


Abstract
Alternative pre-mRNA splicing is key to proteome diversity; however, the biological roles of alternative splicing (AS) in signaling pathways remain elusive. Here, we focus on TEA domain transcription factor 1 (TEAD1), a YAP binding factor in the Hippo signaling pathway. Public database analyses showed that expression of YAP-TEAD target genes negatively correlated with the expression of a TEAD1 isoform lacking exon 6 (TEAD1ΔE6) but did not correlate with overall TEAD1 expression. We confirmed that the transcriptional activity and oncogenic properties of the full-length TEAD1 isoform were greater than those of TEAD1ΔE6, with the difference in transcription related to YAP interaction. Furthermore, we showed that RNA-binding Fox-1 homolog 2 (RBFOX2) promoted the inclusion of TEAD1 exon 6 via binding to the conserved GCAUG element in the downstream intron. These results suggest a regulatory mechanism of RBFOX2-mediated TEAD1 AS and provide insight into AS-specific modulation of signaling pathways.
Graphical Abstract
Graphical Abstract.
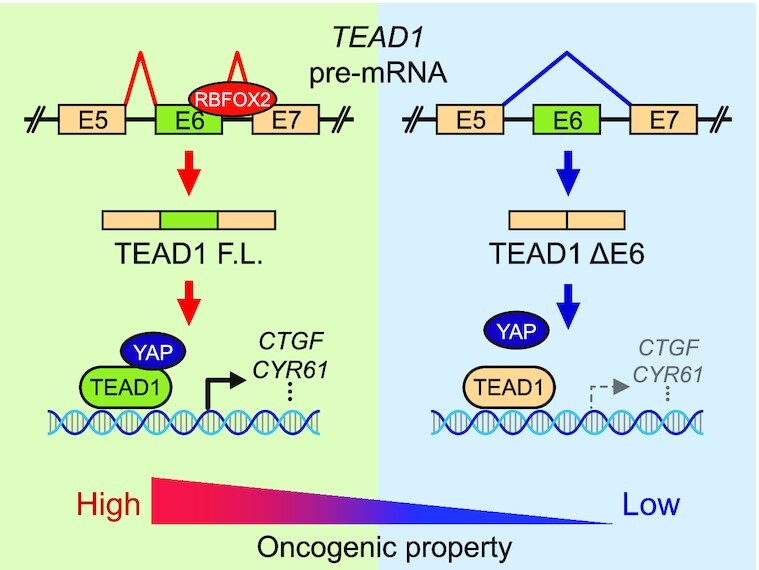
RBFOX2 regulates splicing of TEAD1 exon6, and TEAD1 splicing isoforms induce differential interactions with YAP, affecting the expression level of the YAP-TEAD target gene.
INTRODUCTION
Alternative pre-mRNA splicing (AS) is a post-transcriptional mechanism of gene expression in higher eukaryotes. It plays an important role in expanding proteome diversity by generating multiple functional RNAs from a single gene (1–3). More than 95% of human genes comprising multiple exons express various isoforms through AS (4,5). Abnormal regulation of AS can cause various diseases. According to the Human Gene Mutation Database (HGMD), 15% of genetic diseases, including cancer in humans, are related to abnormal AS regulation by mutation (6,7). Exonic and intronic enhancers and silencers have been defined in pre-mRNAs using various model systems of regulated AS, which identified RNA-binding proteins (RBPs) that could be recruited to these RNA elements, and genome-wide analysis of AS has defined a number of potential cis-elements for splicing regulation (8,9). Moreover, systemic identification of RNA targets of splicing factors enabled prediction of the target-element positions and splicing patterns of RBPs (10,11).
A well-known intronic enhancer element involved in cell-type or tissue-specific regulation of AS is the GCAUG element (12). The RNA-binding Fox (RBFOX) family includes RNA-binding splicing factors (RBFOX1/2/3) harboring an RNA-recognition motif that binds to this GCAUG element and affects the regulation of various AS events (13–17). RBFOX proteins can act as activators and repressors of AS depending on the location of its binding to pre-mRNA based on the alternative exon. Specifically, RBFOX proteins enhance exon inclusion when binding to the GCAUG element located downstream of the alternative exon, whereas binding to this element located upstream of the alternative exon enhances exon skipping (18,19). RBFOX1 exhibits restricted expression in neurons, heart, skeletal muscle myocytes, and RBFOX3 is expressed only in neurons, whereas RBFOX2 is widely expressed in various tissues and cell types (20,21). Many genes that participate in biological processes, including human diseases, reportedly generate alternatively-spliced isoforms through signaling pathways (22–26). Moreover, although some of these genes are candidate targets of RBFOX regulation, the influence of RBFOX-mediated splicing events on signaling pathways in human diseases remains largely unknown.
Maintaining cellular homeostasis in the face of external stimuli requires the regulation of various biological processes, including gene expression, associated with signaling pathways (27,28). Abnormal regulation of signaling pathways can lead to the initiation and/or progression of diseases, such as cancer (29–31). The Hippo signaling pathway is an evolutionarily conserved size-control mechanism that regulates cell survival, growth, and proliferation (32–35). Among the components of this pathway, the TEA domain transcription factor (TEAD) is a DNA-binding protein that plays a pivotal role in regulating the expression of target genes through interaction with YAP/TAZ in the nucleus (36–40). All members of the TEAD family (TEAD1–4) share a highly conserved DNA-binding domain called the TEA domain, which includes a consensus DNA-binding sequence (5′-CATTCCA/T-3′) (41). The TEAD domain contains three α-helices (H1, H2 and H3), with the H3 responsible for DNA-recognition. Additionally, the C-terminus contains the conserved YAP/TAZ-binding domain (42). However, despite its critical role in regulating gene expression, the influence of TEADs in the Hippo signaling pathway is not well understood.
Here, we investigated the role of RBFOX2 in AS regulation in the Hippo signaling pathway. We propose that RBFOX2-dependent AS of TEAD1, which encodes a transcription factor associated with Hippo signaling, drives the cell to acquire YAP-dependent oncogenic properties during cancer progression.
MATERIALS AND METHODS
Cell culture
The human cervical carcinoma cell line HeLa and human embryonic kidney cell line HEK293A were obtained from ATCC (Manassas, VA, USA). LATS1/2-DKO HEK293A cells were generously provided by Dr Kun-Liang Guan, the Department of Pharmacology and Moores Cancer Center at University of California San Diego (43). The cells were maintained in Dulbecco's modified Eagle medium (DMEM; WELGENE, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (FBS; Gibco, Gaithersburg, MD, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (WELGENE). The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Construction of expression plasmids and minigenes
The expression plasmids for Myc-tagged TEAD1 and Myc-tagged RBFOX2 were cloned into the pCS3 + MT vector, and the expression plasmid for C-terminal Flag-tagged RBFOX2 was cloned into pCMV6. The expression plasmid for YAP was purchased from Addgene (#19045; Watertown, MA, USA). The constructs of the TEAD1 minigene contained the sequence required to bind the spliceosome components for RNA splicing. The 5′ splice site of intron 5 contained 107 nucleotides (nts), and the 3′ splice site contained 161 nts. The minigene constructs were obtained by overlap extension (OE)-PCR using four primers (F1, R1, F2 and R2) (Supplementary Table S1). Two fragments containing an overhanging sequence were generated by the first PCR, after which two fragments containing complementary sequences were annealed, followed by a second PCR using minigene F1 and R2 primers. The amplified TEAD1 genomic DNA was cut with EcoRI and SalI restriction enzymes and cloned into the pEGFP-C3 vector. The deletion and mutation constructs of the minigenes were obtained by OE-PCR using four primers (F1, R2, F3 and R3) (Supplementary Table S1). The bold sequences represent the restriction enzyme sites. The underlined nts in the primer sequences targeting TEAD1 (upstream and downstream) represent overhanging nts containing complementary sequences for annealing the PCR products. The underlined nts in the TEAD minigene mutation primers indicate nts for specific mutations. All constructs were verified by sequencing analysis.
Small-interfering (si)RNA and transfection
A non-targeting control siRNA was obtained from Bioneer (Daejeon, Korea). The siRNAs targeting human RBFOX2 were purchased from Dharmacon (Lafayette, CO, USA) and Thermo Fisher Scientific (Waltham, MA, USA). The target sequences for the siRNA duplexes were as follows: siRBFOX2-#1, 5′-GGG AUU CGG GUU CGU AAC U-3′; and siRBFOX2-#2, 5′-AAU GAA CGU GGC UCU AAG GGA UU-3′. Cells were transfected with siRNA using PolyMag transfection reagent (OZ Biosciences, San Diego, CA, USA) according to the manufacturer's instructions. Transfection of plasmids was performed using jetPEI (Polyplus-transfection SA, Illkirch-Graffenstaden, France) according to the manufacturer's instructions. Transfected cells were incubated for 48 h.
Reverse transcription (RT)-PCR and quantitative (q)RT-PCR
Total RNA was extracted using a Hybrid-R RNA extraction kit (GeneAll, Seoul, Korea) according to the manufacturer's instructions and reverse transcribed using M-MLV reverse transcriptase (Promega, Madison, WI, USA) with random hexamers. qRT-PCR was performed using SYBR Green fluorescent dye (GENET BIO, Daejeon, Korea) and the AriaMx PCR system (Agilent Technologies, Santa Clara, CA, USA). Normalization was performed using β-actin as an internal control. RT-PCR was performed with GoldHotStart Taq PCR master mix (Bioneer) using a SimpliAmp thermal cycler PCR machine (Applied Biosystems, Waltham, MA, USA). The specificity of each PCR product was assessed by agarose gel electrophoresis, and band intensities were quantified using ImageJ software (NIH, Bethesda, MD, USA). The ratio of TEAD1 transcripts derived from AS was calculated using the following formula:
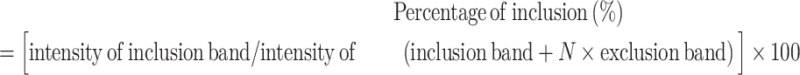 |
where N is the skipped-isoform product size/inclusion isoform product size. The primer set used in this experiment is shown in Supplementary Table S2.
Dual-luciferase assay
Cells were co-transfected with the 8 × GTIIC-Luc reporter plasmid (#34615; Addgene), pRL-null control plasmid (#E2271; Promega), and each indicated plasmid or siRNA. At 48 h post-transfection, luciferase activity was measured using a dual-luciferase reporter assay (Promega) according to the manufacturer's instructions. Luciferase signals were detected using a Luminoskan microplate luminometer (Thermo Fisher Scientific), and the Firefly luciferase signal was normalized to that of Renilla luciferase.
Immunoblot analysis
Whole-cell extracts were prepared with Tris-Triton lysis buffer [10 mM Tris–HCl (pH 7.5), 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 10% glycerol and 0.1% sodium dodecyl sulfate (SDS)] containing a protease inhibitor cocktail (Roche Applied Science, Basel, Switzerland). Protein lysates were denatured and reduced by SDS and β-mercaptoethanol, respectively, followed by separation via SDS-polyacrylamide gel electrophoresis, and transfer to nitrocellulose membranes (Pall Life Science, Port Washington, NY, USA). The membranes were blocked with 5% (w/v) skim milk (Rockland Immunochemicals, West Grove, PA, USA) dissolved in 0.05% Tween-20 in phosphate-buffered saline (PBS), followed by overnight incubation with the primary antibody at 4°C. After washing, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA) for 1 h at 25°C. The proteins were detected using WSE-6200H LuminoGraph II (ATTO, Tokyo, Japan) with a SuperSignal system (Thermo Fisher Scientific). The primary antibodies used in this study were anti-Flag (Sigma-Aldrich, St. Louis, MO, USA), anti-Myc (Invitrogen, Carlsbad, CA, USA), ani-RBFOX2 (Bethyl Laboratories, Montgomery, TX, USA), anti-phospho-YAP(Ser127), anti-YAP (Cell Signaling Technology), anti-TAZ (BD Bioscience, San Jose, CA, USA), and anti-GAPDH (Meridian Life Science, Memphis, TN, USA).
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using a ChIP Express kit (Millipore Biotechnology, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, cells were fixed with 1% formaldehyde for 10 min and then harvested and lysed in SDS lysis buffer (50 mM Tris–HCl pH 8.0, 10 mM EDTA, 1% SDS and 1 mM PMSF) supplemented with protease inhibitor cocktail (Roche Applied Science). Cross-linked cells were sonicated and diluted 10-fold with ChIP dilution buffer [16.7 mM Tris–HCl (pH 8.0), 167 mM NaCl, 1.1% Triton X-100 and 1.2 mM EDTA]. The supernatants were incubated with 5 μg of anti-Myc antibody (Invitrogen) or control mouse IgG (Sigma-Aldrich) for 4 h at 4°C. The antibody/protein-DNA complexes were then incubated at 4°C for 1 h with protein A agarose/salmon sperm DNA. Immunocomplexes were washed with low-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1), and 150 mM NaCl], high-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl (pH 8.1) and 500 mM NaCl], LiCl wash buffer [0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid, 1 mM EDTA and 10 mM Tris (pH 8.1)] and then twice with TE [10 mM Tris–HCl, and 1 mM EDTA (pH 8.0)]. DNA was eluted with elution buffer (1% SDS and 0.1 M NaHCO3), and crosslinks were reversed overnight at 65°C with 200 mM NaCl. After treatment with RNase A and proteinase K, the DNA was purified. qPCR was performed using SYBR Green fluorescent dye and the AriaMx PCR system. The primer set used in this experiment is shown in Supplementary Table S3.
Co-immunoprecipitation (Co-IP) assay
Whole-cell lysates were prepared with IP lysis buffer [25 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 5% glycerol) supplemented with a protease inhibitor cocktail (Roche Applied Science), 1 mM PMSF and 1 mM Na3VO4 (Sigma-Aldrich). For IP using the anti-Myc antibody (Invitrogen), cell lysates containing 1 mg protein were incubated with the anti-Myc antibody or control mouse IgG (Sigma-Aldrich) for 4 h at 4°C, followed by further incubation with Dynabeads Protein G (Invitrogen) for 2 h at 4°C with rotation. The Dynabeads Protein G/antibody/protein complexes were washed six times with IP buffer, followed by the addition of 1× Laemmli sample buffer (Bio-Rad, Hercules, CA, USA) to the immune complexes, which were then subjected to immunoblot analysis.
RNA-IP (RIP)-qPCR
Cells were lysed with RIP buffer [150 mM KCl, 25 mM Tris (pH 7.4), 5 mM EDTA, 0.5 mM DTT, 0.5% NP40 and 100 U/ml RNase inhibitor (Enzynomics, Daejeon, Korea)] containing a protease inhibitor cocktail (Roche Applied Science). Cell lysates containing 600 μg of protein were incubated with the anti-RBFOX2 antibody (Bethyl Laboratories) or control rabbit IgG (Sigma-Aldrich) for 4 h at 4°C, and the lysate was centrifuged at 10,000 × g for 10 min. Dynabeads® Protein G (50 μl; Invitrogen, CA, USA) was washed three times with RIP buffer and incubated with the cell lysate for 2 h at 4°C, after which the beads were washed six times with RIP buffer. Total RNA was extracted using a Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA, USA) and reverse-transcribed using M-MLV reverse transcriptase (Promega) with random hexamers. The primer set for the TEAD1 and TEAD2 intronic region used in this experiment is shown in Supplementary Table S3. RIP-qPCR was performed using SYBR Green fluorescent dye (GENET BIO) and the AriaMx PCR system (Agilent Technologies). Normalization was performed using β-actin as an internal control.
Wound-healing and transwell migration assay
Cell motility was measured using both wound-healing and transwell migration assays. Cells were grown to a confluent monolayer on 6-well plates, and wounds were made on the surface of the cultured cells using a 200-μm micropipette tip. After wounding, the detached cells were removed by washing with PBS. Phase-contrast images of the wound were captured 24 h after wounding using an inverted light microscope (Nikon, Tokyo, Japan). Wound areas were measured using ImageJ software (NIH).
Transwell migration assays were performed using transwell migration chambers (8-μm pore size; BD Falcon, Franklin Lakes, NJ, USA). The transfected cells were treated with trypsin/EDTA solution and resuspended as single-cell solutions in serum-free DMEM. Cells in serum-free DMEM were seeded into the upper chamber of each insert, and DMEM containing 10% FBS was added to the lower chambers. After incubation for 24 h, cells that migrated to the bottom surface of the insert were fixed in 100% methanol for 20 min, stained with Giemsa solution, and rinsed in PBS. Cells on the top surface of the insert were removed by wiping with a cotton swab. Images were captured using an inverted light microscope (Nikon), and the number of cells in each photo field was counted using ImageJ software (NIH).
Colony formation assay
After transfection, cells were plated in 6-well plates at 300 cells/well. After 10 days, the cells were fixed with 4% paraformaldehyde and stained with a staining solution (95% MeOH, 0.5% acetic acid and 0.5% crystal violet). Images were captured under a microscope. The sizes and numbers of the colonies were counted using ImageJ software (NIH).
Cell-free protein expression
The TnT® Quick Coupled Transcription/Translation System (Promega) was used for in vitro cell-free protein expression according to the manufactures’ instructions with DNA containing the SP6 promoter. TEAD1-expressing plasmids (myc-TEAD1-pCS3 + MT) and RBFOX2-expressing plasmids (myc-RBFOX2-pCS3 + MT) were used in this experiment. The reactions were carried out in 25 μl volumes by adding 1 μg of plasmid DNA to the TnT mixture and incubated at 30°C for 90 min.
Electrophoretic mobility shift assay (EMSA) for protein–DNA complexes
Electrophoretic mobility shift assays were performed using the LightShift™ chemiluminescent EMSA kit (Thermo Fisher Scientific) according to the manufacturer's instructions. DNA binding reactions were performed in 20 μl system containing biotinylated DNA probe (1XGT: 5′-[biotin] TTCGATACACTTGTGGAATGTGTTTGATTTGTTAGCCCCG-3′, 20 fmol) and myc-TEAD1 TnT proteins. DNA probes used for EMSA contained double-stranded DNA made of two complementary strands, and sample preparation included binding buffer and 50 ng/μl poly (dI·dC). After incubation for 20 min at 25°C, the reaction mixture was separated by electrophoresis using an 8% native DNA polyacrylamide gel. Thereafter, the protein-DNA complexes were transferred onto a Zeta-Probe® GT Membrane (Bio-Rad) and detected by chemiluminescence.
Pull-down assay
The biotinylated DNA (450 pmol) or RNA (500 pmol) probes and TnT proteins used for EMSA were incubated in NP-40 buffer [50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 0.1% NP-40, and protease inhibitor cocktail] for 3 h at 4°C with gentle rotation. DNA-protein or RNA–protein complexes were further incubated with streptavidin-conjugated agarose beads (Invitrogen) for 1 h at 4°C with gentle rotation. The pulled-down complex was washed 5 times with NP-40 buffer, then separated on an SDS-polyacrylamide gel and analyzed by immunoblot analysis.
In vitro binding assay
The Flag-YAP construct (Addgene, #19045) was overexpressed in LATS1/2-DKO HEK293A cells and then lysed with M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific) containing a protease inhibitor cocktail (Roche Applied Science). The lysate was incubated with ANTI-FLAG® M2 Affinity Gel (Sigma) for 1 h 30 min at 4°C with gentle rotation. After washing 5 times with NP-40 buffer, myc-TEAD1 TnT proteins were added and incubated for 3 h at 4°C with gentle rotation. After that, it was additionally washed 5 times with NP-40 buffer, and the interaction of Flag-YAP and myc-TEAD1 was analyzed by immunoblot analysis.
TCGA data and analysis
Multiple datasets obtained from TCGA were used for cancer patient analysis (Supplementary Excel File. TCGA data set). We downloaded mRNA-expression values and overall survival data from the UCSC XENA data portal (https://xenabrowser.net/datapages/) for TCGA patients. The inclusion ratios of alternative exons were represented as percent spliced in index (PSI) values. PSI values of TEAD1 alternative exons were obtained from the SpliceSeq database (https://bioinformatics.mdanderson.org/TCGASpliceSeq/index.jsp) and used to compare CTGF and CYR61 mRNA-expression levels in individual TCGA patient samples.
Statistical analysis
All in vitro experiments were performed in triplicate. Data are expressed as mean ± SD and compared using GraphPad Prism software (v.5.0; GraphPad Software, San Diego, CA, USA). Comparisons between groups were assessed using unpaired t-tests. Statistical significance was set at P < 0.05.
RESULTS
Correlation in expression between TEAD1 isoforms and YAP-TEAD target genes
The TEAD family includes four highly homologous proteins (TEAD1–4) that share a conserved DNA-binding TEA domain located at the N-terminus of the proteins (36,44). As TEAD family genes (except for TEAD3) include cassette exons, we investigated the effect of each splicing isoform on the Hippo signaling pathway. First, we compared the expression level of each TEAD splicing isoform exhibiting cassette-exon inclusion with the expression profile of connective tissue growth factor (CTGF), a representative YAP-TEAD target gene, according to The Cancer Genome Atlas (TCGA) data. Among the cassette exons, TEAD1 exon 6 exhibited the greatest correlation with CTGF expression level, with correlation coefficients for breast invasive carcinoma (BRCA) and stomach adenocarcinoma (STAD) of 0.43 and 0.47, respectively (Figure 1A). In addition, we compared the expression profile of cysteine-rich angiogenic inducer 61 (CYR61), another YAP-TEAD target gene. As a result, as with CTGF, TEAD1 exon 6 and CYR61 expression level exhibited the greatest correlation coefficients in BRCA and STAD were 0.42 and 0.36, respectively (Figure 1B). Therefore, we focused on the role of TEAD1 exon 6 in the Hippo signaling pathway.
Figure 1.

Expression of an isoform of TEAD1ΔE6 is inversely proportional to the expression of genes related to the Hippo signaling pathway. (A, B) Heatmaps (top) of individual patient samples profiled based on events and according to changes in CTGF (A) and CYR61 (B) mRNA level and the percent spliced in index (PSI) of TEAD family alternative exons. Pearson's correlation coefficient of the PSI versus CTGF and CYR61 expression and statistical analyses are shown at the bottom of the heatmaps. (C, D) Nucleotide-sequence alignments of the regions encoding TEAD1F.L. and TEAD1ΔE6. Red nts representing the exon 6 region. The splicing patterns of TEAD1 exon 6 in human cell lines (C) and mouse tissues (D) were analyzed by RT-PCR. The upper and lower bands represent results for regions encoding TEAD1F.L. and TEAD1ΔE6, respectively. (E, F) Comparison of CTGF and CYR61 mRNA levels versus the ratio of TEAD1 mRNA level to TEAD1 exon 6 inclusion in BLCA and KIRC. (G) Heatmaps of CTGF and CYR61 mRNA levels according to the PSI value of TEAD1 exon 6 or TEAD1 mRNA level in TCGA datasets. Pearson's correlation coefficients are shown at the top of the heatmaps. (H) Kaplan–Meier survival plot showing survival rates of patients with TEAD1 exon 6 high PSI (purple) and low PSI (green) in KIRC and LUAD. BLCA, Bladder Urothelial Carcinoma; BRCA, Breast invasive carcinoma; COAD, Colon adenocarcinoma; ESCA, Esophageal carcinoma; HNSC, Head and Neck squamous cell carcinoma; KIRC, Kidney renal clear cell carcinoma; KIRP, Kidney renal papillary cell carcinoma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; STAD, Stomach adenocarcinoma; THCA, Thyroid carcinoma; UCEC, Uterine corpus endometrial carcinoma.
We examined the expression of the exon 6-inclusion isoform of TEAD1 in various human-derived cell lines using RT-PCR. We used primer pairs designed with sequences located in exons 5 (E5) and 7 (E7) to identify the inclusion or exclusion of exon 6. TEAD1F.L. and TEAD1ΔE6, which indicate the inclusion and exclusion of exon 6, respectively, were expressed in all cell lines (Figure 1C). Additionally, we observed TEAD1F.L. and TEAD1ΔE6 expression in various murine tissues, as well as various human-derived cell lines (Figure 1D). TEAD1, which exhibits DNA-binding activity in the Hippo signaling pathway, regulates the transcription of genes, such as CTGF, CYR61, and ankyrin repeat domain 1 (ANKRD1). To determine the effect of exon 6 inclusion on the transcriptional activity of TEAD1, we classified patients with different tumor types in TCGA according to the PSI value of exon 6 inclusion and analyzed CTGF or CYR61 expression. We found that exon 6 inclusion was proportional to CTGF or CYR61 expression (Figure 1E, F; upper graphs, Supplementary Figure S1A–D; left graphs). Interestingly, TEAD1 expression did not correlate with CTGF or CYR61 expression (Figure 1E, F; lower graphs, Supplementary Figure S1A–D; right graphs). Moreover, in most tumor types, the correlation coefficients between CTGF- or CYR61-expression levels and exon 6 inclusion by TEAD1 were higher than those between CTGF- or CYR61-expression levels and TEAD1 expression (Figure 1G). Moreover, the expression of TEAD1 with exon 6 inclusion did not correlate with TEAD1 levels (Supplementary Figure S1E).
As TEADs are associated with the progression of various types of human cancers, such as colon, lung, breast, and liver (45–47), we investigated the effect of exon 6 inclusion on the survival rate of patients using TCGA data. We found that the survival rate of patients with high exon 6 inclusion was lower than that of patients with low exon 6 inclusion (Figure 1H). These results suggest that the transcriptional activity of TEAD1 in the Hippo-YAP signaling pathway might be correlated with exon 6 inclusion through AS regulation rather than its expression level.
The differential transcription activities and oncogenic properties of two TEAD1 isoforms
Next, we directly confirmed the differential transcription activities of the two TEAD1 splicing isoforms (TEAD1F.L. and TEAD1ΔE6) using the 8 × GT-IIC luciferase reporter containing TEAD response elements (5′-GGAATG-3′) in the HEK293A cells. HEK293A cells are a cell line characterized by being flatter and more contact-inhibited than HEK293 cells (48). As contact inhibition activates large tumor-suppressor kinases (LATS), the HEK293A cell line is the most widely used to study the Hippo signaling pathway (48–52). Immunoblot analysis showed comparable expression between TEAD1ΔE6 and TEAD1F.L., although the increase in reporter activity by TEAD1ΔE6 was significantly lower than that by TEAD1F.L. (Figure 2A, Supplementary Figure S2A). Additionally, overexpression of YAP, a cofactor involved in TEAD1 transcription activity, resulted in markedly lower TEAD1ΔE6 reporter activity (Figure 2A). Moreover, we confirmed that the differences in reporter activity between the two isoforms were similar to observed differences in the expression of endogenous YAP-TEAD target genes (Figure 2B). LATS1/2 are known to regulate upstream signaling in the Hippo signaling pathway and induce YAP phosphorylation, resulting in YAP sequestration from the cytoplasm, and promoting YAP inactivation (53,54). Therefore, LATS1/2 double-knockout (LATS1/2-DKO) blocks phosphorylation of YAP, leading to YAP nuclear translocation and constitutive activation of YAP-TEAD target gene expression (43). Since it was confirmed that the transcriptional activity of the TEAD1 isoform was varied further due to overexpression of YAP, we next conducted an experiment with LATS 1/2-DKO cells where YAP inactivation was blocked. As expected, endogenous mRNA expression levels of CTGF, CYR61 and ANKRD1 were stronger in the LATS1/2-DKO cell line than in the wild-type cell line (Figure 2D). The TEAD1 exon 6 inclusion rate in the LATS1/2-DKO HEK293A cell line was not significantly different from that in WT (Supplementary Figure S2B). We found that the transcriptional activity of TEAD1ΔE6 was significantly lower than that of TEAD1F.L. according to reporter activity and target gene expression LATS1/2-DKO HEK293A cells (Figure 2C, D, Supplementary Figure S2C), indicating that TEAD1F.L. exhibited stronger transcription activity than TEAD1ΔE6. These results suggest that AS-regulated exon 6 inclusion of TEAD1 is a critical post-transcription mechanism regulating YAP-dependent transcription activity.
Figure 2.

The transcriptional activity of TEAD1F.L. is greater than that of TEAD1ΔE6. (A) Transient transfection of HEK293A cells with the 8 × GT-IIC luciferase and the indicated amounts of Myc-TEAD1 constructs in the absence or presence of Flag-YAP. After 48 h, Firefly luciferase activity was normalized to that of Renilla luciferase activity. (B) Transient transfection of HEK293A cells with the indicated amounts of Myc-TEAD1 constructs in the absence or presence of Flag-YAP. After 48 h, mRNA levels of the indicated genes were measured using qRT-PCR and normalized to β-actin level. (C) Transient transfection of WT and LATS1/2-DKO HEK293A cells with 8 × GT-IIC luciferase and the indicated amounts of Myc-TEAD1 constructs. After 48 h, Firefly luciferase activity was normalized to that of Renilla luciferase activity. (D) Transient transfection of WT and LATS1/2-DKO HEK293A cells with the indicated amounts of Myc-TEAD1 constructs. After 48 h, mRNA levels of the indicated genes were measured using qRT-PCR and normalized to β-actin level. All data are expressed as fold values relative to the sample transfected with the mock vector into WT HEK293A cells. (–) in the graph means mock vector transfection. Data represent the mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
TEADs influence oncogenic properties, such as cell proliferation and migration, through their transcription activity during cancer progression (55–57). Therefore, we investigated whether differences in the transcriptional activities of TEAD1ΔE6 and TEAD1F.L. would result in differential cell proliferation and migration. We used LATS1/2-DKO HEK293A cells transfected with Myc-TEAD1F.L. or Myc-TEAD1ΔE6, respectively, to evaluate cell proliferation and perform colony formation assays. The results showed greater increases in cell proliferation due to TEAD1F.L. overexpression compared to TEAD1ΔE6 overexpression (Figure 3A), and colony formation assays revealed that TEAD1F.L. had a greater effect enhanced colony size and number relative to a greater degree than TEAD1ΔE6 (Figure 3B). Moreover, wound-healing and transwell cell-migration assays on LATS1/2-DKO cells revealed significantly faster wound closure and transwell migration following TEAD1F.L. overexpression relative to that observed following TEAD1ΔE6 overexpression (Figure 3C, D). These results suggest that differences in TEAD1 transcription activity mediated by exon 6 inclusion influence cell growth and motility.
Figure 3.

The oncogenic properties of TEAD1F.L. are greater than those of TEAD1ΔE6. (A) Transient transfection of LATS1/2-DKO HEK293A cells with the indicated amounts of Myc-TEAD1 constructs. Cell counts were obtained for each indicated day using a hemocytometer. Data represent the mean ± SD (n = 9). (B) Transient transfection of LATS1/2-DKO HEK293A cells with the indicated amounts of Myc-TEAD1 constructs. Colony-forming efficiency (colony size and number) at day 7 was quantified. Data represent the mean ± SD (n = 4). Data are expressed as fold values relative to the sample transfected with the mock vector into LATS1/2-DKO HEK293A cells. (C) Quantification of cellular migration via measurement of wound closure rate in LATS1/2-DKO HEK293A cells expressing Myc-TEAD1 constructs. Images obtained at 24 h are representative of three independent experiments. Data represent the mean ± SD of nine fields of images from cells expressing Myc-TEAD1 constructs. (D) Quantification of transwell migration of LATS1/2-DKO HEK293A cells expressing Myc-TEAD1 constructs. Images obtained at 24 h are representative of four independent experiments. Data represent the mean ± SD (n = 3). Scale bar, 100 μm. (–) in the data means mock vector transfection. *P < 0.05, **P < 0.01, ***P < 0.001.
The differential YAP interaction of two TEAD1 isoforms
Exon 6 of TEAD1 encodes a four-amino-acid peptide, VTSM, located in the DNA-binding domain (Figure 4A). Therefore, we performed a ChIP assay to investigate binding activity at the promoter region of YAP-TEAD target genes to identify differences in transcriptional activity according to exon 6 inclusion in TEAD1. In LATS1/2-DKO cells transfected with Myc-TEAD1F.L. or -TEAD1ΔE6 constructs, measurement of TEAD1 occupancy by immunoprecipitation using the anti-Myc antibody and qPCR of promoter sites of YAP-TEAD target genes revealed similar Myc-TEAD1F.L. and Myc-TEAD1ΔE6 expression and IP efficiency, and that TEAD1F.L. binding activity was greater than that of TEAD1ΔE6 to the promoter regions of YAP-TEAD target genes (Figure 4B). To confirm whether this result is due to the difference in DNA binding affinity of TEAD1, DNA pull-down assay and EMSA were performed with TEAD1 protein obtained using in vitro transcription and translation in cell-free conditions (TnT). Contrary to our expectation that the DNA binding affinities according to the TEAD1 isoforms would differ, the DNA binding affinities of the two isoforms were similar (Figure 4C, D).
Figure 4.

TEAD1F.L. exhibits greater YAP interaction relative to TEAD1ΔE6. (A) Diagram showing the position of the peptide encoded by TEAD1 exon 6. (B) ChIP assay using LATS1/2-DKO HEK293A cells transiently transfected with Myc-TEAD1 constructs. The anti-Myc antibody was used to precipitate chromatin, and immunoblot analysis of precipitates was performed to determine IP efficiency. Control IgG was used as a negative control. Precipitated DNA fragments were quantified by qPCR using primers targeting the promoter regions of the indicated genes. Data represent the mean ± SD (n = 3). *P < 0.05. (C) Immunoblots show TEAD1 isoform protein binding to biotinylated 1XGT DNA probe (upper blot). Myc-TEAD1 proteins were obtained through in vitro transcription and translation in cell-free conditions (TnT). After precipitation of the biotinylated 1XGT DNA probe with streptavidin, buffers were collected for each washing step and immunoblotting was performed (lower blot). (D) EMSAs performed with the biotinylated 1XGT DNA probe and myc-TEAD1 TnT proteins. For the (-) line, mock vector TnT protein was used instead of myc-TEAD1. The TEAD1-1XGT DNA probe complex (Complex) and the 1XGT DNA probe not bound to TEAD1 (Free probe) are indicated by arrowheads, respectively. Triangles indicate increasing amounts of myc-TEAD1 TnT proteins. (E) The two TEAD1 isoforms show different YAP interaction activities. IP of lysates of LATS1/2-DKO HEK293A cells transiently transfected with Myc-TEAD1 constructs using the anti-Myc antibody and IgG as a control. Immunoprecipitates were subjected to immunoblot analysis using the indicated antibodies. Input comprised 10% of the lysates used for IP analysis. S.E., short exposure; L.E., long exposure. (F) After overexpressing Flag-YAP in LATS1/2-DKO HEK293A, cells were lysed and immunoprecipitated with Flag resin. After that, myc-TEAD1 TnT protein was added and immunoblot analysis was performed using the indicated antibody to confirm the interaction of TEAD1-YAP.
Next, to determine whether the observed TEAD1F.L. binding activity was a consequence of increased nuclear localization relative to that of TEAD1ΔE6, we performed immunofluorescence microscopy analysis of cells overexpressing Myc-TEAD1F.L. or -TEAD1ΔE6, which showed that both TEAD isoforms demonstrated similar nuclear localization (Supplementary Figure S3A). In addition, we overexpressed each of the TEAD1 isoforms in 293A cells, and then fractionated the nucleus and cytoplasm (Supplementary Figure S3C). It was confirmed that TEAD1 isoforms were present in similar amounts in the nuclear and cytoplasmic fractions, and it was verified that there is no difference in intracellular location between TEAD1 isoforms. However, the intracellular location of YAP varied significantly with the TEAD1 isoforms. When TEAD1F.L. was overexpressed, quantification of the immunofluorescence intensity and immunoblot band intensity of protein in the nuclear fraction confirmed that there was more YAP in the nucleus than TEAD1ΔE6 (Supplementary Figure S3A–D). We thought that the retention of YAP in the nucleus could be indirect evidence indicating that the interaction between the TEAD1F.L. and YAP is stronger than that of the TEAD1ΔE6. Therefore, we next investigated the interaction of TEAD1 isoforms with YAP. To confirm that TEAD1 isoforms have differential interacting activity with YAP, myc-TEAD1 was overexpressed in LATS1/2-DKO HEK293A, and then immunoprecipitation was performed using anti-myc antibody. We found that TEAD1F.L. exhibits higher affinity for YAP relative to TEAD1ΔE6 (Figure 4E). The interaction between TEAD1 and YAP was reconfirmed using the TEAD1 TnT protein. After overexpressing Flag-tagged YAP in cells, YAP was purified using Flag resin. After that, the myc-TEAD1 proteins obtained by TnT were bound to YAP. As a result, as seen in Figure 4E, it was confirmed that the interaction between TEAD1F.L. and YAP was stronger than that between TEAD1ΔE6 and YAP (Figure 4F). Although exon 6 encodes part of the N-terminal TEAD1 DNA-binding domain, whether exon 6 exclusion results in conformational change of the YAP/TAZ-binding domain in the C-terminus remains to be verified. We compared the protein structure of the TEAD1 isoforms using the AlphaFold program. As a result, the helix part of the TEAD1ΔE6 was long, but it became flexible due to exon 6 inclusion in the TEAD1F.L. (Supplementary Figure S3E). This conformational change due to exon 6 is expected to facilitate interaction with YAP. These results suggest that exon 6 inclusion affected the interaction with YAP.
RBFOX2 regulates TEAD1 AS
To investigate regulation of TEAD1 cis-regulatory elements by RBPs, we first analyzed the correlation between the PSI value of TEAD1 exon 6 and the expression level of 151 splicing factors in 10 cancer types in the TCGA data set. The top 5 genes with high mean correlation values among 10 cancer types were RBFOX2, KH Domain Containing RNA Binding (QKI), Muscleblind Like Splicing Regulator 1 (MBNL1), NOVA Alternative Splicing Regulator 2 (NOVA2) and AHNAK Nucleoprotein 2 (AHNAK2) (Figure 5A). Next, we investigated the degree of sequence conservation of upstream and downstream intronic regions, including exon 6, according to the UCSC PhastCons conservation score. The results identified two highly conserved elements in the downstream intronic region of exon 6, which contained the GCAUG sequence (Figure 5B). Further analyses identified a GCAUG-binding element as the binding consensus sequence of RBFOX2, which reportedly regulates the AS of many genes via the GCAUG element. Therefore, we focused on RBFOX2 and analyzed its correlation with the YAP-TEAD target gene. In TCGA data pertaining to various cancer types, the expression level of RBFOX2 was not only correlated with the TEAD1 exon 6 PSI value, but also exhibited a high correlation with the expression levels of the YAP-TEAD target genes CTGF and CYR61 (Supplementary Figure S4A). Next, we verified whether this RBFOX2-induced AS regulation was due to binding to the GCAUG element present in the intron region downstream of TEAD1 exon 6. To investigate the interaction between RBFOX2 and TEAD1 pre-mRNA, we performed RIP assays using HEK293A cells and the anti-RBFOX2 antibody. RT-PCR of RNA isolated from RNA–RBFOX2 complexes using primers capable of detecting the flanking regions of the GCAUG elements yielded the predicted products, confirming that RBFOX2 interaction with TEAD1 pre-mRNA (Figure 5C). However, RBFOX2 did not interact with the negative control TEAD2 pre-mRNA. To determine whether the binding of RBFOX2 to the downstream intronic region of TEAD1 exon 6 directly affects exon 6 inclusion, we evaluated the effects of RBFOX2 overexpression and siRNA knockdown of RBFOX2. To maximize the effect of RBFOX2 on TEAD1 exon 6 splicing, the U-2 OS cell line, exhibiting a high TEAD1 exon 6 inclusion ratio, was used for RBFOX2 knockdown evaluation. We found that RBFOX2 overexpression dose-dependently promoted exon 6 inclusion, whereas RBFOX2 depletion decreased exon 6 inclusion. (Figure 5D, E). Then, we used biotinylated RBFOX2 TnT protein and TEAD1 intron 6 RNA in which GCAUG, the RBFOX2 binding site, was mutated, and confirmed that RBFOX2 directly binds to the GCAUG site in the TEAD1 intron 6 sequence through RNA pull-down assay and RNA EMSA (Figure 5F, Supplementary Figure S4B). Next, we investigated which of the two GCAUG elements plays a functional role in exon 6 inclusion using a minigene construct containing exon 6 and flanking exons 5 and 7, as well as TEAD1 introns (Figure 5G). The minigene system is a useful tool for studying AS mechanisms to identify specific cis-regulatory elements and trans-acting factors (RBPs) that bind to these elements. We co-transfected HEK293A cells with the TEAD1 minigene and RBFOX2, followed by RT-PCR using primer pairs targeting the EGFP sequence and exon 7 within the minigene construct. We found that exon 6 inclusion within the minigene construct was increased by RBFOX2 overexpression (Figure 5H). To determine the GCAUG elements targeted by RBFOX2, we mutated the sequences of the RBFOX2-binding motifs using transversion mutations (Figure 5G) and verified changes in exon 6 inclusion by RBFOX2 overexpression. For the Mut2 constructs (mutation of the second GCAUG element to CCTAC), we observed similar exon 6 inclusion between RBFOX2-overexpressing cells and wild-type (WT) cells. In contrast, the Mut1 constructs (mutation of the first GCAUG element to CCTAC) exhibited significantly reduced exon 6 inclusion relative to WT (Figure 5I). For the Mut1/2 constructs (mutation of both GCAUG elements to CCTAC), the increase in exon 6 inclusion by RBFOX2 overexpression was similar to that observed in the Mut1 constructs, showing that the GCAUG element located in close proximity to exon 6 is critical for RBFOX-mediated exon 6 inclusion.
Figure 5.

RBFOX2 enhances TEAD1F.L. expression via binding to the GCAUG element in pre-mRNA. (A) Heatmap of Pearson's correlation coefficients between splicing factors mRNA level and PSI value of TEAD1 exon 6 in TCGA datasets. Splicing factor gene list was obtained from gene ontology (GO:0043484 regulation of RNA splicing). The genes with the top 5 mean correlation value in 10 cancer types are labeled in the heatmap. (B) The UCSC Genome Browser human hg38 assembly showing highly conserved regions across the 100 vertebrate genomes at the downstream intron of TEAD1 exon 6. The red line indicates the amplification region for RIP-PCR. (C) RIP was performed using the anti-RBFOX2 antibody, followed by RT-qPCR. PCR using non-reverse-transcribed RNA confirmed that the extracted RNA was not contaminated with genomic DNA (right). Immunoprecipitated proteins were verified by immunoblot analysis using the anti-RBFOX2 antibody (left). (D) RT-PCR analysis of endogenous TEAD1 exon 6 AS following transfection of the increasing amount of the RBFOX2-Flag construct into HEK293A cells. (E) Transfection of U-2 OS cells with non-targeting control siRNA (–) or two individual siRNAs targeting RBFOX2. At 48 h post-transfection, endogenous TEAD1 exon 6 AS was analyzed by RT-PCR. (F) The biotinylated TEAD1 intron 6 RNA probe (WT) sequence used for RNA pull-down and the RBFOX2 binding site mutated RNA probe (Mut) sequence are shown (upper image). Immunoblots show RBFOX2 protein binding to biotinylated TEAD1 intron 6 RNA probe (lower blot). (G) Diagram showing the TEAD1 minigene constructs, which include the WT or mutant GCAUG elements. (H) RT-PCR analysis of exogenous TEAD1 exon 6 AS following co-transfection of the increasing amount of the RBFOX2-Flag construct with the WT TEAD1 minigene into HEK293A cells. (I) The TEAD1 minigenes, including the mutations described in (G), were co-transfected into HEK293A cells along with RBFOX2-Flag. AS of the TEAD1 minigene was analyzed by RT-PCR. RBFOX2 overexpression or knockdown (D, E, H, I) was confirmed by immunoblot analysis using anti-Flag and anti-RBFOX2 antibodies. (–) in the data (D, H, I) means mock vector transfection. GAPDH served as a loading control. Data represent the mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
RBFOX2 regulates the expression of YAP-TEAD target genes
We then assessed the direct effect of RBFOX2 on the expression of YAP-TEAD target genes in the Hippo signaling pathway. RBFOX2 overexpression in the presence of YAP overexpression caused a dose-dependent increase in TEAD1-mediated transcription activity, as shown in a luciferase reporter assay containing the TEAD consensus binding sequences (Figure 6A). Additionally, we confirmed elevated transcription activities of endogenous YAP-TEAD target genes following RBFOX2 overexpression (Figure 6B, Supplementary Figure S5A). We then determined reporter and transcriptional activities following RBFOX2 knockdown with siRBFOX2 and in the presence of YAP overexpression, revealing decreases in both activities (Figure 6C, D, Supplementary Figure S5B). Furthermore, we confirmed dose-dependent increases in RBFOX2-mediated YAP-TEAD target gene expression in LATS1/2-DKO cells under YAP overexpression conditions. (Figure 6E, F, Supplementary Figure S5C), as well as commensurate decreases in these activities following RBFOX2 silencing (Figure 6G, H, Supplementary Figure S5D). These results suggest that RBFOX2-regulated exon 6 inclusion regulates the expression of YAP-TEAD target genes in the Hippo signaling pathway.
Figure 6.

RBFOX2 regulates the expression of target genes related to the Hippo signaling pathway. (A) Transient transfection of HEK293A cells with the 8 × GT-IIC luciferase construct and the increasing amounts of RBFOX2-Flag in the absence or presence of Flag-YAP. After 48 h, Firefly luciferase activity was normalized to that of Renilla luciferase. (B) Transient transfection of HEK293A cells with the increasing amounts of RBFOX2-Flag in the absence or presence of Flag-YAP. After 48 h, mRNA levels of the indicated genes were measured using qRT-PCR and normalized to β-actin level. (C) Transient transfection of HEK293A cells with 8 × GT-IIC luciferase along with non-targeting control siRNA or two individual siRNAs targeting RBFOX2 in the absence or presence of Flag-YAP. (D) Transient transfection of HEK293A cells with non-targeting control siRNA or two individual siRNAs targeting RBFOX2 in the absence or presence of Flag-YAP. (E) Transient transfection of WT and LATS1/2-DKO HEK293A cells with 8xGT-IIC luciferase with the increasing amount of RBFOX2-Flag. (F) Transient transfection of WT and LATS1/2-DKO HEK293A cells with the increasing amount of RBFOX2-Flag. (G) Transient transfection of WT and LATS1/2-DKO HEK293A cells with 8 × GT-IIC luciferase with non-targeting control siRNA or two individual siRNAs targeting RBFOX2. (H) Transient transfection of WT and LATS1/2-DKO HEK293A cells with non-targeting control siRNA or two individual siRNAs targeting RBFOX2. (–) in the graph means mock vector (A, B, E, F) or non-targeting control siRNA (C, D, G, H) transfection. All data are expressed as fold values relative to the sample transfected with the mock vector into HEK293A WT cells. Data represent the mean ± SD (n = 3); *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
In this study, we identified RBFOX2 as a splicing regulator and demonstrated the mechanism underlying AS regulation in the Hippo signaling pathway. RBFOX2 is an RBP that plays an important role in AS regulation, mRNA stability, and translation (58,59). RBFOX2 exhibits differential AS regulation activity depending on the binding site where the GCAUG element is located. In regulating exon 6 inclusion of TEAD1, RBFOX2 bound to one of two GCAUG elements in the downstream intronic region of exon 6; however, mutation of the functional GCAUG element only partially attenuated RBFOX2-induced exon 6 inclusion. Recent studies report that RNA-binding by RBFOX2 is not limited to the GCAUG sequence (60–62), suggesting that RBFOX2 likely exerts its regulatory activity through other putative binding elements associated with exon 6 inclusion of TEAD1. In addition to RBFOX2, QKI, MBNL1, NOVA2, and ANHAK2 also exhibited a high correlation with TEAD1 exon 6 splicing in TCGA data (Figure 5A). QKI, MBNL1 and NOVA2 are known to compete with or cooperate with the RBFOX family to synergistically regulate alternative splicing (63–69). Since the mechanism controlling splicing is very complex, it is difficult to completely abolish splicing by one splicing factor or one binding site. Therefore, there is also the possibility that splicing of TEAD1 exon 6 might be regulated by other splicing factors.
We used a TEAD1 minigene construct containing a genomic segment of alternatively-spliced exon 6 and its flanking exons and introns to identify an RBFOX2 response element involved in regulating TEAD1 AS. We found that exon 6 inclusion in the TEAD1 minigene following RBFOX2 overexpression was stronger than that observed with endogenous TEAD1. AS is affected by the speed of RNA polymerase II elongation, promoter architecture, the secondary structure of pre-mRNA, and cis-regulatory elements on pre-mRNA and trans-acting splicing factors (70–72). Thus, AS patterns in the endogenous TEAD1 gene mediated by RBFOX2 might differ from those of a minigene comprising a promoter region and genomic segments of specific genes. Although the minigene reporter system does not allow full reproducibility of endogenously-regulated AS for all genes, it remains a useful tool for verifying the cis-regulatory elements and their splicing factors. Therefore, these findings elucidated the role for RBFOX2 in regulating TEAD1 AS through the GCAUG element downstream of exon 6.
TEAD1 exon 6 consists of 12 nts and encodes only four amino acids. These small exons in the range of 3–30 nts are called microexons, and can change protein properties by affecting interactions with partner proteins, enzymatic activity, and protein localization (73). In particular, they tend to be neuron-specific, and have been proposed to play a key regulatory role during brain development. Therefore, research on microexons has primarily focused on the brain (74–77). In this study, we reported for the first time that the interaction with YAP is changed by the inclusion of exon 6, a microexon of TEAD1. The peptide encoded by exon 6 is located in the DNA-binding domain of the TEAD1 N-terminus. In an in vitro DNA binding assay using TEAD1 TnT protein, there was no significant difference in DNA binding activity according to the different TEAD1 isoforms (Figure 4C, D). However, in a ChIP assay using whole cell lysate, more TEAD1F.L. was bound to the promoter regions of the target genes than TEAD1ΔE6 (Figure 4B). Therefore, we cannot exclude the possibility that there may be other external factors that are differentially changed depending on the isoform of TEAD1 bound to DNA. Phosphorylation of TEAD by PKA and PKC is known to inhibit TEAD1 binding to DNA (78,79). There is a possibility that the degree of phosphorylation differs depending on the TEAD1 isoforms, thereby affecting DNA binding. In addition, the binding of partner proteins, including YAP, which is dependent on the TEAD1 isoform, may have influenced the binding of TEAD1 to DNA. The mechanism by which TEAD1 recognizes and binds to DNA is still unclear and thus requires further studies. Contrary to our expectations, exon 6 of TEAD1, which is located in the DNA-binding domain, also affected the interaction with YAP. TEAD1 is known to interact with YAP through the YAP binding domain located in C-terminus. The fact that the interaction with YAP is changed by only four amino acids located in the DNA binding domain is a very interesting result. Based on the present results, we speculate that exon 6 exclusion in TEAD1 also affects YAP interaction via conformational changes in the YAP/TAZ-binding domain. We found that the conformational change of TEAD1 by exon 6 may affect the interaction with YAP using AlphaFold protein structure prediction, but the precise conformational change and detailed mechanism must be elucidated through additional experiments.
Since human multi-exon genes undergo alternative splicing to encode proteins with various functions, most key components in the Hippo signaling pathway also undergo AS regulation (4). The human YAP1 gene consists of nine exons and generates at least 8 splicing variants (80,81). The full-length YAP isoform contains two WW domains, and one of the WW domains is encoded by exon 4. The isoform in which exon 4 of YAP1 is skipped has only one WW domain, and its transcription activity is reduced compared to the isoform containing exon 4. SRSF1 is a splicing factor that regulates alternative splicing of TEAD1 exon 5 (82). Although TEAD1 exon 5 is also an alternative exon, exclusion of exon 5 was not confirmed in the TCGA data or at least in the cell line used in this experiment, and the splicing pattern of exon 5 did not change depending on the expression level of RBFOX2 (unpublished data). TEAD4, a TEAD family member, possesses the exon 3 skipping isoform and generates a truncated protein with an alternative translation start site from exon 6 (83). The exon 3 skipping isoform of TEAD4 lacks an N-terminal DNA-binding domain and exerts a dominant-negative function on YAP activity. Although the transcriptional activity of the Hippo signaling pathway reportedly drives cell proliferation, only a few reports have shown the difference in transcriptional activity of several gene isoforms in the Hippo signaling pathway. To the best of our knowledge, our study is the first to report a correlation between the expression levels of YAP-TEAD target genes and isoform-specific expressions of Hippo pathway components in a human tissue database. Therefore, this study proposes a new paradigm in which AS isoforms rather than total expression levels enable fine-tuning of complex transcription programs.
The Hippo signaling pathway plays an important role in various biological processes, including cancer development (84,85). We found that the expression of Hippo-YAP target genes did not correlate with total TEAD1-expression levels. Transcription factors generally dose-dependently regulate the expression of target genes; however, the expression of TEAD1 isoforms is regulated by AS, subsequently regulating target gene expression and enabling sophisticated regulation of cellular homeostasis via Hippo signaling. These findings indicate the role of AS regulation in modulating the status of diseases related to Hippo signaling. Of note, this approach can potentially be extended to promote a deeper understanding of biological processing by other signaling pathways.
In summary, we determined the functional importance of RBFOX2 in AS regulation of TEAD1 in the Hippo signaling pathway. We demonstrated differences between TEAD1F.L. and TEAD1ΔE6 in promoting target gene transcription, cell growth, and migration. Altogether, these results offer critical insight into RBFOX2-mediated regulation of TEAD1 AS and a broader understanding of the role of Hippo signaling in cancer progression.
DATA AVAILABILITY
For TCGA cohorts, the genomic and clinical data can be retrieved from NCI Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov).
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: H.S.L., E.M.K., W.K. and K.K.K. designed the study. H.S.L., S.C., N.C., I.K., E.M.K., W.K. and K.K.K. performed experiments and analyzed the data. H.S.L., S.C., N.C., E.M.K., W.K. and K.K.K. wrote the manuscript. K.K.K. supervised study.
Contributor Information
Sunkyung Choi, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
Hyo Seong Lee, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
Namjoon Cho, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
Inyoung Kim, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
Seongmin Cheon, School of Biological Sciences and Technology, Chonnam National University, Gwangju 61186, Republic of Korea; Proteomics Core Facility, Biomedical Research Institute, Seoul National University Hospital, Seoul 03080, Republic of Korea.
Chungoo Park, School of Biological Sciences and Technology, Chonnam National University, Gwangju 61186, Republic of Korea.
Eun-Mi Kim, Department of Predictive Toxicology, Korea Institute of Toxicology, Daejeon, 34114, Republic of Korea.
Wantae Kim, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
Kee K Kim, Department of Biochemistry, College of Natural Sciences, Chungnam National University, Daejeon 34134, Republic of Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea [NRF-2020R1A2C4001652 and NRF-2022R1A2C1003870 to K.K.K., and NRF-2021R1A2C4001704 and NRF-2022R1A4A3024551 to W.K.]; Korea Environment Industry & Technology Institute (KEITI) [2022002960005] through the Environment Health Action Program, funded by the Korea Ministry of Environment (MOE to E.M.K.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Black D.L. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000; 103:367–370. [DOI] [PubMed] [Google Scholar]
- 2. Lareau L.F., Green R.E., Bhatnagar R.S., Brenner S.E.. The evolving roles of alternative splicing. Curr Opin. Struct. Biol. 2004; 14:273–282. [DOI] [PubMed] [Google Scholar]
- 3. Nilsen T.W., Graveley B.R.. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010; 463:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J.. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008; 40:1413–1415. [DOI] [PubMed] [Google Scholar]
- 5. Wahl M.C., Will C.L., Luhrmann R.. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009; 136:701–718. [DOI] [PubMed] [Google Scholar]
- 6. Marquez Y., Brown J.W., Simpson C., Barta A., Kalyna M.. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012; 22:1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cui Y., Cai M., Stanley H.E.. Comparative analysis and classification of cassette exons and constitutive exons. Biomed. Res. Int. 2017; 2017:7323508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fu X.D., Ares M. Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014; 15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pan X., Rijnbeek P., Yan J., Shen H.B.. Prediction of RNA–protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics. 2018; 19:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Nostrand E.L., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.Y., Cody N.A.L., Dominguez D.et al.. A large-scale binding and functional map of human RNA-binding proteins. Nature. 2020; 583:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Minovitsky S., Gee S.L., Schokrpur S., Dubchak I., Conboy J.G.. The splicing regulatory element, UGCAUG, is phylogenetically and spatially conserved in introns that flank tissue-specific alternative exons. Nucleic Acids Res. 2005; 33:714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Auweter S.D., Fasan R., Reymond L., Underwood J.G., Black D.L., Pitsch S., Allain F.H.. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 2006; 25:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin Y., Suzuki H., Maegawa S., Endo H., Sugano S., Hashimoto K., Yasuda K., Inoue K.. A vertebrate RNA-binding protein Fox-1 regulates tissue-specific splicing via the pentanucleotide GCAUG. EMBO J. 2003; 22:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jangi M., Boutz P.L., Paul P., Sharp P.A.. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014; 28:637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yeo G.W., Coufal N.G., Liang T.Y., Peng G.E., Fu X.D., Gage F.H.. An RNA code for the FOX2 splicing regulator revealed by mapping RNA–protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009; 16:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim K.K., Nam J., Mukouyama Y.S., Kawamoto S.. Rbfox3-regulated alternative splicing of Numb promotes neuronal differentiation during development. J. Cell. Biol. 2013; 200:443–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang C., Zhang Z., Castle J., Sun S., Johnson J., Krainer A.R., Zhang M.Q.. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008; 22:2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang Z.Z., Zheng S., Nikolic J., Black D.L.. Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol. Cell. Biol. 2009; 29:4757–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Underwood J.G., Boutz P.L., Dougherty J.D., Stoilov P., Black D.L.. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 2005; 25:10005–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim K.K., Kim Y.C., Adelstein R.S., Kawamoto S.. Fox-3 and PSF interact to activate neural cell-specific alternative splicing. Nucleic Acids Res. 2011; 39:3064–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu G., Razanau A., Hai Y., Yu J., Sohail M., Lobo V.G., Chu J., Kung S.K., Xie J.. A conserved serine of heterogeneous nuclear ribonucleoprotein L (hnRNP L) mediates depolarization-regulated alternative splicing of potassium channels. J. Biol. Chem. 2012; 287:22709–22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thorsen K., Mansilla F., Schepeler T., Oster B., Rasmussen M.H., Dyrskjot L., Karni R., Akerman M., Krainer A.R., Laurberg S.et al.. Alternative splicing of SLC39A14 in colorectal cancer is regulated by the Wnt pathway. Mol. Cell. Proteomics. 2011; 10:M110 002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Z., Qiu J., Liu W., Zhou Y., Plocinik R.M., Li H., Hu Q., Ghosh G., Adams J.A., Rosenfeld M.G.et al.. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell. 2018; 71:872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shilo A., Ben Hur V., Denichenko P., Stein I., Pikarsky E., Rauch J., Kolch W., Zender L., Karni R.. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA. 2014; 20:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hollander D., Donyo M., Atias N., Mekahel K., Melamed Z., Yannai S., Lev-Maor G., Shilo A., Schwartz S., Barshack I.et al.. A network-based analysis of colon cancer splicing changes reveals a tumorigenesis-favoring regulatory pathway emanating from ELK1. Genome Res. 2016; 26:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perez-Ortin J.E., Tordera V., Chavez S.. Homeostasis in the central dogma of molecular biology: the importance of mRNA instability. RNA Biol. 2019; 16:1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vargas-Garcia C.A., Ghusinga K.R., Singh A.. Cell size control and gene expression homeostasis in single-cells. Curr. Opin. Syst. Biol. 2018; 8:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yotova I.Y., Quan P., Leditznig N., Beer U., Wenzl R., Tschugguel W.. Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum. Reprod. 2011; 26:885–897. [DOI] [PubMed] [Google Scholar]
- 30. Guo Y.H., Zhang C., Shi J., Xu M.H., Liu F., Yuan H.H., Wang J.Y., Jiang B., Gao F.H.. Abnormal activation of the EGFR signaling pathway mediates the downregulation of miR145 through the ERK1/2 in non-small cell lung cancer. Oncol. Rep. 2014; 31:1940–1946. [DOI] [PubMed] [Google Scholar]
- 31. Chen H., Huang Q., Dong J., Lan Q.. [Cancer initiating cell theory: popularity and controversies]. Ai Zheng. 2006; 25:779–784. [PubMed] [Google Scholar]
- 32. Ma S., Meng Z., Chen R., Guan K.L.. The Hippo pathway: biology and pathophysiology. Annu. Rev. Biochem. 2019; 88:577–604. [DOI] [PubMed] [Google Scholar]
- 33. Zheng Y., Pan D. The Hippo signaling pathway in development and disease. Dev. Cell. 2019; 50:264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim W., Jho E.H.. The history and regulatory mechanism of the Hippo pathway. BMB Rep. 2018; 51:106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu V., Plouffe S.W., Guan K.L.. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell. Biol. 2017; 49:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin K.C., Park H.W., Guan K.L.. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem. Sci. 2017; 42:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ota M., Sasaki H.. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008; 135:4059–4069. [DOI] [PubMed] [Google Scholar]
- 38. Wu S., Liu Y., Zheng Y., Dong J., Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell. 2008; 14:388–398. [DOI] [PubMed] [Google Scholar]
- 39. Zhang L., Ren F., Zhang Q., Chen Y., Wang B., Jiang J.. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell. 2008; 14:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J.D., Wang C.Y., Chinnaiyan A.M.et al.. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008; 22:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi Z., He F., Chen M., Hua L., Wang W., Jiao S., Zhou Z.. DNA-binding mechanism of the Hippo pathway transcription factor TEAD4. Oncogene. 2017; 36:4362–4369. [DOI] [PubMed] [Google Scholar]
- 42. Anbanandam A., Albarado D.C., Nguyen C.T., Halder G., Gao X., Veeraraghavan S.. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:17225–17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plouffe S.W., Meng Z., Lin K.C., Lin B., Hong A.W., Chun J.V., Guan K.L.. Characterization of Hippo pathway components by gene inactivation. Mol. Cell. 2016; 64:993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huh H.D., Kim D.H., Jeong H.S., Park H.W.. Regulation of TEAD transcription factors in cancer biology. Cells. 2019; 8:600–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang C., Nie Z., Zhou Z., Zhang H., Liu R., Wu J., Qin J., Ma Y., Chen L., Li S.et al.. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget. 2015; 6:17685–17697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang W., Gao Y., Li P., Shi Z., Guo T., Li F., Han X., Feng Y., Zheng C., Wang Z.et al.. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014; 24:331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu Y., Wang G., Yang Y., Mei Z., Liang Z., Cui A., Wu T., Liu C.Y., Cui L.. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial-mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. 2016; 35:2789–2800. [DOI] [PubMed] [Google Scholar]
- 48. Zemke N.R., Gou D., Berk A.J.. Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes Dev. 2019; 33:828–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim N.G., Gumbiner B.M.. Cell contact and Nf2/Merlin-dependent regulation of TEAD palmitoylation and activity. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:9877–9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dutta S., Mana-Capelli S., Paramasivam M., Dasgupta I., Cirka H., Billiar K., McCollum D. TRIP6 inhibits Hippo signaling in response to tension at adherens junctions. EMBO Rep. 2018; 19:337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bae S.J., Ni L., Luo X.. STK25 suppresses Hippo signaling by regulating SAV1-STRIPAK antagonism. Elife. 2020; 9:e54863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hansen C.G., Ng Y.L., Lam W.L., Plouffe S.W., Guan K.L.. The Hippo pathway effectors YAP and TAZ promote cell growth by modulating amino acid signaling to mTORC1. Cell Res. 2015; 25:1299–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao B., Wei X., Li W., Udan R.S., Yang Q., Kim J., Xie J., Ikenoue T., Yu J., Li L.et al.. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007; 21:2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao B., Li L., Tumaneng K., Wang C.Y., Guan K.L.. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010; 24:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dey A., Varelas X., Guan K.L.. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020; 19:480–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zanconato F., Cordenonsi M., Piccolo S.. YAP/TAZ at the roots of cancer. Cancer Cell. 2016; 29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Harvey K.F., Zhang X., Thomas D.M.. The Hippo pathway and human cancer. Nat. Rev. Cancer. 2013; 13:246–257. [DOI] [PubMed] [Google Scholar]
- 58. Mitchell S.F., Parker R.. Principles and properties of eukaryotic mRNPs. Mol. Cell. 2014; 54:547–558. [DOI] [PubMed] [Google Scholar]
- 59. Choi S., Sa M., Cho N., Kim K.K., Park S.H.. Rbfox2 dissociation from stress granules suppresses cancer progression. Exp. Mol. Med. 2019; 51:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Damianov A., Ying Y., Lin C.H., Lee J.A., Tran D., Vashisht A.A., Bahrami-Samani E., Xing Y., Martin K.C., Wohlschlegel J.A.et al.. Rbfox proteins regulate splicing as part of a large multiprotein complex LASR. Cell. 2016; 165:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choi S., Park C., Kim K.E., Kim K.K.. An in vitro technique to identify the RNA binding-site sequences for RNA-binding proteins. Biotechniques. 2017; 63:28–33. [DOI] [PubMed] [Google Scholar]
- 62. Begg B.E., Jens M., Wang P.Y., Minor C.M., Burge C.B.. Concentration-dependent splicing is enabled by Rbfox motifs of intermediate affinity. Nat. Struct. Mol. Biol. 2020; 27:901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jacko M., Weyn-Vanhentenryck S.M., Smerdon J.W., Yan R., Feng H., Williams D.J., Pai J., Xu K., Wichterle H., Zhang C.. Rbfox splicing factors promote neuronal maturation and axon initial segment assembly. Neuron. 2018; 97:853–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Conboy J.G. Developmental regulation of RNA processing by Rbfox proteins. Wiley Interdiscip. Rev. RNA. 2017; 8: 10.1002/wrna.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Juan-Mateu J., Rech T.H., Villate O., Lizarraga-Mollinedo E., Wendt A., Turatsinze J.V., Brondani L.A., Nardelli T.R., Nogueira T.C., Esguerra J.L.S.et al.. Neuron-enriched RNA-binding proteins regulate pancreatic beta cell function and survival. J. Biol. Chem. 2017; 292:3466–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li J., Choi P.S., Chaffer C.L., Labella K., Hwang J.H., Giacomelli A.O., Kim J.W., Ilic N., Doench J.G., Ly S.H.et al.. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife. 2018; 7:e37184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Klinck R., Fourrier A., Thibault P., Toutant J., Durand M., Lapointe E., Caillet-Boudin M.L., Sergeant N., Gourdon G., Meola G.et al.. RBFOX1 cooperates with MBNL1 to control splicing in muscle, including events altered in myotonic dystrophy type 1. PLoS One. 2014; 9:e107324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sellier C., Cerro-Herreros E., Blatter M., Freyermuth F., Gaucherot A., Ruffenach F., Sarkar P., Puymirat J., Udd B., Day J.W.et al.. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018; 9:2009–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Venables J.P., Lapasset L., Gadea G., Fort P., Klinck R., Irimia M., Vignal E., Thibault P., Prinos P., Chabot B.et al.. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun. 2013; 4:2480–2489. [DOI] [PubMed] [Google Scholar]
- 70. Cramer P., Pesce C.G., Baralle F.E., Kornblihtt A.R.. Functional association between promoter structure and transcript alternative splicing. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11456–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buratti E., Baralle F.E.. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol. Cell. Biol. 2004; 24:10505–10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Veloso A., Kirkconnell K.S., Magnuson B., Biewen B., Paulsen M.T., Wilson T.E., Ljungman M.. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome. Res. 2014; 24:896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ustianenko D., Weyn-Vanhentenryck S.M., Zhang C.. Microexons: discovery, regulation, and function. Wiley Interdiscip. Rev. RNA. 2017; 8: 10.1002/wrna.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saito Y., Miranda-Rottmann S., Ruggiu M., Park C.Y., Fak J.J., Zhong R., Duncan J.S., Fabella B.A., Junge H.J., Chen Z.et al.. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. Elife. 2016; 5:e14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Johnson V., Junge H.J., Chen Z.. Temporal regulation of axonal repulsion by alternative splicing of a conserved microexon in mammalian Robo1 and Robo2. Elife. 2019; 8:e46042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Coelho M.B., Smith C.W.. Regulation of alternative pre-mRNA splicing. Methods Mol. Biol. 2014; 1126:55–82. [DOI] [PubMed] [Google Scholar]
- 77. Barash Y., Calarco J.A., Gao W., Pan Q., Wang X., Shai O., Blencowe B.J., Frey B.J.. Deciphering the splicing code. Nature. 2010; 465:53–59. [DOI] [PubMed] [Google Scholar]
- 78. Gupta M.P., Kogut P., Gupta M.. Protein kinase-A dependent phosphorylation of transcription enhancer factor-1 represses its DNA-binding activity but enhances its gene activation ability. Nucleic Acids Res. 2000; 28:3168–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang S.W., Dong M., Trujillo M.A., Miller L.J., Eberhardt N.L.. DNA binding of TEA/ATTS domain factors is regulated by protein kinase C phosphorylation in human choriocarcinoma cells. J. Biol. Chem. 2001; 276:23464–23470. [DOI] [PubMed] [Google Scholar]
- 80. Gaffney C.J., Oka T., Mazack V., Hilman D., Gat U., Muramatsu T., Inazawa J., Golden A., Carey D.J., Farooq A.et al.. Identification, basic characterization and evolutionary analysis of differentially spliced mRNA isoforms of human YAP1 gene. Gene. 2012; 509:215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Finch-Edmondson M.L., Strauss R.P., Clayton J.S., Yeoh G.C., Callus B.A.. Splice variant insertions in the C-terminus impairs YAP’s transactivation domain. Biochem. Biophys. Rep. 2016; 6:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Das S., Anczukow O., Akerman M., Krainer A.R.. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012; 1:110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Qi Y., Yu J., Han W., Fan X., Qian H., Wei H., Tsai Y.H., Zhao J., Zhang W., Liu Q.et al.. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016; 7:ncomms11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang M., Dai M., Wang D., Xiong W., Zeng Z., Guo C.. The regulatory networks of the Hippo signaling pathway in cancer development. J. Cancer. 2021; 12:6216–6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pan D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010; 19:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For TCGA cohorts, the genomic and clinical data can be retrieved from NCI Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov).