Weinreb RN, Aung T, and Medeiros FA (2014). The pathophysiology and treatment of glaucoma: a review. JAMA 311, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, and Martin KR (2016). Primary open-angle glaucoma. Nat Rev Dis Primers 2, 16067. [DOI] [PubMed] [Google Scholar]
- Wertz MH, Mitchem MR, Pineda SS, Hachigian LJ, Lee H, Lau V, Powers A, Kulicke R, Madan GK, Colic M, et al. (2020). Genome-wide In Vivo CNS Screening Identifies Genes that Modify CNS Neuronal Survival and mHTT Toxicity. Neuron 106, 76–89 e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag New York: ). [Google Scholar]
- Wiggs JL, and Pasquale LR (2017). Genetics of glaucoma. Hum Mol Genet 26, R21–R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PR, Benowitz LI, Goldberg JL, and He Z (2020). Axon Regeneration in the Mammalian Optic Nerve. Annu Rev Vis Sci 6, 195–213. [DOI] [PubMed] [Google Scholar]
- Winter CC, He Z, and Jacobi A (2022). Axon Regeneration: A Subcellular Extension in Multiple Dimensions. Cold Spring Harb Perspect Biol 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, and Sul HS (2009). A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136, 1056–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortel IMN, van der Meer LT, Kilberg MS, and van Leeuwen FN (2017). Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol Metab 28, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Cho KS, Chen H, Yu D, Wang WH, Luo G, Pang IH, Guo W, and Chen DF (2012). Microbead-induced ocular hypertensive mouse model for screening and testing of aqueous production suppressants for glaucoma. Invest Ophthalmol Vis Sci 53, 3733–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Williams PR, Jacobi A, Wang C, Goel A, Hirano AA, Brecha NC, Kerschensteiner D, and He Z (2019). Elevating Growth Factor Responsiveness and Axon Regeneration by Modulating Presynaptic Inputs. Neuron 103, 39–51 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ATAC-seq metadata, quality control metrics, and annotation of differentially accessible regions (DARs) comparing RGCs injured at day 1 or 3 versus uninjured condition (day 0). Related to Figure 2. (A) ATAC-seq metadata and quality control metrics including percent of duplicated reads after sequencing (DuplicateRate), non-duplicated, uniquely mapped reads (UniqMappedReads), total number of peaks from MACS2 (TotNarrowPeak), TSS enrichment, and fraction of reads in peaks (FRiP). Annotation of injured RGC DARs [absolute log fold changes (logFC) > 0.3 and false discover rate (FDR) < 0.1] comparing day 1 v.s. day 0 (B) or day 3 v.s. day 0 (C).
RNA-seq metadata, quality control metrics, and annotation of differentially expressed genes (DEGs) comparing injured RGCs at indicated time points versus uninjured ones (day 0). Related to Figure 2. (A) RNA-seq metadata and quality control metrics obtained from Picard Tools. Principle component analysis (PCA) was performed on the metrics and the first seven PCs accounting > 95% of variance was kept for further analysis. (B-C) Annotation of injured RGC DEGs (absolute logFC > 0.3 and FDR < 0.1) comparing day 1 v.s. day 0 (B) or day 3 v.s. day 0 (C).
Clustering and annotation of putative peak-gene links. Related to Figure 2. (A) Linked peak-gene pairs. Proximal or distal peaks that are differential accessible are linked to corresponding genes that are differentially expressed at day 1 or day 3 following optic nerve crush. The peak-gene pairs are grouped into two clusters: the 'up' clust er where the accessibility of linked peaks and the expression of corresponding genes are both up-regulated, and the 'down' cluster where both are down -regulated. (B) Gene Ontology of the up-regulated cluster. (C) Gene Ontology of the down-regulated cluster.
TF motif activity in injured RGCs, related to Figure 3. (A) ChromVAR-estimated motif matches in DARs. A list of non-redundant, human or mouse TF motifs (Methods) was used, named in the format of [species]-[database]-[corresponding TF]-[motif ID]. ChromVAR returns variability, which is the standard deviation of the motif z-scores computed across samples, and boostrap confidence intervals and p-value for the variability by resampling samples. These calculations are correlative to motif accessibility changes across samples. (B) Sample-wise correlations of motif activity to the expression levels of corresponding TF.
RNA-seq metadata, quality control metrics and annotation of differentially expressed genes comparing RGCs with or without CRISPR ablation of one of the 4 survival TFs (ATF3, ATF4, C/EBPγ CHOP). Related to Figure 4–5. (A). RNA-seq metadata and quality control metrics obtained from Picard Tools. (B -E) Annotation of DEGs in RGCs with ablation of ATF3, ATF4, C/EBPγ, CHOP, respectively.
Target genes regulated by ATF3/CHOP, or ATF4/C/EBPγ or by all 4 TFs predicted by DNA-footprinting and RNA-seq. Related to Figure 4–5. (A) Genes that are positively or negatively regulated by ATF3/CHOP, but not ATF4/C/EBPγ. (B) Genes that are positively or negatively regulated by ATF4/C/EBPγ, but not ATF3/CHOP. (C) Genest hat are positively or negatively regulated by all four TFs.
Data Availability Statement
RNA-sequencing and ATAC-sequencing data will be available from the Gene Expression Omnibus through series accession number GEO: GSE184547 and GSE190667. All other data referenced in this publication or code used to perform meta-analysis will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request