

Abstract
New series of thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) have been synthesised and assessed for their potential EGFR and VEGFR-2 inhibitory activities. Compounds 10b and 10d exerted potent and selective inhibitory activity towards the two receptor tyrosine kinases; EGFR (IC50 = 40.7 ± 1.0 and 32.5 ± 2.2 nM, respectively) and VEGFR-2 (IC50 = 78.4 ± 1.5 and 43.0 ± 2.4 nM, respectively). The best anti-proliferative activity for the examined thiazolyl-pyrazolines was observed against the non-small lung cancer cells (NSCLC). Compounds 10b and 10d displayed pronounced efficacy against A549 (IC50 = 4.2 and 2.9 µM, respectively) and H441 cell lines (IC50 = 4.8 and 3.8 µM, respectively). Moreover, our results indicated that 10b and 10d were much more effective towards EGFR-mutated NSCLC cell lines (NCI-H1650 and NCI-H1975 cells) than gefitinib. Finally, compounds 10b and 10d induce G2/M cell cycle arrest and apoptosis and inhibit migration in A549 cancerous cells.
Keywords: Anticancer, molecular docking, EGFR inhibitors, VEGFR-2 inhibitors, EGFR-mutated NSCLC, dual kinase inhibitors
Graphical Abstract

1. Introduction
Cancer remains as a major health problem and a life-threatening disease even after decades of fundamental biomedical advances leading to the development of a war chest of anti-tumour therapies. This is largely because of the ability of cancer cells to develop resistance against most antitumor agents1.
Lung cancer is the second most common cancer and the leading cause of cancer-related deaths in both men and women accounting for about 26% of all cancer deaths2. It has been shown that epidermal growth factor receptor (EGFR), the prototypical growth factor receptor tyrosine kinase (RTK)3, is one of the most important key players in the development of many lethal cancers globally4,5 including: colorectal cancers, ovarian cancers6, breast cancers7, and non-small-cell lung cancers (NSCLC) in which EGFR overexpression takes place in approximately 43–89% of all cases and relates with diminutive survival and chemoresistance8. Accordingly, interruption of the EGFR signalling pathway using small-molecule tyrosine kinase inhibitors (TKIs) is considered as a well-established therapeutic approach in cancer treatment since the approval of gefitinib by the United States Food and Drug Administration (FDA) in 20039,10.
Gefitinib and erlotinib (Figure 1) are first-generation EGFR inhibitors that have been approved for the treatment of advanced NSCLC with activating mutations in the EGFR tyrosine kinase domain mainly: L858R (EGFRL858R)11–14. However, some patients develop secondary drug resistance mutations which results in a relapse after nearly one year of treatment15. The “gatekeeper” mutation is the predominant one which is a single point substitution of Thr790 with Met in exon 20 (EGFRT790M)16. Hence, afatinib and dacomitinib (Figure 1) have been developed as irreversible second generation EGFR inhibitors17,18 because they have electrophilic Michael-acceptor systems, such as acrylamide moiety which forms a covalent bond with Cys79719. This resulted in blocking of T790M resistance mutation by increasing target residence time20.
Figure 1.

First-, second- and third generation EGFR inhibitors and dual EGFR/VEGFR-2 inhibitor.
Despite the promising in vitro results in patients with erlotinib-resistant cancers21, they showed poor efficacy at clinically achievable concentration22. This is due to the narrow therapeutic window, as the dose required for the inhibition of mutated EGFRT790M resulted in severe side effects as rash and diarrhoea from inhibiting EGFRwt23. Recently, this problem was solved by the discovery of third generation EGFR inhibitors as osimertinib (Tagrisso®, AZD9291), rociletinib (CO-1686), naquotinib (ASP8273) and nazartinib (EGF816)24,25 (Figure 1), however, a resistance has been developed too26–28. So, exploring more efficient EGFR inhibitors is an urgent and critical demand to surmount the continual evolution of resistance to the current inhibitors.
Besides EGFR, there are other RTKs that have been shown to be important targets in cancer29. For example, the RTK vascular endothelial growth factor receptor-2 (VEGFR-2) stands out as a key target in cancer treatment due to its important role in angiogenesis30. Based on the functional crosstalk of EGFR and VEGFR-2 through shared common downstream signalling pathways31, it is inferred that the simultaneous inhibition of both EGFR and VEGFR is an effective approach for overcoming the reported resistance in NSCLC32. In fact, it has been shown that VEGFR-2 inhibition enhances the cytotoxic effect of EGFR inhibitors, whereas, VEGFR-2 activation results in accelerated tumour growth independent of EGFR signalling, thereby facilitating the emergence of resistance to EGFR inhibitors33. Vandetanib (Caprelsa®) is an example of FDA approved quinazoline-based drug with dual EGFR and VEGFR-2 inhibitory activity34 (Figure 1).
In the current medical era, thiazole is identified as an important heterocyclic motif that emerged as a promising privileged scaffold in the anticancer drug discovery35–37, Several FDA approved anticancer drug incorporate the heterocycle thiazole, such as dabrafenib and dasatinib. Pyrazoline moiety, on the other hand, is a highly active heterocyclic nucleus possessing interesting biological activities38 like; anticancer39–41, anti-inflammatory42, antidepressant43, anticonvulsant44 and antimicrobial activities45.
Recently, some research groups have devoted their efforts to hybridise both the thiazole and pyrazoline into a single frame work, producing novel series based on the thiazolyl-pyrazoline system which thereafter identified as a substantial scaffold in anticancer drug discovery46. Interestingly, there is a large number of reported compounds with potent cytotoxic effect based on thiazolyl-pyrazoline scaffold having EGFR (compounds I, II and III, Figure 2)47–49 and VEGFR-2 (compound IV, Figure 2)50 suppressing potentials. It is worth mentioning that none of these studies have developed dual or multiple kinase inhibitors, in addition they have not evaluated the biological activity against EGFR-mutated cancer cell lines.
Figure 2.

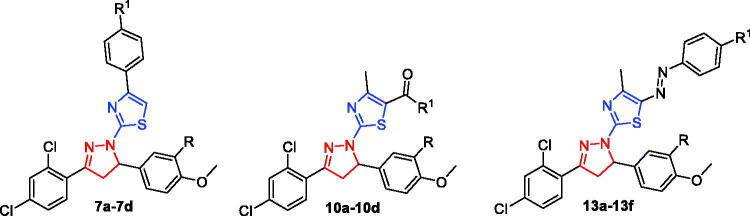
Structure of some reported thiazolyl-pyrazoline based EGFR and VEGFR-2 inhibitors (I–IV), and the target compounds in this study (7a–7d, 10a–10d and 13a–13f).
Motivated by the above mentioned findings, here we sought to develop new different sets of novel thiazolyl-pyrazoline-based small molecules (7a–7d, 10a–10d and 13a–13f) as potential dual EGFR/VEGFR-2 inhibitors with improved anticancer activities based on the molecular hybridisation between thiazole and pyrazoline motifs (Figure 2).
It is worth to mention that the reported pharmacophoric features required for EGFR inhibition include a central flat hetero-aromatic ring scaffold that should occupy the adenine binding pocket, a terminal hydrophobic head that occupies the hydrophobic sub-pocket, as well as a hydrophobic tail to occupy a second hydrophobic region51,52. Additionally, certain pharmacophoric features have been identified for the VEGFR-2 inhibitors including a flat hetero-aromatic moiety to be fitted in the ATP binding region, a terminal hydrophobic motif to achieve several hydrophobic interactions in the allosteric hydrophobic pocket, and the presence of H-bond acceptor (HBA) functionalities53–55. Interestingly, the target thiazolyl-pyrazoline-based small molecules (7a–7d, 10a–10d and 13a–13f) achieved these features required for inhibition of both EGFR and VEGFR-2. Furthermore, the substitution pattern on the pendant phenyl moieties was selected so as to ensure different electronic and lipophilic environments which should manipulate the activity of the target thiazolyl-pyrazolines.
Once created, all derivatives were assessed for their potential inhibitory activity towards EGFR and VEGFR-2 kinases. Also, the inhibitory activities against a panel of 11 kinases (TEK, SYK, EPHB2, ABL1, LCK, CLK1, ROCK1, PKC, AKT1, CDK1, and CDK5) were further investigated to determine the selectivity profile. Then, all compounds were screened for their cytotoxic effect against nine cell lines derived from five tumour subpanels including leukaemia (K562, and KG-1a), breast (MCF-7, BT-549, and HCC70), lung (A549 and H441), colon (HCT116) and liver (HepG2) cancer cell lines. In addition, the anti-proliferative potential of the most potent EGFRWT inhibitors was analysed towards two EGFR mutated cancer cell lines; NCI-H1650 and NCI-H1975. Thereafter, the most efficient cells growth and kinase inhibitors 10b and 10d were selected to explore their possible cellular mechanism of action via cell cycle, apoptosis Annexin-V-FITC and migration assays in NSCLC A549 cells. Finally, in silico studies were carried out to explore the binding interactions of thiazolyl-pyrazolines 10b and 10d within the vicinity of ATP-binding sites on EGFR and VEGFR-2 kinase domains.
2. Results and discussion
2.1. Chemistry
The synthetic pathways adopted for preparation of the intermediates 5a–b and the target thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) were depicted in Schemes 1 and 2. In Scheme 1, chalcones 3a, b were prepared via reaction of 2,4-dichloroacetophenone 1 with the appropriate aromatic aldehydes 2a, b, adopting the base catalysed Claisen–Schmidt condensation reaction as reported56 (Scheme 1). As active intermediates, chalcones 3a, b were used to prepare pyrazolines 5a, b by the reaction with thiosemicarbazide 4 in the presence of sodium hydroxide as a catalyst (Scheme 1). Pyrazoline formation occurs via two steps; the first one is the condensation reaction between the chalcones 3a, b and thiosemicarbazide 4, whereas, the second step is the intramolecular cyclisation through Michael addition of the NH group to the double bond57 (Scheme 1). Structures of compounds 5a, b were confirmed by microanalyses and spectral data. IR spectra of 5a, b showed NH2 bands at 3418, 3444 and 3247, 3322 cm−1, respectively, in addition to two bands at 1236, 1254 and 828, 833 cm−1 referring to C=S. 1H NMR spectra of 5a displayed three doublets of doublets for HA, HM and HX of the pyrazoline ring at δ 3.18, 4.03 and 5.87 ppm, respectively. This AMX pattern confirmed the formation of the pyrazoline ring and the presence of two diastereotopic protons at C-4 (HA and HM) and one single proton at C-5 (HX)58. Furthermore, 1H NMR spectra of 5a, b appeared two D2O exchangeable singlet signals of NH2 around δ 7.75, 7.76 and 8.13 ppm, respectively. In addition, 13C NMR spectra of 5a, b revealed two signals for C-4 and C-5 carbons of pyrazoline ring at δ 45.7 and 63.0, 63.3 ppm, respectively.
Scheme 1.

Synthetic pathway for preparation of intermediates 5a, b and compounds 7a–7d; reagents and reaction conditions: (i) 10% NaOH, EtOH, RT, 4 h; (ii) NaOH, EtOH, reflux, 6 h; (iii) EtOH, reflux, 4 h.
Scheme 2.

Synthetic pathway for preparation of compounds 10a–10d and 13a–13f; reagents and reaction conditions: (i) EtOH, reflux, 4 h; (ii) SO2Cl2, dry Et2O, 0–5 °C, 2 h; (iii) (a) NaNO2-HCl, 0–5 °C, 2 h; (b) NaOAc.3H2O, EtOH, 0–5 °C, 4 h; (iv) EtOH, reflux, 4 h.
Furthermore, reaction of carbothioamide derivatives 5a, b with 1-aryl-2-bromoethanones 6a, b in refluxing ethanol yielded the corresponding 4-(aryl)-2-(3-(2,4-dichlorophenyl)-5-(4-aryl)-4,5-dihydro-1H-pyrazol-1-yl)thiazoles 7a–7d, respectively, via Hantzsch thiazole synthesis54,55 (Scheme 1). Initially, nucleophilic substitution of Br in phenacyl bromide by the S-atom of thioamide generates the isothiourea, which subsequently undergoes cyclocondensation and water elimination to furnish the thiazole ring57 (Scheme 1). The latter synthesised compounds 7a–7d were confirmed based on their elemental analysis and spectral data. IR spectra showed the disappearance of the characteristic band of NH2 in the region of 3418, 3444 and 3247, 3322 cm−1. Their 1H NMR spectra showed an increase in the aromatic integration due to thiazole proton and protons of the extra phenyl ring at δ 6.91–7.82 ppm. 13C NMR spectra of 7b, c showed peak around δ 105 ppm corresponding to C-5 of thiazole ring along with an increase in signals assigned for aromatic carbons at δ 111.6–164.8 ppm.
Similarly, thiazole derivatives 10a–10d were obtained from the reaction of the carbothioamides 5a, b with the appropriate α-chloro-1,3-dicarbonyl compounds 9a–9c in absolute ethanol through the Hantzsch thiazole synthesis49,55 (Scheme 2). Compounds 10a–10d were confirmed based on their elemental analysis and spectral data.
IR spectra of 10a–10d showed a band at 1637–1669 cm−1 due to C=O group, while the spectra of 10c, d revealed a band at 3257 and 3262 cm−1, respectively corresponding to NH group. 1H NMR spectra of 10a–10d showed a signal at δ 2.36–2.54 ppm attributed to the protons of the methyl group at the 4-position of the thiazole ring. Moreover, 1H NMR spectrum of 10a showed a singlet signal at δ 2.40 ppm attributed to CH3 protons of the acetyl group. Furthermore, 1H NMR spectrum of 10b showed two signals at δ 1.35 and 4.29 ppm, respectively attributed to the CH3 and CH2 protons of the ethyl group. Additionally, 1H NMR spectra of 10c, d displayed a D2O exchangeable singlet signal of NH proton at δ 9.72, 8.14 ppm, respectively, in addition to an increase in the aromatic integration at δ 6.78–7.81 ppm assignable to the extra phenyl ring. 13C NMR of compound 10a showed signals assigned to CH3 carbon of the thiazole ring, CH3 and C=O carbons of the acetyl group at δ 19.1, 30.1 and 189.7 ppm, respectively. Additionally, 13C NMR spectrum of 10b displayed signals attributed to ethyl carbons at δ 14.7 and 60.7 ppm along with signal due to ester carbonyl carbon at δ 165.3 ppm. Furthermore, 13C NMR spectrum of 10d revealed signals at δ 18.2 and 163.8 ppm due to CH3 carbon of the thiazole ring and C=O, respectively.
Finally, 2-(3-(2,4-dichlorophenyl)-5-(aryl)-4,5-dihydro-1H-pyrazol-1-yl)-4-mthyl-5-(aryldiazenyl)thiazole derivatives 13a–13f were prepared by heating the carbothioamide derivatives 5a, b with 2-oxo-N-arylpropanehydrazonyl chlorides 12a–12c in absolute ethanol56,57 (Scheme 2). 2-Oxo-N-arylpropanehydrazonyl chlorides 12a–12c were prepared through a coupling reaction between α-chloroacetylacetone and aryl diazonium salts via Japp–Klingeman rearrangement59.
IR spectra of compounds 13a–13f showed a band at 1582–1589 cm−1 referring to (N = N) in addition to the absence of the band corresponding to NH2 group at 3418, 3444 and 3247, 3322 cm−1. 1H NMR spectra of 13b, e, f revealed singlet signals at δ 2.53–2.61 ppm equivalent to CH3 protons of the thiazole ring in addition to an increase in the integration of the aromatic protons at δ 6.78–7.90 ppm. Also, 1H NMR spectra of 13b, e showed additional singlet signals at δ 2.42 and 2.41 ppm attributed to the three protons of 4-CH3, respectively. 13C NMR of 13a–13e showed a signal for CH3 carbon of the thiazole ring at δ 16.1–16.6 ppm along with an increase in the number of signals of aromatic carbons at δ 109.4–165.3 ppm. Also, 13C NMR of 13b, e revealed the presence of a signal at δ 21.4 ppm indicating an additional CH3 carbon at the phenyldiazenyl ring.
2.2. Biological evaluation
2.2.1. EGFR and VEGFR-2 kinase inhibitory activities of thiazolyl-pyrazoline derivatives
In the current study, the potential inhibitory activity of target thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) towards EGFR and VEGFR-2 were examined, with the aim of exploring the plausible mechanism. The IC50 values of the tested compounds were evaluated compared to reference EGFR inhibitor (gefitinib) and VEGFR-2 inhibitor (vandetanib), Table 1.
Table 1.
EGFR and VEGFR-2 kinases inhibitory activity of target thiazolyl-pyrazoline derivatives.
| Compound | R | R1 | IC50 (nM) ± SEM |
|
|---|---|---|---|---|
| EGFR | VEGFR-2 | |||
| 7a | H | H | 145.1 ± 2.0 | 124.9 ± 1.4 |
| 7b | H | Cl | 114.2 ± 0.4 | 195.2 ± 4.6 |
| 7c | OCH3 | H | 511.2 ± 3.2 | 915.2 ± 4.3 |
| 7d | OCH3 | Cl | 132.9 ± 1.2 | 185.5 ± 2.3 |
| 10a | H | CH3 | 274.0 ± 2.3 | 450.3 ± 8.3 |
| 10b | H | OCH2CH3 | 40.7 ± 1.0 | 78.4 ± 1.5 |
| 10c | H | NHC6H5 | 130.7 ± 1.1 | 102.2 ± 4.3 |
| 10d | OCH3 | NHC6H5 | 32.5 ± 2.2 | 43.0 ± 2.4 |
| 13a | H | H | 973.0 ± 2.0 | 1151.2 ± 4.3 |
| 13b | H | CH3 | 179.4 ± 0.0 | 214.3 ± 4.0 |
| 13c | H | Cl | 134.5 ± 1.5 | 173.3 ± 2.6 |
| 13d | OCH3 | H | 93.6 ± 3.0 | 335.8 ± 6.3 |
| 13e | OCH3 | CH3 | 67.7 ± 2.0 | 216.4 ± 4.3 |
| 13f | OCH3 | Cl | 107.13 ± 1.2 | 113.3 ± 1.8 |
| Gefitinib | – | – | 55.74 ± 1.0 | 45.3 ± 1.2 |
| Vandetanib | – | – | 43.14 ± 1.3 | 29.1 ± 1.0 |
Examination of the obtained results (Table 1) hinted out that the target thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) have better kinase inhibitory activity towards EGFR (IC50 range: 32.5–973.0 nM) than VEGFR-2 (IC50 range: 43.0–1151.2 nM), except for compounds 7a and 10c. As shown in Table 1, compounds 10b and 10d displayed the highest inhibitory activity against EGFR (IC50 = 40.7 and 32.5 nM, respectively) as well as VEGFR-2 (IC50 = 78.4 and 43.0 nM, respectively), due to the ability of the carbonyl group in the ester functionality to form hydrogen bonds with the key amino acids in both EGFR and VEGFR-2 active sites. Moreover, compounds 7a, 7b, 7d, 10a, 10c and 13b–13d exhibited moderate inhibitory activity against EGFR (IC50: 67.7–274.0 nM) and VEGFR-2 (IC50: 102.2–450.3 nM). On the other hand, compounds 7c and 13a displayed the lowest activity against EGFR (IC50 = 511.2 and 973.0 nM, respectively) and VEGFR-2 (IC50 = 915.2 and 1151.2 nM, respectively).
The following structure–activity relationships (SARs) can be concluded from the kinases inhibition data displayed in Table 1. Regarding the EGFR inhibitory activity of the first series 7a–7d, it was found that substitution of the pendant phenyl ring at C-4 of the thiazole moiety (compounds 7b and 7d; IC50 = 114.2 ± 0.4 and 132.9 ± 1.2 nM, respectively) elicited an enhancement of effectiveness towards EGFR in comparison to the unsubstituted analogues (compounds 7a and 7c; IC50 = 145.1 ± 2.0 and 511.2 ± 3.2 nM, respectively). In contrast, the 3,4-dimethoxy substitution for the pyrazoline C-5 phenyl ring (compounds 7c and 7d; IC50 = 511.2 ± 3.2 and 132.9 ± 1.2 nM, respectively) resulted in a worsening of inhibitory activity against EGFR in comparison to the 4-methoxy monosubstituted counterparts (compounds 7a and 7b; IC50 = 145.1 ± 2.0 and 114.2 ± 0.4 nM, respectively).
Moreover, the obtained results for the second series 10a–10d towards EGFR revealed that the incorporation of the ester functionality at C-5 of the thiazole moiety (compound 10b; IC50 = 40.7 ± 1.0 nM) was more beneficial for the activity more than the acetyl (compound 10a; IC50 = 274.0 ± 2.3 nM) and amide (compound 10c; IC50 = 130.7 ± 1.1 nM) functionalities. In addition, the 3,4-dimethoxy substitution for the pyrazoline C-5 phenyl ring in the amide-bearing 10d led to the best EGFR inhibition in this study; IC50 = 32.5 ± 2.2 nM. In a similar fashion, incorporation of a 3,4-dimethoxyphenyl moiety within the third series 13a–13f (compounds 13d–13f; IC50 = 93.6 ± 3.0, 67.7 ± 2.0 and 107.13 ± 1.2 nM, respectively) elicited an enhancement of EGFR inhibitory activity in comparison to the monosubstituted analogues (compounds 13a–13c; IC50 = 973.0 ± 2.0, 179.4 ± 0.0 and 134.5 ± 1.5 nM, respectively).
On the other hand, the SAR outcomes for VEGFR-2 inhibition pointed out that utilisation of the ester group at C-5 of the thiazole moiety within the second series 10a–10d (compound 10 b; IC50 = 78.4 ± 1.5 nM) led to an enhanced activity more than the acetyl (compound 10a; IC50 = 450.3 ± 8.3 nM) and amide (compound 10c; IC50 = 102.2 ± 4.3 nM) groups. Similar to the SAR for EGFR inhibition, grafting a second methoxy group for the amide-bearing counterpart 10c (IC50 = 102.2 ± 4.3 nM) produced compound 10d that identified as the most potent VEGFR-2 inhibitor in this work (IC50 = 43.0 ± 2.4 nM). Further analysis of the VEGFR-2 inhibition data for the third series 13, highlighted that both the para-substitution of the appended phenyl ring at C-5 of the thiazole moiety, and the 3,4-dimethoxy substitution for the pyrazoline C-5 phenyl ring resulted in an improvement of the VEGFR-2 inhibitory activity.
2.2.2. Western blotting of EGFR and VEGFR-2 in A549 cells
To confirm the ability of thiazolyl-pyrazolines derivatives to dual inhibit the activity of EGFR and VEGFR-2 kinases, we examined the effect of the most potent compounds 10b and 10d on the expression levels of p-EGFR, EGFR, p-VEGFR-2 and VEGFR-2 proteins in A549 cells using Western blot analysis. As shown in Figure 3, the protein expression levels of p-EGFR and p-VEGFR-2 were significantly downregulated by compounds 10b and 10d.
Figure 3.

Thiazolyl-pyrazoline derivatives downregulated p-EGFR and p-VEGFR-2 protein levels (A) Western blotting analysis of the expression of p-EGFR, EGFR, p-VEGFR-2 and VEGFR-2 in A549 cells treated with 10b or 10d and untreated cells used as a negative control. β-actin served as a loading control. (B) The relative protein expression level of p-EGFR normalised with total EGFR was quantified by the Image J software (C) The relative protein expression level of p-VEGFR-2 normalised with total VEGFR-2. The values are expressed as the mean ± SEM based on three different experiments. ***p < 0.001 indicate a significant difference compared with control.
2.2.3. Kinase selectivity profiling
As compounds 10b and 10d showed high potency against EGFR and VEGFR-2 kinases (Table 1), we further investigated their inhibitory activities against 11 kinases (TEK, SYK, EPHB2, ABL1, LCK, CLK1, ROCK1, PKC, AKT1, CDK1, and CDK5) to determine their selectivity profile (Table 2).
Table 2.
Kinase selectivity profile of 10b and 10d.
| Kinase | IC50 (nM) |
|
|---|---|---|
| 10b | 10d | |
| TEK | >2000 | >2000 |
| SYK | 1120 ± 22 | 870 ± 31 |
| EPHB2 | >2000 | >2000 |
| ABL1 | >2000 | >2000 |
| LCK | >2000 | >2000 |
| CLK1 | 1069 ± 54 | 798 ± 19 |
| ROCK1 | >2000 | >2000 |
| PKC | >2000 | >2000 |
| AKT1 | 816 ± 38 | 702 ± 24 |
| CDK1 | 310 ± 12 | 245 ± 7 |
| CDK5 | 1136 ± 20 | 993 ± 15 |
Interestingly, compounds 10b and 10d displayed moderate inhibition against CDK1 (IC50 = 310 and 245 nM, respectively) and low inhibition against SYK, CLK1, AKT1 and CDK5 (IC50 range: 702–1136 nM). Meanwhile, 10b and 10d did not exhibit measurable inhibition (with IC50 > 2000 nM) against TEK, EPHB2, ABL1, LCK, ROCK1 and PKC kinases, demonstrating significant selectivity of 10b and 10d towards EGFR and VEGFR-2 kinases.
2.2.4. Anti-proliferative activity of thiazolyl-pyrazoline derivatives
To evaluate the anti-proliferative activity of the synthesised thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d, and 13a–13f), a preliminary in vitro one dose (10 µM) anticancer assay was performed. Nine cell lines derived from five tumour subpanels were examined; leukaemia (K562 and KG-1a), breast (MCF-7, BT-549 and HCC70), lung (A549 and H441), colon (HCT116) and liver (HepG2) cancer cell lines. The compounds were added at a single concentration (10 µM) and the cultures were incubated for 24 h, then the per cent growth inhibition (%GI) was calculated.
As shown in Tables 3 and 4, compounds 10b and 10d revealed promising cell growth inhibition activity against almost all the screened cell lines (%GI ranging from 31.0% to 95.3%), especially against the non-small cell lung cancer A549 and H441 cell lines (%GI ranging from 79.1% to 95.3%). Other compounds showed moderate to weak anti-proliferative effect against the tested cell lines; however, the non-small lung cancer cells (A549 and H441) were more sensitive to the target thiazolyl-pyrazoline derivatives.
Table 3.
% Growth inhibition ± SEM of thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) towards leukaemia (K562 and KG-1a) and breast cancer (MCF-7, BT-549 and HCC70) cell lines.
| Compound | K562 | KG-1a | MCF-7 | BT-549 | HCC70 |
|---|---|---|---|---|---|
| 7a | 14.2 ± 2.1 | 18.4 ± 1.3 | 0.07 ± 0.0 | 26.4 ± 3.6 | 27.3 ± 1.0 |
| 7b | 21.5 ± 3.0 | 21.7 ± 4.6 | 25.9 ± 1.2 | 5.2 ± 1.0 | 19.6 ± 0.3 |
| 7c | 5.2 ± 0.5 | 4.3 ± 0.7 | 9.2 ± 0.9 | 3.1 ± 0.6 | 7.3 ± 1.2 |
| 7d | 22.0 ± 1.6 | 25.1 ± 2.0 | 2.2 ± 0.6 | 18.0 ± 2.7 | 17.3 ± 0.6 |
| 10a | 12.2 ± 1.9 | 22.2 ± 1.6 | 13.6 ± 2.9 | 9.4 ± 1.0 | 24.3 ± 2.5 |
| 10b | 54.2 ± 3.8 | 31.0 ± 2.5 | 48.5 ± 2.3 | 47.2 ± 4.9 | 41.5 ± 2.1 |
| 10c | 11.2 ± 1.0 | 9.1 ± 1.0 | 2.5 ± 0.8 | 22.3 ± 0.8 | 18.3 ± 1.6 |
| 10d | 60.2 ± 2.3 | 57.2 ± 3.4 | 60.9 ± 4.3 | 40.1 ± 2.0 | 54.1 ± 2.8 |
| 13a | 2.2 ± 0.2 | 4.1 ± 0.9 | 17.8 ± 1.0 | 10.3 ± 1.1 | 15.3 ± 1.4 |
| 13b | 11.0 ± 0.6 | 7.1 ± 0.2 | 3.4 ± 0.5 | 5.1 ± 0.3 | 8.3 ± 0.9 |
| 13c | 5.2 ± 0.9 | 12.4 ± 1.6 | 10.4 ± 2.9 | 8.7 ± 1.9 | 1.1 ± 0.1 |
| 13d | 18.2 ± 1.8 | 24.1 ± 2.7 | 11.9 ± 1.8 | 14.2 ± 2.2 | 22.3 ± 1.3 |
| 13e | 24.0 ± 2.0 | 15.2 ± 1.8 | 1.8 ± 0.6 | 12.5 ± 1.5 | 16.1 ± 0.9 |
| 13f | 31.5 ± 1.0 | 20.0 ± 2.8 | 0.8 ± 0.2 | 24.6 ± 1.3 | 6.3 ± 1.0 |
Table 4.
% Growth inhibition ± SEM of thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) towards lung (A549 and H441), colon (HCT116) and liver (HepG2) cancer cell lines.
| Compound | A549 | H441 | HepG2 | HCT116 |
|---|---|---|---|---|
| 7a | 34.9 ± 4.3 | 36.1 ± 2.1 | 0.5 ± 0.0 | 0.5 ± 0.1 |
| 7b | 45.2 ± 2.9 | 35.2 ± 1.2 | 34.4 ± 1.4 | 18.8 ± 1.9 |
| 7c | 8.9 ± 1.1 | 16.2 ± 0.3 | 17.1 ± 2.7 | 4.9 ± 0.8 |
| 7d | 31.2 ± 1.4 | 34.8 ± 2.4 | 5.1 ± 1.0 | 0.7 ± 0.0 |
| 10a | 32.3 ± 2.2 | 6.3 ± 0.7 | 28.0 ± 4.3 | 19.0 ± 2.6 |
| 10b | 88.3 ± 4.9 | 79.1 ± 2.6 | 50.6 ± 2.2 | 37.7 ± 3.9 |
| 10c | 28.9 ± 2.0 | 8.7 ± 0.1 | 11.9 ± 1.1 | 7.9 ± 1.1 |
| 10d | 95.3 ± 5.3 | 89.2 ± 3.9 | 65.5 ± 3.9 | 56.5 ± 2.0 |
| 13a | 11.0 ± 0.8 | 22.2 ± 0.9 | 15.8 ± 2.2 | 19.5 ± 1.4 |
| 13b | 16.2 ± 1.3 | 23.3 ± 2.4 | 0.08 ± 0.0 | 0.8 ± 0.2 |
| 13c | 14.8 ± 1.0 | 19.9 ± 1.7 | 14.2 ± 1.0 | 8.7 ± 0.6 |
| 13d | 12.6 ± 2.7 | 38.9 ± 2.4 | 18.6 ± 2.5 | 10.1 ± 3.5 |
| 13e | 30.3 ± 2.4 | 46.9 ± 3.9 | 0.3 ± 0.0 | 9.9 ± 0.8 |
| 13f | 47.5 ± 3.1 | 17.3 ± 0.6 | 0.9 ± 0.3 | 0.2 ± 0.0 |
Overexpression of EGFR and VEGFR-2 is well-reported in the literature to be significantly correlated with the induction of the NSCLC cells proliferation31. Thereafter, the quantitative IC50 values have been determined for thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) towards the most susceptible cell lines at the preliminary assay (non-small lung cancer A549 and H441 cell lines), at testing concentrations of 1, 5, 10, 25, 50 or 100 µM for 24 h. The antitumor drugs gefitinib and vandetanib were used as reference drugs in this MTT cytotoxicity assay (Table 5).
Table 5.
Inhibitory activities of thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d and 13a–13f) against cancerous and normal lung cells. Values represent the mean IC50 values (µM) ± SEM (n = 3) for each drug. Compared IC50s; a compounds versus gefitinib and b compounds versus Vandetanib.
| Compound | IC50 (µM) ± SEM |
||
|---|---|---|---|
| A549 | H441 | WI-38 | |
| 7a | 22.5 ± 1.2a ns, b*** | 38.4 ± 2.3a***, b*** | 88.2 ± 4.2 a***, b*** |
| 7b | 12.0 ± 0.4a***, b*** | 26.5 ± 1.1a***, b*** | 60.6 ± 3.1a***, b*** |
| 7c | >100 | >100 | >100 |
| 7d | 20.2 ± 2.0a ns, b*** | 29.1 ± 3.5a***, b*** | 40.1 ± 2.7a*, b*** |
| 10a | 49.1 ± 3.2a***, b*** | >100 | >100 |
| 10b | 4.2 ± 0.8a***, bns | 4.8 ± 0.1a*, bns | 72.4 ± 7.2a***, b*** |
| 10c | 23.5 ± 1.3a ns, b*** | 45.2 ± 2.3a***, b*** | >100 |
| 10d | 2.9 ± 0.2a***, bns | 3.8 ± 0.5a*, bns | 51.4 ± 3.5a***, b*** |
| 13a | >100 | 94.4 ± 5.5a***, b*** | >100 |
| 13b | 59.1 ± 3.2a***, b*** | 24.2 ± 1.8a***, b*** | >100 |
| 13c | 46.3 ± 2.5a***, b*** | 33.5 ± 3.9a***, b*** | 91.4 ± 8.9a***, b*** |
| 13d | 57.1 ± 4.1a***, b*** | 21.7 ± 1.3a***, b*** | 64.2 ± 2.8a***, b*** |
| 13e | 19.1 ± 1.3a*, b*** | 15.2 ± 3.6a***, b*** | 87.0 ± 4.2a***, b*** |
| 13f | 13.7 ± 2.2a**, b*** | 24.8 ± 1.4a***, b*** | >100 |
| Gefitinib | 22.3 ± 1.4 | 7.1 ± 1.3 | 35.1 ± 2.0 |
| Vandetanib | 3.4 ± 1.0 | 3.9 ± 0.8 | 8.6 ± 0.4 |
*p < 0.05, **p < 0.01, ***p < 0.001 and ns = non-significant.
The results illustrated in Table 5 showed that compounds 10b and 10d have the best anti-proliferative activity, herein reported, against A549 (IC50 = 4.2 and 2.9 µM, respectively) and H441 (IC50 = 4.8 and 3.8 µM, respectively) cell lines, whereas compounds 7c and 13a have non-significant anti-proliferative activity (IC50 more than 100 μM). Other compounds showed moderate to low anti-proliferative effect against A549 (IC50 range: 12.0–59.1 µM) and H441 (IC50 range: 15.2–94.4 µM). Interestingly, the anti-proliferative effect of compounds 10b and 10d was higher than the reference drug gefitinib and was comparable to vandetanib. In addition, compounds 7a, 7b, 7d, 10c, 13e and 13f exerted moderate anticancer activity against A549 cell lines with IC50s values spanning between 12.0 and 23.5 µM, whereas compounds 7b, 7d, 13b and 13d–13f showed moderate growth inhibitory activity towards H441 cells with IC50s spanning in the range 15.2–29.1 µM, Table 5.
It is noteworthy that the synthesised thiazolyl-pyrazoline derivatives (7a–7d, 10a–10d, and 13a–13f) showed weak or non-significant cytotoxicity to human normal WI-38 lung fibroblast cells (IC50 range: 40.1 ± 2.7 – >100 µM) (Table 5). The mean tumour selectivity value calculated for compounds 10b and 10d was found to be 16.1 and 15.3, respectively, which reveals good selectivity towards tumour cells with efficient safety index (Table 6).
Table 6.
Cytotoxic effect of 10b and 10d towards human non-tumorigenic lung WI-38 cells and mean tumour selectivity index (S. I.) (WI-38/A549 and H441).
| Compound | IC50 (µM) ± SEM |
Mean tumour selectivity | ||
|---|---|---|---|---|
| WI-38 | A549 | H441 | ||
| 10b | 72.4 ± 7.2 | 4.2 ± 0.8 | 4.7 ± 0.1 | 16.1 |
| 10d | 51.4 ± 3.5 | 2.9 ± 0.2 | 3.8 ± 0.5 | 15.3 |
2.2.5. Effect of thiazolyl-pyrazoline derivatives on EGFR-mutated NSCLC cell lines
The prolonged use of the TKIs targeting EGFR or VEGFR-2 kinases led to drug resistance due to mutations development and toxicity. In this study, we analysed the anti-proliferative potential of the most potent EGFRWT inhibitors 10b and 10d towards two EGFR mutated cancer cell lines; NCI-H1650 possesses exon19 deletion (delE746-A750) mutation of EGFR and NCI-H1975 carries L858R/T790M double mutation of EGFR. Interestingly, compounds 10b and 10d exhibited significant growth inhibitory activity against NCI-H1650 (IC50 = 5.7 and 3.5 µM, respectively) and NCI-H1975 (IC50 = 6.2 and 4.4 µM, respectively) Table 7 and Figure 4.
Table 7.
Inhibitory activity of thiazolyl-pyrazoline derivatives 10 b and 10d against EGFR-mutated NSCLC cell lines.
| Compound | IC50 (µM) |
|
|---|---|---|
| NCI-H1650 | NCI-H1975 | |
| 10b | 5.7 ± 0.5 | 6.2 ± 0.9 |
| 10d | 3.5 ± 0.2 | 4.4 ± 0.6 |
| Gefitinib | >20 | >20 |
Figure 4.

Effect of thiazolyl-pyrazoline derivatives on EGFR mutated NSCLC cell lines; dose–response curves for 10b and 10d in comparison with gefitinib control. The percentage of viable cells is shown relative to that of untreated control cells. Data were presented as means ± standard error mean (SEM) of three independent experiments.
Moreover, the obtained results indicated that both 10b and 10d were much more sensitive towards NCI-H1650 and NCI-H1975 cells than gefitinib (IC50 > 20 µM towards the two examined cell lines). Accordingly, 10b and 10d may serve as a lead compounds for the treatment of gefitinib-resistant EGFR mutant NSCLC.
2.2.6. Thiazolyl-pyrazoline derivatives inhibited cancer cell migration
As the thiazolyl-pyrazoline derivatives 10b and 10d showed potent inhibitory effect against VEGFR-2 Kinase that plays an important role in cancer cell migration, we investigated the effect of 10b and 10d on the A549 cancer cell migration using wound healing assay. As shown in Figure 5, compounds 10b and 10d were effective in reducing the cancer cell migration compared to the control, highlighting that the VEGFR-2 kinase inhibitory effect of 10b and 10d is the engine for the inhibition of the cancer cell migration.
Figure 5.

Effect of compounds 10b and 10d on A549 cells migration. Scratch wound healing assay was used to analyse the inhibition of cell migration in A549 cells treated for 36 h with either 10b or 10d and the untreated cells served as a negative control. Representative images of scratched areas in confluent A549 cell layers.
2.2.7. Thiazolyl-pyrazoline derivatives induced G2/M cell cycle arrest
In general, the anticancer agents inhibit the proliferation of cancerous cells by arresting cell division at various checkpoints. These checkpoints present at G1/S phase, S-phase and G2/M phases. Therefore, we tested the effect of the most potent compounds (10b and 10d) on cell cycle progression in A549 cell line. Results in Figure 6 obviously indicate that compounds 10b and 10d arrested the cell cycle at a G2/M phase (29.2% and 33.92%, respectively) when compared to the untreated controls (6.06% and 5.93%, respectively). Parallel to these findings, the cell population in G1 and S phases decreased after treatment with 10b and 10d (Figure 6(A,B)).
Figure 6.

Compounds 10b and 10d arrested the cell cycle at a G2/M phase while gefitinib arrested the cell cycle at a G1 phase in A549 cells. (A) Representative cell cycle distribution data are shown by flow cytometric analysis upon incubation with 0 (control), 2.5 or 5 μM of 10b or 10d or with 0 (control), 5 or 20 μM of gefitinib for 24 h. (B) Quantification of cell cycle distribution upon 24 h incubation with 10b or 10d. The values are means ± SEM of three different experiments. *p < 0.05 and **p < 0.01 indicate significant differences compared with control. ns = non-significant.
On the other hand, gefitinib (20 µM) significantly arrested the cell cycle at G1 phase. This difference between the synthesised compounds and gefitinib in the arrested cell cycle phase could be attributed to the moderate inhibitory effect of 10b and 10d on the CDK1 kinase (IC50 = 310 and 245 nM, respectively) that regulates the G2/M phase transition. This result strongly suggests that the synthesised thiazolyl-pyrazoline derivatives inhibit the proliferation of A549 cells by arresting cell cycle at G2/M phase.
2.2.8. Thiazolyl-pyrazoline derivatives induced apoptosis
Furthermore, the pro-apoptotic effect of compounds 10b and 10d was also investigated on A549 cells using the Annexin V-FITC/PI double staining. The proportion of annexin V-positive apoptotic cells increased gradually from 5.4% at control to 16.5% at 5 μM 10b (Figure 7(A,B)) and from 6.3% at control to 28.5% at 5 μM 10d. These data indicate that the synthesised thiazolyl-pyrazoline derivatives treatment provokes the induction of apoptosis in A549 cells.
Figure 7.

Apoptotic effect of compounds 10b and 10d on A549 cells. (A) Representative cytograms of apoptotic A549 cells stimulated with 0 (control), 2.5 or 5 μM 10b or 10d, for 24 h. The lower-right (annexin V + PI- cells) and the upper-right (annexin V + PI + cells) quadrants show early and late apoptotic cells, and the lower-left (annexin V-PI- cells) and the upper-left (annexin V-PI- cells) quadrants represent viable and necrotic cells, respectively. (B) Quantification of annexin V-positive apoptotic cells upon stimulation with 10b or 10d. The values are the means ± SEM of three different experiments. *p < 0.05 and *p < 0.01 indicate significant differences compared with control.
2.3. Molecular docking
In view of the promising kinase inhibitory activity of the thiazolyl-pyrazoline derivatives, herein reported, a molecular docking study was carried out for the most potent derivatives 10b and 10d to provide insights for their potential binding interactions within the examined kinases binding sites. The molecular operating environment (MOE) 2019.02 was implemented in the conduction, analysis and visualisation of the entire docking studies. The 3D structural co-ordinates of EGFR and VEGFR-2 were downloaded from the protein databank PDB IDs 1M17 and 4ASD, respectively. Both the selected PDB IDs have excellent resolution of 2.6 Å and 2.03 for 1M17 and 4ASD, respectively. In addition, 1M17 contains EGFR in complex with potent inhibitor erlotinib, while 4ASD contains VEGFR-2 in complex with the potent inhibitor sorafenib, making them optimum choice for the docking studies. Pose retrieval step of the co-crystallised ligands resulted in values of 0.85 and 0.63 Å between the docked and the co-crystallised poses for erlotinib and sorafenib, respectively, indicating a valid docking protocol (Figure 8).
Figure 8.

Superimposition of the co-crystallised (blue) and the docking pose (pink) of erlotinib in the EGFR (A) and sorafenib in the VEGFR-2 (B) binding sites.
Moreover, the previous step resulted in an energy scores (S) of −11.7 and −14.9 kcal/mol, for erlotinib and sorafenib, respectively. The retrieved docking scores of the co-crystalized ligands were used as comparative means to benchmark the docking score values of compounds 10b and 10d.
2.3.1. Docking of compounds 10b and 10d into EGFR active site
Visual inspection of erlotinib binding with EGFR active site, revealed the formation of two hydrogen bonds with the key residues Leu768 and Met769, in addition, two carbon hydrogen bonds were noticed with Glu738 and Gln767. Through a water molecule, erlotinib was able also to interact with Cys751 and Thr766 (Figure 9).
Figure 9.

2D diagram for the binding of erlotinib in the EGFR active site (PDB: 1M17).
Docking simulations for thiazolyl-pyrazoline derivatives 10b and 10d showed that they fit well into the EGFR active site with good docking scores (−12.9 and −14.1 kcal/mol, respectively) comparable to that of erlotinib (−11.7 kcal/mol). The general binding patterns of compounds 10b and 10d are consistent with crystallographic binding mode of erlotinib within the EGFR-TK active site (PDB: 1M17), Figure 10.
Figure 10.

2D diagram for the binding interactions of compound 10b (A) and compound 10d (B) in the EGFR active site (PDB: 1M17).
For instance, the carbonyl group in the amide functionality of compound 10d was engaged in two hydrogen bonds with the key residues Leu768 and Met769. Similarly, the carbonyl group in the ester functionality of compound 10b was engaged in hydrogen bond interaction with Met769, in addition to three hydrogen bonds with Gln767, Cys751 and Thr766 through a water molecule (Figures 10 and 11).
Figure 11.

3D diagram of compounds 10b (A) and 10d (B) showing their binding interaction with the EGFR active site (PDB: 1M17).
Compounds 10b and 10d formed a carbon hydrogen bond with Cys773 and Val693, respectively. The ortho-chloride atom of compound 10b formed a halogen interaction with Gly695, while the ortho-chloride atom of compound 10d formed a halogen interaction with Lys692. On the other hand, only compound 10d was engaged in arene bond interactions with Leu694, Lys704 and Leu802, which explains the superior activity and docking score of compound 10d over compound 10b. Also, it highlights the important role of the additional phenyl ring attached to the amide linker in compound 10d in EGFR inhibition (Figures 10 and 11).
2.3.2. Docking of compounds 10b and 10d into VEGFR-2 active site
The efficient VEGFR-2 inhibitory activity of compounds 10b and 10d were illustrated by their good docking scores (−13.7 and −14.4 kcal/mol) comparable to that of sorafenib (−14.9 kcal/mol). As depicted in Figure 12, sorafenib exerts its VEGFR-2 inhibition activity through formation of hydrogen bonds with the essential residues; Glu885, Cys1045, Asp1046 and Cys919, in addition to other multiple interactions.
Figure 12.

2D diagram for the binding interactions of sorafenib in the VEGFR-2 active site (PDB: 4ASD).
Interestingly, both compounds 10b and 10d were proven to maintain multiple essential interactions upon docking into VEGFR-2 active site (Figures 13 and 14). For instance, compound 10b was engaged in hydrogen bond interactions with Leu889 and Asp1046 via the nitrogen and sulphur atoms of the thiazole ring, respectively. The nitrogen of pyrazoline ring formed two hydrogen bonds with Cys1045, in addition, the carbonyl group in the ester functional contributed to a hydrogen bond with Phe1047. The ortho-chloride and the para-methoxy substituents contributed to bonding interactions with Ile888 and Glu885, respectively. In a similar manner, all the previous interactions were achieved by compound 10d in addition to an extra hydrogen bond interaction with Lys868, in addition to an arene interaction with Leu1035 (Figures 13 and 14).
Figure 13.

2D diagram of compounds 10b (A) and 10d (B) showing their binding interaction with the VEGFR-2 active site (PDB: 4ASD).
Figure 14.

3D diagram of compounds 10b (A) and 10d (B) showing their binding interaction with the VEGFR-2 active site (PDB: 4ASD).
3. Conclusion
In conclusion, this study reports the facile synthesis of potent anticancer series of thiazolyl-pyrazoline derivatives acting as dual EGFR/VEGFR-2 inhibitors with significant proapoptotic properties. All the synthesised thiazolyl-pyrazolines have been screened for their inhibitory activities against EGFR and VEGFR-2 kinases and for their cytotoxic effect against nine cell lines derived from five tumour subpanels. Compounds 10b and 10d displayed potent and selective inhibitory activity towards EGFR-TK (IC50 = 40.7 ± 1.0 and 32.5 ± 2.2 nM, respectively) and VEGFR-2 (IC50 = 78.4 ± 1.5 and 43.0 ± 2.4 nM, respectively). Furthermore, compounds 10b and 10d showed a promising anticancer activity against non-small lung cell lines as they were the most potent derivatives with IC50 values equal 4.2 and 2.9 µM against A549 cells, and equal 4.8 and 3.8 µM against H441 cells, respectively. Moreover, our results indicated that 10b and 10d were much effective towards EGFR-mutated NSCLC cell lines (NCI-H1650 and NCI-H1975 cells) than gefitinib. Finally, compounds 10b and 10d induce G2/M cell cycle arrest and apoptosis and inhibit migration in A549 cancerous cells. Furthermore, the molecular docking study explored the binding mode and possible different interactions between the target compounds and the active sites of EGFR and VEGFR-2 enzymes. Accordingly, thiazolyl-pyrazoline scaffold can be considered as promising scaffold for further development of more potent dual EGFR and VEGFR-2 inhibitors.
4. Experimental
4.1. Chemistry
4.1.1. General
Melting points were measured with a Stuart melting point apparatus (Stuart Scientific, Redhill, UK) and were uncorrected. Infra-red (IR) spectra were recorded on FT-IR spectrometer using KBr discs (Perkin Elmer, Waltham, MA). Mass spectra (MS) were performed on a Varian electron impact EI mass spectrometer (EI-MS) at 70 eV at Regional Centre for Mycology and Biotechnology, Al-Azhar University utilising Thermo Scientific ISQ LT mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). NMR Spectra were recorded on a Bruker NMR spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) at Center for Drug Discovery Research and Development, Faculty of Pharmacy, Ain Shams University. 1H spectra were run at 400 MHz and 13C spectra were run at 100 MHz in deuterated dimethyl sulfoxide (DMSO-d6) or chloroform CDCl3. Chemical shifts are expressed in δ values (ppm) using the solvent peak as internal standard. All coupling constant (J) values are given in hertz. The abbreviations used are as follows: s, singlet; d, doublet; m, multiplet. Elemental analyses were carried out at the Regional Centre for Mycology and Biotechnology, Al-Azhar University utilising Thermo Scientific Flash 2000 elemental analyser (Thermo Fisher Scientific Inc., Waltham, MA). Analytical thin-layer chromatography (TLC) was employed routinely to follow the course of reactions and to check the purity of products using aluminium sheets pre coated with silica gel (Kieselgel, F254, pore size 60 Å, Merck, Darmstadt, Germany) and observed under a UV lamp (short-wavelength, 254 nm). All reagents and solvents were purified and dried by standard techniques.
4.1.2. General procedure for preparation of 1-(2,4-dichlorophenyl)-3-(aryl) prop-2-en-1-one 3a, b
To a solution of 2,4-dichloroacetophenone 1 (1.76 g, 9.3 mmol) and an appropriate aldehyde derivative 2a, b (9.3 mmol) in absolute ethanol (40 ml), 10% sodium hydroxide solution (15 ml) was added portion wise. The mixture was stirred for 4 h at room temperature. The separated precipitate was filtered, washed with water, dried and crystallised from ethanol to afford compounds 3a, b, respectively.
4.1.2.1. 1-(2,4-Dichlorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (3a)
Yellow powder in 87% yield, m.p. 102–103 °C (reported m.p. 106–107 °C60) IR (KBr, ν cm−1): 1653 (C=O); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.81 (s, 3H, OCH3), 6.99 (d, J = 7.6 Hz, 2H, Ar-H), 7.11 (d, J = 16.8 Hz, 1H, olefinic H), 7.38 (d, J = 15.2 Hz, 1H, olefinic H), 7.58 (s, 2H, Ar-H), 7.72–7.76 (m, 3H, Ar-H); MS (EI) m/z (%): 307.28 (M+, 9.98), 131.17 (100); Anal. Calcd. for C16H12Cl2O2 (307.17): C, 62.56; H, 3.94; Found; C, 62.50; H, 3.97%.
4.1.2.2. 1-(2,4-Dichlorophenyl)-3-(3,4-dimethoxyphenyl)prop-2-en-1-one (3b)
Yellow powder in 96% yield, m.p. 138–139 °C (reported m.p. 134–136 °C61) IR (KBr, ν cm−1): 1657 (C=O); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.81 (s, 6H, 2 (OCH3)), 7.01 (d, J = 8.4 Hz, 1H, Ar-H), 7.16 (d, J = 16 Hz, 1H, olefinic H), 7.31–7.41 (m, 3H, Ar-H + olefinic H), 7.58 (s, 2H, Ar-H), 7.78 (s, 1H, Ar-H); MS (EI) m/z (%): 337.14 (M+, 7.37), 83.13 (100); Anal. Calcd. for C17H14Cl2O3 (337.2): C, 60.55; H, 4.18; Found; C, 60.62; H, 4.13%.
4.1.3. General procedure for preparation of carbothioamide derivatives 5a, b
To a mixture of 3a, b (1 mmol) and thiosemicarbazide 4 (1 mmol) in absolute ethanol (50 ml), sodium hydroxide (0.29 g, 7 mmol) was added. The reaction mixture was heated under reflux with stirring for 6 h. After cooling, the formed product was filtered, washed with ethanol, dried and crystallised from ethanol to give the corresponding pyrazoline derivatives 5a, b, respectively.
4.1.3.1. 3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (5a)
Yellow powder in 70% yield, m.p. 168–169 °C (reported m.p. 175–178 °C62) IR (KBr, ν cm−1): 3418 and 3247 (NH2), 1599 (C = N), 1236 and 828 (C = S); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.18 (dd, J = 2.8, 16 Hz, 1H, HA), 3.73 (s, 3H, OCH3), 4.03 (dd, J = 12.6, 17.2 Hz, 1H, HM), 5.87 (dd, J = 3.2, 12 Hz, 1H, HX), 6.88 (d, J = 10.4 Hz, 2H, Ar-H), 7.08 (d, J = 8.4 Hz, 2H, Ar-H), 7.52 (dd, J = 2.4, 8.4 Hz, 1H, Ar-H), 7.71 (d, J = 1.6 Hz, 1H, Ar-H), 7.75 (s, 1H, NH, D2O exchangeable), 8.00 (d, J = 8 Hz, 1H, Ar-H), 8.13 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 45.7 (CH2 pyrazoline), 55.5 (OCH3), 63.0 (CH pyrazoline), 114.3 (2 C), 127.1 (2 C), 128.1, 129.3, 130.7, 132.7, 133.4, 135.2, 135.7, 153.3, 158.7, 177.0 (C=S); MS (EI) m/z (%): 380.16 (M+, 28.54), 182.72 (100), Anal. Calcd. for C17H15Cl2N3OS (380.29): C, 53.69; H, 3.98; N, 11.05; Found; C, 53.74; H, 3.95; N, 11.13%.
4.1.3.2. 3-(2,4-Dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazole-1-carbothioamide (5b)
Light yellow powder in 98% yield, m.p. 198–200 °C; IR (KBr, ν cm−1): 3444 and 3322 (NH2), 1581 (C=N), 1254 and 833 (C=S); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.19–3.23 (m, 1H, HA), 3.72 (s, 6H, 2OCH3), 3.99–4.07 (dd, J = 17.6, 11.6 Hz, 1H, HM), 5.86–5.89 (m, 1H, HX), 6.65 (d, J = 8.8 Hz, 1H, Ar-H), 6.80 (s, 1H, Ar-H), 6.88 (d, J = 7.6 Hz, 1H, Ar-H), 7.52 (d, J = 9.6 Hz, 1H, Ar-H), 7.71 (s, 1H, Ar-H), 7.76 (s, 1H, NH, D2O exchangeable), 7.96 (d, J = 9.6 Hz, 1H, Ar-H), 8.13 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 45.7 (CH2 pyrazoline), 55.9 (OCH3), 56.0 (OCH3), 63.3 (CH pyrazoline), 110.0, 112.3, 117.6, 128.1, 129.4, 130.7, 132.7, 133.4, 135.5, 135.7, 148.3, 149.2, 153.5 (C=N), 177.1 (C=S); MS (EI) m/z (%): 410 (M+, 24.38), 216.30 (100); Anal. Calculated for C18H17Cl2N3O2S (410): C, 52.69; H, 4.18; N, 10.24; Found; C, 52.73; H, 4.11; N, 10.29%.
4.1.4. General procedure for preparation of compounds 7a–7d
A mixture of carbothioamides 5a, b (1 mmol) and the appropriate 1-aryl-2-bromoethanone 6a, b (1.1 mmol) in absolute ethanol (20 ml) was heated under reflux for 4 h. After cooling, the formed precipitate was filtered and crystallised from ethanol to afford the corresponding compounds 7a–7d.
4.1.4.1. 2-(3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (7a)
Yellow powder in 55% yield, m.p. 72–75 °C; IR (KBr, ν cm−1): 1583 (C=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.38–3.44 (m, 1H, HA), 3.70 (s, 3H, OCH3), 4.05–4.12 (m, 1H, HM), 5.64 (s, 1H, HX), 6.91 (d, J = 8.8, 2H, Ar-H), 7.26–7.35 (m, 6H, 5 Ar-H and thiazole H) , 7.51 (d, J = 12 Hz, 1H, Ar-H), 7.69–7.79 (m, 4H, Ar-H); MS (EI) m/z (%): 480 (M+, 16.80), 76.10 (100); Anal. Calcd. for C25H19Cl2N3OS (480): C, 62.50; H, 3.99; N, 8.75; Found; C, 62.41; H, 4.17; N, 8.82%.
4.1.4.2. 4-(4-Chlorophenyl)-2-(3-(2,4-dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)thiazole (7b)
Yellow powder in 60% yield, m.p. 140–141 °C; IR (KBr, ν cm−1): 1583 (C=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.42–3.46 (m, 1H, HA), 3.72 (s, 3H, OCH3), 4.10 (dd, J = 17.6, 12 Hz, 1H, HM), 5.63 (dd, J = 14, 5.2 Hz, 1H, HX), 6.91 (d, J = 8.8 Hz, 2H, Ar-H), 7.34–7.42 (m, 5H, Ar-H and thiazole H), 7.53 (d, J = 10.4 Hz, 1H, Ar-H), 7.72–7.80 (m, 4H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 45.8 (CH2 pyrazoline), 55.5 (OCH3), 64.3 (CH pyrazoline), 106.0, 114.4 (2 C), 127.7 (2 C), 128.1, 128.5 (2 C), 129.0 (2 C), 129.2, 130.9, 132.2, 132.5, 132.9, 133.7, 133.7, 135.0, 149.8, 150.8, 159.2, 164.8; MS (EI) m/z (%): 514 (M+, 10.28), 89.34 (100); Anal. Calcd. for C25H18Cl3N3OS (514): C, 58.32; H, 3.52; N, 8.16; Found; C, 58.44; H, 3.76; N, 8.41%.
4.1.4.3. 2-(3-(2,4-Dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-phenylthiazole (7c)
Yellow powder in 69% yield, m.p. 120–122 °C; IR (KBr, ν cm−1): 1584 (C=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.50 (dd, J = 17.6, 8 Hz, 1H, HA), 3.72 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 4.12 (dd, J = 17.6, 12.4 Hz, 1H, HM), 5.64 (dd, J = 12.4, 7.2 Hz, 1H, HX), 6.92–7.09 (m, 3H, Ar-H), 7.26–7.38 (m, 4H, 3 Ar-H, thiazole H), 7.56 (dd, J = 9.2, 2.8 Hz, 1H, Ar-H), 7.76–7.78 (m, 3H, Ar-H), 7.82 (d, J = 8.8 Hz, 1H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 45.8 (CH2 pyrazoline), 55.9 (2OCH3), 64.7 (CH pyrazoline), 105.2, 111.6, 112.3, 119.2, 126.0 (2 C), 128.1, 128.2, 129.0, 129.4 (2 C), 130.8, 132.3, 132.9, 134.2, 134.9, 135.0, 148.8, 149.0, 150.8, 151.0, 164.77; MS (EI) m/z (%): 510 (M+, 15.80), 96.39 (100); Anal. Calcd. for C26H21Cl2N3O2S (510): C, 61.18; H, 4.15; N, 8.23; Found; C, 61.02; H, 4.39; N, 8.47%.
4.1.4.4. 4-(4-Chlorophenyl)-2-(3-(2,4-dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)thiazole (7d)
Yellow powder in 79% yield, m.p. 158–159 °C; IR (KBr, ν cm−1): 1584 (C=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.48 (dd, J = 15.2, 8 Hz, 1H, HA), 3.72 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 4.11 (dd, J = 14.8, 9.6 Hz, 1H, HM), 5.62 (dd, J = 15.2, 9.2 Hz, 1H, HX), 6.93 (s, 2H, Ar-H), 7.06 (s, 1H, Ar-H), 7.40–7.43 (m, 3H, 2Ar-H and thiazole H), 7.54 (d, J = 7.6 Hz, 1H, Ar-H), 7.75–7.81 (m, 4H, Ar-H); MS (EI) m/z (%): 544 (M+, 21.11), 218.49 (100); Anal. Calcd. for C26H20Cl3N3O2S (544): C, 57.31; H, 3.70; N, 7.71; Found; C, 57.53; H, 3.86; N, 7.89%.
4.1.5. General procedure for preparation of compounds 10a–10d
A mixture of carbothioamides 5a, b (1 mmol) and 3-chloropentane-2,4-diones 9a–9c (1.1 mmol) in absolute ethanol (30 ml) was heated under reflux for 4 h. After cooling, the formed precipitate was filtered and crystallised from ethanol to afford the corresponding compounds 10a–10d.
4.1.5.1. 1-(2-(3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methylthiazol-5-yl)ethan-1-one (10a)
Yellow powder in 72% yield, m.p. 205–207 °C; IR (KBr, ν cm−1): 1647 (C=O); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.38 (s, 3H, CH3), 2.40 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 4.15 (dd, J = 18.8, 11.6 Hz, 1H, HM) 5.70 (dd, J = 11.6, 5.6 Hz, 1H, HX), 6.90 (d, J = 10.4 Hz, 2H, Ar-H), 7.21 (d, J = 8.8 Hz, 2H, Ar-H), 7.55 (d, J = 8.8 Hz, 1H, Ar-H), 7.77 (s, 1H, Ar-H), 7.81 (d, J = 9.6 Hz, 1H, Ar-H); 13C NMR (DMSO-d6, 100 MHz) δ ppm: 19.1 (CH3), 30.1 (CH3), 46.1 (CH2 pyrazoline), 55.5 (OCH3), 63.1 (CH pyrazoline), 114.6 (2 C), 124.6, 127.8, 128.2, 128.8, 131.0, 132.5, 133.2, 133.3, 135.5, 153.4, 157.5, 159.2, 165.2 (aromatic carbons), 189.7 (C=O); MS (EI) m/z (%): 460 (M+, 15.52), 153.75 (100); Anal. Calcd. for C22H19Cl2N3O2S (460): C, 57.40; H, 4.16; N, 9.13; Found; C, 57.31; H, 4.30; N, 9.25%.
4.1.5.2. Ethyl-2-(3-(2,4-dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methylthiazole-5-carboxylate (10b)
Yellow powder in 63% yield, m.p. 192–195 °C; IR (KBr, ν cm−1): 1669 (C=O); 1H NMR (CDCl3, 400 MHz) δ ppm: 1.35 (t, J = 7.6 Hz, 3H, CH3), 2.54 (s, 3H, CH3), 3.53 (dd, J = 18.4, 8 Hz, 1H, HA), 3.81 (s, 3H, OCH3), 4.11 (dd, J = 20.4, 9.2 Hz, 1H, HM), 4.29 (q, J = 7.6 Hz, 2H, CH2), 5.90 (s, 1H, HX), 6.88 (d, J = 8.4 Hz, 2H, Ar-H), 7.27–7.29 (m, 2H, Ar-H), 7.34 (dd, J = 9.2, 3.6 Hz, 1H, Ar-H), 7.48 (d, J = 2.4 Hz, 1H, Ar-H), 7.85 (d, J = 7.6 Hz, 1H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 14.7 (CH3), 17.7 (CH3), 46.3 (CH2 pyrazoline), 55.5 (OCH3), 60.7 (CH2), 63.2 (CH pyrazoline), 111.4, 114.6, 127.8 (2 C), 128.2, 128.8, 131.0, 132.5, 133.1, 133.3, 135.5, 153.2, 159.2, 159.35, 162.2 (aromatic carbons), 165.3 (C=O); MS (EI) m/z (%): 490 (M+, 10.41), 55.10 (100); Anal. Calcd. for C23H21Cl2N3O3S (490): C, 56.33; H, 4.32; N, 8.57; Found; C, 56.60; H, 4.47; N, 8.78%.
4.1.5.3. 2-(3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-N-phenylthiazole-5-carboxamide (10c)
White powder in 74% yield, m.p. 195–197 °C; IR (KBr, ν cm−1): 3257 (NH), 1637 (C=O); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.36 (s, 3H, CH3), 3.35–3.40 (m, 1H, HA), 3.71 (s, 3H, OCH3), 4.15 (dd, J = 17.6, 12 Hz, 1H, HM) 5.68–5.71 (m, 1H, HX), 6.92 (d, J = 8.8 Hz, 2H, Ar-H), 7.06 (t, J = 8.8 Hz, 1H, Ar-H), 7.23 (d, J = 8.8 Hz, 2H, Ar-H), 7.30 (t, J = 8.8 Hz, 2H, Ar-H), 7.55 (d, J = 10.4 Hz, 1H, Ar-H), 7.62 (d, J = 8.8 Hz, 2H, Ar-H), 7.76 (s, 1H, Ar-H), 7.80 (d, J = 9.6 Hz, 1H, Ar-H), 9.72 (s, 1H, NH, D2O exchangeable); MS (EI) m/z (%): 537 (M+, 36.86), 245.37 (100); Anal. Calcd. for C27H22Cl2N4O2S (537): C, 60.34; H, 4.13; N, 10.42; Found; C, 60.49; H, 4.22; N, 10.68%.
4.1.5.4. 2-(3-(2,4-Dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-N-phenylthiazole-5-carboxamide (10d)
Buff powder in 50% yield, m.p. 166–167 °C; IR (KBr, ν cm−1): 3262 (NH), 1637 (C=O); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.38 (s, 3H, CH3), 3.72 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 4.14 (dd, J = 18.4, 12 Hz, 1H, HM), 5.68 (dd, J = 12, 5.6 Hz, 1H, HX), 6.78 (d, J = 7.6 Hz, 1H, Ar-H), 6.87–6.95 (m, 2H, Ar-H), 7.06 (t, J = 6.4 Hz, 1H, Ar-H), 7.30 (t, J = 7.2 Hz, 2H, Ar-H), 7.55 (d, J = 11.6 Hz, 1H, Ar-H), 7.63 (d, J = 7.2 Hz, 2H, Ar-H), 7.75–7.81 (m, 2H, Ar-H), 8.14 (s, 1H, NH, D2O exchangeable); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 18.2 (CH3), 46.1 (CH2 pyrazoline), 56.0 (2OCH3), 63.7 (CH pyrazoline), 110.7, 112.3, 116.5, 118.3, 120.8, 124.0 (2 C), 128.2, 129.0 (2 C), 130.9, 132.4, 133.0, 133.7, 135.3, 139.5, 148.7, 149.2, 152.3, 154.6, 160.9 (aromatic carbons), 163.8 (C=O); MS (EI) m/z (%): 567 (M+, 34.07), 299.67 (100); Anal. Calcd. for C28H24Cl2N4O3S (567): C, 59.26; H, 4.26; N, 9.87; Found; C, 59.40; H, 4.38; N, 9.82%.
4.1.6. General procedure for preparation of compounds 13a–13f
A mixture of carbothioamides 5a, b (1 mmol) and 2-oxo-N-arylpropanehydrazonoyl chlorides 12a–12c (1.1 mmol) in absolute ethanol (30 ml) was heated under reflux for 4 h. The produced product was filtered and washed with hot ethanol to give the corresponding compounds 13a–13f.
4.1.6.1. 2-(3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-5-(phenyldiazenyl)thiazole (13a)
Red powder in 50% yield, m.p. 168–170 °C; IR (KBr, ν cm−1): 1582 (N = N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.46–3.47 (m, 1H, HA), 3.74 (s, 3H, CH3), 4.19 (dd, J = 18, 12 Hz, 1H, HM), 5.81 (dd, J = 12.4, 5.6 Hz, 1H, HX), 6.93 (d, J = 6.8 Hz, 2H, Ar-H), 7.25 (d, J = 12 Hz, 2H, Ar-H), 7.38 (t, J = 6 Hz, 1H, Ar-H), 7.48 (t, J = 7.6 Hz, 2H, Ar-H), 7.56 (d, J = 7.6 Hz, 1H, Ar-H), 7.69 (d, J = 7.6, 2H, Ar-H), 7.78 (s, 1H, Ar-H), 7.85 (d, J = 9.6 Hz, 1H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 16.5 (CH3 of thiazole), 46.3 (CH2 pyrazoline), 55.6 (OCH3), 63.0 (CH pyrazoline), 114.7 (2 C), 122.1 (2 C), 127.8 (2 C), 128.2 (2 C), 128.6, 129.8, 131.1 (2 C), 132.7, 133.2, 133.3, 135.7, 141.4, 152.6, 154.3, 158.3, 159.3, 165.0; MS (EI) m/z (%): 524 (M+ + 2, 1.17), 522 (M+, 4.86), 40.16 (100); Anal. Calcd. for C26H21Cl2N5OS (522): C, 59.77; H, 4.05; N, 13.40; Found; C, 59.86; H, 4.13; N, 13.62%.
4.1.6.2. 2-(3-(2,4-Dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-5-(p-tolyldiazenyl)thiazole (13b)
Red powder in 80% yield, m.p. 170–171 °C; IR (KBr, ν cm−1): 1582 (N=N); 1H NMR (CDCl3, 400 MHz) δ ppm: 2.42 (s, 3H, CH3), 2.61 (s, 3H, CH3), 3.53 (dd, J = 18, 7.2 Hz, 1H, HA), 3.81 (s, 3H, OCH3), 4.12 (dd, J = 19.2, 9.6 Hz, 1H, HM), 5.77–5.80 (m, 1H, HX), 6.89 (d, J = 9.2 Hz, 2H, Ar-H), 7.23–7.29 (m, 4H, Ar-H), 7.32 (dd, J = 9.2, 2 Hz, 1H, Ar-H), 7.47 (d, J = 2.8 Hz, 1H, Ar-H), 7.67 (d, J = 6.4 Hz, 2H, Ar-H), 7.90 (d, J = 10 Hz, 1H, Ar-H); 13 C NMR (CDCl3, 100 MHz) δ ppm: 16.1 (CH3 of thiazole), 21.4 (CH3), 46.2 (CH2 pyrazoline), 55.3 (OCH3), 63.5 (CH pyrazoline), 114.3 (2 C), 122.0 (2 C), 127.3 (2 C), 127.5 (2 C), 128.7, 129.6, 130.8 (2 C), 131.4, 132.6, 133.5, 136.3, 139.4, 142.2, 150.9, 152.3, 159.4, 164.8; MS (EI) m/z (%): 536 (M+, 32.38), 110.11 (100); Anal. Calcd. for C27H23Cl2N5OS (536): C, 60.45; H, 4.32; N, 13.05; Found; C, 60.31; H, 4.49; N, 13.28%.
4.1.6.3. 5-((4-Chlorophenyl)diazenyl)-2-(3-(2,4-dichlorophenyl)-5-(4-methoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methylthiazole (13c)
Red powder in 80% yield, m.p. 170–171 °C; IR (KBr, ν cm−1): 1582 (N=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.44 (dd, J = 16.8, 6.4 Hz, 1H, HA), 3.73 (s, 3H, CH3), 4.18 (dd, J = 16, 8 Hz, 1H, HM), 5.80 (dd, J = 9.6, 6 Hz, 1H, HX), 6.93 (d, J = 7.2 Hz, 2H, Ar-H), 7.24 (d, J = 9.6 Hz, 2H, Ar-H), 7.51 (d, J = 8.8 Hz, 2H, Ar-H), 7.55 (d, J = 8 Hz, 1H, Ar-H), 7.68 (d, J = 8 Hz, 2H, Ar-H), 7.77 (s, 1H, Ar-H), 7.84 (d, J = 7.6 Hz, 1H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 16.6 (CH3), 46.3 (CH2 pyrazoline), 55.6 (OCH3), 63.0 (CH pyrazoline), 114.7 (2 C), 120.2, 123.6 (2 C), 127.8, 128.3, 128.5, 129.8, 131.1, 132.7 (2 C), 133.1, 133.3, 133.8, 135.8, 141.3, 151.3, 154.7, 159.3, 159.4, 165.3; MS (EI) m/z (%): 556 (M+, 35.77), 311.62 (100); Anal. Calcd. for C26H20Cl3N5OS (556): C, 56.07; H, 3.62; N, 12.58; Found; C, 56.20; H, 3.88; N, 12.61%.
4.1.6.4. 2-(3-(2,4-Dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-5-(phenyldiazenyl)thiazole (13d)
Reddish orange powder in 50% yield, m.p. 187–188 °C; IR (KBr, ν cm−1): 1587 (N=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 3.44–3.50 (m, 1H, HA), 3.72 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.18 (dd, J = 17.2, 11.6 Hz, 1H, HM), 5.80 (dd, J = 11.2, 5.6 Hz, 1H, HX), 6.79 (d, J = 6.4 Hz, 1H, Ar-H), 6.91–6.96 (m, 2H, Ar-H), 7.37 (t, J = 7.6 Hz, 1H, Ar-H), 7.48 (t, J = 8.4 Hz, 2H, Ar-H), 7.55 (d, J = 8.8 Hz, 1H, Ar-H), 7.68 (d, J = 8.4 Hz, 2H, Ar-H), 7.76 (s, 1H, Ar-H), 7.83 (d, J = 7.6 Hz, 1H, Ar-H); 13 C NMR (DMSO-d6, 100 MHz) δ ppm: 16.4 (CH3 of thiazole), 46.2 (CH2 pyrazoline), 55.9 (2OCH3), 63.3 (CH pyrazoline), 110.5 (2 C), 112.3, 118.1, 122.1 (2 C), 128.3 (2 C), 129.8, 131.0 (2 C), 132.6, 133.2 (2 C), 133.4, 135.8 (2 C), 148.8, 149.2 (2 C), 152.6, 165.1; MS (EI) m/z (%): 552 (M+, 15.69), 329.74 (100); Anal. Calcd. for C27H23Cl2N5O2S (552): C, 58.70; H, 4.20; N, 12.68; Found; C, 58.92; H, 3.37; N, 12.68%.
4.1.6.5. 2-(3-(2,4-Dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methyl-5-(p-tolyldiazenyl)thiazole (13e)
Reddish orange powder in 73% yield, m.p. 176–177 °C; IR (KBr, ν cm−1): 1589 (N=N); 1H NMR (CDCl3, 400 MHz) δ ppm: 2.41 (s, 3H, CH3), 2.59 (s, 3H, CH3), 3.55 (dd, J = 18, 4 Hz, 1H, HA), 3.87 (s, 6H, 2OCH3), 4.10 (dd, J = 17.2, 13.2 Hz, 1H, HM), 5.73 (dd, J = 13.2, 4 Hz, 1H, HX), 6.83–6.86 (m, 3H, Ar-H), 7.22–7.34 (m, 3H, Ar-H), 7.46 (s, 1H, Ar-H), 7.66 (d, J = 9.6 Hz, 2H, Ar-H), 7.87 (d, J = 8.8 Hz, 1H, Ar-H); 13 C NMR (CDCl3, 100 MHz) δ ppm: 16.1 (CH3), 21.4 (CH3), 46.1 (CH2 pyrazoline), 55.9 (OCH3), 56.0 (OCH3), 63.7 (CH pyrazoline), 109.4, 111.4, 118.1, 122.0 (2 C), 127.5, 128.7, 129.6 (2 C), 130.8, 131.3, 133.0, 133.5, 136.3, 139.4, 148.8, 149.2, 150.9, 152.3, 156.5, 164.8; MS (EI) m/z (%): 566 (M+, 16.00), 46.07 (100); Anal. Calcd. for C28H25Cl2N5O2S (566): C, 59.36; H, 4.45; N, 12.36; Found; C, 59.46; H, 4.38; N, 12.59%.
4.1.6.6. 5-((4-Chlorophenyl)diazenyl)-2-(3-(2,4-dichlorophenyl)-5-(3,4-dimethoxyphenyl)-4,5-dihydro-1H-pyrazol-1-yl)-4-methylthiazole (13f)
Orange powder in 70% yield, m.p. 190–191 °C; IR (KBr, ν cm−1): 1586 (N=N); 1H NMR (DMSO-d6, 400 MHz) δ ppm: 2.53 (s, 3H, CH3), 3.48 (dd, J = 17.6, 5.6 Hz, 1H, HA), 3.73 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.19 (dd, J = 19.6, 12.8 Hz, 1H, HM), 5.81 (dd, J = 12, 5.6 Hz, 1H, HX), 6.78 (d, J = 7.6 Hz, 1H, Ar-H), 6.92–6.96 (m, 2H, Ar-H), 7.53 (d, J = 9.2 Hz, 2H, Ar-H), 7.57 (d, J = 7.6 Hz, 1H, Ar-H), 7.71 (d, J = 9.2 Hz, 2H, Ar-H), 7.79 (d, J = 3.2 Hz, 1H, Ar-H), 7.85 (d, J = 10 Hz, 1H, Ar-H); MS (EI) m/z (%): 586 (M+, 55.20), 458.78 (100); Anal. Calcd. for C27H22Cl3N5O2S (586): C, 55.25; H, 3.78; N, 11.93; Found; C, 55.43; H, 3.89; N, 12.14%.
4.2. Biological evaluation
4.2.1. Cell culture and reagents
Human leukaemia (K562 and KG-1a), breast (MCF-7, BT-549 and HCC70), lung (A549, H441, NCI-H1650 and NCI-H1975), colon (HCT116), liver (HepG2) cancer cell lines and WI-38 human lung fibroblast cell line were obtained from the American Type Culture Collection (ATCC; Manassas, VA) and were cultured in their suitable media containing 10% foetal bovine serum (FBS; Sigma-Aldrich) in a humidified atmosphere with 5% CO2 at 37 °C. Gefitinib and vandetanib were purchased from Sigma-Aldrich. All chemicals used in this study were analytical or cell-culture grade.
4.2.2. Kinase inhibition assay
The EGFR kinase assay was carried out in 96-well plates coated with PGT (poly l-glutamic acid l-tyrosine, 4:1, Sigma Aldrich, St. Louis, MO) as previously described63. IC50 values were calculated using GraphPad Prism 5. Each experiment was carried out at least three times. The VEGFR-2 kinase assay was carried out in 96-well streptavidin coated plate using recombinant human VEGFR-2/KDR ELISA kit according to manufacturer’s instructions. Moreover, the IC50 values of 10b and 10d on 11 different kinases (TEK, SYK, EPHB2, ABL1, LCK, CLK1, ROCK1, PKC, AKT1, CDK1 and CDK5) were determined using Z'-LYTE® technology which is based on FRET (Invitrogen/Life Technologies).
4.2.3. Western blot analysis
The protein extraction and quantification were done as previously described64.
4.2.4. Anti-proliferative assay
Cytotoxicity was measured using the MTT assay. Human leukaemia (K562 and KG-1a), breast (MCF-7, BT-549 and HCC70), lung (A549, H441, NCI-H1650 and NCI-H1975), colon (HCT116), liver (HepG2) cancer cell lines and WI-38 human lung fibroblast cell line were plated at a density of 1 × 104 cells per well in 96-well plates overnight and then treated with the compounds. After 24 h treatment, 20 μL of MTT solution (2 mg/mL in phosphate-buffered saline [PBS]) was added to each well and the cells were cultured for another 4 h at 37 °C. The medium was aspirated and 150 μL DMSO was added to solubilise MTT formazan crystals. The plates were then shaken, and the optical density was determined at 570 nm using an ELISA plate reader (Model 550, Bio-Rad, Hercules, CA). At least three independent experiments were performed. The IC50 values were calculated using GraphPad Prism 5 (Version 5.01, GraphPad Software, San Diego, CA).
4.2.5. Cell cycle and apoptosis analyses
DNA content was measured using 7-aminoactinmycin D (7AAD) (Biotium, Inc., Hayward, CA) staining and is commonly used for cell cycle phase analysis as previously described65. Apoptosis was evaluated using the annexin V/propidium iodide (PI) staining kit (BioLegend, San Diego, CA) according to the manufacturer’s instructions and as previously described.
4.3. Molecular docking
In this work, all the docking studies were conducted using Molecular Operating Environment (MOE 2019.02) software67,68. The crystal structures of EGFR in complex with erlotinib and VEGFR-2 in complex with sorafenib were downloaded from the protein databank PDB IDs 1M17 and 4ASD, respectively. All the receptors and ligands were prepared using the default parameters of MOE software. The binding sites in the two targets were determined by selecting the pocket surrounding the binding domain of the co-crystallised ligands. Prior commencing the docking of compounds 10b and 10d, a pose-retrieval docking experiments for the X-ray coordinates of the co-crystallised ligands in their corresponding binding sites was carried out. The RMSD values between the co-crystallised pose and the docking pose from the previous step were 0.85 and 0.63 Å for erlotinib and sorafenib, respectively. After the positive indication on the docking validity, compounds 10b and 10d were docked into the two receptors using the same conditions. Finally, the results of the docking were visualised using the 2 D and 3 D interaction diagrams generated by MOE.
4.4. Scratch wound healing assay
A549 cells were seeded and incubated in 6-well cell culture plates till reaching confluence. Then, the confluent monolayers were scratched to form a “wound” using a sterile needle. The cells were treated with either vehicle as a control, 10b or 10d for 36 h. The images were recorded at 0 and 36 h to monitor the migration of cells into the wounded area using a light photomicroscope.
4.5. Statistical analysis
Data were presented as the means ± standard error of mean (SEM). Student’s t-test was performed to determine the statistical significance compared to the vehicle treated control, gefitinib or vandetanib. Statistical significance was defined as *p < 0.05, **p < 0.01 or ***p < 0.001. Data are representative of three independent experiments.
Supplementary Material
Funding Statement
Amer Ali Abd El-Hafeez was supported by an NIH-funded Cancer Therapeutics Training Program [CT2, T32 CA121938] and Pradipta Ghosh by the NIH [CA238042, CA100768 and CA160911].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Howard HA. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol 2013;71:829–42. [DOI] [PubMed] [Google Scholar]
- 2.Dela Cruz CS, Tanoue LT, Matthay RA.. Lung cancer: epidemiology, etiology, and prevention. Clin Chest Med 2011;32:605–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wee P, Wang Z.. “Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) 2017;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roskoski R. The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun 2004;319:1–11. [DOI] [PubMed] [Google Scholar]
- 5.Pines G, Köstler WJ, Yarden Y.. Oncogenic mutant forms of EGFR: lessons in signal transduction and targets for cancer therapy. FEBS Lett 2010;584:2699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheng Q, Liu J.. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br J Cancer 2011;104:1241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdelsalam EA, Zaghary WA, Amin KM, et al. . Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg Chem 2019;89:102985. [DOI] [PubMed] [Google Scholar]
- 8.Gupta R, Dastane AM, Forozan F, et al. . Evaluation of EGFR abnormalities in patients with pulmonary adenocarcinoma: the need to test neoplasms with more than one method. Mod Pathol 2009;22:128–33. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, Yang PL, Gray NS.. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39. [DOI] [PubMed] [Google Scholar]
- 10.Tebbutt N, Pedersen MW, Johns TG.. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer 2013;13:663–73. [DOI] [PubMed] [Google Scholar]
- 11.Koudelakova V, Kneblova M, Trojanec R, et al. . Non-small cell lung cancer - genetic predictors. Biomed Pap 2013;157:125–36. [DOI] [PubMed] [Google Scholar]
- 12.Maemondo M, Inoue A, Kobayashi K, et al. . Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N Engl J Med 2010;362:2380–8. [DOI] [PubMed] [Google Scholar]
- 13.Rosell R, Carcereny E, Gervais R, et al. . Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46. [DOI] [PubMed] [Google Scholar]
- 14.Pao W, Miller V, Zakowski M, et al. . EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci 2004;101:13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niederst MJ, Engelman JA.. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal 2013;6:re6–re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ko B, Paucar D, Halmos B.. EGFR T790M: revealing the secrets of a gatekeeper. Lung Cancer Targets Ther 2017;8:147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li D, Ambrogio L, Shimamura T, et al. . BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008;27:4702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelman JA, Zejnullahu K, Gale CM, et al. . PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007;67:11924–32. [DOI] [PubMed] [Google Scholar]
- 19.Jackson PA, Widen JC, Harki DA, Brummond KM.. Covalent modifiers: a chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via Hetero-Michael addition reactions. J Med Chem 2017;60:839–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yun C-H, Mengwasser KE, Toms AV, et al. . The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci 2008;105:2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reckamp KL, Giaccone G, Camidge DR, et al. . A phase 2 trial of dacomitinib (PF‐00299804), an oral, irreversible pan‐HER (human epidermal growth factor receptor) inhibitor, in patients with advanced non–small cell lung cancer after failure of prior chemotherapy and erlotinib. Cancer 2014;120:1145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ou S-H, Soo R.. Dacomitinib in lung cancer: a ‘lost generation’ EGFR tyrosine-kinase inhibitor from a bygone era? Drug Des Dev Ther 2015;9:5641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camidge DR, Pao W, Sequist V.. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 2014;11:473–81. [DOI] [PubMed] [Google Scholar]
- 24.Tan C-S, Kumarakulasinghe NB, Huang Y-Q, et al. . Third generation EGFR TKIs: current data and future directions. Mol Cancer 2018;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murtuza A, Bulbul A, Shen JP, et al. . Novel third-generation EGFR tyrosine kinase inhibitors and strategies to overcome therapeutic resistance in lung cancer. Cancer Res 2019;79:689–98. [DOI] [PubMed] [Google Scholar]
- 26.Yu HA, Tian SK, Drilon AE, et al. . Acquired resistance of EGFR-mutant lung cancer to a T790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol 2015;1:982–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thress KS, Paweletz CP, Felip E, et al. . Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat Med 2015;21:560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y-C, Zhou Q, Wu Y-L.. Clinical management of third-generation EGFR inhibitor-resistant patients with advanced non-small cell lung cancer: current status and future perspectives. Cancer Lett 2019;459:240–7. [DOI] [PubMed] [Google Scholar]
- 29.Montor WR, Salas AROSE ,de Melo FHM.. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: the current arsenal of inhibitors. Mol Cancer 2018;17:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000;5:3–10. [DOI] [PubMed] [Google Scholar]
- 31.Pennell NA, Lynch TJ.. Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist 2009;14:399–411. [DOI] [PubMed] [Google Scholar]
- 32.Le X, Nilsson M, Goldman J, et al. . Dual EGFR-VEGF pathway inhibition: a promising strategy for patients with EGFR-mutant NSCLC. J Thorac Oncol 2021;16:205–15. [DOI] [PubMed] [Google Scholar]
- 33.Tortora G, Ciardiello F, Gasparini G.. Combined targeting of EGFR-dependent and VEGF-dependent pathways: rationale, preclinical studies and clinical applications. Nat Clin Pract Oncol 2008;5:521–30. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar S, Mazumdar A, Dash R, et al. . ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol Ther 2010;9:592–603. [DOI] [PubMed] [Google Scholar]
- 35.Deng X, Tan X, An T, et al. . Synthesis, characterization, and biological activity of a novel series of benzo[4,5]imidazo[2,1-b]thiazole derivatives as potential epidermal growth factor receptor inhibitors. Molecules 2019;24:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahmoud HK, Farghaly TA, Abdulwahab HG, et al. . Novel 2-indolinone thiazole hybrids as sunitinib analogues: design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity. Eur J Med Chem 2020;208:112752. [DOI] [PubMed] [Google Scholar]
- 37.Amin KM, Barsoum FF, Awadallah FM, Mohamed NE.. Identification of new potent phthalazine derivatives with VEGFR-2 and EGFR kinase inhibitory activity. Eur J Med Chem 2016;123:191–201. [DOI] [PubMed] [Google Scholar]
- 38.Faria JV, Vegi PF, Miguita AGC, et al. . Recently reported biological activities of pyrazole compounds. Bioorg Med Chem 2017;25:5891–903. [DOI] [PubMed] [Google Scholar]
- 39.El-Husseiny WM, El-Sayed MAA, Abdel-Aziz NI, et al. . Synthesis, antitumour and antioxidant activities of novel α,β-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J Enzyme Inhib Med Chem 2018;33:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed MF, Santali EY, El-Haggar R.. Novel piperazine–chalcone hybrids and related pyrazoline analogues targeting VEGFR-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J Enzyme Inhib Med Chem 2021;36:307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alkamaly OM, Altwaijry N, Sabour R, Harras MF.. Dual EGFR/VEGFR2 inhibitors and apoptosis inducers: synthesis and antitumor activity of novel pyrazoline derivatives. Arch Pharm 2021;354:2000351. [DOI] [PubMed] [Google Scholar]
- 42.Viveka S, Shama DP, Nagaraja GK, et al. . Design and synthesis of some new pyrazolyl-pyrazolines as potential anti-inflammatory, analgesic and antibacterial agents. Eur J Med Chem 2015;101:442–51. [DOI] [PubMed] [Google Scholar]
- 43.Kaplancıklı ZA, Özdemir A, Turan-Zitouni G, et al. . New pyrazoline derivatives and their antidepressant activity. Eur J Med Chem 2010;45:4383–7. [DOI] [PubMed] [Google Scholar]
- 44.Beyhan N, Kocyigit-Kaymakcioglu B, Gümrü S, Aricioglu F.. Synthesis and anticonvulsant activity of some 2-pyrazolines derived from chalcones. Arab J Chem 2017;10:S2073–S2081. [Google Scholar]
- 45.Bano S, Alam MS, Javed K, et al. . Synthesis, biological evaluation and molecular docking of some substituted pyrazolines and isoxazolines as potential antimicrobial agents. Eur J Med Chem 2015;95:96–103. [DOI] [PubMed] [Google Scholar]
- 46.Wang H-H, Qiu K-M, Cui H-E, et al. . Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives containing benzodioxole as potential anticancer agents. Bioorg Med Chem 2013;21:448–55. [DOI] [PubMed] [Google Scholar]
- 47.Lv P-C, Li D-D, Li Q-S, et al. . Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents. Bioorg Med Chem Lett 2011;21:5374–7. [DOI] [PubMed] [Google Scholar]
- 48.Yuan J-W, Wang S-F, Luo Z-L, et al. . Synthesis and biological evaluation of compounds which contain pyrazole, thiazole and naphthalene ring as antitumor agents. Bioorg Med Chem Lett 2014;24:2324–8. [DOI] [PubMed] [Google Scholar]
- 49.George RF, Samir EM, Abdelhamed MN, et al. . Synthesis and anti-proliferative activity of some new quinoline based 4,5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg Chem 2019;83:186–97. [DOI] [PubMed] [Google Scholar]
- 50.Mohamed TK, Batran RZ, Elseginy SA, et al. . Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg Chem 2019;85:253–73. [DOI] [PubMed] [Google Scholar]
- 51.Elzahabi HSA, Nossier ES, Alasfoury RA, et al. . Design, synthesis, and anti-cancer evaluation of new pyrido[2,3-d]pyrimidin-4(3H)-one derivatives as potential EGFRWT and EGFRT790M inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem 2022;37:1053–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Warhi T, El Kerdawy AM, Said MA, et al. . Novel 2-(5-Aryl-4,5-dihydropyrazol-1-yl)thiazol-4-one as EGFR inhibitors: synthesis, biological assessment and molecular docking insights. Drug Des Dev Ther 2022;16:1457–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yousef RG, Ibrahim A, Khalifa MM, et al. . Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies. J Enzyme Inhib Med Chem 2022;37:1389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taghour MS, Elkady H, Eldehna WM, et al. . Design and synthesis of thiazolidine-2,4-diones hybrids with 1,2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: in-vitro anticancer evaluation and in-silico studies. J Enzyme Inhib Med Chem 2022;37:1903–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hagras M, Saleh MA, Ezz Eldin RR, et al. . 1,3,4-Oxadiazole-naphthalene hybrids as potential VEGFR-2 inhibitors: design, synthesis, antiproliferative activity, apoptotic effect, and in silico studies. J Enzyme Inhib Med Chem 2022;37:386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sivakumar PM, Prabhakar PK, Doble M.. Synthesis, antioxidant evaluation, and quantitative structure-activity relationship studies of chalcones. Med Chem Res 2011;20:482–92. [Google Scholar]
- 57.Sharifzadeh B, Mahmoodi NO, Mamaghani M, et al. . Facile regioselective synthesis of novel bioactive thiazolyl-pyrazoline derivatives via a three-component reaction and their antimicrobial activity. Bioorg Med Chem Lett 2013;23:548–51. [DOI] [PubMed] [Google Scholar]
- 58.Altıntop MD, Özdemir A, Turan-Zitouni G, et al. . A novel series of thiazolyl–pyrazoline derivatives: synthesis and evaluation of antifungal activity, cytotoxicity and genotoxicity. Eur J Med Chem 2015;92:342–52. [DOI] [PubMed] [Google Scholar]
- 59.Yao HC, Resnick P.. Azo-hydrazone conversion. I. The Japp-Klingemann Reaction. J Am Chem Soc 1962;84:3514–7. [Google Scholar]
- 60.Thirunarayanan G, Thirumurthy K, Vanangamudi G, et al. . Fly-ash : water catalyzed greener synthesis and insect antifeedant activities of some chalcones. Elixir Org Chem 2012;45:7898–905. [Google Scholar]
- 61.Narender T, Papi Reddy K.. A simple and highly efficient method for the synthesis of chalcones by using borontrifluoride-etherate. Tetrahedron Lett 2007;48:3177–80. [Google Scholar]
- 62.Mubeen M, Kini SG, Pai KSR.. Design, synthesis, antioxidant and anticancer activity of novel pyrazole derivatives. Der Pharm Chem 2015;7:215–23. [Google Scholar]
- 63.Fathi MAA, Abd El-Hafeez AA, Abdelhamid D, et al. . “1,3,4-Oxadiazole/chalcone hybrids: design, synthesis, and inhibition of leukemia cell growth and EGFR, Src, IL-6 and STAT3 activities. Bioorg Chem 2019;84:150–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fathy Abdel-Wahab B, Sediek A, Ahmed Mohamed H, Elsayed Ahmed Awad G.. Novel 2-pyrazolin-1-ylthiazoles as potential antimicrobial agents. Lett Drug Des Discov 2012;10:111–8. [Google Scholar]
- 65.Vignon C, Debeissat C, Georget M-T, et al. . Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS One 2013;8:e68425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abd El-Hafeez AA, Fujimura T, Kamei R, et al. . A methoxyflavanone derivative from the Asian medicinal herb (Perilla frutescens) induces p53-mediated G2/M cell cycle arrest and apoptosis in A549 human lung adenocarcinoma. Cytotechnology 2018;70:899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vilar S, Cozza G, Moro S.. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem 2008;8:1555–72. [DOI] [PubMed] [Google Scholar]
- 68.Scholz C, Knorr S, Hamacher K, Schmidt B.. DOCKTITE—a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J Chem Inf Model 2015;55:398–406. [DOI] [PubMed] [Google Scholar]
- 69.El Hassab MA, Ibrahim TM, Al-Rashood ST, et al. . In silico identification of novel SARS-COV-2 2′-O-methyltransferase (nsp16) inhibitors: structure-based virtual screening, molecular dynamics simulation and MM-PBSA approaches. J Enzyme Inhib Med Chem 2021;36:727–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.