

Abstract
In non-mammalian vertebrates, the molecular mechanisms involved in the transformation of haploid germ cells (HGCs) into spermatozoa (spermiogenesis) are largely unknown. Here, we investigated this process in the marine teleost gilthead seabream (Sparus aurata) through the examination of the changes in the transcriptome between cell-sorted HGCs and ejaculated sperm (SPZEJ). Samples were collected under strict quality controls employing immunofluorescence microscopy as well as by determining the sperm motion kinematic parameters by computer-assisted sperm analysis. Deep sequencing by RNA-seq identified a total of 7286 differentially expressed genes (DEGs) (p-value < 0.01) between both cell types, of which nearly half were upregulated in SPZEJ compared to HCGs. In addition, approximately 9000 long non-coding RNAs (lncRNAs) were found, of which 56% were accumulated or emerged de novo in SPZEJ. The upregulated transcripts are involved in transcriptional and translational regulation, chromatin and cytoskeleton organization, metabolic processes such as glycolysis and oxidative phosphorylation, and also include a number of ion and water channels, exchangers, transporters and receptors. Pathway analysis conducted on DEGs identified 37 different signaling pathways enriched in SPZEJ, including 13 receptor pathways, from which the most predominant correspond to the chemokine and cytokine, gonadotropin-releasing hormone receptor and platelet derived growth factor signaling pathways. Our data provide new insight into the mRNA and lncRNA cargos of teleost spermatozoa and uncover the possible involvement of novel endocrine mechanisms during the differentiation and maturation of spermatozoa.
Subject terms: Reproductive biology, Transcriptomics
Introduction
The developmental remodeling of haploid germ cells (HGCs) into highly polarized spermatozoa capable of motility activation and fertilization is an evolutionary conserved mechanism of male gamete formation1. In both mammals and fishes, this spermiogenic differentiation process typically results in the elimination of cytoplasm and organelles together with the morphogenesis of three major compartments, the head containing a condensed haploid nucleus, a mid-section containing the proximal centriole together with one or more mitochondria, and the terminal region composed of an elongated flagellum or flagella2,3. Although a variety of specializations evolved in the different lineages, such as the acystic and cystic cellular environments in which mammalian and piscine spermatogenesis respectively progresses, and the absence of an acrosome in most teleost spermatozoa, the overall spermatazoon bauplan is conserved. Similarly, in both lineages, the major endocrine mediators of spermatogenesis are the pituitary follicle-stimulating (FSH) and luteinizing (LH) gonadotropins, that are induced by hypothalamic gonadotropin releasing hormones (GnRH)4–6. The LH regulates spermiogenesis indirectly through its cognate LH/choriogonadotropin receptor (LHCGR) to stimulate androgen secretion in the somatic Leydig cells, or also directly through the activation of the LHCGR in the spermatids of teleost fishes2,4,7. An understanding of which signaling pathways are activated in the developing germ cells can reveal the conserved or divergent nature of molecular mechanisms regulating vertebrate spermiogenesis and further uncover novel traits associated with spermatozoon formation and fertility.
In mammals, such as boars, bulls, stallions, rodents and primates, transcriptomic studies of spermatozoa or sperm have identified populations of RNAs associated with fertility or that are important for subsequent embryonic development8–14. Intricate single-cell RNA-seq studies of cell-sorted germ cells have also identified specific markers of cell populations with significantly distinct transcriptomes, further revealing the conserved and unique spermatogenic features of mice and human15–17. By contrast, spermatogenic transcriptomic studies in fishes have been conducted on the whole testes, and therefore these studies have not differentiated between the somatic and germ cell RNA populations18–33. As a result, although these works have identified important genes and pathways for spermatogenesis, the genetic network of spermiogenesis remains unexplored.
To gain insight into the molecular regulation of spermiogenesis in fish, the main objective of the present work was to provide a deep characterization of the transcriptome of haploid germ cells (HGC) and ejaculated spermatozoa (SPZEJ) from a modern marine teleost, the gilthead seabream (Sparus aurata), by RNA-seq technologies. To achieve this, we employed cell-sorted HGC and strict quality controls of the SPZEJ. By means of this design, our study offers a reliable set of sperm next-generation sequencing data that enable a deeper understanding of the RNA cargo of fish spermatozoa. In addition, in silico analyses of the data uncovered several novel endocrine signaling pathways that may play important roles during the differentiation and maturation of spermatozoa.
Results
Isolation and characterization of HGC and SPZEJ
The changes in gene expression during the differentiation and maturation of seabream spermatozoa were investigated by whole-transcriptome RNA-seq of HGCs and SPZEJ. The HGCs were isolated by fluorescence-activated cell sorting (FACS) from testis samples. Flow cytometry of the extract from the seabream whole mature testis showed the proportion of diploid and haploid cells to be 16% and 84%, respectively (Fig. 1a). The percentage of diploid cells was lower than expected because the centrifugation steps of the extract before cell sorting partially depleted this population. Flow cytometry identified two subpopulations of haploid cells based on their relative size and SYBR Green I fluorescence intensity: a subpopulation formed by spermatocytes II and spermatids (SPC II and SPD, respectively), which we refer here as HGCs, and another subpopulation formed by intratesticular spermatozoa (SPZI) (Fig. 1a, b). The percentage of HGCs and SPZI in the testicular extracts was of 34 ± 4% and 66 ± 4% (n = 15), respectively (Fig. 1b).
Figure 1.

Isolation of testicular haploid germ cells and ejaculated spermatozoa from the seabream and RNA extraction. (a, b) Representative flow cytometry plots of the seabream cell populations in the whole testis before and after centrifugation generated with the MoFlo XDP cell sorter software (https://www.beckman.es/). In a, the populations of testicular haploid and diploid cells from the testis crude extract before centrifugation are encircled. In b, after centrifugation two subpopulations of haploid germ cells (HGCs) are recovered, one corresponding to a mix of type II spermatocytes (SPC II) and spermatids (SPD), and the other to intratesticular spermatozoa (SPZI). (c) Representative immunostaining of Lys9 acetylated histone 3 (H3K9ac), meiotic recombination protein Spo11 and α-tubulin (Tuba) in sorted HGC shown in b and ejaculated spermatozoa (SPZEJ). For each cell type the brightfield (left panels) and epifluorescence (right panels) images are shown. SPDR, round spermatids; SPDE, elongating spermatids. Scale bars, 2 and 5 µm. (d, e) Electrophoretic size distribution of RNAs in representative samples of SPCII + SPD (d) and SPZEJ (e) populations analyzed and plotted by the Agilent Bioanalyzer 2100 Expert Software (https://www.agilent.com/). For each sample, fluorescence is plotted against mRNA length (in nucleotides, nt). The RNA integrity number (RIN) is indicated for each sample.
Microscopic examination of the HGC-enriched population after FACS confirmed the presence of SPC II, and round and elongating SPD in this fraction (Fig. 1c). Whole-mount immunostaining revealed strong expression of Lys9 acetylated histone 3 (H3K9ac) and meiotic recombination protein Spo11 in SPC II, which progressively decreased in round and elongating SPD, and completely vanished in SPZEJ (Fig. 1c). Immunolocalization of α-tubulin (Tuba) showed that this protein was spread in the cytoplasm in SPC II and round SPD, became detectable in the nascent flagellar region of elongating SPD, and was finally distributed along the flagellum of differentiated SPZEJ (Fig. 1c). These observations indicate a high occurrence of meiotic recombination in SPC II and a progressive DNA condensation during the differentiation of SPC II into SPD and spermatozoa, which are conserved features during vertebrate germ cell development34,35. These data therefore confirmed that the sorted population of cells from the mature seabream testis correspond to HGCs before differentiation into spermatozoa.
The SPZEJ were collected by manual stripping of naturally spermiating males. The kinematic parameters of the spermatozoa were determined by computer-assisted sperm analysis (CASA) at ~ 2 h after time of collection to assure that the sperm employed for RNA-seq analysis was of high quality. The SPZEJ selected for further RNA isolation showed a percentage of motility and progressivity at 5 s postactivation from 88 to 98%, and from 16 to 51%, respectively. The time during which spermatozoa remained motile was also monitored and this ranged from 5 to 8 min.
To evaluate the purity and RNA size distribution profiles of the RNA extracted from the HCG and SPZEJ samples for further RNA-seq analysis, we used the Agilent 2100 Bioanalyzer. The electrophoretic size distribution of extracted RNAs showed that HGC exhibited defined peaks for 18S and 28S ribosomal subunits and high RNA integrity number (RIN) (Fig. 1d, inset). In contrast, in the SPZEJ extracts the 18S and 28S rRNAs appeared strongly reduced, thus showing a low RIN, which is a typical feature of spermatozoa, together with more abundant short-length mRNA species (Fig. 1e, inset). These data, together with immunofluorescence microscopy examination, indicate that these samples were almost devoid of non-sperm cells36.
Transcriptome profiling of HGC and SPZEJ
Four unstranded RNA libraries (biological replicates) for low-input RNA were subsequently constructed for each of the two HGC and SPZEJ cell types. The total RNA from HGC and SZPEJ was extracted from 12 males, and each of the four libraries was constructed from a pool of three different males. Library sequencing rendered 30–62 million reads per library corresponding to a yield of 5–10 Gb per library. From these data, we produced a new integrative S. aurata genome annotation before the RNA-seq analysis. This new annotation was carried out by re-annotating the available S. aurata reference genome37, and by adding 202 de novo assembled transcripts that were not present in the genome assembly. In total, 31,501 protein-coding genes were annotated, which produced 57,396 transcripts (1.82 transcripts per gene) that encode for 51,365 unique protein products. Functional labels were assigned to 62% of the annotated proteins. In addition, 165,898 non-coding transcripts were annotated, of which 159,925 are long non-coding RNAs (lncRNAs) and 5973 correspond to short non-coding RNAs.
Principal component analysis (PCA) of the expression data showed that FACS-purified HGC and SPZEJ formed two relatively well-separated clusters, suggesting that the developmental stage has a large effect on the pattern of gene expression (Fig. 2a). However, while the four HGC biological replicates clustered very closely, those of SPZEJ were more distant, indicating a higher variability in the transcriptome of the SPZEJ replicates. Nevertheless, the RNA-seq analysis revealed a total of 7286 differentially expressed genes (DEGs) (adjusted p-value < 0.01) between both cell types, of which nearly half (3446) were upregulated in SPZEJ when compared to HGCs (Fig. 2b–d). In addition, 239 transcripts were detected only in SPZEJ (Fig. 2d), indicating that they emerged de novo in the spermatozoa during the differentiation and maturation phases. The top ten upregulated mRNAs included transcripts potentially involved in spermatogenesis, sperm capacitation, oxidative stress, or sperm-egg interaction, such as tlr1, zp, tyro3 and alox15b, as well as other genes with potential functions in transcription, cell adhesion, binding and presentation of antigens, or mitochondrial permeability (mafb, apbb1, cdc209c, fhl2, h2-aa and arhgap11b) (Table 1). Finally, we also found a high number of differentially expressed lncRNAs (9059 sequences) of which 5114 were upregulated in SPZEJ (Fig. 2d), which indicates that 56% of the total identified lncRNAs were accumulated in SPZEJ. In addition, most of the upregulated lncRNAs (67%) were unique in SPZEJ.
Figure 2.
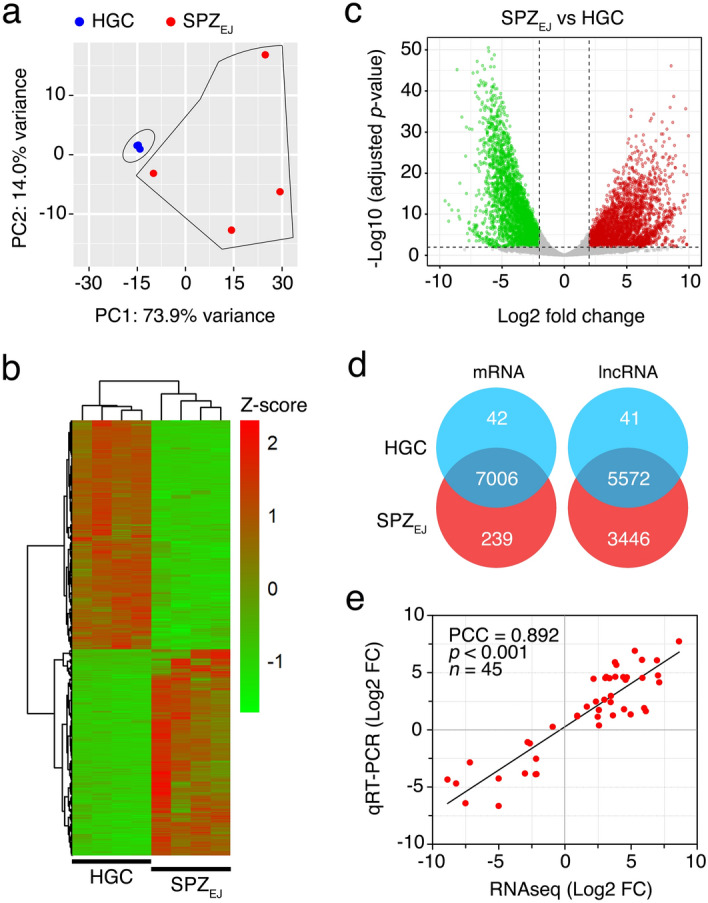
RNA-seq data analysis of seabream HGC and SPZEJ. (a) Principal component analysis (PCA) using the top 500 most variable genes between HGC and SPZEJ (n = 4 pools) and the 'rlog' transformation of the counts. (b) Heatmap generated by unsupervised hierarchical clustering of RNAseq expression z-scores computed for the 7286 differentially expressed genes (DEGs) (p-adj < 0.01; Log2 fold change > 1) between HGC and SPZEJ. The heatmap was generated with the ‘pheatmap’ R package (https://CRAN.R-project.org/package=pheatmap). (c) Volcano plot representation of DEGs in the SPZEJ versus HGC comparison. The x-axis shows Log2 fold changes in expression and the y-axis the negative logarithm of their p-value to base 10. Red and green points mark the genes with significantly increased or decreased expression respectively in SPZEJ compared to HGC (FDR < 0.01). (d) Venn diagrams showing the number of HGCs- and SPZEJ-specific and common mRNAs and lncRNAs (in intersect region) which are differentially expressed between the two stages. The Venn diagrams and volcano plots were performed with the ‘VenDiagramm’ R package (https://CRAN.R-project.org/package=VennDiagram) and ‘ggplot2’ R package (https://ggplot2.tidyverse.org), respectively. (e) Validation of the RNAseq data by qRT-PCR. The plot represents the Pearson's correlation analysis of DEGs in HGC and SPZEJ determined by RNAseq and qRT-PCR. The Pearson’s correlation coefficient (PCC) of the Log2 fold change analyzed by RNAseq (x-axis) and using qRT-PCR (y-axis), the p-value, and the number of DEGs analyzed are indicated. Plots in a, d and e were generated using the GraphPad Prism v8.4.3 (686) software (https://www.graphpad.com/).
Table 1.
Top 10 upregulated mRNAs in SPZEJ, their fold-change in expression with respect to HGC and mean of fragments per kilo base of transcript per million mapped fragments (FPKM), and their potential associated functions according to GeneCards (www.genecards.org).
| Product | Gene | Sequence ID | Log2 FC | Mean FPKM | Reproductive-related function |
|---|---|---|---|---|---|
| Toll-like receptor 1 | tlr1 | XM_030396315.1 | 11.16 | 1698 | Involved in ovulation, sperm capacitation and fertilisation |
| Zona pellucida protein X, partial | zp | AAY21008.1 | 11.02 | 657 | Sperm-egg interaction |
| Transcription factor MafB | mafb | XP_030276052.1 | 10.69 | 558 | Acts as a transcriptional activator or repressor that may be involved in spermatogenesis. Unknown function in spermatozoa |
| Amyloid beta A4 precursor -binding family B member 1 | apbb1 | XP_030254479.1 | 10.08 | 236 | Transcription coregulator that can have both coactivator and corepressor functions. Can bind modified histones and chromatin modifying enzymes, thereby regulating transcription |
| CD209 antigen C | cd209c | XP_030265713.1 | 10.05 | 839 | C-type lectin that functions in cell adhesion and pathogen recognition. Unknown function in spermatozoa |
| Four and a half LIM domain 2 | fhl2 | XP_030299463.1 | 10.03 | 226 | May function as a molecular transmitter linking various signaling pathways to transcriptional regulation. Unknown function in spermatozoa |
| H-2 class II histocompatibility antigen, A-Q alpha chain | h2-aa | XP_030263565.1 | 9.96 | 1644 | Binding and presentation of peptides derived from antigens. Unknown function in spermatozoa |
| Tyrosine-protein kinase receptor TYRO3 | tyro3 | XP_030287857.1 | 9.88 | 308 | Involved in spermatogenesis. Unknown function in spermatozoa |
| Arachidonate 15-lipoxygenase B | alox15b | XP_030274670.1 | 9.83 | 212 | Catalyzes the peroxidation of free and esterified polyunsaturated fatty acids (PUFAs) generating a spectrum of bioactive lipid mediators. Involved in the oxidative stress cascade of spermatozoa |
| Inactive Rho GTPase-activating protein 11B | arhgap11b | XP_030261033.1 | 9.83 | 647 | Inhibits the mitochondrial permeability transition pore (mPTP), thereby increasing mitochondrial Ca2+. Unknown function in spermatozoa |
The quality of the RNA-seq data and the reliability of the DEGs identified were validated on randomly selected 45 DEGs by real-time quantitative reverse transcription PCR (qRT-PCR) in three biological replicates. Fold changes from qRT-PCR were compared with the RNA-seq expression profiles (Fig. 2e). The dynamic expression patterns of all genes were consistent with the RNA-seq analysis, showing a high correlation (Pearson’s correlation coefficient of 0.892) between RNA-seq and qRT-PCR data. These results therefore indicated the reliability of the RNA-seq for mRNA differential expression analysis.
Functional enrichment analysis of DEGs during spermatozoa differentiation and maturation
Gene ontology (GO) term-enriched analysis of DEGs in SPZEJ with significant differences revealed that a large number of biological processes were represented. The five top-ranked GO terms in biological processes were regulation of biological, cellular and metabolic processes, and organic substance and metabolic processes (Suppl. Fig. S1a). Further analysis of GO term distribution indicated that the most represented biological process was the regulation of gene expression, followed by positive regulation of macromolecule and cellular metabolism, regulation of signal transduction, and regulation of cellular biosynthesis (Suppl. Fig. S1b). Interestingly, genes with GO terms such as cellular response to stimulus, cell communication, signal transduction, response to external or chemical stimulus, cell adhesion, and cell surface receptor signaling pathway, were only upregulated in SPZEJ (Suppl. Fig. S1a and S1b). For the GO molecular function, the top enriched terms were binding to ribonucleotides and purine nucleotides, whereas the terms Ca2+, phosphatidylinositol and actin binding, ion channel activity, and transmembrane transport of inorganic cations and organic anions appeared to be only upregulated in SPZEJ (Suppl. Fig. S1c). Taken together these findings indicate the enrichment of gene expression, metabolic and signaling processes in SPZEJ.
To gather more information on genes with a potential impact on spermatozoa function, the DEGs in SPZEJ were manually classified into five functional categories by using GO analysis and the Uniprot database. These categories included transcription, translation and chromatin organization (978 genes), receptors (395 genes), metabolism of proteins, lipids and carbohydrates (469 genes), cytoskeleton and cell movement (487 genes), and channels, exchangers and transporters (303 genes) (Fig. 3a and Suppl. Table S1). The genes upregulated in SPZEJ related to transcription, translation and chromatin organization (365 genes) mainly correspond to transcription factors (41%) and regulators of transcription (21%), followed by structural constituents of ribosomes (12%), regulators of translation (4%), chromatin and RNA binding (4 and 5%, respectively), and histones and histone modification (7%) (Fig. 3b). Most of the highest upregulated genes of this group (log2 fold change > 5) correspond to transcriptional regulators, including transcription factors, but other mRNAs encoding late histone H2A.2.2 and H2B.L4-like (h2a.2.2 and h2b.l4), helicase with zinc finger domain 2 isoform X1 (helz2), or DNA (cytosine-5)-methyltransferase 3B (dnmt3b), were also highly accumulated in SPZEJ (Suppl. Table S1). Interestingly, ~ 25% of these highly upregulated genes are possibly involved in activation rather than repression of transcription, whereas ~ 29% of them can potentially repress or activate transcriptional activity.
Figure 3.

Functional classification of DEGs during sperm differentiation and maturation. (a) Transcriptional regulation of a subpopulation of DEGs classified into five functional categories: transcription and translation and chromatin organization, receptors, metabolism, cytoskeleton and cell movement, and channels, exchangers and transporters. (b–f) Pie charts showing the GO term distribution of upregulated DEGs in SPZEJ included in each of the five functional groups. The numbers are the percentage of genes in each category. Plots were generated using the GraphPad Prism v8.4.3 (686) software (https://www.graphpad.com/).
Most of the receptor-encoding upregulated genes (266 genes) were G protein-coupled receptors (37%), tyrosine phosphatase and kinase receptors (11%), cytokine receptors (7%), as well as other receptors mainly including glycoprotein, Fc, and scavenger receptors (17%) (Fig. 3c). The highest expressed genes in each of these groups were lysophosphatidic acid receptor 6 (lpar6), potentially involved in protection of oxidative stress, receptor-type tyrosine- phosphatase C (ptprc), interleukin-1 receptor type 1 (il1r1), and zona pellucida sperm-binding protein 3 (zp3), likely implicated in the sperm-egg interaction (Suppl. Table S2).
For the upregulated genes encoding metabolic components (230 genes), those related to the metabolism of lipids, proteins and carbohydrates were almost equally represented (39, 32 and 29%, respectively) (Fig. 3d). The most enriched genes in SPZEJ belonging to these groups were those related to glycerophospholipid hydrolysis and fatty acid biosynthesis (18%), such as lysophosphatidylserine lipase ABHD12 (abhd12) and elongation of very long chain fatty acids 1 (elovl1), proteases (17%), such as transmembrane protease serine 9 (tmprss9), and glycolysis and gluconeogenesis (9%), such as triosephosphate isomerase (tpi1), which synthesizes D-glyceraldehyde 3-phosphate from glycerone phosphate at the beginning of the glycolytic pathway (Fig. 3d and Suppl. Table S3).
The genes encoding for proteins involved in cytoskeletal organization (30%), actin binding (26%) and molecular motors (16%) were the most abundant upregulated genes involved in the category of cytoskeleton and cell movement (116 genes) (Fig. 3e). The highest upregulated genes, however, correspond to actin binding, such as myristoylated alanine-rich C-kinase substrate (marcks), and motor proteins or components of the motor-adapter complex, such as kinesin Kif20a (kif20a) and syntabulin (sybu) (Suppl. Table S4).
Finally, in the group including genes encoding for channels, exchangers and transporters (134 genes), more than a half of the upregulated genes encode for ion channels (58%), whereas the rest included water channels (3%) and transporters of peptides and amino acids (17%), carbohydrates (7%), nucleosides and nucleotides (5%), vitamins (4%), neurotransmitters (2%) and bile acids (0.7%) (Fig. 3f). The K+ and metal specific channels (20%), such as potassium voltage-gated channel subfamily K members 6 and 4 (kcnk6 and kcng4) and transmembrane channel 7 (tmc7), as well as cation channels (13%), such as solute carrier family 22 member 5-like (slc22a5), were the most enriched in SPZEJ (Fig. 3f and Suppl. Table S5). Interestingly, the water and glycerol-facilitating channel aquaporin-7 (aqp7), which was previously described in seabream spermatozoa38, and the solute carrier family 43 member 3 (slc43a3), which is a sodium-independent purine-selective nucleobase transporter, were the most upregulated mRNAs in SPZEJ from the whole group of channels, exchangers and transporters (log2 fold change of 9.58 and 8.85, respectively) (Suppl. Table S5), suggesting an important role of these genes during the differentiation and maturation of spermatozoa.
Protein–protein interaction analyses
In an effort to identify specific transcription/translation and carbohydrate metabolic processes enriched in SPZEJ, we built a putative protein interactome network of DEGs classified into these two categories by using the STRING protein–protein interaction (PPI) database for known PPIs39 under very stringent inclusion criteria. As a result, a connected network comprising 766 proteins and 3588 connections was mapped for the proteins encoded by genes involved in transcription and translation (Fig. 4a). These proteins could be divided into five major subclusters based on their known biological functions established through GO analysis, including mitochondrial translation, tRNA aminoacylation, translation initiation, cytosolic ribosomes, and mRNA splicing (Fig. 4a). All of the DEGs grouped into the cytosolic ribosome subunit subcluster, and half of the DEGs belonging to the transfer RNA (tRNA) aminoacylation, mitochondrial translation, and translation initiation subclusters, were upregulated (Fig. 4a). These findings, together with the previous observation of the relatively high abundance of transcription activators among the upregulated genes in SPZEJ, suggest that both transcription and mitochondrial and cytoplasmic translation activity may occur during the differentiation and maturation of spermatozoa.
Figure 4.

Protein–protein interaction (PPI) networks of DEGs. The PPI information of DEGs potentially involved in transcription and translation and chromatin organization (a), and metabolism (b), was obtained through a database search using STRING database v11 with a high confidence score (0.9), and imported into Cytoscape v3.8.2 (https://cytoscape.org/) for network construction and plotting. Proteins and their interactions are shown as nodes (spheres) and edges (lines), respectively. Nodes in red or green color indicate upregulated and downregulated DEGs, respectively. Proteins are grouped based on their known biological functions. OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway; TCA, tricarboxylic acid.
The metabolism interactome showed 379 proteins and 821 connections divided into fifteen subclusters, from which those corresponding to glycolysis/gluconeogenesis, the pentose phosphate (PP) pathway and sphingolipid, galactose, glycogen and glutathione metabolism, were the most upregulated in SPZEJ (Fig. 4b). Further mapping of the 76 DEGs coding for enzymes involved in respiratory pathways, as well as qRT-PCR validation of a few of these transcripts, indicated that most of the genes of the tricarboxylic acid (TCA) cycle, as well as three genes coding for specific enzymes of gluconeogenesis, such as phosphoenol-pyruvate carboxykinase (pck2), fructose 1,6-bisphosphatase (fbp1) and glucose 6-phosphatase (g6pc), were downregulated or not differentially expressed in SPZEJ (Fig. 5a–c). In contrast, half of the genes involved in the PP pathway, such as 6-phosphogluconate dehydrogenase (pgd), transketolase (tkt) and transaldolase (tal), and most of the glycolytic enzyme-encoding genes, including the two key enzymes hexokinase-1 (hk1) and pyruvate kinase (pkm), were upregulated in SPZEJ (Fig. 5a–c). Similarly, many of the genes coding for enzymes catalyzing oxidative phosphorylation (OXPHOS) were also upregulated in SPZEJ (Fig. 5a–c). These data therefore suggest that both glycolysis and OXPHOS are important pathways for ATP generation in seabream spermatozoa.
Figure 5.

Analysis and mapping of DEGs potentially involved in respiratory pathways in seabream spermatozoa. (a) Hierarchical clustering heatmap of RNA-seq expression z-scores computed for genes encoding enzymes potentially involved in carbohydrate metabolism, penthose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (OXPHOS), that are differentially expressed (p-adj < 0.01; Log2 fold change > 1) between HGC and SPZEJ. The map was generated with the ‘pheatmap’ R package (https://CRAN.R-project.org/package=pheatmap). (b) Schematic diagram of the biochemical pathways of glycolysis/gluconeogenesis, PPP, TCA cycle and OXPHOS. Enzyme-coding DEGs in green and red color denotes downregulation and upregulation, respectively, whereas black color indicates no change in the expression levels. The diagram was generated using CorelDRAW Graphics Suite 2021.5 (https://www.coreldraw.com/). (c) Validation of the changes in expression of 11 selected genes classified into respiratory pathways by qRT-PCR. Data from qRT-PCR are the mean ± SEM (n = 3 pools of 3 different fish each) and were plotted using the GraphPad Prism v8.4.3 (686) software (https://www.graphpad.com/).
The cytokine, PDGF and GnRHR signaling pathways are highly upregulated in SPZEJ
In order to identify signaling pathways enriched in SPZEJ, pathway analysis was carried out for the 7287 DEGs using the PANTHER classification system40. The analysis identified a total of 960 transcripts belonging to 37 different signaling pathways, including 13 receptor pathways (Fig. 6). Highly enriched and significant pathways were integrin, epidermal growth factor receptor (EGFR), fibroblast growth factor (FGF), cholecystokinin receptor (CCKR), rat sarcoma virus (Ras), vascular endothelial growth factor (VEGF), and B cell activation. However, amongst the most dominant pathways regulated in SPZEJ in terms of number of genes identified, rich factor and lowest p-values were the inflammation mediated by chemokine and cytokine, PDGF and GnRHR signaling pathways (Fig. 6). Mapping of these DEGs, as well as of other transcripts detected but not regulated in SPZEJ, on the corresponding KEGG pathway and WikiPathways databases showed that most components of the chemokine and cytokine, PDGF and GnRH signaling pathways were expressed in SPZEJ (Suppl. Figs. S2–S4).
Figure 6.
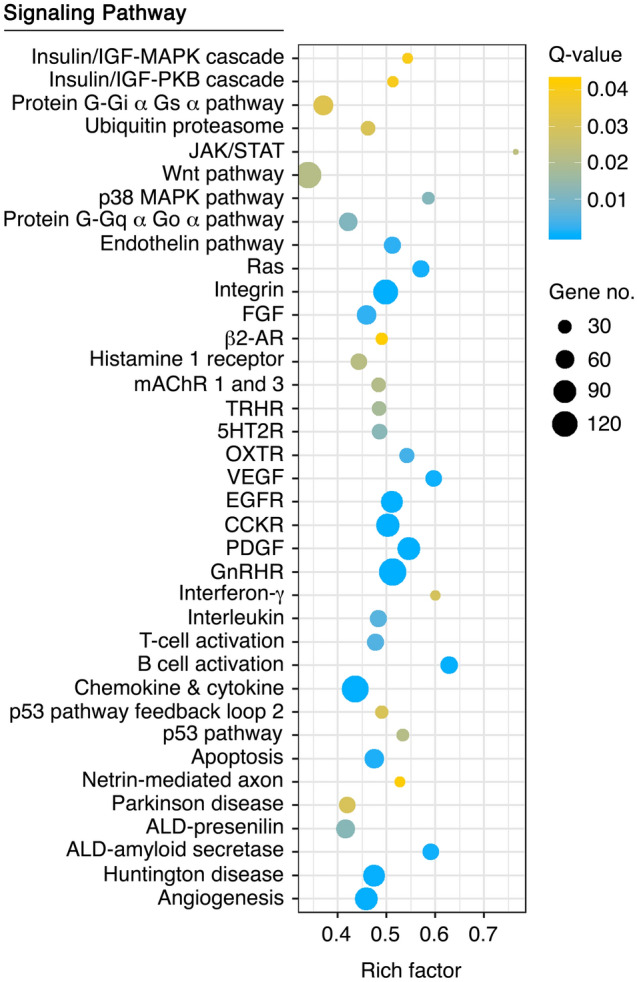
Pathway enrichment analysis among DEGs during spermatozoa differentiation and maturation using the PANTHER Classification System. The plot generated with the ‘ggplot2’ R package (https://ggplot2.tidyverse.org) shows the 37 most highly enriched signaling pathways (FDR < 0.05) in SPZEJ. The left Y-axis shows the PANTHER pathway, whereas the X-axis shows the rich factor. A high Q-value is represented by yellow and a low Q-value is represented by blue.
Hierarchical clustering heatmaps showed that most of the genes related to the chemokine and cytokine, PDGF and GnRHR pathways were upregulated in SPZEJ (Fig. 7a–c). Thus, for the chemokine and cytokine pathway the most accumulated transcripts (log2 fold change > 7) were C-X-C motif chemokine 10 (cxcl10), C-C chemokine receptor type 6 (ccr6), C-C motif chemokine 3 and 19 (ccl3 and ccl19), and ras-related C3 botulinum toxin substrate 2 (rac2) (Fig. 7a). For the PDGF pathway, the Pdgf receptor b (pdgfrb), phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit delta isoform (pik3cd), inhibitor of nuclear factor kappa-B kinase subunit beta (ikbkb), rapidly accelerated fibrosarcoma (RAF) proto-oncogene serine threonine-protein kinase (raf1), and signal transducer and activator of transcription 1 (stat1), were the highest upregulated genes (log2 fold change > 4) (Fig. 7b). Finally, in the GnRHR pathway the highest expressed genes (log2 fold change > 6) were matrix metallopeptidase-2 and -14 (mmp2 and mmp14), mitogen-activated kinase 12 (mapk12), calcium calmodulin-dependent kinase type II delta 1 chain (camk2d1) and adenylate cyclase type 9 (adcy9) (Fig. 7c). These data were validated by qRT-PCR for a number of genes from each pathway, including three Gnrhrs identified in our transcriptome (gnrhr1, gnrhr2 and gnrhr3) for which the RNA-seq did not detect significantly different expression levels (Fig. 7d). The qRT-PCR analysis showed however that both gnrhr2 and gnrhr3 are in fact upregulated in SPZEJ, whereas the gnrhr1 is not (Fig. 7d). Altogether, these data suggest the activation of the chemokine and cytokine, GnRHR and PDGF signaling pathways during seabream spermiogenesis.
Figure 7.

Hierarchical clustering heatmaps of RNA-seq expression for genes potentially involved in the inflammatory chemokine and citokine (a), PDGF (b) and GnRHR (c) signaling pathways. The maps were generated with the ‘pheatmap’ R package (https://CRAN.R-project.org/package=pheatmap). (d) qRT-PCR validation of the changes in expression of selected genes classified into these pathways. Data from qRT-PCR are the mean ± SEM (n = 3 pools of 3 different fish each) and were plotted using the GraphPad Prism v8.4.3 (686) software (https://www.graphpad.com/).
Transcripts expressed de novo in SPZEJ
We finally investigated the 236 transcripts that were only identified in SPZEJ, which were considered as emerging de novo during the differentiation and maturation of spermatozoa. These transcripts were classified into different functional categories by using GO analysis and the Uniprot database, which showed that those potentially involved in recognition, binding or adhesion processes of cells, cell signaling, metabolism, and transcription and translation, were the most abundant (21%, 17%, 14% and 13%, respectively) (Suppl. Fig. S5a). Interestingly, among these transcripts, the CD209 antigen (cd209) (mean FPKM = 231), toll-like receptor 13 (tlr13) (mean FPKM = 38), tetraspanin-13 (tspan13) (mean FPKM = 21), guanine nucleotide-binding G(I) G(S) G(T) subunit beta-2 (gnb2) (men FPKM = 10), tumor necrosis factor alpha-induced protein 8-like protein 3 (tnfaip8l3) (mean FPKM = 17), gamma-glutamyltransferase 5-like (ggt5) (mean FPKM = 14), and transcription factor jun-D-like (jund) (mean FPKM = 12) showed the highest expression in SPZEJ (Suppl. Fig. S5a). Further interactome analysis of the proteins potentially encoded by the de novo transcripts revealed the upregulation of components of the Ca2+ and cAMP, and phosphatidylinositol 3-kinase (PI3K/Akt), signaling pathways in SPZEJ (Suppl. Fig. S5b), suggesting an important role of these pathways for spermatozoon function. In addition, gene set enrichment and pathway analyses among the de novo genes in SPZEJ confirmed the activation of the GnRHR and PDGF signaling pathways, and also suggested a potential role of heterotrimeric G proteins-mediated signal transduction mechanisms in SPZEJ (Suppl. Fig. S6).
Discussion
The present study reports for the first time to our knowledge the molecular signature of spermiogenesis in a teleost fish. By combining flow cytometry and cell sorting with transcriptomics, we were able to identify key candidate genes and functional pathways that may be essential to sperm function and early embryo development. Our data also support the view that the teleost sperm transcriptome is a complex and heterogeneous network of coding and non-coding RNA molecules as previously noted in the human sperm14.
The role of lncRNAs on mammalian spermatogenesis has been predicted, but functional studies exploring their mechanism of action and relevance to sperm function are lacking41. In the human spermatozoon the lncRNA cargo of the is of about 7521 sequences14, thus close to the number of lncRNAs found in seabream SPZEJ in our study (8560). In human, most of the lncRNAs detected in the spermatozoon were categorized as antisense and long intervening/intergenic noncoding RNAs (lincRNAs), which frequently regulate gene expression in cis14,42. The lncRNAs cargo of human and seabream spermatozoa is possibly understimated since the workflow employed in both studies allowed the inclusion of coding and non-coding transcripts with a selection of poly(A) RNAs, which enriched the RNA-seq libraries in polyadenylated antisense lncRNAs43. In mammals, many targets of spermatozoon lncRNAs have been predicted44, and found to be enriched in apoptosis (e.g., PI3K-AKT, p53)45, capacitation-related pathways (e.g., Ca2+, cAMP, and MAPK signaling)46, motility47–49, and first stages of embryo development14. In teleosts, such information is still lacking, but our present findings show that the number of de novo emerging lncRNAs in mature seabream sperm is much higher than that of mRNAs. This observation resembles that reported for mature bull sperm, where the number of differentially expressed lncRNAs is also much greater than that of mRNAs49. It will therefore be interesting to investigate in the future whether such large number of lncRNAs in the seabream mature spermatozoon reflects different roles of these molecules in motility and early embryo development as suggested for mammals.
The transcriptomic analysis carried out here identified many DEGs between HGC and SPZEJ, indicating that substantial changes in gene expression occurred during the transition between these two germ cell stages. The analysis however also revealed a higher heterogenicity in gene expression in SPZEJ than in HGC, suggesting that sperm cells at different stage of maturation may be present in the first population, thereby resulting in different cohorts of motile spermatozoa. In fact, the motility and progressivity of SPZEJ from different males at 5 s postactivation ranged from 88 to 98% and from 16 to 51%, respectively, while the time during which spermatozoa remained motile ranged from 5 to 8 min, which could be the consequence of differences in gene expression. These observations thus raise the possibility that specific transcripts are directly related to sperm motility in teleosts, and may be used as potential targets in fertility biomarkers as in mammals41.
In SPZEJ, different genes involved in the cytoskeleton and ion and water transport were upregulated, which can be expected since sperm cells need to differentiate the flagellum and an intracellular signaling machinery in order to activate and maintain motility in the aquatic environment. The upregulated cystokeletal genes were mostly related to actin remodeling, such as marcks and other actin binding genes, and components of the kinesin motor complex, such as kif20a and sybu, which may be involved in the maturation of spermatozoa50. However, many components of the axoneme, which are likely necessary for flagellar function in piscine spermatozoa as in mammals51–54, such as a number of cilia- and flagella-associated proteins (cfap43, -57, -61, -70), sperm flagellar proteins (spef1 and -2) and different dynein motor proteins, except dynein heavy chain 12 (dnah12) and cytoplasmic dynein 1 intermediate chain 2 (dync1i2), were downregulated in SPZEJ. This observation suggests that these transcripts might be transiently upregulated during the differentiation and/or maturation of spermatozoa in the extratesticular ducts (ETDs) prior to ejaculation, although this needs further confirmation. Regarding membrane channels, aqp7 was the most highly upregulated in SPZEJ, which also accumulated transcripts encoding aqp3a and -11, suggesting that water and/or uncharged solute transport may be important during sperm formation. The ion channels and exchangers upregulated include the transient receptor potential cation channel subfamily V member 1 (trpv1), different potassium voltage-gated ion channels (kcnc4, kcng4, kcnh1, kcnk6) and the sodium potassium calcium exchanger 2 (slc24a2), which may be involved in the activation and maintenance of sperm motility55,56 or in the late stages of spermatogenesis57. Other ion channels potentially implicated in cell volume regulation upon activation of sperm in the hyperosmotic seawater, such as voltage-dependent anion-selective channel protein 1 (vdac1), volume-regulated anion channel subunit Lrrc8d (lrrc8d) and the mechanosensitive ion channel Piezo 1 (piezo1)58,59, were also highly accumulated in SPZEJ.
An important process during the differentiation of spermatozoa is to prepare the cells to become fertilization competent, and therefore genes involved in sperm-egg interaction are likely to be activated. Accordingly, different genes encoding for cell adhesion proteins potentially involved in this mechanism were upregulated in SPZEJ, such as several C-type lectins and cell surface glycoproteins (cd209c, cd209e, cd200r1a) and immunoglobulin receptors (pigr, fcgr2a and -3, ildr1). Interestingly, we also found two transcripts with sequence homology to zona pellucida (ZP) proteins highly upregulated in SPZEJ (log2 fold change > 9), one that seems to be the ortholog of mammalian ZP3 (zp3; XM_030443218.1) and another distantly related to ZP2 (zp2; AY928799.1). The expression of ZP proteins in mammals and fish has been originally thought to be ovary specific, where they are synthesized by the liver under estrogenic regulation and/or in the oocyte, and further deposited in the vitelline envelope separating the oocyte from the somatic cells which will form the chorion60,61. Recent studies, however, have found the expression of ZP3 proteins in spermatogonia, spermatocytes and round and elongated spermatids, but not in spermatozoa, in both human and mouse62. Therefore, our findings are not completely surprising, but they may reflect the presence of remnant mRNAs from the processes of spermatozoa differentiation rather than mRNAs playing a role during sperm-egg recognition. In any event, further studies are necessary to confirm the presence of ZP proteins in fish spermatozoa and elucidate their potential function.
An intriguing observation of the present study was the high number of upregulated transcripts related to transcription and translation found in SPZEJ. Many of these mRNAs encode for transcription factors, transcriptional and translational regulators, and ribosomal proteins, including seven different mitochondrial ribosomes. Vertebrate spermatozoa are believed to be trancriptionally and translationally silent as a result of the degradation of ribosomal RNAs and the gradual replacement of the histones of the DNA-packing nucleosomes by protamines during the spermiogenic differentiation phase63,64. The many types of RNAs still present in the sperm are thus thought to be the remnants of the high transcriptional activity of the spermatocytic and spermatidogenic phases63, or delivered via epididymal extracellular microvesicles, called exosomes or epididysomes65. However, some studies suggest that mitochondrial ribosomal pathways, rather than the canonical cytoplasmic mechanisms, remain active in the spermatozoon and yield paternal factors that are important for sperm maturation, capacitation in the female reproductive tract, fertility, and early zygotic development66–69. As in several lineages of anamniotes70–72, the seabream spermatozoa are devoid of protamines34, which agrees with the failure to detect protamine-encoding mRNAs in our transcriptome. This observation, together with the high accumulation of transcription and translation related genes in seabream SPZEJ, raises the question of whether transcriptional quiescence is a general feature of post-meiotic sperm maturation in teleosts. However, fish also produce exosomes in the ETDs as mammals73, and therefore it is also plausible that these spermatozoon mRNAs, as well as the lncRNAs, are originated from exosomes released from cells lining the ETDs for further function in early embryogenesis. Future studies should investigate the chromatin architecture reorganization and epigenetic marks in seabream spermatozoa that might allow transcription and translation during the maturation phase in the ETDs.
Previous studies in teleosts have shown that the energy-supplying pathways in spermatozoa include glycolysis, phospholipid catabolism and triglyceride metabolism, the TCA cycle, and OXPHOS, the latter two mechanisms being the key metabolic pathways that sustain basal metabolism52,74. Studies in the seabream suggest that lipids are the major substrate for ATP/energy production via the mitochondrial TCA cycle, while cytosolic glycolysis and carbohydrate metabolism seems to have a lesser contribution75. In contrast, upon motility activation OXPHOS seems to be the main ATP source for flagellar movement, which achieves a balance between energy production and consumption76,77. According to this view our interactome analysis showed that enzymes involved in gluconeogenesis were downregulated in SPZEJ, while those playing distinct roles in the TCA cycle and OXPHOS, as well as in triglyceride synthesis and lipid catabolism, were in general upregulated. However, we also found that many glycolytic enzymes were upregulated in SPZEJ, including lactate dehydrogenase (ldha and ldhb) involved in the conversion of pyruvate to lactate. This observation thus suggests that glycolysis may be an important metabolic pathway during the maturation of spermatozoa in the ETDs since this pathway is more effective under anaerobic than aerobic conditions.
Finally, the in silico analysis of DEGs in SPZEJ revealed the enrichment of different signaling pathways, amongst which the cytokine/chemokine, GnRHR and PDGF pathways appear the most predominant. Evidence for the existence of chemotaxis in fish, guiding the spermatozoa to the vicinity of the egg and even orienting the direction of their swimming path toward the egg micropyle, is increasing74,78–81, and therefore our observation of the presence of almost all components of the cytokine/chemokine signaling pathway in SPZEJ is consistent with these findings. The expression of the GnRHR and PDGF pathways in spermatozoa, including mRNAs encoding Gnrh and Pdgf cognate receptors, is however more compelling. Previous studies in teleosts have shown that administration of some hormones, such as progestins, androgens and gonadotropins, can increase the seminal plasma pH in the ETDs, which results in the elevation of intra-sperm cAMP levels, increase hydration, or induce the secretion of sperm-immobilizing ions by the ETD epithelium2,82. However, the cellular sources of these hormones in the ETDs and their potential signal transducing effects in the maturing spermatozoa are completely unknown. Nevertheless, it can be speculated that the expression of the GnRHR and PDGF pathways in seabream spermatozoa may reflect a prior function of these factors as paracrine signals in the ETDs for inducing the maturation and acquisition of full motility of spermatozoa, which might involve the activation of transcription and/or translation of specific genes as discussed above. This challenging hypothesis is not yet proven and merits further investigation.
In summary, the transcriptome dynamics during spermiogenesis described for the first time here provide important insights into the molecular mechanisms underlying sperm differentiation and maturation in non-mammalian vertebrates. Our data uncovered a number of candidate genes, lncRNAs and novel endocrine pathways that may play important roles for the acquisition of the spermatozoon motility and during early embryo development of teleosts. Further studies will be necessary to dissect out the specific functions of these genes during the transition of haploid cells to spermatozoa, as well as during the subsequent maturation in the extratesticular tract.
Methods
Animals and sample collection
Adult gilthead seabream males were raised in captivity at the Institut de Recerca i Tecnologia Agroalimentàries (IRTA) aquaculture facilities in San Carlos de la Rápita (Tarragona, Spain) and maintained in the laboratory as described previously38. Samples of testis and SPZEJ were obtained from males during the natural reproductive season (November-February). The sperm was collected from males sedated with 500 ppm of phenoxyethanol (Merck) by the application of a soft pressure to the abdominal area and removal of sperm from the gonopore with a syringe, while the testes were collected from anaesthezized fish and euthanized by decapitation. Procedures relating to the care and use of animals and sample collection were approved by the Ethics Committee (EC) of Institut de Recerca i Tecnologia Agroalimentàries (IRTA), following the International Guiding Principles for Research Involving Animals (EU 2010/63), and in accordance with ARRIVE guidelines (https://arriveguidelines.org).
Cell cytometry
To isolate HGC by FACS, testicular biopsies were cut into small pieces of ~ 1 g and treated with 0.2% collagenase (Merck type 1A) for 1 h under agitation in non-activating medium (NAM; in mg/ml: 3.5 NaCl, 0.11 KCl, 1.23 MgCl2, 0.39 CaCl2, 1.68 NaHCO3, 0.08 glucose, 1 bovine serum albumine [BSA], pH 7.7; 280 mOsm) (51) supplemented with 200 μg/mL penicillin/streptomycin (Life Technologies Corp.). Samples were centrifuged at 200× g for 1 min to remove cell aggregates, and the supernatant centrifuged again at 400× g for 1 min to enrich in haploid cells. The cells were centrifuged at 400× g for 5 min and the pellet resuspended in 1 ml NAM. The concentration of cells was determined by light microscopy and the ISASv1 software (Proiser), and this was adjusted to 150 × 106 cells/ml. Cells were then stained with 200 nM of a solution of SYBR Green I (SGI) fluorescent nucleic acid stain (Molecular Probes, Life Technologies Corp.) for 45–60 min in the dark at room temperature, just prior to flow cytometry.
FACS was performed with a MoFlo XDP cell sorter (Beckman Coulter) equipped with three lasers (blue solid state of 488 nm, red diode of 635 nm, and argon ion UV laser of 351 nm). Sterilized PBS served as the sheath fluid. The sorter was set in 4-way purify sort mode and with a flow sorting rate of ~ 1500 events/s. The sorted population of HGC was collected in 4 ml of NAM in 15 ml tubes and centrifuged at 200× g for 15 min. The resulting pellet was resuspended in 100 μl of NAM to obtain aliquots of 3 to 5 × 106 cells, which were centrifuged again at 200× g and frozen in liquid nitrogen and stored at − 80 °C.
Immunofluorescence microscopy
Sorted germ cells and SPZEJ were processed as described previously7,38 and attached to UltraStick/UltraFrost Adhesion slides (Electron Microscopy Sciences). Samples were fixed in 4% PFA in PBS for 15 min before antigen retrieval in three consecutive 5-min incubations with boiling citrate (10 mM at pH 6), followed by triton X-100 (0.2% in PBS) for 15 min. After blocking for one hour in PBST with 5% normal goat serum (Merck G9023) and 0.1% BSA, antibodies were applied overnight at 4 °C in a humidified chamber. The primary antibodies were α-tubulin (Merck T9026; 1:1000), H3K9ac (Abcam ab4441; 1:1000), and Spo11 (Santa Cruz Biotechnology sc-33146; 1:1000). Anti-mouse or anti-rabbit IgG coupled with Alexa-555 (Invitrogen A-21422 and Merck AP510C, respectively) were applied for 1 h at room temperature and cells were counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Merck G8294; 1:3000) before mounting with Fluoromount™.
Evaluation of sperm motility by CASA
The percentage of motile and progressive spermatozoa, as well as the time during which the sperm remained motile, was determined by CASA using the Integrated Semen Analysis System (ISASv1, Proiser) software as previously described83.
RNA extraction, library construction, and sequencing
Total RNA from HGC (3 × 107 cells) and SPZEJ (3–30 × 107 cells) was extracted with the RNeasy Plus Mini Kit (Qiagen). The full-spectrum UV–Vis spectro-photometer NanoDropVC 2000 (Thermo Fisher Scientific) was used to determine the purity and concentration of the extracted RNA by measuring their 260/280 nm absorbance ratio. RNA size distribution profiles were analyzed using the Agilent 2100 Bioanalyzer (Agilent Technologies). The RIN values ranged from 8.4 to 7.5 in HGC, whereas these values ranged from 1 to 3.2 in SPZEJ. The absence of peaks corresponding to 28S and 18S rRNAs in SPZEJ was confirmed to verify the absence of non-sperm cells in these samples.
Four unstranded RNA libraries (replicates) for low-input RNA were constructed for each of the HGC and SPZEJ groups; each replicate being a pool of cells collected from three different males. The libraries from the total RNA were prepared following the SMARTseq2 protocol for low-input RNA84 with some modifications. Briefly, reverse transcription with 2 ng RNA was performed using SuperScript II (Invitrogen) in the presence of oligo-dT30VN (1 µM; 5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′), template-switching oligonucleotides (1 µM) and betaine (1 M). The cDNA was amplified using the KAPA Hifi Hotstart ReadyMix (Merck), 100 nM ISPCR primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′) and 12 cycles of amplification. Following purification with Agencourt Ampure XP beads (1:1 ratio; Beckmann Coulter), product size distribution and quantity were assessed on a Bioanalyzer High Sensitvity DNA Kit (Agilent). The amplified cDNA (200 ng) was fragmented for 10 min at 55 °C using Nextera® XT (Illumina) and amplified for 12 cycles with indexed Nextera® PCR primers. The library was purified twice with Agencourt Ampure XP beads (0.8:1 ratio) and quantified on a Bioanalyzer using a High Sensitvity DNA Kit.
The libraries were sequenced on HiSeq2500 (Illumina) in paired-end mode with a read length of 2 × 76 bp using TruSeq SBS Kit v4. We generated more than 30 million paired-end reads for each sample in a fraction of a sequencing v4 flow cell lane, following the manufacturer’s protocol. Image analysis, base calling and quality scoring of the run were processed using the manufacturer’s software Real Time Analysis (RTA 1.18.66.3) and followed by generation of FASTQ sequence files by CASAVA 1.8.
Genome annotation
To improve the gilthead seabream reference genome37 annotation for the differential expression analysis, the genome was reannotated, and a de novo transcriptome assembly was generated from which those transcripts not present in the genome assembly were added to the analysis.
Genome reannotation
Repeats present in the seabream genome assembly were annotated with RepeatMasker v4-0-7 (http://www.repeatmasker.org) using the zebrafish repeat library included in RepeatMasker. The gene annotation was obtained by combining transcript alignments, protein alignments and ab initio gene predictions. First, the RNA-seq reads were aligned to the genome with STAR v-2.5.3a85. Subsequently, transcript models were generated using Stringtie v1.0.486 and PASA assemblies were produced with PASA v2.0.287 by adding also the 114,155 S. aurata ESTs present in NCBI (October 2017). Secondly, the complete Actinopterygii proteomes were downloaded from Uniprot in October 2017 and aligned to the genome using Spaln v2.4.788. Ab initio gene predictions were performed on the repeat masked assembly with three different programs: GeneID v1.489, Augustus v3.2.390 and Genemark-ES v2.3e91 with and without incorporating evidence from the RNA-seq data. The gene predictors were run with trained parameters for human except Genemark that runs on a self-trained manner. Finally, all the data was combined into consensus CDS models using EvidenceModeler-1.1.187. Additionally, UTRs and alternative splicing forms were annotated through two rounds of PASA annotation updates. Functional annotation was performed on the annotated proteins with Blast2go92, using Blastp93 search against the nr database (March 2018) and Interproscan94 to detect protein domains on the annotated proteins.
The annotation of non-coding RNAs was carried out using the following steps. First, the program cmsearch v1.195 included in the Infernal software96 was run against the RFAM v12.0 database of RNA families96. The tRNAscan-SE v1.2397 was also run to detect the transfer RNA genes present in the genome assembly. To detect the lncRNAs we selected those PASA-assemblies that had not been included into the annotation of protein-coding genes in order to get all those expressed genes that were not translated into a protein. Finally, those PASA-assemblies without protein-coding gene annotation that were longer than 200 bp and whose length was not covered at least in an 80% by a small ncRNA were incorporated into the ncRNA annotation as lncRNAs. The resulting transcripts were clustered into genes using shared splice sites or significant sequence overlap as criteria for designation as the same gene.
Complementing the annotation with de novo assembled transcripts
The RNA-seq reads were assembled with Trinity v2.2.098 allowing for trimming and normalization of the reads. Next, Rapclust v0.199 was run, in which the process of pseudoalignment was first performed with Sailfish v0.10.0100, and then Rapclust was used to cluster the assembled sequences into contained isoforms in order to reduce redundancy and to cluster together all the isoforms that are likely to belong to the same gene. For evaluation of the resulting transcriptomes we estimated their completeness with BUSCO v3.0.2101 using an Actinopterygii specific dataset of 4584 genes. After obtaining the reference transcriptome, open reading frames (ORFs) were annotated in the assembled transcripts with Transdecoder102 and functional annotation was performed on the annotated proteins with Blast2GO, as described above. Finally, the assembled transcripts were mapped against the seabream reference genome assembly with GMAP103. Those transcripts for which less than 50% of their length aligned to the genome, and with a complete ORF and functional annotation, were added to the reference genome as separate annotated contigs.
Differential expression analysis
RNA-seq reads were mapped against the improved version of the seabream reference genome with STAR v2.5.3a using ENCODE parameters for long RNAs. Genes were quantified with RSEM v1.3.0104 using the improved annotation. Sample similarities were inspected with a PCA using the top 500 most variable genes and the 'rlog' transformation of the counts. Differential expression analysis was performed with DESeq2 v1.18105 with default options, and genes with a false discovery rate (FDR) < 1% were considered significant. Heatmaps with the ‘rlog’ transformed counts of the DEGs were plotted with the ‘pheatmap’ R v1.0.12 package available at the Rstudio v1.2.1335 (http://www.rstudio.com/). Venn diagrams and volcano plots were performed with the ‘VenDiagramm’ v1.6.20 R package and ‘ggplot2’ v3.1.1 R package, respectively, from Rstudio v1.2.1335.
Gene classification, ontology, and pathway analysis of DEGs
The GO enrichment of DEGs and signaling pathway analyses were performed using the PANTHER v14.1 Classification System and analysis tools (http://www.pantherdb.org/). GO terms and pathways with FDR < 0.05% were considered significant. Scatter plots of pathway analyses were carried out with ‘ggplot2’ R package. Functional category classifications were also done manually using the Uniprot database (https://www.uniprot.org/) and QuickGO browser (http://www.ebi.ac.uk/QuickGO). Interactome analyses were conducted using the STRING database v11.0b39 with a high-confidence interaction score (0.9), and plots were performed using Cytoscape v3.8.2 (https://cytoscape.org/). In some cases, selected transcripts were mapped using the KEGG pathway database106 (https://www.genome.jp/kegg/pathway.html) and WikiPathways (https://www.wikipathways.org).
Validation of gene expression by qRT-PCR
The qRT-PCR were carried out as described previously7,38, except that in this case the cDNA was synthesized from 13 to 20 ng of total RNA using the AccuScript High-Fidelity 1st Strand cDNA Synthesis Kit (Agilent 200,820) following the manufacturer’s instructions. For qRT-PCR, relative gene expression levels with respect to HGC were determined by the 2−ΔΔCt method, using glutathione-specific gamma-glutamylcyclotransferase 1 (chac1) and beta-actin (bactin) as reference genes. The analyses were done on three cDNAs synthesized from three different pools of three animals each using technical duplicates. Primer3 v. 0.4.0 software (https://bioinfo.ut.ee/primer3-0.4.0/) was used for primer design. Primer sequences are listed in Suppl. Table S6.
Supplementary Information
Acknowledgements
This work was supported by the Spanish Ministry of Science and Innovation (MCIN/AEI/10.13039/501100011033) and FEDER “A way of making Europe”, European Union, Grant no. AGL2016-76802-R (to J.C.). F.C. and J.C.A. were supported, respectively, by the “Ramon y Cajal” programe (RYC-2015-17103) and a predoctoral (BES-2017-080778) contract from Spanish MCIN. A.E.C. was funded by ISCIII/MCIN (PT17/0009/0019) and co-funded by FEDER, whereas R.N.F. was supported by the University of Bergen, Norway. We also acknowledge support of the Spanish MICIN through the Instituto de Salud Carlos III and to the EMBL partnership, the Centro de Excelencia Severo Ochoa, the Generalitat de Catalunya through the CERCA Programme, Departament de Salut and Departament d’Empresa i Coneixement, and funds from the European Regional Development Fund (Programa Operatiu FEDER de Catalunya 2014-2020) cofinanced by the Spanish MCIN (Programa Operativo FEDER Plurirregional de España (POPE) 2014-2020).
Abbreviations
- CASA
Computer-assisted sperm analysis
- DEGs
Differentially expressed genes
- ETDs
Extratesticular ducts
- FACS
Fluorescence-activated cell sorting
- GnRHR
Gonadotropin releasing hormone receptor
- GO
Gene ontology
- HGCs
Haploid germ cells
- lncRNA
Long non-coding RNAs
- OXPHOS
Mitochondrial oxidative phosphorylation
- PCA
Principal component analysis
- PDGF
Platelet-derived growth factor
- qRT-PCR
Real-time quantitative reverse transcription PCR
- RIN
RNA integrity number
- SPZEJ
Ejaculated spermatozoa
- SPZI
Intratesticular spermatozoa
- TCA
Tricarboxylic acid cycle
- ZP
Zona pellucida
Author contributions
J.C.: conceptualization. J.G.G., T.A., A.E.C., M.D., J.C.: methodology. J.C.-A., F.C.: investigation. A.E.C., M.D., J.C.-A., J.C.: formal analysis. J.C.-A., R.N.F., J.C.: visualization. J.C.: supervision and funding acquisition. J.C.-A.: writing original draft. T.A., R.N.F., J.C.: writing, review and editing. All authors read and approved the submitted version of the manuscript.
Data availability
The RNA-seq datasets generated in this study have been submitted to Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under accession no. GSE173088. Reannotation data from the seabream genome are available at https://denovo.cnag.cat/Saurata.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-18422-2.
References
- 1.White-Hooper H, Bausek N. Evolution and spermatogenesis. Philos. Trans. R. Soc. B. 2010;365:1465–1480. doi: 10.1098/rstb.2009.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schulz RW, et al. Spermatogenesis in fish. Gen. Comp. Endocrinol. 2010;165:390–411. doi: 10.1016/j.ygcen.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Temple-Smith PD, Ravichandran A, Nunez FEH. (2018) Sperm: comparative vertebrate. In: Skinner MK, editor. Encyclopedia of Reproduction. 2. Academic Press; 2018. pp. 210–220. [Google Scholar]
- 4.Holdcraft RW, Braun RE. Hormonal regulation of spermatogenesis. Int. J. Androl. 2004;27:335–342. doi: 10.1111/j.1365-2605.2004.00502.x. [DOI] [PubMed] [Google Scholar]
- 5.Levavi-Sivan B, Bogerd J, Mañanós EL, Gómez A, Lareyre JJ. Perspectives on fish gonadotropins and their receptors. Gen. Comp. Endocrinol. 2010;165:412–437. doi: 10.1016/j.ygcen.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 6.Rosati L, et al. Spermatogenesis and regulatory factors in the wall lizard Podarcis sicula. Gen. Comp. Endocrinol. 2020;298:113579. doi: 10.1016/j.ygcen.2020.113579. [DOI] [PubMed] [Google Scholar]
- 7.Chauvigné F, Zapater C, Gasol JM, Cerdà J. Germ-line activation of the luteinizing hormone receptor directly drives spermiogenesis in a nonmammalian vertebrate. PNAS. 2014;111:1427–1432. doi: 10.1073/pnas.1317838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feugang JM, et al. Transcriptome analysis of bull spermatozoa: implications for male fertility. Reprod. Biomed. Online. 2010;21:312–324. doi: 10.1016/j.rbmo.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 9.Lalancette C, et al. Identification of human sperm transcripts as candidate markers of male fertility. J. Mol. Med. 2009;87:735–748. doi: 10.1007/s00109-009-0485-9. [DOI] [PubMed] [Google Scholar]
- 10.Gòdia M, et al. A RNA-Seq analysis to describe the boar sperm transcriptome and its seasonal changes. Front. Genet. 2019;10:299. doi: 10.3389/fgene.2019.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alvarez-Rodriguez M, et al. The transcriptome of pig spermatozoa, and its role in fertility. Int. J. Mol. Sci. 2020;21:1572. doi: 10.3390/ijms21051572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Selvaraju S, et al. Deciphering the complexity of sperm transcriptome reveals genes governing functional membrane and acrosome integrities potentially influence fertility. Cell Tissue Res. 2021;385:207–222. doi: 10.1007/s00441-021-03443-6. [DOI] [PubMed] [Google Scholar]
- 13.Sun YH, et al. Single-molecule long-read sequencing reveals a conserved intact long RNA profile in sperm. Nat. Commun. 2021;12:1361. doi: 10.1038/s41467-021-21524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corral-Vazquez C, et al. The RNA content of human sperm reflects prior events in spermatogenesis and potential post-fertilization effects. Mol. Hum. Reprod. 2021;27:gaab035. doi: 10.1093/molehr/gaab035. [DOI] [PubMed] [Google Scholar]
- 15.Green CD, et al. Comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev. Cell. 2018;46:651–667. doi: 10.1016/j.devcel.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki S, Diaz VD, Hermann BP. What has single-cell RNA-seq taught us about mammalian spermatogenesis? Biol. Reprod. 2019;101:617–634. doi: 10.1093/biolre/ioz088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shami AN, et al. Single-cell RNA sequencing of human, macaque, and mouse testes uncovers conserved and divergent features of mammalian spermatogenesis. Dev. Cell. 2020;54:529–547. doi: 10.1016/j.devcel.2020.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, et al. Comparative analysis of the testis and ovary transcriptomes in zebrafish by combining experimental and computational tools. Comp. Funct. Genomics. 2004;5:403–418. doi: 10.1002/cfg.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rolland AD, et al. Expression profiling of rainbow trout testis development identifies evolutionary conserved genes involved in spermatogenesis. BMC Genomics. 2009;10:546. doi: 10.1186/1471-2164-10-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forné I, et al. Transcriptional and proteomic profiling of flatfish (Solea senegalensis) spermatogenesis. Proteomics. 2011;11:2195–2211. doi: 10.1002/pmic.201000296. [DOI] [PubMed] [Google Scholar]
- 21.Das PJ, et al. Stallion sperm transcriptome comprises functionally coherent coding and regulatory RNAs as revealed by microarray analysis and RNA-seq. PLoS ONE. 2013;8(2):e56535. doi: 10.1371/journal.pone.0056535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun F, et al. Male-biased genes in catfish as revealed by RNA-seq analysis of the testis transcriptome. PLoS ONE. 2013;8(7):e68452. doi: 10.1371/journal.pone.0068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao W, et al. Characterization of gonadal transcriptomes from Nile Tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS ONE. 2013;8(5):e63604. doi: 10.1371/journal.pone.0063604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manousaki T, et al. The sex-specific transcriptome of the hermaphrodite sparid sharpsnout seabream (Diplodus puntazzo) BMC Genomics. 2014;15:655. doi: 10.1186/1471-2164-15-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma E, et al. Transcriptome assemblies for studying sex-biased gene expression in the guppy, Poecilia reticulata. BMC Genomics. 2014;15:400. doi: 10.1186/1471-2164-15-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue H, Li C, Du H, Zhang S, Wei Q. Sequencing and de novo assembly of the gonadal transcriptome of the endangered Chinese sturgeon (Acipenser sinensis) PLoS ONE. 2015;10(6):e0127332. doi: 10.1371/journal.pone.0127332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou YF, et al. Testes transcriptome profiles of the anadromous fish Coilia nasus during the onset of spermatogenesis. Mar. Genomics. 2015;2:241–243. doi: 10.1016/j.margen.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Bar I, Cummins S, Elizur A. Transcriptome analysis reveals differentially expressed genes associated with germ cell and gonad development in the Southern bluefin tuna (Thunnus maccoyii) BMC Genomics. 2016;17:217. doi: 10.1186/s12864-016-2397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, et al. Transcriptome analysis of the gonads of olive flounder (Paralichthys olivaceus) Fish Physiol. Biochem. 2016;42:1581–1594. doi: 10.1007/s10695-016-0242-2. [DOI] [PubMed] [Google Scholar]
- 30.Hu F, et al. Different expression patterns of sperm motility-related genes in testis of diploid and tetraploid cyprinid fish. Biol. Reprod. 2017;96:907–920. doi: 10.1093/biolre/iox010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy A, Basak R, Rai UD. novo sequencing and comparative analysis of testicular transcriptome from different reproductive phases in freshwater spotted snakehead Channa punctatus. PLoS ONE. 2017;12(3):e0173178. doi: 10.1371/journal.pone.0173178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang X, et al. Transcriptome dynamics during turbot spermatogenesis predicting the potential key genes regulating male germ cell proliferation and maturation. Sci. Rep. 2018;8:15825. doi: 10.1038/s41598-018-34149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo S, et al. Transcriptome sequencing reveals the traits of spermatogenesis and testicular development in large yellow croaker (Larimichthys crocea) Genes. 2019;10:958. doi: 10.3390/genes10120958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurtz K, Saperas N, Ausió J, Chiva M. Spermiogenic nuclear protein transitions and chromatin condensation: proposal for an ancestral model of nuclear spermiogenesis. J. Exp. Zool. B Mol. Dev. Evol. 2009;312B:149–163. doi: 10.1002/jez.b.21271. [DOI] [PubMed] [Google Scholar]
- 35.Hazzouri M, et al. Regulated hyperacetylation of core histones during mouse spermatogenesis: involvement of histone deacetylases. Eur. J. Cell Biol. 2000;79:950–960. doi: 10.1078/0171-9335-00123. [DOI] [PubMed] [Google Scholar]
- 36.El Fekih S, et al. Sperm RNA preparation for transcriptomic analysis: review of the techniques and personal experience. Andrologia. 2017;49:e12767. doi: 10.1111/and.12767. [DOI] [PubMed] [Google Scholar]
- 37.Pauletto M, et al. Genomic analysis of Sparus aurata reveals the evolutionary dynamics of sex-biased genes in a sequential hermaphrodite fish. Commun. Biol. 2018;1:119. doi: 10.1038/s42003-018-0122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chauvigné F, Boj M, Vilella S, Finn RN, Cerdà J. Subcellular localization of selectively permeable aquaporins in the male germ line of a marine teleost reveals spatial redistribution in activated spermatozoa. Bio. Reprod. 2013;89(2):37. doi: 10.1095/biolreprod.113.110783. [DOI] [PubMed] [Google Scholar]
- 39.Szklarczyk D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47:D419–D426. doi: 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahoo B, Choudhary RK, Sharma P, Choudhary S, Gupta MK. Significance and relevance of spermatozoal RNAs to male fertility in livestock. Front. Genet. 2021;12:768196. doi: 10.3389/fgene.2021.768196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016;17:47–62. doi: 10.1038/nrg.2015.10. [DOI] [PubMed] [Google Scholar]
- 44.Gao Y, et al. Analysis of Long Non-Coding RNA and mRNA expression profiling in immature and mature bovine (Bos taurus) testes. Front. Genet. 2019;10:646. doi: 10.3389/fgene.2019.00646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong S, et al. Dysregulation of lncRNA and circRNA expression in mouse testes after exposure to triptolide. Curr. Drug Metab. 2019;20:665–673. doi: 10.2174/1389200220666190729130020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao F, et al. Dysregulation of long noncoding RNAs in mouse testes and spermatozoa after exposure to cadmium. Biochem. Biophys. Res. Commun. 2017;484:8–14. doi: 10.1016/j.bbrc.2017.01.091. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y, et al. Long noncoding RNA expression profile changes associated with dietary energy in the sheep testis during sexual maturation. Sci. Rep. 2017;7(1):5180. doi: 10.1038/s41598-017-05443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, et al. Expression profiles and characteristics of human lncRNA in normal and asthenozoospermia sperm. Biol. Reprod. 2019;100:982–993. doi: 10.1093/biolre/ioy253. [DOI] [PubMed] [Google Scholar]
- 49.Wang X, et al. Integrated analysis of mRNAs and long noncoding RNAs in the semen from Holstein bulls with high and low sperm motility. Sci. Rep. 2019;9(1):2092. doi: 10.1038/s41598-018-38462-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma DD, Wang DH, Yang WX. Kinesins in spermatogenesis. Biol. Reprod. 2017;96:267–276. doi: 10.1095/biolreprod.116.144113. [DOI] [PubMed] [Google Scholar]
- 51.Sironen A, et al. Loss of SPEF2 function in mice results in spermatogenesis defects and primary ciliary dyskinesia. Bio. Reprod. 2011;85:690–701. doi: 10.1095/biolreprod.111.091132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dzyuba V, Cosson J. Motility of fish spermatozoa: from external signaling to flagella response. Reprod. Biol. 2014;14:165–175. doi: 10.1016/j.repbio.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 53.Li L, Feng F, Wang Y, Guo J, Yue W. Mutational effect of human CFAP43 splice-site variant causing multiple morphological abnormalities of the sperm flagella. Andrologia. 2020;52:e13575. doi: 10.1111/and.13575. [DOI] [PubMed] [Google Scholar]
- 54.Wu S, et al. Motor proteins and spermatogenesis. Adv. Exp. Med. Biol. 2021;1288:131–159. doi: 10.1007/978-3-030-77779-1_7. [DOI] [PubMed] [Google Scholar]
- 55.Majhi RK, et al. Thermosensitive ion channel TRPV1 is endogenously expressed in the sperm of a fresh water teleost fish (Labeo rohita) and regulates sperm motility. Channels (Austin) 2013;7:483–492. doi: 10.4161/chan.25793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y, et al. Sperm motility modulated by Trpv1 regulates zebrafish fertilization. Theriogenology. 2020;151:41–51. doi: 10.1016/j.theriogenology.2020.03.032. [DOI] [PubMed] [Google Scholar]
- 57.Regnier G, et al. Targeted deletion of the Kv6.4 subunit causes male sterility due to disturbed spermiogenesis. Reprod. Fertil. Dev. 2017;29:1567–1575. doi: 10.1071/RD16075. [DOI] [PubMed] [Google Scholar]
- 58.Triphan X, et al. Localisation and function of voltage-dependent anion channels (VDAC) in bovine spermatozoa. Pflugers Arch. 2008;455:677–686. doi: 10.1007/s00424-007-0316-1. [DOI] [PubMed] [Google Scholar]
- 59.Jentsch TJ. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016;17:293–307. doi: 10.1038/nrm.2016.29. [DOI] [PubMed] [Google Scholar]
- 60.Modig C, Raldúa D, Cerdà J, Olsson PE. Analysis of vitelline envelope synthesis and composition during early oocyte development in gilthead seabream (Sparus aurata) Mol. Reprod. Dev. 2008;75:1351–1360. doi: 10.1002/mrd.20876. [DOI] [PubMed] [Google Scholar]
- 61.Moros-Nicolás C, et al. New insights into the mammalian egg zona pellucida. Int. J. Mol. Sci. 2021;22:3276. doi: 10.3390/ijms22063276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pulawska K, et al. Novel expression of zona pellucida 3 protein in normal testis; potential functional implications. Mol. Cell. Endocrinol. 2022;539:111502. doi: 10.1016/j.mce.2021.111502. [DOI] [PubMed] [Google Scholar]
- 63.Ren X, Chen X, Wang Z, Wang D. Is transcription in sperm stationary or dynamic? J. Reprod. Dev. 2017;63:439–443. doi: 10.1262/jrd.2016-093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rathke C, Baarends WM, Awe S, Renkawitz-Pohl R. Chromatin dynamics during spermiogenesis. Biochim. Biophys. Acta. 2014;1839:155–168. doi: 10.1016/j.bbagrm.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 65.James ER, et al. The role of the epididymis and the contribution of epididymosomes to mammalian reproduction. Int. J. Mol. Sci. 2020;21:5377. doi: 10.3390/ijms21155377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gur Y, Breitbart H. Mammalian sperm translate nuclear-encoded proteins by mitochondrial-type ribosomes. Genes Dev. 2006;20:411–416. doi: 10.1101/gad.367606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao C, et al. Role of translation by mitochondrial-type ribosomes during sperm capacitation: an analysis based on a proteomic approach. Proteomics. 2009;9:1385–1399. doi: 10.1002/pmic.200800353. [DOI] [PubMed] [Google Scholar]
- 68.Rajamanickam GD, Kastelic JP, Thundathil JC. Content of testis-specific isoform of Na/K-ATPase (ATP1A4) is increased during bovine sperm capacitation through translation in mitochondrial ribosomes. Cell Tissue Res. 2017;368:187–200. doi: 10.1007/s00441-016-2514-7. [DOI] [PubMed] [Google Scholar]
- 69.Zhu Z, et al. Gene expression and protein synthesis in mitochondria enhance the duration of high-speed linear motility in boar sperm. Front. Physiol. 2019;10:252. doi: 10.3389/fphys.2019.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shimizu Y, Mita K, Tamura M, Onitake K, Yamashita M. Requirement of protamine for maintaining nuclear condensation of medaka (Oryzias latipes) spermatozoa shed into water but not for promoting nuclear condensation during spermatogenesis. Int. J. Dev. Biol. 2000;44:195–199. [PubMed] [Google Scholar]
- 71.Wu SF, Zhang H, Cairns BR. Genes for embryo development are packaged in blocks of multivalent chromatin in zebrafish sperm. Genome Res. 2011;21:578–589. doi: 10.1101/gr.113167.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wike CL, et al. Chromatin architecture transitions from zebrafish sperm through early embryogenesis. Genome Res. 2021;31:1–14. doi: 10.1101/gr.269860.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang B, et al. Seminal plasma exosomes: promising biomarkers for identification of male and pseudo-males in Cynoglossus semilaevis. Mar. Biotechnol. (NY) 2019;21:310–319. doi: 10.1007/s10126-019-09881-2. [DOI] [PubMed] [Google Scholar]
- 74.Dzyuba B, Bondarenko O, Gazo I, Prokopchuk G, Cosson J. Energetics of fish spermatozoa: the proven and the possible. Aquaculture. 2017;472:60–72. doi: 10.1016/j.aquaculture.2016.05.038. [DOI] [Google Scholar]
- 75.Lahnsteiner F, Mansour N, Caberlotto S. Composition and metabolism of carbohydrates and lipids in Sparus aurata semen and its relation to viability expressed as sperm motility when activated. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2010;157:39–45. doi: 10.1016/j.cbpb.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 76.Lahnsteiner F, Caberlotto S. Motility of gilthead seabream Sparus aurata spermatozoa and its relation to temperature, energymetabolism and oxidative stress. Aquaculture. 2012;370–371:76–83. doi: 10.1016/j.aquaculture.2012.09.034. [DOI] [Google Scholar]
- 77.Chauvigné F, Boj M, Finn RN, Cerdà J. Mitochondrial aquaporin-8-mediated hydrogen peroxide transport is essential for teleost spermatozoon motility. Sci. Rep. 2015;5:7789. doi: 10.1038/srep07789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yanagimachi R, et al. Chemical and physical guidance of fish spermatozoa into the egg through the micropyle. Biol. Reprod. 2017;96:780–799. doi: 10.1093/biolre/iox015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kholodnyy V, Dzyuba B, Gadêlha H, Cosson J, Boryshpolets S. Egg-sperm interaction in sturgeon: role of ovarian fluid. Fish Physiol. Biochem. 2021;47:653–669. doi: 10.1007/s10695-020-00852-2. [DOI] [PubMed] [Google Scholar]
- 80.Kholodnyy V, et al. Does the rainbow trout ovarian fluid promote the spermatozoon on its way to the egg? Int. J. Mol. Sci. 2021;22:9519. doi: 10.3390/ijms22179519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Devigili A, Cattelan S, Gasparini C. Sperm accumulation induced by the female reproductive fluid: putative evidence of chemoattraction using a new tool. Cells. 2021;10:2472. doi: 10.3390/cells10092472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marshall WS, Bryson SE, Idler DR. Gonadotropin action on brook trout sperm duct epithelium: ion transport stimulation mediated by cAMP and Ca2+ Gen. Comp. Endocrinol. 1993;90:232–242. doi: 10.1006/gcen.1993.1078. [DOI] [PubMed] [Google Scholar]
- 83.Chauvigné F, et al. A multiplier peroxiporin signal transduction pathway powers piscine spermatozoa. PNAS. 2021;18:e2019346118. doi: 10.1073/pnas.2019346118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 85.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pertea M, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haas BJ, et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iwata H, Gotoh O. Benchmarking spliced alignment programs including Spaln2, an extended version of Spaln that incorporates additional species-specific features. Nucleic Acids Res. 2012;40:e161. doi: 10.1093/nar/gks708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Parra G, Blanco E, Guigó R. GeneID in Drosophila. Genome Res. 2000;10:511–515. doi: 10.1101/gr.10.4.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006;7:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lomsadze A, Burns PD, Borodovsky M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014;42:e119. doi: 10.1093/nar/gku557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 93.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/s0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 94.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cui X, et al. CMsearch: simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics. 2016;32:i332–i340. doi: 10.1093/bioinformatics/btw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nawrocki EP, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43:D130–D137. doi: 10.1093/nar/gku1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019;1962:1–14. doi: 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 102.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 104.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq datasets generated in this study have been submitted to Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under accession no. GSE173088. Reannotation data from the seabream genome are available at https://denovo.cnag.cat/Saurata.