

SUMMARY
Protein fatty acylation regulates numerous cell signaling pathways. Polyunsaturated fatty acids (PUFAs) exert a plethora of physiological effects, including cell signaling regulation, with underlying mechanisms to be fully understood. Herein, we report that docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) regulate PI3K-AKT signaling by modifying PDK1 and AKT2. DHA-administered mice exhibit altered phosphorylation of proteins in signaling pathways. Methylene bridge-containing DHA/EPA acylate δ1 carbon of tryptophan 448/543 in PDK1 and tryptophan 414 in AKT2 via free radical pathway, recruit both the proteins to the cytoplasmic membrane, and activate PI3K signaling and glucose uptake in a tryptophan acylation-dependent but insulin-independent manner in cultured cells and in mice. DHA/EPA deplete cytosolic PDK1 and AKT2 and induce insulin resistance. Akt2 knockout in mice abrogates DHA/EPA-induced PI3K-AKT signaling. Our results identify PUFA’s methylene bridge tryptophan acylation, a protein fatty acylation that regulates cell signaling and may underlie multifaceted effects of methylene-bridge-containing PUFAs.
In brief
Hu et al. find that methylene-bridge-containing polyunsaturated fatty acids EPA and DHA covalently modify surface tryptophan of membrane-targeting proteins, including PDK1 and AKT2, and recruit them to the cytoplasmic membrane to activate AKT signaling. The EPA and DHA actions facilitate glucose uptake but can also induce insulin resistance.
Graphical Abstract
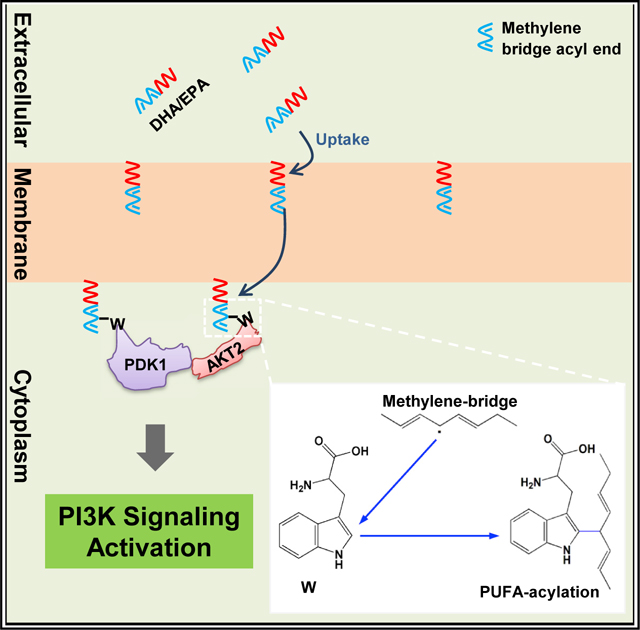
INTRODUCTION
Among a multitude of roles of the fatty acids, including their function as building blocks of membrane and fuel sources of energy metabolism, they are also involved in signaling molecules. Many signaling pathways are regulated by protein fatty acylation. Two major types of fatty acylation are documented in eukaryotic cell signaling regulators. N-myristoylation, catalyzed by N-myristoyltransferases, attaches myristate to glycine via amide bonds in the consensus sequence (M)GXXT/S in proteins (Farazi et al., 2001) to regulate calcium-dependent protein kinases and small GTPases of the ADP-ribosylation factor family (Boutin, 1997). Palmitoylation attaches palmitate to cysteine via a thioester bond (Smotrys and Linder, 2004) to regulate signaling such as epidermal growth factor receptor (EGFR) (Kharbanda et al., 2020), N-ras (Cuiffo and Ren, 2010), and TEA domain (TEAD) transcription factors (Noland et al., 2016). In addition, some other fatty acylation has also been reported (Resh, 2016) that directs the modified proteins to membranes, to exert their effects.
Polyunsaturated fatty acids (PUFAs) are well known for their abilities to alter cell signaling. The discovery of correlations between high PUFA contents in the diet of an Inuit population and low incidences of myocardial infarction, platelet reactivity, and antiatherogenic blood lipid pattern (Bang et al., 1971, 1980; Dyerberg et al., 1978) has promoted the worldwide dietary supplementation of PUFAs, particularly marine omega-3 (ω−3) fatty acids such as docosahexaenoic acid (DHA) (22:6) and eicosapentaenoic acid (EPA) (20:5) to reduce the risks of various human diseases, such as cardiovascular diseases and diabetes (William et al., 2017; De Caterina, 2011; The Ascend Study Collaborative Group, 2018). Despite the numerous beneficial efficacies of DHA and EPA, including their blood-glucose-lowering effects (De Caterina, 2011; Dry Eye Assessment and Management Study Research Group et al., 2018; The Ascend Study Collaborative Group, 2018; Origin Trial Investigators et al., 2012a, 2012b; Roncaglioni et al., 2013; Zambon et al., 1992), their efficacies on glucose disposal-regulating insulin signaling were controversial. Some studies concluded that DHA and EPA sensitize insulin signaling (Fedor and Kelley, 2009; Lalia and Lanza, 2016), consistent with their blood-glucose-lowering efficacies, while some other studies have reported their efficacies in inducing insulin resistance (Ahren et al., 2009; Brown et al., 2019; Lalia and Lanza, 2016; Mostad et al., 2008; Stacpoole et al., 1988; Topping et al., 1987). It has recently been reported that 4-hydroxy-2-hexenal, a byproduct of ω−3 fatty acid peroxidation, induces insulin resistance (Soulage et al., 2018), but contradicts the blood-glucose-lowering effects of DHA and EPA. These discrepancies in insulin signaling regulation by DHA and EPA require in-depth understanding of the mechanisms of actions of DHA and EPA.
Several mechanisms had been proposed for the actions of DHA, EPA, and other PUFAs. PUFAs can be converted to other metabolites with biological functions. For example, nitration of PUFAs can generate nitro-fatty acids that globally suppress inflammation (Woodcock et al., 2006); cyclooxygenase 2 converts EPA into prostaglandin I3, a weak inflammation inducer (Lee et al., 2009b). As integral components of the membrane, PUFAs may alter the properties of lipid rafts and caveolae and thus contribute to membrane fluidity. As signaling molecules, PUFAs may bind to proteins or receptors to alter signaling pathways in cells. For example, ω−3 PUFAs were found to bind to G protein-coupled receptor 120 in monocytes and macrophages and mediate insulin-sensitizing and anti-diabetic effects by repressing macrophage-induced tissue inflammation (Oh et al., 2010b). Although these suggested mechanisms can explain the effects of given PUFAs, they cannot explain common features of PUFAs, such as the number and position of double bonds in PUFAs, which are critical for the efficacies of PUFAs (Simopoulos, 2002a). Whether PUFAs exsert similar efficacies via common mechanisms remains to be answered.
RESULTS
DHA altered membrane protein trafficking-regulated PI3K signaling
Employing a high-throughput phosphoproteomics analysis (Humphrey et al., 2015), we were able to compare the levels of 4205 detected phosphorylation sites in proteins of skeletal muscles of DHA-treated and -untreated 129/C57BL6 mice (Table S1). Among the 326 sites that were altered by more than 2-fold (p < 0.05), 214 sites were upregulated, and 112 sites were downregulated by DHA treatment (Table S1). Bioinformatic analysis revealed that DHA upregulated the phosphorylation of components of PI3K-AKT signaling and its related pathways, such as insulin signaling and insulin resistance signaling (Figure 1A), consistent with the efficacies of DHA activating glucose disposal as reported. DHA also downregulated phosphorylation in pathways such as biosynthesis of antibiotics and carbon metabolism (Figure S1A). Interestingly, each of the identified DHA-upregulated signaling pathways contained at least one component regulated by membrane trafficking (Table S2), suggesting the possible roles of DHA in regulating cell signaling through alteration of protein membrane trafficking.
Figure 1. DHA alters membrane protein trafficking-regulated PI3K signaling.

(A) Top-ranked Gene Ontology (GO) terms of DHA upregulated phosphorylation sites in DHA-treated 129/C57BL6 mice.
(B) DHA and EPA promote GLUT4 membrane enrichment. Total GLUT4 expression and cell surface GLUT4 expression were determined by confocal fluorescence microscopy for mCherry (red) and Myc (green), respectively, in 3T3-L1 cells that were successfully transfected with the Myc-GLUT4-mCherry construct. Cells treated with control, DHA, EPA, and insulin were measured. Scale bars, 25 μm.
(C and D) DHA and EPA enrich PDK1 and AKT2 at the membrane. PDK1 (C) and AKT2 (D) were ectopically expressed in 3T3-L1 cells. The localization of PDK1 and AKT2 was monitored using confocal live-cell images after the cells were treated with DHA, EPA, and insulin. Non-treated cells were used as controls. Results of representative time points are presented. Scale bar, 20 μm. The fraction of responded cells was quantified (right) (n = 50, mean ± SEM).
(E) DHA and EPA promoted PDK1 onto the membrane of AKT2−/− cells. The contents of PDK1 in membrane of AKT2−/− 3T3-L1 cells before and after DHA or EPA treatment were detected.
(F) AKT2 is required for DHA and EPA to induce GLUT4 membrane enrichment. GLUT4 distribution was determined in wild-type (WT) and AKT2−/− 3T3-L1 cells after DHA and EPA treatment. Scale bar, 25 μm.
(G and H) DHA and EPA do not alter PI3K and PIP3. 3T3-L1 cells treated with DHA or EPA and the membrane protein levels of p110α and p110β of PI3K (G), and membrane PIP3 levels (H) were detected. Untreated levels were arbitrarily set as 100% (n = 4, mean ± SEM).
(I) DHA and EPA did not activate insulin signaling. The levels of p-IR(Y1150/1151), p-IRS1(Y895), p-AKT2(T308), p-AKT2(S473), and p-AS160(T642) were detected in DHA-, EPA-, and insulin-treated 3T3-L1 cells.
(J) DHA- and EPA-activated PI3K signaling lasts longer than that activated by insulin treatment. The levels of p-AKT2(T308) were determined at different time points after 3T3-L1 cells were treated with DHA, EPA, or insulin.
Further, to confirm the ability of DHA and EPA to alter protein membrane trafficking, we constructed Myc-GLUT4-mCherry fusion protein (Figure S1B) to evaluate the extracellular (Myc) and intracellular (mCherry) distribution of GLUT4 in cells (Lim et al., 2015). Like insulin, both DHA (Kim et al., 2015; Oh et al., 2010b) and EPA promoted translocation of PDK1 and AKT2 (Figures S1C) and GLUT4 (Figure 1B) to the cytoplasmic membrane of 3T3-L1 mouse adipocytes, and GLUT4 to the cytoplasmic membrane of Chinese hamster ovary CHO-K1 cells (Figure S1D). Moreover, PDK1 and AKT2, two upstream regulators of GLUT4, were time-dependently recruited to the cytoplasmic membrane from the cytoplasm of 3T3-L1 following DHA, EPA, and insulin treatments, with DHA and EPA taking a longer time than insulin to do so (Figures 1C and 1D). The DHA-, EPA-, and insulin-induced membrane accumulation and cytoplasmic depletion of GLUT4, PDK1, and AKT2 were further confirmed by subcellular fractionation (Figure S1E). Notably, DHA and EPA promoted PDK1 (Figure 1E), but not GLUT4, to the membrane of AKT2 knockout (Akt2−/−) 3T3-L1 and CHO-K1 cells (Figures 1F and S1F). On the contrary, DHA and EPA treatments failed to alter the membrane levels of PI3K and phosphatidylinositol-3,4,5-triphosphate (PIP3) that were induced by insulin treatment (Figures 1G and 1H). Similarly, DHA and EPA treatment could not induce the phosphorylation of either tyrosine 1150/1151 of the insulin receptor (IR) or tyrosine 895 of the IR substrate (Figure 1I), the signatures of insulin signaling activation, consistent with DHA and EPA activating AKT2, as assayed by either AKT2 phosphorylation (Figure 1J) or direct enzymatic analysis (Figure S1G). Additionally, the retention of AKT2 (Figure S1H) and activation of PI3K signaling by DHA and EPA lasted over 4 h with no noticeable decay, whereas insulin-activated AKT signaling diminished 2 h after treatment (Figure 1J), and DHA and EPA dose-dependently activated PI3K signaling (Figures S1I and S1J). Collectively, these results suggested that DHA and EPA act on GLUT4 through AKT2, and the mechanism of activation of PI3K-AKT signaling is distinct from that of insulin.
Methylene bridge-containing PUFAs react with tryptophan
To evaluate whether DHA and EPA could covalently attach to target proteins, several purified proteins, namely PDK1, AKT2, phosphoglycerate kinase 1 (PGK1), and pyruvate kinase isozymes M2 (PKM2), were incubated with fatty acids with distinct acyl chains (Figure 2A). The fatty acids containing at least one methylene bridge, i.e., two or more double bonds in the cis configuration separated by a single methylene group, induced slower migration of these proteins in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 2B), in a disulfide bond breaker DTT-independent manner (Figure S2A), indicating a chemical reaction beyond disulfide bond forming happened between methylene bridge-containing PUFAs and proteins. Moreover, incubation with linolenic acid phosphoserine (PS) resulted in no migration shifts of PDK1 and AKT2; however, further treatment of PS-incubated PDK1 and AKT2 with lipase to expose the carboxyl groups of PS via triglycerol removal (Figure S2B) resulted in slower shift of these proteins (Figure 2C). Furthermore, carboxy-methyl/ethyl-DHA/EPA failed to induce mobility shift of AKT2 and PDK1 in vitro (Figure S2C). These results were consistent with the methylene bridge reacting with PDK1 and AKT2 to add negatively charged carboxyl groups and thus slow their migration in SDS-PAGE.
Figure 2. Methylene bridge PUFAs acylate tryptophan.

(A) Chemical structures of palmitic acid, oleic acid, conjugated linoleic acid, linoleic acid, EPA, and DHA. The names of methylene bridges (red circled) containing PUFAs are in blue.
(B) Methylene bridge PUFAs form covalent adducts with proteins. PDK1, AKT2, PGK1, PKM2, and their tryptophan-free mutants (AKT27W/L, PDK16W/L, PGK14W/L, PKM23W/L) were incubated with specific fatty acids. The resulting proteins were resolved by SDS-PAGE.
(C) Addition of carboxyl groups to proteins by acylation caused protein migration shift. Untreated and linolenic acid-PS-incubated AKT2 and PDK1 samples followed by lipase treatment were resolved by SDS-PAGE.
(D) Formation of EPA-acylated peptide was detected by MS and confirmed by MS/MS.
(E and F) Tryptophan inhibits methylene bridge protein acylation. (E) DHA or EPA was used to acylate AKT2 and PDK1 in the absence or presence of tryptophan or alanine. The products were analyzed by SDS-PAGE. (F) DHA, EPA, or linoleic acid was used to acylate a synthetic peptide in the absence or presence of tryptophan or alanine. The products were analyzed by MS (left). The amounts of acylated peptide were quantified (right) by MS signals (n = 3, mean ± SEM).
Subsequently, a series of synthetic N-terminal acetylation-blocked peptides with all 20 amino acids appearing at least once (Figure S2D) were tested for their reactivities with methylene bridges containing EPA, DHA, and linoleic acid to identify the amino acids that react with methylene bridges. Mass spectrometry (MS) identified new species that were formed only in reaction mixes with tryptophan-residue-containing peptides (Figures S2D and S2E), whereas the tandem MS (MS/MS) analysis of the newly formed species revealed that EPA, DHA, and linoleic acid formed methylene acylation adducts (Figures 2D, S2F, and S2G). Moreover, substituting the tryptophan with alanine in the reactive peptides abrogated their reactivities to the fatty acids (Figure S2H), and substituting all tryptophan residues with leucine residues in AKT2, PDK1, PGK1, and PKM2 (AKT27W/L, PDK16W/L, PGK14W/L, PKM23W/L), respectively, prevented linoleic acid, EPA, and DHA from inducing mobility shifts of these proteins (Figure 2B). These, together with tryptophan, but not alanine supplemental to the reaction mix, abrogating EPA, DHA, and linoleic acid to induce slower migration of recombinant PDK1 and AKT2 (Figure 2E) and form adducts with tryptophan-containing peptides (Figure 2F), collectively identified tryptophan as the amino acid that reacts with the methylene bridges.
Cδ1 in tryptophan is the site for methylene bridge fatty acylation
Methylene bridges might form free radicals in vitro (Aliwarga et al., 2017; Davis et al., 2006) and in the presence of other radicals such as reactive oxygen spices (ROS), and these free radicals could attack either Nℇ1 or Cδ1 in tryptophan to form adducts (Figure 3A). Free radical inducer ammonia persulfate promoted DHA, EPA, and linoleic acid to form peptide adducts (Figure 3B) and, conversely, quenching the free radicals in the reaction mix with ascorbic acid (Nimse and Pal, 2015) prevented the formation of adducts (Figure 3C), which substantiated the free radical reaction model. Moreover, on the one hand, replacing tryptophan in the synthetic peptides (Figure S3A) with a Cδ1 methyl-blocked tryptophan analogue (Figure S3B) abrogated the reactivity of the peptide with DHA, EPA, and linoleic acid; on the other hand, replacement of tryptophan with a Nℇ1 methyl-blocked tryptophan analogue (Figure S3B) maintained its reactivity and only decreased its reaction efficiency (Figure 3D). These findings suggested that Cδ1 in tryptophan is modified by methylene bridge-containing PUFAs. Furthermore, the nuclear magnetic resonance (NMR) analysis revealed that EPA, DHA, and linoleic acid reduced the signals of Cδ1 of tryptophan (Figure 3E) and that of Cδ1-containing tryptophan analogues such as tryptamine and 5-hydroxyl-tryptamine (Figures S3B and S3C). Additionally, tryptophan (Figures S1I and S1J), tryptamine, and 5-hydroxyl-tryptamine antagonized DHA and EPA to decrease phosphorylation of AKT2 and AS160 (Figures 3F, 3G, S3D, and S3E). Lastly, probed with DHA or EPA antibodies that recognized the DHA-/EPA-acylated peptides (Figures S3F), or DHA-/EPA-acylation-specific antibodies that were made with DHA-/EPA-treated tryptophan-containing peptides (Figures S3G), DHA and EPA dose-dependently increased acylations on AKT2 and PDK1 (Figures S3H and S3I) in a tryptophan-reversible manner. These findings collectively substantiated the speculation that Cδ1 in tryptophan is the site of methylene bridge acylation.
Figure 3. Cδ1 of tryptophan is the site of methylene bridge acylation.

(A) Proposed mechanisms for methylene bridge-tryptophan adduct formation. After induction by other free radicals, methylene bridge free radicals may attack either Cδ1 and Nɛ1 of tryptophan to form either adduct I or II, respectively.
(B) Free radical inducer promotes tryptophan acylation. The formation of EPA, DHA, and LA adducts in a peptide with or without ammonia persulfate in thereaction mix was analyzed by MS (upper), and adducts levels were measured (lower) (n = 3, mean ± SEM).
(C) Free radical scavenger inhibits tryptophan acylation. The formation of the EPA, DHA, and LA adduct in a peptide with or without ascorbic acid in the reaction mix was analyzed by MS (upper), and adducts levels were measured (lower) (n = 3, mean ± SEM).
(D) Cδ1 of tryptophan is the acylation site by a methylene bridge. A tryptophan-containing peptide (Ac-FTEGAFKDWGYQLA) and peptides with the same sequence but with tryptophan replaced by Nɛ1 or Cδ1 methyl-blocked tryptophan were tested for their abilities to be adducted by linoleic acid, EPA, and DHA. Adduct formation was assayed by MS.
(E) NMR analysis of the reactivities of Cδ1 of tryptophan. The NMR signal of Cδ1 of tryptophan was decreased by LA (left), DHA (middle), and EPA (right).
(F and G) AKT signaling activation by DHA was reversed by tryptamine (F) and 5-hydroxy-tryptamine (5-HT) (G). The levels of p-AKT2(T308) and p-AS160(T642) were determined in 3T3-L1 cells cultured in media supplemented with DHA, DHA/tryptamine, or DHA/5-HT.
DHA and EPA acylate PDK1 and AKT2 and recruit them to membrane
Treating the cells with various levels of DHA or EPA, and estimating their membrane and cytosolic levels by gas chromatography (GC)-MS revealed that the accumulations of DHA and EPA in the membrane fraction were dose and time dependent, whereas they showed a sparse distribution in the cytoplasm (Figures 4A and 4B), as reported earlier (Harris et al., 1984; Kim, 2007; Kim et al., 1999). Moreover, employing living-cell imaging, we found the protein DHA- and EPA-acylation signals were accumulated in cytoplasmic membrane of CHO-K1 cells (Figure S4A), consistent with membrane-embedded DHA and EPA modifying proteins. Further, it was observed that insulin induced membrane accumulation of PDK1 and AKT2 in few minutes; in contrast, DHA and EPA induced their membrane accumulation in about half an hour (Figures 1C and 1D), which was like the time required for DHA and EPA to accumulate in membrane. Moreover, employing chlorpromazine, which inhibits phospholipid synthesis (Novotny et al., 1984), decreased the phosphorylation of AKT2 and AS160, whereas quinacrine, which inhibits the hydrolysis of phospholipids (Chiariello et al., 1987), potentiated DHA and EPA to increase phosphorylation of AKT2 and AS160 (Figure 4C). Furthermore, uptake of DHA and EPA was nearly completely converted into phospholipids (Figure S4B). These results supported that DHA and EPA act on proteins in the forms of phospholipids. Lastly, DHA and EPA also promoted PDK1 and AKT2 cytoplasmic membrane accumulation in skeletal muscle of 129/C57BL6 mice in a tryptophan-inhibitable manner (Figure 4D). Collectively, from these results, it is speculated that EPA and DHA promote membrane enrichment of PDK1 and AKT2 via tryptophan acylation.
Figure 4. Membrane-embedded DHA and EPA acylate and anchor PDK1 and AKT2 to membrane.

(A) Dose-dependent accumulation of DHA and EPA to the cell membrane. Various levels of DHA or EPA were added to the culture media for 1 h. The membrane and cytosolic levels of DHA and EPA were detected by GC-MS (n = 3, mean ± SEM).
(B) Time course accumulation of DHA and EPA to the cell membrane. 100 μM DHA or EPA were used to treat cells; the membrane and cytosolic levels of DHA and EPA were detected by GC-MS (n = 3, mean ± SEM).
(C) PUFA phospholipid formation affected DHA and EPA efficacies. The activation of PI3K-AKT signaling by DHA and EPA was detected in 3T3-L1 cells when phospholipid formation and hydrolyzation were inhibited by chlorpromazine (CPZ) and quinacrine (QA), respectively.
(D) DHA and EPA acylation promote PDK1 and AKT2 enrichment on the cell membrane in vivo. 129/C57BL6 mice were starved for 6 h before gavage with normal saline (control), DHA, DHA/tryptophan, EPA, or EPA/tryptophan. The distribution of PDK1 and AKT2 in the skeletal muscle of treated mice was analyzed by immunofluorescence following sacrifice after 4 h of treatment. Scale bar, 50 μm.
(E) The sites and the acylation levels of PDK1 and AKT2 in cells before and after DHA or EPA treatment. Targeted proteomic analysis was used to search for possible acylated PDK1 and AKT2 in 3T3-L1 adipocytes prior to and after being treated with DHA or EPA. Levels of acylation were estimated by employing synthetic peptides of known concentrations as internal controls (n = 3, mean ± SEM).
(F) DHA and EPA promote AKT2, AKT2W334L, PDK1, PDK1W266/347L, but not the AKT2W414L, PDK1W448/543L enrichment on the cell membrane. The localization of AKT2, AKT2W334L, AKT2W414L, PDK1, PDK1W266/347L, and PDK1W448/543L were monitored in confocal live-cell images after the cells were treated with DHA and EPA. Results of representative time points are presented (left). Scale bar, 20 μm. The fraction of responded cells were quantified (right) (n = 50, mean ± SEM).
AKT2 and PDK1 isolated from DHA-/EPA-treated 3T3-L1 cells are partially acylated, as shown by lipase treatment causing mobility shift of a fraction of PDK1 and AKT2 (Figure S4C), an indication of more negativity of protein, which may change the SDS binding to proteins. To find out which tryptophan residues of PDK1 and AKT2 are acylated in vivo, we searched and quantified their acylated peptides by MS. The acylation levels of W448 and W543 of PDK1 and W414 of AKT2, but not other tryptophan residues in these two proteins, was significantly increased by DHA and EPA treatment as quantified by MS quantitation (Figure 4E). The specific DHA/EPA acylation of PDK1 and AKT2 in cells was supported by removal of W448 and W543 of PDK1 and W414 of AKT2 almost completely abrogating DHA or EPA to increase their DHA/EPA acylation (Figures S4D and S4E). However, only removal of all surface W residues, rather than in vivo acylated W sites, rendered PDK1 and AKT2 mutants irresponsive to DHA-/EPA-induced mobility shifts (Figure S4F), suggesting the DHA-/EPA-acylation specificity is induced in vivo. Notably, DHA and EPA also activated AKT1 signaling (Figure S4G), and replacing W413 in AKT1, the equivalent residue of AKT2 W414, also abrogated AKT1’s DHA/EPA acylation (Figure S4H). These results further confirmed that AKT2 W414 is the major DHA-/EPA-acylation site, suggesting that DHA and EPA effects are beyond AKT2.
Removal of in vivo acylation sites did not prevent DHA and EPA inducing mobility shifts of mutant AKT2 and PDK1 in vitro, which were abrogated only by removal of all surface tryptophan in them (Figures 2B and S4F), suggested that DHA and EPA acylation specificity can only be obtained in vivo and, consistent with replacing W448 and W543 of PDK1 and W414 of AKT2 in PDK1 and AKT2 to leucine (PDK12W/L, AKT2W414L), which had negligible effects on their activities (Figures S4I and S4J), prevented DHA and EPA from enriching PDK1 and AKT2 onto the membrane as analyzed by both immunofluorescence (Figure 4F) and western blotting (Figures S4K and S4L). Lastly, in investigating whether PDK1 and AKT2 DHA/EPA acylation are reversible, we removed DHA or EPA from culture media and found that the AKT2 phosphorylation signals time-dependently disappeared with the decay of acylation signal while the AKT2 protein levels remain unaffected (Figures S4M and S4N). Moreover, cell lysate, but not denatured cell lysate, had the ability to reverse the shifted bands of in vitro DHA-/EPA-acylated AKT2 and PDK1 that were affinity purified from 3T3-L1 cells (Figure S4O). These results indicated that DHA/EPA acylation can possibly be reversed by as-yet unidentified enzymatic mechanisms. Furthermore, AKT2 DHA-/EPA-acylation signal presented in endoplasmic reticulum (ER) after treatment (Figure S4P) suggested that either DHA/EPA acylate ER membrane AKT2, or DHA-/EPA-acylated AKT2 may translocate among membranes.
Membrane-docking proteins are preferably recruited by DHA and EPA
Next, we investigated whether DHA and EPA recruit the cytoplasmic proteins onto membrane unbiasedly, and found that DHA and EPA only increased the membrane contents of membrane-targeting PDK1, AKT2, and TNF receptor-associated factor 6 (TRAF6), but not other proteins such as mitochondria-targeting SIRT3, cytochrome c oxidase subunit 4 (COX4), cytosolic enzymes GAPDH and PGK1, and membrane-embedded Na-K-ATPase (Figure 5A). These results suggested that membrane targeting is a prerequisite of a protein to be recruited onto membrane by DHA and EPA. Replacing arginine 25 in the PH domain of AKT2 with cysteine (AKT2R25C) or arginine 474 in the PH domain of PDK1 with alanine (PDK1R474A) damaged their PIP3 binding (Carpten et al., 2007) but did not prevent them being acylated by DHA or EPA in vitro (Figure S5A and S5B), rendered them unable to be acylated by DHA or EPA in 3T3-L1 cells (Figures 5B and 5C), and destroyed the membrane docking of AKT2R25C (Figure 5D) and PDK1R474A (Figure S5C). This consequently aborted the ability of DHA or EPA to recruit AKT2R25C to the membrane in 3T3-L1 adipocytes; conversely, fusing a PIP3-binding PH domain to cytoplasmic PGK1 (Figure S5D) enabled DHA and EPA to enrich the fusion protein to the membrane and increase its DHA/EPA acylation levels (Figures 5E and 5F), which confirmed that a membrane-targeting protein can be recruited onto membrane by DHA and EPA via its tryptophan residue. Consistently, neither AKT2R25C (Figure 5G) nor PDK1R474A (Figure 5H) was able to restore PI3K signaling responses to DHA/EPA in Akt2−/− or in Pdk1−/− 3T3-L1 cells, respectively. Moreover, increased PIP3 levels with insulin promoted and decreased PIP3 levels with wortmannin inhibited, respectively, AKT2 DHA/EPA acylation and PI3K signaling activation by DHA/EPA (Figures S5E and S5F). Further, DHA (Figure S5G) and EPA (Figure S5H) acylated constitutive membrane anchoring myr-AKT (Kohn et al., 1996) regardless of whether it contained an intact PH domain. These, together with the acylation of PDK1 and AKT2 being time dependent (Figure 5I), and Triton X-100, a known trans-bilayer lipid motion (flip-flop) activator (Pantaler et al., 2000) that may increase the exposure of membrane-embedded DHA and EPA to cytoplasm (Figure S5I), shortened the time of membrane enrichment of AKT2 (Figure 5J) and PDK1 (Figure S5J) by DHA and EPA, all consistent with transient membrane anchoring by PIP3 providing sufficient chance for membrane-embedded DHA and EPA to acylate and anchor PDK1 and AKT2 onto the membrane to activate PI3K-AKT signaling (Figure S5K).
Figure 5. DHA and EPA selectively recruit membrane-docking proteins onto the membrane.

(A) DHA and EPA specifically promote membrane-targeting proteins to the membrane. HepG2 cells were treated with DHA or EPA for 1 h before their membrane fractions were obtained and detected for AKT2, PDK1, TRAF6, SIRT3, COX4, PGK1, GAPDH, and Na-K-ATPase by western blotting.
(B and C) Impaired PIP3 binding abrogates AKT2 and PDK1 acylation. 3T3- L1 adipocytes expressing AKT2-FLAG and AKT2R25C-FLAG (B), or expressing PDK1-FLAG and PDKL1R474A-FLAG (C) were treated with DHA or EPA for 1 h. The DHA- (upper) and EPA-acylation (lower) of these proteins were analyzed.
(D) Impaired PIP3 binding abrogates AKT2 enrichment at the membrane. 3T3-L1 adipocytes expressing AKT2-EGFP and AKT2R25C-EGFP were treated with DHA, EPA, or insulin. The localization of the EGFP was monitored by confocal live-cell imaging. Scale bar, 20 μm. The fraction of response cells was quantified (right) (n = 50, mean ± SEM).
(E) PH domain fusion enables PGK1 enrichment to the membrane by DHA and EPA. 3T3-L1 cells expressing PGK1-PH-EGFP and PGK1-EGFP were treated with DHA or EPA. EGFP localization was monitored by confocal live-cell imaging. Scale bar, 20 μm. The fraction of response cells was quantified (right) (n = 50, mean ± SEM).
(F) PH domain fusion enables PGK1 to be acylated by DHA and EPA. PGK1-PH-FLAG and PGK1-FLAG were expressed in 3T3-L1 cells. The DHA (upper) and EPA acylation (lower) of these proteins were analyzed.
(G and H) Both AKT2 and PDK1 are required for DHA and EPA to activate PI3K signaling. The abilities of DHA, EPA, and insulin to activate PI3K signaling were detected in AKT2−/− cells and AKT2−/− cells that put back either AKT2 or AKT2R25C (G), and in PDK1−/− cells and PDK1−/− cells that put back either PDK1 or PDK1R474A (H).
(I) DHA and EPA time-dependently induce mobility shifts of proteins. Immunoprecipitated PDK1 and AKT2 were incubated with either DHA or EPA. The resulting proteins were purified from the reaction mix after the start of incubation for different times before they were resolved by SDS and blotted by antibodies.
(J) Membrane flip-flop activity promotes DHA and EPA acylation. Confocal live-cell fluorescence images were obtained for AKT2-EGFP-expressing 3T3-L1 cells that were treated with DHA or EPA with or without Triton X-100 (10 nM). Triton X-100 was supplemented prior to EPA and DHA treatment for 1 h. Scale bar, 20 μm. The fraction of response cells was quantified (right) (n = 50, mean ± SEM).
DHA and EPA promote glucose uptake through acylating tryptophan of PDK1 and AKT2
In 3T3-L1 cells, DHA and EPA increased phosphorylation levels of threonine 308 (T308) of AKT2, serine 227 of ribosomal S6 kinase-2 (Jensen et al., 1999), substrates of PDK1, and serine 642 (S642) of AS160 (Alessi et al., 1996) (Figure 1I), serine 9 of glycogen synthase kinase-3 (Cross et al., 1995), threonine 246 of PRAS40 of the mammalian target of rapamycin complex 1 (Sancak et al., 2007), and substrates of AKT2 (Figure 6A), indicating that DHA and EPA could activate PI3K-AKT signaling. However, the increased phosphorylation of AKT2 and AS160 by DHA and EPA in 3T3-L1 cells (Figure 6B) and the increased phosphorylation of AKT2 in adipose and skeletal muscles and in liver (Figures 6C and S6A) of 129/C57BL6 mice with DHA/EPA chow, which brought cell membrane DHA/EPA levels to around 40 mg/g(protein), comparable levels (Figure S6B) with those in other in vivo studies (Wen and Kim, 2004; Zhang et al., 2015), were reversed by the supplementation of tryptophan, but not alanine, in the culture media. These findings supported that tryptophan acylation promoted the abilities of DHA and EPA to activate PI3K-AKT signaling in cultured cells and in vivo.
Figure 6. DHA or EPA tryptophan acylation activate AKT signaling and promotes glucose uptake.

(A) DHA and EPA activate PDK1 and AKT2. Phosphorylation levels of substrates of PDK1 and AKT2 were detected in 3T3-L1 cells treated with DHA and EPA.
(B) AKT signaling activation by DHA or EPA was reversed by tryptophan. The levels of p-AKT2(T308) and p-AS160(T642) were determined in 3T3-L1 cells cultured in media supplemented with DHA, DHA/tryptophan, DHA/alanine, EPA, EPA/tryptophan, or EPA/alanine.
(C) DHA acylation activates PI3K/AKT signaling in vivo. The levels of p-AKT2(T308) were determined in adipose tissue, liver, and muscle of starved 129/C57BL6 mice treated with DHA, DHA/tryptophan, or DHA/alanine for 4 h.
(D) DHA- or EPA-induced glucose uptake was reversed by tryptophan. The 2-DG uptake abilities of 3T3-L1 were determined when cells were cultured in DMEM or DMEM supplemented with DHA, DHA/methyl-tryptophan, DHA/alanine, EPA, EPA/methyl-tryptophan, EPA/alanine, insulin insulin/methyl-tryptophan, or insulin/anine, (n = 3, mean ± SEM).
(E) DHA and EPA promote glucose disposal in vivo. After food removal, the blood glucose levels of 129/C57BL6 mice and DHA-, DHA/tryptophan-, EPA-, EPA/tryptophan, insulin-, or insulin/tryptophan-treated 129/C57BL6 mice were detected (n = 6, mean ± SEM).
(F) DHA and EPA acylation induces glucose tolerance in mice. Glucose tolerance tests were performed in 129/C57BL6 mice and in DHA-, DHA/tryptophan-, EPA-, EPA/tryptophan, insulin-, or insulin/tryptophan-treated 129/C57BL6 mice (n = 6, mean ± SEM).
(G) AKT2 tryptophan acylation is essential for AKT signaling activation. AKT2−/− 3T3-L1 cells were transfected with FLAG-tagged AKT2 and AKT2W414L. The levels of p-AKT2(T308), p-AS160(T642) in the whole cell lysate, and AKT2 in the cytosol were detected by western blotting after the transfected cells were treated with DHA, EPA, or insulin.
(H) Tryptophan acylation accounts for DHA’s and EPA’s abilities to increase glucose uptake. The effects of DHA, EPA, and insulin on 2-DG uptake were analyzed in WT, AKT2−/−, and AKT2−/− transfected with AKT2 or AKT2W414L 3T3-L1 cells (n = 3, mean ± SEM).
(I) DHA, EPA, and insulin cannot increase p-AS160(T642) in Akt2−/− mice. The levels of p-AS160(T642) in Akt2−/− mouse skeletal muscle and adipose were determined.
(J) Overexpression of AKT2, but not AKT2W414L, enabled DHA and EPA to stimulate GLUT4 membrane enrichment. The levels of GLUT4 in membrane fraction of the skeletal muscle of Akt2−/− mice overexpressed with vector, AKT2, and AKT2W414L.
Glucose uptake, measured by 2-deoxy-D-glucose (2-DOG) uptake (Cuendet et al., 1976), was also upregulated by DHA, EPA, and insulin in 3T3-L1 cells; however, only the DHA- and EPA-promoted glucose uptake was reversed by methyl-tryptophan treatment (Figure 6D), which increased intracellular tryptophan levels (Figure S6C). Furthermore, in 129/C57BL6 mice, glucose disposal test and glucose tolerance test were tryptophan-reversibly enhanced by EPA or DHA (Figures 6E and 6F). These results were consistent with DHA and EPA activating the PDK1-AKT2-AS160 axis and consequently glucose uptake via tryptophan acylation.
To further confirm that DHA and EPA function through PDK1 and AKT2, we deleted AKT2 from 129/C57BL6 mice (Akt2−/− mice) (Figure S6D) and investigated their responses to DHA and EPA treatments. AKT2 or PDK1 deletion rendered the phosphorylation levels of AS160 (Figures 6G and S6E) and glucose uptake (Figure 6H) of Akt2−/− or PDK1−/− 3T3-L1 cells irresponsive to the treatments of insulin, DHA, and EPA, consistent with the activation of both AKT2 and PDK1 being required for PI3K signaling activation by DHA, EPA, and insulin. However, putting back tryptophan acylation null AKT2W414L restored the alterations in AS160 phosphorylation and glucose uptake rendered by insulin treatment but failed to restore those caused by DHA and EPA treatments (Figures 6G and 6H), consistent with tryptophan acylation triggering DHA and EPA to activate PI3K signaling and glucose uptake; the PI3K signaling activated by insulin can be rescued by PDK1 and PDK1W448/543L put back, while the PI3K signaling activated by DHA and EPA can only be rescued by PDK1, but not tryptophan acylation null PDK1W448/543L (Figure S6E). These, together with activated AKT2, indicated by p-T308, being associated with high DHA/EPA acylation (Figure S6F), confirmed PI3K signaling activating roles of DHA/EPA acylation. Moreover, phosphorylation of AS160 was not activated by insulin, DHA, and EPA in skeletal muscle and adipose tissue of Akt2−/− mice (Figure 6I). It was also observed that expressing AKT2, but not AKT2W414L, enabled DHA and EPA to stimulate AKT2 and AS160 phosphorylation (Figures 6G and S6G) and Glut4 membrane enrichment (Figure 6J). Overall, these results provided genetic evidence that DHA and EPA promote glucose uptake by acting on AKT2 and PDK1.
DHA and EPA deplete cytoplasmic AKT2 and induce insulin resistance
Akt2−/− mice developed insulin resistance as reported (Cho et al., 2001) (Figures 7A and S7A), suggesting a possibility that DHA and EPA may compete in insulin signaling by competing for cytosolic AKT2 availability and saturating PI3K signaling (Figure S7B). Supporting this hypothesis, DHA and EPA dose-dependently decreased cytosolic AKT2 and abrogated potency of insulin to increase AS160 phosphorylation (Figure 7B) and glucose uptake (Figure 7C), which sustained for at least 24 h after treatment (Figure S7C) in mice skeleton muscle. Moreover, AKT2 but not AKT2W414L putback enabled DHA and EPA to activate AKT signaling but block insulin’s potency in Akt2−/− 3T3-L1 cells (Figures 7D and S7D), and allowed DHA and EPA to recruit AKT2 onto membrane, increase AKT2 phosphorylation (Figure 7E), and decrease blood glucose (Figures 7F and S7E) in Akt2−/− mice.
Figure 7. DHA and EPA induce insulin resistance by depleting cytoplasmic PDK1 and AKT2.

(A) DHA acylation induced insulin resistance. Insulin tolerance tests were performed in DHA-treated Akt2−/− 129/C57BL6 mice and Akt2−/− 129/C57BL6 mice overexpressing AKT2WT or AKT2W414L (n = 6, mean ± SEM).
(B) DHA and EPA antagonize insulin to activate AKT signaling. The increase in whole cell p-AKT2(T308) and p-AS160(T642), the decrease in cytoplasmic AKT2, and the increase in membrane AKT2 by insulin were detected in 3T3-L1 pre-treated with DHA and EPA. Quantification was represented as mean ± SEM based on three independent repeats.
(C) DHA and EPA antagonize insulin’s ability to stimulate glucose uptake. The 2-DG uptake abilities of 3T3-L1 treated with DHA and EPA were determined before and after 10 nM insulin stimulation (n = 3, mean ± SEM).
(D) AKT2 putback enabled DHA to block insulin signaling in AKT2−/− 3T3-L1 cells. The increase in whole-cell p-AKT2(T308), p-AS160(T642), decrease in cytoplasmic AKT2, and increase in membrane AKT2 by insulin were detected in DHA pre-treated AKT2−/− 3T3-L1 cells transfected with vector, AKT2, and AKT2W414L. Insulin was added to DHA/EPA-pre-treated cells. Quantification was represented as mean ± SEM based on three independent repeats.
(E) AKT2 putback allowed DHA and EPA to recruit AKT2 onto membrane, and increase AKT2 phosphorylation in AKT2−/− mice. The increase in skeletal muscle membrane AKT2 and increase of p-AKT2(T308) in skeletal muscle by DHA and EPA were detected in AKT2−/− mice transfected with vector, AKT2, and AKT2 W414L.
(F) AKT2 is required for DHA to decrease blood glucose in mice. Glucose tolerance tests were performed to Akt2−/− mice and Akt2−/− mice forced expressing either AKT2 or AKT2W414L, under absence or presence of DHA treatments (n = 6, mean ± SEM).
(G) DHA and EPA failed to enrich GLUT4 to membrane of skeletal muscle of Akt2—/— mice. The abilities of DHA, EPA, and insulin to enrich GLUT4 onto mice skeletal muscle membrane were detected in both WT and Akt2−/− mice.
(H) DHA tryptophan reversibly deplete cytosolic AKT2 and induces p-AS160(T642). The levels of p-AS160(T642) in the whole-cell lysate and AKT2 in the cytosol were determined in 3T3-L1 cells. Quantification was represented as mean ± SEM based on three independent repeats.
(I) DHA and EPA acylation induce insulin resistance in mice. Insulin tolerance test was performed in 129/C57BL6 mice and DHA-, DHA/tryptophan-, EPA-, or EPA/tryptophan-treated129/C57BL6 mice (n = 6, mean ± SEM).
Although the cytoplasmic AKT2 in skeletal muscles of Akt2−/− mice was restored by the forced expression of both AKT2 and AKT2W414L (Figure S7F), neither insulin, DHA, nor EPA could enrich GLUT4 to membrane of skeletal muscle of Akt2−/− mice (Figure 7G). Moreover, DHA and EPA induced insulin resistance in Akt2−/− mice overexpressing AKT2 but not AKT2W414L (Figures 7A and S7A). These results confirmed that depleting AKT2 from cytoplasm by DHA and EPA blocks insulin signaling in vivo. Furthermore, the insulin signaling blockade (Figures 7H and S7G) and insulin resistance induction (Figure 7I) ability of DHA and EPA was inhibited by tryptophan, but not alanine, supplementation, which confirmed that tryptophan acylation underlines DHA’s and EPA’s ability to induce insulin signaling blockade.
DISCUSSION
Here, we report that protein methylene bridge tryptophan acylation is an undocumented general protein fatty acylation that allows methylene bridge-containing PUFAs, represented by DHA and EPA, to exert their cell signaling regulatory activities, extending fatty acylation residues from glycine of N-myristoylation and cysteine of palmitoylation (Linder and Deschenes, 2007; McIlhinney, 1998; Udenwobele et al., 2017) to tryptophan, a residue whose posttranslational modifications have not yet been reported. PUFA fatty acylation readily occurs in cells for two reasons. First, ROS and other free radicals are widespread in environment and in cells, and they can induce methylene bridge radicals (Aliwarga et al., 2017; Davis et al., 2006) that are required for tryptophan acylation. Second, many proteins, such as PDK1 and AKT2 studied here, have cytoplasm exposed tryptophan; in addition, the δ1 of tryptophan is chemically active and is known to be vulnerable to modifications (Henriot et al., 2001; Perez-Rizquez et al., 2019; Antos and Francis, 2004). Methylene bridge tryptophan acylation regulates protein trafficking, which may be because, like DHA and EPA, insoluble PUFAs accumulate in the cell membranes, and the flip-flop movements of the membrane can expose the methylene bridges of the cytoplasm embedded within the interior of the membrane to anchor proteins. Moreover, methylene bridge tryptophan acylation seems to specifically recruit membrane-targeting proteins because these proteins may transiently remain on the membranes long enough to allow the acylation to occur.
Our findings can be used as references to explain several unexplained observations of PUFAs. All methylene-bridge-containing PUFAs can modify proteins, which is consistent with the fact that PUFAs of distinct chemical structures exert similar efficacies (Pironi et al., 2015). Moreover, methylene bridge-containing PUFAs may generate more methylene bridge radicals to acylate proteins, which explains the observation that PUFAs with high number of methylene bridges, such as DHA and EPA, exert more pronounced bio-efficacies. Methylene bridges closer to the distal acyl ends of PUFAs have higher chances to be exposed to the cytoplasm by membrane flip-flop motion, consistent with the finding that methylene bridges closer to the distal acyl ends, i.e., ω−3 and ω−6, had greater bio-impact on cell function and that ω−3 and ω−6 methylene bridges are vulnerable to being oxidized and forming free radicals (Mika and Sledzinski, 2017; Spickett and Pitt, 2015). Lastly, methylene bridge acylation may occur on many proteins and cause cytoplasmic depletion of many effector proteins; this can be explained by the finding that DHA and EPA have effects in an array of human diseases, including cardiovascular disease (Valagussa et al., 1999), diabetes (Niinisto et al., 2017), stroke (Tanaka et al., 2008), and inflammation (Simopoulos, 2002b), and the outcome may be either beneficial or detrimental.
PUFAs are a large family of fatty acids that are used in our daily lives. Our findings provide suggestions for the preventive and therapeutic use of PUFAs including DHA and EPA. DHA and EPA may decrease blood glucose levels. However, they may have limited benefits in alleviating insulin resistance, because they render weaker insulin efficacy by depleting insulin effectors; i.e., cytosolic PDK1 and AKT2. Moreover, the spontaneous nature of DHA and EPA acylation suggests that it obeys the mass law. This can be supported by the finding that approximately 1 g/day doses of DHA or EPA in clinical trials (De Caterina, 2011; Huang and Czech, 2007; Kim et al., 2015; Lee et al., 2009a; Oh et al., 2010a; Simopoulos, 2002a), which was an order of magnitude lower than that in the Greenland Inuit diet (16 g/day) (Bang et al., 1976, 1980), had very mild efficacies, and, conversely, increased doses of DHA or EPA exhibited increased efficacies (Sorgi et al., 2007). Therefore, significant results can be achieved only if DHA and/or EPA are used at high doses.
Limitations of the study
Our data suggest that the ω−3/ω−6 methylene bridge is the most likely one to react with Trp C δ1 carbon. The modification of tryptophan can be probed by DHA/EPA- or DHA/EPA-acylation antibodies; however, due to the spontaneous nature of the DHA/EPA acylation, site-specific acylation information provided by these antibodies is limited. Moreover, how the membrane-embedded methylene bridge reached its substrates and how this post-translational modification (PTM) is dynamically regulated need to be further clarified. The enhanced PDK1 and AKT2 acylation by flip-flop enhancer Triton X-100 supports that flip-flop movement may expose lipid membrane-embedded PUFAs to cytosolic proteins, but other mechanisms may still exist because Triton X-100 may also increase PUFAs’ cytosolic exposure by disrupting membrane structure. More rigid experiments are needed to confirm the cytosolic exposing mechanisms of membrane-embedded PUFAs. Moreover, unidentified de-acylases that may reverse and dynamically regulate the PUFAs’ tryptophan acylation may exist (Figure S4O). Since a C–C bond is formed during PUFAs’ tryptophan acylation, it is possible that tryptophan 2.3-dioxygenases that attack the Nɛ1-Cδ1 bond of Trp to open the pyrrole ring (Guengerich and Yoshimoto, 2018) could be involved in tryptophan de-acylation. Furthermore, PUFAs can be obtained both from food and synthesized intracellularly by fatty acid desaturases, so the specific physiologic significance of each PUFA tryptophan acylation need to be examined individually.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shi-Min Zhao (zhaosm@fudan.edu.cn).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
Raw phosphoproteomics data and raw targeted proteomics data have been deposited at Mendeley and are publicly available as the date of publication. DOIs are listed in the Key Resource Table.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| phospho-AKT2(T308) | Cell Signaling Technology | Cat# 13038; RRID: AB_2629447 |
| AKT2 | Cell Signaling Technology | Cat# 3063; RRID: AB_2225186 |
| phospho-RSK2(S227) | Cell Signaling Technology | Cat# 3556; RRID: AB_2181465 |
| RSK2 | Cell Signaling Technology | Cat# 5528; RRID: AB_10860075 |
| phospho-AS160(T642) | Cell Signaling Technology | Cat# 8881; RRID: AB_2651042 |
| AS160 | Cell Signaling Technology | Cat# 2670; RRID: AB_2199375 |
| phospho-GSK3β(S9) | Cell Signaling Technology | Cat# 5558; RRID: AB_10013750 |
| GSK3β | Cell Signaling Technology | Cat# 12456; RRID: AB_2636978 |
| phospho-TSC2(S939) | Cell Signaling Technology | Cat# 3615; RRID: AB_2207796 |
| TSC2 | Cell Signaling Technology | Cat# 3990; RRID: AB_2209986 |
| phospho-PRAS40(T246) | Cell Signaling Technology | Cat# 13175; RRID: AB_2798140 |
| PRAS40 | Cell Signaling Technology | Cat# 2691; RRID: AB_2225033 |
| PDK1 | Cell Signaling Technology | Cat# 5662; RRID: AB_10839264 |
| GLUT4 | Cell Signaling Technology | Cat# 2213; RRID: AB_823508 |
| Na-K-ATPase | Cell Signaling Technology | Cat# 3010; RRID: AB_2060983 |
| GAPDH | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| phospho-IR(Y1150/1151) | Cell Signaling Technology | Cat# 3024; RRID: AB_331253 |
| IR | Cell Signaling Technology | Cat# 3020; RRID: AB_2249166 |
| phospho-IRS1(Y895) | Cell Signaling Technology | Cat# 3070; RRID: AB_2127863 |
| IRS1 | Cell Signaling Technology | Cat# 2390; RRID: AB_10692516 |
| phospho-AKT2(S473) | Cell Signaling Technology | Cat# 4060; RRID: AB_2224726 |
| CDK2 | Cell Signaling Technology | Cat# 2546; RRID: AB_2276129 |
| SIRT3 | Cell Signaling Technology | Cat# 5490; RRID: AB_10828246 |
| COX4 | Cell Signaling Technology | Cat# 4884; RRID: AB_2085427 |
| PGK1 | Abcam | Cat# ab38007; RRID: AB_2161220 |
| PI3K p110α | Cell Signaling Technology | Cat# 4255; RRID: AB_659888 |
| PI3K p85 | Cell Signaling Technology | Cat# 4292; RRID: AB_329869 |
| Flag | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| DHA | LSbio | Cat# LS-C714681; |
| EPA | LSbio | Cat# LS-C714683; |
| AKT1 | Abcam | Cat# ab89402; RRID: AB_2049150 |
| phospho-AKT1(S473) | Cell Signaling Technology | Cat# 9018; RRID: AB_2629283 |
| phospho-AKT1(T308) | Abcam | Cat# ab105731; RRID: AB_10900490 |
| phospho-S6K(T389) | Cell Signaling Technology | Cat# 9234; RRID: AB_2269803 |
| S6K | Cell Signaling Technology | Cat# 9202; RRID: AB_331676 |
| BiP | Cell Signaling Technology | Cat# 3177 RRID: AB_2119845 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DHA | Sigma-Aldrich | Cat# D8768 |
| Methyl-DHA | Sigma-Aldrich | Cat# D2659 |
| Ethyl-DHA | Sigma-Aldrich | Cat# D2410 |
| EPA | Sigma-Aldrich | Cat# E6627 |
| Methyl-EPA | Sigma-Aldrich | Cat# CRM47571 |
| Ethyl-EPA | Sigma-Aldrich | Cat# E0085000 |
| linoleic acid | Sigma-Aldrich | Cat# L8134 |
| palmitic acid | Sigma-Aldrich | Cat# P9767 |
| oleic acid | Sigma-Aldrich | Cat# O7501 |
| conjugated linoleic acid | Sigma-Aldrich | Cat# 16413 |
| linolenic acid | Sigma-Aldrich | Cat# L2378 |
| Insulin | Sigma-Aldrich | Cat# 1342106 |
| Tryptophan | Sigma-Aldrich | Cat# V900470 |
| methyl-tryptophan | Sigma-Aldrich | Cat# 447439 |
| Tryptamine | Sigma-Aldrich | Cat# 246557 |
| DTT | Sigma-Aldrich | Cat# D9760 |
| Wortmannin | Sigma-Aldrich | Cat# W1628 |
| Alanine | Sigma-Aldrich | Cat# 05130 |
| 5-hydroxytryptamine | J&K Scientific Ltd. | Cat# 153–98-0 |
| L-α-phosphoatidylserine | Avanti | Cat# 870336C |
| Peptides, see Figure S2C | GL Biochem (Shanghai) Ltd. | N/A |
| Penicillin-Streptomycin | Invitrogen | Cat# 15070063 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse PIP3 ELISA | Shanghai Enzyme-linked Biotechnology Co., LTD. | Cat# ml037511–1 |
| Glucose Uptake-GloTM Assay Kit | Promega | Cat# J1341 |
| AKT kinase activity assay kit | Abcam | Cat# ab139436 |
| PDK1 kinase activity assay kit | Promega | V9681 |
|
| ||
| Deposited data | ||
|
| ||
| Raw phosphoproteomics data | N/A | Mendeley data: https://doi.org/10.17632/rc3djvsgn3.1 |
| Raw targeted proteomics data | N/A | Mendeley data: https://doi.org/10.17632/n4npj2j8rx.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: 3T3-L1 | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNM25 |
| Cricetulus griseus: CHO-K1 | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNHa 4 |
| Human: HepG2 | Stem Cell Bank, Chinese Academy of Sciences | Cat# SCSP-510 |
| Human: HEK293T | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNHu17 |
|
| ||
| Experimental Models: Organisms/strains | ||
|
| ||
| Mouse: 129/C57BL6 | Shanghai SLAC Laboratory Animal Co.,Ltd | N/A |
| Mouse: 129/C57BL6, AKT2 KO | Shanghai SLAC Laboratory Animal Co., Ltd | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Myc-GLUT4-mCherry | This work | N/A |
| AKT2-EGFP | This work | N/A |
| AKT2R25C-EGFP | This work | N/A |
| AKT2R25C-Flag | This work | N/A |
| PDK1-EGFP | This work | N/A |
| PDK1R474A-Flag | This work | N/A |
| PDK1W448/543L-EGFP | This work | N/A |
| PDK1w266/347L-EGFP | This work | N/A |
| PDK1-Flag | This work | N/A |
| PDK16W/L-Flag | This work | N/A |
| PDK1W448/543L-Flag | This work | N/A |
| PDK1W266/347L-Flag | This work | N/A |
| PDK1R474A-Flag | This work | N/A |
| AKT2-Flag | This work | N/A |
| AKT27W/L-Flag | This work | N/A |
| AKT2W414L-Flag | This work | N/A |
| AKT2W414L-EGFP | This work | N/A |
| AKT2W334L-Flag | This work | N/A |
| AKT2W334L-EGFP | This work | N/A |
| AKT1-Flag | This work | N/A |
| AKT1W413L-Flag | This work | N/A |
| PGK1-EGFP | This work | N/A |
| PGK1-Flag | This work | N/A |
| PGK1-PH-EGFP | This work | N/A |
| PGK1-PH-Flag | This work | N/A |
| PKM2-Flag | This work | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| MaxQuants | Max planck institute of biochemistry | https://www.maxquant.org |
| Perseus software | Max planck institute of biochemistry | https://maxquant.net/perseus/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Human embryonic kidney HEK293T cells and human liver hepatocellular carcinoma HepG2 cells were cultured in normal Dulbecco’s Modified Eagle’s Medium (DMEM) (HyClone) supplemented with 10% fatal bovine serum (FBS) (Gibco), 100 units/mL penicillin (Invitrogen) and 100 μg/mL streptomycin (Invitrogen). Chinese hamster ovary CHO-K1 cells were cultured in F12K medium supplemented with 10% FBS. Mice preadipocytes 3T3-L1 were cultured and differentiated into adipocytes by published protocols. In brief, 3T3-L1 fibroblasts were cultured in DMEM supplemented with 10% newborn calf serum at 370C, 10% CO2. Two days after confluence, cells were switched into differentiation medium containing 10% FBS, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone and 1 μM insulin, the cells were then switched to DMEM supplemented with 10% FBS and 167 nM insulin 2 days after differentiation medium culturing and cultured for 2 days before they were maintained in DMEM with 10% FBS. Adipocytes were used for experiments at day 10 post differentiation.
Animals
Male 129/C57BL6 mice of 4 weeks ages were housed in a specific pathogen-free facility and given free access to food and water. All animal procedures were in accordance with the animal care committee at Fudan University, Shanghai, China.
Mice were fed with normal chow (13.5% fat), high-fat diet (60% fat), EPA Chow (high-fat diet containing 16% EPA) and DHA chow (high-fat diet containing 16% DHA) ad libitum as indicated in studies.
METHOD DETAILS
Plasmid construction
Plasmid were constructed by PCR-amplifying using Phanta Max Super-Fidelity DNA polymerase (Vazyme, China). Site-directed mutation of target plasmid was introduced via Mut Express MultiS Fast mutagenesis Kit V2 (Vazyme, China). Primers were designed according to manufacturer instruction.
Cell treatments
DHA or EPA treatments: DHA or EPA were added to culture media to reach final concentrations of 100 μM, cells were harvested for analysis 1 hour (unless specified) after the start of treatment.
Insulin treatment: insulin was added to the culture media to a final concentration of 10 nM, cells were harvested for analysis 20 minutes (unless specified) after start of treatment.
Combined treatments: 1, combined with insulin. Cells were treated by either 100 μM DHA or 100 μM EPA for 1 hour, insulin (10 nM) was then added and the cells were harvested for analysis 20 minutes after the start of insulin treatment. 2, combined with methyl-tryptophan, tryptamine, 5-HT, quinacrine, chlorpromazine and wortmannin. Cells were pre-treated by 1 mM methyl-tryptophan, 1 mM tryptamine, 1 mM 5-HT, 50 μM quinacrine, 50 μM chlopromazine or 100 nM wortmannin for 20 min, then the cells were treated with 100 μM DHA or 100 μM EPA for 1 hour before they were harvested for analysis.
Cellular fractionation
Membrane and cytoplasm isolation kit (Beyotime) was used following the manufacture’s guidance. Briefly, 1×107 cells were washed twice with cold PBS and then were suspended in 1 mL buffer A supplemented with protease inhibitors on ice for 10–15 min. Cells were broken by Dounce homogenizer with 30 strokes on ices. Lysates were centrifuged at 700 rcf for 10 min (4°C) to remove debris. Carefully transfer the supernatant to another centrifuge tube and centrifuge at 14,000 rcf for 30 min (4°C). The membrane fractions were in the pellet and the cytoplasm fractions were in the supernatant.
Mice treatments
Acute DHA treatments: male mice were starved overnight before DHA at 2.5 μg/g were injected through tail vein. The treated mice were sacrificed 3 hours after injection and the skeletal muscle was used for phosphoproteomics analysis.
Insulin signaling related experiments: before DHA or EPA treatments, mice were fed with high fat-diet for 4 weeks before DHA chow or EPA chow were employed to feed mice. Blood samples were taken at time points specified and the tissue samples were obtained from sacrificed mice 8 hours after the start of DHA chow or EPA chow feeding.
Insulin tolerance test
Insulin tolerance tests (ITT) were performed in 129/C57BL6 male mice. For ITT, food was removed from mice 16 hours before the start of the tests. 0.5 U/kg insulin were injected to mice intraperitoneally and 5 μL blood sample was collected via tail nick prior and at time points specified in experiments after injection.
Intramuscular injections
A reported method was adopted (Horton et al., 1999). Briefly, 50 μg of plasmid DNA in 50 μL of saline was injected into the rectus femoris muscle of each hind leg for a total DNA dose of 100 μg. Limit the needle from penetrating further than 2 mm into the rectus femoris muscle.
Glucose disposal test
Male mice were fed with high-fat diet, W chow (high-fat diet containing 1.25% tryptophan), DHA chow (high-fat diet containing 16% DHA), DHA + W chow (high-fat diet containing 16% DHA and 1.25% tryptophan), EPA chow (high-fat diet containing 16% EPA), EPA + W chow (high-fat diet containing 16% EPA and 1.25% tryptophan) for 4 weeks. Food was removed from mice before the start of the tests. 0.05 U/kg insulin were injected to indicated mice intraperitoneally 0.5 h before the start of the tests. Then 5 μL blood sample was collected via tail nick prior end at time points specified in experiments after food removing and glucose was assessed with ROCHE glucose monitoring system.
Glucose tolerance test
Male mice were fed with high-fat diet, W chow (high-fat diet containing 1.25% tryptophan), DHA chow (high-fat diet containing 16% DHA), DHA + W chow (high-fat diet containg 16% DHA and 1.25% tryptophan), EPA chow (high-fat diet containing 16% EPA), EPA + W chow (high-fat diet containing 16% EPA and 1.25% tryptophan) for 4 weeks. Food was removed from mice for 16 hours before the start of the tests. 0.05 U/kg insulin were injected to indicated mice intraperitoneally 0.5 h before the start of the tests. Blood glucose concentrations were assessed before and 15, 30, 60, 90 and 120 min after the injection of 1 g/kg dextrose.
Phosphoproteomics analysis
Protein digestion
The procedures for protein digestion were performed according to the filter-aided sample preparation (FASP) (Wisniewski et al., 2009) technique with slight modification. Briefly, the supernatant was transferred to Vivacon 500 ultrafiltration spin columns (Sartorius AG, Germany) and centrifuged at 14,000 g for 15 min. Then, the samples were washed twice with 100 mM ammonium bicarbonate. The proteins were digested in 100 μL of 100 mM ammonium bicarbonate overnight with 1:100 (w/w, protein/enzyme) trypsin at 37°C. After digestion, the peptide mixtures were collected by centrifugation at 14,000 g for 20 min and kept at −80°C until use.
Phosphopeptide enrichment
Phosphopeptide enrichment was performed with Ti4+-IMAC bead as described previously (Yao et al., 2017). Protein digest were mixed with identical volume of loading buffer (80% ACN/6%TFA), and then the mixture was loaded on solid phase extraction (SPE) column packed with a new Ti4+-IMAC beads synthesized in-house. The SPE column was subsequently washed with 50% ACN/200 mM NaCl/6% TFA, followed by washing with 30% ACN/0.1% TFA. Finally, the bound phosphopeptides were eluted with 10% NH4OH, and concentrated in a SpeedVac. Before mass spectrometry analysis, all the phosphopeptides were desalted.
LC-MS/MS analysis
All LC-MS/MS experiments were carried out on a Q Exactive HF Mass Spectrometer (Thermo Scientific, USA) coupled with a Dionex UltiMate 3000 RSLCnano system (T hermo Scientific, USA). The samples were resuspended in 1% FA/H2O solution and automatically loaded onto a C18 trapping column (3 cm × 150 μm inner diameter) at a flow rate of 5 μL/min with 100% mobile phase A. The analytical capillary column (150 μm i.d.) was packed in-house with 1.9μm C18 ReproSil particles (Dr. Maisch GmbH). Column temperature was maintained at 55°C using a column oven. Peptides were separated with a binary buffer system of 0.1% FA (buffer A) and 80% ACN/0.1% FA (buffer B), at a flow rate of 600 nL/min. The gradient elution was performed as follows: 0–9% mobile phase B for 5 min; 9–25% B for 75 min; 25–45% B for 17 min; 45–90% B for 1 min; 90% B for 10 min; and finally equilibration with mobile phase A for 10 min. The Q Exactive HF Mass Spectrometer was operated in data-dependent MS/MS in data-dependent MS/MS acquisition mode. The full mass scan acquired in the Orbitrap mass analyzer was from m/z 350 to 1750 with a resolution of 60,000 (m/z 200) at AGC target of 3 × 106. The top 20 intense ions from the full scan were fragmented by higher-energy collisional dissociation (HCD). For each MS/MS scan with a resolution of 30,000 at m/z 200, the AGC target was set at 5 × 105 with a max injection time of 120 ms. A spray voltage of 2.6 kV was applied between the spray tip and MS interface. The temperature of the ion transfer capillary was set as 250°C and the normalized collisional energy for HCD was set to 27%. Dynamic exclusion was set as 25 s. System control and data collection were performed by Xcalibur software.
Data processing and analysis
Raw mass spectrometry files were searched using MaxQuantsoftware suite (version 1.6.4.0). Proteins and peptides were identified with parameters previously described (Yao et al., 2019) and quantified using the label-free quantification (LFQ) algorithm integrated in MaxQuant. Searches were performed against a non-redundant UniProt mouse database (containing 16981 reviewed protein sequences, and download from the web site www.uniprot.org), the default reverse and potential contaminant hits were included and any hits to theses excluded from further analysis. The false discovery rate (FDR) was set to 1% at the level of protein, peptide spectrum matches (PSM) and PTM site, other parameters were used the default settings with the following minor changes: carbamidomethyl cysteine (C) was selected as fixed modifications and oxidized methionine (M), acetylation (protein N-term), and phosphorylation of serine (S), threonine (T), and tyrosine (Y) were set as variable modifications minimum peptide length of seven amino acid, and “match between run” (MBR) was enabled with a matching time window of 0.7 min. The class I phosphorylation sites were defined by a combination localization probability of ≥0.75 and ΔPTM score of ≥5. Trypsin was chosen as the proteolytic enzyme with allowing two missed cleavage sites. Data analysis was supported by the robust statistical tool Perseus software (version 1.5.8.5) using default settings including t test.
Proteomic analysis for DHA and EPA acylation sites in PDK1 and AKT2
Targeted proteomic analysis was employed as we described previously (He et al., 2018). Briefly, after cells were homogenized by sonication (20,000–40,000 Hz) in pre-chilled PBS buffer (NaCl 1.37 mM, KCl 27 mM, Na2HPO4 100 mM, KH2PO4 18 mM, pH7.4), the membrane and the cytosolic proteome were precipitated by adding 43 volume −80°C pre-chilled acetone each to the fractions. The resulted proteome was vacuum dried and digested with trypsin. The resulted peptides were resuspended with 25 μL solvent A (A: water with 0.1% formic acid; B: ACN with 0.1% formic acid), and separated by nanoLC (Thermo Scientific Acclaim PepMap C18 column, 100 μm × 2 cm, 3 μm particle size, flow 10 μL/min for 3 min followed on the analytical column Acclaim PepMap C18, 75 μm × 15 cm with a linear gradient, from 5% B to 45% B in 75 minutes), and analyzed by Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). Raw MS files were analyzed by MaxQuant version 1.4.1.2. MS/MS spectra were searched by the Andromeda search engine against the SwissProt-human database (Release, 2014–04-10) containing forward and reverse sequences (total of 40492 entries including forward and reverse sequences). Minimal peptide length was set to seven amino acids and a maximum of four miscleavages was allowed. The false discovery rate (FDR) was set to 0.01 for peptide and protein identifications, all scores of identified peptides were above 40. For quantification of targeted fatty acylated peptides, a published method (He et al., 2018) was employed.
SDS-PAGE and western blot
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot was performed following standard protocols.
In vitro PUFAs acylation
In vitro PUFAs acylation reactions were carried out in a 30 μL reaction mix contains 50 mM HEPES (pH 7.5), 500 mM PUFAs and 0.05 mg/mL synthetic peptide or 1 μM recombinant protein. The reactions were allowed to proceed for 6 hours at 37°C. The resulted peptide was desalted by passing through a C18 ZipTip (Millpore) and subject to analyzation by a MALDI-TOF/TOF mass spectrometer (SCIEX-5800). The resulted proteins were subject to SDS-PAGE analysis.
Preparation of α-DHA/EPA acylation antibody
The tryptophan DHAacylation and EPAacylation antibody was developed by using KLH-conjugated fatty-acylated peptides as antigen according to the reported procedure (He et al., 2018). Briefly, 20 mM synthetic peptides (sequnence:CKKKWGG GWGGGWGGGWGG) was chemically DHA-acylated and EPA-acylated by reaction with 100 mM EPA and DHA in 1 mL PBS buffer at 37°C for 2hours. Reaction efficiency was confirmed by a 4700 MS spectrometry. The resulted fatty-acylated peptides were conjugated to KLH as antigen before subjecting to immunize rabbits. All antibodies were made in Abclonal Shanghai and DHA/EPA-acylation antibodies were affinity purified by EPA/DHA-Trp peptide before experimentation.
Lipase treatment
Flag-tagged PDK1 and AKT2 were ectopically-expressed in 3T3-L1 cells. Cells were treated with 100 μM DHA or EPA for 1h before PDK1 and AKT2 were purified with anti-flag M2 beads (Sigma, Cat A2220) via immunoprecipitation. The flag beads-coupled PDK1 and AKT2 proteins then added in a 200 μL reaction buffer containing 50 mM Tris-HCl (pH 8.0), 4 mM CaCl2, and 150 mM NaCl and 10 mg lipase (Sigma, cat. 54327). The protein samples immunoblotted by α-DHA/EPA and DHA/EPA-acylation antibody were pre-incubated with lipase at 37°C for 3 hours before the resulted proteins are subject to SDS-PAGE analysis.
Isolation of free PUFAs from membrane
A reported method (Szachowicz-Petelska et al., 2007) was adopted. Briefly, the cell membrane was homogenized in 2% acetic acid in ethyl ether (2:1, v/v). The solution was then filtered out with degreased paper filters. The suspensions were centrifuged at 500 rcf for 2 min. The organic and the aqueous phases were separated, the aqueous phase was shaken again with 2% acetic acid in ethyl ether (2:1, v/v) and the phases were separated. The organic phases contained PUFAs were combined and were evaporated to dryness.
Isolation of lipids from membrane
A published method (Bligh and Dyer, 1959) was employed. Briefly, the cell membrane was homogenized for 2 min in solution contained 333 μL chloroform and 666 μL methanol, before another 333 μL chloroform was added and mixed for 30 sec, followed by addition of 333 μL distilled water. After complete mixing for 30 sec, the mixture was centrifuged at 800 rcf for 10 min. The Lipids containing chloroform layer were transferred to another centrifuge tube and were evaporated to dryness.
MS detection of fatty acids
A reported method (Li et al., 2015) was adopted. Briefly, the cytosolic and membrane fractions of 1×107 cells in 1 mL methanol was first homogenized with a tissuelyzer (20 Hz, 90 s) three times. After methyl esterification with acetyl chloride, the methylated fatty acids were resolved with a DB-225 capillary GC column (Agilent, length 10 m, inner diameter 0.1 mm, and film thickness of 0.1 μm) with 1:60 splitter, and measured on a Shimadzu GCMS-QP2010Plus spectrometer (Shimadzu Scientific Instruments) having a flame ionization detector (FID) and a mass spectrometer (MS) with an electron impact (EI) source. Sample injection volume was 1 μL. Helium gas was used as carrier and makeup gas of GC. FID was used with air and hydrogen gas flow rate of 40 and 400 mL/min, respectively. The column temperature was set as follows: initial, 55°C with 1 min hold; ramp, 55°C/min to 205°C (30°C/min) with a 3 min hold, 205 to 230°C (5°C/min). Both the injection port and detector temperatures were set at 230°C. EI-MS spectra were acquired with the EI voltage of 70 eV and the m/z range of 45–450 with helium gas flow rate of 18.4 mL/min. Fatty acid identifications were achieved by comparing data with a mixture of 37 known standards and further confirmed with the mass spectral data from standard databases. Quantification of the methylated fatty acids was done with the FID data using methylated C17:0 and C23:0 as internal standards and quality controls. The results were calculated as micro molar fatty acids per gram for 3T3-L1 cells.
Solid phase peptides synthesis
Peptides that contained designated amino acids or amino acid-derivatives were synthesized by solid phase synthesis. Firstly, Fmocrink Amide resin was used as the starting material. Fmoc protected amino acids was assembled on Rink amide resin by general solid phase synthesis protocol. After washed by DMF (5 mL), DCM (5 mL) and EtOH (5 mL) three times respectively, the resin was dried by pumps. Cleavage of the peptide from the resin was achieved by treatment with a mixture of 4 mL TFA:TIS:H2O (95:2.5:2.5, v/v/v) at room temperature for 3 h. The crude peptide was triturated with diethyl ether chilled at −20°C, then centrifuged at 8000 rpm for 5 min. The crude product was purified by HPLC with a Vydac C18 column using a water (0.1% TFA)-acetonitrile (0.1% TFA) gradient to produce overall yield about 78%. Its homogeneity (>96%) was confirmed by reversed phase analytical HPLC with a C18 column. The successful synthesis was confirmed by ESI mass spectrometry.
Immunofluorescence analysis
Mouse skeletal muscle samples were fixed in 4% paraformaldehyde overnight at 4°C, followed by dehydrated in PBS containing 30% sucrose. Mouse skeletal muscle samples were embedded in paraffin, 10mm-thick paraffin sections were used for immunofluorescence staining. The primary antibodies used were: rabbit anti-AKT (1:100, Cell Signaling Technology, 4685), anti-PDK1 (1:100, Cell Signaling Technology, 5662), anti-GLUT4 (1:100, Abcam, ab654), mouse anti-E-cadherin (1:100, Cell Signaling Technology, 14472). The secondary antibodies (1:1000, Thermo Fisher Scientific) used were: Alexa Fluor 488 donkey anti-mouse and Alexa Fluor 555 donkey anti-rabbit. Sections were examined suing a Zeiss LSM710 confocal microscope as mentioned above.
Confocal live-cell images
Cells were seeded onto 35-mm poly-D-lysine coated glass bottom microwell dishes and transfected with an AKT-PH-EGFP construct (Addgene plasmid 18836) and full-length AKT2-EGFP plasmid or a PDK1-PH-EGFP (residues 409–556) construct and full-length PDK1-EGFP plasmid generated from HEK293T cell cDNA and for 16h, followed by serum starvation and EPA,DHA or Insulin treatment as described earlier. Time-lapse movies were recorded at 15 s intervals for 30 min in a heated chamber at 37°C contain 5% CO2, Images were captured using a spinning disk confocal scan head (CSU-X/M2N, Yokogawa) attached to an inverted microscope (IX81, Olympus) with x60 NA/1.49 ApoN oil-immersion objectives, and an EMCCD camera (DU897BV, Andor). All microscope hardware and image acquisition were controlled by Micro-Manager software and images were analyzed using Fiji image process software.
Myc-GLUT4-mCherry assay
Cells were seeded on poly-D-lysine coated overslips were transfected with Myc-GLUT4-mCherry plasmid by electro transfection. Cells were harvested and fixed with 4% paraformaldehyde for 10 min, then washed with cold PBS two times and blocked with PBS containing 5% donkey serum for 45 minutes at room temperature. Cells were then incubated with primary rabbitanti-Myc antibody overnight at 4°C, followed by Alexa Fluor 488-conjugated Donkey anti-rabbit secondary antibody at least 1 hour at room temperature. After washing, the overslips were mounted with DAKO mounting medium. Mounted samples were subjected to confocal imagining using a Zeiss LSM710 confocal laser microscope system with an x63 NA/1.40CFI Plan APOVC oil-immersion objectives. ZEN 2009 Light Edition software was used for quantitative measurement of GLUT4 translocation. In briefly, cells expressing GLUT4-Myc-mCherry was selected for measurement of fluorescence intensity of both Myc- and mCherry after removal of background fluorescence.
NMR analysis
All the one-dimensional 1H NMR spectra were acquired at 298 K on a Bruker Advance III 600 MHz NMR spectrometer (600.13 MHz for proton frequency) equipped with an inverse cryogenic probe (Bruker Biospin, Germany) using the first increment of the gradient selected NOESY pulse sequence (NOESYGPPR1D: recycle delay-G1–900-T1–900-tm-G2–900-acquisition). 64 transients were collected into 32 k data points with a spectral width of 20 ppm for each sample.
Tryptophan fatty acylation biodegradation activity assay
HEK293T cells were Dounce homogenized in buffer A (10 mM Tris-HCl pH 7.4, 250 mM Sucrose containing protease inhibiters). Lysates were centrifuged at 1,200 g to remove incompletely lysed cells and debris. Lysates were then adjusted to 0.5 mg/mL protein and 100 μL was incubated with 500 ng/mL DHA/EPA pretreated-purified AKT2 or PDK1 for 2 h at 37°C. Control samples were heat denatured by boiling for 10 min before incubation. After reaction, AKT2 and PDK1 were adjusted for Western blot.
Glucose uptake assay
The non-radioactive 2-dexoyglucose uptake was detected using a Glucose Uptake-Glo™ Assay Kit (J1341, Promega) according to manufactured instruction. Briefly, cells were seed on 96-well plates and serum starved for 3h before the assay. Cells were washed with PBS three times then replaced the medium with DMEM without serum or glucose and treatment with 100 nM insulin or 100 μM EPA or DHA for 30 min. Cells were incubated with 1 mM 2-DG in PBS for additional 10 min at 25°C. Cells were then lysed in stop buffer and neutralized with neutralization buffer. Lysates were then centrifuged at 15000 g for 15 min at 4°C, and the supernatant were incubated with 2DG6P detection reagent for 1h at 25°C, then recorded luminescence with 0.3–1 second integration on a luminometer (Glomax96, Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were presented as the mean ± SEM. Number of replicates (n) can be found for each experiment in figure legend. Unpaired two-tailed Student’s t test were used to compare two conditions. Analysis was performed using the statistical software package, Prism 8.0 (Graphpad Software). Differences were considered significant at p < 0.05.
Supplementary Material
Highlights.
The methylene-bridge-containing polyunsaturated fatty acids EPA and DHA modify proteins
Tryptophan is a site of posttranslational modifications by polyunsaturated fatty acids
EPA and DHA activate AKT signaling by modifying PDK1 and AKT2
EPA and DHA promote glucose uptake but induce insulin resistance
ACKNOWLEDGMENTS
This work was supported by grants from the State Key Development Programs of China (nos. 2018YFC1004700, 2018YFA0800300, 2018YFA0801300, 2019YFA0801900), the National Science Foundation of China (nos. 31821002, 31871432, 31930062, 32171298, 81971449, 82171672), the Program of Shanghai Academic Research Leader (21XD1423000), an Innovation-oriented Science and Technology Grant from Key Laboratory of Reproduction Regulation of NHC (CX2017-0X), and a grant from Key Laboratory of Reproduction Regulation of NPFPC.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110509.
REFERENCES
- Ahren B, Mari A, Fyfe CL, Tsofliou F, Sneddon AA, Wahle KW, Winzell MS, Pacini G, and Williams LM (2009). Effects of conjugated linoleic acid plus n-3 polyunsaturated fatty acids on insulin secretion and estimated insulin sensitivity in men. Eur. J. Clin. Nutr 63, 778–786. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, and Hemmings BA (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Aliwarga T, Raccor BS, Lemaitre RN, Sotoodehnia N, Gharib SA, Xu LB, and Totah RA (2017). Enzymatic and free radical formation of cis- and trans-epoxyeicosatrienoic acids in vitro and in vivo. Free Radical Biol. Med 112, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antos JM, and Francis MB (2004). Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc 126, 10256–10257. [DOI] [PubMed] [Google Scholar]
- Bang HO, Dyerberg J, and Nielsen AB (1971). Plasma lipid and lipoprotein pattern in Greenlandic West-coast Eskimos. Lancet 1, 1143–1145. [DOI] [PubMed] [Google Scholar]
- Bang HO, Dyerberg J, and Hjoorne N.(1976). The composition of food consumed by Greenland Eskimos. Acta Med. Scand 200, 69–73. [DOI] [PubMed] [Google Scholar]
- Bang HO, Dyerberg J, and Sinclair HM (1980). The composition of the Eskimo food in north western Greenland. Am. J. Clin. Nutr 33, 2657–2661. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Phys 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Boutin JA (1997). Myristoylation. Cell Signal. 9, 15–35. [DOI] [PubMed] [Google Scholar]
- Brown TJ, Brainard J, Song F, Wang X, Abdelhamid A, Hooper L, Ajabnoor S, Alabdulghafoor F, Brainard J, Brown TJ, et al. (2019). Omega-3, omega-6, and total dietary polyunsaturated fat for prevention and treatment of type 2 diabetes mellitus: systematic review and meta-analysis of randomised controlled trials. BMJ 366, l4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, et al. (2007). A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 448, 439–444. [DOI] [PubMed] [Google Scholar]
- De Caterina R.(2011). n-3 fatty acids in cardiovascular disease. N. Engl. J. Med 364, 2439–2450. [DOI] [PubMed] [Google Scholar]
- Chiariello M, Ambrosio G, Cappellibigazzi M, Nevola E, Perronefilardi P, Marone G, and Condorelli M.(1987). Inhibition of ischemia-induced phospholipase activation by quinacrine protects jeopardized myocardium in rats with coronary-artery occlusion. J. Pharmacol. Exp. Ther 241, 560–568. [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu QW, Crenshaw EB, Kaestner KH, Bartolomei MS, Shulman GI, and Birnbaum MJ (2001). Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 292, 1728–1731. [DOI] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, and Hemmings BA (1995). Inhibition of glycogen-synthase kinase-3 by insulin-mediated by protein-kinase-B. Nature 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Cuendet GS, Loten EG, Jeanrenaud B, and Renold AE (1976). Decreased basal, noninsulin-stimulated glucose uptake and metabolism by skeletal soleus muscle isolated from obese-hyperglycemic (ob/ob) mice. J. Clin. Invest 58, 1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuiffo B, and Ren R.(2010). Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood 115, 3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TA, Gao L, Yin HY, Morrow JD, and Porter NA (2006). In vivo and in vitro lipid peroxidation of arachidonate esters: the effect of fish oil omega-3 lipids on product distribution. J. Am. Chem. Soc 128, 14897–14904. [DOI] [PubMed] [Google Scholar]
- Dry Eye Assessment and Management Study Research Group; Asbell PA, Maguire MG, Pistilli M, Ying GS, Szczotka-Flynn LB, Hardten DR, Lin MC, and Shtein RM (2018). n-3 fatty acid supplementation for the treatment of Dry Eye disease. N. Engl. J. Med 378, 1681–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyerberg J, Bang HO, Stoffersen E, Moncada S, and Vane JR (1978). Eicosapentaenoic acid and prevention of thrombosis and atherosclerosis? Lancet 2, 117–119. [DOI] [PubMed] [Google Scholar]
- Farazi TA, Waksman G, and Gordon JI (2001). The biology and enzymology of protein N-myristoylation. J. Biol. Chem 276, 39501–39504. [DOI] [PubMed] [Google Scholar]
- Fedor D, and Kelley DS (2009). Prevention of insulin resistance by n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. Metab. Care 12, 138–146. [DOI] [PubMed] [Google Scholar]
- The Ascend Study Collaborative Group (ASC Group) (2018). Effects of n-3 fatty acid supplements in diabetes mellitus. N. Engl. J. Med 379, 1540–1550. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, and Yoshimoto FK (2018). formation and cleavage of C-C bonds by enzymatic oxidation reduction reactions. Chem. Rev 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Baxter DM, Mitchell MA, and Hitzemann RJ (1984). Physical properties and lipid composition of brain membranes from ethanol tolerant-dependent mice. Mol. Pharmacol 25, 401–409. [PubMed] [Google Scholar]
- He XD, Gong W, Zhang JN, Nie J, Yao CF, Guo FS, Lin Y, Wu XH, Li F, Li J, et al. (2018). Sensing and transmitting intracellular amino acid signals through reversible lysine aminoacylations. Cell Metab. 27, 151–166.e6. [DOI] [PubMed] [Google Scholar]
- Henriot S, Lepoittevin JP, and Trifilieff E.(2001). Haptenization of ovalbumin with the skin sensitizer methyl octanesulfonate: characterization of the methylated OVA323–339 T-cell epitope at His331. J. Pept. Sci 7, 331–337. [DOI] [PubMed] [Google Scholar]
- Horton HM, Anderson D, Hernandez P, Barnhart KM, Norman JA, and Parker SE (1999). A gene therapy for cancer using intramuscular injection of plasmid DNA encoding interferon alpha. Proc. Natl. Acad. Sci. U S A 96, 1553–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, and Czech MP (2007). The GLUT4 glucose transporter. Cell Metab. 5, 237–252. [DOI] [PubMed] [Google Scholar]
- Humphrey SJ, Azimifar SB, and Mann M.(2015). High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol 33, 990–U142. [DOI] [PubMed] [Google Scholar]
- Origin Trial Investigators; Bosch J, Gerstein HC, Dagenais GR, Diaz R, Dyal L, Jung H, Maggiono AP, Probstfield J, Ramachandran A, et al. (2012a). n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N. Engl. J. Med 367, 309–318. [DOI] [PubMed] [Google Scholar]
- Origin Trial Investigators; Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, Pogue J, Probstfield J, Ramachandran A, et al. (2012b). Basal insulin and cardiovascular and other outcomes in dysglycemia. N. Engl. J. Med 367, 319–328. [DOI] [PubMed] [Google Scholar]
- Jensen CJ, Buch MB, Krag TO, Hemmings BA, Gammeltoft S, and Frodin M.(1999). 90-kDa ribosomal S6 kinase is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem 274, 27168–27176. [DOI] [PubMed] [Google Scholar]
- Kharbanda A, Walter DM, Gudiel AA, Schek N, Feldser DM, and Witze ES (2020). Blocking EGFR palmitoylation suppresses PI3K signaling and mutant KRAS lung tumorigenesis. Sci. Signal 13, eaax2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-Y (2007). Novel metabolism of docosahexaenoic acid in neural cells. J. Biol. Chem 282, 18661–18665. [DOI] [PubMed] [Google Scholar]
- Kim HY, Edsall L, Garcia M, and Zhang H.(1999). The release of polyunsaturated fatty acids and their lipoxygenation in the brain. Adv. Exp. Med. Biol 447, 75–85. [DOI] [PubMed] [Google Scholar]
- Kim N, Lee JO, Lee HJ, Kim HI, Kim JK, Lee YW, Lee SK, Kim SJ, Park SH, and Kim HS (2015). Endogenous ligand for GPR120, docosahexaenoic acid, exerts benign metabolic effects on the skeletal muscles via AMP-activated protein kinase pathway. J. Biol. Chem 290, 20438–20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Summers SA, Birnbaum MJ, and Roth RA (1996). Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem 271, 31372–31378. [DOI] [PubMed] [Google Scholar]
- Lalia AZ, and Lanza IR (2016). Insulin-sensitizing effects of omega-3 fatty acids: lost in translation? Nutrients 8, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SA, Kim HJ, Chang KC, Baek JC, Park JK, Shin JK, Choi WJ, Lee JH, and Paik WY (2009a). DHA and EPA down-regulate COX-2 expression through suppression of NF-kappaB activity in LPS-treated human umbilical vein endothelial cells. Korean J. Physiol. Pharmacol 13, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SA, Kim HJ, Chang KC, Baek JC, Park JK, Shin JK, Choi WJ, Lee JH, and Paik WY (2009b). DHA and EPA down-regulate COX-2 expression through suppression of NF-kappaB activity in LPS-treated human umbilical vein endothelial cells. Korean J. Physiol. Pharmacol 13, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhang LL, Dong FC, Liu Y, Li N, Li HH, Lei HH, Hao FH, Wang YL, Zhu Y, et al. (2015). Metabonomic changes associated with atherosclerosis progression for LDLR−/− mice. J. Proteome Res 14, 2237–2254. [DOI] [PubMed] [Google Scholar]
- Lim C-Y, Bi X, Wu D, Kim JB, Gunning PW, Hong W, and Han W.(2015). Tropomodulin3 is a novel Akt2 effector regulating insulin-stimulated GLUT4 exocytosis through cortical actin remodeling. Nat. Commun 6, 5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder ME, and Deschenes RJ (2007). Palmitoylation: policing protein stability and traffic. Nat. Rev. Mol. Cell Biol 8, 74–84. [DOI] [PubMed] [Google Scholar]
- McIlhinney RA (1998). Membrane targeting via protein N-myristoylation. Methods Mol. Biol 88, 211–225. [DOI] [PubMed] [Google Scholar]
- Mika A, and Sledzinski T.(2017). Alterations of specific lipid groups in serum of obese humans: a review. Obes. Rev 18, 247–272. [DOI] [PubMed] [Google Scholar]
- Mostad IL, Bjerve KS, Lydersen S, and Grill V.(2008). Effects of marine n-3 fatty acid supplementation on lipoprotein subclasses measured by nuclear magnetic resonance in subjects with type II diabetes. Eur. J. Clin. Nutr 62, 419–429. [DOI] [PubMed] [Google Scholar]
- Niinisto S, Takkinen HM, Erlund I, Ahonen S, Toppari J, Ilonen J, Veijola R, Knip M, Vaarala O, and Virtanen SM (2017). Fatty acid status in infancy is associated with the risk of type 1 diabetes-associated autoimmunity. Diabetologia 60, 1223–1233. [DOI] [PubMed] [Google Scholar]
- Nimse SB, and Pal D.(2015). Free radicals, natural antioxidants, and their reaction mechanisms. RSC Adv. 5, 27986–28006. [Google Scholar]
- Noland CL, Gierke S, Schnier PD, Murray J, Sandoval WN, Sagolla M, Dey A, Hannoush RN, Fairbrother WJ, and Cunningham CN (2016). Palmitoylation of TEAD transcription factors is required for their stability and function in hippo pathway signaling. Structure 24, 179–186. [DOI] [PubMed] [Google Scholar]
- Novotny I, Saleh F, and Novotna R.(1984). The effect of chlorpromazine and dibucaine on phospholipid-metabolism in the frog sartorius muscle. Physiol. Bohemoslov. 33, 59–65. [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010a). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, Li PP, Lu WJ, Watkins SM, and Olefsky JM (2010b). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaler E, Kamp D, and Haest CW (2000). Acceleration of phospholipid flip-flop in the erythrocyte membrane by detergents differing in polar head group and alkyl chain length. Biochim. Biophys. Acta 1509, 397–408. [DOI] [PubMed] [Google Scholar]
- Perez-Rizquez C, Abian O, and Palomo JM (2019). Site-selective modification of tryptophan and protein tryptophan residues through PdNP bionanohybrid-catalysed C-H activation in aqueous media. Chem. Commun 55, 12928–12931. [DOI] [PubMed] [Google Scholar]
- Pironi L, Agostini F, and Guidetti M.(2015). Intravenous lipids in home parenteral nutrition. World Rev. Nutr. Diet 112, 141–149. [DOI] [PubMed] [Google Scholar]
- Resh MD (2016). Fatty acylation of proteins: the long and the short of it. Prog. Lipid Res 63, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncaglioni MC, Tombesi M, Avanzini F, Barlera S, Caimi V, Longoni P, Marzona I, Milani V, Silletta MG, Tognoni G, et al. (2013). n-3 fatty acids in patients with multiple cardiovascular risk factors. N. Engl. J. Med 368, 1800–1808. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, and Sabatini DM (2007). PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 25, 903–915. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP (2002a). The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother 56, 365–379. [DOI] [PubMed] [Google Scholar]
- Simopoulos AP (2002b). Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr 21, 495–505. [DOI] [PubMed] [Google Scholar]
- Smotrys JE, and Linder ME (2004). Palmitoylation of intracellular signaling proteins: regulation and function. Annu. Rev. Biochem 73, 559–587. [DOI] [PubMed] [Google Scholar]
- Sorgi PJ, Hallowell EM, Hutchins HL, and Sears B.(2007). Effects of an open-label pilot study with high-dose EPA/DHA concentrates on plasma phospholipids and behavior in children with attention deficit hyperactivity disorder. Nutr. J 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulage CO, Puig LS, Soulere L, Zarrouki B, Guichardant M, Lagarde M, and Pillon NJ (2018). Skeletal muscle insulin resistance is induced by 4-hydroxy-2-hexenal, a by-product of n-3 fatty acid peroxidation. Diabetologia 61, 688–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spickett CM, and Pitt AR (2015). Oxidative lipidomics coming of age: advances in analysis of oxidized phospholipids in physiology and pathology. Antioxidants Redox Signal. 22, 1646–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacpoole PW, Alig J, Kilgore LL, Ayala CM, Herbert PN, Zech LA, and Fisher WR (1988). Lipodystrophic diabetes-mellitus - investigations of lipoprotein metabolism and the effects of omega-3 fatty-acid administration in 2 patients. Metab., Clin. Exp 37, 944–951. [DOI] [PubMed] [Google Scholar]
- Szachowicz-Petelska B, Sulkowski S, and Figaszewski ZA (2007). Altered membrane free unsaturated fatty acid composition in human colorectal cancer tissue. Mol. Cell Biochem 294, 237–242. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ishikawa Y, Yokoyama M, Origasa H, Matsuzaki M, Saito Y, Matsuzawa Y, Sasaki J, Oikawa S, Hishida H, et al. (2008). Reduction in the recurrence of stroke by eicosapentaenoic acid for hypercholesterolemic patients - subanalysis of the JELIS trial. Stroke 39, 2052–2058. [DOI] [PubMed] [Google Scholar]
- Topping DL, Trimble RP, and Storer GB (1987). Failure of insulin to stimulate lipogenesis and triacylglycerol secretion in perfused livers from rats adapted to dietary fish oil. Biochim. Biophys. Acta 927, 423–428. [DOI] [PubMed] [Google Scholar]
- Udenwobele DI, Su RC, Good SV, Ball TB, Shrivastav SV, and Shrivastav A.(2017). Myristoylation: an important protein modification in the immune response. Front. Immunol 8, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valagussa F, Franzosi MG, Geraci E, Mininni N, Nicolosi GL, Santini M, Tavazzi L, Vecchio C, Marchioli R, Bomba E, et al. (1999). Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet 354, 447–455. [PubMed] [Google Scholar]
- Wen ZM, and Kim HY (2004). Alterations in hippocampal phospholipid profile by prenatal exposure to ethanol. J. Neurochem 89, 1368–1377. [DOI] [PubMed] [Google Scholar]
- William TC, MGeorge B, Lawrence B, Andrew JM, David D, Mary G, Eddie LG, Frank BH, Steven EK, Derek L, et al. (2017). Standards of medical care in diabetes-2017: summary of revisions. Diabetes care 40, S4–S5. [DOI] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, and Mann M.(2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–U360. [DOI] [PubMed] [Google Scholar]
- Woodcock SR, Marwitz AJV, Bruno P, and Branchaud BP (2006). Synthesis of nitrolipids. All four possible diastereomers of nitrooleic acids: (E)- and (Z)-, 9- and 10-nitro-octadec-9-enoic acids. Org. Lett 8, 3931–3934. [DOI] [PubMed] [Google Scholar]
- Yao Y, Dong J, Dong M, Liu F, Wang Y, Mao J, Ye M, and Zou H.(2017). An immobilized titanium (IV) ion affinity chromatography adsorbent for solid phase extraction of phosphopeptides for phosphoproteome analysis. J. Chromatogr. A 1498, 22–28. [DOI] [PubMed] [Google Scholar]
- Yao Y, Wang Y, Wang S, Liu X, Liu Z, Li Y, Fang Z, Mao J, Zheng Y, and Ye M.(2019). One-step SH2 superbinder-based approach for sensitive analysis of tyrosine phosphoproteome. J. Proteome Res 18, 1870–1879. [DOI] [PubMed] [Google Scholar]
- Zambon S, Friday KE, Childs MT, Fujimoto WY, Bierman EL, and Ensinck JW (1992). Effect of glyburide and omega 3 fatty acid dietary supplements on glucose and lipid metabolism in patients with non-insulin-dependent diabetes mellitus. Am. J. Clin. Nutr 56, 447–454. [DOI] [PubMed] [Google Scholar]
- Zhang WT, Liu J, Hu XM, Li PY, Leak RK, Gao YQ, and Chen J.(2015). n-3 polyunsaturated fatty acids reduce neonatal hypoxic/ischemic brain injury by promoting phosphatidylserine formation and Akt signaling. Stroke 46, 2943–2950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw phosphoproteomics data and raw targeted proteomics data have been deposited at Mendeley and are publicly available as the date of publication. DOIs are listed in the Key Resource Table.
This paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| phospho-AKT2(T308) | Cell Signaling Technology | Cat# 13038; RRID: AB_2629447 |
| AKT2 | Cell Signaling Technology | Cat# 3063; RRID: AB_2225186 |
| phospho-RSK2(S227) | Cell Signaling Technology | Cat# 3556; RRID: AB_2181465 |
| RSK2 | Cell Signaling Technology | Cat# 5528; RRID: AB_10860075 |
| phospho-AS160(T642) | Cell Signaling Technology | Cat# 8881; RRID: AB_2651042 |
| AS160 | Cell Signaling Technology | Cat# 2670; RRID: AB_2199375 |
| phospho-GSK3β(S9) | Cell Signaling Technology | Cat# 5558; RRID: AB_10013750 |
| GSK3β | Cell Signaling Technology | Cat# 12456; RRID: AB_2636978 |
| phospho-TSC2(S939) | Cell Signaling Technology | Cat# 3615; RRID: AB_2207796 |
| TSC2 | Cell Signaling Technology | Cat# 3990; RRID: AB_2209986 |
| phospho-PRAS40(T246) | Cell Signaling Technology | Cat# 13175; RRID: AB_2798140 |
| PRAS40 | Cell Signaling Technology | Cat# 2691; RRID: AB_2225033 |
| PDK1 | Cell Signaling Technology | Cat# 5662; RRID: AB_10839264 |
| GLUT4 | Cell Signaling Technology | Cat# 2213; RRID: AB_823508 |
| Na-K-ATPase | Cell Signaling Technology | Cat# 3010; RRID: AB_2060983 |
| GAPDH | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| phospho-IR(Y1150/1151) | Cell Signaling Technology | Cat# 3024; RRID: AB_331253 |
| IR | Cell Signaling Technology | Cat# 3020; RRID: AB_2249166 |
| phospho-IRS1(Y895) | Cell Signaling Technology | Cat# 3070; RRID: AB_2127863 |
| IRS1 | Cell Signaling Technology | Cat# 2390; RRID: AB_10692516 |
| phospho-AKT2(S473) | Cell Signaling Technology | Cat# 4060; RRID: AB_2224726 |
| CDK2 | Cell Signaling Technology | Cat# 2546; RRID: AB_2276129 |
| SIRT3 | Cell Signaling Technology | Cat# 5490; RRID: AB_10828246 |
| COX4 | Cell Signaling Technology | Cat# 4884; RRID: AB_2085427 |
| PGK1 | Abcam | Cat# ab38007; RRID: AB_2161220 |
| PI3K p110α | Cell Signaling Technology | Cat# 4255; RRID: AB_659888 |
| PI3K p85 | Cell Signaling Technology | Cat# 4292; RRID: AB_329869 |
| Flag | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| DHA | LSbio | Cat# LS-C714681; |
| EPA | LSbio | Cat# LS-C714683; |
| AKT1 | Abcam | Cat# ab89402; RRID: AB_2049150 |
| phospho-AKT1(S473) | Cell Signaling Technology | Cat# 9018; RRID: AB_2629283 |
| phospho-AKT1(T308) | Abcam | Cat# ab105731; RRID: AB_10900490 |
| phospho-S6K(T389) | Cell Signaling Technology | Cat# 9234; RRID: AB_2269803 |
| S6K | Cell Signaling Technology | Cat# 9202; RRID: AB_331676 |
| BiP | Cell Signaling Technology | Cat# 3177 RRID: AB_2119845 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DHA | Sigma-Aldrich | Cat# D8768 |
| Methyl-DHA | Sigma-Aldrich | Cat# D2659 |
| Ethyl-DHA | Sigma-Aldrich | Cat# D2410 |
| EPA | Sigma-Aldrich | Cat# E6627 |
| Methyl-EPA | Sigma-Aldrich | Cat# CRM47571 |
| Ethyl-EPA | Sigma-Aldrich | Cat# E0085000 |
| linoleic acid | Sigma-Aldrich | Cat# L8134 |
| palmitic acid | Sigma-Aldrich | Cat# P9767 |
| oleic acid | Sigma-Aldrich | Cat# O7501 |
| conjugated linoleic acid | Sigma-Aldrich | Cat# 16413 |
| linolenic acid | Sigma-Aldrich | Cat# L2378 |
| Insulin | Sigma-Aldrich | Cat# 1342106 |
| Tryptophan | Sigma-Aldrich | Cat# V900470 |
| methyl-tryptophan | Sigma-Aldrich | Cat# 447439 |
| Tryptamine | Sigma-Aldrich | Cat# 246557 |
| DTT | Sigma-Aldrich | Cat# D9760 |
| Wortmannin | Sigma-Aldrich | Cat# W1628 |
| Alanine | Sigma-Aldrich | Cat# 05130 |
| 5-hydroxytryptamine | J&K Scientific Ltd. | Cat# 153–98-0 |
| L-α-phosphoatidylserine | Avanti | Cat# 870336C |
| Peptides, see Figure S2C | GL Biochem (Shanghai) Ltd. | N/A |
| Penicillin-Streptomycin | Invitrogen | Cat# 15070063 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse PIP3 ELISA | Shanghai Enzyme-linked Biotechnology Co., LTD. | Cat# ml037511–1 |
| Glucose Uptake-GloTM Assay Kit | Promega | Cat# J1341 |
| AKT kinase activity assay kit | Abcam | Cat# ab139436 |
| PDK1 kinase activity assay kit | Promega | V9681 |
|
| ||
| Deposited data | ||
|
| ||
| Raw phosphoproteomics data | N/A | Mendeley data: https://doi.org/10.17632/rc3djvsgn3.1 |
| Raw targeted proteomics data | N/A | Mendeley data: https://doi.org/10.17632/n4npj2j8rx.1 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: 3T3-L1 | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNM25 |
| Cricetulus griseus: CHO-K1 | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNHa 4 |
| Human: HepG2 | Stem Cell Bank, Chinese Academy of Sciences | Cat# SCSP-510 |
| Human: HEK293T | Stem Cell Bank, Chinese Academy of Sciences | Cat# GNHu17 |
|
| ||
| Experimental Models: Organisms/strains | ||
|
| ||
| Mouse: 129/C57BL6 | Shanghai SLAC Laboratory Animal Co.,Ltd | N/A |
| Mouse: 129/C57BL6, AKT2 KO | Shanghai SLAC Laboratory Animal Co., Ltd | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| Myc-GLUT4-mCherry | This work | N/A |
| AKT2-EGFP | This work | N/A |
| AKT2R25C-EGFP | This work | N/A |
| AKT2R25C-Flag | This work | N/A |
| PDK1-EGFP | This work | N/A |
| PDK1R474A-Flag | This work | N/A |
| PDK1W448/543L-EGFP | This work | N/A |
| PDK1w266/347L-EGFP | This work | N/A |
| PDK1-Flag | This work | N/A |
| PDK16W/L-Flag | This work | N/A |
| PDK1W448/543L-Flag | This work | N/A |
| PDK1W266/347L-Flag | This work | N/A |
| PDK1R474A-Flag | This work | N/A |
| AKT2-Flag | This work | N/A |
| AKT27W/L-Flag | This work | N/A |
| AKT2W414L-Flag | This work | N/A |
| AKT2W414L-EGFP | This work | N/A |
| AKT2W334L-Flag | This work | N/A |
| AKT2W334L-EGFP | This work | N/A |
| AKT1-Flag | This work | N/A |
| AKT1W413L-Flag | This work | N/A |
| PGK1-EGFP | This work | N/A |
| PGK1-Flag | This work | N/A |
| PGK1-PH-EGFP | This work | N/A |
| PGK1-PH-Flag | This work | N/A |
| PKM2-Flag | This work | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| MaxQuants | Max planck institute of biochemistry | https://www.maxquant.org |
| Perseus software | Max planck institute of biochemistry | https://maxquant.net/perseus/ |