

Abstract
Background
Anaemia occurs in chronic kidney disease (CKD) and is more prevalent with lower levels of kidney function. Anaemia in CKD is associated with death related to cardiovascular (CV) disease and infection. Established treatments include erythropoiesis‐stimulating agents (ESAs), iron supplementation and blood transfusions. Oral hypoxia‐inducible factors (HIF) stabilisers are now available to manage anaemia in people with CKD.
Objectives
We aimed to assess the benefits and potential harms of HIF stabilisers for the management of anaemia in people with CKD.
Search methods
We searched the Cochrane Kidney and Transplant Register of Studies up to 22 November 2021 through contact with the Information Specialist using search terms relevant to our review. Studies in the Register are identified through searches of CENTRAL, MEDLINE, EMBASE, conference proceedings, the International Clinical Trials Register (ICTRP) Search Portal, and ClinicalTrials.gov.
Selection criteria
Randomised and quasi‐randomised studies evaluating hypoxia‐inducible factors stabilisers compared to placebo, standard care, ESAs or iron supplementation in people with CKD were included.
Data collection and analysis
Five authors independently extracted data and assessed the risk of bias. Treatment estimates were summarised using random effects pair‐wise meta‐analysis and expressed as a relative risk (RR) or mean difference (MD), with a corresponding 95% confidence interval (CI). Evidence certainty was assessed using GRADE.
Main results
We included 51 studies randomising 30,994 adults. These studies compared HIF stabilisers to either placebo or an ESA.
Compared to placebo, HIF stabiliser therapy had uncertain effects on CV death (10 studies, 1114 participants): RR 3.68, 95% CI 0.19 to 70.21; very low certainty evidence), and nonfatal myocardial infarction (MI) (3 studies, 822 participants): RR 1.29, 95% CI 0.31 to 5.36; I² = 0%; very low certainty evidence), probably decreases the proportion of patients requiring blood transfusion (8 studies, 4329 participants): RR 0.51, 95% CI 0.44 to 0.60; I² = 0%; moderate certainty evidence), and increases the proportion of patients reaching the target haemoglobin (Hb) (10 studies, 5102 participants): RR 8.36, 95% CI 6.42 to 10.89; I² = 37%; moderate certainty evidence).
Compared to ESAs, HIF stabiliser therapy may make little or no difference to CV death (17 studies, 10,340 participants): RR 1.05, 95% CI 0.88 to 1.26; I² = 0%; low certainty evidence), nonfatal MI (7 studies, 7765 participants): RR 0.91, 95% CI 0.76 to 1.10; I² = 0%; low certainty evidence), and nonfatal stroke (5 studies, 7285 participants): RR 1.06, 95% CI 0.71 to 1.56; I² = 8%; low certainty evidence), and had uncertain effects on fatigue (2 studies, 3471 participants): RR 0.80, 95% CI 0.56 to 1.16; I² = 0%; very low certainty evidence). HIF stabiliser therapy probably decreased the proportion of patients requiring blood transfusion (11 studies, 10,786 participants): RR 0.87, 95% CI 0.76 to 1.00; I² = 25%; moderate certainty evidence), but may make little or no difference on the proportion of patients reaching the target Hb (14 studies, 4601 participants): RR 1.00, 95% CI 0.93 to 1.07; I² = 70%; low certainty evidence), compared to ESA.
The effect of HIF stabilisers on hospitalisation for heart failure, peripheral arterial events, loss of unassisted dialysis vascular access patency, access intervention, cancer, infection, pulmonary hypertension and diabetic nephropathy was uncertain.
None of the included studies reported life participation. Adverse events were rarely and inconsistently reported.
Authors' conclusions
HIF stabiliser management of anaemia had uncertain effects on CV death, fatigue, death (any cause), CV outcomes, and kidney failure compared to placebo or ESAs. Compared to placebo or ESAs, HIF stabiliser management of anaemia probably decreased the proportion of patients requiring blood transfusions, and probably increased the proportion of patients reaching the target Hb when compared to placebo.
Plain language summary
Are hypoxia‐inducible factor stabilisers effective for management of anaemia among people with chronic kidney disease?
What is the issue?
Anaemia (reduced levels of circulating red blood cells) is common in people with chronic kidney disease (CKD). Anaemia is linked to cardiovascular disease, infection and death. Hypoxia‐inducible factors (HIF) stabilisers have now become available to manage anaemia and can be taken by mouth, thus avoiding injections.
What did we do?
We evaluated whether HIF stabilisers are beneficial for children and adults with CKD to manage anaemia. We evaluated all clinical studies for hypoxia‐inducible factor stabilisers and summarised the results. We evaluated how certain we could be about the evidence related to hypoxia‐inducible factors stabiliser using a system called "GRADE".
What did we find?
We included 51 studies randomising 30,994 adult patients. Patients in the studies were given a HIF stabiliser, a sugar pill (placebo), or erythropoietin treatment. The treatment they got was decided by random chance. The studies were generally short‐term (over a few weeks). There were no studies in children or people who had received a kidney transplant.
HIF stabilisers decreased blood transfusions for people with CKD when compared to placebo or erythropoietin treatment. HIF stabilisers increased the number of patients reaching their haemoglobin target level when compared to placebo. HIF stabilisers have uncertain effects on life expectancy and the chance of heart disease in people with CKD.
Conclusions
HIF stabilisers decreased the need for a blood transfusion for people with CKD and increased the number of patients reaching their haemoglobin target level. We are not sure whether hypoxia‐inducible factor stabilisers have any impact on life expectancy or life quality in people with CKD when compared to a placebo or other treatments for anaemia.
Summary of findings
Summary of findings 1. Hypoxia‐inducible factor (HIF) stabilisers versus placebo for people with chronic kidney disease (CKD).
| HIF stabilisers versus placebo for people with CKD | ||||||
|
Patient or population: people with CKD (including HD and PD) Settings: multinational Intervention: HIF stabilisers Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | HIF stabilisers | |||||
|
Cardiovascular death Median follow‐up: 16 weeks |
Low risk population (CKD) |
RR 3.68 (0.19 to 70.21) |
1114 (10) | ⊕⊝⊝⊝ very low1,2,3 | Studies were not designed to measure effects of HIF stabiliser management of anaemia on CV death compared with placebo in CKD and HD | |
| No events | 3/607** | |||||
| High risk population (HD) | ||||||
| No events | No events | |||||
| Fatigue | Not reported | Not reported | ‐‐ | ‐‐ | ‐‐ | No studies reported this outcome |
| Life participation | Not reported | Not reported | ‐‐ | ‐‐ | ‐‐ | No studies reported this outcome |
|
Nonfatal myocardial infarction Median follow‐up: 24 weeks |
Low risk population (CKD) |
RR 1.29 (0.31 to 5.36) |
822 (3) | ⊕⊝⊝⊝ very low1,2,4 | The effects of HIF stabiliser management of anaemia on nonfatal MI were uncertain compared with placebo in CKD | |
| 8 per 1000 |
2 more per 1000 (from 6 fewer to 35 more) |
|||||
|
Nonfatal stroke Median follow‐up: 21 weeks |
Low risk population (CKD) | Not estimable | 228 (2) | ⊕⊝⊝⊝ very low1,2,4 | Studies were not designed to measure effects of HIF stabiliser management of anaemia on nonfatal stroke compared with placebo in CKD | |
| No events | No events | |||||
|
Proportion of patients requiring blood transfusion Median follow‐up: 18 weeks |
Low risk population (CKD) | RR 0.51 (0.44 to 0.60) | 4329 (8) | ⊕⊕⊕⊝ moderate1 | HIF stabiliser management of anaemia probably decreases the proportion of patients requiring blood transfusion compared to placebo in CKD and HD | |
| 200 per 1000 | 96 fewer per 1000 (from 112 fewer to 80 fewer) | |||||
| High risk population (HD) | ||||||
| 214 per 1000 | 169 fewer per 1000 (206 fewer to 30 more) | |||||
|
Proportion reaching target haemoglobin Median follow‐up: 16 weeks |
Low risk population (CKD) | RR 8.36 (6.42 to 10.89) | 5102 (10) | ⊕⊕⊕⊝ moderate1 | HIF stabiliser management of anaemia probably increases the proportion of patients reaching their Hb target compared to placebo in CKD and HD | |
| 83 per 1000 | 594 more per 1000 (424 more to 821 more) | |||||
| High risk population (CKD and HD) | ||||||
| No events | 63/141** | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ** Event rate derived from the raw data. A 'per thousand' rate is non‐informative in view of the scarcity of evidence and zero events in the control group HD: haemodialysis; PD: peritoneal dialysis; CI: confidence interval; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Evidence certainty was downgraded by one level due to study limitations. Some studies had unclear risks for sequence generation and/or allocation concealment and the majority or all of them were not blinded (participant/investigator and/or outcomes assessor). All studies reported sources of funding
2 Evidence certainty was downgraded by one level due to imprecision
3 Evidence certainty was downgraded by one level due to indirectness in the study population
4 Evidence certainty was downgraded by one level due to imprecision (optimal information size was not met and the included studies reported zero events)
Summary of findings 2. Hypoxia‐inducible factor (HIF) stabilisers versus erythropoiesis‐stimulating agent (ESA) for people with chronic kidney disease (CKD).
| HIF stabilisers versus ESA for people with CKD | ||||||
|
Patient or population: people with CKD (including HD and PD) Settings: multinational Intervention: HIF stabilisers Comparison: ESA | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ESA | HIF stabilisers | |||||
|
Cardiovascular death Median follow‐up: 28 weeks |
Low risk population (CKD) |
RR 1.05 (0.88 to 1.26) |
10,340 (17) | ⊕⊕⊝⊝ low1,2 | HIF stabiliser management of anaemia may have little or no difference on CV death compared with ESA in CKD | |
| 34 per 1000 |
6 more per 1000
(from 3 fewer to 18 more) |
|||||
| High risk population (HD and PD) | ||||||
| 77 per 1000 |
3 fewer per 1000
(from 19 fewer to 17 more) |
|||||
|
Fatigue Median follow‐up: 57 weeks |
Low risk population (CKD) |
RR 0.80 (0.56 to 1.16) |
3471 (2) | ⊕⊝⊝⊝ very low1,2,3 | HIF stabiliser management of anaemia had uncertain effects on fatigue compared with ESA in CKD | |
| 36 per 1000 |
7 fewer per 1000
(from 16 fewer to 6 more) |
|||||
| Life participation | Not reported | Not reported | ‐‐ | ‐‐ | ‐‐ | No studies reported this outcome |
|
Nonfatal myocardial infarction Median follow‐up: 26 weeks |
Low risk population (CKD) |
RR 0.91 (0.76 to 1.10) |
7765 (7) | ⊕⊕⊝⊝ low1,2 | HIF stabiliser management of anaemia may have little or no difference on nonfatal MI compared with ESA in CKD, HD and PD | |
| 46 per 1000 |
3 more per 1000
(from 9 fewer to 18 more) |
|||||
| High risk population (HD and PD) | ||||||
| 80 per 1000 | 16 fewer per 1000 (from 30 fewer to 2 more) | |||||
|
Nonfatal stroke Median follow‐up: 28 weeks |
Low risk population (CKD) |
RR 1.06 (0.71 to 1.56) |
7285 (5) | ⊕⊕⊝⊝ low1,2 | HIF stabiliser management of anaemia may have little or no difference on nonfatal stroke compared with ESA in CKD, HD and PD | |
| 10 per 1000 |
5 more per 1000
(from 1 fewer to 16 more) |
|||||
| High risk population (HD) | ||||||
| 24 per 1000 | 5 fewer per 1000 (from 12 fewer to 8 more) | |||||
|
Proportion of patients requiring blood transfusion Median follow‐up: 52 weeks |
Low risk population (CKD) | RR 0.87 (0.76 to 1.00) | 10786 (11) | ⊕⊕⊕⊝ moderate1 | HIF stabiliser management of anaemia probably decreases the proportion of patients requiring blood transfusion compared to ESA in CKD, HD and PD | |
| 121 per 1000 | 4 fewer per 1000 (19 fewer to 16 more) | |||||
| High risk population (HD) | ||||||
| 154 per 1000 | 31 fewer per 1000 (55 fewer to 2 more) | |||||
|
Proportion reaching target haemoglobin Median follow‐up: 27 weeks |
Low risk population (CKD) | RR 1.00 (0.93 to 1.07) | 4601 (14) | ⊕⊕⊝⊝ low1,2 | HIF stabiliser management of anaemia may have little or no difference on the proportion of patients reaching their Hb target compared to ESA in CKD, HD and PD | |
| 793 per 1000 | 16 more per 1000 (79 fewer to 127 more) | |||||
| High risk population (HD and PD) | ||||||
| 540 per 1000 | 11 fewer per 1000 (49 fewer to 32 more) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). HD: haemodialysis; PD: peritoneal dialysis; CI: confidence interval; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Evidence certainty was downgraded by one level due to study limitations. Some studies had unclear risks for sequence generation and/or allocation concealment and the majority or all of them were not blinded (participant/investigator and/or outcomes assessor). All studies reported sources of funding
2 Evidence certainty was downgraded by one level due to imprecision
3 Evidence certainty was downgraded by one level because similar studies assessed the outcomes
Background
Description of the condition
Chronic kidney disease (CKD), reduced kidney function or structural changes in kidney tissue lasting longer than three months, affects approximately 0.7 billion people globally with 20 million additional people affected each year (Global Burden of Disease 2017). Reduced kidney function and raised levels of albumin in the urine are important risk factors for cardiovascular (CV) disease. CKD increases CV risk approximately two‐ to four‐fold in excess of traditional CV risk factors (Gansevoort 2013). CKD is associated with fatigue (Mathias 2020) and lower quality of life (QoL), and incurred 61.3 million disability‐affected years worldwide in 2017 (GBD Kidney Disease 2017; Global Burden of Disease 2017; Wyld 2019).
Anaemia (reduced levels of circulating red blood cells (RBC)) occurs as a result of the progression of CKD due to impaired kidney erythropoietin secretion, lower absorption of iron, macrophage sequestration of iron by uraemic inflammation, and shortened RBC survival (Babitt 2012). Anaemia is a critically important outcome for people with CKD (SONG 2017) and may worsen the impact of CKD on health‐related (HR) QoL including decreased work productivity (van Haalen 2020). Anaemia prevalence is higher at lower levels of kidney function, affecting one in five people with moderate CKD (estimated glomerular filtration rate (eGFR) 30 to 59 mL/min/1.73 m²) (El‐Achkar 2005). Anaemia in CKD is associated with increased death, including death related to CV disease and infection (Ma 1999).
Description of the intervention
Treatments for anaemia caused by CKD include erythropoiesis‐stimulating agents (ESAs), iron supplementation and blood transfusion. Clinical practice guidelines suggest that iron deficiency is corrected prior to initiation of ESA therapy, minimising RBC transfusions especially to avoid allosensitization, except when rapid correction of anaemia is required (KDIGO Clinical Practice Guideline Anemia 2012). ESAs to target higher haemoglobin (Hb) levels (> 130 g/L) in people with CKD increase the risk of death and adverse CV events (Phrommuntikul 2007), leading to clinical practice guidance that suggests ESA therapy is used to avoid Hb concentrations below 90 g/L (KDIGO Clinical Practice Guideline Anemia 2012). Hypoxia‐inducible factors (HIF) are promising orally administered drugs to treat anaemia in people with CKD (Haase 2021).
How the intervention might work
HIF are transcription factors present in cells formed through binding of HIF‐α and β subunits (Semenza 2011). The HIF‐β subunit is expressed constitutively, while the HIF‐α subunit is regulated through hydroxylation at proline residues by HIF‐prolyl‐hydroxylases. During tissue hypoxia, the HIF‐prolyl‐hydroxylase is inhibited, stabilising HIF‐1 and HIF‐2, which act to up‐regulate expression of many genes, including those that promote erythropoiesis and angiogenesis as well as metabolic processes. HIF stabilisers inhibit HIF‐prolyl‐hydroxylase activity and stimulate erythropoiesis in people with CKD (Bernhardt 2010).
Oral HIF stabilisers correct anaemia in people with CKD in a dose‐dependent manner (Provenzano 2016c). HIF suppresses hepcidin production, which is the main regulator of systemic iron homeostasis (Nemeth 2009), enabling ferroportin stabilisation and promoting intestinal uptake and iron mobilisation from the reticuloendothelial system (Liu 2012; Renassia 2019; Schwartz 2019). Although HIF stabilisation has potentially pleiotropic cellular effects, changes is vascular endothelial growth factor (VEG‐F) have not been seen at doses used in randomised controlled trials (RCTs). Potential adverse consequences of HIF stabiliser treatment include tumour activity and angiogenesis (LaGory 2016). HIF stabilisers provide a potential oral therapy for sustained correction of anaemia in CKD, less dependent on iron (particularly intravenous (IV) supplementation. Although oral treatment adherence in a dialysis setting is still not clearly defined among nephrologists, oral HIF stabiliser therapy may be more acceptable to patients, including the potential to avoid the known adverse consequences of treatments with ESAs and blood transfusions. Several HIF stabilisers are available including roxadustat, vadadustat, daprodustat, desidustat, enarodustat and molidustat.
Why it is important to do this review
Evidence for HIF stabilisers to treat anaemia in people with CKD is emerging in RCTs. With data presented from phase 2 studies for competitive HIF stabilisers and preliminary data from phase 3 studies on roxadustat in patients requiring dialysis, sufficient evidence was available to determine the efficacy and safety of HIF stabilisers compared to other treatment strategies, including ESAs therapy. This Cochrane review evaluated the benefits and potential harms of HIF stabilisers in CKD and provide a summary of the certainty of available evidence for decision‐makers including clinicians, patients, and policy‐makers.
Objectives
We aimed to assess the benefits and potential harms of HIF stabilisers for the management of anaemia in people with CKD.
Methods
Criteria for considering studies for this review
Types of studies
All RCTs and quasi‐RCTs (RCTs in which treatment allocation was obtained by alternation, use of alternate medical records, date of birth or other predictable methods) looking at the effects of HIF stabilisers versus other anaemia therapies, placebo or standard care in people with CKD were included.
Types of participants
Inclusion criteria
Adults and children with CKD were included. We defined CKD as those who are receiving any form of kidney replacement therapy (KRT), have a functioning kidney transplant, have impaired kidney function defined as a reduced eGFR < 60 mL/min/1.73 m², or the presence of other markers of kidney damage such as proteinuria (KDOQI stages 1‐5) (KDIGO Clinical Practice Guideline CKD 2012), or elevated serum creatinine (SCr) (> 120 mmol/L), or as defined by study authors.
Types of interventions
We evaluated the following treatment comparisons:
HIF stabiliser versus placebo
HIF stabiliser versus standard care
HIF stabiliser versus ESA
HIF stabiliser versus iron supplementation
We evaluated HIF stabiliser therapy given orally at any frequency. We included RCTs regardless of the target Hb used to guide dose and frequency.
We investigated studies comparing different doses and phase 1 and 2 studies using subgroup analysis.
We excluded studies assessing head‐to‐head comparisons of HIF stabilisers.
We excluded studies with follow‐up of less than eight weeks.
Types of outcome measures
We did not exclude studies based on non‐reporting of outcomes of interest.
The outcomes selected included the relevant SONG core outcome sets as specified by the Standardised Outcomes in Nephrology initiative (SONG 2017).
Primary outcomes
CV death
Life participation
Fatigue
Secondary outcomes
CV disease (nonfatal myocardial infarction (MI), nonfatal stroke, peripheral arterial event, hospitalisation for heart failure (HF))
Proportion of patients requiring blood transfusion
Vascular access (including vascular access failure, early thrombosis (< eight weeks), loss of unassisted patency (combined data for stenosis/occlusions), access failure to attain suitability for dialysis, and need for access intervention (combined data for surgically or by radiological guided angioplasty))
Cancer
Kidney failure
Infection
Graft health (including graft loss, graft function, acute rejection and chronic rejection)
Peritoneal dialysis (PD) infection
PD failure
Proportion of patients reaching the target Hb
Adverse events (including pulmonary hypertension, deterioration of diabetic retinopathy, kidney and liver cysts and hyperkalaemia)
Search methods for identification of studies
Electronic searches
We searched the Cochrane Kidney and Transplant Register of Studies up to 22 November 2021 through contact with the Information Specialist using search terms relevant to our review. The Register contains studies identified from the following sources.
Monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL)
Weekly searches of MEDLINE OVID SP
Searches of kidney and transplant journals and the proceedings and abstracts from major kidney and transplant conferences
Searching the current year of EMBASE OVID SP
Weekly current awareness alerts for selected kidney and transplant journals
Searches of the International Clinical Trials Register (ICTRP) Search Portal and ClinicalTrials.gov
Studies contained in the Register are identified through searches of CENTRAL, MEDLINE, and EMBASE based on the scope of Cochrane Kidney and Transplant. Details of search strategies and a list of handsearched journals, conference proceedings and current awareness alerts are available on the Cochrane Kidney and Transplant website.
See Appendix 1 for search terms used in strategies for this review.
Searching other resources
Reference lists of review articles, relevant studies, and clinical practice guidelines
Contacting relevant individuals/organisations seeking information about unpublished or incomplete studies
Grey literature sources (e.g. abstracts, dissertations, and theses), in addition to those already included in the Cochrane Kidney and Transplant Register of Studies, were also searched
Data collection and analysis
Selection of studies
The search strategies described were used to obtain titles and abstracts of studies that may be relevant to the review. The titles and abstracts were screened by five authors (PN, EH, MR, DH, VS) working independently, who discarded studies that were not applicable, however, studies and reviews that might include relevant data or information on studies were retained initially. Five authors (PN, EH, MR, DH, VS) independently assessed retrieved abstracts and, if necessary, the full text of these studies to determine which studies satisfy the inclusion criteria. Disagreements were resolved in consultation with another author (SP).
Data extraction and management
Data extraction was carried out independently by five authors (PN, EH, MR, DH, VS) using standard data extraction forms. Disagreements were resolved in consultation with another author (SC). Studies reported in non‐English language journals were translated before assessment. Where more than one publication of one study existed, reports were grouped together, and the publication with the most complete data were used in the analyses. Where relevant outcomes were only published in earlier versions these data were used. Any discrepancy between published versions was highlighted.
Assessment of risk of bias in included studies
Five authors (PN, EH, MR, DH, VS) independently assessed the following items using the risk of bias assessment tool (Higgins 2020) (see Appendix 2).
Was there adequate sequence generation (selection bias)?
Was allocation adequately concealed (selection bias)?
-
Was knowledge of the allocated interventions adequately prevented during the study?
Participants and personnel (performance bias)
Outcome assessors (detection bias)
Were incomplete outcome data adequately addressed (attrition bias)?
Are the study reports free of suggestion of selective outcome reporting (reporting bias)?
Was the study free of other problems that could put it at risk of bias?
Measures of treatment effect
For dichotomous outcomes (death, CV disease, blood transfusion, vascular access, cancer, hospitalisation for HF, kidney failure, infection, graft health, PD infection, PD failure, proportion reaching Hb target, adverse events) results were expressed as risk ratio (RR) with 95% confidence intervals (CI). Where continuous scales of measurement were used to assess the effects of treatment (life participation, fatigue) the mean difference (MD) was used, or the standardised mean difference (SMD) if different scales have been used. Studies analysing change scores were included in meta‐analyses together with studies including endpoint outcome data. Missing standard deviations were imputed.
Unit of analysis issues
For cross‐over studies, we extracted data for the end of the first period of treatment.
Dealing with missing data
We requested any further information required from the original author by written correspondence (e.g. emailing corresponding author/s) and any relevant information obtained in this manner were included in the review. Evaluation of important numerical data such as screened, randomised patients, as well as intention‐to‐treat, as‐treated and per‐protocol population, were carefully performed. Attrition rates, for example, drop‐outs, losses to follow‐up and withdrawals were investigated. Issues of missing data and imputation methods (e.g., last‐observation‐carried‐forward) were critically appraised (Higgins 2020).
Assessment of heterogeneity
We first assessed the heterogeneity by visual inspection of the forest plot. We quantified statistical heterogeneity using the I² statistic, which describes the percentage of total variation across studies due to heterogeneity rather than sampling error (Higgins 2003). A guide to the interpretation of I² values was as follows.
0% to 40%: might not be important
30% to 60%: may represent moderate heterogeneity
50% to 90%: may represent substantial heterogeneity
75% to 100%: considerable heterogeneity.
The importance of the observed value of I² depends on the magnitude and direction of treatment effects and the strength of evidence for heterogeneity (e.g. P‐value from the Chi² test or a CI for I²) (Higgins 2020).
Assessment of reporting biases
If possible, funnel plots were used to assess the potential existence of small study bias (Higgins 2020). We planned to generate funnel plots if at least 10 studies examining the same treatment comparison were included in the review and comment on whether any asymmetry in the funnel plot was due to publication bias or methodological or clinical heterogeneity of the studies.
Data synthesis
Data were pooled using the random‐effects model, but the fixed‐effect model was also used to ensure the robustness of the model chosen and susceptibility to outliers.
Subgroup analysis and investigation of heterogeneity
We used subgroup analyses to explore possible sources of heterogeneity. Heterogeneity among participants could be related to the stage of kidney disease (stage 3‐5 not requiring KRT, dialysis, kidney transplant) and the presence of comorbidities (CV disease, diabetes). Heterogeneity in treatments could be related to a prior agent(s) used, the Hb target during therapy, the type, and the frequency and the duration of therapy. Adverse effects were tabulated and assessed with descriptive techniques, as they were likely to be different for the various agents used. Where possible, the risk difference (RD) with 95% CI was calculated for each adverse effect, either compared to no treatment or another agent. Studies comparing different doses and phase 1 and 2 studies were investigated using subgroup analysis.
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the following factors on effect size:
Repeating the analysis, excluding unpublished studies
Repeating the analysis taking into account the risk of bias, as specified
Repeating the analysis, excluding any very long or large studies to establish how much they dominate the results
Repeating the analysis excluding studies using the following filters: diagnostic criteria, language of publication, source of funding (industry versus other), and country.
Summary of findings and assessment of the certainty of the evidence
We presented the main results of the review in 'Summary of findings' tables. These tables present key information concerning the certainty of the evidence, the magnitude of the effects of the interventions examined, and the sum of the available data for the main outcomes (Schunemann 2020a). The 'Summary of findings' tables also included an overall grading of the evidence related to each of the main outcomes using the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) approach (GRADE 2008; GRADE 2011). The GRADE approach defines the certainty of a body of evidence as the extent to which one can be confident that an estimate of effect or association is close to the true quantity of specific interest. This was assessed by three authors (PN, EH, MR). The certainty of a body of evidence involves consideration of the within‐trial risk of bias (methodological quality), directness of evidence, heterogeneity, the precision of effect estimates and risk of publication bias (Schunemann 2020b). We reported the following outcomes in the 'Summary of findings' tables.
CV death
Fatigue
Life participation
Nonfatal MI
Nonfatal stroke
Proportion of patients requiring blood transfusion
Proportion of patients reaching the target Hb
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies
Results of the search
After searching the Specialised Register, we identified 173 records. After screening titles, abstracts, and undertaking full‐text review, 51 studies (117 records) were included, and 31 studies (47 records) were excluded. Eight ongoing studies were identified (ASCEND‐FBF 2018; CTRI/2019/06/019635; DREAM‐D 2019; NCT04027517; NCT04134026; NCT04313153; PER‐038‐14; SLCTR‐2019‐032) and one study was completed prior to publication; however, no results are as yet available (FO2RWARD‐2 2019). These nine studies will be assessed in a future update of this review (Figure 1).
1.
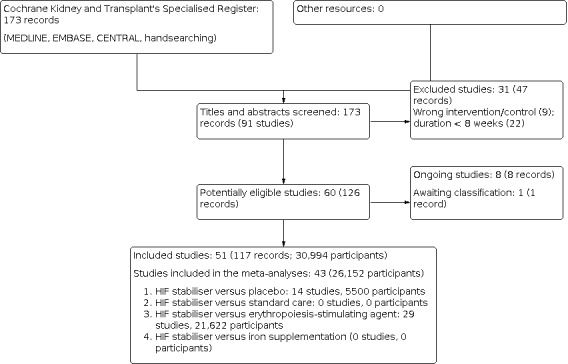
Flow diagram.
Included studies
We included 51 studies (117 records), randomising 30,994 participants. The characteristics of the participants and the interventions used are detailed in the Characteristics of included studies.
Study design, setting and characteristics
Study duration varied from 8 to 108 weeks, with a median of 28 weeks. No study had a cross‐over or cluster‐randomised design. Studies were conducted from 2013 to 2021 in China (Chen 2019; Chen 2019a; Chen DD 2017; Chen NDD 2017), Japan (Akizawa 2017; Akizawa 2019; Akizawa 2020a; Akizawa 2020c; Akizawa 2020f; Akizawa 2021; Hou 2021; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; SYMPHONY HD 2021; SYMPHONY ND 2021), and the USA (Besarab 2015; Pergola 2016; Provenzano 2008; Provenzano 2016; Provenzano 2016a), or were multinational (ASCEND‐D 2021; ASCEND‐ID 2021; ASCEND‐ND 2021; ASCEND‐NHQ 2021; ASCEND‐TD 2021; ALPS 2021; ANDES 2021; Brigandi 2016; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; INNO2VATE 2020; INNO2VATE 2020a; Meadowcroft 2019; OLYMPUS 2021; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; PYRENEES 2021; ROCKIES 2019; SIERRAS 2021). All but three studies (Akizawa 2020f; Hou 2021; SYMPHONY ND 2021) received at least some funding from pharmaceutical companies. No studies were phase 1 studies, 19 studies (Akizawa 2017; Akizawa 2019; Besarab 2015; Brigandi 2016; Chen DD 2017; Chen NDD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; Holdstock 2019; Holdstock 2019a; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; Pergola 2016; Provenzano 2008; Provenzano 2016; Provenzano 2016a; SIERRAS 2021) were phase 2 studies, and 32 studies were phase 3 studies.
Study participants
The sample size varied from 51 (NDD‐CKD 2020) to 3872 participants (ASCEND‐ND 2021) (median of 223 participants). The mean study age ranged from 48 years (Chen 2019) to 72 years (MIYABI ND‐C 2019; Nangaku 2021a) (median 63 years). No studies evaluated treatment in children or in recipients of a kidney transplant.
Twenty‐five studies in people with CKD stages 3 to 5 not treated with dialysis (Akizawa 2019; Akizawa 2020f; ALPS 2021; ANDES 2021; ASCEND‐ND 2021; ASCEND‐NHQ 2021; Besarab 2015; Chen 2019a; Chen DD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; DOLOMITES 2021; Holdstock 2019; Holdstock 2019a; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021a; Nangaku 2021b; NDD‐CKD 2020; OLYMPUS 2021; Pergola 2016; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; Provenzano 2008; SYMPHONY ND 2021), one study in people with CKD stages 3‐5 including 5D (Brigandi 2016), 14 studies in people treated with haemodialysis (HD) (Akizawa 2017; Akizawa 2020a; Akizawa 2020c; ASCEND‐TD 2021; Chen DD 2017; DIALOGUE 4 2019; Meadowcroft 2019; MIYABI HD‐M 2019; Nangaku 2021; NCT01888445; NDD‐CKD 2020a; Provenzano 2016; Provenzano 2016a; SYMPHONY HD 2021), one study in people with PD (Hou 2021), and 10 studies (ASCEND‐D 2021; ASCEND‐ID 2021; Chen 2019; HIMALAYAS 2021; INNO2VATE 2020; INNO2VATE 2020a; HIMALAYAS 2021; PYRENEES 2021; ROCKIES 2019; SIERRAS 2021) included people treated with HD and PD.
Seventeen studies reported information regarding the baseline eGFR in participants (Akizawa 2019; Akizawa 2021; ANDES 2021; Besarab 2015; Chen 2019a; Chen NDD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; Holdstock 2019; Holdstock 2019a; Nangaku 2021a; NDD‐CKD 2020; OLYMPUS 2021; Pergola 2016; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; SYMPHONY ND 2021).
Fourteen studies enrolled people who were prescribed concomitant ESA (Akizawa 2020a; Akizawa 2020c; Akizawa 2020f; ASCEND‐D 2021; ASCEND‐ID 2021; ASCEND‐ND 2021; ASCEND‐TD 2021; DIALOGUE 2 2019; DIALOGUE 4 2019; Hou 2021; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; SIERRAS 2021), three studies enrolled people who were prescribed iron supplements (Besarab 2015; Chen 2019a; NCT01888445), and four studies enrolled people who were also prescribed ESA, iron supplements or both (Akizawa 2017; PRO2TECT‐CORRECTION 2021; SYMPHONY HD 2021; SYMPHONY ND 2021). One study enrolled people who did not receive ESA (ASCEND‐NHQ 2021).
The target Hb levels in the included studies were as follows:
One study reported the Hb target level ≥ 10 g/dL or an increase of at least 1.0 g/dL in people with a baseline Hb of 8.0 g/dL or more or an increase of at least 2.0 g/dL in people with a baseline Hb of less than 8.0 g/dL (Chen 2019a)
Four studies reported the Hb target increase ≥ 1 g/dL (Besarab 2015; Chen 2019; Chen DD 2017; Provenzano 2008)
One study reported either an increase in Hb ≥ 1 g/dL or Hb target ≥ 11.0 g/dL (Chen NDD 2017)
One study reported an increase in Hb of 0.5 to 1.0 g/dL (Brigandi 2016)
One study reported the Hb target increase of at least 0.5 to 2.0 g/dL (Akizawa 2017)
Five studies reported the Hb target was10 to 11 g/dL (ASCEND‐D 2021; ASCEND‐ID 2021; ASCEND‐ND 2021; ASCEND‐TD 2021; DIALOGUE 4 2019)
Two studies reported the Hb target was 10 to 11.5 g/dL (INNO2VATE 2020; Meadowcroft 2019)
Seventeen studies reported the Hb target was 10 to 12 g/dL (Akizawa 2019; Akizawa 2020a; Akizawa 2020c; Akizawa 2021; DIALOGUE 2 2019; DOLOMITES 2021; MIYABI HD‐M 2019; MIYABI ND‐M 2019; Nangaku 2021; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; OLYMPUS 2021; PYRENEES 2021; ROCKIES 2019; SYMPHONY HD 2021; SYMPHONY ND 2021)
One study reported the Hb target was 10 to 12 g/dL but also an increase of at least 1 g/dL (SIERRAS 2021)
Two studies reported the Hb target was 11 g/dL and an increase of 1 g/dL if baseline Hb was > 8 g/dL or an increase of 2 g/dL if baseline Hb was < 8 g/dL (ANDES 2021; HIMALAYAS 2021)
One study reported both an increase in Hb ≥ 1 g/dL or Hb target ≥ 11.0 g/dL (ALPS 2021)
Five studies reported the Hb target 11 to 13 g/dL (MIYABI ND‐M 2019; Nangaku 2021a; Nangaku 2021b; Provenzano 2016; Provenzano 2016a)
One study reported the Hb target was 11 to 12 g/dL and an increase of 1 g/dL (ASCEND‐NHQ 2021)
One study reported the Hb target was 8 to 11 g/dL (Holdstock 2019a)
One study reported the Hb target was 9 to 10.5 g/dL (Holdstock 2019)
One study reported the Hb target was ≥ 11.0 g/dL (Pergola 2016)
Three studies reported the Hb target in the USA was ≥ 10 to 11 g/dL and in the non‐USA countries was ≥ 10 to 12 g/dL (INNO2VATE 2020a; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021)
Three studies did not report a Hb target (Akizawa 2020f; ASCEND‐D 2021; DIALOGUE 1 2019).
Interventions
One study (Chen NDD 2017) compared three arms, including different doses of HIF stabiliser and placebo
Seven studies (Akizawa 2019; Chen DD 2017; DIALOGUE 2 2019; Holdstock 2019; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a) included different doses of HIF stabiliser and ESA (Chen DD 2017; DIALOGUE 2 2019; Holdstock 2019; NCT01888445), or different doses of HIF stabiliser and placebo (Akizawa 2019; NDD‐CKD 2020; NDD‐CKD 2020a)
Five studies (Akizawa 2017; Besarab 2015; Brigandi 2016; DIALOGUE 4 2019; Provenzano 2016) compared five arms, including different doses of HIF stabiliser and ESA (DIALOGUE 4 2019; Provenzano 2016), or different doses of HIF stabiliser and placebo (Akizawa 2017; Besarab 2015; Brigandi 2016)
One study (DIALOGUE 1 2019) compared six arms, including different doses of HIF stabiliser and placebo
One study (Provenzano 2016a) compared seven arms, including different doses of HIF stabiliser and ESA
Forty‐three studies (Akizawa 2017; Akizawa 2019; Akizawa 2020a; Akizawa 2020c; Akizawa 2021; ASCEND‐D 2021; ASCEND‐ND 2021; ALPS 2021; ANDES 2021; Besarab 2015; Brigandi 2016; Chen 2019; Chen 2019a; Chen DD 2017; Chen NDD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; Hou 2021; INNO2VATE 2020; INNO2VATE 2020a; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; OLYMPUS 2021; Pergola 2016; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; Provenzano 2008; PYRENEES 2021; SIERRAS 2021; SYMPHONY HD 2021; SYMPHONY ND 2021) (26,152 participants) were included in the meta‐analyses.
HIF stabilisers versus placebo
Sixteen studies (6330 participants) compared HIF stabiliser to placebo; 14 studies (5500 participants) could be meta‐analysed.
Daprodustat (2 studies, 147 participants) (Akizawa 2017; Brigandi 2016)
FG2216 (1 study, 142 participants) (Provenzano 2008)
Molidustat (1 study, 121 participants) (DIALOGUE 1 2019)
Roxadustat (7 studies, 4769 participants) (Akizawa 2019; ALPS 2021; ANDES 2021; Besarab 2015; Chen 2019a; Chen NDD 2017; OLYMPUS 2021)
Vadadustat (3 studies, 321 participants) (NDD‐CKD 2020; NDD‐CKD 2020a; Pergola 2016)
HIF stabilisers versus standard care
No studies compared HIF stabilisers to standard care.
HIF stabilisers versus erythropoiesis‐stimulating agent
Thirty‐four studies (23,141 participants) compared HIF stabilisers to ESA; 29 studies (21,406 participants) could be meta‐analysed.
Daprodustat versus not specified EPO (2 studies, 252 participants) (Holdstock 2019; Holdstock 2019a)
Daprodustat versus darbepoetin alfa (3 studies, 4759 participants) (Akizawa 2020c; ASCEND‐ND 2021; DOLOMITES 2021)
Daprodustat versus darbepoetin alfa or EPO alfa (1 study, 2964 participants) (ASCEND‐D 2021)
Daprodustat versus mircera (1 study, 299 participants) (Nangaku 2021b)
Enarodustat versus darbepoetin alfa (2 studies, 389 participants) (SYMPHONY HD 2021; SYMPHONY ND 2021)
Molidustat versus epoetin alfa and beta (1 study, 199 participants) (DIALOGUE 4 2019)
Molidustat versus darbepoetin alfa (4 studies, 679 participants) (DIALOGUE 2 2019; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019)
Roxadustat versus epoetin alfa (4 studies, 2176 participants) (Chen 2019; Chen DD 2017; HIMALAYAS 2021; SIERRAS 2021)
Roxadustat versus darbepoetin alfa (3 studies, 696 participants) (Akizawa 2020a; Akizawa 2021; NCT01888445)
Roxadustat versus epoetin alfa and darbepoetin alfa (1 study, 838 participants) (PYRENEES 2021)
Roxadustat versus not specified ESA (1 study, 129 participants) (Hou 2021)
Vadadustat versus darbepoetin alfa (6 studies, 8026 participants) (INNO2VATE 2020; INNO2VATE 2020a; Nangaku 2021; Nangaku 2021a; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021).
HIF stabilisers versus iron supplementation
No studies compared HIF stabilisers with iron supplementation.
Excluded studies
We excluded 31 studies. The reasons for exclusion were:
Follow‐up less than eight weeks (22 studies: Akizawa 2019a; Akizawa 2019b; ASCEND:Fe 2018; ASCEND‐BP 2017; Bailey 2019; Buch 2014; DD‐CKD 2020; EudraCT2012‐004049‐34; EudraCT2012‐004050‐29; EudraCT2015‐004790‐32; Frohna 2007; Hartman 2014; Holdstock CKD 2016; Holdstock HD 2016; Martin 2017; NCT01971164; NCT03992066; Pai 2015; Parmar 2019; Provenzano 2011; Provenzano 2011a; Wiecek 2005)
Wrong interventions (9 studies: Akizawa 2015a; Akizawa 2020g; Akizawa 2020; Akizawa 2020b; Besarab 2016; Haase 2016; NCT01679587; NCT04059913; Provenzano 2016b).
Ongoing studies
Our search identified eight studies that have yet to be completed.
Daprodustat versus darbepoetin alfa (ASCEND‐FBF 2018)
Desidustat versus darbepoetin (CTRI/2019/06/019635; SLCTR‐2019‐032)
Desidustat versus epoetin alfa (DREAM‐D 2019)
Enarodustat versus darbepoetin alfa (NCT04027517)
Roxadustat versus epoetin alfa (NCT04134026; PER‐038‐14)
Vadadustat versus darbepoetin alfa (NCT04313153)
Risk of bias in included studies
The risk of bias for studies overall are summarised in Figure 2 and the risk of bias in each study is shown in Figure 3.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
Methods for generating the random sequence were at low risk of bias in 22 studies (Akizawa 2019; Akizawa 2020a; Akizawa 2020c; ANDES 2021; ASCEND‐D 2021; ASCEND‐ND 2021; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; Meadowcroft 2019; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021a; Nangaku 2021b; NCT01888445; OLYMPUS 2021; SIERRAS 2021; SYMPHONY ND 2021). The method for generating the random sequence was unclear in 29 studies.
Allocation concealment
Allocation concealment was at low risk of bias in 19 studies (Akizawa 2020a; Akizawa 2020c; ANDES 2021; ASCEND‐D 2021; ASCEND‐ND 2021; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; Meadowcroft 2019; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021b; OLYMPUS 2021; SIERRAS 2021; SYMPHONY ND 2021). The risk of bias for allocation concealment was unclear in 32 studies.
Blinding
Performance bias
Six studies (Akizawa 2019; ANDES 2021; DIALOGUE 1 2019; Meadowcroft 2019; MIYABI HD‐M 2019; OLYMPUS 2021) included blinding to treatment allocation for participants and investigators. Thirty‐two studies (Akizawa 2020f; Akizawa 2021; ASCEND‐D 2021; ASCEND‐ID 2021; ASCEND‐ND 2021; Besarab 2015; Brigandi 2016; Chen 2019; Chen DD 2017; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; Hou 2021; INNO2VATE 2020; INNO2VATE 2020a; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021a; Nangaku 2021b; NCT01888445; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; Provenzano 2008; Provenzano 2016; Provenzano 2016a; PYRENEES 2021; ROCKIES 2019; SIERRAS 2021; SYMPHONY ND 2021) were not blinded to treatment allocation for participants and investigators. The risk of performance bias was unclear in 13 studies.
Detection bias
Eight studies (ASCEND‐ND 2021; DOLOMITES 2021; Holdstock 2019; Holdstock 2019a; Meadowcroft 2019; Nangaku 2021b; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021) assessed outcomes based on objective laboratory assessments and were at low risk of bias. Thirty‐seven studies (Akizawa 2017; Akizawa 2019; Akizawa 2020a; Akizawa 2020c; Akizawa 2020f; Akizawa 2021; ALPS 2021; ANDES 2021; ASCEND‐ID 2021; ASCEND‐NHQ 2021; ASCEND‐TD 2021; Besarab 2015; Brigandi 2016; Chen 2019; Chen 2019a; Chen DD 2017; Chen NDD 2017; HIMALAYAS 2021; Hou 2021; INNO2VATE 2020; INNO2VATE 2020a; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; OLYMPUS 2021; Pergola 2016; Provenzano 2008; PYRENEES 2021; ROCKIES 2019; SIERRAS 2021; SYMPHONY HD 2021; SYMPHONY ND 2021) were at high risk of bias for blinding of outcome assessment in reporting patient‐centred outcomes, including adverse events. Six studies were considered at unclear risk of bias.
Incomplete outcome data
Twelve studies (Akizawa 2020c; ANDES 2021; ASCEND‐ID 2021; Besarab 2015; Brigandi 2016; Chen 2019; Chen 2019a; Chen NDD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; NDD‐CKD 2020) were at low risk of attrition bias. Seventeen studies (Akizawa 2019; Akizawa 2020f; Akizawa 2021; ALPS 2021; ASCEND‐NHQ 2021; ASCEND‐TD 2021; Chen DD 2017; Holdstock 2019; Holdstock 2019a; Meadowcroft 2019; NCT01888445; NDD‐CKD 2020a; Provenzano 2008; Provenzano 2016; Provenzano 2016a; PYRENEES 2021; ROCKIES 2019) were at high risk of attrition bias as there was a differential loss to follow‐up between treatment groups and/or high attrition rates in both treatment groups. Loss to follow‐up was commonly due to withdrawal from the study or adverse events. The risk of attrition bias was unclear in 22 studies.
Selective reporting
Twenty studies (Akizawa 2019; Akizawa 2020a; ASCEND‐D 2021; ASCEND‐ND 2021; Chen 2019; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; Holdstock 2019; Holdstock 2019a; Hou 2021; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021a; NCT01888445; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; SIERRAS 2021) reported expected and clinically‐relevant outcomes and were at low risk of bias. Thirty‐one studies did not report patient‐centred outcomes of death or adverse events.
Other potential sources of bias
One study (Hou 2021) was assessed to be at low risk of bias, 49 studies (Akizawa 2017; Akizawa 2019; Akizawa 2020a; Akizawa 2020c; Akizawa 2021; ALPS 2021; ANDES 2021; ASCEND‐D 2021; ASCEND‐ID 2021; ASCEND‐ND 2021; ASCEND‐NHQ 2021; ASCEND‐TD 2021; Besarab 2015; Brigandi 2016; Chen 2019; Chen 2019a; Chen DD 2017; Chen NDD 2017; DIALOGUE 1 2019; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; INNO2VATE 2020; INNO2VATE 2020a; Meadowcroft 2019; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; NCT01888445; NDD‐CKD 2020; NDD‐CKD 2020a; OLYMPUS 2021; Pergola 2016; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; Provenzano 2008; Provenzano 2016; Provenzano 2016a; PYRENEES 2021; ROCKIES 2019; SIERRAS 2021; SYMPHONY HD 2021; SYMPHONY ND 2021) were assessed to be at high risk of bias due to the potential role of funding, and one study was assessed as unclear risk of bias for this domain (Akizawa 2020f).
Effects of interventions
HIF stabiliser versus placebo
Fourteen studies (Akizawa 2017; Akizawa 2019; ALPS 2021; ANDES 2021; Besarab 2015; Brigandi 2016; Chen 2019a; Chen NDD 2017; DIALOGUE 1 2019; NDD‐CKD 2020; NDD‐CKD 2020a; OLYMPUS 2021; Pergola 2016; Provenzano 2008) compared HIF stabiliser management of anaemia versus placebo in patients with CKD (stages 3, 4 or 5), including patients undergoing HD, during a median follow‐up of 17 weeks. The certainty of the evidence was mainly low or very low (Table 1).
Primary outcomes
Cardiovascular death
Compared to placebo, HIF stabiliser therapy had uncertain effects on CV death (Analysis 1.1 (10 studies, 1114 participants): RR 3.68, 95% CI 0.19 to 70.21; very low certainty evidence) in people with CKD or undergoing HD.
1.1. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 1: Cardiovascular death
Secondary outcomes
Death (any cause)
Compared to placebo, HIF stabiliser therapy may make little or no difference to death (any cause) (Analysis 1.2 (12 studies, 4469 participants): RR 1.12, 95% CI 0.97 to 1.30; I² = 0%; low certainty evidence) in people with CKD or undergoing HD.
1.2. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 2: Death (any cause)
Myocardial infarction
The effect of HIF stabiliser treatment on nonfatal MI was uncertain (Analysis 1.3 (3 studies, 822 participants): RR 1.29, 95% CI 0.31 to 5.36; I² = 0%; very low certainty evidence) compared with placebo in people with CKD.
1.3. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 3: Nonfatal myocardial infarction
When MI was reported as fatal or nonfatal events, HIF stabiliser therapy may make little or no difference to the numbers with fatal or nonfatal MI (Analysis 1.4 (5 studies, 4499 participants): RR 1.06, 95% CI 0.59 to 1.90; I² = 0%; low certainty evidence) compared with placebo in people with CKD.
1.4. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 4: Fatal or nonfatal myocardial infarction (overall)
Stroke
HIF stabiliser treatment had uncertain effects on nonfatal stroke (Analysis 1.5: 2 studies, 228 participants), as no events were reported in these two studies.
1.5. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 5: Nonfatal stroke
When stroke was reported as a fatal or nonfatal event, the effects of HIF stabiliser therapy on fatal or nonfatal stroke were uncertain (Analysis 1.6 (3 studies, 822 participants): RR 2.08, 95% CI 0.23 to 18.46; very low certainty evidence) compared with placebo in people with CKD.
1.6. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 6: Fatal or nonfatal stroke (overall)
Peripheral arterial events
DIALOGUE 1 2019 reported HIF stabilisers had uncertain effects on peripheral arterial events (Analysis 1.7 (1 study, 121 participants): RR 0.20, 95% CI 0.01 to 3.04) compared with placebo in people with CKD.
1.7. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 7: Peripheral arterial events
Proportion of patients requiring blood transfusion
HIF stabiliser treatment probably decreases the proportion of patients requiring blood transfusion (Analysis 1.8 (8 studies, 4329 participants): RR 0.51, 95% CI 0.44 to 0.60; I² = 0%; moderate certainty evidence) compared with placebo in people with CKD or undergoing HD.
1.8. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 8: Transfusion
Proportion of patients reaching the target haemoglobin
HIF stabiliser therapy probably increases the proportion of patients reaching the target Hb (Analysis 1.9 (10 studies, 5102 participants): RR 8.36, 95% CI 6.42 to 10.89; I² = 37%; moderate certainty evidence) compared to placebo in people with CKD or undergoing HD.
1.9. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 9: Proportion reaching target haemoglobin
Kidney failure
HIF stabiliser therapy may make little or no difference to kidney failure (Analysis 1.10 (8 studies, 2228 participants): RR 1.22, 95% CI 0.98 to 1.51; I² = 0%; low certainty evidence) compared to placebo in people with CKD.
1.10. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 10: Kidney failure
Thrombosis
HIF stabiliser therapy may increase thrombosis (Analysis 1.11 (3 studies, 3452 participants): RR 2.36, 95% CI 1.19 to 4.66; I² = 0%; low certainty evidence) compared to placebo in people with CKD or undergoing HD.
1.11. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 11: Thrombosis
Loss of unassisted dialysis vascular access patency
The effect of HIF stabiliser therapy on the loss of unassisted dialysis vascular access patency was uncertain (Analysis 1.12 (2 studies, 157 participants): RR 1.18, 95% CI 0.13 to 10.31; I² = 0%; very low certainty evidence) compared to placebo in people undergoing HD.
1.12. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 12: Loss of unassisted patency (stenosis)
Hyperkalaemia
HIF stabiliser therapy may increase hyperkalaemia (Analysis 1.13 (7 studies, 4845 participants): RR 1.29, 95% CI 1.01 to 1.64; I² = 18%; low certainty evidence) compared to placebo in people with CKD.
1.13. Analysis.

Comparison 1: Hypoxia‐inducible factor (HIF) stabiliser versus placebo, Outcome 13: Hyperkalaemia
Subgroup analyses for HIF stabiliser versus placebo
Additional analyses were performed stratifying by stage of CKD.
-
CV death
CKD: Analysis 2.1.1 (7 studies, 850 participants: RR 3.68, 95% CI 0.19 to 70.21; very low certainty evidence)
HD: Analysis 2.1.2 (2 studies, 157 participants: no reported events)
CKD and HD: Analysis 2.1.3 (1 study, 107 participants: no reported events)
-
Nonfatal MI
CKD: Analysis 2.2.1 (3 studies, 822 participants: RR 1.29, 95% CI 0.31 to 5.36; I² = 0%; very low certainty evidence)
-
Proportion of patients requiring blood transfusion
CKD: Analysis 2.3.1 (7 studies, 4271 participants: RR 0.52, 95% CI 0.44 to 0.60; I² = 0%; moderate certainty evidence)
HD: Analysis 2.3.2 (1 study, 58 participants: RR 0.21, 95% CI 0.04 to 1.14, very low certainty evidence)
-
Proportion of patients reaching the target Hb
CKD: Analysis 2.4.1 (8 studies, 4931 participants: RR 8.18, 95% CI 6.13 to 10.93; I² = 50%, low certainty evidence)
CKD and HD: Analysis 2.4.2 (2 studies, 171 participants: RR 14.35, 95% CI 2.07 to 99.61; I² = 0%, very low certainty evidence)
2.1. Analysis.

Comparison 2: Analyses for SOF table 1 stratifying by CKD stage (HIF versus placebo), Outcome 1: Cardiovascular death
2.2. Analysis.

Comparison 2: Analyses for SOF table 1 stratifying by CKD stage (HIF versus placebo), Outcome 2: Nonfatal myocardial infarction
2.3. Analysis.

Comparison 2: Analyses for SOF table 1 stratifying by CKD stage (HIF versus placebo), Outcome 3: Transfusion
2.4. Analysis.

Comparison 2: Analyses for SOF table 1 stratifying by CKD stage (HIF versus placebo), Outcome 4: Proportion reaching target haemoglobin
Other subgroup analyses were not possible due to the limited number of studies and data.
Sensitivity analysis for HIF stabiliser versus placebo
Sensitivity analyses did not provide substantively different results or were not possible due to few data and studies.
HIF stabiliser versus erythropoiesis‐stimulating agent
Twenty‐nine studies (Akizawa 2020a; Akizawa 2020c; Akizawa 2021; ASCEND‐D 2021; ASCEND‐ND 2021; Chen 2019; Chen DD 2017; DIALOGUE 2 2019; DIALOGUE 4 2019; DOLOMITES 2021; HIMALAYAS 2021; Holdstock 2019; Holdstock 2019a; Hou 2021; INNO2VATE 2020; INNO2VATE 2020a; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; NCT01888445; PYRENEES 2021; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; SIERRAS 2021; SYMPHONY HD 2021; SYMPHONY ND 2021) compared HIF stabiliser versus ESA management of anaemia in adults with CKD (stages 3, 4 or 5), including patients undergoing HD and PD, during a median follow‐up of 52 weeks. The certainty of the evidence was mainly moderate or low (Table 2).
Primary outcomes
Cardiovascular death
HIF stabiliser therapy may make little or no difference to CV death (Analysis 3.1 (17 studies, 10,340 participants): RR 1.05, 95% CI 0.88 to 1.26; I² = 0%; low certainty evidence) compared to ESA in people with CKD, or those undergoing HD or PD.
3.1. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 1: Cardiovascular death
Fatigue
The effect of HIF stabiliser management of anaemia on fatigue was uncertain (Analysis 3.2 (2 studies, 3471 participants): RR 0.80, 95% CI 0.56 to 1.16; I² = 0%; very low certainty evidence) compared with ESA in people with CKD.
3.2. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 2: Fatigue
Secondary outcomes
Death (any cause)
HIF stabiliser therapy probably makes little or no difference to death (any cause) (Analysis 3.3 (29 studies, 21,370 participants): RR 0.98, 95% CI 0.91 to 1.06; I² = 0%; moderate certainty evidence) compared to ESA in people with CKD, or those undergoing HD or PD.
3.3. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 3: Death (any cause)
Myocardial infarction
HIF stabiliser treatment may make little or no difference to nonfatal MI (Analysis 3.4 (7 studies, 7765 participants): RR 0.91, 95% CI 0.76 to 1.10; I² = 0%; low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.4. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 4: Nonfatal myocardial infarction
When MI was reported as fatal or nonfatal events, HIF stabiliser treatment probably makes little or no difference to fatal or nonfatal MI (Analysis 3.5 (15 studies, 14,183 participants): RR 0.95, 95% CI 0.80 to 1.12; I² = 0%; moderate certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.5. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 5: Fatal or nonfatal myocardial infarction (overall)
Stroke
HIF stabiliser treatment may make little or no difference to nonfatal stroke (Analysis 3.6 (5 studies, 7285 participants): RR 1.06, 95% CI 0.71 to 1.56; I² = 8%; low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.6. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 6: Nonfatal stroke
When stroke was reported as a fatal or nonfatal event, HIF stabiliser treatment may make little or no difference to fatal or nonfatal stroke (Analysis 3.7 (7 studies, 8025 participants): RR 0.95, 95% CI 0.64 to 1.40; I² = 23%; low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.7. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 7: Fatal or nonfatal stroke (overall)
Hospitalisation for heart failure
The effect of HIF stabiliser therapy on nonfatal hospitalisation for heart failure was uncertain (Analysis 3.8 (2 studies, 6836 participants): RR 1.23, 95% CI 1.00 to 1.52; I² = 0%; very low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.8. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 8: Nonfatal hospitalisation for heart failure
The effect of HIF stabiliser therapy on fatal or nonfatal hospitalisation for heart failure was uncertain (Analysis 3.9 (3 studies, 7452 participants): RR 1.15, 95% CI 0.97 to 1.36; I² = 0%; very low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.9. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 9: Fatal or nonfatal hospitalisation for heart failure
Peripheral arterial events
HIF stabiliser management of anaemia had uncertain effects on peripheral arterial events (Analysis 3.10: 2 studies, 323 participants), as no events were reported in the included studies.
3.10. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 10: Peripheral arterial event
Proportion of patients requiring blood transfusion
HIF stabiliser treatment probably decreases the proportion of patients requiring blood transfusion (Analysis 3.11 (11 studies, 10,786 participants): RR 0.87, 95% CI 0.76 to 1.00; I² = 25%; moderate certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD.
3.11. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 11: Transfusion
Proportion of patients reaching the target haemoglobin
HIF stabiliser treatment may make little or no difference to the proportion of patients reaching the target Hb (Analysis 3.12 (14 studies, 4601 participants): RR 1.00, 95% CI 0.93 to 1.07; I² = 70%; low certainty evidence) compared with ESA in people with CKD, or those undergoing HD or PD. There was moderate to high heterogeneity among the studies.
3.12. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 12: Proportion reaching target haemoglobin
Kidney failure
HIF stabiliser treatment may make little or no difference to kidney failure (Analysis 3.13 (9 studies, 7312 participants): RR 1.02, 95% CI 0.91 to 1.15; I² = 0%; low certainty evidence) compared with ESA in people with CKD.
3.13. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 13: Kidney failure
Thrombosis
HIF stabiliser therapy may make little or no difference to thrombosis (Analysis 3.14 (11 studies, 17,026 participants): RR 1.09, 95% CI 0.86 to 1.39; I² = 46%; low certainty evidence) compared with ESA in people with CKD, HD and PD. There was moderate heterogeneity among the studies.
3.14. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 14: Thrombosis
Loss of unassisted dialysis access patency
HIF stabiliser therapy may have little or no difference on the loss of unassisted dialysis access patency (including stenosis and occlusions) (Analysis 3.15 (8 studies, 2945 participants): RR 1.16, 95% CI 0.85 to 1.59; I² = 0%; low certainty evidence) compared with ESA in people undergoing HD.
3.15. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 15: Loss of unassisted patency (occlusion/stenosis)
Access intervention
MIYABI ND‐C 2019 reported that HIF stabiliser therapy made no difference to access interventions (Analysis 3.16 (1 study, 161 participants): RR 0.58, 95% CI 0.14 to 2.34) compared with ESA in people with CKD.
3.16. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 16: Access intervention
Cancer
HIF stabiliser therapy may make little or no difference to the number with cancer (Analysis 3.17 (7 studies, 1687 participants): RR 0.83, 95% CI 0.43 to 1.59; I² = 8%; low certainty evidence) compared with ESA in people with CKD or those undergoing HD.
3.17. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 17: Cancer
Infection
Chen 2019 reported that HIF stabiliser treatment made no difference in the number with infection (Analysis 3.18 (1 study, 304 participants): RR 0.82, 95% CI 0.20 to 3.35) compared with ESA in people with HD and PD.
3.18. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 18: Infection
Hyperkalaemia
Twenty‐one studies (Akizawa 2020a; Akizawa 2020c; Akizawa 2021; ASCEND‐D 2021; ASCEND‐ND 2021; Chen 2019; DOLOMITES 2021; HIMALAYAS 2021; Hou 2021; INNO2VATE 2020; INNO2VATE 2020a; MIYABI HD‐M 2019; MIYABI ND‐C 2019; MIYABI ND‐M 2019; Nangaku 2021; Nangaku 2021a; Nangaku 2021b; PRO2TECT‐CONVERSION 2021; PRO2TECT‐CORRECTION 2021; PYRENEES 2021; SIERRAS 2021) reported hyperkalaemia without providing a clear definition.
HIF stabiliser treatment probably makes little or no difference to hyperkalaemia (Analysis 3.19 (21 studies, 20,177 participants): RR 0.92, 95% CI 0.82 to 1.04; I² = 10%; moderate certainty evidence) compared with ESA in people with CKD or those undergoing HD or PD.
3.19. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 19: Hyperkalaemia
Pulmonary hypertension
The effect of HIF stabiliser treatment on pulmonary hypertension was uncertain (Analysis 3.20 (7 studies, 8641 participants): RR 1.06, 95% CI 0.56 to 2.01; I² = 11%; very low certainty evidence) compared with ESA in people with CKD and HD.
3.20. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 20: Pulmonary hypertension
Diabetic retinopathy
HIF stabiliser therapy may make little or no difference to the number with diabetic retinopathy (Analysis 3.21 (8 studies, 5036 participants): RR 1.26, 95% CI 0.71 to 2.22; I² = 0%; low certainty evidence) compared with ESA in people with CKD or those undergoing HD.
3.21. Analysis.

Comparison 3: Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), Outcome 21: Diabetic retinopathy
Subgroup analyses for HIF stabiliser versus ESA
Additional analyses to compare HIF stabiliser management versus ESA were performed stratifying by stage of CKD.
-
CV death
CKD: Analysis 4.1.1 (7 studies, 5591 participants: RR 1.19, 95% CI 0.91 to 1.55; I² = 0%; low certainty evidence)
HD: Analysis 4.1.2 (7 studies, 1352 participants: RR 0.98, 95% CI 0.23 to 4.19; I² = 0%; low certainty evidence)
PD: Analysis 4.1.3 (1 study, 129 participants: RR 0.50, 95% CI 0.03 to 7.80)
HD and PD: Analysis 4.1.4 (2 studies, 3268 participants: RR 0.96, 95% CI 0.75 to 1.23; low certainty evidence)
-
Fatigue
CKD: Analysis 4.2.1 (2 studies, 3471 participants: RR 0.80, 95% CI 0.56 to 1.16; I² = 0%; very low certainty evidence)
-
Nonfatal MI
CKD: Analysis 4.3.1 (2 studies, 3996 participants: RR 1.05, 95% CI 0.80 to 1.39; low certainty evidence)
HD: Analysis 4.3.2 (2 studies, 372 participants: RR 1.97, 95% CI 0.22 to 17.59; I² = 0%; very low certainty evidence)
PD: Analysis 4.3.3 (1 study, 129 participants: RR 1.52, 95% CI 0.06 to 36.48)
HD and PD: Analysis 4.3.4 (2 studies, 3268 participants: RR 0.80, 95% CI 0.62 to 1.03; I² = 0%; low certainty evidence)
-
Nonfatal stroke
CKD: Analysis 4.4.1 (3 studies, 4122 participants: RR 1.43, 95% CI 0.82 to 2.48; low certainty evidence)
HD: Analysis 4.4.2 (1 study, 199 participants: RR 1.36, 95% CI 0.07 to 27.82)
HD and PD: Analysis 4.4.3 (1 study, 2964 participants: RR 0.82, 95% CI 0.51 to 1.34)
-
Proportion of patients requiring blood transfusion
CKD: Analysis 4.5.1 (5 studies, 4933 participants: RR 0.97, 95% CI 0.84 to 1.13; I² = 0%; low certainty evidence)
HD and PD: Analysis 4.5.2 (6 studies, 5853 participants: RR 0.80, 95% CI 0.64 to 1.01; I² = 46%; very low certainty evidence)
-
Proportion of patients reaching the target Hb
CKD: Analysis 4.6.1 (6 studies, 1369 participants: RR 1.02, 95% CI 0.90 to 1.16; I² = 79%; very low certainty evidence)
HD and PD: Analysis 4.4.2 (8 studies, 3232 participants: RR 0.98, 95% CI 0.91 to 1.06; I² = 59%; very low certainty evidence).
4.1. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 1: Cardiovascular death
4.2. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 2: Fatigue
4.3. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 3: Nonfatal myocardial infarction
4.4. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 4: Nonfatal stroke
4.5. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 5: Transfusion
4.6. Analysis.

Comparison 4: Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA), Outcome 6: Proportion reaching target haemoglobin
Other subgroup analyses were not possible due to the limited number of studies and data.
Subgroup analyses for proportion reaching Hb target: stratifying by stage of CKD
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.77), suggesting that different stages of CKD do not modify the effect of HIF stabiliser management of anaemia on the proportion reaching the Hb target (Analysis 5.1). However, a smaller number of participants contributed data to CKD and HD than to HD and PD subgroups, meaning that the analysis may not be able to detect subgroup differences.
5.1. Analysis.

Comparison 5: Subgroup analysis: stage CKD (HIF versus ESA), Outcome 1: Proportion reaching target haemoglobin
Subgroup analyses for thrombosis: stratifying by stage of CKD
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.99), suggesting that different stages of CKD do not modify the effect of HIF stabiliser management of anaemia on thrombosis (Analysis 5.2). However, a smaller number of participants and events contributed data to CKD and HD than to HD and PD subgroup, meaning that the analysis may not be able to detect subgroup differences.
5.2. Analysis.

Comparison 5: Subgroup analysis: stage CKD (HIF versus ESA), Outcome 2: Thrombosis
Subgroup analyses for proportion reaching Hb target: stratifying by the duration of therapy
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.78), suggesting that different duration of therapy does not modify the effect of HIF stabiliser management of anaemia on the proportion reaching the Hb target (Analysis 6.1). However, a smaller number of participants and events contributed data to the duration of therapy from 8 to 23 weeks and at least 54 weeks than from the 24 to 53 weeks subgroup, meaning that the analysis may not be able to detect subgroup differences.
6.1. Analysis.

Comparison 6: Subgroup analysis: duration of therapy (HIF versus ESA), Outcome 1: Proportion reaching target haemoglobin
Subgroup analyses for thrombosis: stratifying by the duration of therapy
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.44), suggesting that different duration of therapy does not modify the effect of HIF stabiliser management of anaemia on thrombosis (Analysis 6.2). However, a smaller number of participants and events contributed data to the duration of therapy from 24 to 53 weeks subgroup than at least 54 weeks subgroup, meaning that the analysis may not be able to detect subgroup differences.
6.2. Analysis.

Comparison 6: Subgroup analysis: duration of therapy (HIF versus ESA), Outcome 2: Thrombosis
Subgroup analyses for proportion reaching Hb target: stratifying by frequency of HIF stabiliser administration
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.43), suggesting that different frequency of HIF stabiliser administration does not modify the effect of HIF stabiliser management of anaemia on proportion reaching the Hb target (Analysis 7.1). However, a smaller number of participants and events contributed data to once/day administration than to three times/week administration subgroup, meaning that the analysis may not be able to detect subgroup differences.
7.1. Analysis.

Comparison 7: Subgroup analysis: frequency of administration (HIF versus ESA), Outcome 1: Proportion reaching target haemoglobin
Subgroup analyses for thrombosis: stratifying by frequency of HIF stabiliser administration
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.17), suggesting that different frequency of HIF stabiliser administration does not modify the effect of HIF stabiliser management of anaemia on thrombosis (Analysis 7.2). However, a smaller number of participants and events contributed data to three times/week administration than the once/day administration subgroup, meaning that the analysis may not be able to detect subgroup differences.
7.2. Analysis.

Comparison 7: Subgroup analysis: frequency of administration (HIF versus ESA), Outcome 2: Thrombosis
Subgroup analyses for proportion reaching Hb target: stratifying by type of study (phase 2 versus phase 3)
The test for subgroup differences indicates that there is no statistically significant subgroup effect (P = 0.26), suggesting that type of study does not modify the effect of HIF stabiliser management of anaemia on the proportion reaching the Hb target (Analysis 8.1). However, a smaller number of participants and events contributed data to phase 2 studies than phase 3 studies subgroup, meaning that the analysis may not be able to detect subgroup differences.
8.1. Analysis.

Comparison 8: Subgroup analysis: phase 2 versus phase 3 studies (HIF versus ESA), Outcome 1: Proportion reaching target haemoglobin
Subgroup analyses for thrombosis: stratifying by type of study (phase 2 versus phase 3)
The test for subgroup differences indicates no statistically significant subgroup effect (P = 0.96), suggesting that type of study does not modify the effect of HIF stabiliser management of anaemia on thrombosis (Analysis 8.2). However, a smaller number of participants and events contributed data to phase 2 studies than phase 3 studies subgroup, meaning that the analysis may not be able to detect subgroup differences.
8.2. Analysis.

Comparison 8: Subgroup analysis: phase 2 versus phase 3 studies (HIF versus ESA), Outcome 2: Thrombosis
Sensitivity analysis for HIF stabiliser versus ESA
Sensitivity analyses did not provide substantively different results or were not possible due to few data and studies.
HIF stabiliser versus standard care
No studies were designed to compare HIF stabiliser management of anaemia versus standard care.
HIF stabiliser versus iron supplementation
No studies were designed to compare HIF stabiliser management of anaemia with iron supplementation.
Discussion
Summary of main results
We identified 51 studies randomising 30,994 adults evaluating HIF stabilisers for the treatment of anaemia in CKD (stages 3‐5), including people receiving HD or PD. No studies were performed in children. No studies compared HIF stabiliser with standard care or iron supplementation. Most studies (29 studies randomising 21,622 participants) compared a HIF stabiliser management of anaemia with ESA for a median of 52 weeks. Risks of bias in the included studies were often high or unclear, and these risks combined with imprecision in effect estimates frequently led to low or very low certainty evidence.
HIF stabiliser management of anaemia had uncertain effects on CV death, fatigue and death (any cause). It is uncertain whether HIF stabiliser management of anaemia reduces nonfatal MI or nonfatal stroke. HIF stabilise management of anaemia probably decreased the proportion of patients requiring blood transfusion compared to placebo or ESA. HIF stabiliser management of anaemia probably increased the proportion of patients reaching their Hb target compared to placebo. The effects of HIF stabiliser management of anaemia on hospitalisation for HF, kidney failure, peripheral arterial event, thrombosis, loss of unassisted dialysis access patency, cancer, infection, pulmonary hypertension, and diabetic nephropathy were very uncertain. Subgroup analyses suggested that different stages of CKD, duration of therapy, frequency of HIF stabiliser administration and type of study (phase 2 or phase 3 studies) did not modify the effects of HIF stabiliser management of anaemia either on the proportion reaching the target Hb, or thrombosis. Adverse events were rarely reported; specifically, the definition of hyperkalaemia was not clearly stated.
Overall completeness and applicability of evidence
Evidence from the existing studies was frequently of low or very low certainty and not available to inform clinical care or policy. About half of the included studies were in people with CKD not treated with dialysis. Many studies compared different treatment doses. No studies were conducted on recipients of a kidney transplant or children.
Recently, the Food and Drug Administration (FDA) did not approve the use of roxadustat for the treatment of anaemia in people with CKD, including those requiring dialysis, due to the higher risk of thrombosis shown among HIF stabilisers compared to placebo or ESA (FDA Briefing Document 2021). HIF stabilisers could be considered second‐choice drugs to treat anaemia in people undergoing dialysis who have been resistant to other drugs for anaemia (FDA Briefing Document 2021). In our review, most studies compared a HIF stabiliser management of anaemia with ESA, and clinically important outcome data were rarely reported. Nineteen studies were phase 2 studies that addressed drug efficacy rather than key clinical outcomes, such as death and adverse events. The majority of studies had a small sample size, were of short‐term duration, had methodological limitations, or were primarily designed to evaluate surrogate measures of effect. Due to the included studies' short duration, no studies reported outcome data for life participation, PD infection, or PD technique longevity. Fatigue, hospitalisation due to HF, and infection were rarely reported. Adverse events related to treatment were not systematically reported (Appendix 3), preventing an adequate safety evaluation between HIF stabilisers and placebo or ESA.
No studies compared HIF stabilisers with standard care or iron supplementation.
Future studies on HIF stabilisers for treating anaemia should evaluate treatment outcomes as prioritised by stakeholders (patients, caregivers and health professionals) (SONG 2017) to inform clinical practice and decision making.
Quality of the evidence
We used standard risk of bias domains within the Cochrane tool and GRADE methodology (GRADE 2008) to assess the quality of study evidence. Based on low or very low certainty evidence for the majority of outcomes assessed, future studies might provide different results.
Some studies were at high or unclear risks of bias for most of the risk of bias domains assessment. The majority of studies did not report adequate allocation concealment, blinding or attrition. All but three studies received funding from pharmaceutical companies. Relevant clinical outcomes were rarely available for many of the included studies.
Heterogeneity in the reporting methods of many outcomes constrained data synthesis by meta‐analysis. The targeted Hb values were wide heterogeneous. The limited number of studies prevented the exploration of other potential sources of heterogeneity in the analyses. Some subgroup and sensitivity analyses could not be conducted to explore for heterogeneity owing to insufficient data. Due to the limited number of studies, the assessment of adverse events was not possible.
Potential biases in the review process
Our review was carried out using standard Cochrane methods. Each step was completed independently by at least two authors including a selection of studies, data management, and risk of bias assessment to minimise the risks of misclassification and adjudication of evidence. A highly sensitive search of the Cochrane Kidney Transplant specialised register was undertaken in November 2021, without language restriction and including grey literature. Where possible, we contacted study authors to obtain further data. Many studies did not report key outcomes in a format suitable for meta‐analysis.
Potential bias identified in our review included: 1) phase 2 studies may not be generalisable to phase 3 or real‐world experience because they were of limited duration and not adequately controlled, and tested treatment strategies that may not then be subsequently used in phase 3 studies aimed at approval from regulatory agencies; 2) we pooled different HIF stabilisers agents which have yet to be established in significance; 3) we reported different rate of rise for each HIF stabiliser agent; 4) we pooled different frequency of administration; 5) we pooled major adverse CV events (MACE)‐driven studies and not MACE‐driven studies; 6) we precluded heterogeneity between treatment interventions due to the small number of data observations; 7) we did not exclude poor quality studies due to the small number of included studies; 8) the limited number of studies was a constraint on our ability to assess for potential reporting bias and selective outcome reporting; 9) the definition of Hb target varied across the eligible studies; 10) the effects of HIF stabilisers management of anaemia on longer‐term outcomes were uncertain and the treatment endpoints were principally surrogate outcomes (e.g. blood pressure and laboratory parameters); 11) adverse events were rarely and inconsistently reported due to the short duration of the included studies; 12) no clear definition was provided for hyperkalaemia preventing the evaluation on different threshold; 13) the included studies were conducted in different geographical areas which translates to differences in underlying morbidity for patients.
Visual inspection of funnel plots for HIF stabilisers versus ESA did not suggest any evidence of small study effects indicating possible publication bias for CV death (Figure 4).
4.

Funnel plot of comparison: 2 Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA), outcome: 2.1 Cardiovascular death.
Agreements and disagreements with other studies or reviews
Few studies have examined the efficacy of HIF stabilisers in people with CKD, and the number of meta‐analyses published in this field is limited.
Wang 2020 performed a meta‐analysis of RCTs including only 2804 patients with CKD. The outcomes assessed included mainly surrogate outcomes and adverse events (hypertension, hyperkalaemia, CV events, vascular access thrombosis, headache, vomiting, nasopharyngitis, nausea, and diarrhoea). Our findings confirmed that HIF stabiliser management of anaemia may increase the risk of hyperkalaemia compared with placebo. Compared to our review, Wang 2020 did not investigate death in the included studies, excluded non‐English studies and grey literature, and did not use GRADE to evaluate evidence certainty.
Zhong 2018 carried out a systematic review and meta‐analysis of nine RCTs to assess the efficacy of HIF stabilisers in the treatment of anaemia in CKD. However, due to the limited studies and short follow‐up, Zhong 2018 could not detect relevant clinical outcomes, including death and CV events, and focused mainly on surrogate outcomes (Hb, ferritin, hepcidin and total iron‐binding capacity), preventing comparisons with our review. In addition, Zhong 2018 did not use GRADE to evaluate evidence certainty.
Li 2020 reviewed the findings from three clinical trials on roxadustat for treating kidney anaemia. Data were reported only for surrogate outcomes. The main differences with our review are the exclusion of other HIF stabilisers, such as daprodustat, molidustat and vadadustat, and the lack of data on mortality and safety outcomes.
Authors' conclusions
Implications for practice.
HIF stabilisers have uncertain effects on CV death, fatigue, death (any cause), nonfatal MI, nonfatal stroke, and kidney failure compared with placebo or ESA in people with CKD. HIF stabiliser management of anaemia probably decreases the proportion of patients requiring blood transfusion compared with placebo or ESA. HIF stabilisers probably increase the proportion of patients reaching their Hb target compared to placebo. There is scant evidence to inform decision‐making in children and kidney transplant recipients. Adverse events were rarely and inconsistently provided.
Implications for research.
In the near future, well‐designed and adequately powered RCTs will be included to assess the benefits and harms of HIF stabilisers to treat anaemia in all stages of CKD, including people requiring any form of dialysis and kidney transplant recipients. The potential use of oral HIF stabilisers for anaemia may lead to higher compliance with the treatment in people with CKD and reduce the need for blood transfusion, although unwanted side effects could be a major concern in this population. Future real‐world experience studies will assess the effects of HIF stabiliser regimen on the progression of CKD, and blood pressure and define the proper dose in people with and without iron supplementation. Future studies will evaluate whether HIF stabiliser effects are dependent on the CKD stage.
Our review has identified several ongoing and promising RCTs that could help to increase our knowledge and confidence in the findings, focusing on important outcomes, such as death and CV events. Further research is likely to change the estimated effects of treatments for anaemia in CKD and provide relevant information on HRQoL and patient‐reported outcomes (including life participation and fatigue). Larger studies should be conducted in PD to address specific outcomes related to this population, including PD infection and PD technique longevity.
Future HIF stabiliser studies compared with standard care or iron supplementation will increase our certainty of the evidence based on the paucity of evidence in CKD.
Evaluation of cost‐effectiveness for HIF stabilisers in the treatment of anaemia would assist decision‐making by policy‐makers and health care providers in this population.
What's new
| Date | Event | Description |
|---|---|---|
| 26 August 2022 | Amended | Acknowledgements updated |
History
Protocol first published: Issue 10, 2020
Notes
Acknowledgement section updated
Acknowledgements
The authors are grateful to the following peer reviewers for their time and comments: Anatole Besarab MD (Stanford University School of Medicine), Joshua M. Kaplan (Rutgers‐New Jersey Medical School), Takeshi Hasegawa, MD, PH, PhD (Showa University), Ana Cabrita MD (Nephrology Department, Centro Hospitalar Universitário do Algarve‐Faro).
Appendices
Appendix 1. Electronic search strategies
| Database | Search terms |
| CENTRAL |
|
| MEDLINE |
|
| EMBASE |
|
Appendix 2. Risk of bias assessment tool
| Potential source of bias | Assessment criteria |
|
Random sequence generation Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence |
Low risk of bias: Random number table; computer random number generator; coin tossing; shuffling cards or envelopes; throwing dice; drawing of lots; minimisation (minimisation may be implemented without a random element, and this is considered to be equivalent to being random). |
| High risk of bias: Sequence generated by odd or even date of birth; date (or day) of admission; sequence generated by hospital or clinic record number; allocation by judgement of the clinician; by preference of the participant; based on the results of a laboratory test or a series of tests; by availability of the intervention. | |
| Unclear: Insufficient information about the sequence generation process to permit judgement. | |
|
Allocation concealment Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment |
Low risk of bias: Randomisation method described that would not allow investigator/participant to know or influence intervention group before eligible participant entered in the study (e.g. central allocation, including telephone, web‐based, and pharmacy‐controlled, randomisation; sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes). |
| High risk of bias: Using an open random allocation schedule (e.g. a list of random numbers); assignment envelopes were used without appropriate safeguards (e.g. if envelopes were unsealed or non‐opaque or not sequentially numbered); alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. | |
| Unclear: Randomisation stated but no information on method used is available. | |
|
Blinding of participants and personnel Performance bias due to knowledge of the allocated interventions by participants and personnel during the study |
Low risk of bias: No blinding or incomplete blinding, but the review authors judge that the outcome is not likely to be influenced by lack of blinding; blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding; blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Blinding of outcome assessment Detection bias due to knowledge of the allocated interventions by outcome assessors. |
Low risk of bias: No blinding of outcome assessment, but the review authors judge that the outcome measurement is not likely to be influenced by lack of blinding; blinding of outcome assessment ensured, and unlikely that the blinding could have been broken. |
| High risk of bias: No blinding of outcome assessment, and the outcome measurement is likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding. | |
| Unclear: Insufficient information to permit judgement | |
|
Incomplete outcome data Attrition bias due to amount, nature or handling of incomplete outcome data. |
Low risk of bias: No missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not enough to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes not enough to have a clinically relevant impact on observed effect size; missing data have been imputed using appropriate methods. |
| High risk of bias: Reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk enough to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardized difference in means) among missing outcomes enough to induce clinically relevant bias in observed effect size; ‘as‐treated’ analysis done with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation. | |
| Unclear: Insufficient information to permit judgement | |
|
Selective reporting Reporting bias due to selective outcome reporting |
Low risk of bias: The study protocol is available and all of the study’s pre‐specified (primary and secondary) outcomes that are of interest in the review have been reported in the pre‐specified way; the study protocol is not available but it is clear that the published reports include all expected outcomes, including those that were pre‐specified (convincing text of this nature may be uncommon). |
| High risk of bias: Not all of the study’s pre‐specified primary outcomes have been reported; one or more primary outcomes is reported using measurements, analysis methods or subsets of the data (e.g. sub‐scales) that were not pre‐specified; one or more reported primary outcomes were not pre‐specified (unless clear justification for their reporting is provided, such as an unexpected adverse effect); one or more outcomes of interest in the review are reported incompletely so that they cannot be entered in a meta‐analysis; the study report fails to include results for a key outcome that would be expected to have been reported for such a study. | |
| Unclear: Insufficient information to permit judgement | |
|
Other bias Bias due to problems not covered elsewhere in the table |
Low risk of bias: The study appears to be free of other sources of bias. |
| High risk of bias: Had a potential source of bias related to the specific study design used; stopped early due to some data‐dependent process (including a formal‐stopping rule); had extreme baseline imbalance; has been claimed to have been fraudulent; had some other problem. | |
| Unclear: Insufficient information to assess whether an important risk of bias exists; insufficient rationale or evidence that an identified problem will introduce bias. |
Appendix 3. Studies reporting adverse events
| Study ID | Intervention | Control | Adverse events in the intervention arm | Adverse events in the control arm | Comments |
| Akizawa 2017 | Daprodustat (4, 6, 8, 10 mg) | Placebo | Death (0/78), nasopharyngitis (5/78), hypertension (2/78), cerebral haemorrhage (0/78), pyoderma gangrenosum (1/78) Further data were available on the NCT02019719: any AE (26/78), SAE (0/78), cerebral haemorrhage (0/78), ear discomfort (1/78), gastritis atrophic (1/78), stomatitis (1/78), vomiting (1/78), pyrexia (1/78), peripheral oedema (1/78), seasonal allergy (1/78), nasopharyngitis (5/78), pharyngitis (1/78), pharyngitis bacterial (1/78), upper respiratory tract infection (1/78), AVF thrombosis (1/78), burns second degree (1/78), injury (1/78), procedural hypotension (1/78), shunt stenosis (1/78), decreased appetite (1/78), back pain (2/78), arthralgia (1/78), lumbar spinal stenosis (1/78), dyspnoea 0(/78), dermatitis (2/78), blister rupture (1/78), drug eruption (1/78), haemorrhage subcutaneous (1/78), rash (0/78), hypertension (2/78) |
Death (0/19), nasopharyngitis (0/19), hypertension (0/19), cerebral haemorrhage (1/19) pyoderma gangrenosum (0/19) Further data were available on the NCT02019719: any AE (5/19), SAE (1/19), cerebral haemorrhage (1/19), ear discomfort (0/19), gastritis atrophic (0/19), stomatitis (0/19), vomiting (0/19), pyrexia (1/19), peripheral oedema (0/19), seasonal allergy (0/19), nasopharyngitis (0/19), pharyngitis (0/19), pharyngitis bacterial (0/19), upper respiratory tract infection (0/19), AVF thrombosis (0/19), burns second degree (0/19), injury (0/19), procedural hypotension (0/19), shunt stenosis (0/19), decreased appetite (1/19), back pain (0/19), arthralgia (0/19), lumbar spinal stenosis (0/19), dyspnoea (1/19), dermatitis (1/19), blister rupture (0/19), drug eruption (0/19), haemorrhage subcutaneous (0/19), rash (1/19), hypertension (0/19) |
"On‐therapy AEs were reported in a total of 32 subjects. All AEs were reported as single instances in each treatment group, except for nasopharyngitis, which was reported in 1 subject in the 4 mg group and 2 subjects each in the 8 and 10 mg groups. Two non‐serious AEs of hypertension, in 1 subject each in the 6 and 10 mg groups, were assessed as treatment‐related by the investigator. There were 2 serious on‐therapy AEs reported in the study: cerebral haemorrhage in the placebo group and pyoderma gangrenosum in the 10 mg group. The cerebral haemorrhage was assessed as treatment‐related by the investigator. No deaths occurred during the study." |
| Akizawa 2019 | Roxadustat (50, 70, 100 mg) | Placebo | SAE (11/80), death (0/80), MI (0/80), stroke (0/80), GI (22/80), diarrhoea (8/80), nausea (5/80), infections/infestations (32/80), nasopharyngitis (21/80), renal/urinary disorders (5/80), declining kidney function (5/80) musculoskeletal and connective tissue disorders (11/80), muscle spasms (1/80), respiratory, thoracic, and mediastinal disorders (5/80), cough (1/80), initiation of dialysis (4/80) | SAE (2/27), death (0/27), MI (0/27), stroke (0/27), GI (2/27), diarrhoea (1/27), nausea (0/27), infections/infestations (9/27), nasopharyngitis (6/27), renal/urinary disorders (1/27), declining kidney function (1/27), musculoskeletal and connective tissue disorders (4/27), muscle spasms (2/27), respiratory, thoracic, and mediastinal disorders (2/27), cough (2/27), initiation of dialysis (0/27) | "The incidence of overall TEAEs ranged between 70.4% (placebo) and 88.5% (70 mg roxadustat). No major adverse cardiac events (i.e., myocardial infarction, stroke, death) occurred in roxadustat‐treated patients. Two cases of congestive heart failure (one with 50 mg TIW and one with placebo) occurred during the study; the case in the placebo‐treated patient was considered drug‐related. In terms of clinically relevant arrhythmias, two cases of atrial fibrillation occurred. A total of six cases of declining kidney function were reported (placebo TIW, n = 1; roxadustat 50 mg TIW, n = 4; roxadustat 100 mg TIW, n = 1)ed (one with placebo and one with 70 mg TIW). Discontinuation due to progressive disease requiring initiation of dialysis occurred in three patients (11.1%) in the roxadustat 50‐mg TIW group and one patient (3.7%) in the roxadustat 100‐mg TIW group. Two cases of hepatic dysfunction occurred in patients treated with placebo TIW." |
| Akizawa 2020a | Roxadustat | Darbepoetin alfa | SAE (31/150), angina pectoris (5/150), acute MI (1/150), aortic valve stenosis (0/150), atrioventricular block complete (0/150), bradycardia (1/150), cardiac failure (0/150), cardiac failure congestive (1/150), coronary artery stenosis (1/150), myocardial ischemias (0/150), sudden hearing loss (1/150), GI haemorrhage (1/150), vascular stent occlusion (1/150), cellulitis (2/150), UTI (1/150), shunt stenosis (6/150), shunt occlusion (3/150), joint dislocation (1/150), subcutaneous hematoma (0/150), spinal column injury (1/150), lumbar spinal stenosis (1/150), basal cell carcinoma (0/150), gastric cancer (1/150), malignant neoplasm of renal pelvis (0/150), transitional cell carcinoma (0/150), lip and/or oral cavity cancer (0/150), cerebral infarction (1/150), psychiatric disorders (0/150), asthma (1/150), pulmonary oedema (0/150), surgical and medical procedures (1/150), DVT (2/150), orthostatic hypotension (1/150), venous occlusion (1/150), peripheral arterial occlusive disease (0/150), subclavian vein stenosis (1/150), retinal haemorrhage (5/150), dental caries (5/150), nasopharyngitis (52/150), contusion (10/150), wound (3/150), procedural hypotension (5/150), hyperkalaemia (5/150), back pain (3/150), skin exfoliation (6/150), internal haemorrhage (2/150), vertigo (1/150), eye disorders (5/150), faeces soft (0/150), general disorders and administration site conditions (3/150), nervous system disorders (2/150), psychiatric disorders (2/150), eczema (0/150) | SAE (22/152), angina pectoris (2/152), acute MI (0/152), aortic valve stenosis (1/152), atrioventricular block complete (1/152) bradycardia (0/152), cardiac failure (1/152), cardiac failure congestive (0/152), coronary artery stenosis (0/152), myocardial ischemias (1/152), sudden hearing loss (0/152), GI haemorrhage (0/152), vascular stent occlusion (0/152), cellulitis (0/152), UTI (0/152), shunt stenosis (6/152), shunt occlusion (2/152), joint dislocation (0/152), subcutaneous hematoma (1/152), spinal column injury (0/152), lumbar spinal stenosis (0/152), basal cell carcinoma (1/152), gastric cancer (0/152), malignant neoplasm of renal pelvis (4/152), transitional cell carcinoma (4/152), lip and/or oral cavity cancer (4/152), cerebral infarction (0/152), psychiatric disorders (1/152), asthma (0/152), pulmonary oedema (1/152), surgical and medical procedures (2/152), DVT (0/152), orthostatic hypotension (0/152), venous occlusion (0/152), peripheral arterial occlusive disease (1/152), subclavian vein stenosis (0/152), retinal haemorrhage (6/152), dental caries (0/152), nasopharyngitis (40/152), contusion (10/152), wound (5/152), procedural hypotension (1/152), hyperkalaemia (1/152), back pain (7/152), skin exfoliation (2/152), internal haemorrhage (6/152), vertigo (2/152,) eye disorders (4/152), faeces soft (1/152), general disorders and administration site conditions (1/152), nervous system disorders (2/152), psychiatric disorders (0/152), eczema (1/152) | "The proportion of patients who reported TEAEs was similar in the roxadustat (129/150; 86.0%) and DA (126/152; 82.9%) groups. The incidence of serious TEAEs was 20.7% (31/150) and 14.5% (22/152) in the roxadustat and DA groups, respectively. Serious TEAEs considered potentially drug related were reported in 3.3%(5/150) and 3.9%(6/152) of patients in the roxadustat and DA groups, respectively. The incidence of TEAEs leading to withdrawal of treatment and, in turn, withdrawal from the study, was 8.7% (13/150) and 5.3% (8/ 152) in the roxadustat and DA groups, respectively. Two deaths were reported during the study, both of which occurred in the roxadustat group. TEAEs included nasopharyngitis, shunt stenosis, diarrhoea, contusion, and vomiting. The incidences of nasopharyngitis and vomiting were higher in the roxadustat group than in the DA group. Retinal haemorrhage was reported as a TEAE in 3.3% (5/150) and 3.9%(6/152) of patients in the roxadustat and DA groups, respectively." |
| Akizawa 2020c | Daprodustat | Darbepoetin alfa | Death (0/136), nasopharyngitis (57/136), pharyngitis (10/136), gastroenteritis (7/136), shunt stenosis (19/136), contusion (17/136), skin abrasion (10/1360, procedural hypotension (11/136), diarrhoea (20/136), vomiting (15/136), nausea (9/136), constipation (8/136), abdominal discomfort (3/136), back pain (6/136), arthralgia (5/136), muscle spasms (7/136), pain in extremity (1/136), headache (8/136), pyrexia (7/136), hypertension (5/136), arteriosclerosis coronary artery (0/136), cardiac failure (1/136), cerebral infarction (1/136), pulmonary oedema (0/136), retinal vein occlusion (2/136), retinal artery occlusion (0/136), shunt events (4/136), venous occlusion (1/136), gastritis erosive (2/136), chronic gastritis (0/136), duodenal perforation (1/136), gastric ulcer (0/136), haemorrhagic erosive gastritis (0/136), macular oedema (2/136), anterior chamber angle neovascularization (2/136), retinal haemorrhage (0/136), hypertensive cardiomyopathy (0/136), pulmonary hypertension (1/136), pancreatic carcinoma (0/136), rheumatoid arthritis (0/136), thrombosis and/or tissue ischemias secondary to excessive erythropoiesis (0/136) | Death (1/135), nasopharyngitis (57/135), pharyngitis (10/135), gastroenteritis (7/135), shunt stenosis (19/135), contusion (17/135), skin abrasion (10/135), procedural hypotension (11/135), diarrhoea (20/135), vomiting (15/135), nausea (9/135), constipation (8/135), abdominal discomfort (3/135), back pain (6/135), arthralgia (5/135), muscle spasms (7/135), pain in extremity (1/135), headache (8/135), pyrexia (7/135), hypertension (5/135), arteriosclerosis coronary artery (1/135), cardiac failure (2/135), cerebral infarction (1/135), pulmonary oedema (1/135), retinal vein occlusion (1/135), retinal artery occlusion (1/135), shunt events (4/135), venous occlusion (0/135), gastritis erosive (2/135), chronic gastritis (1/135), duodenal perforation (0/135), gastric ulcer (1/135), haemorrhagic erosive gastritis (1/135), macular oedema (2/135), anterior chamber angle neovascularization (1/135), retinal haemorrhage (1/135), hypertensive cardiomyopathy (1/135), pulmonary hypertension (0/135), pancreatic carcinoma (1/135), rheumatoid arthritis (1/135), thrombosis and/or tissue ischemias secondary to excessive erythropoiesis (0/135) | "Most participants in both treatment groups experienced one or more AEs (93% daprodustat, 97% darbepoetin alfa). Of the AEs occurring in $5% of participants in any group (Table 4), contusion and diarrhoea were more frequent ($5% difference) in the daprodustat group, whereas nasopharyngitis and extremity pain was more frequent ($5% difference) in the darbepoetin alfa group. Hyperkalaemia was reported in 3% of participants in the daprodustat group versus 1% in the darbepoetin alfa group. SAEs occurred in 15% (daprodustat) and 27% (darbepoetin alfa) of participants. SAEs reported in two or more participants ($1%) in any treatment group were shunt stenosis (3% daprodustat, 4% darbepoetin alfa), shunt occlusion (,1% daprodustat, 2% darbepoetin alfa), shunt malfunction (0% daprodustat, 1% darbepoetin alfa), pneumonia (,1% daprodustat, 1% darbepoetin alfa), sepsis (0% daprodustat, 1% darbepoetin alfa), and cardiac failure congestive (0% daprodustat, 1% darbepoetin alfa). No deaths were reported in the daprodustat group. One fatal event (sepsis) was reported during the follow‐up period in the darbepoetin alfa group." |
| Akizawa 2020f | Roxadustat | Darbepoetin alfa | The incidence of TEAEs was 78.6% in roxadustat | The incidence of TEAEs was 70.2% in darbepoetin alfa. No other clear information were provided | "The incidence of TEAEs observed during the 24‐week treatment period was 78.6% in roxadustat (CG), 70.2% in DA (CG), and 77.1% in roxadustat (RG). Common TEAEs included nasopharyngitis, CKD, hyperkalaemia, and hypertension; rates of these were comparable between groups. The incidence of serious TEAEs (TESAEs) was 17.6% (23/131 patients) in the roxadustat (comparative) group, 13.0% (17/131 patients) in the DA (comparative) group." |
| Akizawa 2021 | Roxadustat | Darbepoetin alfa | TEAE (103/131), serious TEAE (23/131) Overall (103/131); GI disorders (32/131); constipation (5/131); diarrhoea (3/131); dental caries (3/131); nausea (5/131); general disorders and administration site conditions (13/131); oedema, peripheral (5/131); pyrexia (5/131); infections and infestations (42/131); nasopharyngitis (25/131); pneumonia (4/131); gastroenteritis (5/131); cystitis (0/131); pharyngitis (0/131); Injury, poisoning and procedural complications (15/131); contusion (4/131); investigations (8/131); blood potassium increased (0/131); metabolism and nutrition disorders (15/131); hyperkalaemia (5/131); hypoglycaemia (1/131); musculoskeletal and connective tissue disorders (12/131); back pain (4/131); nervous system disorders (16/131); headache (3/131); renal and urinary disorders (15/131); CKD (9/131); skin and subcutaneous tissue disorders (7/131); eczema (1/131); vascular disorders (5/131); hypertension (3/131) |
TEAE (92/131), serious TEAE (17/131) Overall (92/131); GI disorders (19/131); constipation (4/131); diarrhoea (5/131); dental caries (1/131); nausea (1/131); general disorders and administration site conditions (13/131); oedema, peripheral (4/131); pyrexia (4/131); infections and infestations (48/131); nasopharyngitis (34/131); pneumonia (4/131); gastroenteritis (1/131); cystitis (2/131); pharyngitis (1/131); injury, poisoning and procedural complications (10/131); contusion (2/131); investigations (6/131); blood potassium increased (4/131); metabolism and nutrition disorders (16/131); hyperkalaemia (5/131); hypoglycaemia (0/131); musculoskeletal and connective tissue disorders (13/131); back pain (5/131); nervous system disorders (9/131); headache (4/131); renal and urinary disorders (13/131); CKD (9/131); skin and subcutaneous tissue disorders (11/131); eczema (3/131); vascular disorders (6/131); hypertension (5/131) |
"In the SAF, the incidence of TEAEs was comparable across treatment arms. The incidence of TEAEs was 78.6% (103/131 patients) in the roxadustat (comparative) group, 70.2% (92/131 patients) in the DA (comparative) group." |
| ALPS 2021 | Roxadustat | Placebo | All AE (343/391), SAE (61.6%), death (45/391), kidney failure (135/391), hypertension (87/391), oedema peripheral (45/391), GFR decreased (43/391), hyperkalaemia (39/391), viral upper respiratory tract infection (38/391), nausea (37/391), diarrhoea (33/391), pneumonia (28/391), iron deficiency (26/391), anaemia (24/391), headache (21/391), AVF thrombosis (20/391), pruritus (20/391), asthenia (19/391), hyperuricaemia (9/391), azotaemia (10/391), AKI (1/391), sepsis (6/391), peritonitis (5/391), pyelonephritis chronic (5/391), device related infection (4/391), MI (8/391), acute MI (6/391), atrial fibrillation (3/391), cardiac failure chronic (3/391), coronary artery disease (1/391), hip fracture (4/391), DVT (4/391), hypertensive crisis (4/391), cerebrovascular accident (5/391), Ischaemic stroke (4/391), diabetic neuropathy (0/391), pleural effusion (3/391), pulmonary oedema (3/391), hydrothorax (0/391), diabetic metabolic decompensation (1/391), asthenia (1/391), multiple organ dysfunction syndrome (1/391), catheter site haemorrhage (0/391), peripheral swelling (0/391) | All AE (176/203), SAE (56.7%), death (20/203), kidney failure (62/203), hypertension (28/203), oedema peripheral (21/203), GFR decreased (23/203), hyperkalaemia (15/203), viral upper respiratory tract infection (9/203), nausea (6/203), diarrhoea (7/203), pneumonia (14/203), iron deficiency (8/203), anaemia (37/203), headache (11/203), AVF thrombosis (2/203), pruritus (2/203), asthenia (12/203), hyperuricaemia (11/203), azotaemia (8/203), AKI (2/203), sepsis (0/203), peritonitis (1/203), pyelonephritis chronic (2/203), device related infection (0/203), MI (3/203), acute MI (2/203), atrial fibrillation (3/203), cardiac failure chronic (3/203), coronary artery disease (2/203), hip fracture (0/203), DVT (0/203), hypertensive crisis (5/203), cerebrovascular accident (0/203), Ischaemic stroke (1/203), diabetic neuropathy (2/203), pleural effusion (3/203), pulmonary oedema (2/203), hydrothorax (2/203), diabetic metabolic decompensation (2/203), asthenia (2/203), multiple organ dysfunction syndrome (2/203), catheter site haemorrhage (2/203), peripheral swelling (2/203) |
"The overall incidence of TEAEs was comparable between groups: 87.7% of patients in the roxadustat group and 86.7% of patients in the placebo group were reported to have experienced TEAEs. A comparable proportion of patients in both groups (47.3% roxadustat, 43.3% placebo) had TEAEs ≥ Grade 3 in severity. A greater proportion of patients in the roxadustat group (20.7%) experienced TEAEs considered related to treatment by the investigator compared with the placebo group (13.3%). In the roxadustat group 61.6% of patients compared with 56.7% in the placebo group had serious TEAEs; 6.4% and 2.0% of patients respectively had serious TEAEs considered related to treatment by the investigator. A total of 45 patients in the roxadustat group and 20 in the placebo group experienced death. The most common TEAE in both groups were end stage renal disease, hypertension, oedema peripheral and glomerular filtration rate decreased." |
| ANDES 2021 | Roxadustat | Placebo | Hyperkalaemia (111/611), constipation (105/611), viral upper respiratory tract infection (98/611), hypertension (95/611), oedema peripheral (89/611), nausea (85/611), upper respiratory tract infection (79/611), diarrhoea (78/611), UTI (68/611), kidney failure (67/611), headache (66/611), insomnia (63/611), dizziness (58/611), cough (57/611), back pain (55/611), CKD (54/611), pruritus (54/611), vomiting (54/611), hypoglycaemia (53/611), AKI (49/611), oedema (48/611), arthralgia ( 45/611), pneumonia (44/611), decreased appetite (41/611), muscle spasms (41/611), hyperphosphataemia (40/611), dyspepsia (39/611), pain in extremity (39/611), pyrexia (39/611), abdominal pain (35/611), bronchitis (34/611), dyspnoea (34/611), cellulitis (32/611), gout (32/611), asthenia (31/611), hypotension (31/611), metabolic acidosis (29/611), anaemia (17/611), cardiac failure congestive (23/611), hyponatraemia (15/611), azotaemia (13/611), hypoglycaemia (13/611), peritonitis (13/611), pulmonary oedema (4/611), arthralgia (32/611), diabetes mellitus inadequate control (6/611), acute MI (5/611), fluid overload (3/611), gastroenteritis (2/611), chronic obstructive pulmonary disease (1/611) | Hyperkalaemia (41/305), constipation (34/305), viral upper respiratory tract infection (40/305), hypertension (27/305), oedema peripheral (28/305), nausea (29/305), upper respiratory tract infection (48/305), diarrhoea (31/305), UTI (29/305), kidney failure (18/305), headache (26/305), insomnia (9/305), dizziness (32/305), cough (28/305), back pain (18/305), CKD (21/305), pruritus (19/305), vomiting (20/305), hypoglycaemia (15/305), AKI (11/305), oedema (9/305), arthralgia (24/305), pneumonia (18/305), decreased appetite (8/305), muscle spasms (9/305), hyperphosphataemia (10/305), dyspepsia (12/305), pain in extremity (14/305), pyrexia (9/305), abdominal pain (13/305), bronchitis (13/305), dyspnoea (23/305), cellulitis (7/305), gout (20/305), asthenia (11/305), hypotension (10/305), metabolic acidosis (18/305), anaemia (44/305), cardiac failure congestive (9/305), hyponatraemia (3/305), azotaemia (5/305), hypoglycaemia (4/305), peritonitis (1/305), pulmonary oedema (6/305), arthralgia (19/305), diabetes mellitus inadequate control (0/305), acute MI (3/305), fluid overload (3/305), gastroenteritis (4/305), chronic obstructive pulmonary disease (3/305) | "The incidences of TEAEs and TESAEs were comparable. [...] TEAEs were reported by 92.3% (564/611) of roxadustat and 89.5% (273/305) of placebo treated patients, corresponding to incidence rates of 554.4 and 594.5/100 PEY. The most common TEAEs in the roxadustat or placebo group were hyperkalaemia (13.6 vs. 12.5/100 PEY), constipation (12.2 vs. 10.3/100 PEY), viral upper respiratory tract infection (16.9 vs. 15.4/100 PEY), upper respiratory tract infection (12.8 vs. 18.0/100 PEY), and hypertension (11.3 vs. 10.1/100 PEY) (Table 2). The incidence rates of TESAEs were 74.2 and 66.0/100 PEY among roxadustat‐ vs. placebo‐treated patients. Incidence rates for fatal TESAEs were 3.8 and 2.9, respectively." |
| ASCEND‐D 2021 | Daprodustat |
Darbepoetin alfa (PD patients) ESA (HD patients) |
SAE (773/1482); AE (1307/1482); thrombosis or tissue ischemias due to excessive erythropoiesis (20/1482); cardiomyopathy (15/1482); pulmonary‐artery hypertension (9/1482); cancer‐related death or tumour progression or recurrence (47/1482); oesophageal or gastric erosions (60/1482); proliferatives retinopathy, macular oedema, or choroidal neovascularization (38/1482); exacerbation of rheumatoid arthritis (2/1482); worsening of hypertension (293/1482) | Data were reported on 1474 participants SAE (748/1474); AE (1252/1474); thrombosis or tissue ischemias due to excessive erythropoiesis (11/1474); cardiomyopathy (16/1474); pulmonary‐artery hypertension (12/1474); cancer‐related death or tumour progression or recurrence (51/1474);oesophageal or gastric erosions (81/1474); proliferatives retinopathy, macular oedema, or choroidal neovascularization (35/1474); exacerbation of rheumatoid arthritis (1/1474); worsening of hypertension (302/1474) |
"Serious adverse events during the trial were reported in 773 patients (52.2%) in the daprodustat group and in 748 (50.7%) in the ESA group." |
| ASCEND‐ID 2021 | Daprodustat | Darbepoetin alfa | Worsening of hypertension: 24% Rates of AE: 76% Rescue treatment: 3% |
Worsening of hypertension: 19% Rates of AE: 72% Rescue treatment: 3% |
"Rescue treatment was the same in both groups (3%). While the number of subjects with worsening of hypertension (24% Dapro vs 19% Darbe) was numerically higher, the overall effect of daprodustat on BP was similar to Darbe. Rates of AEs were similar (76% Dapro vs 72% Darbe)." |
| ASCEND‐ND 2021 | Daprodustat | Darbepoetin alfa | SAE (850/1937); AE (1545/1937); thrombosis or tissue ischemias due to excessive erythropoiesis (5/1937); cardiomyopathy (6/1937); pulmonary‐artery hypertension (15/1937); cancer‐related death or tumour progression or recurrence (72/1937); oesophageal or gastric erosions (70/1937); proliferatives retinopathy, macular oedema, or choroidal neovascularization (54/1937); exacerbation of rheumatoid arthritis (2/1937); worsening of hypertension (344/1937) | SAE (703/1933); AE (1487/1933); thrombosis or tissue ischemias due to excessive erythropoiesis (13/1933); cardiomyopathy (7/1933); pulmonary‐artery hypertension (9/1933); cancer‐related death or tumour progression or recurrence (49/1933); oesophageal or gastric erosions (41/1933); proliferatives retinopathy, macular oedema, or choroidal neovascularization (44/1933); exacerbation of rheumatoid arthritis (4/1933); worsening of hypertension (363/1933) | "Serious adverse events that started or worsened after the initiation of trial treatment were reported in 850 of 1937 patients in the daprodustat group (43.9%) and 703 of 1933 patients in the darbepoetin alfa group (36.4%)." |
| Besarab 2015 | Roxadustat (0.7, 1, 1.5, 2 mg/kg) | Placebo | Any treatment‐related AE: 52/88 Diarrhoea (8/88), headache (6/88), back pain (4/88), fatigue (4/88), hyperkalaemia (4/88), peripheral oedema (3/88), dizziness (2/88), insomnia (2/88), seasonal allergy (1/88), UTI (1/88), SAE (4/88), death (0/88), constipation (1/88), gastro oesophageal reflux disease (2/88), nausea (1/88), abdominal discomfort (0/88), abdominal pain (1/88), abdominal pain upper (1/88), ascites (1/88), dyspepsia (1/88), gastritis (1/88), GI disorder (1/88), lip swelling (1/88), influenza (1/88), bronchitis (1/88), candidiasis (0/88), cystitis (1/88), onychomycosis (0/88), sinusitis (1/88), tooth abscess (1/88), tooth infection (1/88), upper respiratory tract infection (1/88), hyperuricaemia (2/88), acidosis (1/88), anorexia (1/88), decreased appetite (1/88), diabetes mellitus (1/88), gout (1/88), hyperglycaemia (1/88), hypernatraemia (1/10), hyperphosphataemia (1/88), hypoglycaemia (1/88), metabolic acidosis (1/88), type 2 diabetes mellitus (1/88), vitamin D deficiency (1/88), muscle spasms (1/88), myalgia (2/88), arthralgia (1/88), neck pain (0/88), osteoarthritis (0/88), osteoporosis (1/88), oedema peripheral (3/88), asthenia (0/88), chills (1/88), non‐cardiac chest pain (1/88), oedema (1/88), pyrexia (1/88), neuropathy peripheral (1/88), drug eruption (1/88), erythema (1/88), increased tendency to bruise (1/88), intertrigo (1/88), pruritus (1/88), rash (1/88), skin lesion (1/88), skin ulcer (1/88), stasis dermatitis (0/88), sinus bradycardia (0/88), bradycardia (1/88), bundle branch block right (0/88), cardiac failure congestive (1/88), pericarditis (0/88), ventricular extrasystoles (1/88), AVF site complication (1/88), excoriation (1/88), femoral neck fracture (1/88), foot fracture (1/88), laceration (1/88), procedural pain (1/88), skin laceration (1/88), immune system disorders (1/88), depression (1/88), renal impairment (2/88), micturition urgency (1/88), AKI (0/88), allergic sinusitis (1/88), cough (1/88), dyspnoea (1/88), postnasal drip (1/88), wheezing (1/88), hot flush (1/88), hypertension (1/88), hypotension (0/88), vasculitis (0/88), anaemia (2/88), hyperparathyroidism secondary (1/88), hypothyroidism (1/88), diabetic retinopathy (1/88), eyelid oedema (1/88), breast cyst (1/88), prostatitis (1/88), ear pain (1/88), gallbladder polyp (1/88), benign breast neoplasm (1/88), nail operation (1/88) |
Any treatment‐related AE: 13/28 Diarrhoea (2/28), headache (1/28), back pain (1/28), fatigue (0/28), hyperkalaemia (0/28), peripheral oedema (0/28), dizziness (0/28), insomnia (1/28), seasonal allergy (2/28), UTI (3/28), SAE (4/28), death (0/28), constipation (1/28), gastro oesophageal reflux disease (0/28), nausea (1/28), abdominal discomfort (1/28), abdominal pain (1/28), abdominal pain upper (0/28), ascites (0/28), dyspepsia (0/28), gastritis (0/28), GI disorder (0/28), lip swelling (0/28), influenza (1/28), bronchitis (0/28), candidiasis (1/28), cystitis (0/28), onychomycosis (1/28), sinusitis (0/28), tooth abscess (0/28), tooth infection (0/28), upper respiratory tract infection (0/28), hyperuricaemia (0/28), acidosis (0/28), anorexia (0/28), decreased appetite (0/28), diabetes mellitus (0/28), gout (0/28), hyperglycaemia (0/28), hypernatraemia (0/10), hyperphosphataemia (0/28), hypoglycaemia (0/28), metabolic acidosis (0/28), type 2 diabetes mellitus (0/28), vitamin D deficiency (0/28), muscle spasms (1/28), myalgia (1/28), arthralgia (0/28), neck pain (0/28), osteoarthritis (1/28), osteoporosis (1/28), oedema peripheral (0/28), asthenia (0/28), chills (1/28), non‐cardiac chest pain (1/28), oedema (1/28), pyrexia (0/28), neuropathy peripheral (0/28), drug eruption (0/28), erythema (0/28), increased tendency to bruise (0/28), intertrigo (0/28), pruritus (0/28), rash (0/28), skin lesion (0/28), skin ulcer (0/28), stasis dermatitis (1/28), sinus bradycardia (2/28), bradycardia (0/28), bundle branch block right (1/28), cardiac failure congestive (0/28), pericarditis (1/28), ventricular extrasystoles (0/28), AVF site complication (0/28), excoriation (0/28), femoral neck fracture (0/28), foot fracture (0/28), laceration (0/28), procedural pain (0/28), skin laceration (0/28), immune system disorders (3/28), depression (0/28), renal impairment (0/28), micturition urgency (0/28), AKI (1/28), allergic sinusitis (0/28), cough (0/28), dyspnoea (0/28), postnasal drip (0/28), wheezing (0/28), hot flush (0/28), hypertension (0/28), hypotension (1/28), vasculitis (1/28), anaemia (0/28), hyperparathyroidism secondary (0/28), hypothyroidism (0/28), diabetic retinopathy (0/28), eyelid oedema (0/28), breast cyst (0/28), prostatitis (0/28), ear pain (0/28), gallbladder polyp (0/28), benign breast neoplasm (0/28), nail operation (0/28) |
"AEs were reported by 52 (59%) roxadustat‐treated and 13 (46%) placebo subjects; the most common AEs were expected in CKD and did not differ clinically differ between groups. Serious AEs (SAEs) were reported by four (5%) roxadustat‐treated subjects and one (4%) placebo patient; the SAEs in roxadustat treated subjects included vascular access complications, femoral neck fracture, noncardiac chest pain and dyspnoea. The vascular access complication was reported in a patient with graft infection 4 days after arteriovenous (AV) graft placement whose baseline eGFR was 18.4 mL/min. Initial Hb was 9.3 g/dL. Despite a good Hb response for 6 weeks, a left arm AV graft was placed to prepare for future dialysis. Four days later, the patient presented with signs and symptoms compatible with ‘graft’ infection and was treated with clindamycin and vancomycin. No CV SAEs and no death occurred during study. The incidences of seizure, thromboembolic and CV events during roxadustat treatment are of special interest in this population; no such AEs were reported. Two episodes of moderate exacerbation of hypertension were reported by one site investigator as AEs in the same patient who had a prior history of hypertension, received the lowest roxadustat dose 0.7 mg/kg BIW and gained excessive fluid weight. No hypertension exacerbation or other AEs of special interest were reported in the higher dose groups, and overall, no safety signal was detected from the ABPM. No evidence of liver toxicity or sustained increases of liver enzymes or serum bilirubin were reported in this study." |
| Brigandi 2016 | GSK1278863 | Placebo |
CKD participants: nausea (6/61), dyspepsia (2/61), vomiting (3/61), hypotension (3/61), abdominal pain upper (2/61), dizziness (2/61), insomnia (2/61), idiopathic thrombocytopenic purpura (1/61), acute coronary syndrome (1/61), abdominal discomfort (1/61), diarrhoea (1/61), pancreatitis acute (1/61), chest pain (1/61), malaise (1/61), carbon dioxide abnormal (1/61), anorexia (1/61), dehydration (1/61), hyperuricaemia (1/61), arthritis (1/61), headache (1/61), lethargy (1/61), AKI (1/61), reduced kidney function (1/61), death (0/61) SAE: acute coronary syndrome, AKI, diabetic ketoacidosis, ketoacidosis, hyperglycaemia, hypotension, lower respiratory tract infection, AKI, dehydration, respiratory failure, pneumonia (no information on number of patients were reported). HD participants: abdominal pain (1/31), nausea (0/31), decreased appetite (0/31), death (0/31) SAE: atrial fibrillation, hepatitis acute, peripheral arterial occlusive disease (no information on number of patients were reported) |
CKD participants: nausea (0/9), dyspepsia (1/9), vomiting (0/9), hypotension (0/9), abdominal pain upper (0/9), dizziness (0/9), insomnia (0/9), idiopathic thrombocytopenic purpura (0/9), acute coronary syndrome (0/9), abdominal discomfort (0/9), diarrhoea (0/9), pancreatitis acute (0/9), chest pain (0/9), malaise (0/9), carbon dioxide abnormal (0/9), anorexia (0/9), dehydration (0/9), hyperuricaemia (0/9), arthritis (0/9), headache (0/9), lethargy (0/9), AKI (0/9), reduced kidney function (0/9), death (0/9) SAE: impaired liver functions, sciatica, pyrexia, peripheral arterial occlusive disease (no information on number of patients were reported). HD participants: abdominal pain (0/6), nausea (1/6), decreased appetite (1/6), death (0/6) SAE (0/6) |
“AEs, regardless of causality, were reported by 35 of 61 (57%) patients with CKD‐3/4/5 and 15 of 31 (48%) patients with CKD‐5D receiving GSK1278863. Overall, the frequency of AE reports was similar in the placebo arms in both groups. The most common AE in the CKD‐3/4/5 group was nausea (n = 9 [13%]; 3 [21%] with 25 mg and 6 [40%] with 100 mg). In the CKD‐3/4/5 group, 16 of 70 (23%) patients had investigator‐assessed drug‐related AEs, of which nausea was the most common (reported by 6 of 70 [9%] of patients). In the CKD‐5D group, the most commonly reported AEs were anaemia and hypotension, each occurring in 2 (5%) patients. In this population, a total of 2 patients reported investigator‐assessed drug‐related AEs of abdominal pain in 1 patient (10‐mg dose) and nausea and decreased appetite, both in 1 patient receiving placebo. Early termination from the study due to AEs was reported by 6 (9%) and 3 (8%) patients in the CKD‐3/ 4/5 and CKD‐5D groups, respectively, and none was considered drug‐related by the investigator. In the CKD‐3/4/5 group, serious AEs were reported for 7 individuals. Only 2 patients, both in the 100‐mg group, had possibly related serious AEs. In the CKD‐5D group, 3 individuals had serious AEs. No deaths were reported during the study." |
| Chen 2019 | Roxadustat | EPO alfa | Any AE during treatment (96/204), upper respiratory tract infection (37/204), hypertension (25/204), hyperkalaemia (15/204), chest discomfort (13/204), vomiting (12/204), asthenia (12/204), alanine aminotransferase increased (11/204), dizziness (10/204), hypotension (10/204), muscle spasms (5/204), any SAE during treatment (29/204), blood or lymphatic system disorder (1/204), cardiac disorder (5/204), endocrine disorder (1/204), GI disorder (2/204), hepatobiliary disorder (2/204), immune system disorder (2/204), infection or infestation (5/204), injury, poisoning, or procedural complication (7/204), metabolism or nutrition disorder (1/204), nervous system disorder (3/204), product issue (0/204), renal or urinary disorder (4/204), reproductive system or breast disorder (1/204), vascular disorder (2/204), death (0/204) | Any AE during treatment (36/100), upper respiratory tract infection (11/100), hypertension (16/100), hyperkalaemia (1/100), chest discomfort (0/100), vomiting (2/100), asthenia (2/100), alanine aminotransferase increased (4/100), dizziness (6/100), hypotension (6/100), muscle spasms (5/100), any SAE during treatment (10/100), blood or lymphatic system disorder (0/100), cardiac disorder (1/100), endocrine disorder (0/100), GI disorder (0/100), hepatobiliary disorder (0/100), immune system disorder (0/100), infection or infestation (3/100), injury, poisoning, or procedural complication (5/100), metabolism or nutrition disorder (0/100), nervous system disorder (0/100), product issue (1/100), renal or urinary disorder (0/100), reproductive system or breast disorder (0/100), vascular disorder (0/100), death (0/100) | “A total of 159 of 204 patients (77.9%) treated with roxadustat and 63 of 100 patients (63.0%) treated with epoetin alfa reported having at least one adverse event during treatment. The most frequently reported event was upper respiratory infection, which occurred in 37 patients (18.1%) in the roxadustat group and in 11 (11.0%) in the epoetin alfa group. A total of 29 patients (14.2%) treated with roxadustat and 10 (10.0%) treated with epoetin alfa reported having at least one serious adverse event during treatment. The most frequently reported serious adverse event was vascular‐access complication, which occurred in similar proportions of the treatment groups (6 patients [2.9%] in the roxadustat group and 3 patients [3.0%] in the epoetin alfa group). No deaths occurred during the reporting period. Adverse events that occurred in at least 5% of the patients in either group. Hyperkalemia was reported more frequently in the roxadustat group than in the epoetin alfa group in this open‐label trial. The proportion of patients with potassium values within categories from 5.5 mmol/L or less, more than 5.5 to 6.0 mmol/L, more than 6.0 to 6.5 mmol per liter, and more than 6.5 mmol/L at baseline and at weeks 13 and 27 were generally similar in the treatment groups." |
| Chen 2019a | Roxadustat | Placebo | Any AE (37/101), hyperkalaemia (16/101), metabolic acidosis (12/101), anaemia (0/101), diarrhoea (0/101), peripheral oedema (7/101), pyrexia (2/101), upper respiratory tract infection (5/101), gout (1/101), back pain:0/101), dizziness (1/101), hypertension (6/101), any SAE (9/101), serious anaemia (0/101), corona‐artery disease (0/101), GI haemorrhage (1/101), acute cholecysts (0/101), cholelithiasis (0/101), lung infection (1/101), serious hyperkalaemia (2/101), hypokalaemia (1/101), serious metabolic acidosis (1/101), kidney failure (1/101), chronic GN (1/101), azotaemia (0/101), renal impairment (0/101), dysfunctional uterine bleeding (0/101), acute respiratory failure (0/101), rash (1/101), hypertension (1/101) | Any AE (25/51), hyperkalaemia (4/51), metabolic acidosis (1/51), anaemia (3/51), diarrhoea (3/51), peripheral oedema (3/51), pyrexia (3/51), upper respiratory tract infection (4/51), gout (3/51), back pain (3/51), dizziness (4/51), hypertension (2/51), any SAE (6/51), serious anaemia (1/51), corona‐artery disease (1/51), GI haemorrhage (0/51), acute cholecysts (1/51), cholelithiasis (1/51), lung infection (1/51), serious hyperkalaemia (0/51), hypokalaemia (0/51), serious metabolic acidosis (0/51), kidney failure (0/51), chronic GN (0/51), azotaemia (1/51), renal impairment:1/51), dysfunctional uterine bleeding (1/51), acute respiratory failure (1/51), rash (0/51), hypertension (0/51) | "During the randomized phase, at least one adverse event was reported in 69 of 101 patients (68%) in the roxadustat group (14.4 patient‐years) and in 38 of 51 patients (75%) in the placebo group. Hyperkalemia and metabolic acidosis were reported more often in the roxadustat group than in the placebo group (in 16 patients [16%] in the roxadustat group and in 4 patients [8%] in the placebo group for hyperkalaemia and in 12 patients [12%] and 1 patient [2%], respectively, for metabolic acidosis). Serious adverse events, which were consistent with those generally seen in patients with chronic kidney disease, were reported in 9 patients (9%) in the roxadustat group and in 6 patients (12%) in the placebo group. There were no deaths during the randomized phase of the trial. An increase in the level of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) occurred in 2 patients in the roxadustat group and 1 patient in the placebo group (2% in each group)" |
| Chen DD 2017 | Roxadustat | EPO alfa | Death (0/60), SAE (0/60) | Death (0/22), SAE (0/22) | "No SAEs occurred during the DD study. No deaths or major adverse cardiac events occurred in FG‐4592 subjects. In the DD study, 32 subjects (43%) among the total of 74 FG‐4592‐treated subjects and 4 subjects (18%) among the total of 22 epoetin alfa‐treated subjects reported having at least one TEAE. One adverse event of rash in the mid‐dose FG‐4592 arm in the DD study led to treatment discontinuation.” |
| Chen NDD 2017 | Roxadustat | Placebo | Death (0/61), SAE (0/61), muscle spasms (1/61), diarrhoea (2/61), vomiting (2/61), abdominal discomfort (0/61), nausea (4/61), dizziness (3/61), headache (2/61), hypertension (4/61), hyperkalaemia (6/61), liver injury (0/61), decreased appetite (0/61), TSAT decreased (8/61), CKD (4/61), nasopharyngitis (2/61), upper respiratory tract infection (3/61 | Death (0/30), SAE (0/30), muscle spasms (0/30), diarrhoea (1/30), vomiting (0/30), abdominal discomfort (1/30), nausea (1/30), dizziness (3/30), headache (0/30), hypertension (0/30), hyperkalaemia (2/30), liver injury (1/30), decreased appetite (0/30), TSAT decreased (1/30), CKD (0/30), nasopharyngitis (0/30), upper respiratory tract infection (3/30 | "Treatment‐emergent serious adverse events (SAEs) were reported in four (13.3%) placebo‐treated subjects and eight (13.1%) FG‐4592‐treated subjects in the blinded NDD study; no SAEs were deemed related to FG‐4592. One cardiovascular SAE of unstable angina was reported in a placebo‐treated subject but no such events were reported in FG‐4592‐treated subjects. No deaths or major adverse cardiac events occurred in FG‐4592 subjects. Nineteen (63%) placebo‐treated subjects and 36 (59%) FG‐4592 subjects reported at least one treatment emergent adverse event (TEAE) in the NDD study. One placebo‐treated but no FG4592‐treated subjects had elevations 3> upper limit of normal of either ALT or AST. Two adverse events of urinary tract infection and worsening chronic renal failure in the low‐dose FG4592 arm in the NDD study (a decline in eGFR from 11 mL/ min at screening to 8 mL/min at EOT) led to treatment discontinuation.” |
| DIALOGUE 1 2019 | Molidustat (25, 50, 75, 100 mg) | Placebo | Hyperparathyroidism secondary (1/101), constipation (5/101), diarrhoea (4/101), vomiting (0/101), nasopharyngitis (7/101), UTI (4/101), hyperkalaemia (4/101), dizziness (5/101), hypertension (10/101), acute MI (1/101), stroke (0/101), arterial occlusive disease (1/101), peripheral arterial occlusive disease (0/101), peripheral artery thrombosis (1/101), peripheral venous disease (0/101), death (0/101) | Hyperparathyroidism secondary (3/20), constipation (1/20), diarrhoea (1/20), vomiting (1/20), nasopharyngitis (2/20), UTI (3/20), hyperkalaemia (3/20), dizziness (3/20), hypertension (5/20), acute MI (0/20), stroke (1/20), arterial occlusive disease (0/20), peripheral arterial occlusive disease (1/20), peripheral artery thrombosis (0/20), peripheral venous disease (1/20), death (0/20) | "The incidences of treatment‐emergent adverse events (TEAEs) and serious TEAEs in the molidustat combined‐dose group were numerically lower than those seen in the placebo group. One increase in alanine transaminase levels in the placebo group of DIALOGUE 1 was reported." |
| DIALOGUE 2 2019 | Molidustat (25, 50, 75, mg) | Darbepoetin alfa | Diarrhoea (5/92), peripheral oedema (8/92), CKD (10/92), hypertension (14/92), acute MI (10/92), stroke (0/92), arterial occlusive disease (0/92), peripheral arterial occlusive disease (0/92), peripheral artery thrombosis (1/92), peripheral venous disease (0/92), death (1/92) | Diarrhoea (1/32), peripheral oedema (2/32), CKD (0/3), hypertension (4/32), acute MI (0/32), stroke (1/32), arterial occlusive disease (0/32), peripheral arterial occlusive disease (0/32), peripheral artery thrombosis (0/32), peripheral venous disease (0/32), death (1/32) | "The incidence of TEAEs in DIALOGUE 2 was numerically higher in the molidustat combined‐dose group than in the darbepoetin group, and the incidence of study drug‐related TEAEs in both DIALOGUE 2 and DIALOGUE 4 was numerically higher in the molidustat groups than in the darbepoetin/epoetin groups. Study drug‐related serious TEAEs occurred in only one and two patients in the molidustat groups of DIALOGUE 2 and DIALOGUE 4, respectively (hyponatraemia, prolonged QT interval, and hypotension)." |
| DIALOGUE 4 2019 | Molidustat (25, 50, 75, 150 mg) | Epoetin alfa or beta | Diarrhoea (12/157), nausea (8/157), vomiting (6/157), nasopharyngitis (9/157), Hb decreased (10/157), Hb increased (13/157), hypertension (17/157), acute MI (2/157), stroke (2/157), arterial occlusive disease (0/157), peripheral arterial occlusive disease (0/157), peripheral artery thrombosis (0/157), peripheral venous disease (0/157), venous occlusion (1/157), death (1/157) | Diarrhoea (2/42), nausea (2/42), vomiting (0/42), nasopharyngitis (1/42), Hb decreased (2/42), Hb increased (2/42), hypertension (8/42), acute MI (0/42), stroke (0/42), arterial occlusive disease (0/42), peripheral arterial occlusive disease (0/42), peripheral artery thrombosis (0/42), peripheral venous disease (0/42), death (0/42) | "The incidence of study drug‐related TEAEs in both DIALOGUE 2 and DIALOGUE 4 was numerically higher in the molidustat groups than in the darbepoetin/epoetin groups. Study drug‐related serious TEAEs occurred in only one and two patients in the molidustat groups of DIALOGUE 2 and DIALOGUE 4, respectively (hyponatraemia, prolonged QT interval, and hypotension)" |
| DOLOMITES 2021 | Roxadustat | Darbepoetin | Total SAEs (209/323). The complete list is available in the results posted in the NCT02021318 Acute MI (5/323), cardiac arrest (3/323), coronary artery disease (1/323), MI (3/323), glaucoma (0/323), abdominal pain (1/323), diarrhoea (3/323), vomiting (1/323), cardiac death (2/323), chest pain (2/323), sudden death (14/323), liver injury (0/323), kidney transplant failure (0/323), catheter site infection (0/323), renal cyst infection (2/323), AVF site complication (3/323), arterial bypass occlusion (1/323), shunt occlusion (0/323), tibia fracture (0/323), vascular access site thrombosis (0/323), diabetes mellitus (2/323), cerebral ischemias (1/323), Ischaemic stroke (0/323), depression (1/323), AKI (7/323), CKD (1/323), kidney failure (108/323), GFR decreased (33/323), pulmonary hypertension (1/323), thrombosis (1/323) Other AEs (not including serious) GFR decrease: 33/323 Overall data > 5% reported in the Astellas website AVF thrombosis (16/323), hyperkalaemia (38/323) |
Total SAEs (181/293). The complete list is available in the results posted in the NCT02021318 Acute MI (8/293), cardiac arrest (3/293) coronary artery disease (3/293), MI (0/293), glaucoma (1/293), abdominal pain (1/293), diarrhoea (2/293), vomiting (0/293), cardiac death (0/293), chest pain (1/293), sudden death (1/293), liver injury (1/293), renal transplant failure (1/293), catheter site infection (1/293), renal cyst infection (0/293), AVF site complication (1/293), arterial bypass occlusion (0/293), shunt occlusion (1/293), tibia fracture (1/293), vascular access site thrombosis (1/293), diabetes mellitus (0/293), cerebral ischemias (0/293), Ischaemic stroke (3/293), depression (0/293), AKI (7/293), CKD (0/293), kidney failure (106/293), GFR decreased (28/293), pulmonary hypertension (3/293), thrombosis (0/293) Other AEs (not including serious) GFR decrease: 28/293 Overall data > 5% reported in the Astellas website AVF thrombosis (10/293), hyperkalaemia (42/293) |
"Common TEAEs in both groups were end‐stage renal disease, hypertension, decreased eGFR, and peripheral edema." |
| HIMALAYAS 2021 | Roxadustat | EPO alfa | The following are related > 5% AEs Hypertension (99/522), diarrhoea (72/522), muscle spasms (60/522), AVF thrombosis (59/522), headache (57/522), hypotension (54/522), hyperphosphataemia (52/522), nausea (45/522), pneumonia (42/522), constipation (35/522), vomiting (32/522), AVF site complication (31/522), pruritus (30/522), fluid overload (29/522), cough (28/522), dizziness (28/522), hyperkalaemia (26/522), procedural hypotension (26/522), hyperparathyroidism, secondary (25/522), back pain (18/522) AEs < 1% were reported in table 8 of Provenano 2021 |
The following are related to > 5% AEs Hypertension (88/517), diarrhoea (38/517), muscle spasms (39/517), AVF thrombosis (46/517), headache (44/517), hypotension (35/517), hyperphosphataemia (35/517), nausea (30/517), pneumonia (40/517), constipation (23/517), vomiting (17/517), AVF site complication (43/517), pruritus (22/517), fluid overload (28/517), cough (21/517), dizziness (24/517), hyperkalaemia (36/517), procedural hypotension (31/517), hyperparathyroidism, secondary (27/517), back pain (27/517) AEs < 1% were reported in table 8 of Provenano 2021 |
"More than 85%of patients in the roxadustat and epoetin alfa groups experienced one or more TEAE during treatment. The most frequently reported TEAE in the roxadustat group was hypertension, which occurred in 19.0%of patients in the roxadustat group and 17.0% in the epoetin alfa group. Hyperkalemia rates were lower in the roxadustat versus epoetin alfa group (5.0% versus 7.0%). In the roxadustat and epoetin alfa groups, 44.8% and 42.2%, respectively, experienced one or more TESAE during treatment (Table 8). There were 63 (12.1%) fatal TEAEs in the roxadustat group and 59 (11.4%) in the epoetin alfa group." |
| Holdstock 2019 | Daprodustat | EPO | AEs and frequency of MACE and component endpoints in the combined daprodustat (both Holdstock 2019 and Holdstock 2019a) Note: cumulative data for all‐cause death, MI, stroke, blood transfusion and hospitalisation due to heart failure were reported |
AEs and frequency of MACE and component endpoints in the combined control group (both Holdstock 2019 and Holdstock 2019a) To note that cumulative data for all‐cause death, MI, stroke, blood transfusion and hospitalisation due to heart failure were reported |
"Other adverse events included sequelae of excessive erythropoiesis, fatal CV and thromboembolic events, cardiomyopathy, pulmonary artery hypertension, cancer, oesophageal and gastric erosions, exacerbation of rheumatoid arthritis, and retinal and choroidal neovascularization. The proportion of these events was the same in both groups (8%). One participant with advanced CKD and gout who was randomized to daprodustat 1 mg met the protocol‐defined liver stopping criteria 2 weeks after treatment initiation, with alanine aminotransferase (ALT) increased." |
| Holdstock 2019a | Daprodustat | EPO | AEs and frequency of MACE and component endpoints in the combined daprodustat (both Holdstock 2019 and Holdstock 2019a) | AEs and frequency of MACE and component endpoints in the combined control group (both Holdstock 2019 and Holdstock 2019a) | "Other adverse events included sequelae of excessive erythropoiesis, fatal CV and thromboembolic events, cardiomyopathy, pulmonary artery hypertension, cancer, oesophageal and gastric erosions, exacerbation of rheumatoid arthritis, and retinal and choroidal neovascularization. The proportion of these events was the same in both groups (8%). One participant with advanced CKD and gout who was randomized to daprodustat 1 mg met the protocol‐defined liver stopping criteria 2 weeks after treatment initiation, with alanine aminotransferase (ALT) increased." |
| Hou 2021 | Roxadustat | ESA | Upper respiratory tract infection (2/86), hypertension (5/86), hyperkalaemia (8/86), chest discomfort (2/86), vomiting (3/86), asthenia (2/86), alanine aminotransferase increased (2/86), dizziness (0/86), hypotension (1/86), muscle spasms (0/86), constipation (1/86), pruritus (1/86), peritonitis (1/86), ostealgia (2/86), headache (3/86), insomnia (5/86) | Upper respiratory tract infection (1/43), hypertension (3/43), hyperkalaemia (2/43), chest discomfort (1/43), vomiting (1/43), asthenia (1/43), alanine aminotransferase increased (0/43), dizziness (1/43), hypotension (1/43), muscle spasms (1/43), constipation (0/43), pruritus (0/43), peritonitis (1/43), ostealgia (0/43), headache (2/43), insomnia (0/43) | "Common adverse events included hyperkalaemia, hypertension, and insomnia." "During treatment, serious adverse events (SAEs) were reported in 2 (2%) roxadustat‐treated subjects and 1 (2%) ESAs‐treated patient. One SAE of MI was reported in a roxadustat‐treated subject, and he discontinued the trial. A 66‐year‐old patient in the roxadustat group with hypertension died in the intensive care unit (death cause: acute heart failure) during follow‐up. A 61‐year‐old patient with diabetes in the ESAs group died on day 84 due to heart failure." "Overall, 38 of 86 patients (44%) treated with roxadustat and 15 of 43 patients (35%) treated with ESAs incurred at least one adverse event during treatment." |
| INNO2VATE 2020 | Vadadustat | Darbepoetin alfa | Any AE: 150/179 Hypertension (29/179), diarrhoea (18/179), pneumonia (13/179), hyperkalaemia (8/179), fluid overload (13/179), fall (11/179), headache (8/179), hypotension (7/179), nausea (14/179), vomiting (13/179), UTI (11/179), dialysis‐related complication (8/179), cough (11/179), AVF site complication (8/179), dyspnoea (13/179), upper respiratory tract infection (5/179), pain in extremity (8/179), sepsis (6/179), AVF thrombosis (6/179), nasopharyngitis (10/179), back pain (7/179), hypoglycaemia (5/179), atrial fibrillation (5/179), bronchitis (5/179), procedural hypotension (11/179) |
Any AE: 159/186 Hypertension (24/186), diarrhoea (18/186), pneumonia (15/186), hyperkalaemia (10/186), fluid overload (6/186), fall (9/186), headache (11/186), hypotension (16/186), nausea (13/186), vomiting (10/186), UTI (16/186), dialysis‐related complication (11/186), cough (5/186), AVF site complication (9/186), dyspnoea (10/186), upper respiratory tract infection (9/186), pain in extremity (6/186), sepsis (9/186), AVF thrombosis (10/186), nasopharyngitis (8/186), back pain (4/186), hypoglycaemia (9/186), atrial fibrillation (6/186), bronchitis (7/186), procedural hypotension (12/186) |
"In the incident DD‐CKD trial, 83.8% of the patients in the vadadustat group and 85.5% of the patients in the darbepoetin alfa group had at least one adverse event after the start of treatment. The incidence of serious adverse events was 49.7% in the vadadustat group and 56.5% in the darbepoetin alfa group." |
| INNO2VATE 2020a | Vadadustat | Darbepoetin alfa | Any AE: 1562/1768 Hypertension (187/1768), diarrhoea (230/1768), pneumonia (195/1768), hyperkalaemia (160/1768), fluid overload (156/1768), fall (150/1768), headache (160/1768), hypotension (146/1768), nausea (149/1768), vomiting (120/1768), UTI (110/1768), dialysis‐related complication (99/1768), cough (99/1768), AVF site complication (94/1768), dyspnoea (92/1768), upper respiratory tract infection (99/1768), pain in extremity (91/1768), sepsis (89/1768), AVF thrombosis (106/1768), nasopharyngitis (92/1768), back pain (76/1768), hypoglycaemia (92/1768), atrial fibrillation (69/1768), bronchitis (67/1768), procedural hypotension (69 |
Any AE: 1580/1769 Hypertension (244/1769), diarrhoea (178/1769), pneumonia (172/1769), hyperkalaemia (191/1769), fluid overload (173/1769), fall (159/1769), headache (135/1769), hypotension (141/1769), nausea (134/1769), vomiting (124/1769), UTI (117/1769), dialysis‐related complication (122/1769), cough (121/1769), AVF site complication (120/1769), dyspnoea (119/1769), upper respiratory tract infection (112/1769), pain in extremity (117/1769), sepsis (101/1769), AVF thrombosis (78/1769), nasopharyngitis (84/1769), back pain (99/1769), hypoglycaemia (78/1769), atrial fibrillation (95/1769), bronchitis (95/1769), procedural hypotension (74/1769) |
"In the prevalent DD‐CKD trial, 88.3% of the patients in the vadadustat group and 89.3% of the patients in the darbepoetin alfa group had at least one adverse event after the start of treatment. The incidence of serious adverse events was 55.0% and 58.3%, respectively." |
| Meadowcroft 2019 | Daprodustat | Placebo | AE: diarrhoea (16/177), nasopharyngitis (15/177), nausea (13/177), headache (9/177), hypertension (9/177), hyperkalaemia (8/177), back pain (7/177), any MACE (7/177), death (any cause) (5/177), MI (3/177), stroke (0/177), hospitalisation due to heart failure (5/177) SAE (31/177), cardiac arrest (3/177), thrombosis related to vascular access (3/177), cancer (3/177), oesophageal and gastric erosions (3/177), pulmonary artery hypertension (2/177), elevations of estimated sPAP (2/177), macular oedema in (2/177), exacerbation of RA (1/177) |
AE: diarrhoea (2/39), nasopharyngitis (5/39), nausea (0/39), headache (2/39), hypertension (1/39), hyperkalaemia (0/39), back pain (4/39), any MACE (0/39), death (any cause) (0/39), MI (0/39), stroke (0/39), hospitalisation due to heart failure (1/39) SAE (10/39), cardiac arrest (0/177), thrombosis related to vascular access (2/39), cancer (0/39), oesophageal and gastric erosions (0/39), pulmonary artery hypertension (0/39), elevations of estimated sPAP (0/39), macular oedema in (0/39), exacerbation of RA (0/39) |
"On the AEs most frequently reported, diarrhoea, nausea, hypertension and hyperkalaemia were reported more often in the combine daprodustat group, while nasopharyngitis and back pain were reported more often in the control group. [...] Serious adverse events were reported in 31 participants in the daprodustat group and 10 participants in the control group. the most common adverse events. The most common Serious AEs in the intervention group were myocardial infarction and cardiac arrest, in 3 participants each, followed by unstable angina, cardiac failure, hypertensive crisis, lobar pneumonia and pulmonary oedema in 2 participants each. In the control group no specific serious AEs were reported in more than one participant." "Thrombosis events related to vascular access were reported in 6 (3%) daprodustat participants and 2 (5%) control participants. Also reported for the combined daprodustat group were cancer progression or recurrence in 3 (2%) participants (2 retrospectively identified as pre‐existing but undiagnosed, and 1 basal cell carcinoma); oesophageal and gastric erosions in 3 (2%) participants; pulmonary artery hypertension, reported as elevations of estimated sPAP, in 2 (1%) participants; macular edema in 2 (1%) participants, based on review of ophthalmology exam data; and an exacerbation of RA in 1 (<1%) participants with pre‐existing RA" |
| MIYABI HD‐M 2019 | Molidustat | Darbepoetin alfa | AE (TEAEs) (95.4%), serious TEAEs (24.2%), TEAEs with an outcome of death (1.3%) or AEs of special interest (4.6%) | AE (TEAEs) (94.7%), serious TEAEs (18.4%), TEAEs with an outcome of death (2.6%) or AEs of special interest (3.9%) | "There were no apparent between‐group differences in incidence of treatment‐emergent adverse events (TEAEs) (molidustat, 95.4%; darbepoetin alfa, 94.7%), serious TEAEs (24.2%; 18.4%), TEAEs with an outcome of death (1.3%; 2.6%) or AEs of special interest (4.6%; 3.9%)." |
| MIYABI ND‐C 2019 | Molidustat | Darbepoetin alfa | AEs: 84.1%; the most common AEs were nasopharyngitis (20.7%) and worsening CKD (13.4%) Constipation (10/82), dental caries (1/82), diarrhoea (8/82), nausea (6/82), stomatitis (1/82), pyrexia (1/82), bronchitis (5/82), nasopharyngitis (26/82), pneumonia (6/82), contusion (10/82), hyperkalaemia (10/82), back pain (8/82), muscle spasms (2/82), CKD worsening (16/82), cough (2/82), eczema (3/82), AVF operation (3/82), hypertension (8/82) |
AEs: 91.1%; the most common AEs were nasopharyngitis (25.3%) and worsening CKD (6.3%) Constipation (8/82), dental caries (4/82), diarrhoea (3/82), nausea (4/82), stomatitis (4/82), pyrexia (5/82), bronchitis (3/82), nasopharyngitis (21/82), pneumonia (2/82), contusion (3/82), hyperkalaemia (9/82), back pain (3/82), muscle spasms (5/82), CKD worsening (9/82), cough (4/82), eczema (4/82), AVF operation (5/82), hypertension (4/82) |
"AEs were experienced in 84.1% of patients in the MO group and in 91.1% of patients in the DA group up to 36 weeks. The most common AEs occurred ≥ 5% of patients in any group were nasopharyngitis (20.7% and 25.3%, respectively) and worsening of chronic kidney disease (13.4% and 6.3%, respectively)." "Severe TEAEs were also observed in 17.1% of patients for molidustat and 7.6% for darbepoetin. The most common TEAEs were nasopharyngitis (31.7% for molidustat and 26.6% for darbepoetin) and worsening of CKD (19.5% for molidustat and 11.4% for darbepoetin)." |
| MIYABI ND‐M 2019 | Molidustat | Darbepoetin alfa | Constipation (7/82), diarrhoea (7/82), large intestine polyp (0/82), oedema, peripheral (7/82), influenza (6/82), nasopharyngitis (28/82), pneumonia (4/82), hyperkalaemia (2/82), hypoglycaemia (6/82), back pain (6/82), insomnia (5/82), CKD worsening (15/82), hypertension (2/82) | Constipation (7/82), diarrhoea (10/82), large intestine polyp (5/82), oedema, peripheral (2/82), influenza (1/82), nasopharyngitis (33/82), pneumonia (5/82), hyperkalaemia (7/82), hypoglycaemia (0/82), back pain (3/82), insomnia (4/82), CKD worsening (8/82), hypertension (5/82) | "The proportion of patients who reported at least 1 treatment‐emergent adverse event (TEAE) was 92.7% for molidustat and 96.3% for darbepoetin." |
| Nangaku 2021 | Vadadustat | Darbepoetin alfa | Nasopharyngitis (19.8%), diarrhoea (10.5%), and shunt stenosis (8.0%) The incidence rates of SAE were 13.0% CV event, cardiac failure (13/162), cerebral infarction (1/162), carotid artery stenosis (1/162), cerebellar infarction (1/162), intracranial aneurysm (1/162), lacunar infarction (1/162), thrombotic cerebral infarction (1/162), subarachnoid haemorrhage (0/162), cardiac failure congestive (2/162), angina pectoris (1/162), coronary artery stenosis (1/162), myocardial ischemias (1/162), angina unstable (1/162), arteriosclerosis coronary artery (1/162), cardiac failure (1/162), pulmonary oedema (2/162), subdural hematoma (1/162), coronary artery restenosis (0/162), retinal disorder (21/162), retinal haemorrhage (16/162), diabetic retinopathy (2/162), macular oedema (1/162), retinal oedema (1/162), retinal vein occlusion (1/162), vitreous floaters (1/162), cystoid macular oedema (1/162), chorioretinopathy (1/162), retinal detachment (0/162), retinal vascular disorder (0/162), vitreous detachment (0/162), vitreous haemorrhage (0/162), retinal aneurysm (0/162), age‐related macular degeneration (0/162), malignancy (7/162), breast cancer (1/162), gastric cancer (1/162), seborrhoeic keratosis (1/162), cholesteatoma (1/162), laryngeal papilloma (1/162), squamous cell carcinoma of skin (1/162), uterine leiomyoma (1/162), pyogenic granuloma (0/162), thymoma (0/162), prostate cancer (0/162), pancreatic neoplasm (0/162), urethral neoplasm (0/162), renal cell carcinoma (0/162), GI submucosal tumour (0/162), hyperkalaemia (1/162), thromboembolism (12/162), cerebral infarction (1/162), cerebellar infarction (1/162), lacunar infarction (1/162), thrombotic cerebral infarction (1/162), retinal vein occlusion (1/162), peripheral arterial occlusive disease (3/162), thrombophlebitis (0/162), peripheral artery occlusion (0/162), shunt occlusion (4/162), shunt thrombosis (1/162), AVF thrombosis (0/162), pulmonary hypertension (0/162) |
Nasopharyngitis (28.6%), diarrhoea (9.9%), and shunt stenosis (12.4%) The incidence rates of SAEs were 10.6% CV event, cardiac failure (15/161), cerebral infarction (5/161), carotid artery stenosis (1/161), cerebellar infarction (1/161), intracranial aneurysm (0/161), lacunar infarction (0/161), thrombotic cerebral infarction (0/161), subarachnoid haemorrhage (1/161), cardiac failure congestive (0/161), angina pectoris (3/161), coronary artery stenosis (2/161), myocardial ischemias (1/161), angina unstable (0/161), arteriosclerosis coronary artery (0/161), cardiac failure (0/161), pulmonary oedema (1/161), subdural hematoma (0/161), coronary artery restenosis (1/161), retinal disorder (16/161), retinal haemorrhage (10/161), diabetic retinopathy (1/161), macular oedema (3/161), retinal oedema (0/161), retinal vein occlusion (0/161), vitreous floaters (0/161), cystoid macular oedema (0/161), chorioretinopathy (0/161), retinal detachment (1/161), retinal vascular disorder (1/161), vitreous detachment (1/161), vitreous haemorrhage (1/161), retinal aneurysm (1/161), age‐related macular degeneration (1/161), malignancy (9/161), breast cancer (1/161), gastric cancer (1/161), seborrhoeic keratosis (1/161), cholesteatoma (0/161), laryngeal papilloma (0/161), squamous cell carcinoma of skin (0/161), uterine leiomyoma (0/161), pyogenic granuloma (1/161), thymoma (1/161), prostate cancer (1/161), pancreatic neoplasm (1/161), urethral neoplasm (1/161), renal cell carcinoma (1/161), GI submucosal tumour (1/161), hyperkalaemia (1/161), thromboembolism (14/161), cerebral infarction (5/161), cerebellar infarction (1/161), lacunar infarction (0/161), thrombotic cerebral infarction (0/161), retinal vein occlusion (0/161), peripheral arterial occlusive disease (3/161), thrombophlebitis (1/161), peripheral artery occlusion (1/161), shunt occlusion (4/161), shunt thrombosis (0/161), AVF thrombosis (1/161), pulmonary hypertension (0/161) |
"A similar proportion of patients reported at least one AE during the 52‐week treatment period: 95.1% (154 of 162 patients) and 98.1% (158 of 161 patients) in the vadadustat and darbepoetin alfa groups, respectively, as presented in Table 3. During the 52‐week treatment period, 11.1% and 3.7% of patients in the vadadustat and darbepoetin alfa groups, respectively, reported at least one ADR, and 25.3 and 27.3% of those in the vadadustat and darbepoetin alfa groups, respectively, reported at least one serious AE; however, none of the serious events was considered to be related to study treatment." |
| Nangaku 2021a | Vadadustat | Darbepoetin alfa | AE: 72.2% The most common AEs in the VDT group were nasopharyngitis (VDT: 14.6%, DA: 12.4%), diarrhoea (VDT: 10.6%, DA: 3.3%), and constipation (VDT: 5.3%, DA: 3.9%) The incidence rate of SAEs was 13.9% Nasopharyngitis (37/151), diarrhoea (18/151), constipation (14/151), contusion (11/151), peripheral oedema (11/151), vomiting (10/151), CKD (9/151), renal impairment (8/151), pyrexia (8/151), pruritus (7/151), cystitis (6/151), eczema (5/151), hypertension (2/151), CV events (9/151), cardiac failure, chronic (3/151), cardiac failure, congestive (2/151), intracranial aneurysm (1/151), cardiac failure (1/151), myocardial ischemias (1/151), subarachnoid haemorrhage (1/151), cardiac failure, acute (0/151), lacunar infarction (0/151), cerebral infarction (0/151), retinal disorders (1/151), retinal haemorrhage (2/151), macular oedema (1/151), retinal exudates (1/151), retinal vein occlusion (1/151), age‐related macular degeneration (1/151), diabetic retinopathy (0/151), scintillating scotoma (0/151), vitreous floaters (0/151), retinal aneurysm (0/151), macular fibrosis (0/151), malignancy (2/151), colon adenoma (1/151), oral papilloma (1/151), basal cell carcinoma (0/151), gastric cancer (0/151), keratoacanthoma (0/151), renal cancer (0/151), seborrhoeic keratosis (0/151), skin papilloma (0/151), renal cancer metastatic (0/151), kidney angiomyolipoma (0/151), hyperkalaemia (1/151), thromboembolism (1/151), retinal vein occlusion (1/151), lacunar infarction (0/151), cerebral infarction (0/151), acute MI (0/151), pulmonary embolism (0/151), shunt occlusion (0/151), pulmonary hypertension (0/151) |
AE: 73.2% The incidence rate of SAEs was 14.4% Nasopharyngitis (43/153), diarrhoea (8/153), constipation (11/153), contusion (7/153), peripheral oedema (5/153), vomiting (3/153), CKD (14/153), renal impairment (8/153), pyrexia (1/153), pruritus (8/153), cystitis (9/153), eczema (8/153), hypertension (11/153), CV events (5/153), cardiac failure, chronic (0/153), cardiac failure, congestive (0/153), intracranial aneurysm (1/153), cardiac failure (0/153), myocardial ischemias (0/153), subarachnoid haemorrhage (0/153), cardiac failure, acute (2/153), lacunar infarction (2/153), cerebral infarction (1/153), retinal disorders (0/153), retinal haemorrhage (5/153), macular oedema (0/153), retinal exudates (0/153), retinal vein occlusion (0/153), age‐related macular degeneration (0/153), diabetic retinopathy (3/153), scintillating scotoma (1/153), vitreous floaters (1/153), retinal aneurysm (1/153), macular fibrosis (1/153), malignancy (6/153), colon adenoma (0/153), oral papilloma (0/153), basal cell carcinoma (1/153), gastric cancer (1/153), keratoacanthoma (1/153), renal cancer (1/153), seborrhoeic keratosis (1/153), skin papilloma (1/153), renal cancer metastatic (1/153), kidney angiomyolipoma (1/153), hyperkalaemia (5/153), thromboembolism (6/153), retinal vein occlusion (0/153), lacunar infarction (2/153), cerebral infarction (1/153), Acute MI (1/153), pulmonary embolism (1/153), shunt occlusion (1/153), pulmonary hypertension (0/153) |
"At least one adverse event (AE) was seen in 72.2% (VDT) and 73.2% (DA) subjects. The most common AEs in the VDT group were nasopharyngitis (VDT: 14.6%, DA: 12.4%), diarrhoea (VDT: 10.6%, DA: 3.3%), and constipation (VDT: 5.3%, DA: 3.9%). The incidence rates of serious AEs were 13.9% (VDT) and 14.4% (DA). [...] In the vadadustat group, nine patients reported a CV event or cardiac failure, four reported at least one retinal disorder event, two reported a malignancy, one reported an event of hyperkalaemia, and one reported a thromboembolism. There were no patients with pulmonary hypertension. All AEs of special interest, except one retinal haemorrhage, were considered to be not related to vadadustat." |
| Nangaku 2021b | Daprodustat | Mircera | Nasopharyngitis (49/149), constipation (10/149), back pain (12/149), renal impairment (9/149), hyperkalaemia (12/149), pruritus (12/149), CKD (6/149), influenza (8/149), contusion (5/149), diarrhoea (5/149), BP increased (8/149), hypertension (4/149), muscle spasms (4/149) | Nasopharyngitis (56/150), constipation (18/150), back pain (11/150), renal impairment (13/150), hyperkalaemia (8/150), pruritus (5/150), CKD (10/150), influenza (8/150), contusion (8/150), diarrhoea (7/150), BP increased (4/150), hypertension (8/150), muscle spasms (7/150) | "There was no meaningful difference in the frequencies of adverse events." |
| NCT01888445 | Roxadustat (dose 50, 75, 100 mg) | Darbepoetin alfa | Treatment 1 (33 participants), treatment 2 (32 participants), treatment 3 (32 participants) TEAEs: treatment 1 (24, 72.7%), treatment 2 (26. 81.3%), treatment 3 (27, 84.4%) Drug‐related TEAEs: treatment 1 (8, 24.2%), treatment 2 (7. 21.9%), treatment 3 (12, 37.5%) Deaths: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Serious TEAEs: treatment 1 (4, 12.1%), treatment 2 (7, 21.9%), treatment 3 (4. 12.5%) Drug‐related serious TEAEs: treatment 1 (0), treatment 2 (2, 6.3%), treatment 3 (1, 3.1%) Eye disorders: treatment 1 (3, 9.1%), treatment 2 (2, 6.3%), treatment 3 (8, 25.0%) Retinal haemorrhage: treatment 1 (2, 6.1%), treatment 2 (2, 6.3%), treatment 3 (5, 15.6%) GI disorders: treatment 1 (7, 21.2%), treatment 2 (11, 34.4%), treatment 3 (13, 40.6%) Constipation: treatment 1 (1. 3.0%), treatment 2 (2, 6.3%), treatment 3 (2, 6.3%) Diarrhoea: treatment 1 (2, 6.1%), treatment 2 (1, 3.1%), treatment 3 (2, 6.3%) Nausea treatment: 1 (3, 9.1%), treatment 2 (2, 6.3%), treatment 3 (4, 12.5%) Vomiting: treatment 1 (2, 6.1%), treatment 2 (2, 6.3%), treatment 3 (7, 21.9%) Infections and infestations: treatment 1 (12, 36.4%), treatment 2 (12, 37.5%), treatment 3 (9, 28.1%) Nasopharyngitis: treatment 1 (10, 30.3%), treatment 2 (10, 31.3%), treatment 3 (8, 25.0%) Cardiac disorders: treatment 1 (2, 6.1%), treatment 2 (0), treatment 3 (2, 6.3%) Cardiac failure, congestive: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (2, 6.3%) Myocardial ischemias: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) GI disorders: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (1, 3.1%) Gastric ulcer: treatment 1 (0), treatment 2 (0), treatment 3 (0) Ileus: treatment 1 (0), treatment 2 (0): treatment 3 (1, 3.1%) Vomiting: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) General disorders and administration site conditions: treatment 1 (0), treatment 2 (0), treatment 3 (1, 3.1%) Gait disturbance: treatment 1 (0), treatment 2 (0), treatment 3 (1, 3.1%) Pneumonia bacterial: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Viral upper respiratory tract infection: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Injury, poisoning and procedural complications: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Vascular graft occlusion: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Investigations: treatment 1 (1, 3.0%), treatment 2 (1, 3.1%), treatment 3 (0) Hb decreased: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Liver function test abnormal: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Metabolism and nutrition disorders: treatment 1 (0): treatment 2 (0), treatment 3 (1, 3.1%) Decreased appetite: treatment 1 (0), treatment 2 (0), treatment 3 (1, 3.1%) Nervous system disorders: treatment 1 (0), treatment 2 (2, 6.3%), treatment 3 (1, 3.1%) Cerebral infarction: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Dizziness: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Lacunar infarction: treatment 1 (0), treatment 2 (0), treatment 3 (1, 3.1%) Respiratory, thoracic and mediastinal disorders: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Pneumonia aspiration: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Surgical and medical procedures: treatment 1 (0), treatment 2 (2, 6.3%), treatment 3 (0) Gastric polypectomy: treatment 1 (0), treatment 2 (1, 3.1%), treatment 3 (0) Intestinal polypectomy: treatment 1 (0), treatment 2 (2, 6.3%), treatment 3 (0) Vascular disorders: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Embolism, venous: treatment 1 (1, 3.0%), treatment 2 (0), treatment 3 (0) Haematoma: treatment 1 (0), treatment 2 (0), treatment 3 (0) |
Control: 32 participants. TEAEs: 25 (78.1%) Drug‐related TEAEs: 2 (6.3%) Deaths: 0 Serious TEAEs: 2 (6.3%) Drug‐related serious TEAEs: 0 Eye disorders: 4 (12.5%) Retinal haemorrhage: 0 GI disorders: 9 (28.1%) Constipation: 2 (6.3%) Diarrhoea: 2 (6.3%) Nausea: 1 (3.1%) Vomiting: 2 (6.3%) Infections and infestations: 15 (46.9%) Nasopharyngitis: 14 (43.8%) Cardiac disorders: 0 Cardiac failure congestive: 0. Myocardial ischemias control 0 GI disorders: 1 (3.1%). Gastric ulcer: 1 (3.1%) Ileus: 0 Vomiting: 0 General disorders and administration site conditions: 0 Gait disturbance: 0 Pneumonia bacterial: 0 Viral upper respiratory tract infection: 0 Injury, poisoning and procedural complications: 0 Vascular graft occlusion: 0 Investigations treatment: 0 Hb decreased: 0 Liver function test abnormal: 0 Metabolism and nutrition disorders: 0 Decreased appetite: 0 Nervous system disorders: 0 Cerebral infarction: 0 Dizziness: 0 Lacunar infarction: 0 Respiratory, thoracic and mediastinal disorders: 0 Pneumonia aspiration: 0 Surgical and medical procedures: 0 Gastric polypectomy: 0 Intestinal polypectomy: 0 Vascular disorders: 1 (3.1%) Embolism venous: 0 Haematoma: 1 (3.1%) |
"The proportion of patients experiencing drug‐related TEAEs and serious TEAEs in the ASP1517 groups was higher than that in the darbepoetin alfa group. The common (incidence > 5%) TEAEs in the pooled ASP1517 group included nasopharyngitis, vomiting, nausea, retinal haemorrhage, constipation, and diarrhoea. The common (incidence > 5%) TEAEs in the darbepoetin alfa group included nasopharyngitis, procedural hypotension, hyperparathyroidism secondary, constipation, diarrhoea, vomiting, excoriation, and back pain. The common (incidence > 1%) drug‐related TEAEs included vomiting, nausea, retinal haemorrhage, hypertension and blood pressure increased in the pooled ASP1517 group and macular fibrosis and hypertension in the darbepoetin alfa group. One patient in the ASP1517 50 mg group died of embolism venous and the event was considered unrelated to the study treatment. Other than this patient, the outcome of patients with serious TEAEs was reported as recovered or recovering. Drug‐related TEAEs leading to discontinuation of study treatment were reported in 2 (6.3%) patients each in the ASP1517 70 mg and 100 mg groups. A potential drug‐induced liver injury case was reported in 1 patient, which was considered unrelated to the study treatment because both AST and ALT remained within normal ranges during the study treatment and the event occurred approximately 2 weeks after discontinuation of the study treatment. No notable safety concerns were reported in other clinical laboratory evaluations, vital signs, ECGs or other safety‐related observations. Due to an apparent imbalance between the occurrences of retinal haemorrhage events reported as adverse events, it was decided to conduct a descriptive aggregate analysis of the same ophthalmological images following centralized and masked grading by experienced graders. When interpreting treatment outcomes, one needs to consider that the ASP1517 and darbepoetin treatment groups were generally well balanced in terms of factors related to the efficacy of ASP1517 at baseline. Differences between groups were present in terms of factors that predicted new retinal pathology including longer dialysis vintage duration and a higher number of subjects with a status of laser therapy prior to treatment in the darbepoetin treated patients. At screening retinal haemorrhages were seen in 15.5% of the study population prior to any study intervention in the analysis. During treatment, a total of 9 (10.1%) patients in the pooled ASP1517 group and 2 (6.5%) patients in the darbepoetin group displayed new or worsening retinal haemorrhage. For the group of patients without haemorrhages at baseline, the incidence of new or worsening retinal haemorrhages after the start of treatment was comparable between pooled ASP1517 and darbepoetin treatment groups, 8.0% vs 7.1%, respectively. When new or worsening retinal haemorrhage assessments were presented by baseline status, low patient numbers in the group with retinal haemorrhages at baseline made it difficult to interpret the data but the incidence prior to and during treatment appeared similar. No change in retinal thickness was observed over the course of the study in the ASP1517 and darbepoetin alfa treatment groups." |
| NDD‐CKD 2020 | Vadadustat (150, 300, 600 mg) | Placebo | Death (0/37), SAE (7/37), duodenal ulcer haemorrhage (0/37), hepatic function abnormal (1/37), influenza (0/37), lung infection (1/37), spinal compression1 fracture (0/37), AKI (1/37), kidney failure (1/37), renal impairment (0/37), asthma (0/37), interstitial lung disease (1/37), arteriovenous shunt procedure (3/37) | Death (0/14), SAE (4/14), duodenal ulcer haemorrhage (1/14), hepatic function abnormal (0/14), influenza (0/14), lung infection (0/14), spinal compression fracture (10/14), AKI (0/14), kidney failure (0/14), renal impairment (1/14), asthma (1/14), interstitial lung disease (0/14), arteriovenous shunt procedure (0/14) | "During the primary efficacy period, the incidence of treatment‐emergent adverse events (AEs) with placebo and vadadustat 150, 300 and 600mg was 36, 33, 58 and 54% (NDD‐CKD) and 40, 53, 73 and 40% (DDCKD), respectively. The most common AEs during the primary efficacy period were nausea and hypertension (NDD‐CKD) and diarrhoea, nasopharyngitis and shunt stenosis (DD‐CKD). Of 23 serious AEs in 18 patients, 1 was deemed related (hepatic function abnormal); no deaths were reported." |
| NDD‐CKD 2020a |
Vadadustat (150, 300, 600 mg) |
Placebo | Death (0/45), SAE (6/45), pericarditis (1/45), cholecystitis acute (1/45), enteritis infectious (1/45), AFVS complication (0/45), shunt stenosis (1/45), toxic encephalopathy (1/45), gastric ulcer haemorrhage (1/45), shunt stenosis (1/45), cerebral haemorrhage (1/45), anxiety (1/45) | Death (0/15), SAE (1/15), pericarditis (0/15), cholecystitis acute (0/15), enteritis infectious (0/15), AFVS complication (1/15), shunt stenosis (0/15), toxic encephalopathy (0/15), gastric ulcer haemorrhage (0/15), shunt stenosis (0/15), cerebral haemorrhage (0/15), anxiety (0/15) | "During the primary efficacy period, the incidence of treatment‐emergent adverse events (AEs) with placebo and vadadustat 150, 300 and 600mg was 36, 33, 58 and 54% (NDD‐CKD) and 40, 53, 73 and 40% (DDCKD), respectively. The most common AEs during the primary efficacy period were nausea and hypertension (NDD‐CKD) and diarrhoea, nasopharyngitis and shunt stenosis (DD‐CKD). Of 23 serious AEs in 18 patients, 1 was deemed related (hepatic function abnormal); no deaths were reported." |
| Pergola 2016 | Vadadustat | Placebo | SAE (33/138), initiation of dialysis (11/138), diarrhoea (14/138), nausea (14/138), constipation (5/138), GI haemorrhage (0/138), fatigue (12/138), oedema peripheral (10/138), UTI (9/138), upper respiratory tract infection (2/138), hyperkalaemia (7/138), headache (8/138), dizziness (7/138), AKI (10/138), CKD (7/138), dyspnoea (6/138), hypertension (11/138), hypotension (6/138), death (3/138) | SAE 11 (15.3%), initiation of dialysis (7/72), diarrhoea (3/72), nausea (3/72), constipation (4/72), GI haemorrhage (4/72), fatigue (5/72), oedema peripheral (7/72), UTI (6/72), upper respiratory tract infection (5/72), hyperkalaemia (0/72), headache (2/72), dizziness (3/72), AKI (4/72), CKD (3/72), dyspnoea (4/72), hypertension (2/72), hypotension (4/72), death (0/72) | "The percentage of patients who experienced at least 1 adverse events was comparable between the vadadustat and placebo groups (74.6% vs. 73.6%). Occurrence of at least 1 drug related AE was reported in 25.4% of vadadustat‐treated patients (35 of 138) and 11.1% of placebo‐treated patients (8 of 72). Most commonly reported drug‐related AEs in the vadadustat group included diarrhoea (4.3%) and nausea (4.3%), whereas diarrhoea (2.8%) was the most commonly reported drug‐related AE in the placebo group. Hypertension was reported as an AE in 8.0% of vadadustat‐treated patients (11 of 138) and 2.8% of those treated with placebo (2 of 72). A total of 33 vadadustat‐treated patients (23.9%) reported at least 1 serious adverse event (SAE), as did 11 placebo treated patients (15.3%); the higher incidence of SAEs was primarily due to a higher incidence of renal‐related SAEs in the vadadustat group (10.1%) compared with the placebo group (2.8%). The requirement for initiation of dialysis was evenly balanced between the vadadustat (11 of 138, 8.0%) and placebo (7 of 72, 9.7%) groups." |
| PRO2TECT‐CONVERSION 2021 | Vadadustat | Darbepoetin alfa | Any AE (767/878), death (139/878), diarrhoea (119//878), kidney failure (237/878), fall (69/878), hyperkalaemia (81/878), hypertension (124/878), peripheral oedema (85/878), pneumonia (86/878), UTI (105/878), nausea (73/878) | All data were related to 861 participants: Any AE (756/861), death 139/861), diarrhoea (76/861), kidney failure (245/861), fall (65/861), hyperkalaemia (85/861), hypertension (128/861), peripheral oedema (87/861), pneumonia (84/861), UTI (125/861), nausea (58/861) |
"The full list of AEs is provided in the supplementary materials." |
| PRO2TECT‐CORRECTION 2021 | Vadadustat | Darbepoetin alfa | Any AE (798/870), death (180/870), diarrhoea (122/870), kidney failure (305/870), fall (84/870), hyperkalaemia (108/870), hypertension (155/870), peripheral oedema (110/870), pneumonia (86/870), UTI (113/870), nausea (88/870) | Any AE (797/862), death (168/862), diarrhoea (87/862), kidney failure (306/862), fall (87/862), hyperkalaemia (136/862), hypertension (192/862), peripheral oedema (91/862), pneumonia (75/862), UTI (104/862), nausea (71/862) | "The full list of AEs is provided in the supplementary materials." |
| Provenzano 2008 | FG2216 (375, 625, 1250 mg) | Placebo | Death (1/128) (not reported in which treatment group) | Death (0/14) | "There were 41 SAE in 25 participants, 2 were assessed as possibly related to FG2216, including 1 death due to fulminant hepatitis." |
| PYRENEES 2021 | Roxadustat | EPO alfa | Hypertension (74/414), AVF thrombosis (50/414), headache (36/414), diarrhoea (35/414), bronchitis (33/414), hypotension (33/414), iron deficiency (30/414), nausea (29/414), viral upper respiratory tract infection (29/414), pneumonia (23/414), AVF site complication (23/414), hyperparathyroidism, secondary (22/414), anaemia (21/414), atrial fibrillation (20/414), muscle spasms (15/414), upper respiratory tract infection (14/414), fall (13/414), peritonitis (10/414), sepsis (8/414), bronchitis (5/414), gangrene (5/414), UTI (4/414), gastroenteritis (0/414), femur fracture (2/414), shunt thrombosis (1/414), atrial fibrillation (12/414), acute MI (9/414), cardiac failure (8/414), angina pectoris (5/414), cardiac failure, congestive (5/414), cardiac arrest (4/414), myocardial ischemias (4/414), MI (1/414), supraventricular tachycardia (1/414), DVT (4/414), peripheral arterial occlusive disease (2/414), peripheral ischemias (2/414), respiratory, thoracic and mediastinal disorders (29/414), pleural effusion (6/414), pulmonary oedema (6/414), dyspnoea (4/414), pulmonary embolism (4/414), general disorders and administration site conditions (28/414), pyrexia (4/414), duodenal ulcer (4/414), GI haemorrhage (0/414), nervous system disorders 15/414), cerebral infarction (0/414), metabolism and nutrition disorders (12/414), hyperkalaemia (4/414), product Issues (6/414), device malfunction (4/414), blood and lymphatic system disorders (5/414) | Hypertension (79/420), AVF thrombosis (31/420), headache (29/420), diarrhoea (35/420), bronchitis (29/420), hypotension (27/420), iron deficiency (51/420), nausea (8/420), viral upper respiratory tract infection (39/420), pneumonia (27/420), AVF site complication (21/420), hyperparathyroidism, secondary (16/420), anaemia (16/420), atrial fibrillation (25/420), muscle spasms (33/420), upper respiratory tract infection (22/420), fall (21/420), peritonitis (3/420), sepsis (9/420), bronchitis (3/420), gangrene (4/420), UTI (0/420), gastroenteritis (6/420), femur fracture (5/420), shunt thrombosis (14/420), atrial fibrillation (8/420), acute MI (11/420), cardiac failure (9/420), angina pectoris (6/420), cardiac failure, congestive (1/420), cardiac arrest (8/420), myocardial ischemias (4/420), MI (6/420), supraventricular tachycardia (5/420), DVT (0/420), peripheral arterial occlusive disease (4/420), peripheral ischemias (4/420), respiratory, thoracic and mediastinal disorders (21/420), pleural effusion (2/420), pulmonary oedema (2/420), dyspnoea (4/420), pulmonary embolism (1/420), general disorders and administration site conditions (20/420), pyrexia (4/420), duodenal ulcer (0/420), GI haemorrhage (6/420), nervous system disorders (21/420), cerebral infarction (4/420), metabolism and nutrition disorders (12/420), hyperkalaemia (3/420), product Issues (1/420), device malfunction (0/420), blood and lymphatic system disorders (8/420) | "The overall incidence of TEAEs and TEAEs PEY during the safety emergent period was comparable between treatment groups, with the overall event profile largely driven by events in the Infections and Infestations (most commonly viral upper respiratory tract infections and bronchitis in both treatment groups), Injury, Poisoning and Procedural Complications (with a greater incidence of arteriovenous (AV) fistula thrombosis in the roxadustat treatment group, mainly in the subgroup of patients receiving roxadustat compared with epoetin), Vascular Disorders (most commonly hypertension in both treatment groups) and gastrointestinal Disorders (with a greater incidence of diarrhoea and nausea in the roxadustat treatment group) MedDRA SOCs. TEAEs with an increased incidence in the roxadustat treatment group were mostly in the gastrointestinal Disorders, Skin and Subcutaneous Disorders, Nervous System Disorders and General Disorders and Administration Site Conditions SOCs. There were imbalances in the overall incidence of nausea and arteriovenous fistulae thrombosis, with a greater number in the roxadustat treatment group, and upper respiratory tract/viral upper respiratory tract infections, iron deficiency and muscle spasms occurring in a greater number of patients in the ESA treatment group. Hypertension and hypotension were seen in both treatment groups in comparable amounts. Differences were apparently due to the increased incidences of deaths and serious TEAEs over time in patients receiving roxadustat compared with epoetin alfa." |
| Provenzano 2016 | Roxadustat (part 1 and part 2) | EPO alfa | Data were reported considering all participants in the intervention group both in part 1 and part 2. Separate data were not reported | Data were reported considering all participants in the control group both in part 1 and part 2. Separate data were not reported | "AE and SAE rates were consistent with background disease of this ESRD population. In the safety population, 69 of 108 (63.9%) roxadustat treated and 22 of 36 (61%) epoetin alfa–treated participants had at least 1 AE. Thirty‐two of 144 (22.2%) participants in the safety population had a total of 50 treatment‐emergent SAEs. Of roxadustat‐treated participants, 26 of 108 (24.1%) had at least 1 SAE. The only SAE considered as possibly related to roxadustat treatment was acute pancreatitis. Of epoetin alfa–treated participants, 6 of 36 (17%) had at least 1 SAE. Three roxadustat‐treated participants died during the study." |
| SIERRAS 2021 | Roxadustat | EPO alfa | Nausea (63/370), hypertension (62/370), vomiting (60/370), hyperkalaemia (60/370), AVF site complication (58/370), dyspnoea (56/370), diarrhoea (54/370), cough (50/370), pain in extremity (49/370), constipation (44/370), upper respiratory tract infection (43/370), pneumonia (42/370), hypotension (41/370), headache (42/370), anaemia (40/370), fluid overload (40/370), back pain (39/370), non‐cardiac chest pain (37/370), AVF thrombosis (37/370), fall (37/370), acute MI (34/370), abdominal pain (33/370), pyrexia (33/370), UTI (32/370), arteriovenous graft thrombosis (31/370), cardiac failure, congestive (30/370), asthenia (27/370), viral upper respiratory tract infection (26/370), arthralgia (26/370), bronchitis (25/370), cellulitis (25/370), sepsis (25/370), hypoglycaemia (25/370), dizziness (25/370), oedema, peripheral (23/370), pruritus (22/370), cardiac arrest (21/370), tachycardia (20/370), atrial fibrillation (19/370), abdominal pain, upper (19/370), limb injury (19/370), acute respiratory failure (19/370), peripheral swelling (18/370), coronary artery disease (17/370), muscle spasms (17/370), syncope (17/370), anxiety (16/370), pain (15/370), musculoskeletal pain (15/370), insomnia (15/370), bradycardia (14/370), contusion (14/370), pleural effusion (14/370), GI haemorrhage (13/370), iron deficiency (12/370), angina pectoris (11/370), face oedema (10/370), vascular graft complication (10/370), pulmonary oedema (7/370) | Nausea (60/370), hypertension (47/370), vomiting (57/370), hyperkalaemia (56/370), AVF site complication (72/370), dyspnoea (67/370), diarrhoea (70/370), cough (69/370), pain in extremity (59/370), constipation (49/370), upper respiratory tract infection (40/370), pneumonia (52/370), hypotension (43/370), headache (40/370), anaemia (54/370), fluid overload (47/370), back pain (39/370), non‐cardiac chest pain (41/370), AVF thrombosis (42/370), fall (58/370), acute MI (26/370), abdominal pain (31/370), pyrexia (34/370), UTI (31/370), arteriovenous graft thrombosis (28/370), cardiac failure, congestive (33/370), asthenia (21/370), viral upper respiratory tract infection (26/370), arthralgia (37/370), bronchitis (29/370), cellulitis (30/370), sepsis (27/370), hypoglycaemia (30/370), dizziness (30/370), oedema, peripheral (34/370), pruritus (21/370), cardiac arrest (23/370), tachycardia (19/370), atrial fibrillation (26/370), abdominal pain, upper (18/370), limb injury (17/370), acute respiratory failure (29/370), peripheral swelling (26/370), coronary artery disease (22/370), muscle spasms (25/370), syncope (21/370), anxiety (21/370), pain (19/370), musculoskeletal pain (20/370), insomnia (25/370), bradycardia (21/370), contusion (20/370), pleural effusion (24/370), GI haemorrhage (19/370), iron deficiency (23/370), angina pectoris (23/370), face oedema (19/370), vascular graft complication (24/370), pulmonary oedema (24/370) | "At least 1 TEAE was experienced by 91.6% (event rate/100 PEY: 728.1) and 91.4% (event rate/100 PEY: 728.5) of patients in the roxadustat and epoetin alfa groups." |
| SYMPHONY HD 2021 | Enarodustat | Darbepoetin alfa | Retinal disorders (6/87), retinal haemorrhage (3/87), chorioretinopathy (1/87), diabetic retinopathy (1/87), macular oedema (1/87), vitreous haemorrhage (1/87), malignant or unspecified tumours (2/87), malignant neoplasm of renal pelvis (0/87), neoplasm skin (1/87), renal cancer (1/87), hypertension (4/87), embolic and thrombotic events (6/87), shunt occlusion (4/87), acute MI (1/87), arterial occlusive disease (0/87), lacunar infarction (1/87), pulmonary embolism (1/87) | Retinal disorders (3/86), retinal haemorrhage (3/86), chorioretinopathy (0/86), diabetic retinopathy (0/86), macular oedema (0/86), vitreous haemorrhage (0/86), malignant or unspecified tumours (1/86), malignant neoplasm of renal pelvis (1/86), neoplasm skin (0/86), renal cancer (0/86), hypertension (2/86), embolic and thrombotic events (5/86), shunt occlusion (4/86), acute MI (0/86), arterial occlusive disease (1/86), lacunar infarction (0/86), pulmonary embolism (0/86) | "In this study, 76/87 subjects (87.4%) in the enarodustat arm and 72/86 subjects (83.7%) in the DA arm experienced at least 1 AE. No death occurred in this study. Serious AEs occurred in 13 subjects (14.9%) in the enarodustat arm and 12 subjects (14.0%) in the DA arm, none of which were judged to be related to the study drug. Four subjects (4.6%) in the enarodustat arm and 3 subjects (3.5%) in the DA arm discontinued the study due to AE. AEs that occurred in ≥5% of subjects in any arm are listed in Table 2. The most frequent AE was viral upper respiratory tract infection in each arm. Vomiting and gastroenteritis in the enarodustat arm were observed at least twice as often as in the DA arm, but were considered unrelated to the study drug in both arms." |
| SYMPHONY ND 2021 | Enarodustat | Darbepoetin alfa | Any AEs (70/107), viral upper respiratory tract infection (19/107), diarrhoea (3/107), upper respiratory tract inflammation (2/107), contusion (1/107), embolic and thrombotic events (0/107), hypertension (5/107), BP increased (4/107), hypertension (1/107), malignant or unspecified tumours (0/107), malignant neoplasm of renal pelvis (0/107), gastric cancer (0/107), soft tissue neoplasm (0/107), retinal disorders (4/107), retinal haemorrhage (2/107), retinal tear (1/107), retinal detachment (1/107), macular oedema (1/107), diabetic retinal oedema (0/107) | Any AEs (90/109), viral upper respiratory tract infection (25/109), diarrhoea (9/109), upper respiratory tract inflammation (7/109), contusion (6/109), embolic and thrombotic events (0/109), hypertension (5/109), BP increased (2/109), hypertension (3/109), malignant or unspecified tumours (3/109), malignant neoplasm of renal pelvis (1/109), gastric cancer (1/109), soft tissue neoplasm (1/109), retinal disorders (1/109), retinal haemorrhage (0/109), retinal tear (0/109), retinal detachment (0/109), macular oedema (0/109), diabetic retinal oedema (1/109) | "There were no apparent differences in the incidence of adverse events between arms (65.4% [enarodustat], 82.6% [DA])." "Except for death, 15 serious adverse events (SAEs) occurred in 13 subjects in the enarodustat arm, and 14 SAEs occurred in 11 subjects in the DA arm. Four SAEs (fluid retention, hyperkalaemia, edema, and pneumonia) were judged to be related to enarodustat. Overall, 65.4% of subjects receiving enarodustat and 82.6% of subjects receiving DA experienced at least one AE." |
Footnotes:AE ‐ adverse events; AKI ‐ acute kidney injury; AVF ‐ arteriovenous fistula; BP ‐ blood pressure; CKD ‐ chronic kidney disease; DVT ‐ deep vein thrombosis; EPO ‐ erythropoietin; GFR ‐ glomerular filtration rate: MACE ‐ major adverse cardiovascular events; MI ‐ myocardial infarction; SAE ‐ serious adverse events; TEAE ‐ treatment‐emergent adverse event; TSAT ‐ transferrin saturation; UTI ‐ urinary tract infection
Data and analyses
Comparison 1. Hypoxia‐inducible factor (HIF) stabiliser versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Cardiovascular death | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1.1 Low‐dose HIF | 8 | 349 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.1.2 Medium‐dose HIF | 8 | 563 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.19, 70.21] |
| 1.1.3 High‐dose HIF | 9 | 508 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.1.4 Overall dose HIF | 10 | 1114 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.19, 70.21] |
| 1.2 Death (any cause) | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.2.1 Low‐dose HIF | 8 | 349 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.2.2 Medium‐dose HIF | 10 | 3918 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.97, 1.30] |
| 1.2.3 High‐dose HIF | 9 | 508 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.2.4 Overall dose HIF | 12 | 4469 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.97, 1.30] |
| 1.3 Nonfatal myocardial infarction | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.3.1 Low‐dose HIF | 2 | 93 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.3.2 Medium‐dose HIF | 3 | 707 | Risk Ratio (M‐H, Random, 95% CI) | 1.56 [0.32, 7.65] |
| 1.3.3 High‐dose HIF | 2 | 116 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.06, 34.46] |
| 1.3.4 Overall dose HIF | 3 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.31, 5.36] |
| 1.4 Fatal or nonfatal myocardial infarction (overall) | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Low‐dose HIF | 2 | 93 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.4.2 Medium‐dose HIF | 5 | 4384 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.60, 1.96] |
| 1.4.3 High‐dose HIF | 2 | 116 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.06, 34.46] |
| 1.4.4 Overall dose HIF | 5 | 4499 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.59, 1.90] |
| 1.5 Nonfatal stroke | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5.1 Low‐dose HIF | 2 | 93 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.5.2 Medium‐dose HIF | 2 | 113 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.5.3 High‐dose HIF | 2 | 116 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.5.4 Overall dose HIF | 2 | 228 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.6 Fatal or nonfatal stroke (overall) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.6.1 Low‐dose HIF | 2 | 93 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.6.2 Medium‐dose HIF | 3 | 707 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.23, 18.46] |
| 1.6.3 High‐dose HIF | 2 | 116 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.6.4 Overall dose HIF | 3 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 2.08 [0.23, 18.46] |
| 1.7 Peripheral arterial events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.7.1 Low‐dose HIF | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.35 [0.02, 8.10] |
| 1.7.2 Medium‐dose HIF | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.03, 7.59] |
| 1.7.3 High‐dose HIF | 1 | 62 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.01, 3.83] |
| 1.7.4 Overall dose HIF | 1 | 121 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.01, 3.04] |
| 1.8 Transfusion | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.8.1 Low‐dose HIF | 5 | 205 | Risk Ratio (M‐H, Random, 95% CI) | 0.79 [0.23, 2.79] |
| 1.8.2 Medium‐dose HIF | 7 | 4077 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.38, 0.62] |
| 1.8.3 High‐dose HIF | 5 | 229 | Risk Ratio (M‐H, Random, 95% CI) | 0.58 [0.15, 2.21] |
| 1.8.4 Overall dose HIF | 8 | 4329 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.44, 0.60] |
| 1.9 Proportion reaching target haemoglobin | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.9.1 Low‐dose HIF | 4 | 197 | Risk Ratio (M‐H, Random, 95% CI) | 4.16 [2.28, 7.59] |
| 1.9.2 Medium‐dose HIF | 8 | 4698 | Risk Ratio (M‐H, Random, 95% CI) | 9.36 [8.06, 10.86] |
| 1.9.3 High‐dose HIF | 5 | 351 | Risk Ratio (M‐H, Random, 95% CI) | 7.16 [3.62, 14.14] |
| 1.9.4 Overall dose HIF | 10 | 5102 | Risk Ratio (M‐H, Random, 95% CI) | 8.36 [6.42, 10.89] |
| 1.10 Kidney failure | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.10.1 Low‐dose HIF | 4 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.12 [0.46, 9.82] |
| 1.10.2 Medium‐dose HIF | 6 | 1855 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [0.94, 1.62] |
| 1.10.3 High‐dose HIF | 5 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 1.63 [0.43, 6.24] |
| 1.10.4 Overall dose HIF | 8 | 2228 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.98, 1.51] |
| 1.11 Thrombosis | 3 | 3452 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.19, 4.66] |
| 1.11.1 Medium‐dose HIF | 3 | 3452 | Risk Ratio (M‐H, Random, 95% CI) | 2.36 [1.19, 4.66] |
| 1.12 Loss of unassisted patency (stenosis) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.12.1 Low‐dose HIF | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 1.12.2 Medium‐dose HIF | 2 | 127 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.16, 14.05] |
| 1.12.3 High‐dose HIF | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 3.00 [0.13, 68.26] |
| 1.12.4 Overall dose HIF | 2 | 157 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.13, 10.31] |
| 1.13 Hyperkalaemia | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.13.1 Low‐dose HIF | 2 | 99 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.21, 3.42] |
| 1.13.2 Medium‐dose HIF | 5 | 4541 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [1.04, 1.56] |
| 1.13.3 High‐dose HIF | 3 | 275 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.26, 3.84] |
| 1.13.4 Overall dose HIF | 7 | 4845 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [1.01, 1.64] |
Comparison 2. Analyses for SOF table 1 stratifying by CKD stage (HIF versus placebo).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Cardiovascular death | 10 | 1114 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.19, 70.21] |
| 2.1.1 CKD | 7 | 850 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.19, 70.21] |
| 2.1.2 HD | 2 | 157 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 2.1.3 CKD and HD | 1 | 107 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 2.2 Nonfatal myocardial infarction | 3 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.31, 5.36] |
| 2.2.1 CKD | 3 | 822 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.31, 5.36] |
| 2.3 Transfusion | 8 | 4329 | Risk Ratio (M‐H, Random, 95% CI) | 0.51 [0.44, 0.60] |
| 2.3.1 CKD | 7 | 4271 | Risk Ratio (M‐H, Random, 95% CI) | 0.52 [0.44, 0.60] |
| 2.3.2 HD | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.04, 1.14] |
| 2.4 Proportion reaching target haemoglobin | 10 | 5102 | Risk Ratio (M‐H, Random, 95% CI) | 8.36 [6.42, 10.89] |
| 2.4.1 CKD | 8 | 4931 | Risk Ratio (M‐H, Random, 95% CI) | 8.18 [6.13, 10.93] |
| 2.4.2 CKD and HD | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 14.35 [2.07, 99.61] |
Comparison 3. Hypoxia‐inducible factor (HIF) stabiliser versus erythropoiesis‐stimulating agent (ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Cardiovascular death | 17 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 Low‐dose HIF | 6 | 981 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.14, 6.41] |
| 3.1.2 Medium‐dose HIF | 7 | 7442 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.88, 1.27] |
| 3.1.3 High‐dose HIF | 9 | 2067 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.31, 3.96] |
| 3.1.4 Overall dose HIF | 17 | 10340 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.88, 1.26] |
| 3.2 Fatigue | 2 | 3471 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.56, 1.16] |
| 3.2.1 Medium‐dose HIF | 2 | 3471 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.56, 1.16] |
| 3.3 Death (any cause) | 29 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.3.1 Low‐dose HIF | 9 | 1295 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.33, 2.75] |
| 3.3.2 Medium‐dose HIF | 15 | 15586 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.88, 1.08] |
| 3.3.3 High‐dose HIF | 13 | 4745 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.63, 1.37] |
| 3.3.4 Overall dose HIF | 29 | 21370 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.91, 1.06] |
| 3.4 Nonfatal myocardial infarction | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.4.1 Low‐dose HIF | 2 | 148 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.4.2 Medium‐dose HIF | 5 | 7153 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.72, 1.16] |
| 3.4.3 High‐dose HIF | 4 | 612 | Risk Ratio (M‐H, Random, 95% CI) | 1.90 [0.31, 11.54] |
| 3.4.4 Overall dose HIF | 7 | 7765 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.76, 1.10] |
| 3.5 Fatal or nonfatal myocardial infarction (overall) | 15 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.5.1 Low‐dose HIF | 2 | 148 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.5.2 Medium‐dose HIF | 9 | 10949 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.78, 1.10] |
| 3.5.3 High‐dose HIF | 8 | 3234 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.68, 2.19] |
| 3.5.4 Overall dose HIF | 15 | 14183 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.80, 1.12] |
| 3.6 Nonfatal stroke | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.6.1 Low‐dose HIF | 3 | 336 | Risk Ratio (M‐H, Random, 95% CI) | 4.78 [0.24, 96.68] |
| 3.6.2 Medium‐dose HIF | 3 | 6918 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.62, 1.83] |
| 3.6.3 High‐dose HIF | 1 | 115 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.6.4 Overall dose HIF | 5 | 7285 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.56] |
| 3.7 Fatal or nonfatal stroke (overall) | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.7.1 Low‐dose HIF | 4 | 398 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [0.11, 17.51] |
| 3.7.2 Medium‐dose HIF | 4 | 6980 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.70, 1.50] |
| 3.7.3 High‐dose HIF | 3 | 795 | Risk Ratio (M‐H, Random, 95% CI) | 0.49 [0.16, 1.53] |
| 3.7.4 Overall dose HIF | 7 | 8025 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.64, 1.40] |
| 3.8 Nonfatal hospitalisation for heart failure | 2 | 6836 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [1.00, 1.52] |
| 3.8.1 Medium‐dose HIF | 2 | 6836 | Risk Ratio (M‐H, Random, 95% CI) | 1.23 [1.00, 1.52] |
| 3.9 Fatal or nonfatal hospitalisation for heart failure | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.9.1 Medium‐dose HIF | 2 | 6836 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.98, 1.39] |
| 3.9.2 High‐dose HIF | 1 | 616 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.59, 1.79] |
| 3.9.3 Overall dose HIF | 3 | 7452 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.97, 1.36] |
| 3.10 Peripheral arterial event | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.10.1 Low‐dose HIF | 2 | 148 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.10.2 Medium‐dose HIF | 2 | 144 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.10.3 High‐dose HIF | 2 | 179 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.10.4 Overall dose HIF | 2 | 323 | Risk Ratio (M‐H, Random, 95% CI) | Not estimable |
| 3.11 Transfusion | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.11.1 Low‐dose HIF | 3 | 192 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.23, 4.75] |
| 3.11.2 Medium‐dose HIF | 7 | 7343 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.81, 1.02] |
| 3.11.3 High‐dose HIF | 7 | 3443 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.62, 1.16] |
| 3.11.4 Overall dose HIF | 11 | 10786 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.76, 1.00] |
| 3.12 Proportion reaching target haemoglobin | 14 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.12.1 Low‐dose HIF | 7 | 861 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.91, 1.10] |
| 3.12.2 Medium‐dose HIF | 4 | 507 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.69, 1.29] |
| 3.12.3 High‐dose HIF | 9 | 3425 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.91, 1.12] |
| 3.12.4 Overall dose HIF | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 3.13 Kidney failure | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.13.1 Low‐dose HIF | 2 | 361 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.20, 6.43] |
| 3.13.2 Medium‐dose HIF | 7 | 6647 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.91, 1.16] |
| 3.13.3 High‐dose HIF | 2 | 368 | Risk Ratio (M‐H, Random, 95% CI) | 6.88 [0.89, 53.20] |
| 3.13.4 Overall dose HIF | 9 | 7312 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.91, 1.15] |
| 3.14 Thrombosis | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.14.1 Medium‐dose HIF | 7 | 14532 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.71, 1.37] |
| 3.14.2 High‐dose HIF | 4 | 2494 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.99, 1.83] |
| 3.14.3 Overall dose HIF | 11 | 17026 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.86, 1.39] |
| 3.15 Loss of unassisted patency (occlusion/stenosis) | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.15.1 Low‐dose HIF | 1 | 271 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.53, 1.69] |
| 3.15.2 Medium‐dose HIF | 3 | 800 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.29, 1.47] |
| 3.15.3 High‐dose HIF | 4 | 1874 | Risk Ratio (M‐H, Random, 95% CI) | 1.51 [0.99, 2.30] |
| 3.15.4 Overall dose HIF | 8 | 2945 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.85, 1.59] |
| 3.16 Access intervention | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.16.1 High‐dose HIF | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.17 Cancer | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.17.1 Low‐dose HIF | 2 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.10, 3.35] |
| 3.17.2 Medium‐dose HIF | 3 | 641 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.24, 1.74] |
| 3.17.3 High‐dose HIF | 2 | 531 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.13, 5.85] |
| 3.17.4 Overall dose HIF | 7 | 1687 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.43, 1.59] |
| 3.18 Infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.18.1 High‐dose HIF | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.19 Hyperkalaemia | 21 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.19.1 Low‐dose HIF | 2 | 570 | Risk Ratio (M‐H, Random, 95% CI) | 1.72 [0.77, 3.85] |
| 3.19.2 Medium‐dose HIF | 10 | 15152 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.82, 1.00] |
| 3.19.3 High‐dose HIF | 9 | 4455 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.68, 1.50] |
| 3.19.4 Overall dose HIF | 21 | 20177 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.82, 1.04] |
| 3.20 Pulmonary hypertension | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.20.1 Low‐dose HIF | 2 | 570 | Risk Ratio (M‐H, Random, 95% CI) | 2.98 [0.12, 72.46] |
| 3.20.2 Medium‐dose HIF | 4 | 7455 | Risk Ratio (M‐H, Random, 95% CI) | 1.13 [0.51, 2.47] |
| 3.20.3 High‐dose HIF | 1 | 616 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.03, 2.89] |
| 3.20.4 Overall dose HIF | 7 | 8641 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.56, 2.01] |
| 3.21 Diabetic retinopathy | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.21.1 Low‐dose HIF | 1 | 299 | Risk Ratio (M‐H, Random, 95% CI) | 2.01 [0.18, 21.97] |
| 3.21.2 Medium‐dose HIF | 6 | 4435 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.65, 2.15] |
| 3.21.3 High‐dose HIF | 1 | 302 | Risk Ratio (M‐H, Random, 95% CI) | 3.04 [0.12, 74.03] |
| 3.21.4 Overall dose HIF | 8 | 5036 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.71, 2.22] |
Comparison 4. Analyses for SOF table 2 stratifying by CKD stage (HIF versus ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Cardiovascular death | 17 | 10340 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.88, 1.26] |
| 4.1.1 CKD | 7 | 5591 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.91, 1.55] |
| 4.1.2 HD | 7 | 1352 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.23, 4.19] |
| 4.1.3 PD | 1 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.03, 7.80] |
| 4.1.4 HD and PD | 2 | 3268 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.75, 1.23] |
| 4.2 Fatigue | 2 | 3471 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.56, 1.16] |
| 4.2.1 CKD | 2 | 3471 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.56, 1.16] |
| 4.3 Nonfatal myocardial infarction | 7 | 7765 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.76, 1.10] |
| 4.3.1 CKD | 2 | 3996 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.80, 1.39] |
| 4.3.2 HD | 2 | 372 | Risk Ratio (M‐H, Random, 95% CI) | 1.97 [0.22, 17.59] |
| 4.3.3 PD | 1 | 129 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [0.06, 36.48] |
| 4.3.4 HD and PD | 2 | 3268 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.62, 1.03] |
| 4.4 Nonfatal stroke | 5 | 7285 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.71, 1.56] |
| 4.4.1 CKD | 3 | 4122 | Risk Ratio (M‐H, Random, 95% CI) | 1.43 [0.82, 2.48] |
| 4.4.2 HD | 1 | 199 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.07, 27.82] |
| 4.4.3 HD and PD | 1 | 2964 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.51, 1.34] |
| 4.5 Transfusion | 11 | 10786 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.76, 1.00] |
| 4.5.1 CKD | 5 | 4933 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.84, 1.13] |
| 4.5.2 HD and PD | 6 | 5853 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.64, 1.01] |
| 4.6 Proportion reaching target haemoglobin | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 4.6.1 CKD | 6 | 1369 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.90, 1.16] |
| 4.6.2 HD and PD | 8 | 3232 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.91, 1.06] |
Comparison 5. Subgroup analysis: stage CKD (HIF versus ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Proportion reaching target haemoglobin | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 5.1.1 CKD | 6 | 1369 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.90, 1.16] |
| 5.1.2 HD | 5 | 1150 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.88, 1.06] |
| 5.1.3 HD and PD | 3 | 2082 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.86, 1.15] |
| 5.2 Thrombosis | 11 | 17026 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.86, 1.39] |
| 5.2.1 CKD | 4 | 7959 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.59, 1.86] |
| 5.2.2 HD | 1 | 323 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.06, 15.75] |
| 5.2.3 HD and PD | 6 | 8744 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.81, 1.46] |
Comparison 6. Subgroup analysis: duration of therapy (HIF versus ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Proportion reaching target haemoglobin | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 6.1.1 From 8 to 23 weeks | 3 | 405 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.67, 1.55] |
| 6.1.2 From 24 to 53 weeks | 9 | 3392 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.92, 1.06] |
| 6.1.3 At least 54 weeks | 2 | 804 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.89, 1.25] |
| 6.2 Thrombosis | 11 | 17026 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.86, 1.39] |
| 6.2.1 From 24 to 53 weeks | 4 | 5239 | Risk Ratio (M‐H, Random, 95% CI) | 1.26 [0.87, 1.81] |
| 6.2.2 At least 54 weeks | 7 | 11787 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.75, 1.43] |
Comparison 7. Subgroup analysis: frequency of administration (HIF versus ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Proportion reaching target haemoglobin | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 7.1.1 Once a day | 7 | 1315 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.88, 1.06] |
| 7.1.2 Three times a week | 7 | 3286 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.93, 1.12] |
| 7.2 Thrombosis | 11 | 17026 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.86, 1.39] |
| 7.2.1 Once daily | 7 | 14532 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.71, 1.37] |
| 7.2.2 Three times a week | 4 | 2494 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.99, 1.83] |
Comparison 8. Subgroup analysis: phase 2 versus phase 3 studies (HIF versus ESA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Proportion reaching target haemoglobin | 14 | 4601 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.93, 1.07] |
| 8.1.1 Phase 2 | 6 | 1356 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.90, 1.34] |
| 8.1.2 Phase 3 | 8 | 3245 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.91, 1.05] |
| 8.2 Thrombosis | 11 | 17026 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.86, 1.39] |
| 8.2.1 Phase 2 | 1 | 740 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.60, 2.02] |
| 8.2.2 Phase 3 | 10 | 16286 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.83, 1.42] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Akizawa 2017.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low‐dose)*
Treatment group 2 (medium‐dose)
Treatment group 3 (medium dose)
Treatment group 4 (high‐dose)
Control group
Co‐interventions
*Note: If data were not available to report HIF considering the dose (low, medium and high dose), the analyses have been performed considering the average dose > dose assessed according to Meadowcroft 2019 |
|
| Outcomes |
Primary outcome
Secondary outcomes
*CFB was calculated by subtracting the baseline value from the post‐dose value at week 4 |
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind." Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. In the treatment groups were reported side effects that participants and/or investigators could know to be specific for one of the interventions. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "A total of 97 subjects were randomized, of whom 86 (89%) subjects completed the study. Across treatment groups, the predominant reasons for premature withdrawal were reaching protocol‐defined stopping criteria and AEs." ITT: treatment group 1 (19/19); treatment group 2 (20/20); treatment group 3 (19/19); treatment group 4 (20/20); control group (19/18) Safety population: treatment group 1 (19/19); treatment group 2 (20/20); treatment group 3 (19/19); treatment group 4 (20/20); control group (19/19) |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov were not measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Akizawa 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Dynamic allocation was conducted using a biased‐coin minimisation approach with the following factors for the first randomisation: study site, average Hb level at screening assessment and screening period, estimated glomerular filtration rate (eGFR) at screening assessment. For the second randomisation, the allocation factors were study site, roxadustat dose immediately before the second randomisation, and Hb." |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "A double‐blind procedure for participants, care providers, and those assessing outcomes ensured that oral roxadustat and placebo capsules were indistinguishable in appearance and all treatments were coded." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "TEAEs were coded using the Medical Dictionary for Regulatory Activities version 15.1 terminology." Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "A total of 83 (77.6%) patients completed the 24‐week study. The overall discontinuation rate was 40.7% for the placebo TIW group and 16.3% for the roxadustat TIW pooled group (50 mg, n = 9 [33.3%]; 70 mg, n = 0; 100 mg, n = 4 [14.8%]). Patients discontinued as a result of a TEAE (n = 7, 8.8% roxadustat TIW pooled; n = 2, 7.4% placebo TIW), progressive disease (n = 4, 5.0% roxadustat TIW pooled), Hb level\8 g/dL (n = 5, 18.5% placebo TIW), lack of efficacy (n = 4, 14.8% placebo TIW), withdrawal by patient (n = 1, 1.3% roxadustat TIW pooled), or other reason (n = 1, 1.3% roxadustat TIW pooled)." Lost to follow‐up: > 5% with differences between groups |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Akizawa 2020a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Assignment was implemented by a web‐based randomisation system." |
| Allocation concealment (selection bias) | Low risk | Quote: "Assignment was implemented by a web‐based randomisation system." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double blind." Quote: "To maintain blinding, a double‐dummy design was used; only the drug assignment manager and designated staff had access to the randomisation code." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "303 were randomized to receive either roxadustat (n=151) or DA (n=152). A total of 250 (82.5%) patients (roxadustat, n=119 [78.8%]; DA, n=131 [86.2%]) completed the study and 53 (17.5%) patients (roxadustat, n=32 [21.2%]; DA, n=21 [13.8%]) discontinued. Across all randomised patients, the leading reasons for discontinuation were adverse events (roxadustat, n=12 [7.9%]; DA, n=8 [5.3%]), protocol deviations (roxadustat, n=7 [4.6%]; DA, n=4 [2.6%]), and withdrawal by the patient (roxadustat, n=5 [3.3%]; DA, n=4 [2.6%])." Patients analysed in the intervention group were: SAF (150), FAS (150), PPS (114); patients analysed in the control group were: SAF (1520, FAS (151), PPS (131) |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Akizawa 2020c.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (low dose)*
Control group
Co‐interventions
*Note: dose assessed according to Meadowcroft 2019 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Company‐validated system." Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent. In addition, a validated system could be considered at low risk of bias |
| Allocation concealment (selection bias) | Low risk | Quote: "A biostatistician generated the randomisation codes using a company‐validated system." Quote: "Interactive Web Response System." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind study." Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. In the treatment groups were reported side effects that participants and/or investigators could know to be specific for one of the interventions. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 332 participants screened, 271 participants were randomized (136 daprodustat, 135 darbepoetin alfa). A total of 115 participants (85%) in the daprodustat group and 120 (89%) in the darbepoetin alfa group completed the study. The most common reasons for study withdrawal in both treatment groups were AEs (ten daprodustat, eight darbepoetin alfa)." Quote: "All 271 participants were included in the safety population, 267 participants (133 daprodustat, 134 darbepoetin alfa) were included in the ITT population, and 245 participants (120 daprodustat, 125 darbepoetin alfa) were included in the modified ITT population." Lost to follow‐up: < 5% |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov were not measured or reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
Akizawa 2020f.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not reported in sufficient detail to permit judgement |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | Unclear risk | Baseline characteristics, or different non‐randomised co‐interventions were not reported between groups Funder and conflicts of interest were not reported |
Akizawa 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 Note: patients receiving rHuEPO or darbepoetin alfa before conversion were randomised to roxadustat or darbepoetin alpha (comparative arms). Patients who had used EBP before conversion were allocated to roxadustat (reference arm) ‐ in this review we focused only on patients randomised reported in the comparative group |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not reported. However, due to the difference in the interventions, blinding was unlikely |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "At the end of Week 24, 109 [82.6%] participants in the roxadustat [comparative] group, 121 [92.4%] in the DA [comparative] group, completed the 24‐week treatment period." Loss to follow‐up: > 5% |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov were not measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
ALPS 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Of 597 patients randomized into 1 of 2 treatment groups: 394 to the roxadustat group and 203 to the placebo group, 3 patients in the roxadustat treatment group were excluded due to GCP violations. Of 391 patients that received roxadustat, 114 discontinued during the first year and 146 discontinued up to 2 years with a total of 245 patients completing 2 years of treatment. Of the 203 patients that received placebo, 87 discontinued during the first year and 114 discontinued up to 2 years with a total of 89 patients completed 2 years of treatment." Quote: "Overall, 18.5% of patients discontinued study treatment with the reason given as “withdrawal by patient” (14.8% roxadustat, 25.6% placebo), 9.3% of patients overall were considered to have discontinued treatment due to the event of death (10.0% roxadustat, 7.9% placebo) and 5.1% withdrew due to AEs (5.4% roxadustat, 4.4% placebo). The incidence of treatment discontinuations with time was lower overall for patients in the roxadustat treatment group compared with the placebo treatment group." ITT analyses were not reported, some analyses were performed on 389/391 participants in the intervention group versus 203/203 in the placebo group (lost to follow‐up > 5%, differences between groups) |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov were not measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were not reported |
ANDES 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization schedules were prospectively prepared, and automated randomisation and treatment assignments were provided by an interactive web response system." |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization schedules were prospectively prepared, and automated randomisation and treatment assignments were provided by an interactive web response system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind study" Quote: "The investigator, study site staff, patient, sponsor, and designees were all blinded to the study drug assignment—but not the dose—which was achieved by using identical roxadustat and placebo |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "In the roxadustat group, 43.3% (267/616) of patients discontinued treatment, while 68.0% (208/306) of placebo‐treated patients discontinued. This between‐group difference in discontinuations was largely due to the lack of efficacy among patients in the placebo group. The primary reasons for discontinuations in the placebo group were withdrawal of consent and lack of efficacy. The primary reasons for discontinuations in the roxadustat group which occurred a lesser rates were adverse events or death and withdrawal of consent." All participants were included in the ITT analysis |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov were not measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Quote: "FibroGen employees and subcontractors had a role in study design, data collection, data analysis, data interpretation, and writing of the manuscript." Funder influenced data analysis and study reporting or interpretation Authors declared conflicts of interest |
ASCEND‐D 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed according to Meadowcroft 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were stratified by dialysis type of HD [including hemodiafiltration (HDF) and hemofiltration (HF)] or PDg, by region, and by participation in the ambulatory blood pressure (BP) monitoring sub‐study. Following stratification, patients were randomized 1:1 to receive oral daprodustat or rhEPO control. A central randomisation approach was used to protect against selection bias due to the open‐label design." Quote: "Investigators used an interactive voice‐ or Web‐response system to determine treatment assignments." |
| Allocation concealment (selection bias) | Low risk | Quote: "Investigators used an interactive voice‐ or Web‐response system to determine treatment assignments." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The safety of trial patients was overseen by an independent data monitoring." Not clearly stated if the data monitoring was blinded to the treatment assigned |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT population Quote: "Eight patients (5 in the daprodustat group and 3 in the ESA group) were excluded from the safety analyses because they did not receive the randomised treatment" |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Quote: "The trial drug was stopped prematurely." Quote: "The sponsor conducted the analysis." Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funder influenced data analysis and study reporting or interpretation. Conflicts of interest were reported |
ASCEND‐ID 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Overall 99% (155/157) pts on Dapro and 97% (151/155) on Darbe completed the study" Loss to follow‐up: < 5% |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Baseline characteristics, or different non‐randomised co‐interventions were not reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were not reported |
ASCEND‐ND 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed according to Meadowcroft 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The investigators used an interactive voice‐response or Web‐response system to determine the treatment assignments. Randomizsation was stratified according to use or nonuse of an ESA, geographic region, and participation or nonparticipation in an ambulatory blood‐pressure monitoring sub‐study." |
| Allocation concealment (selection bias) | Low risk | Quote: "The investigators used an interactive voice‐response or Web‐response system to determine the treatment assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" Quote: "Although the investigators and patients were aware of the treatment assignments, the sponsor and steering committee remained unaware of the aggregate treatment assignments throughout the trial." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Blinded adjudication of cardiovascular outcomes." Quote: "An independent data monitoring committee over‐ saw the safety of the patients" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Nineteen patients (<1.0%) in the daprodustat group and 12 patients (<1.0%) in the darbepoetin alfa group had unknown vital status at the end of the trial." ITT analysis |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Quote: "The sponsor, GlaxoSmithKline, and an academic steering committee designed and oversaw the trial conduct and analysis." Quote: "Daprodustat was discontinued prematurely for reasons other than death in 571 of 1937 patients (29.5%), and darbepoetin alfa was discontinued prematurely for reasons other than death in 560 of 1935 patients (28.9%)." Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funder influenced data analysis and study reporting or interpretation Conflicts of interest were not reported |
ASCEND‐NHQ 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not reported in sufficient detail to perform adjudication |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Baseline characteristics, or different non‐randomised co‐interventions were not reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were not reported |
ASCEND‐TD 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not reported in sufficient detail to perform adjudication |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Baseline characteristics, or different non‐randomised co‐interventions were not reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were not reported |
Besarab 2015.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Eligible patients were sequentially enrolled into one of four dose cohorts: 1.0, 1.5, 2.0 and 0.7 mg/Kg” Quote: “The first 35 patients at treatment sites were enrolled 7:7:2:2 and at 2:2:1:1 at PK sites to roxadustat BIW, TIW or placebo BIW, TIW. Remaining 82 patients were enrolled 10:10:3:3 at treatment sites only” Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: “This was a multicenter randomized study (single blind), placebo‐controlled, with sequential dose escalation and evaluation of administration” |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | According to Figure 1 (Patient disposition), two patients in Roxadustat groups and one in placebo group discontinued treatment because of adverse events (unrelated to medication). One patient in Roxadustat group withdrew consent. Seven patients in Roxadustat group and one in placebo group discontinued dosing by sponsor’s decision” 88/88 and 28/29 participants completed the study according to the safety population. 78/88 and 26/28 completed dosing (89.7%). 73/23 (83%) provided efficacy evaluable population. 76/26 (88%) completed study. I could not find reason for sponsor’s decisions to withdraw patients All patients were included in the intention‐to treat analysis |
| Selective reporting (reporting bias) | High risk | The planned outcomes (efficacy, safety) on ClincialTrials.gov have been measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported Data on fatigue, diabetic retinopathy, cancer, renal impairment and hyperkalaemia were reported but no data were reported for the different doses |
| Other bias | High risk | Quote: "All authors except sponsor contributed participants to study." There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported. Funders designed study so could influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Brigandi 2016.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "This phase 2A, multicenter (Australia, New Zealand, India, and Russia), single‐blind, randomized, placebo‐controlled, parallel‐group study." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants were included into the analysis Quote: “Adverse events responsible for withdrawal in 6 non dialysis and 3 dialysis patients. 32/70 withdrawn from CKD group & 11/37 withdrawn from HD group.” |
| Selective reporting (reporting bias) | High risk | Not all the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report. No reasoning provided. No reporting of results of SF36 surveys that were listed as outcome in Clinical Trial Registration Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Chen 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Randomisation was performed centrally in sequence, stratified according to the dose of epoetin alfa at baseline (<8000 IU or ≥8000 IU per week) and dialysis method (haemodialysis or peritoneal dialysis).” Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "This trial (FGCL‐4592‐806) was a randomized, open‐label, active‐controlled, phase 3 trial." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “A total of 48 patients (42 in the roxadustat group and 6 in the epoetin alfa group) discontinued the assigned medication. A total of 256 patients (162 in the roxadustat group and 94 in the epoetin alfa group) completed treatment.” 256/304 (84%) participants completed the study according to the safety population. Reason for discontinuations were provided that seemed unrelated with the treatment assigned Lost to follow‐up: < 5% 305 patients underwent randomisation (204 patients to the roxadustat group and 101 to the epoetin alfa group). One patient in the epoetin alfa group did not receive treatment, so 304 patients were included in the full analysis set (ITT population) All participants were included in the ITT analysis |
| Selective reporting (reporting bias) | Low risk | Expected outcomes reported and correlate with the planned outcomes on ClincialTrials.gov. Clinically‐relevant outcomes that would be expected for this type of intervention were reported (death and CV events). |
| Other bias | High risk | Quote: “The trial was designed by the first two authors and the sponsor (FibroGen). The sponsor provided financial support and was responsible for data collection and analysis.” There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Chen 2019a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Eligible patients were randomly assigned in a 2:1 ratio to receive roxadustat or placebo. Randomization was performed centrally and was stratified according to the use or nonuse of an erythropoiesis‐stimulating agent within 12 weeks before randomisation and according to the estimated glomerular filtration rate (GFR) (<20 ml or ≥20 ml per minute per 1.73 m2 of body‐surface area)." Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind phase" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. In the treatment groups were reported side effects that participants and/or investigators could know to be specific for one of the interventions. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "One patient in each group discontinued participation in the trial because of hyperkalaemia." "2 [participants] did not receive a trial regimen. One patient in the safety population took one dose of placebo and was lost to follow‐up" 151/154 participants completed the study according to the intention‐to‐treat population and 152/154 participants completed the study according to the safety population Loss to follow‐up: < 5%, without differences between groups Reason for discontinuations were provided that seemed unrelated with the treatment assigned |
| Selective reporting (reporting bias) | High risk | Not all of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report. No reasoning provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Quote: "The first two authors designed the trial in collaboration with representatives of the sponsor, FibroGen; company representatives were responsible for the collection and analysis of the data." There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
Chen DD 2017.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low dose)*
Treatment group 2 (medium dose)
Treatment group 3 (high dose)
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The open‐label DD study." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Of 65 subjects, 5 that were randomized to FG‐4592 withdrew from the study within 2 weeks, including a subject who incorrectly received a single dose of epoetin‐alfa and a subject who developed a Grade 1rash. The other three early terminations withdrew consent. Therefore, the pre‐specified EE population" 22/22 participants in treatment group 1, 18/22 participants in treatment group 2, 20/22 participants in treatment group 3, 22/22 participants in control group were included into the analysis (< 5% lost to follow‐up, with differences between groups) Reasons for discontinuations were reported as follows: "Withdrawn due to having had one dose of epoetin alfa (prohibited medication) administered in error, brash (hypersensitivity)." |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups FibroGen Inc. was the study sponsor that designed the study in consultation with the Principal Investigators (N.C. and J.Q.) FibroGen was responsible for data collection and analysis Authors declared conflicts of interest |
Chen NDD 2017.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low dose)*
Treatment group 2 (high dose)
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: “Qualified subjects were randomized 2:1 to FG‐4592 or placebo orally thrice weekly (TIW) sequentially into first low‐ (1.1–1.75 mg/kg) and then high‐dose (1.50–2.25 mg/kg) FG‐4592 cohorts using weight tiered dosing (40–60 kg, >60 to 80 kg or >80 to 100 kg), and were treated for 8 weeks.” Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “The double‐blinded NDD study.” Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. In the treatment groups were reported side effects that participants and/or investigators could know to be specific for one of the interventions. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. Reporting of some outcomes (adverse effects) were unlikely to be biased because outcome assessors were blinded to treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: “The intent‐to‐treat (ITT) populations included all randomized subjects in each study. The safety populations in both studies included all subjects that had received at least one dose of study drug (FG‐4592, placebo or active comparator)”. Quote: "Two adverse events in the low dose FG‐4592 arm were urinary tract infection and worsening chronic renal failure. The one placebo subject was discontinued for adverse event of worsening anaemia (and received rescue therapy)." Quote: “91 subjects were randomized from December 2011 to August 2012 to receive either FG‐4592 (n = 61) or placebo (n = 30) at 11 study sites in China (Figure 1A), constituting both ITT and safety populations” All ITT populations analysed in groups to which they were allocated. 4/2/3 discontinued treatment but all included in the analyses |
| Selective reporting (reporting bias) | High risk | All of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups FibroGen Inc. was the study sponsor that designed the study in consultation with the Principal Investigators (N.C. and J.Q.). FibroGen was responsible for data collection and analysis Authors declared conflicts of interest |
DIALOGUE 1 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low dose)*
Treatment group 2 (medium dose)
Treatment group 3 (high dose)
Treatment group 4 (medium dose)
Treatment group 5 (high dose)
Control group
Co‐interventions
*Note: this study was considered as a reference for HIF dosage |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind." Macdougall 2019 quote: "Both, patients and physicians were blinded to treatment allocation." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Macdougall 2019 quote: "An independent adjudication committee assessed all deaths and any serious AEs of severe arrhythmias, thromboembolic events, syncope or symptomatic hypotension, or heart failure." It was not reported if the independent adjudication committee was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Akizawa 2019 (Nephron) quote: "Of 121 patients randomized in D1, all (101 molidustat, 20 placebo) were included in the FAS and ITT population, and 60 patients (59.5%) receiving molidustat and 2 patients (10.0%) receiving placebo discontinued the study. Of those who discontinued molidustat, the majority discontinued by the last 4 weeks and had a blood Hb concentration above the upper limit of 13 g/dL or an increase in blood Hb concentration of > 1.0 g/dL in 2 weeks." All participants were included in FAS and ITT analyses |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
DIALOGUE 2 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low dose)*
Treatment group 2 (medium dose)
Treatment group 3 (high dose)
Control group
Co‐interventions
*Note: dose assessed according to DIALOGUE 1 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Macdougall 2019 quote: "An independent adjudication committee assessed all deaths and any serious AEs of severe arrhythmias, thromboembolic events, syncope or symptomatic hypotension, or heart failure." It was not reported if the independent adjudication committee was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Akizawa 2019 (Nephron) quote: "In D2, all of the 124 randomized patients (92 molidustat, 32 darbepoetin) were included in the FAS and ITT population, and 20 patients (21.7%) receiving molidustat and 5 patients (15.6%) receiving darbepoetin discontinued the study." All participants were included in FAS and ITT analyses |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
DIALOGUE 4 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (low dose)*
Treatment group 2 (medium dose)
Treatment group 3 (high dose)
Treatment group 4 (high dose)
Control group
Co‐interventions
*Note: dose assessed according to DIALOGUE 1 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Low risk | Macdougall 2019 quote: "At the randomizations visit, the investigator had to check the patient’s eligibility and stratification factor levels; an interactive response system subsequently randomly assigned the treatment group according to computer generated randomisation lists." No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Macdougall 2019 quote: "An independent adjudication committee assessed all deaths and any serious AEs of severe arrhythmias, thromboembolic events, syncope or symptomatic hypotension, or heart failure." It was not reported if the independent adjudication committee was blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Akizawa 2019 (Nephron) quote: "All of the 199 patients randomized in D4 (157 molidustat, 42 epoetin) were included in the FAS and ITT population, and 53 patients (33.8%) receiving molidustat and 3 patients (7.1%) receiving epoetin discontinued the study." All participants were included in FAS and ITT analyses |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interest |
DOLOMITES 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An Independent Review Committee was established to adjudicate cardiovascular events in a blinded manner." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of 616 randomized pts (roxadustat, 323; DA, 293), 424 completed 2 years of treatment (roxadustat, 215; DA, 209)." Quote: "A total of 424 [roxadustat group, n= 215 (66.6%); DA group, n= 209 (71.3%)] patients completed 2 years of treatment, whereas 33.4and 28.7% of patients discontinued treatment in the roxadustat and DA groups, respectively. Primary reasons for discontinuation in the roxadustat and DA groups were death [n= 27 (8.4%) versus n= 30 (10.2%)], withdrawal by patient [n= 32 (9.9%) versus n= 20 (6.8%)], progressive disease [n= 8 (2.5%) versus n= 15 ([5.1%)] and AEs [n= 21 (6.5%) versus n= 8] (2.7%)." ITT on 322 participants in the intervention group and 292 participants in the control group |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Baseline characteristics, or different non‐randomised co‐interventions were not reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were not reported |
HIMALAYAS 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions:
*Note: dose assessed as high according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Provenzano 2021 quote: "The randomisation code for patients was generated by a third party vendor in a blinded fashion based a permuted block design in a 1:1 ratio (oral roxadustat: parenteral epoetin alfa), stratified by the randomisation stratification factors specified in the protocol (mean qualifying screening haemoglobin (≤8.0 vs. >8.0 g/dL), history of cardiovascular (CV), cerebrovascular, or thromboembolic disease (yes/no), and geographical region (US vs. Ex‐US). Then the randomisation code was uploaded into an Interactive Response Technology (IRT) system accessible by all eligible sites. Each site would screen patients and log onto the secured IRT system to randomised eligible patients and get the treatment assignment. Eligible patients were randomized by sites sequentially across all sites according to the randomisation code in the IRT system. The randomisation code was concealed in the IRT system managed by the third party vendor." |
| Allocation concealment (selection bias) | Low risk | Provenzano 2021 quote: "The randomisation code for patients was generated by a third party vendor in a blinded fashion based a permuted block design in a 1:1 ratio (oral roxadustat: parenteral epoetin alfa), stratified by the randomisation stratification factors specified in the protocol (mean qualifying screening haemoglobin (≤8.0 vs. >8.0 g/dL), history of cardiovascular (CV), cerebrovascular, or thromboembolic disease (yes/no), and geographical region (US vs. Ex‐US). Then the randomisation code was uploaded into an Interactive Response Technology (IRT) system accessible by all eligible sites. Each site would screen patients and log onto the secured IRT system to randomised eligible patients and get the treatment assignment. Eligible patients were randomized by sites sequentially across all sites according to the randomisation code in the IRT system. The randomisation code was concealed in the IRT system managed by the third party vendor. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐Label." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT population Provenzano 2021: 304/522 in the intervention group and 306/521 completed the study |
| Selective reporting (reporting bias) | High risk | ClincialTrials.gov information was not reported. Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Authors' disclosure was not reported |
Holdstock 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1 (rHuEPO‐naive participants) (low dose)*
Treatment group 2 (rHuEPO‐naive participants) (low dose)
Treatment group 3 (rHuEPO‐naive participants) (low dose)
Control group (rHuEPO‐naive participants)
Co‐interventions
*Note: dose assessed as low according to Akizawa 2017 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Separate randomisation lists were generated for the two groups (rhEPO naive and rhEPO users) using the GlaxoSmithKline randomisation system RandAll." |
| Allocation concealment (selection bias) | Low risk | Quote: "Separate randomisation lists were generated for the two groups (rhEPO naive and rhEPO users) by a GlaxoSmithKline statistician using the GlaxoSmithKline randomisation system RandAll." Quote: "Participants were assigned a randomisation number by an interactive voice/web response system." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An internal GlaxoSmithKline Safety Review Team reviewed blinded safety data instream and an independent data monitoring committee periodically reviewed the same safety data, but it was unblinded." Quote: "Other adverse events of interest, based on the mechanism of action or pharmacological activity of hypoxia‐inducible factor‐prolyl hydroxylase inhibitors, were monitored and evaluated by blinded review based on individual case details during the study." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Two hundred fifty‐two participants were randomized to either daprodustat (n=172) or control (n=80). Of those randomized, 250 (>99%) were included in the safety population and 235 (93%) in the ITT population, with 222 (88%) completing the study (i.e. participants who completed the Week 24 visit regardless of whether they remained on study treatment) [148 (86%) in the daprodustat group and 74 (93%) in the control group]. The most common reasons for premature withdrawal were an AE in the daprodustat group (5%) and withdrawal by participant in the control group (3%). Similar proportions of participants (20% randomized to daprodustat;18% randomized to control) discontinued study treatment prematurely. The most common reasons for treatment discontinuation were an AE in the daprodustat group (10%) and reaching the protocol‐defined stopping criteria in the control group (8%) [included renal transplant, increased systolic pulmonary artery pressure (sPAP) of 20mmHg, drop of left ventricular ejection fraction (LVEF) 10% from baseline and <50% and blood transfusion]." Only 235/252 performed ITT > 5% lost to follow‐up with discrepancies between group |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were reported (death and CV events) |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
Holdstock 2019a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (rHuEPO‐users participants) (low dose)*
Control group (rHuEPO‐users participants)
Co‐interventions
*Note: dose assessed as low according to Akizawa 2017 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Separate randomisation lists were generated for the two groups (rhEPO naive and rhEPO users) using the GlaxoSmithKline randomisation system RandAll." |
| Allocation concealment (selection bias) | Low risk | Quote: "Separate randomisation lists were generated for the two groups (rhEPO naive and rhEPO users) by a GlaxoSmithKline statistician using the GlaxoSmithKline randomisation system RandAll." Quote: "Participants were assigned a randomisation number by an interactive voice/web response system." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An internal GlaxoSmithKline Safety Review Team reviewed blinded safety data instream and an independent data monitoring committee periodically reviewed the same safety data, but it was unblinded." Quote: "Other adverse events of interest, based on the mechanism of action or pharmacological activity of hypoxia‐inducible factor‐prolyl hydroxylase inhibitors, were monitored and evaluated by blinded review based on individual case details during the study." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Two hundred fifty‐two participants were randomized to either daprodustat (n=172) or control (n=80). Of those randomized, 250 (>99%) were included in the safety population and 235 (93%) in the ITT population, with 222 (88%) completing the study (i.e. participants who completed the Week 24 visit regardless of whether they remained on study treatment) [148 (86%) in the daprodustat group and 74 (93%) in the control group]. The most common reasons for premature withdrawal were an AE in the daprodustat group (5%) and withdrawal by participant in the control group (3%). Similar proportions of participants (20% randomized to daprodustat;18% randomized to control) discontinued study treatment prematurely. The most common reasons for treatment discontinuation were an AE in the daprodustat group (10%) and reaching the protocol‐defined stopping criteria in the control group (8%) [included renal transplant, increased systolic pulmonary artery pressure (sPAP) of 20mmHg, drop of left ventricular ejection fraction (LVEF) 10% from baseline and <50% and blood transfusion]." Only 235/252 performed ITT > 5% lost to follow‐up with discrepancies between group |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were reported (death and CV events) |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
Hou 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed as low according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not reported. However, it was likely that participants and/or investigators could be aware of treatment assigned |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of those randomized, 129 patients were enrolled in the ITT population (86 in the roxadustat group and 43 in the ESAs group) and the safety population (86 in the roxadustat group and 43 in the ESAs group). In total, 74 patients in the roxadustat group and 38 patients in the ESAs group completed treatment" ITT analyses |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were reported (death and CV events) |
| Other bias | Low risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups There was nothing to declare in the disclosure Funder was unlikely to influence data analysis and study reporting or interpretation |
INNO2VATE 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed as medium according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label study" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Ecardt 2021: cumulative data were reported for INNO2VATE 2020 and INNO2VATE 2020a: overall 3902/3923 participants were included into the analyses |
| Selective reporting (reporting bias) | High risk | Not all of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report. No reasoning provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
INNO2VATE 2020a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions:
*Note: dose was considered medium according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label study" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Ecardt 2021: cumulative data were reported for INNO2VATE 2020 and INNO2VATE 2020a: overall 3902/3923 participants were included into the analyses |
| Selective reporting (reporting bias) | High risk | Not all of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report. No reasoning provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
Meadowcroft 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Participants were assigned to study treatment in accordance with a randomisation schedule that utilized central randomisation. The randomisation schedule was computer generated using the randomisation system RandAll" Quote: "Eligible participants were stratified by region (Japan versus non‐Japan) and prior rhEPO dose and then randomized 2:2:2:2:1:2." |
| Allocation concealment (selection bias) | Low risk | Quote: "Participants randomized to daprodustat had automatic dose adjustments through an interactive voice/web response system based on a prespecified dose‐adjustment algorithm," Quote: "The response system managed by Perceptive, Nottingham, UK" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "For the first 4 weeks, treatment was fully blinded to treatment group with control participants receiving placebo QD. Thereafter, only the dose of daprodustat was blinded, with control participants receiving standard‐of‐care open‐label rhEPO (epoetin or their biosimilars, or darbepoetin alfa) as required for the remaining 20 weeks to achieve haemoglobin within the target range (10‒11.5 g/dL)." For the scope of this review, we considered only the first 4 weeks, where the comparison was clearly HIF vs placebo. The first 4 weeks were conducted in double blind |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An internal GlaxoSmithKline Safety Review Team reviewed blinded safety data in stream and an independent data monitoring committee periodically reviewed the same safety data but unblinded." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not reported in sufficient detail to perform adjudication in the first 4 weeks of the treatment. Overall, ITT performed on 171/177 participants in the intervention group. and 39/39 participants in the control group. Imbalance between the two groups |
| Selective reporting (reporting bias) | High risk | All pre‐specified outcomes were not reported, for the end of the 4th week where the intervention were compared only with placebo Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Stopped early due to some data‐dependent process (including a formal‐stopping rule) Funder was likely to influence data analysis and study reporting or interpretation Authors declared conflicts of interests |
MIYABI HD‐M 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed as high according to DIALOGUE 1 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Treatment allocation was conducted via an interactive voice/web response system (IxRS), and the computer‐prepared randomisation list was provided to the IxRS supplier by the sponsor." |
| Allocation concealment (selection bias) | Low risk | Quote: "Treatment allocation was conducted via an interactive voice/web response system (IxRS), and the computer‐prepared randomisation list was provided to the IxRS supplier by the sponsor." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind" Quote: "Investigators and patients were blinded to treatment allocation." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of 229 patients randomized to molidustat (n=153) or darbepoetin alfa (n=76), 180 completed 52 weeks of treatment (n=115 and 65)." 151/153 participants in the intervention group and 74/76 participants in the control group completed the follow‐up period, as reported in Figure 1 Some analyses were performed on the ITT population |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
MIYABI ND‐C 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: assessed as medium‐dose according to DIALOGUE 1 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were then randomized 1:1 using an interactive voice/web response system to receive either molidustat or darbepoetin treatment for 52 weeks." |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were then randomized 1:1 using an interactive voice/web response system to receive either molidustat or darbepoetin treatment for 52 weeks." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Following screening, 162 patients were randomized to receive molidustat (n = 82) or darbepoetin (n = 80) (online suppl. Fig. 2). All randomized patients received the assigned study drug except for 1 in the darbepoetin group. In total, 135 patients completed treatment up to week 36 (63 [76.8%] for molidustat and 72 [90.0%] for darbepoetin), and 118 patients completed treatment up to week 52 (53 [64.6%] for molidustat and 65 [81.3%] for darbepoetin)." Some analyses were performed on the ITT population |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
MIYABI ND‐M 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed as medium‐dose according to DIALOGUE 1 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Interactive voice/web response system" |
| Allocation concealment (selection bias) | Low risk | Quote: "Interactive voice/web response system" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "In total, 164 patients were randomized to molidustat (n = 82) or darbepoetin (n = 82). Of these, 133 patients completed treatment up to week 36 (65 [79.3%] in the molidustat group and 68 [82.9%] in the darbepoetin group) and 120 patients completed treatment up to week 52 (57 [69.5%] in the molidustat group and 63 [76.8%] in the darbepoetin group)." ITT analyses |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
Nangaku 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: assessed as medium dose according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind phase" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of the 323 randomized patients, 120 and 135 completed the 52‐week treatment period in the vadadustat and darbepoetin alfa groups, respectively." Reasons for discontinuation were provided and some of them were related to the intervention ITT analyses were performed for some outcomes |
| Selective reporting (reporting bias) | High risk | Not all of the planned outcomes on ClincialTrials.gov have been measured and reported on in the final report. No reasoning provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions were reported between groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation |
Nangaku 2021a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed as medium according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were enrolled by the investigator and randomized 1:1 (using a web‐based system) to oral vadadustat (Akebia Therapeutics Inc., Cambridge, MA) or subcutaneous darbepoetin alfa (Kyowa Kirin Co., Ltd, Tokyo, Japan)." |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "Of the 432 patients who gave informed consent, 304 were randomized to vadadustat (151 patients) or darbepoetin alfa (153 patients) (Figure 2).Of these, 271 patients (130 in the vadadustat group, 141 in the darbepoetin alfa group) completed the 24‐week treatment period and 234 patients (111 in the vadadustat group, 123 in the darbepoetin alfa group) completed the 52‐week treatment period. The most common reason for withdrawal from both groups was initiation of kidney replacement therapy, including chronic haemodialysis/peritoneal dialysis or kidney transplant." Data were reported on the ITT population |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
Nangaku 2021b.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
*Note: in this review we have reported data only on CKD patients because no other publications were identified |
|
| Interventions |
Treatment group (cohort 1) (low dose)
Control group (cohort 1)
Treatment group (cohort 3) (low dose)*
Control group (cohort 3)
Co‐interventions
*Note: assessed as low dose according to Meadowcroft 2019 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A biostatistician generated the randomisation codes using a company‐validated system. A random permutation of treatment assignments within blocks (block size was set to 4) stratified by current ESA use/nonuse and the haemoglobin level (ESA‐naïve: ≤9.5 g/dL, >9.5 g/dL, ESA users: <11.0 g/dL, ≥11.0 g/dL) was used for the randomisation sequence. Investigators accessed the Inter‐ active Web Response System to obtain the assigned interventions." |
| Allocation concealment (selection bias) | Low risk | Quote: "A biostatistician generated the randomisation codes using a company‐validated system. A random permutation of treatment assignments within blocks (block size was set to 4) stratified by current ESA use/nonuse and the haemoglobin level (ESA‐naïve: ≤9.5 g/dL, >9.5 g/dL, ESA users: <11.0 g/dL, ≥11.0 g/dL) was used for the randomisation sequence. Investigators accessed the Inter‐ active Web Response System to obtain the assigned interventions." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Team blinded to treatment assignment conducted periodic case reviews to evaluate which events constituted AEs of special interest." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 111/149 participants in the intervention group and 113/150 participant in the control group completed the study ITT analyses was performed |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were not reported |
| Other bias | High risk | Similar baseline characteristics, or different non‐randomised co‐interventions between groups Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
NCT01888445.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Web registration system." |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The investigator(s) prescribed ASP1517, under double‐blind conditions to patients assigned to Treatment Arms 1, 2 and 3 based on the drug number that was randomly assigned to Treatment Arms 1 to 4 and notified by the web registration system at the time of second registration. The investigator(s) administered darbepoetin alfa to patients assigned to Treatment Arm 4 under open‐label conditions." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Of the 130 randomized patients, 129 (99.2%) were included in the safety analysis set (SAF), 127 (97.7%) were included in the full analysis set (FAS) and pharmacokinetic analysis set (PKAS) and 86 (66.2%) were included in the per protocol set (PPS)." Quote: "A total of 130 patients were randomized. Of these, 129 patients were treated with the study treatment and 1 patient in the ASP1517 100 mg group discontinued before the first dose of study treatment due to the withdrawal of consent. A total of 80 (61.5%) patients completed the study and 50 (38.5%) patients discontinued. A total of30 (23.1%) patients discontinued during the fixed dose period and most of them (27 patients) discontinued within first 1 week in the fixed dose period. The frequency of study discontinuation during the fixed dose period was higher in the pooled ASP1517 group (27.6%) compared with the darbepoetin alfa group (9.4%). The frequency of study discontinuation during the titration period was similar between the pooled ASP1517(14.3%) and darbepoetin alfa (15.6%) groups. The most common reason for the study discontinuation was the discontinuation criterion of Hb being < 8.0 g/dL (31 patients, 23.8%) and most of them (27 patients) were reported during the fixed dose period. A total of 6 (4.6%) patients discontinued due to AEs and all of them were reported in the ASP1517 groups. Most of study discontinuation due to AEs (5 patients) were reported in the titration period." 127/130 participants completed the full analysis (>5% lost to follow‐up), with differences between groups. Reasons were provided |
| Selective reporting (reporting bias) | Low risk | All of the planned outcomes on ClincialTrials.gov were measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were reported (death and CV events) |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation |
NDD‐CKD 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
Note 1 from DD study: active treatment indicated treatment with an anticoagulant (blood thinner), such as heparin, enoxaparin, warfarin, rivaroxaban, apixaban, edoxaban, argatroban, and fondaparinux. Aspirin was not considered active treatment for DVT or pulmonary embolism Note 2: subjects on kidney transplant wait list, or with a history of failed kidney transplant, corneal transplants, or stem cell therapy for knee arthritis were not excluded |
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "All participants were included in the safety and ITT populations." |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov have been measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
NDD‐CKD 2020a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities (DD patients)
Note 1 from DD study: active treatment indicated treatment with an anticoagulant (blood thinner), such as heparin, enoxaparin, warfarin, rivaroxaban, apixaban, edoxaban, argatroban, and fondaparinux. Aspirin was not considered active treatment for DVT or pulmonary embolism Note 2: subjects on kidney transplant wait list, or with a history of failed kidney transplant, corneal transplants, or stem cell therapy for knee arthritis were not excluded |
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "All participants were included in the safety population and 58 were included in the ITT population." 58/68 participants completed the full analysis (>5% lost to follow‐up), with differences between groups. Reasons were provided |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov have been measured and reported on in the final report Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
OLYMPUS 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed as medium according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A total of approximately 2600 patients will be randomized at 1:1 ratio to roxadustat or placebo via an Interactive Web Response System (IWRS)/ Interactive Voice Response System (IVRS)." |
| Allocation concealment (selection bias) | Low risk | Quote: "A total of approximately 2600 patients will be randomized at 1:1 ratio to roxadustat or placebo via an Interactive Web Response System (IWRS)/ Interactive Voice Response System (IVRS)." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This is a double blind, placebo‐controlled study. The investigator, study site staff and the patient, are blinded to study treatment, but not to the dose or dosing frequency. The Sponsor and designees except the personnel analysing the pharmacokinetic (PK) samples are blinded to study treatment, dose and dosing frequency. Sponsor study team members responsible for IWRS system are blinded to study treatment and can in special cases be unblinded to dose and dosing frequency." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote Fishbane 2021: "2781 patients were randomized to roxadustat (1393) or placebo (1388). Twenty patients were excluded due to incorrect randomisation (4) or significant GCP violations (16) in obtaining or recording the data that might affect the validity of the data; therefore, the ITT population comprised 1384 and 1377 patients receiving roxadustat and placebo, respectively. The FAS included 2728 patients (roxadustat 1371; placebo 1357) and the OT+28 population included 2760 patients (roxadustat 1384; placebo 1376)." ITT population was not reported for all patients but loss was < 5% |
| Selective reporting (reporting bias) | High risk | All planned outcomes reported in the study protocol were not reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | It was not possible to assess if there was imbalance between intervention groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation |
Pergola 2016.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: assessed as medium dose according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind" Although author reported that the study used a double‐blind design, information about blinding of participants and investigators were not clearly stated. However, since interventions were different, it was possible that investigators and/or participants were aware of treatment allocation. Possible deviations from the intended intervention that arose from the trial context were not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "The intent‐to‐treat population included 210 patients who received the study drug (vadadustat, n= 138; placebo, n= 72) and were included in the safety analyses. The modified intent‐to‐treat population, used for all efficacy analyses, comprised 208 patients who had a baseline and at least 1 post baseline Hb and red blood cell measurement (vadadustat, n= 136; placebo, n= 72)." In the modified ITT analysis 208/210 participants were included into the analysis (< 5% lost to follow‐up, slight imbalance between the two groups) Quote: "There were 160 patients who qualified for the per‐protocol population." |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov were not measured and reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
PRO2TECT‐CONVERSION 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: assessed as medium dose according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent clinical end‐points committee, whose members were unaware of the treatment assignments, adjudicated MACE." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | In the pooled data from NCT02648347 + NCT02680574 3471/3476 participants were included in the analyses Some outcomes were reported in 821/879 participants in the intervention groups and 811/872 participants in the control group |
| Selective reporting (reporting bias) | Low risk | All planned outcomes on ClincialTrials.gov were measured and reported Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
PRO2TECT‐CORRECTION 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: assessed as medium dose according to NDD‐CKD 2020 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent clinical end‐points committee, whose members were unaware of the treatment assignments, adjudicated MACE." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | In the pooled data from NCT02648347 + NCT02680574 3471/3476 participants were included in the analyses Some outcomes were reported in 821/879 participants in the intervention groups and 811/872 participants in the control group |
| Selective reporting (reporting bias) | Low risk | All planned outcomes on ClincialTrials.gov were measured and reported Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported. Funder was likely to influence data analysis and study reporting or interpretation |
Provenzano 2008.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Single blind" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 96/142 participants completed the study |
| Selective reporting (reporting bias) | High risk | Protocol was not reported Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | It was not possible to assess if there was imbalance between intervention groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation |
Provenzano 2016.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Independent data monitoring committee." Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment It was not reported if the Independent data monitoring committee was blind to the treatments assigned |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Reasons for discontinuation from the study were lack of efficacy (10), withdrawal of consent (4), AE/serious AE (6; including 3 deaths), 3 protocol violations, and 3 others (leaving canter, prolonged hospitalisation, and kidney transplantation)." Not reported in sufficient detail to perform adjudication for part 1 and 2 separately |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov were not measured and reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Quote: "FibroGen was the study sponsor and designed the study in consultation with the principal investigators." There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
Provenzano 2016a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Treatment group 4
Treatment group 5
Treatment group 6
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "Independent data monitoring committee." Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment It was not reported if the Independent data monitoring committee was blind to the treatments assigned |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Reasons for discontinuation from the study were lack of efficacy (10), withdrawal of consent (4), AE/serious AE (6; including 3 deaths), 3 protocol violations, and 3 others (leaving canter, prolonged hospitalisation, and kidney transplantation)." Not reported in sufficient detail to perform adjudication for part 1 and 2 separately |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov were not measured and reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | Quote: "FibroGen was the study sponsor and designed the study in consultation with the principal investigators." There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding and authors' disclosure were reported Funder was likely to influence data analysis and study reporting or interpretation |
PYRENEES 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed as high (mean dose 210 mg) according to NCT01888445 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. There were no imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐Label." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "All data from site (2 patients randomized to the ESA treatment group) are excluded due to Good Clinical Practice (GCP) violations; therefore a total of 836 patients were considered randomized for analysis: 415 to the roxadustat treatment group and 421 to ESA." Quote: "A total of 558 (66.7%) patients completed the study up to 2 years of treatment, 249 (60.0%)in the roxadustat treatment group and 309 (73.4%) in the ESA treatment group. Overall, 40.0% of patients in the roxadustat treatment group and 26.6% of patients in the ESA treatment group discontinued treatment up to 2 years. A total of 13.0% of patients withdrew due to death (14.9% roxadustat vs 11.2% ESA) and 9.1% withdrew by patient(12.0% vs 6.2%)." Analyses were performed on different number of participants (> 5% lost to follow‐up with imbalance between the two groups) |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov were not measured and reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation Conflicts of interest were reported |
ROCKIES 2019.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. It was not possible to assess if there was imbalance between intervention groups |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open‐label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Not reported in sufficient detail to perform adjudication |
| Selective reporting (reporting bias) | High risk | All planned outcomes on ClincialTrials.gov were not measured and reported on in the final report. Reasons were not provided Clinically‐relevant outcomes that would be expected for this type of intervention were not reported |
| Other bias | High risk | It was not possible to assess if there was imbalance between intervention groups Funding was reported and authors' disclosure were not reported Funder was likely to influence data analysis and study reporting or interpretation |
SIERRAS 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (high dose)*
Control group
Co‐interventions
*Note: dose assessed as high according to NCT01888445 |
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Automated randomisation and treatment assignments were performed using an Interactive Web Response System." |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomisation code was concealed in the IRT system managed by the third party vendor. In this setting, the sponsor, a site or a patient would not know the treatment assignment beforehand and could not predict the treatment assignment for the next patient in line." Quote: "Automated randomisation and treatment assignments were performed using an Interactive Web Response System." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Open label" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Objective and subjective outcomes were reported |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT population 334/370 participants in the intervention group and 337/371 participants in the control group The number of participants included in the safety population, full analysis set and per protocol set varied |
| Selective reporting (reporting bias) | Low risk | All planned outcomes on ClincialTrials.gov were measured and reported Clinically‐relevant outcomes that would be expected for this type of intervention (death and CV events) were reported |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Funding influenced data analysis and study reporting or interpretation Conflicts of interest were reported. |
SYMPHONY HD 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (medium dose)*
Control group
Co‐interventions
*Note: dose assessed as medium according to SYMPHONY ND 2021 |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Interactive Web Response System was contacted by the site and randomly assigned the eligible subject with permutated block at a 1:1 ratio to receive once‐daily oral enarodustat tablet." |
| Allocation concealment (selection bias) | Low risk | Quote: "Interactive Web Response System was contacted by the site and randomly assigned the eligible subject with permutated block at a 1:1 ratio to receive once‐daily oral enarodustat tablet." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double blind" |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "In total, 173 subjects were randomized into the study and administered the study drug (87 subjects in the enarodustat arm and 86 subjects in the DA arm). All subjects were included in the SAF. Overall, 172 subjects were included in the FAS because 1 subject in the enarodustat arm had <2 efficacy measurements. In the enarodustat arm, 79 subjects completed the treatment period, and 78 subjects were included in the PPS because 1 subject experienced protocol deviation. In the DA arm, 80 subjects completed the treatment period, and all were included in the PPS." ITT analyses |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported (death and CV events) |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Conflict of interest were reported Funder was likely to influence data analysis and study reporting or interpretation Kyowa Kirin Co administered DA |
SYMPHONY ND 2021.
| Study characteristics | ||
| Methods |
|
|
| Participants |
General information
Baseline characteristics
Comorbidities
|
|
| Interventions |
Treatment group (low dose)
Control group
Co‐interventions
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation methods were not reported in sufficient detail to permit judgement. However, no imbalance between intervention groups was apparent |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment was not reported in sufficient detail to permit judgement. No imbalance between intervention groups was apparent |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label study |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcomes were principally laboratory measures and were at low risk of detection bias regardless of whether blinding of investigators or outcome assessors occurred. However, some outcomes (adverse events) could be influenced by knowledge of treatment assignment |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "In total, 216 subjects (102 ESA‐naïve and 114 ESA‐ treated subjects) were randomly assigned to the enarodustat arm (n ¼ 107; 50 ESA‐naïve and 57 ESA‐treated subjects) or DA arm (n ¼ 109; 52 ESA‐naïve and 57 ESA‐treated subjects) and 195 subjects completed the study. The PPS included 193 subjects after the exclusion of 23 subjects (10 in the enarodustat arm and 13 in the DA arm) with <3 Hb measurements during the evaluation period. Overall, 212 subjects were included in the full analysis set after the exclusion of four subjects (2 in the enarodustat arm and 2 in the DA arm) with <2efficacy measurements from week 4 onward." All 216 subjects (107 in the enarodustat arm and 109 in the DA arm) were included the safety population |
| Selective reporting (reporting bias) | High risk | Clinically‐relevant outcomes that would be expected for this type of intervention were not reported (death and CV events) |
| Other bias | High risk | There was no evidence of different baseline characteristics, or different non‐randomised co‐interventions between groups Conflict of interest were reported Funder was not reported |
ALP ‐ alkaline phosphatase; ALT ‐ alanine aminotransferase; APD ‐ automated peritoneal dialysis; Apo ‐ apolipoproteins; AST ‐ aspartate aminotransferase; AVF ‐ arteriovenous fistula; AUC ‐ area under the curve; AZA ‐ azathioprine; BMI ‐ body mass index; BP ‐ blood pressure; CAPD ‐ continuous ambulatory peritoneal dialysis; CFB ‐ change from baseline; CHF ‐ chronic heart failure; CHr ‐ reticulocyte haemoglobin; CKD ‐ chronic kidney disease; CKD‐EPI ‐ CKD Epidemiology Collaboration; Cmax ‐ maximum concentration; CRP ‐ C‐reactive protein; CV ‐ cardiovascular; DBP ‐ diastolic blood pressure; DVT ‐ deep vein thrombosis; EBP ‐ epoetin beta pegol; ECG ‐ electrocardiogram; eGFR ‐ estimated glomerular filtration rate; EPO ‐ erythropoietin; EQ‐5D‐5L ‐ EuroQol 5 Dimension 5 Level Health Utility Index; ESA ‐ erythropoietin‐stimulating agent; FACT ‐ Functional Assessment of Cancer Therapy; FAS ‐ full analysis set; GI ‐ gastrointestinal; GN ‐ glomerulonephritis; Hb ‐ haemoglobin; HCT ‐ haematocrit; HbA1c ‐ haemoglobin A1c (glycated); HBsAg ‐ hepatitis B surface antigen; HCV Ab ‐ hepatitis C virus antibody; HD ‐ haemodialysis; HF ‐ haemofiltration; HDF ‐ haemodiafiltration; HDL ‐ high‐density lipoprotein; HIF ‐ hypoxia‐inducible factor; HIF‐PHI ‐ Hypoxia‐inducible factor prolyl hydroxylase inhibitor; HIV ‐ human immunodeficiency virus; HRQoL ‐ heath‐related quality of life; HRT ‐ hormone replacement therapy; HUS ‐ haemolytic uraemic syndrome; ITT ‐ intention to treat; IU ‐ international units; IV ‐ intravenous; KDOQI ‐ Kidney Disease Outcomes Quality Initiative; KRT ‐ kidney replacement therapy; LDL ‐ low‐dose lipoprotein; LFT ‐ liver function test/s; LLN ‐ lower limit of normal; MACE ‐ major adverse cardiovascular event (composite of death (any cause), non‐fatal MI, and non‐fatal stroke); MAP ‐ mean arterial pressure; MI ‐ myocardial infarction; NYHA ‐ New York Heart Association; PCKD ‐ polycystic kidney disease; PD ‐ peritoneal dialysis; PGI‐S ‐ Patient Global Impression of Severity Scale; PPS ‐ per protocol set; PRCA ‐ pure red cell aplasia; PTH ‐ parathyroid hormone; QtcB ‐ Bazett's corrected QT interval; RBC ‐ red blood cell; RCT ‐ randomised controlled trial; rHuEPO ‐ recombinant human EPO; SAS ‐ safety analysis set; SBP ‐ systolic blood pressure; SCr ‐ serum creatinine; SGOT ‐ serum glutamic oxaloacetic transaminase; SGPT ‐ serum glutamic pyruvic transaminase; SLE ‐ systemic lupus erythematosus; SC ‐ subcutaneously; sPAP ‐ systolic pulmonary artery pressure; TCM ‐ traditional Chinese medicine; TIA ‐ transient ischaemic attack; TIBC ‐ total iron‐binding capacity; TSAT ‐ transferrin saturation; UACR ‐ urinary albumin:creatinine ratio; UF ‐ ultrafiltration; UIBC ‐ unbound iron binding capacity; ULN ‐ upper limit of normal; URR ‐ urea reduction ratio; VAS ‐ Visual Analogue Scale; VEGF ‐ vascular endothelial growth factor; WBC ‐ white blood cell; WPAI ‐ Work Productivity and Activity Impairment
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Akizawa 2015a | Wrong intervention/control: different dosing regimens of JTZ‐951 |
| Akizawa 2019a | Duration of follow‐up < 8 weeks Phase 1: patients randomised to different doses of enarodustat versus placebo for 6 weeks Phase 2: all participants, including those in the placebo group, took enarodustat until the end of the follow‐up period. It was not clearly stated if the second phase was randomised |
| Akizawa 2019b | Duration of follow‐up < 8 weeks Phase 1: patients randomised to different doses of enarodustat versus placebo for 6 weeks Phase 2: all participants, including those in the placebo group, took enarodustat until the end of the follow‐up period. It was not clearly stated if the second phase was randomised |
| Akizawa 2020 | Wrong intervention: patients not previously receiving ESA were randomised to roxadustat at a starting dose of 50 or 70 mg 3 times/week; patients previously receiving ESA switched from ESA to roxadustat 70 or 100 mg 3 times/week depending on the prior ESA dose |
| Akizawa 2020b | Wrong intervention/control: different dosing regimens of roxadustat |
| Akizawa 2020g | Wrong intervention: patients not previously receiving ESA were randomised to roxadustat at a starting dose of 50 or 70 mg 3 times weekly; patients previously receiving ESA switched from ESA to roxadustat 70 or 100 mg three times weekly depending on the prior ESA dose |
| ASCEND:Fe 2018 | Duration of follow‐up < 8 weeks: protocol reporting that at day 28 participants will be crossed over |
| ASCEND‐BP 2017 | Duration of follow‐up < 8 weeks: protocol reporting that at day 28 participants will be crossed over |
| Bailey 2019 | Duration of follow‐up < 8 weeks |
| Besarab 2016 | Wrong intervention/control: roxadustat + no iron, roxadustat + oral iron, roxadustat + IV iron |
| Buch 2014 | Duration of follow‐up < 8 weeks |
| DD‐CKD 2020 | Duration of follow‐up < 8 weeks: 2 RCTs were included, with dialysis or CKD participants. In both studies in the first 6 weeks patients were randomised to vadadustat 150, 300 or 600 mg versus placebo. For the following 10 weeks vadadustat dose adjustments to achieve target Hb level of 10.0 to 12.0 g/dL, and placebo patients switched to vadadustat 150, 300 or 600 mg. It was not clearly stated if the second phase was randomised for a second time |
| EudraCT2012‐004049‐34 | Duration of follow‐up < 8 weeks |
| EudraCT2012‐004050‐29 | Duration of follow‐up < 8 weeks |
| EudraCT2015‐004790‐32 | Duration of follow‐up < 8 weeks |
| Frohna 2007 | Duration of follow‐up < 8 weeks |
| Haase 2016 | Wrong intervention/control: different dosing regimens of vadadustat |
| Hartman 2014 | Duration of follow‐up < 8 weeks |
| Holdstock CKD 2016 | Duration of follow‐up < 8 weeks |
| Holdstock HD 2016 | Duration of follow‐up < 8 weeks |
| Martin 2017 | Duration of follow‐up < 8 weeks |
| NCT01679587 | Wrong intervention/control: different dosing regimens of molidustat |
| NCT01971164 | Duration of follow‐up < 8 weeks |
| NCT03992066 | Duration of follow‐up < 8 weeks |
| NCT04059913 | Wrong intervention/control: different dosing regimens of roxadustat |
| Pai 2015 | Duration of follow‐up < 8 weeks |
| Parmar 2019 | Duration of follow‐up < 8 weeks |
| Provenzano 2011 | Duration of follow‐up < 8 weeks |
| Provenzano 2011a | Duration of follow‐up < 8 weeks |
| Provenzano 2016b | Wrong intervention/control: different dosing regimens of roxadustat |
| Wiecek 2005 | Duration of follow‐up < 8 weeks |
CKD ‐ chronic kidney disease; ESA ‐ erythropoietin‐stimulating agent; Hb ‐ haemoglobin; IV ‐ intravenous; RCT ‐ randomised controlled trial
Characteristics of studies awaiting classification [ordered by study ID]
FO2RWARD‐2 2019.
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group 1
Treatment group 2
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Notes |
|
ALP ‐ alkaline phosphatase; AST ‐ aspartate aminotransferase; CKD ‐ chronic kidney disease; DVT ‐ deep vein thrombosis; ESA ‐ erythropoietin‐stimulating agent; Hb ‐ haemoglobin; HD ‐ haemodialysis; HIF‐PHI ‐ Hypoxia‐inducible factor prolyl hydroxylase inhibitor; IV ‐ intravenous; LLN ‐ lower limit of normal; MI ‐ myocardial infarction; NYHA ‐ New York Heart Association; PRCA ‐ pure red cell hyperplasia; RBC ‐ red blood cell; RCT ‐ randomised control trial; SGOT ‐ serum glutamic oxaloacetic transaminase; SGPT ‐ serum glutamic pyruvic transaminase; TSAT ‐ transferrin saturation; ULL ‐ upper limit of normal
Characteristics of ongoing studies [ordered by study ID]
ASCEND‐FBF 2018.
| Study name | Anemia study in chronic kidney disease (CKD): erythropoiesis via a novel prolyl hydroxylase inhibitor (PHI) daprodustat ‐forearm blood flow (ASCEND‐FBF) |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | January 2019 |
| Contact information | GSKClinicalSupportHD@gsk.com [mailto:GSKClinicalSupportHD%40gsk.com?subject=NCT03446612, 205767, Anemia Study in Chronic Kidney Disease (CKD) : Erythropoiesis Via a Novel Prolyl Hydroxylase Inhibitor (PHI) Daprodustat ‐Forearm Blood Flow (ASCEND‐FBF)] |
| Notes |
|
CTRI/2019/06/019635.
| Study name | Desidustat in the treatment of anaemia in chronic kidney disease (CKD) |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | June 2019 |
| Contact information | kevinkumar.kansagra@zyduscadila.com |
| Notes |
|
DREAM‐D 2019.
| Study name | Desidustat in the treatment of anaemia in CKD |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | December 2019 |
| Contact information | kevinkumarkansagra@zyduscadila.com |
| Notes |
|
NCT04027517.
| Study name | A study to evaluate efficacy and safety of JTZ‐951 compared to darbepoetin alfa in Korean renal anemia patients receiving hemodialysis |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | January 2019 |
| Contact information | cslimjy@gmail.com [mailto:cslimjy%40gmail.com?subject=NCT04027517, JWP‐JTZ‐301, A Study to Evaluate Efficacy and Safety of JTZ‐951 Compared to Darbepoetin Alfa in Korean Renal Anemia Patients Receiving Hemodialysis.] |
| Notes |
|
NCT04134026.
| Study name | Evaluate the efficacy and safety of roxadustat for the treatment of anemia and risks of cardiovascular and cerebrovascular events in ESRD newly initiated dialysis patients |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | October 2019 |
| Contact information | liuh0618@163.com [mailto:liuh0618%40163.com?subject=NCT04134026, CSU‐SXH‐CT‐2019‐015, Evaluate the Efficacy and Safety of Roxadustat for the Treatment of Anemia and Risks of Cardiovascular and Cerebrovascular Events in ESRD Newly Initiated Dialysis Patients] |
| Notes |
|
NCT04313153.
| Study name | Trial evaluating the efficacy and safety of oral vadadustat once daily (QD) and three times weekly (TIW) for the maintenance treatment of anemia in hemodialysis subjects converting from erythropoiesis‐stimulating agents (ESAs) |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group 1
Treatment group 2
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcome
|
| Starting date | May 2020 |
| Contact information | trials@akebia.com |
| Notes |
|
PER‐038‐14.
| Study name | A phase 3, multicenter, randomized, open‐label active‐controlled study of the efficacy and safety of FG‐4592 in the treatment of anaemia in incident‐dialysis patients |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group 1
Treatment group 2
Treatment group 3
Control group
Co‐interventions
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | December 2014 |
| Contact information | angela.flores@iconplc.com |
| Notes |
|
SLCTR‐2019‐032.
| Study name | A phase 3, multicenter, multi‐country, open‐label, randomized, active‐controlled clinical trial to evaluate the efficacy and safety of desidustat versus darbepoetin for the treatment of anemia in patients with chronic kidney disease (CKD) who are not on dialysis |
| Methods |
|
| Participants |
General information
|
| Interventions |
Treatment group
Control group
Co‐interventions
|
| Outcomes |
Primary outcome
Secondary outcomes
|
| Starting date | August 2018 |
| Contact information | ercmed@kln.ac.lk |
| Notes |
|
ALP ‐ alkaline phosphatase; ALT ‐ alanine aminotransferase; AST ‐ aspartate aminotransferase; AVF ‐ arteriovenous fistula; BP ‐ blood pressure; CKD ‐ chronic kidney disease; CKD‐EPI ‐ CKD Epidemiology Collaboration; CRP ‐ C‐reactive protein; CV ‐ cardiovascular; DBP ‐ diastolic blood pressure; DVT ‐ deep vein thrombosis; ECG ‐ electrocardiogram; eGFR ‐ estimated glomerular filtration rate; EPO ‐ erythropoietin; ESA ‐ erythropoietin‐stimulating agent; FBF ‐ forearm blood flow; GI ‐ gastrointestinal; Hb ‐ haemoglobin; HCT ‐ hematocrit; HbA1c ‐ haemoglobin A1c (glycated); HBsAg ‐ hepatitis B surface antigen; HCV Ab ‐ hepatitis C virus antibody; HD ‐ haemodialysis; HDF ‐ haemodiafiltration; HIF‐PHI ‐ Hypoxia‐inducible factor prolyl hydroxylase inhibitor; HIV ‐ human immunodeficiency virus; HRT ‐ hormone replacement therapy; ITP ‐ idiopathic thrombocytopenic purpura; IU ‐ international units; IV ‐ intravenous; KRT ‐ kidney replacement therapy; LDL ‐ low‐dose lipoprotein; LLN ‐ lower limit of normal; MI ‐ myocardial infarction; NYHA ‐ New York Heart Association; PD ‐ peritoneal dialysis; PRCA ‐ pure red cell aplasia; QoL ‐ quality of life; QtcB ‐ Bazett's corrected QT interval; RBC ‐ red blood cell; RCT ‐ randomised controlled trial; rHuEPO ‐ recombinant human EPO; SBP ‐ systolic blood pressure; SCr ‐ serum creatinine; SGOT ‐ serum glutamic oxaloacetic transaminase; SGPT ‐ serum glutamic pyruvic transaminase; SLE ‐ systemic lupus erythematosus; SC ‐ subcutaneously; TCM ‐ traditional Chinese medicine; TIA ‐ transient Ischaemic attack; TSAT ‐ transferrin saturation; ULN ‐ upper limit of normal; VEGF ‐ vascular endothelial growth factor; WBC ‐ white blood cell
Differences between protocol and review
We included death (any cause), fatal or nonfatal MI, fatal or nonfatal stroke and thrombosis as outcomes.
We have combined data for loss of unassisted patency (including both stenosis/occlusions), and need for access intervention (including both surgically or by radiological guided angioplasty).
We have performed subgroup analyses considering the frequency of HIF stabilisers therapy.
Due to the short follow‐up, we have added the proportion of patients requiring blood transfusion and the proportion of patients reaching the target Hb in the Summary of Findings tables, instead of cancer and infection.
Contributions of authors
Draft the protocol: PN, AT, MR, SP
Study selection: PN, EH, MR, DH, VS
Extract data from studies: PN, EH, MR, DH, VS
Enter data into RevMan: PN, SP
Carry out the analysis: PN, EH, SP
Interpret the analysis: all authors
Draft the final review: PN, EH, SP
Disagreement resolution: SP, GS
Update the review: PN, SP, JC, GS
Sources of support
Internal sources
No sources of support provided
External sources
No sources of support provided
Declarations of interest
Patrizia Natale: no relevant interests were disclosed
Suetonia C Palmer: no relevant interests were disclosed
Allison Jaure: no relevant interests were disclosed
Elisabeth M Hodson: no relevant interests were disclosed
Marinella Ruospo: no relevant interests were disclosed
Tess E Cooper: no relevant interests were disclosed
Deirdre Hahn: no relevant interests were disclosed
Valeria Saglimbene: no relevant interests were disclosed
Jonathan Craig: no relevant interests were disclosed
Giovanni FM Strippoli: no relevant interests were disclosed
New
References
References to studies included in this review
Akizawa 2017 {published data only}
- Akizawa T, Tsubakihara Y, Nangaku M, Endo Y, Nakajima H, Kohno T, et al. Effects of daprodustat, a novel hypoxia-inducible factor prolyl hydroxylase inhibitor on anemia management in Japanese hemodialysis subjects. American Journal of Nephrology 2017;45(2):127-35. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Endo Y, Kohno T, Imai Y, Kawase N, Hara K, Lepore JJ, et al. A 4 -week dose response study of the hypoxia inducible factor-prolyl hydroxylase inhibitor GSK1278863 in Japanese anemic haemodialysis subjects [abstract no: SA-PO819]. Journal of the American Society of Nephrology 2015;26(Abstract Suppl):818A. [Google Scholar]
- NCT02019719. Study to evaluate the safety, efficacy and pharmacokinetics of GSK1278863 in Japanese hemodialysis-dependent subjects with anemia associated with chronic kidney disease [A 4-week, phase ii, randomized, double-blind, dose-ranging, placebo-controlled, parallel-group, multi-center study to evaluate the safety, efficacy and pharmacokinetics of GSK1278863 in Japanese hemodialysis-dependent subjects with anemia associated with chronic kidney disease]. clinicaltrials.gov/show/NCT02019719 (first received 24 December 2013).
Akizawa 2019 {published data only}
- Akizawa T, Iwasaki M, Otsuka T, Reusch M, Misumi T. Roxadustat for the treatment of chronic kidney disease-associated anemia in Japanese patients not on dialysis [abstract no: FR-PO1142]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):7B. [Google Scholar]
- Akizawa T, Iwasaki M, Otsuka T, Reusch M, Misumi T. Roxadustat treatment of chronic kidney disease-associated anemia in Japanese patients not on dialysis: a phase 2, randomized, double-blind, placebo-controlled trial. Advances in Therapy 2019;36(6):1438-54. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Akizawa 2020a {published data only}
- Akizawa T, Iwasaki M, Yamaguchi Y, Majikawa Y, Reusch M. Authors' Reply. Journal of the American Society of Nephrology 2021;32(4):1005-7. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Iwasaki M, Yamaguchi Y, Majikawa Y, Reusch M. Phase 3, randomized, double-blind, active-comparator (darbepoetin alfa) conversion study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan [abstract no: TH-PO1151]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Iwasaki M, Yamaguchi Y, Majikawa Y, Reusch M. Phase 3, randomized, double-blind, active-comparator (darbepoetin alfa) study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan. Journal of the American Society of Nephrology 2020;31(7):1628-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Yamaguchi Y, Majikawa Y, Reusch M. Factors affecting the doses of roxadustat vs darbepoetin alfa for anemia treatment in hemodialysis patients. Therapeutic Apheresis & Dialysis 2021;25(5):575-85. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astellas1517-CL-0307. A study of intermittent oral dosing of ASP1517 in hemodialysis chronic kidney disease patients with anemia [A phase 3, multi-center, randomized, 2-arm parallel, double-blind, active-comparator (darbepoetin alfa) conversion study of intermittent oral dosing of ASP1517 in hemodialysis chronic kidney disease patients with anemia]. www.clinicaltrials.astellas.com/study/?pid=1517-CL-0307 (accessed 4 May 2022).
- Liu F, Wang J, Ye Q, Fu H, Mao J. Roxadustat for renal anemia in ESRD from PKD patients: is it safe enough? Journal of the American Society of Nephrology 2021;32(4):1005. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen QD, Sepah YJ, Yamaguchi Y, Otsuka T, Majikawa Y, Reusch M, et al. Ophthalmological effects of roxadustat in the treatment of anemia in dialysis-dependent and non-dialysis-dependent CKD patients: findings from two phase 3 studies [abstract no: PO0267]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):134. [EMBASE: 633698979] [Google Scholar]
- Sepah Y, Nguyen QD, Yamaguchi Y, Majikawa Y, Reusch M, Akizawa T. Ophthalmological effects of roxadustat in the treatment of anaemia in chronic kidney disease patients on dialysis in a phase 3, randomised, double-blind, active-comparator conversion study [abstract no: MO002]. Nephrology Dialysis Transplantation 2020;35(Suppl 3):iii102. [EMBASE: 633421367] [Google Scholar]
Akizawa 2020c {published data only}
- Akizawa T, Nangaku M, Yonekawa T, Okuda N, Kawamatsu S, Onoue T, et al. Efficacy and safety of daprodustat compared with darbepoetin alfa in Japanese hemodialysis patients with anemia: a randomized, double-blind, phase 3 trial. Clinical Journal of the American Society of Nephrology: CJASN 2020;15(8):1155-65. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozeki H, Akizawa T, Nangaku M, Yonekawa T, Okuda N, Kawamatsu S, et al. Efficacy and safety of daprodustat compared with darbepoetin alfa in Japanese hemodialysis patients with anemia: A randomized, double-blind, phase 3 trial [abstract no: SaO036]. Nephrology Dialysis Transplantation 2019;34(Suppl 1):a350. [EMBASE: 631305259] [DOI] [PMC free article] [PubMed] [Google Scholar]
Akizawa 2020f {published data only}
- Akizawa T, Iwasaki M, Otsuka T, Yamaguchi Y, Reusch M. A phase 3, multicenter, randomized, open-label, active comparator conversion study of roxadustat in non-dialysis-dependent (NDD) patients with anemia in chronic kidney disease (CKD) [abstract no: POS-244]. Kidney International Reports 2021;6(4 Suppl):S103. [EMBASE: 2011682459] [Google Scholar]
- Akizawa T, Iwasaki M, Otsuka T, Yamaguchi Y, Reusch M. A phase 3, multicenter, randomized, open-label, active comparator conversion study of roxadustat in non-dialysis-dependent (NDD) patients with anemia in CKD [abstract no: PO0269]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):134. [EMBASE: 633699036] [Google Scholar]
- Nguyen QD, Sepah YJ, Yamaguchi Y, Otsuka T, Majikawa Y, Reusch M, et al. Ophthalmological effects of roxadustat in the treatment of anemia in dialysis-dependent and non-dialysis-dependent CKD patients: findings from two phase 3 studies [abstract no: PO0267]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):134. [EMBASE: 633698979] [Google Scholar]
Akizawa 2021 {published data only}
- Akizawa T, Iwasaki M, Otsuka T, Yamaguchi Y, Reusch M. Phase 3 study of roxadustat to treat anemia in non-dialysis-dependant CKD. Kidney International Reports 2021;6(7):1810-28. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Tanaka-Amino K, Otsuka T, Yamaguchi Y. Factors affecting doses of roxadustat versus darbepoetin alfa for anemia in nondialysis patients. American Journal of Nephrology 2021;52(9):702-13. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
ALPS 2021 {published data only}
- Astellas 1517-CL-0608. Roxadustat in the treatment of anemia in chronic kidney disease patients not requiring dialysis [A phase 3, randomized, double-blind, placebo controlled study of the efficacy and safety of roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis]. www.trialsummaries.com/Study/StudyDetails?id=14384&tenant=MT_AST_9011 2019. [DOI] [PMC free article] [PubMed]
- Esposito C, Csiky B, Tataradze A, Reusch M, Han C, Sulowicz W. Two phase 3, multicenter, randomized studies of intermittent oral roxadustat in anemic CKD patients on (PYRENEES) and not on (ALPS) dialysis [abstract no: SA-PO225]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):822. [EMBASE: 633767739] [Google Scholar]
- Pecoits-Filho R, Chan TM, Hardy E, Yu KH, Fishbane S. Roxadustat treatment results in consistent improvements in hemoglobin (Hb) vs. placebo: an analysis of three multinational randomized clinical trials in patients with non-dialysis-dependent CKD (NDD-CKD) [abstract no: TH-OR05]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):2. [EMBASE: 633697943] [Google Scholar]
- Rastogi A, Pecoits-Filho R, Fishbane S, Little DJ, Hardy E, Yu KH, et al. Roxadustat treatment corrects anemia to hemoglobin (HB) values ≥10 g/dL in the majority of patients with non-dialysis-dependent chronic kidney disease (NDD-CKD) [abstract no: PO0264]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):133. [EMBASE: 633698803] [Google Scholar]
- Shutov E, Sulowicz W, Esposito C, Tataradze A, Andric B, Reusch M, et al. Roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis: a phase 3, randomized, double-blind, placebo-controlled study (ALPS). Nephrology Dialysis Transplantation 2021;36(9):1629-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
ANDES 2021 {published data only}
- Coyne DW, Roger SD, Chou W, Besarab A, Leong R, Lee TT, et al. Roxadustat favorably modifies iron indices in patients with non-dialysis-dependent CKD-related anemia [abstract no: PO0262]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):132. [EMBASE: 633698686] [Google Scholar]
- Coyne DW, Roger SD, Shin S, Kim SG, Cadena AA, Moustafa MA, et al. Roxadustat for CKD-related anemia in non-dialysis patients. Kidney International Reports 2021;6(3):624-35. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne DW, Roger SD, Shin SK, Kim SG, Cadena AA, Moustafa MA, et al. ANDES: A phase 3, randomized, double-blind, placebo controlled study of the efficacy and safety of roxadustat for the treatment of anemia in CKD patients not on dialysis [abstract no: SA-PO0228]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):822-3. [EMBASE: 633767955] [Google Scholar]
- Pecoits-Filho R, Chan TM, Hardy E, Yu KHP, Fishbane S. Roxadustat treatment results in consistent improvements in hemoglobin (Hb) vs. placebo: an analysis of three multinational randomized clinical trials in patients with non-dialysis-dependent CKD (NDD-CKD) [abstract no: TH-OR05]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):2. [EMBASE: 633697943] [Google Scholar]
- Rastogi A, Pecoits-Filho R, Fishbane S, Little DJ, Hardy E, Yu KH, et al. Roxadustat treatment corrects anemia to hemoglobin (HB) values ≥10 g/dL in the majority of patients with non-dialysis-dependent chronic kidney disease (NDD-CKD) [abstract no: PO0264]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):133. [EMBASE: 633698803] [Google Scholar]
ASCEND‐D 2021 {published data only}
- Singh AK, Blackorby A, Cizman B, Carroll K, Cobitz AR, Davies R, et al. Study design and baseline characteristics of patients on dialysis in the ASCEND-D trial. Nephrology Dialysis Transplantation 2021;37(5):960-72. [MEDLINE: ] [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Blackorby A, Cizman B, Cobitz AR, Godoy SA, Jha V, et al. Regional variation of erythropoietin-stimulating agent hyporesponsiveness in the global daprodustat dialysis study (ASCEND-D) [abstract no: TH-OR08]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):3. [EMBASE: 633698062] [Google Scholar]
- Singh AK, Carroll K, Perkovic V, Solomon S, Jha V, Johansen KL, et al. Daprodustat for the treatment of anemia in patients undergoing dialysis. New England Journal of Medicine 2021;385(25):2325-35. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
ASCEND‐ID 2021 {published data only}
- Singh AK. Daprodustat is noninferior to darbepoetin alfa in treating anemia in incident dialysis patients [abstract no: PO0465]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):184-5. [EMBASE: 636331207] [Google Scholar]
ASCEND‐ND 2021 {published data only}
- Perkovic V, Blackorby A, Cizman B, Carroll K, Cobitz A, Davies R, et al. ASCEND-ND: Study design and baseline characteristics. Nephrology Dialysis Transplantation 2021;36(Suppl 1):i334-5. [EMBASE: 635918331] [Google Scholar]
- Singh AK, Carroll K, McMurray JJ, Solomon S, Jha V, Johansen KL, et al. Daprodustat for the treatment of anemia in patients not undergoing dialysis. New England Journal of Medicine 2021;385(25):2313-24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
ASCEND‐NHQ 2021 {published data only}
- Johansen KL, Cobitz AR, Lopes RD, Singh AK, Obrador GT, Cizman B, et al. Effects of daprodustat on hemoglobin and quality of life in patients with CKD: results of the ascend-NHQ randomized, double-blind, placebo-controlled trial [abstract no: FR-OR53]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):36. [EMBASE: 636330656] [Google Scholar]
ASCEND‐TD 2021 {published data only}
- Coyne DW, Singh AK, Lopes RD, Bailey CK, Dimino TL, Huang C, et al. ASCEND-TD: A randomized, double-blind, active-controlled study of daprodustat administered three times weekly in hemodialysis patients [abstract no: PO0487]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):191. [EMBASE: 636329480] [Google Scholar]
Besarab 2015 {published data only}
- Besarab A, Hutler HN, Klaus S, Lee TT, Lilienfeld DE, Neff TB, et al. FG-4592, a novel oral HIF prolyl hydroxylase inhibitor, elevates hemoglobin in anemic stage 3/4 CKD patients [abstract no: SA-FC416]. Journal of the American Society of Nephrology 2010;21(Abstract Suppl):95A. [Google Scholar]
- Besarab A, Provenzano R, Hertel J, Zabaneh R, Klaus SJ, Lee T, et al. Randomized placebo-controlled dose-ranging and pharmacodynamics study of roxadustat (FG-4592) to treat anemia in nondialysis-dependent chronic kidney disease (NDD-CKD) patients. Nephrology Dialysis Transplantation 2015;30(10):1665-73. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brigandi 2016 {published data only}
- Brigandi RA, Johnson B, Oei C, Westerman M, Olbina G, Zoysa J, et al. A novel hypoxia-inducible factor-prolyl hydroxylase inhibitor (GSK1278863) for anemia in CKD: a 28-day, phase 2a randomized trial. American Journal of Kidney Diseases 2016;67(6):861-71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Brigandi RA, Johnson B, Oei C, Westerman ME, Olbina G, Kumar S, et al. Induction of erythropoiesis in anemic patients by prolylhydroxylase inhibitor in a repeat dose, randomized placebo controlled trial [abstract no: SA-PO117]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):662A. [Google Scholar]
- Brigandi RA, Westerman ME, Olbina G, Oei C, Russ SF, Kumar S. Modulation of hepcidin by prolylhydroxylase inhibitor in randomized trials [abstract no: SA-PO118]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):662A. [Google Scholar]
Chen 2019 {published data only}
- Chen N, Hao C, Liu B, Lin HL, Caili W, Xing CY, et al. A phase 3, randomized, open-label, active-controlled study of efficacy and safety of roxadustat for treatment of anemia in subjects with CKD on dialysis [abstract no: TH-PO1152]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):B5. [Google Scholar]
- Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. New England Journal of Medicine 2019;381(11):1011-22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Chen 2019a {published data only}
- Chen N, Hao C, Peng X, Lin H, Yin A, Hao L, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. New England Journal of Medicine 2019;381(11):1001-10. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chen N, Hao C, Peng X, Lin HL, Yin A, Hao L, et al. A phase 3, randomized, double-blind, placebo-controlled study of efficacy and safety of roxadustat (FG-4592) for treatment of anemia in subjects with CKD not on dialysis [abstract no: TH-PO1153]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):B5. [Google Scholar]
Chen DD 2017 {published data only}
- Chen N, Qian J, Chen J, Yu X, Mei C, Hao C, et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrology Dialysis Transplantation 2017;32(8):1373-86. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen NDD 2017 {published data only}
- Chen N, Qian J, Chen J, Yu X, Mei C, Hao C, et al. Phase 2 studies of oral hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for treatment of anemia in China. Nephrology Dialysis Transplantation 2017;32(8):1373-86. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian JQ, Chen N, Chen J, Hao C, Lin HL, Ni D, et al. A randomized, double-blind, placebo controlled trial of FG-4592 for correction of anemia in subjects with chronic kidney disease in China [abstract no: FR-OR011]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):38A. [Google Scholar]
DIALOGUE 1 2019 {published data only}
- Akizawa T, Macdougall IC, Berns JS, Bernhardt T, Taguchi M, Ogura E, et al. Iron regulation by molidustat, bay 85-3934, a daily oral hypoxia-inducible factor prolyl hydroxylase inhibitor in patients with chronic kidney disease [abstract no: SP334]. Nephrology Dialysis Transplantation 2018;33(Suppl 1):i457. [EMBASE: 622606520] [Google Scholar]
- Akizawa T, Macdougall IC, Berns JS, Yamamoto H, Taguchi M, Iekushi K, et al. Iron regulation by molidustat, a daily oral hypoxia-inducible factor prolyl hydroxylase inhibitor, in patients with chronic kidney disease. Nephron 2019;143(4):243-54. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Akizawa T, Berns J, Lentini S, Bernhardt T. Molidustat increases haemoglobin in erythropoiesis stimulating agents (ESA)-naive anaemic patients with chronic kidney disease not on dialysis (CKD-ND) [abstract no: SO036]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i16. [EMBASE: 72325954] [Google Scholar]
- Macdougall IC, Akizawa T, Berns JS, Bernhardt T, Krueger T. Effects of molidustat in the treatment of anemia in CKD [Erratum in: Clin J Am Soc Nephrol. 2019 Oct 7;14(10):1524]. Clinical Journal of the American Society of Nephrology: CJASN 2019;14(1):28-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Berns JS, Akizawa T, Fishbane S, Bernhardt T. DIALOGUE phase 2 program for BAY85-3934 a HIF-PH inhibitor with daily oral treatment in anemic patients suffering from CKD/ESRD [abstract no: FR-PO219]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):413A. [Google Scholar]
DIALOGUE 2 2019 {published data only}
- Akizawa T, Macdougall IC, Berns JS, Yamamoto H, Taguchi M, Iekushi K, et al. Iron regulation by molidustat, a daily oral hypoxia-inducible factor prolyl hydroxylase inhibitor, in patients with chronic kidney disease. Nephron 2019;143(4):243-54. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Akizawa T, Berns J, Lentini S, Bernhardt T, Kruger T. Safety and efficacy of molidustat in erythropoiesis stimulating agents (ESA) pre-treated anaemic patients with chronic kidney disease not on dialysis (CKD-ND) [abstract no: SP309]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i193. [EMBASE: 72326411] [Google Scholar]
- Macdougall IC, Akizawa T, Berns J, Lentini S, Bernhardt T. Molidustat increases haemoglobin in erythropoiesis stimulating agents (ESA)-naive anaemic patients with chronic kidney disease not on dialysis (CKD-ND) [abstract no: SO036]]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i16. [EMBASE: 72325954] [Google Scholar]
- Macdougall IC, Akizawa T, Berns JS, Bernhardt T, Krueger T. Effects of molidustat in the treatment of anemia in CKD [Erratum in: Clin J Am Soc Nephrol. 2019 Oct 7;14(10):1524]. Clinical Journal of the American Society of Nephrology: CJASN 2019;14(1):28-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Berns JS, Akizawa T, Fishbane S, Bernhardt T. DIALOGUE phase 2 program for BAY85-3934 a HIF-PH inhibitor with daily oral treatment in anemic patients suffering from CKD/ESRD [abstract no: FR-PO219]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):413A. [Google Scholar]
DIALOGUE 4 2019 {published data only}
- Akizawa T, Macdougall IC, Berns JS, Yamamoto H, Taguchi M, Iekushi K, et al. Iron regulation by molidustat, a daily oral hypoxia-inducible factor prolyl hydroxylase inhibitor, in patients with chronic kidney disease. Nephron 2019;143(4):243-54. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Akizawa T, Berns J, Lentini S, Bernhardt T. Molidustat increases haemoglobin in erythropoiesis stimulating agents (ESA)-naive anaemic patients with chronic kidney disease not on dialysis (CKD-ND) [abstract no: SO036]. Nephrology Dialysis Transplantation 2016;31(Suppl 1):i16. [EMBASE: 72325954] [Google Scholar]
- Macdougall IC, Akizawa T, Berns JS, Bernhardt T, Krueger T. Effects of molidustat in the treatment of anemia in CKD [Erratum in: Clin J Am Soc Nephrol. 2019 Oct 7;14(10):1524]. Clinical Journal of the American Society of Nephrology: CJASN 2019;14(1):28-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall IC, Berns JS, Akizawa T, Fishbane S, Bernhardt T. DIALOGUE phase 2 program for BAY85-3934 a HIF-PH inhibitor with daily oral treatment in anemic patients suffering from CKD/ESRD [abstract no: FR-PO219]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):413A. [Google Scholar]
DOLOMITES 2021 {published data only}
- Barratt J, Andric B, Tataradze A, Schoemig M, Reusch M, Valluri U, et al. Roxadustat for the treatment of anemia in CKD patients not on dialysis (NDD): a phase 3, randomized, open-label, active-controlled study [abstract no: TH-OR02]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):1. [EMBASE: 633697812] [Google Scholar]
- Barratt J, Andric B, Tataradze A, Schomig M, Reusch M, Valluri U, et al. Roxadustat for the treatment of anaemia in chronic kidney disease patients not on dialysis: a phase 3, randomised, open-label, active-controlled study (DOLOMITES). Nephrology Dialysis Transplantation 2021;36(9):1616-28. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt J, Andric B, Tataradze A, Schomig M, Reusch M, Valluri U, et al. Roxadustat for the treatment of anaemia in chronic kidney disease patients not on dialysis: a phase 3, randomised, open-label, active controlled study [abstract no: MO001]. Nephrology Dialysis Transplantation 2020;35(Suppl 3):iii101. [EMBASE: 633423023] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt J, Andric B, Tataradze A, Schomig M, Reusch M, Valluri U, et al. Roxadustat for the treatment of anemia in chronic kidney disease (CKD) patients not on dialysis (NDD): a phase 3, randomized, open-label, active-controlled study [abstract no: Pos-247]. Kidney International Reports 2021;6(4 Suppl):S104. [EMBASE: 2011683542] [Google Scholar]
HIMALAYAS 2021 {published data only}
- Provenzano M, Evgeny S, Liubov E, Korneyeva S, Kathresal AA, Poole L, et al. HIMALAYAS: a phase 3, randomized, open-label, active-controlled study of the efficacy and safety of roxadustat in the treatment of anemia in incident-dialysis patients [abstract no: TH-OR021]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):5. [EMBASE: 633771140] [Google Scholar]
- Provenzano R, Shutov E, Eremeeva L, Korneyeva S, Poole L, Saha G, et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrology Dialysis Transplantation 2021;36(9):1717-30. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Holdstock 2019 {published data only}
- Cizman B, Ackert J, Meadowcroft A, Biswas N, Kelly D, Kleinman D, et al. Ocular safety profile of daprodustat: results of two 24 week studies [abstract no: SAO004]. Nephrology Dialysis Transplantation 2018;33(Suppl 1):i316. [EMBASE: 622604997] [Google Scholar]
- Holdstock L, Cizman B, Meadowcroft AM, Biswas N, Johnson BM, Jones D, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants with chronic kidney disease. Clinical Kidney Journal 2019;12(1):129-38. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdstock L, Cizman B, Meadowcroft AM, Biswas N, Jones D, Johnson BM, et al. Daprodustat, a HIF-prolyl-hydroxylase inhibitor, increases and maintains hemoglobin over 24 weeks in anemic chronic kidney disease subjects [abstract no: TH-PO908]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):304A. [Google Scholar]
Holdstock 2019a {published data only}
- Cizman B, Ackert J, Meadowcroft A, Biswas N, Kelly D, Kleinman D, et al. Ocular safety profile of daprodustat: results of two 24 week studies [abstract no: SAO004]. Nephrology Dialysis Transplantation 2018;33(Suppl 1):i316. [EMBASE: 622604997] [Google Scholar]
- Holdstock L, Cizman B, Meadowcroft AM, Biswas N, Johnson BM, Jones D, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants with chronic kidney disease. Clinical Kidney Journal 2019;12(1):129-38. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdstock L, Cizman B, Meadowcroft AM, Biswas N, Jones D, Johnson BM, et al. Daprodustat, a HIF-prolyl-hydroxylase inhibitor, increases and maintains hemoglobin over 24 weeks in anemic chronic kidney disease subjects [abstract no: TH-PO908]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):304A. [Google Scholar]
Hou 2021 {published data only}
- Hou YP, Mao XY, Wang C, Xu ZH, Bu ZH, Xu M, et al. Roxadustat treatment for anemia in peritoneal dialysis patients: a randomized controlled trial. Journal of the Formosan Medical Association 2021;121(2):529-38. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
INNO2VATE 2020 {published data only}
- Chertow GM, Boudville N, Chowdhury P, Gonzalez C, Kooienga L, Luo W, et al. Vadadustat for treatment of anemia in patients with dialysis-dependent CKD receiving peritoneal dialysis [abstract no: PO0464]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):184. [EMBASE: 636331136] [Google Scholar]
- Eckardt KU, Agarwal R, Aswad A, Awad A, Block GA, Bacci MR, et al. Safety and efficacy of vadadustat for anemia in patients undergoing dialysis. New England Journal of Medicine 2021;384(17):1601-12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Agarwal R, Farag YM, Jardine AG, Khawaja Z, Koury MJ, et al. Global Phase 3 programme of vadadustat for treatment of anaemia of chronic kidney disease: rationale, study design and baseline characteristics of dialysis-dependent patients in the INNO2VATE trials. Nephrology Dialysis Transplantation 2020;36(11):2039-48. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt KU. Global phase 3 clinical trials of vadadustat vs. darbepoetin alfa for treatment of anemia in patients with dialysis-dependent CKD [abstract no: TH-OR01]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):1. [EMBASE: 633697781] [Google Scholar]
- Parfrey PS, Luo W, Maroni B, Anders R, Vargo D, McCullough PA. Thromboembolic events with vadadustat vs. darbepoetin alfa for anemia treatment in patients with dialysis-dependent CKD [abstract no: PO0463]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):184. [EMBASE: 636331080] [Google Scholar]
- Winkelmayer W, Tumlin JA, Fishbane S, Farag Y, Vargo D, Luo W, et al. Hematologic efficacy of vadadustat for anemia in patients with kidney failure on dialysis [abstract no: MO529]. Nephrology Dialysis Transplantation 2021;36(Suppl 1):i325-6. [EMBASE: 635917983] [Google Scholar]
- Winkelmayer WC, Fishbane S, Tumlin JA, Farag YM, Luo W, Anders R, et al. Iron-related outcomes in patients with dialysis-dependent CKD randomized to vadadustat vs. darbepoetin alfa [abstract no: PO0457]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):182. [EMBASE: 636330773] [Google Scholar]
INNO2VATE 2020a {published data only}
- Eckardt KU, Agarwal R, Aswad A, Awad A, Block GA, Bacci MR, et al. Safety and efficacy of vadadustat for anemia in patients undergoing dialysis. New England Journal of Medicine 2021;384(17):1601-12. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Eckardt KU, Agarwal R, Farag YM, Jardine AG, Khawaja Z, Koury MJ, et al. Global Phase 3 programme of vadadustat for treatment of anaemia of chronic kidney disease: rationale, study design and baseline characteristics of dialysis-dependent patients in the INNO2VATE trials. Nephrology Dialysis Transplantation 2020;36(11):2039-48. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt KU. Global phase 3 clinical trials of vadadustat vs. darbepoetin alfa for treatment of anemia in patients with dialysis-dependent CKD [abstract no: TH-OR01]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):1. [EMBASE: 633697781] [Google Scholar]
- Winkelmayer WC, Fishbane S, Tumlin JA, Farag YM, Luo W, Anders R, et al. Iron-related outcomes in patients with dialysis-dependent CKD randomized to vadadustat vs. darbepoetin alfa [abstract no: PO0457]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):182. [EMBASE: 636330773] [Google Scholar]
Meadowcroft 2019 {published data only}
- Cizman B, Ackert J, Meadowcroft A, Biswas N, Kelly D, Kleinman D, et al. Ocular safety profile of daprodustat: results of two 24 week studies [abstract no: SAO004]. Nephrology Dialysis Transplantation 2018;33(Suppl 1):i316. [EMBASE: 622604997] [Google Scholar]
- Cobitz AR, Meadowcroft AM, Cizman B, Holdstock L, Biswas N, Jones D, et al. Daprodustat, a HIF-prolyl-hydroxylase inhibitor, maintains hemoglobin levels over 24 weeks in anemic hemodialysis subjects switching from recombinant human erythropoietin. [abstract no: SA-OR114]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):93A. [Google Scholar]
- Meadowcroft AM, Cizman B, Holdstock L, Biswas N, Johnson BM, Jones D, et al. Daprodustat for anemia: a 24-week, open-label, randomized controlled trial in participants on hemodialysis. Clinical Kidney Journal 2019;12(1):139-48. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
MIYABI HD‐M 2019 {published data only}
- Akizawa T, Taguchi M, Matsuda Y, Iekushi K, Yamada T, Yamamoto H. Molidustat for the treatment of renal anaemia in patients with dialysis-dependent chronic kidney disease: Design and rationale of three phase III studies. BMJ Open 2019;9(6):e026602. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Yamada T, Nobori K, Matsuda Y, Hayashi Y, Hayasaki T, et al. Molidustat for Japanese patients with renal anemia receiving dialysis. Kidney International Reports 2021;6(10):2604–16. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Yamada T, Nobori K, Matsuda Y, Hayashi Y, Hayasaki T, et al. Results from a phase 3 study comparing the efficacy and safety of molidustat vs. Darbepoetin Alfa in patients receiving hemodialysis and treated with erythropoiesis-stimulating agents (ESAS) [abstract no: PO22623]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):B4. [EMBASE: 633697376] [Google Scholar]
MIYABI ND‐C 2019 {published data only}
- Yamamoto H, Nobori K, Matsuda Y, Hayashi Y, Hayasaki T, Akizawa T. Efficacy and safety of molidustat for anemia in ESA-naive nondialysis patients: a randomized, phase 3 trial. American Journal of Nephrology 2021;52(10-11):871-83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Taguchi M, Matsuda Y, Iekushi K, Yamada T, Akizawa T. Molidustat for the treatment of renal anaemia in patients with non-dialysis-dependent chronic kidney disease: design and rationale of two phase III studies. BMJ Open 2019;9(6):e026704. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Taguchi M, Nobori K, Matsuda Y, Iekushi K, Akizawa T. To investigate the efficacy and safety of molidustat in non-dialysis patients with renal anemia who are not treated with erythropoiesis stimulating agents: MIYABI ND-C [abstract no: P1866]. Nephrology Dialysis Transplantation 2020;35(Suppl 3):iii2177. [EMBASE: 633422996] [Google Scholar]
MIYABI ND‐M 2019 {published data only}
- Yamamoto H, Nobori K, Matsuda Y, Hayashi Y, Hayasaki T, Akizawa T. Molidustat for renal anemia in nondialysis patients previously treated with erythropoiesis-stimulating agents: a randomized, open-label, phase 3 study. American Journal of Nephrology 2021;52(10-11):884-93. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Taguchi M, Matsuda Y, Iekushi K, Yamada T, Akizawa T. Molidustat for the treatment of renal anaemia in patients with non-dialysis-dependent chronic kidney disease: design and rationale of two phase III studies. BMJ Open 2019;9(6):e026704. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Taguchi M, Nobori K, Matsuda Y, Iekushi K, Akizawa T. To investigate the efficacy and safety of molidustat in non-dialysis patients with renal anemia who are treated with erythropoiesis stimulating agents: Miyabi ND-M [Abstract no: P1868]. Nephrology Dialysis Transplantation 2020;35(Suppl 3):iii2179. [EMBASE: 633423006] [Google Scholar]
Nangaku 2021 {published data only}
- Nangaku M, Kondo K, Ueta K, Kokado Y, Kaneko G, Matsuda H, et al. Efficacy and safety of vadadustat compared with darbepoetin alfa in Japanese anemic patients on hemodialysis: a phase 3, multicenter, randomized, double-blind study. Nephrology Dialysis Transplantation 2021;36(9):1731-41. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Kondo K, Ueta K, Kokado Y, Kaneko G, Matsuda H, et al. Randomized, double-blinded, active-controlled (darbepoetin alfa), phase 3 study of vadadustat in CKD patients with anemia on hemodialysis in Japan [abstract no: TH-OR024]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):6. [EMBASE: 633771357] [Google Scholar]
Nangaku 2021a {published data only}
- Nangaku M, Kondo K, Kokado Y, Ueta K, Kaneko G, Shiosaka M, et al. Randomized, open-label, active-controlled (darbepoetin alfa), phase 3 study of vadadustat for treating anemia in non-dialysis-dependent CKD patients in Japan [abstract no: SA-PO229]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):823. [EMBASE: 633767995] [Google Scholar]
- Nangaku M, Kondo K, Kokado Y, Ueta K, Kaneko G, Tandai T, et al. Phase 3 randomized study comparing vadadustat with darbepoetin alfa for anemia in Japanese patients with nondialysis-dependent CKD. Journal of the American Society of Nephrology 2021;32(7):1779-90. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nangaku 2021b {published data only}
- GSK201753. A 52-week, Phase III, open-label, multi-center study to evaluate efficacy and safety of GSK1278863 in Japanese non-dialysis and peritoneal dialysis subjects with anemia associated with chronic kidney disease [study protocol]. www.clinicaltrials.gov/ProvidedDocs/63/NCT02791763/Prot_000.pdf (accessed 9 May 2022).
- Nangaku M, Hamano T, Akizawa T, Tsubakihara Y, Nagai R, Okuda N, et al. Daprodustat compared with epoetin beta pegol for anemia in Japanese patients not on dialysis: a 52-week randomized open-label phase 3 trial. American Journal of Nephrology 2021;52(1):26-35. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01888445 {published data only}
- Astellas1517-CL-0304. A study to investigate the effect of ASP1517 after intermittent oral dosing in dialysis chronic kidney disease patients with anemia compared with darbepoetin as a reference drug [A Japanese, phase 2, multicenter, randomized, 4-arm parallel, double-blind (arms 1-3), open-label (arm 4), active-comparator (darbepoetin alfa) study of intermittent oral dosing of asp1517 in hemodialysis-dependent chronic kidney disease patients with anemia [1517-CL-0304 clinical study results report]]. www.trialsummaries.com/Study/StudyDetails?id=14175&tenant=MT_AST_9011; www.clinicaltrials.gov/ct2/show/NCT01888445 (first received 6 August 2018).
NDD‐CKD 2020 {published data only}
- Nangaku M, Farag YM, deGoma E, Luo W, Vargo D, Khawaja Z. Vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, for treatment of anemia of chronic kidney disease: two randomized Phase 2 trials in Japanese patients. Nephrology Dialysis Transplantation 2020 Jul 20 [epub ahead of print]. [DOI: 10.1093/ndt/gfaa060] [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nangaku M, Khawaja Z, Luo W, Garafola S, De Goma E, Komatsu Y. Randomized, placebo-controlled phase 2 trials of vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI), to treat anemia of CKD [abstract no: TH-PO228]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):171. [EMBASE: 633737146] [Google Scholar]
- Solinsky C, Farag YM, Khawaja Z, Anders R, Luo W, Chavan A, et al. Impact of vadadustat on iron regulation in Japanese subjects with chronic kidney disease (CKD) [abstract no: 352]. American Journal of Kidney Diseases 2020;75(4):638-9. [EMBASE: 2005716872] [Google Scholar]
NDD‐CKD 2020a {published data only}
- Nangaku M, Farag YM, deGoma E, Luo W, Vargo D, Khawaja Z. Vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, for treatment of anemia of chronic kidney disease: two randomized Phase 2 trials in Japanese patients. Nephrology Dialysis Transplantation 2020 Jul 20 [epub ahead of print]. [DOI: 10.1093/ndt/gfaa060] [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nangaku M, Khawaja Z, Luo W, Garafola S, deGoma E, Komatsu Y. Randomized, placebo-controlled phase 2 trials of vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI), to treat anemia of CKD [abstract no: TH-PO228]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):171. [EMBASE: 633737146] [Google Scholar]
- Solinsky C, Farag YM, Khawaja Z, Anders R, Luo W, Chavan A, et al. Impact of vadadustat on iron regulation in Japanese subjects with chronic kidney disease (CKD) [abstract no: 352]. American Journal of Kidney Diseases 2020;75(4):638-9. [EMBASE: 2005716872] [Google Scholar]
OLYMPUS 2021 {published data only}
- AstraZeneca. A phase 3, multicenter, randomized, double-blind, placebo-controlled study evaluating the safety and efficacy of roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis [clinical study protocol V6; 31 August 2018]. www.clinicaltrials.gov/ProvidedDocs/27/NCT02174627/Prot_000.pdf (accessed 9 May 2022).
- Eleftheriadis T, Pissas G, Liakopoulos V, Stefanidis I. On the increased event rate of urinary tract infection and pneumonia in CKD patients treated with roxadustat for anemia. Journal of the American Society of Nephrology 2021;32(6):1537. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbane S, El-Shahawy MA, Pecoits-Filho R, Van BP, Houser MT, Frison L, et al. OLYMPUS: a phase 3, randomized, double-blind, placebo-controlled, international study of roxadustat efficacy in patients with non-dialysis-dependent (NDD) CKD and anemia [abstract no: TH-OR023]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):6. [EMBASE: 633771285] [Google Scholar]
- Fishbane S, El-Shahawy MA, Pecoits-Filho R, Van BP, Houser MT, Frison L, et al. Roxadustat for treating anemia in patients with CKD not on dialysis: results from a randomized phase 3 study. Journal of the American Society of Nephrology 2021;32(3):737-55. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbane S, El-Shahawy MA, Van BP, Little DJ. Authors' Reply. Journal of the American Society of Nephrology 2021;32(6):1537-8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecoits-Filho R, Chan TM, Hardy E, Yu KH, Fishbane S. Roxadustat treatment results in consistent improvements in hemoglobin (Hb) vs. placebo: an analysis of three multinational randomized clinical trials in patients with non-dialysis-dependent CKD (NDD-CKD) [abstract no: TH-OR05]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):2. [EMBASE: 633697943] [Google Scholar]
- Rastogi A, Pecoits-Filho R, Fishbane S, Little DJ, Hardy E, Yu KH, et al. Roxadustat treatment corrects anemia to hemoglobin (HB) values ≥10 g/dL in the majority of patients with non-dialysis-dependent chronic kidney disease (NDD-CKD) [abstract no: PO0264]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):133. [EMBASE: 633698803] [Google Scholar]
Pergola 2016 {published data only}
- Haase VH, Spinowitz BS, Pergola PE, Farmer T, Maroni BJ, Hartman CS. AKB-6548 demonstrates controlled hemogloblin (HGB) response in a phase 2b study for the treatment of anemia in patients with chronic kidney disease not on dialysis (ND-CKD) [abstract no: TH-PO649]. Journal of the American Society of Nephrology 2015;26(Abstract Suppl):237A. [Google Scholar]
- Haase VH, Spinowitz BS, Pergola PE, Khawaja Z, Chan J, Zuraw Q, et al. Efficacy and dose requirements of vadadustat are independent of systemic inflammation and prior erythropoiesis-stimulating agent(ESA) dose in patients with non-dialysis dependent chronic kidney disease (NDD-CKD) [abstract no: TH-PO907]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):304A. [Google Scholar]
- Pergola PE, Spinowitz B, Haase VH, Hartman CS, Farmer TM, Polu KR, et al. AKB-6548, a novel hypoxia-inducible factor prolyl-hydroxylase inhibitor (HIF-PHI) for the treatment of anemia in patients with chronic kidney disease not on dialysis (ND-CKD) [abstract no: FO015]. Nephrology Dialysis Transplantation 2015;30(Suppl 3):iii8. [EMBASE: 72206296] [Google Scholar]
- Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney International 2016;90(5):1115-22. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Spinowitz BS, Pergola PE, Haase VH, Farmer TM, Hartman CS, Maroni BJ. Hemoglobin (HGB) response in a phase 2b study of AKB-6548 for the treatment of anemia in patients with non-dialysis dependant chronic kidney disease (NDD-CKD). [abstract no: TH-OR038]. Journal of the American Society of Nephrology 2015;26(Abstract Suppl):11A. [Google Scholar]
PRO2TECT‐CONVERSION 2021 {published data only}
- Bunn HF. Vadadustat for anemia in patients with dialysis-dependent or non-dialysis-dependent chronic kidney disease. New England Journal of Medicine 2021;385(16):e56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM, Pergola PE, Agarwal R, Block GA, Farag YM, Jardine AG, et al. Cardiovascular safety and efficacy of vadadustat for the treatment of anemia in non-dialysis-dependent CKD: design and baseline characteristics. American Heart Journal 2021;235(5):1-11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM, Pergola PE, Farag YM, Agarwal R, Arnold S, Bako G, et al. Vadadustat in patients with anemia and non-dialysis-dependent CKD. New England Journal of Medicine 2021;384(17):1589-600. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM. Global phase 3 clinical trials of vadadustat vs. darbepoetin alfa for treatment of anemia in patients with non-dialysis-dependent CKD [abstract no: FR-OR54]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):B2. [EMBASE: 633697273] [Google Scholar]
- Koury M, Pergola PE, Roy-Chaudhury P, Farag YM, Luo W, Anders R, et al. Iron-related outcomes in patients with non-dialysis-dependent CKD randomized to vadadustat vs. darbepoetin Alfa [abstract no: PO0482]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):190. [EMBASE: 636329134] [Google Scholar]
- McCullough PA, Luo W, Anders R, Vargo D, Parfrey PS. Assessment of thromboembolic events with vadadustat vs. darbepoetin alfa for treatment of anemia in patients with non-dialysis-dependent CKD [abstract no: PO0462]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):184. [EMBASE: 636331039] [Google Scholar]
PRO2TECT‐CORRECTION 2021 {published data only}
- Bunn HF. Vadadustat for anemia in patients with dialysis-dependent or non-dialysis-dependent chronic kidney disease. New England Journal of Medicine 2021;385(16):e56. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM, Pergola PE, Agarwal R, Block GA, Farag YM, Jardine AG, et al. Cardiovascular safety and efficacy of vadadustat for the treatment of anemia in non-dialysis-dependent CKD: design and baseline characteristics. American Heart Journal 2021;235(5):1-11. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM, Pergola PE, Farag YM, Agarwal R, Arnold S, Bako G, et al. Vadadustat in patients with anemia and non-dialysis-dependent CKD. New England Journal of Medicine 2021;384(17):1589-600. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Chertow GM. Global phase 3 clinical trials of vadadustat vs. darbepoetin alfa for treatment of anemia in patients with non-dialysis-dependent CKD [abstract no: FR-OR5]. Journal of the American Society of Nephrology 2020;31(Abstract Suppl):B2. [EMBASE: 633697273] [Google Scholar]
- Koury M, Pergola PE, Roy-Chaudhury P, Farag YM, Luo W, Anders R, et al. Iron-related outcomes in patients with non-dialysis-dependent CKD randomized to vadadustat vs. darbepoetin alfa [abstract no: PO0482]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):190. [EMBASE: 636329134] [Google Scholar]
- McCullough PA, Luo W, Anders R, Vargo D, Parfrey PS. Assessment of thromboembolic events with vadadustat vs. darbepoetin alfa for treatment of anemia in patients with non-dialysis-dependent CKD [abstract no: PO0462]. Journal of the American Society of Nephrology 2021;32(Abstract Suppl):184. [EMBASE: 636331039] [Google Scholar]
Provenzano 2008 {published data only}
- Provenzano R, Fadda G, Bernardo M, James C, Kochendoerfer G, Lee T, et al. FG2216, a novel oral HIF-PHI, stimulates erythropoiesis and increases hemoglobin concentration in patients with non-dialysis CKD [abstract no: 212]. American Journal of Kidney Diseases 2008;51(4):A80. [Google Scholar]
- Provenzano R, Hulter H, Agarwal A, Klaus S, Lee T, Yu P, et al. Hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitor FG-2216 increases Hb without FE depletion in the absence of IV Fe [abstract no: 249]. American Journal of Kidney Diseases 2013;61(4):A78. [EMBASE: 71024073] [Google Scholar]
Provenzano 2016 {published data only}
- Provenzano R, Besarab A, Dua SL, Nguyen PV, Wright SH, Zeig S, et al. FG-4592, a novel oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI), maintains hemoglobin levels and lowers cholesterol in hemodialysis patients: phase 2 comparison with epoetin alfa [abstract no: FR-PO257]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):428A. [Google Scholar]
- Provenzano R, Besarab A, Wright S, Dua S, Zeig S, Nguyen P, et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. American Journal of Kidney Diseases 2016;67(6):912-24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Provenzano 2016a {published data only}
- Provenzano R, Besarab A, Dua SL, Nguyen PV, Wright SH, Zeig S, et al. FG-4592, a novel oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI), maintains hemoglobin levels and lowers cholesterol in hemodialysis patients: phase 2 comparison with epoetin alfa [abstract no: FR-PO257]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):428A. [Google Scholar]
- Provenzano R, Besarab A, Wright S, Dua S, Zeig S, Nguyen P, et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. American Journal of Kidney Diseases 2016;67(6):912-24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
PYRENEES 2021 {published data only}
- Astellas 1517-CL-0613. Roxadustat in the treatment of anemia in end stage renal disease (ESRD) patients on stable dialysis [A phase 3, randomized, open-label, active-controlled study to evaluate the efficacy and safety of roxadustat in the maintenance treatment of anemia in end stage renal disease patients on stable dialysis]. www.trialsummaries.com/Study/StudyDetails?id=14380&tenant=MT_AST_9011 (accessed 9 May 2022).
- Csiky B, Schomig M, Esposito C, Barratt J, Reusch M, Valluri U, et al. Roxadustat for the maintenance treatment of anemia in patients with end-stage kidney disease on stable dialysis: a European phase 3, randomized, open-label, active-controlled study (PYRENEES). Advances in Therapy 2021;38(10):5361-80. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito C, Csiky B, Tataradze A, Reusch M, Han C, Sulowicz W. Two phase 3, multicenter, randomized studies of intermittent oral roxadustat in anemic CKD patients on (PYRENEES) and not on (ALPS) dialysis. Swiss Medical Weekly 2020;150(Suppl 248):21-2S. [EMBASE: 634241523] [Google Scholar]
- Esposito C, Csiky B, Tataradze A, Reusch M, Han C, Sulowicz W. Two phase 3, multicenter, randomized studies of intermittent oral roxadustat in anemic CKD patients on (PYRENEES) and not on (ALPS) dialysis [abstract no: SA-PO225]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):822. [EMBASE: 633767739] [Google Scholar]
ROCKIES 2019 {published data only}
- AstraZeneca. A phase 3, multicenter, randomized, open-label, active-controlled study of the safety and efficacy of roxadustat in the treatment of anemia in dialysis patients [Clinical study protocol v8.0 19 September 2018]. www.clinicaltrials.gov/ProvidedDocs/31/NCT02174731/Prot_000.pdf (accessed 10 May 2022).
- Fishbane S, Pollock CA, El-Shahawy MA, Escudero ET, Rastogi A, Van BP, et al. ROCKIES: an international, phase 3, randomized, open-label, active-controlled study of roxadustat for anemia in dialysis-dependent CKD patients [abstract no: TH-OR022]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):6. [EMBASE: 633771219] [Google Scholar]
SIERRAS 2021 {published data only}
- Charytan C, Manllo-Karim R, Martin ER, Steer D, Bernardo M, Dua SL, et al. A randomized trial of roxadustat in anemia of kidney failure: SIERRAS study. Kidney International Reports 2021;6(7):1829-39. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charytan C, Manllo-Karim R, Martin ER, Steer D, Bernardo M, Dua SL, et al. SIERRAS: A phase 3, open-label, randomized, active-controlled study of the efficacy and safety of roxadustat in the maintenance treatment of anemia in subjects with ESRD on stable dialysis [abstract no: SA-PO227]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):822. [EMBASE: 633767862] [Google Scholar]
SYMPHONY HD 2021 {published data only}
- Akizawa T, Maeda K, Miyazawa Y, Koretomo R. Phase 3 study to compare the efficacy and safety of enarodustat (JTZ-951), an oral HIF-PH inhibitor, with darbepoetin alfa in anemic patients with CKD receiving maintenance hemodialysis [abstract no: TH-PO1186]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):B4. [Google Scholar]
- Akizawa T, Nangaku M, Yamaguchi T, Koretomo R, Maeda K, Miyazawa Y, et al. A phase 3 study of enarodustat (JTZ-951) in Japanese hemodialysis patients for treatment of anemia in chronic kidney disease: SYMPHONY HD study. Kidney Diseases 2021;7(6):494-502. [EMBASE: 2013576033] [DOI] [PMC free article] [PubMed] [Google Scholar]
SYMPHONY ND 2021 {published data only}
- Akizawa T, Nangaku M, Yamaguchi T, Koretomo R, Maeda K, Miyazawa Y, et al. A phase 3 study of enarodustat in anemic patients with CKD not requiring dialysis: the SYMPHONY ND study. Kidney International Reports 2021;6(7):1840-9. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Akizawa 2015a {published data only}
- Akizawa T, Hanaki K, Arai M. JTZ-951, an oral novel HIF-PHD inhibitor, elevates hemoglobin in Japanese anemic patients with chronic kidney disease receiving maintenance hemodialysis [abstract no: FO019]. Nephrology Dialysis Transplantation 2015;30(Suppl 3):iii10. [EMBASE: 72206300] [Google Scholar]
Akizawa 2019a {published data only}
- Akizawa T, Miyazawa Y, Matsui A, Koretomo R, Arai M. Enarodustat (JTZ-951) , an oral HIF-PH inhibitor, elevates and maintains hemoglobin levels over 30 weeks in Japanese anemic patients with CKD not on dialysis [abstract no: TH-PO225]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):171. [EMBASE: 633737010] [Google Scholar]
- Akizawa T, Nangaku M, Yamaguchi T, Arai M, Koretomo R, Matsui A, et al. A placebo-controlled, randomized trial of enarodustat in patients with chronic kidney disease followed by long-term trial. American Journal of Nephrology 2019;49(2):165-74. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Akizawa 2019b {published data only}
- Akizawa T, Miyazawa Y, Maeda K, Koretomo R, Arai M. Enarodustat (JTZ-951), an oral HIF-PH inhibitor, maintains hemoglobin levels switching from ESAs over 30 weeks in Japanese anemic patients with CKD receiving maintenance hemodialysis [abstract no: TH-PO226]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):171. [EMBASE: 633737068] [Google Scholar]
- Akizawa T, Nangaku M, Yamaguchi T, Arai M, Koretomo R, Maeda K, et al. Enarodustat, conversion and maintenance therapy for anemia in hemodialysis patients: a randomized, placebo-controlled phase 2b trial followed by long-term trial. Nephron 2019;143(2):77-85. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Akizawa 2020 {published data only}
- Akizawa T, Otsuka T, Reusch M, Ueno M. Intermittent oral dosing of roxadustat in peritoneal dialysis chronic kidney disease patients with anemia: a randomized, phase 3, multicenter, open-label study. Therapeutic Apheresis & Dialysis 2020;24(2):115-25. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akizawa T, Otsuka T, Reusch M, Ueno M. Phase 3, multicenter, open-label study of intermittent oral roxadustat in peritoneal dialysis CKD patients with anemia [abstract no: SA-OR075]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):99. [EMBASE: 633736837] [Google Scholar]
Akizawa 2020b {published data only}
- Akizawa T, Otsuka T, Yamaguchi Y, Reusch M. Phase 3, multicenter, randomized, open-label, non-comparative study of intermittent oral roxadustat in ESA-naive CKD patients not on dialysis in Japan [abstract no: SA-PO226]. Journal of the American Society of Nephrology 2019;30(Abstract Suppl):822. [EMBASE: 633767826] [Google Scholar]
- Akizawa T, Yamaguchi Y, Otsuka T, Reusch M. A phase 3, multicenter, randomized, two-arm, open-label study of intermittent oral dosing of roxadustat for the treatment of anemia in Japanese erythropoiesis-stimulating agent-naive chronic kidney disease patients not on dialysis. Nephron 2020;144(8):372-82. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Akizawa 2020g {published data only}
- Akizawa T, Ueno M, Shiga T, Reusch M. Oral roxadustat three times weekly in ESA-naive and ESA-converted patients with anemia of chronic kidney disease on hemodialysis: Results from two phase 3 studies. Therapeutic Apheresis & Dialysis 2020;24(6):628-41. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
ASCEND:Fe 2018 {published data only}
- NCT03457701. Anemia studies in CKD: erythropoiesis via a novel prolyl hydroxylase inhibitor (PHI) daprodustat- iron (ASCEND: Fe) [A repeat dose, open label, two period, randomized, cross over study to compare the effect of daprodustat to recombinant, human erythropoietin (rhEPO) on oral iron absorption in adult participants with anemia associated with chronic kidney disease who are not on dialysis]. www.clinicaltrials.gov/show/NCT03457701 (first received 7 March 2018).
ASCEND‐BP 2017 {published data only}
- NCT03029247. Anemia study in chronic kidney disease (CKD): erythropoiesis via a novel prolyl hydroxylase inhibitor (PHI) daprodustat-blood pressure (ASCEND-BP) [A randomized, open-label study to evaluate the effect of daprodustat on blood pressure in subjects with anemia associated with chronic kidney disease on hemodialysis switched from a stable dose of an erythropoiesis-stimulating agent]. www.clinicaltrials.gov/show/NCT03029247 (first received 24 January 2017).
Bailey 2019 {published data only}
- Bailey CK, Caltabiano S, Cobitz AR, Huang C, Mahar KM, Patel V, et al. A 29-day safety, efficacy, and pharmacodynamic study of a hypoxia-inducible factor prolyl hydroxylase inhibitor, daprodustat, administered TIW in anemic subjects on hemodialysis (HD) [abstract no: SA-PO811]. Journal of the American Society of Nephrology 2017;28(Abstract Suppl):889. [EMBASE: 633698132] [Google Scholar]
- Bailey CK, Caltabiano S, Cobitz AR, Huang C, Mahar KM, Patel VV. A randomized, 29-day, dose-ranging, efficacy and safety study of daprodustat, administered three times weekly in patients with anemia on hemodialysis. BMC Nephrology 2019;20(1):372. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Besarab 2016 {published data only}
- Besarab A, Chan DT, Dua SL, Franco M, Henry E, Leong R, et al. Hypoxia inducing factor prolyl hydroxylase inhibitor FG-4592 corrects anemia in peritoneal dialysis [abstract no: SA-OR087]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):91A. [Google Scholar]
- Besarab A, Chernyavskaya E, Motylev I, Shutov E, Kumbar LM, Gurevich K, et al. Roxadustat (FG-4592): correction of anemia in incident dialysis patients. Journal of the American Society of Nephrology 2016;27(4):1225-33. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besarab A, Chernyavskaya EN, Motylev I, Evgeny S, Yampolskiy AF, Kumbar LM, et al. FG-4592, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, corrects anemia without iron supplementation in incident dialysis patients [abstract no: TH-OR096]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):21A. [Google Scholar]
- Besarab A, Szczech L, Yu KH, Neff TB. Impact of iron regimen on iron indices and hepcidin during roxadustat anemia correction in incident dialysis patients [abstract no: TH-PO847]. Journal of the American Society of Nephrology 2014;25(Abstract Suppl):304a. [Google Scholar]
Buch 2014 {published data only}
- Buch A, Farmer TM, Hartman C, Alcorn H, Shalwitz R. Hemodialysis has minimal impact on the pharmacokinetics of AKB-6548, a once-daily oral inhibitor of hypoxia inducible factor prolyl-hydroxylases (HIFPHs) for the treatment of anemia related to chronic kidney disease (CKD) [abstract no: FR-PO952]. Journal of the American Society of Nephrology 2014;25(Abstract Suppl):590A. [Google Scholar]
DD‐CKD 2020 {published data only}
- Nangaku M, Farag YM, deGoma E, Luo W, Vargo D, Khawaja Z. Vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor, for treatment of anemia of chronic kidney disease: two randomized Phase 2 trials in Japanese patients. Nephrology Dialysis Transplantation 2020 Jul 28 [epub ahead of print]. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Nangaku M, Khawaja Z, Luo W, Garafola S, deGoma E, Komatsu Y. Randomized, placebo-controlled phase 2 trials of vadadustat, an oral hypoxia-inducible factor prolyl hydroxylase inhibitor (HIF-PHI), to treat anemia of CKD [abstract no: TH-PO228]. Journal of the American Society of Nephrology 2018;29(Abstract Suppl):171. [EMBASE: 633737146] [Google Scholar]
- Solinsky C, Farag YM, Khawaja Z, Anders R, Luo W, Chavan A, et al. Impact of vadadustat on iron regulation in Japanese subjects with chronic kidney disease (CKD) [abstract no: 352]. American Journal of Kidney Diseases 2020;75(4):638-9. [EMBASE: 2005716872] [Google Scholar]
EudraCT2012‐004049‐34 {published data only}
- 2012-004049-34. A four-week, phase IIa, randomized, active-controlled, parallel-group, multi-center study to evaluate the safety, efficacy and pharmacokinetics of switching subjects from a stable dose of recombinant human erythropoietin to GSK1278863 in hemodialysis-dependent subjects with anaemia associated with chronic kidney disease - 4 week switch study in HD subjects. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2012-004049-34 2013.
- GSK116582. A 4 week phase IIa, randomized, active-controlled, parallel-group, multi-center study to evaluate the safety, efficacy and pharmacokinetics of switching subjects from a stable dose of recombinant human erythropoietin to GSK1278863 in hemodialysis-dependent subjects with anemia associated with chronic kidney disease. www.clinicaltrialsregister.eu/ctr-search/trial/2012-004049-34/results 2013.
EudraCT2012‐004050‐29 {published data only}
- GSK116581. A four-week phase IIa, randomized, double-blind, placebo controlled, parallel-group, multi-center study to evaluate the safety, efficacy and pharmacokinetics of GSK1278863 in subjects with anemia associated with chronic kidney disease who are not taking recombinant human erythropoietin and are not undergoing dialysis. www.clinicaltrialsregister.eu/ctr-search/trial/2012-004050-29/results 2012.
EudraCT2015‐004790‐32 {published data only}
- 2015-004790-32. A 29-day, randomized, double-blinded, placebo-controlled, parallel-group, multi-center study to evaluate the efficacy, safety and pharmacokinetics of three-times weekly dosing of GSK1278863 in hemodialysis-dependent subjects with anemia associated with chronic kidney disease who are switched from a stable dose of an erythropoiesis-stimulating agent. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2015-004790-32 2016 Jan 13.
Frohna 2007 {published data only}
- Frohna PA, Milwee S, Pinkett J, Lee T, Moore-Perry K, Chou J, et al. Preliminary results from a randomized, single-blind, placebo-controlled trial of FG-4592, a novel hypoxia inducible factor prolyl hydroxylase inhibitor, in CKD anemia [abstract no: SU-PO806]. Journal of the American Society of Nephrology 2007;18(Abstracts Issue):763A. [Google Scholar]
Haase 2016 {published data only}
- Haase VH, Khawaja Z, Chan J, Zuraw Q, Farzaneh-Far R, Maroni BJ, et al. Vadadustat maintains hemoglobin (Hb) levels in dialysis-dependent chronic kidney disease (DD-CKD) patients independent of systemic inflammation or prior dose of erythropoiesis-stimulating agent (ESA) [abstract no: TH-PO960]. Journal of the American Society of Nephrology 2016;27(Abstract Suppl):318A. [Google Scholar]
Hartman 2014 {published data only}
- Hartman CS, Shalwitz IR, Shalwitz RA. Controlled hemoglobin response in a double-blind, placebo-controlled trial of AKB-6548 in subjects with chronic kidney disease [abstract no: SO051]. Nephrology Dialysis Transplantation 2014;29(Suppl 3):iii22. [EMBASE: 71491516] [Google Scholar]
- Shalwitz R, Hartman C, Flinn C, Shalwitz I, Peters KG, Besarab A, et al. AKB-6548, a new hypoxia-inducible factor prolyl hydroxylase inhibitor, increases hemoglobin in chronic kidney disease patients without increasing basal erythropoietin levels [abstract no: FR-OR116]. Journal of the American Society of Nephrology 2012;23(Abstract Suppl):56A. [Google Scholar]
Holdstock CKD 2016 {published data only}
- Holdstock L, Meadowcroft AM, Maier R, Johnson BM, Jones D, Rastogi A, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. Journal of the American Society of Nephrology 2016;27(4):1234-44. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Holdstock HD 2016 {published data only}
- Holdstock L, Meadowcroft AM, Maier R, Johnson BM, Jones D, Rastogi A, et al. Four-week studies of oral hypoxia-inducible factor-prolyl hydroxylase inhibitor GSK1278863 for treatment of anemia. Journal of the American Society of Nephrology 2016;27(4):1234-44. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadowcroft AM, Holdstock L, Maier R, Jones D, Johnson B, Cobitz AR, et al. Four-week safety, efficacy and pharmacodynamic study of hypoxia-inducible factor (HIF)-prolyl inhibitor GSK1278863 in anemic hemodialysis subjects switching from recombinant human erythropoietin [abstract no: SA-PO1092]. Journal of the American Society of Nephrology 2013;24(Abstract Suppl):6B. [Google Scholar]
Martin 2017 {published data only}
- Martin ER, Smith MT, Maroni BJ, Zuraw QC, deGoma EM. Clinical trial of vadadustat in patients with anemia secondary to stage 3 or 4 chronic kidney disease. American Journal of Nephrology 2017;45(5):380-8. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01679587 {published data only}
- NCT01679587. Dose escalation study to investigate safety, tolerability, pharmacodynamics, and pharmacokinetics of BAY85-3934 in subjects with chronic kidney disease (CKD) [A multicenter, randomized, single-blind, placebo-controlled, combined 2-fold cross-over and group-comparison, dose-escalation study to investigate safety, tolerability, pharmacodynamics, and pharmacokinetics of single oral doses of BAY 85-3934 in subjects with chronic kidney disease (CKD)]. www.clinicaltrials.gov/show/NCT01679587 (first received 6 September 2012).
NCT01971164 {published data only}
- Yamamoto H. Safety, tolerability, PK & PD study of JTZ-951 in anemic subjects with end-stage renal disease [Randomized, single-blind, placebo-controlled, multiple ascending dose study to evaluate safety, tolerability, pharmacokinetics & pharmacodynamics of JTZ-951 administered once daily for 15 days in anemic subjects with end-stage renal disease]. www.clinicaltrials.gov/show/NCT01971164 (first received 29 October 2013).
NCT03992066 {published data only}
- NCT03992066. Study to evaluate the pharmacokinetics, pharmacodynamics, and safety of vadadustat in hemodialysis subjects with anemia associated with chronic kidney disease [A phase 1b, randomized, open-label study to evaluate the pharmacokinetics, pharmacodynamics, and safety of vadadustat in hemodialysis subjects with anemia associated with chronic kidney disease]. www.clinicaltrials.gov/show/NCT03992066 (first received 19 June 2019).
NCT04059913 {published data only}
- NCT04059913. Evaluate the efficacy and safety of multiple roxadustat dosing regimens for the treatment of anemia in dialysis subjects with chronic kidney disease [A prospective, randomized, open-label, multi-center study to evaluate the efficacy and safety of multiple roxadustat dosing regimens for the treatment of anemia in dialysis subjects with chronic kidney disease]. www.clinicaltrials.gov/show/NCT04059913 (first received 16 August 2019).
Pai 2015 {published data only}
- Pai S, Koretomo R, Tamaki S, Berg J, Marbury T, Galloway C, et al. JTZ-951, a novel HIF-PHD inhibitor, demonstrates increases in hemoglobin, iron mobilization, reproducible pharmacokinetics, and safety following once daily administration for 15 days in patients with anemia receiving hemodialysis [abstract no: FP658]. Nephrology Dialysis Transplantation 2015;30:iii293-4. [EMBASE: 72207075] [Google Scholar]
Parmar 2019 {published data only}
- Parmar D, Kansagra K. A phase II trial to assess safety, tolerability and efficacy of phd-2 inhibitor (desidustat-zyan1) in the treatment of anemia in pre-dialysis chronic kidney disease patients [abstract no: MON-318]. Kidney International Reports 2019;4(7 Suppl):S430. [EMBASE: 2002179966] [Google Scholar]
- Parmar DV, Kansagra KA, Patel JC, Joshi SN, Sharma NS, Shelat AD, et al. Outcomes of desidustat treatment in people with anemia and chronic kidney disease: a phase 2 study. American Journal of Nephrology 2019;49(6):470-8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Provenzano 2011 {published data only}
- Provenzano R, Goodkin D, Klaus S, Linde P, Kazazi F, Lee T, et al. Evaluation of FG-4592, a novel oral hypoxia-inducible factor prolyl hydroxylase inhibitor, to treat anemia in hemodialysis patients [abstract no: 253]. American Journal of Kidney Diseases 2011;57(4):A80. [EMBASE: 70379812] [Google Scholar]
Provenzano 2011a {published data only}
- Provenzano R, Tumlin J, Zabaneh R, Chou J, Hemmerich S, Neff TB, et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for treatment of anemia in chronic kidney disease: a placebo-controlled study of pharmacokinetic and pharmacodynamic profiles in hemodialysis patients. Journal of Clinical Pharmacology 2020;60(11):1432-40. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano R, Tumlin J, Zabaneh R, Linde P, Chou J, Zhong M, et al. Pharmacokinetics of oral FG-4592 to treat anemia in hemodialysis (HD) patients (PTS) [abstract no: 254]. American Journal of Kidney Diseases 2011;57(4):A80. [EMBASE: 70379813] [Google Scholar]
Provenzano 2016b {published data only}
- Besarab A, Belo D, Diamond S, Martin E, Sun C, Lee T, et al. Evaluation of hypoxia-inducible factor prolyl hydroxylase inhibitor FG-4592 for hemoglobin correction and maintenance in nondialysis chronic kidney disease patients for 16 and 24 weeks [abstract no: FP215]. Nephrology Dialysis Transplantation 2012;27(Suppl 2):ii144-5. [EMBASE: 70765709] [Google Scholar]
- Besarab A, Provenzano R, Fishbane S, Sun CH, Belo DS, Neff TB, et al. FG-4592 oral hypoxia-inducible factor prolyl hydroxylase inhibitor corrects anemia in nondialysis CKD patients without IV iron [abstract no: TH-PO364]. Journal of the American Society of Nephrology 2011;22(Abstract Suppl):196A. [Google Scholar]
- Provenzano R, Besarab A, Sun CH, Diamond SA, Durham JH, Cangiano JL, et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clinical Journal of the American Society of Nephrology: CJASN 2016;11(6):982-91. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiecek 2005 {published data only}
- Wiecek A, Piecha G, Ignacy W, Schmidt R, Neumayer HH, Scigalla P, et al. Pharmacological stabilization of HIF increased hemoglobin concentration in anemic patients with chronic kidney disease [abstract no: MO24]. Nephrology Dialysis Transplantation 2005;20(Suppl 5):v195. [Google Scholar]
References to studies awaiting assessment
FO2RWARD‐2 2019 {published data only}
- NCT03799627. Study of vadadustat in hemodialysis patients with anemia switching from epoetin alfa (FO2RWARD-2) [Phase 2, randomized, open-label, active-controlled, efficacy, safety, pharmacokinetics, and pharmacodynamics study of oral vadadustat for the treatment of anemia in hemodialysis subjects converting from epoetin alfa (FO2RWARD-2)]. www.clinicaltrials.gov/show/NCT03799627 (first received 10 January 2019).
References to ongoing studies
ASCEND‐FBF 2018 {published data only}
- NCT03446612. Anemia study in chronic kidney disease (CKD) : erythropoiesis via a novel prolyl hydroxylase inhibitor (PHI) daprodustat -forearm blood flow (ASCEND-FBF) [A randomized, repeat dose, open label, parallel group, multi-center study to evaluate the effect of daprodustat compared to darbepoetin alfa on forearm blood flow in participants with anemia of chronic kidney disease that are not dialysis dependent]. www.clinicaltrials.gov/show/NCT03446612 (first received 27 February 2018).
CTRI/2019/06/019635 {published data only}
- Kansagra K. Desidustat in the treatment of anemia in chronic kidney disease (CKD) [A phase 3, multicenter, multicountry, open-label, randomized, active-controlled clinical trial to evaluate the efficacy and safety of desidustat versus darbepoetin for the treatment of anemia in patients with chronic kidney disease (CKD) who are not on dialysis.]. ctri.nic.in/Clinicaltrials/showallp.php?mid1=33329&EncHid=&userName=CTRI/2019/06/019635 (first received 12 June 2019).
DREAM‐D 2019 {published data only}
- Kansagra K. Desidustat in the treatment of anemia in CKD. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=36915&EncHid=&userName=CTRI/2019/12/022312 (first received 11 December 2019).
NCT04027517 {published data only}
- Lee JW. A study to evaluate efficacy and safety of JTZ-951 compared to darbepoetin alfa in Korean renal anemia patients receiving hemodialysis [A multi-center, randomized, open-label, active-controlled, parallel-group, phase iii study to compare the efficacy and safety of JTZ-951 with darbepoetin alfa in anemic patients with chronic kidney disease receiving maintenance hemodialysis]. www.clinicaltrials.gov/show/NCT04027517 (first received 22 July 2019).
NCT04134026 {published data only}
- Liu H. Evaluate the efficacy and safety of roxadustat for the treatment of anemia and risks of cardiovascular and cerebrovascular events in ESRD newly initiated dialysis patients [Phase 4 multicenter, randomized, open-lable, active-controlled study of the efficacy and safty of roxadustat for the treatment of anemia and risks of cardiovascular and cerebrovascular events in incident-dialysis patients]. www.clinicaltrials.gov/show/NCT04134026 (first received 21 October 2019).
NCT04313153 {published data only}
- NCT04313153. Trial evaluating the efficacy and safety of oral vadadustat once daily (QD) and three times weekly (TIW) for the maintenance treatment of anemia in hemodialysis subjects converting from erythropoiesis-stimulating agents (ESAs) [Phase 3b, randomized, open-label, active-controlled trial evaluating the efficacy and safety of oral vadadustat once daily (QD) and three times weekly (TIW) for the maintenance treatment of anemia in hemodialysis subjects converting from erythropoiesis-stimulating agents (ESAs)]. www.clinicaltrials.gov/show/NCT04313153 (first received 18 March 2020).
PER‐038‐14 {published data only}
- PER-038-14. A phase 3, multicenter, randomized, open-label active-controlled study of the efficacy and safety of FG-4592 in the treatment of anemia in incident-dialysis patients. www.ins.gob.pe/ensayosclinicos/rpec/recuperarECPBNuevoEN.asp?numec=038-14 (first received 11 December 2014).
SLCTR‐2019‐032 {published data only}
- Nazar AL. A randomized, active-controlled clinical trial to evaluate the efficacy and safety of desidustat versus darbepoetin for the treatment of anemia in patients with chronic kidney disease (CKD) who are not on dialysis [A phase 3, multicenter, multi-country, open-label, randomized, active-controlled clinical trial to evaluate the efficacy and safety of desidustat versus darbepoetin for the treatment of anemia in patients with chronic kidney disease (CKD) who are not on dialysis]. slctr.lk/trials/slctr-2019-032 (first received 13 August 2019).
Additional references
Babitt 2012
- Babitt JL, Lin HY. Mechanisms of anemia in CKD. Journal of the American Society of Nephrology 2012;23(10):1631-4. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bernhardt 2010
- Bernhardt WM, Wiesener MS, Scigalla P, Chou J, Schmeider RE, Günzler V, et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. Journal of the American Society of Nephrology 2010;21(12):2151-6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Achkar 2005
- El-Achkar TM, Ohmit SE, McCullogh PA, Crook ED, Brown WW, Grimm R, et al. Higher prevalence of anemia with diabetes mellitus in moderate kidney insufficiency: The Kidney Early Evaluation Program. Kidney International 2005;67(4):1483-8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
FDA Briefing Document 2021
- US Food & Drug Administration. Cardiovascular and Renal Drugs Advisory Committee Meeting July 15, 2021 Roxadustat. www.fda.gov/media/150728/download (accessed 10 May 2022).
Gansevoort 2013
- Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJ, Mann JF, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 2013;382(9889):339-52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
GBD Kidney Disease 2017
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020;395(10225):709-33. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Global Burden of Disease 2017
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017 [Erratum in: Lancet. 2019 Jun 22;393(10190):e44]. Lancet 2018;392(10159):1789-858. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383-94. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Haase 2021
- Haase VH. Hypoxia-inducible factor-prolyl hydroxylase inhibitors in the treatment of anemia of chronic kidney disease. Kidney Int Suppl (2011) 2021;11(1):8-25. doi: 10.1016/j.kisu.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327(7414):557-60. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2020
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
KDIGO Clinical Practice Guideline Anemia 2012
- Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney International Supplements 2012;2(4):279-335. [https://kdigo.org/wp-content/uploads/2016/10/KDIGO-2012-Anemia-Guideline-English.pdf] [Google Scholar]
KDIGO Clinical Practice Guideline CKD 2012
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney International Supplements 2013;3(1):1-150. [https://kdigo.org/guidelines/ckd-evaluation-and-management/] [DOI] [PMC free article] [PubMed] [Google Scholar]
LaGory 2016
- LaGory EL, Giaccia AJ. The ever expanding role of HIF in tumour and stromal biology. Nature Cell Biology 2016;18(4):356-65. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020
- Li ZL, Tu Y, Liu BC. Treatment of renal anemia with roxadustat: advantages and achievement. Kidney Diseases 2020;6(2):65–73. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2012
- Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. Journal of Clinical Investigation 2012;122(12):4635–44. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ma 1999
- Ma JZ, Ebben J, Xia H, Collins AJ. Hematocrit level and associated mortality in hemodialysis patients. Journal of the American Society of Nephrology 1999;10(3):610-9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mathias 2020
- Mathias SD, Blum SI, Sikirica V, Johansen KL, Colwell HH, Okoro T. Symptoms and impacts in anemia of chronic kidney disease. Journal of Patient-Reported Outcomes 2020;4(1):64. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nemeth 2009
- Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematologica 2009;122(2-3):78–86. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Phrommuntikul 2007
- Phrommintikul A, Haas SJ, Elsik M, Krum H. Mortality and target haemoglobin concentrations in anaemic patients with chronic kidney disease treated with erythropoietin: a meta-analysis. Lancet 2007;369(9559):381-8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Provenzano 2016c
- Provenzano R, Besarab A, Sun CH, Diamond SA, Durham JH, Cangiano JL, et al. Oral hypoxia-inducible factor prolyl hydroxylase inhibitor Roxadustat (FG-4592) for the treatment of anemia in patients with CKD. Clinical Journal of The American Society of Nephrology: CJASN 2016;11(6):982-91. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Renassia 2019
- Renassia C, Peyssonnaux P. New insights into the links between hypoxia and iron homeostasis. Current Opinion in Hematology 2019;26(3):125-30. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schunemann 2020a
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. www.training.cochrane.org/handbook.
Schunemann 2020b
- Schünemann HJ, Vist GE, Higgins JP, Santesso N, Deeks JJ, Glasziou P, et al. Chapter 15: Interpreting results and drawing conclusions. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.1 (updated September 2020). Cochrane, 2020. Available from www.training.cochrane.org/handbook.
Schwartz 2019
- Schwartz AJ, Das NK, Ramakrishnan SK, Jain C Jurkovic MT, Wu J, et al. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. Journal of Clinical Investigation 2019;129(1):336-48. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Semenza 2011
- Semenza GL. Oxygen sensing, homeostasis, and disease [Erratum in: N Engl J Med. 2011 Sep 8;365(10):968]. New England Journal of Medicine 2011;365(6):537-47. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
SONG 2017
- SONG Initiative. The SONG Handbook Version 1.0. www.songinitiative.org/reports-and-publications/ 2017.
van Haalen 2020
- Haalen H, Jackson J, Spinowitz B, Milligan G, Moon R. Impact of chronic kidney disease and anemia on health-related quality of life and work productivity: analysis of multinational real-world data. BMC Nephrology 2020;21(1):88. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020
- Wang B, Yin Q, Han YC, Wu M, Li ZL, Tu Y, et al. Effect of hypoxia-inducible factor-prolyl hydroxylase inhibitors on anemia in patients with CKD: a meta-analysis of randomized controlled trials including 2804 patients. Renal Failure 2020;42(1):912-25. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wyld 2019
- Wyld ML, Morton RL, Calyton P, Wong MG, Jardine M, Polkinghorne K, et al. The impact of progressive chronic kidney disease on health-related quality of life: A 12-year community cohort study. Quality of Life Research 2019;28(8):2081-90. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Zhong 2018
- Zhong H, Zhou T, Li H, Zhong Z. The role of hypoxia-inducible factor stabilizers in the treatment of anemia in patients with chronic kidney disease. Drug Design, Development & Therapy 2018;12:3003-11. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Natale 2020
- Natale P, Palmer SC, Tong A, Ruospo M, Hodson EM, Cooper TE, et al. Hypoxia-inducible factor stabilisers for the anaemia of chronic kidney disease. Cochrane Database of Systematic Reviews 2020, Issue 10. Art. No: CD013751. [DOI: 10.1002/14651858.CD013751] [DOI] [PMC free article] [PubMed] [Google Scholar]