

Abstract
Covalent drugs have been used to treat diseases for more than a century, but tools that facilitate the rational design of covalent drugs have emerged more recently. The purposeful addition of reactive functional groups to existing ligands can enable potent and selective inhibition of target proteins, as demonstrated by the covalent epidermal growth factor receptor (EGFR) and Bruton’s tyrosine kinase (BTK) inhibitors used to treat various cancers. Moreover, the identification of covalent ligands through ‘electrophile-first’ approaches has also led to the discovery of covalent drugs, such as covalent inhibitors for KRAS(G12C) and SARS-CoV-2 main protease. In particular, the discovery of KRAS(G12C) inhibitors validates the use of covalent screening technologies, which have become more powerful and widespread over the past decade. Chemoproteomics platforms have emerged to complement covalent ligand screening and assist in ligand discovery, selectivity profiling and target identification. This Review showcases covalent drug discovery milestones with emphasis on the lessons learned from these programmes and how an evolving toolbox of covalent drug discovery techniques facilitates success in this field.
Subject terms: Medicinal chemistry, Small molecules, Drug discovery and development
The rational discovery of covalent drugs depends on an expanding toolset of techniques. Here, Daniel Nomura and colleagues highlight covalent drugs that have achieved success over the past decade and discuss the tools and strategies that facilitate their discovery, describing two complementary approaches, namely, ligand-first and electrophile-first strategies.
Introduction
Covalent drugs incorporate a mildly reactive functional group that forms a covalent bond with protein targets to confer additional affinity beyond the non-covalent interactions involved in drug binding1. Historically, concerns about the interference of these reactive molecules with biological assays and potential lack of selectivity often discouraged further investigation2,3. Many early covalent drugs were discovered serendipitously and bind active sites to inhibit enzymatic activity4. These drugs often mimic a substrate transition state to enable covalent modification of a catalytic amino acid residue. Over the past 30 years, the rational design of covalent drugs has garnered increased interest, and covalently targeting non-conserved amino acids to increase selectivity has become commonplace2,5. The prolonged target engagement of covalent drugs can provide distinct pharmacodynamic profiles and exceptional potency6.
The potential benefits of covalency have inspired medicinal chemists to explore the covalent drug space despite concerns about reactivity. In many cases, compromises between reactivity, selectivity and potency have produced safe and effective drugs. Key examples that we discuss here (Fig. 1 and Table 1) include the Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib (AbbVie) and the epidermal growth factor receptor (EGFR) inhibitor osimertinib (AstraZeneca), with sales totalling US$8.43 billion and $4.33 billion in 2020, respectively7,8. Moreover, potent inhibition through covalent modification has enabled targeting of traditionally ‘undruggable’ proteins, exemplified by the approval of sotorasib (Amgen), which is an inhibitor of mutant KRAS(G12C), a GTPase that resisted decades of drug discovery efforts9,10 (Fig. 1). At the same time, more traditional covalent targeting of protease active sites has continued to yield valuable drugs, such as nirmatrelvir (Pfizer), which inhibits the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro)11 (Fig. 1).
Fig. 1. Timeline of the development of major covalent drugs.

Each covalent drug is classified according to the drug type or type of disease it treats. Unless otherwise indicated, the date refers to the first approval by the US Food and Drug Administration. NSAID, non-steroidal anti-inflammatory drug.
Table 1.
Key examples of covalent drugs
| Drug (company; former compound name) | Structure | Target | Approvala | Refs. |
|---|---|---|---|---|
| Osimertinib (AstraZeneca; AZD9291) | 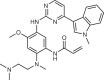 |
Mutant-selective EGFR inhibitor | 2015 for treatment of NSCLC | 44,45,48,49 |
| Ibrutinib (AbbVie; PCI-32765) | 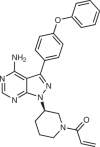 |
BTK inhibitor | 2013 for treatment of mantle-cell lymphoma and, subsequently, many other B cell malignancies | 55,56,60 |
| Sotorasib (Amgen; AMG-510) | 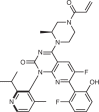 |
KRAS(G12C) inhibitor | 2021 for treatment of NSCLC with KRASG12C mutation | 93–95 |
| Nirmatrelvir (Pfizer; PF-07321332) | 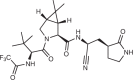 |
SARS-CoV-2 main protease inhibitor | Authorized for emergency use by FDA in 2021 for treatment of COVID-19b | 103,105 |
| Voxelotor (Global Blood Therapeutics; GBT-440) | 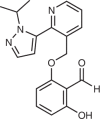 |
Mutant-haemoglobin modulator | 2019 for treatment of sickle cell anaemia | 142,143,145 |
BTK, Bruton’s tyrosine kinase; COVID-19, coronavirus disease 2019; EGFR, epidermal growth factor receptor; NSCLC, non-small-cell lung cancer; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2. aUnless otherwise indicated, the date of the first approval by one of the major regulatory agencies is provided, which was the FDA in each case. bNirmatrelvir is approved for use in combination with ritonavir (Paxlovid). The FDA’s emergency use authorization preceded approval by other agencies, but Paxlovid has not yet been fully approved by the FDA.
Targeted covalent inhibitors are often discovered through structure-guided design by incorporating an electrophile into a ligand that would otherwise reversibly bind the target protein. The incorporated electrophile binds irreversibly to an amino acid on the target protein, introducing a covalent interaction in addition to the reversible interactions already at play.
Covalent ligand screening is another ligand discovery approach that is becoming more common, whereby various methods are used to discover covalent ligands from libraries of electrophilic compounds7,12,13. This ‘electrophile-first’ approach is partly facilitated by the development of chemoproteomic platforms that enable rapid target identification and selectivity profiling of covalent ligands14–18. Combining the advances in covalent ligand screening and chemoproteomics with structural biology to empower medicinal chemistry has the potential to generate molecules that selectively bind challenging protein targets.
In this Review, we start by briefly highlighting historical examples of covalent drugs and their mechanisms of action. We then elaborate on milestones in covalent drug discovery over the past decade, categorizing our discussion on the basis of the discovery approach taken. Finally, we summarize the toolbox of emerging covalent drug discovery techniques, with emphasis on screening strategies and selectivity profiling.
History of covalent drugs
Compounds that contain protein-reactive functional groups have often been avoided in medicinal chemistry and excluded from compound screening collections owing to their potential for assay interference and off-target promiscuity. Many historical examples of covalent drugs were discovered to act through covalent mechanisms after their use was already widespread. One of the most prominent among these is the non-steroidal anti-inflammatory drug (NSAID) aspirin, which has been marketed since 1899 (ref.19) (Fig. 1). Aspirin’s mechanism of action was unknown until 1971, when it was discovered to exert its anti-inflammatory effects by acetylating Ser529 in the substrate-binding channel of cyclooxygenase 1, preventing conversion of the substrate arachidonic acid into prostaglandins20.
Early covalent drugs also tend to be derived from or inspired by natural sources. β-Lactam antibiotics such as penicillin (Fig. 1), produced by Penicillium fungi, bind to penicillin-binding proteins (PBPs), which are involved in bacterial cell wall synthesis21. All PBPs contain active-site serine residues that can be acylated by penicillin, inhibiting PBP activity and leading to cell membrane rupture21. Another covalent antibiotic is the epoxide-containing fosfomycin (Fig. 1), which is produced by some Streptomyces bacteria and acts by reacting with the catalytic cysteine of UDP-N-acetylglucosamine-enolpyruvyl transferase (MurA) to disrupt peptidoglycan synthesis and induce membrane rupture22–24.
Some covalent drugs are prodrugs with thiol-containing metabolites that form disulfide bonds to inactivate their targets25. The proton pump inhibitor omeprazole (Fig. 1), approved by the US Food and Drug Administration (FDA) in 1988 to treat gastro-oesophageal reflux disease, is an example of this and is also a drug that was brought to market before its mechanism of action was understood to be covalent. Both omeprazole and clopidogrel (Fig. 1), an antiplatelet medication used to prevent strokes and heart attacks, are activated by cytochrome P450 enzymes in the liver to produce bioactive thiol metabolites26.
Covalent drugs have also been historically significant in cancer therapy. The pyrimidine nucleoside analogues 5-fluorouracil27,28 and gemcitabine29 are prodrugs used to inhibit thymidylate synthase and ribonucleotide reductase I, respectively, to treat a wide range of cancers (Fig. 1). Bortezomib (Fig. 1), a dipeptide boronic acid that covalently binds to and inhibits a catalytic threonine residue of the 26S proteasome, was approved by the FDA in 2003 to treat patients with multiple myeloma30.
Covalent drugs have been used to treat a variety of diseases. However, focusing on covalency from the outset of a project, instead of discovering a covalent mechanism of action after the fact, provides opportunities to improve drug design. Recent work in this field showcases how covalent drug discovery tools present solutions to otherwise intractable drug discovery challenges.
Discoveries by ligand-first approaches
Major milestones of covalent drug discovery have been reached over the past decade, including the FDA approval of the first covalent EGFR inhibitor, afatinib, in 2013, the BTK inhibitor, ibrutinib, in 2013 and the discovery of other kinase inhibitors. To discover these compounds, mildly reactive electrophilic functional groups were incorporated into known reversible ligands to enhance the inhibition of protein function. These examples offer lessons for future programmes, as each compound must balance reactivity, potency and selectivity.
Covalent EGFR inhibitors
Overactivity of the receptor tyrosine kinase EGFR drives the progression of non-small-cell lung cancer (NSCLC), making EGFR a key drug target in oncology31. During clinical development in the early 2000s, the reversible, first-generation EGFR inhibitors gefitinib and erlotinib (Fig. 2a) were discovered to be effective against tumours harbouring somatic activating mutations in EGFR, either deletions in exon 19 or the L858R point mutation, which occur in 10–30% of patients with NSCLC31–33. However, the disease in these patients eventually still progressed; in 60% of cases this was due to the acquisition of the T790M ‘gatekeeper mutation’34,35. This mutation of the gatekeeper residue in the ATP-binding site of EGFR not only decreases the binding affinity of many reversible inhibitors for EGFR but also increases the binding affinity of EGFR for ATP36.
Fig. 2. Progression of EGFR inhibitor structures.

Progression from first-generation (part a) to second-generation (part b) epidermal growth factor receptor (EGFR) inhibitors involved the addition of a reactive acrylamide electrophile (highlighted in red) to covalently bind a cysteine residue (Cys797) in EGFR. In the progression from second-generation to third-generation (part c) EFGR inhibitors, the quinazoline moiety is replaced with a pyrimidine unit to provide selectivity for the T790M mutant.
To overcome this problem, covalent second-generation inhibitors were strategically designed with acrylamide Michael acceptors to react with a cysteine residue (Cys797) in EGFR (Fig. 2b). Cys797 is located adjacent to the ATP-binding site, and irreversible binding of EGFR ligands to EGFR partially restores activity against the T790M gatekeeper mutant33. In addition to modest activity against T790M, covalent second-generation inhibitors provided prolonged suppression of EGFR signalling, suggesting that these covalent EGFR inhibitors could be more efficacious than reversible first-generation inhibitors such as erlotinib33. Afatinib (Boehringer Ingelheim) (Figs. 1 and 2b) was approved by the FDA in 2013 as a first-line treatment for patients with metastatic NSCLC with activating mutations in EGFR37,38. Despite the increased potency that covalent engagement brought against the disease target, the dose-limiting toxicity caused by inhibition of wild-type EGFR likely prevented afatinib from increasing overall survival when compared head-to-head with platinum-based chemotherapy in treating cancers bearing the T790M gatekeeper mutation39. Other second-generation inhibitors include neratinib (Puma), which potently inhibits HER2 by covalently binding Cys805 (a cysteine residue homologous to Cys797 on EGFR) and was approved by the FDA for treatment of HER2+ breast cancer in 2017, and dacomitinib (Pfizer) (Fig. 2b), which was approved by the FDA to treat NSCLC in 2018 (refs.40–42).
A third generation of EGFR inhibitors followed afatinib; these covalent inhibitors selectively target the T790M mutant over wild-type EGFR, and include WZ4002 (Dana–Farber Cancer Institute)43, osimertinib44,45 and rociletinib (Clovis Oncology; CO-1686)46 (Fig. 2c). These compounds maintain the acrylamide group to covalently bind Cys797 but exchange the quinazoline moiety of first-generation and second-generation compounds for a pyrimidine to promote selectivity for mutant EGFR(T790M) (ref.47). Higher affinity for T790M over wild-type EGFR not only results in efficacy in cancers with the EGFR gatekeeper mutation but also contributes to an improved safety profile and enables a higher recommended dose for osimertinib than for afatinib48. Osimertinib was granted accelerated approval by the FDA in 2015 as a second-line treatment for NSCLC, and was approved as a first-line treatment for metastatic NSCLC in 2018 (ref.49). However, osimertinib depends on Cys797 for covalent binding, and C797X mutations account for 15% of cases of resistance to second-line osimertinib50–52. Generally, drugs whose efficacy relies on covalent binding to a specific nucleophilic amino acid are vulnerable to mutations at that site, which could lead to drug resistance.
The success of covalent EGFR inhibitors has validated the approach of covalently engaging non-catalytic, non-conserved cysteines adjacent to kinase active sites to increase the potency and modulate the pharmacodynamics of initially reversible ligands. Development of these drugs has shown that the acrylamide electrophile is reactive enough to engage a cysteine adjacent to an ATP-binding site but not so reactive as to induce haptenization and an adverse immune response. Incorporating covalent binding in EGFR inhibitors also enables selectivity between kinases through an interaction with a non-conserved cysteine instead of highly conserved active-site residues that typically interact with ATP.
Covalent BTK inhibitors
The discovery of covalent BTK inhibitors shares several themes with the discovery of covalent EGFR inhibitors, including the ligand-first approach and the use of Michael acceptor electrophiles. BTK became a target of interest in chronic lymphocytic leukaemia owing to its crucial role downstream of the B cell receptor53. Activation of the B cell receptor induces phosphorylation of BTK through Lyn and Syk kinases, and eventually activates transcription factors related to B cell proliferation, differentiation, cell migration and adhesion54. This key role in B cell development indicated that BTK was a relevant target for B cell malignancies.
In the early 2000s, scientists at Celera Genomics who were interested in using BTK inhibitors to treat rheumatoid arthritis used a structure-based approach to discover an acrylamide-containing inhibitor of the BTK kinase domain that could be used as a tool compound to fluorescently label BTK55. It was subsequently discovered that the tool compound itself, later named ibrutinib (Table 1), had sufficient activity and suitable physicochemical properties to advance into clinical studies56–58. Ibrutinib was approved by the FDA for the treatment of mantle-cell lymphoma in 2013 and subsequently for chronic lymphocytic leukaemia (CLL), Waldenstrom’s macroglobulinaemia and chronic graft versus host disease59–63.
Similarly to EGFR inhibitors, ibrutinib binds to a cysteine residue (Cys481) adjacent to the ATP-binding site in BTK, and because only a few kinases have a homologous cysteine, ibrutinib should exhibit a degree of selectivity for BTK over other kinases64. The rapid clearance of ibrutinib (which has a half-life of 2–3 h) could also enable kinase selectivity; ibrutinib should maintain activity against BTK owing to prolonged covalent engagement, while the reversible inhibition of off-targets is minimized65. This combination of fast covalent engagement of BTK with rapid clearance might allow for selectivity in vivo despite the off-target kinase inhibition observed in biochemical assays.
Several other covalent BTK inhibitors have been approved or are currently in clinical trials and some of these highlight the variety of Michael acceptors that can be used as alternative electrophiles to acrylamides66–68. Most prominent among these is acalabrutinib (AstraZeneca), approved by the FDA in 2019 to treat CLL, which contains a butyramide electrophile instead of an acrylamide69. The butyramide electrophile is less reactive than an acrylamide, which, in addition to other substitutions, is hypothesized to account for the superior selectivity of acalabrutinib compared with ibrutinib for BTK and could be responsible for the reduced number of adverse cardiovascular events70,71. Further work has examined the use of cyanoacrylamides as electrophiles to design reversible covalent BTK inhibitors, which would ideally show increased potency and lower covalent off-target reactivity than irreversible covalent BTK inhibitors72–74. The long, tunable off-rates of reversible covalent inhibitors highlights the grey area that exists between reversible inhibition and irreversible covalent mechanisms.
Overcoming historical concerns relating to the potential toxicity of covalent drugs, the success of ibrutinib demonstrates that rationally designed covalent drugs can achieve acceptable safety profiles and blockbuster status. Ibrutinib, and covalent EGFR inhibitors, demonstrate that kinase inhibitors that target non-conserved cysteines adjacent to the ATP-binding site can be developed into selective and potent drugs. The pharmacokinetic and pharmacodynamic properties of ibrutinib allow for prolonged BTK blockade while reducing off-target kinase inhibition through rapid clearance in vivo. Notably, the performance of ibrutinib in treating B cell malignancies emphasizes that molecules once considered chemical biology tool compounds can become effective drugs.
Other covalent kinase inhibitors
Covalent inhibitors have been used to selectively target kinases other than EGFR and BTK with non-conserved cysteine residues adjacent to their ATP-binding sites75,76. One example is Janus kinase 3 (JAK3), a non-receptor tyrosine kinase primarily expressed in leukocytes and involved in cytokine signalling77. Covalent targeting of the non-conserved Cys909 of JAK3 has yielded inhibitors selective for JAK3 over other JAK family members for the treatment of autoimmune diseases77–81. One of these inhibitors, ritlecitinib (Pfizer; PF-06651600), has shown promising results for patients with rheumatoid arthritis in a phase II clinical trial82. Several covalent inhibitors of fibroblast growth factor receptor 4 (FGFR4) target the non-conserved Cys552 residue in FGFR4 to confer selectivity over FGFR1, FGFR2 and FGFR3, as well as to overcome mutations that confer resistance to reversible FGFR inhibitors in hepatocellular carcinoma (HCC)83,84. The acrylamide-containing FGFR4 inhibitor fisogatinib (Blueprint Medicines; BLU-554) is currently the subject of a phase II clinical trial (NCT04194801). Aldehyde-containing roblitinib (Novartis; FGF401), which is a reversible covalent FGFR4 inhibitor that also reacts with Cys552, is also under clinical investigation (NCT02325739)83,85. Overall, the rational design of covalent kinase inhibitors that target non-conserved cysteines adjacent to the ATP-binding site has become a routine approach to enhancing the potency and selectivity of kinase inhibitors.
Discoveries by electrophile-first approaches
Covalent drugs are also discovered through electrophile-first approaches, meaning that the initial discovery process is rooted in finding a covalent ligand from the outset, instead of incorporating covalency into a known reversible ligand. Key examples of drugs discovered through this approach include the KRAS(G12C) inhibitor sotorasib and the SARS-CoV-2 Mpro inhibitor nirmatrelvir (Fig. 1).
Covalent KRAS(G12C) inhibitors
The discovery and development of covalent KRAS(G12C) inhibitors is one of the most exciting discovery-to-clinic stories featuring covalent drugs. KRAS is a GTPase-encoding oncogene that is mutated in about 25% of all cancers, most notably in pancreatic, colorectal and lung cancers86. Wild-type KRAS is carefully regulated between the active GTP-bound state and inactive GDP-bound state, but many KRAS mutations attenuate GTPase activity, leading to low rates of GTP hydrolysis and elevated RAS signalling, driving tumorigenesis87. Since the discovery of the role of KRAS in cancer nearly 30 years ago, attempts to drug it directly using traditional drug discovery methods have been unsuccessful9,10. KRAS does not have accessible pockets for reversible inhibitors to bind to, competitive inhibitors would need to overcome the picomolar binding affinities of GTP and GDP, and inhibitors active against wild-type KRAS could show on-target toxicity86,88.
Covalent KRAS inhibitors against the G12C mutant are appealing for several reasons. First, targeting mutant KRAS could allow for selective cytotoxicity to cancer cells. Second, the affinity enabled by covalent binding would be advantageous as KRAS lacks easily ligandable pockets. Third, 12–14% of KRAS mutations in NSCLC are KRASG12C, presenting a promising patient group that would directly benefit from KRAS(G12C) inhibition89. Finally, position 12 in KRAS sits closely beneath the effector-binding region and the nucleotide-binding pocket, suggesting that covalent KRAS(G12C) ligands might affect KRAS function87.
In 2013, researchers at the University of California, San Francisco reported the first mutant-selective covalent KRAS(G12C) inhibitor. The inhibitor (compound 12 in their study) was discovered through a disulfide-fragment screening approach known as tethering, whereby a library of 480 disulfide fragments was screened against KRAS(G12C) in the GDP-bound state using intact protein mass spectrometry (MS)88,90. Co-crystal structures of KRAS(G12C) showed that hit compounds bound to the switch II region, and subsequent medicinal chemistry efforts to exchange the disulfide moiety for acrylamide and vinyl sulfonamide electrophiles yielded KRAS(G12C) inhibitors that were active in vitro, including compound 12. Binding of compound 12 to the switch II pocket impaired KRAS signalling by shifting nucleotide affinity from favouring GTP to GDP and led to the accumulation of KRAS in its inactive state91.
This novel mechanism for selective KRAS(G12C) inhibition set the stage for the development of clinical covalent KRAS(G12C) inhibitors. In 2016, Wellspring Biosciences disclosed ARS-853, which is a selective covalent inhibitor of KRAS(G12C) with in cellulo efficacy in the low micromolar range92. Structure-guided optimization of compound 12 and use of a cellular liquid chromatography with tandem mass spectrometry (LC–MS/MS)-based assay to determine the degree of KRAS(G12C) engagement in H358 cells, yielded ARS-853 (ref.92). ARS-853 treatment in KRAS(G12C)-dependent cell lines decreased the amount of active KRAS(G12C), inhibited downstream RAS signalling and induced apoptosis92. Although KRAS(G12C) had been thought to be constitutively active, the selective binding of ARS-853 to GDP-bound, inactive KRAS(G12C) provided evidence that KRAS mutants cycle between GTP-bound and GDP-bound states92.
The discovery of clinical KRAS(G12C) inhibitors continued with ARS-1620, which was the result of an effort to overcome metabolic stability and bioavailability limitations of ARS-853 to facilitate in vivo studies of KRAS(G12C) inhibition87. ARS-1620 is based on a novel quinazoline core scaffold, designed to better occupy the switch II pocket and, thus rigidify a more favourable conformation for covalent reaction between the acrylamide electrophile and cysteine87. Ultimately, ARS-1620 was identified as the first KRAS(G12C) inhibitor suitable for in vivo studies and showed efficacy in KRAS(G12C) patient-derived xenograft models treated at 200 mg kg−1 once per day or twice per day87. The increased potency of this series of KRAS(G12C) inhibitors and success in in vivo models indicated that it might be possible to design clinically efficacious drugs.
Sotorasib (AMG-510) (Table 1) was the first selective KRAS(G12C) inhibitor to enter clinical trials in 2018 and was developed by Amgen, building on discoveries from a partnership with Carmot Therapeutics in which a custom library of small molecules that covalently bind cysteine were screened against KRAS(G12C)93. Molecules identified through this collaboration led to the discovery of a previously unknown pocket on KRAS (a cryptic pocket), which Amgen scientists exploited to discover sotorasib through structure-based design94. Sotorasib was designed to occupy the cryptic pocket by interacting with His95, Tyr96 and Gln99 (ref.94) (Fig. 3). A phase II clinical trial investigating sotorasib was successfully completed in 2020, and was followed by FDA approval for the treatment of adults with KRASG12C-mutated locally advanced or metastatic NSCLC in May 2021 (ref.95). Other covalent KRAS(G12C) inhibitors are entering clinical trials. Adagrasib (MRTX849) emerged from a joint drug discovery collaboration between Mirati Therapeutics and Array BioPharma, in which irreversible covalent inhibitors of KRAS(G12C) were identified; Mirati Therapeutics subsequently used structure-based design approaches to optimize adagrasib, which entered clinical trials in January 2019 (refs.96,97) (Fig. 3). JNJ-74699157 (ARS-3248; J&J and Wellspring Biosciences) was being investigated in patients with several types of advanced solid tumour that express KRASG12C, including NSCLC and colorectal cancer, but its clinical trials have been terminated98.
Fig. 3. Aligned structures of KRAS(G12C) co-crystallized with adagrasib (MRTX849) and sotorasib (AMG-510).
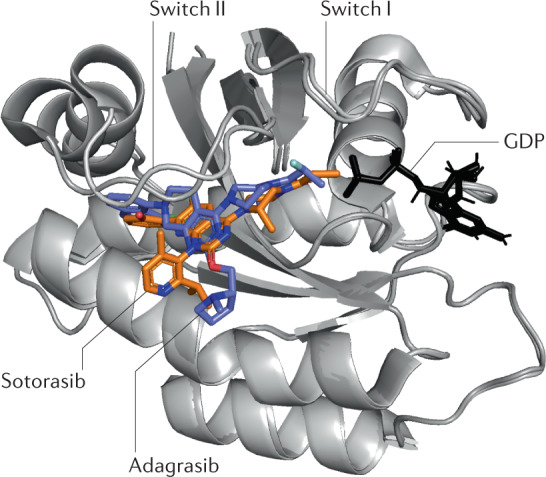
The covalent inhibitors adagrasib (PDB ID: 6UT0) and sotorasib (PDB ID: 6OIM) are bound to the switch II pocket, which is adjacent to the GDP-binding pocket93,96.
Designing small-molecule covalent KRAS(G12C)-selective inhibitors provides an elegant solution to drugging an undruggable cancer target. Before KRAS(G12C) inhibitors, recently discovered targeted covalent inhibitors in oncology were mostly identified using ligand-first approaches. The success of covalent KRAS(G12C) inhibitors validates an electrophile-first approach to covalent drug discovery and affirms the importance of covalent fragment screening techniques (discussed below). Optimization of initial hit compounds that emerge from covalent screening platforms, such as compound 12, can subsequently lead to programmes that produce potent covalent inhibitors such as sotorasib. In addition, the sotorasib story suggests that in other diseases in which a key protein target undergoes substitution of an amino acid to a cysteine residue, covalent inhibitors present an increasingly validated method to potentially provide precision therapy for patients.
SARS-CoV-2 main protease inhibitors
Vaccines against coronavirus disease 2019 (COVID-19) were developed at unprecedented speeds, and similar research momentum has led to the development of therapeutics that will benefit patients with COVID-19. In December 2021, the FDA issued an Emergency Use Authorization for Pfizer’s Paxlovid (a combination of nirmatrelvir and ritonavir) to treat mild-to-moderate COVID-19 (caused by SARS-CoV-2) in adults and some paediatric patients, marking the first approved oral treatment for the disease99. Nirmatrelvir (Table 1) covalently inhibits the Mpro of SARS-CoV-2 (ref.3). This programme highlights how the adaptation of relatively inactive peptidomimetics into potent and selective cysteine protease inhibitors can be accomplished by the addition of cysteine-reactive covalent functional groups to target the protease active site and by structurally informed medicinal chemistry efforts.
SARS-CoV-2 is a virus with a single-stranded RNA genome that encodes two polyproteins (pp1a and pp1ab) as well as structural and accessory proteins100. Viral replication depends on successful cleavage of pp1a and pp1ab by the Mpro (also referred to as 3CLpro), which is a cysteine protease, into functional viral proteins100. The discovery of covalent inhibitors against SARS-CoV-2 Mpro emerged from extensive work on protease inhibitors for SARS-CoV-1, which is the causative virus for severe acute respiratory syndrome coronavirus 1 (SARS1)100. During the 2002–2003 SARS1 outbreak, researchers used a crystal structure of the homologous porcine transmissible gastroenteritis coronavirus (TGEV) Mpro bound to a hexapeptidyl chloromethylketone covalent cysteine-reactive inhibitor to provide a foundation for the design of covalent inhibitors against SARS-CoV-1 Mpro (ref.101). Because of the homology across all Mpro, this work enabled the discovery of rupintrivir (AG7088), which is a mechanism-based inhibitor of the human rhinovirus (HRV) Mpro (ref.101).
Because the SARS-CoV-1 outbreak subsided, work into developing coronavirus Mpro inhibitors slowed until the emergence of SARS-CoV-2 in 2019. SARS-CoV-2 Mpro shares 96% sequence identity with SARS-CoV-1 Mpro, and there is 100% sequence overlap of the catalytic sites102. Renewed interest in improving on previous chloromethylketone inhibitors motivated researchers to adapt rupintrivir into a SARS-CoV-2 Mpro inhibitor potent enough to obtain a co-crystal structure. This discovery in turn enabled identification of the α-hydroxymethylketone-containing antiviral PF-00835231, which demonstrated potent SARS-CoV-2 Mpro inhibition in an activity assay based on fluorescence resonance energy transfer (FRET), activity in antiviral cell-based assays, stability in plasma and low clearance in vivo103.
In subsequent studies, the oral bioavailability of PF-00835231 was improved by replacing the α-hydroxymethylketone moiety with a nitrile group, which can also act as an electrophile103. Nitriles can covalently bind particularly reactive nucleophiles; however, the ease of thiol elimination from the thioimidate adduct makes nitriles more reversible than some other electrophiles, such as acrylamides104. Optimization from PF-00835231 eventually yielded PF-07321332, named nirmatrelvir (Fig. 4 and Table 1), which is a highly potent SARS-CoV-2 reversible covalent inhibitor that displayed potent inhibition in a FRET-based assay across all human coronaviruses, while no inhibitory effects were seen against human cysteine or serine proteases. The in vivo efficacy of nirmatrelvir was demonstrated in a mouse-adapted SARS-CoV-2 (SARS-CoV-2 MA10) model.
Fig. 4. Nirmatrelvir in complex with SARS-CoV-2 main protease.
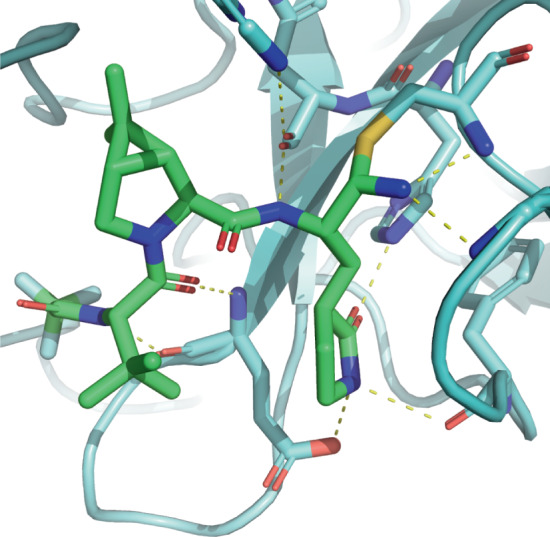
The nitrile group of nirmatrelvir (shown in green) reacts with Cys145 of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) main protease (shown in light blue) to form a covalent thioimidate adduct. Extensive hydrogen-bond interactions (yellow dashed lines) occur throughout the pocket (PDB ID: 7RFW)103.
In phase II/III clinical trial data released in November 2021, Paxlovid was shown to be highly effective at preventing progression to severe COVID-19 in symptomatic patients105. The emergence of this orally bioavailable drug for COVID-19 will help to ameliorate illness for non-hospitalized patients in high-risk groups106. Overall, SARS-CoV-2 Mpro covalent inhibitors provide a promising avenue for the treatment of coronavirus infections either as monotherapies or in combination with other antiviral drugs.
The quick adaptation of previous protease inhibitors to selectively target SARS-CoV-2 Mpro is an example of using structure-based design while taking into account the valuable properties of covalent drugs. Researchers started with an electrophile-containing peptide and optimized both the peptide and electrophile to obtain a highly potent covalent inhibitor. In some ways, this story mirrors that of the discovery and optimization of JAK3 kinase inhibitors, in which EGFR inhibitors were adapted to target Cys909 of JAK3 (ref.79). Both JAK3 and mutant EGFR(T790M) contain a methionine gatekeeper, as well as a homologous cysteine adjacent to the ATP-binding site. These shared features enabled the discovery of potent JAK3-selective covalent inhibitors using an EGFR inhibitor as a starting point, similar to how rupintrivir provided the starting structure for the discovery of nirmatrelvir. Generally, when reactive cysteine residues are shared across a protein family, structurally guided adaptation of previously studied cysteine-reactive covalent inhibitors can lead to selective and potent drugs for other proteins with this feature.
HCV NS3/4a protease inhibitors
α-Ketoamide-based covalent inhibitors have been developed to treat hepatitis C virus (HCV) infection through inhibiting NS3/4a — a serine protease that cleaves the HCV polyprotein into multiple non-structural proteins required for replication107. Although HCV had been treated with a combination of PEGylated interferon-α and ribavirin, modest response rates and notable adverse events prompted studies that resulted in the discovery of NS3/4a protease inhibitors to treat HCV108. On the basis of initial observations that hexapeptide cleavage products could inhibit NS3/4a, the linear peptidomimetic inhibitors boceprevir (Merck)109,110 (Fig. 1) and telaprevir (Vertex)111,112 were designed. These compounds, along with narlaprevir113, use a ketoamide electrophile to covalently engage the catalytic serine of NS3/4a. This covalent interaction is relatively reversible owing to elimination of the serine alcohol group from the protein–inhibitor adduct114. Boceprevir and telaprevir were effective in treating HCV and were approved by the FDA in 2011 after successful trials115,116. However, telaprevir was withdrawn in 2014 owing to adverse events, and boceprevir was discontinued by Merck in 2015 owing to the superiority of newer direct-acting antivirals, in particular the ledipasvir–sofosbuvir combination (Gilead), which targets the HCV polymerases NS5a and NS5b117–119. Nevertheless, the success of ketoamide-based NS3/4a inhibitors in increasing the efficacy of interferon–ribavirin therapy emphasizes the utility of the ketoamide group as a serine-reactive electrophile in designing covalent antivirals.
Covalent proteasome inhibitors
Bortezomib (Takeda) was the first boron-containing drug to be approved by the FDA, and was approved for treatment of multiple myeloma in 2003 (ref.120). Bortezomib was discovered through optimization of an aldehyde-containing proteasome substrate peptide, and set a precedent for the discovery of HCV NS3/4a inhibitors such as telaprevir121–124. Exchange of the aldehyde electrophile for a boronic acid substantially increased the potency of proteasome inhibition121. The effect of bortezomib comes from the boronic acid covalently binding to the hydroxy group of the β5 subunit N-terminal threonine of the 20S proteasome, leading to inhibition of the proteasome’s chymotrypsin-like activity125. Bortezomib takes advantage of the increased sensitivity of haematological cancers such as multiple myeloma and mantle-cell lymphoma to proteasome inhibition123.
The development of bortezomib validated the proteasome as a cancer target, encouraging the discovery of other proteasome inhibitors. Medicinal chemistry efforts transformed the natural product epoxomicin into carfilzomib (Amgen), which was approved by the FDA in 2012 (ref.126) (Fig. 1). The epoxyketone moiety in carfilzomib forms a morpholino ring with the catalytic N-terminal threonine of the 20S proteasome, and this mechanism has been proposed to confer selectivity because most proteases do not have nucleophilic side chains at their N terminus127. Although the epoxyketone could have additional off-targets that bortezomib does not, this proposed mechanism of carfilzomib illustrates how covalency can help to drive selectivity through highly specific mechanisms.
Further work has been done to identify orally bioavailable proteasome inhibitors to improve upon bortezomib, which is administered intravenously128. Ixazomib (Takeda) is a second-generation proteasome inhibitor that also contains a boronic acid group129. This drug is administered orally as the prodrug ixazomib citrate, a boronic ester that hydrolyses upon exposure to aqueous media or plasma130,131. Oprozomib (Amgen), a second-generation epoxyketone-containing proteasome inhibitor, can also be orally administered132.
The discovery of these covalent proteasome inhibitors illustrates the utility of boronic acid electrophiles for targeting protease active sites and demonstrates how a covalent drug discovery project can use an unoptimized electrophile as a starting point and subsequently introduce an alternative electrophile. Just as a chloromethylketone group in a SARS-CoV-2 ligand was optimized to the nitrile in nirmatrelvir, bortezomib was discovered through optimization from an aldehyde. Although targeting especially nucleophilic protease active sites might provide greater flexibility in terms of electrophile choice, using highly reactive electrophiles to gain an initial foothold can be beneficial.
Other boron-containing drugs
Several additional boron-containing drugs inhibit serine hydrolases beyond the proteasome through formation of covalent adducts between catalytic serine residues and boron133,134. Most of these drugs contain benzoxaborole groups and have been discovered through screening of compound collections that contain boron-based electrophiles135. For example, the antifungal tavaborole (Pfizer) was discovered through focused screening of boron-containing compounds previously investigated as antibacterials, and was approved by the FDA to treat onychomycosis in 2014 (refs.136,137). Tavaborole covalently binds to the 2′ and 3′ hydroxy groups on the 3′ terminus of leucyl-tRNA, trapping the tRNA–tavaborole adduct in the editing site of leucyl-tRNA synthetase to block protein synthesis138. Crisaborole (Pfizer), a phosphodiesterase 4 inhibitor, was approved by the FDA to treat psoriasis in 2016, and the β-lactamase inhibitor vaborbactam (Rempex) was approved to treat various bacterial infections in 2017 (refs.139,140). These boron-based drugs target serine hydrolases, and the weak boron–sulfur bond potentially provides selectivity for serine over cysteine hydrolases121. The reversibility of the serine–boron bond hinders chemoproteomic profiling experiments commonly used to characterize the selectivity of covalent ligands. But as the serine hydrolase family is rich with potential drug targets, the numerous boron-containing drugs in clinical trials hold promise across a wide variety of disease types141.
Mutant-haemoglobin modulators
Voxelotor (Global Blood Therapeutics) is a lysine-targeting covalent drug used to treat sickle cell anaemia, and the discovery of voxelotor (Table 1) was dependent on knowledge of heightened lysine side chain reactivity. Sickle cell anaemia is caused by a single mutation in the gene encoding the β-haemoglobin chain that induces polymerization of mutant haemoglobin (HbS) under hypoxic conditions142. An aldehyde-containing natural product and several synthetic aldehyde analogues were found to prevent polymerization by increasing the affinity of HbS for oxygen143. These aldehydes bind in a reversible covalent manner, forming a Schiff base with the N-terminal valine of the α-haemoglobin chain143. Almost 50 years ago, this N-terminal amine was discovered to have a particularly low pKa of 6.9, indicating that it is primarily unprotonated under physiological conditions and, thus, more nucleophilic144. Based on earlier aldehydes, voxelotor was discovered through a structure-guided effort to discover compounds that increase the oxygen affinity of HbS, and was designed to bind the HbS tetramer in a 1:1 stoichiometry, unlike the 2:1 ratio of earlier compounds143,145. With a remarkable red blood cell to plasma ratio of ~150 that likely reduces off-target effects, voxelotor was approved by the FDA in 2019 with a recommended dose of 1.5 g daily, which is an unusually high dose for a covalent drug142,146. The success of voxelotor is similar to that of ibrutinib in demonstrating that covalent drugs can be dosed at high amounts given favourable absorption, distribution, metabolism and excretion (ADME) properties. Furthermore, the discovery of voxelotor shows how the identification of unusually reactive amino acid residues, such as the α-haemoglobin N-terminus, provides opportunities for drug discovery.
The covalent drug discovery toolbox
Many covalent drug discovery programmes, including those for covalent EGFR and BTK inhibitors, have involved the addition of reactive functional groups to previously identified ligands. However, emerging technologies make it possible to approach covalent ligand discovery from an electrophile-first perspective, in which covalent ligands against protein targets of interest are identified before structure-based optimization. For example, this type of approach proved successful in drugging KRAS(G12C) and has facilitated the rapid discovery of E3 ligase ligands for targeted protein degradation applications. Activity-based protein profiling (ABPP) approaches have transformed the characterization of electrophilic compounds, facilitating selectivity profiling and target identification experiments. Special considerations must also be made in evaluating the binding affinity and reactivity of covalent ligands (Box 1). In this section, we discuss the toolbox of emerging techniques that covalent drug discovery relies on.
Box 1 Characterizing the potency and reactivity of covalent ligands.
Although half-maximal inhibitory concentration (IC50) values are often used to measure the potency of covalent inhibitors, the kinact/Ki second-order rate constant (where Ki is the inhibition constant and kinact is the rate of enzyme inactivation) is preferred because it describes inhibitor potency in a time-independent manner212,213. The ratio kinact/Ki can be determined by measuring total occupancy over time in a binding assay, calculating the first-order rate constant of inhibition, kobs, and in turn kinact/Ki (ref. 212). One example of the direct measurement of kinact/Ki involved a time-resolved fluorescence energy transfer assay in which competitive fluorescent probes were used to determine kinact/Ki values versus epidermal growth factor receptor (EGFR)214. Because this kinetic parameter can be resource-intensive to measure, researchers have formulated models that relate IC50 values to kinact/Ki (refs.215–217). For example, a model has been developed to estimate Ki and kinact from time-dependent IC50 data, from work on CYP450 enzyme inhibitors216. Moreover, a method has been designed that uses a competitive covalent probe with a known kinact/Ki value to facilitate the rapid calculation of inhibitor kinact/Ki values based on standard end point IC50 data217. Scientists working on a covalent JAK3 inhibitor programme at Pfizer, however, suggested that fixed-time-point IC50 values can serve as a valuable surrogate for kinact/Ki (ref. 218). When using IC50 as a surrogate, extra care should be taken to confirm that target engagement is covalent (such as by measuring off-rates). However, with an appropriate time-point choice, IC50 values can be a useful tool to rapidly assess potency for covalent inhibitors. Regardless, determination of kinact/Ki provides the most complete information to medicinal chemists about the binding affinity and kinetics of covalent target engagement.
To ensure that potent covalent inhibitors are not promiscuous ligands that lack selectivity, electrophilic compounds are frequently tested for reactivity, usually with respect to thiols. This measurement is often carried out using a glutathione (GSH) reactivity assay, whereby a compound is incubated with excess GSH and consumption of the compound is observed using liquid chromatography–mass spectrometry to determine a rate constant69,219,220. Alternatively, plate-based 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB; also known as Ellman’s reagent) and NMR-based GSH assays have also been used156,221. In a helpful resource for acrylamide-focused projects, two related studies used an NMR-based assay to explore the effects of various substitutions on acrylamide GSH reactivity222,223. Assaying for reactivity is a crucial step, particularly for key molecules and for compound series containing novel electrophiles.
Screening platforms
Many emerging covalent ligand screening platforms involve MS-based detection, enabled by covalent bond formation between the compound and protein. Other phenotypic, DNA-encoded or computational approaches have also been used, which are then paired with MS-based validation, ABPP-based experiments to inform selectivity and structural biology to enable medicinal chemistry. The most prominent methods for covalent screening are summarized in Table 2. The growth of commercial libraries of electrophilic fragment-like compounds is a key factor contributing to the rise of these electrophile-first discovery strategies.
Table 2.
Comparison of screening methods for covalent drug discovery
| Method | Key features | Limitations | Refs. |
|---|---|---|---|
| Intact protein MS | MS detection; compounds can be pooled; provides information on binding stoichiometry; high-throughput | Requires a large amount of purified protein; electrophile must be swapped when using disulfide tethering approaches | 88,156 |
| MS-based ABPP | MS detection; provides information on the selectivity of the ligand; can be done in complex biological systems (such as lysate or tissue) | Lower throughput than intact protein MS; sensitivity affected by peptide abundance and digestion conditions | 180,183 |
| Gel- or plate-based ABPP | Easy detection of hits through fluorescence of competitive reactive probe; can be paired with ABPP proteomics to assess selectivity of hit ligands | Lower throughput compared with assays that are not gel based; complicated by the presence of multiple reactive amino acid residues within a protein; requires purified protein | 189 |
| Phenotypic | ABPP enables rapid identification of covalent hits; potentially high-throughput; can use identical assays for discovering reversible ligands | Does not take advantage of known reactive hotspots identified in chemoproteomics data; requires control counter screens to eliminate untenable (for example, promiscuous or toxic) hit ligands | 186,211 |
| Covalent docking | Advanced software is widely available (such as DOCKovalent); scoring assesses non-covalent interactions | Difficult to model covalent bond formation (relies on assumptions about ligand–target interactions) | 167,169 |
| Covalent DNA-encoded libraries | Affords a massive library size | Enrichment complicated by covalent binding; limited library availability | 165 |
ABPP, activity-based protein profiling; MS, mass spectrometry.
Intact protein MS
Originally, MS-based compound screening grew out of ‘tethering’, which is a technique employed since 2000 that uses libraries of compounds linked to disulfides to identify fragments that bind cysteine-adjacent pockets90,147. Molecules that bind undergo disulfide exchange with the cysteine (which could be endogenous or engineered) to form an adduct with the protein, and pooled screening with MS detection can identify the bound compound. Binding fragments can be combined or grown in a fragment-based approach to identify high-affinity ligands. This strategy was originally designed to identify reversible ligands for challenging targets, but covalent binding can be maintained by replacing the disulfide with an electrophile such as an acrylamide, as in the case of the initial covalent KRAS(G12C) inhibitors88. This approach has been used to discover compounds that modulate protein–protein interactions148,149, and a more recent study employed a similar tethering strategy that uses aldehydes to form imines with lysine residues150.
Over the past decade, covalent ligand discovery has shifted towards screening more drug-like electrophilic fragments13. In 2012, researchers curated 177 electrophilic compounds from the Pfizer compound collection and used an MS-based primary screening strategy to identify covalent inhibitors of the interaction between hypoxia-inducible factor 1α (HIF1α) and aryl hydrocarbon receptor nuclear translocator (ARNT)151. This approach was informed by determining an X-ray crystal structure of the HIF1α–ARNT complex151. Around the same time, the concept of tethering was expanded by using a small set of acrylamides and an MS-based assay to identify thymidylate synthase inhibitors, an approach termed kinetic template-guided tethering152. Building on these studies, an acrylate functionality was appended to 100 fragments to identify non-peptidic inhibitors of the cysteine protease papain153. Electrophilic fragments were pooled, and screening through electrospray ionization (ESI) MS led to the identification of hit compounds out of pooled experiments. Use of a similar library of acrylates enabled the discovery of covalent inhibitors for the HECT E3 ligase NEDD4-1 (ref.154). As with the HIF1α–ARNT protein–protein interaction inhibitors discovered at Pfizer, co-crystal structures were crucial to understanding the mechanism of these NEDD4-1 inhibitors, which prevent association of ubiquitin with the E3 ligase and thus induce a switch from a processive to a distributive mechanism. In another example, acrylate-based inhibitors of the RBR E3 HOIP were also discovered using MS-based screening, highlighting how covalent fragment screening approaches can be useful for protein classes that have been challenging to discover ligands against, such as E3 ligases155.
More recently, a commercial library of 993 acrylamides and chloroacetamides was screened using intact MS to identify ligands of the deubiquitinase OTUB2 and the pyrophosphatase NUDT7 (ref.156). The authors used co-crystal structures with OTUB2 and NUDT7 in complex with the hit compounds to inform fragment growing to increase potency. Although previous studies had paired MS-based screening with structural information to identify cysteine residues in functional sites or to understand the mechanism of inhibition, in this case, the pairing of MS-based screening and structure-guided fragment-based drug discovery supported optimization of the potency of the hit compounds.
The same compound collection was also used to screen against the peptidyl-proline cis–trans isomerase Pin1, which is overexpressed or activated in several tumour types but has been challenging to target selectively157. The resulting chloroacetamide sulfopin was shown to be selective for Pin1 in a covalent inhibitor target-site identification (CITe-Id) chemoproteomics experiment and was effective in regressing neuroblastoma growth in mice157. This result suggests that although chloroacetamides have disadvantages, including rapid metabolism, they can be valuable tool compounds with which to assess target relevance in various disease models.
One particularly powerful example of covalent ligand screening is the discovery of the initial compounds in the series that led to the first approved KRAS(G12C) inhibitor, sotorasib. A library of 3,300 acrylamides was screened in three assays: a thiol reactivity assay, a RAF-coupled nucleotide exchange assay and an intact MS assay158. Combined with crystallographic data that showed how ligand binding revealed the presence of previously closed sub-pockets, this effort provided the basis for the rapid discovery of KRAS(G12C) inhibitors discussed above.
Covalent DNA-encoded libraries
DNA-encoded libraries (DELs) present an alternative approach that enables the screening of massive libraries of small molecules. Unlike MS or ABPP approaches, there is no specialized advantage of covalency in enabling DEL screening, but the throughput of DELs allows for the screening of much larger covalent libraries through a workflow of immobilization, enrichment, amplification and sequencing. The first reports of electrophilic protein–nucleic acid-encoded libraries described the targeting of protease active sites159, but over the past 5 years, cysteine-targeted DNA-encoded or protein–nucleic acid-encoded libraries have been used to identify covalent ligands for bromodomains, including PCAF and BRD4, as well as for JNK1, MEK2 and HER2 (refs.160–162). Further work has explored improvements in the enrichment step of the covalent DEL screening workflow, which differs because covalent engagement prevents elution of DNA-tagged molecules163. A covalent ligand of mitogen-activated protein kinase kinase 6 (MAP2K6) was also identified serendipitously through screening of a DEL against a DNA-encoded protein library164. More recently, even larger covalent DELs (with approximately 100,000,000 members) have been developed and used to identify acrylamide-based and epoxide-based BTK inhibitors with novel scaffolds165. In this study, similar screening results were obtained after storing the library at –80 °C for several years, suggesting that the electrophilic compounds are sufficiently stable in this context. Expansion of the covalent DEL library size and the increasing commercial availability of DELs represent an exciting development, and although unique considerations with respect to enrichment workflow and compound stability must be kept in mind, DELs that contain electrophilic molecules may become more widespread in covalent ligand discovery.
Covalent docking
The advantages of various covalent docking methods in different covalent docking scenarios have been described elsewhere166,167. Most computational programs for covalent docking rely on directly linking models to model conformations under the constraint of a predefined bond between a ligand and a corresponding amino acid site. The covalent docking platform GOLD relies on this assumption, and an example of its use was in the virtual screening for covalent inhibitors of the NEDD8-activating enzyme (NAE), in which three of the hits were confirmed as novel NAE inhibitors168. Development of the DOCKovalent method for virtual screening facilitated the discovery of boronic acid AmpC β-lactamase inhibitors, as well as cyanoacrylamide inhibitors of JAK3 (ref.169). DOCKovalent uses non-covalent docking methods to pre-generate conformations and states for an electrophilic virtual library and then samples each state against the target nucleophile. The same method has been used to identify new covalent inhibitors of the kinase MKK7 (ref.170), as well as compounds that bind KRAS(G12C) to destabilize the protein and accelerate nucleic acid exchange171.
Apart from screening, covalent docking is a useful tool for investigating binding modes of known covalent ligands. For example, GOLD has been used to model the binding modes of the aldehyde-containing proteasome inhibitor MG132 (ref.172). AutoDock uses a flexible side chain approach, whereby the covalent ligand is treated as an amino acid side chain and poses of this flexible ‘side chain’ are scored, using a physics-based scoring function that evaluates the energetics of ligand–protein interactions, as the remainder of the protein is held rigid166. Another method, called CovDock, is based on the Schrӧdinger Glide docking algorithm and Prime structure refinement methodology. CovDock uses traditional non-covalent docking approaches to dock a ligand to a protein target, and then models the covalent bond attachment and refines the complex166. This approach does not consider the reactivity of the electrophile, which can limit the ability to virtually study the differences in docking ligands with different electrophilic functional groups167.
Overall, covalent docking software provides useful information on ligand–protein interactions when key assumptions can be made — namely, when the reactivity of the electrophile and the site of modification is known.
Chemoproteomics-enabled discovery
Chemoproteomic platforms enable the identification of covalent compounds and their corresponding ligandable sites on target proteins directly in complex biological systems. Advances in chemoproteomics have facilitated the discovery of covalent ligands against undruggable disease targets and enabled the selectivity profiling of covalent ligands across the proteome to identify targets and off-targets of these ligands. Here, we discuss the chemoproteomics profiling of reactive ligandable hotspots, ABPP screening platforms and target identification within the context of recent work relevant to drug discovery. Chemoproteomics experiments can provide key information on ligand selectivity and give early guidance for selecting targets for covalent drug discovery programmes.
Chemoproteomics profiling of reactive ligandable hotspots
ABPP facilitates the discovery of covalently ligandable sites and the corresponding ligands in complex biological samples. ABPP was pioneered by Cravatt and Bogyo using active-site-directed chemical probes that covalently target catalytic residues of various enzyme classes, including hydrolases, proteases and kinases173,174. This technique, often using gel-based assays, was employed to gain functional readouts of active enzymes in biological contexts14,173,175. ABPP probes contain a warhead that covalently reacts with nucleophilic amino acids (such as cysteine) and a reporter handle to monitor probe binding, such as a fluorophore, biotin or alkyne moiety for subsequent click chemistry-enabled applications173 (Fig. 5a).
Fig. 5. Isotopic tandem orthogonal proteolysis–activity-based protein profiling.

a | Example of a reactive probe for activity-based protein profiling (ABPP) designed with a broadly reactive electrophilic warhead linked to an analytical handle. b | Schematic of the competitive isoTOP-ABPP (isotopic tandem orthogonal proteolysis–activity-based protein profiling) methodology. Treatment of cells or lysate with a protein-reactive compound prevents subsequent binding of the pan-reactive probe, and this competitive ligand binding can be detected by tandem mass spectrometry (MS/MS) after an enrichment step, to indirectly identify covalent protein targets for the compound of interest. TEV, tobacco etch virus.
Instead of focusing on active sites, more recent ABPP approaches use MS and broadly reactive chemical probes to also map allosteric sites176. In the first reports of the isoTOP-ABPP (isotopic tandem orthogonal proteolysis–activity-based protein profiling) approach, an iodoacetamide probe functionalized with an alkyne handle was used to identify hyper-reactive cysteines across the proteome176,177 (Fig. 5b). The alkyne handle can be used to link the probe-modified protein to a tobacco etch virus (TEV) protease-cleavable tag that contains an azide group and biotin moiety separated by either an isotopically light or heavy valine (Fig. 5b). These functionalities enable enrichment of probe-modified peptides and tandem analysis of the light and heavy samples with MS, which, after controlling for run-to-run variability, allows for quantitative comparisons between samples, including competitive analysis of covalent compounds (Fig. 5b). Using this approach, it was discovered that the hyper-reactivity of cysteines predicts their functionality in catalysis and at sites of post-translational modifications177. Recent adaptations of isoTOP-ABPP have been developed to increase the coverage of cysteines across the proteome and to increase the throughput. For example, optimization of the sample preparation steps (namely single-pot, solid-phase-enhanced sample preparation (SP3)) and combination of this workflow with off-line fractionation and field asymmetrical waveform ion mobility spectrometry (FAIMS) allow for additional separation before MS detection, and enabled the identification of more than 30,000 reactive cysteines across a panel of tumour cell lines178. Additionally, as new probes are developed for other nucleophilic amino acids, reactivity profiling of other amino acid hotspots (such as lysines) across the proteome will allow for the expansion of this technology beyond cysteine.
Overall, reactivity profiling generates large quantities of information across thousands of proteins, ultimately providing a relatively unbiased picture of nucleophilic (usually cysteine) amino acid reactivity. This information can be used to either select for appropriate protein targets in drug discovery programmes or to identify allosteric sites on proteins of interest that may have previously been considered un-ligandable or undruggable. As an example of targeting a traditionally undruggable protein with a covalent molecule, ABPP was paired with a MYC transcription factor activity assay to identify a covalent MYC ligand, EN4 (ref.179). EN4 targets Cys171, which is located within a predicted intrinsically disordered region of MYC, and showed selectivity on a proteome-wide scale in profiling of more than 1,500 cysteines using competitive isoTOP-ABPP. Cys171 was initially identified as a ligandable hotspot on MYC through analysis of compiled cysteine-reactive chemoproteomics data, and this information spurred the subsequent search for a selective covalent ligand against that cysteine.
Activity-based protein profiling screening platforms
The use of competitive isoTOP-ABPP was expanded to identify proteome-wide targets of a small covalent fragment library by competing individual acrylamide and chloroacetamide fragments against an iodoacetamide–alkyne probe180. In this study, more than 700 ligandable cysteines were identified, and information was provided about the proteome-wide selectivity of each covalent fragment in the library. The covalent ligands discovered with this approach and their corresponding ligandable sites were used to help elucidate the role of caspase 8 and caspase 10 in extrinsic apoptosis in T cells, showing that this approach can rapidly identify compounds that target proteins of biological interest. Several studies have built on this work, using isoTOP-ABPP to find new covalent ligands. For example, cysteine reactivity and ligandable sites were mapped in mutant Kelch-like ECH-associated protein 1 (KEAP1) and compared with those in wild-type KEAP1 NSCLC lines. The authors discovered compounds that bind to a ligandable cysteine on the nuclear receptor NR0B1, which is regulated by NRF2, a substrate of KEAP1 (ref.181). Cysteine ligandability has also been explored in activated T cells through the use of promiscuous fragment-like compounds, termed ‘scout fragments’, to map ligandability and functional assays to identify more structurally complex electrophilic compounds that suppress T cell activity182. With exciting implications for drug discovery, this approach was also used to identify several proteins that could be targeted covalently to impair T cell activity, including BIRC2 and BIRC3, the nucleosome remodelling deacetylase (NuRD) complex, and the kinases ITK and CYTIP.
To dramatically increase sample throughput, a tandem mass tag (TMT)-based streamlined cysteine activity-based protein profiling (SLC-ABPP) methodology was designed and used to profile an electrophilic fragment library at an impressive depth of more than 8,000 reactive cysteine sites with a total instrument time of 18 min per compound183. As competitive isoTOP-ABPP becomes more high throughput, comprehensive selectivity and reactivity information will rapidly become available for diverse covalent reactive libraries within a wide context of biological disease states. This information will enable rapid identification of covalent ligands against reactive hotspots, along with providing selectivity information on each ligand. When paired with a parallel phenotypic screen against a desired outcome (such as cancer cell death), this methodology facilitates the identification of functional covalent ligands and their corresponding protein targets in a high-throughput manner.
Chemoproteomics platforms for target identification
IsoTOP-ABPP can also be used to identify the protein targets of known electrophilic drugs. As an example, this approach was applied to dimethyl fumarate, which is used to treat autoimmune disease. Although dimethyl fumarate has been used for three decades to treat psoriasis and was approved by the FDA in 2013 for the treatment of multiple sclerosis, the direct covalent targets of dimethyl fumarate remained unclear until more recently. In separate studies, chemoproteomic approaches were used to identify protein kinase Cθ (PKCθ) and IRAK4 as targets of dimethyl fumarate184,185. In both cases, covalent engagement of a cysteine residue disrupted a protein–protein interaction to modulate immune cell function. Disrupting the interaction of PKCθ with the costimulatory receptor CD28 reduced T cell activation, and disrupting the IRAK4–MYD88 interaction suppressed the production of interferon-α in plasmacytoid dendritic cells184,185.
ABPP can also be used to identify off-targets and generally assess the selectivity of covalent molecules. For example, SLC-ABPP was used to analyse spleen tissue extracted from C57BL/6 mice treated with ibrutinib183. Of ~9,200 cysteine sites identified, BTK Cys481 was one of the cysteines most liganded by ibrutinib. Cys313 on B lymphocyte kinase (BLK), which, analogous to BTK, contains a cysteine within the ATP-binding pocket, was also identified as an off-target of ibrutinib183.
Novel screening platforms are also often easily paired with isoTOP-ABPP target identification experiments. For example, a multiplexed in vivo screening platform was developed in which barcoded pancreatic ductal adenocarcinoma lines were pretreated with electrophilic compounds and injected into mice to observe the compound-dependent decrease in metastatic potential186. IsoTOP-ABPP experiments also enabled the identification of the lipase ABHD6 as the target of hit compounds from this screen, even though ABHD6 was not previously known to have a role in metastasis or cancer progression. Beyond identifying the lipase ABHD6 as crucial for metastatic fitness, this approach enabled screening in a biological context more relevant to the disease state through adaptation of covalent ligand screening to a multiplexed in vivo phenotypic assay. In general, target identification experiments using chemoproteomic platforms are crucial in investigating the mechanism of action of electrophilic compounds discovered through phenotypic assays.
Covalent ligand discovery for induced proximity modalities
Covalent ligands are not only useful as functional inhibitors, but also have important roles in emerging induced proximity modalities. Covalent drug discovery platforms have facilitated the expansion of targeted protein degradation approaches by enabling the discovery of covalent recruiters against E3 ubiquitin ligases14,187–193. Although most bifunctional degrader molecules (proteolysis-targeting chimeras (PROTACs)) recruit the E3 ligases cereblon (CRBN) or von Hippel–Lindau (VHL) protein to degrade target proteins, there are more than 600 E3 ligases with varying substrate scopes. Since 2019, covalent recruiters have been used to validate a large proportion of the E3 ligases that have been harnessed for targeted protein degradation, including RING finger protein 114 (RNF114), RNF4, DDB1 and CUL4-associated factor 16 (DCAF16), DCAF11, KEAP1 and, most recently, fem-1 homologue B (FEM1B)187–193. In 2019, isoTOP-ABPP was used to identify RNF114 as the target of the enone-containing natural product nimbolide, which was used to make bifunctional degraders of bromodomain-containing protein 4 (BRD4) and BCR–ABL187. In a separate study, scout fragments were used to construct bifunctional FKBP12 and BRD4 degraders, and the authors identified DCAF16 as the target E3 ligase responsible for degradation190. These discoveries led to the variety of covalent E3 recruiters now available, which have been reviewed elsewhere194. On the basis of analyses of chemoproteomic data sets assessing cysteine reactivity, 97% of E3 ligases possess reactive cysteines, suggesting that covalent approaches to harness more E3 ligases could continue to be successful194.
Beyond degradation, the identification of non-inhibitory covalent ligands has the potential to contribute to the discovery of novel induced proximity modalities. For example, a targeted protein stabilization platform, termed deubiquitinase-targeting chimeras (DUBTACs) has been developed using a covalent deubiquitinase recruiter195. Through analysis of chemoproteomic data and an ABPP-based screen, a covalent OTUB1 recruiter was discovered that could be incorporated into bifunctional compounds that stabilize mutant cystic fibrosis transmembrane conductance regulator (CFTR), the degradation of which drives cystic fibrosis. In general, the identification of non-inhibitory, allosteric ligands through covalent ligand screening has the potential to facilitate recruitment of other enzymes (such as kinases and deacetylases) to target proteins and direct protein function to neosubstrates for therapeutic benefit.
Nucleophilic covalent ligands
Most protein-reactive covalent drugs tend to be electrophilic, to enable reactions with nucleophilic amino acids. By contrast, nucleophilic drugs can react with electrophilic cofactors and post-translational modifications. Several hydrazine-containing compounds act as mechanism-based inhibitors of monoamine oxidase (MAO) A and B, whereby activation by MAO enables alkylation of a flavin cofactor, inhibiting the enzyme196. ABPP principles and ‘reverse-polarity’ probes have been employed to study electrophilic post-translational modifications, such as N-terminal pyruvoyl and glyoxylyl modifications, that can react with hydrazine-containing compounds197. Hydrazine-based probes have also been used to help identify and characterize ligands for proteins with electrophilic cofactors or post-translational modifications198,199.
Lysine-directed covalent ligands
The low abundance of cysteine enables selectivity but limits opportunities for covalently targeting specific proteins of interest. This problem has driven scientists to investigate the targeting of other nucleophilic amino acids, particularly lysine. Owing to the low nucleophilicity of the ε-amino group of lysine under physiological conditions, the discovery of efficient lysine-targeting covalent ligands requires identification of unusually reactive lysines. IsoTOP-ABPP experiments using several lysine-directed probes have proved powerful in profiling lysine reactivity across the proteome200,201. Through isoTOP-ABPP experiments, more than 9,000 ligandable lysines were identified and more elaborated pentafluorophenol-containing or N-hydroxysuccinimide-ester-containing compounds could selectively label specific proteins of interest200,201. Building on these experiments, a library of approximately 180 electrophiles was assembled and isoTOP-ABPP experiments then performed to assess the selectivity of different chemotypes and identify lysines ligandable with small molecules202. This study yielded more broadly reactive electrophiles, such as dicarboxaldehydes, that could be used for further lysine profiling experiments, but also identified less-reactive electrophiles, including N-acyl-N-alkyl sulfonamides, which had been previously used as tools for bioconjugation in cells202,203.
Aside from voxelotor, which was discovered through optimization from fragment-like aldehydes, most lysine-targeting ligands have been designed using structure-based methods from an existing ligand, often through rationally placing an electrophilic sulfonyl fluoride, fluorosulfate or vinyl sulfone in an appropriate orientation to react with an ε-amino group of a lysine adjacent to an established binding site. Such ligands include a kinetic transthyretin stabilizer204, an isoform-selective PI3Kδ inhibitor205 and inhibitors of cyclin-dependent kinase 2 (CDK2)206 and Hsp90 (ref.207). A sulfonyl fluoride-bearing promiscuous kinase inhibitor that targets a conserved lysine in the ATP-binding site was also used as a probe to profile kinase inhibitor selectivity in live cells208. However, sulfonyl fluoride-based probes are not completely selective for lysine and have also been used to target tyrosine residues209. A recent preprint reported the use of a chemoproteomic approach to profile the amino acid reactivity preference of 54 different electrophiles, which will prove to be a great resource for covalent ligand discovery210. Combining comprehensive electrophile profiling, lysine-directed chemoproteomics and structure-guided approaches will enable scientists to leverage the abundance of lysine residues adjacent to ligand binding sites to enhance covalent drug discovery.
Outlook
Over the past decade, advances in covalent drug discovery have led to successful drugs, including inhibitors of EGFR, BTK, KRAS(G12C) and SARS-CoV-2 Mpro. The approvals of these drugs represent milestones that showcase the evolution of covalent drug discovery from a serendipitous effort to a field with established roadmaps for success.
Adoption of electrophile-first discovery strategies represents a notable shift in the field. Ligand-first strategies will continue to be highly applicable for designing covalent drugs against proteins when existing reversible ligands are already known to bind near a nucleophilic amino acid such as cysteine. However, we anticipate that electrophile-first approaches will be increasingly employed, especially when the discovery of reversible ligands proves challenging. Electrophile-first approaches will be facilitated, in part, by chemoproteomics experiments that profile amino acid reactivity across the proteome, leading to the identification of novel ligandable cysteines, for example, that could be targeted with electrophilic compounds.
Additionally, we expect to see an increase in research that explores the use of reversible covalent mechanisms that strike a balance between potency and selectivity in various contexts. Reversible covalent compounds with long off-rates might achieve a desired therapeutic effect while minimizing covalent off-target effects, and the use of more varied electrophiles will allow for increasingly tailored reactivity. We also look forward to further improvements in chemoproteomic workflows that enable additional multiplexing in MS experiments, which will be valuable for assessing compound selectivity and target engagement.
Covalent drug discovery overcomes obstacles in designing ligands against otherwise undruggable protein targets. We expect that the unique features of covalent ligands will continue to spur the discovery of covalent drugs.
Acknowledgements
The authors thank the members of the Nomura Research Group for critical reading of the manuscript. This work was supported by the Nomura Research Group and the Mark Foundation for Cancer Research ASPIRE Award. This work was also supported by grants from the US NIH (award no. R01CA240981 for D.K.N. and no. F31CA268278 for N.J.H.), and the US National Science Foundation Graduate Fellowship (for L.B.).
Glossary
- Selectivity profiling
With respect to covalent drugs, this refers to the process of mapping amino acid sites within the proteome that react covalently with a ligand of interest with the purpose of identifying the targets and off-targets of the ligand in a biological context.
- Chemoproteomics
Collection of methods used to study small molecule–protein interactions on a proteome-wide scale.
- Gatekeeper mutation
Mutation that alters an amino acid residue at a specific location in the ATP-binding site of a kinase, known as the gatekeeper residue; alterations at this position have a particularly strong effect on binding of certain kinase inhibitors.
- Michael acceptors
Electrophiles that can react with a nucleophile through Michael addition, often containing an α, β-unsaturated carbonyl and an electron-withdrawing group.
- Cryptic pocket
A 3D pocket that forms only when a ligand is bound to a protein; these pockets are hidden when the ligand is not bound to the protein.
- Activity-based protein profiling
A chemical proteomics technique used to characterize enzyme function and amino acid reactivity in native biological systems that uses chemical probes to react with specific classes of proteins and amino acids.
- Fragment growing
The medicinal chemistry process of optimizing a fragment hit to a higher-affinity ligand through increasing the size and complexity of the molecule.
- DNA-encoded libraries
Collections of molecules linked to DNA sequences that serve as amplifiable barcodes, carrying information about each library member; this technology enables screening of massive libraries of compounds.
- Covalent docking
A group of methods used to predict the orientation of a covalent ligand when bound to a protein target.
Author contributions
The authors contributed equally to all aspects of the article.
Peer review
Peer review information
Nature Reviews Drug Discovery thanks Megan Matthews, who co-reviewed with Xie Wang, and the other, anonymous, reviewers for their contribution to the peer review of this work.
Competing interests
D.K.N. is a co-founder, shareholder and adviser for Frontier Medicines and Vicinitas Therapeutics. D.K.N. is on the board of directors of Vicinitas Therapeutics. D.K.N. is also on the scientific advisory board of The Mark Foundation for Cancer Research and Photys Therapeutics. D.K.N. is a consultant for Droia Ventures. L.B. and N.J.H. are employees of Vicinitas Therapeutics.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Lydia Boike, Nathaniel J. Henning.
References
- 1.Tuley A, Fast W. The taxonomy of covalent inhibitors. Biochemistry. 2018;57:3326–3337. doi: 10.1021/acs.biochem.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Samanta I, Mondal A, Liu WR. Covalent inhibition in drug discovery. ChemMedChem. 2019;14:889–906. doi: 10.1002/cmdc.201900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Cesco S, Kurian J, Dufresne C, Mittermaier AK, Moitessier N. Covalent inhibitors design and discovery. Eur. J. Med. Chem. 2017;138:96–114. doi: 10.1016/j.ejmech.2017.06.019. [DOI] [PubMed] [Google Scholar]
- 5.Sutanto F, Konstantinidou M, Dömling A. Covalent inhibitors: a rational approach to drug discovery. RSC Med. Chem. 2020;11:876–884. doi: 10.1039/d0md00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang T, Hatcher JM, Teng M, Gray NS, Kostic M. Recent advances in selective and irreversible covalent ligand development and validation. Cell Chem. Biol. 2019;26:1486–1500. doi: 10.1016/j.chembiol.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lonsdale R, Ward RA. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018;47:3816–3830. doi: 10.1039/c7cs00220c. [DOI] [PubMed] [Google Scholar]
- 8.Sagonowsky, E. The top 20 drugs by worldwide sales in 2020. Fierce Pharmahttps://www.fiercepharma.com/special-report/top-20-drugs-by-2020-sales (2021).
- 9.Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov. 2020;19:533–552. doi: 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021;6:386. doi: 10.1038/s41392-021-00780-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vandyck K, Deval J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021;49:36–40. doi: 10.1016/j.coviro.2021.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lagoutte R, Patouret R, Winssinger N. Covalent inhibitors: an opportunity for rational target selectivity. Curr. Opin. Chem. Biol. 2017;39:54–63. doi: 10.1016/j.cbpa.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 13.Lu W, et al. Fragment-based covalent ligand discovery. RSC Chem. Biol. 2021;2:354–367. doi: 10.1039/d0cb00222d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spradlin JN, Zhang E, Nomura DK. Reimagining druggability using chemoproteomic platforms. Acc. Chem. Res. 2021;54:1801–1813. doi: 10.1021/acs.accounts.1c00065. [DOI] [PubMed] [Google Scholar]
- 15.Roberts AM, Ward CC, Nomura DK. Activity-based protein profiling for mapping and pharmacologically interrogating proteome-wide ligandable hotspots. Curr. Opin. Biotechnol. 2017;43:25–33. doi: 10.1016/j.copbio.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drewes G, Knapp S. Chemoproteomics and chemical probes for target discovery. Trends Biotechnol. 2018;36:1275–1286. doi: 10.1016/j.tibtech.2018.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Moellering RE, Cravatt BF. How chemoproteomics can enable drug discovery and development. Chem. Biol. 2012;19:11–22. doi: 10.1016/j.chembiol.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maurais AJ, Weerapana E. Reactive-cysteine profiling for drug discovery. Curr. Opin. Chem. Biol. 2019;50:29–36. doi: 10.1016/j.cbpa.2019.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandrasekharan N, Simmons DL. The cyclooxygenases. Genome Biol. 2004;5:241. doi: 10.1186/gb-2004-5-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 21.Nicola G, Tomberg J, Pratt RF, Nicholas RA, Davies C. Crystal structures of covalent complexes of β-lactam antibiotics with Escherichia coli penicillin-binding protein 5: toward an understanding of antibiotic specificity. Biochemistry. 2010;49:8094–8104. doi: 10.1021/bi100879m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dijkmans AC, et al. Fosfomycin: pharmacological, clinical and future perspectives. Antibiotics. 2017;6:24. doi: 10.3390/antibiotics6040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hendlin D, et al. Phosphonomycin, a new antibiotic produced by strains of Streptomyces. Science. 1969;166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 24.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. The mechanism of action of fosfomycin (phosphonomycin) Ann. NY Acad. Sci. 1974;235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 25.Olbe L, Carlsson E, Lindberg P. A proton-pump inhibitor expedition: the case histories of omeprazole and esomeprazole. Nat. Rev. Drug Discov. 2003;2:132–139. doi: 10.1038/nrd1010. [DOI] [PubMed] [Google Scholar]
- 26.Savi P, et al. Identification and biological activity of the active metabolite of clopidogrel. Thromb. Haemost. 2000;84:891–896. [PubMed] [Google Scholar]
- 27.Thomas D, Zalcberg J. 5-Fluorouracil: a pharmacological paradigm in the use of cytotoxics. Clin. Exp. Pharmacol. Physiol. 1998;25:887–895. doi: 10.1111/j.1440-1681.1998.tb02339.x. [DOI] [PubMed] [Google Scholar]
- 28.Danenberg PV, Langenbach RJ, Heidelberger C. Fluorinated pyrimidines. Structures of reversible and irreversible complexes of thymidylate synthetase and fluorinated pyrimidine nucleotidest. Biochemistry. 1974;13:926–933. doi: 10.1021/bi00702a016. [DOI] [PubMed] [Google Scholar]
- 29.Xu H, Faber C, Uchiki T, Racca J, Dealwis C. Structures of eukaryotic ribonucleotide reductase I define gemcitabine diphosphate binding and subunit assembly. Proc. Natl Acad. Sci. USA. 2006;103:4028–4033. doi: 10.1073/pnas.0600440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curran MP, McKeage K. Bortezomib: a review of its use in patients with multiple myeloma. Drugs. 2009;69:859–888. doi: 10.2165/00003495-200969070-00006. [DOI] [PubMed] [Google Scholar]
- 31.Lynch TJ, Okimoto RA, Supko JG, Settleman J. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 32.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 33.Ou S-HI. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a better mousetrap? A review of the clinical evidence. Crit. Rev. Oncol. Hematol. 2012;83:407–421. doi: 10.1016/j.critrevonc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Yu HA, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR -mutant lung cancers. Clin. Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat. Rev. Clin. Oncol. 2018;15:694–708. doi: 10.1038/s41571-018-0081-4. [DOI] [PubMed] [Google Scholar]
- 36.Yun C-H, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl Acad. Sci. USA. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soria J-C, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16:897–907. doi: 10.1016/S1470-2045(15)00006-6. [DOI] [PubMed] [Google Scholar]
- 38.Yu HA, Pao W. Afatinib — new therapy option for EGFR-mutant lung cancer. Nat. Rev. Clin. Oncol. 2013;10:551–552. doi: 10.1038/nrclinonc.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harvey RD, Adams VR, Beardslee T, Medina P. Afatinib for the treatment of EGFR mutation-positive NSCLC: a review of clinical findings. J. Oncol. Pharm. Pract. 2020;26:1461–1474. doi: 10.1177/1078155220931926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsou H-R, et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005;48:1107–1131. doi: 10.1021/jm040159c. [DOI] [PubMed] [Google Scholar]
- 41.Deeks ED. Neratinib: first global approval. Drugs. 2017;77:1695–1704. doi: 10.1007/s40265-017-0811-4. [DOI] [PubMed] [Google Scholar]
- 42.Shirley M. Dacomitinib: first global approval. Drugs. 2018;78:1947–1953. doi: 10.1007/s40265-018-1028-x. [DOI] [PubMed] [Google Scholar]
- 43.Zhou W, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cross DAE, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finlay MRV, et al. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014;57:8249–8267. doi: 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- 46.Walter AO, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Butterworth S, Cross DAE, Finlay MRV, Ward RA, Waring MJ. The structure-guided discovery of osimertinib: the first U.S. FDA approved mutant selective inhibitor of EGFR T790M. Med. Chem. Commun. 2017;8:820–822. doi: 10.1039/c7md90012k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jänne PA, et al. AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N. Engl. J. Med. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 49.Mok TS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N. Engl. J. Med. 2016;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Passaro A, Jänne PA, Mok T, Peters S. Overcoming therapy resistance in EGFR-mutant lung cancer. Nat. Cancer. 2021;2:377–391. doi: 10.1038/s43018-021-00195-8. [DOI] [PubMed] [Google Scholar]
- 51.Leonetti A, et al. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer. 2019;121:725–737. doi: 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thress KS, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 2015;21:560–562. doi: 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pal Singh S, Dammeijer F, Hendriks RW. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer. 2018;17:57. doi: 10.1186/s12943-018-0779-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol. Rev. 2009;175:120–127. [PubMed] [Google Scholar]
- 55.Pan Z, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 56.Honigberg LA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl Acad. Sci. USA. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Herman SEM, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–6296. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burger JA, Buggy JJ. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) Leuk. Lymphoma. 2013;54:2385–2391. doi: 10.3109/10428194.2013.777837. [DOI] [PubMed] [Google Scholar]
- 59.Wang ML, et al. Targeting BTK with Ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Byrd JC, et al. Targeting BTK with Ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Byrd JC, et al. Ibrutinib treatment for first-line and relapsed/refractory chronic lymphocytic leukemia: final analysis of the pivotal phase Ib/II PCYC-1102 study. Clin. Cancer Res. 2020;26:3918–3927. doi: 10.1158/1078-0432.CCR-19-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Treon SP, et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015;372:1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- 63.Miklos D, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood. 2017;130:2243–2250. doi: 10.1182/blood-2017-07-793786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davids MS, Brown JR. Ibrutinib: a first in class covalent inhibitor of Bruton’s tyrosine kinase. Future Oncol. 2014;10:957–967. doi: 10.2217/fon.14.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Advani RH, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narita Y, et al. Phase I/II study of tirabrutinib, a second-generation Bruton’s tyrosine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro-Oncol. 2021;23:122–133. doi: 10.1093/neuonc/noaa145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guo Y, et al. Discovery of zanubrutinib (BGB-3111), a novel, potent, and selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 2019;62:7923–7940. doi: 10.1021/acs.jmedchem.9b00687. [DOI] [PubMed] [Google Scholar]
- 68.Caldwell RD, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J. Med. Chem. 2019;62:7643–7655. doi: 10.1021/acs.jmedchem.9b00794. [DOI] [PubMed] [Google Scholar]
- 69.Barf T, et al. Acalabrutinib (ACP-196): a covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 2017;363:240–252. doi: 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- 70.Byrd JC, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Byrd JC, et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: results of the first randomized phase III trial. J. Clin. Oncol. 2021;39:3441–3452. doi: 10.1200/JCO.21.01210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Serafimova IM, et al. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miller RM, Paavilainen VO, Krishnan S, Serafimova IM, Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 2013;135:5298–5301. doi: 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bradshaw JM, et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015;11:525–531. doi: 10.1038/nchembio.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao Z, Bourne PE. Progress with covalent small-molecule kinase inhibitors. Drug Discov. Today. 2018;23:727–735. doi: 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- 76.Liu Q, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thorarensen A, et al. Design of a Janus kinase 3 (JAK3) specific inhibitor 1-((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) allowing for the interrogation of JAK3 signaling in humans. J. Med. Chem. 2017;60:1971–1993. doi: 10.1021/acs.jmedchem.6b01694. [DOI] [PubMed] [Google Scholar]
- 78.Goedken ER, et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J. Biol. Chem. 2015;290:4573–4589. doi: 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tan L, et al. Development of selective covalent Janus kinase 3 inhibitors. J. Med. Chem. 2015;58:6589–6606. doi: 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kempson J, et al. Discovery of highly potent, selective, covalent inhibitors of JAK3. Bioorg. Med. Chem. Lett. 2017;27:4622–4625. doi: 10.1016/j.bmcl.2017.09.023. [DOI] [PubMed] [Google Scholar]
- 81.Telliez J-B, et al. Discovery of a JAK3-selective inhibitor: functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016;11:3442–3451. doi: 10.1021/acschembio.6b00677. [DOI] [PubMed] [Google Scholar]
- 82.Robinson MF, et al. Efficacy and safety of PF-06651600 (ritlecitinib), a novel JAK3/TEC inhibitor, in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. 2020;72:1621–1631. doi: 10.1002/art.41316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim RD, et al. First-in-human phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in hepatocellular carcinoma. Cancer Discov. 2019;9:1696–1707. doi: 10.1158/2159-8290.CD-19-0555. [DOI] [PubMed] [Google Scholar]
- 84.Tan L, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl Acad. Sci. USA. 2014;111:E4869–E4877. doi: 10.1073/pnas.1403438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weiss A, et al. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol. Cancer Ther. 2019;18:2194–2206. doi: 10.1158/1535-7163.MCT-18-1291. [DOI] [PubMed] [Google Scholar]
- 86.Goebel L, Müller MP, Goody RS, Rauh D. KRasG12C inhibitors in clinical trials: a short historical perspective. RSC Med. Chem. 2020;11:760–770. doi: 10.1039/d0md00096e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Janes MR, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172:578–589.e17. doi: 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 88.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov. 2022;12:924–937. doi: 10.1158/2159-8290.CD-21-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Erlanson DA, Wells JA, Braisted AC. Tethering: fragment-based drug discovery. Annu. Rev. Biophys. Biomol. Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 91.Lito P, Solomon M, Li L-S, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351:604–608. doi: 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Patricelli MP, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6:316–329. doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 93.Canon J, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 94.Lanman BA, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem. 2020;63:52–65. doi: 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- 95.Skoulidis F, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 2021;384:2371–2381. doi: 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fell JB, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J. Med. Chem. 2020;63:6679–6693. doi: 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- 97.Hallin J, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang J, et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist. 2022;27:536–e553. doi: 10.1093/oncolo/oyab080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.US FDA. Coronavirus (COVID-19) Update: FDA Authorizes First Oral Antiviral for Treatment of COVID-19. FDAhttps://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19 (2021).
- 100.Qiao J, et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 102.Hoffman RL, et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Owen DR, et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 104.Bandyopadhyay A, Gao J. Targeting biomolecules with reversible covalent chemistry. Curr. Opin. Chem. Biol. 2016;34:110–116. doi: 10.1016/j.cbpa.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hammond J, et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 2022;386:1397–1408. doi: 10.1056/NEJMoa2118542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mahase E. Covid-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;375:n2713. doi: 10.1136/bmj.n2713. [DOI] [PubMed] [Google Scholar]
- 107.de Leuw P, Stephan C. Protease inhibitor therapy for hepatitis C virus-infection. Expert. Opin. Pharmacother. 2018;19:577–587. doi: 10.1080/14656566.2018.1454428. [DOI] [PubMed] [Google Scholar]
- 108.Hézode C, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 2009;360:1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 109.Venkatraman S, et al. Discovery of (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (SCH 503034), a selective, potent, orally bioavailable hepatitis C virus NS3 protease inhibitor: a potential therapeutic agent for the treatment of hepatitis C infection. J. Med. Chem. 2006;49:6074–6086. doi: 10.1021/jm060325b. [DOI] [PubMed] [Google Scholar]
- 110.Venkatraman S. Discovery of boceprevir, a direct-acting NS3/4A protease inhibitor for treatment of chronic hepatitis C infections. Trends Pharmacol. Sci. 2012;33:289–294. doi: 10.1016/j.tips.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 111.Lin C, Kwong A, Perni R. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets. 2006;6:3–16. doi: 10.2174/187152606776056706. [DOI] [PubMed] [Google Scholar]
- 112.Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat. Biotechnol. 2011;29:993–1003. doi: 10.1038/nbt.2020. [DOI] [PubMed] [Google Scholar]
- 113.Arasappan A, et al. Discovery of narlaprevir (SCH 900518): a potent, second generation HCV NS3 serine protease inhibitor. ACS Med. Chem. Lett. 2010;1:64–69. doi: 10.1021/ml9000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McCauley JA, Rudd MT. Hepatitis C virus NS3/4a protease inhibitors. Curr. Opin. Pharmacol. 2016;30:84–92. doi: 10.1016/j.coph.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 115.Kwo PY, et al. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376:705–716. doi: 10.1016/S0140-6736(10)60934-8. [DOI] [PubMed] [Google Scholar]
- 116.Sherman KE, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med. 2011;365:1014–1024. doi: 10.1056/NEJMoa1014463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ramsay, I. D., Lestner, J. M. & Barker, C. I. S. in Side Effects of Drugs Annual (ed. Aronson, J. K.) vol. 35 503–550 (Elsevier, 2014).
- 118.Merck & Co., Inc. Merck Voluntarily Discontinuing VICTRELIS. https://www.fda.gov/files/drugs/published/Dear-Pharmacy-Professional-Letter.pdf (2015).
- 119.Lawitz E, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383:515–523. doi: 10.1016/S0140-6736(13)62121-2. [DOI] [PubMed] [Google Scholar]
- 120.Bross PF, et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004;10:3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- 121.Adams J, et al. Potent and selective inhibitors of the proteasome: dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998;8:333–338. doi: 10.1016/s0960-894x(98)00029-8. [DOI] [PubMed] [Google Scholar]
- 122.Hideshima T, et al. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001;61:3071–3076. [PubMed] [Google Scholar]
- 123.Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteasome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control. 2003;10:361–369. doi: 10.1177/107327480301000502. [DOI] [PubMed] [Google Scholar]
- 124.Arkwright R, Pham TM, Zonder JA, Dou QP. The preclinical discovery and development of bortezomib for the treatment of mantle cell lymphoma. Expert. Opin. Drug Discov. 2017;12:225–235. doi: 10.1080/17460441.2017.1268596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cromm PM, Crews CM. The proteasome in modern drug discovery: second life of a highly valuable drug target. ACS Cent. Sci. 2017;3:830–838. doi: 10.1021/acscentsci.7b00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kim KB, Crews CM. From epoxomicin to carfilzomib: chemistry, biology, and medical outcomes. Nat. Prod. Rep. 2013;30:600. doi: 10.1039/c3np20126k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin:20S proteasome reveals a molecular basis for selectivity of α‘,β‘-epoxyketone proteasome inhibitors. J. Am. Chem. Soc. 2000;122:1237–1238. [Google Scholar]
- 128.Kirk CJ. Discovery and development of second-generation proteasome inhibitors. Semin. Hematol. 2012;49:207–214. doi: 10.1053/j.seminhematol.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 129.Chauhan D, et al. In vitro and In vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer Res. 2011;17:5311–5321. doi: 10.1158/1078-0432.CCR-11-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kupperman E, et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010;70:1970–1980. doi: 10.1158/0008-5472.CAN-09-2766. [DOI] [PubMed] [Google Scholar]
- 131.Smolewski P, Rydygier D. Ixazomib: an investigational drug for the treatment of lymphoproliferative disorders. Expert. Opin. Invest. Drugs. 2019;28:421–433. doi: 10.1080/13543784.2019.1596258. [DOI] [PubMed] [Google Scholar]
- 132.Zhou H-J, et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047) J. Med. Chem. 2009;52:3028–3038. doi: 10.1021/jm801329v. [DOI] [PubMed] [Google Scholar]
- 133.Diaz DB, Yudin AK. The versatility of boron in biological target engagement. Nat. Chem. 2017;9:731–742. doi: 10.1038/nchem.2814. [DOI] [PubMed] [Google Scholar]
- 134.Plescia J, Moitessier N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 2020;195:112270. doi: 10.1016/j.ejmech.2020.112270. [DOI] [PubMed] [Google Scholar]
- 135.Adamczyk-Woźniak A, Borys KM, Sporzyński A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 2015;115:5224–5247. doi: 10.1021/cr500642d. [DOI] [PubMed] [Google Scholar]
- 136.Baker SJ, et al. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2690), for the potential treatment of onychomycosis. J. Med. Chem. 2006;49:4447–4450. doi: 10.1021/jm0603724. [DOI] [PubMed] [Google Scholar]
- 137.Benkovic SJ, et al. Identification of borinic esters as inhibitors of bacterial cell growth and bacterial methyltransferases, CcrM and MenH. J. Med. Chem. 2005;48:7468–7476. doi: 10.1021/jm050676a. [DOI] [PubMed] [Google Scholar]
- 138.Rock FL, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 139.Akama T, et al. Discovery and structure–activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg. Med. Chem. Lett. 2009;19:2129–2132. doi: 10.1016/j.bmcl.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 140.Hecker SJ, et al. Discovery of a cyclic boronic acid β-lactamase inhibitor (RPX7009) with utility vs class A serine carbapenemases. J. Med. Chem. 2015;58:3682–3692. doi: 10.1021/acs.jmedchem.5b00127. [DOI] [PubMed] [Google Scholar]
- 141.Das BC. Boron-containing heterocycles as promising pharmacological agents. Bioorg. Med. Chem. 2022;63:116748. doi: 10.1016/j.bmc.2022.116748. [DOI] [PubMed] [Google Scholar]
- 142.Vichinsky E, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N. Engl. J. Med. 2019;381:509–519. doi: 10.1056/NEJMoa1903212. [DOI] [PubMed] [Google Scholar]
- 143.Metcalf B, et al. Discovery of GBT440, an orally bioavailable r-state stabilizer of sickle cell hemoglobin. ACS Med. Chem. Lett. 2017;8:321–326. doi: 10.1021/acsmedchemlett.6b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Garner MH, Bogardt RA, Gurd FR. Determination of the pK values for the alpha-amino groups of human hemoglobin. J. Biol. Chem. 1975;250:4398–4404. [PubMed] [Google Scholar]
- 145.Oksenberg D, et al. GBT 440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br. J. Haematol. 2016;175:141–153. doi: 10.1111/bjh.14214. [DOI] [PubMed] [Google Scholar]
- 146.Blair HA. Voxelotor: first approval. Drugs. 2020;80:209–215. doi: 10.1007/s40265-020-01262-7. [DOI] [PubMed] [Google Scholar]
- 147.Erlanson DA, et al. Site-directed ligand discovery. Proc. Natl Acad. Sci. USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Arkin MR, et al. Binding of small molecules to an adaptive protein-protein interface. Proc. Natl Acad. Sci. USA. 2003;100:1603–1608. doi: 10.1073/pnas.252756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Keedy DA, et al. An expanded allosteric network in PTP1B by multitemperature crystallography, fragment screening, and covalent tethering. eLife. 2018;7:e36307. doi: 10.7554/eLife.36307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wolter M, et al. Fragment-based stabilizers of protein–protein interactions through imine-based tethering. Angew. Chem. Int. Ed. 2020;59:21520–21524. doi: 10.1002/anie.202008585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cardoso R, et al. Identification of Cys255 in HIF-1α as a novel site for development of covalent inhibitors of HIF-1α/ARNT PasB domain protein-protein interaction: covalent allosteric inhibitors of HIF-1α/ARNT PasB PPI. Prot. Sci. 2012;21:1885–1896. doi: 10.1002/pro.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nonoo RH, Armstrong A, Mann DJ. Kinetic template-guided tethering of fragments. ChemMedChem. 2012;7:2082–2086. doi: 10.1002/cmdc.201200404. [DOI] [PubMed] [Google Scholar]
- 153.Kathman SG, Xu Z, Statsyuk AV. A fragment-based method to discover irreversible covalent inhibitors of cysteine proteases. J. Med. Chem. 2014;57:4969–4974. doi: 10.1021/jm500345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kathman SG, et al. A small molecule that switches a ubiquitin ligase from a processive to a distributive enzymatic mechanism. J. Am. Chem. Soc. 2015;137:12442–12445. doi: 10.1021/jacs.5b06839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Johansson H, et al. Fragment-based covalent ligand screening enables rapid discovery of inhibitors for the RBR E3 ubiquitin ligase HOIP. J. Am. Chem. Soc. 2019;141:2703–2712. doi: 10.1021/jacs.8b13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Resnick E, et al. Rapid covalent-probe discovery by electrophile-fragment screening. J. Am. Chem. Soc. 2019;141:8951–8968. doi: 10.1021/jacs.9b02822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dubiella C, et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 2021;17:954–963. doi: 10.1038/s41589-021-00786-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shin Y, et al. Discovery of N-(1-acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as covalent inhibitors of KRASG12C. ACS Med. Chem. Lett. 2019;10:1302–1308. doi: 10.1021/acsmedchemlett.9b00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Debaene F, Da Silva JA, Pianowski Z, Duran FJ, Winssinger N. Expanding the scope of PNA-encoded libraries: divergent synthesis of libraries targeting cysteine, serine and metallo-proteases as well as tyrosine phosphatases. Tetrahedron. 2007;63:6577–6586. [Google Scholar]
- 160.Zimmermann G, et al. A specific and covalent JNK-1 ligand selected from an encoded self-assembling chemical library. Chem. Eur. J. 2017;23:8152–8155. doi: 10.1002/chem.201701644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zambaldo C, Daguer J-P, Saarbach J, Barluenga S, Winssinger N. Screening for covalent inhibitors using DNA-display of small molecule libraries functionalized with cysteine reactive moieties. Med. Chem. Commun. 2016;7:1340–1351. [Google Scholar]
- 162.Daguer J-P, et al. Identification of covalent bromodomain binders through DNA display of small molecules. Angew. Chem. Int. Ed. 2015;54:6057–6061. doi: 10.1002/anie.201412276. [DOI] [PubMed] [Google Scholar]
- 163.Zhu Z, et al. Development of a selection method for discovering irreversible (covalent) binders from a DNA-encoded library. SLAS Discov. 2019;24:169–174. doi: 10.1177/2472555218808454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chan AI, McGregor LM, Jain T, Liu DR. Discovery of a covalent kinase inhibitor from a DNA-encoded small-molecule library × protein library selection. J. Am. Chem. Soc. 2017;139:10192–10195. doi: 10.1021/jacs.7b04880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Guilinger JP, et al. Novel irreversible covalent BTK inhibitors discovered using DNA-encoded chemistry. Bioorg. Med. Chem. 2021;42:116223. doi: 10.1016/j.bmc.2021.116223. [DOI] [PubMed] [Google Scholar]
- 166.Sotriffer C. Docking of covalent ligands: challenges and approaches. Mol. Inf. 2018;37:1800062. doi: 10.1002/minf.201800062. [DOI] [PubMed] [Google Scholar]
- 167.Scarpino A, Ferenczy GG, Keserű GM. Comparative evaluation of covalent docking tools. J. Chem. Inf. Model. 2018;58:1441–1458. doi: 10.1021/acs.jcim.8b00228. [DOI] [PubMed] [Google Scholar]
- 168.Zhang S, et al. Effective virtual screening strategy toward covalent ligands: identification of novel NEDD8-activating enzyme inhibitors. J. Chem. Inf. Model. 2014;54:1785–1797. doi: 10.1021/ci5002058. [DOI] [PubMed] [Google Scholar]
- 169.London N, et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014;10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Shraga A, et al. Covalent docking identifies a potent and selective MKK7 inhibitor. Cell Chem. Biol. 2019;26:98–108.e5. doi: 10.1016/j.chembiol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 171.Nnadi CI, et al. Novel K-Ras G12C switch-II covalent binders destabilize Ras and accelerate nucleotide exchange. J. Chem. Inf. Model. 2018;58:464–471. doi: 10.1021/acs.jcim.7b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang S, et al. Covalent complexes of proteasome model with peptide aldehyde inhibitors MG132 and MG101: docking and molecular dynamics study. J. Mol. Model. 2009;15:1481–1490. doi: 10.1007/s00894-009-0515-0. [DOI] [PubMed] [Google Scholar]
- 173.Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 174.Edgington LE, Verdoes M, Bogyo M. Functional imaging of proteases: recent advances in the design and application of substrate-based and activity-based probes. Curr. Opin. Chem. Biol. 2011;15:798–805. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc. Natl Acad. Sci. USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Weerapana E, Speers AE, Cravatt BF. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP) — a general method for mapping sites of probe modification in proteomes. Nat. Protoc. 2007;2:1414–1425. doi: 10.1038/nprot.2007.194. [DOI] [PubMed] [Google Scholar]
- 177.Weerapana E, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yan T, et al. SP3-FAIMS chemoproteomics for high-coverage profiling of the human cysteinome. ChemBioChem. 2021;22:1841–1851. doi: 10.1002/cbic.202000870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Boike L, et al. Discovery of a functional covalent ligand targeting an intrinsically disordered cysteine within MYC. Cell Chem. Biol. 2020;28:4–13. doi: 10.1016/j.chembiol.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Backus KM, et al. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bar-Peled L, et al. Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell. 2017;171:696–709.e23. doi: 10.1016/j.cell.2017.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vinogradova EV, et al. An activity-guided map of electrophile-cysteine interactions in primary human T cells. Cell. 2020;182:1009–1026.e29. doi: 10.1016/j.cell.2020.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kuljanin M, et al. Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 2021;39:630–641. doi: 10.1038/s41587-020-00778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Blewett MM, et al. Chemical proteomic map of dimethyl fumarate–sensitive cysteines in primary human T cells. Sci. Signal. 2016;9:rs10. doi: 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zaro BW, et al. Dimethyl fumarate disrupts human innate immune signaling by targeting the IRAK4–MyD88 complex. J. Immunol. 2019;202:2737–2746. doi: 10.4049/jimmunol.1801627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grüner BM, et al. An in vivo multiplexed small-molecule screening platform. Nat. Methods. 2016;13:883–889. doi: 10.1038/nmeth.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Spradlin JN, et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019;15:747–755. doi: 10.1038/s41589-019-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Luo M, et al. Chemoproteomics-enabled discovery of covalent RNF114-based degraders that mimic natural product function. Cell Chem. Biol. 2021;28:559–566.e15. doi: 10.1016/j.chembiol.2021.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ward CC, et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem. Biol. 2019;14:2430–2440. doi: 10.1021/acschembio.8b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019;15:737–746. doi: 10.1038/s41589-019-0279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang X, et al. DCAF11 supports targeted protein degradation by electrophilic proteolysis-targeting chimeras. J. Am. Chem. Soc. 2021;143:5141–5149. doi: 10.1021/jacs.1c00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tong B, et al. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020;10:15543. doi: 10.1038/s41598-020-72491-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Henning NJ, et al. Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J. Am. Chem. Soc. 2022;44:701–708. doi: 10.1021/jacs.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Belcher BP, Ward CC, Nomura DK. Ligandability of E3 ligases for targeted protein degradation applications. Biochemistry. 2021 doi: 10.1021/acs.biochem.1c00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Henning NJ, et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 2022;18:412–421. doi: 10.1038/s41589-022-00971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Binda C, et al. Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry. 2008;47:5616–5625. doi: 10.1021/bi8002814. [DOI] [PubMed] [Google Scholar]
- 197.Matthews ML, et al. Chemoproteomic profiling and discovery of protein electrophiles in human cells. Nat. Chem. 2017;9:234–243. doi: 10.1038/nchem.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang X, et al. Discovery of potent and selective inhibitors against protein-derived electrophilic cofactors. J. Am. Chem. Soc. 2022;144:5377–5388. doi: 10.1021/jacs.1c12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lin Z, et al. Activity-based hydrazine probes for protein profiling of electrophilic functionality in therapeutic targets. ACS Cent. Sci. 2021;7:1524–1534. doi: 10.1021/acscentsci.1c00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hacker SM, et al. Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017;9:1181–1190. doi: 10.1038/nchem.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ward CC, Kleinman JI, Nomura DK. NHS-esters as versatile reactivity-based probes for mapping proteome-wide ligandable hotspots. ACS Chem. Biol. 2017;12:1478–1483. doi: 10.1021/acschembio.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Abbasov ME, et al. A proteome-wide atlas of lysine-reactive chemistry. Nat. Chem. 2021;13:1081–1092. doi: 10.1038/s41557-021-00765-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tamura T, et al. Rapid labelling and covalent inhibition of intracellular native proteins using ligand-directed N-acyl-N-alkyl sulfonamide. Nat. Commun. 2018;9:1870. doi: 10.1038/s41467-018-04343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Grimster NP, et al. Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J. Am. Chem. Soc. 2013;135:5656–5668. doi: 10.1021/ja311729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Dalton SE, et al. Selectively targeting the kinome-conserved lysine of PI3Kδ as a general approach to covalent kinase inhibition. J. Am. Chem. Soc. 2018;140:932–939. doi: 10.1021/jacs.7b08979. [DOI] [PubMed] [Google Scholar]
- 206.Anscombe E, et al. Identification and characterization of an irreversible inhibitor of CDK2. Chem. Biol. 2015;22:1159–1164. doi: 10.1016/j.chembiol.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cuesta A, Wan X, Burlingame AL, Taunton J. Ligand conformational bias drives enantioselective modification of a surface-exposed lysine on Hsp90. J. Am. Chem. Soc. 2020;142:3392–3400. doi: 10.1021/jacs.9b09684. [DOI] [PubMed] [Google Scholar]
- 208.Zhao Q, et al. Broad-spectrum kinase profiling in live cells with lysine-targeted sulfonyl fluoride probes. J. Am. Chem. Soc. 2017;139:680–685. doi: 10.1021/jacs.6b08536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hett EC, et al. Rational targeting of active-site tyrosine residues using sulfonyl fluoride probes. ACS Chem. Biol. 2015;10:1094–1098. doi: 10.1021/cb5009475. [DOI] [PubMed] [Google Scholar]
- 210.Zanon PRA, et al. Profiling the proteome-wide selectivity of diverse electrophiles. ChemRxiv. 2021 doi: 10.26434/chemrxiv.14186561.v1. [DOI] [Google Scholar]
- 211.Chung CY-S, et al. Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 2019;15:776–785. doi: 10.1038/s41589-019-0308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Strelow JM. A perspective on the kinetics of covalent and irreversible inhibition. SLAS Discov. 2017;22:3–20. doi: 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]
- 213.McWhirter, C. in Annual Reports in Medicinal Chemistry (eds Ward, R. A. & Grimster, N. P.) vol. 56 1–31 (Elsevier, 2021).
- 214.Schnute ME, et al. Aminopyrazole carboxamide Bruton’s tyrosine kinase inhibitors. Irreversible to reversible covalent reactive group tuning. ACS Med. Chem. Lett. 2019;10:80–85. doi: 10.1021/acsmedchemlett.8b00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Maurer TS, Tabrizi-Fard MA, Fung H. Impact of mechanism-based enzyme inactivation on inhibitor potency: implications for rational drug discovery. J. Pharm. Sci. 2000;89:1404–1414. doi: 10.1002/1520-6017(200011)89:11<1404::aid-jps4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 216.Krippendorff B-F, Neuhaus R, Lienau P, Reichel A, Huisinga W. Mechanism-based inhibition: deriving KI and kinact directly from time-dependent IC50 values. J. Biomol. Screen. 2009;14:913–923. doi: 10.1177/1087057109336751. [DOI] [PubMed] [Google Scholar]
- 217.Miyahisa I, Sameshima T, Hixon MS. Rapid determination of the specificity constant of irreversible inhibitors (kinact/KI) by means of an endpoint competition assay. Angew. Chem. Int. Ed. 2015;54:14099–14102. doi: 10.1002/anie.201505800. [DOI] [PubMed] [Google Scholar]
- 218.Thorarensen A, et al. The advantages of describing covalent inhibitor in vitro potencies by IC50 at a fixed time point. IC50 determination of covalent inhibitors provides meaningful data to medicinal chemistry for SAR optimization. Bioorg. Med. Chem. 2021;29:115865. doi: 10.1016/j.bmc.2020.115865. [DOI] [PubMed] [Google Scholar]
- 219.Flanagan ME, et al. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J. Med. Chem. 2014;57:10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- 220.Ward RA, et al. Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR) J. Med. Chem. 2013;56:7025–7048. doi: 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- 221.Keeley A, Ábrányi-Balogh P, Keserű GM. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. Med. Chem. Commun. 2019;10:263–267. doi: 10.1039/c8md00327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Cee VJ, et al. Systematic study of the glutathione (GSH) reactivity of N-arylacrylamides: 1. Effects of aryl substitution. J. Med. Chem. 2015;58:9171–9178. doi: 10.1021/acs.jmedchem.5b01018. [DOI] [PubMed] [Google Scholar]
- 223.Birkholz A, et al. Systematic study of the glutathione reactivity of N-phenylacrylamides: 2. Effects of acrylamide substitution. J. Med. Chem. 2020;63:11602–11614. doi: 10.1021/acs.jmedchem.0c00749. [DOI] [PubMed] [Google Scholar]