

Abstract
Background:
Cellular rejection after heart transplantation imparts significant morbidity and mortality. Current immunosuppressive strategies are imperfect, target recipient T-cells, and have adverse effects. The innate immune response plays an essential role in the recruitment and activation of T-cells. Targeting the donor innate immune response would represent the earliest interventional opportunity within the immune response cascade. There is limited knowledge regarding donor immune cell types and functions in the setting of cardiac transplantation and no current therapeutics exist for targeting these cell populations.
Methods:
Using genetic lineage tracing, cell ablation, and conditional gene deletion, we examined donor mononuclear phagocyte diversity and macrophage function during acute cellular rejection of transplanted hearts in mice. We performed single cell RNA sequencing on donor and recipient macrophages and monocytes at multiple timepoints after transplantation. Based on our imaging and single cell RNA sequencing data, we evaluated the functional relevance of donor CCR2+ and CCR2− macrophages using selective cell ablation strategies in donor grafts prior to transplant. Finally, we performed functional validation that donor macrophages signal through MYD88 to facilitate cellular rejection.
Results:
Donor macrophages persisted in the rejecting transplanted heart and co-existed with recipient monocyte-derived macrophages. Single cell RNA sequencing identified donor CCR2+ and CCR2− macrophage populations and revealed remarkable diversity amongst recipient monocytes, macrophages, and dendritic cells. Temporal analysis demonstrated that donor CCR2+ and CCR2− macrophages were transcriptionally distinct, underwent significant morphologic changes, and displayed unique activation signatures after transplantation. While selective depletion of donor CCR2− macrophages reduced allograft survival, depletion of donor CCR2+ macrophages prolonged allograft survival. Pathway analysis revealed that donor CCR2+ macrophages are activated through MYD88/NF-ĸβ signaling. Deletion of MYD88 in donor macrophages resulted in reduced antigen presenting cell recruitment, reduced ability of antigen presenting cells to present antigen to T-cells, decreased emergence of allograft reactive T-cells, and extended allograft survival.
Conclusions:
Distinct populations of donor and recipient macrophages co-exist within the transplanted heart. Donor CCR2+ macrophages are key mediators of allograft rejection and deletion of MYD88 signaling in donor macrophages is sufficient to suppress rejection and extend allograft survival. This highlights the therapeutic potential of donor heart-based interventions.
Keywords: Donor, Macrophages, Heart, Transplant, Rejection, Single Cell RNA-sequencing, CCR2, MyD88
Introduction:
Heart transplantation is the definitive treatment for end-stage heart failure with over 3000 transplants performed annually in the United States1, 2. Given the complexities of donor-recipient matching and imperfect immunosuppressive regimens, approximately 40% of transplanted patients suffer rejection within their first year after transplant leading to adverse short and long-term outcomes3, 4. Current immunosuppression strategies target recipient T-cells and impose harmful consequences of systemic immunosuppression including infection and malignancy5. The ultimate goal of transplant immunology is allograft tolerance where the recipient does not reject the graft in the absence of ongoing immunosuppression. The induction and maintenance of tolerance remains elusive.
T-cell activation by antigen presenting cells, including macrophages, monocytes, and dendritic cells, is a requisite step for allograft rejection6–12. Limiting the emergence and proliferation of alloreactive T-cells by targeting macrophages, monocytes, and/or dendritic cells may mitigate the incidence of allograft rejection while avoiding adverse outcomes associated with systemic T-cell depletion or inhibition. While recipient-derived macrophages and dendritic cells have an established role in heart transplant rejection13–18, the precise role of donor-derived immune cells remains largely unexplored.
Donor hearts contain abundant populations of resident macrophages and fewer dendritic cells19. Targeting cardiac graft-resident macrophage activation represents an attractive approach to reduce rejection while eschewing systemic immunosuppression15. Interestingly, early activation of macrophages within the allograft can result in a prolonged activated inflammatory state that persists in the absence of antigen or other stimuli, a property termed “trained immunity”20. This suggests that early activation of donor macrophages may have long-term functional effects on allograft outcomes that are independent of ongoing immunosuppression. As donor immune cells represent the earliest targetable cell in the rejection cascade, inhibiting the activation of donor macrophages may enable a reduction of systemic immunosuppression.
We have previously reported that mouse and human hearts contain at least two subsets of resident macrophages with divergent origins and functions that can be distinguished based on the expression of C-C chemokine receptor 2 (CCR2)21, 22. CCR2+ macrophages are derived from adult hematopoietic progenitors, are replenished through ongoing monocyte recruitment, and orchestrate inflammatory responses including monocyte and neutrophil infiltration. CCR2− macrophages are derived from embryonic hematopoietic progenitors, are maintained independent of monocyte input, suppress inflammation, and promote tissue repair7, 19, 21–24. The role of donor CCR2+ and CCR2− macrophages in the context of heart transplantation rejection remains to be elucidated25.
In the present study, we utilize a murine heart transplantation model to precisely interrogate the roles of donor macrophages during acute rejection26, 27. By combining genetic lineage tracing with single cell RNA sequencing, we examine transcriptional signatures of donor and recipient macrophages and monocytes and identify putative signaling mechanisms underlying donor CCR2+ and CCR2− macrophage activation. We use genetic lineage tracing to demonstrate that donor CCR2+ and CCR2− macrophages persist after heart transplantation but are ultimately lost due to ongoing rejection. Targeted cell depletion strategies show that reduction of donor CCR2+ macrophages result in prolonged allograft survival with reduced inflammation whereas reduction of donor CCR2− macrophages conversely leads to rapid rejection with increased inflammation. We show that donor CCR2+ macrophages signal in part through MYD88 and inhibition of this signaling either in the donor graft or donor macrophages suppresses immune infiltration and modulates recipient immune alloreactivity resulting in prolongation of allograft survival. Together, these findings establish donor CCR2+ macrophages and/or their signaling as a viable therapeutic target to ameliorate or prevent heart transplant rejection.
Materials and Methods:
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
All scripts used for single-cell data analysis are available from GitHub (https://github.com/jamrute/2021_ACR_Kopecky_Lavine).
Animal Models
Mice were bred and maintained at the Washington University School of Medicine. IRB approval was obtained, and all procedures were in accordance with institutional guidelines and were performed in accordance with the animal use oversight committee. Mouse strains utilized included CD169DTR/+ 28, CCR2DTR/+ 29, MyD88f/f 30, LysMCre/+ 31, CSF1rertCre/+ 32, MyD88−/− 33, CX3CR1GFP/+ CCR2RFP/+ 34, 35. C.Cg-Tg(DO11.10)/10Dlo/J “DO.11” mice were purchased from The Jackson Laboratories. All donor mice were on the C57BL/6 (B6) background and genotyped according to established protocols36. Recipient mice were age- and gender-matched BALB/c mice between 6–8 weeks of age, except in the setting of a syngeneic transplant when the recipient was a B6 mouse. Equal numbers of male and female mice were included in all experiments. Diphtheria toxin receptor (DTR) and control mice were given 200 ng intraperitoneal injections of diphtheria toxin (Sigma Cat #D0564) on three consecutive days prior to transplant. Tamoxifen food pellets (500 mg/kg diet, Envigo Teklad Diets 500 TD130857) were provided for two weeks prior to transplantation.
Heterotopic Heart Transplantation
Heart grafts were harvested from donor mice and transplanted heterotopically into the abdomen of recipients following 1 hour of cold (4°C) ischemia, as previously described26. For low-dose immunosuppression, 200 μg CTLA4-Ig (Bio X Cell Cat BE0099) was administered intraperitoneally on post-transplant days 0, 2, 4, and 6. For high-dose immunosuppression, 1.25 mg CTLA4-Ig was administered on post-transplant days 0, 4, and 14. For syngeneic transplantation, B6 was used for both donor and recipient and no immunosuppression was administered. After transplantation, allografts were palpated daily. Cessation of a palpable heartbeat, confirmed by visual inspection, indicated rejection of the cardiac allograft. Perioperative graft loss (within 72 hours) was excluded from the analysis.
Histology
Immunofluorescence
Alloreactivity Assay
10 days after transplant, recipient BALB/c splenocytes were frozen using C.T.L.-Cryo ABC Media (ImmunoSpot Cat#CTLC-ABC). In brief, fresh spleens were pressed through a 40 μm cell strainer and rinsed with 1X C.T.L. wash (ImmunoSpot Cat# CTLW-010). 1:1 CTL-C and CTL-A/B were added. Up to 10 million splenocytes /mL were aliquoted into 1 mL cyrovials, which were subsequently stored in liquid nitrogen. Responder splenocytes were thawed using C.T.L. anti-aggregate wash (ImmunoSpot Cat# CTL-AA-005), T-cells were isolated (Miltenyi Pan T Cell isolation kit II (130-095-130)) and used for ELISPOT assay. Irradiated (20cGy) B6 (stimulator-allogeneic) and BALB/c (stimulator-syngeneic) splenocytes were added to responder T-cells in C.T.L. Test media (ImmunoSpot Cat# CTLT-010) at a ratio of 600,000 stimulators to 150,000 responder T-cells overnight at 37°C. IFN-γ spots were detected per C.T.L. ELISPOT protocol (ImmunoSpot Mouse IFN-γ Single-Color ELISPOT). Plates were analyzed and quality controlled by a blinded third-party using CTL-ImmunoSpot S6 University Analyzer. Positive (ConA) and negative controls were included. All samples were performed in technical triplicates and each experimental condition consisted of at least six independent samples.
Antigen Presentation Assay
Post-transplant day 3–4 antigen presenting cells (CD45+, CD11b+, Ly6c+ and CD64+) were sorted from cardiac grafts into C.T.L. Test media. Antigen presenting cells were cultured with 1mg/1mL OVA (Sigma Cat # O1641) overnight at 37°C. T-cells from DO.11 mice were isolated (Miltenyi Pan T Cell isolation kit II (130-095-130)) and cultured for 3 days with OVA-loaded antigen presenting cells on ELISPOT plates (5000 APC:100,000 T-cells) and analyzed as above.
Flow Cytometry
Single Cell RNA Library Construction
FACS sorted CD45+CD11b+Ly6G−CD64+ Ly6c−, CD45+CD11b+Ly6G−CD64+ Ly6c+ and CD45+CD11b+Ly6G−CD64− Ly6c+ cells were sorted by the presence (donor library) or absence (recipient library) of GFP and then processed and encapsulated with barcoded oligo-dT containing gel beads with the 10X Genomics Chromium controller. Library preparation was performed as per manufacturer recommended protocols at the McDonnel Genome Institute at Washington University. Single cell libraries were multiplexed into a single lane and were sequenced at a target read depth of 100,000 reads/cell using a NovaSeq sequencer (Illumina).
Single Cell Analysis
Statistical Analysis
Data were analyzed by using the statistical software (Prism, V9.0; GraphPad, La Jolla, CA). The Kaplan-Meier method was used to estimate survival curves. The Log-rank (Mantel-Cox) was used to compare survival between groups with a p-value <0.05 considered statistically significant. Differences between groups were examined by using non-parametric Mann-Whitney U test. Multiple means were compared by using Kruskal-Wallis one-way analysis of variance with the Dunn’s test for multiple comparisons. A p< 0.05 (two-sided) was indicative of a statistically significant difference. Bonferroni correction was performed when multiple hypotheses were tested. Gene set scores were analyzed with a 2-way ANOVA with multiple comparisons including: (CCR2+ pre vs CCR2+ post; CCR2− pre vs CCR2− post; and CCR2+ post vs CCR2− post). Data are presented as dot plots or box whisker plots generated in Prism. Power calculations were performed to ensure adequate sample size (n = number of animals). The exact sample size used to calculate statistical significance is stated in the appropriate figure legend.
Results:
Donor Macrophages Initially Persist after Heart Transplantation
To assess the dynamics of donor macrophages after heart transplantation in the setting of acute rejection, we transplanted B6 CX3CR1GFP/+ CCR2RFP/+ donor hearts into BALB/c recipients that received low-dose CTLA4-Ig. Low-dose CTLA4-Ig (200 μg on post-operative days 0, 2, 4, 6) is commonly used to study rejection as it sub-optimally suppresses the recipient immune system and does not induce tolerance to the donor graft27. Donor CCR2− macrophages (GFP+RFP−CD68+), donor CCR2+ macrophages (GFP+RFP+CD68+), and recipient monocyte-derived macrophages (GFP−RFP−CD68+) were quantified by immunofluorescence in naïve hearts 1, 3, 7, and 14 days after transplantation. There were more donor CCR2− than CCR2+ macrophages in the heart at all timepoints (Figure 1A–C). Donor CCR2− and CCR2+ macrophages persisted at early time points (days 1, 3) following transplantation. By post-transplant day 7, donor macrophages (CCR2−: 85 macrophages/mm2; CCR2+: 9.7 macrophages/mm2) were outnumbered by infiltrating recipient macrophages (704 recipient macrophages/mm2) (Figure 1D). Few donor macrophages (CCR2−: 1.6 macrophages/mm2; CCR2+: 1.1 macrophages/mm2) remained in heart grafts on post-transplant day 14.
Figure 1: Persistence of Donor Macrophages after Heart Transplantation.
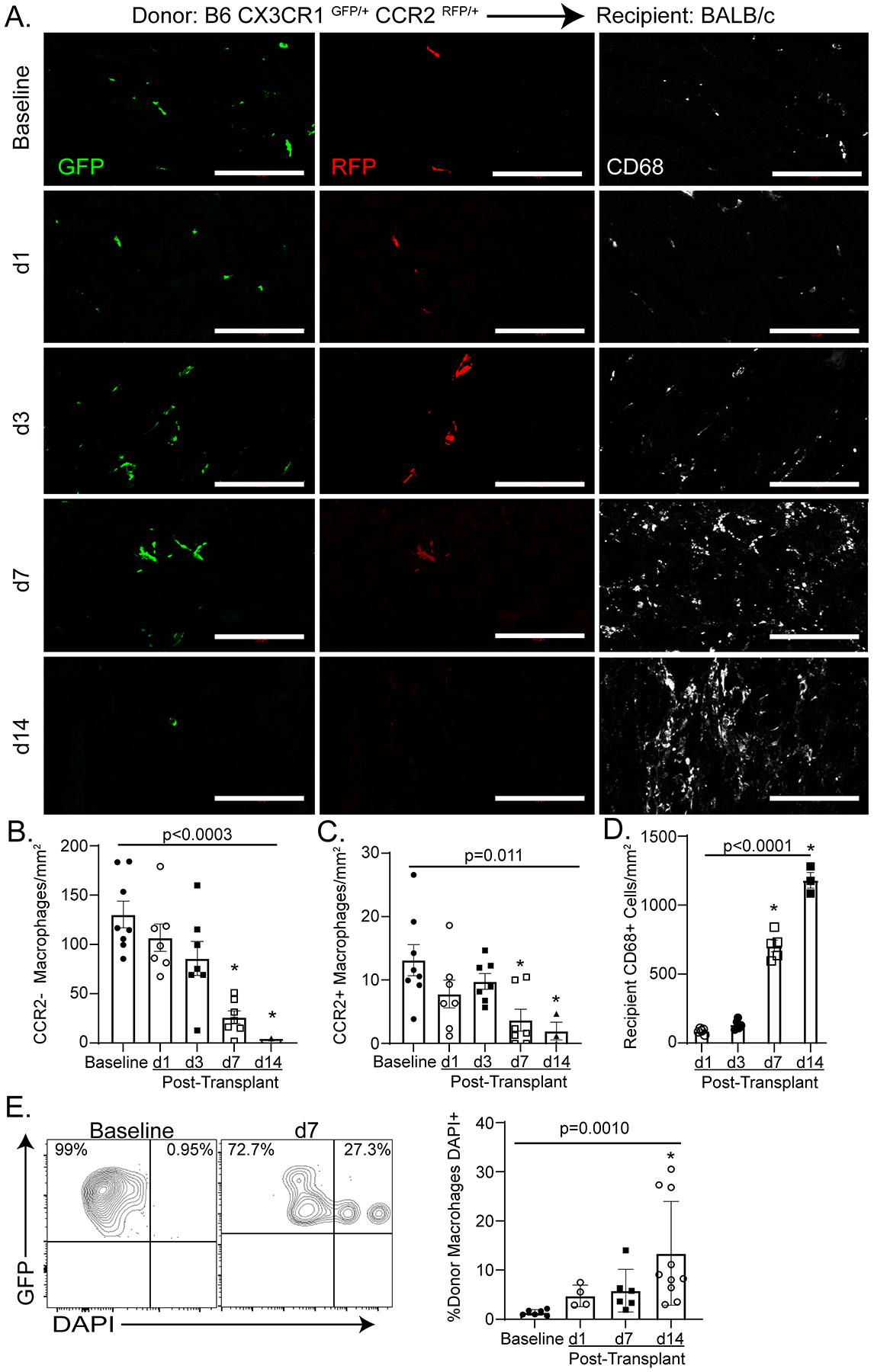
A-D) At baseline, all macrophages co-express GFP and CD68. A subset of GFP+ CD68+ macrophages express RFP and are identified as donor CCR2+ macrophages. Donor CCR2− macrophages are identified as CD68+ GFP+ RFP−. At post-transplant day 1 (d1), donor CCR2− and CCR2+ macrophages co-exist with recipient monocyte derived macrophages (CD68+ only). By post-transplant day 14 (d14), only rare donor macrophages are identified. B) Donor CCR2− macrophages significantly decrease over time from baseline to d14 (Kruskal-Wallis; p < 0.0003). Specifically, there is a decrease in donor CCR2− macrophages between baseline and post-transplant day 7 (d7, n = 7) (Dunn’s test for multiple comparisons; p = 0.0015) and baseline and d14 (Dunn’s test for multiple comparisons; p = 0.0014) (baseline n = 8, d1 = 7, d3 = 7, d 7 = 7, d14 = 3). C) There is a significant decrease of donor CCR2+ macrophages over time from baseline to d14 (Kruskal-Wallis; p = 0.0111) and specifically between baseline and d7 (Dunn’s test for multiple comparisons; p = 0.0197) and baseline and d14 (Dunn’s test for multiple comparisons; p = 0.0215) (baseline n = 8, d1 = 7, d3 = 7, d 7 = 7, d14 = 3). D) There is a significant increase in recipient CD68+ cells during the course of rejection (Kruskal-Wallis; p < 0.0001) and specifically between post-transplant day 1(d1) and d7 (Dunn’s test for multiple comparisons; p = 0.0134) and baseline and d14 (Dunn’s test for multiple comparisons; p = 0.0015) (d1 = 5, d3 = 5, d 7 = 5, d14 = 3). E) DAPI+ donor macrophages (GFP+) were considered to have undergone cell death. There was a significant increase in donor macrophages cell death after transplant (Kruskal-Wallis; p = 0.0010) and from baseline to d7 (Dunn’s test for multiple comparisons; p = 0.002) (baseline n = 6, d1 = 4, d 7 = 6, d14 = 10). Scale bar = 100 μm.
To ensure that CCR2 expression remained stable in donor macrophages after transplantation, we performed genetic lineage tracing of CCR2+ cells. Tamoxifen chow was provided to B6 CCR2ertCre/+Rosa26tdTomatoCCR2GFP/+ CD45.2 mice for two weeks prior to transplantation into CD45.1 BALB/c recipients to permanently label donor CCR2+ macrophages (tdTomato+). Flow cytometry performed 7 days after transplantation revealed that donor macrophages (CD45.2+CD64+) were either tdTomato−GFP− (89.2%) or tdTomato+GFP+ (9.2%). Few donor macrophages were either tdTomato−GFP+ or tdTomato+GFP− indicating that CCR2 expression is stable in donor macrophages after transplantation (Figure S1A).
To explore underlying mechanisms driving donor macrophage loss following transplantation, we measured donor macrophage cell death, proliferation, and emigration from the heart. Flow cytometry and TUNEL staining revealed a significant increase in donor macrophage cell death (1.3% DAPI+ GFP+ at baseline versus 13.3% DAPI + GFP + at post-transplant day14) after transplantation (p = 0.0010) (Figure 1E, Figure S1B–E). We next assessed both hematogenous and lymphatic emigration from the heart. Evaluation of donor macrophages (CX3CR1GFP/+ CCR2RFP/+ into BALB/c with CTLA4-Ig) within the recipient spleen revealed less than 20 donor GFP+ macrophages per spleen, suggestive of minimal hematogenous trafficking after transplantation (Figure S2A)37. Lymphatic connections to the heart are severed after surgery and re-establishment of lymphatic drainage from the graft may take several weeks; however, several reports identify donor immune cells trafficking to the recipient mediastinal lymph nodes38–40. We observed rare donor CCR2− but no CCR2+ macrophages within the mediastinal lymph nodes (Figure S2B). Proliferation has been reported to contribute to cardiac macrophage persistence23, 41. Immunofluorescence revealed that 4–5% of donor CCR2− and CCR2+ macrophages were proliferating (Ki67+) at baseline. Following transplantation, we observed an increase in the relative percentage of Ki67+ donor CCR2− macrophages (4.3% versus 11.0% at day 7, p = 0.035) (Figure S2C–D). A high percentage of recipient CD68+ cells (5.8%) were Ki67+ at post-transplant day 7 (Figure S2D).
To determine whether allograft rejection or reperfusion injury42 was responsible for donor macrophage cell death, we transplanted B6 CX3CR1GFP/+ CCR2RFP/+ donor hearts into BALB/c recipients and administered high-dose CTLA4-Ig (1.25 mg on post-operative days 0, 4, and 14)27, 43 (Figure S2E). This regimen has been shown to prevent cellular rejection with continued treatment but does not induce tolerance as the heart rejects upon cessation of therapy27. High-dose CTLA4-Ig prevented the loss of donor CCR2− macrophages at post-transplant day 14. Cessation of high-dose CTLA4-Ig at post-transplant day 14 resulted in the loss of donor CCR2− macrophages by day 28 post-transplant suggesting that allograft rejection leads to the elimination of donor CCR2− macrophages. High-dose CTLA4-Ig did not prevent the loss of donor CCR2+ macrophages (Figure S2F). To isolate the effect of reperfusion injury in the absence of rejection on donor macrophage survival, we performed syngeneic transplant of B6 CX3CR1GFP/+ CCR2RFP/+ donor hearts into B6 recipients without administration of immunosuppression and again noted the persistence of donor CCR2− macrophages (but not donor CCR2+ macrophages) at post-transplant day 14 (Figure S2F). These data indicate that rejection contributes to the loss of donor CCR2− macrophages and reperfusion injury leads to the loss of donor CCR2+ macrophages.
Donor and Recipient Macrophages are Distinct and Evolve Over Time after Heart Transplantation
We utilized single cell RNA sequencing to investigate the cellular and transcriptional landscape of donor and recipient macrophages after heart transplantation. B6 CX3CR1GFP/+ CCR2RFP/+ donor hearts were transplanted into BALB/c recipients and received low-dose CTLA4-Ig (Figure 2A). From the transplanted heart, donor (CD45+CD11b+CD64+GFP±) and recipient (CD45+CD11b+CD64+GFP−) cells were sorted separately at baseline (donor) and 1 (donor and recipient), 7 (donor and recipient) and 14 (recipient) days after transplantation and underwent single cell RNA sequencing (Figure 2B). We recovered approximately 4,194 donor and 10,590 recipient cells across all time points and detected 3000–5000 genes per cell (Figure S 3). We performed Harmony integration of donor and recipient libraries and noted that donor and recipient transcriptional signatures were unique and the cell populations were readily distinguished based on GFP expression (Figure 2C–E). Differential gene expression analysis identified discrete transcriptional signatures within donor and recipient populations. Donor cells expressed markers of tissue resident macrophages including Cbr2, F13a1, Folr2, and Egr1. Recipient cells expressed Ly6c2, Ly6a, Plac8, Ccl5, S100a8, and S100a9 consistent with monocyte and monocyte-derived macrophage identity (Figure 2F–G). Donor and recipient macrophages showed distinct time evolving transcriptional signatures after transplantation (Figure 2G). Pathways enriched in donor macrophages include MAPK, Myc signaling and ribosomal protein synthesis. In contrast, glycolysis, electron transport chain, type II interferon signaling, proteasome degradation, and TYROBP/DAP-12 signaling pathways were enriched in recipient macrophages and monocytes (Figure 2H–I).
Figure 2: Donor and recipient immune cells are distinct.
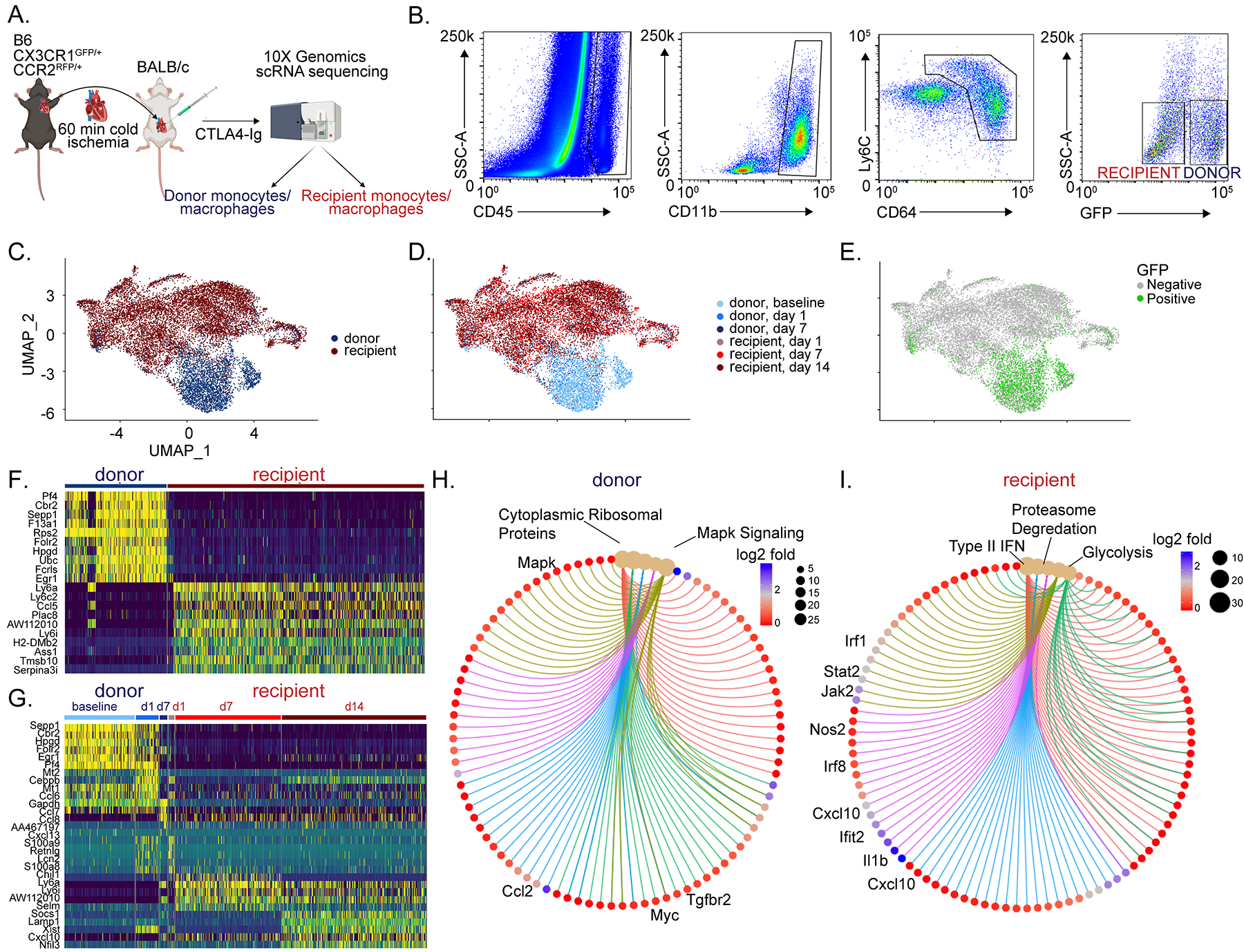
A) Schematic of single cell RNA sequencing workflow. B) Post-transplant day 1 gating scheme. Donor macrophages and monocytes are CD45+ CD11b+ Ly6c+ CD64+ GFP+ whereas recipient macrophages and monocytes are CD45+ CD11b+ Ly6c+ CD64+ GFP−. UMAP embedding plot of aggregated dataset split by C) donor and recipient, D) donor and recipient split by different timepoints, and E) GFP expression showing donor vs recipient demarcation, confirming robustness of FACS gating strategy. Heatmaps of normalized counts for differentially expressed marker genes between F) donor versus recipient and G) unique markers enriched in donor and recipients at different time points. Gene-Concept Network Plot for upregulated pathways with key genes annotated in H) donors and I) recipients using statistically significant genes (adjusted p-value < 0.05, Bonferroni correction) from differential gene testing.
Recipient Monocyte and Macrophage Diversity
Following transplantation, recipient monocytes infiltrate the donor heart and are known to promote allograft rejection44. To investigate the cell fates of infiltrating monocytes following transplantation, we performed an integrated analysis of recipient monocytes and macrophages (Figure 3). Cell clustering revealed distinct cell states of monocytes (Mo1, Mo2), macrophages (Mac1, Mac2, Mac3, Mac4, Mac5, Mac6), and dendritic-like (DC) cells with unique transcriptional signatures (Figure 3A–B, Figure S4). Monocytes were prevalent across all time points after transplantation suggestive of their ongoing recruitment. Mac5 and Mac6 macrophages decreased and Mac1, Mac2, Mac3, and Mac4 macrophages increased in frequency over time suggesting a temporal relationship amongst macrophage subsets (Figure 3C–D). Palantir trajectory analysis indicated that the extent of cell entropy reduced over time after transplantation (Figure S5D) indicating that recipient monocytes and macrophages enter the heart and differentiate over the course of rejection. Mo1, Mac1, Mac4, and DCs showed the greatest degree of entropy and lowest pseudotime values suggesting that they represent relatively undifferentiated states. In contrast, Mo2, Mac2, Mac5, and Mac6 displayed reduced entropy and higher pseudotime values suggesting that these subsets are more differentiated (Figure Ss 5–6).
Figure 3: Macrophage diversity in integrated recipient and donor hearts post heart transplantation.
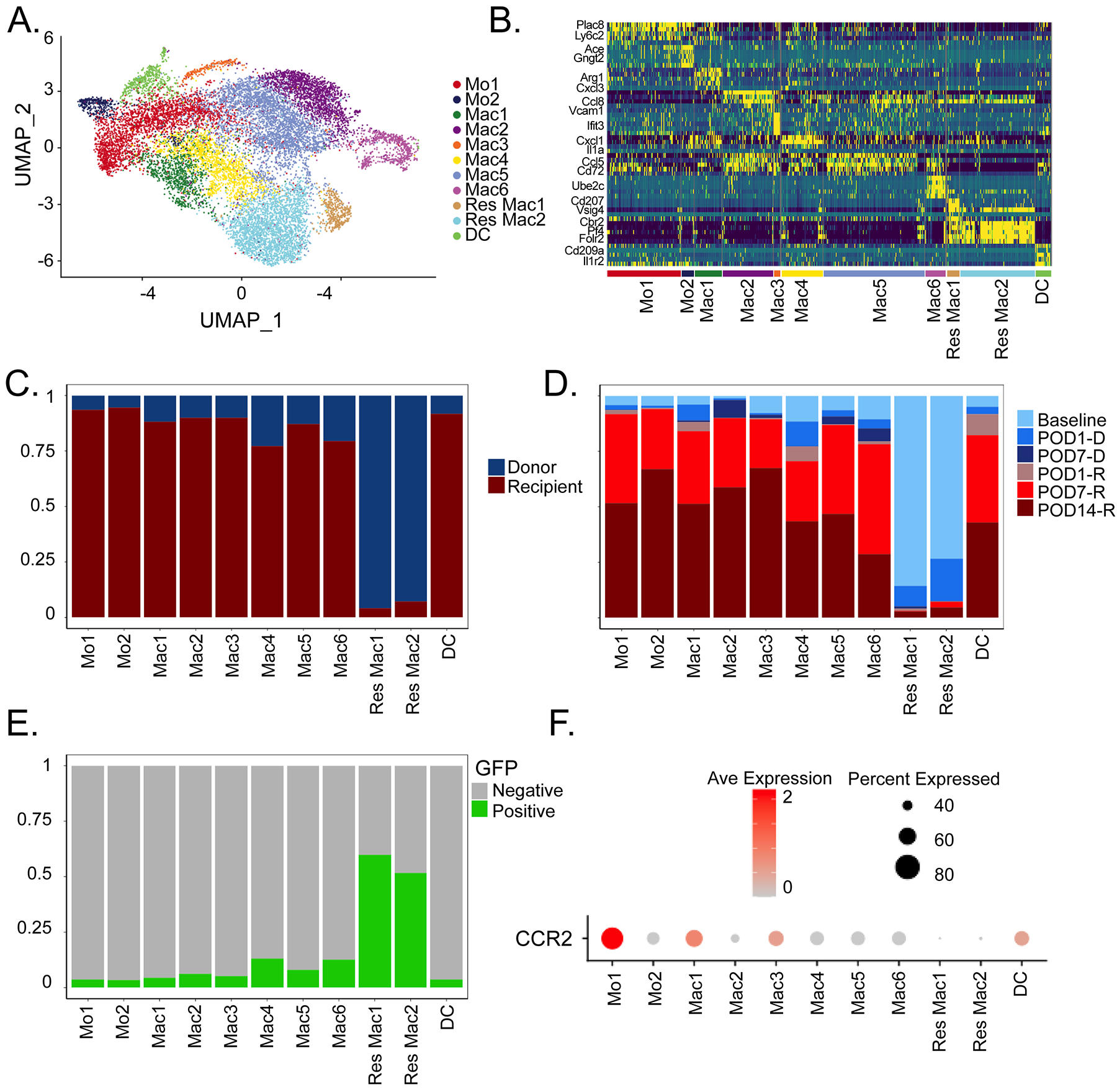
A) UMAP embedding plot of Harmony integrated donor and recipient libraries colored by transcriptionally distinct cell states. B) Heatmap of top 5 differentially expressed genes across populations using statistically significant genes (adjusted p-value < 0.05, Bonferroni correction), with representative genes shown. C) Relative donor and recipient contribution to cell states in (A). D) Cell composition of donor/recipient cells at different time points among the different myeloid cell states. E) Percent GFP positive cells in myeloid cell states from (A). (F) DotPlot of CCR2 expression in different cell states.
Transcriptional Diversity of Donor CCR2− and CCR2+ Macrophages
The composition of donor macrophage populations was assessed using our single cell RNA sequencing dataset. Donor macrophages were identified based on GFP expression and library indexed as separate libraries were constructed for donor and recipient macrophages. Res Mac1 and Res Mac2 contained GFP+ cells (Figure 3E). CCR2 was expressed in both donor and recipient populations with highest expression in the recipient (recruited) cells (Figure 3F).
We next focused on donor macrophages (Figure 4A). We have previously categorized cardiac macrophages based on MHC-II and CCR2 expression19, 22, 23. Consistent with these findings, we detected MHC-IIhigh CCR2−, MHC-IIlow CCR2−, MHC-IIhigh CCR2+, and MHC-IIlow CCR2+ populations (Figure 4B–C, D). Density mapping showed shifts in donor macrophage states after transplantation (Figure 4E–F).
Figure 4: Donor macrophages change their transcriptional state and are diminished post-transplantation.
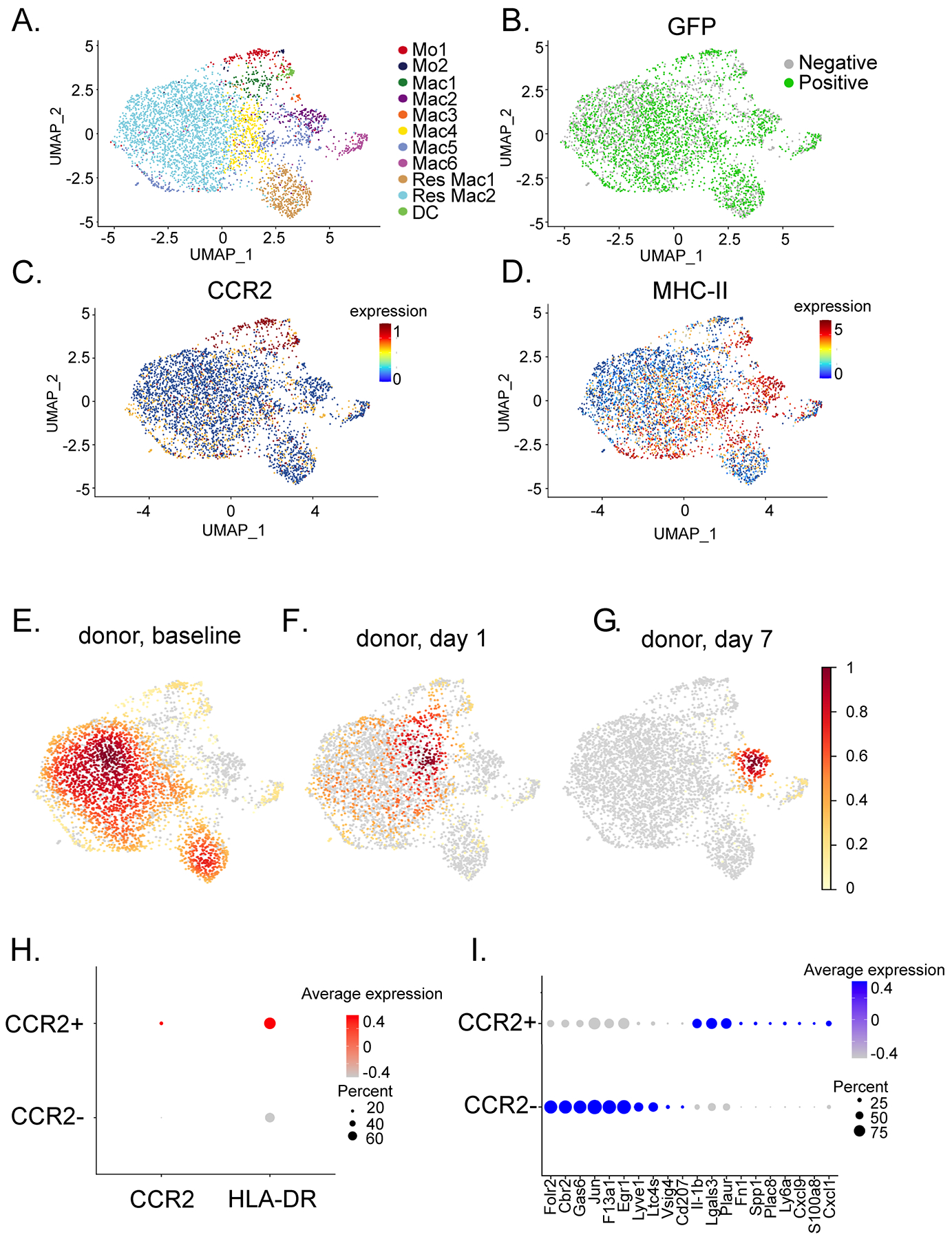
UMAP embedding plot of A) donor macrophages annotated by integrated object annotation B) GFP (51.7% positive) positive cells, C) Ccr2 and D) H2-Aa expression in donor myeloid cells. Gaussian kernel density embedding plots showing density of E) baseline, F) post-transplant day 1, and G) post-transplant day 7 donor myeloid cells in the UMAP space. H) DotPlot of CCR2 and HLA-DR expression in CCR2− (Res Mac1, Res Mac2, Mac2) and CCR2+ (Mo1, Mo2, Mac1, Mac3-6, DC) populations. I) Top differentially expressed genes between donor CCR2+ and CCR2− macrophages using statistically significant genes (adjusted p-value < 0.05, Bonferroni correction).
To delineate transcriptomic differences between donor CCR2− and CCR2+ macrophages, we performed a differential expression analysis. Donor CCR2− macrophages differentially expressed tissue resident macrophages genes including Folr2, Cbr2, Vsig4, Ccl8, and Ccl12. In contrast, donor CCR2+ macrophages expressed inflammatory genes (Figure 4H). Collectively, these data indicate that donor CCR2− and donor CCR2+ macrophages represent distinct populations at the transcriptional level.
Donor Macrophages are Essential for Allograft Survival after Heart Transplantation
To elucidate the functional roles of donor macrophages in allogenic heart transplantation, we utilized CD169DTR/+ 28 and CCR2DTR/+ 29 mice to deplete CCR2− and CCR2+ macrophages from the cardiac graft, respectively. We have previously established the specificity and efficacy of these murine strains in depleting CCR2− and CCR2+ macrophages from the heart19, 24. To this end, we transplanted B6 control, CD169DTR/+, or CCR2DTR/+ hearts into BALB/c recipients that received low-dose CTLA4-Ig. Diphtheria toxin (DT) (200 ng) was administered to the donor mouse daily for three days prior to transplant and three times per week to the recipient after transplantation to maintain depletion. Allografts lacking donor CCR2+ macrophages (CCR2DTR/+) had longer survival than littermate controls (35 versus. 26 days, p=0.0013; log-rank HR 0.287, 0.079–1.031). In contrast, allografts lacking donor CCR2− macrophages (CD169DTR/+) had shorter survival times compared to littermate controls (16 versus. 28 days, p=0.0004; log-rank HR 4.334, 1.147–16.39) (Figure 5A).
Figure 5: Donor macrophages differentially mediate allograft survival.
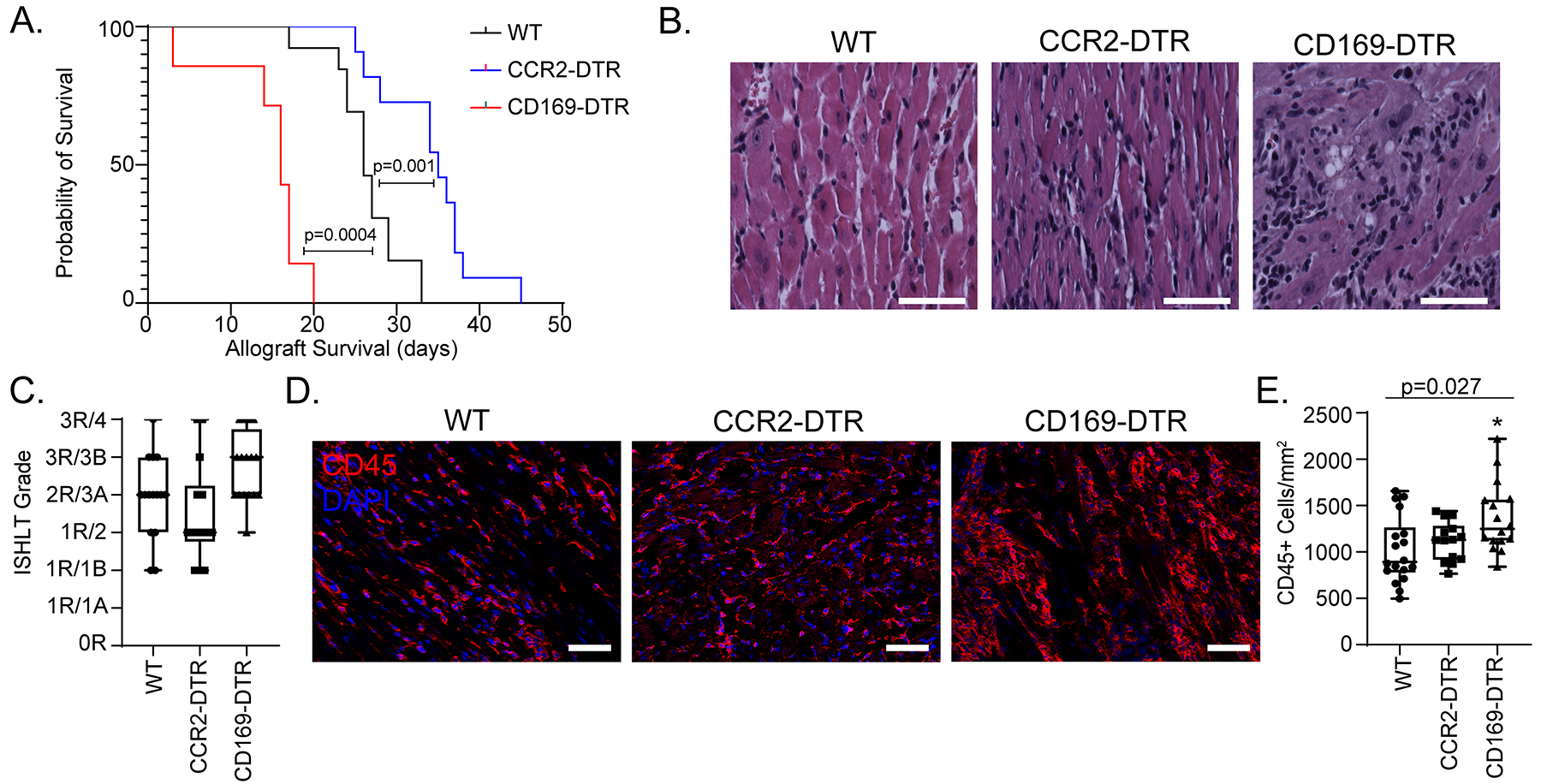
A) Kaplan-Meier survival curve of CD169DTR/+ (n = 7) vs CCR2DTR/+ (n = 11) vs littermate controls (n = 13) (Log-rank). B) H & E stain on transplanted hearts collected at post-transplant day 10. C) At least 3 random regions were evaluated by a trained cardiac pathologist and scored based on the 1990/2004 ISHLT cellular rejection grading guidelines (n=16 hearts for each cohort). D) Post-transplant day 10 hearts were stained with CD45 (red) and DAPI (blue). E) Number of CD45+ cell/mm2 heart was quantified. There was a significant difference among the three groups (Kruskal-Wallis; p = 0.027) and between WT and CD169DTR/+ (Dunn’s test for multiple comparisons; p=0.014). There was no difference noted between WT and CCR2DTR/+ (Dunn’s test for multiple comparisons; p = 0.58). n=16 for each cohort. Scale bar = 50 μm.
To investigate whether donor macrophages influence allograft inflammation, we harvested donor hearts 10 days after transplantation and performed histological analysis (Figure 5B–C). Compared to controls, allografts lacking donor CCR2− macrophages had more severe cellular rejection (3R/3B versus 2R/3A), while allografts lacking donor CCR2+ macrophages had milder cellular rejection (1R/2 versus 2R/3A). Immunostaining revealed increased CD45+ leukocytes in allografts lacking donor CCR2− macrophages compared to controls (1367 versus 1021 CD45+ cells/mm2, p = 0.014). Allografts lacking donor CCR2+ macrophages had a similar number of CD45+ leukocytes compared to controls (1116 versus 1021 CD45+ cells/mm2, p = 0.58) (Figure 5D–E). These data support the conclusion that donor CCR2− macrophages confer protection against acute allograft rejection whereas donor CCR2+ macrophages promote allograft rejection.
Donor Macrophage Activation following Transplantation
To examine the phenotypic behavior of donor macrophages after heart transplantation, we performed high-resolution imaging of donor macrophages at baseline and 1-, 3-, and 7-days post-transplant (Figure 6A–C, Figure S7A–B). At baseline, donor CCR2− and CCR2+ macrophages were relatively small, homogenously distributed within the heart, and displayed few cellular projections. Three-dimensional reconstruction of 30 μm heart sections (Figure 6D–E), revealed that both donor CCR2− and CCR2+ macrophages increase significantly in surface area (CCR2−: baseline 202.6 μm2 versus day 7 1121 μm2, p<0.0001; CCR2+: baseline 277.6μm2 versus day 7 1431 μm2, p<0.0001), volume (CCR2−: baseline 120.1 μm3 versus day 7 941.9 μm3, p<0.0001; CCR2+: baseline 245.1 μm3 versus day 7 1431 μm3, p<0.0001, and number of projections (CCR2−: baseline 0.637 projections/macrophage versus day 7 3.76 projections/macrophage, p<0.0001; CCR2+: baseline 0.43 projections/macrophage versus day 7 4.388 projections/macrophage, p<0.0001) (Figure 6F–K). There was no significant difference in morphologic quantification between donor macrophages at each time point (p > 0.05), except for CCR2− macrophage volume at baseline compared to that of CCR2+ macrophages (p = 0.006) (Figure S7C–E).
Figure 6: Donor macrophages dynamically respond after heart transplantation.
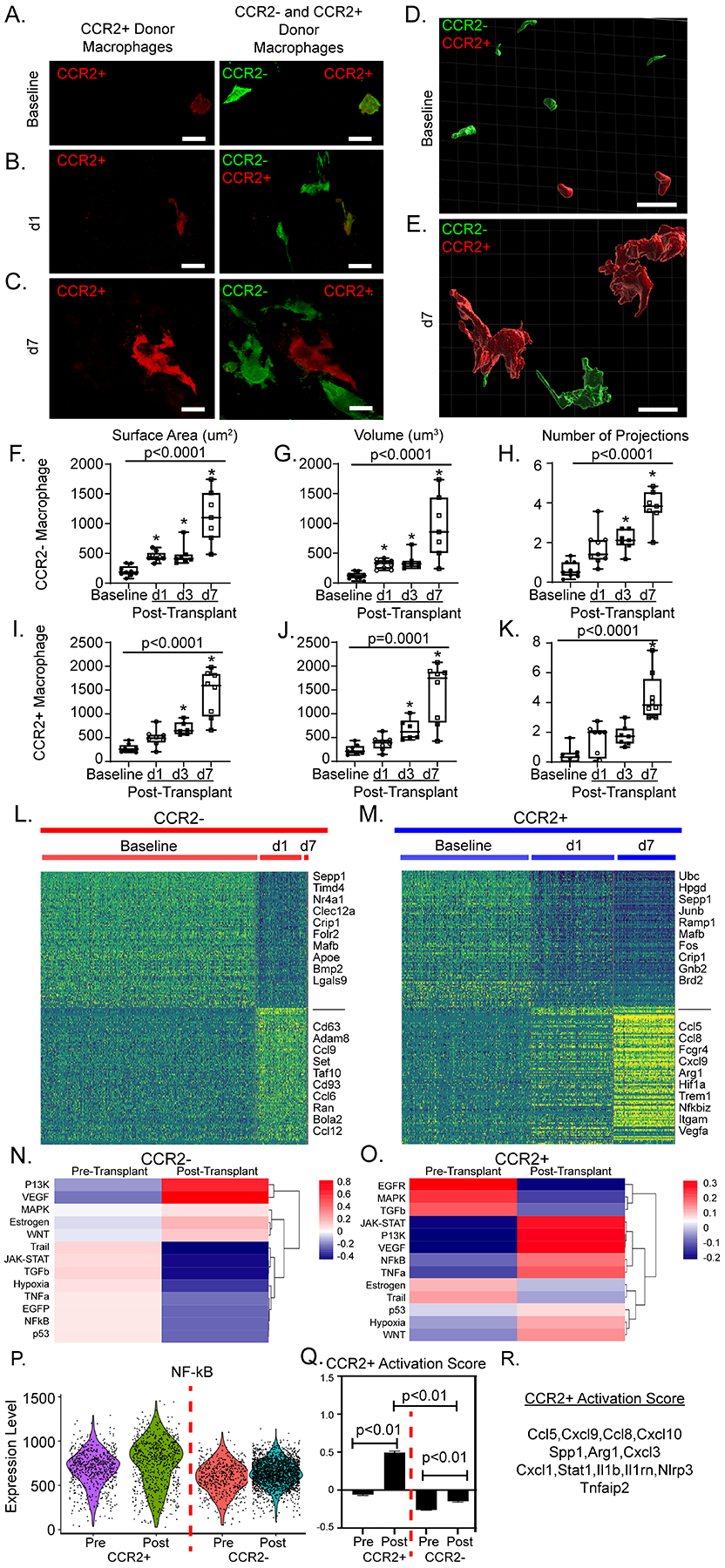
40X magnification z-stacks were obtained of dual reporter mice at A) baseline, B) post-transplant day 1 (d1), and C) post-transplant day 7 (d7). 30 μm thick sections were reconstructed with Imaris software to obtain volumetric reconstructions of donor macrophages at D) baseline and E) at d7. From reconstructions, quantitative measurements of donor CCR2− macrophages F) surface area (μm2), G) volume (μm3) and H) number of projections were obtained. For each timepoint, each data point represents the average of 10–20 macrophages from at least two regions of interest in at least two separate sections per heart. Identical measurements were performed in donor CCR2+ macrophages (I-K) across time. Statistical analyses were computed with Kruskal-Wallis (noted p-value) and Dunn’s test for multiple comparisons (* when < 0.05 compared to baseline) (baseline n = 10, d1 = 9, d3 = 7, d7 = 8). Heatmaps of normalized counts for differentially expressed marker genes between baseline and post-transplant for L) donor CCR2− macrophages and M) donor CCR2+ macrophages. PROGENy pathway analysis at basleine and post-transplant in N) CCR2− and O) CCR2+ macrophages. P) PROGENy derived NF-қB pathway enrichment score violin plot across 4 CCR2− and CCR2+ macrophages at baseline and post-transplant. Q) Gene set score for CCR2+ macrophage activation post-transplant shown in CCR2+ and CCR2− macrophages pre- and post-transplant (p < 0.01). Gene set score p-values calculated with 2-way ANOVA with multiple comparisons. R) Genes used to calculate gene set score. Scale bar = 10 μm.
Differential gene expression analysis demonstrated that donor CCR2− and CCR2+ macrophages displayed distinct activation signatures after transplant (Figure 6L, M). Donor CCR2− macrophages displayed enriched PI3K and VEGF signaling post-transplantation (Figure 6N). Compared to baseline, donor CCR2+ macrophages expressed markedly increased levels of genes associated with enhanced MYD88/NF-κβ signaling post-transplant (Figure 6O–R). Taken together, these data indicate that both donor CCR2− and CCR2+ macrophages are activated through distinct mechanisms following transplantation.
Inhibition of MYD88 Signaling in Donor Macrophages Prevents Allograft Rejection
We have previously demonstrated that donor CCR2+ macrophages orchestrate leukocyte trafficking following syngeneic transplantation through a MYD88 dependent mechanism19, 24. To determine whether inhibition of MYD88 signaling in donor macrophages presents a possible therapeutic approach for preventing or reducing graft rejection, we used donor mice that lack MyD88 either globally or selectively in macrophages. We transplanted B6 control, MyD88−/−, MyD88f/f LysMCre/+, or MyD88f/f CSF1rertCre/+donor hearts into BALB/c recipients, which were subsequently treated with low-dose CTLA4-Ig. Global deletion of MYD88 in the donor heart and conditional deletion of MYD88 in donor macrophages both resulted in significantly prolonged allograft survival compared to controls (all p < 0.009) (Figure 7A).
Figure 7: Donor macrophages signal through MyD88.
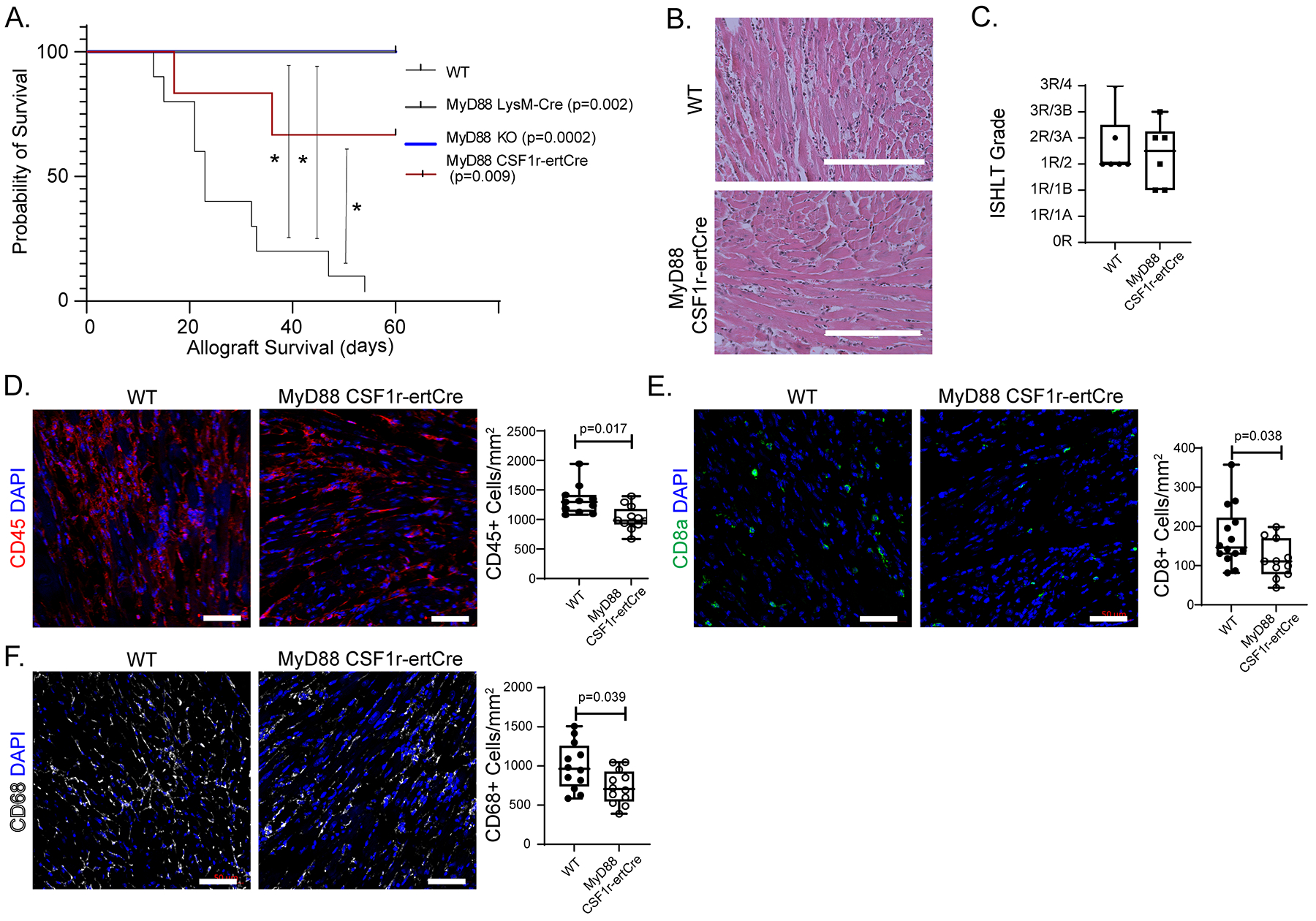
A) Kaplan-Meier survival curve with control (n = 10) versus MyD88f/f LysMCre/+ (Log-rank; p = 0.002, n = 4), MyD88f/f CSF1rertCre/+ (Log-rank; p = 0.009, n = 6), or MyD88−/− (“KO” Log-rank; p = 0.0002; n = 8) donor allografts (censored at 60 days). B) H & E staining of WT and MyD88f/f CSF1rertCre/+ allografts collected at post-transplant day 10. C) ISHLT cellular rejection scores comparing WT (n=12) and MyD88f/f CSF1rertCre/+ (n=12) hearts. D) CD45+ immunofluorescent staining of WT (n=12) and MyD88f/f CSF1rertCre/+ (n=12) hearts with quantification showing significant reduction in CD45+ cells/mm2 in MyD88f/f CSF1rertCre/+ hearts (1022 cells vs 1328 cells/mm2; Mann-Whitney U Test; p = 0.0017,) compared to littermate control. E) CD8a+ immunofluorescent staining of WT (n=14) and MyD88f/f CSF1rertCre/+ (n=11) hearts with quantification showing significant reduction in CD8a+ cells/mm2 in MyD88f/f CSF1rertCre/+ hearts (110 cells vs 146 cells/mm2; Mann-Whitney U Test; p = 0.038) compared to littermate control. F) CD68+ immunofluorescent staining of WT (n=12) and MyD88f/f CSF1rertCre/+ (n=12) hearts with quantification showing significant reduction in CD68+ cells/mm2 in MyD88f/f CSF1rertCre/+ hearts (705 cells vs 964 cells/mm2; Mann-Whitney U Test; p = 0.039,) compared to littermate control. Scale bar: B= 200 μm; D=50 μm.
To assess graft inflammation, we performed histological analyses and CD45+ immunostaining on control and MyD88f/f CSF1rertCre/+donor hearts 10 days after transplantation. Compared to controls, allografts lacking MYD88 in donor macrophages had only mild cellular rejection (1R/2 versus 2R/3A) (Figure 7B–C) decreased CD45+ cells abundance (1022 versus 1328 CD45+ cells/mm2, p = 0.017) (Figure 7D), decreased CD8a+ cell abundance (110 versus 146 CD8a+ cells/mm2, p = 0.038 (Figure 7E). and decreased CD68+ cell abundance (705 versus 964 CD68+ cell/mm2, p = 0.039) (Figure 7F).
Deletion of MYD88 affects Antigen Presentation, Type II Interferon Signaling, and T-cell Activation
To delineate the role of donor macrophage MyD88 signaling on recipient monocyte fate specification, we performed single cell RNA sequencing of recipient monocytes, macrophages, and dendritic cells (CD45+ CD11b+ Ly6c+/− CD64+/−) isolated from WT and MyD88f/f CSF1rertCre/+ donor hearts at post-transplant day 3. We recovered 6,207 WT and 3,598 MyD88f/f CSF1rertCre/+ cells and detected 3,000 to 5,000 genes per cell (Figure S8A). We projected the data onto our donor and recipient monocyte, macrophage, and dendritic cell reference object and obtained high confidence mapping scores (Figure 8A, Figure S8B). Cell composition analysis showed increased proportion of Mo1 in WT allografts and increase Mac1 and Mac6 in MyD88f/f CSF1rertCre/+ donor hearts. We did not detect any differences among other monocyte, macrophage, or dendritic cell states (Figure 8A). Few cells mapped to donor macrophage populations indicating the majority of recovered cells were of recipient origin.
Figure 8: Donor MyD88 depletion leads to modulation of recipient immune cell gene expression.
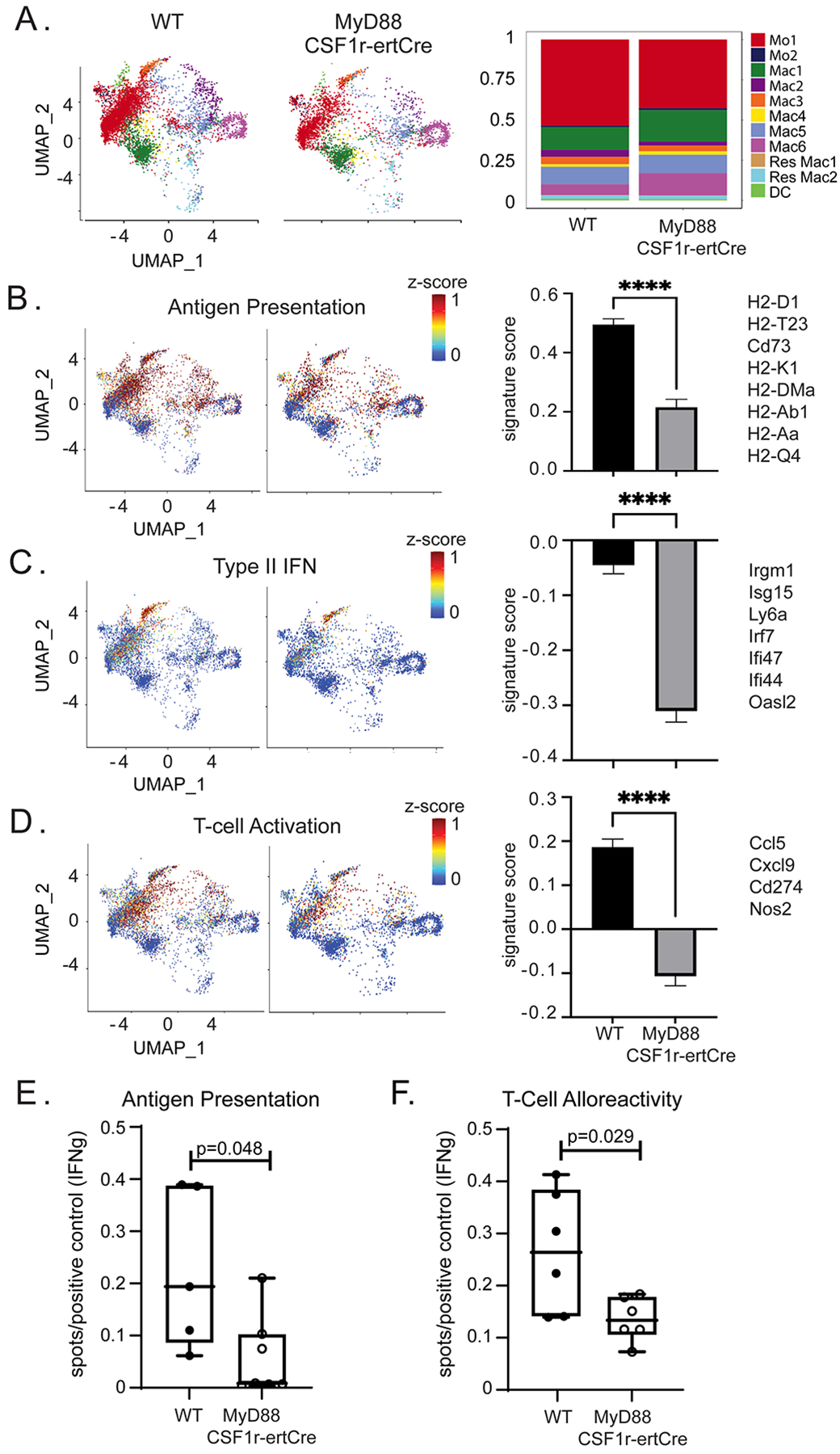
UMAP embedding plot of A) post-transplant day 3 WT and post-transplant day 3 MyD88f/f CSF1rertCre/+ mapped onto integrated donor/recipient UMAP (left). Composition plot showing frequency of each population (right) in WT and MyDf/f CSF1rertCre/+. Combined z-scores for top genes and calculated gene set scores for B) antigen presentation (0.569 vs 0.211, p<0.0001), C) interferon signaling (−0.0032 vs −0.324, p<0.0001), and D) T-cell activation (0.1744 vs −0.1803 p< 0.0001) gene set scores with genes on the right. Statistical test Mann-Whitney U Test. E) Number of IFN-γ spots per conA positive control of DO.11 T-cells co-cultured with OVA-loaded post-transplant days 3–4 macrophages and monocytes (Antigen presentation assay) with a significant reduction in MyD88f/f CSF1rertCre/+ cohort (0.0084 vs 0.19 spots/conA treated T-cell control; Mann-Whitney U Test; p = 0.048; n = 5 WT, n = 6 MyD88f/f CSF1rertCre/+). F) Number of IFN-γ spots per conA positive control of recipient T-cells co-cultured with irradiated donor antigen (T-cell alloreactivity assay) with a significant reduction in MyD88f/f CSF1rertCre/+ cohort (0.13 vs 0.26 spots/conA treated T-cell control; Mann-Whitney U Test; p = 0.029; n = 6 each group).
We detected global differences in gene expression between recipient cells recruited to control and MyD88f/f CSF1rertCre/+ donor hearts. Decreased expression of genes associated with antigen presentation (0.22 versus 0.50, p < 0.001) (Figure 8B), type II interferon signaling (−3.11 versus −0.045, p < 0.001) (Figure 8C), and T-cell activation (−0.107 versus 0.187, p < 0.001) (Figure 8D) were detected in cells recruited to MyD88f/f CSF1rertCre/+ donor hearts. Antigen presentation, type II IFN, and T-cell activation signaling were cell type specific and prominent among the CCR2+ monocyte/macrophage subsets with less impact in the CCR2− macrophages (Figure S8C).
These data suggest that deletion of MyD88 in donor macrophages reduces alloreactive T-cell generation and decreased antigen presentation capacity in infiltrating recipient macrophages as a potential mechanism. We tested the possibility that inhibition of MyD88 in donor macrophages diminished the ability of recipient antigen presenting cells to present antigen to T-cells. Compared to control hearts, recipient antigen presenting cells from donor hearts lacking MyD88 had significantly less ability to present antigen to T-cells (0.0085 versus 0.19 spots/T-cell positive control, p = 0.048) (Figure 8E). Furthermore, compared to controls, recipients transplanted with MyD88f/f CSF1rertCre/+ hearts had significantly fewer T-cells in their spleens that produced IFN-γ following exposure to donor antigen (0.14 versus 0.27 spots/T-cell positive control, p = 0.029). (Figure 8F). Collectively, these findings indicate that MYD88 signaling in donor macrophages is an important regulator of antigen presentation in recipient antigen presenting cells and alloreactive T-cell priming.
Discussion:
To the best of our knowledge, this is the first study to investigate the transcriptional signature and dynamics of donor and recipient myeloid cells following allogenic heart transplantation at a single cell resolution. We defined the functional importance of donor CCR2+ and CCR2− macrophages in allograft rejection and identified donor CCR2+ macrophages as a key cell type that potentiates rejection. Finally, we implicated MYD88 signaling in donor macrophages as a potential therapeutic target to modulate the recipient cell function, reduce the generation of alloreactive T-cells, decrease rejection, and extend allograft survival.
Using genetic lineage tracing and single cell RNA sequencing, we longitudinally dissected the dynamics of donor macrophages after heart transplantation. We show that in the setting of cellular rejection, donor macrophages persisted for approximately 1–2 weeks and are subsequently lost from the graft. Intriguingly, suppression of allograft rejection using high-dose CTLA4-Ig immunosuppression or using a syngeneic transplant model was sufficient to preserve donor CCR2− macrophages, but not donor CCR2+ macrophages. This observation raises the possibility that alloreactive T-cells play an active role in eliminating donor CCR2− macrophages. Future studies will be required to identify the precise population of T-cells that targets donor CCR2− macrophages and to dissect the molecular mechanisms and cell death pathways involved.
Single cell RNA sequencing demonstrated that donor and recipient macrophages are transcriptionally distinct and that these populations dynamically evolve over the course of rejection. Consistent with transcriptional evidence of cell activation, donor macrophages show significant morphologic changes including increased surface area, volume, and cellular projections. Differential gene expression analysis identified signatures of MYD88/NF-κβ signaling in donor CCR2+ macrophages after transplantation. Either depletion of donor CCR2+ macrophages or conditional deletion of MYD88 signaling in donor macrophages was sufficient to reduce rejection and extend allograft survival. These data build on our prior observation that donor CCR2+ macrophages direct infiltration of recipient neutrophils and monocytes into the heart through the expression of MYD88 dependent chemokines and cytokines19, 24. Depletion of donor macrophage MYD88 signaling led to infiltration of recipient cells with lower expression of antigen presentation, type II interferon, and T-cell activation gene expression signatures, which we confirmed experimentally. Based on the robust MYD88/NF- κβ signature observed in donor CCR2+ macrophages after transplantation and improved outcomes seen in donor hearts that lack MyD88 in macrophages, we posit that MYD88 signaling in donor CCR2+ macrophages contributes to allograft rejection. However, we cannot exclude the possibility that MYD88 signaling in donor CCR2− macrophages may also contribute to some of the observed phenotypes. Collectively, these findings provide the first evidence that targeting specific donor macrophage populations may serve as a therapeutic approach to prevent cellular rejection. Targeting donor specific populations could be done using ex-situ perfusion devices prior to implantation and holds promise of ameliorating graft rejection45, 46.
Our data indicated an opposing role for donor CCR2− macrophages. Depletion of donor CCR2− macrophages resulted in rapid rejection and increased immune cell infiltration into the allograft, indicating that donor CCR2− macrophages have a protective function. Following transplantation, we observed patches of donor CCR2− macrophages that appeared to physically interact with and clustered around donor CCR2+ macrophages. It is possible that donor CCR2− macrophages prevent the activation of donor CCR2+ macrophages and other immune cells. Consistent with this hypothesis, tissue resident macrophages have previously been shown to cloak areas of injury and prevent inflammatory activation of innate immune cells. We hypothesize that donor CCR2− macrophage are protective potentially through masking pro-inflammatory signaling from donor CCR2+ macrophages and their depletion could lead to more rapid rejection and increased inflammatory infiltration.
Finally, our single cell RNA sequencing data revealed that donor and recipient macrophages are distinct and shed new light on the surprising diversity of recipient monocyte, macrophage, and dendritic cell populations. Using a probabilistic model to predict the trajectories of differentiating monocytes, we show evidence that recipient monocytes have the capacity to differentiate into several unique macrophage and dendritic cell-like populations. The environmental cues and molecular signals responsible for these fate decisions are of considerable interest. Future studies will focus on the contribution of each recipient monocyte, macrophage, and dendritic cell population to allograft rejection.
Limitations and Conclusions.
Our study is not without limitations. Mouse models of heterotopic heart transplantation are imperfect as they are mechanically unloaded and thus may not fully mirror the clinical scenario. We additionally recognize genetic manipulation of donor macrophage composition and signaling may not be fully recapitulated by pharmacological interventions. We have previously reported that human donor macrophages persist in the allograft following heart transplantation in the absence of clinical rejection21. An important next step is to determine whether transcriptionally similar states of monocytes, macrophages, and dendritic cells infiltrate the transplanted human heart in the context of acute cellular rejection. Additionally, while we used the best available Cre lines to delete MyD88 in macrophages, we cannot exclude a function of MyD88 in dendritic cells. Our gating schemes focused on macrophages, monocytes, and classic dendritic cell 2 and did not specifically interrogate all dendritic cell subsets. Nonetheless, our findings establish donor macrophages as a potential therapeutic target to improve transplant outcomes and identify inhibition of MyD88 signaling in the donor heart as a therapeutic approach to suppress donor CCR2+ macrophage activation and blunt alloimmune responses.
Supplementary Material
Clinical Perspective.
What Is New?
Donor macrophages persist after heart transplantation and have a unique transcriptional signature when compared to recipient macrophages.
Depletion of donor CCR2- and CCR2+ macrophages prior to heart transplantation results in opposing effects on allograft survival benefit.
Interfering with donor CCR2+ macrophage activation by deletion of MyD88 results in extended allograft survival through suppression of recipient antigen presenting cell function and alloreactive T-cell generation.
What Are the Clinical Implications?
These findings suggest that donor macrophages represent a potential therapeutic target prior to transplantation.
Therapies focused on suppressing donor CCR2+ macrophage activation or augmenting CCR2- macrophage function may represent new approaches to reduce alloimmune responses and extend organ survival following heart transplantation.
Acknowledgements:
We thank Geetika Bajpai and Inessa Lokshina for technical expertise and mouse husbandry. We would also like to thank Dr. Burkhard Becher for kindly contributing CCR2ert2Cre/+ mice.
Sources of Funding:
B.K. was supported by the Principles in Cardiovascular Research Training Grant (T32 HL007081), NHLBI K08 HL159359, and the Washington University Physician Scientist Training Program. K.L. is supported by funding provided from the NHLBI (R01 HL138466, R01HL139714, R01 HL151078, R35 HL161185), Leducq Foundation Network (#20CVD02), Burroughs Welcome Fund (1014782), Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CH-II-2015-462, CH-II-2017-628, PM-LI-2019-829), and Foundation of Barnes-Jewish Hospital (8038-88). D.K. is supported by NIH (P01AI116501 and R01 HL094601), Veterans Administration Merit Review (1I01BX002730), and the Foundation for Barnes-Jewish Hospital. We acknowledge the McDonnel Genome Institute, DDRCC histology core (P30 DK52574), and the Washington University Center for Cellular Imaging (WUCCI), which is supported in part by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505 and CDI-CORE-2019-813) and the Foundation for Barnes-Jewish Hospital (3770) for access to imaging resources and valuable technical assistance. J.A.J.F. is supported by the Chan Zuckerberg Initiative as a CZI Imaging Scientist (2020-225726).
Non-standard Abbreviations and Acronyms
- CCR2
C-C chemokine Receptor 2
- CD169
Cluster of Differentiation 169
- CX3CR1
C-X3-C motif Chemokine Receptor 1
- DTR
Diphtheria Toxin Receptor
- ELISPOT
Enzyme-Linked Immune absorbent SPOT
- Ert2-Cre
Estrogen Receptor 2- Cre
- GFP
Green Fluorescent Protein
- RFP
Red Fluorescent Protein
- MyD88
Myeloid Differentiation Primary Response Protein 88
Footnotes
Disclosures:
The authors have nothing to disclose.
References
- 1.Colvin M, Smith JM, Hadley N, Skeans MA, Carrico R, Uccellini K, Lehman R, Robinson A, Israni AK, Snyder JJ, et al. OPTN/SRTR 2016 Annual Data Report: Heart. Am J Transplant. 2018;18 Suppl 1:291–362. [DOI] [PubMed] [Google Scholar]
- 2.Moayedi Y, Fan CPS, Cherikh WS, Stehlik J, Teuteberg JJ, Ross HJ and Khush KK. Survival Outcomes After Heart Transplantation: Does Recipient Sex Matter? Circ Heart Fail. 2019;12:e006218. [DOI] [PubMed] [Google Scholar]
- 3.Chang DH, Kittleson MM and Kobashigawa JA. Immunosuppression following heart transplantation: prospects and challenges. Immunotherapy. 2014;6:181–94. [DOI] [PubMed] [Google Scholar]
- 4.Alba AC, bain e, ng n, stein m, o’brien k, fortoutan f and ross h. Complications after Heart Transplantation: Hope for the Best, but Prepare for the Worst. International Journal of Transplantation Research and Medicine. 2016;2. [Google Scholar]
- 5.McDonald-Hyman C, Turka LA and Blazar BR. Advances and challenges in immunotherapy for solid organ and hematopoietic stem cell transplantation. Sci Transl Med. 2015;7:280rv2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millington TM and Madsen JC. Innate immunity and cardiac allograft rejection. Kidney Int Suppl. 2010:S18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kopecky BJ, Frye C, Terada Y, Balsara KR, Kreisel D and Lavine KJ. Role of donor macrophages after heart and lung transplantation. Am J Transplant. 2020;20:1225–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Egashira N and Masuda S. Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costanzo MR, Dipchand A, Starling R, Anderson A, Chan M, Desai S, Fedson S, Fisher P, Gonzales-Stawinski G, Martinelli L, et al. The International Society of Heart and Lung Transplantation Guidelines for the care of heart transplant recipients. J Heart Lung Transplant. 2010;29:914–56. [DOI] [PubMed] [Google Scholar]
- 10.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–29. [DOI] [PubMed] [Google Scholar]
- 11.Hunt SA and Haddad F. The changing face of heart transplantation. J Am Coll Cardiol. 2008;52:587–98. [DOI] [PubMed] [Google Scholar]
- 12.Costello JP, Mohanakumar T and Nath DS. Mechanisms of chronic cardiac allograft rejection. Tex Heart Inst J. 2013;40:395–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Conde P, Rodriguez M, van der Touw W, Jimenez A, Burns M, Miller J, Brahmachary M, Chen HM, Boros P, Rausell-Palamos F, et al. DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance. Immunity. 2015;42:1143–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schetters STT, Kruijssen LJW, Crommentuijn MHW, Kalay H, Ochando J, den Haan JMM, Garcia-Vallejo JJ and van Kooyk Y. Mouse DC-SIGN/CD209a as Target for Antigen Delivery and Adaptive Immunity. Front Immunol. 2018;9:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Bosch TP, Kannegieter NM, Hesselink DA, Baan CC and Rowshani AT. Targeting the Monocyte-Macrophage Lineage in Solid Organ Transplantation. Front Immunol. 2017;8:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Li C, Zhuang Q, Peng B, Zhu Y, Ye Q and Ming Y. The Evolving Roles of Macrophages in Organ Transplantation. J Immunol Res. 2019;2019:5763430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tse GH and Hughes J. Macrophages and transplant rejection: a novel future target? Transplantation. 2013;96:946–8. [DOI] [PubMed] [Google Scholar]
- 18.Chen S, Lakkis FG and Li XC. The many shades of macrophages in regulating transplant outcome. Cell Immunol. 2020;349:104064. [DOI] [PubMed] [Google Scholar]
- 19.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res. 2019;124:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ochando J, Fayad ZA, Madsen JC, Netea MG and Mulder WJM. Trained immunity in organ transplantation. Am J Transplant. 2020;20:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ and Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111:16029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epelman S, Lavine KJ and Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ and Kreisel D. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pietra BA, Wiseman A, Bolwerk A, Rizeq M and Gill RG. CD4 T cell-mediated cardiac allograft rejection requires donor but not host MHC class II. J Clin Invest. 2000;106:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corry RJ, Winn HJ and Russell PS. Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation. 1973;16:343–50. [DOI] [PubMed] [Google Scholar]
- 27.Schwarz C, Unger L, Mahr B, Aumayr K, Regele H, Farkas AM, Hock K, Pilat N and Wekerle T. The Immunosuppressive Effect of CTLA4 Immunoglobulin Is Dependent on Regulatory T Cells at Low But Not High Doses. Am J Transplant. 2016;16:3404–3415. [DOI] [PubMed] [Google Scholar]
- 28.Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M and Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest. 2007;117:2268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M and Pamer EG. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hou B, Reizis B and DeFranco AL. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clausen BE, Burkhardt C, Reith W, Renkawitz R and Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77. [DOI] [PubMed] [Google Scholar]
- 32.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA and Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K and Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–50. [DOI] [PubMed] [Google Scholar]
- 34.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A and Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM and Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA and Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques. 2000;29:52, 54. [DOI] [PubMed] [Google Scholar]
- 37.Larsen CP, Morris PJ and Austyn JM. Donor dendritic leukocytes migrate from cardiac allografts into recipients’ spleens. Transplant Proc. 1990;22:1943–4. [PubMed] [Google Scholar]
- 38.Edwards LA, Nowocin AK, Jafari NV, Meader LL, Brown K, Sarde A, Lam C, Murray A and Wong W. Chronic Rejection of Cardiac Allografts Is Associated With Increased Lymphatic Flow and Cellular Trafficking. Circulation. 2018;137:488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soong TR, Pathak AP, Asano H, Fox-Talbot K and Baldwin WM 3rd. Lymphatic injury and regeneration in cardiac allografts. Transplantation. 2010;89:500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown K, Badar A, Sunassee K, Fernandes MA, Shariff H, Jurcevic S, Blower PJ, Sacks SH, Mullen GE and Wong W. SPECT/CT lymphoscintigraphy of heterotopic cardiac grafts reveals novel sites of lymphatic drainage and T cell priming. Am J Transplant. 2011;11:225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res. 2016;119:853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Young JS, Khiew SH, Yang J, Vannier A, Yin D, Sciammas R, Alegre ML and Chong AS. Successful Treatment of T Cell-Mediated Acute Rejection with Delayed CTLA4-Ig in Mice. Front Immunol. 2017;8:1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, Rothstein DM and Lakkis FG. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest. 2014;124:3579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chew HC, Macdonald PS and Dhital KK. The donor heart and organ perfusion technology. J Thorac Dis. 2019;11:S938–S945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, MacGowan GA, Ali S and Dark JH. Ex situ heart perfusion: The past, the present, and the future. J Heart Lung Transplant. 2021;40:69–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.