

Abstract
The reverse orthogonal strategy was invented in 2011 in an attempt to address drawbacks of other strategies for glycan assembly. Different from the classical orthogonal approach that relies on the orthogonality of leaving groups, the reverse strategy is based on orthogonal protecting groups that could be removed during the glycosylation step. This strategy remained largely unexplored due to only one combination of orthogonal protecting groups that would fit into this concept. Reported herein are new orthogonal combinations of leaving and protecting groups that help to streamline the glycan assembly. Also reported is further refinement of the previously reported reaction conditions.
Graphical Abstract
INTRODUCTION
Chemical synthesis provides an immensely powerful means to obtain natural and unnatural glycans for the study of their properties and roles. However, even with significant progress, the chemical synthesis of glycans and glycoconjugates remains very challenging.1 Traditional oligosaccharide synthesis comprises a stepwise linear approach according to which first glycosylation takes place between two monosaccharide building blocks, a glycosyl donor equipped with a suitable leaving group (LG) and glycosyl acceptor carrying a free hydroxyl group. Upon glycosylation, a disaccharide derivative is obtained (Scheme 1). The latter is then converted into a glycosyl acceptor of the second generation via liberation of a specific hydroxyl group. This is typically performed as a separate synthetic step involving chemoselective removal of a strategically placed temporary protecting group (PG) and may also include additional chromatographic purification. The disaccharide acceptor is then reacted with a new glycosyl donor, resulting in the formation of a trisaccharide. The deprotection–glycosylation sequence is then reiterated to yield a tetrasaccharide and others. The requirement to perform an additional deprotection step or even multiple steps between each glycosylation is the major disadvantage of the conventional linear approach. However, since the monosaccharide donor is used at every step, the rates of glycosylations are easy to maintain, and the yields typically remain high, even with longer oligosaccharide acceptors.2 Essentially, the same strategic blueprint was used for the automated polymersupported synthesis of 30-mer3 and 50-mer4 mannans by Seeberger et al.
Scheme 1.

Traditional Linear Synthesis of Glycans
Chemoselective or selective activation of leaving groups forms the basis for many modern expeditious strategies for glycan synthesis.5 Regardless of the underpinning principles for differentiating or tuning the reactivity of building blocks, all of these strategies help to eliminate protecting group manipulations between coupling steps. This, in turn, leads to streamlined glycan assembly. Among these strategies is the orthogonal concept invented by Kanie et al.6,7 This method relies on the differential reactivity (orthogonality) of two leaving groups (LGA and LGB, Scheme 2, S-phenyl and fluoride in the original study).
Scheme 2.

Orthogonal Oligosaccharide Synthesis
Availability of two complementary (orthogonal) sets of reaction conditions that would independently activate one leaving group, but not the other, is the key for success of the orthogonal strategy. For example, activator A will selectively activate LGA of the glycosyl donor, and LGB installed at the anomeric center of the glycosyl acceptor will remain intact under these reaction conditions. Conversely, activator B will selectively activate LGB, whereas LGA remains intact. This set of two orthogonal reaction conditions is the key feature of the orthogonal approach that hypothetically allows for an unlimited number of reiterations of leaving groups. This sets the orthogonal approach apart from other approaches based on selective activation of different leaving groups, whereas the more reactive leaving group is activated over the less reactive one and does not permit further reiterations that are available only in the orthogonal approach.
While the orthogonal approach aims to become an ideal way to construct larger glycans, in practice, it has been unable to reach this efficiency. The yields of glycosylations decrease rapidly with the increase in the bulk of the glycosyl donor: di (85%) → tri (72%) → tetra (66%).6 In our related study, wherein S-thiazolinyl (STaz) versus SEt orthogonal activation was achieved, a similar observation was made, and the following decrease in yields was recorded: di (98%) → tri (93%) → tetra (77%) → penta (59%).8
Aiming at improving the state-of-the-art of glycan synthesis, previously, we communicated a new concept that we named the reverse orthogonal strategy.9 This strategy looked to employ the advantages of both traditional synthesis (high and consistent yields) and the orthogonal strategy (less steps) into one superior platform. Different from Ogawa’s orthogonal approach that relies on the orthogonality of leaving groups, we based our approach on orthogonal protecting groups. This resulted in the change of the direction of the glycan chain elongation, the reverse approach. Thus, while the glycan chain elongation during orthogonal activation takes at the reducing end (left to right), the elongation during the reverse approach proceeds at the non-reducing end, from right to left, just like in the conventional, linear assembly.
This reverse strategy requires two orthogonal protecting groups (PGA and PGB) that could be removed during the glycosylation step, and, in principle, the couplings can be executed with only one type of a leaving group (Scheme 3). Thus, in the first step, the glycosyl donor bearing PGB was reacted with a glycosyl acceptor bearing PGA under conditions A. During this step, PGA is removed, and the liberated hydroxyl is glycosylated to form the respective disaccharide derivative bearing PGB that remains intact during this step.
Scheme 3.

Reverse Orthogonal Strategy
In the second step, a new glycosyl donor bearing PGA was set to react with a disaccharide acceptor bearing PGB under conditions B. During this step, PGB is removed, and the liberated hydroxyl is glycosylated to form the corresponding trisaccharide derivative bearing PGA that remains intact during this step. These deprotection/glycosylation steps can then be reiterated until a glycan of the desired length has been assembled.
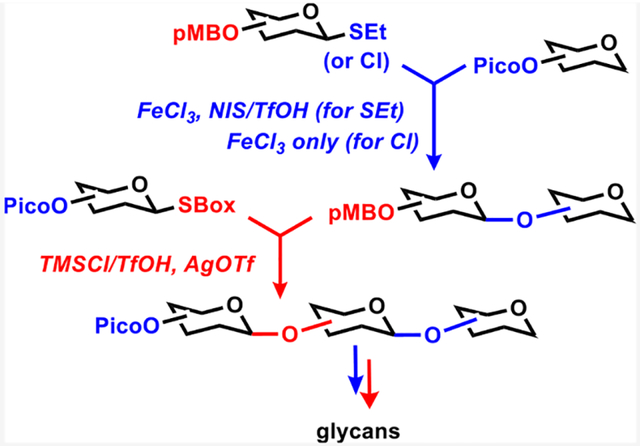
To execute this concept, we identified an orthogonal protecting group combination that comprised pentenoyl (Pent, PGA) and p-methoxybenzyl (pMB) groups. Pent could be deprotected followed by glycosylation of the liberated hydroxyl in the presence of NIS/TfOH/H2O. Conversely, p-methoxybenzyl (pMB) could be deprotected followed by glycosylation of the liberated hydroxyl in the presence of TMSI/AgOTf. We have also matched two different leaving groups, S-ethyl and S-benzoxazolyl (SBox). While not strictly necessary, the two leaving groups provided the best outcome in terms of reaction rates and product yields. As a result of this preliminary study, building blocks A and B shown in Scheme 3 allowed us to synthesize a 1 → 6-linked pentasaccharide with good efficiency and consistent yields: di (81%) → tri (82%) → tetra (71%) → penta (75%). One remaining downside to the reverse orthogonal approach is a limited scope because it remained applicable to only this combination of protecting (and leaving) groups.
RESULTS AND DISCUSSION
With a goal of extending the reverse approach to other orthogonal combinations and hence expanding the scope of the reverse orthogonal strategy, reported herein are new promising orthogonal PG-LG combinations.10 This is not to be confused with the concept of “orthogonal protecting groups” that are commonly referred to as orthogonally removable protecting groups.11–14 A few other concepts for protecting group-based glycosylations are known. Kochetkov and others demonstrated that triphenylmethyl (trityl) ether-substituted glycosyl acceptors can be glycosylated directly with cyanoethylidene,15,16 thiocyanate,17,18 or thioglycoside donors.19,20 Approaches involving “glycodesilylation” of TMS or TBDMS-protected glycosyl acceptors have also been developed.21–27
The premise for the development of new orthogonal PG-LG combinations described herein is our recent studies dedicated to iron catalysis in carbohydrate synthesis. On one hand, we showed that iron(III) chloride (FeCl3) could be used for the activation of glycosyl chloride donors.28 On the other hand, we discovered that FeCl3 could also be used to selectively cleave the picoloyl (Pico) protecting group.29 Because both of these methods, chloride LG activation and Pico PG removal, needed the same reagent FeCl3, we hypothesized that this offers a possibility for establishing another leaving-protecting group combination that would fit into the reverse orthogonal concept.
To evaluate our theory, we synthesized two glycosyl acceptors 1 and 2 depicted in Scheme 4. Compound 1 was obtained via a one-step protection of a common 6-OH precursor30 using the standard picoloylation protocol.31 Compound 2 was obtained as previously reported.9 We have also obtained two glycosyl donors 3 and 4, as shown in Scheme 4. The synthesis of 6-pMB protected donor 3 originated from known p-methoxybenzylidene-protected compound 5,32 which was subjected to the reductive acetal opening with NaCNBH3 in the presence of trifluoroacetic acid to afford 6-pMB derivative 6 with high regioselectivity in 95% yield. Subsequent benzoylation of the liberated 4-OH group afforded compound 7. The latter was converted into the desired glycosyl chloride donor 3 in a high yield via a conventional two-step thioglycoside hydrolysis with NBS in wet acetone followed by chlorination of the intermediate hemiacetal with oxalyl chloride.28 The synthesis of 6-Pico substituted SBox donor 4 originated from known thioglycoside precursor 8,33 which was protected with picolinic acid in the presence of EDC and DMAP to afford compound 9 in 98% yield. The latter was then converted into the desired SBox glycosyl donor 4 in 94% yield over a two-step procedure involving bromination with bromine followed by the SBox leaving group introduction using KSBox in the presence of 18-crown-6 in acetone.
Scheme 4.

New Building Blocks 1–4 for the Reverse Orthogonal Activation
With building blocks 1–4 in hand, we investigated the respective orthogonal coupling reactions. Acceptor 1 and SBox donor 4 are each equipped with the 6-Pico group, whereas acceptor 2 and chloride donor 3 are each equipped with the 6-pMB protecting group. In accordance with our design, glycosyl chloride donor 3 will pair with 6-Pico building blocks 1 and 4 in the presence of anhydrous FeCl3 to deprotect the Pico group and activate the chloride LG, thereby allowing for the intended deprotection/glycosylation in a single step. SBox donor 4 will then pair with pMB-protected building blocks 2 and 3. In this case, deprotection/glycosylation will be affected in the presence of TMSI (to remove pMB) and AgOTf (to activate SBox), as shown in our previous study.9
With these considerations, we first investigated the reaction of glycosyl acceptor 1 with glycosyl donor 3. A thorough preliminary experimentation brought us to a realization that we could not perform these reactions in a one-pot manner. Due to the Pico group cleavage requiring the presence of a nucleophile, normally methanol, we had to do the deprotection/glycosylation with the intermediary removal of methanol. This was executed as follows. Removal of the Pico group from acceptor 1 using 30 mol % of anhydrous FeCl3 in MeOH:CH2Cl2 (9:1, v/v) occurred in 15 min (Scheme 5). The reaction mixture was then concentrated and dried in vacuo for 1.5 h. The residue was redissolved in dichloromethane, molecular sieves (4 Å) were added, and the resulting mixture was stirred for 1 h. Donor 3 was then added along with another portion (30 mol %) of FeCl3. Based on preliminary experimentation, the additional FeCl3 was required to drive this reaction to completion. As a result, disaccharide 10 was obtained in 70% yield. Generally satisfied with the outcome of this coupling, we carried out the next glycosylation using glycosyl donor 4. Applying similar reaction conditions to those previously developed, TMSI was used to selectively cleave off the p-methoxybenzyl group from disaccharide 10 followed by the activation of donor 4 using silver triflate. This resulted in the formation of trisaccharide 11 in 65% yield. When this coupling was repeated in the presence of TMSCl instead of the iodide, trisaccharide 11 was obtained in an improved 70% yield. The first step was then repeated with 6″-Pico protected trisaccharide acceptor 11 and glycosyl chloride donor 3 to obtain the desired tetrasaccharide 12 in 58% yield.
Scheme 5.
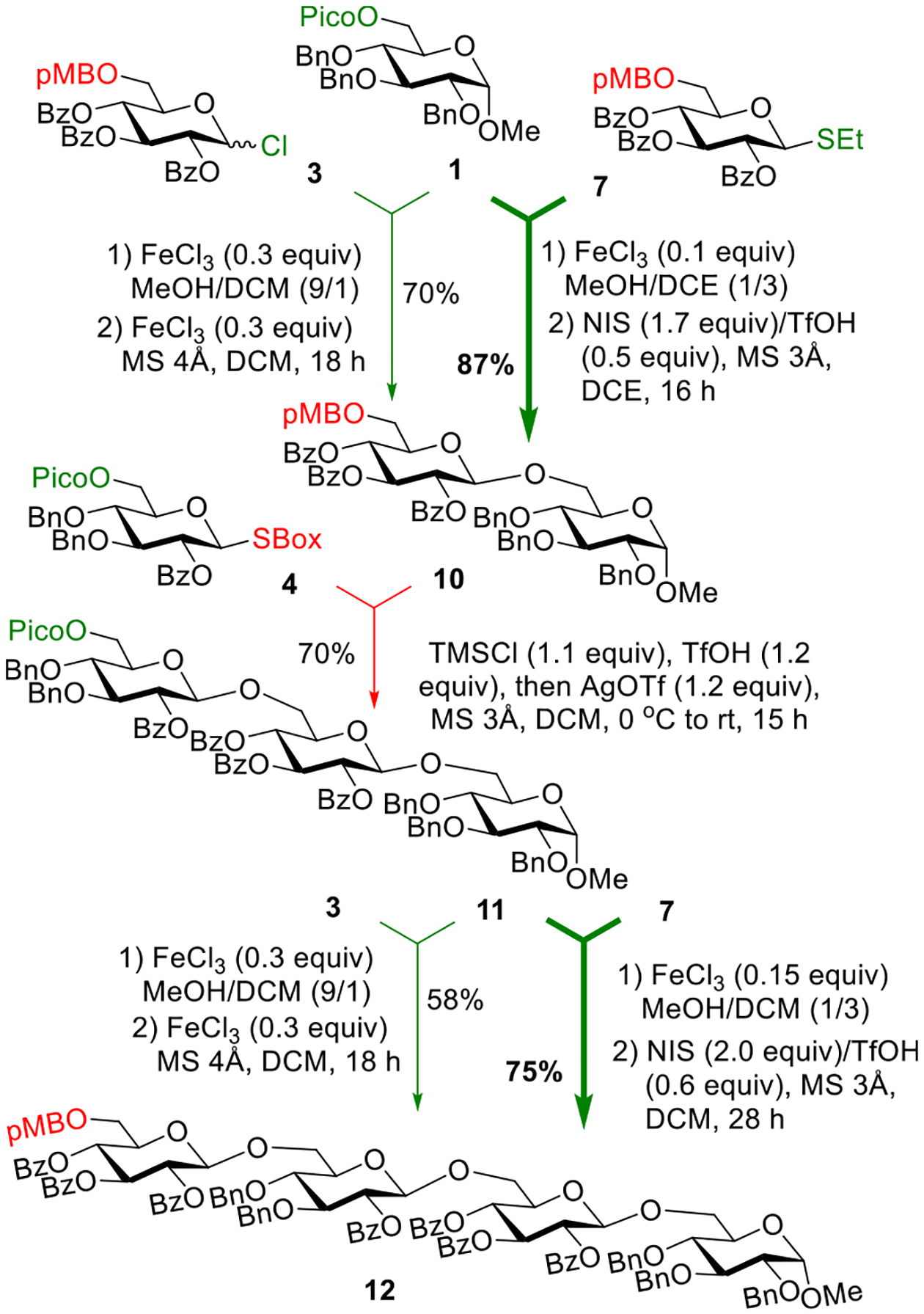
Three-Step Assembly of Tetrasaccharide 12 Using the Reverse Orthogonal Strategy
In a subsequent attempt to enhance the outcome of this synthesis, we have also investigated 6-pMB-protected thioglycoside donor 7 instead of chloride 3 for the first and the third steps of the assembly. This resulted in a significant enhancement of reaction yields for these steps. Thus, when donor 7 was activated for reaction with depicoloylated acceptor 1 in the presence of NIS/TfOH, disaccharide 10 was isolated in 87% yield (vs 70% yield obtained from chloride donor 3). When donor 7 was activated for reaction with depicoloylated trisaccharide acceptor 11 in the presence of NIS/TfOH, tetrasaccharide 12 was isolated in 75% yield (vs 57% yield obtained from chloride donor 3). Another possible solution for this would have been application of the NIS/FeCl3 promoter system.34
Following the general success of the original scheme, we sought to see whether reversing the synthetic steps would offer any benefit. Starting with acceptor 2 and donor 4, we first performed the deprotection/glycosylation using TMSI and AgOTf (Scheme 6). As a result, disaccharide 13 was obtained in 58% yield. Replacing TMSI with TMSCl and adding TfOH led to a swift pMB group deprotection (10 min). Subsequent glycosylation that was affected by the addition of AgOTf produced disaccharide 13 in an excellent yield of 90%. Continuing the synthesis, trisaccharide 14 was produced from glycosyl chloride donor 3 and 6′-Pico protected disaccharide 13 in 48% yield using FeCl3 mediated deprotection/glycosylation reactions, as detailed for the synthesis of 10 (vide supra). Like in the previous synthesis, we have also investigated 6-pMB-protected thioglycoside donor 7 instead of chloride 3. This resulted in a significant enhancement of the reaction yield. Thus, when donor 7 was activated for the reaction with depicoloylated acceptor 13 in the presence of NIS/TfOH, trisaccharide 14 was isolated in 73% yield.
Scheme 6.
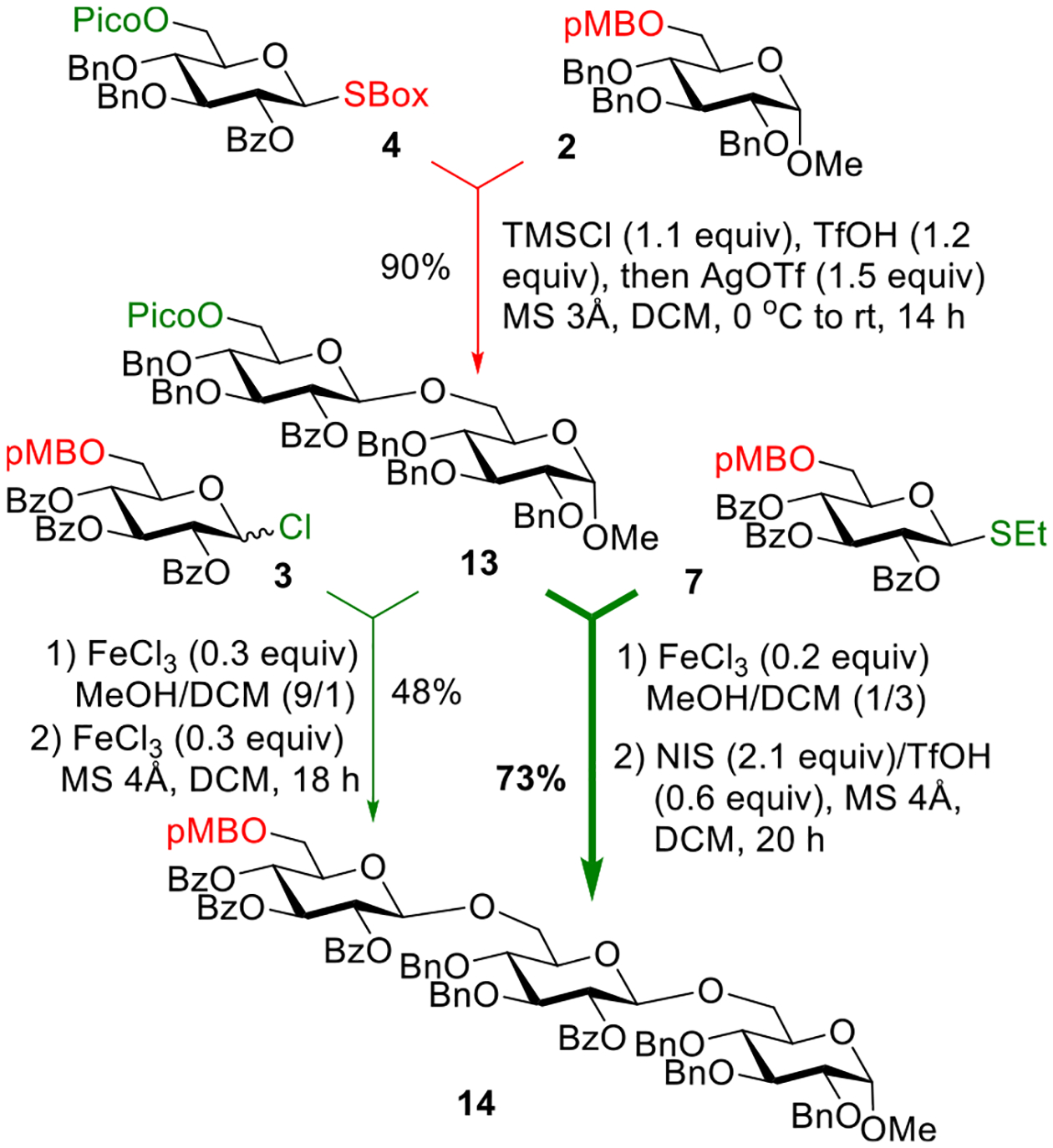
Synthesis of Trisaccharide 14 Using an Alternative Sequence
CONCLUSIONS
It has been demonstrated that the reverse orthogonal strategy is a useful strategy for synthetic carbohydrate chemists to help reduce the number of steps compared to traditional linear synthetic routes. It however was limited in scope due to the restriction to only two certain LG-PG combinations. We have successfully shown that the scope of the reverse orthogonal strategy can be expanded to new LG-PG combinations comprising glycosyl chloride or thioglycoside donors and picoloylated acceptors. These new reaction conditions were shown to pair well with the existing LG-PG pair comprising the SBox glycosyl donor and p-methoxybenzyl protected glycosyl acceptor. The efficacy of this coupling was enhanced by developing modified reaction conditions comprising TMSCl/TfOH and AgOTf. These new orthogonal combinations were evaluated in application to the syntheses of a tetrasaccharide and trisaccharide that were obtained in good-to-excellent yields using conventions of the reverse orthogonal strategy.
EXPERIMENTAL SECTION
General.
Column chromatography was performed on silica gel 60 (70–230 mesh), and reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 and ClCH2CH2Cl (1,2-DCE) were distilled from CaH2 directly prior to application. Pyridine was dried by refluxing with CaH2, then distilled, and stored over molecular sieves (3 Å). Anhydrous DMF was used as it is. Molecular sieves (3 or 4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Optical rotations were measured using a Jasco polarimeter. 1H NMR spectra were recorded in CDCl3 at 400 or 700 MHz. 13C{1H} NMR spectra were recorded in CDCl3 at 100 or 175 MHz. The 1H NMR chemical shifts are referenced to tetramethyl silane (TMS, δH = 0 ppm) or CDCl3 (δH = 7.26 ppm) for 1H NMR spectra for solutions in CDCl3. The 13C{1H} NMR chemical shifts are referenced to the central signal of CDCl3 (δC = 77.00 ppm) for solutions in CDCl3. Structural assignments were made with additional information from COSY and HSQC experiments. Compound purity or compound ratios were accessed or calculated by comparing of the integration intensities of the relevant signals in their 1H NMR spectra. Accurate mass spectrometry determinations were performed using an Agilent 6230 ESI TOF LCMS mass spectrometer.
Synthesis of Monosaccharide Building Blocks.
Methyl 2,3,4-Tri-O-benzyl-6-O-picoloyl-α-d-glucopyranoside (1).
Picolinic acid (168.1 mg, 1.35 mmol), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, 258.8 mg, 1.35 mmol), and 4-dimethylaminopyridine (DMAP, 16.6 mg, 0.14 mmol) were added to a solution of methyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside30 (314.6 mg, 0.68 mmol) in dichloromethane (10 mL), and the resulting mixture was stirred under argon for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~75 mL) and washed with water (2 × 15 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to give the title compound as a colorless foam in 95% yield (366.5 mg, 0.64 mmol). Analytical data for 1: Rf = 0.3 (ethyl acetate/hexane, 1/1, v/v); [α]D23 48.7 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.74 (m, 1H, aromatic), 8.01 (d, 1H, aromatic), 7.79 (m, 1H, aromatic), 7.45 (m, 1H, aromatic), 7.38−7.20 (m, 15H, aromatic), 5.01 (d, 1H, 2J = 10.7 Hz, CHPh), 4.91 (d, 1H, 2J = 11.0 Hz, CHPh), 4.84 (d, 1H, 2J = 10.7 Hz, CHPh), 4.80 (d, 1H, 2J = 12.1 Hz, CHPh), 4.67 (d, 1H, 2J = 12.1 Hz, CHPh), 4.63 (d, 1H, 2J = 11.0 Hz, CHPh), 4.62 (d, 1H, J1,2 = 3.3 Hz, H-1), 4.59−4.53 (m, 2H, H-6a, 6b), 4.05 (t, 1H, J3,4 = 9.2 Hz, H-3), 3.98 (m, 1H, J5,6a = 4.8, J5,6b = 2.6 Hz, H-5), 3.61 (dd, 1H, J4,5 = 9.2 Hz, H-4), 3.57 (dd, 1H, J2,3 = 9.2 Hz, H-2), 3.38 (s, 3H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 164.7 (COPh, Pico), 150.0, 147.8, 138.5, 138.0, 137.8, 136.8, 128.5 (×2), 128.4 (×2), 128.1 (×2), 128.0 (×4), 127.9, 127.8, 127.7, 126.8, 97.9 (C-1), 82.1 (C-3), 80.0 (C-2), 77.6 (C-4), 75.9, 75.0, 73.4 (3 × CH2Ph), 68.6 (C-5), 64.2 (C-6), 55.2 (OCH3) ppm; HRMS (ESI) m/z: [M + H]+ Calcd for C34H36NO7 570.2492; Found: 570.2495.
Methyl 2,3,4-Tri-O-benzyl-6-O-p-methoxybenzyl-α-d-glucopyranoside (2).
This was synthesized from methyl 2,3,4-tri-O-benzyl-α-d-glucopyranoside,30,35 and its analytical data was in accordance with that previously reported.9
Ethyl 2,3-Di-O-benzoyl-6-O-p-methoxybenzyl-1-thio-β-d-lucopyranoside (6).
A mixture containing ethyl 2,3-di-O-benzoyl-4,6-O-p-methoxybenzylidene-1-thio-β-d-glucopyranoside32 (5, 1.83 g, 3.34 mmol) and molecular sieves (4 Å, 1.20 g) in dimethylformamide (DMF, 40 mL) was stirred under argon for 1.5 h at rt. NaCNBH3 (1.05 g, 16.7 mmol) was added, and the resulting mixture was cooled to 0 °C. Trifluoroacetic acid (TFA, 3.82 g, 33.5 mmol) was added dropwise over a period of 1 h, and the resulting mixture was allowed to warm to rt and stirred for 18 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with DCM. The combined filtrate (~200 mL) was washed with sat. aq. NaHCO3 (3 × 40 mL), and the aqueous layer was additionally extracted with dichloromethane (2 × 150 mL). The combined organic phase was dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford the title compound as a colorless foam in 95% yield (1.75 g, 3.17 mmol). Analytical data for 6: Rf = 0.5 (ethyl acetate/hexane, 2/3, v/v); [α]D23 48.1 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.02−7.91 (m, 4H, aromatic), 7.58−7.45 (m, 2H, aromatic), 7.43−7.31 (m, 4H, aromatic), 7.31−7.23 (m, 2H, aromatic), 6.89−6.87 (m, 2H, aromatic), 5.48 (dd, 1H, J3,4 = 9.0 Hz, H-3), 5.44 (dd, 1H, J2,3 = 9.0 Hz, H-2), 4.69 (d, 1H, J1,2 = 9.0 Hz, H-1), 4.56 (d, 1H, 2J = 11.6 Hz, CHPh), 4.52 (d, 1H, 2J = 11.6 Hz, CHPh), 3.96 (m, 1H, J4,5 = 9.3 Hz, H-4), 3.83−3.79 (m, 2H, H-6a, 6b), 3.80 (s, 3H, OCH3), 3.70 (m, 1H, J5,6a = J5,6b = 4.7 Hz, H-5), 3.25 (d, 1H, J4,OH = 3.1 Hz, OH), 2.75 (m, 2H, SCH2CH3), 1.27 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 167.0, 165.4 (2 × COPh), 159.4, 133.4, 133.2, 129.9, 129.8, 129.7, 129.5, 129.3, 129.1, 128.4 (×2), 113.9, 83.6 (C-1), 78.5 (C-5), 77.6 (C-3), 73.5 (CH2Ph), 71.3 (C-4), 70.2 (C-2), 70.0 (C-6), 55.3 (PhOCH3), 24.2 (SCH2CH3), 14.9 (SCH2CH3); HRMS (ESI) m/z: [M + Na]+ Calcd for C30H32O8SNa 575.1716; Found: 575.1741.
Ethyl 2,3,4-Tri-O-benzoyl-6-O-p-methoxybenzyl-1-thio-β-d-glucopyranoside (7).
DMAP (180.8 mg, 1.5 mmol) was added to a solution of compound 6 (1.64 g, 3.0 mmol) in pyridine (30 mL) followed by a dropwise addition of benzoyl chloride (1.25 g, 8.9 mmol), and the resulting mixture was stirred under argon for 16 h at 40 °C. The reaction mixture was cooled to rt, MeOH (20 mL) was added, and the resulting mixture was stirred for 30 min. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene. The residue was dissolved in dichloromethane (~200 mL) and washed with water (40 mL), 1 N aq. HCl (40 mL), sat. aq. NaHCO3 (40 mL), and water (2 × 40 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford the title compound as a colorless foam in 94% yield (1.85 g, 2.8 mmol). Analytical data for 79: [α]D23 13.4 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.96−7.93 (m, 2H, aromatic), 7.89−7.56 (m, 2H, aromatic), 7.82−7.80 (m, 2H, aromatic), 7.52−7.48 (m, 2H, aromatic), 7.44−7.33 (m, 5H, aromatic), 7.29−7.25 (m, 2H, aromatic), 7.17−7.15 (m, 2H, aromatic), 6.76−6.73 (m, 2H, aromatic), 5.85 (dd, 1H, J3,4 = 9.7 Hz, H-3), 5.55 (dd, 1H, J4,5 = 9.6 Hz, H-4), 5.52 (dd, 1H, J2,3 = 9.7 Hz, H-2), 4.79 (d, 1H, J1,2 = 9.9 Hz, H-1), 4.48 (d, 1H, 2J = 11.5 Hz, CHPh), 4.43 (d, 1H, 2J = 11.5 Hz, CHPh), 3.97 (m, 1H, J5,6a = 4.8, J5,6b = 3.6 Hz, H-5), 3.74 (s, 3H, OCH3), 3.68−3.67 (m, 2H, H-6a, 6b), 2.74 (m, 2H, SCH2CH3), 1.29 (t, 3H, SCH2CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 165.82, 165.26, 165.20 (3 × COPh), 159.2, 133.2 (×3), 129.9 (×2), 129.8 (×2), 129.7 (×2), 129.4 (×2), 129.3, 129.1, 129.0, 128.6, 128.5 (×2), 128.4 (×3), 128.3 (×2), 113.7, 83.8 (C-1), 78.1, 77.2, 74.4, 73.2, 70.1, 69.9, 55.2 (OCH3), 24.3 (SCH2CH3), 14.9 (SCH2CH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C37H36O9SNa 679.1978; Found: 679.1987.
2,3,4-Tri-O-benzoyl-6-O-p-methoxybenzyl-α/β-d-glucopyranosyl chloride (3).
N-Bromosuccinimide (2.34 g, 13.1 mmol) was added to a solution of compound 7 (1.73 g, 2.6 mmol) in acetone-water (150 mL, 9/1, v/v), and the resulting mixture was stirred for 15 min at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in dichloromethane (~200 mL) and washed with water (40 mL), sat. aq. NaHCO3 (40 mL), and water (2 × 40 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to give 2,3,4-tri-O-benzoyl-6-O-p-methoxybenzyl-α/β-d-glucopyranose (15, 1.55 g, 2.5 mmol) as a clear syrup in 98% yield (α/β = 4/1). Selected analytical data for α-15: Rf = 0.4 (ethyl acetate/hexane, 2/3, v/v); 1H NMR (400 MHz, CDCl3): δ 7.99−7.95 (m, 2H, aromatic), 7.91−7.83 (m, 4H, aromatic), 7.59−7.16 (m, 12H, aromatic), 6.72−6.70 (m, 1H, aromatic), 6.19 (t, 1H, J3,4 = 9.9 Hz, H-3), 5.75 (dd, 1H, J1,2 = 3.7, J1,OH = 3.8 Hz, H-1), 5.57 (dd, 1H, J4,5 = 9.9 Hz, H-4), 5.29 (dd, 1H, J2,3 = 9.9 Hz, H-2), 4.51 (m, 1H, J5,6a = 5.4, J5,6b = 3.1 Hz, H-5), 3.80 (s, 3H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 90.5 (C-1), 72.2 (C-2), 70.2 (C-3), 69.6 (C-4), 69.5 68.8 (C-5), 68.6 (C-6), 56.1 (OCH3) ppm; HRMS (ESI) m/z: [M + K]+ Calcd for C35H32O10K 651.1633; Found: 651.1661.
A solution of oxalyl chloride (0.96 g, 7.6 mmol) in dichloromethane (15 mL) was added dropwise to a stirring solution of compound 15 (1.55 g, 2.5 mmol) in dichloromethane (50 mL) and DMF (5.0 mL), and the resulting mixture was stirred under argon for 30 min at 0 °C. The external cooling was then removed, and the reaction mixture was allowed to slowly warm to rt and then stirred for additional 1 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in a mixture of ethyl acetate and hexane (40 mL, 1/1, v/v) and passed through a pad of silica gel (40 g) that was additionally eluted with a mixture of ethyl acetate and hexane (150 mL, 1/1, v/v). The combined eluate was concentrated under reduced pressure and dried in vacuo to afford the title compound as a clear syrup in 81% yield (1.23 g, 2.0 mmol) (α/β = 1/4). Selected analytical data for β-3: Rf = 0.8 (ethyl acetate/hexane, 2/3, v/v); 1H NMR (400 MHz, CDCl3): δ 7.96−7.81 (m, 6H, aromatic), 7.57−7.27 (m, 11H, aromatic), 7.17 (m, 1H, aromatic), 6.76−6.71 (m, 1H, aromatic), 5.69−5.61 (m, 2H, H-2, 4), 5.60 (d, 1H, J1,2 = 9.1 Hz, H-1), 4.07 (m, 1H, H-5), 3.81 (s, 3H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 88.0 (C-1), 74.0 (C-2), 73.1 (C-3), 69.0 (C-4), 68.4 (C-6), 56.2 (OCH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C35H31O9ClNa 653.1554; Found: 653.1557.
Ethyl 2-O-Benzoyl-3,4-di-O-benzyl-6-O-picoloyl-1-thio-β-d-glucopyranoside (9).
Picolinic acid (1.90 g, 15.2 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 2.90 g, 15.2 mmol), and 4-dimethylaminopyridine (DMAP, 185.9 mg, 1.5 mmol) were added to a solution of ethyl 2-O-benzoyl-3,4-di-O-benzyl-1-thio-β-d-glucopyranoside33,36 (8, 3.90 g, 7.6 mmol) in dichloromethane (100 mL), and the resulting mixture was stirred under argon for 1 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~200 mL) and washed with water (2 × 75 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (acetone-hexane gradient elution) to give the title compound (4.6 g, 7.4 mmol) as a colorless foam in 98% yield. Analytical data for 9: Rf = 0.45 (acetone/hexane, 2/3, v/v); [α]D23 33.9 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.79−8.77 (m, 1H, aromatic), 8.06−8.03 (m, 3H, aromatic), 7.85−7.81 (m, 1H, aromatic), 7.60−7.56 (m, 1H, aromatic), 7.49−7.43 (m, 3H, aromatic), 7.36−7.21 (m, 5H, aromatic), 7.16−7.13 (m, 5H, aromatic) 5.34 (dd, 1H, J2,3 = 8.4 Hz, H-2), 4.89 (d, 1H, 2J = 10.9 Hz, CHPh), 4.77 (d, 1H, 2J = 10.9 Hz, CHPh), 4.69 (d, 1H, 2J = 10.9 Hz, CHPh), 4.67 (dd, H-6a), 4.65 (d, 1H, 2J = 10.9 Hz, CHPh), 4.60 (d, 1H, J1,2 = 9.5 Hz, H-1), 4.58 (dd, 1H, H-6b), 3.91 (dd, 1H, J3,4 = 8.4 Hz, H-3), 3.83−3.76 (m, 2H, J4,5 = 8.3 Hz, H-4, 5), 2.72−2.62 (m, 2H, SCH2CH3), 1.17 (t, J = 7.4 Hz, 3H, SCH2CH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 165.3, 164.6, 150.1, 147.8, 137.5, 137.4, 136.9, 133.2, 129.8 (×2), 128.5, 128.4, 128.3, 128.2, 128.0 (×2), 127.9, 126.9, 125.2, 84.5 (C-3), 83.6 (C-1), 77.6 (C-4),a 77.2 (C-5),a 75.5, 75.1 (2 × CH2Ph), 77.40 (C-2), 64.3 (C-6), 24.0 (SCH2CH3), 14.9 (SCH2CH3) ppm; HRMS (ESI) m/z: [M + H]+ Calcd for C35H36NO7S 614.2213; Found: 614.2215.
Benzoxazolyl 2-O-Benzoyl-3,4-di-O-benzyl-6-O-picoloyl-1-thio-β-d-glucopyranoside (4).
A mixture containing compound 9 (2.58 g, 4.07 mmol) and molecular sieves (3 Å, 1.0 g) in dry dichloromethane (60 mL) was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C, a 0.6 M solution of bromine in dichloromethane (19.0 mL) was added, and the resulting mixture was stirred for 10 min at 0 °C. Then, the reaction mixture was neutralized with triethylamine (1.3 mL), the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene (2 × 3 mL). The resulting residue was dried in vacuo for 40 min. The crude residue containing the glycosyl bromide intermediate was dissolved in dry acetone (70 mL), 18-crown-6 (0.25 g) and KSBox/KHCO3 (2.60 g, 1/1, w/w) were added, and the resulting mixture was stirred under argon for 8 h at rt. After that, the volatiles were removed under reduced pressure. The residue was dissolved in ethyl acetate, filtered through a pad of Celite, and washed successively with ethyl acetate. The combined filtrate (~200 mL) was washed with water (100 mL), 1% aq. NaOH (50 mL), and water (2 × 100 mL). The organic phase was separated, dried with sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to give the title compound as a colorless foam in 94% yield (2.68 g, 3.82 mmol). Analytical data for 4: Rf = 0.55 (ethyl acetate/hexane, 1/1, v/v); [α]D23−52.9 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.70 (d, 1H, aromatic), 8.01−7.98 (m, 2H, aromatic), 7.89 (d, 1H, aromatic), 7.61−7.57 (m, 3H, aromatic), 7.56−7.13 (m, 16H, aromatic), 5.83 (d, 1H, J1,2 = 10.3 Hz, H-1), 5.54 (dd, 1H, J2,3 = 9.0 Hz, H-2), 4.91 (d, 1H, 2J = 11.0, CHPh), 4.81 (d, 1H, 2J = 10.9 Hz, CHPh), 4.74 (d, 1H, 2J = 10.9 Hz, CHPh), 4.67 (dd, 1H, H-6a), 4.55 (dd, 1H, H-6b), 4.07 (dd, 1H, J3,4 = 8.9 Hz, H-3), 4.04 (m, 1H, J5,6a = 5.6, J5,6b = 2.0 Hz, H-5), 3.87 (dd, 1H, J4,5 = 9.0 Hz, H-4) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 165.3, 164.4, 161.6, 151.8, 149.9, 147.5, 141.5, 137.3, 136.8, 133.5, 129.9 (×2), 129.0, 128.5 (×2), 128.4 (×4), 128.1 (×4), 127.9, 126.8, 125.1, 124.3, 124.2, 118.6, 110.0, 84.1 (C-3), 83.6 (C-1), 77.7 (C-5), 77.2 (C-4), 75.6, 75.1 (2 × CH2Ph), 72.1 (C-2), 64.0 (C-6) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C40H34N2O8SNa 725.1934; Found: 725.1954.
Synthesis of Oligosaccharides.
Methyl 6-O-(2,3,4-Tri-O-benzoyl-6-O-p-methoxybenzyl-β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (10).
Method A.
Anhydrous FeCl3 (1.5 mg, 0.009 mmol) was added to a solution of compound 1 (17.8 mg, 0.031 mmol) in MeOH (0.9 mL) and dichloromethane (0.1 mL), and the resulting mixture was stirred for 30 min at rt. After that, the reaction mixture was concentrated under reduced pressure and the residue was dried in vacuo for 1.5 h. The resulting residue was dissolved in dichloromethane (1.0 mL), molecular sieves (4 Å, 150 mg) were added, and the resulting mixture was stirred under argon for 1 h at rt. Glycosyl chloride 3 (40.0 mg, 0.059 mmol) and FeCl3 (2.9 mg, 0.018 mmol) were added, and the resulting mixture was stirred under argon for 16 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with dichloromethane, and the combined filtrate (~40 mL) was washed with sat. aq. NaHCO3 (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene, gradient elution) to give the title compound (22.8 mg, 0.022 mmol) as a colorless foam in 70% yield. Analytical data for 10:9 [α]D23 7.0 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.88−7.86 (m, 4H, aromatic), 7.82−7.79 (m, 2H, aromatic), 7.52−7.48 (m, 1H, aromatic), 7.42−7.26 (m, 15H, aromatic), 7.25−7.18 (m, 6H, aromatic), 7.16−7.15 (m, 2H, aromatic), 7.13−7.05 (m, 2H, aromatic), 7.04 (m, 2H, aromatic), 6.73−6.71 (m, 2H, aromatic), 5.82 (dd, 1H, J3′,4′ = 9.7 Hz, H-3′), 5.54 (dd, 1H, J2′,3′ = 9.6 Hz, H-2′), 5.50 (dd, 1H, J4′,5′ = 10.0 Hz, H-4′), 4.89 (d, 1H, 2J = 10.9 Hz, CHPh), 4.78 (d, 1H, J1′,2′ = 7.8 Hz, H-1′), 4.73 (d, 1H, 2J = 12.1 Hz, CHPh), 4.69 (d, 1H, 2J = 10.9 Hz, CHPh), 4.59 (d, 1H, 2J = 12.1 Hz, CHPh), 4.51 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.48 (d, 1H, 2J = 12.1 Hz, CHPh), 4.48 (d, 1H, 2J = 10.4 Hz, CHPh), 4.45 (d, 1H, 2J = 11.7 Hz, CHPh), 4.42 (d, 1H, 2J = 11.7 Hz, CHPh), 4.30 (d, 1H, 2J = 11.0 Hz, CHPh), 4.18 (d, 1H, J = 8.6 Hz, H-6a), 3.96−3.90 (m, 1H, H-5′), 3.91−3.86 (dd, 1H, J3,4 = 9.5 Hz, H-3), 3.79−3.61 (m, 4H, H-5, 6a′, 6b, 6b′), 3.71 (s, 3H, OCH3), 3.44 (dd, 1H, J3,4 = 9.5 Hz, H-3), 3.39 (dd, 1H, J4,5 = 9.5 Hz, H-4), 3.21 (s, 3H, OCH3); 13C{1H} NMR (100 MHz, CDCl3): δ 165.8, 165.3, 164.9, 159.1, 138.8, 138.2 (×2), 133.2, 133.1, 133.0, 101.1 (C-1′), 98.0 (C-1), 81.9, 79.8, 77.4, 75.5. 74.7, 74.0, 73.4, 73.2, 73.1, 71.9, 70.0, 69.5, 69.0, 68.2, 55.2, 55.0 ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C63H62O15Na 1081.3986; Found: 1081.4015.‴
Method B.
Iron chloride (14 mg, 87 μmol) was added to a solution of glycosyl donor 7 (654 mg, 0.995 mmol) and compound 1 (420 mg, 0.737 mmol) in dry MeOH/1,2-DCE (20 mL, 1/3, v/v), and the resulting mixture was stirred for 1 h at rt. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene and then dried in vacuo for 1.5 h. The resulting residue was dissolved in dry 1,2-DCE (15 mL), molecular sieves (4 Å, 0.15 g) were added, and the resulting mixture was stirred under argon for 40 min at rt. After that, NIS (27 mg, 1.22 mmol) and TfOH (30 μL, 0.35 mmol) were added, and the resulting mixture was stirred under argon for 16 h at rt. After that, triethylamine (~0.5 mL) was added, and solids were filtered off through a pad of Celite and rinsed successively with DCM. The combined filtrate (~100 mL) was washed with 10% aq. NaS2O3 (25 mL), sat aq. NaHCO3 (25 mL), and water (2 × 40 mL). The organic phase was separated, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene, gradient elution) to give the title compound 10 (678 mg, 0.64 mmol) in 87% yield.
Methyl O-(3,4-Di-O-benzyl-2-O-benzoyl-6-O-picoloyl-β-d-glucopyranosyl)-(1 → 6)-O-(2,3,4-tri-O-benzoyl-β-d-glucopyranosyl)-(1 → 6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (11).
Compounds 10 (40.1 mg, 0.038 mmol) and 4 (34.5 mg, 0.049 mmol) were dissolved in 1,2-dichloromethane (4.0 mL), molecular sieves (3 Å, 100 mg) were added, and the resulting mixture was stirred under argon for 1 h at rt. The reaction was then cooled to 0 °C, TMSCl (5.3 μL, 0.042 mmol) and triflic acid (4 μL, 0.041 mmol) were added, and the resulting mixture was stirred for 10 min at 0 °C. Then, silver triflate (AgOTf, 14.5 mg, 0.057 mmol) was added, and the resulting mixture was stirred under argon for 15 h at rt. After that, the reaction was quenched by addition of triethylamine (~10 μL), and the solids were filtered off through a pad of Celite and rinsed successively with ethyl acetate. The combined filtrate (~40 mL) was washed with water (10 mL), sat. aq. NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene, 2% gradient elution) to give the title compound as a colorless foam in 70% yield (39.5 mg, 26.5 mmol). Analytical data for 11: Rf = 0.80 (ethyl acetate/dichloromethane, 1/4, v/v); [α]D23 14.2 (c = 1, CHCl3); 1H NMR (700 MHz, CDCl3): δ 8.78 (d, 1H, aromatic), 8.03−7.99 (m, 3H, aromatic), 7.84−7.80 (m, 5H, aromatic), 7.74−7.73 (m, 2H, aromatic), 7.52−7.44 (m, 3H, aromatic), 7.39−7.21 (m, 26H, aromatic), 7.16−7.11 (m, 7H, aromatic), 6.98−6.96 (m, 2H, aromatic), 5.70 (dd, 1H, J3′4′ = 10.0 Hz, H-3′), 5.41 (dd, 1H, J4′,5′ = 8.0 Hz, H-4′), 5.29 (dd, 1H, J2″,3″ = 8.0 Hz, H-2″), 5.25 (dd, J2′,3′ = 10.0 Hz, 1H, H-2′), 4.87 (d, 1H, 2J = 11.0 Hz, CHPh), 4.84 (d, 1H, 2J = 11.0 Hz, CHPh), 4.72 (d, 1H, 2J = 11.0 Hz, CHPh), 4.72 (d, 1H, J1″,2″ = 6.0 Hz, H-1″), 4.66 (d, 1H, 2J = 11.0 Hz, CHPh), 4.65 (d, 1H, 2J = 11.0 Hz, CHPh), 4.61 (d, 1H, 2J = 11.0 Hz, CHPh), 4.59 (dd, 1H, H-6a 4.56 (d, 1H, 2J = 11.0 Hz, CHPh), 4.52 (m, 1H, H-6b″), 4.51 (d, 1H, J1,2 = 3.4 Hz, H-1), 4,49 (d, 1H, J1′,2′ = 7.9 Hz, H-1′), 4.37 (d, 1H, 2J = 11.2 Hz, CHPh), 4.16 (d, 1H, 2J = 11.2 Hz, CHPh), 3.91 (dd, 1H, H-6a′), 3.85 (m, 1H, J5′,6a′ = 1.8, J5′,6b′ = 3.9 Hz, H-5a′), 3.84 (m, 1H, J3″,4″ = 8.3 Hz, H-3 3.84−3.81 (m, 2H, H-4″, 6a), 3.77 (dd, 1H, H-6b′), 3.74 (dd, 1H, J3,4 = 9.8 Hz, H-3), 3.71 (m, 1H, J5″,6a″ = 2.0, J5″,6b″ = 4.9 Hz, H-5″), 3.45−3.42 (m, 1H, H-5), 3.42 (dd, 1H, H-6b), 3.35 (dd, 1H, J2,3 = 9.7 Hz, H-2), 3.32 (dd, J4,5 = 8.9 Hz, 1H, H-4), 3.23 (s, 3H, OCH3) ppm; 13C{1H} NMR (175 MHz, CDCl3): δ 165.7, 165.3, 165.0, 164.8, 164.5, 150.1, 147.6, 138.9, 138.3, 138.1, 137.4 (×2), 137.0, 133.4, 133.2, 133.1, 133.0, 125.3, 101.1 (C-1″), 100.6 (C-1′), 98.1 (C-1), 82.8 (C-3″), 81.8 (C-3), 79.7 (C-2), 77.5 (C-4″), 76.9 (C-4), 75.4, 75.2, 75.1, 74.4 (4 × CH2Ph), 74.2 (C-5′), 73.7 (C-2″), 73.4 (CH2Ph), 73.2 (C-5″), 72.8 (C-3′), 71.7 (C-4′), 69.6 (C-2′), 69.4 (C-5), 68.1 (C-6′), 67.3 (C-6), 64.0 (C-6″), 55.3 (OCH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C88H83NO21Na 1512.5355; Found: 1512.5347.
Methyl O-(2,3,4-Tri-O-benzoyl-6-O-p-methoxybenzyl-β-d-glucopyranosyl)-(1→6)-O-(3,4-di-O-benzyl-2-O-benzoyl-β-d-glucopyranosyl)-(1 → 6)-O-(2,3,4-tri-O-benzoyl-β-d-glucopyranosyl)-(1 → 6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (12).
Method A.
FeCl3 (1.7 mg, 0.008 mmol) was added to a solution of compound 11 (37.7 mg, 0.025 mmol) in MeOH (0.9 mL) and dichloromethane (0.1 mL), and the resulting mixture was stirred for 30 min at rt. After that, the reaction mixture was concentrated under reduced pressure and the residue was dried in vacuo for 1.5 h. The resulting residue was dissolved in dichloromethane (1.0 mL), molecular sieves (4 Å, 150 mg) were added, and the resulting mixture was stirred under argon for 1 h at rt. Glycosyl chloride 3 (47.6 mg, 0.075 mmol) and FeCl3 (3.7 mg, 0.022 mmol) were added, and the resulting mixture was stirred under argon for 16 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with dichloromethane, and the combined filtrate (~40 mL) was washed with sat. aq. NaHCO3 (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene, gradient elution) to give the title compound as a colorless foam in 58% yield (29.0 mg, 0.015 mmol). Analytical data for 12: Rf = 0.7 (ethyl acetate/toluene, 3/7, v/v); [α]D23 1.7 (c = 1, CHCl3); 1H NMR (700 MHz, CDCl3): δ 7.97−7.96 (m, 2H, aromatic), 7.92−7.89 (m, 4H, aromatic), 7.86−7.83 (m, 6H, aromatic), 7.76−7.75 (m, 2H, aromatic), 7.50−7.45 (m, 2H, aromatic), 7.44−7.37 (m, 3H, aromatic), 7.34−7.27 (m, 25H, aromatic), 7.22−7.21 (m, 2H, aromatic), 7.19−7.08 (m, 12H, aromatic), 7.02−6.98 (m, 4H, aromatic), 6.72−6.71 (m, 2H, aromatic), 5.93 (dd, 1H, J3‴,4‴ = 9.7 Hz, H-3‴), 5.74 (dd, 1H, J3′,4′ = 9.6 Hz, H-3′), 5.55 (dd, 1H, J4‴,5‴ = 9.7 Hz, H-4‴), 5.52 (dd, 1H, J2‴,3‴ = 9.7 Hz, H-2‴), 5.46 (dd, 1H, J2′,3′ = 9.6 Hz, H-2′), 5.36 (dd, 1H, J4′,5′ = 9.6 Hz, H-4′), 5.15 (dd, 1H, J2″,3″ = 9.2 Hz, H-2″), 4.88 (d, 1H, J1‴,2‴ = 8.1 Hz, H-1‴), 4.86 (d, 1H, 2J = 11.0 Hz, CHPh), 4.69 (d, 1H, 2J = 12.0 Hz, CHPh), 4.66 (d, 1H, 2J = 11.0 Hz, CHPh), 4.58 (d, 1H, 2J = 11.0 Hz, CHPh), 4.55 (d, 1H, 2J = 12.0 Hz, CHPh), 4.53 (d, 1H, J1″,2″ = 7.3 Hz, H-1″), 4.50 (d, 1H, J1,2 = 3.8 Hz, H-1), 4.49 (d, 1H, 2J = 11.0 Hz, CHPh), 4.47 (d, 1H, J1′,2′ = 7.9 Hz, H-1′), 4.45 (d, 1H, 2J = 11.3 Hz, CHPh), 4.40 (d, 1H, 2J = 11.3 Hz, CHPh), 4.39 (d, 1H, 2J = 11.3 Hz, CHPh), 4.19 (d, 1H,2aJ = 11.3 Hz, CHPh), 4.14 (m, 1H, H-6a″), 3.99 (m, 1H, H-5‴), 3.94 (dd, 1H, H-6a′), 3.85 (dd, 1H, H-6a), 3.82 (dd, 1H, J3,4 = 9.4 Hz, H-3), 3.78−3.73 (m, 1H, J5′,6a′ = 2.0 Hz, H-5′), 3.74 (m, 1H, H-6b″), 3.71 (s, 3H, OCH3), 3.70−3.69 (m, 1H, H-6b′), 3.68−3.63 (m, 2H, H-6a‴, 6b‴), 3.64 (dd, 1H, J3″,4″ = 9.2 Hz, H-3″), 3.51 (dd, 1H, J4″,5″ = 9.2 Hz, H-4″), 3.45−3.43 (m, 2H, J5,6a = 2.7, J5,6b = 3.3 Hz, H-5, 5″), 3.41 (dd, 1H, H-6b), 3.34 (dd, 1H, J2,3 = 9.0 Hz, H-2), 3.32 (dd, 1H, J4.5 = 9.5 Hz, H-4), 3.22 (s, 3H, OCH3) ppm; 13C{1H} NMR (175 MHz, CDCl3): δ 165.8, 165.7, 165.4, 165.3, 165.0, 164.9, 164.8, 159.1, 138.9, 138.3, 138.2, 137.8, 137.7, 113.7, 101.3 (C-1‴), 101.0 (C-1″), 100.8 (C-1′), 98.1 (C-1), 82.6 (C-3″), 81.8 (C-3), 79.8 (C-2), 77.4 (C-4″), 77.0 (C-4), 75.4 (CH2Ph), 74.9 (C-5),b 74.7, 74.6, 74.5 (3 × CH2Ph), 74.3 (C-5′), 73.8 (C-5‴), 73.7 (CH2Ph), 73.4 (C-2″), 73.2 (CH2Ph), 73.1 (C-3‴), 72.9 (C-3′), 72.0 (C-2‴), 71.8 (C-2′), 70.0 (C-4‴), 69.9 (C-4′), 69.4 (C-5″),b 68.9 (C-6‴), 68.2 (C-6″), 68.0 (C-6′), 67.4 (C-6), 55.3 (OCH3), 55.2 (PhOCH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C117H110O29Na 2001.7030; Found: 2001.7038.
Method B.
Iron chloride (0.5 mg, 3.2 μmol) was added to a solution of compounds 7 (21 mg, 0.032 mmol) and 11 (32 mg, 0.021 mmol) in dry MeOH/DCM (1.2 mL, 1/3, v/v), and the resulting mixture was stirred for 1 h at rt. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene and dried in vacuo for 1.5 h. To the resulting residue was dissolved in dry DCM (1.2 mL), molecular sieves (4 Å, 30 mg) were added, and the resulting mixture was stirred under argon for 40 min at rt. After that, NIS (9.6 mg, 0.043 mmol) and TfOH (1 μL, 0.013 mmol) were added, and the resulting mixture was stirred under argon for 28 h at rt. After that, triethylamine (5 μL) was added, and the solids were filtered off through a pad of Celite and rinsed successively with DCM. The combined filtrate (~30 mL) was washed with 10% aq. NaS2O3 (10 mL), sat. aq. NaHCO3 (10 mL), and H2O (2 × 10 mL). The organic phase was separated, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane, 2% gradient elution) to give the title compound 12 in 75% yield (32 mg, 0.0161 mmol).
Methyl 6-O-(3,4-Di-O-benzyl-2-O-benzoyl-6-O-picoloyl-β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (13).
Compounds 2 (31.0 mg, 0.053 mmol) and 4 (44.8 mg, 0.064 mmol) were dissolved in 1,1-dichloroethane (2.0 mL), molecular sieves (3 Å, 70 mg) were added, and the resulting mixture was stirred under argon for 1 h at rt. The mixture was cooled to 0 °C, TMSCl (7.5 μL, 0.058 mmol) and triflic acid (5.5 μL, 0.063 mmol) were added, and the resulting mixture was stirred for 10 min at 0 °C. After that, silver triflate (AgOTf, 24.5 mg, 0.096 mmol) was added, and the resulting mixture was stirred under argon for 14 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with ethyl acetate. The combined filtrate (~40 mL) was washed with water (10 mL), sat. NaHCO3 (10 mL), and water (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane, gradient elution) to give the title compound as a colorless foam in 90% yield (43 mg, 0.048 mmol). Analytical data for 13: Rf = 0.3 (ethyl acetate/toluene, 3/7, v/v); [α]D23 24.2 (c = 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.75 (d, 1H, aromatic), 8.05−8.03 (m, 1H, aromatic), 7.95−7.93 (m, 2H, aromatic), 7.80−7.76 (m, 1H, aromatic), 7.47−7.43 (m, 2H, aromatic), 7.31−7.20 (m, 20H, aromatic), 7.15−7.12 (m, 5H, aromatic), 7.02−6.99 (m, 2H, aromatic), 5.39 (dd, J2′,3′ = 9.2 Hz, H-2′), 4.89−4.84 (m, 2H, 2 × CHPh), 4.77−4.67 (m, 5H, 5 × CHPh), 4.66 (m, 1H, H-6a′), 4.61−4.56 (m, 2H, H-6b′, CHPh), 4.57 (d, 1H, J1′,2′ = 8.6 Hz, H-1′), 4.45 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.40 (d, 1H, 2J = 11.0 Hz, CHPh), 4.24 (d, 1H, 2J = 11.0 Hz, CHPh), 4.08 (m, 1H, H-6a), 3.88 (dd, 1H, J3′4′ = 8.9 Hz, H-3′), 3.83 (dd, 1H, J3,4 = 9.6 Hz, H-3), 3.81−3.78 (m, 1H, H-4), 3.76 (m, 1H, H-5′), 3.65−3.60 (m, 2H, H-5, 6b), 3.40 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.3 (dd, 1H, J4,5 = 9.6 Hz, H-4), 3.15 (s, 3H, OCH3) ppm; 13C{1H} NMR (100 MHz, CDCl3): δ 165.0, 164.6 (2 × C=O), 150.0, 147.7, 138.8, 138.2 (×2), 137.5, 137.4, 136.9, 133.0, 129.7, 128.5, 128.4 (×2), 128.3, 128.2 (×2), 128.1 (×3), 128.0, 127.8 (×3), 127.5, 127.4 (×2), 126.9, 125.3, 101.2 (C-1′), 97.8 (C-1), 82.9 (C-3′), 81.8 (C-3), 79.7 (C-2), 77.6 (C-4), 77.4 (C-4′), 75.4, 75.3, 75.1, 74.6, 73.6 (C-2′), 73.3, 73.2, 69.3 (C-5), 68.0 (C-6), 64.1 (C-6′), 54.9 (OCH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C61H61NO13Na 1038.4041; Found: 1038.4036.
Methyl O-(2,3,4-Tri-O-benzoyl-6-O-p-methoxybenzyl-β-d-glucopyranosyl)-(1 → 6)-O-(3,4-di-O-benzyl-2-O-benzoyl-β-d-glucopyranosyl)-(1 → 6)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (14).
Method A.
Anhydrous FeCl3 (1.2 mg, 0.008 mmol) was added to a solution of compound 13 (25.4 mg, 0.025 mmol) in MeOH (0.9 mL) and dichloromethane (0.1 mL), and the resulting mixture was stirred for 30 min at rt. After that, the reaction mixture was concentrated under reduced pressure and the residue was dried in vacuo for 1.5 h. The resulting residue was dissolved in dichloromethane (1.0 mL), molecular sieves (4 Å, 150 mg) were added, and the resulting mixture was stirred under argon for 1 h at rt. Glycosyl chloride 3 (39.9 mg, 0.063 mmol) and FeCl3 (3.1 mg, 0.019 mmol) were added, and the resulting mixture was stirred under argon for 16 h at rt. After that, the solids were filtered off through a pad of Celite and rinsed successively with dichloromethane, and the combined filtrate (~40 mL) was washed with sat. aq. NaHCO3 (2 × 10 mL). The organic phase was separated, dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-toluene, gradient elution) to give the title compound as a colorless foam in 48% yield (18.2 mg, 0.012 mmol). Analytical data for 14: Rf = 0.45 (ethyl acetate/toluene, 1/4, v/v); [α]D23 5.6 (c = 1, CHCl3); 1H NMR (700 MHz, CDCl3): δ 7.89−7.87 (m, 4H, aromatic), 7.84−7.80 (m, 4H, aromatic), 7.51−7.48 (m, 1H, aromatic), 7.42−7.36 (m, 3H, aromatic), 7.35−7.27 (m, 16H, aromatic), 7.24−7.21 (m, 8H, aromatic), 7.21−7.20 (m, 2H, aromatic), 7.16−7.14 (m, 2H, aromatic), 7.11−7.05 (m, 5H, aromatic), 6.94−6.93 (m, 2H, aromatic), 6.72−6.71 (m, 2H, aromatic), 5.78 (dd, 1H, J3″,4″ = 9.8 Hz, H-3″), 5.56 (dd, 1H, J4″,5″ = 9.8 Hz, H-4″), 5.50 (dd, 1H, J2″,3″ = 9.8 Hz, H-2″), 5.28 (dd, 1H, J2′,3′ = 9.5 Hz, H-2′), 4.92 (d, 1H, J1″,2″ = 7.8 Hz, H-1″), 4.86 (d, 1H, 2J = 11.5 Hz, CHPh), 4.70 (d, 1H, 2J = 11.5 Hz, CHPh), 4.66 (d, 1H, 2J = 11.5 Hz, CHPh), 4.65 (d, 1H, 2J = 11.5 Hz, CHPh), 4.64 (d, 1H, 2J = 11.5 Hz, CHPh), 4.57 (d, 1H, 2J = 11.5 Hz, CHPh), 4.57 (d, 1H, 2J = 11.5 Hz, CHPh), 4.54 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.48 (d, 1H, 2J = 11.5 Hz, CHPh), 4.47 (d, 1H, 2J = 11.5 Hz, CHPh), 4.41 (d, 1H, 2J = 11.5 Hz, CHPh), 4.35 (d, 1H, J1′,2′ = 8.0 Hz, H-1′), 4.31 (d, 1H, 2J = 11.5 Hz, CHPh), 4.18 (dd, 1H, H-6a′), 4.15 (d, 1H, 2J = 11.5 Hz, CHPh), 3.96 (dd, 2J = 10.7 Hz, 1H, H-6a), 3.91 (m, 1H, J5″,6a″ = 5.5, J5″,6b″ = 3.2 Hz, H-5″), 3.84 (dd, 1H, J3,4 = 9.3 Hz, H-3), 3.77 (dd, 1H, H-6b′), 3.71 (dd, 1H, J3′,4′ = 9.5 Hz, H-3′), 3.69 (s, 3H, OCH3), 3.68 (dd, 1H, H-6a″), 3.64 (dd, 1H, H-6b″), 3.55 (m, 1H, J5,6a = J5,6b = 2.6 Hz, H-5), 3.55−3.50 (m, 2H, J5′,6a′ = 1.4 Hz, J5′,6b′ = 5.8 Hz, H-4′, 5′), 3.46 (dd, 1H, H-6b), 3.41 (dd, 1H, J2,3 = 9.3 Hz, H-2), 3.36 (dd, 1H, J4,5 = 10.0 Hz, H-4), 3.28 (s, 3H, OCH3) ppm; 13C{1H} NMR (175 MHz, CDCl3): δ 165.8, 165.2, 164.9, 164.8, 159.1, 139.0, 138.3, 138.2, 137.7, 137.6, 133.2, 133.1 (×2), 132.9, 129.7 (×4), 129.6, 129.4, 129.1, 128.9, 128.4 (×2), 128.3 (×3), 128.2 (×3), 128.1, 128.0, 127.9, 127.8 (×2), 127.7, 127.6, 127.4 (×2), 127.3, 113.6, 101.3 (C-1″), 100.8 (C-1′), 98.1 (C-1), 82.6 (C-3′), 81.9 (C-3), 79.7 (C-2), 77.8 (C-4′), 77.1 (C-4), 75.4 (C-5), 75.4, 74.9, 74.8, 74.5 (4 × CH2Ph), 73.8 (C-5″), 73.5 (C-2), 73.4, 73.2 (2 × CH2Ph), 73.1 (C-3″), 72.1 (C-2″), 69.8 (C-4″), 69.3 (C-5), 68.6 (C-6″), 68.3 (C-6′), 67.4 (C-6), 55.2 (OCH3), 55.1 (PhOCH3) ppm; HRMS (ESI) m/z: [M + Na]+ Calcd for C90H88O21Na 1527.5716; Found: 1527.5698.
Method B.
Iron chloride (1.9 mg, 11.7 μmol) was added to a solution of compounds 7 (54 mg, 0.083 mmol) and 13 (49.3 mg, 0.055 mmol) in MeOH/DCM (1.2 mL, 1/3, v/v), and the resulting mixture was stirred for 1 h at rt. After that, the volatiles were removed under reduced pressure, and the residue was co-evaporated with toluene and dried in vacuo for 2 h. To the resulting residue was dissolved in DCM (1.2 mL), molecular sieves (4 Å, 40 mg) were added, and the mixture was stirred under argon for 40 min at rt. After that, NIS (26.4 mg, 0.117 mmol) and TfOH (3 μL, 0.034 mmol) were added, and the resulting mixture was stirred under argon for 20 h at rt. After that, triethylamine (5 μL) was added, and the solids were filtered off through a pad of Celite and rinsed successively with DCM. The combined filtrate (~100 mL) was washed with 10% aq. Na2S2O3 (20 mL), sat. aq. NaHCO3 (20 mL), and water (2 × 30 mL). The organic phase was separated, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (ethyl acetate-hexane, 2% gradient elution) to give the title compound 14 as a colorless foam in 73% yield (60.5 mg, 0.0402 mmol).
Supplementary Material
ACKNOWLEDGMENTS
A.V.D. thanks the National Institute of General Medical Sciences for support of this work (GM111835).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.2c00905
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.2c00905.
Spectra for all compounds (PDF)
The authors declare no competing financial interest.
The assignment of these signals can be interchanged.
The assignment of these signals can be interchanged.
Contributor Information
Scott A. Geringer, Department of Chemistry and Biochemistry, University of Missouri – St. Louis, St. Louis, Missouri 63121, United States
Gustavo A. Kashiwagi, Department of Chemistry and Biochemistry, University of Missouri – St. Louis, St. Louis, Missouri 63121, United States; Department of Chemistry, Saint Louis University, St. Louis, Missouri 63103, United States
Alexei V. Demchenko, Department of Chemistry and Biochemistry, University of Missouri – St. Louis, St. Louis, Missouri 63121, United States; Department of Chemistry, Saint Louis University, St. Louis, Missouri 63103, United States.
REFERENCES
- (1).Panza M; Pistorio SG; Stine KJ; Demchenko AV Automated chemical oligosaccharide synthesis: novel approach to traditional challenges. Chem. Rev 2018, 118, 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Love KR; Andrade RB; Seeberger PH Linear synthesis of a protected h-type II pentasaccharide using glycosyl phosphate building blocks. J. Org. Chem 2001, 66, 8165–8176. [DOI] [PubMed] [Google Scholar]
- (3).Calin O; Eller S; Seeberger PH Automated polysaccharide synthesis: assembly of a 30mer mannoside. Angew. Chem., Int. Ed 2013, 52, 5862–5865. [DOI] [PubMed] [Google Scholar]
- (4).Naresh K; Schumacher F; Hahm HS; Seeberger PH Pushing the limits of automated glycan assembly: synthesis of a 50mer polymannoside. Chem. Commun 2017, 53, 9085–9088. [DOI] [PubMed] [Google Scholar]
- (5).Smoot JT; Demchenko AV Oligosaccharide synthesis: from conventional methods to modern expeditious strategies. Adv. Carbohydr. Chem. Biochem 2009, 62, 161–250. [DOI] [PubMed] [Google Scholar]
- (6).Kanie O; Ito Y; Ogawa T Orthogonal glycosylation strategy in oligosaccharide synthesis. J. Am. Chem. Soc 1994, 116, 12073–12074. [Google Scholar]
- (7).Ito Y; Kanie O; Ogawa T Orthogonal glycosylation strategy for rapid assembly of oligosaccharides on a polymer support. Angew. Chem., Int. Ed 1996, 35, 2510–2512. [Google Scholar]
- (8).Pornsuriyasak P; Demchenko AV S-Thiazolinyl (STaz) glycosides as versatile building blocks for convergent selective, chemoselective, and orthogonal oligosaccharide synthesis. Chem. – Eur. J 2006, 12, 6630–6646. [DOI] [PubMed] [Google Scholar]
- (9).Fujikawa K; Ganesh NV; Tan YH; Stine KJ; Demchenko AV Reverse orthogonal approach to oligosaccharide synthesis. Chem. Commun 2011, 10602–10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Geringer SA Doctoral Dissertation Univestrity of Missouri - St. Louis, 2020. [Google Scholar]
- (11).Debenham JS; Madsen R; Roberts C; Fraser-Reid B Two new orthogonal amine-protecting groups that can be cleaved under mild or neutral conditions. J. Am. Chem. Soc 1995, 117, 3302–3303. [Google Scholar]
- (12).Bowers SG; Coe DM; Boons GJ Application of the 2,5-dimethylpyrrole group as a new and orthogonal amine-protecting group in oligosaccharide synthesis. J. Org. Chem 1998, 63, 4570–4571. [Google Scholar]
- (13).Wu X; Grathwohl M; Schmidt RR Efficient solid-phase synthesis of a complex, branched N-glycan hexasaccharide: use of a novel linker and temporary-protecting-group pattern. Angew. Chem., Int. Ed 2002, 41, 4489–4493. [DOI] [PubMed] [Google Scholar]
- (14).Prabhu A; Venot A; Boons GJ New set of orthogonal protecting groups for the modular synthesis of heparan sulfate fragments. Org. Lett 2003, 5, 4975–4978. [DOI] [PubMed] [Google Scholar]
- (15).Bochkov AF; Kochetkov NK A new approach to the synthesis of oligosaccharides. Carbohydr. Res 1975, 39, 355–357. [Google Scholar]
- (16).Demchenko AV; Boons GJ A highly convergent synthesis of a complex oligosaccharide derived from group B type III Streptococcus. J. Org. Chem 2001, 66, 2547–2554. [DOI] [PubMed] [Google Scholar]
- (17).Kochetkov NK; Klimov EM; Malysheva NN; Demchenko AV A new stereospecific method for 1,2-cis-glycosylation. Carbohydr. Res 1991, 212, 77–91. [DOI] [PubMed] [Google Scholar]
- (18).Kaeothip S; Akins SJ; Demchenko AV On the stereoselectivity of glycosidation of thiocyanates, thiomidates, and thioglycosides. Carbohydr. Res 2010, 345, 2146–2150. [DOI] [PubMed] [Google Scholar]
- (19).Tsvetkov YE; Kitov PI; Backinowsky LV; Kochetkov NK Unusual regioselective glycosylation of sugar secondary trityloxy function in the presence of primary one. Tetrahedron Lett 1993, 34, 7977–7980. [Google Scholar]
- (20).Tsvetkov YE; Kitov PI; Backinowsky LV; Kochetkov NK Highly regioselective glycosylation of a secondary position in sugar primary-secondary ditrityl ethers. J. Carbohydr. Chem 1996, 15, 1027–1050. [Google Scholar]
- (21).Raghavan S; Kahne D A one step synthesis of ciclamycin trisaccharide. J. Am. Chem. Soc 1993, 115, 1580–1581. [Google Scholar]
- (22).Ziegler T The glycosylation of silylated alcohols. J. Prakt. Chem 1998, 340, 204–213. and references therein. [Google Scholar]
- (23).Boons GJ; Bowers S; Coe DM Trityl ethers in oligosaccharide synthesis: a novel strategy for the convergent assembly of oligosaccharides. Tetrahedron Lett 1997, 38, 3773–3776. [Google Scholar]
- (24).Zhu T; Boons GJ A two directional glycosylation strategy for the convergent assembly of oligosaccharides. Tetrahedron Lett 1998, 39, 2187–2190. [Google Scholar]
- (25).Gandolfi-Donadío L; Santos M; de Lederkremer RM; Gallo-Rodriguez C Synthesis of arabinofuranose branched galactofuran tetrasaccharides, constituents of mycobacterial arabinogalactan. Org. Biomol. Chem 2011, 9, 2085–2097. [DOI] [PubMed] [Google Scholar]
- (26).Huang T-Y; Zulueta MML; Hung S-C Regioselective one-pot protection, protection-glycosylation and protection-glycosylation-glycosylation of carbohydrates: a case study with D-glucose. Org. Biomol. Chem 2014, 12, 376–382. [DOI] [PubMed] [Google Scholar]
- (27).Kulkarni SS; Wang CC; Sabbavarapu NM; Podilapu AR; Liao PH; Hung SC ″One-Pot″ Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev 2018, 118, 8025–8104. [DOI] [PubMed] [Google Scholar]
- (28).Geringer SA; Demchenko AV Iron(III) chloride-catalyzed activation of glycosyl chlorides. Org. Biomol. Chem 2018, 16, 9133–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Geringer SA; Mannino MP; Bandara MD; Demchenko AV Picoloyl protecting group in synthesis: focus on a highly chemoselective catalytic removal. Org. Biomol. Chem 2020, 18, 4863–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ranade SC; Kaeothip S; Demchenko AV Glycosyl alkoxythioimidates as complementary building blocks for chemical glycosylation. Org. Lett 2010, 12, 5628–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mannino MP; Demchenko AV Synthesis of β-glucosides with 3-O-picoloyl-protected glycosyl donors in the presence of excess triflic acid: a mechanistic study. Chem. – Eur. J 2020, 26, 2927–2937. [DOI] [PubMed] [Google Scholar]
- (32).Choudhury AK; Ray AK; Roy N Synthesis of Tetrasaccharide Repeating Unit of the K-Antigen from Klebsiella Type-16. J. Carbohydr. Chem 1995, 14, 1153–1163. [Google Scholar]
- (33).Daragics K; Fügedi P Synthesis of glycosaminoglycan oligosaccharides. Part 5: Synthesis of a putative heparan sulfate tetrasaccharide antigen involved in prion diseases. Tetrahedron 2010, 66, 8036–8046. [Google Scholar]
- (34).Mukherjee MM; Ghosh R Synthetic Routes toward Acidic Pentasaccharide Related to the O-Antigen of E. coli 120 Using One-Pot Sequential Glycosylation Reactions. J. Org. Chem 2017, 82, 5751–5760. [DOI] [PubMed] [Google Scholar]
- (35).Kuester JM; Dyong I Partially benzylated carbohydrates, 2. Synthesis of all methyl mono-, di-, and tri-O-benzyl-α-D-glucopyranosides. Justus Liebigs Ann. Chem 1975, 2179–2189. [Google Scholar]
- (36).Daragics K; Fügedi P (2-Nitrophenyl)acetyl: A new, selectively removable hydroxyl protecting group. Org. Lett 2010, 12, 2076–2079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.