

Abstract
Purpose of the Review
Mitochondrial dysfunction has long been proposed to play a crucial role in the pathogenesis of a considerable number of disorders, such as neurodegeneration, cancer, cardiovascular, and metabolic disorders, including obesity-related insulin resistance and non-alcoholic fatty liver disease (NAFLD). Mitochondria are highly dynamic organelles that undergo functional and structural adaptations to meet the metabolic requirements of the cell. Alterations in nutrient availability or cellular energy needs can modify their formation through biogenesis and the opposite processes of fission and fusion, the fragmentation, and connection of mitochondrial network areas respectively. Herein, we review and discuss the current literature on the significance of mitochondrial adaptations in obesity and metabolic dysregulation, emphasizing on the role of hepatocyte mitochondrial flexibility in obesity and NAFLD.
Recent Findings
Accumulating evidence suggests the involvement of mitochondrial morphology and bioenergetics dysregulations to the emergence of NAFLD and its progress to non-alcoholic steatohepatitis (NASH).
Summary
Most relevant data suggests that changes in liver mitochondrial dynamics and bioenergetics hold a key role in the pathogenesis of NAFLD. During obesity and NAFLD, oxidative stress occurs due to the excessive production of ROS, leading to mitochondrial dysfunction. As a result, mitochondria become incompetent and uncoupled from respiratory chain activities, further promoting hepatic fat accumulation, while leading to liver inflammation, insulin resistance, and disease’s deterioration. Elucidation of the mechanisms leading to dysfunctional mitochondrial activity of the hepatocytes during NAFLD is of predominant importance for the development of novel therapeutic approaches towards the treatment of this metabolic disorder.
Keywords: Liver, NAFLD, NASH, Mitochondrial dysfunction, Mitochondrial bioenergetics, Energy metabolism
Introduction
The liver has been characterized as the organism’s metabolic center, since it is responsible for regulating a number of biological processes, including the maintenance of energy homeostasis, production of several biomolecules such as bile and vitamins, and scavenging of harmful endogenous and exogenous metabolites [1, 2]. The orchestration of such procedures demands excessive energy requirements; therefore, the presence of operative mitochondria in hepatocytes is indispensable [1]. Indeed, hepatocytes, constitute 70–85% of the total liver mass and among cell populations residing in the liver, they are the most prone to cellular impairment. Disruption of their capacity to operate properly can lead to numerous pathological conditions, including obesity-related insulin resistance and non-alcoholic fatty liver disease (NAFLD) [1–4]. To abstain such afflictions, hepatocytes require a sufficient amount of adenosine triphosphate (ATP), necessary for the proper execution of biological processes. Hence, mitochondria are found in great numbers in hepatocytes and consist a crucial part in the metabolism of hepatocytes, as they constitute the principal site for oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO), leading to ATP synthesis [2]. Due to their highly dynamic nature, mitochondria can undergo functional and structural adaptations to meet the metabolic requirements of the cell through biogenesis, namely the growth and split of preexisting mitochondria, as well as through the opposite processes of fission and fusion, viz the separation and merge of mitochondria respectively [5]. Disruption in mitochondrial function tends to provoke and aggravate obesity-related metabolic dysregulation and likely contributes to the advancement of NAFLD [6]. Besides, NAFLD is thought as the metabolic manifestation of obesity-related metabolic dysregulation in the liver [7].
Evidence suggests that the emergence of obesity can be attributed to dysregulation of mechanisms of energy homeostasis, rather than simply evolving from excess caloric intake, implying that both obesity and NAFLD could be considered as “mitochondrial diseases” [8, 9]. NAFLD is strongly associated with obesity, with the latter acting as an autonomous risk factor for developing the first. A 3.5-fold higher risk for NAFLD advancement has been proposed for obese individuals compared to non-obese, while body mass index (BMI) appears to be responsible for NAFLD susceptibility in a “dose-dependent” manner [10]. The pathophysiology of NAFLD is guided by the implications on lipid metabolism and other metabolic pathways present in obesity, which result in the accumulation of intrahepatic fat (liver steatosis). Epidemiologic data presented in numerous studies confirm the correlation between obesity and NAFLD. Accordingly, NAFLD prevalence in obese individuals is greater than that of the general population (25–30%), although it varies in different studies with respect to age and other predisposing factors, including type 2 diabetes [11].
In this review, we discuss the recent literature on the impact of mitochondrial adaptations in metabolic disorders, emphasizing on the role of hepatocyte mitochondrial flexibility in obesity and the initiation and advancement of NAFLD. To that end, the current review specifically focuses on liver mitochondrial adaptations across the spectrum of obesity initiated NAFLD, covering the fundamental as well as the latest research on this topic.
Mitochondrial Adaptations: Mitochondrial Dynamics and Bioenergetics
Mitochondria are involved in various substrate catabolism processes, namely FAO (or β-oxidation) and tricarboxylic acid cycle (TCA) [2, 12]. Their primary physiological function is to generate energy in the form of ATP by performing OXPHOS, a process carried out in the electron transport chain (ETC) complexes. Specifically, important cellular metabolites such as pyruvate, amino-acids, and fatty-acids are carried forward into the TCA cycle for production of the reducing equivalents nicotinamide-adenine dinucleotide (NADH) and flavine-adenine dinucleotide (FADH2), which sequentially transfer their electrons to oxygen at the ETC [13]. Importantly, during this process, approximately 0.25–11% of the oxygen consumed by the mitochondrion is converted into superoxide and hydrogen, also known as reactive oxygen species (ROS), contributing to mitochondrial injury in several metabolic pathologies [14].
Mitochondria are assembled in a network of interconnected organelles, able to undergo morphological and functional adaptations to meet the metabolic requirements of the cell, following environmental signals [15]. These morphological changes can arise either as mitochondrial fission (fragmentation) or as fusion (connection). The equilibrium among these two opposite processes regulates mitochondrial number, size and positioning of mitochondria in the cytoplasm, is known as “mitochondrial dynamics” and is regulated by the membrane-remodeling proteins. During fission, dynamin-like/related protein 1 (Drp1) and mitochondrial fission 1 protein (FIS1) compress and separate mitochondrial tubules [16–19]. Oppositely, during fusion, mitofusins 1 and 2 (Mfn1/2) and optic atrophy-1(OPA1) integrate the outer (OMM) and inner (IMM) membrane of mitochondria, respectively [16, 18, 19]. Mitochondrial fusion is triggered from energy requirements and stress leading to an upregulation of metabolic competence and the repair of damaged mitochondrial fragments. In contrast, fission, triggered under relaxed conditions, facilitates uncoupled respiration, thus being related to reduce ATP synthesis. Fission is essential for mitochondrial degradation, where damaged parts of the mitochondria are in turn removed through mitochondrial autophagy, namely mitophagy [17, 20, 21] (Fig. 1). Importantly, mitophagy is of substantial significance in sustaining mitochondrial and cellular homeostasis under stress conditions, while dysregulated mitophagy is thought to have a pathological impact on the development of NAFLD [22–24].
Fig. 1.
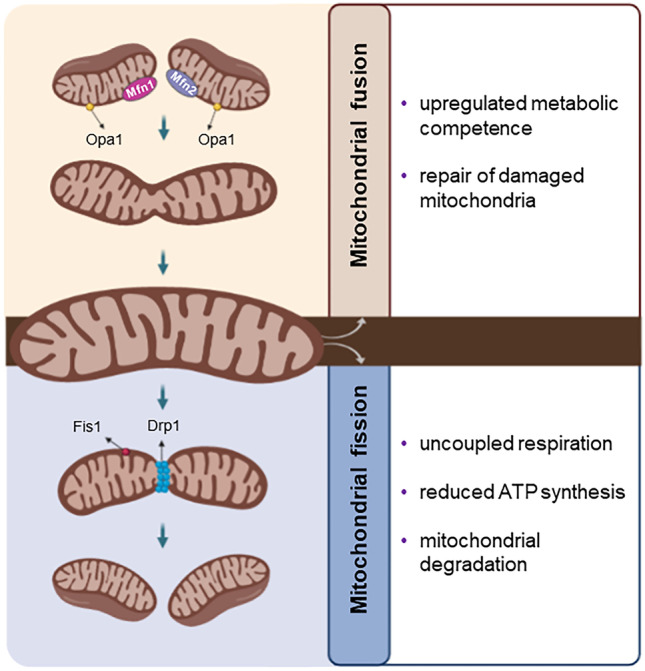
Mitochondrial dynamics: the process of fission and fusion. Mitochondrial fission is regulated by Drp1 and FIS1, which compress and separate mitochondrial tubules. Under conditions of low energy demand, fission facilitates uncoupled respiration, resulting to reduced ATP synthesis. Oppositely, during fusion, Mfn1/2 and OPA1 integrate the OMMs and IMMs. Mitochondrial fusion is stimulated by energy demand and stress and leads to upregulation of metabolic competence. Drp1, dynamin-like/related protein 1; FIS1, mitochondrial fission 1 protein; Mfn1, mitofusin 1; Mfn2, mitofusin 2; OPA1, optic atrophy 1; OMM; outer mitochondrial membrane; IMM, inner mitochondrial membrane
Mitochondrial Plasticity Assessment Methods
Mitochondrial plasticity, another term for mitochondrial adaptation, is directly associated with metabolic flexibility and can be characterized as an adaptive alteration in the mitochondrial content and function, depending on bioenergetic environmental conditions. The fundamental methodologies applied to study mitochondrial plasticity allow for the assessment of the ATP content under resting conditions or ATP production levels caused by metabolic alterations in whole tissues or cells. In principle, such methods involve measuring phosphorus metabolites primarily ATP or phosphocreatine via phosphorus (31P) magnetic resonance spectroscopy for measuring 31P-MRS in resting conditions or following induced metabolic alterations [25]. The OXPHOS rate constant is calculated by quantifying cellular oxygen depletion levels or isolated mitochondria under specific conditions of oxygen and existing energy substrate [26, 27]. In the liver, the submaximal ADP-stimulated OXPHOS activity can be quantified through 31P-MRS following intravenous or oral fructose intake, which triggers an intracellular ATP reduction via the activity of fructokinase [28–30]. Additionally, key observations can be attained via in vitro quantification of the activity of the ETC enzymes, ROS, and lipid peroxidation derivatives as a result of mitochondrial respiration, and antioxidant enzymes including catalase activity, decreased/oxidized glutathione ratio [12, 31, 32].
Methods applied for measuring the basal and maximum OXPHOS activity following ADP stimulation essentially require for high-resolution respirometry. The oxidative capability can be measured under specific energy conditions involving noncanonical, saturating energy substrate flows in the β-oxidation and Krebs cycle [33, 34]. This method allows for fundamental physiological properties of the mitochondrial respiratory chain to be calculated without the contradictory outcomes of differential oxygen or energy substrate transfer which could affect specific in vivo procedures. However, it relies its efficiency on the number of available biopsies which allow to accumulate substantial amounts of tissue to enable systematic mitochondrial analyses.
The evaluation of mitochondrial plasticity in the context of bioenergetics, as well as in the context of dynamics, can be achieved using quantitative PCR (qPCR) to quantify the expression levels of genes implicated to either mitochondrial bioenergetic and metabolic signaling or fission and fusion [35–41]. The morphology of mitochondria is not only fundamental for the maintenance of their optimal functionality but also critically associated to the metabolism of energy and OXPHOS function. Mitochondria morphology and dynamics can be assessed through the examination of tissue sections via electron microscopy (EM). Such methods allow for the visualization of ultrastructural alterations in mitochondrial characteristics, including loss of inner membrane and cristae can be observed, along with changes in mitochondria size (larger or shorter mitochondria) or granules, peroxisome proliferation, and mitochondrial fission or fusion [37, 39, 42–45]. Mitochondrial fission and fusion can also be evaluated using Immunofluorescence and Immunoblot analysis of fusogenic mediators including Mfn1, Mfn2, and OPA1 and fission operators such as Drp1 [46].
Mitochondrial Adaptations in Health and Disease
General Aspects
Mitochondrial adaptability to cellular needs and stress conditions is achieved via activation of advanced intracellular mechanisms. The mitochondrial nuclear crosstalk is of predominant importance in this response and involves the generation of various mitochondrial stress signals and nuclear stress response pathways. Mitochondria release metabolites such as acetyl-CoA and “mito-stress” signals, namely ROS, with a form of retrograde signaling, namely a mitochondria-to-nucleus signaling process that causes alterations in nuclear gene expression. The nucleus responds to these signals by activating stress-induced transcriptional programs, such as the AMP-activated protein kinase (AMPK) signaling. This in turn activates peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) and other transcriptional regulators, including cAMP-response element binding protein (CREB), nuclear factor-κB (NF-κB), and p53. These specific nuclear transcriptional responses are responsible for the repair or removal of damaged mitochondrial DNA (mtDNA) by fusion or fission and a switch towards glycolytic/lactate metabolism. In that way, ATP production is adjusted to meet the energy requirements of the cell [47–49]. For instance, when ATP levels decrease, the aforementioned “mitochondrial quality control” reinforces the cellular bioenergetic competence by enhancing the mitochondrial network via mitochondrial biogenesis and fusion, thus provoking the production of more ATPs [47, 50, 51].
Any dysfunction in this mitochondrial response mechanism is possible to affect cellular function rendering the cell more vulnerable to exogenous stressors, including oxidative stress as well as hypoxic conditions [47, 50, 52]. Gene mutations necessary for mitochondrial fusion, such as OPA1 or Mfn2, can affect multiple tissues, leading to diseases such as autosomal dominant optic atrophy (DOA) and Charot-Marie-Tooth type 2A (CMT2A) respectively [53–55]. Besides, abnormalities in mitochondrial structure and functionality affect the emergence and progression of neuromuscular and neurodegenerative disorders, in particular Parkinson’s, Alzheimer’s, and Huntington’s diseases [56]. Additionally, mitochondrial oxidative dysfunctions occur as secondary effect in the course of several diseases, including cardiovascular, gastrointestinal, skin disorders, and cancer [47, 57–59]. Importantly, mitochondria are associated with cancer advancement, since they enable the capacity of cancer cells to respond to the rapidly changing environmental conditions [47, 52, 59]. Finally, several germline and somatic mtDNA mutations are thought to play a key role in tumor enlargement in hemopoietic, prostate, breast, and renal cancer [60].
Mitochondrial Adaptations in Obesity and Metabolic Dysregulation
During obesity, the excess nutrient input as well as the increased basal lipolysis taking place in adipocytes supplies hepatocytes with increased amounts of free-fatty acids (FFAs), subsequently leading to increased but still insufficient FAO and OXPHOS [61, 62]. This process of oversaturated FAO and OXPHOS inevitably induces mitochondrial dysfunction with an increase of ROS formation and endoplasmic reticulum (ER) stress, provoking mtDNA damage, lipid peroxidation, and likely cell death. Additionally, the stressed hepatocytes secrete inflammatory cytokines and chemokines as well as lipid peroxidation derivatives including malondialdehyde (MDA), resulting to the recruitment of immune cells and triggering hepatic inflammation and insulin resistance [58, 63, 64].
Several studies have reported abnormal mitochondrial activity during obese and/or insulin-resistant conditions. Reduced biogenesis and insufficient oxidative mitochondrial capacity are previously reported in adipose tissue of rodents and individuals with obesity. Houstis et al. have demonstrated that ROS production triggers insulin resistance in cultured 3T3-L1 adipocytes treated with dexamethasone or recombinant mouse TNF and in leptin-deficient ob/ob mice [65, 66]. Analysis of isolated mitochondria from rodents that followed a high-fat diet (HFD) was presented with diminished respiratory capacity along with elevated oxidative stress [67–69]. In addition, obese diabetic db/db and ob/ob mice displayed weakened respiratory capacity of mitochondrial complexes in liver homogenates [70, 71]. Data from obese and diabetic individuals indicate that skeletal muscle mitochondria exhibit lower muscle energy production capacities, decreased FAO and impaired mitochondrial OXPHOS [72, 73]. Moreover, smaller and shorter mitochondria along with elevated mitochondrial fission were observed in the skeletal muscle of obese mice, confirming the correlation between altered mitochondrial fission and insulin resistance in the skeletal muscle [74]. Mitochondrial impairment has also been implicated with elevated fission processes associated with ROS formation in the liver of db/db mice, in rat liver cell lines and H9c2 rat myoblasts treated with high glucose [70, 75]. On the contrary, other studies displayed unaltered or augmented hepatic oxidative activity of mitochondria in adult ob/ob mice as well as in insulin-resistant diabetic Goto-Kakizaki (GK) rats [76, 77]. The justification for these opposing results remains unidentified but might be associated with the variety of diets used, the range of obesity, and different experimental approaches for the evaluation of mitochondrial function.
Hepatocyte Mitochondrial Adaptations in Obesity-Related NAFLD/NASH
Hepatocytes, Obesity, and NAFLD Pathogenesis
The liver, as a regulator of lipid homeostasis, orchestrates the biosynthesis of new fatty acids and sterols, along with their subsequent allocation to other tissues, and their utilization as substrates for energy production [78]. When the balance between lipid gain and disposition is disrupted, fat accumulates in the liver, leading to hepatocyte metabolic dysregulation and liver steatosis. Hepatic fat accumulation is controlled by four important processes, namely the uptake of circulating lipids, FAO, de novo lipogenesis (DNL), and the release of lipids into the circulation in the form of very low-density lipoproteins (VLDL). When one or more of these pathways are dysregulated, hepatic lipid aggregation is induced, contributing to obesity and NAFLD pathogenesis [79].
The transport of circulating fatty acids into the hepatocytes is mainly facilitated by fatty acid transport proteins (FATP), cluster of differentiation 36 (CD36), as well as plasma membrane caveolins. Studies in mice have shown that knockdown of FATP2 or FATP5, two major hepatic FATP isoforms, along with selected deletion of fatty acid translocase protein CD36 in mouse genetic and HFD-induced steatosis models, results in hepatic lipid uptake reduction and amelioration of hepatic steatosis [80–82]. Similarly, when caveolin-1 (CAV1), a structural caveolae protein in the plasma membrane related to hepatic lipid translocation, is ablated in HFD mice, liver steatosis is reduced while hepatic gluconeogenesis is elevated [83]. The second pathway, namely FAO, takes place in mitochondria. It is controlled by peroxisome proliferator-activated receptor α (PPARα) and following its activation, FAO-related genes are transcripted [84]. In ob/ob mice, knockout of PPARα augmented obesity and hepatic steatosis due to decreased FAO [85]. Regarding hepatic lipid aggregation, Zhang et al. demonstrated that the lack of long chain acylCoA dehydrogenase (LCAD), an essential enzyme for mitochondrial FAO, predisposes mice to liver steatosis and insulin resistance [86]. Furthermore, in response to increased food intake, hepatic DNL is induced, which in turn is activated by upregulated insulin signaling and augmented glucose concentrations [87]. Finally, as TGs export from hepatocytes, mainly in the form of VLDL, hepatic lipid load decreases. During obesity and NAFLD, an increased export of VLDL occurs at the beginning of the disease. The latter is plateaued as the disease progresses, leading to escalated fat accumulation further perpetuating hepatic steatosis [88, 89].
Hepatocyte Mitochondrial Bioenergetics in Obesity and NAFLD
Hepatic energy homeostasis is mainly mediated by mitochondrial metabolic processes, encompassing β-oxidation, TCA cycle, ketogenesis, ETC activity, and ATP formation [90, 91]. During NAFLD, FFAs and TGs rapidly accumulate in the liver via the circulation and/or DNL and are associated with enhanced FAO and ROS formation, further contributing to NAFLD progression [2, 92, 93]. Specifically, under physiological conditions, the FFAs present in the cytoplasm are initially transformed into fatty acyl-CoA, which either remains in the cytoplasm to be used for esterification into TGs and can be secreted as VLDLs into the circulation or relocated to the mitochondria. Therein, disintegration follows through β-oxidation to compose acetyl-CoA [38, 94]. In obesity, increased FFA influx due to excess caloric intake leads to TGs aggregation in the form of lipid droplets in hepatocytes, a feature of NAFLD pathology [38, 90]. Besides, heightened FFA availability upregulates β-oxidation and consecutive production of NADH and FADH2 that serve as co-enzymes in the mitochondrial ETC, acting as electron transporters [38]. Hence, the electron flow within the ETC is overloaded and subsequently disrupted due to upregulation of proton leak, an over-reduction of the respiratory chain along with ROS generation (Fig. 2). This overproduction of ROS triggers the secretion of proinflammatory cytokines such as IL-1β and TNF by the hepatocytes, starting an endless loop of immune cell recruitment and inflammation, perpetuation of steatosis, and activation of hepatic stellate cells into profibrotic cells [6, 93, 95]. Besides, genomic- and mitochondrial-DNA damage can occur during this process, provoking hepatocyte senescence, which has been shown to aggravate NAFLD and progression to non-alcoholic steatohepatitis (NASH) [96, 97].
Fig. 2.

Hepatic mitochondrial adaptations in non-alcoholic fatty liver disease (NAFLD). In NAFLD, rapid accumulation of triglycerides (TGs) in the liver, due to high availability of free fatty acids (FFAs), and/or de novo lipogenesis (DNL) is associated with an elevated mitochondrial oxidative activity. The cytoplasmic FFAs are first converted into fatty acyl-CoA which is relocated to mitochondria to be decomposed via β-oxidation and produce acetyl-CoA. Increased FFA influx leads to insufficient hepatic β-oxidation and therefore, lipotoxic intermediates accumulate, triggering inflammation and disrupting insulin signaling. In contrast, the utilization of acetyl-CoA by the mitochondrial tricarboxylic acid (TCA) cycle continues unabated to meet the energetic demands of gluconeogenesis. Mitochondrial β-oxidation generates NADH and flavine-adenine dinucleotide (FADH2), the electrons (e-) of which are transferred to the electron transport chain (ETC). Disruption of the electron flow within the ETC induces leakage of electrons and the generation of reactive oxygen species (ROS), contributing to NAFLD progression, mainly by triggering hepatocyte stress and damage. Furthermore, when hepatocytes are exposed to excessive nutrient overload and FFAs, mitochondria become disintegrated through increased fission. DNL, de novo lipogenesis; TGs, triglycerides; OMM; outer mitochondrial membrane; IMM, inner mitochondrial membrane; ATP, adenosine triphosphate; Drp1, dynamin-like/related protein 1. Pathways and procedures that are increased are designated by ↑
Several mitochondrial genomic and transcriptomic alterations have been reported during the whole phenotypic spectrum of NAFLD in both humans and rodents affecting the metabolic machinery of the mitochondria. Sookoian et al. have performed mitochondrial genome sequencing in the liver of NAFLD patients across the entire spectrum of the disease. Their findings revealed that the hepatic mtDNA of NAFLD patients harbors intricate mitochondrial genomes with a notably elevated mutation rate as compared with controls. Moreover, analysis of the entire hepatic mitochondrial genomes of patients with progressed fibrosis indicated that the severity of the disease is associated with increased number of hepatic mtDNA mutation-carrying variants encoding for proteins implicated in OXPHOS [98]. Other studies demonstrate downregulated expression of genes encoding for members of the OXPHOS system, resulting from epigenetic alterations taking place at the hepatic mtDNA [36, 37]. For instance, Pirola et al. provided evidence that increased hepatic mtDNA methylation affecting the transcriptional activity of mitochondrially encoded NADH dehydrogenase 6 (MT-ND6) participates in modulation of the histological severity of NAFLD leading to NASH [37]. Regarding the unrelated to obesity phenotype of NAFLD, a clear correlation of this lean phenotype and mitochondrial adaptations is missing [99]. However, Komatsu et al. described that hepatic steatosis occurs in patients afflicted with citrullinemia type 2 (CTLN2), an autosomal recessive disorder caused by a mutation in the gene encoding mitochondrial aspartate glutamate carrier 2 (SLC25A13) and these patients were not categorized as obese [100]. Furthermore, a study by Hakim et al. associates mitochondrial mutations with idiopathic NAFLD, as they report that non-obese NAFLD individuals harbor mutations in NADH dehydrogenase (ubiquinone) 1 beta subcomplex 3, (NDUFB3) gene, which encodes a subunit of respiratory chain complex I [101]. When NAFLD progresses to NASH, the metabolic flexibility of hepatic mitochondria including oxidative capacity and redox defenses ultimately fades [102, 103]. Koliaki et al. reported upregulation of OXPHOS effectiveness in hepatic mitochondria obtained from patients at early stages of NAFLD and significant downregulation in NASH patients [104]. Furthermore, it is demonstrated that NASH is correlated to genetic changes of hepatic cellular respirasome, involving high cytochrome b variations along with mtDNA damage, leading to severe respirasome supercomplex inadequacy and consequently resulting to cell death in high energy-demanding tissues [105]. Besides, other studies regarding obesity reported reduced hepatic ATP concentration in ob/ob mice and those of obese and diabetic individuals [106–108]. Similarly, Nair et al. demonstrated an inverted correlation between ATP hepatic stores and BMI, although the ATP reduction and recovery rates did not differ among obese and lean individuals upon fructose injection [29]. Additional data suggest that mice and patients with NASH exhibit downregulated ketogenesis, diminished mitochondrial respiration, mitochondrial rupture, and leakage [109]. On the contrary, during this progressive disease stage, the mitochondrial TCA cycle is hyperactive in order to respond to the high energy requirements [102, 104, 110]. Recent evidence of Grossini et al. demonstrates that the exposure of primary human hepatocytes to plasma from NAFLD patients leads to reduced hepatocyte viability and mitochondrial membrane potential in these cells as compared to those treated with plasma from healthy individuals, along with increased ROS and H2O2 production and triglyceride accumulation. Their results also presented that the plasma of NAFLD patients induced increased hepatocyte expression of peroxisome-proliferator-activating-ligand-receptor-γ (PPARγ), sterol-regulatory-element-binding-protein-1c (SREBP-1c), nuclear-factor-kappa-light-chain-enhancer of activated B cells (NF-kB), and NADPH oxidase 2 (NOX2), which are implicated to mitochondrial bioenergetic and metabolic signaling [35]. Ajaz et al. followed a mitochondrial functional and metabolomic approach in peripheral mononuclear blood cells (PBMCs) from patients with NAFLD at different fibrotic stages versus healthy individuals. They demonstrated that the progression of NAFLD is associated to mitochondrial abnormalities related to changes in metabolites of the urea cycle; specifically, reduced hepatic mitochondrial respiratory capacity and significant changes in five out of fourteen metabolites involved in urea cycle were observed in patients presenting with progressed fibrosis in comparison to mild/moderate fibrosis [111].
Although the findings above implicate mitochondrial dysfunction in NAFLD progression, opposing evidence exists in the literature. Studies examining the liver, muscle, and adipose tissue of mice with muscle- and liver-specific ablation of mitochondrial flavoprotein apoptosis inducing factor (AIF), inducing mitochondrial OXPHOS defects, have shown that abnormalities in mitochondrial respiratory activity may induce an insulin-sensitive metabolic condition, which protects against the adipogenic and diabetogenic effects of HFD [112]. Such evidence agrees with the phenotype reported in mice that specifically lack the mitochondrial transcription factor-A (TFAM) in muscle and adipose tissue which exhibited upregulated insulin susceptibility in both tissues, regardless of the inadequate mitochondrial respiratory system therein [113, 114]. Therefore, these reports impose that mitochondrial impairment might not be a trigger of NAFLD, suggesting instead that mitochondrial impairment might be a derivative, outcome of the disease’s advancement [115].
Another important component of mitochondrial bioenergetic adaptation in NAFLD is the transfer of a portion of excess energy intermediates in the ineffective cycle of uncoupled respiration. In preclinical studies, mitochondrial uncoupled respiration appeared increased due to the upregulation of the uncoupling protein 2 (UCP-2) in hepatocytes obtained from an obese rodent model. The in vitro exposure of rat hepatocytes to lipid emulsions resulted in augmented UCP-2 expression, possibly through mechanisms mediated by excess ROS production [106, 116]. Morris et al. demonstrated that a short-term HFD treatment on rats predisposed to obesity can lead to decreased metabolic adaptability and liver mitochondrial respiratory capacity which is effectively averted by hepatic overexpression of peroxisomal proliferator-activated receptor-γ coactivator-1α (PGC-1α) [117]. In human NAFLD, this phenomenon culminates in the established NASH environment and is also mediated by upregulated UCP-2 expression, which is consistent with indications of severe oxidative stress and injury [104, 118]. This illustrates that heightened uncoupled respiration in progressed NAFLD may act in a protective manner against the energetic overstrain and subsequent redox imbalance of ETC complex. Nevertheless, this comes at the cost of decreased bioenergetic effectiveness, possibly accounting for the diminished energy-producing capacity of the liver in NASH, making hepatocytes more susceptible to severe energy-demand challenges, such as ischemic injury [118, 119].
Hepatocyte Mitochondrial Dynamics in Obesity and NAFLD
Hepatocyte mitochondrial dynamics is a fundamental mechanism of the adjustment of a cell to its metabolic requirements. When nutrient availability changes occur, mitochondria are subjected to coordinated fission and fusion cycles to sustain energy homeostasis. In rich-nutrient environmental conditions, mitochondria fragment to prevent energy waste, while reducing bioenergetic efficiency and augmenting mitochondrial uncoupling, which results to a simultaneous increase of nutrient storage [38, 74, 120]. On the contrary, under nutrient deprivation conditions, hepatic mitochondria remain elongated through the process of increased fusion [18]. Of note, when hepatocytes are exposed to high levels of cellular stress along with excessive nutrient overload and FFAs, a common phenomenon in obesity and NAFLD, the mitochondrial network becomes more disintegrated through increased fission [18, 19] (Fig. 2). The segregated mitochondria are frequently depolarized, a phenomenon caused by diminished respiratory capacity, enabling mitophagy to occur, which is a highly selective autophagic clearance of defective and dysfunctional mitochondria [121].
Numerous in vivo and in vitro studies highlight changes pertinent to mitochondrial fusion and fission during obesity and NAFLD. Evidence from in vitro studies suggests that when hepatocytes are treated with palmitate, their mitochondria become fragmented, lose their transmembrane potential, while there is a cytochrome c release in the cytoplasm and elevated ROS activity [122, 123]. It has been also demonstrated that protein expression of Drp1 is increased in animal models of NAFLD, indicating mitochondrial fragmentation [38–40]. Analysis made with electron microscopy exhibited higher mitochondrial fission in the livers of animals subjected to HFD, as well as heightened hepatocyte lipolysis [39, 42]. Moreover, by using HFD-feeding in mice that express the dominant-negative fission mutant DLP1-K38A in a doxycycline-inducible manner, Galloway et al. showed that transgenic inhibition of mitochondrial fission was protective against liver steatosis, alleviating HFD-induced oxidative stress and hepatic damage as well. This suggests a mechanistic role of mitochondrial fission in controlling hepatic lipid regulation and oxidative stress implicated with NAFLD [39]. Takeichi et al. showed that hepatic deletion of mitochondrial fission factor (MFF) stimulates ER stress and diminishes the secretion of triaglycerol in the liver both in vivo and in vitro. They also presented that mice lacking MFF in hepatocytes (MffLiKO mice) are more prone to NASH phenotypes caused by HFD than control mice, mainly due to the hepatic activation of apoptosis from ER stress and the suppression of triglyceride excretion from hepatocytes. Thus, their research brings new information for the implication of mitochondrial fission in NASH development [124].
Oppositely, a decline in mitochondrial fusion has been reported during NAFLD in vitro and in vivo. The expression of Mfn1 is reduced in hepatocytes of HFD-fed mice and is associated with steatohepatitis [41]. Importantly, a pro-inflammatory factor, CXCR3 expressed by hepatocytes, stellate cells along with a plethora of immune cells of the liver microenvironment induces a reduction of the protein levels of Mfn1, thereby inducing mitochondrial deterioration in animal models of NASH [40]. Palmitate treatment in hepatocytes induces downregulation of Mfn2 in both transcript and protein levels [123]. When Mfn2 was ablated specifically in mouse livers, hepatic ER activity was increased, leading to metabolic abnormalities, along with deteriorated glucose tolerance and insulin resistance in mice subjected to HFD [125]. Moreover, diminished Mfn2 levels are detected in the liver of NASH individuals and in NAFLD/NASH murine models. Interestingly, hepatic Mfn2 deletion promoted inflammation, triglyceride concentration, fibrosis, and hepatic cancer in mouse models of NASH, while adenovirus-mediated re-expression of Mfn2 in liver-specific Mfn2 knockout mice led to disease alleviation [126, 127]. These data demonstrate the importance of Mfn2 in NAFLD pathophysiology.
Regarding the mitochondrial structural morphology, it has been reported that hepatic mitochondria from NAFLD patients have circuit geometry, due to cristae loss. As the disease progresses, patients develop megamitochondria with paracrystalline inclusion bodies, indicating either a protective or a degenerative response to injury [43, 44]. These morphological deformations are a consequence of mitochondrial dynamics’ dysregulation in NAFLD patients, mainly attributed to disturbed ATP homeostasis due to impaired function of the mitochondrial ETC [44]. Similar modifications were observed in animal models of diet-induced obesity, where hepatic mitochondria appeared to have a rounder and shorter shape, with lack of cristae, while indications of swelling and matrix compression were also present [45]. Of note, mitochondrial ultrastructural modifications were prevented in HFD mice following a systematic strength training program versus sedentary HFD mice [128]. Although there is evidence associating alterations in mitochondrial integrity and elevated oxidative stress with obesity and NAFLD pathogenesis, additional investigation is necessary for the elucidation of the precise implication of mitochondrial pathophysiology into disease initiation and advancement. Tables 1 and 2 present major animal and human studies on mitochondrial adaptations in NAFLD.
Table 1.
Major human studies on mitochondrial adaptation in NAFLD
| First author, year, reference | Cohorts |
Tissue samples Cell lines |
Methods | Key results | Challenges/limitations |
|---|---|---|---|---|---|
| Sookoian, 2016 [98] |
-Controls (8) and NAFLD patients (20) -Patients with NASH at different stages (10) and patients with moderate of advanced fibrosis (14) -Unrelated patients with NAFLD (NAFL = 62, NASH = 76) and healthy individuals (100) |
Liver biopsies Serum samples PBMCs |
Deep coverage whole genome sequencing in hepatic mitochondrial genome with NGS |
-Hepatic mtDNA of NAFLD patients harbors intricate mitochondrial genomes with a notably ↑ mutation rate versus controls -In patients with ↑ fibrosis the severity of NAFLD is associated with ↑number of OXPHO-related hepatic mtDNA mutation-carrying variants |
-Using NGS, the number of reads could probably limit the sensitivity to detect low frequency heteroplasmic variants and somatic mutations -Mitochondrial genetics differ from nuclear genetic -No functional data to support the effect of missence mutations |
| Pirola, 2013 [37] |
Controls (18) NAFLD patients (45) |
Liver biopsies |
-Biochemical evaluation -Histopathological evaluation -Bisulfite treatment of DNA and methylation specific PCR -qPCR |
-↑ hepatic mtDNA methylation affecting the transcriptional activity of MT-ND6 participates in modulation of histological severity of NAFLD leading to NASH | None |
| Koliaki, 2015 [104] |
Obese IR patients w/o NAFLD (18) Obese IR patients with NAFLD (16) Obese IR patients with NASH (17) Lean individuals (12) |
Liver biopsies Serum samples |
-Metabolic characterization -Histology -Immunoblotting -Mitochondrial respiration evaluation -Oxidative stress evaluation -qPCR |
-↑ OXPHOS effectiveness in liver mitochondria obtained from early stages of NAFLD patients -↓ OXPHOS effectiveness in liver mitochondria of NASH patients |
-Insulin infusion -FCCP-induced maximum uncoupled respiration -ADP-stimulated respiration -There is no standard method for associating mitochondrial function to mitochondrial content |
| Pirola, 2021 [105] | NAFLD patients (252) | Liver biopsies |
-MT-CYB sequencing and analysis of mtDNA damage -Global liver transcriptome analysis |
-NASH correlation with genetic changes of liver cellular respirasome (↑ cytochrome b variations, mtDNA damage) resulting to severe respirasome supercomplex inadequacy and cell death | Modest sample size |
| Grossini, 2021 [35] |
Controls (12) NAFLD patients (12) |
Liver biopsies Plasma samples Huh7.5 cell line |
-Hepatocyte isolation from human liver biopsies -Co-culture of Huh7.5/primary human hepatocytes by using Transwell inserts -Mitochondrial ROS, ROS-Glo H202 quantification -Immunoblotting |
-Exposure of primary human hepatocytes to plasma from NAFLD patients results to ↓ hepatocyte viability and ↓ mitochondrial membrane potential, ↑ ROS, ↑ H202 production compared to those treated with plasma from healthy individuals | None |
| Ajaz, 2021 [111] |
Controls (10) NAFLD patients with fibrosis (10) NAFLD patients with severe fibrosis (10) |
PBMCs |
-Mitochondrial functional analysis with Seahorse XFp In PBMCs -Global metabolomics -ELISA |
-NAFLD progression is associated with mitochondrial dysfunctions related to changes in metabolites of the urea cycle (↓ hepatic mitochondrial respiratory capacity, significant changes in 5 out of 14 metabolites of the urea cycle in patients with progressed fibrosis in comparison to mild/moderate fibrosis) |
-Limited number of samples -Lack of liver tissue to measure gene expression of the urea cycle enzymes |
| Nair, 2013 [29] | Healthy individuals with varying BMI (19) | Liver biopsies | -MR Spectrometry | -↓ liver ATP stores are more common in overweight and obese individuals | Modest sample size |
Table 2.
Major Animal studies on mitochondrial adaptation in NAFLD
| First author, year, reference | Animal models |
Tissue, samples Cell lines |
Methods | Key results | Challenges/limitations |
|---|---|---|---|---|---|
| Gao, 2015 [85] |
-ob/ob mice -PPARa-deficient ob/ob mice |
Liver |
-Glucose tolerance and insulin resistance -Biochemical assays -Histology, immunohistochemistry -Immunoblotting -Northen blotting and qPCR |
-Knockout of PPARa ↑ obesity and ↑ hepatic steatosis due to ↓ FAO | None |
| Zhang, 2007 [86] |
LCAD-/- mice WT mice |
Liver |
-Hyperinsulinemic-Euglycemic clamp -Biochemical assays -qPCR |
-Lack of LCAD predispose mice to liver steatosis and insulin resistance | None |
| Pospisilik, 2007 [112] | Liver- and Muscle-Specific AIF -/- mice |
Liver muscle Adipose tissue |
-Glucose and insulin tolerance tests -Hyperinsulinemic clamp -Indirect calorimetry -Immunoblotting -Mircoarray analyses -Determination of intracellular metabolites -Histology -Mitochondrial DNA quantification |
-Mitochondrial dysfunctions in respiratory activity may induce insulin-sensitive metabolic condition, protecting against the adipogenic and diabetogenic effects of HFD in muscle- and liver-specific AIF knockout mice | None |
| Galloway, 2014 [39] | Transgenic mice expressing DLP1-K38A in a doxycycline-inducible manner | Liver |
-Histology and immunohistochemistry -Mitochondrial morphology assessment -Isolation of Primary hepatocyte and treatments -Oxygen consumption analyses |
-Transgenic inhibition of mitochondrial fission is protective against hepatic steatosis, ameliorating HFD-induced oxidative stress, and liver damage, suggesting a mechanistic role of mitochondrial fission in regulating hepatic lipid regulation and oxidative stress associated with NAFLD | None |
| Takeichi, 2021 [124] | Mice with hepatocyte-specific deletion of MFF (MffLi -/- mice) | Liver |
-Isolation of primary hepatocytes isolated from MffLiKO -Metabolic and biochemical evaluations -qPCR -Immunoblotting -Mitochondria isolation experiments |
-Hepatic deletion of mitochondrial fission factor (MFF) stimulates ER stress and ↓ the secretion of triglycerides in the liver -MffLiKO mice are more susceptable to NASH phenotypes caused by HFD than control mice |
Further studies needed to delineate the correlation of mitochondrial fission with the regulation of glucose metabolism |
| Du, 2017 [40] |
CXCR3-/- mice WT mice |
-TEM -Mitochondria isolation from liver, Mitochondria membrane potential, mtDNA damage analysis -Immunoblotting, Immunofluorescence -FACS -qPCR -siRNA transfection for CXCR3 knockdown -ATP measurement |
-CXCR3 ↓ the protein levels of Mfn1, thereby causing mitochondrial deterioration in animal models of NASH | None | |
| Sebastian, 2012 [125] | Liver-specific Mfn2 KO mice |
Liver Skeletal muscle Adipose tissue |
-Isolation of mouse hepatocytes -High-resolution respirometry with the Oxygraph-2 k and Seahorse Bioscience XF |
-Specific hepatic ablation of Mfn2 resulted to ↑ hepatic ER activity, leading to metabolic dysregulations, deteriorated glucose tolerance and insulin resistance in HFD-mice -Mfn2 coordinates mitochondria and ER function, leading to modulation of insulin signaling and glucose homeostasis in vivo |
None |
| Piacentini, 2018 [42] |
TG2-/- mice WT mice |
Liver Serum samples |
-Metabolic and biochemical evaluations -Histopathological analysis -Transmission electron ultrastructural analysis -Immunoblotting |
-↑ mitochondrial fission when subjected to HFD -TG2 activation may offer protection in the context of NAFLD |
None |
Conclusion and Future Perspectives
Emerging evidence illustrates that alterations in hepatic mitochondrial dynamics and bioenergetics hold a substantial role in the pathogenesis of NAFLD [2, 21, 104, 129]. When the fatty acid influx is elevated in hepatocytes, as it occurs during obesity and NAFLD, oxidative stress takes place as consequence of ROS overproduction, causing mitochondrial dysfunction. As a result, mitochondria become ineffective and disengaged from respiratory chain activity and ATP formation, further favoring hepatic fat accumulation, while leading to liver inflammation and insulin resistance in a ROS-dependent manner. These phenomena lead to subsequent hepatocyte dysfunction or damage and trigger the transition of NAFLD to NASH by parallel activation of stellate cells [39, 122, 123, 130, 131].
Therapeutic strategies diminishing the oxidative burden are able to reinforce the mitochondrial capacity for ATP generation and can suppress mitochondrial fission, thus attenuating or even reversing the advancement of NAFLD [1, 39]. Such approaches include anti-diabetic drugs such as thiazolidinediones, which are synthetic peroxisome proliferator-activated receptor gamma (PPAR-γ) ligands [132, 133]. For instance, pioglitazone, a widely known PPARγ agonist, inhibits cytochrome c leakage, normalizes the mitochondrial transmembrane potential, prevents ROS production, and induces the activation of the ETC complexes, and therefore could be used for NAFLD treatment as well [134]. Along this line, metformin, a first-line diabetic drug, has the capacity to stimulate the AMPK signaling pathway and enhance mitochondrial fission in mice following HFD, leading to upregulated mitochondrial respiration, stabilized mitochondrial membrane potential and increased ATP levels [135, 136]. Besides, angiotensin II type 1 receptor (ATIR) antagonists have been observed to have beneficial impact in rodent models of NAFLD, as these agents could augment mitochondrial biogenesis or protect mitochondria from oxidative stress [137]. Additionally, irisin, a recently identified hormone secreted by muscle cells, and resolvin D1 (RvD1), an anti-inflammatory and antioxidant lipid mediator, hinder mitochondrial fission and protect against liver ischemia–reperfusion injury [138, 139].
Therapeutic approaches directly targeting the mitochondria machinery have shown great potential against NAFLD. Specifically, therapeutic targeting of methylation-controlled J protein (MCJ) in the liver, an endogenous negative regulator of the respiratory chain complex I, efficiently ameliorated liver lipid accumulation and fibrosis in multiple NAFLD mouse models. Specifically, blockage of MCJ by utilizing nanoparticle- and GalNAc-formulated siRNA heightened hepatocyte capacity to mediate FAO and decreased lipid accumulation, resulting in diminished hepatocyte damage and fibrosis [140]. Moreover, pharmacological inhibition of Drp1 blocks mitochondrial fragmentation and mitochondrial release of cytochrome c and apoptosis. Mdivi-1, a derivative of quinazolinone acting as a selective inhibitor of Drp1 [141], blocks apoptotic cell death and drastically reduces the expression of cytochrome c [142, 143]. Overall, treatments targeting and manipulating cellular processes specifically associated with hepatic mitochondrial adaptation and bioenergetics may be implemented in the future to alleviate NAFLD and hence should be rigorously investigated for their therapeutic potential.
Abbreviations
- NAFLD
Non-alcoholic fatty liver disease
- ATP
Adenosine triphosphate
- OXPHOS
Oxidative phosphorylation
- FAO
Fatty acid oxidation
- BMI
Body mass index
- TCA
Tricarboxylic acid cycle
- ETC
Electron transport chain
- ROS
Reactive oxygen species
- IMM
Inner mitochondrial membrane
- OMM
Outer mitochondrial membrane
- Drp1
Dynamin-like/related protein 1
- mtDNA
Mitochondrial DNA
- FFA
Free fatty acid
- TGs
Triglycerides
- DNL
De novo lipogenesis
- FATP
Fatty acid transport proteins
- ER
Endoplasmic reticulum
- HFD
High-fat diet
- VLDL
Very low-density lipoproteins
- NASH
Non-alcoholic steatohepatitis
Author Contribution
A-IL carried out literature search, wrote, edited the manuscript, and designed the figures; IIM performed literature search and contributed to the writing of the manuscript; MS performed literature search and assisted with the writing of the manuscript; GP performed literature search; R-IV performed literature search, contributed to the writing, and editing of the manuscript; AC had the idea of the article, designed, edited, supervised, and revised the manuscript. All authors have read and approved the final manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. Supported by grants from the Deutsche Forschungsgemeinschaft (CH 1862/3–1 to AC) and the Hellenic Foundation for Research and Innovation (HFRI).
Data Availability
Not applicable.
Code Availability
Not applicable.
Declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All authors have read and approved the final manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Footnotes
This article is part of the Topical Collection on Metabolism
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Aigli-Ioanna Legaki, Email: elinalegaki@med.uoa.gr.
Ioannis I. Moustakas, Email: ioamoustakas@med.uoa.gr
Michalina Sikorska, Email: sikorskamichalina@gmail.com.
Grigorios Papadopoulos, Email: grpapad@med.uoa.gr.
Rallia-Iliana Velliou, Email: rallia-iliana.velliou.16@ucl.ac.uk.
Antonios Chatzigeorgiou, Email: achatzig@med.uoa.gr.
References
- 1.Auger C, Alhasawi A, Contavadoo M, Appanna VD. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol. 2015;3:40. doi: 10.3389/fcell.2015.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei Y, Rector RS, Thyfault JP, Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. 2008;14:193–199. doi: 10.3748/wjg.14.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auger C, Sivayoganathan T, Abdullahi A, Parousis A, Jeschke MG. Hepatic mitochondrial bioenergetics in aged C57BL/6 mice exhibit delayed recovery from severe burn injury. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2705–2714. doi: 10.1016/j.bbadis.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Degli Esposti D, Hamelin J, Bosselut N, Saffroy R, Sebagh M, Pommier A, Martel C, Lemoine A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem Res Int. 2012;2012:387626. doi: 10.1155/2012/387626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simoes ICM, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2018;95:93–99. doi: 10.1016/j.biocel.2017.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz MW, Seeley RJ, Zeltser LM, Drewnowski A, Ravussin E, Redman LM, Leibel RL. Obesity pathogenesis: an endocrine society scientific statement. Endocr Rev. 2017;38:267–296. doi: 10.1210/er.2017-00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Li L, Liu DW, Yan HY, Wang ZY, Zhao SH, Wang B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes Rev. 2016;17:510–519. doi: 10.1111/obr.12407. [DOI] [PubMed] [Google Scholar]
- 11.Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: from pathophysiology to therapeutics. Metabolism. 2019;92:82–97. doi: 10.1016/j.metabol.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O'Rourke B, Paolocci N, Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol. 2012;139:479–491. doi: 10.1085/jgp.201210772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 16.Galloway CA, Yoon Y. Mitochondrial morphology in metabolic diseases. Antioxid Redox Signal. 2013;19:415–430. doi: 10.1089/ars.2012.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seitz S, Kwon Y, Hartleben G, Jülg J, Sekar R, Krahmer N, Najafi B, Loft A, Gancheva S, Stemmer K, Feuchtinger A, Hrabe de Angelis M, Müller TD, Mann M, Blüher M, Roden M, Berriel Diaz M, Behrends C, Gilleron J, Herzig S, Zeigerer A. Hepatic Rab24 controls blood glucose homeostasis via improving mitochondrial plasticity. Nat Metab. 2019;1:1009–1026. doi: 10.1038/s42255-019-0124-x. [DOI] [PubMed] [Google Scholar]
- 18.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Zhang Y, Chang X, Zhang X. Imbalance in mitochondrial dynamics induced by low PGC-1alpha expression contributes to hepatocyte EMT and liver fibrosis. Cell Death Dis. 2020;11:226. doi: 10.1038/s41419-020-2429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnasamy Y, Gooz M, Li L, Lemasters JJ, Zhong Z. Role of mitochondrial depolarization and disrupted mitochondrial homeostasis in non-alcoholic steatohepatitis and fibrosis in mice. International journal of physiology, pathophysiology and pharmacology. 2019;11:190–204. [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Liu X, Nie J, Zhang J, Kimball SR, Zhang H, Zhang WJ, Jefferson LS, Cheng Z, Ji Q, Shi Y. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology. 2015;61:486–496. doi: 10.1002/hep.27420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang L, Liu K, Liu D, Lv F, Zang Y, Xie F, Yin J, Shi Y, Wang Y, Chen D. Differential effects of reticulophagy and mitophagy on nonalcoholic fatty liver disease. Cell Death Dis. 2018;9:90. doi: 10.1038/s41419-017-0136-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang NP, Liu XJ, Xie L, Shen XZ, Wu J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab Invest. 2019;99:749–763. doi: 10.1038/s41374-018-0177-6. [DOI] [PubMed] [Google Scholar]
- 25.Valkovic L, Chmelik M, Krssak M. In-vivo(31)P-MRS of skeletal muscle and liver: a way for non-invasive assessment of their metabolism. Anal Biochem. 2017;529:193–215. doi: 10.1016/j.ab.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmid AI, Szendroedi J, Chmelik M, Krssak M, Moser E, Roden M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care. 2011;34:448–453. doi: 10.2337/dc10-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oberhaensli RD, Galloway GJ, Taylor DJ, Bore PJ, Radda GK. Assessment of human liver metabolism by phosphorus-31 magnetic resonance spectroscopy. Br J Radiol. 1986;59:695–699. doi: 10.1259/0007-1285-59-703-695. [DOI] [PubMed] [Google Scholar]
- 29.Nair S, V PC, Arnold C and Diehl AM, Hepatic ATP reserve and efficiency of replenishing: comparison between obese and nonobese normal individuals. Am J Gastroenterol. 2003;98:466–470. doi: 10.1111/j.1572-0241.2003.07221.x. [DOI] [PubMed] [Google Scholar]
- 30.Bawden SJ, Stephenson MC, Ciampi E, Hunter K, Marciani L, Macdonald IA, Aithal GP, Morris PG, Gowland PA. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: a (31)P MRS study. Clin Nutr. 2016;35:645–649. doi: 10.1016/j.clnu.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 31.Adjeitey CN, Mailloux RJ, Dekemp RA, Harper ME. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am J Physiol Endocrinol Metab. 2013;305:E405–E415. doi: 10.1152/ajpendo.00057.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Hafidi M, Franco M, Ramirez AR, Sosa JS, Flores JAP, Acosta OL, Salgado MC, Cardoso-Saldana G. Glycine increases insulin sensitivity and glutathione biosynthesis and protects against oxidative stress in a model of sucrose-induced insulin resistance. Oxid Med Cell Longev. 2018;2018:2101562. doi: 10.1155/2018/2101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gnaiger E. Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol. 2009;41:1837–1845. doi: 10.1016/j.biocel.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 34.Ojuka E, Andrew B, Bezuidenhout N, George S, Maarman G, Madlala HP, Mendham A, Osiki PO. Measurement of beta-oxidation capacity of biological samples by respirometry: a review of principles and substrates. Am J Physiol Endocrinol Metab. 2016;310:E715–E723. doi: 10.1152/ajpendo.00475.2015. [DOI] [PubMed] [Google Scholar]
- 35.Grossini E, Garhwal DP, Calamita G, Romito R, Rigamonti C, Minisini R, Smirne C, Surico D, Bellan M, Pirisi M. Exposure to plasma from non-alcoholic fatty liver disease patients affects hepatocyte viability, generates mitochondrial dysfunction, and modulates pathways involved in fat accumulation and inflammation. Front Med (Lausanne) 2021;8:693997. doi: 10.3389/fmed.2021.693997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sookoian S, Rosselli MS, Gemma C, Burgueño AL, Fernández Gianotti T, Castaño GO, Pirola CJ. Epigenetic regulation of insulin resistance in nonalcoholic fatty liver disease: impact of liver methylation of the peroxisome proliferator–activated receptor γ coactivator 1α promoter. Hepatology. 2010;52:1992–2000. doi: 10.1002/hep.23927. [DOI] [PubMed] [Google Scholar]
- 37.Pirola CJ, Gianotti TF, Burgueño AL, Rey-Funes M, Loidl CF, Mallardi P, Martino JS, Castaño GO, Sookoian S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013;62:1356–1363. doi: 10.1136/gutjnl-2012-302962. [DOI] [PubMed] [Google Scholar]
- 38.Stevanovic J, Beleza J, Coxito P, Ascensao A, Magalhaes J. Physical exercise and liver “fitness”: role of mitochondrial function and epigenetics-related mechanisms in non-alcoholic fatty liver disease. Mol Metab. 2020;32:1–14. doi: 10.1016/j.molmet.2019.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galloway CA, Lee H, Brookes PS, Yoon Y. Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. American journal of physiology. Gastrointestinal and liver physiology. 2014;307:G632–G641. doi: 10.1152/ajpgi.00182.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du J, Zhang X, Han J, Man K, Zhang Y, Chu ESH, Nan Y, Yu J. Pro-inflammatory CXCR3 impairs mitochondrial function in experimental non-alcoholic steatohepatitis. Theranostics. 2017;7:4192–4203. doi: 10.7150/thno.21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong F, Gao L, Ding T. IDH2 protects against nonalcoholic steatohepatitis by alleviating dyslipidemia regulated by oxidative stress. Biochem Biophys Res Commun. 2019;514:593–600. doi: 10.1016/j.bbrc.2019.04.069. [DOI] [PubMed] [Google Scholar]
- 42.Piacentini M, Baiocchini A, Del Nonno F, Melino G, Barlev NA, Rossin F, D'Eletto M, Falasca L. Non-alcoholic fatty liver disease severity is modulated by transglutaminase type 2. Cell Death Dis. 2018;9:257–257. doi: 10.1038/s41419-018-0292-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 44.Caldwell SH, Chang CY, Nakamoto RK, Krugner-Higby L. Mitochondria in nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8(595–617):x. doi: 10.1016/j.cld.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Lionetti L, Mollica MP, Donizzetti I, Gifuni G, Sica R, Pignalosa A, Cavaliere G, Gaita M, De Filippo C, Zorzano A, Putti R. High-lard and high-fish-oil diets differ in their effects on function and dynamic behaviour of rat hepatic mitochondria. PLoS ONE. 2014;9:e92753. doi: 10.1371/journal.pone.0092753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazumder S, De R, Debsharma S, Bindu S, Maity P, Sarkar S, Saha SJ, Siddiqui AA, Banerjee C, Nag S, Saha D, Pramanik S, Mitra K, Bandyopadhyay U. Indomethacin impairs mitochondrial dynamics by activating the PKCzeta-p38-DRP1 pathway and inducing apoptosis in gastric cancer and normal mucosal cells. J Biol Chem. 2019;294:8238–8258. doi: 10.1074/jbc.RA118.004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herst PM, Rowe MR, Carson GM, Berridge MV. Functional mitochondria in health and disease. Front Endocrinol (Lausanne) 2017;8:296. doi: 10.3389/fendo.2017.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.da Cunha FM, Torelli NQ, Kowaltowski AJ. Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev. 2015;2015:482582. doi: 10.1155/2015/482582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cagin U, Enriquez JA. The complex crosstalk between mitochondria and the nucleus: What goes in between? Int J Biochem Cell Biol. 2015;63:10–15. doi: 10.1016/j.biocel.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 50.Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J. 2017;284:183–195. doi: 10.1111/febs.13820. [DOI] [PubMed] [Google Scholar]
- 51.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 52.Singh K, Modica-napolitano J. Special issue: mitochondria in cancer. Semin Cancer Biol. 2017;47:iv–vi. doi: 10.1016/j.semcancer.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Zuchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schroder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 54.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 55.Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–210. doi: 10.1038/79936. [DOI] [PubMed] [Google Scholar]
- 56.Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh KK and Modica-Napolitano JS. Special Issue: Mitochondria in Cancer. Semin Cancer Biol. 2017;47:iv-vi. 10.1016/j.semcancer.2017.10.013 [DOI] [PubMed]
- 58.Govindaraj P, Khan NA, Rani B, Rani DS, Selvaraj P, Jyothi V, Bahl A, Narasimhan C, Rakshak D, Premkumar K, Khullar M, Thangaraj K. Mitochondrial DNA variations associated with hypertrophic cardiomyopathy. Mitochondrion. 2014;16:65–72. doi: 10.1016/j.mito.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Weir HJ, Lane JD, Balthasar N. SIRT3: a central regulator of mitochondrial adaptation in health and disease. Genes Cancer. 2013;4:118–124. doi: 10.1177/1947601913476949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Gisbergen MW, Voets AM, Starmans MH, de Coo IF, Yadak R, Hoffmann RF, Boutros PC, Smeets HJ, Dubois L, Lambin P. How do changes in the mtDNA and mitochondrial dysfunction influence cancer and cancer therapy? Challenges, opportunities and models. Mutat Res Rev Mutat Res. 2015;764:16–30. doi: 10.1016/j.mrrev.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 61.de Mello AH, Costa AB, Engel JDG, Rezin GT. Mitochondrial dysfunction in obesity. Life Sci. 2018;192:26–32. doi: 10.1016/j.lfs.2017.11.019. [DOI] [PubMed] [Google Scholar]
- 62.Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab. 2012;23:435–443. doi: 10.1016/j.tem.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song K, Zhang Y, Ga Q, Bai Z, Ge RL. High-altitude chronic hypoxia ameliorates obesity-induced non-alcoholic fatty liver disease in mice by regulating mitochondrial and AMPK signaling. Life Sci. 2020;252:117633. doi: 10.1016/j.lfs.2020.117633. [DOI] [PubMed] [Google Scholar]
- 64.Nati M, Haddad D, Birkenfeld AL, Koch CA, Chavakis T, Chatzigeorgiou A. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH) Rev Endocr Metab Disord. 2016;17:29–39. doi: 10.1007/s11154-016-9339-2. [DOI] [PubMed] [Google Scholar]
- 65.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440:944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 66.Yin X, Lanza IR, Swain JM, Sarr MG, Nair KS, Jensen MD. Adipocyte mitochondrial function is reduced in human obesity independent of fat cell size. J Clin Endocrinol Metab. 2014;99:E209–E216. doi: 10.1210/jc.2013-3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iossa S, Lionetti L, Mollica MP, Crescenzo R, Botta M, Barletta A, Liverini G. Effect of high-fat feeding on metabolic efficiency and mitochondrial oxidative capacity in adult rats. Br J Nutr. 2003;90:953–960. doi: 10.1079/bjn2003000968. [DOI] [PubMed] [Google Scholar]
- 68.Raffaella C, Francesca B, Italia F, Marina P, Giovanna L, Susanna I. Alterations in hepatic mitochondrial compartment in a model of obesity and insulin resistance. Obesity (Silver Spring) 2008;16:958–964. doi: 10.1038/oby.2008.10. [DOI] [PubMed] [Google Scholar]
- 69.Mantena SK, Vaughn DP, Andringa KK, Eccleston HB, King AL, Abrams GA, Doeller JE, Kraus DW, Darley-Usmar VM, Bailey SM. High fat diet induces dysregulation of hepatic oxygen gradients and mitochondrial function in vivo. Biochem J. 2009;417:183–193. doi: 10.1042/BJ20080868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holmstrom MH, Iglesias-Gutierrez E, Zierath JR, Garcia-Roves PM. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am J Physiol Endocrinol Metab. 2012;302:E731–E739. doi: 10.1152/ajpendo.00159.2011. [DOI] [PubMed] [Google Scholar]
- 71.Holmstrom MH, Tom RZ, Bjornholm M, Garcia-Roves PM, Zierath JR. Effect of leptin treatment on mitochondrial function in obese leptin-deficient ob/ob mice. Metabolism. 2013;62:1258–1267. doi: 10.1016/j.metabol.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 72.Fleischman A, Kron M, Systrom DM, Hrovat M, Grinspoon SK. Mitochondrial function and insulin resistance in overweight and normal-weight children. J Clin Endocrinol Metab. 2009;94:4923–4930. doi: 10.1210/jc.2009-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritov VB, Menshikova EV, Azuma K, Wood R, Toledo FG, Goodpaster BH, Ruderman NB, Kelley DE. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab. 2010;298:E49–58. doi: 10.1152/ajpendo.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32:309–319. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brady LJ, Brady PS, Romsos DR, Hoppel CL. Elevated hepatic mitochondrial and peroxisomal oxidative capacities in fed and starved adult obese (ob/ob) mice. Biochem J. 1985;231:439–444. doi: 10.1042/bj2310439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ferreira FM, Palmeira CM, Seica R, Santos MS. Alterations of liver mitochondrial bioenergetics in diabetic Goto-Kakizaki rats. Metabolism. 1999;48:1115–1119. doi: 10.1016/s0026-0495(99)90124-5. [DOI] [PubMed] [Google Scholar]
- 78.Nguyen P, Leray V, Diez M, Serisier S, Le Bloc'h J, Siliart B, Dumon H. Liver lipid metabolism. J Anim Physiol Anim Nutr (Berl) 2008;92:272–283. doi: 10.1111/j.1439-0396.2007.00752.x. [DOI] [PubMed] [Google Scholar]
- 79.Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75:3313–3327. doi: 10.1007/s00018-018-2860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Falcon A, Doege H, Fluitt A, Tsang B, Watson N, Kay MA, Stahl A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2010;299:E384–E393. doi: 10.1152/ajpendo.00226.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doege H, Grimm D, Falcon A, Tsang B, Storm TA, Xu H, Ortegon AM, Kazantzis M, Kay MA, Stahl A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J Biol Chem. 2008;283:22186–22192. doi: 10.1074/jbc.M803510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilson CG, Tran JL, Erion DM, Vera NB, Febbraio M, Weiss EJ. Hepatocyte-specific disruption of CD36 attenuates fatty liver and improves insulin sensitivity in HFD-fed mice. Endocrinology. 2016;157:570–585. doi: 10.1210/en.2015-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Asterholm IW, Mundy DI, Weng J, Anderson RG, Scherer PE. Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab. 2012;15:171–185. doi: 10.1016/j.cmet.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kersten S, Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. 2017;136:75–84. doi: 10.1016/j.biochi.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 85.Gao Q, Jia Y, Yang G, Zhang X, Boddu PC, Petersen B, Narsingam S, Zhu Y-J, Thimmapaya B, Kanwar YS, Reddy JK. PPARα-deficient ob/ob obese mice become more obese and manifest severe hepatic steatosis due to decreased fatty acid oxidation. Am J Pathol. 2015;185:1396–1408. doi: 10.1016/j.ajpath.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, Cline GW, Wood PA, Shulman GI. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A. 2007;104:17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sanders FW, Griffin JL. De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc. 2016;91:452–468. doi: 10.1111/brv.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 89.Fabbrini E, Mohammed BS, Magkos F, Korenblat KM, Patterson BW, Klein S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–431. doi: 10.1053/j.gastro.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 91.Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116–141. doi: 10.1016/j.freeradbiomed.2020.02.025. [DOI] [PubMed] [Google Scholar]
- 92.Iozzo P, Bucci M, Roivainen A, Nagren K, Jarvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, Virtanen K, Savunen T, Salvadori PA, Ferrannini E, Knuuti J and Nuutila P. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139:846–56, 856 e1–6. 10.1053/j.gastro.2010.05.039 [DOI] [PubMed]
- 93.Katsarou A, Moustakas II, Pyrina I, Lembessis P, Koutsilieris M, Chatzigeorgiou A. Metabolic inflammation as an instigator of fibrosis during non-alcoholic fatty liver disease. World J Gastroenterol. 2020;26:1993–2011. doi: 10.3748/wjg.v26.i17.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen Z, Yu Y, Cai J, Li H. Emerging molecular targets for treatment of nonalcoholic fatty liver disease. Trends Endocrinol Metab. 2019;30:903–914. doi: 10.1016/j.tem.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 95.Peverill W, Powell LW, Skoien R. Evolving concepts in the pathogenesis of NASH: beyond steatosis and inflammation. Int J Mol Sci. 2014;15:8591–8638. doi: 10.3390/ijms15058591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Papatheodoridi A-M, Chrysavgis L, Koutsilieris M, Chatzigeorgiou A. The role of senescence in the development of nonalcoholic fatty liver disease and progression to nonalcoholic steatohepatitis. Hepatology. 2020;71:363–374. doi: 10.1002/hep.30834. [DOI] [PubMed] [Google Scholar]
- 97.Moustakas II, Katsarou A, Legaki A-I, Pyrina I, Ntostoglou K, Papatheodoridi A-M, Gercken B, Pateras IS, Gorgoulis VG, Koutsilieris M, Chavakis T, Chatzigeorgiou A. Hepatic senescence accompanies the development of NAFLD in non-aged mice independently of obesity. Int J Mol Sci. 2021;22:3446. doi: 10.3390/ijms22073446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sookoian S, Flichman D, Scian R, Rohr C, Dopazo H, Gianotti TF, Martino JS, Castaño GO, Pirola CJ. Mitochondrial genome architecture in non-alcoholic fatty liver disease. J Pathol. 2016;240:437–449. doi: 10.1002/path.4803. [DOI] [PubMed] [Google Scholar]
- 99.Wattacheril J, Sanyal AJ. Lean NAFLD: an underrecognized outlier. Curr Hepatol Rep. 2016;15:134–139. doi: 10.1007/s11901-016-0302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Komatsu M, Yazaki M, Tanaka N, Sano K, Hashimoto E, Takei Y, Song YZ, Tanaka E, Kiyosawa K, Saheki T, Aoyama T, Kobayashi K. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease. J Hepatol. 2008;49:810–820. doi: 10.1016/j.jhep.2008.05.016. [DOI] [PubMed] [Google Scholar]
- 101.Hakim A, Zhang X, DeLisle A, Oral EA, Dykas D, Drzewiecki K, Assis DN, Silveira M, Batisti J, Jain D, Bale A, Mistry PK, Vilarinho S. Clinical utility of genomic analysis in adults with idiopathic liver disease. J Hepatol. 2019;70:1214–1221. doi: 10.1016/j.jhep.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, Browning JD. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight. 2019 doi: 10.1172/jci.insight.127737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morris EM, Rector RS, Thyfault JP, Ibdah JA. Mitochondria and redox signaling in steatohepatitis. Antioxid Redox Signal. 2011;15:485–504. doi: 10.1089/ars.2010.3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21:739–746. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 105.Pirola CJ, Garaycoechea M, Flichman D, Castaño GO, Sookoian S. Liver mitochondrial DNA damage and genetic variability of cytochrome b – a key component of the respirasome – drive the severity of fatty liver disease. J Intern Med. 2021;289:84–96. doi: 10.1111/joim.13147. [DOI] [PubMed] [Google Scholar]
- 106.Chavin KD, Yang S, Lin HZ, Chatham J, Chacko VP, Hoek JB, Walajtys-Rode E, Rashid A, Chen CH, Huang CC, Wu TC, Lane MD, Diehl AM. Obesity induces expression of uncoupling protein-2 in hepatocytes and promotes liver ATP depletion. J Biol Chem. 1999;274:5692–5700. doi: 10.1074/jbc.274.9.5692. [DOI] [PubMed] [Google Scholar]
- 107.Fritsch M, Koliaki C, Livingstone R, Phielix E, Bierwagen A, Meisinger M, Jelenik T, Strassburger K, Zimmermann S, Brockmann K, Wolff C, Hwang JH, Szendroedi J, Roden M. Time course of postprandial hepatic phosphorus metabolites in lean, obese, and type 2 diabetes patients. Am J Clin Nutr. 2015;102:1051–1058. doi: 10.3945/ajcn.115.107599. [DOI] [PubMed] [Google Scholar]
- 108.Szendroedi J, Chmelik M, Schmid AI, Nowotny P, Brehm A, Krssak M, Moser E, Roden M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology. 2009;50:1079–1086. doi: 10.1002/hep.23093. [DOI] [PubMed] [Google Scholar]
- 109.Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Mendez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, Reinhard MK, McDougall DJ, Rocca JR, Yost RA, Cusi K, Garrett TJ, Sunny NE. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab. 2016;310:E484–E494. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ajaz S, McPhail MJ, Gnudi L, Trovato FM, Mujib S, Napoli S, Carey I, Agarwal K. Mitochondrial dysfunction as a mechanistic biomarker in patients with non-alcoholic fatty liver disease (NAFLD) Mitochondrion. 2021;57:119–130. doi: 10.1016/j.mito.2020.12.010. [DOI] [PubMed] [Google Scholar]
- 112.Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, Esterbauer H, Kozlov A, Kahn CR, Kroemer G, Rustin P, Burcelin R, Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 113.Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson N-G. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin Matthew J, An D, Lee Kevin Y, Ilkayeva Olga R, Zingaretti Cristina M, Emanuelli B, Smyth G, Cinti S, Newgard Christopher B, Gibson Bradford W, Larsson N-G, Kahn CR. Adipose-specific deletion of tfam increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16:765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.García-Ruiz C, Fernández-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatology communications. 2018;2:1425–1439. doi: 10.1002/hep4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cortez-Pinto H, Zhi Lin H, Qi Yang S, Odwin Da Costa S, Diehl AM. Lipids up-regulate uncoupling protein 2 expression in rat hepatocytes. Gastroenterology. 1999;116:1184–1193. doi: 10.1016/s0016-5085(99)70022-3. [DOI] [PubMed] [Google Scholar]
- 117.Morris EM, Jackman MR, Meers GM, Johnson GC, Lopez JL, MacLean PS, Thyfault JP. Reduced hepatic mitochondrial respiration following acute high-fat diet is prevented by PGC-1alpha overexpression. Am J Physiol Gastrointest Liver Physiol. 2013;305:G868–G880. doi: 10.1152/ajpgi.00179.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, Sastre J, Vendemiale G, Altomare E. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut. 2008;57:957–965. doi: 10.1136/gut.2007.147496. [DOI] [PubMed] [Google Scholar]
- 119.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 120.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 121.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47:1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang L, Seitz LC, Abramczyk AM, Chan C. Synergistic effect of cAMP and palmitate in promoting altered mitochondrial function and cell death in HepG2 cells. Exp Cell Res. 2010;316:716–727. doi: 10.1016/j.yexcr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Takeichi Y, Miyazawa T, Sakamoto S, Hanada Y, Wang L, Gotoh K, Uchida K, Katsuhara S, Sakamoto R, Ishihara T, Masuda K, Ishihara N, Nomura M, Ogawa Y. Non-alcoholic fatty liver disease in mice with hepatocyte-specific deletion of mitochondrial fission factor. Diabetologia. 2021;64:2092–2107. doi: 10.1007/s00125-021-05488-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sebastian D, Hernandez-Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P, Oresic M, Pich S, Burcelin R, Palacin M, Zorzano A. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci U S A. 2012;109:5523–5528. doi: 10.1073/pnas.1108220109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hernández-Alvarez MI, Sebastián D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, Plana N, Veiga SR, Hernández V, Vasconcelos N, Peddinti G, Adrover A, Jové M, Pamplona R, Gordaliza-Alaguero I, Calvo E, Cabré N, Castro R, Kuzmanic A, Boutant M, Sala D, Hyotylainen T, Orešič M, Fort J, Errasti-Murugarren E, Rodrígues CMP, Orozco M, Joven J, Cantó C, Palacin M, Fernández-Veledo S, Vendrell J, Zorzano A. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell. 2019;177:881–895.e17. doi: 10.1016/j.cell.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 127.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 128.Goncalves IO, Passos E, Rocha-Rodrigues S, Diogo CV, Torrella JR, Rizo D, Viscor G, Santos-Alves E, Marques-Aleixo I, Oliveira PJ, Ascensao A, Magalhaes J. Physical exercise prevents and mitigates non-alcoholic steatohepatitis-induced liver mitochondrial structural and bioenergetics impairments. Mitochondrion. 2014;15:40–51. doi: 10.1016/j.mito.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 129.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iozzo P, Bucci M, Roivainen A, Någren K, Järvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, Virtanen K, Savunen T, Salvadori PA, Ferrannini E, Knuuti J, Nuutila P. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology. 2010;139:846–856.e6. doi: 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 131.Leveille M and Estall JL. Mitochondrial dysfunction in the Transition from NASH to HCC. 2019;9. 10.3390/metabo9100233 [DOI] [PMC free article] [PubMed]
- 132.Bogacka I, Xie H, Bray GA, Smith SR. Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes. 2005;54:1392–1399. doi: 10.2337/diabetes.54.5.1392. [DOI] [PubMed] [Google Scholar]
- 133.Chatzigeorgiou A, Kandaraki E, Papavassiliou AG, Koutsilieris M. Peripheral targets in obesity treatment: a comprehensive update. Obes Rev. 2014;15:487–503. doi: 10.1111/obr.12163. [DOI] [PubMed] [Google Scholar]
- 134.Grattagliano I, Montezinho LP, Oliveira PJ, Fruhbeck G, Gomez-Ambrosi J, Montecucco F, Carbone F, Wieckowski MR, Wang DQ, Portincasa P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem Pharmacol. 2019;160:34–45. doi: 10.1016/j.bcp.2018.11.020. [DOI] [PubMed] [Google Scholar]
- 135.Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, Radovick S, Hussain M, Maheshwari A, Wondisford FE, O'Rourke B, He L. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep. 2019;29(1511–1523):e5. doi: 10.1016/j.celrep.2019.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kim J-a, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rosselli MS, Burgueño AL, Carabelli J, Schuman M, Pirola CJ, Sookoian S. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis. 2009;206:119–126. doi: 10.1016/j.atherosclerosis.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 138.Bi J, Zhang J, Ren Y, Du Z, Li Q, Wang Y, Wei S, Yang L, Zhang J, Liu C, Lv Y, Wu R. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019;20:296–306. doi: 10.1016/j.redox.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kang JW, Choi HS, Lee SM. Resolvin D1 attenuates liver ischaemia/reperfusion injury through modulating thioredoxin 2-mediated mitochondrial quality control. Br J Pharmacol. 2018;175:2441–2453. doi: 10.1111/bph.14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Barbier-Torres L, Fortner KA, Iruzubieta P, Delgado TC, Giddings E, Chen Y, Champagne D, Fernandez-Ramos D, Mestre D, Gomez-Santos B, Varela-Rey M, de Juan VG, Fernandez-Tussy P, Zubiete-Franco I, Garcia-Monzon C, Gonzalez-Rodriguez A, Oza D, Valenca-Pereira F, Fang Q, Crespo J, Aspichueta P, Tremblay F, Christensen BC, Anguita J, Martinez-Chantar ML, Rincon M. Silencing hepatic MCJ attenuates non-alcoholic fatty liver disease (NAFLD) by increasing mitochondrial fatty acid oxidation. Nat Commun. 2020;11:3360. doi: 10.1038/s41467-020-16991-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang N, Wang S, Li Y, Che L, Zhao Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett. 2013;535:104–109. doi: 10.1016/j.neulet.2012.12.049. [DOI] [PubMed] [Google Scholar]
- 142.Hasnat M, Yuan Z, Ullah A, Naveed M, Raza F, Baig M, Khan A, Xu D, Su Y, Sun L, Zhang L, Jiang Z. Mitochondria-dependent apoptosis in triptolide-induced hepatotoxicity is associated with the Drp1 activation. Toxicol Mech Methods. 2020;30:124–133. doi: 10.1080/15376516.2019.1669247. [DOI] [PubMed] [Google Scholar]
- 143.Basalay MV, Davidson SM, Gourine AV, Yellon DM. Neural mechanisms in remote ischaemic conditioning in the heart and brain: mechanistic and translational aspects. Basic Res Cardiol. 2018;113:25. doi: 10.1007/s00395-018-0684-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.
Not applicable.