

Abstract
Objective:
We previously reported a characterization of the HCC immune contexture and described an immune-specific class. We now aim to further delineate the immunogenomic classification of HCC to incorporate features that explain responses/resistance to immunotherapy.
Design:
We performed RNA and whole-exome sequencing, TCR sequencing, multiplex immunofluorescence and immunohistochemistry in a novel cohort of 240 HCC patients and validated our results in other cohorts comprising 660 patients.
Results:
Our integrative analysis led to define: a) the Inflamed class of HCC (37%), which includes the previously reported immune subclass (22%) and a new immune-like subclass (15%) with high interferon signaling, cytolytic activity, expression of immune-effector cytokines and a more diverse T-cell repertoire. A 20-gene signature was able to capture ~90% of these tumors and is associated with response to immunotherapy. Proteins identified in liquid biopsies recapitulated the Inflamed class with an AUC of 0.91; b) The Intermediate class, enriched in TP53 mutations (49 vs 29%, p=0.035), and chromosomal losses involving immune-related genes and; c) the Excluded class, enriched in CTNNB1 mutations (93 vs 27%, p<0.001) and PTK2 overexpression due to gene amplification and promoter hypomethylation. CTNNB1 mutations outside the Excluded class led to weak activation of the Wnt-βcatenin pathway or occurred in HCCs dominated by high interferon signaling and type I antigen presenting genes.
Conclusion:
We have characterized the immunogenomic contexture of HCC and defined Inflamed and Non-inflamed tumors. Two distinct CTNNB1 patterns associated with a differential role in immune evasion are described. These features may help predict immune response in HCC.
Keywords: Hepatocellular carcinoma, Liver cancer, Molecular classes, Immune Classification, Microenvironment, Immune checkpoint inhibitors, Immune therapies, CTNNB1
INTRODUCTION
Liver cancer incidence is increasing worldwide with more than 1 million annual cases expected by year 2025[1,2]. Hepatocellular carcinoma (HCC) accounts for 90% of all primary liver cancers and is mainly caused by chronic hepatitis B virus (HBV), hepatitis C virus (HCV) infection, alcohol abuse, and non-alcoholic steatohepatitis (NASH)[1,2].
It is estimated that around 50–60% of HCC patients will receive systemic treatments[3], where the combination of the immune checkpoint inhibitor (ICI) atezolizumab with bevacizumab (anti-VEGF) has recently become the new first-line treatment of advanced HCC[4,5]. This combination has triggered a breakthrough in HCC management[6], although only one third of patients clearly respond. Thus, a refined understanding of the immune landscape of HCC to predict outcomes after ICI therapy is still lacking and there is an unmet need to define the factors determining tumor immunogenicity. Evidence in other cancer types suggests that an inherently inflamed tumor microenvironment (TME) can be leveraged by ICI therapy to elicit better outcomes, whereas immune excluded tumors are prone to resistance[7].
We previously described the Immune class of HCC (~25% of patients), characterized by a high immune infiltration and molecular features resembling melanoma patients who respond to ICIs[8]. In that report, we suggested that further studies should refine the immune traits of the remaining ~75% of HCC cases. Moreover, recent findings have suggested that mutations in CTNNB1 (βcatenin) and subsequent activation of the Wnt-βcatenin pathway could be implicated in driving an excluded phenotype[9–11], although discordant results on its predictive potential in HCC suggest the need for further analysis[12–14].
In the current integrative analysis using 240 newly collected HCC samples and cutting-edge genomic technology, we define the Inflamed class of HCC in ~35% of cases, including the previously reported Immune subclass (22%) and a newly identified Immune-like subclass (15%). In addition, we describe the non-inflamed classes which we characterize as Intermediate and Excluded classes with distinct molecular and immune traits. Finally, we decipher the impact of CTNNB1 mutations in HCC and establish that while it is associated with immune exclusion in most cases, in some instances Wnt-βcatenin activated tumors harbor strong interferon signaling leading to an inflamed microenvironment.
Overall, our findings hold great potential to guide the discovery of clinically useful biomarkers and lay the groundwork for the development of new combination-based therapeutic strategies.
MATERIALS AND METHODS
Patients and samples
For the purpose of this study, we collected a total of 240 clinically annotated matched tumor and non-tumoral liver samples from HCC patients undergoing resection at several institutions[15]. Samples encompassed the most common etiologies (90 HCV-infected, 75 HBV-infected and 75 non-viral) and were collected upon approval of the Institutional Review Board at each Institution. A second cohort of 71 tissue samples with matched baseline blood samples was used for the liquid biopsy-based biomarker analysis[16]. Additional cohorts were used to validate our findings, including the TCGA-LIHC dataset[17] and the Heptromic cohort[18] (Supplementary Data). Figure 1 describes the flow chart of the study.
Figure 1: Flow chart of the study.
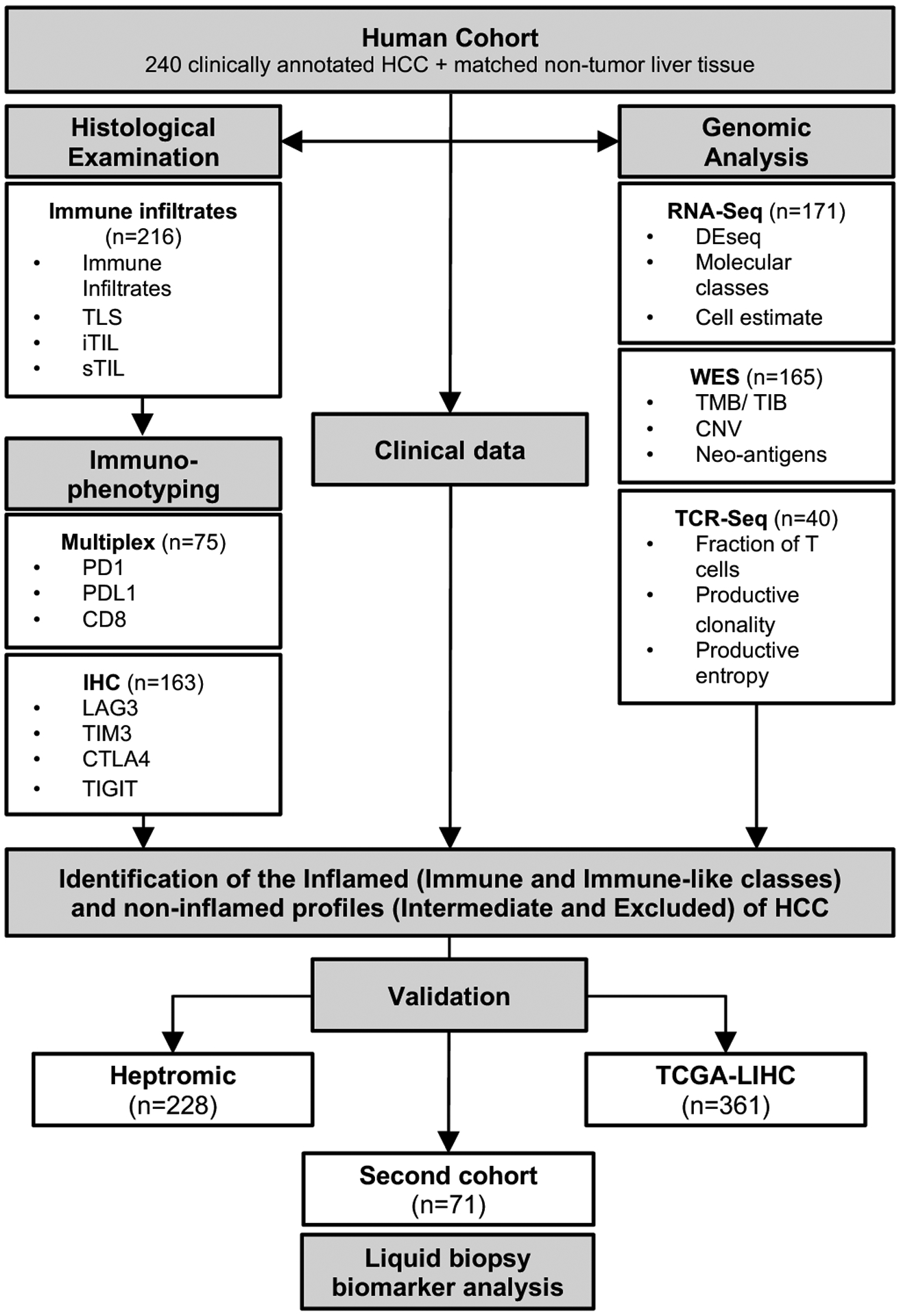
A total of 240 clinically annotated HCC and matched non-tumour tissue samples were used in this study as the discovery cohort. findings were then validated in two additional datasets comprising 589 additional patients and a new cohort of 71 patients with baseline tissue and blood samples. CD8, cluster of differentiation 8; CTLA4, cytotoxic T-lymphocyte associated protein 4; HCC, hepatocellular carcinoma; iTIL, intratumoural TILs; sTIL, stromal tumour infiltrating lymphocytes; IHC, immunohistochemistry; LAG3, lymphocyte-activation gene 3; PD1, programmed cell death protein 1; PDL1, programmed death-ligand 1; TCGA-LIHC, The Cancer Genome Atlas Liver Hepatocellular Carcinoma; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM3, T-cell immunoglobulin domain and mucin domain 3; TMB, tumour mutational burden.
Methodological details on histological assessment, immunohistochemical analyses, multiplex immunophenotyping, TCR sequencing, RNA sequencing, whole exome sequencing and statistical analysis are described in the Supplementary Data File.
RESULTS
We first characterized the distinct immune-related classes of HCC in a new cohort of 240 HCC samples. The main clinico-pathological traits of this cohort are depicted in Supplementary Table 1; with a median follow up of 55.1 months, median overall survival (mOS) was 70 months and median recurrence-free survival (mRFS) was 26.9 months (Supplementary Figure 1A,B).
Defining inflamed and non-inflamed HCCs
We first validated the Immune class of HCC (22%) and its two components, the Immune Active and Exhausted subclasses, in line with our previous findings[8]. The Immune Active type presented a significantly better survival when compared with the rest of the cohort in both univariate and multivariate analysis (mOS 95 vs 51 months, p=0.01, Supplementary Figure 1, Supplementary Table 2), while the Immune Exhausted subclass predicted worse RFS (HR 2.84, 95% CI 1.22–6.64, p=0.016) (Supplementary Table 2). Then, we aimed to characterize the remaining non-immune samples (78%). Amongst those, we depicted a subset of patients that presented high interferon signaling despite not being captured by the Immune class signature (Supplementary Figure 2A). Based on the expression of interferon genes, we clustered these samples into a separate class which we referred to as Immune-like subclass (15%) (Figure 2, Supplementary Figure 2A). Due to the commonalities in the immune traits between the Immune and Immune-like classes, including 37% of the cohort, we named these cases as Inflamed class (Figure 2). The remaining 63% of HCC cases depicted non-inflamed profiles. Among those, a subset of tumors showed low immune infiltration associated with the CTNNB1 class, consistent with our previous description[8]. Using a Wnt-βcatenin signature that captures activation due to CTNNB1 mutations or other mutations in the pathway[19,20] we were able to identify this group of tumors (20%) and are referred to as the Excluded class (Figure 2 and Supplementary Figure 2B, Supplementary Table 3). The remaining non-inflamed patients were grouped in a separate class termed Intermediate (43%) (Figure 2). We then focused on characterizing the driver traits that substantiate the uniqueness of each identified class.
Figure 2: Heatmap representation of the main molecular and immune features of the distinct immune-related profiles.

*P-values shown are calculated by Student’s T test for continuous variables or Fisher’s exact test for categorical variables. Unless otherwise indicated, it represents differences between the inflamed and non-inflamed classes; aCompares Excluded vs rest of the cohort. bCompares Exhausted vs rest of the cohort; cCompares Intermediate vs rest of the cohort.
Inflamed class
The Inflamed class has three components: our previously described Immune Active and Immune Exhausted subclasses along with a newly identified Immune-like subclass, which has been further characterized (Figure 2). We were able to design a 20-gene signature that captured ~90% of cases of the Inflamed class and identify blood-based biomarkers recapitulating this class.
Immune-like subclass of HCC
Tumors from patients with the Immune-like subclass harbored high interferon signaling, a higher immune enrichment score (p=6.59×10−14), an enrichment in signatures capturing CD8 T cells (p=1.26×10−15), M1 macrophages (p=2.09×10−27) and tertiary lymphoid structures (p=3.09×10−20, Figure 2) when compared with the non-inflamed profiles. We observed that the Immune-like subclass presented a higher enrichment in PD-1 signaling pathway (62 vs 13%, p=1.25×10−6) and expression of checkpoint molecules including CTLA4, PD-1 and PD-L1 (p<1×10−3, Figure 2). Furthermore, there was a significant enrichment in signatures involved in response to immunotherapy, such as interferon signaling, the HCC IFNAP signature, cytolytic activity and the HCC inflammatory signature (p<1×10−3, Figure 2). Of note, the immune features of this class, including high expression of checkpoint molecules, inflammation-related pathways and enrichment in signatures of response to immunotherapy, were similar to those of the Immune subclass[8] (Figure 2). The Immune-like subclass patients did not have differences in survival when compared to the non-inflamed profiles –as opposed to the Immune Active type (Supplementary Figure 1) – or any clinical or pathological differences when compared to the other immune types (Supplementary Table 4).
We identified striking molecular differences[19,21], however, between the Immune and Immune-like subclasses. The Immune-like was enriched in Hoshida S2 (19 vs 3%, p=0.04) and Chiang CTNNB1 classes (46 vs 3%, p=2.87×10−5) and presented a significant exclusion of the Hoshida S1 (8 vs 68%, p=1.6×10−6) and Chiang IFN class (0 vs 32%, p=9.17×10−4) when compared with the Immune subclass (Figure 2, Supplementary Table 5 and 6). Also, we observed an enrichment in the liver-related Wnt-βcatenin signaling pathway (54 vs 3%, p=2.35×10−6) and a significant exclusion of Wnt-TGFβ signaling (4 vs 55%, p=1.11×10−5) in the Immune-like subclass compared with the Immune (Figure 2). Altogether, these data suggest that our newly identified Immune-like subclass captures a subset of tumors with similar immune features but distinct molecular traits compared to the other immune types.
Characterization of the inflamed class immunophenotype
We next explored the immune landscape of the Inflamed class by performing transcriptomic deconvolution, TCR sequencing, multiplex immunophenotyping, histological assessment and IHC. Histologically, patients in the Inflamed class showed a higher immune infiltration when compared with the non-inflamed profiles (53 vs 32%, p=0.01, Figure 3A, Supplementary Table 7), higher presence of stromal tumor infiltrating lymphocytes (sTILs, 42 vs 18%, p=0.01), intratumoral TILs (iTILs, 33 vs 16%, p=0.07) and tertiary lymphoid structure (high TLS density, 27 vs 12%, p=0.09, Figure 3B–D, Supplementary Figure 3–4, Supplementary Table 8).
Figure 3: The Inflamed class shows high immune infiltration and features of inflammation.
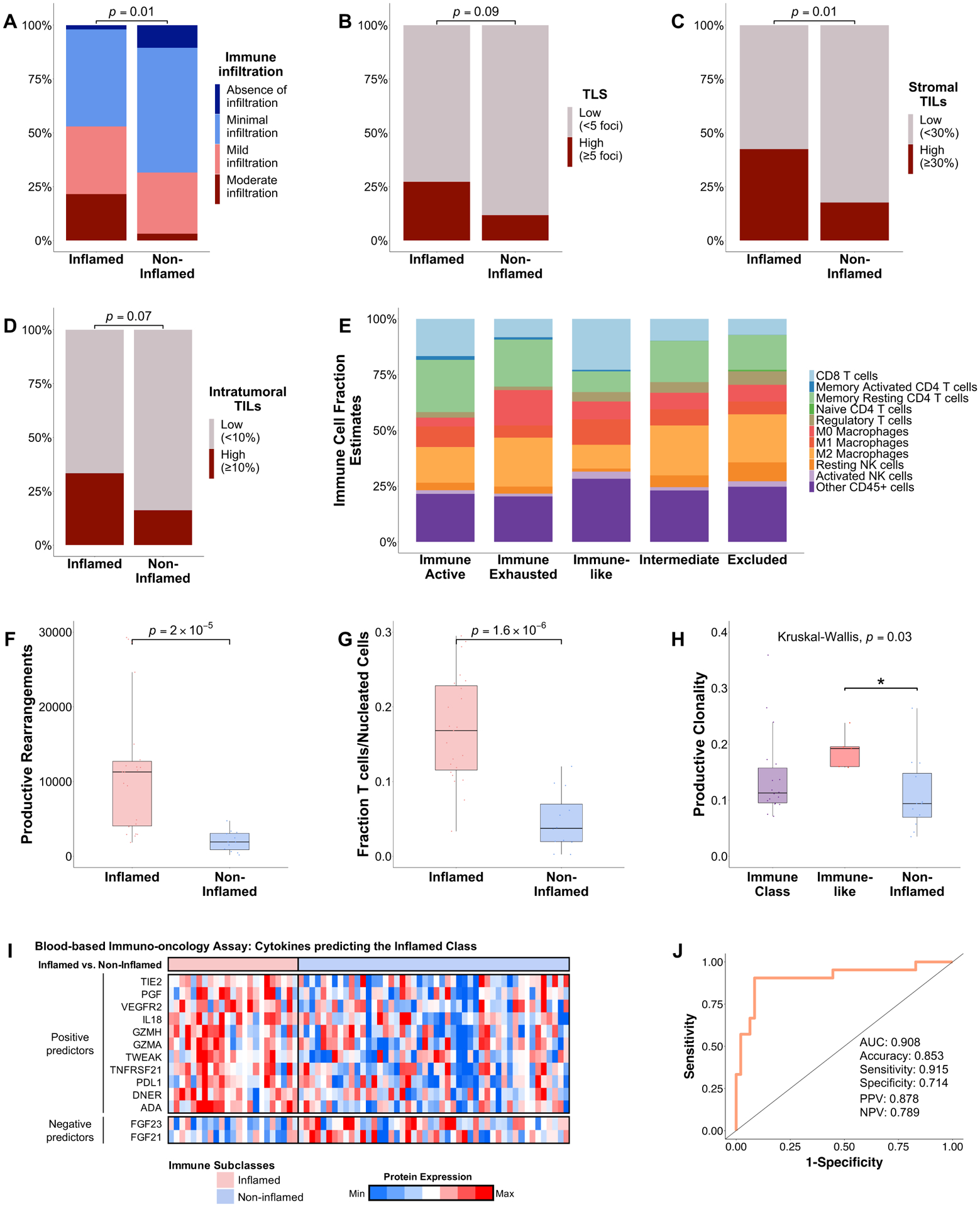
(A–D) Barplot depicting (A) the richness of the immune infiltrate, (B) density of TLS, (C) the stromal TILs and (D) the intratumoural TILs as assessed by H&E examination. (E) Stacked barplot depicting the fraction of 22 immune cell types inferred by CIBERSORTx. (F–H) TCR sequencing results showing (F) the number of productive rearrangements, (G) fraction of T cells and the (H) productive clonality. (I) Heatmap representation of 11 cytokines which positively predict the inflamed class and two cytokines which were enriched in the non-inflamed class. (J) AUC showing the performance of the devised 13-protein signature in capturing the Inflamed class. P value are calculated by (A–D) Fisher’s exact test, (F–G) Wilcoxon’s rank-sum test and (H) Kruskal-Wallis test with post hoc Dunn’s test. *P < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. AUC, area under the ROC curve; NPV, negative predictive value; PPV, positive predictive value; TILs, tumour infiltrating lymphocytes; TLS, tertiary lymphoid structures.
We additionally performed IHC for CTLA4, LAG3, TIGIT and TIM3. The Inflamed class presented a significant enrichment for LAG3 (39 vs 19%, p=0.049) while the Immune class presented a higher presence of CTLA4 (58 vs 29%, p=0.03, Supplementary Figure 4) when compared with the non-inflamed profiles. No differences were observed amongst the immune-related subclasses for TIM3 and TIGIT.
To further assess the tumor microenvironment, we performed multiplex immunophenotyping of CD8, PD-1 and PD-L1 in a subset of tumors. Consistent with our transcriptomic assessment, we observed an enrichment of intratumoral CD8 T cells (CD8≥1%, 58 vs 30%, p=0.08) and PD-L1 (PD-L1≥1%, 21 vs 4%, p=0.19) in the inflamed profiles when compared with the non-inflamed (Supplementary Figure 3–4).
Next, we applied CIBERSORTx to characterize the immune microenvironment. We observed a significantly higher fraction of CD8 T cells (p=3.51×10−7) and M1 macrophages (p=1.82×10−4) in the Inflamed class when compared with the non-inflamed profiles and a significant exclusion of M2 macrophages in the Immune-like subclass (p=1.78×10−6, Figure 3E). We further validated these findings by applying xCell deconvolution (Supplementary Figure 5).
Subsequently, we performed TCR sequencing to better characterize the TCR repertoire. Consistent with our previous analysis, the Inflamed class contained a higher fraction of T cells, a higher number of productive rearrangements and a richer and more diverse T cell repertoire when compared with the non-inflamed profiles (Figure 3F–G, Supplementary Figure 6). Overall, TCR clonality was low (range 0.04–0.36) indicating that HCC samples at baseline are mostly polyclonal. Interestingly, cases belonging to the immune-like subclass showed a higher productive clonality (p=0.01) when compared with the other classes, indicating a probable oligoclonal expansion of T cells (Figure 3H). When we examined the clones with the highest productive frequency, no shared sequences were observed across groups and within each group, suggesting that the top clones are unique in each patient.
Overall, this suggests that Inflamed HCC tumors, in addition to containing a higher infiltration of T cells and CD8 T cells, display a more diverse T-cell repertoire.
Discovery of the inflamed gene signature
In order to provide a tool to accurately capture HCC cases belonging to the Inflamed class, we designed and validated a 20-gene signature that we refer to as “Inflamed signature” (Supplementary Table 9). The most characteristic gene components of the Inflamed signature were related to T-cell signaling and activation (CD3D, CD2, LCP2), lymphocyte chemotaxis (CXCL9, CCL5) and cytolytic activity (GZMA, GZMB) (Supplementary Figure 7A). Similarly, they were enriched in pathways related to antigen presentation, T cell signaling and B cell activation (Supplementary Figure 7B). In the discovery cohort of 240 cases, the signature had an accuracy of 89%, sensitivity and specificity of 80 and 95% and a positive and negative predictive value (NPV) of 89% and 91%, respectively (Supplementary Figure 7C, Supplementary Table 10). Increasing the number of genes in the signature did not increase its accuracy (Supplementary Figure 7D).
We validated our Inflamed signature in the Heptromic[8] and the TCGA-LIHC cohorts[22]. In both cohorts, the signature had an accuracy of ~90% (Supplementary Figure 7C and Supplementary Table 10). Additionally, we observed that in both validation cohorts, the Inflamed class showed similar molecular traits to our discovery cohort, with increased interferon signaling, higher immune infiltration and higher cytolytic activity, amongst other features (Supplementary Figure 8). When tested in an external cohort of advanced HCC patients treated with ICIs[23], patients who responded showed a significant overexpression of the Inflamed signature when compared with non-responders (p=0.047, Supplementary Figure 9), suggesting it could accurately predict response to ICIs.
A liquid-biopsy based biomarker recapitulating the Inflamed class
To identify patients belonging to the Inflamed class by using liquid biopsy-based biomarkers, we assembled a new cohort of 71 HCC samples with matched tumor tissue, non-tumor tissue and baseline blood samples in 68 cases (Supplementary Figure 10). We measured 92 protein biomarkers in peripheral blood and built a predictive model to recapitulate the Inflamed class. This allowed us to devise a 13-protein signature that captured the Inflamed class with an AUC of 0.91, an accuracy of 85% and a sensitivity of 92% (Figure 3, I–J).
Non-inflamed classes
Overall, the non-inflamed classes (~63% of cases) depict immune features that are significantly distinct from the inflamed class (Figure 2). Nonetheless, the non-inflamed cases were also heterogeneous in immune and molecular characteristics, allowing us to differentiate two distinct classes based on the underlying mechanisms of immune evasion: the Intermediate and the Excluded classes.
The Intermediate Class
In terms of immune traits, the Intermediate class presented a mild infiltration of immune cells as assessed by the ESTIMATE and xCell tools. Immune infiltration was significantly decreased when compared with the Inflamed class (p=9.31×10−24) but significantly higher than in the Excluded class (p=5.46×10−7) (Figure 2).
In terms of somatic mutations, tumors belonging to the Intermediate class were enriched in TP53 mutations when compared with the Inflamed class (49 vs 29%, p=0.04) and the rest of the cohort (49 vs 31%, p=0.04, Figure 2, Supplementary Figure 11). We next explored the status of chromosomal aberrations, which have been associated with immune evasion[24,25], by using the CNApp algorithm[26] to infer copy-number scores and genomic imbalances. We first calculated a single-sample score for broad and focal events, which accounts for the number, amplitude and length of the broad and focal segments, respectively. Interestingly, there was a significant increase in broad and focal scores in both the Intermediate and Excluded classes (Figure 4A–B). To further validate these findings, we applied the GISTIC algorithm, revealing an increased number of focal deletions in the Intermediate and Excluded classes compared with the Inflamed class and an increased burden of broad deletions in the Intermediate class (Supplementary Figure 12). There was a significantly increased number of losses in genomic regions that harbored genes involved in antigen presentation or interferon signaling in the Intermediate class compared with the rest of the cohort, including subcytobands 16p13.13 (harboring CIITA[27,28], 43 vs 16%, p=6.45×10−4), 4q21.1 (harboring CXCL9, CXCL10, CXCL11, 54 vs 19%, p=1.53×10−5) and 4q35.1 (harboring IRF2[29,30], 56 vs 18%, p=2.74×10−6) (Figure 4C, Supplementary Figure 13, Supplementary Table 11–13). These deletions also had an impact on the gene mRNA expression (Supplementary Figure 14).
Figure 4: Genomic overview of the distinct Immune classes of HCC.
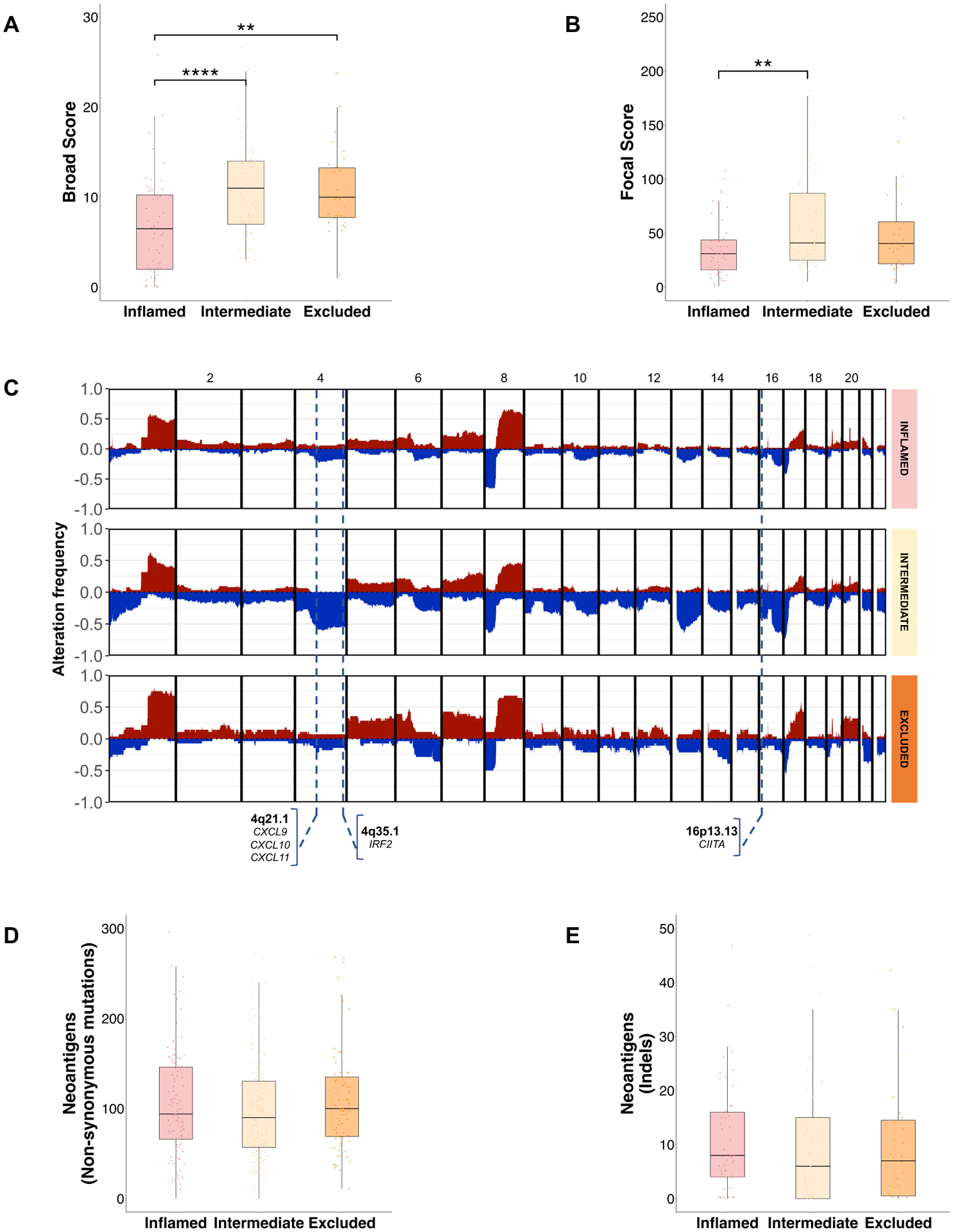
(A, B) Boxplot depicting the distribution of (A) broad and (B) focal chromosomal aberration as assessed by the CNApp algorithm. Kruskal-Wallis, p=9.5×10−5 and Kruskal-Wallis, p=0.02, respectively. (C) Genomic overview showing the percentage of samples with copy number events among each immune class. Blue represent deletions, red represent gains, the Y axis depicts the percentage of these events across each immune class. Potentially Impactful subcytobands in immune evasion are indicated with a dotted line. (D) Boxplot showing the distribution of inferred neoantigens and (E) neoantigens from insertions and deletions. Kruskal-Wallis, p=0.87 and Kruskal- Wallis, p=0.3, respectively. For (A, B, D, E), p values are calculated by Kruskal-Wallis test with post hoc Dunn’s test. *P < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. HCC, hepatocellular carcinoma.
Finally, we explored the tumor mutational burden (TMB) and neoantigen burden, which have been associated with inflammation and immune response[31,32]. Overall, the median number of non-synonymous mutations was 77 (range 14–226) and median TMB was 2.6 mutation/Mb (range 0.2–8.2), with no differences among classes. A median of 35 (range 4–164) immunogenic mutations (defined as those mutations that were predicted to generate at least one HLA-binding epitope; ~45% of the total mutations) were identified, with no differences among classes. The median number of expressed neoantigens and high-affinity neoantigens (defined as those with a rank binding affinity <0.5%) across the whole cohort was 99 (range 4–456) and 27 (range 1–124), respectively. No differences were found amongst all classes in this discovery cohort (Figure 4D, Supplementary Figure 15A–B) and in TCGA-LIHC[33] (Supplementary Figure 15C). Also, we found no differences amongst classes in the distribution of neoantigens derived from indels in our cohort (Figure 4E, Supplementary Figure 15D–E) and TCGA-LIHC[33] (Supplementary Figure 15F).
Altogether, these data show that the Intermediate class is characterized by an enrichment in TP53 mutations and higher levels of chromosomal instability, with frequent deletions in genes related to antigen presentation or interferon signaling. These structural features are associated with a significant decrease in immune infiltration and immune score observed in the Intermediate class (Figure 2).
The Excluded Class
The Excluded class (20% n=34/171) presented the lowest immune infiltration (p=2.82×10−10) (Figure 2) and was characterized by a significant enrichment of CTNNB1 mutations when compared with the Intermediate class (93 vs 17%, p=5.55×10−12) and the rest of the cohort (93 vs 27%, p=1.06×10−10, Figure 5A). As different CTNNB1 genotypes result in different levels of activation of the Wnt-βcatenin pathway[34], we further analyzed the type of CTNNB1 mutations in our cohort. Most CTNNB1 mutations were located on exon 3 (91.2%) (Supplementary Figure 16A). We classified these mutations in high, moderate and low activation according to a previously established classification[34] (Supplementary Figure 16B) and observed that 88% of mutations in the Excluded class caused high or moderate activation of the Wnt-βcatenin pathway (Figure 5A–B). In addition, we also observed a higher presence of missense variants in APC compared to the Intermediate class (11 vs 0%, p=0.03), which also activates the Wnt-βcatenin pathway. Conversely, no differences were observed with AXIN1 variants (3.6 vs 13%, p=0.27), which is consistent with previously published data showing a lack of activation of the Wnt-βcatenin pathway as a result of these mutations[35,36]. We further characterized the Excluded class and observed a significant overexpression of PTK2 when compared with the Intermediate (p=0.02) and the Inflamed class (p=1.13×10−5) which was validated by IHC (Supplementary Figure 17A–D). To understand the mechanisms underlying PTK2 overexpression, we identified those samples that had an expression value of the gene that was at least 2-fold-times higher than the mean expression in non-tumoral tissue. According to this definition, 32% of samples in our cohort overexpressed PTK2, which was similar to the one in TCGA-LIHC (27%). We found that 70% of overexpressing samples harboured a gene-amplification (>3 copies of the 8q24.3 subcytoband) (Supplementary Figure 17E), which increased to 83.5% in TCGA-LIHC. When incorporating promoter region hypomethylation of PTK2 in TCGA-LIHC, almost 95% of the samples had an identifiable mechanism of overexpression (Supplementary Figure 17F). This suggests that both gene amplification and promoter region methylation are major drivers of PTK2 overexpression in HCC. Overall, the Excluded class is characterized by the presence of highly activating CTNNB1 mutations and overexpression of PTK2.
Figure 5: Mutational landscape of CTNNB1 mutations across the Immune class.
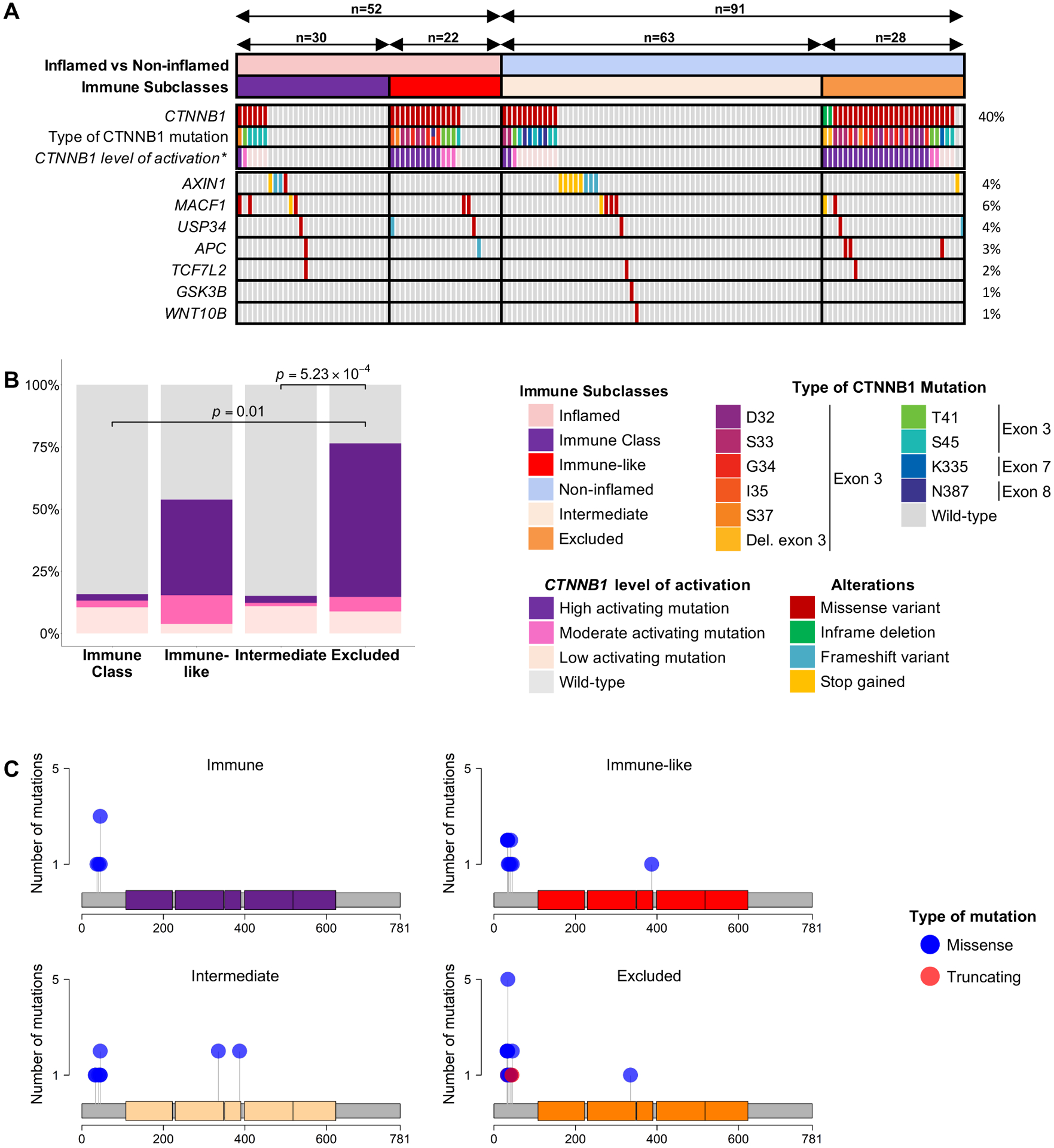
(A) Heatmap representation of the distribution of mutations of key genes involved in the Wnt-βcatenin pathway. (B) Stacked barplot showing the distribution of the type of CTNNB1 mutations across immune classes. (C) Lollipop plots showing the distribution of CTNNB1 mutations in the distinct immune classes. P values are calculated by (B) Student’s t-test and (C) Fisher’s exact test. *CTNNB1 level of activation is based on Rebouissou S, Franconi A, Calderaro J, et al.[34]
CTNNB1 mutations are associated with two distinct types of immune activation
Moderate or high-activating CTNNB1 mutations in the Intermediate and Immune class were significantly less prevalent compared with the Excluded class (88 vs 27%, p=5.23×10−4; 88 vs 33%, p=0.01, respectively) (Figure 5A–B). Even though most mutations in the Immune subclass were low-activating mutations, 93% of mutations in the Immune-like subclass were either moderate or high-activating mutations, which was no different than the Excluded class (93 vs 88%, p>0.05) (Figure 5B–C). This is consistent with our previous observation showing an enrichment of the Wnt-βcatenin activation pathway in the Immune-like when compared with the Immune subclass (Figure 2). Therefore, we next focused on understanding the implications Wnt-βcatenin activation, which occurred in 49 of our samples (Figure 6A). In two thirds of cases Wnt activation was associated with non-inflamed classes (n=34, 69%) - described as a potential mechanism of immune exclusion[10,11], but in one third it was identified within the Inflamed class (n=15, 31%). While the former group was clearly enriched in the Excluded class and dominated by a paucity of immune infiltrate, the latter group was dominated by a significant enrichment of immune infiltration, activated CD8+ and CD4+ T cells, signatures of immune response, inflammation and interferon signaling (Figure 6A). Differential gene expression analysis between inflamed and non-inflamed Wnt-βcatenin tumors (Figure 6B, Supplementary Table 14) and subsequent pathway enrichment analysis (Supplementary Figure 16C, Supplementary Table 15), confirmed that most of the enriched pathways in the inflamed profiles were immune-related. Histological analysis of the immune infiltrates further confirmed these findings. We observed a higher rate of immune cell infiltration (54 vs 22%, p=0.07) and a higher density of TLS (≥ 5 foci, 50 vs 0%, p=3.42×10−3) in the inflamed profiles when compared with the non-inflamed (Figure 6C–D). Moreover, there was a significant enrichment of sTILs (sTILs ≥30%, 63 vs 5%, p=3.05×10−3) and a trend in iTILs (iTILs ≥10%, 38 vs 5%, p=0.06) (Supplementary Figure 16D). Collectively, these findings suggest that two thirds of Wnt-βcatenin activated tumors are characterized by immune paucity and exclusion, while one third of tumors with this activation are counterbalanced by mechanisms leading to high immune infiltration and inflammation. Gene set enrichment analysis (GSEA) revealed enhanced immune-related pathways in the inflamed profiles, such as IFNγ signaling, allograft rejection, complement activation and inflammatory response, amongst others (Supplementary Figure 16E, Supplementary Table 16). Conversely, there was no differences in the mechanism responsible for activation of the Wnt-βcatenin pathway (assessed by 19 gene signatures capturing canonical and non-canonical Wnt-βcatenin activation) (Supplementary Table 17 and 18).
Figure 6: Two distinct profiles of Wnt-βcatenin activated tumours are identified based on immune features.
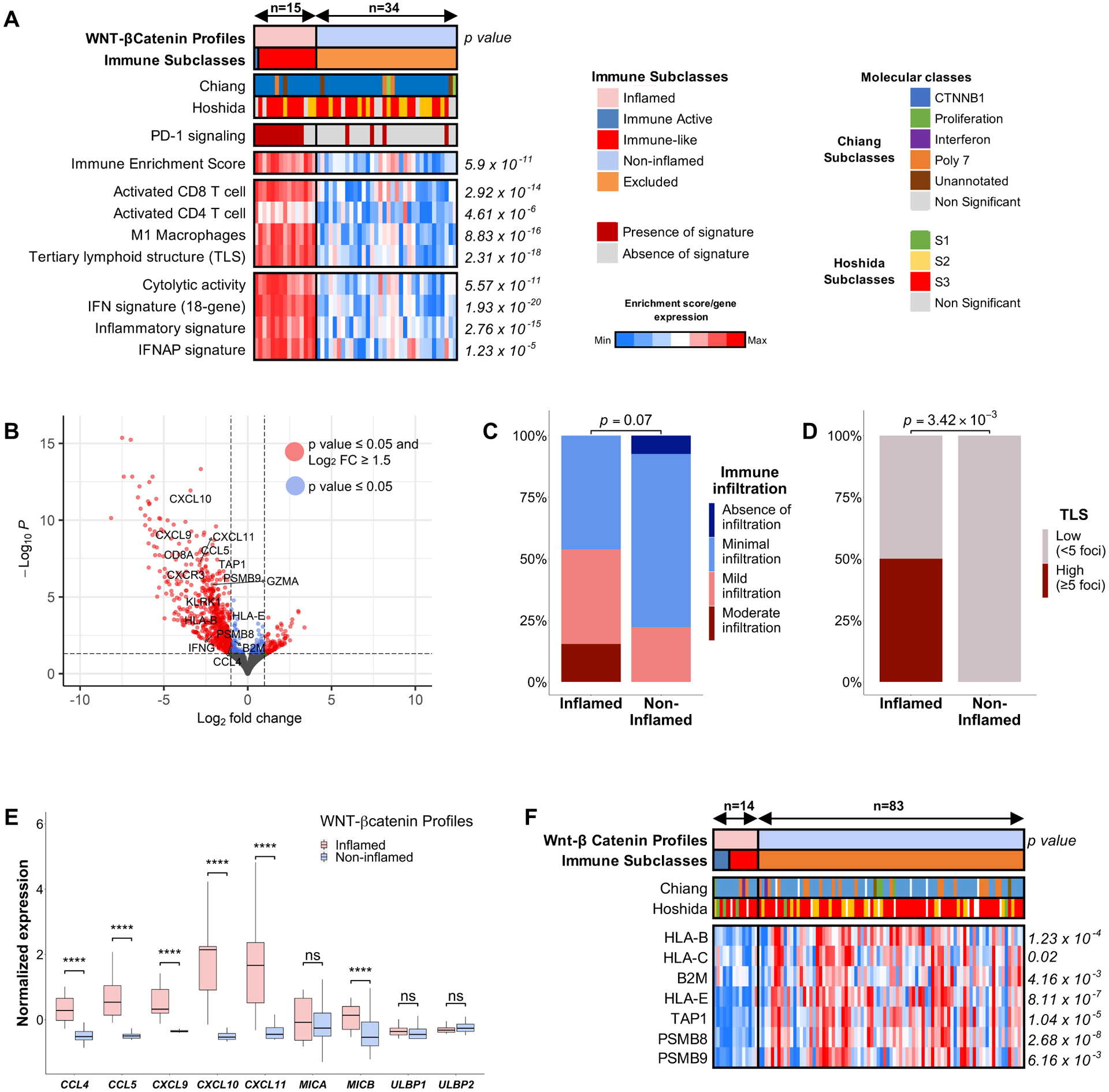
(A) Heatmap representation of the main immune features of the distinct profiles. (B) Volcano plot showing the differentially expressed genes between inflamed and non-inflamed profiles. (C, D) Barplot representation of (C) the richness of the immune infiltrate and (D) TLS density as assessed by H&E examination. (E) Boxplot comparing the expression of cytokines and ligands repressed by Wnt-βcatenin pathway. (F) Heatmap comparing the methylation of genes involved in antigen type I presentation in the TCGA cohort. P values are calculated by (A, F) Student’s t-test, (C, D) Fisher’s exact test and (E) Wilcoxon rank-sum test. *P < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. ns, not significant. TCGA, The Cancer Genome Atlas Liver Hepatocellular Carcinoma; TLS, tertiary lymphoid structures.
Since evidence suggests that Wnt-βcatenin drives immune exclusion in HCC by interfering with dendritic cell and lymphocyte chemotaxis (through CCL5 and CCL4[10,11]) and NK cell chemotaxis (through NKG2D ligands[37]), we investigated whether these mechanisms were preserved in these two distinct profiles. The inflamed profiles expressed more CCL5, CCL4 and other major cytokines involved in lymphocyte chemotaxis, such as CXCL9, CXCL10 and CXCL11 (Figure 6E). Consistently, CIBERSORTx deconvolution analysis revealed higher proportions of CD8+ T cells in inflamed profiles compared to non-inflamed profiles (Supplementary Figure 16F). Conversely, we did not find differences in the expression of most NKG2D ligands between the inflamed and non-inflamed profiles, including MICA, ULPB1 and ULPB2 (Figure 6E), a finding that was consistent with CIBERSORTx and xCell deconvolution results that showed no differences in infiltration of activated NK cells (Supplementary Figure 16G–H). These data suggest that the inflamed profiles include enhanced CCL5 expression and T cell recruitment despite activation of the Wnt-β catenin pathway.
We further analyzed the differential gene expression data and observed there was a significant upregulation of genes involved in antigen type I presentation in the inflamed profiles, including HLA-B, B2M and TAP1, amongst others. Defects in the antigen presentation machinery, including copy-number deletions and epigenetic modifications such as hypermethylation, have been related to immune escape[38]. We found no enrichment in copy-number losses in antigen presenting genes. To analyze possible epigenetic changes, we explored the TCGA-LIHC cohort. There was significant hypermethylation of genes involved in antigen type I presentation including TAP1, B2M, HLA-B and HLA-C (Figure 6F) in Wnt-βcatenin HCCs from the non-inflamed group, which, as expected, inversely correlated with their expression levels (Supplementary Figure 18)
Overall, these data suggests that while ~70–85% of tumors with activation of the Wnt-β catenin pathway show features of immune exclusion and immune cell paucity, ~15–30% of tumors with activation of this pathway express features of inflammation and immune activation. Importantly, these tumors overexpress CCL5 and have enhanced CD8+ T cell infiltration. Complementary mechanisms, such as hypermethylation of antigen-presenting genes, may contribute to these phenotypic differences.
DISCUSSION
The development of ICIs has transformed the field of immuno-oncology and revolutionized the management of cancer. However, only a small fraction of patients (~20%) present a durable response in HCC. Therefore, there is an ongoing unmet need to identify biomarkers that accurately predict which patients will benefit from this form of therapy. Recently, significant differences have been uncovered in the clinical outcomes following ICIs based on underlying liver disease etiology, with a significantly increased benefit in viral-related HCC compared with non-viral[39]. This finding adds a new layer of complexity to the already daunting field of biomarker discovery. Thus, a thorough understanding of the HCC immune microenvironment and its molecular underpinnings is imperative to guide the rational discovery of clinically useful biomarkers.
In this study, we have built upon our previous findings[8] to provide an in-depth characterization of the HCC immunological classes (Figure 7). First, we identify a cluster of tumors harboring immune traits –the so-called Immune-like subclass– resembling our previously described Immune subclass[8] but not captured by the immune signature and presenting significant activation of the Wnt-βcatenin pathway due to CTNNB1 mutations. Overall, the proposed Inflamed class accounts for ~35% of HCC cases and presents high interferon signaling, PD-1 signaling and overexpression of genes related to lymphocyte chemotaxis such as CXCL9 and CXCL10. We have therefore developed a novel Inflamed signature capable of identifying these tumors with an accuracy of ~90% and have validated it in three additional cohorts. Additionally, we provide preliminary evidence supporting the role of liquid biopsy in identifying this class. Importantly, these inflamed HCC tumors were enriched in two recently reported inflammatory signatures[14,40] predicting ICI response in advanced HCC patients and was enriched in responders to ICIs in a small published cohort[41]. These findings strongly support the notion that inflamed HCC tumors represent potential responders to ICI therapies, a feature consistent with a report where HCCs responding to ICI after sorafenib progression present an inflamed phenotype and higher infiltrate of cytotoxic T lymphocytes[42]. Finally, we propose a diagnostic algorithm which aims to implement this classification in clinical practice to further evaluate the predictive potential of the Inflamed class in ICI-treated HCC cohorts (Supplementary Figure 19).
Figure 7: Graphic representation of the distinct Immune profiles of HCC.
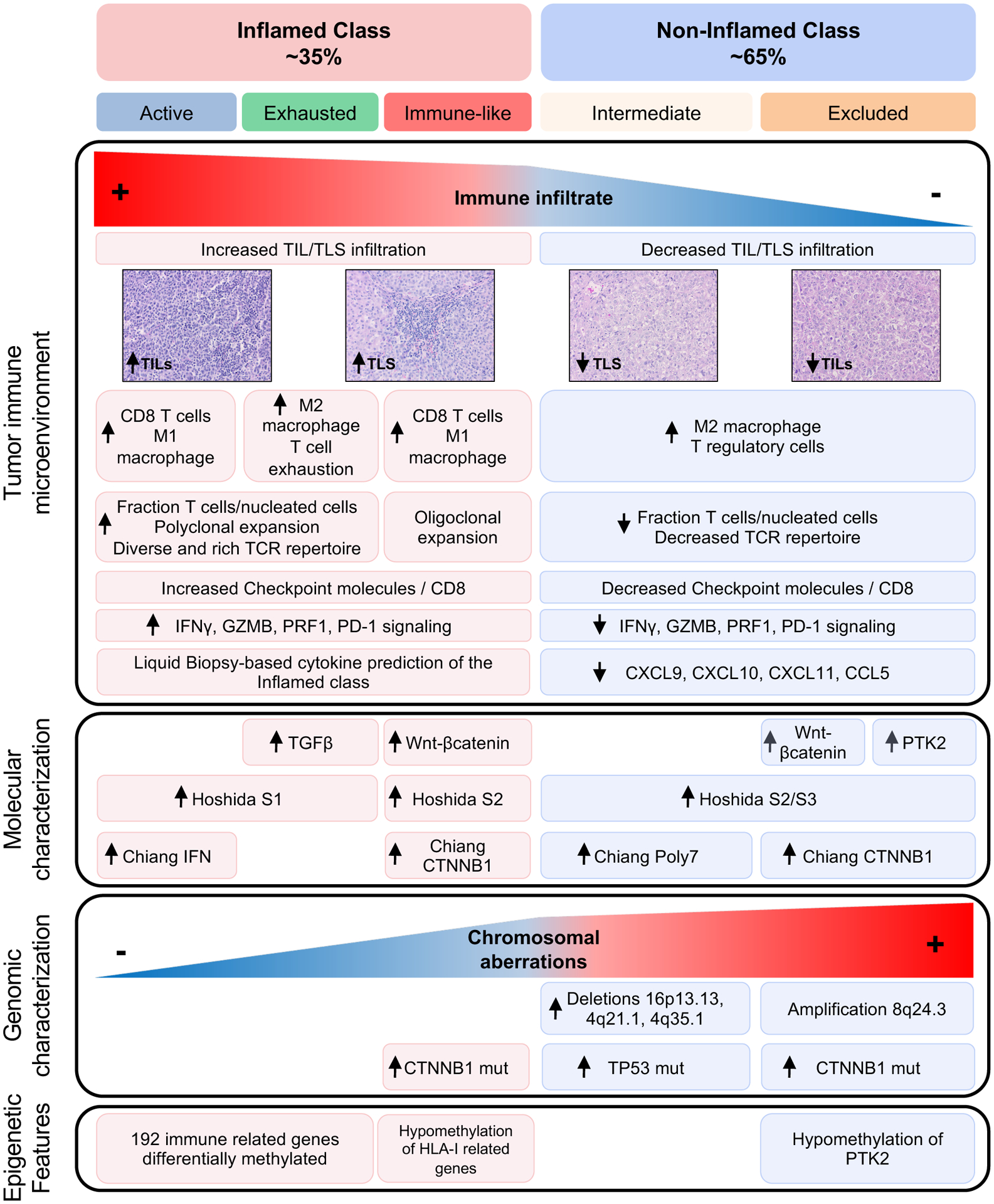
The figure summarizes the main molecular and histopathological features according to current findings and data previously published[3,8]. HCC, hepatocellular carcinoma; TIL, tumor infiltrating lymphocyte.
Tumors immunologically classified as Intermediate showed enrichment of TP53 mutations and losses in genes related to antigen presentation and interferon signaling. Interestingly, loss of TP53 in cancer cells promotes the recruitment of immune suppressive cells and attenuates the response of both cytotoxic and T-helper cells[43,44]. Therefore, the increased rate of TP53 mutations seen in the intermediate class could be key in promoting an immunosuppressive microenvironment. Furthermore, we show that these non-inflamed profiles harbor more chromosomal aberrations, with significantly more broad and focal chromosomal alterations, which in line with previous pan-cancer analyses[24,25,45]. We find significantly higher deletions in genes related to interferon signaling and antigen presentation in the Intermediate class, which could potentially drive this non-inflamed phenotype [28–30,46]. Functional studies are needed to validate these findings.
Compelling experimental evidence in an immunocompetent animal model of HCC has established that activation of the Wnt-βcatenin pathway may promote immune exclusion through a defective recruitment of lymphocytes and dendritic cells[11]. In the current study, deep immunophenotyping combining multiplex immunofluorescence and transcriptomic analysis of the non-inflamed profiles has revealed that ~20% of HCC patients present activation of Wnt-βcatenin and low immune infiltration and an overexpression of PTK2. We coined this group the Excluded class. An intriguing finding in our study is the observation that ~15–30% of tumors with high activation of the Wnt-βcatenin pathway belong to the Inflamed class. When compared with the Excluded class, these tumors express more CCL5 and CCL4, two cytokines regulating lymphocyte chemotaxis that are usually repressed by the Wnt-βcatenin pathway[10,11]. Conversely, there are no differences in the expression of the NKG2D ligands MICA, ULPB1 and ULPB2, involved in cancer immunosurveillance and also downregulated by the Wnt-βcatenin pathway[37]. Furthermore, there is an upregulation of MHC-I related molecules in the inflamed Wnt-βcatenin activated profiles, accompanied by a significant demethylation of these genes[47]. Therefore, these data suggest that the immune landscape of Wnt-βcatenin activated tumors is heterogeneous and the immune phenotype could be a result of an altered balance between the immunosuppressive function of Wnt-βcatenin and the pro-inflammatory interferon pathway. Importantly, the distinction between these two profiles could partly clarify the discrepancies observed in the predictive value of CTNNB1 mutations in in HCC[12–14].
Overall, we have defined a new Inflamed profile of HCC with high interferon signaling despite presenting an enrichment in CTNNB1 mutations. Furthermore, we have described new potential mechanisms of immune evasion in HCC including TP53 mutations, deletions in genes related to interferon signaling and antigen presentation and hypermethylation of genes involved in antigen type I presentation. We foresee that this classification will enable a better stratification of patients, although further studies are needed to establish its predictive value.
Supplementary Material
Significance of this study.
What is already known on this subject?
We previously reported the immune class of HCC, present in ~25% of patients. However, the immune traits of the remaining ~75% of HCCs are ill-defined. Further, the association of response (15–20% of HCC cases) or resistance to immune checkpoint inhibitors and the role CTNNB1 mutations is unclear.
What are the new findings?
By using an integrative genomic approach, we have now refined the Inflamed class (~35% of cases), which includes the immune subclass and the newly described immune-like subclass. A 20-gene signature captures this class and is associated with response to immunotherapy. Also, we characterize non-inflamed profiles and classes and decipher the potential dual role of CTNNB1 mutations with response and evasion.
How might it impact on clinical practice in the foreseeable future?
This revised immunogenomic classification of HCC unveils several novel mechanisms of immune response and evasion and may help to better predict the distinct patterns of outcome associated with immunotherapy in HCC. A 20-gene signature capturing the Inflamed class can be tested as a direct biomarker of response.
ACKNOWLEDGEMENTS:
Genomic analyses were run at the New York Genomic Center and at the Genomics Core Facility from the Icahn School of Medicine. We would like to thank the Biorepository and Pathology Core of the Icahn School of Medicine at Mount Sinai for their services, especially to Rachel Brody, Frances Avila, Alan Soto, Tin Htwe Thin, Anastasiya Dzhun, and Rafael Cabal. This study has been developed in part in the Centre Esther Koplowitz from IDIBAPS / CERCA Programme / Generalitat de Catalunya.
FUNDING:
This study was partially funded by a grant of Bristol-Myers Squibb to JML at Mount Sinai Liver Cancer Programme, Division of Liver Diseases, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, USA. CM is supported by a Rio Hortega grant from Instituto de Salud Carlos III (ISCIII), Fondo Social Europeo, ID code (CM19/00039). FC is supported by a AECC Clínico Junior grant, ID code (CLJUN20007CAST). PKH is supported by the fellowship grant of the German Research Foundation (DFG: HA 8754/1–1). SLF is supported by grants from the NIDDK (R01-56621,R01-128289) and US Department of Defence (CA150272P3). DS is supported by the Tisch Cancer Institute, the Dr. Franklin M. Klion Young Scientist Research Award and the PhD Scientist Innovative Research Award. JML is supported by National Cancer Institute (P30-CA196521), NIDDK (R01 DK128289), US Department of Defence (CA150272P3), European Commission/ Horizon 2020 Programme (HEPCAR, Ref. 6 67 273-2), Accelerator Award (CRUK, AECC, AIRC) (HUNTER, Ref. C9380/A26813), Samuel Waxman Cancer Research Foundation, Generalitat de Catalunya/AGAUR (SGR-1358), Plan Estratégico de Investigación e Innovación en Salud (PERIS) (SLT017/20/000206), and the Acadèmia de Ciències Mèdiques i de la Salut de Catalunya i de Balears, Ministerio de Economia y Competitividad (MINECO) proyectos de I+D+i PID2019-105378RB-100.
COMPETING INTERESTS:
Dr. Jeff Anderson and Jaclyn Neely are staff scientists from Bristol-Myers Squibb. J.M.L. is receiving research support from Bayer HealthCare Pharmaceuticals, Eisai Inc, Bristol-Myers Squibb, Boehringer-Ingelheim and Ipsen, and consulting fees from Eli Lilly, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Eisai Inc, Celsion Corporation, Exelixis, Merck, Ipsen, Genentech, Roche, Glycotest, Nucleix, Sirtex, Mina Alpha Ltd and AstraZeneca. The remaining co-authors have nothing to disclose related to this manuscript.
Footnotes
DATA TRANSPARENCY STATEMENT: The RNAseq and Whole-exome sequencing data for the primary cohort have been deposited at the European Genome-Phenome Archive (EGA), which is hosted by The European Bioinformatics Institute (EBI) and the Centre for Genomic Regulation (CRG) (Accession code EGAS00001005364). The TCRseq and any other relevant data can be obtained from the corresponding authors upon reasonable request. The data used for liquid-biopsy analysis in a second cohort has been deposited at GEO with accession number GSE174570.
REFERENCES
- 1.Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nature Reviews Disease Primers 2021;7:7. doi: 10.1038/s41572-021-00245-6 [DOI] [PubMed] [Google Scholar]
- 2.Villanueva A Hepatocellular Carcinoma. New England Journal of Medicine 2019;380:1450–62. doi: 10.1056/NEJMra1713263 [DOI] [PubMed] [Google Scholar]
- 3.Llovet JMM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nature reviews Clinical oncology 2021;:Online ahead of print. doi: 10.1038/S41571-021-00573-2 [DOI] [PubMed] [Google Scholar]
- 4.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. New England Journal of Medicine 2020;382:1894–905. doi: 10.1056/NEJMoa1915745 [DOI] [PubMed] [Google Scholar]
- 5.Cheng A-L, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. Journal of Hepatology Published Online First: December 2021. doi: 10.1016/j.jhep.2021.11.030 [DOI] [PubMed] [Google Scholar]
- 6.Castet F, Willoughby CE, Haber PK, et al. Atezolizumab plus Bevacizumab: A Novel Breakthrough in Hepatocellular Carcinoma. Clinical Cancer Research 2021;27:1827–9. doi: 10.1158/1078-0432.CCR-20-4706 [DOI] [PubMed] [Google Scholar]
- 7.Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nature Reviews Cancer 2020;20:662–80. doi: 10.1038/s41568-020-0285-7 [DOI] [PubMed] [Google Scholar]
- 8.Sia D, Jiao Y, Martinez-Quetglas I, et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017;153:812–26. doi: 10.1053/j.gastro.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luke JJ, Bao R, Sweis RF, et al. WNT/β-catenin Pathway Activation Correlates with Immune Exclusion across Human Cancers. Clinical Cancer Research 2019;25:3074–83. doi: 10.1158/1078-0432.CCR-18-1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. doi: 10.1038/nature14404 [DOI] [PubMed] [Google Scholar]
- 11.de Galarreta MR, Bresnahan E, Molina-Sánchez P, et al. β-catenin activation promotes immune escape and resistance to anti–PD-1 therapy in hepatocellular carcinoma. Cancer Discovery 2019;9:1124–41. doi: 10.1158/2159-8290.CD-19-0074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Felden J, Craig AJ, Garcia-Lezana T, et al. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene 2021;40:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harding JJ, Nandakumar S, Armenia J, et al. Prospective genotyping of hepatocellular carcinoma: Clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clinical Cancer Research 2019;25:2116–26. doi: 10.1158/1078-0432.CCR-18-2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haber PK, Torres-Martín M, Andreu-Oller C, et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. Gastroenterology 2021;:(under revision). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.[Dataset]Montironi C, Castet F, Haber PK, et al. Data from: Inflamed and non-inflamed classes of HCC: a revised immunogenomic classification. European Genome Phenome Archive. https://ega-archive.org/studies/EGAS00001005364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.[Dataset]Esteban-Fabró R, Willoughby CE, Piqué-Gili M, et al. Data from: Cabozantinib enhances anti-PD1 efficacy and elicits a neutrophil-based immune response in murine models: implications for human HCC. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE174570 [Google Scholar]
- 17.[Dataset]Ally A, Balasundaram M, Carlsen R, et al. Data from: Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. NCI Genomic Data Commons. 2017.https://portal.gdc.cancer.gov/legacy-archive/search/f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.[Dataset]Villanueva A, Portela A, Sayols S, et al. Data from: DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Gene Expression Omnibus. 2015.https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiang DY, Villanueva A, Hoshida Y, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Research 2008;68:6779–88. doi: 10.1158/0008-5472.CAN-08-0742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachenmayer A, Alsinet C, Savic R, et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18:4997–5007. doi: 10.1158/1078-0432.CCR-11-2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshida Y, Nijman SMB, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer research 2009;69:7385–92. doi: 10.1158/0008-5472.CAN-09-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research Network. Electronic address: wheeler@bcm.edu; Cancer Genome Atlas Research. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.[Dataset]Hsu C-L, Ou D-L, Bai L-Y, et al. Data from: Exploring Markers of Exhausted CD8 T Cells to Predict Response to Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Gene Expression Omnibus. 2021.https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE140901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bassaganyas L, Pinyol R, Esteban-Fabro R, et al. Copy-Number Alteration Burden Differentially Impacts Immune Profiles and Molecular Features of Hepatocellular Carcinoma. Clinical Cancer Research 2020;26:6350–61. doi: 10.1158/1078-0432.CCR-20-1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davoli T, Uno H, Wooten EC, et al. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. doi: 10.1126/science.aaf8399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franch-Expósito S, Bassaganyas L, Vila-Casadesús M, et al. CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications. eLife 2020;9:1–22. doi: 10.7554/eLife.50267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Axelrod ML, Cook RS, Johnson DB, et al. Biological Consequences of MHC-II Expression by Tumor Cells in Cancer. Clinical Cancer Research 2019;25:2392–402. doi: 10.1158/1078-0432.CCR-18-3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson AM, Bullock BL, Neuwelt AJ, et al. Cancer Cell–Intrinsic Expression of MHC Class II Regulates the Immune Microenvironment and Response to Anti–PD-1 Therapy in Lung Adenocarcinoma. The Journal of Immunology 2020;204:2295–307. doi: 10.4049/jimmunol.1900778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019;35:559–572.e7. doi: 10.1016/j.ccell.2019.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kriegsman BA, Vangala P, Chen BJ, et al. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. The Journal of Immunology 2019;203:1999–2010. doi: 10.4049/jimmunol.1900475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Losic B, Craig AJ, Villacorta-Martin C, et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nature Communications 2020;11:1–15. doi: 10.1038/s41467-019-14050-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nature Medicine 2019;25:389–402. doi: 10.1038/s41591-019-0382-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thorsson VV, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rebouissou S, Franconi A, Calderaro J, et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ß-catenin activity associated with liver tumor progression. Hepatology 2016;64:2047–61. doi: 10.1002/HEP.28638 [DOI] [PubMed] [Google Scholar]
- 35.Abitbol S, Dahmani R, Coulouarn C, et al. AXIN deficiency in human and mouse hepatocytes induces hepatocellular carcinoma in the absence of β-catenin activation. Journal of Hepatology 2018;68:1203–13. doi: 10.1016/j.jhep.2017.12.018 [DOI] [PubMed] [Google Scholar]
- 36.Zucman-Rossi J, Benhamouche S, Godard C, et al. Differential effects of inactivated Axin1 and activated β-catenin mutations in human hepatocellular carcinomas. Oncogene 2007;26:774–80. doi: 10.1038/sj.onc.1209824 [DOI] [PubMed] [Google Scholar]
- 37.Cadoux M, Caruso S, Pham S, et al. Expression of NKG2D ligands is downregulated by β-catenin signalling and associates with HCC aggressiveness. Journal of Hepatology 2021;74:1386–97. doi: 10.1016/j.jhep.2021.01.017 [DOI] [PubMed] [Google Scholar]
- 38.Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nature Reviews Cancer Published Online First: March 2021. doi: 10.1038/s41568-021-00339-z [DOI] [PubMed] [Google Scholar]
- 39.Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021;592:450–6. doi: 10.1038/s41586-021-03362-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sangro B, Melero I, Wadhawan S, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. Journal of Hepatology 2020;73:1460–9. doi: 10.1016/j.jhep.2020.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu CL, Ou DL, Bai LY, et al. Exploring Markers of Exhausted CD8 T Cells to Predict Response to Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Liver Cancer Published Online First: 2021. doi: 10.1159/000515305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yong Hong J, Jin Cho H, Sa JK, et al. Hepatocellular carcinoma patients with high circulating cytotoxic T cells and intra-tumoral immune signature benefit from pembrolizumab: results from a single-arm phase 2 trial. doi: 10.1186/s13073-021-00995-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blagih J, Zani F, Chakravarty P, et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Reports 2020;30:481–496.e6. doi: 10.1016/j.celrep.2019.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou X, Singh M, Sanz Santos G, et al. Pharmacologic Activation of p53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discovery 2021;11:3090–105. doi: 10.1158/2159-8290.CD-20-1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tamborero D, Rubio-Perez C, Muiños F, et al. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clinical Cancer Research 2018;24:3717–28. doi: 10.1158/1078-0432.CCR-17-3509 [DOI] [PubMed] [Google Scholar]
- 46.Klement JD, Paschall AV., Redd PS, et al. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. Journal of Clinical Investigation 2018;128:5549–60. doi: 10.1172/JCI123360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berglund A, Mills M, Putney RM, et al. Methylation of immune synapse genes modulates tumor immunogenicity. Journal of Clinical Investigation 2020;130:974–80. doi: 10.1172/JCI131234 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.