

Abstract
The accumulation of amyloid beta (Aβ) plaques in the brain is a hallmark of Alzheimer’s disease (AD) pathology. Microglial activation-mediated neuroinflammation has been implicated in the pathogenesis of AD and the expression levels of interleukin-6 (IL-6) were increased in the brains of AD patients. However, the mechanisms by which IL-6 expression is regulated in human microglia are incompletely understood. Here, we show that Aβ1–40 oligomers (Aβ40) dose-dependently stimulate IL-6 expression in HMC3 human microglial cells. Treatment with Aβ40 promotes the transcription of IL-6 and tumor necrosis factor α (TNFα) mRNAs in both HMC3 and THP-1 cells. Mechanistic studies reveal that Aβ40-induced increase of IL-6 secretion is associated with the activation of p38 mitogen-activated protein kinase (p38 MAPK). Inhibition of p38 MAPK by BIRB 796 or SB202190 abrogates Aβ40-induced increase of IL-6 production. Through analyzing brain specimens, we found that the immunoreactivity for IL-6 and phosphorylated (the activated form) p38 MAPK were markedly higher in microglia of AD patients than in age-matched control subjects. Moreover, our studies identified the co-localization of IL-6 with phosphorylated p38 MAPK in microglia in the cortices of AD patients. Taken together, these results indicate that p38 MAPK is a major regulator of Aβ-induced IL-6 production in human microglia, which suggests that targeting p38 MAPK may represent a new approach to ameliorate Aβ accumulation-induced neuroinflammation in AD.
Keywords: Amyloid beta, p38 MAPK, Microglia, Interleukin-6, Neuroinflammation, Alzheimer’s disease
Introduction
Alzheimer’s disease (AD), the most common cause of dementia, is characterized by the early accumulation of extracellular amyloid-beta peptide (Aβ) in neuritic plaques and followed by hyperphosphorylated tau-containing intracellular neurofibrillary tangles, along with the progressive loss of memory and cognitive functions [1, 2]. In response to Aβ accumulation, microglia are activated to facilitate the clearance of Aβ aggregates, but unfortunately this process can trigger a neuroinflammatory response that could be detrimental to neurons [3, 4]. The neuroinflammatory responses include the elevated production of pro-inflammatory cytokines, such as interleukin (IL)-1, IL-6 and tumor necrosis factor alpha (TNFα), which are associated with various neurodegenerative disorders, including AD, Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis and HIV-associated dementia [4–9]. Increased IL-6 expression levels have been observed in brain specimens from AD patients [10] and are linked to diffuse plaques representing the early stage of plaque formation [11]. Moreover, it has been shown that human cognitive functions adversely correlate with serum IL-6 levels [12]. An epidemiologic meta-analysis indicated that IL-6 levels may be a useful biological marker to correlate with the severity of cognitive impairments in AD patients [13]. Preclinical models provided further support and showed that cerebral overexpression of IL-6 resulted in transgene dose- and animal age-dependent deficits in avoidance learning as well as progressive neurodegenerative pathologies [14]. These results suggest that the elevated production of IL-6 may play a critical role in neurodegenerative disorders. Nevertheless, the molecular mechanisms by which IL-6 expression is regulated in human microglial cells remain poorly understood.
Aβ peptides consist of 38 to 43 amino acids and are generated from a sequential proteolytic cleavage of the amyloid precursor protein (APP) by β- and γ-secretase [15]. Aβ monomers can bind to one another to form Aβ oligomers, polymers, insoluble fibrils, and eventually senile plaques. Aβ oligomers are soluble and exhibit the strongest toxicity to neurons [16], which can cause neuronal cell damage, impairment of synaptic plasticity and memory loss [16–18]. The Aβ1–40 (Aβ40) and Aβ1–42 (Aβ42) peptides are two of the most abundant amyloid species in senile plaques [19]. Aβ40 oligomers can be delivered to the parenchyma via the glymphatic transport system and co-localize with senile plaques, suggesting an important role of Aβ40 in plaque formation [20]. Although the impact of Aβ on microglia-mediated neuroinflammation has been reported in previous studies using animal models or rodent cell lines [21–25], the neuroinflammatory response of human microglia to Aβ stimulation has not been investigated previously. Moreover, a recent study reported that treatment with a selective p38 inhibitor alleviated neuropathology and cognitive impairments in a mouse model of AD [26]. However, the molecular mechanisms by which p38 inhibition is beneficial for AD are not completely understood. In the present study, we show that Aβ40 selectively induces the activation of p38 MAPK (p38 hereafter), but not the Erk and JNK MAPKs, in HMC3 human microglial cells. Inhibition of p38 by BIRB 796 (BIRB) or SB202190 (SB) is sufficient to block Aβ40-induced increase in IL-6 production. Moreover, our studies have confirmed the colocalization of IL-6 with phosphorylated p38 (p-p38) in microglia in the cortices of AD patients. Collectively, these results pinpoint a major role of the p38 pathway in modulating Aβ-induced IL-6 production in human microglia, suggesting that targeted inhibition of p38 may represent a new therapeutic approach to curb Aβ accumulation-induced neuroinflammation in AD.
Materials and Methods
Materials
Aβ40 and scrambled Aβ40 (Scr-Aβ40) peptides were purchased from rPeptide (Athens, GA). Brefeldin A was purchased from Biolegend (San Diego, CA). BIRB 796 and SB202190 were purchased from Selleckchem (Houston, TX). LPS was purchased for Sigma (E coli O55:B5, cat# L2880, St. Louis, MO). Rabbit anti-human p-p38, p38, p-p44 (pErk), p44 (Erk), IL-6, IL-10, TGF-β and JNK1 monoclonal antibodies, and mouse anti-human p-JNK1 monoclonal antibodies were purchased from Cell Signaling (Danvers, MA). Mouse anti-human CD68 monoclonal antibody was obtained from ThermoFisher (Cat# 14-0688-82, clone: KP1). Mouse anti-human TMEM119 monoclonal antibody (#400111) was obtained from Synaptic Systems (Gottingen, Germany). Mouse anti-human α-tubulin monoclonal antibody was obtained from Santa Cruz Biotechnology (Santa cruz, CA). Human IL-6 DuoSet ELISA (DY206) and human TNF-α DuoSet ELISA (DY210) kits were purchased from R&D Systems (Minneapolis, MN). EMEM medium was purchased from American Type Culture Collection (ATCC, cat# 30–2003). RPMI-1640 medium was obtained from Gibco (Gibco, cat# 11875093).
Preparation of Aβ oligomers
The soluble Aβ40 oligomers were prepared as previously described [27–29] with minor modifications. Briefly, lyophilized Aβ40 peptide powder was dissolved in 1% (w/v) NH4OH to prepare a stock solution of 231 mM. The stock was diluted to 100 μM solution using serum-free RPMI-1640 medium and was aliquoted and stored in a −80°C freezer. Aβ40 solution was incubated at 4°C for 24 h prior to use.
Human Brain Specimens
A total of 11 formalin-fixed and paraffin-embedded human brain tissue blocks were obtained from the Carroll A. Campbell, Jr. Brain Bank at the Medical University of South Carolina (MUSC). The specimens included 6 brain tissue blocks isolated from AD patients and another 5 brain samples isolated from age-matched non-demented control subjects (Table 1). Tissue sections (6-μm-thick) were prepared by the Biorepository Core facility of Hollings Cancer Center at MUSC.
Table 1.
Demographic data and clinical characteristics of the brain specimens used for this study
| Subject ID | Category | Age | Gender | ApoE Genotype | Clinical Diagnosis | Braak stage |
|---|---|---|---|---|---|---|
| HB82 | Control | 85 | F | E3/E3 | Stroke | 0 |
| HB76 | Control | 63 | F | E3/E3 | HTN | 0 |
| HB7 | Control | 80 | F | E3/E3 | Cancer | 0 |
| HB80 | Control | 81 | M | E3/E3 | Cancer | 0 |
| HB77 | Control | 81 | F | E3/E3 | Stroke | 0 |
| HB135 | AD | 87 | F | Unknown | AD | IV/VI |
| HB140 | AD | 90 | M | Unknown | AD | V/VI |
| HB138 | AD | 77 | M | Unknown | AD | V/VI |
| HB186 | AD | 76 | M | Unknown | AD | V/VI |
| HB122 | AD | 78 | F | E3/E3 | AD | V/VI |
| HB112 | AD | 59 | F | E3/E4 | AD | II/III |
Confocal microscopy
Immunofluorescent staining and confocal microscopic assays were performed to determine IL-6 and p-p38 levels in microglia in the brain tissues of AD patients. Briefly, tissue sections were deparaffinized using xylene and rehydrated in gradient ethanol. Antigen retrieval was achieved by boiling with 10 mM Sodium Citrate buffer (pH 6.0). Then tissue sections were permeabilized with 0.3% Triton X-100 for 15 min and blocked with 5% goat serum in 0.1% Triton X-100/PBS at room temperature (RT) for 1 h. Primary antibodies chicken anti-IBA1 (1:1000, SYSY, #234009), rabbit anti-human IL-6 (1:200, Cell Signaling, #12153) and mouse anti-phosphorylated p38 (1:200, Cell Signaling, #58970) were incubated overnight at 4°C. Secondary antibodies Alexa Fluor-594 labeled goat anti-chicken IgG (1:500, Invitrogen, #A10680), Alexa Fluor-488 labeled goat anti-rabbit IgG (1:500, Invitrogen, #A10680) and Alexa Fluor-647 labeled goat anti-mouse IgG (1:500, Invitrogen, #A21244) were incubated at RT for 2 h. Nuclei were counterstained with DAPI and slides were mounted with anti-fade reagent (Invitrogen, P36930). Images were captured and analyzed using an LSM 880 Airyscan super-resolution confocal microscope (Carl Zeiss, Oberkochen, Germany). Mean fluorescence intensity (MFI) was quantified using ImageJ software (NIH). Area of IL-6 or p-p38 positive staining was measured using the Analyze Particles plugin of ImageJ.
Cell Culture
The HMC3 human microglia, Raw 264.7 mouse macrophage and THP-1 human monocyte cell lines were obtained from ATCC. HMC3 cells were cultured using EMEM medium containing 10% FBS, 2 mM L-glutamine and 100 μg/mL of penicillin-streptomycin (Invitrogen). The BV-2 microglial cell line was generously provided by Dr. Sanjay Maggirwar (University of Rochester Medical Center, Rochester, NY, USA). THP-1, BV-2 and Raw 264.7 cells were cultured with RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine and 100 μg/mL of penicillin-streptomycin (Invitrogen). All the cells were cultured in a humidified incubator with 5% CO2 and 95% air at 37°C.
Cell viability assay
The MTT assays were performed to determine cell viability. Briefly, HMC3 cells were seeded into 96-well plates at a density of 3,000 cells per well. One day after cell plating, cells were treated with various concentrations of Aβ40 or Scr- Aβ40 for 48 h. Then MTT reagent was added to the medium (0.5 mg/mL, final concentration) and incubated at 37°C, 5% CO2 for 4 h. The medium was removed and 100 μl DMSO was added to each well to dissolve formazan precipitate thoroughly for 15 min. The absorbance at 570 nm and 650 nm was read using a Synergy H1 microplate reader (BioTek).
CD68 Immunofluorescence
CD68 expression levels were assessed using immunofluorescence and confocal microscopy. Briefly, HMC3 cells were cultured in 4-well CELLview™ dishes (Greiner Bio-One North America) and treated with Aβ40 or Scr-Aβ40 for 48 h. Cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min. Next, cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) for 5 min and then incubated with blocking buffer (5% goat serum and 0.1% Triton X-100 in PBS) at RT for 1 h. Cells were incubated with mouse anti-human CD68 (1:100, ThermoFisher Cat# 14-0688-82, clone: KP1) and rabbit anti-Iba1 (1:500, FUJIFILM, #019–19741) antibodies overnight at 4°C. Then, cells were incubated with Alexa Fluor-488-labeled goat anti-rabbit (1:500, Invitrogen, #A11008) and Alexa Fluor-647-labeled goat anti-mouse (1:500, Invitrogen, #A32728) secondary antibodies at RT for 2 h. Nuclei were counterstained with 1 μg/mL DAPI, and cells were mounted with ProLong Gold anti-fade reagent (Invitrogen, Cat# 36930). Images were captured and analyzed using an LSM 880 Airyscan super-resolution confocal microscope (Carl Zeiss, Oberkochen, Germany). Mean fluorescence intensity (MFI) was quantified using ImageJ (NIH).
Real-time reverse transcriptase-PCR (RT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen). Quality and quantity of RNA samples were assessed using a Synergy H1 microplate reader (BioTek). First-strand cDNA was synthesized from 1 μg of total RNA using an iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. The PCR primers used for this study are listed in Table 2. Expression levels of human IL-6, IL-1β and TNF-α mRNA were measured using iTaq Universal SYBR® Green Supermix (Bio-Rad) on a LightCycler® 480 System (Roche). GAPDH expression levels were used as an internal reference to calculate the changes in target mRNA expression levels using the 2−ΔΔCT method as described previously [30].
Table 2.
PCR Primers used in this study
| Genes | Forward primers | Reverse primers |
|---|---|---|
| IL-6 | 5′-GAAGGCAGCAGGCAACAC-3′ | 5′-TGAACTCCTTCTCCACAAGCG-3′ |
| IL-1β | 5′-AACCTCTTCGAGGCACAAGG-3′ | 5′-AGCCATCATTTCACTGGCGA-3’ |
| TNF-α | 5′-CTTCTGCCTGCTGCACTTTG-3’ | 5′-GTCACTCGGGGTTCGAGAAG-3′ |
| GAPDH | 5′-GACAGTCAGCCGCATCTTCT-3′ | 5′-GCGCCCAATACGACCAAATC-3′ |
Western blotting
Western blot analyses were performed as previously described [31]. Briefly, protein samples were prepared using cell lysis buffer (Cell Signaling) supplemented with a cocktail of proteinase inhibitors (Sigma). Protein concentrations were quantified using a Bio-Rad Dc protein assay kit (Bio-Rad Laboratories, Hercules, CA). Proteins (30 μg) were resolved on 4 – 20% Mini-Protean TGX gels (Bio-Rad) and transferred onto 0.2 μm PVDF membrane (Millipore). Blots were blocked with 5% non-fat milk at RT for 1 h before incubating with primary antibodies at 4°C overnight. After extensive washing with TBS-T, blots were incubated with appropriate HRP-conjugated secondary antibodies at RT for 1.5 h. Protein bands were detected using an ECL Plus Western Blotting Detection System (GE Healthcare Life Science) and an Odyssey Fc Imaging System (LI-COR Biosciences).
Cytokine measurements
Enzyme-linked immunosorbent assay (ELISA) was employed to measure pro-inflammatory cytokines in supernatants of HMC3 and THP-1 cultures. HMC3 cells were seeded in 12-well plates at 60,000 cells per well, while THP-1 cells were seeded in 12-well plates at 300,000 cells per well. One day after cell plating, cells were treated with Aβ40 in serum-free medium with or without p38 inhibitors. Cell culture supernatants were collected, and cells debris were removed by centrifuging at 4000 rpm for 5 min. IL-6 and TNFα concentrations were measured using a human IL-6 DuoSet ELISA kit (DY206) and a human TNF-α DuoSet ELISA kit (DY210), respectively, according to the manufacturer’s instructions (R&D Systems).
Mitochondrial ROS Analysis
Mitochondrial ROS (mROS) levels were determined using a published protocol [32]. Briefly, HMC3 cells were cultured in 4-well CELLview™ cell culture dishes (Greiner Bio-One, North America). One day after cell plating, cells were treated with Aβ40 (2 μM) for 6 h. Then, cells were incubated with 2.5 μM MitoSox Red probe in serum-free EMEM medium at 37°C for 30 minutes. Nuclei were counterstained with DAPI. Images were captured using an LSM 880 laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany). The levels of mROS florescence intensity were analyzed using ImageJ software (NIH).
Flow cytometric analysis
MitoSox Red staining and flow cytometric assays were performed to detect mitochondrial superoxide (mROS) levels using a published protocol [33]. Briefly, THP-1 cells were treated with various doses of Aβ40 for a duration as indicated in the figure legends. Then, cells were incubated with serum-free RPMI-1640 medium containing 2.5 μM MitoSox Red at 37°C for 30 min. The levels of mROS were analyzed by measuring the mean fluorescence intensity (MFI) of MitoSox using a CytoFLEX Flow Cytometer (Beckman Coulter, Brea, CA).
Nitric oxide assay
Levels of nitric oxide (NO) were evaluated using a Griess nitrite measurement kit (Thermofisher, MA). Briefly, HMC3 and Raw 264.7 cells were seeded in 12-well plates at 60,000 cells and 200,000 cells per well, respectively. Cells were then stimulated with LPS (0.1 μg/mL) or Aβ40 (5 μM) for 24 h. Equal volumes of supernatants and Griess reagent were mixed and incubated in 96-well plates at RT for 30 min. Then the absorbance was measured at 548 nm using a Synergy H1 microplate reader (BioTek). NO concentrations in cell culture supernatants were calculated using the sodium nitrite standard curve.
Phagocytosis assay
HMC3 and BV-2 cells were plated in 4-well CELLview™ cell culture dishes (Greiner Bio-One North America) at 1 × 104 and 3 × 104 cells/well, respectively. One day after cell plating, cells were pretreated with 2 μM BIRB for 1 h prior to incubation with 1 μM fluorescence-labeled FAM-488-Aβ42 at 37°C for 2 h. Cells were then washed with PBS to remove the non-phagocytized Aβ peptides and fixed with 4% paraformaldehyde. Cells were then incubated with blocking buffer (5% goat serum, 0.1% Triton X-100 in PBS) for 1 h and followed with rabbit anti-Iba1 (1:500, FUJIFILM, #019–19741) at 4°C overnight. Alexa Fluor-647-labeled goat anti-rabbit (1:500, Invitrogen, #A21244) secondary antibody was incubated for 2 h to visualize Iba1 staining. Slides were mounted with anti-fade reagent (Invitrogen, P36930). Images were captured and processed using a Zeiss LSM 880 laser scanning confocal microscope. Imaging data were analyzed using ImageJ software (NIH).
Statistical analysis
Data is presented as mean ± SEM. Comparisons between two groups were carried out using Student’s t-test. Multiple comparisons were performed using one-way analysis of variance (ANOVA). Differences were considered statistically significant at p < 0.05. All analyses were carried out using GraphPad Prism software (GraphPad Software, Inc. San Diego, CA).
Results
Aβ40 stimulates IL-6 production in HMC3 human microglial cells
We investigated the effects of Aβ40 on IL-6 expression in HMC3 cells. The data showed that Aβ40 treatment resulted in a dose-dependent increase of IL-6 production in HMC3 cells (Fig. 1A, Supplemental Fig. S1A). In contrast, treatment with an equal amount of Scr-Aβ40 did not significantly affect IL-6 production in HMC3 cells (Fig. 1B). We also determined the effects of Aβ40 treatment on the expression of other inflammatory mediators, including several M1 and M2 markers, in HMC3 cells. Our data indicate that treatment with Aβ40 increases iNOS expression but has no significant effects on TGF-β expression. In contrast, LPS treatment led to a marked increase in expression levels of iNOS and TGF-β. Moreover, our data showed that IL-10 levels were not detectable in HMC3 cells (Supplemental Fig. S1B). Additionally, we also examined the impact of Aβ on human microglial cell proliferation. The results indicated that treatment with Aβ40 slightly inhibited the proliferation of HMC3 cells, whereas Scr-Aβ40 did not show any such effects (Supplemental Fig. S1C).
Fig. 1. Aβ40 dose-dependently stimulates IL-6 production in human microglia.

(A, B) HMC3 cells were treated with various doses of Aβ40 or Scr-Aβ40, brefeldin A (0.5 μg/mL) was added to the medium during the last 4 h of culture. IL-6 levels were assessed by immunoblotting at 8 h after treatment. Tubulin was probed as loading control. (C) IL-6 Levels in supernatants of HMC3 cells were determined using ELISA at 8 h after Aβ40 treatment. Data indicates that Aβ40 dose-dependently stimulates IL-6 secretion. (D) Time course studies depict that Aβ40 (2 μM) promotes IL-6 secretion in a time-dependent manner. (E) HMC3 cells were treated with 2 μM Aβ40 or Scr-Aβ40. Immunofluorescence and confocal microscopic assays were performed to assess CD68 expression levels. Representative images of confocal microscopic analysis of CD68 expression are shown. (F) Mean fluorescence intensity (MFI) of CD68 immunostaining was quantified using ImageJ software and the results show that Aβ40 increases CD68 expression in HMC3 cells. Data are presented as mean +/− SEM of three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001.
To further determine if Aβ40 affects IL-6 secretion from human microglia, we measured IL-6 levels in cell culture supernatants using ELISA assays. The results showed that Aβ40 stimulated the secretion of IL-6 by HMC3 cells in a dose- and time-dependent fashion (Fig. 1C & D). Given that the increased production of inflammatory cytokines is a well-documented feature of microglial activation [3, 4], these new findings indicate that Aβ40 stimulation may activate HMC3 human microglia. CD68 is thought to be a marker of activated microglia [34, 35], which prompted us to determine the influence of Aβ40 on CD68 expression. As shown in Fig. 1E & F, immunofluorescence and confocal microscopic assays unveil that treatment with Aβ40, but not Scr-Aβ40, significantly increases CD68 expression levels in HMC3 cells.
Treatment with Aβ40 promotes the transcription of IL-6 and TNF mRNAs in HMC3 cells
To further determine whether Aβ40 affects the expression of other pro-inflammatory cytokines, we analyzed the expression levels of IL-6, TNFα and IL-1β mRNAs in HMC3 cells at various time points after Aβ40 treatment. Our data show that Aβ40 treatment increases the transcription of both IL-6 and TNFα mRNAs in a time-dependent fashion (Fig. 2A & B). The peak for Aβ-induced up-regulation of IL-6 and TNFα mRNA expression was at 3 h after treatment. Although there was an increase in TNFα transcription following Aβ40 treatment, we had difficulty in measuring TNFα cytokine in supernatant using ELISA, which is consistent with a recent report showing that HMC3 cells are not efficient at secreting TNFα [36]. Furthermore, we did not observe any significant changes in IL-1β mRNA expression levels in response to Aβ40 treatment (Fig. 2C). This result is consistent with a previous report indicating that IL-1β expression was not detectable in HMC3 cells [37].
Fig. 2. Aβ40 promotes the transcription of IL-6 and TNF-α in HMC3 cells.

HMC3 cells were treated with Aβ40 (2 μM) and RNA samples were prepared at different time points after treatment. mRNA expression levels for IL-6 (A), TNF-α (B) and IL-1β (C) were determined using real-time RT-PCR assays and were normalized to fold change (FC) relative to that in control cells. Data are presented as mean +/− SEM of three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001.
Aβ40 treatment stimulates the transcription of IL-6, TNFα and IL-1β in THP-1 cells
Since microglia are the resident monocytes/macrophage of the central nervous system [38], we decided to examine the effects of Aβ40 on the transcription of IL-6, TNFα and IL-1β mRNAs in THP-1 human monocytes. Quantitative RT-PCR analyses revealed that treatment with either LPS or Aβ40 leads to a significant increase in IL-1β, IL-6, and TNFα mRNA levels in THP-1 cells (Fig. 3A–C). Furthermore, we assessed the protein levels of IL-6 and TNFα in supernatants of THP-1 cell cultures. The results demonstrate that Aβ40 dose-dependently stimulates the secretion of IL-6 and TNFα by THP-1 cells (Fig. 3D & E).
Fig. 3. Aβ40 increases expression levels of IL-6, TNF-α and IL-1 mRNAs in THP-1 cells.
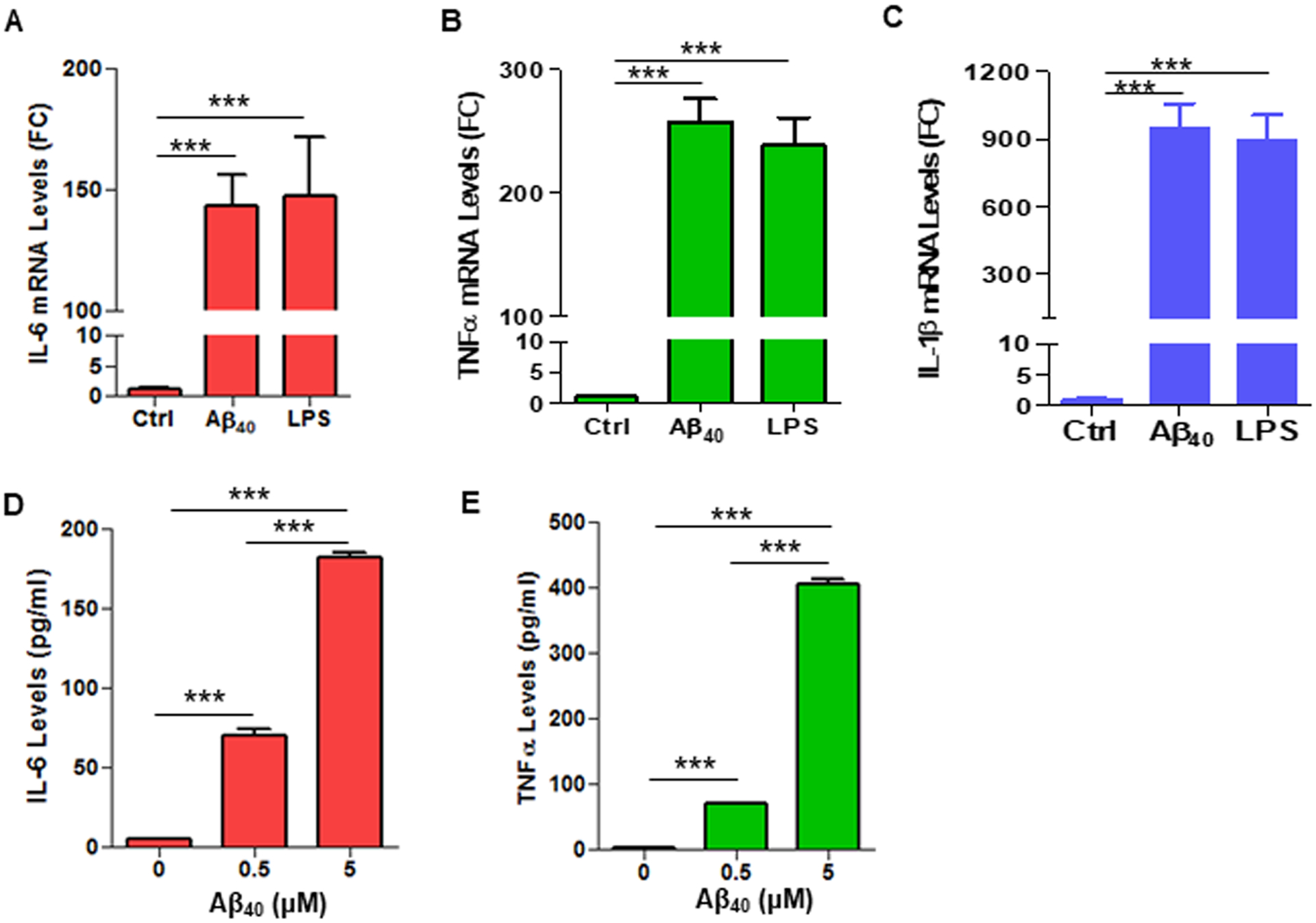
THP-1 cells were treated with Aβ40 (2 μM) or LPS (0.1 μg/mL) and mRNA expression levels for IL-6 (A), TNF-α (B) and IL-1β (C) were determined using real-time RT-PCR. (D, E) THP-1 cells were treated with indicated doses of Aβ40. Protein levels of IL-6 (D) and TNF-α (E) in supernatants of THP-1 cell cultures were analyzed using ELISA. Data are presented as mean +/− SEM of three independent experiments. *** p < 0.001.
Aβ40 selectively induces p38 activation in HMC3 and THP-1 cells
The p38 signaling pathway has been implicated in modulating stress and inflammatory responses in various model systems [39–41]. However, the role of p38 in regulating the production of IL-6 in human microglia has yet to be determined. Our data indicated that Aβ40 treatment selectively activated p38, but not the ErK and JNK, MAPK signaling pathways, in HMC3 cells (Fig. 4A). The inability of Aβ40 to induce JNK activation was further confirmed by the observation that UV exposure strongly induced JNK phosphorylation in 293T cells, whereas Aβ40 treatment had no significant effects on JNK phosphorylation in HMC3 cells (Fig. 4B). Interestingly, we found that exposure to UV significantly increased the levels of phosphorylated JNK (Supplemental Fig. S2), suggesting that UV exposure may activate the JNK pathway in HMC3 cells.
Fig. 4. Aβ40 selectively induces p38 activation in HMC3 and THP-1 cells.

(A) HMC3 cells were treated with indicated doses of Aβ40 for 4 h. Expression levels of p-p38, p38, p-Erk, Erk, p-JNK, and JNK were assessed by immunoblotting. (B) UV irradiated 293T cells were used as a positive control for p-JNK immunoblotting. The data indicate that Aβ40 selectively induces p38, but not Erk and JNK, activation in HMC3 cells. (C) Immunoblotting assays indicate that Aβ40 (5 μM) time-dependently stimulates p38 activation in HMC3 cells. (D) Immunoblotting results show that Aβ40 induces p38 activation in THP-1 cells in a dose- and time-dependent fashion. Data represent two independent experiments with similar results.
In addition, our data showed that Aβ40 induced p38 activation in a dose- and time-dependent manner and the peak for Aβ40-induced p38 activation was about 2 h after treatment (Fig. 4C). Notably, levels of phosphorylated (the activated form) p38 remain significantly higher even 24 h after Aβ40 treatment (Fig. 4C), suggesting that Aβ40 treatment may result in a sustained p38 activation in human microglia. In contrast, phosphorylated p65 (p-p65) levels were only transiently and slightly increased at 2 h after Aβ treatment (Fig. 4C). These findings suggest that p38 signaling, but not the NF-κB signaling pathway may play a major role in modulating Aβ-induced increases in the production of pro-inflammatory cytokines such as IL-6 and TNFα.
Next, we investigated whether Aβ40 influences the activation of p38, Erk and JNK kinases in macrophages. Our data showed that Aβ40 dose-dependently stimulated the activation of the p38 signaling pathway, but not the Erk and JNK pathways in THP-1 cells (Fig. 4D). Furthermore, a time course study indicated that Aβ40 stimulated p38 activation within 30 min after treatment, the peak of Aβ40-induced p38 activation was approximately at 1 h after treatment and this activation was persistent for more than 24 h (Fig. 4D). Taken together, our data demonstrate for the first time that Aβ40 dose-dependently stimulates p38 activation in both HMC3 and THP-1 cells.
Inhibition of p38 abrogates the Aβ40-induced increase in IL-6 secretion
Based on the observation that Aβ40 selectively stimulates p38 activation (Fig. 4), we hypothesized that inhibition of p38 activation could attenuate Aβ-induced neuroinflammatory response. To test this hypothesis, we investigated if pharmacologic inhibition of p38 by BIRB 796 (BIRB), a potent p38 inhibitor [42], could ameliorate Aβ40-induced increase of IL-6 production. The results indicate that BIRB dose-dependently inhibits Aβ40-induced p38 activation in HMC3 cells (Fig. 5A). Our data also show that pre-incubation with BIRB prevents Aβ40-induced p38 activation in THP-1 cells (Fig. 5B). Furthermore, immunoblotting assays demonstrate that inhibition of p38 by BIRB abrogates Aβ-induced up-regulation of IL-6 in HMC3 cells (Fig. 5C).
Fig. 5. Inhibition of p38 abolishes Aβ40-induced IL-6 production in HMC3 and THP-1 cells.
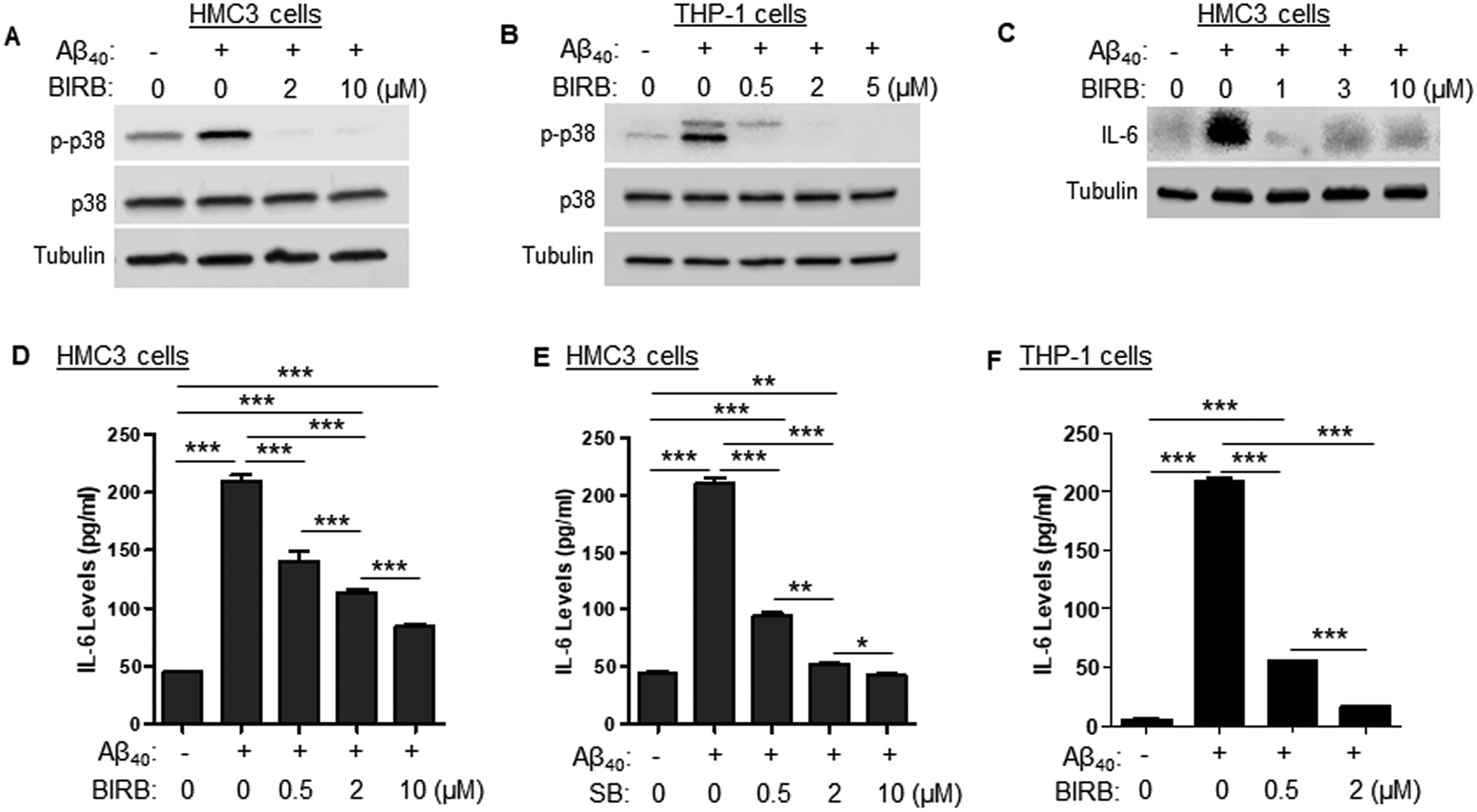
(A) HMC3 cells were pre-incubated with BIRB or DMSO as vehicle control (drug concentration was marked as 0) 30 min prior to Aβ40 (2 μM) treatment. Expression levels of p-p38 and total p38 were determined by Western blotting at 4 h after Aβ40 treatment. (B) THP-1 cells were pre-incubated with BIRB 30 min prior to Aβ40 (2 μM) treatment or DMSO. Expression levels of p-p38 and total p38 were determined by immunoblotting at 2 h after Aβ treatment. (C) HMC3 cells were incubated with BIRB or DMSO as vehicle control 30 min prior to Aβ40 (2 μM) treatment. Brefeldin A (0.5 μg/mL) was added to the medium during the last 4 h of culture. IL-6 expression levels were assessed by immunoblotting at 8 h after Aβ40 treatment. (D) ELISA data indicates that BIRB dose-dependently attenuates Aβ-induced increase in IL-6 secretion. (E) ELISA data depicts that SB202190 (SB) dose-dependently inhibits Aβ-induced IL-6 secretion by HMC3 cells. (F) ELISA reveals that BIRB dose-dependently attenuates Aβ-induced increase in IL-6 secretion by THP-1 cells. Data are presented as mean +/− SEM of three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. control.
Next, we investigated the effects of p38 inhibition on Aβ40-induced IL-6 secretion. Levels of IL-6 in cell culture supernatants were analyzed using ELISA. The data reveal that inhibition of p38 by BIRB inhibits the Aβ40-induced increase of IL-6 secretion by HMC3 cells (Fig. 5D). The ability of p38 inhibition to block Aβ40-induced IL-6 secretion was further confirmed by using another structurally and mechanistically different p38 inhibitor, SB202190 (SB) (Fig. 5E). Additionally, we show that inhibition of p38 by BIRB dose-dependently inhibits Aβ40-induced secretion of IL-6 by THP-1 cells (Fig. 5F). Collectively, these results demonstrate that inhibition of p38 by SB or BIRB is sufficient to abolish Aβ40-induced increase in IL-6 secretion.
Aβ40-induced p38 activation is associated with increased mROS production
It has been shown that mitochondria and the generation of mitochondrial ROS (mROS) are critical to T cell activation and LPS-induced pro-inflammatory response in mouse microglia [43, 44]. However, the role of mROS in Aβ40-induced p38 activation in human microglia remains largely unknown. We thus examined the effects of Aβ40 treatment on mROS production. Our data indicate that Aβ40 treatment markedly increases the generation of mROS in THP-1 cells in a time- and dose-dependent manner (Fig. 6A–C). Furthermore, using confocal microscopy, we found that Aβ40 treatment significantly increases the levels of mROS in HMC3 microglial cells (Fig. 6D & E). Together with the observation that Aβ40 treatment selectively induces p38 activation in both HMC3 and THP-1 cells (Fig. 4), our studies demonstrate that Aβ40-induced p38 activation is associated with an increase in mROS production. These findings are consistent with previous reports showing that oxidative stress is a potent inducer of p38 activation, and that oxidative stress-induced p38 activation has been implicated in Aβ- and tau-induced toxicities in neurons [45–49]. Furthermore, it was reported that Aβ can stimulate the production of nitrogen oxide (NO) in rodent glial cells [50–52]. However, our data show that Aβ40 has no significant effects on NO production in HMC3 cells, whereas LPS markedly increases NO production in Raw 264.7 cells (Supplemental Fig. S3).
Fig. 6. Aβ40 stimulates the production of mROS in HMC3 and THP-1 cells.
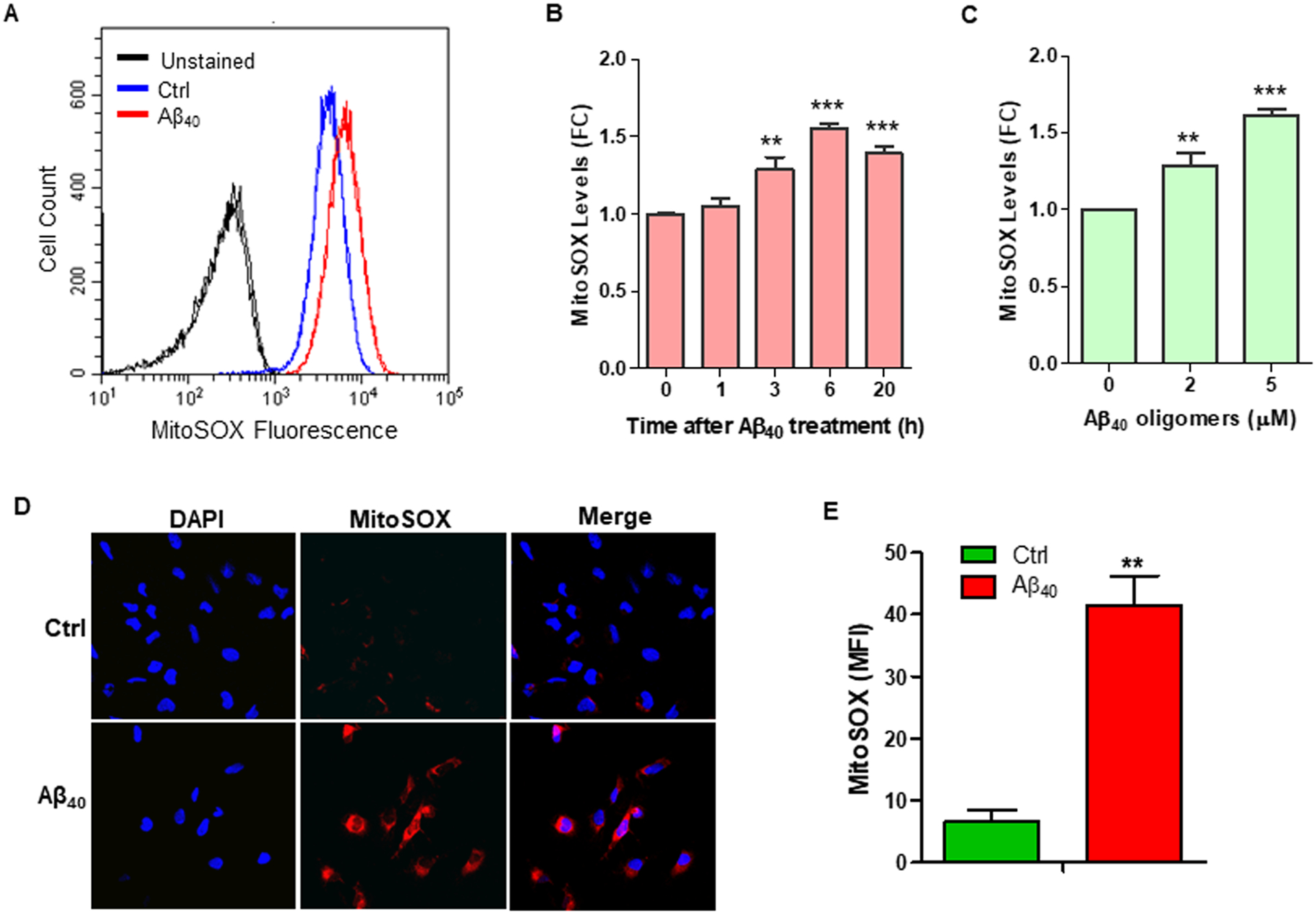
(A) MitoSox staining and flow cytometric assays were performed to measure mROS levels in THP-1 cells at 6 h after Aβ40 (2 μM) treatment. Representative flow cytometric graphs are shown, and the results indicate that Aβ40 increases mROS production in THP-1 cells. (B) Treatment with Aβ40 (2 μM) increases mROS production in THP-1 cells in a time-dependent manner. (C) Aβ40 dose-dependently promotes mROS production in THP-1 cells. (D) MitoSox staining and confocal microscopy were employed to measure mROS in HMC3 cells at 6 h after Aβ40 (2 μM) treatment. (E) Mean fluorescence intensity (MFI) of MitoSOX staining was quantified using ImageJ software and the quantified results are graphed. Data are presented as mean +/− SEM of three independent experiments. ** p < 0.01, *** p < 0.001 vs. control.
Inhibition of p38 diminishes Aβ phagocytosis by microglia
Microglia were thought to be a major neural cell type involved in clearing of Aβ deposition in the brain [53, 54]. However, whether p38 is involved in regulating the phagocytosis of Aβ in human microglia has not been studied previously. Our confocal microscopic data indicate that blocking of p38 by BIRB markedly inhibits the phagocytosis of Aβ by HMC3 cells (Fig. 7A & B). Similar results were observed using BV-2 mouse microglial cells (Fig. 7C & D). These results demonstrate a role of p38 in modulating the phagocytosis of Aβ by microglia, suggesting that BIRB-mediated blockade of Aβ uptake may be a possible mechanism by which p38 inhibition attenuates Aβ-induced IL-6 production in HMC cells.
Fig. 7. Inhibition of p38 attenuates the phagocytosis of Aβ by microglia.
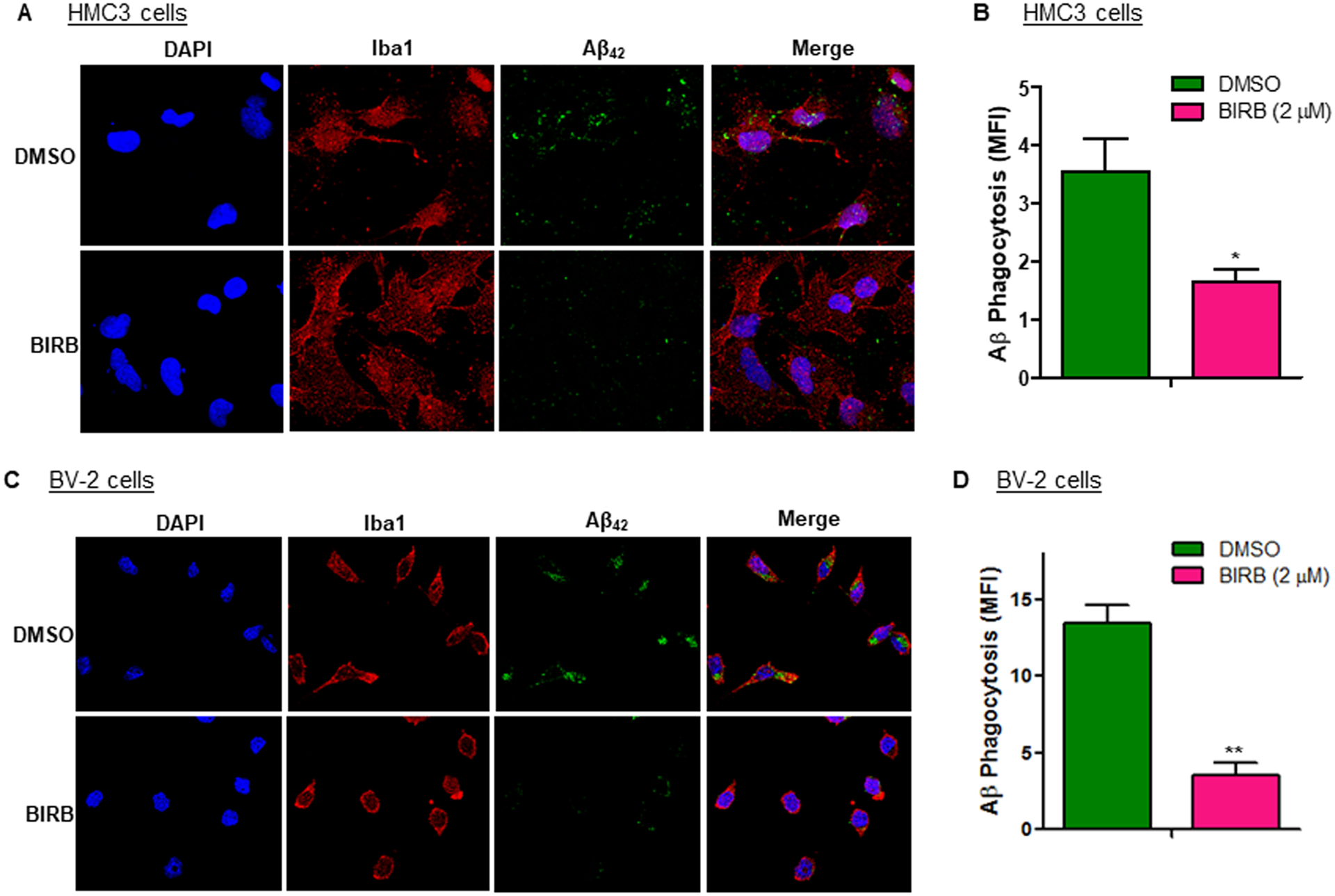
(A) HMC3 cells were treated with BIRB (2 μM) at 1 h before incubation with 1 μM FAM-488 labeled-Aβ42 for 2h. Cells were fixed with 4% paraformaldehyde and stained with an IBA1-specific antibody to label microglia. Nuclei were counterstained with DAPI. Levels of Aβ42 uptake were assessed using confocal microscopy. (B) Confocal imaging data were analyzed and quantified using ImageJ. Levels of Aβ phagocytosis were presented as MFI of FAM-488 labeled-Aβ42. (C) BV-2 cells were treated and analyzed using the same protocol as described in A. Shown are representative confocal microscopic images of Aβ phagocytosis. (D) Confocal imaging data were analyzed and quantified using ImageJ. Levels of Aβ phagocytosis were presented as MFI of FAM-488 labeled-Aβ42. Data are presented as mean +/− SEM of three independent experiments. * p < 0.05, ** p < 0.01 vs. DMSO control.
IL-6 colocalized with phosphorylated p38 in human microglia of AD patients
It was reported that IL-6 expression levels were elevated in the cortices of AD patients [10, 11]. However, the cellular sources for the increased IL-6 in AD remain to be determined. Using immunofluorescence and Airyscan super-resolution confocal microscopy, we investigated if IL-6 expression levels were altered in microglia of AD patients. Consistent with the observation of a recently published study [55], we confirmed that the morphologic characteristics of human microglia were markedly altered in the cortices of AD patients, which displayed the pathological feature of shortened and de-ramified processes along with discontinuity and puncta IBA1 immunostaining (Fig. 8A). Quantification of the confocal microscopic imaging data demonstrated that both IL-6 and phosphorylated p38 (p-p38) expression levels were dramatically increased in microglia of AD patients compared with those in age-matched control subjects (Fig. 8B). Transmembrane protein 119 (TMEM119) is a cell-surface protein highly expressed in both mouse and human microglia and is thought to be a microglia-specific marker [56]. Using a TMEM-specific antibody, we further confirmed that most microglia, especially those with de-ramified and amoeboid characteristics of activated microglia, expressed high levels of IL-6. In contrast, a few ramified microglia with smaller size did not show immunoreactivity for IL-6 (Supplemental Fig. S4). Our studies also included the secondary antibody only controls to test the specificity of the antibodies (Supplemental Fig. S5).
Fig. 8. Increased expression and co-localization of IL-6 with p-p38 in microglia in the cortices of AD patients.

(A) IL-6 and IBA1 co-labeling and confocal microscopy were performed to assess IL-6 expression levels in microglia in human brain tissues isolated from AD patients and age-matched control subjects. (B) Quantification of confocal imaging data unveiled that the fluorescent intensity and positively stained area for IL-6 and p-p38 were substantially higher in microglia from AD patients than those from age-matched control subjects. (C) Representative confocal microscopic images depict the elevated expression and co-localization of IL-6 with p-p38 in human AD microglia. Scale bar = 20 μm. * p < 0.05, ** p < 0.01, *** p < 0.001 vs. control subjects (Ctrl).
Although activated microglia are considered as the major source of inflammatory cytokines, it is worth noting that astrocytes and microglia can work collaboratively to regulate neuroinflammation in AD [3]. Moreover, there are conflicting reports regarding whether astrocytes produce IL-6 or not [57, 58]. To examine if reactive astrocytes express IL-6 in AD brains, we performed a confocal microscopic analysis of AD patient samples. Consistent with a recently published observation [58], we found that a portion of the astrocytes showed immunoreactivity for IL-6 in the brain tissues of AD patients (Supplemental Fig. S6).
To determine the role of the p38 pathway in regulating IL-6 expression in AD microglial cells, we investigated if there is a link between increased IL-6 levels and the activated form of p38 protein in postmortem brain specimens isolated from AD patients. Our data have confirmed the colocalization of IL-6 with p-p38 in microglia in the cortices of AD patients (Fig. 8C). Taken together, the results support the hypothesis that p38 may play a major role in modulating IL-6 production in microglia of AD patients.
Discussion
AD and AD-related dementias (ADRD) affect more than 6 million senior Americans aged 65 or older, but effective therapies have yet to be developed [1, 2]. One of the major roadblocks toward the success of AD prevention and treatment is that we still do not fully understand the pathogenic mechanisms of this devastating disease. The chronic deposition of Aβ peptides in senile plaques stimulates a sustained inflammatory response, leading to a persistent increase in the production of pro-inflammatory cytokines in the AD brain, which is referred to as neuroinflammation [3]. Since pro-inflammatory cytokines have been shown to affect the neuropathology and cognitive function in neurodegenerative disease animal models, Aβ-induced neuroinflammation is thought to be an important contributing factor to the pathogenesis of AD [10–13]. In agreement with this concept, it has been shown that increased levels of inflammatory cytokine production can cause synaptic dysfunction, neuronal death, and neurodegenerative disorders [6, 7, 10, 14]. Notably, IL-6 expression levels are increased in the brains of AD patients [10, 11]. Moreover, a growing body of evidence suggests that increased levels of IL-6 production may play a causal role in mediating inflammatory stress-induced neurodegenerative pathologies and cognitive declines [6, 11, 14]. However, the mechanisms by which IL-6 expression is regulated in brain tissues of AD patients have not been clearly elucidated. In the present study, we show that Aβ40 dose-dependently stimulates IL-6 production in HMC3 human microglial cells. We also show that Aβ treatment promotes the transcription of IL-6 and TNFα mRNAs in both HMC3 and THP-1 cells. Furthermore, our data reveal that Aβ selectively induces p38 activation and pharmacological inhibition of p38 by BIRB or SB abrogates the Aβ-induced increase in IL-6 secretion. These results suggest that activation of the p38 signaling pathway may play a critical role in Aβ-induced IL-6 production in human microglia.
Microglial activation has been found in the pre-plaque stage in transgenic animal models of AD [59, 60]. Reactive microglia were also observed in the brains of AD patients and were colocalized with Aβ plaques [61, 62]. The sequential emergence of amyloid accumulation and microglial activation and release of cytokines suggests that microglia may play a significant role in mediating Aβ-induced neural cell death, neuroinflammation and subsequent tau pathology, all of which are involved in AD pathogenesis. Mitochondrial ROS (mROS) can function as signaling intermediates and are required for T cell activation [43]. Moreover, the production of mROS is required for LPS-induced inflammatory response in mouse microglia [44]. Nevertheless, the role of mROS in Aβ-induced p38 activation and IL-6 production in human microglia has not been explored previously. Our data reveal that Aβ40 treatment increases the levels of mROS in both THP-1 and HMC3 cells. These results demonstrate a link between Aβ40-induced p38 activation and the increased production of mROS in human microglial cells. Consistent with these findings, previous studies indicated that oxidative stress could induce p38 activation [45, 46].
The accumulation of Aβ plaques in the brain is thought to be a critical and initial event in the pathogenesis of AD, which is the main concept of the Aβ hypothesis of AD [63]. However, therapeutics that block Aβ-induced neuroinflammation have yet to be developed. Recently, the anti-human Aβ antibody, Aduhelm (aducanumab), was granted a fast-track approval by the U.S. Food and Drug Administration (FDA), for AD treatment. AD patients who received aducanumab treatment displayed a dose- and time-dependent reduction of Aβ plaques, while patients in the control arm did not show such changes [64, 65]. These results not only support the Aβ hypothesis of AD, but also suggest that targeting Aβ by Aduhelm is a viable therapeutic approach for AD treatment. Although p38 has been found to be a key mediator of various stress responses [39–41], the role of p38 in modulating IL-6 production in human microglia was largely unknown prior to our present study. Our data demonstrate that activation of p38 is critical for Aβ40-induced increase of IL-6 production in human microglia, suggesting that blocking of p38 may represent an alternative therapeutic approach for AD via targeting the downstream effector of the Aβ-induced pathogenic process. In agreement with this, there is evidence that p38 is involved in regulating Aβ-induced synaptic dysfunction and that inhibition of p38 ameliorates tau pathology, leading to decreases in hyperphosphorylated tau levels in microglia [66–69]. Moreover, it has been shown that genetic depletion of p38 attenuates AD pathology in a mouse model of AD [69].
Although previous studies have shown that IL-6 levels were elevated in the brains of AD patients [10, 11], it remains unclear about the exact brain cell types where the elevated levels of IL-6 come from. More importantly, a link between the elevated IL-6 expression levels and increased immunostaining for the activated form of p38 (p-p38) in microglia in the brains of AD patients has yet to be established. In the present study, we found that the immunoreactivities for both IL-6 and p-p38 were markedly higher in microglia from AD patients than those from age-matched control subjects. More importantly, our studies have demonstrated for the first time that IL-6 colocalize with p-p38 in microglia in the cortices of AD patients.
The role of p38 signaling in Aβ-induced alterations in neuroinflammatory cytokines such IL-6 in human microglia was largely unknown prior to the present study. This is an important knowledge gap because it has been clearly shown that there are species-specific differences in gene expression patterns between mouse and human microglia [70]. Our data show that treatment with Aβ40 leads to persistent activation of p38 in HMC3 human microglia. In contrast, only a transient increase of phosphorylated p65 was observed in HMC3 cells following Aβ40 treatment. These results suggest that p38 is likely a key regulator of Aβ-induced neuroinflammatory response in human microglia. In agreement with this idea, our data have confirmed that pharmacological inhibition of p38 by SB or BIRB is sufficient to abrogate Aβ-induced increase in IL-6 production in HMC3 cells.
In summary, our studies offer the first experimental evidence that Aβ40 stimulates IL-6 production in human microglia in a dose- and time-dependent fashion. The Aβ40-induced increase in IL-6 production is associated with the activation of the p38 pathway. Inhibition of p38 abrogates Aβ40-induced increase in IL-6 production. An analysis of clinical brain specimens revealed that the immunoreactivities for IL-6 and p-p38 were markedly higher in microglia of AD patients than in age-matched control subjects. Moreover, our studies have confirmed the co-localization of IL-6 with p-p38 in microglia in the cortices of AD patients. These results demonstrate a critical role for p38 in mediating Aβ-induced neuroinflammatory response such as increased IL-6 production from microglia in AD.
Supplementary Material
Acknowledgements
The authors thank Dr. Yuan Shao in the Biorepository Core facility at the Medical University of South Carolina (MUSC) for assistance with the preparation of brain tissue sections. We thank Dr. Sandeep Kumar Mishra for excellent technical assistance. The authors also wanted to thank the Cell and Molecular Imaging Shared Resource at MUSC for technical assistance with confocal imaging. The Cell and Molecular Imaging Core at MUSC is supported in part by MUSC Cancer Center Support Grant (P30 CA138313), the SC COBRE in Oxidants, Redox Balance, and Stress Signaling (P20 GM103542), and the NIH Shared Instrumentation Grants S10 OD018113 and S10 OD028663.
JJ was supported by the Distinguished Experts Special Funds (Grant# 2019B12) and the Medical High Level Talents Training Program (Grant# G202002005) from Guangxi Province in China.
Funding
This study was supported in part by the National Institute on Aging (NIA) of the National Institutes of Health (NIH) under Award Number R01AG068286.
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Ethics approval
The use of human brain specimens for this study was reviewed by the Medical University of South Carolina (MUSC) Institutional Review Board and approved as meeting the “Not Human Research” criteria set forth by the Code of Federal Regulations (45CFR46).
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP (2013) The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9:63–75. [DOI] [PubMed] [Google Scholar]
- 2.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer’s disease. Lancet 388:505–17. [DOI] [PubMed] [Google Scholar]
- 3.Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17:157–172. [DOI] [PubMed] [Google Scholar]
- 4.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL et al. (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26:349–54. [DOI] [PubMed] [Google Scholar]
- 6.Hüll M, Strauss S, Berger M, Volk B, Bauer J (1996) The participation of interleukin-6, a stress-inducible cytokine, in the pathogenesis of Alzheimer’s disease. Behav Brain Res 78:37–41. [DOI] [PubMed] [Google Scholar]
- 7.Nagatsu T, Sawada M (2005) Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des 11:999–1016. [DOI] [PubMed] [Google Scholar]
- 8.Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P et al. (2018) A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun 9: 2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C (2002) The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci 202:13–23. [DOI] [PubMed] [Google Scholar]
- 10.Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M (1991) Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett 285:111–4. [DOI] [PubMed] [Google Scholar]
- 11.Huell M, Strauss S, Volk B, Berger M, Bauer J (1995) Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol 89:544–51. [DOI] [PubMed] [Google Scholar]
- 12.Cojocaru IM, Cojocaru M, Miu G, Sapira V (2011) Study of interleukin-6 production in Alzheimer’s disease. Rom J Intern Med 49:55–8. [PubMed] [Google Scholar]
- 13.Lai KSP, Liu CS, Rau A, Lanctôt KL, Köhler CA, Pakosh M, Carvalho AF, Herrmann N (2017) Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry 88:876–882. [DOI] [PubMed] [Google Scholar]
- 14.Heyser CJ, Masliah E, Samimi A, Campbell IL, Gold LH (1997) Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc Natl Acad Sci U S A 94:1500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283:29615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selkoe DJ (2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192:106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MJ Lemere CA, Regan CM Walsh DM, Sabatini BL Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14:837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baranello RJ, Bharani KL, Padmaraju V, Chopra N, Lahiri DK, Greig NH, Pappolla MA, Sambamurti K (2015) Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr Alzheimer Res 12:32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng W, Achariyar TM, Li B et al. (2016) Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis 93:215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore Z, Mobilio F, Walker FR, Taylor JM, Crack PJ (2020) Abrogation of type-I interferon signaling alters the microglial response to Aβ1–42. Sci Rep 10:3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jana M, Palencia CA, Pahan K (2008) Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol 181:7254–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong L, Jiang X, Zhu Z et al. (2019) Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia 67:498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jian M, Kwan JS, Bunting M, Ng RC, Chan KH (2019) Adiponectin suppresses amyloid-β oligomer (AβO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling pathway. J Neuroinflammation. 16:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bachstetter AD, Xing B, de Almeida L, Dimayuga ER, Watterson DM, Van Eldik LJ (2011) Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Aβ). J Neuroinflammation 8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gee MS, Son SH, Jeon SH, Do J, Kim N, Ju YJ, Lee SJ, Chung EK, Inn KS, Kim NJ, Lee JK (2020) A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment and modulates microglia function in 5XFAD mouse. Alzheimers Res Ther 12:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hellstrand E, Boland B, Walsh DM, Linse S (20100 Amyloid β-protein aggregation produces highly reproducible kinetic data and occurs by a two-phase process. ACS Chem Neurosci 1:13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson RD, Schauerte JA, Wisser KC, Gafni A, Steel DG (2011) Direct observation of single amyloid-β (1–40) oligomers on live cells: binding and growth at physiological concentrations. PLoS One 6: e23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan TM, Caine J, Mertens HD, et al. (2013) Ammonium hydroxide treatment of Aβ produces an aggregate free solution suitable for biophysical and cell culture characterization. Peer J 1: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo H, Yang A, Schulte BA, Wargovich MJ, Wang GY (2013) Resveratrol Induces Premature Senescence in Lung Cancer Cells via ROS-Mediated DNA Damage. PLoS One 8:e60065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang A, Qin S, Schulte BA, Ethier SP, Tew KD, Wang GY (2017) MYC Inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res 77:6641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin S, He X, Lin H, Schulte BA, Zhao M, Tew KD, Wang GY (2021) Nrf2 inhibition sensitizes breast cancer stem cells to ionizing radiation via suppressing DNA repair. Free Radic Biol Med 169:238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P (2007) Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2:2295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopperton KE, Mohammad D, Trépanier MO, Giuliano V, Bazinet RP (2018) Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: a systematic review. Mol Psychiatry 23:177–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumura A, Suzuki S, Iwahara N, Hisahara S, Kawamata J, Suzuki H, Yamauchi A, Takata K, Kitamura Y, Shimohama S (2015) Temporal changes of CD68 and α7 nicotinic acetylcholine receptor expression in microglia in Alzheimer’s disease-like mouse models. J Alzheimers Dis 44:409–23. [DOI] [PubMed] [Google Scholar]
- 36.Hankittichai P, Lou HJ, Wikan N, Smith DR, Potikanond S, Nimlamool W (2020) Oxyresveratrol Inhibits IL-1β-Induced Inflammation via Suppressing AKT and ERK1/2 Activation in Human Microglia, HMC3. Int J Mol Sci 21: 6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dello Russo C, Cappoli N, Coletta I, Mezzogori D, Paciello F, Pozzoli G, Navarra P, Battaglia A (2018) The human microglial HMC3 cell line: where do we stand? A systematic literature review. J Neuroinflammation 15:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benakis C, Garcia-Bonilla L, Iadecola C, Anrather J (2015) The role of microglia and myeloid immune cells in acute cerebral ischemia. Front Cell Neurosci 8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashwell JD (2006) The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol 6:532–40. [DOI] [PubMed] [Google Scholar]
- 40.Morganti JM, Goulding DS, Van Eldik LJ (2019) Deletion of p38α MAPK in microglia blunts trauma-induced inflammatory responses in mice. J Neuroinflammation 16:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA (2009) p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pargellis C, Tong L, Churchill L et al. (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol 9:268–72. [DOI] [PubMed] [Google Scholar]
- 43.Sena LA, Li S, Jairaman A et al. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park J, Min JS, Kim B, Chae UB, Yun JW, Choi MS, Kong IK, Chang KT, Lee DS (2015) Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett 584:191–6. [DOI] [PubMed] [Google Scholar]
- 45.Giraldo E, Lloret A, Fuchsberger T, Viña J (2014) Aβ and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol 2:873–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tormos AM, Taléns-Visconti R, Nebreda AR, Sastre J (2013) p38 MAPK: a dual role in hepatocyte proliferation through reactive oxygen species. Free Radic Res 47:905–16. [DOI] [PubMed] [Google Scholar]
- 47.Vallés SL, Borrás C, Gambini J, Furriol J, Ortega A, Sastre J, Pallardó FV, Viña J (2008) Oestradiol or genistein rescues neurons from amyloid beta-induced cell death by inhibiting activation of p38. Aging Cell 7:112–8. [DOI] [PubMed] [Google Scholar]
- 48.Choi WS, Eom DS, Han BS et al. (2004) Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and −9-mediated apoptotic pathways in dopaminergic neurons. J Biol Chem 279:20451–60. [DOI] [PubMed] [Google Scholar]
- 49.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO (2011) Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J Signal Transduct 2011:792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ii M, Sunamoto M, Ohnishi K, Ichimori Y (1996) beta-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Res 720:93–100. [DOI] [PubMed] [Google Scholar]
- 51.Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ (1998) Amyloid-beta peptide activates cultured astrocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res 785:195–206. [DOI] [PubMed] [Google Scholar]
- 52.Akama KT, Albanese C, Pestell RG, Van Eldik LJ (1998) Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci U S A 95:5795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heckmann BL, Teubner BJW, Tummers B (2019) LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178:536–551.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee J, Kim DE, Griffin P, Sheehan PW, Kim DH, Musiek ES, Yoon SY (2020) Inhibition of REV-ERBs stimulates microglial amyloid-beta clearance and reduces amyloid plaque deposition in the 5XFAD mouse model of Alzheimer’s disease. Aging Cell 19: e13078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davies DS, Ma J, Jegathees T, Goldsbury C (2017) Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol 27:795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett ML, Bennett FC, Liddelow SA, et al. (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113: E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishijima T, Nakajima K (2021) Inflammatory cytokines TNFα, IL-1β, and IL-6 are induced in endotoxin- stimulated microglia through different signaling cascades. Sci Prog 104: 368504211054985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lyra E Silva NM, Gonçalves RA, Pascoal TA, et al. (2021) Pro-inflammatory interleukin-6 signaling links cognitive impairments and peripheral metabolic alterations in Alzheimer’s disease. Transl Psychiatry 11:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanzel CE, Pichet-Binette A, Pimentel LS et al. (2014) Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol Aging 35:2249–62. [DOI] [PubMed] [Google Scholar]
- 60.Heneka MT, Sastre M, Dumitrescu-Ozimek L et al. (2005) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGeer PL, McGeer EG (2015) Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets 19:497–506. [DOI] [PubMed] [Google Scholar]
- 62.McGeer PL, Itagaki S, Tago H, McGeer EG (1987) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79:195–200. [DOI] [PubMed] [Google Scholar]
- 63.Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–6. [DOI] [PubMed] [Google Scholar]
- 64.Sevigny J, Chiao P, Bussière T, et al. (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537: 50–6. [DOI] [PubMed] [Google Scholar]
- 65.Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M (2020) Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther 12:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N (2020) Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int J Mol Sci 21:5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sun A, Liu M, Nguyen XV, Bing G (2003) p38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol 183:394–405. [DOI] [PubMed] [Google Scholar]
- 68.Origlia N, Righi M, Capsoni S et al. (2008) Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-beta-mediated cortical synaptic dysfunction. J Neurosci 28:3521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schnöder L, Gasparoni G, Nordström K et al. (2020) Neuronal deficiency of p38α-MAPK ameliorates symptoms and pathology of APP or Tau-transgenic Alzheimer’s mouse models. FASEB J 2020; 34:9628–9649. [DOI] [PubMed] [Google Scholar]
- 70.Masuda T, Sankowski R, Staszewski O, et al. (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566:388–392. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.