

Abstract
Poly(A)‐binding protein (PABP) is an essential element of cellular translational machinery. Recent studies have revealed that poly(A) tail modifications can modulate mRNA stability and translational potential, and that oligoadenylate‐derived PABP ligands can act as effective translational inhibitors with potential applications in pain management. Although extensive research has focused on protein‐RNA and protein‐protein interactions involving PABPs, further studies are required to examine the ligand specificity of PABP. In this study, we developed a microscale thermophoresis‐based assay to probe the interactions between PABP and oligoadenylate analogs containing different chemical modifications. Using this method, we evaluated oligoadenylate analogs modified with nucleobase, ribose, and phosphate moieties to identify modification hotspots. In addition, we determined the susceptibility of the modified oligos to CNOT7 to identify those with the potential for increased cellular stability. Consequently, we selected two enzymatically stable oligoadenylate analogs that inhibit translation in rabbit reticulocyte lysates with a higher potency than a previously reported PABP ligand. We believe that the results presented in this study and the implemented methodology can be capitalized upon in the future development of RNA‐based biological tools.
Keywords: chemical modifications, microscale thermophoresis, mRNA, PABP, poly(A)
Oligonucleotide mRNA poly(A) tail analogs containing various nucleotide modifications were evaluated as binders for poly(A) binding protein (PABP) using a microscale thermophoresis‐based assay. Their susceptibility to specific degradation by CNOT7 deadenylase was also examined in order to identify best stabilizing modifications. Analogs exhibiting highest affinity and stability were tested in rabbit reticulocyte lysate system and proved to act as efficient translational inhibitors with potential applications in mRNA‐related research.

Introduction
Most eukaryotic mRNAs are modified during maturation by the addition of 5’ and 3’ regulatory elements known as the 7‐methylguanosine (m7G) cap and poly(A) tail, respectively. These elements protect mRNA from premature degradation[ 1 , 2 ] and play important roles in nucleocytoplasmic transport and translation through various RNA‐protein and protein‐protein interactions. [3] The poly(A) tail at the 3’ end of mRNA is directly recognized by poly(A)‐binding proteins (PABPs) [4] to form an elongated multimeric structure. [5] PABP also interacts with the scaffold protein, eIF4G, which is a key component of the m7G‐cap‐binding multiprotein complex, eIF4F. It is thought that these interactions bring mRNA ends together to form a “closed‐loop”,[ 6 , 7 ] which facilitates translation initiation [8] and stabilizes mRNA. However, increasing evidence has suggested that this simple model may not reflect the full complexity and dynamics of the mRNA‐protein interaction network. [9]
The general structure of eukaryotic cytoplasmic PABPs is highly conserved [10] and consists of four RNA recognition motif (RRM) domains and a C‐terminal PABC domain.[ 11 , 12 ] Monomeric PABP binds to a 24–27 nt fragment of the poly(A) tail; therefore, the total number of PABPs in multimeric complexes depends on poly(A) tail length.[ 5 , 13 ] Previous studies have reported that the complete structure of PABP (RRM 1‐4‐PABC) is not necessary for its biological functions, including eIF4G recognition[ 14 , 15 ] which can be carried out by two N‐terminal domains, RRM1 and RRM2. [15] In recombinant PABP, these domains align to create a narrow RNA binding crevice that can accommodate 11‐or 12‐nt oligoadenylate chains. [11]
The poly(A) tail is one of the primary regulators of mRNA fate. Deadenylation is the enzyme‐catalyzed shortening of the poly(A) tail, and is the first step in both major mRNA decay pathways (5’‐3’ and 3’‐5’). [16] The binding of PABP to the poly(A) tail promotes translation initiation and protects the poly(A) tail from degradation by deadenylases.[ 17 , 18 ] Recently, chemical and genetically‐encoded poly(A) modifications have been investigated intensively for mRNA labeling and to increase the translational efficiency of in vitro‐transcribed mRNAs for therapeutic use.[ 19 , 20 , 21 , 22 ] However, the impact of these modifications on PABP binding has not yet been systematically studied.
PABP has recently been linked to ‘pain receptor’‐mediated rapid translational initiation. [23] In particular, translational inhibition using a chemically modified poly(A) analog (SPOT‐ON) targeting PABP was shown to reduce behavioral responses to pain in mice. [23] The poly(A) analog (SPOT‐ON) contained multiple nucleotide modifications to ensure sufficient stability and biological activity. Oligoadenylate analogs (OAs) could therefore be added to the ever‐growing range of RNA‐based therapeutics, which includes antisense RNAs, splice‐correcting RNAs, siRNAs, miRNAs, and mRNAs.[ 24 , 25 ] Cost‐ and time‐effective methods to determine the structure‐activity relationship in PABP‐poly(A) interactions could significantly facilitate further studies on poly(A)‐modified therapeutic mRNAs and the development of next‐generation PABP‐targeting translational inhibitors. Although several methods, such as the electrophoretic mobility shift assay (EMSA) [26] and the selection/amplification assay, [27] have already been used to investigate the PABP‐poly(A) interaction, these methods are limited by relatively low resolution, high sample consumption, and semiquantitative results.
In this study, we developed a microscale thermophoresis (MST)‐based assay that enables the straightforward quantitative evaluation of the binding affinity between various modified oligonucleotide analogs and recombinant PABP. Using this assay, we evaluated a synthetic library of 28 OAs containing various modifications. We found that PABP binding affinity is highly dependent on both the type of modification and its position in the oligo, allowing us to identify hotspots that can be modified without obstructing the interaction with PABP. We also performed enzymatic experiments with recombinant deadenylase CNOT7 to establish how these modifications affect susceptibility to enzymatic degradation. We subsequently designed two OAs combining the identified beneficial features and studied their potential as translational inhibitors in a rabbit reticulocyte lysate (RRL) in vitro system. Notably, these compounds had superior translational inhibitory activity in RRL relative to SPOT‐ON.
Results and Discussion
Development of a competition assay to evaluate PABP ligands
To investigate the impact of RNA modifications on PABP binding affinity, we developed a competition assay based on MST, a biophysical method that enables observation of changes in the thermophoretic mobility of a fluorescently labeled molecule (e. g., small molecule, nucleic acid, protein) when it forms a complex with another entity (e. g., ion, small molecule, protein). [28] MST allows the precise determination of K D in the nM‐mM range with very low sample consumption. [29] Quantitative methods such as EMSA [26] and the selection/amplification assay [27] have approximated the K D of full‐length PABP with 25 nucleotide poly(A) fragments as 7 nM and qualitative methods have been used to study the affinities of PABP recombinants.[ 30 , 31 ] However, few in‐depth studies have examined the ligand specificity of PABP beyond its general affinity for polyadenylated RNA. We concluded that an MST‐based method would suit our needs, as the potential determination of nanomolar K D values was necessary, as well as precise detection of minor affinity changes due to small structural differences of the ligands, both of which MST is capable of. [32]
To avoid potential problems associated with PABP fluorescent labeling, we developed the assay in a competitive binding mode (Figure 1A). First, we selected an appropriate PABP construct and designed a fluorescently labeled oligoadenylate probe. We chose a shortened version of human PABP (PABP1−190 consisting of RRM1/2), which exhibits a high affinity for 11–12 nt poly(A) fragments (Figure 1B). [11] As a PABP‐binding probe, we employed a 12‐mer poly(A) analog labeled at the 5’ end with fluorescein (5’‐FAM‐A12). An A12 oligo equipped with a phosphohexynyl handle at the 5’ end (5’‐HexA12) was obtained by solid phase synthesis and labeled with 5’‐FAM azide via a CuAAC click reaction (Figure S1).
Figure 1.

Development of MST‐based competition binding assay to assess the influence of oligoadenylate modification on PABP interactions. A) Assay overview. B) Structure of PABP1−190 (RRM1/2; blue) in complex with poly(A) chain (yellow); PDB entry 4F02 by Safaee et al. [14] eIF4G protein PABP‐binding site is marked in green. C) Binding of 5’‐FAM−A12 by PABP. Top: MST traces from a typical direct binding experiment. Blue and red indicate cold and hot regions used for further analysis (temp. jump mode). Bottom: binding curve fitted to the MST data using a 1 : 1 binding model. D) Competition experiment; A12 ligand replaces 5’‐FAM‐A12 probe in the binding pocket of PABP1−190 binding pocket, resulting in a reversed curve.
To determine the K D of PABP1−190 for the probe, we performed a direct binding assay with varying PABP1−190 and constant 5’‐FAM‐A12 concentrations (Figure 1C). Assuming a simple 1 : 1 binding model, a K D value of 12±2 nM was determined for the complex, which is slightly higher than the 7 nM K D value for full‐length PABP and A25. [26] Next, we tested whether the unmodified A12 ligand could displace the probe from PABP1−190. A close‐to‐saturating PAPB concentration was mixed with 5’‐FAM‐A12 and varying concentrations of A12 (Figure 1D). The MST binding curve obtained by plotting the relative MST mobility as a function of ligand concentration enabled the determination of an apparent K D value (K D app ) for A12 of 780±130 nM, which was considered a benchmark for future experiments. Using a previously reported global fitting procedure, [33] we calculated an equilibrium K D value for the PABP1−190‐A12 complex of 18±4 nM (Figure S2).
Evaluation of PABP binding with differently modified OAs
Next, we prepared a set of 12‐mer OAs analogs with different modification types and placements (Figure 2). The selected modifications were interchain phosphorothioates (Aps or PS), 2’‐O‐methyladenosine (Am) and 2’‐O‐fluoroadenosine (AF), N6‐methyladenosine (m6A), and guanosine for adenosine substitution (G). These specific modifications were selected based on their potential ability to stabilize or modulate RNA properties, or because they occur naturally in poly(A). PS linkages are known to increase RNA stability and ribonuclease resistance,[ 34 , 35 ] and mRNAs containing PS modifications have been studied in prokaryotic and eukaryotic systems with different outcomes depending on modification density and sequence placement.[ 22 , 36 ] In eukaryotic cells, mRNAs containing oligo(A) tails with up to 30 % randomly distributed PS bonds displayed decreased susceptibility to deadenylation without affecting translation, whereas a higher proportion of PS disturbed translational activity. [22] Moreover, extensive PS modifications can lead to cytotoxicity and should be introduced strategically to avoid adverse effects. [37]
Figure 2.

Overview of oligoadenylate modifications evaluated in this study and their placement within the A12 chain. A) Structures of five evaluated nucleotide modifications. B) Modification placements and their combinations within the chain.
2’‐O‐methylation is commonly employed to ensure RNA stability against nucleases. [38] Unfortunately, this type of modification can also negatively affect RNA recognition. [39] OH‐to‐F substitution at the 2’‐position confers similar properties to 2’‐O‐methylation, providing RNAs with increased thermal stability and half‐life; [40] however, their effects on mRNA properties when present in poly(A) tails has not yet been studied. m6A is the most common natural internal mRNA modification and is found in poly(A) tails[ 41 , 42 ] at an estimated rate of one per 700–800 nucleotides in HeLa cells. [43] Since m6A plays an important role in pre‐mRNA maturation and transport, [44] it is important to consider its effect on mRNA binding processes. In some organisms, poly(A) tails are subdivided by short inserts consisting of other nucleotides with preference for G. [45] For example, in Arabidopsis thaliana, guanosine residues are found in approximately 10 % of poly(A) tails and can constitute up to 28 % of their structure. [46] By dividing poly(A) tails into pure‐adenosine fragments of varying lengths, guanosines interposed into the tail structure can alter PABP binding affinity and thus the translational efficiency of mRNAs. [46] These insertions can also increase mRNA stability by hindering certain deadenylases. [47] Here, we first introduced the selected modifications at precise positions near either end or in the middle of the oligoadenylate chain (Figure 2B).
The complete results of the competition assay for all 28 OAs are shown in Figure 3 and Table 1. The K D app varied significantly depending on the modification and its position. Most modifications placed closer to the middle of the oligonucleotide chain had an adverse effect on binding affinity (OAs: Am3, AF3, G3, and m6A2; Figures 3 and S2) compared to unmodified A12, whereas 3’ or 5’ terminal nucleotide modifications had little effect. Significant differences were also observed based on the type of modification. Although modifications in the middle part of the chain significantly decreased binding affinity, the extent of this effect varied greatly for different modifications (Figure S2). For example, AF2, Am2, G2, and m6A2 had K D app approximately 4‐, 7‐, 10‐, and 12‐fold higher, respectively, compared to unmodified A12, whereas the K D app of Aps7 and Aps8 remained similar to that of A12. Only one type of modification increased PABP binding affinity in some cases: m6A modification at the 3’ and 5’ ends of the m6A1 and m6A3 OAs, respectively, slightly increased PABP affinity compared to unmodified A12.
Figure 3.

Apparent binding affinity values (K D app ) for complexes between PABP and modified poly(A) analogs determined by an MST competition assay. Data sets represent different nucleotide modifications/substitutions. A) PS modification. B) Am modification. C) m6A modification. D) Guanosine for adenosine substitution. E) 2’‐F modification. F) Table of abbreviations. G) Compilation of all K D app values, from highest to lowest.
Table 1.
Binding affinity values (K D app ) between PABP and modified poly(A) analogs
|
Entry |
Abbreviation |
Oligonucleotide |
K D app [μM] |
SD [μM] |
|---|---|---|---|---|
|
1 |
A12 |
A12 |
0.73 |
0.13 |
|
2 |
Aps1 |
A9ApsA2 D1 |
1.59 |
0.18 |
|
3 |
Aps2 |
A9ApsA2 D2 |
0.52 |
0.10 |
|
4 |
Aps3 |
A10ApsA D1 |
1.28 |
0.16 |
|
5 |
Aps4 |
A10ApsA D2 |
0.66 |
0.12 |
|
6 |
Aps5 |
ApsA11 D1 |
0.58 |
0.70 |
|
7 |
Aps6 |
ApsA11D2 |
1.00 |
0.20 |
|
8 |
Aps7 |
A5ApsA6 D1 |
0.70 |
0.13 |
|
9 |
Aps8 |
A5ApsA6D2 |
0.80 |
0.30 |
|
10 |
Aps9 |
Aps11 A |
1.50 |
0.20 |
|
11 |
Aps10 |
ApsA9ApsA |
1.22 |
0.13 |
|
12 |
A11 |
A11 |
3.10 |
0.60 |
|
13 |
A13 |
A13 |
0.66 |
0.12 |
|
14 |
G1 |
A11G |
2.70 |
0.60 |
|
15 |
G2 |
A5GA6 |
13.00 |
3.00 |
|
16 |
G3 |
GA11 |
0.90 |
0.20 |
|
17 |
Am1 |
A10AmA |
1.00 |
0.20 |
|
18 |
Am2 |
A5AmA6 |
4.70 |
0.80 |
|
19 |
Am3 |
AmA11 |
1.10 |
0.30 |
|
20 |
Am4 |
Am11A |
46.0 |
5.0 |
|
21 |
Am5 |
AmA11Am |
0.83 |
0.18 |
|
22 |
Am6 |
AmpsAm9AmpsAm |
31.0 |
3.0 |
|
23 |
Am7 |
A11Am |
1.50 |
0.40 |
|
24 |
m6A1 |
A10m6A |
0.41 |
0.09 |
|
25 |
m6A2 |
A5m6A6 |
9.2 |
1.3 |
|
26 |
m6A3 |
m6A‐A11 |
0.48 |
0.06 |
|
27 |
AF1 |
A10AFA |
0.73 |
0.15 |
|
28 |
AF2 |
A5AFA6 |
2.90 |
0.40 |
|
29 |
AF3 |
AFA11 |
1.43 |
0.20 |
K D app – apparent dissociation constant value, determined via MST competition assay. SD – standard deviation of the KD, app values. Color‐coding: Aps – OAs with Aps modifications; A11 – OAs with G substitution and different chain lengths; Am – OAs with Am modifications; m6A – OAs with m6A modifications; AF – OAs with AF modifications
Based on these results, we designed analogs combining multiple modifications to further examine the impact of modification type and position on PABP affinity. The number of modified nucleotides consistently increased the K D app for Aps9, Aps10, Am4, and Am6 (Figure 3A, B). Moreover, OAs containing PS modifications (Aps9 and Aps10) performed better than their Am counterparts (Am4, Am5, and Am6/SPOT‐ON). Interestingly, there was little difference in binding affinity between the OA fully modified with PS (Aps9) and the OA modified only at the ends (Aps10). Conversely, the corresponding OAs containing Am modifications (Am5, Am4) performed differently, with Am5 (end modification only) retaining similar affinity to A12, and fully modified Am4 exhibiting a K D app that was two orders of magnitude higher. SPOT‐ON oligo containing multiple Am and PS modifications (Am6) had one of the lowest binding affinities for PABP among the tested oligos.
AF‐modified OAs exhibited similar patterns of affinity to PS, Am, and m6A OAs, as well as G‐modified OAs (Figure 3E). In particular, OAs modified at the ends showed either no change (AF1) or a small increase in K D app (AF3) compared to A12, while the one modified in the middle (AF2) displayed a more prominent increase. However, these changes were less significant than in OAs containing Am, m6A, or G modifications. Consequently, AF modifications were deemed more acceptable for PABP than the other modifications introduced in our study. Overall, PS modification was the least disruptive regardless of the position and number of modified subunits. Other modifications performed similarly to each other, with little or no effect on binding affinity compared to A12 when introduced at either end of the chain and with a significant increase in the K D app when introduced in the middle or throughout the chain.
Determination of deadenylation susceptibility
To evaluate their susceptibility to enzymatic degradation, the same 12‐mer poly(A) analogs were subjected to deadenylation assays using the CNOT7 catalytic subunit of the CCR4NOT deadenylase complex,[ 48 , 49 ] which has 3’→5’ poly(A) exoribonuclease activity. The compounds were incubated with CNOT7 for 30 min total at 37 °C. During incubation, aliquots from the reactions were taken at 5 min intervals, starting at 0 min. The samples were separated by high‐resolution polyacrylamide gel electrophoresis (Figure S3) with a CNOT7‐free sample as a negative control. Based on band intensity, the remaining fraction of the oligonucleotide substrate was calculated at each time point to determine the degradation rate (Figure 4I). PS and Am modifications provided full protection against CNOT7 deadenylase activity if located at the 3’ end (Figure 4A, B, H), whereas some modifications in the middle of the chain resulted in the accumulation of partial degradation products (Figure 4G, H). Regardless of the type, 5’ end modification provided no protection against degradation as it was the last subunit degraded by CNOT7 (Figure 4H). Interestingly, AF modification provided similar levels of resistance as Am and PS modifications; however, since AF subunits were situated at least one position away from the 3’ end due to synthetic reasons, CNOT7 partially degraded these analogs until it encountered the modified nucleotide (e. g., AF1, AF2, Figure 4C, G, H). Neither m6A modification nor G substitution substantially reduced susceptibility to degradation (Figure 4E, F) and both shorter and longer unmodified poly(A) chains were degraded at a similar rate to the unmodified A12 analog (Figure 4D).
Figure 4.

Degradation of modified poly(A) analogs by CNOT7 deadenylase. Degradation products were separated using urea polyacrylamide gel electrophoresis and the gel was visualized with SYBR Gold nucleic acid stain. Data points represent the normalized band intensity of the degradation substrate. Degradation rate heavily depended on modification type and position: A) Phosphorothioate modification; B) Am modification; C) AF modification; D) poly(A) analog length; E) Guanosine substitution; F) m6A nucleobase modification; G) increase in concentration of CNOT7‐resistant degradation products of primary substrates containing specific modifications in the middle of the oligonucleotide chain. H) Images of polyacrylamide gels after electrophoretic separation of CNOT7‐treated oligonucleotide analogs; I) initial degradation rates. No bar indicates no poly(A) analog degradation after 30 min incubation with CNOT7 deadenylase.
Poly(A) analogs as translational inhibitors
Finally, we evaluated the OAs with high affinity for PABP1−190 and low susceptibility to deadenylation as inhibitors of cap‐dependent translation in an RRL model. Both AmA10Am (Am5) and ApsA9ApsA (Aps10) exhibited complete resistance to CNOT7 degradation and relatively low KD app values for PABP binding (0.83 and 1.22 μM, respectively). The inhibitory activity of these nucleotides was compared to that of a cap‐derived translation inhibitor (m7GpSppG) [50] and a previously reported poly(A) analog, SPOT‐ON (Am6; AmpsAm10AmpsA). [23] Both Am5 and Aps10 were potent translation inhibitors (Figure 5A) with IC50 values (12 and 25 μM, respectively) higher than that of m7GpSppG (1 μM) but lower than the IC50 of SPOT‐ON (108 μM).
Figure 5.
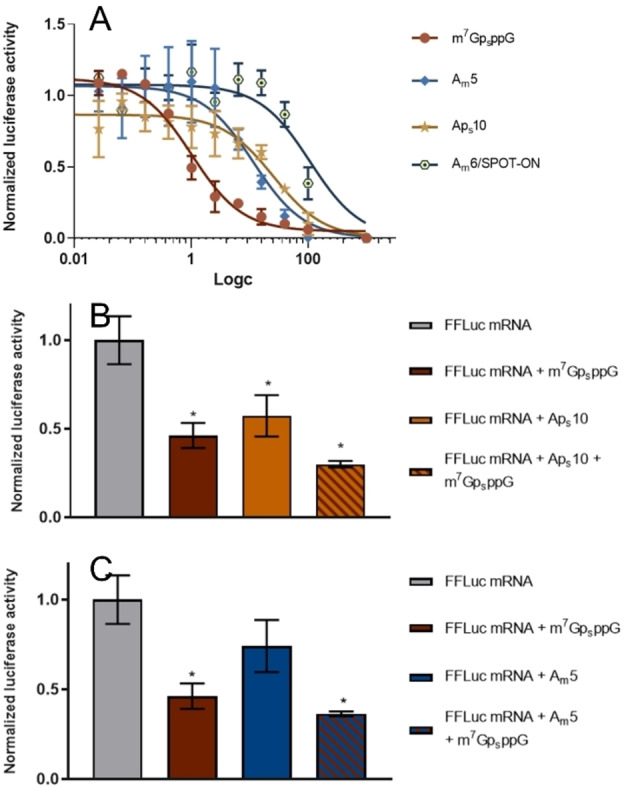
Influence of poly(A) analogs on the translation efficiency of mRNAs with poly(A) tails. The measured luminescence of the reaction product catalyzed by firefly luciferase is assumed to correlate with mRNA translation efficiency. Data represent the mean values ± standard deviation (SD) obtained from three experiments of A) translation inhibition by poly(A) analogs (Am5, Aps10 or Am6/SPOT‐ON) and m7GpsppG plotted as a function of ligand concentration or co‐inhibition by m7GpsppG at 1.03 μM and B) Aps10 or C) Am5 at 28 μM normalized to the translation efficiency of mRNA alone. Statistical significance: (*) P<0.05, (**) P<0.01, (***) P<0.001, (****) P<0.0001 (one‐way analysis of variance [ANOVA] with Dunnett's multiple comparison test).
The Am6 analog, SPOT‐ON, was previously shown to be very stable and bind to PABP with high affinity. [23] Since we found that multiple oligonucleotide modifications had a negative effect on binding affinity, we decided to measure the binding affinity and inhibitory potential of SPOT‐ON using the same methodology as that used to study Am5 and Aps10 inhibitors. As indicated previously (Figure 3B), SPOT‐ON exhibited much lower affinity for PABP (K D app 31 μM) than the unmodified OA A12 (K D app 0.73 μM). Translation inhibition assays also supported these initial findings, revealing that SPOT‐ON had higher IC50 (108 μM) for PABP than the other two inhibitors. Thus, reducing the number of modifications and introducing them at strategic positions resulted in much stronger PABP inhibition in the RRL system. Further experiments confirmed the additive potency of our PABP inhibitors (Am5 and Aps10) and a known eIF4E binder – m7GpsppG (Figure 5B, C), suggesting that PABPs could be targeted to inhibit translation alongside existing approaches. The apparent correlation between PABP affinity and inhibition coefficients in RRL strongly suggest that our inhibitors reduce translation efficiency by specifically targeting PABP and not through non‐specific interactions with translation machinery. Moreover, it is important to note that RRL system contains full‐length PABP, and thus, the qualitative agreement between MST and in vitro experiments in RRL suggests that the results obtained for PABP1−190 are translatable to more complex biological systems. However, it is also important to underline several limitations of the RRL system, compared to cell culture and in vivo studies, in which SPOT‐ON has been previously evaluated. RLL contains practically only translational machinery, while it lacks degradation machinery and other cellular mechanisms important for RNA activity, and has relatively weak poly(A)‐dependence. [51] Therefore, verification of our findings in more complex biological models will be required in the future.
Conclusion
In summary, we have developed a highly effective methodology for studying the interactions between shortened human PABP variant (PABP1−190 , RRM1/2) and its oligoadenylate ligands. Our MST‐based assay allowed us to determine the binding affinities between PABP1−190 and OAs while registering changes caused by minute structural differences within the ligands. We also prepared a small library of Oas with different nucleotide modifications and substitutions which allowed us to identify 5’ and 3’ ends as modification hotspots, i. e., sites that can be modified without disrupting the binding affinity for PABP. Moreover, we observed significant differences based on the type of modification introduced into the Oas: primarily, PS and AF modifications were the least disruptive and most acceptable for PABP1−190 binding. For other modification types, the position of the modified subunit within the oligoadenylate chain was the most crucial factor. Generally, Oas modified at 3’, 5’, or both ends retained a similar binding affinity to unmodified A12. Together, these findings improve our understanding of the structure‐activity relationship of PABP‐poly(A) interactions and ligand specificity of PABP and contribute toward the design of future high‐affinity inhibitors. To the best of our knowledge this is the first study looking at quantitative differences in PABP binding affinities evoked by chemical modification of oligoadenylate chains.
By performing enzymatic deadenylation experiments featuring CNOT7 deadenylase, we were able to identify Oas that were completely resistant to this type of specific degradation. Both the position and type of modification affected these results; for instance, neither G nor m6A modifications suppressed deadenylation, but PS, Am, and AF modifications effectively stopped CNOT7 deadenylase, especially when introduced at the 3’ end of the OA chain.
Finally, we selected two OAs with high affinity for PABP1−190 and full resistance to specific deadenylation and evaluated them as translation inhibitors in the RRL system alongside the poly(A) analog SPOT‐ON [23] and a known cap‐derived eIF4E inhibitor. Although all three OAs had inhibitory potential, our results strongly suggested that introducing a small number of modifications at specific positions can be more beneficial than fully modifying the oligoadenylate chain, as is the case with SPOT‐ON. Indeed, in our MST‐based binding affinity assay, SPOT‐ON exhibited a much lower affinity for PABP1−190 than our Am5 and Aps10 OAs (which had affinity close to unmodified A12) and had an IC50 value in RRL that was one order of magnitude higher than those of our inhibitors. However, this hypothesis requires further verification in cell culture and in vivo models.
The field of RNA‐based therapeutics is ever‐expanding and poly(A) tail modification has attracted increasing interest as a potential way to improve mRNA stability and translational efficiency. Although more in‐depth research is necessary to fully evaluate chemically‐modified OAs or mRNAs carrying them as potential therapeutic agents, we believe that our implemented methodology and findings can be capitalized upon for the future development of RNA‐based biological tools.
Experimental Section
Chemical synthesis
General information on oligonucleotide solid‐phase synthesis procedures
Solid‐phase synthesis was performed using AKTA Oligopilot synthesizer. Reagents and solvents were sourced commercially. Five phosphoramidite equivalents were used for each coupling step. Oligonucleotides were synthesized on a 10 μmol scale.
Cleavage from the solid support with simultaneous deprotection was performed using AMA (1 : 1 concentrated aqueous ammonia and concentrated aqueous methylamine) at 50 °C for 1 h, followed by filtration (to remove solid support particles) and freeze‐drying. Final deprotection of the 2’‐OH group was performed using triethylamine trihydrofluoride in DMSO at 60 °C for 4 h. After precipitation in n‐butanol with sodium acetate (final concentration 75 μM), analogs were purified using an Agilent Tech. Series 1200 HPLC with a Clarity Oligo‐RP C‐18 column (150×10 mm, flow rate 5 mL/min) in triethylamonium acetate (TEAAc) buffer with a linear acetonitrile gradient, coupled with UV‐Vis detection at 260 nm. The structure and homogeneity of the synthesized oligonucleotides were confirmed using high‐resolution mass spectrometry with negative electrospray ionization.
A12 : Ribo A300 CPG 1000/110 support (34 mg, 10 μmol) was suspended in dry acetonitrile. 2’‐TBDSilyl adenosine (n‐acetyl) CED phosphoramidite (642 mg, 650 μmol; LinkTech, Biosearch Technologies) was dissolved in acetonitrile to a final concentration of 0.2 M. The synthesis product was cleaved, deprotected, and purified according to the general procedure.
5’‐Hex‐A12 : Synthesis was performed using a similar method as for A12 synthesis, with additional final coupling with 5′‐hexynyl phosphoramidite (29.8 mg, 100 μmol; Glen Research), which was also dissolved in dry acetonitrile to a final concentration of 0.2 M.
Am, m6A, AF and G oligonucleotides: Modified polyA analogs were synthesized using a similar protocol as for the A12 but with particular coupling steps performed using dry acetonitrile solutions of 2’‐O‐methyl‐rA(bz) phosphoramidite, N6‐methyl‐A‐CE phosphoramidite, and 2‐F−Ac‐C‐CE phosphoramidite or Ac−G‐CE phosphoramidite (LinkTech, Biosearch Technologies).
Phosphorothioate‐modified oligonucleotides: Oligonucleotides containing phosphorothioate linkages were obtained using the method used for A12 synthesis with certain couplings followed by sulfurization with 3‐ethoxy‐1,2,4‐dithiazoline‐5‐one (0.05 M solution in acetonitrile) to form a phosphorothioate (PS) linkage.
5’‐FAM‐A12 : 5’‐Hex‐A12 (1 mg, 234 nmol) was dissolved in 9.5 μL of water to afford 25 mM solution and then 1 μL of 94 mM aqueous solution of CuSO4 (0.4 equiv.) and 1 μL of 0.94 M aqueous solution of sodium ascorbate (4 equiv.) were added. Next, 5‐FAM‐azide (3.5 μL, 100 mM solution in DMSO, 1.5 equiv.; Lumiprobe) was added to the final volume of 15 μL. The final concentrations of the 5’‐HEX‐A12, CuSO4, sodium ascorbate and 5‐FAM‐azide substrates were as follows: 15.8 mM, 6.3 mM, 63 mM, 23.3 mM. The reaction was stirred at room temperature for approximately 1 h until full conversion was achieved. The product was purified using reverse‐phase high‐performance liquid chromatography (HPLC) and the collected eluate was freeze‐dried to afford 0.8 mg (178 nmol) of solid 5’‐FAM‐A12 (yield: 76 %). The structure and homogeneity of the synthesized oligonucleotide probe was confirmed using high‐resolution mass spectrometry with negative electrospray ionization. HRMS ESI (‐) m/z calculated for C149H169N64O79P12 − [M‐2H]2− 2244.90326, found 2244.89680.
MST binding assay
Sixteen solutions with decreasing PABP concentrations were prepared in MST buffer (50 mM HEPES, 100 mM KCl, 1 mM EDTA, 0.2 % Tween20, pH 7.4) by serial half‐log dilution, starting from the highest PABP concentration of 10 μM. Each solution was mixed with an equal volume of 50 nM 5’‐FAM‐A12 solution in the same buffer (final concentration of the probe in the sample was 25 nM). After 15 min incubation at room temperature, Monolith NT.115 Capillaries were filled with each solution and MST was measured (25 °C, blue detector, 20 % laser power). Thermophoresis data was analyzed using Palmist software (version 1.5.8). [33] Data points for the binding curve were acquired using Temperature Jump (TJ) mode. The binding curve was fitted using 1 : 1 direct binding model and the K D value was calculated as an average value ± S.D. from at least three independent measurements. Raw data from all MST measurements (MST traces) and binding curves can be found in the Supporting Information file.
MST competition assay
PABP (600 nM) and 5’‐FAM‐A12 (50 nM) were incubated for 15 min at room temperature in MST buffer. Sixteen solutions with decreasing concentrations of the poly(A) analog in the same buffer were prepared by serial half‐log dilution, starting from the highest concentration of 150 μM. Equal volumes of the PABP/5’‐FAM‐A12 solution was then added to each poly(A) analog sample and incubated for 15 min at room temperature (the final concentrations in the assay of PABP, 5’‐FAM‐A12, and poly(A) analog were 300 nM, 25 nM, 75 μM–23 nM, respectively). Sixteen Monolith NT.115 Capillaries were filled with each of the prepared solutions and MST was measured for each sample (25 °C, blue detector, 20 % laser power). Thermophoresis data was analyzed using Palmist software. The data points for the binding curve were acquired using Temperature Jump (TJ) mode. The binding curve was fitted using 1 : 1 direct binding model and the K D app values were calculated as an average value ± S.D. from at least three independent measurements. Raw data from all MST measurements (MST traces) and binding curves can be found in the Supporting Information file.
CNOT7 degradation assay
The reaction was initiated by mixing 30 μL of 6.25 ng/mL CNOT7 [22] solution in a reaction buffer (Tris 25 mM, 25 mM DTT, 12.5 mM MgCl2, 125 mM KCl, pH 8.0) with 7.5 μL aqueous solution of poly(A) analog (100 μM) to afford following final concentrations: 5 ng/mL CNOT7, 20 μM poly(A) analog, 20 mM Tris, 20 mM DTT, 10 mM MgCl2, 100 mM KCl. Reactions were incubated for total time of 30 min at 37 °C, 750 rpm. During the incubation, an aliquot (3.75 μL) from each reaction was taken every 5 min and stopped by mixing with 4 μL of loading dye and aqueous EDTA (25 mM) mixture and put on ice. A control sample was prepared by mixing poly(A) analog and buffer solutions at the same concentrations without CNOT7.
High resolution urea‐polyacrylamide gel electrophoresis
First, 36 mL of 18 % polyacrylamide and 8 M urea solution in Tris‐Borate‐EDTA (TBE) buffer were mixed with 360 μL of 10 % ammonium persulfate (APS) and 14.4 μL of Tetramethylethylenediamine (TEMED). After polymerization in a gel‐casting chamber (20 cm×22 cm×1 mm), a 30 min pre‐run (23 W) was performed to heat the gel. Samples (3.5 μL) were then loaded and electrophoretic separation was performed for 1.5 h. The gel was stained for 10 min using SYBR gold dye solution in water and imaged using a Typhoon Gel Imaging System. Band intensity was determined using ImageJ software.
PABP1−190 expression and purification
The sequence encoding the N‐terminal region of the human PABP1 protein (Gene ID: 26986) containing the RRM1 and RRM2 motifs (PABP1−190) was amplified from the pET11_PABP1 plasmid using PABP1_hs_FL_SL_for (5'‐GAAGTCTACCAGGAACAAACCGGTGGATCCATGAACCCCAGTGCCCCCAG‐3') PABP1_hs_190_SL_rev (5'‐CTCAGTGGTGGTGGTGGTGGTGGTGCTCGAGTTATTAGAATTCTTTTGCCCTAGCTCC‐3') primers. Amplified sequence was further subcloned using the SLIC approach into a pET28‐derived expression vector linearized with BamHI and XhoI restriction enzymes. The fusion protein was overexpressed in an E. coli BL21(DE3) CodonPlus‐RIL (Stratagene) expression strain in LB medium supplemented with kanamycin (50 μg/mL) and chloramphenicol (32 μg/mL). 6×His‐SUMO‐PABP1−190 expression was induced with 1 mM IPTG at an optical density (OD600) of ∼0.6. Cells were cultured for a further 16 h at 18 °C, spun down at 5000×g at 4 °C for 15 min, and stored at −20 °C. For purification, cells from 250 mL of culture were thawed, resuspended in 30 mL of lysis buffer (100 mM NaCl, 30 mM Tris‐HCl pH 8.5, 10 % glycerol, 5 mM β‐mercaptoethanol, and 10 mM imidazole) supplemented with protease inhibitors (aprotinin, leupeptin, pepstatin, and PMSF) and lysozyme (1 mg/mL) and incubated on ice for 30 min. After the NaCl concentration in the lysis solution had been increased to 500 mM, cells were incubated on ice for 15 min and disrupted by sonication. The cell extract was clarified by centrifugation at 15000 rpm at 4 °C and filtered using syringe filters with a 0.45 μm PVDF membrane. Clarified lysates were loaded onto a 5 mL HisTrap FF column (GE Healthcare) equilibrated with buffer containing 500 mM NaCl, 30 mM Tris‐HCl pH 8.5, 10 % glycerol, 5 mM β‐mercaptoethanol, and 10 mM imidazole. The 6×His‐SUMO‐PABP1−190 protein was eluted using equilibration buffer supplemented with 300 mM imidazole. To remove the N‐terminal 6×His‐SUMO tag, 5 μL His6_Ulp1 Sumo protease (MCLAB) was added to 10 mL of the eluted 6×His‐SUMO‐PABP1−190 protein. The mixture was dialyzed overnight at 4 °C against a buffer containing 500 mM NaCl, 30 mM Tris‐HCl pH 8.5, 5 % glycerol, 0.5 mM EDTA, 0.5 mM DTT, and 10 mM imidazole. After being filtered using a 0.45 μm PVDF membrane, the mixture was loaded onto a 5 mL HisTrap FF column (GE Healthcare) equilibrated with dialysis buffer. The flow‐through fraction containing PABP1−190 was filtered, concentrated using an Amicon Ultra‐15 (Millipore) filtration unit, and loaded onto a Hi Load 26/600 Superdex 75 gel filtration column (GE Healthcare), equilibrated with buffer containing 100 mM NaCl, 30 mM Tris‐HCl pH 7.5, 5 % glycerol, 0.5 mM EDTA, and 0.5 mM DTT. Proteins collected after size‐exclusion chromatography were analyzed using 12 % PAGE and selected fractions were concentrated using an Amicon Ultra‐4 10 kDa MWCO (Millipore) filtration unit to 2.26 mg/mL (105 μM), aliquoted, flash‐frozen with liquid nitrogen, and stored at −80 °C. The final A260/280 ratio of ∼0.6 confirmed that PABP1−190 was not contaminated with RNA.
Preparation of firefly luciferase‐coding mRNA
The mRNAs for studying translation inhibition were prepared by in vitro transcription. The final transcript contained a short 5’ UTR, ORF, two consecutive H. sapiens β‐globin 3’ UTRs, and poly(A)35 tail. [52] The reaction (30 μL) was conducted for 4 h at 40 °C in transcription buffer (40 mM Tris‐HCl pH 7.9, 10 mM MgCl2, 1 mM DTT, and 2 mM spermidine), 10 U/μL SP6 HC RNA polymerase (ThermoFisher Scientific), 2 U/μL RiboLock RNase inhibitor, 2 mM ATP, CTP, and UTP, 0.25 mM GTP, 2.5 mM of “anti‐reverse” cap analog (ARCA) m2 7,3′−OGpppG, [53] and 100 ng/μL of the PCR‐amplified template. Crude mRNA was purified using NucleoSpin RNA Clean‐up XS (Macherey‐Nagel) and then by RP HPLC using an Agilent Technologies Series 1200 HPLC with an RNASepTM Prep (ADS Biotec) column at 55 °C, applying a 40–60 % linear gradient of buffer B (0.1 M triethylammonium acetate pH 7.0 and 25 % acetonitrile) in buffer A (0.1 M triethylammonium acetate pH 7.0) over 25 min at 0.9 mL/min. mRNA was recovered by precipitation with NaOAc pH 5.2 (final concentration 0.3 M) and one volume of isopropanol, followed by two washing steps with 0.4 mL of 70 % ethanol. The mRNA concentration was determined from the extinction coefficient predicted for the actual mRNA sequence using DNA Calculator software (www.molbiotools.com) and the spectrophotometric absorbance measurement at 260 nm. mRNA purity was analyzed by 1× TBE 1 % agarose gel electrophoresis.
Translation efficiency assay
An RRL system (Promega) was used to study the influence of the poly(A) analogs on the translation efficiency of firefly luciferase‐coding mRNA. RRL (4 μL) was diluted with 4 μL of translation buffer containing 100 μM amino acid mixture, 475 mM KOAc, and 2.5 mM MgOAc, and incubated for 1 h at 30 °C. Next, 1 μL of 0.5 ng/μL HPLC‐purified mRNA mixed with 1 μL of poly(A) analog or m7GpsppG dilution (0.0262–100 μM) was added to 8 μL of the translation mixture and incubated for 1 h at 30 °C. The reaction was stopped by freezing in liquid N2. To detect luminescence, 50 μL of Luciferase Assay Reagent (0.47 mM luciferin (VivoGlo™ Luciferin, in vivo grade; Promega), 20 mM tricine, 3.55 mM Mg(HCO3)2, 2.66 mM MgSO4, 0.1 mM EDTA, 33.3 mM DTT, 0.53 mM ATP, and 0.27 mM coenzyme A) was added to 10 μL of the translation mixture, thawed on ice, and diluted 2× with H2O. Luminescence was measured using a Synergy H1 (BioTek) microplate reader. To compare the effect of poly(A) analogs on translation efficiency, the mean luminescence values were normalized to the luminescence signal obtained for samples without ligands.
Co‐inhibition by poly(A) analogs and m7GpsppG
One microliter of 0.5 ng/μL HPLC‐purified mRNA mixed with either 1 μL of 280 μM poly(A) analog (Am5 or Aps10), 1 μL of 10.3 μM m7GpsppG, or both were added to the translation mixture (4 μL RRL mixed with 4 μL translation buffer), preincubated for 1 h at 30 °C and incubated for 1 h after ligand addition. Luminescence was measured as described above. To compare the translation efficiencies of firefly luciferase‐coding mRNA in the presence of m7GpsppG, poly(A) analogs, or both, mean values were normalized to the luminescence of samples without ligands.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Mariusz Czarnocki‐Cieciura (International Institute of Molecular and Cell Biology, Warsaw, Poland) for providing purified recombinant human CNOT7 deadenylase and professor Megerditch Kiledjian (Rutgers University, Piscataway, USA) for providing plasmid pET11_PABP1. The research was financially supported by the National Science Centre, Poland (2019/33/B/ST4/01843 to J.J. and 2018/31/B/ST5/03821 to J.K.).
O. Perzanowska, M. Smietanski, J. Jemielity, J. Kowalska, Chem. Eur. J. 2022, 28, e202201115.
Contributor Information
Prof. Jacek Jemielity, Email: j.jemielity@cent.uw.edu.pl.
Dr. Joanna Kowalska, Email: jkowalska@fuw.edu.pl.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Ramanathan A., Robb G. B., Chan S.-H., Nucleic Acids Res. 2016, 44, 7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dreyfus M., Régnier P., Cell 2002, 111, 611–613. [DOI] [PubMed] [Google Scholar]
- 3. Liu S., Li B., Liang Q., Liu A., Qu L., Yang J., WIREs RNA 2020, 11, e1601. [DOI] [PubMed] [Google Scholar]
- 4. Bernstein P., Ross J., Trends Biochem. Sci. 1989, 14, 373–377. [DOI] [PubMed] [Google Scholar]
- 5. Sawazaki R., Imai S., Yokogawa M., Hosoda N., Hoshino S., Mio M., Mio K., Shimada I., Osawa M., Sci. Rep. 2018, 8, 1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vicens Q., Kieft J. S., Rissland O. S., Mol. Cell 2018, 72, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Archer S. K., Shirokikh N. E., Hallwirth C. V., Beilharz T. H., Preiss T., RNA Biol. 2015, 12, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Costello J., Castelli L. M., Rowe W., Kershaw C. J., Talavera D., Mohammad-Qureshi S. S., Sims P. F. G., Grant C. M., Pavitt G. D., Hubbard S. J., Ashe M. P., Genome Biol. 2015, 16, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thompson M. K., Gilbert W. V., Curr. Genet. 2017, 63, 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mangus D. A., Evans M. C., Jacobson A., Genome Biol. 2003, 4, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deo R. C., Bonanno J. B., Sonenberg N., Burley S. K., Cell 1999, 98, 835–845. [DOI] [PubMed] [Google Scholar]
- 12. Kozlov G., Trempe J.-F., Khaleghpour K., Kahvejian A., Ekiel I., Gehring K., Proc. Nat. Acad. Sci. 2001, 98, 4409 LP – 4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin J., Fabian M., Sonenberg N., Meller A., Biophys. J. 2012, 102, 1427–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Safaee N., Kozlov G., Noronha A. M., Xie J., Wilds C. J., Gehring K., Mol. Cell 2012, 48, 375–386. [DOI] [PubMed] [Google Scholar]
- 15.S. H. Kessler, A. B. Sachs, Mol. Cell. Biol. 1998, 18, 51–57. [DOI] [PMC free article] [PubMed]
- 16. Chen C.-Y. A., Shyu A.-B., WIREs RNA 2011, 2, 167–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nicholson A. L., Pasquinelli A. E., Trends Cell Biol. 2019, 29, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jalkanen A. L., Coleman S. J., Wilusz J., Semin. Cell Dev. Biol. 2014, 34, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aditham A., Shi H., Guo J., Zeng H., Zhou Y., Wade S. D., Huang J., Liu J., Wang X., ACS Chem. Biol. 2022, DOI: 10.1021/acschembio.1c00569. [DOI] [PubMed] [Google Scholar]
- 20. Westerich K. J., Chandrasekaran K. S., Gross-Thebing T., Kueck N., Raz E., Rentmeister A., Chem. Sci. 2020, 11, 3089–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mamot A., Sikorski P. J., Siekierska A., de Witte P., Kowalska J., Jemielity J., Nucleic Acids Res. 2022, 50, e3–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strzelecka D., Smietanski M., Sikorski P. J., Warminski M., Kowalska J., Jemielity J., RNA 2020, 26, 1815–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barragán-Iglesias P., Lou T.-F., Bhat V. D., Megat S., Burton M. D., Price T. J., Campbell Z. T., Nat. Commun. 2018, 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuijper E. C., Bergsma A. J., Pijnappel W. W. M. P., Aartsma-Rus A., J. Inherited Metab. Dis. 2021, 44, 72–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nissim L., Wu M.-R., Pery E., Binder-Nissim A., Suzuki H. I., Stupp D., Wehpaun C., Tabach Y., Sharp P. A., Lu T. K., Cell 2017, 171, 1138–1150.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kühn U., Pieler T., J. Mol. Biol. 1996, 256, 20–30. [DOI] [PubMed] [Google Scholar]
- 27. Görlach M., Burd C. G., Dreyfuss G., Exp. Cell Res. 1994, 211, 400–407. [DOI] [PubMed] [Google Scholar]
- 28. Jerabek-Willemsen M., André T., Wanner R., Roth H. M., Duhr S., Baaske P., Breitsprecher D., J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar]
- 29. Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S., Nat. Commun. 2010, 1, 100. [DOI] [PubMed] [Google Scholar]
- 30. Melo E. O., Dhalia R., de Sa C. M., Standart N., de Melo Neto O. P., J. Biol. Chem. 2003, 278, 46357–46368. [DOI] [PubMed] [Google Scholar]
- 31. Burd C. G., Matunis E. L., Dreyfuss G., Mol. Cell. Biol. 1991, 11, 3419–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jerabek-Willemsen M., Wienken C. J., Braun D., Baaske P., Duhr S., Assay Drug Dev. Technol. 2011, 9, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scheuermann T. H., Padrick S. B., Gardner K. H., Brautigam C. A., Anal. Biochem. 2016, 496, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Volk D. E., Lokesh G. L. R., Biomed. 2017, 5, 41. [Google Scholar]
- 35. Duschmalé J., Hansen H. F., Duschmalé M., Koller E., Albaek N., Møller M. R., Jensen K., Koch T., Wengel J., Bleicher K., Nucleic Acids Res. 2020, 48, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kawaguchi D., Kodama A., Abe N., Takebuchi K., Hashiya F., Tomoike F., Nakamoto K., Kimura Y., Shimizu Y., Abe H., Angew. Chem. Int. Ed. 2020, 59, 17403–17407; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 17556–17560. [Google Scholar]
- 37. Amarzguioui M., Holen T., Babaie E., Prydz H., Nucleic Acids Res. 2003, 31, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hernandez F. J., Stockdale K. R., Huang L., Horswill A. R., Behlke M. A., McNamara 2nd J. O., Nucleic Acid Ther. 2012, 22, 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choung S., Kim Y. J., Kim S., Park H.-O., Choi Y.-C., Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [DOI] [PubMed] [Google Scholar]
- 40. Piao X., Wang H., Binzel D. W., Guo P., RNA 2018, 24, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.J. A. Bokar, in (Ed.: H. Grosjean), Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 141–177.
- 42. Roundtree I. A., Evans M. E., Pan T., He C., Cell 2017, 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lavi U., Fernandez-Muñoz R., Darnell J. E. J., Nucleic Acids Res. 1977, 4, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Finkel D., Groner Y., Virology 1983, 131, 409–425. [DOI] [PubMed] [Google Scholar]
- 45. Liu Y., Nie H., Liu H., Lu F., Nat. Commun. 2019, 10, 5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao T., Huan Q., Sun J., Liu C., Hou X., Yu X., Silverman I. M., Zhang Y., Gregory B. D., Liu C.-M., Qian W., Cao X., Genome Biol. 2019, 20, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lim J., Kim D., Lee Y., Ha M., Lee M., Yeo J., Chang H., Song J., Ahn K., Kim V. N., Science 2018, 361, 701 LP – 704. [DOI] [PubMed] [Google Scholar]
- 48. Wahle E., Winkler G. S., Biochim. Biophys. Acta 2013, 1829, 561–570. [DOI] [PubMed] [Google Scholar]
- 49. Mostafa D., Takahashi A., Yanagiya A., Yamaguchi T., Abe T., Kureha T., Kuba K., Kanegae Y., Furuta Y., Yamamoto T., Suzuki T., RNA Biol. 2020, 17, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kowalska J., Lewdorowicz M., Zuberek J., Grudzien-Nogalska E., Bojarska E., Stepinski J., Rhoads R. E., Darzynkiewicz E., Davis R. E., Jemielity J., RNA 2008, 14, 1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wakiyama M., Futami T., Miura K., Biochimie 1997, 79, 781–785. [DOI] [PubMed] [Google Scholar]
- 52. Warminski M., Sikorski P. J., Warminska Z., Lukaszewicz M., Kropiwnicka A., Zuberek J., Darzynkiewicz E., Kowalska J., Jemielity J., Bioconjugate Chem. 2017, 28, 1978–1992. [DOI] [PubMed] [Google Scholar]
- 53. Stepinski J., Waddell C., Stolarski R., Darzynkiewicz E., Rhoads R. E., RNA 2001, 7, 1486–1495. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.