

Abstract
We have cloned and sequenced three genes from Rhizobium meliloti (Sinorhizobium meliloti) that are involved in sulfate activation for cysteine biosynthesis. Two of the genes display homology to the Escherichia coli cysDN genes, which code for an ATP sulfurylase (EC 2.7.7.4). The third gene has homology to the E. coli cysH gene, a 3′-phosphoadenosine-5′-phosphosulfate (PAPS) reductase (EC 1.8.99.4), but has greater homology to a set of genes found in Arabidopsis thaliana that encode an adenosine-5′-phosphosulfate (APS) reductase. In order to determine the specificity of the R. meliloti reductase, the R. meliloti cysH homolog was histidine tagged and purified, and its specificity was assayed in vitro. Like the A. thaliana reductases, the histidine-tagged R. meliloti cysH gene product appears to favor APS over PAPS as a substrate, with a Km for APS of 3 to 4 μM but a Km for PAPS of >100 μM. In order to determine whether this preference for APS is unique to R. meliloti among members of the family Rhizobiaceae or is more widespread, cell extracts from R. leguminosarum, Rhizobium sp. strain NGR234, Rhizobium fredii (Sinorhizobium fredii), and Agrobacterium tumefaciens were assayed for APS or PAPS reductase activity. Cell extracts from all four species also preferentially reduce APS over PAPS.
Sulfur is an important biological element essential for life in all organisms (12). It is used in the synthesis of the amino acids cysteine and methionine, from which it can be transferred to other sulfur-containing molecules (31); It is incorporated into some polysaccharides and lipids by certain plants and animals; and in mammalian systems, several sulfuryl derivatives are important for the inflammation response as well as the detoxification of endogenous and exogenic biomolecules such as phenolics and steroids (23).
The most abundant form of sulfur present under the oxidizing conditions of the atmosphere is sulfate (31). However, sulfate alone is fairly unreactive and must first be converted to a more reactive form in order to be used by the cell. Sulfate is activated by coupling to a nucleoside to make high-energy nucleoside phosphosulfates via a pathway that appears to be similar in most organisms. The first step in this pathway is the sulfation of ATP by an ATP sulfurylase (EC 2.7.7.4) to produce adenosine-5′-phosphosulfate (APS):
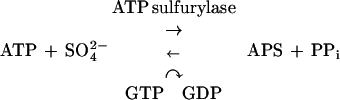 |
In this reaction, the free energy of hydrolysis of the phosphosulfate bond is higher than that of the pyrophosphate linkage, making APS production energetically unfavorable. In order to drive the ATP sulfurylase reaction in the forward direction, (i) sulfation of ATP is coupled with GTP hydrolysis and (ii) APS is phosphorylated at the 3′ position by an APS kinase (EC 2.7.1.25) to produce 3′-phosphoadenosine-5′-phosphosulfate (PAPS):
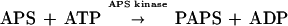 |
GTP hydrolysis shifts the mass ratio of the sulfation reaction toward APS by (5.4 × 106)-fold (20), and the free energy of APS phosphorylation, which is highly negative, further drives the two-step reaction forward. In most eukaryotes, PAPS is used as a sulfate donor in reactions in which the sulfate is directly transferred to a second molecule. In cysteine biosynthesis, PAPS is reduced to sulfite through the action of a PAPS reductase (EC 1.8.99.4), followed by production of sulfide by a sulfite reductase (EC 1.8.1.2). The sulfide is reacted with O-acetyl serine to produce cysteine via cysteine synthase (EC 4.2.99.8). A variation of this pathway has recently been found in the plant Arabidopsis thaliana (9, 37). Several A. thaliana cDNAs contain homologies to the Escherichia coli PAPS reductase gene cysH and can functionally complement cysH mutants in vivo. However, biochemical analysis showed that the protein products of these genes are reductases that use APS as a substrate more efficiently than PAPS.
Members of the genera Rhizobium, Bradyrhizobium, Azorhizobium, and Sinorhizobium are bacteria which form a symbiotic relationship with plants of the legume family (7, 21, 38, 40). The bacteria invade the roots of these plants and induce the formation of nodules, which the bacteria colonize. Rhizobium meliloti requires sulfur both for a symbiosis-specific synthetic pathway (sulfation of the Nod factor, a chemical signal that initiates nodulation) (8, 17, 27) and for general metabolic processes (biosynthesis of cysteine and sulfation of lipopolysaccharides [LPSa]) (5). The three genes involved in sulfation of the Nod factor, nodP and nodQ (which each exist in two copies) (27) and nodH (3, 8), were found as part of the nod regulon. Together, nodP and nodQ code for both ATP sulfurylase and APS kinase activities. nodP is homologous to the E. coli gene cysD (an ATP sulfurylase subunit), and nodQ appears to represent a fusion of cysN (an ATP sulfurylase subunit)- and cysC (APS kinase)-homologous sequences (34). NodP and NodQ together carry out the activation of sulfate by catalyzing the formation of PAPS from ATP and free sulfate. The nodH gene product is a sulfotransferase, which transfers the sulfate group from PAPS directly to the nascent Nod factor (8).
Despite their homologies to the E. coli cysDN genes, nodPQ1 and nodPQ2 are not necessary for cysteine biosynthesis (35). A strain in which both copies of nodPQ were deleted was prototrophic. A third locus, designated saa (for sulfur amino acid) (26) was identified by auxotrophic phenotype as responsible for activation of sulfate for cysteine synthesis. We have cloned, sequenced, and biochemically characterized the saa locus and found that it contains homologs to the E. coli cysDN (ATP sulfurylase) and cysH (PAPS reductase) genes, but no homolog to the E. coli cysC gene (APS kinase). We show that an R. meliloti extract from a strain with deletions of both nodPQ loci produces only APS, confirming the lack of an APS kinase activity outside the nod regulon. The lack of an APS kinase activity at this locus is correlated with the altered specificity of the R. meliloti cysH homolog, which we show reduces the sulfate on APS preferentially over PAPS during the production of free sulfite.
In addition, we asked whether the ability of R. meliloti to use APS in the production of free sulfite is correlated with the existence of a symbiosis-specific PAPS-dependent reaction (Nod factor sulfation), or if APS reduction is a more general property of the family Rhizobiaceae. Three other rhizobia were examined—R. leguminosarum, Rhizobium sp. strain NGR234, and R. fredii—as well as the closely related plant pathogen Agrobacterium tumefaciens. We show that cell extracts of all four members of the family Rhizobiaceae utilize APS as a substrate for the production of free sulfite, although only two of the species display symbiosis-specific PAPS synthetic pathways.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this study are described in Table 1.
TABLE 1.
Characteristics of the strains used in this study
| Strain, phage, or plasmid | Relevant genotype | Source or reference |
|---|---|---|
| Strains | ||
| Rhizobium meliloti | ||
| Rm1021 | Wild type | 22 |
| DsAux7 | Cysteine auxotroph, Tn5-233 | J. Glazebrook and G. Walker |
| JSS27 | nodPQ1 and nodPQ2 deletion | 35 |
| MW26 | cysDRm::Tn5 transduced into JSS27 | This study |
| Escherichia coli | ||
| XL1-Blue | Wild type | Stratagene |
| JM96 | cysH56 | E. coli Genetic Stock Center |
| JM226 | cysH57 | E. coli Genetic Stock Center |
| CAG12182 | cysC3152::Tn10kan | E. coli Genetic Stock Center |
| Rhizobium leguminosarum 8401/pRL1 | Wild type | J. A. Downie |
| Rhizobium sp. strain NGR234 | Wild type | W. Broughton |
| Rhizobium fredii USDA257 | Wild type | S. Pueppke |
| Agrobacterium tumefaciens A136 | Wild type | 42 |
| λ clones | ||
| λ2-3 | λ clone containing Tn5-233 insertion of DsAux7 | This study |
| λ211 | λ clone containing wild-type DNA corresponding to the DsAux7 insertion site | This study |
| Plasmids | ||
| pALTER | Tcr site-directed mutagenesis vector | Promega |
| pQE-30 | Apr, 6-histidine protein-tagging and expression vector | Qiagen |
| pBluescript SK+/− | Apr ColE1, cloning vector | Stratagene |
| pSW213 | Tcr IncP, broad-host-range vector | S. C. Winans |
| pMW121 | pBluescript carrying one Tn5-233 end plus surrounding genomic DNA from λ2-3 | This study |
| pMW122 | Same as pMW121, but carrying the opposite Tn5-233 end | This study |
| pMW126 | pSW213 carrying an 8-kb BamHI fragment of R. meliloti DNA encoding all of cysN and part of cysD | This study |
| pMW163 | pBluescript carrying a 6-kb PstI-XhoI fragment containing the full-length R. meliloti cysHDN gene | This study |
| pMW179 | pALTER carrying a 4.3-kb HindIII fragment containing nodPQ2 | This study |
| pMW179-Kpn | pMW179 with a KpnI site introduced at the translation start site of nodQ2 | This study |
| pMW183 | pQE-30 carrying the KpnI-HindIII fragment of pMW179-Kpn containing nodPQ2 | This study |
| pProEX-1 | Apr, 6-histidine protein-tagging and expression vector | Gibco BRL |
| pPIA8 | pProEx-1 carrying the R. meliloti cysH gene | This study |
R. meliloti DsAux7 is a cysteine auxotroph caused by a Tn5-233 insertion in the chromosome. In order to clone the genes surrounding the Tn5-233 insertion, a BamHI λ library with DNA from DsAux7 was constructed and probed with a BglI fragment from the Tn5-233 inverted repeat. One λ clone, λ2-3, was isolated and further subcloned into pBluescript as two SacI fragments containing the two ends of Tn5-233, generating pMW121 and pMW122. Partial sequence analysis with primers reading out from the Tn5-233 end showed that the insertion was in an open reading frame with good homology to the R. meliloti gene nodP and the E. coli gene cysD. The inserts in pMW121 and pMW122 were used to screen a wild-type R. meliloti strain, Rm1021, genomic BamHI λ library. Several clones were obtained that contained the same 8-kb fragment, which was recloned into pSW213 to make pMW126. This 8-kb insert was discovered to be incomplete, so a partial Sau3AI λ library from Rm1021 was constructed and probed with the inserts from pMW121 and pMW122. A single positive clone, λ211, was obtained, although it too was found to be incomplete. A full-length fragment was constructed by fusing a 5-kb PstI fragment from λ211 to a 0.75-kb PstI-XhoI fragment from pMW126 to generate pMW163.
NodQ2 was fused at the N terminus to six histidines (His6) to facilitate purification. A HindIII fragment containing nodPQ2 was cloned into pALTER, resulting in pMW179. A KpnI site was engineered at the ATG of nodQ2 by using the Altered Sites kit (Promega), making pMW179-Kpn. nodQ2 was excised from pMW179-Kpn as a KpnI-HindIII fragment and inserted into pQE-30, creating pMW183. pMW183 has a T5 promoter driving expression of a His6-NodQ2 fusion.
A PCR product of the R. meliloti cysH gene was fused at its N terminus to 6 histidine residues plus a spacer region by placement into pPro-EX1 (Gibco BRL) to make pPIA8.
DNA sequencing.
DNA sequencing was done by the dideoxynucleotide chain-termination method (29) by using the Sequenase 2.0 kit (U.S. Biochemicals). Most of the sequencing was completed with exonuclease III deletions, which were constructed in pBluescript as previously described (28). For regions not covered by the deletions, several subclones of pMW163 were constructed in pBluescript. Single-stranded DNA was produced as described earlier (41). Sequence analysis was performed with the University of Wisconsin Genetics Computer Group sequence analysis programs.
A putative CysB binding site located at −122 to −76 bp upstream of the major cysH transcription start site (SS1) was identified by sequence homology to the published consensus sequence TTA . . T . c . . tT . . . . . . T . . # . . . AT . . . . . Aa . C . . T … T (11), where uppercase letters denote identical nucleotides in six out of six CysB binding sites, lowercase letters denote identical nucleotides in five out of six CysB binding sites, and # denotes a gap in four out of six CysB binding sites. The binding site consists of two 19-bp half-sites separated by 1 or 2 bp. In R. meliloti, the putative CysB binding site has two 19-bp half-sites separated by 9 bp.
RNA isolation.
Total RNA was isolated as described by Chomczynski and Sacchi (6) with modifications as described below. One hundred-milliliter cultures of bacteria were grown to an optical density at 600 nm (OD600) of 0.7 in either M9 sucrose or M9 sucrose with 40 μg of cysteine per ml and 40 μg of methionine per ml, pelleted, and frozen at −80°C. The pellet was resuspended in 8 ml of Trizol (Bethesda Research Laboratories), vortexed with 0.5-μm glass beads for 5 min, and incubated at room temperature for 5 min. The homogenate was spun at 12,000 × g for 10 min at 4°C to remove glass beads and cell debris, and the supernatant was removed and extracted with 1.6 ml of chloroform. Nucleic acids were precipitated with 4 ml of isopropanol and resuspended in 0.6 ml of water. RNA was precipitated with 0.2 ml of 8 M LiCl, resuspended in 0.4 ml of water, reprecipitated with 40 μl of 3 M sodium acetate and 1 ml of 95% ethanol, and resuspended in water. RNA samples were quantitated by spectrophotometry and stored at −80°C.
Primer extension.
Oligonucleotide primers 35 nucleotides in length and homologous to the region of the cysH gene just downstream of the translation start site were synthesized and gel purified by Integrated DNA Technologies, Inc. Primers were end labeled with [γ-32P]ATP (Amersham) (2) and purified on Sephadex G-25 Quick Spin columns (Boehringer Mannheim). One to 20 μg of total cellular RNA was annealed to 1.25 ng of primer in 30 μl of hybridization buffer (0.01 M Tris [pH 8.5], 0.15 M KCl, 0.1 mM EDTA). The nucleic acids were precipitated, resuspended in 25 μl of elongation mix (0.5 mM deoxynucleoside triphosphates, 1× Superscript II buffer, 10 mM dithiothreitol [DTT], 40 U of RNasin), and preincubated at 50°C for 2 min. Two hundred units of Superscript II reverse transcriptase (Bethesda Research Laboratories) was added, and the reaction mixture was incubated at 50°C for a further 30 min. The reaction was stopped by addition of EDTA to 20 mM. The RNA was removed by incubating the reaction mixture for 30 min at 37°C with 1 μg of RNase. The reaction mixture was extracted with an equal volume of phenol-chloroform (1:1), and the extension products were precipitated, resuspended in loading buffer (54% formamide, 11 mM EDTA, 0.03% bromophenol blue, 0.03% xylene cyanol FF), and analyzed on a 7% polyacrylamide sequencing gel.
Protein purification and cell extract preparation.
The histidine-tagged cysH gene product was overexpressed from plasmid pPIA8 in E. coli XL1-Blue cells. The cells were grown at 37°C in Luria broth (LB) and 50 μg of ampicillin per ml to an OD600 of 0.5, induced with 0.6 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h, and harvested. After harvesting, the cells were broken by passing them once through a Bio-Nebulizer (Biology Department, Indiana University), and then they were clarified by centrifugation. The supernatant was bound in batch to Ni-nitrilotriacetic acid resin (GIBCO BRL) for 2 h at 4°C and then packed into a column. The column was washed with 100 column volumes of binding buffer (50 mM NaPO4 [pH 8.0], 300 mM NaCl, 20 mM imidazole, 10% glycerol) and eluted with 20 column volumes of eluted buffer (50 mM NaPO4 [pH 8.0], 300 mM NaCl, 500 mM imidazole, 10% glycerol), with 0.5-ml fractions collected. His6-CysHRm eluted in the first two fractions which were then dialyzed overnight against a mixture of 40 mM Tris-HCl (pH 8.0), 20 mM MgCl2, and 10 mM DTT. After dialysis, an equal volume of 60% glycerol was added to each fraction and the fractions were stored at −80°C.
APS kinase was produced by overexpression of the histidine-tagged nodQ2 gene on pMW183 in E. coli by using Qiagen’s Qiaexpress kit. Cells were grown in 1 liter of LB at 37°C to stationary phase. Expression was induced with 20 μM IPTG for 1 h at 30°C, and the cells were spun down at 5,000 × g for 10 min at 4°C. The pellet was washed with 10 mM MgSO4 and repelleted. The cells were broken as described above, and an S30 extract was prepared. His6-NodQ2 was purified on Ni-nitrilotriacetic acid agarose as recommended by the manufacturer.
For the preparation of cell extracts, R. meliloti was grown in M9 sucrose and E. coli was grown in M9 glucose with appropriate antibiotics and amino acids, omitting cysteine. R. leguminosarum, Rhizobium sp. strain NGR 234, and R. fredii were grown in RDM sucrose, and A. tumefaciens was grown in RDM glucose with appropriate antibiotics and amino acids, again omitting cysteine. The cells were grown to an OD600 of ∼1.0, washed twice in 10 mM MgSO4, and resuspended in breaking buffer (66.7 mM Tris-HCl [pH 8.0], 130 mM NaCl, 13.3 mM MgAc, 1.33 mM EDTA, 0.12 mM DTT, 13.3% glycerol). The cells were broken by passing them through a Bio-Nebulizer three times and then were clarified by centrifugation. The amount of protein in the cell extracts was quantitated by using the Bradford assay, and all cell extracts, except for the R. fredii extract, were brought to a protein concentration of 0.70 mg/ml with breaking buffer. The R. fredii extract was concentrated to 1.75 mg of protein per ml. Each cell extract was then passed over a Centri-Sep spin column (Princeton Separations) to remove endogenous ATP.
ATP sulfurylase and APS kinase enzyme assays.
ATP sulfurylase and APS kinase activities were measured by a method modified from that of Leyh et al. (13). Aliquots of protein extracts were incubated with the following mixture at 30°C for 10 min: 20 mM Tris (pH 8.0), 30 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10% glycerol, 2.5 μCi of [35S]Na2SO4 per μl, 4 mM Na2SO4, 25 mM ATP, 5 mM GTP, and 0.1 U of pyrophosphatase per μl. The reaction mixtures were boiled for 1.5 min, and spun in a microcentrifuge for 2 min. Polyethyleneimine-cellulose thin-layer chromatography (TLC) plates (J.T. Baker) were washed for 5 min and air dried. Aliquots of each reaction mixture were loaded onto a washed TLC plate, and the products were resolved with 0.9 M LiCl. The plates were air dried, covered in plastic wrap, and analyzed by autoradiography or phosphorimaging.
His6-CysHRm enzyme assays and kinetic analysis.
35S-labeled APS and PAPS were prepared as described previously (18, 33) by incubating [35S]Na2SO4, ATP, ATP sulfurylase (Sigma), pyrophosphatase (Sigma), and His6-NodQ2 (omitting His6-NodQ2 if making APS) together in buffer B (50 mM Tris-HCl [pH 8.0], 30 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 10% glycerol), with 10.2 mM GTP as a cofactor for the ATP sulfurylase, at 30°C for 3 h. Purified protein (0.312 mg/ml) or cell extracts (1/10 final reaction volume) were incubated at 30°C with various amounts of [35S]APS or [35S]PAPS in reaction buffer (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 25 mM NaF, 5 mM DTT, and 45 μg of E. coli thioredoxin per ml) for either 1 h (cell extracts) or various amounts of time (kinetic experiments). Reaction mixtures were heat killed at 95°C for 3 min and placed on ice. Products were analyzed by spotting aliquots of the reaction mixture onto prewashed polyethyleneimine-cellulose TLC plates and developing them in 1 M LiCl. The TLC plates were exposed to a phosphorimager plate (Bio-Rad), and the intensity of the various bands was quantitated by using the program Molecular Analyst (Bio-Rad). Kinetic analysis was done with the program Winzyme.
Nucleotide sequence accession number.
The nucleotide sequence of the cysHDN locus has been submitted to GenBank under accession no. AF158023.
RESULTS
Cloning and sequencing of cysHDN.
R. meliloti DsAux7 is a cysteine auxotroph caused by a Tn5-233 insertion in the genome (Table 1). The cysteine auxotrophy can be rescued by the addition of either cysteine or methionine to the growth medium or by a plasmid expressing nodPQ1 from the lac promoter (35). This suggests that the Tn5-233 insertion is in the structural gene encoding either ATP sulfurylase or APS kinase. The region around the Tn5-233 insertion in DsAux7 was cloned and sequenced and was found to contain three open reading frames with good homology to cysteine biosynthetic genes in E. coli and other organisms (Fig. 1). The two downstream open reading frames are homologous to the E. coli cysD and cysN genes, which together encode an ATP sulfurylase (18). However, unlike the E. coli operon in which the third downstream open reading frame is cysC (APS kinase), no open reading frames were found for several kilobases downstream of the cysN homolog. Instead, a third open reading frame was found upstream of cysD with homology to the E. coli cysH gene, which encodes a PAPS reductase (16). The stop and start codons for the cysD and cysN homologs overlap, whereas 47 bp separate the cysH and cysD homologs.
FIG. 1.

Organization of sulfate activation genes in R. meliloti and E. coli. Regions of homology are blocked off with dotted lines. Domains that make up enzyme activities are bracketed and named at the bottom of the figure.
Primer extension analysis identified two transcription start sites, both upstream of the cysH homolog (Fig. 2), making it likely that all three open reading frames are in a single operon. In E. coli, transcription of the cysDNC operon is driven by the activator CysB, which binds upstream of cys regulon promoters and is subject to feedback inhibition by cysteine. In R. meliloti, the major transcription start site (SS1) is just downstream from a putative CysB binding site (Fig. 2), and the transcript generated from this start site is repressed in the presence of cysteine (Fig. 2, lane 5). A minor, constitutive start site (SS2) is located approximately 172 nucleotides upstream of the cysH translational start site. The presence of SS2, which lies within the putative CysB binding site, is noteworthy, since CysB and other LysR-type transcriptional regulators typically occupy their DNA binding sites even in the absence of inducer (10, 24, 25, 30). At present, nothing is known about the mechanism of transcription initiation at either SS1 or SS2. One possible model is that transcription at SS2 might initiate at times when a CysB-like protein does not occupy or is displaced from the putative CysB binding site. Another possibility is that the region from −122 to −76 bp upstream of SS1 is not a binding site for a CysB-like regulator. At this time, no CysB homolog has been identified in R. meliloti, and further experiments need to be done to understand the mechanism of transcription regulation of the cysHDN locus.
FIG. 2.

Transcription start site of cysHRm. Primer extension experiments reveal two transcription start sites, SS1 and SS2. (a) Sequencing ladder (lanes 1 to 4), with primer extension reactions using Rm1021/pMW205 RNA grown in M9 minimal medium plus cysteine and methionine (lane 5) or M9 minimal medium alone (lane 6). (b) Region upstream of the ATG for cysHRm. The solid arrows represent SS1 and SS2, while the putative CysB binding site, the putative ribosome binding site (RBS), and the −10 and −35 regions are boxed and labeled. SS2 is upstream of the putative CysB binding site. (c) Structure of cysHDNRm. The small solid rectangle represents the putative CysB binding site.
BLAST analysis shows that the R. meliloti cysH homolog has greater sequence similarity to genes from A. thaliana—Prh-19, Prh-26, and Prh-43 (9) and APR1, APR2, and APR3 (37)—than to the E. coli cysH gene (Fig. 3). These A. thaliana genes have been shown to code for proteins which utilize APS rather than PAPS as a substrate in the formation of free sulfite. The high sequence similarity between the R. meliloti cysH homolog and the A. thaliana sequences, coupled with the lack of an APS kinase homolog at the R. meliloti cysHDN locus, suggests that R. meliloti may utilize a sulfate activation and reduction pathway distinct from that previously defined for bacteria. Therefore we investigated the biochemical activities associated with the R. meliloti cysHDN locus.
FIG. 3.

Alignment of the deduced amino acid sequences of the R. meliloti cysH gene product, the first 345 residues of the A. thaliana prh43 gene product, and the E. coli cysH gene product. Identical residues are boxed, and gaps are indicated by dashes. Sequences were aligned by the Clustal method in the program Lasergene.
The two nodQ loci contain the only APS kinase activity in R. meliloti.
Although the cysHDNRm locus lacked a gene corresponding to an APS kinase homolog, it was possible that such a gene was located elsewhere. We directly tested protein extracts from R. meliloti JSS27, which contains deletions of both nodPQ1 and nodPQ2, to determine whether a third APS kinase activity was present. When JSS27 was grown on methionine or glutathione, which does not result in feedback inhibition of the cys regulon in other systems (13–15), the resulting cell extract was able to produce only APS (Fig. 4, lanes 5 and 6). APS production was inhibited by cysteine (Fig. 4, lanes 3 and 4) and was not present in a cell extract from MW26, a strain with Tn5 insertions in all three sulfate activation loci (Fig. 4, lane 7). Under the same conditions, an extract from a wild-type R. meliloti strain produced both APS and PAPS (Fig. 4, lane 2). These results show that R. meliloti does not possess an APS kinase activity other than at the two nodQ loci and imply that APS is the final activated sulfate moiety in R. meliloti cysteine biosynthesis.
FIG. 4.

Sulfate activation from the R. meliloti cysteine regulon. (a) TLC plate analysis of several R. meliloti extracts for production of APS and PAPS. Lane 1 is a control reaction with no protein extract. The protein extracts were isolated from Rm1021 in M9 minimal medium (lane 2), JSS27 in M9 medium plus cysteine and methionine (lane 3), JSS27 in M9 medium plus cysteine (lane 4), JSS27 in M9 medium plus methionine (lane 5), JSS27 in M9 medium plus glutathione (lane 6), and MW26 in M9 medium plus methionine (lane 7). (b) Quantitation of the data from two experiments as in panel a.
His6-CysHRm from R. meliloti reduces APS preferentially over PAPS.
The presence of a CysHRm reductase homolog in the same putative operon encoding a CysD-CysN complex predicted to produce only APS suggested that the CysHRm reductase might be APS specific. Therefore we analyzed the preference of CysHRm for APS or PAPS. We constructed a six-histidine-tagged CysHRm protein and purified the fusion protein over an Ni-nitrilotriacetic acid column to homogeneity (Fig. 5a). The activity of this fusion was verified in vivo by its ability to complement two E. coli cysH mutant strains, JM96 and JM226 (data not shown). The substrate preference of the purified His6-CysHRm protein was assayed in vitro with either [35S]APS (Fig. 5b, lanes 1 to 7) or [35S]PAPS (Fig. 5b, lanes 8 to 14) as a substrate and with the appearance of free sulfite measured over time. Only when APS was present did the free sulfite band appear, showing that under the conditions used, APS rather than PAPS is the preferred substrate.
FIG. 5.

(a) Coomassie-stained gel of the His6-CysHRm protein purification. Lanes 1 and 2 are elution fractions 1 and 2. The arrow indicates the His6-CysHRm protein. (b) Radioactive TLC plate imaged with a phosphoimager, showing the preferential reduction of APS versus PAPS by the His6-CysHRm protein. Either APS (lanes 1 to 7) or PAPS (lanes 8 to 14) was used as the substrate for His6-CysHRm for 0, 1, 5, 10, 30, 60, or 120 min, respectively.
Kinetic analysis further supports this result. When APS was used as a substrate, the Km was 3 to 4 μM and the Vmax was 5 to 7 nmol min−1 mg of protein−1. These values are comparable to those obtained for the E. coli (Km = 10 μM, Vmax = 94 to 99 μmol min−1 mg of protein−1) and S. cerevisiae (Km = 3 to 4 μM, and Vmax = 4 to 7 nmol min−1 mg of protein−1) PAPS reductases when PAPS was used as a substrate. In contrast, when the R. meliloti His6-CysH is presented with PAPS as a substrate, the Km was >100 μM and the Vmax was <0.21 pmol min−1 mg of protein−1.
In order to show that APS is directly reduced to free sulfite, either 5′-AMP or 3′-AMP was added to the APS reduction reaction. If APS is directly reduced, the products of the reaction should be 5′-AMP and free sulfite. Thus, addition of 5′-AMP, but not 3′-AMP, to the reaction should be inhibitory if APS is reduced directly, but will have no effect on the reaction if APS is first converted to another molecule. Inclusion of 5′-AMP in the reaction does inhibit the reduction of APS, whereas inclusion of 3′-AMP does not (Fig. 6).
FIG. 6.

Inhibition of His6-CysHRm (0.312 mg/ml)-mediated reduction of 83 nM [35S]APS by 5′-AMP (○) but not by 3′-AMP (✻). The standard reaction mixture was incubated for 1 h at 30°C, heat killed at 95°C for 3 min, and analyzed on a polyethyleneimine-cellulose TLC plate developed in 1 M LiCl. The intensity of each spot was quantitated with a phosphoimager. The percent sulfite was calculated as [(amount of sulfite signal)/(amount of sulfite signal + APS signal)] × 100.
As with the E. coli and S. cerevisiae PAPS reductases, the R. meliloti His6-CysH protein requires thioredoxin for optimal activity (Fig. 7). In the absence of a thioredoxin cofactor, the R. meliloti His6-CysH activity drops approximately fivefold. Glutathione cannot replace thioredoxin in the reaction, strengthening arguments that APS reduction by CysHRm is not due to a side reaction of APS kinase, as had been found previously in plant cytosolic extracts (1, 19, 32, 39), and implies further that APS is the bona fide substrate.
FIG. 7.

Requirement for thioredoxin but not glutathione in the His-CysH-mediated reduction of APS.
Other members of the Rhizobiaceae also produce sulfite by APS reduction.
In order to determine if the use of APS as a direct substrate for sulfite production is restricted to R. meliloti or occurs in other closely related bacteria, we assayed for APS or PAPS reductase activities in cell extracts of R. leguminosarum, Rhizobium sp. strain NGR234, R. fredii, A. tumefaciens, and E. coli (Table 2). We chose these species because of their differential incorporation of sulfate in either Nod factor or LPS (Table 2) (26). We speculated that if the presence of PAPS-dependent sulfation pathways for production of Nod factor and extracellular carbohydrates provided evolutionary pressure to channel sulfate for cysteine biosynthesis by reduction of APS rather than PAPS, then only those species showing sulfated carbohydrates would display the APS-dependent cysteine pathway. Our assays showed that the cell extracts from R. leguminosarum, R. sp. strain NGR234, R. fredii, and A. tumefaciens all reduced APS and not PAPS, while the E. coli control uniquely reduced PAPS as predicted (Table 2). Therefore, the APS-dependent CysH enzyme is common to all species of Rhizobiaceae tested and does not correlate with the presence of PAPS-dependent sulfurylation pathways for symbiosis.
TABLE 2.
Differential use of sulfate among species of Rhizobiaceae and E. coli and their ability to reduce APS versus PAPS
DISCUSSION
Unlike the previously characterized enteric bacteria, R. meliloti and several related species in the family Rhizobiaceae reduce APS rather than PAPS for sulfite production during cysteine biosynthesis. The kinetic properties of the purified R. meliloti His6-CysHRm protein (His6-APS reductase) and data showing inhibition of the reduction reaction by 5′-AMP but not 3′-AMP (Fig. 6) are consistent with a preference for APS.
An in vivo test for heterologous function of CysHRm in E. coli did not result in complementation of an E. coli strain deficient in APS kinase (data not shown). This is in contrast to the APS-reducing cysH homologs found in Arabidopsis (9, 26) which can complement an E. coli APS kinase mutant. The Arabidopsis cysH homologs contain a thioredoxin-like domain, raising the possibility that the inability of the His6-cysHRm construct to complement the E. coli APS kinase mutant could be due to reduced affinity of the R. meliloti APS reductase for the E. coli thioredoxin. A parallel may be seen in the case of Saccharomyces cerevisiae: the affinity of purified S. cerevisiae PAPS reductase for the E. coli thioredoxin (Km = 1.4 μM) is less than its affinity for the homologous S. cerevisiae thioredoxin (Km = 0.6 μM) (36). Thus, it is possible that in vivo, the R. meliloti His6-APS reductase is physiologically inefficient in interacting with the E. coli thioredoxin. The reaction is able to occur in vitro because a large excess of E. coli thioredoxin is added, but in vivo, it is possible that the amount of thioredoxin is limiting. A second possibility is that the CysHRm protein forms a complex with the products of the cysDN genes and that this complex is absent due to either the histidine tag, the heterologous nature of the partners, or, perhaps, interference caused by binding of the mutated CysC protein.
The use of APS rather than PAPS by R. meliloti in cysteine biosynthesis is noteworthy, given that ATP sulfurylase and APS kinase activities are fused into a single peptide, NodQ, in the sulfate activation pathway for Nod factor sulfation (23). The presence of ATP sulfurylase and APS kinase activities in the same polypeptide may be advantageous: it could allow the APS produced by NodPQ to be rapidly converted to PAPS for Nod factor sulfation, perhaps keeping the APS generated by NodPQ sequestered from the CysHRm APS reductase activity used in cysteine biosynthesis. The use of two different substrates for sulfate activation could provide one example of how the cell can keep processes needed for general metabolism from interfering with those needed for symbiosis and vice versa.
This use of two distinct pathways for sulfate activation in R. meliloti led us to ask if other members of the family Rhizobiaceae also reduce APS for cysteine biosynthesis and if APS reduction correlated with the presence of symbiosis-dependent sulfurylation reactions. We asked whether the loss of a CysC-like APS kinase and specialization of CysH occurred in evolution as a response to the acquisition of a NodPQ-NodH pathway for PAPS use, and thus is only present in R. meliloti, or whether the APS-dependent pathway existed broadly in the Rhizobiaceae group. We assayed for APS or PAPS reduction in cell extracts from several other members of the Rhizobiaceae, chosen for their differential incorporation of sulfate in either Nod factor or LPS (Table 2). We found that each of the species of Rhizobiaceae tested reduced APS. Therefore, the ability to reduce APS directly for cysteine biosynthesis does not appear to be correlated with use of sulfurylation pathways for symbiotic processes, but rather seems to be more widespread among members of the family Rhizobiaceae.
ACKNOWLEDGMENTS
A.P.A. and M.G.W. contributed equally to this work.
We thank Audrey Southwick for assistance with PAPS synthesis and assays and the other members of our laboratory for useful discussions and critical reading of the manuscript. We are grateful to Bradley Ruehs for providing data in advance of publication and for helpful discussions. We thank G. Walker, J. A. Downie, W. Broughton, S. Pueppkem and S. C. Winans for the gifts of strains and for discussions of Rhizobium biochemistry.
S.R.L. is an investigator of the Howard Hughes Medical Institute (HHMI). Additional support for this research was provided by the Department of Energy (DE-FG03-90ER200120). A.P.A. was supported by HHMI. M.G.W. was supported by an NIH training grant to Stanford University. R.C.W. was supported by a summer undergraduate research award from Stanford University and by HHMI.
REFERENCES
- 1.Arz H E, Gisselmann G, Schiffmann S, Schwenn J D. A cDNA for adenylyl sulphate (APS)-kinase from Arabidopsis thaliana. Biochim Biophys Acta. 1994;1218:447–452. doi: 10.1016/0167-4781(94)90203-8. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current protocols in molecular biology. New York, N.Y: Greene Publishing Associates, John Wiley & Sons, Inc.; 1994. [Google Scholar]
- 3.Banfalvi Z, Kondorosi A. Production of root hair deformation factors by Rhizobium meliloti nodulation genes in Escherichia coli: hsnD (nodH) is involved in the plant host-specific modification of the nodABC factor. Plant Mol Biol. 1989;13:1–12. doi: 10.1007/BF00027330. [DOI] [PubMed] [Google Scholar]
- 4.Carlson R W, Price N P J, Stacey G. The biosynthesis of rhizobial lipo-oligosaccharide nodulation signal molecules. Mol Plant-Microbe Interact. 1994;7:684–695. doi: 10.1094/mpmi-7-0684. [DOI] [PubMed] [Google Scholar]
- 5.Cedergren R A, Lee J, Ross K L, Hollingsworth R I. Common links in the structure and cellular localization of Rhizobium chitolipooligosaccharides and general Rhizobium membrane phospholipid and glycolipid components. Biochemistry. 1995;34:4467–4477. doi: 10.1021/bi00013a040. [DOI] [PubMed] [Google Scholar]
- 6.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 7.Dénarié J, Truchet G, Promé J C. Lipo-oligosaccharide signalling: the mediation of recognition and nodule organogenesis induction in the legume-Rhizobium symbiosis. Biochem Soc Symp. 1994;60:51–60. [PubMed] [Google Scholar]
- 8.Ehrhardt D W, Atkinson E M, Faull K F, Freedberg D I, Sutherlin D P, Armstrong R, Long S R. In vitro sulfotransferase activity of NodH, a nodulation protein of Rhizobium meliloti required for host-specific nodulation. J Bacteriol. 1995;177:6237–6245. doi: 10.1128/jb.177.21.6237-6245.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutierrez-Marcos J F, Roberts M A, Campbell E I, Wray J L. Three members of a novel small gene-family from Arabidopsis thaliana able to complement functionally an Escherichia coli mutant defective in PAPS reductase activity encode proteins with a thioredoxin-like domain and “APS reductase” activity. Proc Natl Acad Sci USA. 1996;93:13377–13382. doi: 10.1073/pnas.93.23.13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hryniewicz M M, Kredich N M. The cysP promoter of Salmonella typhimurium: characterization of two binding sites for CysB protein, studies of in vivo transcription initiation, and demonstration of the anti-inducer effects of thiosulfate. J Bacteriol. 1991;173:5876–5886. doi: 10.1128/jb.173.18.5876-5886.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hryniewicz M M, Kredich N M. Hydroxyl radical footprints and half-site arrangements of binding sites for the CysB transcriptional activator of Salmonella typhimurium. J Bacteriol. 1995;177:2343–2353. doi: 10.1128/jb.177.9.2343-2353.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huxtable R J. Biochemistry of sulfur. New York, N.Y: Plenum Press; 1986. [Google Scholar]
- 13.Jones-Mortimer M C. Positive control of sulphate reduction in Escherichia coli. Biochem J. 1968;110:597–602. doi: 10.1042/bj1100597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones-Mortimer M C, Wheldrake J F, Pasternak C A. The control of sulphate reduction in Escherichia coli by O-acetyl-L-serine. Biochem J. 1968;107:51–53. doi: 10.1042/bj1070051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kredich N M. Regulation of L-cysteine biosynthesis in Salmonella typhimurium. J Biol Chem. 1971;246:3474–3484. [PubMed] [Google Scholar]
- 16.Krone F A, Westphal G, Schwenn J D. Characterisation of the gene cysH and of its product phospho-adenylylsulphate reductase from Escherichia coli. Mol Gen Genet. 1991;225:314–319. doi: 10.1007/BF00269864. [DOI] [PubMed] [Google Scholar]
- 17.Lerouge P, Roche P, Faucher C, Maillet F, Truchet G, Promé J C, Dénarié J. Symbiotic host-specificity of Rhizobium meliloti is determined by a sulphated and acylated glucosamine oligosaccharide signal. Nature (London) 1990;344:781–784. doi: 10.1038/344781a0. [DOI] [PubMed] [Google Scholar]
- 18.Leyh T S, Taylor J C, Markham G D. The sulfate activation locus of Escherichia coli K12: cloning, genetic, and enzymatic characterization. J Biol Chem. 1988;263:2409–2416. [PubMed] [Google Scholar]
- 19.Li J Y, Schiff J A. Purification and properties of adenosine 5′-phosphosulphate sulphotransferase from Euglena. Biochem J. 1991;274:355–360. doi: 10.1042/bj2740355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Wang R, Varlamova O, Leyh T S. Regulating energy transfer in the ATP sulfurylase-GTPase system. Biochemistry. 1998;37:3886–3892. doi: 10.1021/bi971989d. [DOI] [PubMed] [Google Scholar]
- 21.Long S R. Rhizobium symbiosis: Nod factors in perspective. Plant Cell. 1996;8:1885–1898. doi: 10.1105/tpc.8.10.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meade H M, Long S R, Ruvkun G B, Brown S E, Ausubel F M. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J Bacteriol. 1982;149:114–122. doi: 10.1128/jb.149.1.114-122.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitchell S C. Biology of sulfur. In: Mitchell S C, editor. Biological interactions of sulfur compounds. Bristol, Pa: Taylor & Francis, Inc.; 1996. pp. 20–41. [Google Scholar]
- 24.Monroe R S, Ostrowski J, Hryniewicz M M, Kredich N M. In vitro interactions of CysB protein with the cysK and cysJIH promoter regions of Salmonella typhimurium. J Bacteriol. 1990;172:6919–6929. doi: 10.1128/jb.172.12.6919-6929.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrowski J, Kredich N M. In vitro interactions of CysB protein with the cysJIH promoter of Salmonella typhimurium: inhibitory effects of sulfide. J Bacteriol. 1990;172:779–785. doi: 10.1128/jb.172.2.779-785.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reuhs, B. L. Personal communication.
- 27.Roche P, Debellé F, Maillet F, Lerouge P, Faucher C, Truchet G, Dénarié J, Promé J C. Molecular basis of symbiotic host specificity in Rhizobium meliloti: nodH and nodPQ genes encode the sulfation of lipo-oligosaccharide signals. Cell. 1991;67:1131–1143. doi: 10.1016/0092-8674(91)90290-f. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 29.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schell M A. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993;47:597–626. doi: 10.1146/annurev.mi.47.100193.003121. [DOI] [PubMed] [Google Scholar]
- 31.Schiff J A, Fankhauser H. Assimilatory sulfate reduction. In: Bothe H, Trebst A, editors. Biology of inorganic nitrogen and sulfur. New York, N.Y: Springer-Verlag; 1981. pp. 153–168. [Google Scholar]
- 32.Schmidt A. Sulfate reduction in a cell-free system of Chlorella. The ferredoxin dependent reduction of a protein-bound intermediate by a thiosulfonate reductase. Arch Mikrobiol. 1973;93:29–52. [PubMed] [Google Scholar]
- 33.Schwedock J, Long S R. ATP sulphurylase activity of the nodP and nodQ gene products of Rhizobium meliloti. Nature (London) 1990;348:644–647. doi: 10.1038/348644a0. [DOI] [PubMed] [Google Scholar]
- 34.Schwedock J S, Liu C, Leyh T S, Long S R. Rhizobium meliloti NodP and NodQ form a multifunctional sulfate-activating complex requiring GTP for activity. J Bacteriol. 1994;176:7055–7064. doi: 10.1128/jb.176.22.7055-7064.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwedock J S, Long S R. Rhizobium meliloti genes involved in sulfate activation: the two copies of nodPQ and a new locus, saa. Genetics. 1992;132:899–909. doi: 10.1093/genetics/132.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwenn J D, Krone F A, Husmann K. Yeast PAPS reductase: properties and requirements of the purified enzyme. Arch Microbiol. 1988;150:313–319. doi: 10.1007/BF00408300. [DOI] [PubMed] [Google Scholar]
- 37.Setya A, Murillo M, Leustek T. Sulfate reduction in higher plants: molecular evidence for a novel 5′-adenylylsulfate reductase. Proc Natl Acad Sci USA. 1996;93:13383–13388. doi: 10.1073/pnas.93.23.13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spaink H P. The molecular basis of the host specificity of the Rhizobium bacteria. Antonie Leeuwenhoek. 1994;65:81–98. doi: 10.1007/BF00871750. [DOI] [PubMed] [Google Scholar]
- 39.Tsang M L, Schiff J A. Assimilatory sulfate reduction in an Escherichia coli mutant lacking thioredoxin activity. J Bacteriol. 1978;134:131–138. doi: 10.1128/jb.134.1.131-138.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Rhijn P, Vanderleyden J. The Rhizobium-plant symbiosis. Microbiol Rev. 1995;59:124–142. doi: 10.1128/mr.59.1.124-142.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vieira J, Messing J. Production of single-stranded plasmid DNA. Methods Enzymol. 1987;153:3–11. doi: 10.1016/0076-6879(87)53044-0. [DOI] [PubMed] [Google Scholar]
- 42.Watson B, Currier T C, Gordon M P, Chilton M-D, Nester E W. Plasmid required for virulence of Agrobacterium tumefaciens. J Bacteriol. 1975;123:255–264. doi: 10.1128/jb.123.1.255-264.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]