

ABSTRACT
Ferroptosis is a form of inflammatory cell death for which key mediators remain obscure. Here, we report that the proteoglycan decorin (DCN) is released by cells that are dying from ferroptosis and then acts as an alarm signal to trigger innate and adaptive immune responses. The early release of DCN during ferroptosis is an active process that involves secretory macroautophagy/autophagy and lysosomal exocytosis. Once released, extracellular DCN binds to the receptor advanced glycosylation end-product-specific receptor (AGER) on macrophages to trigger the production of pro-inflammatory cytokines in an NFKB/NF-κB-dependent manner. Pharmacological and genetic inhibition of the DCN-AGER axis protects against ferroptotic death-related acute pancreatitis and limits the capacity of ferroptotic cancer cells to induce a tumor-protective immune response. Thus, DCN is an essential mediator of the inflammatory and immune consequences of ferroptosis.
KEYWORDS: Ferroptosis, DAMP, autophagy, inflammation, macrophages
Introduction
Physiological cell death acts as a homeostatic mechanism that maintains cell populations and tissue size during development [1–4]. In contrast, excessive cell death may cause inflammatory diseases because it leads to the release of danger/damage-associated molecular patterns (DAMPs) that act on surrounding cells [5,6]. Distinct cellular components, including proteins and non-proteinaceous molecules, can act as DAMPs to trigger inflammation and immune responses [7–9]. For example, the release of the nuclear protein HMGB1 (high mobility group box 1) occurs in various cell death modalities, including apoptosis [10], necrosis [11], pyroptosis [12], and ferroptosis [13]. Extracellular HMGB1 then exerts pro-inflammatory effects upon binding to pattern recognition receptors including advanced glycosylation end-product-specific receptor (AGER) and toll-like receptor 4 (TLR4) [14]. Thus, neutralization of HMGB1 may mitigate cell death-related inflammation [15]. However, identification of a specific DAMP for a particular type of cell death still remains a challenge in translational medicine.
Ferroptosis is a type of iron-dependent oxidative cell death that was initially characterized when erastin was discovered as a selective cytotoxic agent that kills cancer cells with RAS mutations [16,17]. Erastin-mediated activation of mitochondrial voltage-dependent anion channels (VDACs) or inhibition of SLC7A11/system xc− (solute carrier family 7 member 11) contributes to the induction of ferroptosis [18,19]. Ferroptosis also occurs in cells or tissues subjected to environmental stresses, such as cysteine depletion [20], heat damage [21], or ionizing radiation [22,23]. In addition to increasing reported oxidative and antioxidant systems, the susceptibility to drug-induced ferroptosis is fine-tuned by gene transcription, protein degradation, and metabolic reprogramming [17]. As a highly inflammatory form of cell death, ferroptotic cells may not be effectively eliminated by macrophage-mediated efferocytosis [24]. Consequently, ferroptotic death-mediated sterile inflammation has been implicated in the pathogenesis of several diseases, including cancer [25], ischemia-reperfusion damage [26], and pancreatitis [27]. However, the molecular and cellular mechanisms that govern specific ferroptosis-dependent immune response remain poorly understood [28].
In this study, we utilized an antibody array technique to assay ferroptosis-relevant DAMPs. We identified DCN (decorin) as a DAMP that is released during the early phase of ferroptosis (but not apoptosis or necroptosis) and then acts on AGER. Inhibition of the DCN-AGER axis prevents ferroptosis-related pancreatitis and curtails the capacity of ferroptotic cancer cells to stimulate an antitumor immune response.
Results
The selectively release of DCN during ferroptosis
We used a multi-antibody array chip containing 507 target proteins (including cytokines, chemokines, and other immune molecules) to analyze the protein levels in the supernatant of two human cancer cell lines (HT1080 and HeLa) in the absence or presence of erastin. Compared to the human cervical cancer cell line HeLa, the human fibrosarcoma cell line HT1080 was more sensitive to erastin-induced cell death (Figure 1A), thereby releasing a larger quantity of proteins (Figure 1B). The proteoglycan DCN was identified as one of the top 10 erastin-induced proteins released in both HT1080 and HeLa cells (Figure 1B,C). Quantification of DCN by an enzyme-linked immunosorbent assay (ELISA) further confirmed that erastin and other ferroptosis activators, including SLC7A11/system xc− inhibitors (sulfasalazine and sorafenib), cysteine depletion, and GPX4 inhibitors (RSL3, ML162, ML210, and FIN56), induced DCN release in human or mouse cell lines, including HT1080 (Figure 1D), PANC1 (Figure S1A), mouse embryonic fibroblasts (MEFs) (Figure S1B), and KPC (Figure S1C) cells. Of note, the release of DCN was reversed by the ferroptosis inhibitor ferrostatin-1 (an aromatic amine that specifically binds with lipid ROS and protects cells against lipid peroxidation [16,29]), rather than the apoptosis inhibitor Z-VAD-FMK (a pan-caspase inhibitor) or the necroptosis inhibitor necrosulfonamide (an inhibitor of MLKL [mixed lineage kinase domain like pseudokinase [30]) (Figure 1D and FigureS1A–S1C). In contrast to the effects of ferroptosis activators, the ability of staurosporine, an apoptosis inducer [31], or CCT137690, a necroptosis inducer [32], to release DCN (Figure 1D and Figure S1A–S1C) was significantly reduced.
Figure 1.
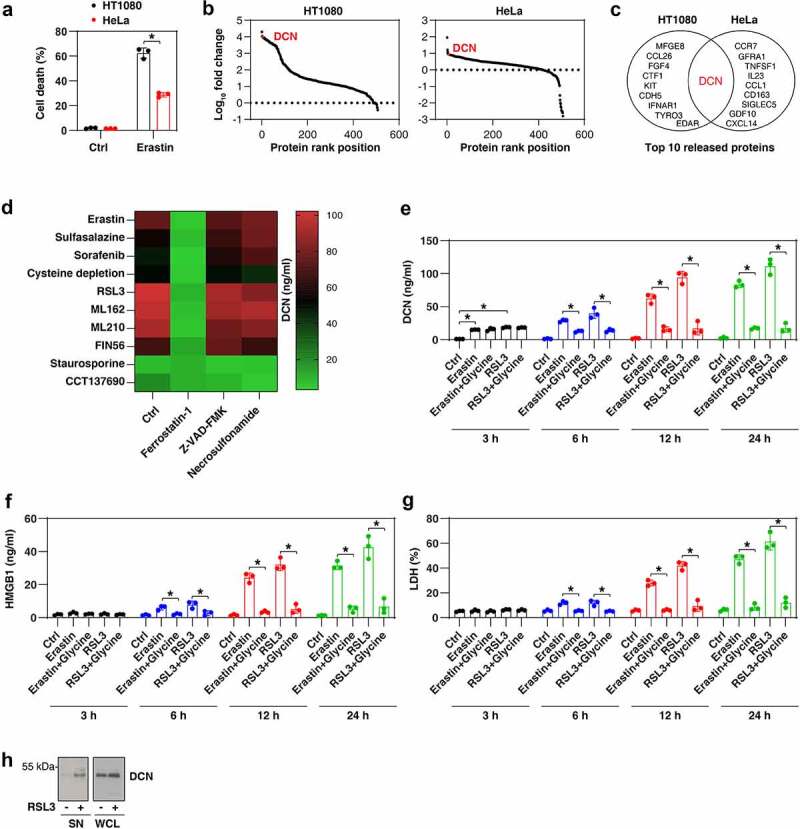
DCN is a DAMP released during ferroptosis. (A) Cell death of HT1080 and HeLa cells following treatment with erastin (5 μM) for 24 h (n = 3 biologically independent samples; *P < 0.05, one-tailed t test; data are presented as means ± SD). (B) An antibody array technology-based screening of protein release from HT1080 and HeLa cells following treatment with erastin (5 µM) for 24 h. The data are presented as the rank of fold change of protein release. (C) Top 10 released proteins in the setting of panel B. (D) ELISA assay of DCN release in HT1080 cells following treatment with erastin (5 μM), sulfasalazine (500 μM), sorafenib (10 μM), cysteine depletion, RSL3 (0.5 μM), ML162 (0.5 μM), ML210 (5 μM), FIN56 (2.5 μM), staurosporine (200 nM), or CCT137690 (5 μM) in the absence or presence of ferrostatin-1 (1 µM), Z-VAD-FMK (10 µM), or necrosulfonamide (1 μM) for 24 h (data are shown in a heat map as the means of three biologically independent samples). (E–G) HT1080 cells were treated with erastin (5 μM) or RSL3 (5 μM) in the absence or presence of glycine for 3–24 h and the release of DCN, HMGB1, and LDH were assayed by ELISA (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (H) Western blot assay of DCN in secreted proteins (SN) and whole cell lysates (WCL) in HT1080 cells following RSL3 (5 μM) treatment for 3 h.
To further analyze the sensitivity of DCN release in monitoring ferroptosis, we compared the release dynamics of DCN, HMGB1 (a universal DAMP [14]), and LDH (lactate dehydrogenase; an indicator of plasma membrane rupture and necrosis [33]) from HT1080 cells responding to erastin or RSL3, which are the most commonly used pharmacological ferroptosis inducers. The release of DCN was observed within 3 h, i.e., earlier than HMGB1 or LDH (Figure 1E–G). Glycine, an amino acid used to prevent rapid membrane destruction in lytic cell death [34], failed to inhibit erastin- or RSL3-induced DCN release at 3 h (Figure 1E–G). In contrast, glycine inhibited the release of DCN, HMGB1, and LDH at 6–24 h (Figure 1E–G). To rule out the possibility that the release of DCN is associated with the cleavage of this protein [35,36], we compared the molecular weight of DCN in whole-cell lysate and supernatants. Immunoblot analysis revealed no change in the molecular weight of secreted DCN compared to intracellular DCN (Figure 1H).
Autophagy favors DCN secretion during ferroptosis
Ferroptosis is considered as a type of autophagy-dependent cell death coupled to lysosomal dysfunction [37]. We previously demonstrated that autophagy-inducible cancer cells (as monitored by the detection of MAP1LC3B [microtubule-associated protein 1 light chain 3 beta]-II) are sensitive to ferroptosis induction in 60 cancer cell lines [38]. To identify the molecular basis for increased DCN secretion during the early phase of ferroptosis, we investigated the relationship between autophagy induction and DCN secretion. When measuring the release of DCN from the entire panel of 60 cancer cell lines treated with erastin or RSL3 for 3 h, we observed a significant positive correlation between the autophagosome marker MAP1LC3B-II and the secretion of DCN (Figure 2A). Indeed, in contrast to MAP1LC3B-II-inducible cell lines (e.g., HT1080 and PANC1), MAP1LC3B-II-non-inducible cell lines (e.g., BX-PC3 and NCI-H460) were resistant to both the induction of cell death and DCN secretion by erastin or RSL3 (Figure 2B,C).
Figure 2.
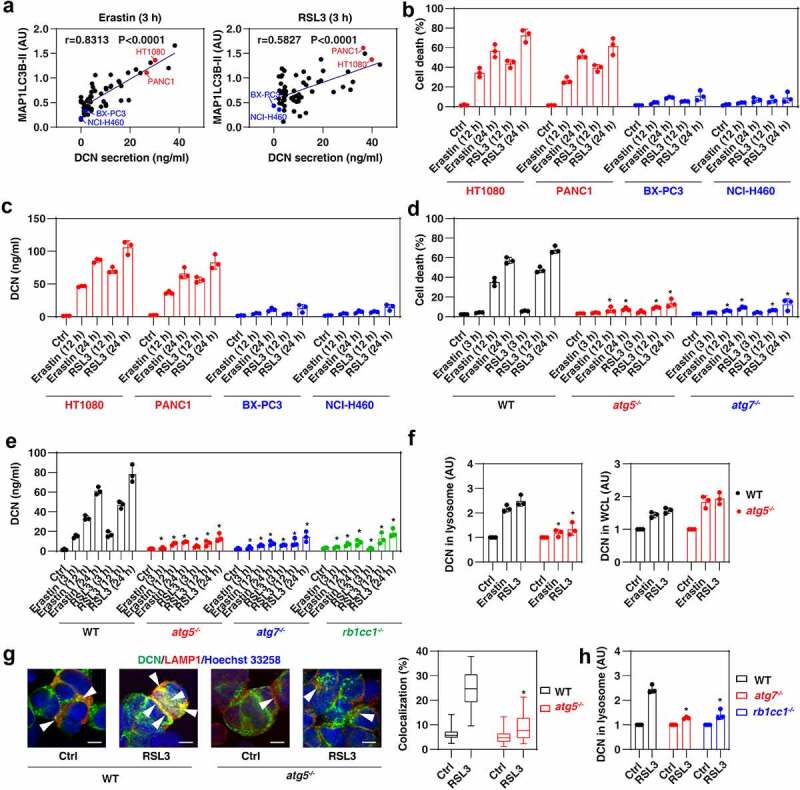
Autophagy mediates DCN secretion during ferroptosis. (A) The Pearson correlation coefficient was used to determine the relationship between the expression of MAP1LC3B-II and the release of DCN in 60 cancer cell lines after treatment with erastin or RSL3 for 3 h. The original western blot data and quantification of MAP1LC3B-II were shown in our previous study [38]. (B,C) The indicated human cancer cell lines were treated with erastin (5 μM) or RSL3 (0.5 μM) for 12 and 24 h and then cell death and DCN release were assayed (n = 3 biologically independent samples; data are presented as means ± SD). (D,E) The indicated MEFs were treated with erastin (5 μM) or RSL3 (0.5 μM) for 3–24 h and then cell death and DCN release were assayed (n = 3 biologically independent samples; *P < 0.05 versus WT group, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (F) ELISA analysis of DCN expression in the lysosomal fraction or whole-cell extracts (WCL) in WT and atg5−/− MEFs following treatment with erastin (5 μM) or RSL3 (0.5 μM) for 3 h (n = 3 biologically independent samples; *P < 0.05 versus WT group, one-tailed t test; data are presented as means ± SD). (G) Image analysis of the colocalization between DCN and lysosomes in WT and atg5−/− MEFs following treatment with RSL3 (0.5 μM) for 3 h. The data are presented as box-and-whisker plots from 10 fields. Boxes represent the median and the 25th and 75th percentiles. *P < 0.05 versus WT group, one-tailed t test. Bar = 15 μm. (H) ELISA analysis of DCN expression in the lysosomal fraction in WT, atg7−/− and rb1cc1−/− MEFs following treatment with RSL3 (0.5 μM) for 3 h (n = 3 biologically independent samples; *P < 0.05 versus WT group, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD).
To confirm the relationship between autophagy and DCN release during ferroptosis, we used MEFs that lacked either of two genes essential for the formation of autophagosomes (atg5−/− or atg7−/−) [39,40]. Compared to wild-type (WT) cells, the induction of cell death determined at 3, 12, and 24 h (Figure 2D) and the release of DCN measured at the same time points (Figure 2E) were blocked in atg5−/− and atg7−/− cells. The hypothesis that autophagy mediates active secretion of DCN during ferroptosis was further confirmed using human fibrosarcoma HT1080 cells in which either ATG5 or ATG7 were knocked down [38] (Figure S2). Moreover, the loss of Rb1cc1/Fip200 (RB1-inducible coiled-coil 1), a regulator of the initiation of autophagosome formation, limited erastin- or RSL3-induced DCN release in rb1cc1−/− MEFs (Figure 2E).
Because lysosomes are acidic organelles that integrate autophagy and the secretory pathway [41], we hypothesized that DCN would accumulate in this compartment. ELISA analysis of the lysosomal fraction of MEFs revealed the accumulation of DCN in lysosomes after treatment with erastin or RSL3 for 3 h, and this process was diminished in atg5−/− MEFs (Figure 2F). In contrast, the deletion of Atg5 did not affect the upregulation of DCN protein in the whole-cell lysate of MEFs. Immunofluorescence microscopy confirmed that Atg5 is required for erastin- or RSL3-induced accumulation of DCN in the lysosomes of MEFs (Figure 2G). Moreover, RSL3-induced accumulation of DCN in lysosomes was also reduced in atg7−/− or rb1cc1−/− MEFs (Figure 2H). These data indicate that the formation of autophagosomes contributes to the sort of DCN to lysosomes during ferroptosis.
The knockdown of Mcoln1 (mucolipin 1) by two different shRNAs (Figure S3A), a key gene encoding a lysosomal ion channel involved in lysosomal exocytosis [42], blocked erastin- or RSL3-induced DCN secretion at 3 h (Figure S3B), but did not interfere with the accumulation of DCN in lysosomes (Figure S3C) and in whole-cell lysates (Figure S3D). Immunoprecipitation revealed an interaction between DCN and MCOLN1 that was increased by RSL3 or erastin, but not by the apoptosis inducer staurosporine or the necroptosis activator CCT137690 (Figure S3E). MCOLN1 is also involved in promoting autophagosome formation induced by oxidative stress [43]. The formation of MAP1LC3B puncta induced by erastin or RSL3 was inhibited by the knockdown of Mcoln1 in MEFs (Figure S3F). As expected, the knockdown of Mcoln1 increased the accumulation of the lysosomal cysteine protease CTSB (cathepsin B) (Figure S3G), a mediator of lysosomal dysfunction during ferroptosis [44]. The levels of DCN in lysosome is affected by many factors, including gene transcription, protein degradation, and release. During RSL3 treatment, the deletion of Atg5 or Mcoln1 in MEFs failed to affect Dcn gene transcription (Figure S3H) and protein half-life (Figure S3I). Overall, these assays establish a ferroptosis-related DCN secretion pathway that involves autophagy machinery-mediated lysosomal accumulation of DCN and its subsequent MCOLN1-dependent exocytosis.
DCN drives inflammation caused by ferroptotic cells
Inflammation is the primary tissue damage response, and extracellular DCN has been reported to be a mediator of septic inflammation, pancreatitis, and renal inflammation [45–47]. To determine whether the ferroptosis-associated release of DCN might be an immediate trigger for an inflammatory response, we challenged primary wild-type mouse bone marrow-derived macrophages (BMDMs) with ferroptotic dcn−/− or WT MEFs. Ferroptotic WT cells triggered the production of proinflammatory cytokines (e.g., tumor necrosis factor [TNF] and interleukin 6 [IL6]) much more efficiently than dcn−/− ferroptotic cells (Figure 3A). Similarly, ferroptotic atg5−/− or Mcoln1-knockdown MEFs showed a reduced ability to activate BMDMs to produce TNF and IL6 (Figure 3A). DCN-knockdown ferroptotic HT1080 cells were also ineffective in activating primary human peripheral blood monocytes (HPBMs), human peripheral blood dendritic cells (HPBDs), or peripheral blood mononuclear cells (PBMCs) (Figure S4), supporting the hypothesis that DCN released from ferroptotic cells plays a broad role in activating different myeloid cell types.
Figure 3.
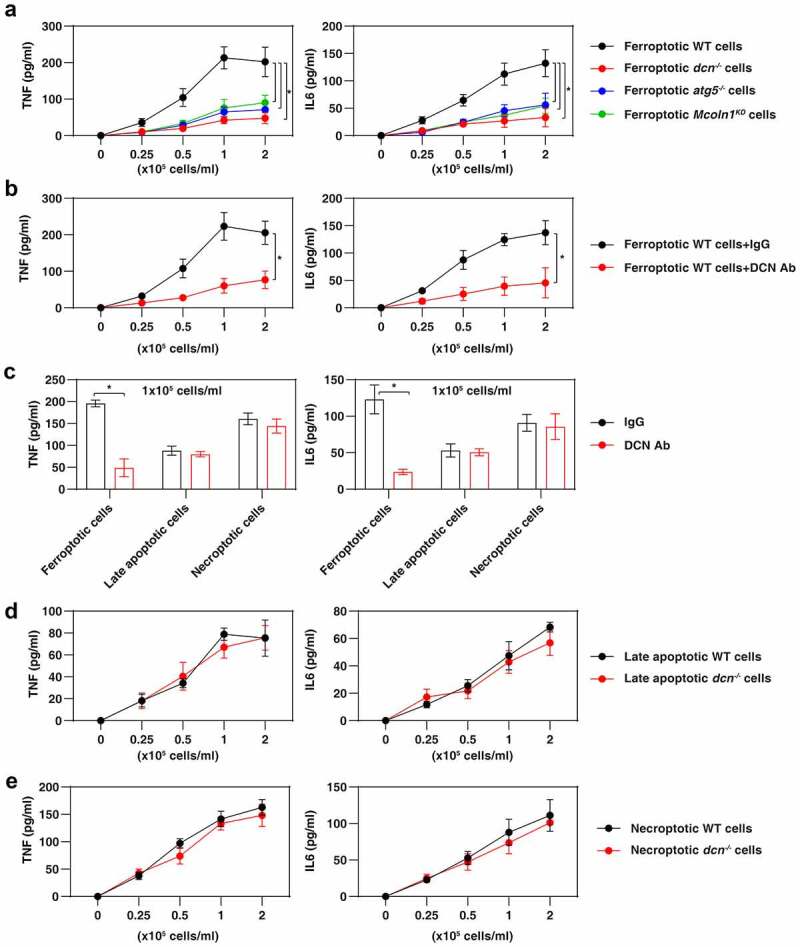
DCN mediates inflammation caused by ferroptotic cells. (A) Ferroptotic cells lacking Dcn, Atg5, or Mcoln1 do not elicit the production of the pro-inflammatory TNF and IL6 cytokine by 1 × 106 BMDMs (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (B,C) Anti-DCN neutralizing antibody (1 mg/ml) inhibits ferroptotic cells, but not late apoptotic cell- or necroptotic cell-induced TNF and IL6 release in BMDMs (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (D,E) Lack of Dcn in late apoptotic or necroptotic cells fails to affect the production of TNF and IL6 by BMDMs (n = 3 biologically independent samples; data are presented as means ± SD).
Neutralization of DCN by means of a monoclonal antibody attenuated the secretion of TNF and IL6 by BMDMs cultured with WT ferroptotic cells (Figure 3B). In contrast, anti-DCN antibody was ineffective in attenuating the release of TNF and IL6 from BMDMs expose to late apoptotic cells or necroptotic cells (Figure 3C), meaning that DCN is an essential DAMP only in the context of ferroptosis. In line with this notion, WT and dcn−/− late apoptotic cells or necroptotic cells were similarly efficient in eliciting the production of TNF and IL6 by BMDMs (Figure 3D,Figure 3E). Altogether, these findings provide compelling evidence that DCN is one of the main diffusible signals of ferroptosis, but not apoptosis or necroptosis.
AGER mediates DCN activity during ferroptosis
Several receptors have been implicated in extracellular DCN activity in immune and nonimmune cells. These putative DCN receptors include epidermal growth factor receptor (EGFR) [48], kinase insert domain receptor (KDR/VEGFR-2) [49], (transforming growth factor beta receptor 1 (TGFBR1) [50], MET proto-oncogene, receptor tyrosine kinase (MET) [51], insulin-like growth factor 1 receptor (IGF1R) [52], toll-like receptor 2 (TLR2) [45], and TLR4 [45]. In addition, AGER reportedly mediates HMGB1 activity during ferroptotic death [13]. We used lentiviral shRNA-mediated RNAi to suppress the expression of these eight receptors in BMDMs (Figure 4A). We found that Ager shRNA, but none of the other shRNAs, diminished ferroptotic cell-induced secretion of TNF and IL6 by BMDMs (Figure 4B). Consistently, as compared to WT BMDMs, BMDMs from ager−/− mice secreted less TNF and IL6 upon exposure to ferroptotic cells (Figure 4C). Moreover, antibody-mediated neutralization of AGER, but not TLR4, limited the secretion of TNF and IL6 by WT BMDMs (Figure 4D).
Figure 4.
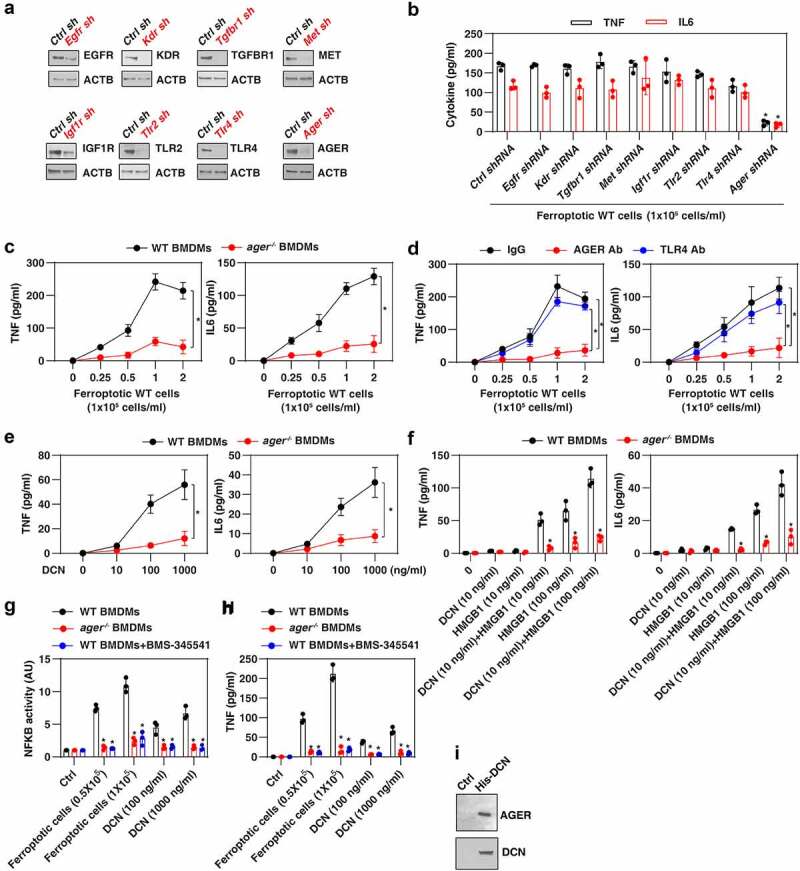
AGER mediates the response to DCN. (A) Western blot analysis of protein expression by BMDMs after transfection with the indicated shRNAs. (B) ELISA analysis of TNF and IL6 release in the indicated gene knockdown BMDMs following treatment with ferroptotic MEFs (n = 3 biologically independent samples; *P < 0.05 versus control shRNA group, one-tailed t test; data are presented as means ± SD). (C) Lack of Ager in BMDMs blocks ferroptotic cell-induced the production of the pro-inflammatory TNF and IL6 cytokines (n = 3 biologically independent samples; two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (D) Anti-AGER neutralizing antibody (Ab; 1 mg/ml), but not anti-TLR4 neutralizing antibody (1 mg/ml), inhibits ferroptotic cell-induced TNF and IL6 release in BMDMs (n = 3 biologically independent samples; *P < 0.05, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (E, F) Lack of Ager in BMDMs inhibits DCN-induced TNF and IL6 release in the absence or presence of HMGB1 (n = 3 biologically independent samples; *P < 0.05 versus WT group, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (G, H) Analysis of NFKB activity and TNF release in the indicated BMDMs following treatment with ferroptotic cells or DCN in the absence or presence of the NFKB pathway inhibitor BMS-345541 (n = 3 biologically independent samples; *P < 0.05 versus WT group, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD). (I) His-tag affinity pull-down analysis of the binding of DCN to AGER.
To ensure that DCN is a direct activator of AGER, we used purified DCN protein generated by NS0 mouse myeloma cells, which are frequently used for the production of monoclonal antibodies and other recombinant proteins [53]. Recombinant DCN protein dose-dependently elicited TNF and IL6 secretion by WT, but not ager−/− BMDMs (Figure 4E). Moreover, the production of TNF and IL6 could be synergistically induced by low-dose DCN and HMGB1 in WT, but not in ager−/− BMDMs (Figure 4F).
Given that AGER-mediated inflammatory cytokine production requires activation of nuclear factor kappa B (NFKB/NF-κB) [54,55], we measured the interaction of NFKB with a consensus-binding site (5ʹ-GGGACTTTCC-3ʹ) [56,57]. Ferroptotic cells or DCN induced the transcriptional activity of NFKB in WT, but not in ager−/− BMDMs (Figure 4G). BMS-345541, an NFKB pathway inhibitor [58], blocked ferroptotic cell- or DCN-induced transcriptional activity of NFKB (Figure 4G) and subsequent TNF production (Figure 4H). A His-tag affinity pull-down assay confirmed a direct interaction between DCN and AGER proteins in vitro (Figure 4I). Altogether, these data underscore a role for AGER in driving NFKB-mediated inflammation caused by DCN.
The DCN-AGER axis mediates ferroptosis-initiated inflammation and immune responses in vivo
The conditional knockout of Gpx4 within the pancreas of mice (termed gpx4−/− mice) reportedly accelerates cerulein-induced acute pancreatitis by activating ferroptotic damage [59]. We used this model to evaluate whether targeting the DCN-AGER axis would protect against ferroptosis-induced acute pancreatitis. Histological assessment of pancreatic damage revealed exaggerated acinar cell death, leukocyte infiltration, and interstitial edema in the gpx4−/− mice compared to WT controls (Figure 5A). These histological changes were accompanied by signs of exacerbated pancreatitis, including increased serum AMY (amylase) (Figure 5B), pancreatic trypsin activity (Figure 5C), and pancreatic MPO (myeloperoxidase) activity (Figure 5D), as well as serum DCN (Figure 5E), serum TNF (Figure 5F), and serum IL6 (Figure 5G). Similar to the administration of the ferroptosis inhibitor liproxstatin-1 [60], the administration of anti-DCN antibody or anti-AGER antibody protected against cerulein-induced pancreatitis especially in gpx4−/− mice (Figure 5A–Figure 5G). The depletion of ager also protected against cerulein-induced acute pancreatitis in WT and gpx4−/− mice (Figure S5). These preclinical studies indicate a role for the DCN-AGER pathway in mediating ferroptosis-related pancreatitis.
Figure 5.
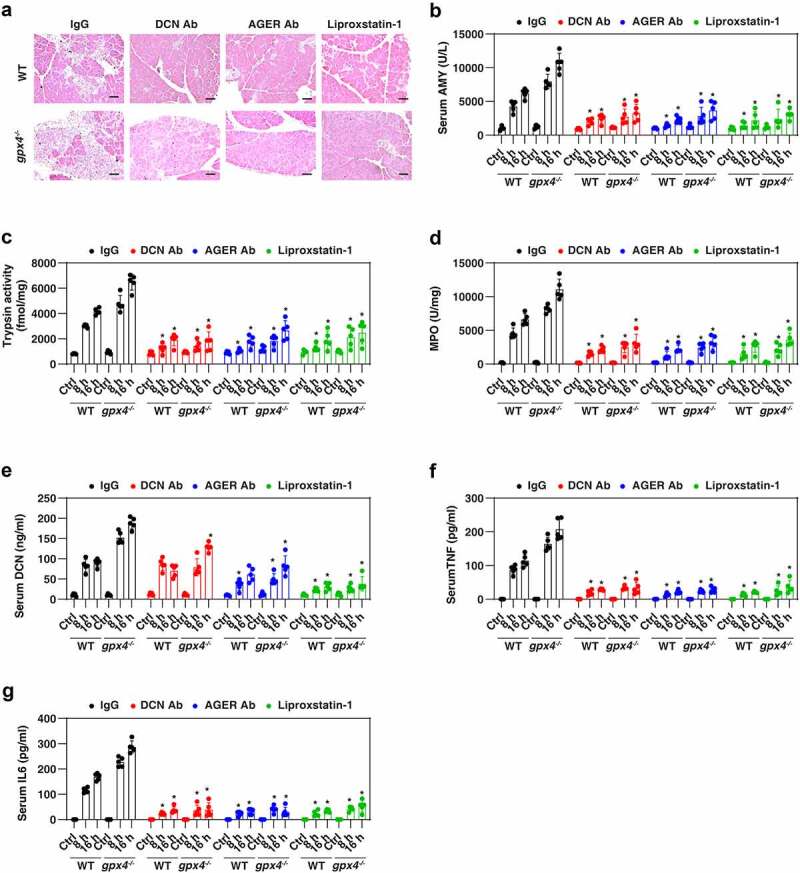
Inhibiting the DCN-AGER pathway protects against cerulein-induced acute pancreatitis. WT and pancreatic gpx4−/− mice received anti-DCN Ab (20 mg/kg; monoclonal rat IgG2A; R&D Systems, 161026), anti-AGER Ab (20 mg/kg; monoclonal rat IgG2A; R&D Systems, 175410), control IgG (20 mg/kg; R&D Systems) or liproxsatin-1 (10 mg/kg) by i.p. injection 2 h after completion of the cerulein-induced pancreatitis protocol. (A) Pancreatic sections were stained with H&E at 16 h (bar: 100 μm). (B–G) Serum AMY (amylase), pancreatic trypsin activity, pancreatic MPO activity, serum DCN, serum TNF, and serum IL6 were assayed at 8 and 16 h (n = 5 mice/group; *P < 0.05 versus IgG group, two-way ANOVA with Tukey’s multiple comparisons test; data are presented as means ± SD).
Ferroptosis can be considered as a variant of immunogenic cell death (ICD) in that early ferroptotic cancer cells injected into mice elicit a specific immune response that protects against subsequent rechallenge with live cancer cells of the same kind [61,62]. To evaluate the role of the DCN-AGER axis in ferroptosis-initiated adaptive immunity, we took advantage of this prophylactic tumor vaccination model [63]. RSL3-treated pancreatic ductal adenocarcinoma KPC cells (C57BL/6J background) were injected subcutaneously into the right flank of immunocompetent C57BL/6J mice. Such ferroptotic KPC cells were able to protect the majority of mice against rechallenge with live KPC cells injected into the opposite flank 1 week later (Figure 6A). However, RSL3-treated KPC cells co-administered together with anti-DCN or anti-AGER antibody (but not anti-TLR4 antibody) failed to elicit a protective anticancer immune response (Figure 6A). This contrasts with the observation that anti-TLR4 (but not anti-DCN or anti-AGER) antibody limited the cancer-preventive activity of a vaccine composed of oxaliplatin-treated KPC cells (Figure 6B). These results confirm that TLR4 is essential for the response to oxaliplatin-induced apoptotic ICD [64], but dispensable for RSL3-induced ICD. As a control, the vaccination efficacy of RSL3-induced ferroptotic cell death was lost in immune-deficient (rag2−/−) mice (Figure 6C), supporting the notion that dying cells drive an adaptive antitumor immune response [65].
Figure 6.
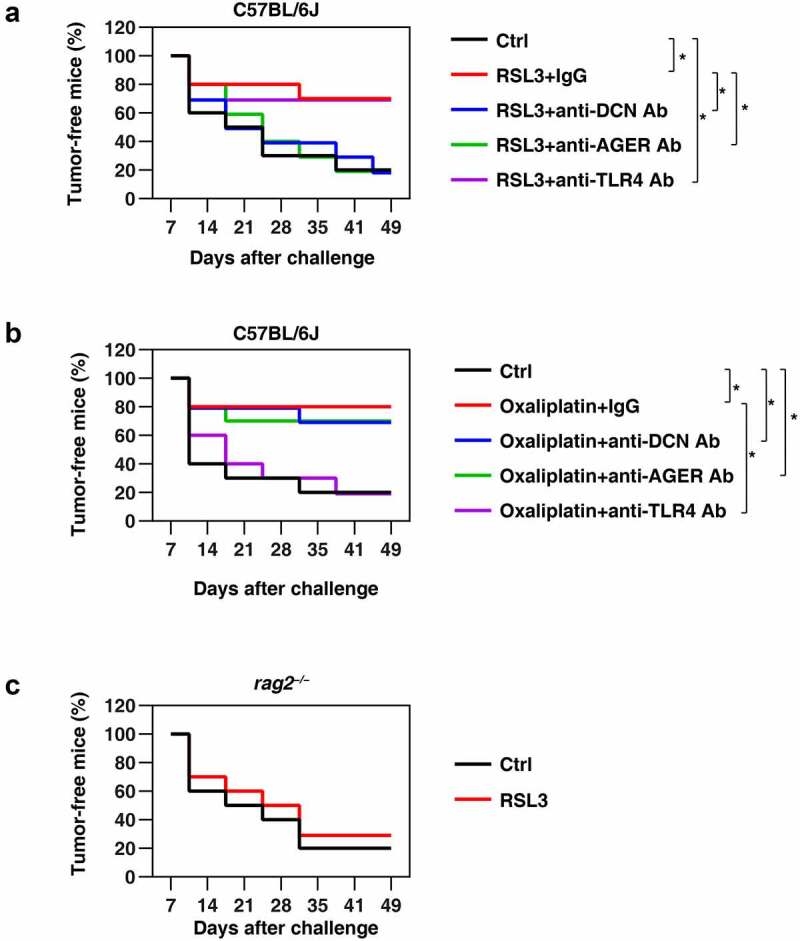
The DCN-AGER pathway drives antitumor immunity induced by vaccination with ferroptotic cancer cells. (A) The inhibition of DCN and AGER (but not TLR4) with specific blocking antibodies (Ab; 20 mg/kg) abolished the capacity of RSL3-treated tumor cells to vaccinate against KPC cells in C57BL/6 J mice. The percentage of tumor-free mice is indicated (n = 10 mice/group, *P < 0.05, two-way ANOVA test). (B) The inhibition of TLR4 (but not DCN or AGER) with specific blocking antibodies (20 mg/kg) abolished the capacity of oxaliplatin-treated tumor cells to vaccinate against KPC cells in C57BL/6 J mice. The percentage of tumor-free mice is indicated (n = 10 mice/group, *P < 0.05, two-way ANOVA test). (C) The vaccination effect of RSL3-induced ferroptotic cell death in KPC cells was not observed in immune-deficient (rag2−/−) mice (n = 10 mice/group).
Discussion
Recent years have yielded profound insights into the molecular mechanisms, pharmacological modulation, and functional implication of ferroptosis in health and disease [66]. However, the mediators of its immunological consequences remain poorly characterized. In this study, we report a key role for DCN as a mediator of ferroptosis-initiated innate and adaptive immune response that acts on AGER (Figure 7). These findings not only contribute to our basic understanding of the “danger theory,” proposed by Polly Matzinger to explain how the immune system distinguishes between self and non-self [67], but also might pave the way for the development of more effective immunotherapy strategies [28].
Figure 7.
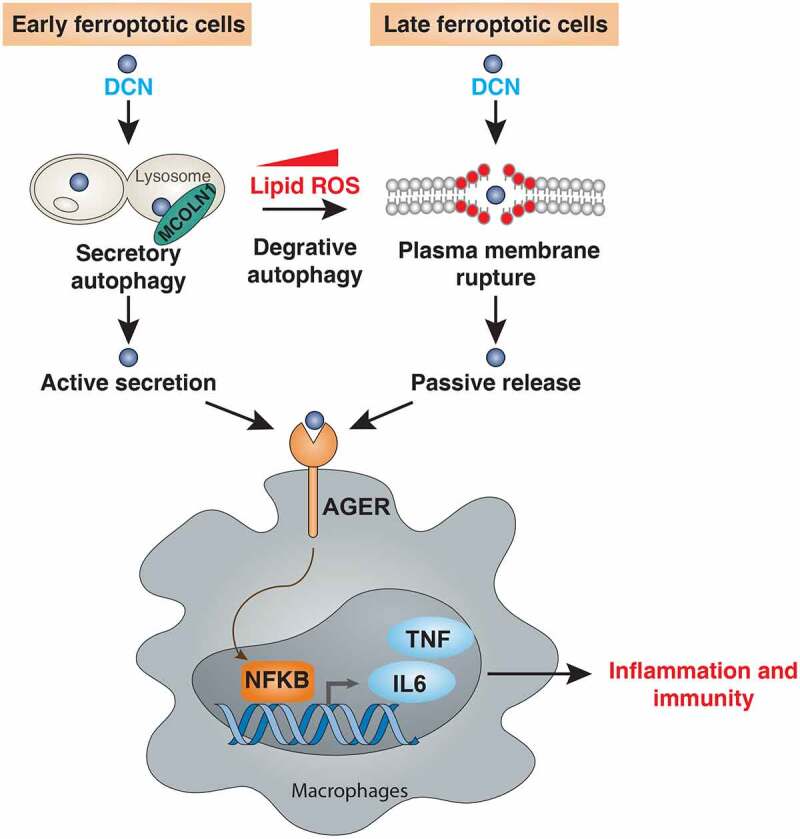
A model illustrating the role of DCN in the communication between ferroptotic cells and macrophages. DCN can be actively secreted during the early phase of ferroptosis through MCOLN1-dependent secretory autophagy. Increased ROS can further stimulate autophagy, which causes the degradation of anti-ferroptotic proteins (e.g., ferritin [80], GPX4 [81], ARNTL [82], and SLC40A1/ferroportin-1 [83]) or organelles (e.g., lipid droplets [84]), eventually leading to the rupture of the plasma membrane and passive release of DCN. Once released, DCN can bind AGER on macrophages to activate NFKB-dependent cytokine production, thus igniting inflammatory and immune responses.
The danger theory claims that damaged or dying cells can release endogenous molecules, namely DAMPs, to alarm the immune system by activation of pattern recognition receptors (PRRs) [7]. DAMPs play a significant role in promoting wound healing. However, in excess, they result in inflammation and immune dysfunction [8]. Some highly abundant proteins or molecules (such as HMGB1, ATP, and DNA) are released from cells succumbing to various types of death [68]. Thus, ferroptosis, which is a type of lytic cell death characterized by plasma membrane rupture, is accompanied by the release of these DAMPs [13,69,70]. Our analysis reveals that DCN may be a relatively specific DAMP that is released by ferroptotic cells. This release may be linked to the formation of a protein complex between DCN and MCOLN1 during ferroptosis, rather than apoptosis and necroptosis. DCN apparently precedes the release of other DAMPs, including HMGB1. Compared with other reported biomarkers at the gene or protein level (such as acyl-CoA synthetase long chain family member 4 [ACSL4] [71] or transferrin receptor [TFRC] [72]), the detection of DCN release in culture medium or serum by ELISA may offer a convenient way to monitor ferroptotic responses.
The autophagy pathway acts as an integration hub for stress signals and provides adaptive responses to determine cell fate [73–75]. Compared with the pro-survival effect of autophagy, the signals and mechanisms of autophagy-dependent cell death remain largely elusive [76,77]. Increased autophagy usually inhibits apoptosis and necroptosis [78,79]. However, excessive autophagy contributes to the induction and execution of ferroptosis through the selective degradation of proteins (e.g., ferritin [80], GPX4 [81], ARNTL [82], and SLC40A1/ferroportin-1 [83]) or organelles (e.g., lipid droplets [84]), which are repressors of lipid peroxidation. Our current study suggests that the autophagy-mediated protein secretion pathway (also known as secretory autophagy [85]), but not degradative autophagy, mediates DCN release in early ferroptotic cells through lysosomal exocytosis (Figure 7). This autophagy-dependent unconventional secretion is different from Golgi-dependent protein secretion. Secretory autophagy plays a role in the release of DAMPs (e.g., HMGB1 [86]) or cytokines (e.g., IL1B/IL1β [87]). In addition, lipid peroxidation mediated by degradative autophagy in late-stage ferroptotic cells can lead to the rupture of the plasma membrane [88], indicating the possibility that autophagy machinery also contributes to the passive release of DCN during ferroptosis (Figure 7). In necroptosis, which is another well-characterized lytic cell death process [89], the accumulation of lipid ROS is less obvious than in ferroptosis [16], correlating with a reduced release of DCN. Our future analyses will focus on the question as to how cells can distinguish and classify cargo to route it toward degradative versus secretory autophagy.
Our study also demonstrates the implication of AGER as a multi-ligand PRR responding to ferroptosis. AGER is a transmembrane receptor from the immunoglobulin superfamily and is expressed by various cell types involved in innate and adaptive immunity [90]. In addition to the well-known AGER ligands (such as advanced glycation end-products, HMGB1, S100 family proteins, and DNA) [90], DCN interacts with, and activates, AGER. Once released by ferroptotic cells, extracellular DCN binds to AGER on macrophages and other immune cells to trigger pro-inflammatory cytokine production. We also found that HMGB1 enhances the inflammatory response mediated by DCN, indicating that different DAMPs may coordinately mediate the immune response during ferroptosis. Genetic or pharmacological inhibition of AGER reduces the host inflammatory response to sterile and infectious threats [91–93]. In the current study, we used two mouse models (pancreatitis and prophylactic tumor vaccination) to demonstrate that the DCN-AGER axis (rather than the TLR4 pathway) is necessary for the innate and adaptive immune response induced by ferroptotic death.
In summary, the inflammatory and immune response elicited by ferroptosis involves DCN as an essential DAMP that acts on the PRR AGER. Given the potent and wide role of AGER in mediating the response to various ferroptosis-associated DAMPs (including DCN, HMGB1, and KRAS) [13,94], blockade of AGER may constitute a valid strategy for mitigating systemic responses to ferroptosis.
Materials and methods
Reagents
The antibodies to DCN (MAB1060), TGFBR1 (MAB2401), and AGER (MAB1179) were purchased from R&D Systems. The antibodies to EGFR (54359), KDR (9698), MET (82202), IGF1R (3027), TLR2 (13744), TLR4 (14358), and ACTB (3700) were purchased from Cell Signaling Technology (Danvers, MA, USA). The antibodies to MAP1LC3B (NB100-2220) and MCOLN1 (NB110-82375) were purchased from NOVUS (Centennial, CO, USA). The antibodies to DCN (PA5-13538) and LAMP1 (MA1-164) were purchased from Thermo Fisher Scientific (Pittsburgh, PA, USA). The antibody to TLR4 (117607) was purchased from BioLegend (San Diego, CA, USA). Rat IgG2A isotype control (MAB006) was purchased from R&D Systems (San Diego, CA, USA).
Erastin (S7242), RSL3 (S8155), liproxstatin-1 (S7699), ferrostatin-1 (S7243), Z-VAD-FMK (S7023), FIN56 (S8254), necrosulfonamide (S8251), sulfasalazine (S1576), cycloheximide (S7418), sorafenib (S7397), CCT137690 (S2744), oxaliplatin (S1224), BMS-345541 (S8044), and desferoxamine (S5685) were purchased from Selleck Chemicals (Houston, TX, USA). Cerulein (C9026) was purchased from Sigma-Aldrich (Houston, CA, USA). ML162 (20455), ML210 (23282), and staurosporine (25096) were purchased from Cayman Chemical (Ann Arbor, MI, USA). Puromycin (A1113802) and polybrene (TR1003 G) were purchased from Thermo Fisher Scientific.
Cell culture and treatment
The HT1080 (CCL-121), PANC1 (CRL-1469), BX-PC3 (CRL-1687), HeLa (CCL-2), and NCI-H460 (HTB-177) cell lines were obtained from the American Type Culture Collection (ATCC). The atg5−/−, atg7−/−, and WT MEF cell lines were a gift from Noboru Mizushima (University of Tokyo, Japan). The dcn−/− MEF cell line was a gift from Lizhi Cao (Central South University, China). The mouse pancreatic ductal adenocarcinoma cancer cell line KPC was a gift from Herbert Zeh (UT Southwestern, USA). Immortalized BMDMs were a gift from Kate Fitzgerald (University of Massachusetts Medical School, USA). Primary BMDMs were isolated from ager−/− mice [95]. Primary HPBMs (70034), HPBDs (70041), or PBMCs (70025) were obtained from STEMCELL Technologies. Cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, 11995073) or RPMI 1640 (Thermo Fisher Scientific, 11875119) supplemented with 10% heat-inactivated fetal bovine serum (Millipore, TMS-013-B) and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070-063) at 37°C, 95% humidity, and 5% CO2. All cells were mycoplasma-free and authenticated using short tandem repeat DNA profiling analysis.
Dimethyl sulfoxide (DMSO; VWR International, IC0219605525) was used to prepare the stock solution of drugs. The final concentration of DMSO in the drug working solution in the cells was <0.01%. DMSO of 0.01% was used as a vehicle control in all cell culture assays. RSL3 (0.5 μM, 3 h), staurosporine (200 nM, 16 h), and CCT137690 (5 μM, 16 h) were used to generate ferroptotic, apoptotic, and necroptotic cells, respectively. Dying cells (0.25–2 × 105/ml) were used to stimulate the indicated immune cells for 1 h and then the release of TNF and IL6 was assayed. In addition, mouse DCN full-length recombinant protein with a His tag at the C terminus (purity of >95%; lipopolysaccharide <0.10 EU per 1 μg) was obtained from R&D Systems (1060-DE-100), which was generated from mouse myeloma cell line NS0. All treatments by DCN protein were carried out in serum-free Opti-MEM I medium (Thermo Fisher Scientific, 31985070).
Animal models
WT (000664), rag2−/− (008449), and Pdx1-Cre (014647) mice on the C57BL/6J background were obtained from the Jackson Laboratory. ager−/− mice on the C57BL/6J background were a gift from Angelika Bierhaus (deceased; University of Heidelberg, Germany). Gpx4flox/flox mice on the C57BL/6 background were a gift from Qitao Ran (University of Texas Health Science Center at San Antonio, USA). Pdx1-Cre and Gpx4flox/flox mice were crossed to generate Pdx1-Cre;gpx4−/− mice [59]. Mice were housed on a regular 12-h light and dark cycle (7:00–19:00 light period; room temperature: 20–25°C; relative humidity: 40–60%). Food and water were available ad libitum. Experiments were carried out under pathogen-free conditions and the health status of mouse lines was routinely checked by veterinary staff. No wild animals were used in the study. Experiments were carried out with randomly chosen littermates of the same sex and matched by age and body weight. We conducted all animal care and experimentation in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from institutional animal care and use committees.
Pancreatitis model: mice (6–8 weeks old; male) received seven hourly i.p. injections of 50 μg/kg cerulein (Sigma-Aldrich, C9026) in sterile saline, while controls were given saline as described previously [96]. Animals were sacrificed at the indicated time by CO2 asphyxiation, and a blood sample and tissue were collected. Blood samples were collected and centrifuged at 10,000 × g for 10 min at 4°C. Following centrifugation, the serum was aspirated and used for measurement of level or activity of AMY (amylase), DCN, TNF, and IL6 by ELISA. Tissue samples were collected, snap frozen in liquid nitrogen, and stored at −80°C for analysis of the activity of trypsin or MPO. Formalin-fixed pancreas samples were processed, and 5-μm-thick paraffin sections were stained with hematoxylin and eosin (H&E) for histological analysis. Histological images were acquired using an EVOS FL auto cell imaging system (Thermo Fisher Scientific).
Tumor vaccination model: A total of 5 × 106 KPC cells (C57BL/6J background), untreated or treated with either oxaliplatin (50 µM, 24 h) or RSL3 (0.5 µM, 3 h), were inoculated subcutaneously in 200 µl phosphate-buffered saline (PBS) into the lower flank of C57BL/6J mice (6–8 weeks old; male:female = 1:1), whereas 5 × 105 untreated control cells were inoculated into the contralateral flank 7 days later [97]. The percentage of tumor-free mice was monitored every week.
Cytotoxicity assay
The level of cell death was assayed using a LIVE/DEAD cell viability/cytotoxicity assay kit (Thermo Fisher Scientific, L3224) according to the manufacturer’s protocol.
Cell fractionation and organelle isolation
Supernatants of cells were concentrated using a Microcon-10kDa Centrifugal Filter Unit with Ultracel-10 membrane (Millipore, MRCPRT010). The lysosome isolation kit (Abcam, ab234047) was used to isolate lysosomal fractions from cultured cells by differential centrifugation followed by density gradient centrifugation. Finally, the purified lysosomal fraction was obtained using an ultracentrifuge (145,000 × g) for 2 h at 4°C.
Biochemical assay
Commercially available ELISA kits were used to measure the concentrations or activity of HMGB1 (Shino Test Corporation, ST51011), DCN (R&D Systems, DY143, DY1060), IL6 (R&D Systems, M6000B, D6050), TNF (R&D Systems, MTA00B, DTA00D), NFKB (Active Motif, 43,296), MPO (Abcam, ab105136), LDH (Abcam, ab102526), trypsin (Abcam, ab102531), CTSB (Abcam, ab119585), and AMY (Abcam, ab102523) in the indicated samples.
RNAi
All shRNA constructs in a lentiviral format as described in Table S1 were purchased from Sigma-Aldrich. We seeded 1 × 105 cells in each well of a 12-well plate in 500 μl of complete medium and transduced by lentiviral vectors at a multiplicity of infection (MOI) of 10:1. Transduction was carried out in the presence of polybrene (8 μg/ml; Thermo Fisher Scientific, TR1003G) in an antibiotic-free medium. After recovering with complete culture medium, puromycin (5 μg/ml; Thermo Fisher Scientific, A1113802) was used for the selection of transduced cells.
qPCR
Total RNA was extracted using an RNeasy plus kit (QIAGEN, 74034) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 1 µg of RNA using the iScript cDNA synthesis kit (Bio-Rad, 1708890). Briefly, 20-μl reactions were prepared by combining 4 μl of iScript select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20×, 2 μM), and 12 μl of RNA diluted in RNase-free water. Quantitative real-time PCR (qPCR) was carried out using synthesized cDNA, primers as described in Table S1, and SsoFast EvaGreen supermix (Bio-Rad, 172-5204). The data were normalized to Rn18s RNA, and the fold change was calculated via the 2−ΔΔCt method. Relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1.
Western blot analysis
Cells were lysed in cell lysis buffer (Cell Signaling Technology, 9803) with protease inhibitor cocktail (Thermo Fisher Scientific, 78429). Cleared lysates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; Bio-Rad, 3450124) and then transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad, 1704273). The membranes were blocked with Tris-buffered saline Tween 20 (TBST; Cell Signaling Technology, 9997) containing 5% skim milk (Cell Signaling Technology, 9999) for 1 h at room temperature and then incubated with the indicated primary antibodies (1:1000) overnight at 4°C. After being washed with TBST, the membranes were incubated with an HRP-linked anti-mouse IgG secondary antibody (Cell Signaling Technology, 7076; 1:1000) or HRP-linked anti-rabbit IgG secondary antibody (Cell Signaling Technology, 7074; 1:1000) for 1 h at room temperature. The membranes were washed three times in TBST and then visualized and analyzed with a ChemiDoc touch imaging system (Bio-Rad, 1708370) or x-ray films.
Immunoprecipitation analysis
Cells were lysed at 4°C in ice-cold radio-immunoprecipitation assay (RIPA) lysis buffer (Millipore, 20-188) and cell lysates were cleared by a brief centrifugation (12,000 × g, 10 min) [98]. Concentrations of proteins in the supernatant were determined by bicinchoninic acid (BCA) assay. Prior to immunoprecipitation, samples containing equal amounts of proteins were pre-cleared with protein A/G sepharose beads (4°C, 3 h) from Abcam (ab193262) and subsequently incubated with irrelevant IgG or specific antibodies (4 µg/ml) in the presence of protein A/G sepharose beads overnight at 4°C with gentle shaking. Following incubation, protein A/G sepharose beads were washed extensively with PBS (Cell Signaling Technology, 9808) and proteins eluted by boiling in 2 × SDS sample buffer before SDS-PAGE electrophoresis.
Antibody array analysis
To assess a relatively wide range of secretion levels for several different proteins in HT1080 and HeLa cells after treatment with erastin (5 μM) for 24 h, cell culture supernatants were collected and concentrated using a centricon (Millipore, MRCPRT010). Concentrated media from each group of three mixed samples was then analyzed using a RayBio L-series human antibody array 507 glass slide kit (Raybiotech, AAH-BLG-1-4) in order to detect 507 proteins, as per the manufacturer’s instructions. The first step was to biotinylate the primary amine groups of the proteins in the samples. The glass slide arrays were then blocked, and the biotin-labeled sample was added onto the glass slide, which is preprinted with capture antibodies. The slide was incubated to allow binding of target proteins. Streptavidin-conjugated fluorescent dye (Cy3 equivalent) was then applied to the array. Finally, the glass slide was dried, and laser fluorescence scanning was used to visualize the signals. To normalize signal intensity data, one sub-array was defined as “reference” to which the other arrays were normalized.
Immunofluorescence analysis
Cells were cultured on glass coverslips and fixed in 3% formaldehyde for 30 min at room temperature prior to detergent extraction with 0.1% Triton X-100 (Cell Signaling Technology, 39487) for 10 min at 25°C. Coverslips were saturated with 2% bovine serum albumin (Cell Signaling Technology, 9998) in PBS for 1 h at room temperature and processed for immunofluorescence with primary antibodies, followed by Cy3- (1:500; Thermo Fisher Scientific, A10521) or Alexa Fluor 488-conjugated IgG (1:500; Thermo Fisher Scientific, A21206). Nuclear morphology was analyzed with the fluorescent dye Hoechst 33258 (Thermo Fisher Scientific, H1398). Images were taken with a ZEISS LSM 800 confocal microscope.
Affinity pull-down assay
We used MagZ™ Particles System (Promega, V8830) to evaluate the direct binding between the bait protein and the prey protein. In brief, 10 µg His-DCN bait protein was added to 30 µl MagZ™ binding particles and incubated for 15 min on a shaker. The MagZ™ particles were washed three times with 200 µl of 20 mM sodium phosphate (pH 7.4) and resuspended with 30 µl of MagZ™ binding/wash buffer. Also, 5 µl of particles was transferred to new tubes. Particles were resuspended in 175 µl of the MagZ™ binding/wash buffer. Then 2 µg AGER protein was added to the prepared bait His-DCN/MagZ™ particles and incubated for 60 min on a shaker. The particles were washed three times in the same final wash buffer used in the immobilization followed by an additional wash of 500 mM imidazole. Then 20 µl SDS buffer was added to the particles and incubated for 5 min with shaking, and the samples were collected for SDS-PAGE electrophoresis.
Protein stability assay
The protein degradation rate of DCN was tested in the indicated MEFs after treatment with RSL3 (0.5 µM) in the absence or presence of 10 µM cycloheximide for indicated times. Whole-cell lysates were analyzed for DCN and ACTB by western blot analyses.
Statistical analysis
GraphPad Prism 8.4.3 was used to collect and analyze data. Unpaired Student’s t tests were used to compare the means of two groups. A one-way or two-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used for comparison among the different groups. The Pearson correlation was used to assess the relationship between the expression of MAP1LC3B-II protein and the release of DCN. A P value of <0.05 was considered statistically significant.
Supplementary Material
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Gudipaty SA, Conner CM, Rosenblatt J, et al. unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu Rev Cell Dev Biol. 2018;34(1):311–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tang D, Kang R, Berghe, TV.. The molecular machinery of regulated cell death. Cell Death Differ. 2019;29:347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ghose P, Shaham S. Cell death in animal development. Development. 2020;. 147: ;dev191882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schwartz LM. Autophagic cell death during development - ancient and mysterious. Front Cell Dev Biol. 2021;9:656370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li Y, Klein C, Kotlarz D. Dysregulation of cell death in human chronic inflammation. Cold Spring Harb Perspect Biol. 2020;. 12: ;a037036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Anderton H, Wicks IP, Silke J. Cell death in chronic inflammation: breaking the cycle to treat rheumatic disease. Nat Rev Rheumatol. 2020;16:496–513. [DOI] [PubMed] [Google Scholar]
- [7].Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- [8].Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20:95–112. [DOI] [PubMed] [Google Scholar]
- [9].Tang D, Kang R, Coyne CB, et al. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bell CW, Jiang W, Reich CF 3rd, et al. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291(6):C1318–25. [DOI] [PubMed] [Google Scholar]
- [11].Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418(6894):191–195. [DOI] [PubMed] [Google Scholar]
- [12].Lamkanfi M, Sarkar A, Vande Walle L, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wen Q, Liu J, Kang R, et al. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–283. [DOI] [PubMed] [Google Scholar]
- [14].Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;. 17: 2054–2081 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yagoda N, Von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Distefano AM, Martin MV, Cordoba JP, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216:463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lei G, Zhang Y, Koppula P, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ye LF, Chaudhary KR, Zandkarimi F, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kloditz K, Fadeel B. Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019;5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;. 18: 280–296 . [DOI] [PubMed] [Google Scholar]
- [26].Yan HF, Tuo QZ, Yin QZ, et al. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool Res. 2020;41:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu K, Liu J, Zou B, et al. Trypsin-mediated sensitization to ferroptosis increases the severity of pancreatitis in mice. Cell Mol Gastroenterol Hepatol. 2021;. 13: 483–500 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;. 218: ;e20210518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. [DOI] [PubMed] [Google Scholar]
- [31].Malsy M, Bitzinger D, Graf B, et al. Staurosporine induces apoptosis in pancreatic carcinoma cells PaTu 8988t and Panc-1 via the intrinsic signaling pathway. Eur J Med Res. 2019;24:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xie Y, Zhu S, Zhong M, et al. Inhibition of aurora kinase a induces necroptosis in pancreatic carcinoma. Gastroenterology. 2017;153:1429–43 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chan FK, Moriwaki K, De Rosa MJ. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol. 2013;979:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Weinberg JM, Bienholz A, Venkatachalam MA. The role of glycine in regulated cell death. Cell Mol Life Sci. 2016;73:2285–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mimura T, Chang JH, Kim TI, et al. MT1-MMP cleavage of the antiangiogenic proteoglyca0n decorin: role in corneal angiogenesis. Cornea. 2011;30(Suppl 1):S45–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [36].Ang LS, Boivin WA, Williams SJ, et al. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. 2011;2:e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Sem Cancer Biol. 2020;. 66: 89–100 . [DOI] [PubMed] [Google Scholar]
- [38].Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;. 17: ;3361–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mizushima N, Noda T, Yoshimori T, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. [DOI] [PubMed] [Google Scholar]
- [40].Suzuki K, Kirisako T, Kamada Y, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yim WW, Mizushima N. Lysosome biology in autophagy. Cell Discov. 2020;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dong XP, Cheng X, Mills E, et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature. 2008;455:992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang X, Cheng X, Yu L, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kuang F, Liu J, Li C, et al. Cathepsin B is a mediator of organelle-specific initiation of ferroptosis. Biochem Biophys Res Commun. 2020;533:1464–1469. [DOI] [PubMed] [Google Scholar]
- [45].Merline R, Moreth K, Beckmann J, et al. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci Signal. 2011;4:ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koninger J, Giese NA, Bartel M, et al. The ECM proteoglycan decorin links desmoplasia and inflammation in chronic pancreatitis. J Clin Pathol. 2006;59:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nastase MV, Janicova A, Roedig H, et al. Small leucine-rich proteoglycans in renal inflammation: two sides of the coin. J Histochem Cytochem. 2018;66:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Iozzo RV, Moscatello DK, McQuillan DJ, et al. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem. 1999;274:4489–4492. [DOI] [PubMed] [Google Scholar]
- [49].Khan GA, Girish GV, Lala N, et al. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol. 2011;25:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–284. [DOI] [PubMed] [Google Scholar]
- [51].Goldoni S, Humphries A, Nystrom A, et al. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol. 2009;185:743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schonherr E, Sunderkotter C, Iozzo RV, et al. Decorin, a novel player in the insulin-like growth factor system. J Biol Chem. 2005;280:15767–15772. [DOI] [PubMed] [Google Scholar]
- [53].de La Luz-hernandez KR, Rojas-del Calvo L, Rabasa-Legon Y, et al. Metabolic and proteomic study of NS0 myeloma cell line following the adaptation to protein-free medium. J Proteomics. 2008;71:133–147. [DOI] [PubMed] [Google Scholar]
- [54].Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274:19919–19924. [DOI] [PubMed] [Google Scholar]
- [55].Sousa MM, Yan SD, Stern D, et al. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab Invest. 2000;80:1101–1110. [DOI] [PubMed] [Google Scholar]
- [56].Mors K, Horauf JA, Kany S, et al. Ethanol decreases inflammatory response in human lung epithelial cells by inhibiting the canonical NF-kB-pathway. Cell Physiol Biochem. 2017;43:17–30. [DOI] [PubMed] [Google Scholar]
- [57].Lan J, Heneghan AF, Sano Y, et al. Parenteral nutrition impairs lymphotoxin beta receptor signaling via NF-kappaB. Ann Surg. 2011;253:996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Burke JR, Pattoli MA, Gregor KR, et al. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278:1450–1456. [DOI] [PubMed] [Google Scholar]
- [59].Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11:6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Efimova I, Catanzaro E, Van der Meeren L, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;. 8: ;e001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tang D, Kepp O, Kroemer G. Ferroptosis becomes immunogenic: implications for anticancer treatments. Oncoimmunology. 2020;10:1862949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;. 8: ;e000337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. [DOI] [PubMed] [Google Scholar]
- [65].Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- [66].Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. [DOI] [PubMed] [Google Scholar]
- [68].Murao A, Aziz M, Wang H, et al. Release mechanisms of major DAMPs. Apoptosis. 2021;26:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Pedrera L, Espiritu RA, Ros U, et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021. 28: 1644–1657 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS Protein. Autophagy. 2020;. 16: 2069–2083 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–1343. [DOI] [PubMed] [Google Scholar]
- [72].Feng H, Schorpp K, Jin J, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30:3411–23 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–364. [DOI] [PubMed] [Google Scholar]
- [76].Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J Cell Sci. 2018. 131: ;jcs215152. [DOI] [PubMed] [Google Scholar]
- [77].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021;17:1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med. 2017;214:3687–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hou W, Han J, Lu C, et al. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010;6:891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116:2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Li J, Liu J, Xu Y, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy. 2021;. 17: 3361–3374 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003. [DOI] [PubMed] [Google Scholar]
- [85].New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy. 2019;15:1682–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang Z, Zhou H, Zheng H, et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin in fl ammation. Autophagy. 2021;17:529–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Dupont N, Jiang S, Pilli M, et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18:1106–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Hudson BI, Lippman ME. Targeting RAGE Signaling in Inflammatory Disease. Annu Rev Med. 2018;69:349–364. [DOI] [PubMed] [Google Scholar]
- [91].Liliensiek B, Weigand MA, Bierhaus A, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hong J, Wang X, Zhang N, et al. D-ribose induces nephropathy through RAGE-dependent NF-kappaB inflammation. Arch Pharm Res. 2018;41:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Kang R, Chen R, Xie M, et al. The receptor for advanced glycation end products activates the AIM2 inflammasome in acute pancreatitis. J Immunol. 2016;196:4331–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Li Z, Scott MJ, Fan EK, et al. Tissue damage negatively regulates LPS-induced macrophage necroptosis. Cell Death Differ. 2016;23:1428–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Kang R, Zhang Q, Hou W, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146:1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yang M, Li C, Zhu S, et al. TFAM is a novel mediator of immunogenic cancer cell death. Oncoimmunology. 2018;7:e1431086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.