

Abstract
Background & aims:
Chronic hepatitis C viral (HCV) infection is a leading etiologic driver of cirrhosis and ultimately hepatocellular carcinoma (HCC). Of the approximately 71 million individuals chronically infected with HCV, 10–20% are expected to develop severe liver complications in their lifetime. Epigenetic mechanisms including DNA methylation and histone modifications become profoundly disrupted in disease processes including liver disease.
Methods:
To understand how HCV infection influences the epigenome and whether these events remain as ‘scars’ following cure of chronic HCV infection, we mapped genome-wide DNA methylation, four key regulatory histone modifications (H3K4me3, H3K4me1, H3K27ac, and H3K27me3) and open chromatin by ATAC-seq in parental and HCV-infected immortalized hepatocytes and the Huh7.5 HCC cell line, along with DNA methylation and gene expression analyses following elimination of HCV in these models through treatment with interferon-α or a direct-acting antiviral (DAA).
Results:
Our data demonstrate that HCV infection profoundly impacts the epigenome (particularly enhancers), HCV shares epigenetic targets with interferon-α targets, an overwhelming majority of epigenetic changes induced by HCV remain as ‘scars’ on the epigenome following viral cure Similar findings are observed in primary human patient samples cured of chronic HCV infection. Supplementation of interferon-α/DAA antiviral regimens with DNA methyltransferase inhibitor 5-aza-2’deoxycytidine synergizes in reverting aberrant DNA methylation induced by HCV. Finally, both HCV-infected and cured cells displayed a blunted immune response, demonstrating a functional effect of epigenetic scarring.
Conclusions:
Integration of epigenetic and transcriptional data elucidates key gene deregulation events driven by HCV infection and how this may underpin the long-term elevated risk for HCC in patients cured of HCV due to epigenome scarring.
Keywords: Epigenomics, DNA methylation, histone code, hepatitis C, sustained virologic response, carcinoma, hepatocellular
Introduction
Hepatitis C Virus (HCV) infection is a global pandemic affecting more than 180 million people(1). Chronic HCV infection is a major etiologic driver for liver cirrhosis that leads to a 15–20-fold increased risk of progression to hepatocellular carcinoma (HCC), with a 1–3% transformation rate over 30 years(2). The introduction of direct-acting antivirals (DAAs) has increased optimism for eradicating HCV, yet sustained virologic response (SVR) does not always reverse accumulated liver damage including steatosis and fibrosis. While elevated risk for HCC is reduced, it is not eliminated in patients following SVR suggesting that viral infection leads to long-term genetic and/or epigenetic damage in the liver that is not reversed following SVR(3).
Epigenetic marks, including DNA methylation and histone modifications, collaborate to influence gene expression and ultimately manifest cellular phenotypes. DNA methylation (5mC) is driven by three members of the DNA methyltransferase (DNMT) family of proteins: DNMT1, DNMT3A, and DNMT3B. These enzymes are critical regulators of normal homeostasis and gene expression via the marks they write at enhancers and promoters. Regulation of histone modifications is considerably more complex with respect to the machinery involved, with multiple histone readers, writers, and erasers working together to modulate gene expression. Specific combinations of histone marks collaborate to partition the genome into repressive (e.g. H3K27me3) or activating/enhancer (e.g. H3K27ac/H3K4me1) epigenomic states(4).
Cirrhosis, considered the key pre-neoplastic risk state of HCC, is typically a decades-long process involving chronic hepatitis regulated by both genetic and epigenetic mechanisms. DNA methylation changes preceding HCC are thought to be drivers of the disease, characterized by genome-wide hypomethylation and locus-specific hypermethylation(5, 6). Moreover, specific DNA methylation patterns have been linked to grade, stage, and survival outcomes, suggesting these defects are mechanistically involved(7). Histone modifications have also been intimately linked to cancer initiation and progression. Expression of the H3K27me3 methyltransferase EZH2 correlated with grade and outcome in liver cancer(8). Identifying alterations in different facets of the epigenetic landscape is essential to defining initiation and progression mechanisms of liver disease, while simultaneously providing novel drug targets.
To better define how chronic HCV infection impacts the epigenome, and determine to what extent pharmacologic cure of HCV reverts HCV-induced changes, we profiled DNA methylation, histone modifications, and gene expression in two isogenic cell line models of chronic HCV infection and cure with a DAA or high-dose interferon (IFN) treatment. Our results demonstrate that HCV profoundly impacts the epigenome, both at the level of DNA methylation and histone modifications, and that these marks collaborate to mediate gene expression changes. HCV-induced epigenetic changes are largely unaffected by HCV cure, leaving persistent alterations that perpetuate disrupted gene transcription programs. Our data also pinpoint genes central to pathogenic processes linked to cirrhosis and HCC that may serve as therapeutic targets in an HCV-cure scenario. Primary liver samples from patients that achieved SVR following DAA treatment mirror in vitro findings, segregating samples into HCV-naïve and HCV-infected groups based on DNA methylation profiles. Interestingly, much of the HCV infection-induced epigenetic landscape is shared with that of interferon treatment; IFN-treated DNA methylation and expression landscapes phenocopy HCV infection and are linked to key biological pathways including apoptosis. Significantly, treatment with a DNA methylation inhibitor reverses most of the pathologic epigenetic landscape, suggesting that such agents could act as adjuvants to further reduce risk for HCC following SVR.
Methods
Cell lines and patient samples
Two human liver-derived cell lines were used in this study to model the effects of HCV infection and SVR in the absence of confounding immune cell infiltration: Hu1545 and Huh7.5 (Supplemental Figs. S1–3). Additional details about Hu1545/Huh7.5 derivation and characterization are in the Supplemental Methods.
Normal liver samples were obtained post-mortem from the National Disease Research Interchange (NDRI), while primary patient diseased liver samples were obtained from Rutgers University (IRB# Pro20160000683) from liver transplant surgeries (Supplemental Table S1). All HCV-positive patients had been treated with Harvoni (ledipasvir/sofosbuvir), achieved SVR, and were determined to be HCV-negative for >1.5 years before undergoing liver transplantation. Primary normal liver samples were confirmed HCV negative by qRT-PCR.
DNA methylation
Genomic DNA was isolated from cell lines and primary human tissues by standard procedures. All samples were run in duplicate on the Infinium MethylationEPIC BeadChip arrays (850k) and were processed as previously described using ‘minfi’ v1.30.0(9, 10). Unless otherwise noted, a change in absolute methylation level of 10% (Δβ > |0.1|) and a false discovery adjusted p-value of <0.05 were considered significant. All array data has been deposited in NCBI GEO (GSE168186).
Gene expression
RNA-seq was performed as previously described in duplicate(6). The resulting data was processed using Salmon v1.4.0(11) for quantification and ‘DESeq2’ v1.24.0(12) for downstream analysis. Expression levels were confirmed for a subset of genes (Supplemental Table S2) using qRT-PCR (Supplemental Figs. S4–6).
Histone modifications and chromatin accessibility
ChIP-seq was performed as previously described(6) using the following antibodies: H3K4me1 (ab8896, Abcam), H3K27ac (ab4729, Abcam), H3K4me3 (ab8580, Abcam), and H3K27me3 (#9733, CST). Peaks were called using SICER and MACS. ATAC-seq was performed using standard protocols(13) and sequenced/processed as for ChIP-seq. Locus-specific ChIP-qPCR (Supplemental Table S2) confirmation is shown in Supplemental Figs. S5–6.
Software and statistics
Gene ontology was performed using Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA). A pruned 12 state (from 10–15 states) ChromHMM model was used to construct histone modification state models, as previously described(6). Statistical comparisons were considered significant below p<0.05 with adjustment for multiple testing where applicable.
Results
Isogenic model systems of chronic HCV infection and HCV cure
To define the role of hepatitis C viral infection in altering the epigenome and transcriptome, we made use of two isogenic hepatocyte-derived models, Hu1545 (surrogate normal) and Huh7.5 (HCC) as described in the supplemental methods and Supplemental Fig. S1. To model the epigenetic changes after a patient is cured of HCV infection (SVR), we electroporated full-length HCV JFH-1 RNA into these lines and selected with G418 for two weeks. Stable HCV replication was confirmed using qRT-PCR and immunofluorescence for HCV NS5A, and stably replicating clones isolated (Supplemental Fig. S3). Long-term culture (>6 months) demonstrated sustained HCV replication status. We then treated each HCV-replicating cell line with either interferon-α (IFN) or a direct-acting antiviral (DAA; telaprevir) and confirmed complete loss of HCV (cured condition, Supplemental Fig. S3). Parental cell lines were similarly treated with IFN or telaprevir to control for drug effects independent of HCV. Cell lines were also treated with the DNA demethylating agent 5-aza-2’-deoxycytidine (5-azadC) (Supplemental Fig. S1) to examine reversibility of 5mC changes. Five primary human liver samples with SVR for greater than 1.5 years (average 3.3) were also profiled. Isogenic cell lines and primary tissues were subjected to DNA methylation analysis using the Illumina MethylationEPIC BeadChip (850k), gene expression by RNA-seq, histone modifications by ChIP-seq, and chromatin accessibility by ATAC-seq, as indicated in Supplemental Fig. S1.
HCV infection leads to global DNA hypomethylation
Principal component analysis showed that stable HCV replication redefined the DNA methylation landscape in both Hu1545 (Fig. 1A, Supplemental Table S3) and Huh7.5 cells (Supplemental Fig. S7). The primary separation was driven by HCV replication, independent of drug treatment, while interferon treatment of parental cells lacking HCV also significantly altered the epigenome. Importantly, replicates for each of the cell lines and drug treatments clustered together, demonstrating reproducibility of treatments and epigenetic profiling methods (Figs. 1A–B).
Fig. 1: HCV infection leads to genome-wide loss of DNA methylation and locus-specific hypermethylation.
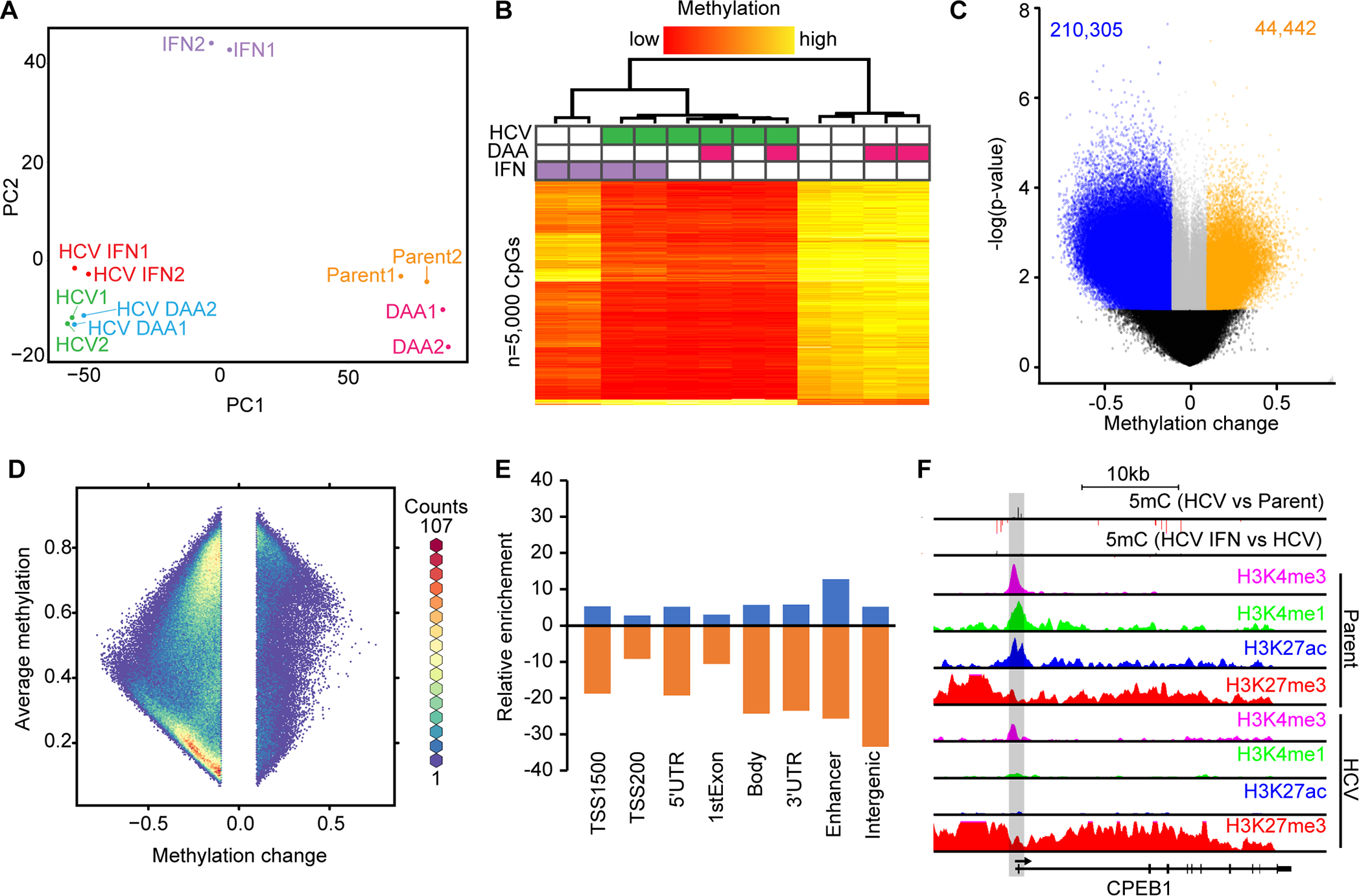
(A) PCA of Hu1545 and its derivatives used in this study. Each condition has two biological replicates. (B) Unsupervised hierarchical clustering of the 5,000 most variable CpGs across the six biological conditions indicated (in duplicate). A color bar is shown. (C) A volcano plot of DNA methylation changes against significance in HCV-infected Hu1545 cells compared to parent. Statistically significant hypermethylation (orange) and hypomethylation (blue) events are shown. (D) A hexbin plot of changes in methylation in HCV infected cells against the average methylation of all samples (Hu1545 Parent + HCV). (E) Bar plot depicting the distribution of HCV-induced DNA methylation changes across features on the 850k array. Enhancers are regions marked by both H3K4me1 and H3K27ac. (F) UCSC browser view of a representative locus (CPEB1) demonstrating promoter hypermethylation in HCV-infected cells. The second track depicts methylation changes induced by interferon treatment in HCV-infected cells. Also shown are histone modifications by ChIP-seq for H3K4me3, H3K27ac, H3K4me1, and H3K27me3 in parent (top four) and HCV (bottom four) Hu1545 cells.
We then compared HCV infected cells with each corresponding parental cell line to identify CpGs altered by persistent active HCV infection (Fig. 1C, Huh7.5 in Supplemental Figs. S7B–C). Globally, DNA hypomethylation events dominated HCV-induced 5mC changes with approximately five times as many hypomethylated (n=210,305) as hypermethylated CpGs (n=44,442) at a methylation change of 10% or greater (Supplemental Table S3). The general hypomethylation phenotype in the presence of HCV replication is consistent in the Huh7.5 cell model, albeit to a lesser extent (Supplemental Table S3). This reduced effect is likely due to the fact that Huh7.5 cells were previously infected with HCV and cured, thus they may already manifest epigenetic scarring (Supplemental Table S3, Supplemental Fig. S7D).
Within the global hypomethylation associated with HCV infection, a significant portion of changes involved lowly methylated CpG sites (~10–20% methylation level) in parental cells becoming completely unmethylated in isogenic HCV infected cells (Fig. 1D). We expect that these fully demethylated sites are functionally relevant for gene expression, especially those that reside in key regulatory regions like promoters and enhancers (Fig. 1E; enhancers are defined as H3K4me1+H3K27ac without H3K4me3). Further examination of these fully demethylated CpGs revealed that they were enriched in regions with characteristics of enhancers in parental Hu1545 cells. One potential mechanism underpinning the global loss of methylation is through alterations in the DNA methylation machinery. Indeed, we observe significant downregulation of DNMT1 and DNMT3A in HCV-infected cells (Supplemental Fig. S6A). We identified many genes showing consistent changes across multiple CpGs within their regulatory regions such as CPEB1 (Fig. 1F), ACSL1, CYP27A1, and APOC1 (Supplemental Fig. S5), which become aberrantly methylated following infection with HCV, and lead to altered expression of the gene. Altogether, HCV infection induced significant genome-wide changes in DNA methylation with enrichment in regulatory regions.
HCV infection leads to a globally altered gene expression profile
As shown in the principal component analysis in Fig. 2A, active HCV replication significantly alters global transcriptional profiles. Comparable to observations from DNA methylation, samples clustered based on whether they had been infected with HCV or were HCV naïve (with the caveat that Huh7.5 cells have been previously infected with HCV and cured, Fig. 2B). Consistent with genome-wide hypomethylation events being enriched in regulatory regions, HCV infection largely resulted in upregulation of gene expression (Figs. 2C–D).Gene ontology demonstrated that infection with HCV affects pathways linked to “cancer”, “organ injury”, and “gastrointestinal disease”, among others (Fig. 2E). Many genes also display corresponding changes at the epigenome level, such as CYP27A1, whose promoter becomes hypermethylated accompanied by decreased gene expression following HCV infection (Fig. 2F, Supplemental Fig. S5B). Indeed, there is a relatively strong negative correlation between gene expression and DNA methylation in promoter regions in Hu1545 cells, and a strong positive correlation in gene body associated CpGs and expression (Supplemental Fig. S8). Thus, HCV infection impacts the hepatocyte at both the epigenetic and transcriptional levels, and a significant subset of altered DNA methylation events lead to transcriptional changes.
Fig. 2: HCV infection induces upregulation of disease-relevant pathways in immortalized liver cells.
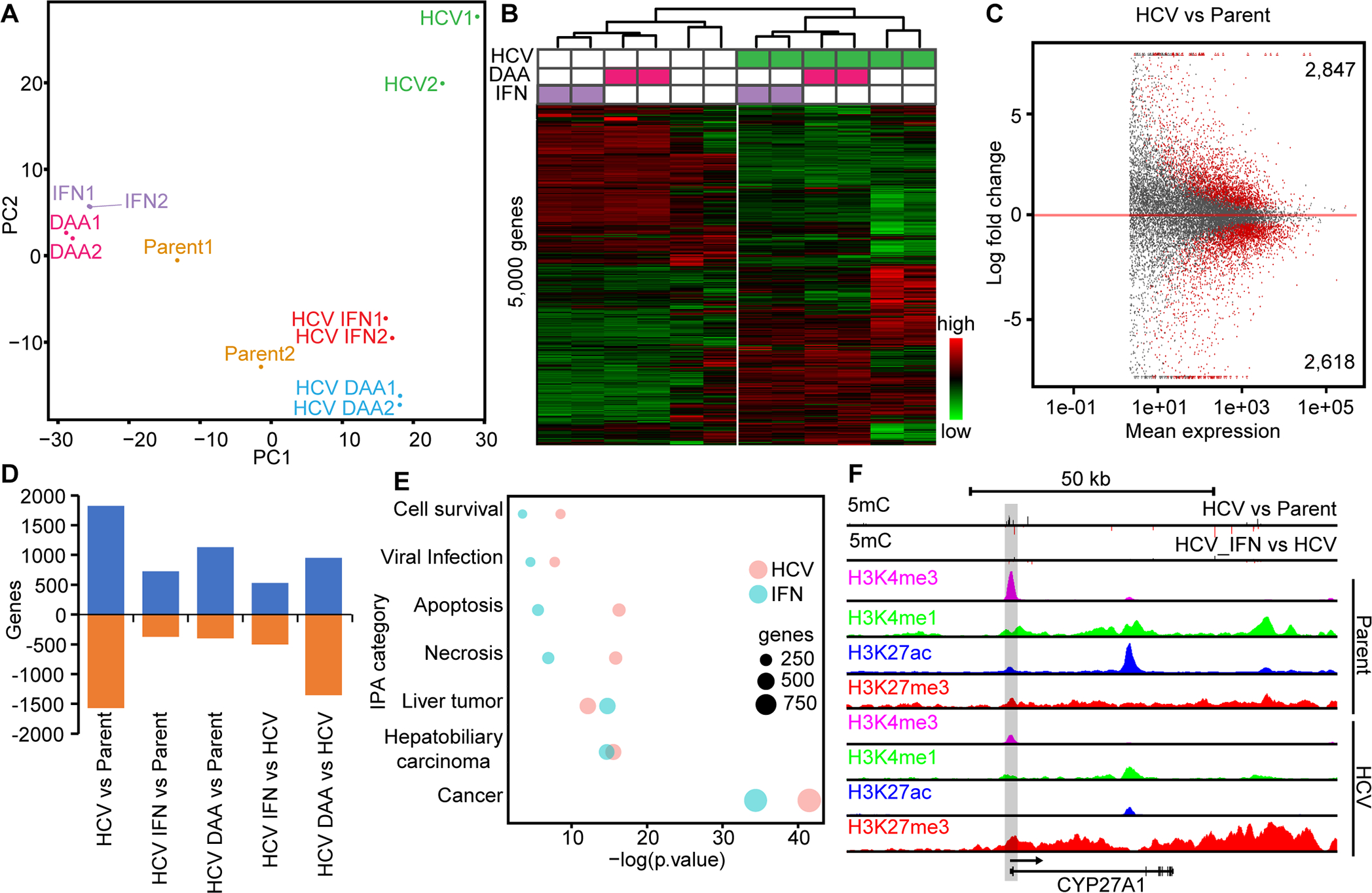
(A) PCA of Hu1545 and its derivatives assayed by RNA-seq. Each condition has two biological replicates. (B) Unsupervised hierarchical clustering of the 5,000 most variable genes across the six biological conditions (in duplicate). A color bar is shown. (C) An MA plot of gene expression changes against mean expression in HCV-infected Hu1545 cells compared to parent. Significant events are colored in red (adjusted p-value <0.05). (D) A bar chart depicting the changes in gene expression between paired treatments of Hu1545 with HCV, DAA, Interferon, or a combination. (E) Ingenuity pathway analysis summary of disease and biological pathways impacted by gene expression changes induced by HCV infection (red) or IFN treatment (blue) of Hu1545 cells. The top 1,000 most differentially expressed genes were used for each group (FDR <0.05, log2FC > |1|). (F) UCSC browser shot of the CYP27A1 locus that is downregulated in Hu1545-HCV cells with a corresponding decrease in H3K4me3 (promoter mark) and increase in promoter hypermethylation. Histone modifications by ChIP-seq are shown for H3K4me3, H3K27ac, H3K4me1, and H3K27me3 in parent (top four) and HCV (bottom four) Hu1545 cells.
Deregulation of enhancers in HCV infection
We profiled four histone modifications (H3K4me3 –promoter, H3K4me1 - enhancer, H3K27ac –promoter/enhancer activation, and H3K27me3 – repression) by ChIP-seq, and chromatin accessibility indicative of active regulatory regions by ATAC-seq. Examining H3K4me1 and H3K27ac, we observed an increase in the total number of enhancer and activating mark peaks (active enhancers) in HCV-infected cells relative to the parental cell line, and a concomitant decrease in repressive H3K27me3 genome-wide (Fig. 3A). This is consistent with the global increase in gene expression unveiled by RNA-seq and with the modest increase in total peaks obtained from ATAC-seq (Figs. 2, 3A). While the total number of H3K4me1 and H3K27ac peaks increases in HCV-infected cells, a cohort of regions whose epigenetic marks do not change in parental with respect to HCV-infected cells is also observed (Fig. 3B). We also identify regions marked by H3K4me1 in parental cells that become more robustly marked with H3K27ac in HCV-infected cells (Fig. 3C, clusters 1 and 3). By contrast, H3K4me3 is relatively consistent in terms of peak number between parent and HCV-infected cells, suggesting that enhancers play a larger role in modulating gene expression than promoters.
Fig. 3: Enhancers are enriched targets for epigenetic changes induced by HCV.
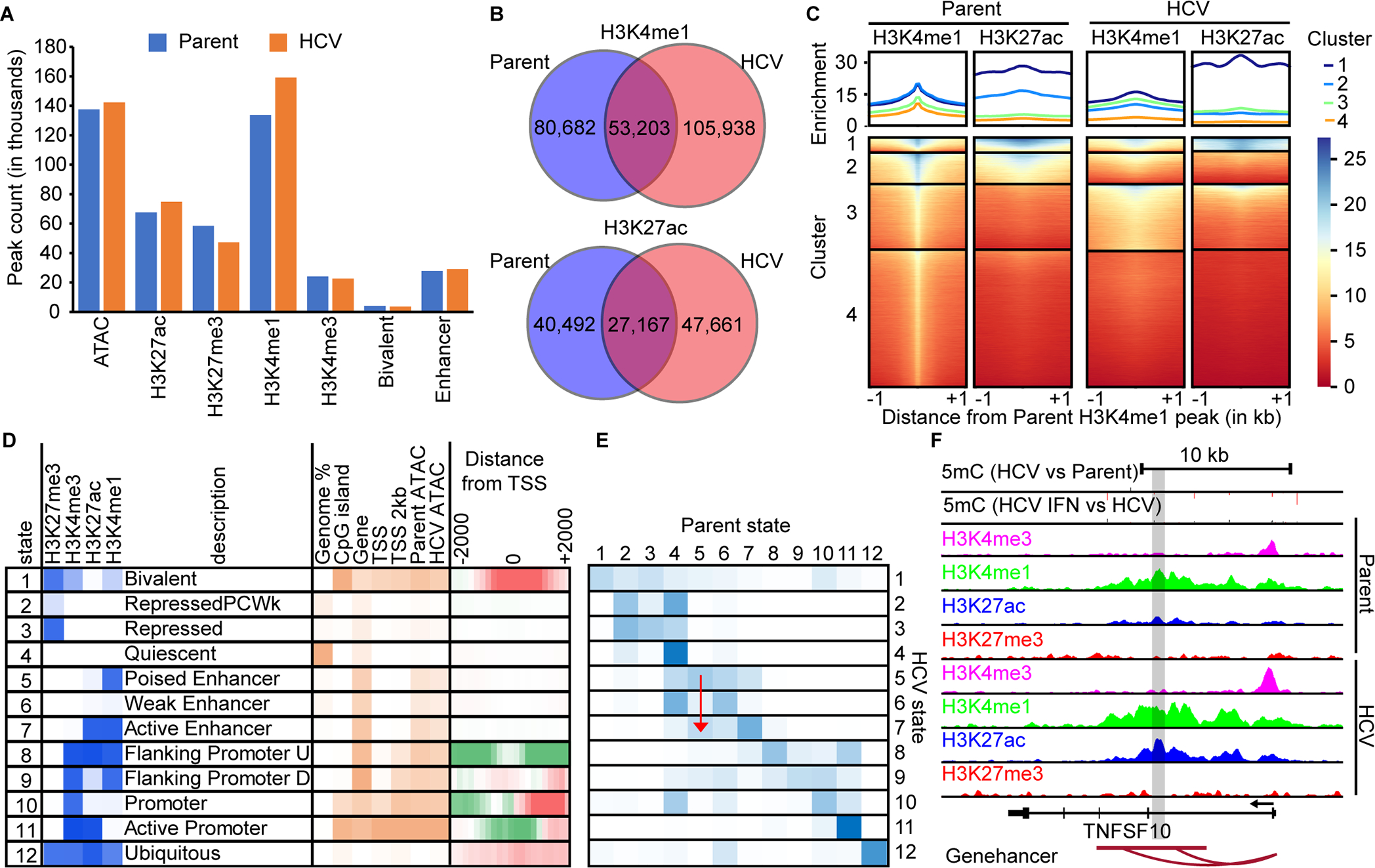
(A) A bar chart of MACS called peaks for H3K4me3, H3K4me1, H3K27ac, and H3K27me3. (B) Venn diagrams demonstrating the overlap between H3K4me1 and H3K27ac peaks in parent versus HCV infected Hu1545 cells. (C) A deeptools heatmap of enhancer regions defined by combined parent and HCV peak summits for H3K4me1. Four clusters are shown based on the combination of histone marks across parent (left two panels) and HCV (right two panels). Higher enrichment for the noted histone mark is colored in blue. (D) ChromHMM modeling of four histone marks with twelve states with a description of their regulatory function. Darker blue represents higher enrichment of that mark within the called state. (E) A heatmap displaying the changes in ChromHMM states between parent and HCV Hu1545 cells in the called states in part D. The blue color represents high conservation between the given states. A red arrow is shown to highlight the transition from State 5 (poised enhancer) in the parental line to State 7 (active enhancer) in HCV-infected cells. (F) A UCSC browser shot of the TNSF10 locus demonstrating a gain of active enhancers (as denoted by both gains in H3K4me1 and H3K27ac as well as the Genehancer annotation) identified by ChromHMM.
Due to the number of potential combinations of histone modifications, and the need to integrate them with expression and DNA methylation, we employed ChromHMM to distill histone modifications genome-wide into specific combinations most prevalent in our cell lines and of functional relevance. Using data from both parent and HCV-infected cells, we derived a twelve-state model using H3K4me3, H3K4me1, H3K27ac, and H3K27me3 (Fig. 3D). These states correspond to well-defined regulatory classes including bivalent (State 1), active enhancers (State 7), and active promoters (State 11). Of note, the generated states are consistent with expected epigenetic and transcriptional interactions, such as low 5mC and high expression in Hu1545 cells in regions corresponding to state 11 (active enhancer, Supplemental Fig. S9). Using this model, we compared the genome-wide distribution of states between HCV-infected and naïve cells to understand how the epigenome responds to HCV infection. As can be seen (Fig. 3D), much of the epigenome (~66% of the genome), remains unchanged between parent and HCV-infected Hu1545 cells. As for specific states, we observe higher conservation between the parent and HCV-infected lines in promoter-linked states (e.g. 69% of state 11), while enhancer related states are more dynamic (e.g. 41% of state 7, Fig. 3D). Comparison of parental and HCV Hu1545 cells in the vicinity of the transcription start site shows a significant depletion of bivalent regions (State 1) and a corresponding increase in active promoters (State 11, Fig. 3D). Many regions of the genome display altered configurations of epigenetic marks between parent and HCV-infected cells (Fig. 3E). These regions are linked to genes like TNFSF10 (Fig. 3F), which transitions from State 5 (poised enhancer) to State 7 (active enhancer) by gaining H3K27ac with a corresponding increase in gene transcription (red arrow in Fig. 3E).
HCV cure does not revert aberrant epigenetic and transcriptional changes
Residual epigenetic changes induced by HCV but not reversed following cure could continue to influence expression programs even in the absence of active HCV infection, and potentially contribute to long-term elevated risk for HCC in this setting. Therefore, we treated HCV-infected Hu1545 cells with either interferon or DAA to fully ablate HCV from the cultures (Figs. 4A–C, Supplemental Figs. S1, S3A–B). Both DNA methylation and gene expression programs (Figs. 1A–B, Figs. 2A–B, respectively) segregate into HCV-naïve and HCV-infected groups for the Hu1545 cell model. Interestingly, while treatment of HCV-infected cells with either IFN or DAA induced DNA methylation changes, the hyper- and hypo-methylation events were of significantly lower frequency and magnitude than those caused by HCV infection itself (e.g. top left quadrant indicates hypermethylation reversal, while middle left is hypermethylated even following drug treatment; Fig. 4A). This suggests that cells robustly retain an epigenetic memory of prior HCV infection. Moreover, at the level of gene expression, samples further clustered (independent of the anti-HCV treatment) into two sub-trees consisting of either IFN/DAA or no-treatment, indicating that interferon and DAA have overlapping targets (Fig. 2B). Taken together these data demonstrate that attainment of HCV cure is not sufficient to restore Hu1545 cells to their baseline transcriptional and epigenetic programs, and that HCV leaves a pathologic ‘scar’ on the epigenome that continues to influence transcription.
Fig. 4: Epigenetic marks coordinate to leave scars following HCV infection in immortalized human liver cells.
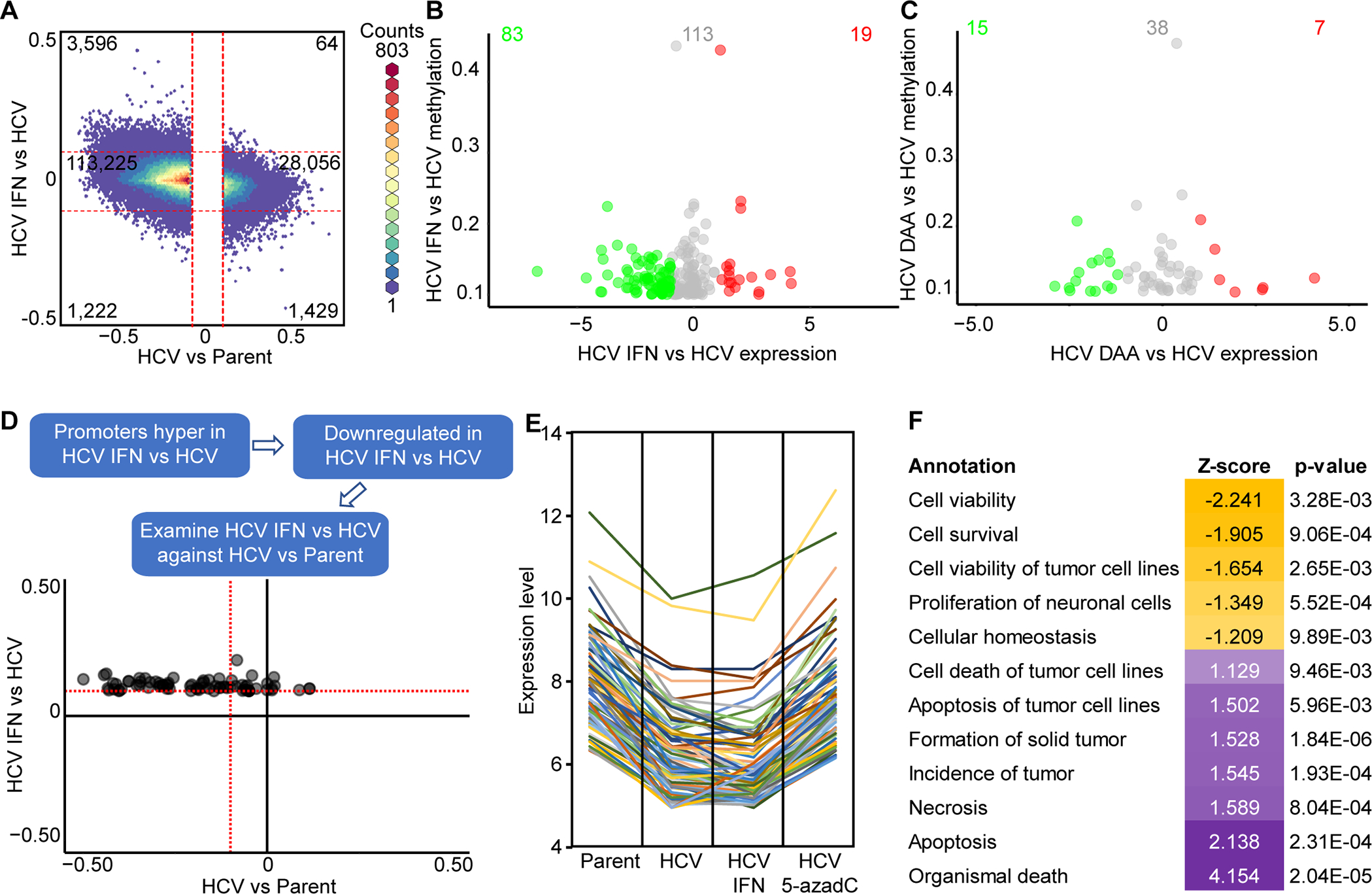
(A) A hexbin plot of changes induced by HCV and subsequent changes induced by clearance of HCV by IFN. Scatterplot of DNA methylation changes against gene expression in promoter regions induced by IFN (B) or DAA (C) treatment of HCV infected Hu1545 cells. (D) A scatterplot of IFN-induced DNA methylation changes and how they behave with HCV infection. CpGs were selected based on IFN-driven methylation change and corresponding change in expression (from C). (E) Spaghetti plot of genes downregulated in Hu1545 HCV cells that remain repressed following IFN treatment but are re-expressed following 5-azadC treatment. (F) Gene ontology of the 80 genes identified in (E) that are upregulated by 5-azadC treatment, demonstrating enrichment for apoptotic and tumorigenic terms.
As shown in Fig. 4A, DNA methylation changes induced by HCV infection are predominantly unaffected by IFN treatment (or DAA treatment, Supplemental Fig. S10). There are, however, a subset of CpGs altered by either IFN or DAA-treatment in HCV-infected cells, and these HCV cure-associated reversal events are enriched in regulatory regions associated with genes whose expression is also reversed upon cure (Fig. 4B, Supplemental Fig. S10B–C). In the proximal promoter region, hypermethylation in IFN-treated HCV cells was linked to downregulation (Fig. 4B; DAA in Supplemental Fig. S10C), suggesting that most of these IFN/DAA-induced changes have a functional impact on expression. Importantly, this set of 5mC changes were chiefly reversal events (Fig. 4D, Supplemental Fig. S10D) rather than new or worsening aberrant DNA methylation changes.
As a significant portion of 5mC alterations induced by chronic HCV-infection remain following HCV elimination with two drugs that act through distinct mechanisms against HCV, we asked whether treatment with DNA methyltransferase inhibitor 5-azadC could revert aberrant methylation in HCV-infected cells and return them closer to the infection naive state (Supplemental Fig. S11). Chronic HCV-infection induced 78,464 hypermethylation events at a threshold of 10% or greater (Δβ > 0.1). Treatment with interferon alone reversed 5,239 of those events, or just under 7%. However, treatment with IFN followed by 5-azadC reversed methylation at 36,606 of the HCV-scarred CpGs (~47%). Interestingly, a similar trend is observed for hypomethylation events, with 5-azadC treatment having an even greater effect on methylation scar reversal than interferon treatment (14.2% vs 1.3%, Supplemental Fig. S11). Treatment with DAA showed a very similar phenotype as IFN, reversing <15% of HCV infection-induced hypermethylation events (Supplemental Fig. S11). As shown in Fig. 4E, a distinct cohort of genes is downregulated in HCV infected cells that remain unchanged following HCV cure, but which become re-expressed with 5-azadC treatment. Indeed, many of these genes are linked to apoptosis and cell viability pathways, suggesting they are therapeutically relevant targets (Fig. 4F). Importantly, we maintained cells post-HCV cure from both the IFN and DAA-treated groups for six months in culture to determine if more epigenetic events reverted with time. While there was a modest number of additional reversion events during this period, the majority of HCV-induced epigenetic scars remained after more than half a year post-HCV cure and continuous growth (>80% for both IFN and DAA, data not shown). Altogether, this data shows that most chronic HCV-infection induced epigenetic scars become permanent fixtures within the epigenome that exert an ongoing functional impact on gene expression. These scars therefore have implications for disease pathogenesis and long-term risk for HCC. They further indicate that use of 5-azadC as an adjuvant therapy in individuals cured of their chronic HCV infection may be beneficial for mitigating HCC risk by promoting greater scar reversal and/or restoring apoptotic capacity to previously HCV infected cells.
Primary human tissue data corroborates cell line findings
Patients who achieve SVR following chronic HCV infection remain at elevated risk for disease progression, including HCC(3). As such, we examined whether our findings derived from cell culture models translated into patients that were functionally cured of HCV, typically prior to liver transplant. We profiled DNA methylation in four normal livers (no cancer, no cirrhosis/fibrosis), 5 cirrhotic patients with SVR, and 9 cirrhotic patients with active HCV infection. Based on DNA methylation alone, normal liver segregates from patients with active or previous HCV infection on PC1, while PC2 separates HCV-positive from HCV-negative liver (Fig. 5A). When we select CpGs that are significantly different between HCV SVR tissues and active HCV, HCV SVR samples cluster more closely with normal liver, demonstrating that at least a subset of CpGs are reversed with DAAs (Fig. 5B). Since we cannot examine epigenetic scarring in the same manner as with cell lines within patients because do not have pre- and post-SVR samplings from the same individual, we integrated our Hu1545 cell line model. Using all CpGs that were driven by HCV infection in Hu1545 (n=131,408), we observed that normal liver clustered independently from HCV SVR primary tissue (Fig. 5C). Moreover, when we selected for the most robust changes (>20% methylation change) in our cell line model and primary tissue, we identified a cohort of CpGs that retain a signature of HCV infection despite DAA-induced SVR in these patients for >1.5 years (Fig. 5D). This finding indicates that hundreds of genes remain epigenetically scarred in primary tissue. One gene of interest, ACSL1, was identified as a target of HCV epigenetic scarring in both the cell line model and primary tissue, showing acquisition of both robust promoter hypermethylation and a repressive chromatin state with HCV infection (Fig. 5E). TCGA LIHC data for ACSL1 demonstrated that hypermethylation of one of the same CpGs scarred in primary and cultured cells is significantly associated with poor HCC patient outcome, and a similar trend is observed for downregulated expression of this gene in HCC patients (Fig. 5F). Taken together, this data demonstrates that a multifactorial epigenetic landscape coordinates to alter the expression of disease-relevant genes in HCV, and this signature does not revert following SVR.
Fig. 5: Primary human SVR liver tissues reflect observations in cell culture models.
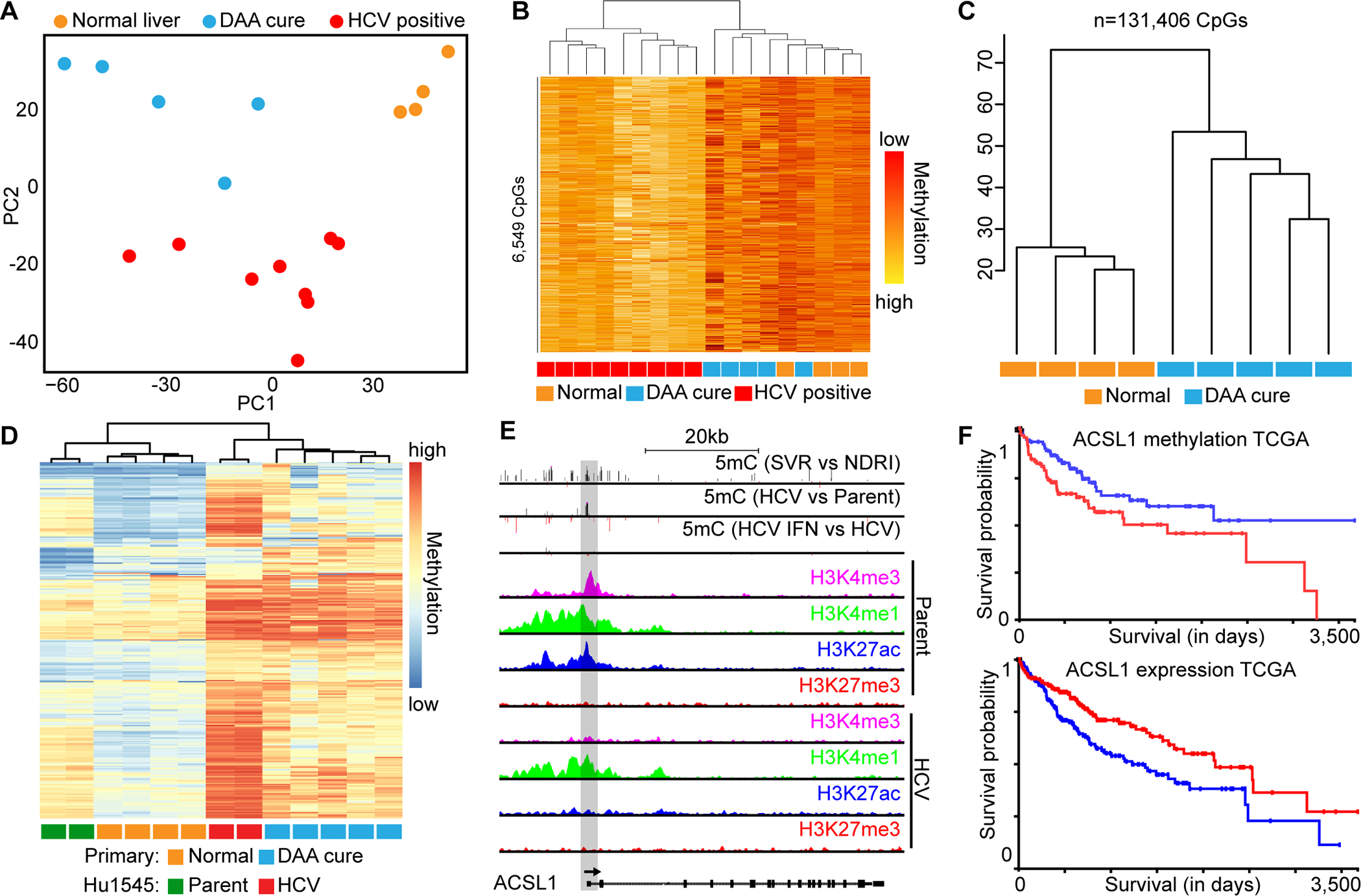
(A) PCA plot depicting separation of normal primary liver tissue from cirrhotic (PC1) and HCV-positive from HCV-negative tissues (PC2). (B) Hierarchical clustering of differentially methylated CpGs between HCV SVR and actively HCV infected primary liver tissue. Group membership, where orange is normal liver, red is HCV-positive, and blue are HCV-SVR primary patient samples, is shown. (C) Hierarchical clustering of primary normal liver and HCV SVR patient samples based on 131,406 CpGs differentially methylated between parental and HCV infected Hu1545 cells. (D) Supervised hierarchical clustering of robustly hypermethylated CpGs in HCV infection of cell lines and epigenetic scars in primary tissue. (E) A UCSC browser shot of ACSL1, a gene derived from D, showing promoter hypermethylation in HCV SVR primary liver versus normal liver tissue, as well as in HCV vs parent Hu1545 cells. Also shown are histone modifications in parental and HCV infected Hu1545 cells for H3K4me3, H3K4me1, H3K27ac, and H3K27me3. (F) Kaplan-Meier survival curves for DNA methylation (top) and gene expression (bottom) from TCGA LIHC data for ASCL1.
Signatures of HCV infection overlap with interferon treatment
Observations derived from both DNA methylation and gene expression landscapes hinted at overlapping patterns between chronic HCV infection and interferon treatment of HCV-naive Hu1545 cells (Figs. 1A–B). Indeed, the majority of DNA methylation changes (>85%) were shared between HCV-infected and interferon-treated parental Hu1545 cells (Fig. 6A, Huh7.5-based results in Supplemental Fig. S12A). A similar relationship was observed for gene expression (Fig. 6B, Supplemental Fig. S12B). Genes upregulated in both HCV and interferon settings also display an enrichment for hypomethylation in their promoters, indicating the expression changes are due, at least in part, to 5mC changes driven by these two ‘exposures’. Unsupervised hierarchical clustering demonstrates that HCV infection and IFN treatment conditions segregate independently of untreated or DAA-treated cells (Fig. 6C). Altogether, this finding suggests that HCV infection to a major extent phenocopies the methylation changes induced by interferon treatment of HCV naive hepatocytes.
Fig. 6: HCV infection phenocopies IFN treatment at the transcriptomic and epigenomic levels.
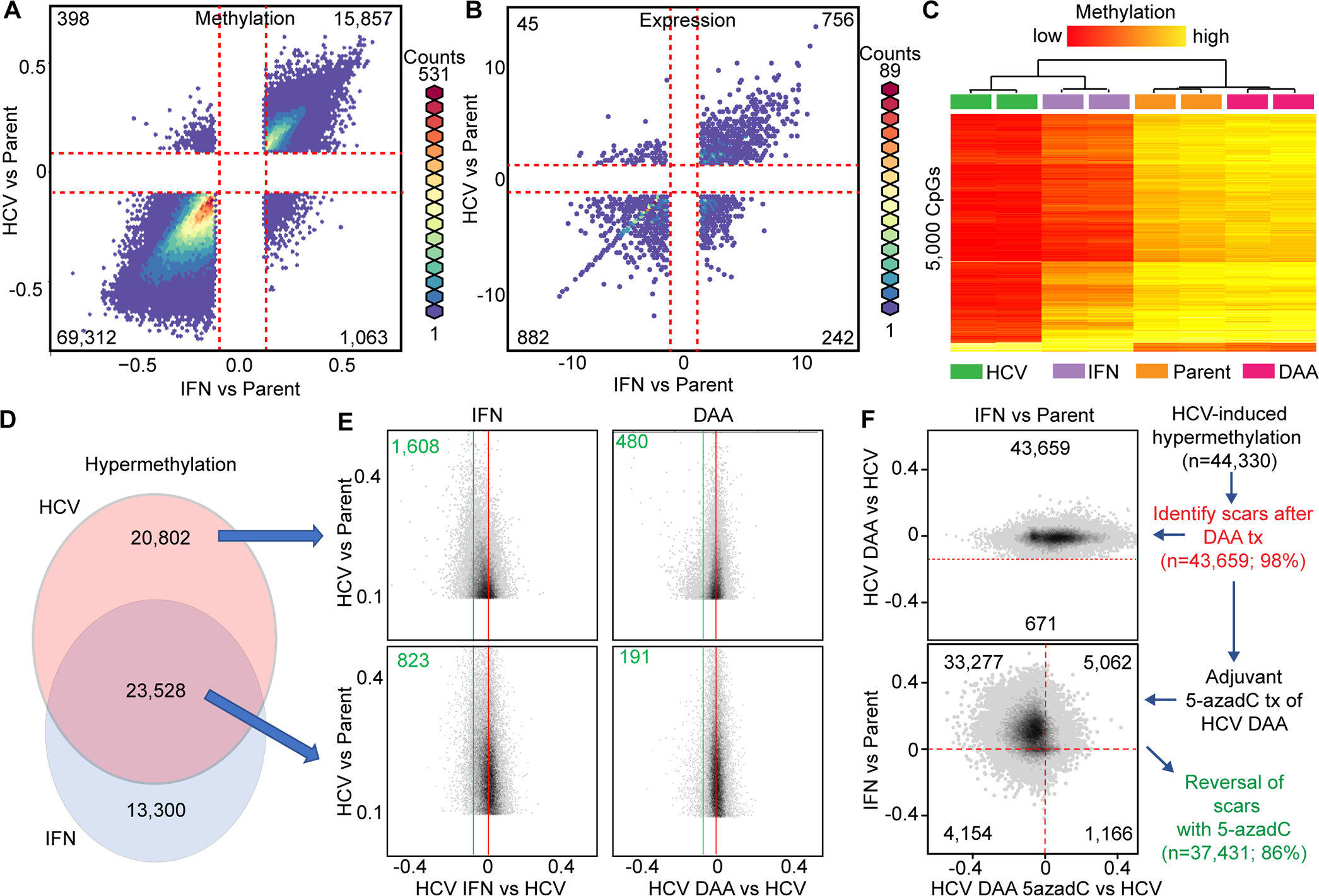
Hexbin plots of DNA methylation (A) and gene expression (B) comparing changes induced by either HCV (y-axis) or IFN (x-axis) relative to parental Hu1545 cells. (C) A heatmap of the 5,000 most variable CpGs between Hu1545 parental, IFN-alone, DAA-alone, and Hu1545-HCV cell lines. (D) Overlap of hypermethylation events between HCV and IFN relative to parent is shown as a Venn diagram. (E) Sub-setting CpGs into HCV-unique (top) and HCV/IFN-shared (bottom) demonstrates the relative reversal of aberrant methylation changes with IFN (left) or DAA (right). (F) Hexbin plots of HCV scars following DAA treatment (top) and the effects of 5-azadC effects on those HCV scars in the context of IFN (bottom).
This result also indicated that it was important to further parse out overlapping HCV and interferon driven changes to identify DNA methylation events that are truly unique to HCV infection, rather than those driven by innate immune responses induced by interferon in response to viral infection. Since chronic inflammation (regulated in part through interferon and its regulated pathways) acts as a driver of tumor initiation(14), we examined DNA methylation changes induced by both HCV infection and interferon alone, and after HCV cure in Hu1545 cells. More than half of all CpGs hypermethylated by HCV infection of Hu1545 cells were also hypermethylated by interferon treatment of parental Hu1545 cells (Fig. 6D). Interestingly, when HCV infected cells were treated with IFN or DAA, a greater proportion (roughly twice as many: 1,608 and 480 vs 823 and 191 in IFN and DAA, respectively) of HCV infection-specific DNA methylation changes revert, compared to those induced by either exposure alone (Fig. 6E). Aberrant DNA methylation changes shared between HCV and IFN, however, were particularly sensitive to 5-azadC treatment, with the majority of hypermethylation events being reversed following 5-azadC treatment (Fig. 6F, 86% of all HCV-induced hypermethylation reversed with 5-azadC, 89% of which are also driven, at least in part, by IFN). Collectively, these findings indicate that not only do HCV epigenetics scars significantly overlap with CpGs altered by interferon treatment alone, but also that these changes are reversible with DNMT inhibitors, providing a potential avenue for mitigating the impact of epigenetic scars on disease initiation and progression.
Due to the phenocopying of altered DNA methylation and gene expression profiles induced by interferon and HCV-infection, we sought to better understand the functional effects of IFN deregulation in HCV-infected Hu1545 cells. We profiled gene expression for key IFN pathway genes. As expected, we observed elevated IFNα following chronic HCV infection which, surprisingly, persisted following cure (Fig. 7A). However, we also noted a striking repression of TLR3, a key pathogen sensor of dsRNA including HCV, whose activation leads to apoptosis of virus-infected cells. TLR3 is epigenetically silenced in HCV-infected Hu1545 cells through loss of promoter H3K27 acetylation and a corresponding increase in the repressive H3K27me3 mark across the entire gene locus (Fig. 7B, Supplemental Fig. S13). Functionally, we confirmed that TLR3 effects are abrogated in HCV-infected and cured cells, as stimulation with poly(I:C) induces a potent increase in TLR3 in parental Hu1545 cells, which is blocked in HCV cells with and without IFN or DAA treatment (Fig. 7A, lowest panel). This finding indicates that active HCV infection and post-cure scarring directly alter TLR3 expression via epigenetic mechanisms to suppress the innate immune response, evade apoptosis, and predispose towards hepatocarcinogenesis through a reduced sensitivity of damaged hepatocytes to apoptotic signals. Indeed, TLR3 is expressed at lower levels in HCC (Fig. 7C), suggesting that chronic HCV infection downregulates TLR3 to escape apoptosis, a phenomenon that is not reversed with HCV cure, and which may set the stage for immune evasion in cellular transformation.
Fig. 7: Epigenetic scarring of innate immunity regulators and links to HCC.
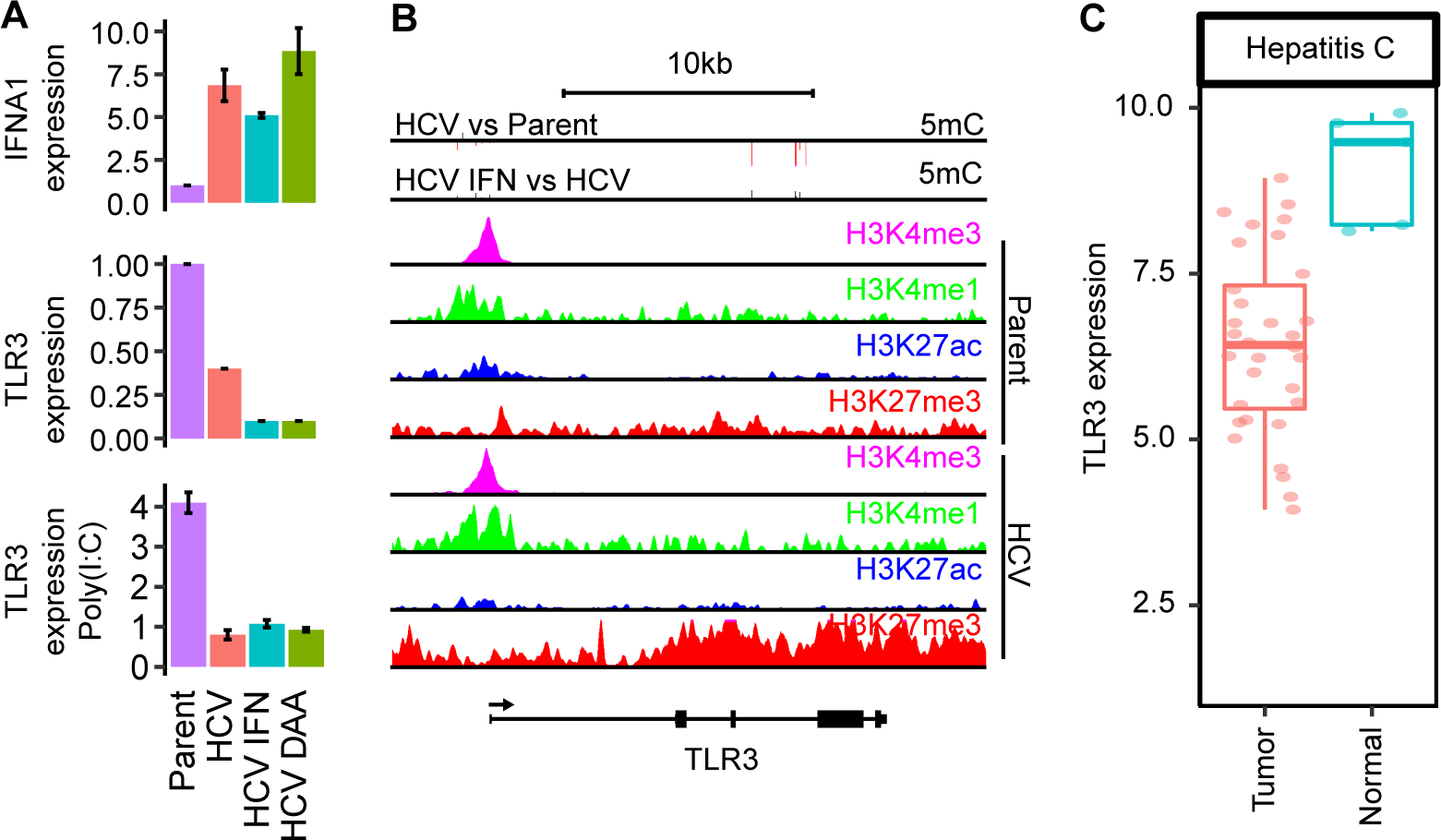
(A) Gene expression by real-time qRT-PCR of IFNA1 (top) and TLR3 (middle), as well as TLR3 expression following 5 mg/ml poly(I:C) treatment (bottom). (B) A UCSC genome browser view of the TLR3 locus. (C) Gene expression of TLR3 in normal and tumor tissues from TCGA LIHC.
Discussion
Hepatitis C viral infection is one of the most frequent etiologic drivers of cirrhosis and HCC worldwide. Treatment of patients with pegylated-interferon or with direct acting antivirals including telaprevir and boceprevir, leads to HCV clearance and SVR in approximately 46% and 75% percent of cases, respectively(15). However, individuals with previous HCV infection remain at elevated risk for cirrhosis and HCC(3, 16), suggesting that chronic HCV exposure leaves behind some mark or scar after SVR. While there is evidence that this scar could be genetic in nature(17), we focused our studies here on epigenetic processes since they are reversible and may thus be more amenable to pharmacologic intervention. We demonstrate that the epigenome is profoundly disrupted by HCV, and this disruption is only marginally reversed following HCV elimination with interferon or DAA treatment. A similar trend is also observed at the gene expression level, with enrichment for correlative changes in DNA methylation and expression at key genes linked to liver disease. Epigenetic changes induced by HCV are shared by those driven by interferon treatment in the absence of HCV. This interferon-driven landscape of epigenetic scarring is significantly reversed by treatment with DNA methyltransferase inhibitors. SVR in primary human tissues with differing HCV therapies also results in alterations to the epigenome, which are not restored to an HCV-naïve state. Epigenetic scars are distributed throughout the epigenome targeting disease-relevant pathways and genes that may explain, at least in part, why patients who no longer have active HCV infection remain at risk for developing HCC. In particular, HCV appears to target the innate immune pathway to deregulate key genes including TLR3, which primes the immune landscape for cellular transformation.
Recent studies have shown that chronic HCV infection alters the H3K27ac landscape, and following SVR with DAA or interferon-based therapies, many of these aberrant activating histone marks remain(18). A similar study examining four histone modifications (H3K4me3, H3K9ac, H3K9me3, and H3K27me3), but not DNA methylation, showed that >50% of HCV-induced histone mark alterations fail to revert following treatment with DAAs in Huh7.5 cells(19). An interesting observation to arise from our work was that the Hu1545 immortalized hepatocyte cell line model had a markedly higher frequency of epigenetic changes compared to that of the traditional Huh7.5 model that has been used extensively. We hypothesize that this is due to the fact that Huh7.5 is not HCV-naïve, as it was clonally derived from an HCV-infected cell line cured with IFN, and as such is already marked by epigenetic scars induced by prior HCV infection(20). However, HCV still induces many epigenetic and transcriptional changes in Huh7.5 cells. Moreover, elucidation of events that occur before transformation are critical to understanding how an HCV epigenetic signature contributes to persistent risk of HCC following SVR. As such, results related to HCV-driven effects on the epigenome generated with the Huh7.5 model should be interpreted with some caution. Regardless, our Hu1545 cells display a similar phenotype at several levels (e.g. global hypomethylation, gene upregulation, similar overlapping-IFN phenotypes, epigenetic scarring in both models), giving further confidence to the functional effects of epigenetic scarring in the liver and demonstrating the utility of this new model for HCV research.
We observe an interesting interplay between HCV infection and the interferon pathway at both the epigenetic and transcriptional levels. It stands to reason that a chronic interferon response driven by HCV infection could contribute to the processes of cirrhosis and transformation to HCC in vivo. This interferon/HCV axis may also have implications for disease processes following SVR in the patient setting. For example, prolonged type I interferon signaling alters the JAK-STAT signaling pathway(21), which reduces the antiviral innate immune response. Long-term exposure to type I interferon may lead to PD-1 blockade, potentially reducing the effectiveness of immune checkpoint inhibitors that are approved for HCC(22), as well as T-cell exhaustion driven by HCV infection, which is not fully restored with IFN/DAA cure(23). Type I interferons also act as immunosuppressants and lead to acquisition of cancer stem cell-like properties(24). Chronic activation of the interferon pathway due to epigenetic scars induced by HCV infection, therefore, could lead to reduced efficacy of HCC treatment, impaired host tumor immunity, and pro-tumorigenic pathway activation (e.g. TLR3 downregulation). Indeed, TLR3 activation exerts anti-tumor effects, and recent reports demonstrate that suppression of TLR3 promotes hepatocarcinogenesis through reduced responsiveness to pro-apoptotic signals(25). TLR3 is epigenetically regulated through histone acetylation during development(26), supporting our findings that H3K27ac/me3 epigenetic scars have the potential to functionally suppress TLR3-mediated immune and apoptosis pathways. While it has been known for some time that HCV induces an interferon-α response during the acute phase of infection(27), the similarity between chronic HCV infection- and IFN-induced DNA methylation landscapes has not been reported previously. Moreover, our results demonstrating that the FDA-approved DNA methyltransferase inhibitor 5-azadC efficiently reverses both the aberrant hyper- and hypomethylation scars driven by chronic HCV infection, suggest that it, or some other epigenetic agent (since not all HCV-induced epigenetic changes work through DNA methylation) could serve as an adjuvant therapy in the setting of HCV SVR. Such a treatment could reprogram the epigenome of damaged hepatocytes to one that is more responsive to normal liver homeostatic signals (e.g. apoptotic signals in damaged hepatocytes) and reduce the risk of progression to HCC within the SVR population. Indeed, recent work demonstrated that in a NASH mouse model of HCC, treatment with the epigenetic inhibitor JQ1 reduced frequency of tumor nodule formation(28). Since the HCV SVR population is expected to grow significantly in the coming years(29), such treatments could have a major impact on liver disease management.
Our study identifies a large set of epigenetically regulated genes that are intimately linked to the pathogenesis of liver disease. Many of these genes (e.g. ACLS1, CYP27A1, and CPEB1) show coordinated disruption of DNA methylation and histone modifications culminating in aberrant expression. These loci, when deregulated alone or in combination, could contribute to deregulated hepatocyte homeostasis and increased risk of transformation. For example, downregulation of ACSL1 is linked to poor prognosis and promotion of HCC growth(30), CPEB1 is downregulated in HCC and its silencing promotes cancer stemness and chemoresistance(31), while decreased expression of CYP27A1 is associated with venous invasion in HCC(32). Thus, while treatment with DNA methyltransferase inhibitors has the potential to re-sensitize tumors to the immune system, it could have a multimodal impact by also reverting aberrant silencing of tumor suppressor/growth-modulatory genes to improve prognosis or delay disease progression.
Front-line treatments for unresectable HCC include multikinase inhibitors (sorafenib and lenvatinib), PD-L1 checkpoint inhibitors (nivolumab and ipilimumab), and PD-L1/VEGF immunotherapy (atezolizumab and bevacizumab), which only improve survival by several months. HCC has well-established driver mutations including hTERT and CTNNB1. Unfortunately, none of the most frequently mutated genes in HCC are clinically targetable, leaving an unmet need for improvements to HCC therapy and agents that have efficacy in the growing SVR population. Even more desirable would be treatments that target the preneoplastic phase of liver damage and prevent HCC from developing. This study implicates the epigenome in a multimodal fashion in promoting liver disease pathogenesis through epigenetic scarring of potential HCC drivers and by repressing the innate immune system through the IFN-TLR3 axis.
Supplementary Material
Acknowledgements
We thank the University of Minnesota Genomics Core (UMGC), the Mayo Clinic Medical Genome Facility (MGF), and the Mayo Clinic Epigenomics Development Laboratory (EDL) for epigenomic and DNA sequencing services. We would also like to thank the National Disease Research Interchange (NDRI) for providing normal human liver tissue. This work was supported by NIH grant R01DK110024 (to CL and KDR), and an AASLD Clinical and Translational Research Award (RAH).
Abbreviations
- 5-azadC
5-aza-2’-deoxycytidine
- 5mC
DNA 5-methylcytosine
- 850k
Infinium MethylationEPIC BeadChip
- DAA
Direct-acting antiviral
- DNMT
DNA methyltransferase
- GSEA
Gene set enrichment analysis
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- IFN
Interferon
- IPA
Ingenuity pathway analysis
- NDRI
National Disease Research Interchange
- SVR
Sustained virologic response
- TCGA LIHC
The Cancer Genome Atlas Liver Hepatocellular Carcinoma
Footnotes
Conflict of interest: None.
References
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013;57:1333–1342. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118–1127. [DOI] [PubMed] [Google Scholar]
- 3.Ioannou GN, Green PK, Berry K. HCV eradication induced by direct-acting antiviral agents reduces the risk of hepatocellular carcinoma. J Hepatol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, Gouysse G, McKay-Chopin S, Tavtigian SV, Scoazec JY, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One 2010;5:e9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hlady RA, Sathyanarayan A, Thompson JJ, Zhou D, Wu Q, Pham K, Lee JH, et al. Integrating the Epigenome to Identify Drivers of Hepatocellular Carcinoma. Hepatology 2019;69:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J, Imbeaud S, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015;61:1945–1956. [DOI] [PubMed] [Google Scholar]
- 8.Hung SY, Lin HH, Yeh KT, Chang JG. Histone-modifying genes as biomarkers in hepatocellular carcinoma. Int J Clin Exp Pathol 2014;7:2496–2507. [PMC free article] [PubMed] [Google Scholar]
- 9.Hlady RA, Zhao X, Pan X, Yang JD, Ahmed F, Antwi SO, Giama NH, et al. Genome-wide discovery and validation of diagnostic DNA methylation-based biomarkers for hepatocellular cancer detection in circulating cell free DNA. Theranostics 2019;9:7239–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol 2012;13:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 2017;14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenkiewicz E, Malasi S, Hogenson TL, Flores LF, Barham W, Phillips WJ, Roesler AS, et al. Genomic and Epigenomic Landscaping Defines New Therapeutic Targets for Adenosquamous Carcinoma of the Pancreas. Cancer Res 2020;80:4324–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coussens LM, Werb Z. Inflammation and cancer. Nature 2002;420:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geddawy A, Ibrahim YF, Elbahie NM, Ibrahim MA. Direct Acting Anti-hepatitis C Virus Drugs: Clinical Pharmacology and Future Direction. J Transl Int Med 2017;5:8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ioannou GN. HCC surveillance after SVR in patients with F3/F4 fibrosis. J Hepatol 2021;74:458–465. [DOI] [PubMed] [Google Scholar]
- 17.Brunner SF, Roberts ND, Wylie LA, Moore L, Aitken SJ, Davies SE, Sanders MA, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019;574:538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamdane N, Juhling F, Crouchet E, El Saghire H, Thumann C, Oudot MA, Bandiera S, et al. HCV-Induced Epigenetic Changes Associated With Liver Cancer Risk Persist After Sustained Virologic Response. Gastroenterology 2019;156:2313–2329 e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez S, Kaspi A, Domovitz T, Davidovich A, Lavi-Itzkovitz A, Meirson T, Alison Holmes J, et al. Hepatitis C virus leaves an epigenetic signature post cure of infection by direct-acting antivirals. PLoS Genet 2019;15:e1008181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 2002;76:13001–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu H, Nelson DR, Crawford JM, Liu C. Defective Jak-Stat activation in hepatoma cells is associated with hepatitis C viral IFN-alpha resistance. J Interferon Cytokine Res 2005;25:528–539. [DOI] [PubMed] [Google Scholar]
- 22.Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, Ferrere G, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res 2019;29:846–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luxenburger H, Neumann-Haefelin C, Thimme R, Boettler T. HCV-Specific T Cell Responses During and After Chronic HCV Infection. Viruses 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017;6:e1314424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonnin M, Fares N, Testoni B, Estornes Y, Weber K, Vanbervliet B, Lefrancois L, et al. Toll-like receptor 3 downregulation is an escape mechanism from apoptosis during hepatocarcinogenesis. J Hepatol 2019;71:763–772. [DOI] [PubMed] [Google Scholar]
- 26.Porras A, Kozar S, Russanova V, Salpea P, Hirai T, Sammons N, Mittal P, et al. Developmental and epigenetic regulation of the human TLR3 gene. Mol Immunol 2008;46:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A 2002;99:15669–15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Juhling F, Hamdane N, Crouchet E, Li S, El Saghire H, Mukherji A, Fujiwara N, et al. Targeting clinical epigenetic reprogramming for chemoprevention of metabolic and viral hepatocellular carcinoma. Gut 2021;70:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zator ZA, Chung RT. After the cure: management of HCV after achievement of SVR. Curr HIV/AIDS Rep 2013;10:428–435. [DOI] [PubMed] [Google Scholar]
- 30.Sun Y, Wang Q, Zhang Y, Geng M, Wei Y, Liu Y, Liu S, et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via miR-27a-3p. J Hepatol 2020;73:603–615. [DOI] [PubMed] [Google Scholar]
- 31.Xu M, Fang S, Song J, Chen M, Zhang Q, Weng Q, Fan X, et al. CPEB1 mediates hepatocellular carcinoma cancer stemness and chemoresistance. Cell Death Dis 2018;9:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsunedomi R, Iizuka N, Hamamoto Y, Uchimura S, Miyamoto T, Tamesa T, Okada T, et al. Patterns of expression of cytochrome P450 genes in progression of hepatitis C virus-associated hepatocellular carcinoma. Int J Oncol 2005;27:661–667. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.