

Graphical abstract
Keywords: Hepatocellular carcinoma, Cholesterol metabolism, Prognostic signature, Therapeutic response
Abstract
Hepatocellular carcinoma (HCC) represents the most important type of liver cancer, the 5-year survival rate for advanced HCC is 2%. The heterogeneity of HCC makes previous models fail to achieve satisfactory results. The role of Cholesterol-based metabolic reprogramming in cancer has attracted more and more attention. In this study, we screened cholesterol metabolism-related genes (CMRGs) based on a systematical analysis from TCGA and GEO database. Then, we constructed a prognostic signature based on the screened 5 CMRGs: FDPS, FABP5, ANXA2, ACADL and HMGCS2. The clinical value of the five CMRGs was validated by TCGA database and HPA database. HCC patients were assigned to the high-risk and low-risk groups on the basis of median risk score calculated by the five CMRGs. We evaluated the signature in TCGA database and validated in ICGC database. The results revealed that the prognostic signature had good prognostic performance, even among different clinicopathological subgroups. The function analysis linked CMRGs with KEGG pathway, such as cell adhesion molecules, drug metabolism-cytochrome P450 and other related pathways. In addition, patients in the high-risk group exhibited characteristics of high TP53 mutation, high immune checkpoints expression and high immune cell infiltration. Furthermore, based on the prognostic signature, we identified 25 most significant small molecule drugs as potential drugs for HCC patients. Finally, a nomogram combined risk score and TNM stage was constructed. These results indicated our prognostic signature has an excellent prediction performance. This study is expected to provide a potential diagnostic and therapeutic strategies for HCC.
1. Introduction
Liver cancer is a common malignant digestive system cancer type, in which ranks sixth in morbidity and third in mortality worldwide. Hepatocellular carcinoma (HCC), as the main type of liver cancer, comprises 75–85% of liver cancer cases [1]. The 5-year survival rate of early-stage HCC exceeds 70%, while that of advanced-stage HCC is 2% [2], [3]. Unfortunately, only a few HCC patients can be diagnosed in their early stage [4]. Surgery and liver transplantation remain the most effective treatment for HCC. However, the high metastasis/recurrence rate (∼50%–70%) within five years after operation restricts the outcome of patients [5]. Classical clinical models use TNM staging, microvascular invasion and other indicators to predict the prognosis of HCC, but the heterogeneity of HCC makes these models fail to achieve satisfactory results [6]. Therefore, new models are needed for the therapy and prognosis of HCC patients.
Metabolic reprogramming has become one of the important signs of cancer. Metabolic enzymes and metabolites are involved in various aspects of tumor formation [7]. Cholesterol-based metabolic reprogramming has been ignored in previous studies, but in recent years, more and more studies regard it as an integral part of tumor progression [8]. Cholesterol is essential for maintaining tumor cell homeostasis and critical cellular structure. Studies have demonstrated that cholesterol metabolism is associated with the growth and migration of cancer cells [9], [10]. In addition, studies have shown that cholesterol was involved in tumor-associated macrophage (TAM) polarization and T cell exhaustion [11], [12]. Actually, previous studies have shown that statins, as a class of compounds inhibiting HMG-CoA reductase, have good anti-tumor effects in mouse models [13]. However, the effect of statin in clinical trials is still controversial [14], [15]. Therefore, elucidating the mechanism of cholesterol in tumors will help to better understand the tumorigenesis, guide the therapy and improve the prognosis of tumor patients.
Here, we identified 5 prognostic cholesterol metabolism-related genes (CMRGs) in HCC. We constructed a signature based on CMRGs, and further validated the clinical application value and related mechanisms of the signature. Our findings are expected to provide a latent diagnostic and therapeutic strategies for HCC.
2. Materials and methods
2.1. Data collection pre-processing
A total of 5 databases were incorporated into this study. The mRNA expression data from GEO database was downloaded through 3 microarray data sets (GSE14520, GSE62232, GSE102079). The mRNA expression data were processed by GEO2R, an online analysis tool based on R (Version 3.2.3). Clinical and expression data from TCGA-LIHC cohort was regarded as training set. Clinical and expression data from ICGC-LIRI-JP cohort was regarded as validation set. TCGA-LIHC cohort was downloaded from TCGA (https://portal.gdc.cancer.gov/). ICGC-LIRI-JP cohort was downloaded from HCCDB (https://lifeome.net/database/hccdb/home.html). The CMRGs (n = 140) were downloaded from the Molecular Signatures Database (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp) [16], [17]. For details of the GEO datasets and gene list, see Supplementary Table S1 and S2.
2.2. Differentially expressed CMRGs
Differentially expressed genes (DEGs) were screened from GEO dataset with threshold of |log2FC| > 1 and adjusted P < 0.05. Differentially expressed CMRGs (DECMRGs) were defined as the intersection of DEGs and CMRGs. Further cox analysis with P < 0.05 and differential expression was performed in TCGA-LIHC to screen final CMRGS for subsequent analysis.
2.3. Gene-Gene and Protein–Protein interaction network for CMRGs
GeneMANIA (https://genemania.org/) is a website for functional analysis of genes [18]. In this study, GeneMANIA was used to perform gene-gene interaction network for CMRGs. The protein–protein interaction network was obtained from the STRING database [19].
2.4. Identification and validation of prognostic signature
We used least absolute shrinkage and selection operator (LASSO) regression algorithm [20] to construct a signature of CMRGs in TCGA-LIHC cohort by the R package “glmnet”. The coefficients corresponding to the optimal λ value were incorporated into the prognostic signature formula as following:
. Coefi is the coefficient, Genei is the transcripts per million reads (TPM) value of each selected gene. HCC Patients with a risk score above the median risk score were assigned to the high-risk group, and those with a risk score below the median risk score were assigned to the low-risk group. Survival analysis was conducted by R packages “survival” and “survminer”. The nomogram model was constructed by R package “rms”. The receiver operating characteristic (ROC) curve, calibration curve and detrended correspondence analysis (DCA) were performed to evaluate the performance of the signature. Immunohistochemistry information of CMRGs was downloaded from HPA (https://www.proteinatlas.org/), a pathology atlas of the human cancer transcriptome [21].
2.5. Function and somatic mutations analysis
Gene set enrichment analysis (GSEA) was used to explore the intrinsic function associated with risk score by R package “clusterProfiler” [22]. In GSEA, p-value (P) < 0.05 and false discovery rate (FDR) < 0.05 were considered statistically significant. Somatic mutation analysis was conducted to explored the mutation rates associated with risk score by R package “maftools” [23].
2.6. Immune infiltration analysis
The evaluation of tumor immune infiltration includes immune cell infiltration, immune-related score and immune checkpoint expression. Immune cell infiltration is calculated by xCell [24] and MCPCOUNTER [25] algorithms. The immune-related score was calculated by xCell algorithm, and the immune checkpoint was from the published literature [26]. The results of this section are downloaded from TIMER 2.0 website [27].
2.7. Therapeutic response and drug prediction
The immunophenoscore (IPS) obtained from The Cancer Immune Atlas (TCIA) was used to assess the response to immunotherapy [28]. For the purpose of drug prediction, the DEGs was identified from the high-risk and low-risk groups. Then the DEGs were uploaded to L1000FWD (https://maayanlab.cloud/L1000FWD/), a web application that provides interactive visualization of drug induced gene expression signatures [29]. The structure of drug was obtained from PubChem (pubchem.ncbi.nlm.nih.gov).
2.8. Statistical analysis
Statistical Analysis were conducted by R (Version 4.0.5).The unpaired Student’s t-test and Wilcoxon test was used for comparing normal or nonnormal data, respectively. The log-rank test was used for Kaplan-Meier curve. The clinical characteristics among groups were assessed by χ2 test or Fisher’s exact test. In this study, P < 0.05. was considered as statistically significant.
3. Results
3.1. Identification of differentially expressed prognostic CMRGs
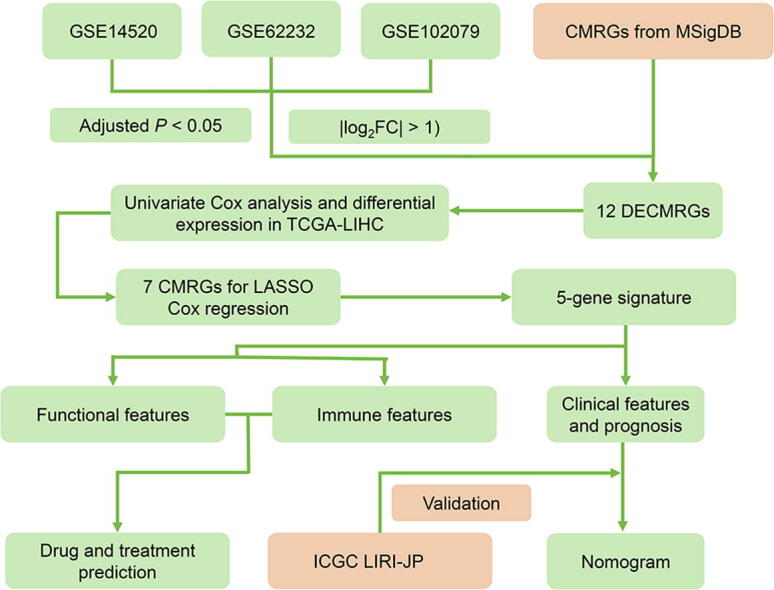
The workflow of the present study was illustrated in Fig. S1. Table 1 summarized the baseline characteristics of the HCC patients enrolled in present study. DEGs were screened from three GEO datasets with threshold of |log2FC| > 1 and adjusted P < 0.05 (Fig. 1A). Then 12 DECMRGs were obtained in the intersection of the DEGs and 140 CMRGs (Fig. 1B). Further cox analysis with P < 0.05 and differential expression was performed in TCGA-LIHC to screen final CMRGS for differentially expressed prognostic CMRGs (Fig. 1C, D). The protein–protein interaction network and correlation network among these differentially expressed prognostic CMRGs were shown in Fig. 1E, F.
Table 1.
The baseline characteristics of the HCC patients enrolled in this study.
| Characteristic | TCGA-LIHC | ICGC LIRI-JP |
|---|---|---|
| Total | 374 | 203 |
| Age, median (rage) | 61 (16, 90) | 69(31,86) |
| Sex, n (%) | ||
| Female | 121 (32.4 %) | 50 (24.6 %) |
| Male | 253 (67.6 %) | 153 (75.4 %) |
| TNM stage, n (%) | ||
| I | 172 (46.0 %) | 33 (16.3 %) |
| II | 88 (23.5 %) | 96 (47.3 %) |
| III | 85 (22.7 %) | 59 (29.1 %) |
| IV | 5 (1.3 %) | 15 (7.4 %) |
| Unknown | 24 (6.4 %) | 0 (0 %) |
| Histologic grade, n (%) | ||
| G1 | 55 (14.7 %) | NA |
| G2 | 178 (47.6 %) | NA |
| G3 | 124 (33.2 %) | NA |
| G4 | 12 (3.2 %) | NA |
| Unknown | 5 (1.3 %) | NA |
Fig. 1.

Identification of Differentially Expressed Prognostic CMRGs. (A) Screening of DEGs in three GEO datasets. (B) The 12 overlapping genes differentially expressed in all three datasets. Cox analysis revealed 8 prognostic related genes (C), and 7 genes were differentially expressed in TCGA-LIHC (D). (E, F) The protein–protein interaction network and correlation network among these differentially expressed prognostic CMRGs. Ns: not significant, * P < 0.05, ** P < 0.01, *** P < 0.001.
3.2. Establishment of prognostic signature based on CMRGs in TCGA-LIHC
Based on 7 differentially expressed CMRGs, we developed a prognostic signature by LASSO regression analysis and finally screened five genes: FDPS, FABP5, ANXA2, ACADL and HMGCS2 (Fig. 2A, B). The coefficients and correlation network of the five CMRGs were displayed in Fig. 2C, D. The risk score of patients was calculated by the formula: risk score = (0.1216*FABP5) + (0.0757*ANXA2) + (0.1984*FDPS) + (−0.0979*HMGCS2) + (−0.0142*ACADL). The prognostic value of the five CMRGs was evaluated in TCGA-LIHC cohort. The prognosis of HCC patients with low expression of FDPS, FABP5 and ANXA2 was better than that of HCC patients with high expression of FDPS, FABP5 and ANXA2. While the prognosis of HCC patients with highly expressed HMGCS2 and ACADL was better than that of HCC patients with lowly expressed HMGCS2 and ACADL (Fig. 2E). In the HPA database, the expressions of ACADL and HMGCS2 in tumor tissues were lower compared with those in normal tissues. While the expressions of FDPS, FABP5 and ANXA2 were significantly higher in tumor tissues compared with those in normal tissues (Fig. 2F).
Fig. 2.

Establishment of a prognostic signature based on CMRGs in TCGA-LIHC. (A, B, C) Identification of nonzero coefficient genes by LASSO regression analysis. (D) Correlation network of 5 genes. (E) Survival analysis of 5 genes. (F) Immunohistochemical analysis of 5 genes.
3.3. Evaluation and validation of prognostic signature
We divided the HCC patients in TCGA-LIHC cohort into high-risk (n = 187) and low-risk (n = 187) groups based on median risk score (1.3013). Similarly, HCC patients in the validation cohort ICGC LIRI-JP were separated into high-risk (n = 101) and low-risk (n = 102) groups based on median risk score (1.6224). The risk survival status chart showed that survival time of HCC patients in the low-risk group was longer than that of HCC patients in the high-risk group (Fig. 3A, D). The 2-year, 3-year and 4-year area under curve (AUC) values were calculated to evaluate the sensitivity and specificity of the signature. They were 0.660, 0.667 and 0.657 in TCGA-LIHC (Fig. 3B) and 0.650,0.687 and 0.680 in ICGC LIRI-JP (Fig. 3D). The 5-year survival probability of HCC patients in the low-risk group significantly higher than that of HCC patients in the high-risk group (Fig. 3C, F). In addition, we investigated the universality of the signature in different clinical subgroups (Fig. 4A, B). we found that advanced HCC (TNM stage III or IV) predominate in high-risk patients, while early HCC (TNM stage I or II) predominate in low-risk patients (Fig. 4C, E). Compared with the low-risk group, patients in the high-risk group had significantly higher expression level of FDPS, FABP5 and ANXA2 and significantly lower expression level of HMGCS2 and ACADL (Fig. 4D, F). Finally, we performed a clinical stratified analysis on different clinical factors including age, sex and TNM stage. We found that even in different clinical subgroups, the survival probability of HCC patients in the high-risk group was significantly lower than that of HCC patients in the low-risk group (Fig. 5). The results above revealed that our signature had high specificity and sensitivity in predicting the prognosis of HCC patients.
Fig. 3.

Evaluation and validation of prognostic signature. Evaluation the prognostic signature in TCGA-LIHC by (A) risk survival status chart, (B) ROC curve, and (C) Kaplan-Meier curve. Validation the prognostic signature in ICGC LIRI-JP by (D) risk survival status chart, (E) ROC curve, and (F) Kaplan-Meier curve.
Fig. 4.

Explore the relationship between risk score and different clinical features. Heatmap presented the correlation between risk score and sex, TNM stage, grade, age and AFP in (A) TCGA-LIHC, and (B) ICGC LIRI-JP cohort. (C, E) The distribution of TNM stage in two risk groups was showed in the histogram. (D, F)Expression levels of the 5 CMRGs between two risk groups. * P < 0.05, ** P < 0.01, *** P < 0.001.
Fig. 5.

Stratified analysis of the signature by TCGA-LIHC and ICGC LIRI-JP cohort. Kaplan-Meier curve under different clinical subgroups: (A, D) age, (B, E) Sex, and (C, F) TNM stage.
3.4. Function and somatic mutations analysis of signature
To elucidate the potential function and pathways related to our prognostic signature, we performed GSEA analysis which annotated by KEGG databases in the TCGA cohort. Altogether 12,066 DEGs, identified between the high-risk and low-risk groups, were included in the GSEA analysis (Table S3). The results showed that the high-risk group was mainly enriched in Cell adhesion molecules, Cytokine-cytokine receptor interaction, ECM-receptor interaction and other pathways (Fig. 6A), while the low-risk group was mainly enriched in Drug metabolism-cytochrome P450, Fatty acid degradation, PPAR signaling pathway and other pathways (Fig. 6B). Table S4 listed the detailed results of GSEA analysis.
Fig. 6.

Function analysis, somatic mutations and immune-related score in TCGA-LIHC. (A) Main pathways enriched in the high-risk group. (B) Main pathways enriched in the low-risk group. (C) Significantly mutated genes in the high-risk group. (D) Significantly mutated genes in the low-risk group. (E) Stromal score of both groups. (F) Immune score of both groups. (G) Microenvironment score of both groups. * P < 0.05, ** P < 0.01, *** P < 0.001.
Next, we performed somatic mutations analysis to reveal the relationship between risk score and mutation. We identified the top 15 genes with the highest mutation rate in both groups, and missense mutations were the most common mutation type (Fig. 6C, D). The gene with the highest mutation rate was TP53 in the high-risk group, while in the low-risk group was TTN. The mutation rates of TP53, TTN and CTNNB1 were > 20 % in both groups.
3.5. Immune infiltration analysis
To investigate the immune infiltration in between the high-risk and low-risk groups, we first performed xCell algorithm to calculate the immune-related score in TCGA-LIHC. The results showed that compared with the low-risk group, the high-risk group had significantly higher stromal score (Fig. 6E, P < 0.001), immune score (Fig. 6F, P < 0.05) and microenvironment score (Fig. 6G, P < 0.001).
Next, we used xCell (Fig. 7A) and MCPCOUNTER (Fig. 7B) algorithms to evaluate the immune cell infiltration in TCGA-LIHC. We found that compared with the low-risk group, the high-risk group had significantly higher infiltration abundances of myeloid dendritic cell, monocyte, B cell, memory CD4+ T cell, CD8+ T cell etc. and significantly lower infiltration abundances of endothelial cell, macrophage M2, T cell regulatory (Tregs). Finally, we explored the differential expression of immune checkpoints between the high-risk and low-risk groups (Fig. 7C). The results showed that there was no difference in CTLA4 and TIGIT expression between the high-risk and low-risk groups, while the expression of OX40 (P < 0.001), PD-L1 (P < 0.001), TIM3 (P < 0.001), LAG3 (P < 0.001) and PD-1 (P < 0.001) in the high-risk group were significantly higher than those in the low-risk group.
Fig. 7.

The tumor immune infiltration between high-risk and low-risk groups in TCGA-LIHC. (A) Comparison of immune cell abundance by xCell algorithm. (B) Comparison of immune cell abundance by MCPCOUNTER algorithm. (C) The differential expression of immune checkpoints between the low-risk and high-risk groups. Ns: not significant, P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001.
3.6. Therapeutic response and drug prediction
Given that mutation and immune infiltration characteristics were different between the high-risk and low-risk groups, we further compared the response to immunotherapy between the two groups by TCIA, a database providing immune analysis based on cancer patients in TCGA. To our surprise, there was no difference in IPS-CTLA4 blocker score, IPS-PD1/PD-L1/PD-L2 blocker score and IPS-CTLA4 plus PD1/PD-L1/PD-L2 blocker score between the two groups (Fig. 8B-D). However, we found that the total IPS score in the low-risk group was significantly higher than that in the high-risk group (Fig. 8A, P < 0.01). These results suggested that risk score may be related to immunotherapy.
Fig. 8.

The immunotherapy response and potential drugs between high-risk and low-risk groups in TCGA-LIHC. (A-D) IPS score between two groups. (E) The potential drugs for HCC treatment. (F) The 2D structure of HLI-373. (G) The 3D structure of HLI-373. Ns: not significant, P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001.
In order to predict potential drugs for HCC, we uploaded the top 150 DEGs identified from the high-risk and low-risk groups to the L1000FWD database. We identified 25 most significant small molecule drugs as potential drugs (Fig. 8E). HLI-373 was predicted as the most potential drug. The 2D and 3D structure of HLI-373 were visualized by the PubChem website (Fig. 8F, G).
3.7. Construction and validation of the risk Score-Related nomogram
To construct a nomogram for predicting the survival probability of HCC patients, we performed univariate Cox regression in TCGA-LIHC and the parameters of P < 0.05 were included for multivariate Cox regression. The results indicated that only TNM stage and risk score were independent risk factors for overall survival (Table S5). Then, we constructed a nomogram model combining with TNM stage and risk score to predict the survival probability of HCC patients (Fig. 9A). The 1-year, 3-year and 5-year AUC values of the nomogram model in TCGA-LIHC were 0.750, 0.731 and 0.709, respectively (Fig. 9B). The 1-year, 3-year and 5-year AUC values of the nomogram model in ICGC LIRI-JP were 0.935, 0.952 and 1.000, respectively (Fig. 9E). We further performed calibration plots and DCA curve to evaluate the nomogram model in TCGA-LIHC (Fig. 9C, D) and ICGC LIRI-JP (Fig. 9F, G), respectively. The results indicated that risk score was a better prognostic indicator than traditional TNM stage, even better performance was obtained by combining with TNM stage and risk score.
Fig. 9.

Construction and validation of the risk score-related nomogram. (A) The nomogram to predict the 1-year, 3-year, and 5-year overall survival of HCC patients. The AUC curve of time-dependent ROC curves verified the prognostic performance of the nomogram in (B) TCGA-LIHC cohort, or (E) ICGC LIRI-JP cohort. Calibration plots of the nomogram in predicting the 1-year, 3-year, and 5-year overall survival of HCC patients in (C) TCGA-LIHC cohort, or (F) ICGC LIRI-JP cohort. DCA curves to assess the ability of TNM stage, risk score, and their combination to predict the 1-year, 3-year, and 5-year overall survival of HCC patients in (D) TCGA-LIHC cohort, or (G) ICGC LIRI-JP cohort.
4. Discussion
Liver cancer is a common malignant tumor which ranks third in cancer-related mortality worldwide [1]. HCC is the main type of liver cancer. Despite significant progress has been achieved in recent years, the prognosis of HCC remains poor. Therefore, it is urgent to find new strategies to guide the diagnosis and treatment of HCC. Cholesterol, as a structural component of cells, has attracted more and more attention in recent years. A previous study showed that cholesterol accumulation can promote the progress of HCC [30]. Preclinical studies have shown that the combination therapy of statins and sorafenib has synergistic antitumor effects on HCC cells [31]. To date, the specific role of cholesterol metabolism in HCC remains unclear. In this context, the present study aimed to construct a novel cholesterol metabolism-related prognostic signature for HCC patients.
In this study, we screened 7 CMRGs based on a systematical analysis from TCGA and GEO database. Then, we constructed a novel prognostic signature through LASSO regression analysis and finally identified five genes: FDPS, FABP5, ANXA2, ACADL and HMGCS2. The expression and prognostic value of the five genes was validated by TCGA database and HPA database. Next, We divided HCC patients above median risk score into high risk group and HCC patients below median risk score into low risk group. We evaluated the signature in TCGA-LIHC cohort and validated in ICGC-LIRI-JP cohort. The results of ROC curve and Kaplan-Meier curve revealed that the prognostic signature has good performance in predicting the prognosis of HCC patients. More importantly, the signature still yielded significant prognostic performance among the patients with different clinicopathological factors. Further function analysis revealed that the DEGs are enriched in Cell adhesion molecules, Cytokine-cytokine receptor interaction, Drug metabolism-cytochrome P450, Fatty acid degradation and other related pathways. The somatic mutations analysis showed that there were different mutation modes between the high-risk and low-risk groups. We found that immune cell infiltration in the high-risk group were higher than those in the low-risk group. Furthermore, based on the prognostic signature, we identified 25 most significant small molecule drugs as candidate drugs for HCC patients. Finally, we developed a risk score-related nomogram for predicting the prognosis of HCC patients. These results indicated our prognostic signature has an excellent prediction performance.
In present study, the prognostic signature integrated 5 CMRGs, including FDPS, FABP5, ANXA2, HMGCS2 and ACADL. Among them, FDPS, FABP5 and ANXA2 were associated with poor prognosis of HCC, while HMGCS2 and ACADL were associated with good prognosis of HCC. FDPS (farnesyl diphosphate synthase) is a pleiotropic enzyme which is involved in cholesterol biosynthesis. Previous studies have found that FDPS is highly expressed in various tumors and associated with several malignancies, including prostate cancer, pancreatic, colon cancer, and glioma [32], [33], [34], [35]. However, its role in HCC remains unclear. FABP5 (fatty acid binding protein 5) transports lipids intracellularly for storage purposes. Wu et al. found that knocking down of FABP5 in retinal pigment epithelial cells leads to a decrease in cellular cholesterol levels [36]. Another study showed that circulating FABP5 may directly regulate HDL function independently of HDL cholesterol level and decrease cholesterol efflux in macrophages [37]. FABP5 was shown to promote angiogenesis in HCC [38]. These studies suggest a potential role of FABP5 in cholesterol homeostasis and HCC. However, how FABP5 affects HCC through cholesterol metabolism is still unclear, which deserves further study. ANXA2 is a member of annexin family, functions of which include vesicle transport, cell proliferation and cell growth [39]. Multi-omics analysis showed that ANXA2 was involved in the development of HCC [40]. Kitamura et al. found that ANXA2 possesses an essential function for the basal LDLR expression in HepG2 cells [41]. Of course, more studies are needed to elucidate the specific mechanism by which ANXA2 regulates cholesterol metabolism. HMGCS2 (3-hydroxy-3-methylglutaryl CoA synthase 2) is a mitochondrial enzyme primarily involved in the ketogenesis pathway [42]. Also, it is essential in converting acetoacetyl-CoA to HMG-CoA, a cholesterol precursor [43]. HMGCS2 was found to be downregulated in HCC and acts as a tumor suppressor [44]. Wang et al. found that HCC cells with HMGCS2 downregulation possess altered lipid metabolism that increases cholesterol synthesis [45]. Therefore, we hypothesized that HMGCS2 may affect HCC progression through cholesterol synthesis, but further studies are needed to confirm this hypothesis. ACADL (Long-chain acyl-CoA dehydrogenase) is a mitochondrial enzyme that catalyzes fatty acid oxidation. Studies have shown that it also plays an important role in the regulation of triglycerides and cholesterol [46]. ACADL acts as a tumor suppressor, which inhibits the cell growth of HCC by targeting YAP pathway [47]. The YAP pathway is involved in HCC through cholesterol metabolism [48]. This study further elucidates the important role of these 5 CMRGs in HCC, and the prognostic signature constructed by them has good sensitivity and specificity in predicting the survival probability of HCC patients.
GSEA analysis suggested that KEGG pathways, including Cell adhesion molecules, Cytokine-cytokine receptor interaction, Drug metabolism-cytochrome P450, IL-17 signaling pathway, PPAR signaling pathway, Primary bile acid biosynthesis and other related pathways associated with cholesterol metabolism. In fact, cholesterol serves as a crucial component of lipid rafts, is involved in downstream processes such as proliferation, differentiation, adhesion and apoptosis [49]. Ma et al. found that IL-17 signaling pathway is involved in the activation of SREBP1/2 to promote cholesterol synthesis and progression of HCC [50]. Peroxisome proliferator-activated receptors α (PPARα) and γ (PPARγ) played an vital role in inhibiting cholesterol biosynthesis [51]. Previous studies have shown that PPARα and PPARγ are involved in HCC progression [52], [53]. Thus, PPARα and PPARγ are considered as potential drug targets for cancer patients [54].
In this study, TP53 had the highest mutation rate in the high-risk group, while TTN had the highest mutation rate in the low-risk group. Indeed, TP53 is involved in all aspects of cholesterol synthesis and transport [55]. Genome-wide analysis also indicated that TP53 can affect cholesterol metabolism in HCC [56]. Lonetto et al. have shown that TP53 mutations can affect tumor progression by mediating metabolic disturbance. Therefore, high TP53 mutations may be an important cause of poor prognosis in high-risk patients. TTN also plays an important role in the progression of HCC [57], but to our surprise, the mutation rate of TTN in the two groups is similar.
Immunotherapy has become the most promising treatment of cancer, but the effect of immunotherapy is closely related to immune infiltration [58]. CD8+ T cells are the main lymphocytes that kill HCC cells. Lack of recovery of CD8+ T cells is associated with poor survival in HCC patients [59]. Tregs, a kind of tolerance-inducing cells, are enriched in HCC microenvironment. Sasaki et al. found that the high number of tumor-infiltrating Tregs are associated with poor prognosis of HCC patients after hepatic resection [60]. Consistent with these findings, our study revealed that the high-risk group has higher CD8+ T cell infiltration and lower Tregs infiltration than the low-risk group. In addition, we found that immune checkpoint molecules are highly expressed in the high-risk group. Recently, encouraging progress has been made in the immunotherapy targeting immune checkpoint molecules [61]. These results indicate that HCC patients in the high-risk group may benefit from immunotherapy.
We identified 25 most significant small molecule drugs for HCC treatment. The top 3 of them were HLI-373, medrysone and irbesartan. Irbesartan has been found to have an anti-tumor effect in colorectal cancer [62]. HLI373 is a Hdm2 ubiquitin ligase (E3) inhibitor which blocks Hdm2-mediated ubiquitylation, proteasomal degradation of p53 and activates p53-dependent transcription. It was found to induce apoptosis in tumor cell lines that express wild type p53 [63]. Therefore, we speculate that HLI-373 inhibit HCC by inhibiting Hdm2 and reducing the degradation of p53. As for medrysone, there is currently few evidences of its effectiveness in tumors. In short, we identified a series of potential drugs for HCC patients, but the actual effects of these drugs need to be further proved by more clinical trials.
Our study proposed a new prognostic signature based on CMRGs in HCC. However, some limitations are inevitable in present research. Firstly, this study was based on public database, more prospective clinical studies are needed to verify the accuracy of our model. But we have used external data to verify the reliability of our model as much as possible. Secondly, some clinical data are missing in the cohort, such as the etiology, grade and AFP level, which may affect the predictive ability of our model. Thirdly, the value of 5 CMRGs we identified need more in vitro and in vivo experiments to verify.
5. Conclusions
In summary, we constructed and validated a prognostic signature based on 5 CMRGs, which can be used for survival prediction, reflecting the tumor mutation and immune infiltration of HCC patients. What’s more, it may play a critical role in HCC immunotherapy. This study is expected to provide new insights into the precision diagnosis and treatment of HCC.
Authors contributions
XX designed the study. XW, QW and KW helped to revise the manuscript. ZQ, JZ and GF were responsible for data collection. RC and RW were responsible for data analysis. LT was responsible for data analysis and writing manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by grants from the National Key Research and Development Program of China (Grant number: 2021YFA1100500) and The Major Research Plan of the National Natural Science Foundation of China (Grant number: 92159202).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.07.030.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Tang L., Chen R., Xu X. Synthetic lethality: a promising therapeutic strategy for hepatocellular carcinoma. Cancer Lett. 2020;476:120–128. doi: 10.1016/j.canlet.2020.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Wang W., Wei C. Advances in the early diagnosis of hepatocellular carcinoma. Genes Dis. 2020;7:308–319. doi: 10.1016/j.gendis.2020.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao T., Bai D., Qian J., Zhang C., Jin S. The growth rate of hepatocellular carcinoma is different with different TNM stages at diagnosis. Hepatobiliary Pancreat Dis Int. 2021;20:330–336. doi: 10.1016/j.hbpd.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Yang W.J., Sun Y.F., Jin A.L., Lv L.H., Zhu J., Wang B.L., et al. BCL11B suppresses tumor progression and stem cell traits in hepatocellular carcinoma by restoring p53 signaling activity. Cell Death Dis. 2020;11 doi: 10.1038/s41419-020-03115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang T., Nie Y., Gu J., Cai K., Chen X., Li H., et al. Identification of mitochondrial-related prognostic biomarkers associated with primary bile acid biosynthesis and tumor microenvironment of hepatocellular carcinoma. Front Oncol. 2021;11:1–18. doi: 10.3389/fonc.2021.587479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/J.CELL.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Riscal R., Skuli N., Simon M.C. Even cancer cells watch their cholesterol! Mol Cell. 2019;76:220–231. doi: 10.1016/j.molcel.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villa G.R., Hulce J.J., Zanca C., Bi J., Ikegami S., Cahill G.L., et al. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell. 2016;30:683–693. doi: 10.1016/j.ccell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullen P.J., Yu R., Longo J., Archer M.C., Penn L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16:718–731. doi: 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- 11.Ma X., Bi E., Lu Y., Su P., Huang C., Liu L., et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goossens P., Rodriguez-Vita J., Etzerodt A., Masse M., Rastoin O., Gouirand V., et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019;29:1376–1389.e4. doi: 10.1016/j.cmet.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 13.Li J., Liu J., Liang Z., He F., Yang L., Li P., et al. Simvastatin and atorvastatin inhibit DNA replication licensing factor MCM7 and effectively suppress RB-deficient tumors growth. Cell Death Dis. 2017;8:1–12. doi: 10.1038/cddis.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sehdev A., Shih Y.A.C.T., Huo D., Vekhter B., Lyttle C., Polite B. The role of statins for primary prevention in non-elderly colorectal cancer patients. Anticancer Res. 2014;34:5043–5050. doi: 10.1200/jco.2013.31.15_suppl.1513. [DOI] [PubMed] [Google Scholar]
- 15.Gray R.T., Loughrey M.B., Bankhead P., Cardwell C.R., McQuaid S., O’Neill R.F., et al. Statin use, candidate mevalonate pathway biomarkers, and colon cancer survival in a population-based cohort study. Br J Cancer. 2017;116:1652–1659. doi: 10.1038/bjc.2017.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011 doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franz M., Rodriguez H., Lopes C., Zuberi K., Montojo J., Bader G.D., et al. GeneMANIA update 2018. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky311. W60–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D s, al g, d l, a j, s w, j h-c, m s, nt d, jh m, p b, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets Nucleic Acids Res 2019 47:D607–D613 10.1093/NAR/GKY1131. [DOI] [PMC free article] [PubMed]
- 20.Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw (2010) 33:1. Available at: /pmc/articles/PMC2929880/ [Accessed January 10, 2022]. [PMC free article] [PubMed]
- 21.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. A pathology atlas of the human cancer transcriptome. Science (80-) (2017) 357: doi:10.1126/science.aan2507. [DOI] [PubMed]
- 22.Yu G., Wang L.G., Han Y., He Q.Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. Omi A J Integr Biol. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayakonda A., Lin D.C., Assenov Y., Plass C., Koeffler H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017 181 (2017) 18:1–14. doi:10.1186/S13059-017-1349-1. [DOI] [PMC free article] [PubMed]
- 25.Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;171:1–20. doi: 10.1186/S13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T-HO, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. The Immune Landscape of Cancer. Immunity (2018) 48:812-830.e14. doi:10.1016/J.IMMUNI.2018.03.023 [DOI] [PMC free article] [PubMed]
- 27.Li T., Fu J., Zeng Z., Cohen D., Li J., Chen Q., et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020 doi: 10.1093/NAR/GKAA407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang Z, Lachmann A, Keenan AB, Ma’Ayan A. L1000FWD: Fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics (2018) 34:2150–2152. doi:10.1093/bioinformatics/bty060. [DOI] [PMC free article] [PubMed]
- 30.Lu M., Hu X.H., Li Q., Xiong Y., Hu G.J., Xu J.J., et al. A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol Cell Biol. 2013;5:404–415. doi: 10.1093/jmcb/mjt039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou T.Y., Zhuang L.H., Hu Y., Zhou Y.L., Lin W.K., Wang D.D., et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells article. Sci Rep. 2016;6:1–12. doi: 10.1038/srep30483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Souchek J.J., Baine M.J., Lin C., Rachagani S., Gupta S., Kaur S., et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139–1149. doi: 10.1038/bjc.2014.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao S., Soares F., Wang S., Wong C.C., Chen H., Yang Z., et al. CRISPR screens identify cholesterol biosynthesis as a therapeutic target on stemness and drug resistance of colon cancer. Oncogene. 2021;40:6601–6613. doi: 10.1038/s41388-021-01882-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seshacharyulu P., Rachagani S., Muniyan S., Siddiqui J.A., Cruz E., Sharma S., et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene. 2019;38:5265–5280. doi: 10.1038/s41388-019-0791-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Chen G, Zhao H. FDPS promotes glioma growth and macrophage recruitment by regulating CCL20 via Wnt / β -catenin signalling pathway. (2020)9055–9066. doi:10.1111/jcmm.15542. [DOI] [PMC free article] [PubMed]
- 36.Wu T., Tian J., Cutler R.G., Telljohann R.S., Bernlohr D.A., Mattson M.P., et al. Knockdown of FABP5 mRNA decreases cellular cholesterol levels and results in decreased apoB100secretion and triglyceride accumulation in ARPE-19 cells. Lab Investig. 2010;90:906–914. doi: 10.1038/labinvest.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furuhashi M., Ogura M., Matsumoto M., Yuda S., Muranaka A., Kawamukai M., et al. Serum FABP5 concentration is a potential biomarker for residual risk of atherosclerosis in relation to cholesterol efflux from macrophages. Sci Rep. 2017;7:1–10. doi: 10.1038/s41598-017-00177-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan L., Xiao H., Liao R., Chen Q., Peng C., Zhang Y., et al. Biomedicine & Pharmacotherapy Fatty acid binding protein 5 promotes tumor angiogenesis and activates the IL6 / STAT3 / VEGFA pathway in hepatocellular carcinoma. Biomed Pharmacother. 2018;106:68–76. doi: 10.1016/j.biopha.2018.06.040. [DOI] [PubMed] [Google Scholar]
- 39.Mussunoor S, Murray GI. The role of annexins in tumour development and progression. (2008)131–140. doi:10.1002/path [DOI] [PubMed]
- 40.Shen Y, Xiong W, Gu Q, Zhang Q, Yue J, Liu C, Wang D. Multi-Omics Integrative Analysis Uncovers Molecular Subtypes and mRNAs as Therapeutic Targets for Liver Cancer. (2021) 8:1–13. doi:10.3389/fmed.2021.654635. [DOI] [PMC free article] [PubMed]
- 41.Kitamura K., Okada Y., Okada K., Kawaguchi Y., Nagaoka S. Epigallocatechin gallate induces an up-regulation of LDL receptor accompanied by a reduction of PCSK9 via the annexin A2-independent pathway in HepG2 cells. Mol Nutr Food Res. 2017;61:1–12. doi: 10.1002/mnfr.201600836. [DOI] [PubMed] [Google Scholar]
- 42.Shafqat N., Turnbull A., Zschocke J., Oppermann U., Yue W.W. Crystal structures of human HMG-CoA synthase isoforms provide insights into inherited ketogenesis disorders and inhibitor design. J Mol Biol. 2010;398:497–506. doi: 10.1016/j.jmb.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 43.Puchalska P., Crawford P.A. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262–284. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J., Li M. SLC38A4 functions as a tumour suppressor in hepatocellular carcinoma through modulating Wnt / β -catenin / MYC / HMGCS2 axis. Br J Cancer. 2021 doi: 10.1038/s41416-021-01490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Suk F, Liao Y. Loss of HMGCS2 Enhances Lipogenesis and Attenuates the Protective Effect of the Ketogenic Diet in Liver Cancer. Cancers (Basel) (2020) 12: doi: 10.3390/cancers12071797. [DOI] [PMC free article] [PubMed]
- 46.Shen L., He J., Zhao Y., Niu L., Chen L., Tang G., et al. MicroRNA-126b-5p exacerbates development of adipose tissue and diet-induced obesity. Int J Mol Sci. 2021;22 doi: 10.3390/ijms221910261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao X, Qin W, Jiang Y, Yang Z, Yuan B, Dai R, Shen H, Chen Y, Fu J. OPEN ACADL plays a tumor-suppressor role by targeting Hippo / YAP signaling in hepatocellular carcinoma. npj Precis Oncol (2020) doi:10.1038/s41698-020-0111-4. [DOI] [PMC free article] [PubMed]
- 48.Guo Y., Peng Q., Hao L., Ji J., Zhang Z., Xue Y., et al. Dihydroartemisinin promoted FXR expression independent of YAP1 in hepatocellular carcinoma. FASEB J. 2022;36:1–14. doi: 10.1096/fj.202200171R. [DOI] [PubMed] [Google Scholar]
- 49.Zalba S., Hagen T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017;52:48–57. doi: 10.1016/j.ctrv.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma H.Y., Yamamoto G., Xu J., Liu X., Karin D., Kim J.Y., et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J Hepatol. 2020;72:946–959. doi: 10.1016/j.jhep.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giacomini I., Gianfanti F., Desbats M.A., Orso G., Berretta M., Prayer-Galetti T., et al. Cholesterol metabolic reprogramming in cancer and its pharmacological modulation as therapeutic strategy. Front Oncol. 2021;11:1–23. doi: 10.3389/fonc.2021.682911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z., Wang Y., Dou C., Sun L., Li Q., Wang L., et al. MicroRNA-1468 promotes tumor progression by activating PPAR-γ-mediated AKT signaling in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2018;37:1–14. doi: 10.1186/s13046-018-0717-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Chen S., Ling Y., Yu L., Song Y., Chen X., Cao Q., et al. 4-phenylbutyric acid promotes hepatocellular carcinoma via initiating cancer stem cells through activation of PPAR-α. Clin Transl Med. 2021;11 doi: 10.1002/ctm2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gou Q., Gong X., Jin J., Shi J., Hou Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget. 2017;8:60704–60709. doi: 10.18632/oncotarget.19610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen L.L., Wang W.J. P53 regulates lipid metabolism in cancer. Int J Biol Macromol. 2021;192:45–54. doi: 10.1016/j.ijbiomac.2021.09.188. [DOI] [PubMed] [Google Scholar]
- 56.Jiang J., Zheng Q., Zhu W., Chen X., Lu H., Chen D., et al. Alterations in glycolytic/cholesterogenic gene expression in hepatocellular carcinoma. Aging (Albany NY) 2020;12:10300–10316. doi: 10.18632/aging.103254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jhunjhunwala S., Jiang Z., Stawiski E.W., Gnad F., Liu J., Mayba O., et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014;15:436. doi: 10.1186/s13059-014-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishida N, Kudo M. Immune phenotype and immune checkpoint inhibitors for the treatment of human hepatocellular carcinoma. Cancers (Basel) (2020) 12: doi: 10.3390/cancers12051274. [DOI] [PMC free article] [PubMed]
- 59.Carr B.I., Metes D.M. Peripheral blood lymphocyte depletion after hepatic arterial 90Yttrium microsphere therapy for hepatocellular carcinoma. Int J Radiat Oncol Biol Phys. 2012;82:1179–1184. doi: 10.1016/j.ijrobp.2010.10.042. [DOI] [PubMed] [Google Scholar]
- 60.Sasaki A., Tanaka F., Mimori K., Inoue H., Kai S., Shibata K., et al. Prognostic value of tumor-infiltrating FOXP3+ regulatory T cells in patients with hepatocellular carcinoma. Eur J Surg Oncol. 2008;34:173–179. doi: 10.1016/j.ejso.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 61.Yau T., Kang Y.K., Kim T.Y., El-Khoueiry A.B., Santoro A., Sangro B., et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6:2–9. doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones M.R., Schrader K.A., Shen Y., Pleasance E., Ch’ng C., Dar N., Yip S., Renouf D.J., Schein J.E., Mungall A.J., et al. Response to angiotensin blockade with irbesartan in a patient with metastatic colorectal cancer. Ann Oncol. 2016;27:801–806. doi: 10.1093/annonc/mdw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitagaki J., Agama K.K., Pommier Y., Yang Y., Weissman A.M. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol Cancer Ther. 2008;7:2445–2454. doi: 10.1158/1535-7163.MCT-08-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.