

Abstract
Objectives
Elevated plasma levels of sCD14 predict all-cause mortality in people with HIV (PWH). Epigenetic regulation plays a key role in infection and inflammation. To reveal the epigenetic relationships between sCD14, immune function and disease progression among PWH, we conducted an epigenome-wide association study (EWAS) of sCD14 and investigated the relationship with mortality.
Design and Methods
DNA methylation (DNAm) levels of peripheral blood samples from PWH in the Veterans Aging Cohort Study (VACS) were measured using the Illumina Infinium Methylation 450K (n=549) and EPIC (850K) BeadChip (n=526). Adjusted for covariates and multiple testing, we conducted an epigenome-wide discovery, replication, and meta-analysis to identify significant associations with sCD14. We then examined and replicated the relationship between the principal epigenetic sites and survival using Cox regression models.
Findings
We identified 118 DNAm sites significantly associated with sCD14 in the meta-analysis of 1,075 PWH. The principal associated DNAm sites mapped to genes (e.g., STAT1, PARP9, IFITM1, MX1, and IFIT1) related to inflammation and antiviral response. Adjusting for multiple testing, 10 of 118 sCD14-associated DNAm sites significantly predicted survival time conditional on sCD14 levels.
Conclusions
The identification of DNAm sites independently predicting survival may improve our understanding of prognosis and potential therapeutic targets among PWH.
Keywords: Epigenome-wide Association Study (EWAS), DNA-methylation, inflammation, survival, HIV
Introduction
Human Immunodeficiency Virus (HIV) infection is characterized by chronic inflammation and immune activation which persists even in patients who are virologically suppressed by antiretroviral therapy (ART). Biological markers of inflammation have been associated with chronic non-infectious comorbid conditions and increased mortality for people with HIV (PWH)1. Soluble Cluster of Differentiation-14 (sCD14) is an important marker in the pathogenesis of HIV, with many studies linking elevated plasma levels of sCD14 to poor clinical outcomes2.
CD14 is a human protein which is primarily produced by monocytes and macrophages and to a lesser degree by neutrophils. It is the main ligand for lipopolysaccharide (LPS), a pathogen associated molecular pattern found in the outer membrane of gram-negative bacteria3. CD14 exists in two forms; membrane-CD14 anchored to the cell membrane by a glycosylphosphatidylinositol (GPI) tail and sCD14 which appears in plasma after shedding from CD14 or is directly secreted from intracellular vesicles3. The gastrointestinal tract is one of the earliest and primary sites of inflammation and immune activation during HIV infection. Severe depletion of gut CD4+ T-cells and increased production of proinflammatory cytokines and chemokines by CD8+ T-cells can damage the integrity of the gastrointestinal mucosa leading to the translocation of microbial products including LPS into the blood stream.
Elevated plasma levels of sCD14 have been shown to be predictive of all-cause mortality in PWH2 and associated with more rapid progression of disease in primary HIV infection4. In addition, sCD14 is independently associated with impaired neurocognitive testing, coronary calcification, subclinical atherosclerosis, liver, and kidney dysfunction in PWH. The pleiotropic associations of this inflammatory biomarker and others with HIV pathogenesis and organ dysfunction highlight the crucial role of inflammation and immune activation in diseases associated with chronic HIV infection. Identifying genetic factors and epigenetic modifications which determine inter-individual variations of circulating levels of inflammatory markers presents a unique opportunity to inform the mechanisms of disease, identify new treatment targets, and better predict individual patient outcomes. Epigenetic changes have the potential to continuously contribute to the pathophysiology of inflammation in the setting of HIV infection, and in turn, can be modified by inflammatory mediators.
A small number of studies have characterized epigenetic factors associated with levels of circulating inflammatory markers5–7. Epigenome-wide association studies (EWAS) of inflammation markers including C-reactive protein (CRP), interleukin (IL)-1β, IL-4, IL-6, IL-8, and IL-10, interferon (IFN)-γ and tumor necrosis factor (TNF)-α have also been conducted; however, the results were not consistent across studies and may not reflect the epigenetic associations for PWH8,9. Differential DNA methylation (DNAm) has been associated with HIV infection10, diabetes, chronic renal disease, frailty, and mortality for PWH11 providing unique insights into pathogenesis. Despite the many associations between sCD14 and various aspects of HIV disease, little is known about the genetic and environmental determinants of sCD14 in PWH.
The epigenetic profile of leukocytes could capture cumulative risk exposures and physiological responses that mediate the inflammatory process among PWH. An EWAS examines epigenome-wide markers at the population level to scan epigenetic markers associated with a trait12,13. In the present study, we conducted an EWAS of sCD14 levels among PWH, and performed replication and meta-analyses to identify genomic regions, genes, and pathways linked to CD14 through epigenetic modifications and their relationship to survival.
Methods
Study sample
The Veterans Aging Cohort Study (VACS) is a prospective, cohort study of veterans in care at the department of Veterans Affairs Medical Centers (VAMC) across the United States. The aim of this cohort is to study the associations between various diseases and clinical outcomes for veterans with and without HIV infection matched on age, race/ethnicity, and sex and enrolled from eight Veterans Affairs (VA) facilities 10. Clinical and laboratory information was collected from the electronic health record. Total white blood cell counts and CD4+ T-cell subsets were analyzed at the time of peripheral blood sample collection. A sub-cohort was recruited to collect biospecimens for molecular and genetic assays. As previously described, the measure of sCD14 was conducted with an ELISA (Quantikine sCD14 immunoassay, R&D Systems). The sCD14 detectable range was 40–3200 ng/ml, using a standard 200-fold sample dilution and 4 controls14. The phenotypes involved in the analysis included alcohol abuse, hepatitis B status, hepatitis C status, and plasma HIV-1 RNA viral load (VL). Alcohol abuse was categorized as not current, non-hazardous, hazardous, or abuse. Hepatitis B status and hepatitis C status were both defined as positive or negative. VL was dichotomized as ≤75 or >75 copies/ml. The study was approved by the respective Institutional Review Boards and Veterans Affairs Research and Development Committees and for participating sites.
DNA methylation profiling and data processing
The epigenome-wide DNA methylation levels were measured in two mutually exclusive subsets from previously published studies using the Illumina Infinium Methylation 450K and EPIC (850K) BeadChip, respectively15,16. Following the standard protocols, bisulfite-converted DNA samples were whole-genome amplified, enzymatically fragmented, purified and hybridized to the arrays, which were then fluorescently stained, scanned, and assessed for fluorescence intensities. Quality control data normalization and batch correction using control-probe adjustment of the intensity data was performed, as previously described11. We used a quantile normalization approach in the R package “minfi” for processing Methylation 450K and EPIC (850K) data to correct for methylation signals, and to generate adjusted β-values for the associated analyses. After all quality control procedures, a total of 526 samples from the 850K dataset and 549 samples from the 450K dataset were included for the analysis. The DNAm sites measured by the EPIC and 450K chips were mapped to Genome Research Consortium human build 37 (GRCh37).
Statistical Analysis
We processed and analyzed the 450K and 850K datasets separately. After quality control procedures, 412,583 autosomal and 11,232 X chromosomal sites from 450K dataset (n=549), and 812,583 autosomal and 20,232 X chromosomal sites from 850K dataset (n=526) remained for EWAS analysis. We calculated cell type proportions (CD4+ T cells, CD8+ T cells, NK cells, B cells, monocytes, and granulocytes) using cell-type specific DNA methylation in the blood17. These leukocyte proportions were then modeled as covariates to adjust for one of the most important confounders in EWAS. We calculated the inflation factor from the EWAS summary statistics to evaluate the level of global inflation. We calculated the Bonferroni corrected p-value based on the number of tests to adjust for multiple testing.
Replication and Meta-analysis
After conducting EWAS of 450K and 850K data separately, we pursued bi-directional replication of epigenome-wide significant DNAm sites (Bonferroni corrected p-value <0.05) between the two subsets considering the directionality of effect and multiple significance thresholds. The 850K chip builds upon the Infinium HumanMethylation450 (450K) BeadChip with > 90% of the original CpGs plus an additional 350,000 CpGs in enhancer regions18. Because the overlapping content covered by both platforms, the 850K BeadChip can be used to replicate findings from the 450K BeadChip, in addition to novel discovery due to expanded coverage of CpG sites19. Among 366,197 autosomal and X chromosomal DNAm sites examined in both 450K and 850K EWAS of sCD14, we performed inverse-variance weighted fixed-effects meta-analysis. We reported the epigenome-wide significant (Bonferroni p-value <0.05, nominal p-value < 1.40×10−7) DNAm sites considering a total of 366,197 tests and their heterogeneity between two EWAS subsets. We also examined the epigenetic associations with sCD14 stratified by VL (≤75 or >75 copies/ml) for 118 significant DNAm sites using the meta-analysis approach to combine summary statistics from the 450K and 850K subsets. The difference of beta coefficients between the VL categories were assessed using two-sample t-test.
Survival Analysis
Among the 118 significant DNAm sites from the meta-analysis, we conducted survival analysis using a Cox proportional-hazards model to analyze the association between survival time and DNA methylation level, adjusted for, age, BMI, smoking, VL, alcohol abuse, race/ethnicity, HBV, HCV, sCD14, and cell type proportions. We reported the Bonferroni-corrected p-value significant DNAm sites. As a comparison, we also reported results for models without adjustment for sCD14. We performed a sensitivity analysis among treated individuals (N = 781) to assess the impact of HIV treatment on our results.
Pathway Enrichment Analysis
We conducted pathway enrichment analysis based on annotated genes from the significant DNAm sites from meta-analysis results. The analysis was completed using the online tool DAVID Functional Annotation Bioinformatics Microarray Analysis 20.
Results
Table 1 summarizes the baseline statistics and clinical features of the two subsets with DNAm measured by EPIC (n=526) and 450K (n=549) arrays. Most of the study participants were of Black race, had a history of cigarette smoking and uncontrolled viral load in the EPIC and 450K datasets respectively. The levels of sCD14 were similar between the two subsets, 1.79±0.53 and 1.82±0.50 μg/ml in the EPIC and 450K chip subset, respectively. Using two-sample t-test or chi-squared test, the distribution of age, BMI, VL control, and HCV were significantly different between the EPIC chip and the 450K chip subsets.
Table 1.
Characteristics of study samples in the EWAS of EPIC and 450K subsets.
| Variable | EPIC (n=526) N (%) or Mean (SD) | 450K (n=549) N (%) or Mean (SD) |
|---|---|---|
| Race | ||
| White, % | 48 (9.13) | 57 (10.38) |
| Black, % | 423 (80.42) | 464 (84.52) |
| Hispanic, % | 40 (7.60) | 15 (2.73) |
| Other, % | 15 (2.85) | 13 (2.37) |
| Smoking, yes, % | 405 (77.00) | 438 (79.78) |
| Viral Load ≤ 75 copies/ml, % | 156 (29.66) | 124 (22.71) |
| Age (years) | 51.50 (7.75) | 52.68 (7.78) |
| BMI (kg/m2) | 26.19 (5.06) | 25.58 (4.75) |
| Alcohol abuse, % | 230 (43.73) | 240 (43.96) |
| HBV Positive, % | 29 (5.54) | 41 (7.54) |
| HCV Positive, % | 139 (26.43) | 216 (39.34) |
| ART Use, % | 437(83.24) | 461(83.97) |
| sCD14 (μg/ml) | 1.79 (0.53) | 1.82 (0.50) |
Abbreviations: SD-standard deviation; HBV-Hepatitis B; HCV: Hepatitis C virus; sCD14: Soluble Cluster of differentiation 14, BMI- Body Mass Index, ART, ART-Antiretroviral therapy
Adjusting for age, race/ethnicity, BMI, smoking status, alcohol abuse, hepatitis B status, hepatitis C status, VL, and cell type proportions we identified 71 and 4 DNAm sites significantly associated with sCD14 (Bonferroni p-value < 0.05) among PWH, in the subset of EPIC chip (Supplementary Figure 1) and 450K chip (Supplementary Figure 2), respectively. The most significant sCD14-associated DNAm site from the EPIC subset was cg07839457 (−7.99, 95% CI −10.01 to −5.96, p-value 1.22×10−13); however, this site was not measured on the 450K chip. The epigenome-wide significant associations from the EPIC chip analysis were replicated in the 450K chip analysis when the DNAm sites were measured on both platforms (34 out of 71 DNA sites). Supplementary Table 1 summarizes the consistency in directionality of associations (100%) and significance at nominal (88%) and multiple testing corrected threshold (79%). Among four DNAm sites having epigenome-wide significance in the 450K EWAS, all associations with sCD14 had consistent direction in beta coefficients and were significant at nominal or multiple testing corrected threshold (Supplementary Table 1).
A meta-analysis of the two subsets identified 118 DNAm sites (Supplementary Table 2) significantly associated with sCD14 levels (Bonferroni corrected p-value < 0.05, nominal p-value < 1.37×10−7). These sCD14 associated sites were located in 18 autosomes (Figure 1A). No epigenome-wide significant association was identified on the X chromosome. The distribution of p-values from the sCD14 meta-analysis had moderate inflation of 1.16 (Figure 1B). The top DNAm site, cg00676801 (STAT1), was negatively associated with sCD14 levels (p-value = 8.43×10−19) in both EPIC and 450K subsets (Table 2). A total of two DNAm sites in the STAT1 gene region were significantly associated with sCD14 levels. Additionally, IFITM1 harbored 7 DNAm sites while MX1, IRF7 and PSMB8 harbored four sCD14-associated DNAm sites, each. All 118 DNAm sites showed consistent associations with sCD14 levels between two subsets, with strong correlation between beta coefficients (Figure 2, r= 0.93). Notably, 98 out of 118 (83.1%) identified DNAm sites were negatively associated sCD14 levels, indicating that hypomethylation of these DNAm sites link to increased level of sCD14.
Figure 1.
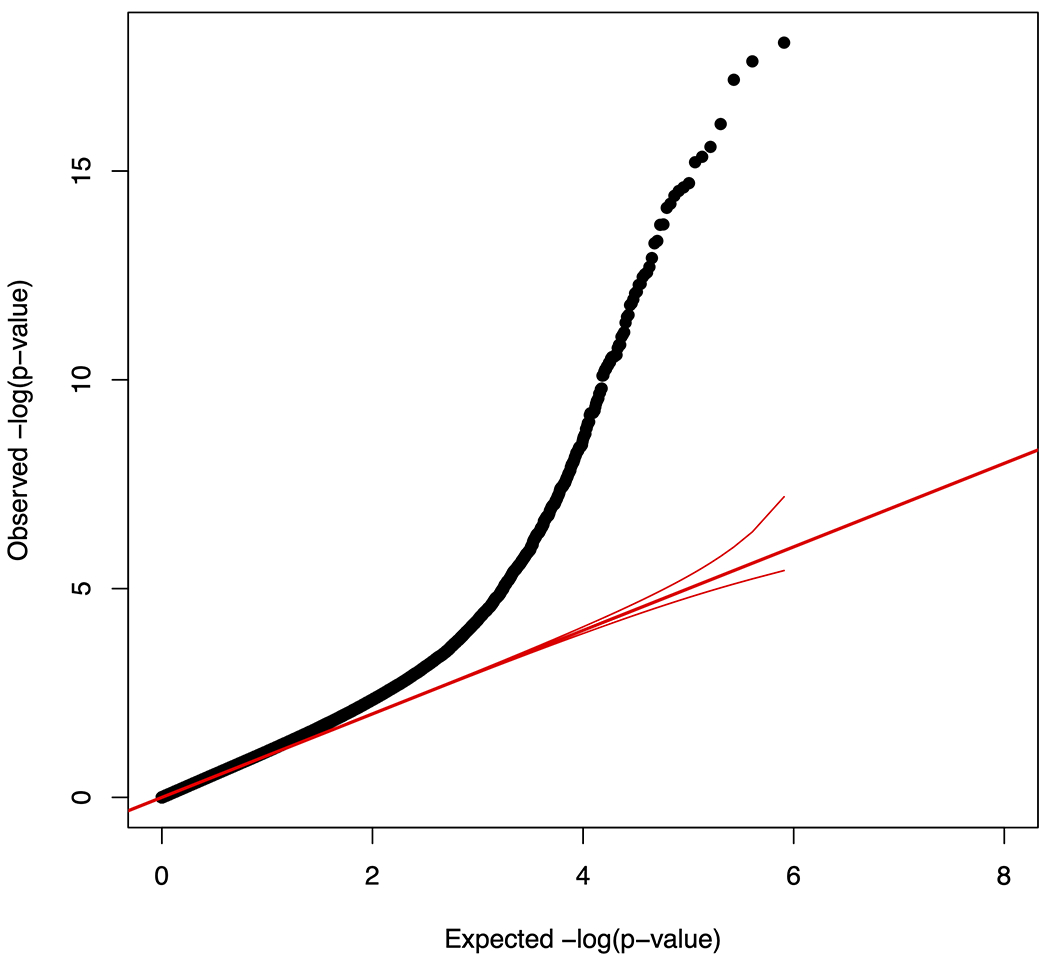
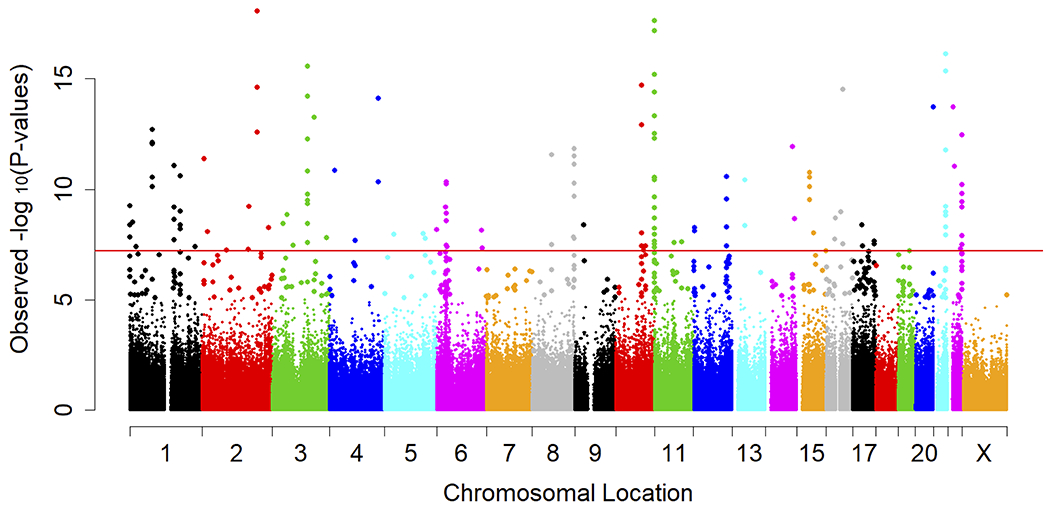
Meta-analysis summary of the sCD14 EWAS. A: Quantile-quantile (Q-Q) plot of 366,197 CpG sites; X axis shows the expected −log10 (p-value) while Y axis shows the observed −log10 (p-value); Red line in the middle is a diagonal line; B: Manhattan plot showing observed −log10 (p-value) of each CpG site, organized by chromosomal positions including the X-chromosome; The horizontal red line represents the genome-wide significant threshold (Bonferroni corrected p-value of 0.05, nominal p-value of 1.40×10−7).
Table 2.
Summary statistics of top 20 sCD14-associated DNAm sites from the meta-analysis of EPIC and 450K EWAS
| Meta-analysis | EPIC (850K) Subset | 450K Subset | Chromosomal Location | Annotated Gene Name | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| CpG ID | BETA | SE | P-value | BETA | SE | P-value | BETA | SE | P-value | Chr | Basepair | |
| cg00676801 | −2.49 | 0.28 | −8.43×10−19 | −3.47 | 0.47 | 7.74×10−13 | −1.93 | 0.35 | 8.23×10−8 | 2 | 191876673 | STAT1 |
| cg03038262 | −4.66 | 0.53 | 2.37×10−18 | −5.43 | 0.76 | 5.22×10−12 | −3.91 | 0.75 | 2.92×10−7 | 11 | 315262 | IFITM1 |
| cg23570810 | −5.38 | 0.62 | 6.54×10−18 | −6.49 | 0.90 | 4.14×10−12 | −4.36 | 0.86 | 6.57×10−7 | 11 | 315102 | IFITM1 |
| cg22862003 | −6.83 | 0.82 | 7.50×10−17 | −8.02 | 1.21 | 1.14×10−10 | −5.81 | 1.12 | 3.17×10−7 | 21 | 42797588 | MX1 |
| cg08122652 | −7.00 | 0.85 | 2.64×10−16 | −8.65 | 1.28 | 6.10×10−11 | −5.67 | 1.14 | 1.15×10−6 | 3 | 122281939 | PARP9; DTX3L |
| cg21549285 | −9.68 | 1.19 | 4.57×10−16 | −11.32 | 1.71 | 1.24×10−10 | −8.12 | 1.67 | 1.63×10−6 | 21 | 42799141 | MX1 |
| cg01971407 | −3.25 | 0.40 | 6.17×10−16 | −3.56 | 0.55 | 2.61×10−10 | −2.88 | 0.59 | 1.67×10−6 | 11 | 313624 | IFITM1 |
| cg05552874 | −5.49 | 0.69 | 1.96×10−15 | −6.53 | 1.02 | 4.50×10−10 | −4.60 | 0.94 | 1.55×10−6 | 10 | 91153143 | IFIT1 |
| cg14951497 | −2.36 | 0.30 | 2.47×10−15 | −3.31 | 0.51 | 3.56×10−10 | −1.87 | 0.37 | 4.98×10−7 | 2 | 191875807 | STAT1 |
| cg07839457 | −5.99 | 0.76 | 3.01×10−15 | −7.99 | 1.03 | 1.22×10−13 | −3.65 | 1.12 | 1.19×10−3 | 16 | 57023022 | NA |
| cg10552523 | −3.95 | 0.50 | 3.92×10−15 | −4.78 | 0.70 | 3.17×10−11 | −3.04 | 0.72 | 3.34×10−5 | 11 | 313478 | IFITM1 |
| cg22930808 | −8.19 | 1.05 | 6.06×10−15 | −9.79 | 1.52 | 3.92×10−10 | −6.73 | 1.45 | 4.90×10−6 | 3 | 122281881 | PARP9; DTX3L |
| cg05883128 | −5.33 | 0.69 | 7.62×10−15 | −6.76 | 1.07 | 8.61×10−10 | −4.34 | 0.89 | 1.69×10−6 | 4 | 169239131 | DDX60 |
| cg01190666 | −2.63 | 0.34 | 1.90×10−14 | −3.25 | 0.49 | 1.16×10−10 | −2.03 | 0.48 | 3.41×10−5 | 20 | 62204908 | HELZ2 |
| cg14293575 | −5.47 | 0.72 | 1.94×10−14 | −6.37 | 1.10 | 1.64×10−8 | −4.82 | 0.94 | 4.66×10−7 | 22 | 18635460 | USP18 |
| cg09026253 | −2.89 | 0.38 | 4.73×10−14 | −3.34 | 0.57 | 1.27×10−8 | −2.53 | 0.52 | 1.47×10−6 | 11 | 313267 | IFITM1 |
| cg06981309 | −5.24 | 0.70 | 5.39×10−14 | −6.44 | 1.02 | 7.85×10−10 | −4.18 | 0.95 | 1.52×10−5 | 3 | 146260954 | PLSCR1 |
| cg06188083 | −5.57 | 0.75 | 1.21×10−13 | −6.38 | 1.08 | 7.62×10−9 | −4.80 | 1.05 | 6.23×10−6 | 10 | 91093005 | IFIT3 |
| cg03607951 | −5.49 | 0.75 | 1.98×10−13 | −6.27 | 1.06 | 9.18×10−9 | −4.74 | 1.05 | 8.45×10−6 | 1 | 79085586 | IFI44L |
| cg03110996 | −2.18 | 0.30 | 2.69×10−13 | −2.93 | 0.49 | 5.08×10−9 | −1.73 | 0.38 | 5.49×10−6 | 2 | 191883483 | NA |
Figure 2.
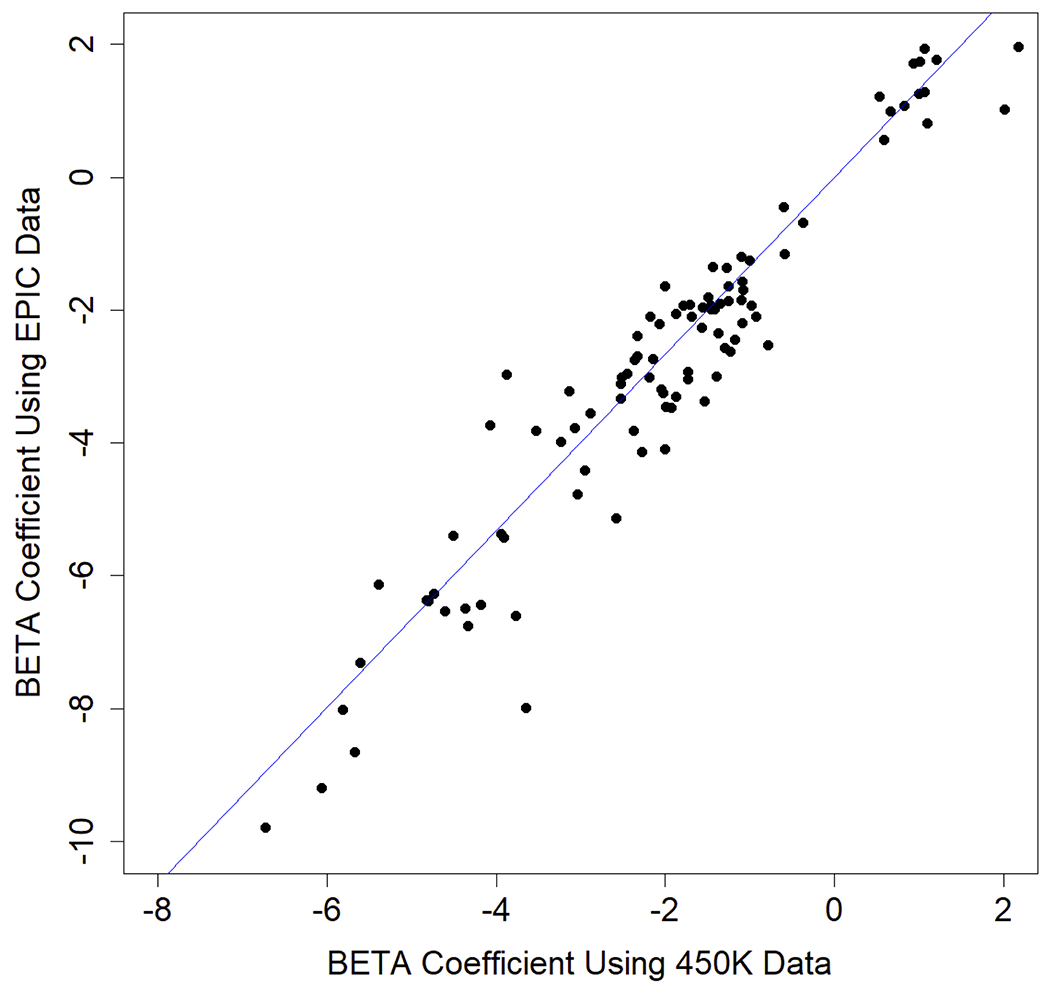
Comparison of significant epigenetic associations from the EPIC vs 450K chip subsets. X axis shows estimated beta coefficients from the 450K chip subsets and Y axis shows estimated beta coefficients from EPIC chip subsets. Blue line in the middle is a diagonal line.
To examine if the VL modifies the epigenetic associations with sCD14, we conducted meta-analysis among PWH with controlled (≤75 copies/ml) and uncontrolled (>75 copies/ml) from 450K and 850K subsets, and compared the beta coefficients of 118 significant DNAm sites (Supplementary Table 3 and 4). Although the beta coefficients between controlled and uncontrolled VL groups were highly correlated (correlation coefficient=0.69, p<2.2×10−16), they were mostly higher in the uncontrolled VL group. After Bonferroni correction of 118 tests, the sCD14 association of 14 DNAm sites were statistically different between two groups (nominal p<0.05/118=4.24×10−4) while 73 DNAm sites had nominal p<0.05. For the sensitivity analysis among treated patients, effect sizes of the top sCD14-associated DNAm sites from the main analysis and the sensitivity analysis (treated only) are almost identical. P-values of the sCD14-associated DNAm sites from the sensitivity analysis were higher than in the main analysis (Supplementary Figure 3)
Using meta-analysis of Cox regression models adjusted for age, BMI, smoking, VL control, alcohol abuse, ethnicity, HBV, HCV, 58 out of the 118 sites were significantly associated with time to mortality after multiple testing correction (nominal p-value<4.2×10−4, Supplementary Table 5). With additional adjustment for sCD14, ten CpG sites remained significant (nominal p-value<5.1×10−4, Table 3). The most significant site cg23560388 (TIAM2) had hazard ratio (HR) of 1.14 (95% CI 1.07-1.21, p-value of 5.06×10−5) per 1% increase of the beta value, which was independent from the sCD14 levels. The HR were 1.17 (95% CI 1.08-1.28) and 1.10 (95% CI 1.00-1.21) in the EPIC and 450K subsets, respectively. Three DNAm sites in the IFITM1 region were significantly associated with mortality in the meta-analysis (Table 3, Supplementary Figure 4). All ten CpG sites had consistent associations with survival time in both EPIC and 450K subsets. Hypermethylation of three CpG sites were associated with increased hazard, while hypomethylation of the other seven sites were associated with increased hazard.
Table 3.
Summary statistics of ten sCD14-associated DNAm sites significantly predicting survival after multiple testing correction.
| CpG ID | Meta-analysis | EPIC (850K) Subset | 450K Subset | Chromosomal Location | Gene Name | ||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| HR (95% CI) | P-value | HR (95% CI) | P-value | HR (95% CI) | P-value | Chr | Base pair | ||
| cg23560388 | 1.14 (1.07, 1.21) | 5.06×10−5 | 1.17 (1.08, 1.28) | 1.95×10−4 | 1.10 (1.00, 1.21) | 0.059 | 6 | 155542639 | TIAM2 |
| cg11849692 | 1.11 (1.05, 1.17) | 1.06×10−4 | 1.10 (1.02, 1.18) | 0.011 | 1.13 (1.04, 1.23) | 0.003 | 10 | 103875969 | LDB1 |
| cg23570810 | 0.97 (0.96, 0.99) | 1.13×10−4 | 0.98 (0.96, 1.00) | 0.035 | 0.97 (0.95, 0.99) | 7.67×10−4 | 11 | 315102 | IFITM1 |
| cg14750551 | 0.96 (0.93, 0.98) | 1.21×10−4 | 0.97 (0.94, 1.00) | 0.069 | 0.94 (0.91, 0.97) | 2.93×10−4 | 3 | 122401343 | PARP14 |
| cg03038262 | 0.97 (0.95, 0.98) | 1.27×10−4 | 0.98 (0.95, 1.00) | 0.036 | 0.96 (0.94, 0.98) | 9.33×10−4 | 11 | 315262 | IFITM1 |
| cg01971407 | 0.96 (0.94, 0.98) | 1.56×10−4 | 0.97 (0.94, 1.01) | 0.11 | 0.95 (0.92, 0.97) | 2.99×10−4 | 11 | 313624 | IFITM1 |
| cg00598235 | 0.94 (0.91, 0.97) | 1.76×10−4 | 0.96 (0.92, 1.01) | 0.093 | 0.91 (0.86, 0.95) | 1.55×10−4 | 4 | 17580680 | LAP3 |
| cg00533183 | 0.94 (0.90, 0.97) | 2.86×10−4 | 0.94 (0.89, 0.99) | 0.011 | 0.93 (0.89, 0.98) | 0.010 | 6 | 32810742 | PSMB8; PSMB8-AS1 |
| cg21448870 | 1.09 (1.04, 1.14) | 3.09×10−4 | 1.11 (1.04, 1.18) | 1.00×10−3 | 1.06 (0.99, 1.14) | 0.081 | 3 | 72492399 | RYBP |
| cg24898914 | 0.92 (0.88, 0.96) | 3.72×10−4 | 0.92 (0.86, 0.98) | 0.012 | 0.91 (0.85, 0.98) | 0.012 | 6 | 32810706 | PSMB8; PSMB8-AS1 |
HR: hazard ratio; CI: confidence interval; Chr: chromosome; POS: position
Cox Regression Model: association between DNA methylation level and overall survival time, adjusted for sCD14, age, BMI, smoking, virus load, alcohol abuse, ethnicity, HBV, HCV
Human Genome Built Version of EPIC and 450K Chip: GRCh37
Discussion
In this first EWAS of sCD14, an important marker of gut microbial translocation, monocyte activation and inflammation for PWH, we identified DNAm sites and genes associated with sCD14 and mortality. We identified 118 DNAm sites significantly associated with sCD14 in the meta-analysis of 1,075 PWH. The principal associated DNAm sites mapped to genes (e.g., STAT1, PARP9, IFITM1, MX1, and IFIT1) related to inflammation and antiviral response. Adjusting for multiple testing, 10 of 118 sCD14-associated DNAm sites significantly predicted survival time conditional on sCD14 levels.
The leading DNAm sites and genes (e.g., STAT1, PARP9, IFITM1, MX1, and IFIT1) associated with sCD14 are implicated in inflammation and antiviral response IFN-stimulated pathways. The pathway enrichment analysis based on the 118 significant sCD14-associated DNAm sites confirms these biological pathways by identifying gene clusters in antiviral, interferon-I stimulated, and innate immunity pathways. Figure 3 provides an overview of key pathways of inflammation in HIV and the role of sCD14 in these pathways.
Figure 3:
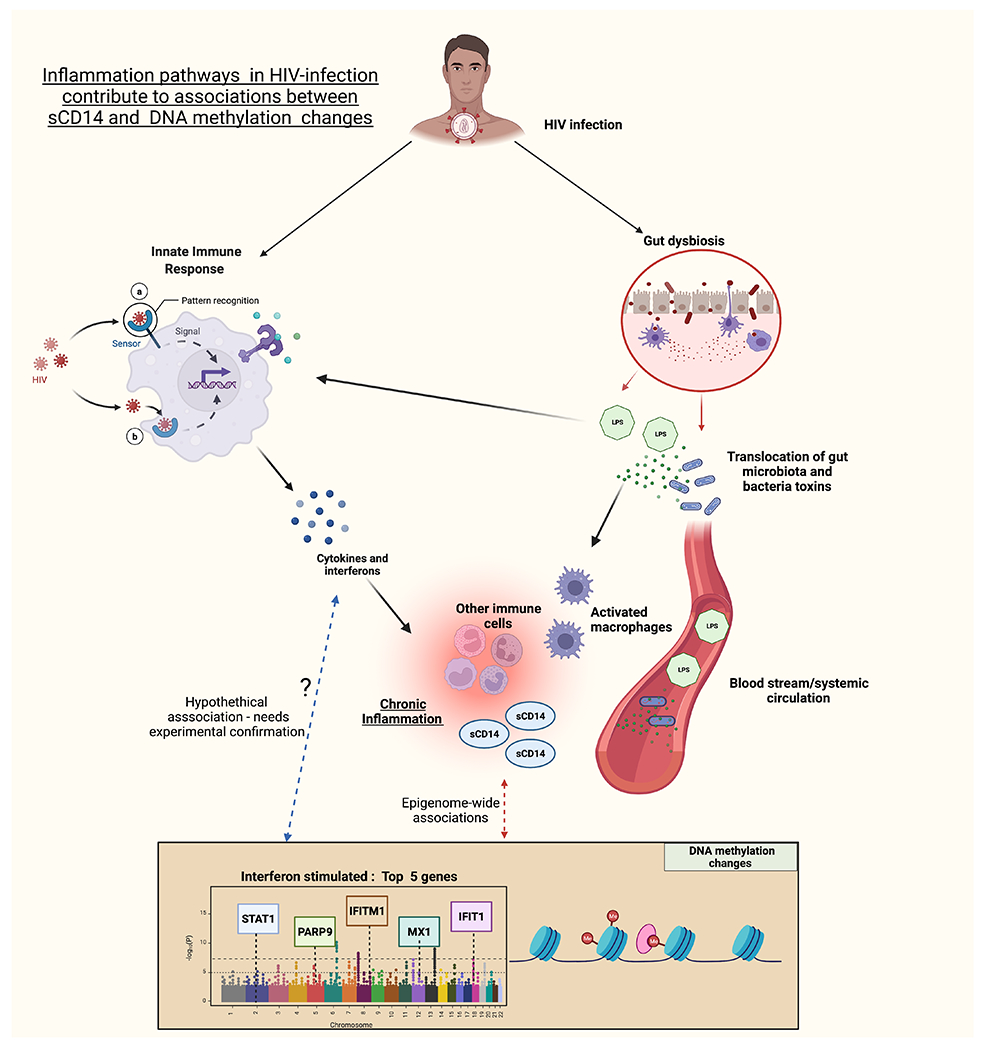
HIV-infection, triggers innate immune responses which contribute to inflammation in people with HIV (PWH). Gut dysbiosis engendered by destruction and loss of gut immune cells during HIV-infection leads to translocation of gut microbiota and bacterial toxins which further stimulate a proinflammatory immune response (including type 1 interferons) by binding to toll-like receptors on innate immune cells. As part of this response, soluble CD14 (sCD14) is produced primarily by monocytes and macrophages and is an important marker of inflammation in HIV pathogenesis. This study demonstrates a strong association between sCD14 and DNA methylation changes with principal associated sites mapping to interferon stimulated genes. Determining whether a pro-inflammatory state in chronic HIV-infection via biomarkers such as sCD14 leads to differential DNAm or whether DNA methylation changes in the setting of HIV contribute to sustaining chronic inflammatory pathways and elevate levels of relevant biomarkers need to be verified and confirmed by experimental studies.
STAT1 (Signal Transducer and Activator of Transcription-1) promotes an antiviral state in response to type I and type II interferons. IFN-γ/STAT1 pathway is as an important regulator of macrophage activation and polarization and controls the synthesis of multiple cytokines relevant for HIV pathogenesis (including IL-6), nitric oxide (NO), and reactive oxygen intermediates implicated in inflammation and tissue repair21. PARP9, a member of the Poly-ADP-Ribose Polymerase (PARP) gene family, plays a significant role in viral infections, inflammation, and aging by promoting pro-inflammatory activation of macrophages. PARP9 coregulates pro-inflammatory activation of human macrophages with PARP14. In vivo and in vitro studies pertaining to IFN-γ signaling in macrophages suggest that PARP14 mitigates proinflammatory phosphorylated STAT1 via ADP-ribosylation, and that PARP9 may act to inhibit PARP14’s enzymatic activity22. A recent transcriptomics study of brain tissue in simian immunodeficiency virus (SIV)+ macaques has identified the role of PARPs in SIV-associated neurogenerative disease23 which closely mimics HIV-associated neurocognitive disorders. PARP9 was found to be upregulated in macaque frontal cortices with detectable SIV in tissue23. Targeting PARP and STAT-mediated pathways may provide new therapeutic avenues for HIV-associated chronic inflammation and its multi-organ effects.
IFITM1, MX1, and IFIT1 are interferon induced genes which encode proteins with a broad spectrum of antiviral activity. IFITM1 (Interferon-induced transmembrane protein-1) interferes with virus endosomal fusion24. IFITM1 is strongly induced by IFN-α and has potent anti-HIV activity. Although the mechanism by which it restricts HIV is not fully understood, it may interfere with endocytosis leading to elimination of virus particles before established infection25. Recently in vivo models of HIV latency have shown that IFITM1 is overexpressed in resting latently infected CD4+ T-cells25. These findings suggest a plausible role of IFITM1 as a biomarker of latency and a therapeutic target for the elimination of HIV-1 reservoirs in PWH25.
A more recent in vitro study has shown that expression of IFITM1 reduces HIV-1 viral protein synthesis by preferentially excluding viral mRNA transcripts from translation and thereby restricting viral production26.
Human MX1 (myxovirus resistance-1) restricts a range of RNA and DNA viruses but does not restrict retroviruses such as HIV-127. Persistent higher expression of MX1 has however been demonstrated in a clinical cohort of women with HIV 28 and may contribute at least in part to the increased immune activation of chronic HIV disease. IFIT1 (Interferon-induced protein with tetratricopeptide repeat-1) acts in complex with other IFIT proteins to restrict HIV replication in macrophages29. This may contribute to persistent non-cytopathic infection of macrophages and thus influence the size of the HIV reservoir in macrophages in vivo29. Unsurprisingly, epigenetic association with sCD14 was more strongly correlated in viremic individuals, likely a reflection of persistent gut microbial translocation and immune activation in individuals with uncontrolled HIV infection.
It is remarkable that all top five genes identified in our sCD14 EWAS are IFN-stimulated genes. IFN-I blockade in conjunction with antiretroviral therapy has been suggested as a plausible approach for targeting the chronic immune activation associated with HIV infection. In an in-vivo mouse study, blockade of IFN-I signaling during chronic HIV-infection reduced immune activation, decreased expression of markers of T-cell exhaustion, restored HIV-specific CD8+ T- cell function and reduced viral replication30. Our study strengthens the key role of the IFN-signaling pathway in sustaining chronic inflammation and immune activation during HIV infection.
We demonstrate robust replication between the 450K and EPIC (850K) datasets supporting the strength of our observations and the high likelihood that the identified sCD14 -associated DNAm sites are true positives. Due to the difference in content and technologies, a substantial proportion of sCD14-associated DNAm sites, including the most significant site cg07839457, from the EPIC chip cannot be replicated in the 450K chip. Improved coverage and expanded replication samples would likely increase the discovery of sCD14-associated DNAm sites and potential utility of the epigenetic findings.
The survival analysis identified a significant association between DNAm level and multiple genes notably TIAM2, LDB1, IFITM1, and PARP14. T-cell lymphoma invasion and metastases 2 (TIAM2) and lim domain binding protein 1 (LDB1) have been associated with the progression and survival prognostication for several cancers31,32, in which immune dysregulation and chronic inflammation may play a role. IFITM1 and PARP14 genes have been implicated in inflammatory and antiviral pathways as previously discussed. This highlights the potential role of sCD14-associatiated DNAm in identifying important predictors for survival among PWH. Conditional on sCD14 levels in this large EWAS to characterize DNAm patterns in the host genome, we also identified ten DNAm sites independently predicting survival. Differential methylation signals associated with survival were observed in genes important for inflammation and interferon-induced antiviral responses and provide a crucial step towards understanding the mechanisms underpinning chronic inflammation in PWH and how these may impact on survival.
The generalizability of our findings is limited by several factors. Firstly, our study sample includes only male veterans, limiting our ability to explore epigenetic associations with sCD14 in women with HIV. Since the DNAm sites were measured as the mean methylation levels across all leukocyte subtypes, we were also limited to examine the functional roles of the sCD14-associated DNAm sites in different subtypes of leukocytes. Also, we cannot infer whether the relationships between sCD14 and DNA methylation and mortality are causal or surrogates for yet to be determined factors. Future studies investigating the regulatory mechanisms of identified methylation alteration in targeted cell types, particularly related to HIV infection, immunological response and inflammation could provide additional insight into the epigenetic mechanisms of chronic disease outcome and mortality among PWH.
Conclusion
The potential of using epigenetic and epigenomic profiles to inform disease pathogenesis, identify new therapeutic targets, and predict disease progression and survival remains relatively unexplored in the field of HIV. Our study demonstrates statistically robust epigenetic associations and provides a first step in examining the role of epigenetic modifications in the pathogenesis of inflammation and its implications for survival and chronic disease in HIV.
Supplementary Material
Funding:
This work was supported by the following grants:
BKT, ZW, QH, VCM and YVS received support from the National Institute of Health (R01DK125187), VCM and YVS received support from Emory CFAR (P30AI050409), Xu received support from the National Institute on Drug Abuse (R01DA042691, R01DA047820, R01DA047063), KS received support from NIH K01HL134147, VACS - NIH NIAAA U24 AA020794, U01 AA020790, U10 AA013566 completed.
Declaration of Competing Interest
V.C.M. has received investigator-initiated research grants (to the institution) and consultation fees from Lilly, Bayer, Gilead Sciences and ViiV. Other coauthors have no conflicts of interest to disclose.
References
- 1.Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013;39:633–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandler NG, Wand H, Roque A, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 2011;203:780–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Granucci F, Zanoni I. Role of CD14 in host protection against infections and in metabolism regulation. [DOI] [PMC free article] [PubMed]
- 4.Krastinova E, Lecuroux C, Leroy C, et al. High Soluble CD14 Levels at Primary HIV-1 Infection Predict More Rapid Disease Progression. [DOI] [PubMed]
- 5.de Vries PS, Chasman DI, Sabater-Lleal M, et al. A meta-analysis of 120 246 individuals identifies 18 new loci for fibrinogen concentration. Hum Mol Genet 2016;25:358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matteini AM, Li J, Lange EM, et al. Novel gene variants predict serum levels of the cytokines IL-18 and IL-1ra in older adults. Cytokine 2014;65:10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith NL, Huffman JE, Strachan DP, et al. Genetic predictors of fibrin D-dimer levels in healthy adults. Circulation 2011;123:1864–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahsan M, Ek WE, Rask-Andersen M, et al. The relative contribution of DNA methylation and genetic variants on protein biomarkers for human diseases. PLOS Genetics 2017;13:e1007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun YV, Lazarus A, Smith JA, et al. Gene-specific DNA methylation association with serum levels of C-reactive protein in African Americans. PLoS One 2013;8:e73480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Justice AC, Hu Y, et al. Epigenome-wide differential DNA methylation between HIV-infected and uninfected individuals. Epigenetics 2016;11:750–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Huang Y, Hui Q, et al. Epigenetic Associations With Estimated Glomerular Filtration Rate Among Men With Human Immunodeficiency Virus Infection. Clin Infect Dis 2020;70:667–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun YV. The Influences of Genetic and Environmental Factors on Methylome-wide Association Studies for Human Diseases. Curr Genet Med Rep 2014;2:261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun YV, Hu YJ. Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases. Adv Genet 2016;93:147–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Justice AC, Freiberg MS, Tracy R, et al. Does an index composed of clinical data reflect effects of inflammation, coagulation, and monocyte activation on mortality among those aging with HIV? Clin Infect Dis 2012;54:984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solomon O, MacIsaac J, Quach H, et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics 2018;13:655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Hu Y, Justice AC, et al. DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty. 2017;8:2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016;8:389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barcelona V, Huang Y, Brown K, et al. Novel DNA methylation sites associated with cigarette smoking among African Americans. Epigenetics 2019;14:383–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan MH. STAT signaling in inflammation. JAKSTAT 2013;2:e24198–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fehr AR, Singh SA, Kerr CM, Mukai S, Higashi H, Aikawa M. The impact of PARPs and ADP-ribosylation on inflammation and host-pathogen interactions. Genes Dev 2020;34:341–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mavian C, Ramirez-Mata AS, Dollar JJ, et al. Brain tissue transcriptomic analysis of SIV-infected macaques identifies several altered metabolic pathways linked to neuropathogenesis and poly (ADP-ribose) polymerases (PARPs) as potential therapeutic targets. J Neurovirol 2021:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Li J, Winkler CA, An P, Guo J-T. IFITM Genes, Variants, and Their Roles in the Control and Pathogenesis of Viral Infections. [DOI] [PMC free article] [PubMed]
- 25.Lu J, Pan Q, Rong L, Liu S-L, Liang C. The IFITM Proteins Inhibit HIV-1 Infection. 2011;85:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee WJ, Fu RM, Liang C, Sloan RD. IFITM proteins inhibit HIV-1 protein synthesis. Sci Rep 2018;8:14551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofer U. Interfering with HIV infection. [DOI] [PubMed]
- 28.Chang JJ, Woods M, Lindsay RJ, et al. Higher expression of several interferon-stimulated genes in HIV-1-infected females after adjusting for the level of viral replication. [DOI] [PMC free article] [PubMed]
- 29.Nasr N, Alshehri AA, Wright TK, et al. Mechanism of Interferon-Stimulated Gene Induction in HIV-1-Infected Macrophages. 2017;91:e00744–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhen A, Rezek V, Youn C, et al. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest 2017;127:260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li S, Ou Y, Liu S, et al. The Fibroblast TIAM2 Promotes Lung Cancer Cell Invasion and Metastasis. J Cancer 2019;10:1879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simonik EA, Cai Y, Kimmelshue KN, et al. LIM-Only Protein 4 (LMO4) and LIM Domain Binding Protein 1 (LDB1) Promote Growth and Metastasis of Human Head and Neck Cancer (LMO4 and LDB1 in Head and Neck Cancer). PLoS One 2016;11:e0164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.