

Abstract
Alzheimer's disease (AD) is a progressive neurodegenerative disease that primarily manifests as memory deficits and cognitive impairment and has created health challenges for patients and society. In AD, amyloid β-protein (Aβ) induces Toll-like receptor 4 (TLR4) activation in microglia. Activation of TLR4 induces downstream signaling pathways and promotes the generation of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and interleukin-1β (IL-1β), which also trigger the activation of astrocytes and influence amyloid-dependent neuronal death. Therefore, TLR4 may be an important molecular target for treating AD by regulating neuroinflammation. Moreover, TLR4 regulates apoptosis, autophagy, and gut microbiota and is closely related to AD. This article reviews the role of TLR4 in the pathogenesis of AD and a range of potential therapies targeting TLR4 for AD. Elucidating the regulatory mechanism of TLR4 in AD may provide valuable clues for developing new therapeutic strategies for AD.
1. Introduction
Dementia is the major cause of disability and death in people over 65 worldwide [1]. Approximately 50 million people worldwide have dementia, and this number will surpass 131 million by 2050 [2]. Alzheimer's disease (AD) is a complex age-related neurodegenerative disease and a major cause of dementia [3]. AD is characterized by progressive memory loss and learning deficits and is usually accompanied by language dysfunction, personality and behavioral changes, such as emotional apathetic state, depression and anxiety [4–7]. The AD incidence rate will markedly increase in the elderly with population ageing. Epidemiological studies have shown that the incidence of AD is about 10% in people over 65, rising to almost one-half in people over 85 [8]. This trend poses enormous challenges to AD patients and creates a heavy social burden [9].
AD is classified into two main forms: sporadic form and familial form. Nearly 90% of AD patients are sporadic. The condition impacts people at any age, but most individuals are over 65 years, often categorized as late-onset AD (LOAD) [10]. However, the cause of sporadic AD is still unclear. Familial AD usually occurs early and has a hereditary character. Genes encoding Aβ precursor protein (APP), presenilin 1 (PSEN1), and PSEN2 are causative genes [11]. The exact pathogenesis of AD is still unclear. Risk factors are aging, family history, unhealthy lifestyle, nutrition deficiency, and diabetes [12–14]. Among them, aging is the most well-known hazardous factor for AD [15]. Aging-associated cognitive dysfunction has become a predictable health threat in the elderly population. With society aging, AD could result in serious public health issues in the future.
The main pathological features of AD consist of amyloid plaques formed by abnormal aggregation of Aβ in the brain and neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein in neuronal cells [16, 17], which is associated with a range of neurodegenerative events involving microglia cell activation, neurite dystrophy, neuroinflammation, oxidative injury, and mitochondrial disorder [17–19]. Although several studies have focused on AD therapy, clinical researches on treatments targeting Aβ or tau aggregation have produced unsatisfactory results [20]. The approved agents for AD treatment generally alleviate symptoms rather than address the underlying cause or pathogenesis [21]. Thus, it is urgent to investigate the molecular mechanisms and discover prevention and treatment strategies.
Toll-like receptors (TLRs) are transmembrane pattern-recognition receptors (PRRs) of the innate immune system that identify pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides (LPS), and damage-associated molecular patterns (DAMPs), such as high-mobility family protein box 1 (HMGB1) and Aβ [3, 22]. Stimulation of TLRs by injury factors results in severe inflammation via the release of proinflammatory cytokines, including IL-1β and interleukin-18 (IL-18) [23]. So far, studies have confirmed 10 functional TLRs (TLR1-10) in humans and 12 in mice (TLR1–9, 11–13) [24]. TLRs 1-9 can be expressed in human microglia [25]. Besides, TLRs are produced in various immune-associated cells, including monocytes, macrophages, microglia, and astrocytes, and nonimmune cells, such as endothelial and epithelial cells, in the brain parenchyma [26, 27]. TLR activation by pathogens and damaged cells triggers the phagocytic property of phagocytes/microglia to remove pathogens, injured tissues, and accumulated wastes [28–31]. TLR4 is the first TLR discovered in humans and the most studied member of the TLR family [32, 33]. As a neuroinflammatory receptor, TLR4 is expressed on astrocytes, microglia, and neurons in the brain and plays a key role in neuroinflammation in AD progression by recognizing exogenous and endogenous ligands [34, 35]. Aβ, a well-known exogenous ligand of TLR4, is reported to bind to TLR4 on the surface of microglia and astrocytes to trigger the release of proinflammatory factors [36, 37]. In AD mouse models, activation of TLR4 can enhance the clearance of amyloid plaques through phagocytosis of glial cells, thereby decreasing amyloid plaque load [38, 39]. In summary, the TLR4 signaling pathway plays an important role in the pathological mechanisms of AD. This review summarizes the relationship between TLR4 and AD to provide effective therapeutic targets for future research of AD.
2. Toll-Like Receptor 4 Signaling Pathway
As is shown in Figure 1, TLR4 is activated mainly through two pathways. One is dependent on the myeloid differentiation factor 88 (MyD88) signaling pathway, and the other is dependent on the Toll/interleukin-1 receptor- (TIR-) domain-containing adaptor inducing interferon-β (TRIF) signaling pathway. MyD88 is a common connector molecule that activates the inflammatory response. The MyD88-dependent signaling pathway is an essential stimulator of NF-κB, which affects the subsequent expression of NF-κB [40]. Before ligand-induced signaling occurs, TLR4 first needs to associate with its extracellular binding partner, myeloid differentiation factor 2 (MD-2) [41, 42]. The TLR4-MD-2 complex, on binding to the ligand, may recruit another TLR4-MD-2 pair and create a homodimeric state [40]. Then, the receptor multimer transmits intracellular signals by the TIR structural domain of TLR4 [43, 44]. TLR4-MD2 dimerization recruits TIR domain-containing adaptor protein (TIRAP) and MyD88. Signals from MyD88 are transmitted to the interleukin-1 receptor-associated kinase (IRAK) family of protein kinases via the interaction between MyD88 and the IRAK4 death domain [45] to recruit the tumor necrosis factor receptor-associated factor 6 (TRAF-6)[46–48]. Together with ubiquitin-conjugating enzyme 13 (Ubc13) and ubiquitin-conjugating enzyme E2 variant 1 (Uev1A), TRAF6 triggers the activation of a complex composed of transforming growth factor-β-activated kinase (TAK1), TAK1-binding protein 1 (TAB1), and TAB2, causing the phosphorylation of TAK1 and TAB2/3 [49, 50]. One part of the TAK1 pathway is the activation of the IκB kinase (IKK) complex, which is an inhibitor of NF-κB, including IKK-α, IKK-β, and IKK-γ. NF-κB is composed of p65 and p50 dimers and is inactive if present in the cytoplasm with IκB [51]. NF-κB is activated, and proinflammatory factors (TNF-α and IL-1β) are released. The other part of the TAK1 pathway is the activation of mitogen-activated protein kinases (MAPKs), which involves p38, ERK, and JNK. MAPKs induce nuclear translocation of transcription factor complex AP-1, leading to the expression of cytokine genes [23, 52–54]. Moreover, the suppressor of cytokine signaling (SOCS1) is a cytokine-induced protein that negatively modulates cytokine signaling and directly downregulates TLRs signaling [55, 56]. SOCS1 can suppress TLR signaling by affecting NF-κB, MAPK activity, and p65 phosphorylation. The TLR4 pathway through MyD88 is ‘early stage' NF-κB activation, while TRIF-dependent activation is designated as ‘late stage' NF-κB activation. The coordination of ‘early' and ‘late' signaling is specific to TLR4. The complex composed of TLR4-LPS causes the formation of the endosome, which leads to the translocation of TRAM into the cytoplasm and activation of TRIF-dependent signaling pathways [23]. The N terminus of TRIF activates the TNF-receptor-related factor 3 (TRAF3) and TANK-binding kinase/IκB kinase (TBK1/IKKi) complex, resulting in phosphorylation of interferon regulatory factor 3 (IRF3) and IRF7, which triggers type I IFN gene expression, such as IFNβ. There is crosstalk between TRIF-dependent and MyD88-dependent pathways [57]. TRAF6 is another downstream activation target of TRIF. The C-terminus of TRIF interacts with receptor-interacting serine/threonine-protein kinase 1 (RIP1). TRAF6 is activated by RIP1, which activates the TAK1 complex and NF-κB [50, 57, 58]. In conclusion, TLR4 can cooperatively activate NF-κB through TRIF-dependent and MyD88-dependent pathways, but type I IFN can only be generated by activating TRIF-dependent pathways [59, 60].
Figure 1.
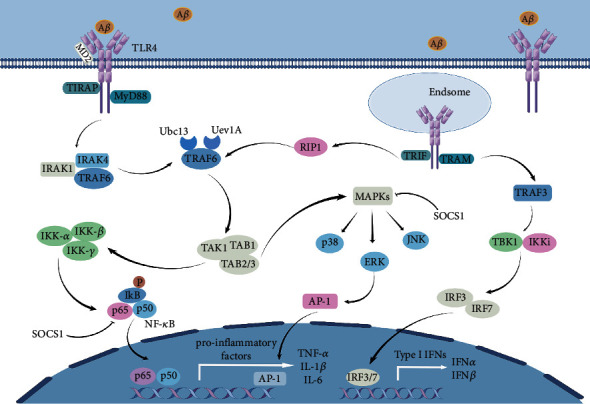
TLR4 binds to ligands and activates downstream pathways in both MyD88-dependent and MyD88-independent pathways. TLR4-MD2 recruits TIRAP and MyD88 and then signals to IRAKs. Then recruit TRAF-6, which with Ubc13 and Uev1A initiate the complex consisting of TAK1, TAB1, and TAB2/3 activation. The complexes of IKK-α, IKK-β, and IKK-γ are activated, promoting NF-κB entry into the nucleus and leading to the release of proinflammatory factors, such as TNF-α, IL-1β, and IL-6. In addition, MAPKs are activated, and MAPKs-induced p38, ERK and JNK lead to AP-1 nuclear translocation. SOCS1 inhibits the TLR4 signaling pathway by affecting NF-κB, MAPK activity, and p65 phosphorylation. TLR4-MD2 leads to endosome formation, resulting in TRAM translocation into the cytoplasm and activation of the TRIF-dependent signaling pathway. TRIF activates TRAF3 and the TBK1/IKKi complex, leading to phosphorylation of the interferon regulatory factors IRF3 and IRF7, which induce type I IFN gene expressions, such as IFNα and IFNβ. Besides, TRIF interacts with RIP1, which activates the TAK1 complex and NF-κB (this figure is made using the Figdraw).
3. Evidence for the Involvement of TLR4 in AD
Emerging studies have reported that TLR4 plays a crucial role in the pathogenesis of AD. Genetic profiling of the human brain after death showed the upregulated TLR4 expression in the frontal cortex of AD compared with age-matched controls [61]. Moreover, TLR4 agonist, LPS, was identified in hippocampal lysates and neocortex of AD patients. The levels were two to three times higher than in age-matched control groups. In particular, LPS control levels were even 26 times higher in some cases of advanced AD [22]. These findings indicate that LPS from the microbiome or bacterial infections may accumulate in the brain, leading to AD. APP/PS1 double transgenic mice mimic progressive cognitive deficits and neuropathological changes in humans, which are dependable, easy to operate, and commonly used in AD studies. Higher p-Tau levels and Aβ aggregation in APP/PS1 mouse brains were related to increased IL-1β, IL-6, TNF-α, NF-κB, and TLR4[62]. Likewise, TLR4 levels and IL-1β gene expression were significantly increased in hippocampal differentiated mice models without amyloidosis (i.e., entorhinal cortex damaged mice) compared with pseudodamaged mice [61]. The Aβ1-42 injection model is useful specifically in AD to explore the effect of inflammation [63]. Lateral ventricular injection of Aβ triggered inflammation, resulting in neuronal death, synaptic loss and cognitive dysfunction in WT mice but not in TLR4 knockout mice. Moreover, a selective TLR4 receptor antagonist inhibited Aβ-oligomer-induced microglial activation and memory dysfunction, which was not present in TLR4-deficient mice [64]. Therefore, the role of TLR4 in AD has attracted extensive attention.
TLR4 is closely associated with the occurrence of AD. Genetic association researches of TLR4 suggested that single nucleotide polymorphisms (SNPs) of TLR4 were associated with susceptibility to AD [65]. Asp299Gly polymorphism in the TLR4 gene could reduce inflammatory responses and prevent the development of sporadic AD. In the Italian cohort, a coding variant of TLR4 (rs4986790) was demonstrated to extend lifespan and reduce the risk of AD. Preclinical-stage familial AD (FAD) cases showed that the TLR4 variant was associated with a reduced risk of AD, better visuospatial and structural skills, and stable levels of IL-1β in cerebrospinal fluid over time [66–68]. Some TLR4 SNPs, such as rs10759930, rs12377632, rs7037117, and rs7045953, had neuroprotective effects in the Chinese Han population. rs11367 and rs1927907 were associated with an increased risk of AD [3]. Nevertheless, the exact function in AD of most of these gene variants has not been determined and still requires further research.
4. The Regulatory Mechanisms of TLR4 in AD
As is shown in Figure 2, TLR4 regulates AD progression by modulating inflammation, apoptosis, autophagy, and gut microbiota homeostasis.
Figure 2.
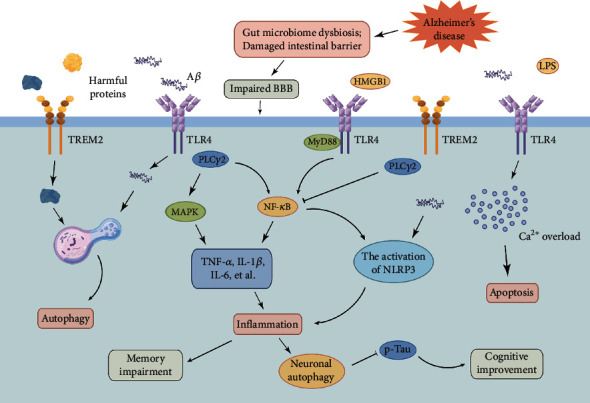
TLR4 mediates AD pathology by regulating inflammation, apoptosis, autophagy, and gut microbiota (this figure is made using the Figdraw).
4.1. TLR4 Activation Contributes to Inflammation
From a physiological point of view, inflammatory reactions exert a protective effect on the brain, but excessive inflammation is detrimental. Neuroinflammation is closely related to the pathogenesis of AD [69]. The characteristic of neuroinflammation observed in AD is activated microglial cells around Aβ deposits [70]. Currently, microglia perform a double-edged role in the pathogenesis of AD. On the one hand, microglia may exert beneficial effects by removing Aβ plaques. They consume harmful extracellular proteins by TLRs and TREM2 and decompose them further by lysosomal-dependent means, including autophagy. Microglia promote the transformation of the ‘resting' type into an anti-inflammatory phenotype, including homeostasis, regeneration, and neuroprotection [71]. On the other hand, following the disruption of microglial phagocytosis and degradation, the persistence of Aβ plaques and the release of inflammatory factors convert microglia from an anti-inflammatory phenotype to a proinflammatory phenotype related to inflammation, neuronal injury, and death [72, 73]. Abnormal activation of microglia can liberate various proinflammatory or cytotoxic cytokines, such as TNF-α, IL-1β, IL-6, nitric oxide (NO), reactive oxygen species (ROS), cyclooxygenase-2 (COX-2), and prostaglandin E2 (PGE2), which can significantly accelerate neuroinflammatory and neurotoxic reactions. Neuroinflammation ultimately results in neuronal cell death, synaptic degeneration, and cognitive impairment [74–78]. Currently, targeting neuroinflammation has become one of the essential therapeutic goals for AD, and understanding the mechanism underlying neuroinflammation in AD is a crucial breakthrough point for exploring the pathogenesis of AD. Researches have shown that TLR4 plays a key role in the pathogenesis of inflammation. The regulation mechanism of TLR4 on inflammation in AD is described below.
4.1.1. The Relationship Between Aβ and TLR4 in AD
In AD, insoluble Aβ macrofibrils and oligomers can be recognized by the innate immune system in the brain as a dangerous signal, resulting in activation of the innate immune response [79]. Aβ, the main toxic protein in AD patients, can trigger microglial activation and release of inflammatory cytokines in the brain, finally leading to AD-related neuroinflammation [80]. Studies have shown that Aβ combines with TLR4 on the surface of microglia and astrocytes and then activates the NF-κB and MAPK signaling pathways and triggers the release of proinflammatory factors, such as TNF-α, IL-1β, and IL-6 [81–83]. Overexpression of TLR4 was triggered by stimulation of aging rat neurons with Aβ oligomer, which made cells sensitive to subsequent TLR4 stimulation. In vitro, supernatant from Aβ-stimulated microglial cells was added to primary murine neurons, and TLR4 was found to be involved in Aβ-regulated microglia neurotoxicity and upregulating TNF-α, IL-1β, IL-10, and interleukin-17 (IL-17)[84]. In addition, LPS-cultured microglia incubated with Aβ42 for 24 h reduced Aβ42 in the culture medium by 50%, suggesting that TLR4 increased Aβ clearance [38]. This finding also implies that microglia can be activated through the TLR4 pathway to suppress Aβ deposition in the early stages of β-amyloidosis, thus protecting neurons against Aβ-mediated neurotoxicity [85]. Following the disease progression continued exposure of microglia to Aβ mitigates TLR4 response and activated microglia cannot clear Aβ deposition [86]. In cocultured neuron-glia cells, Aβ oligomers- (Aβo-) driven neuronal cell death was primarily attributed to the Aβ-sensitized TLR4 pathway of astrocytes and microglia through autocrine/paracrine mechanisms, suggesting another mechanism through TLR4-mediated Aβ toxicity. Studies showed that TLR4 was involved in Aβo-mediated memory loss in AD. A single lateral ventricle injection of Aβo in C57BL/6J mice activated glial cells, resulting in increased expression of pro-inflammatory factors and significantly impaired recognition and memory. TLR4 receptor antagonists eliminated the harmful effects of Aβo on memory, and Aβo had no effect on glial cell activation or memory in TLR4-knockout mice [64]. In summary, Aβ is involved in the progression of inflammation in AD through the TLR4 signaling pathway and may impact memory.
4.1.2. The Relationship Between HMGB1 and TLR4 in AD
High-mobility family protein box 1 (HMGB1) is a typical DAMP released by necrotic or excitatory neurons. HMGB1 protein is involved in initiating and activating neuroinflammation under pathologic conditions and the pathogenesis of neurodegenerative disorders, such as AD. Research suggested that the expression of HMGB1, RAGE, TLR4-NF-κB, and inflammatory cytokines increased in the nucleus and cytoplasm of hippocampal neurons in Aβ25-35-induced AD-associated neuroinflammation models [87]. HMGB1 exerted its biological property by directly combining with receptors for advanced glycation end products (RAGE) and TLR4, acting as a chemoattractant or proinflammatory factor [88]. HMGB1 gene silencing alleviated inflammation induced by Aβ in hippocampal neuron cultures [87]. Extracellular HMGB1 acted as a chaperone of Aβ, decreased microglial Aβ clearance, and interacted with RAGE and TLR4, which participated in microglial Aβ phagocytosis, leading to AD progression [38, 89, 90]. Notably, subcutaneous injections of anti-HMGB1 antibodies prevented neurodegeneration and reversed cognitive deficits, even in the presence of Aβ plaques [91].
Disruption of learning and memory are important features of AD. In the new object recognition test (NORT), a decrease in the new object preference index demonstrated the effect of HMGB1 injection on memory encoding. When HMGB1 was injected into control mice and TLR4 and RAGE knockout mice, the damage to memory coding was similar [92]. In addition, TLR4 antagonists blocked the amnesic role of HMGB1 in RAGE knockout mice [92]. These studies suggest that HMGB1-induced memory dysfunction is regulated by RAGE and TLR4, but the exact mechanisms are still unclear. Myristoylated alanine-rich C-kinase substrate (MARCKS), a submembrane protein, helps stabilize the actin network. Studies have shown that HMGB1 released by necrotic or overexcited neurons binds to TLR4 and activates MAPKs, inducing MARCKS phosphorylation to initiate neurite degeneration, resulting in impaired memory function [91]. Thus, these results suggest that HMGB1 is involved in AD progression through the TLR4 pathway.
4.1.3. The Relationship Between TREM2 and TLR4 in AD
TREM2 is a transmembrane receptor (one of the members of the TREM family) expressed on the surface of several myeloid cells, including microglia, monocytes, and macrophages. It is an important innate immune receptor in the brain, which regulates microglia survival, proliferation, biosynthesis, and factor release; moreover, it has a protective effect on Aβ pathology [93, 94]. Reports indicated that TREM2 was expressed at higher levels in the brains of AD patients than in normal controls [95]. Polymorphisms in TREM2 genes were associated with a higher risk of LOAD [96]. Recent epidemiological surveys suggested that serum levels of TREM2 might serve as a prospective new predictive biomarker for the incidence of dementia [97, 98]. Studies have shown that TREM2 plays an important role in the protective mechanisms of AD, in which TLRs play a key role. Reports showed that TREM2 negatively regulated TLR4-mediated inflammation [99, 100]. The APP/PS1 mice had higher levels of TLR4 and TREM2 in their brains; TLR4 showed continuous upregulation, and TREM2 levels significantly decreased in APP/PS1 mice after LPS treatment, suggesting that LPS-induced TLR4 hyperactivity inhibited the negative regulation of inflammation in TREM2[100]. Moreover, overexpression of TREM2 in microglia inhibited TLR4 levels, leading to altered expression of downstream effectors of TLR4 (ERK, p38, and p65) and proinflammatory factors (IL-6, IL-1β, and TNF-α), whereas silenced TREM2 gene elevated TLR4 levels [101]. These results suggest that TREM2 inhibits neuroinflammatory responses by downregulating the TLR4 pathway.
Phospholipase Cγ2 (PLCγ2) is an intracellular enzyme that can cleave membrane phosphatidylinositol-4,5-diphosphate (PIP2). The variation of the PLCγ2 gene is associated with AD [102]. Recent studies have shown that PLCγ2 regulates various microglial functions via numerous upstream molecules, such as TREM2 and TLR ligands. TREM2 alleviated PLCγ2-mediated inflammation, and TLR4/PLCγ2-reliant inflammatory signals were amplified without TREM2 [2]. The exact relationship between TREM2 and TLR4 in AD needs further study.
4.1.4. The Relationship Between NLRP3 and TLR4 in AD
The activation of nod-like receptor protein 3 (NLRP3) inflammasome is an early pathogenic AD event. Extracellular fiber Aβ activated the typical inflammasome pathway by triggering the TLR4/MyD88/NF-κB signal pathway. Besides, oligomers and fiber Aβ could interact directly with NLRP3 and ASC, leading to activation of NLRP3 inflammasome [103]. Microglia activation was induced by injections of fibrin Aβ into the striatum of mice, whereas microglia activation was inhibited in MyD88-deficient or ASC-deficient mice, indicating that aggregated Aβ produced a signaling cascade, including MyD88 and NLRP3 inflammasome [104]. Furthermore, MyD88 deletion reduced microglial activation and brain Aβ load and improved behavioral dysfunction in APP/PS1 mice [105, 106]. These findings indicate that the TLR4/MyD88 pathway participates in the initiation of NLRP3 activation in AD mouse models.
4.2. TLR4 Activation Contributes to Neuronal Apoptosis in AD
Aβo facilitates Ca2+ entry and mitochondrial Ca2+ overload, resulting in neuronal cell death. As a TLR4 receptor agonist, LPS enhanced the concentration of cytoplasmic Ca2+, resulting in apoptosis. Studies have found that LPS or Aβo exposure for 48 h could not lead to apoptosis of young cultured cells. The combination of Aβo and LPS enhanced Ca2+ response and neuronal death in cultured young rat neurons, and this effect significantly increased in aged neurons [107]. These data suggested that Ca2+ signals activated by TLR4 and induced by Aβo might crosstalk with each other, thereby boosting cell death in the neurons, especially in senescence. This finding may be related to Aβo-induced changes in TLR4 expression. In conclusion, the TLR4 signaling pathway is closely associated with AD by triggering apoptosis.
4.3. TLR4 Activation Contributes to Neuronal Autophagy in AD
Autophagy is a mechanism that promotes the clearance of damaged organelles or abnormal intracellular protein aggregation related to multiple CNS diseases involving tau and Aβ in AD. Transcription of autophagy-associated genes, such as Beclin1, ATG5 and ATG7, was downregulated in the elderly brain [108]. Studies demonstrated that promoting autophagy could prolong the longevity of numerous organisms, from yeast to mice, and attenuate the progression of age-related disorders. Therefore, autophagy may provide protective roles, especially on aging neurons. There is increasing evidence of impaired autophagy in AD [109–111]. Histological analysis revealed the accumulation of autophagosomes and autolysosomes in AD brain neurons [112]. Studies showed that TLR4 ligand promoted or suppressed autophagy, which might had opposite roles on protein and organelle turnover. Qin et al. found that chronic mild TLR4 stimulation might boost neuronal autophagy, reduce brain p-tau protein levels, and ameliorate cognitive dysfunction in transgenic AD mice [39]. Contrarily, reports showed that chronic TLR4 activation activated premature organelle autophagy, leading to impaired neuronal signal and cell death [113]. Moreover, sustained IL-4-induced activation of TREM2 led to increased lysosomal and LC3II/I expression, increased microglial autophagy, and impaired CARD9-TLR4 pathway, contributing to improved cognitive function in AD mice. Thus, the TLR4 signaling pathway is closely related to AD by regulating autophagy.
4.4. TLR4 Activation Regulates Gut microbiota in AD
About 37 trillion microbes reside in the human body; 70% of these microbes are found in the gut [114]. These microorganisms can protect the host from pathogen invasion, facilitate digestion and absorption of the host, and modulate drug metabolism [115]. Recent research has shown that histological and behavioural presentations of AD are associated with dysbiosis of the gut microbiome. In an AD mouse model, antibiotic-induced perturbations of gut microbial diversity affected neuroinflammation and amyloidosis. Changes in gut microbiome composition due to aging may result in chronic low-grade inflammation, a hazardous factor associated with cognitive decline in older adults. Brain amyloidosis in cognitively impaired older adults was associated with proinflammatory intestinal flora and markers of peripheral inflammation. AD was associated with damage to the intestinal barrier, through which LPS and proinflammatory factors produced by pathogens entered the body, disrupted the integrity of the blood-brain barrier (BBB), enhanced the uptake of Aβ and α-syn in the brain, and activated microglia-induced immune reactions through the LPS/TLR4/NF-κB pathway, resulting in neuronal loss [116, 117]. Studies have suggested that the effects of aging gut microbiome dysregulation on cognitive functioning may be related to the LPS-activated TLR4/NF-κB signaling and neuroinflammation in the brain. These findings suggest that inflammation, apoptosis, autophagy, and gut microbiota are closely related to AD via triggering TLR4 signaling. Therefore, a better understanding of the regulatory mechanism of TLR4 in AD may help identify potential therapeutic approaches to prevent AD.
5. Application of Therapies Targeting TLR4 in AD
As represented in Table 1, studies showed that multiple AD therapies targeting TLR4 exerted protective effects primarily by inhibiting the expression of TLR4 signaling pathway molecules, suppressing microglia activation, reducing neuronal death, and improving learning and memory function.
Table 1.
Summary of AD therapeutic approaches targeting TLR4.
| Intervention | Animal model | Treatment | Mechanism | Reference |
|---|---|---|---|---|
| Hesperetin | Aβ1-42-induced AD model in C57BL/6N mice | 50 mg/kg treatment for 6 weeks | (a) Inhibiting oxidative stress by reducing LPO and ROS and increasing Nrf2 and HO-1 (b) Inhibiting neuroinflammation by reducing TLR4, p-NF-κB, TNF-α, and IL-1β (c) Inhibiting apoptosis by reducing Bax, caspase-3, and PARP-1 (d) Reducing memory dysfunction by increasing the levels of syntaxin, SNAP-25, PSD-95, Syp, and SNAP-23 |
[129] |
|
| ||||
| Soybean isoflavone (SIF) | Aβ1-42-induced AD model in Wistar rats | 80 mg/kg treatment for 14 days | (a) Improving learning and memory skills (b) Inhibiting levels of proinflammatory factors TNF-α and IL-1β (c) Inhibiting Aβ-induced elevation of TLR4 levels and NF-κB expression in the nucleus |
[131] |
|
| ||||
| GX-50 | APP transgenic AD model | 1 mg/kg/day for 2 months at 5 months of age | (a) Inhibiting the expression of TNF-α, IL-1β, NO, PGE2, and iNOS and COX-2 in microglia of Aβ-treated rats (b) Inhibiting microglia activation and the expression of IL-1β, iNOS, and COX-2 in APP transgenic mice (c) Inhibiting the activation of NF-κB and MAPK cascades (d) Reducing TLR4, MyD88, and TRAF6 expressions in vitro and in vivo |
[73] |
|
| ||||
| ProBiotic-4 | 9-month-old senescence-accelerated mouse prone 8 (SAMP8) mice | 2 × 109 CFU once daily for 12 weeks | (a) Reducing IL-6 and TNF-α levels, plasma, and brain LPS concentrations, TLR4 expression and NF-kB nuclear translocation in the brain, resulting in improved cognitive dysfunction in aged mice | [153] |
|
| ||||
| MG136-pMG36e-Glucagon-like peptide-1 (GLP-1) | LPS (0.25 mg/kg) for 7 days in male C57BL/6 mice | Administered in drinking water for 14 days | (a) Attenuating neuroinflammation and improving LPS-induced memory impairment by downregulating the TLR4/NF-κB pathway | [159] |
|
| ||||
| TAK-242 | Male APP/PS1 transgenic mice | 2 mg/kg/day for 28 successive days | (a) Inhibiting TLR4 and Bax levels, significantly improves neurological function (b) Promoting the conversion of microglia from the M1 to the M2 phenotype (c) Inhibiting MyD88/NF-κB and NLRP3 signaling pathways |
[33] |
|
| ||||
| Cattle encephalon glycoside and ignotin (CEGI) | Male APP/PS1 transgenic mice; C57BL/6J mice | 6.6 ml/kg/day CEGI for 30 days | (a) Inhibiting TLR4 expression and NF-κB p65 phosphorylation to exert antineuroinflammatory effects | [173] |
|
| ||||
| Fasudil | Male APP/PS1 transgenic mice | 25 mg/kg/day for 2 months | (a) Inhibiting microglia activation and promoting their conversion to an anti-inflammatory phenotype by suppressing TLR4, MyD88, and NF-κB expression, and promoting astrocyte conversion from A1 to A2 phenotype | [179] |
|
| ||||
| Atorvastatin | Aβ1-42-induced AD model in Sprague-Dawley male rats | 5 and 10 mg/kg from 3 weeks before to 6 days after Aβ1–42 injections | (a) Reducing TLR4, TRAF6, and NF-κB levels, inhibiting microglia and astrocyte activation, and improving spatial learning ability and memory impairment | [37] |
|
| ||||
| Tetrandrine | APP/PS1 transgenic 5XFAD mice; Aβ1-42-induced BV2 cells | 10, 20, and 40 mg/kg every 2 days from the age of 5 months to 7 months | (a) Dose-dependently improving cognitive performance in mice (b) Promoting reduced amyloid plaque deposition and hippocampal apoptosis in the brain (c) Inhibiting the expression of inflammation-related genes (TNFα, IL-1β, and IL-6) and TLR4, p65, iNOS, and COX-2 |
[193] |
|
| ||||
| Pinoresinol diglucoside (PDG) | Aβ1-42-induced AD model in BALB/c mice | 5 and 10 mg/kg every day for 3 weeks | (a) Reversing Aβ1-42-induced memory impairment in mice (b) Inhibiting the release of proinflammatory cytokines (TNF-α and IL-1β), ROS, and MDA and promotes the activity of antioxidant enzymes (SOD and CAT) (c) Upregulating the ratio of Bcl-2/Bax and downregulates the expression of Cyt C and cleaved caspase-3, thereby inhibiting neuronal apoptosis (d) Reducing TLR4 expression and NF-κB p65 activation and promotes Nrf2 and HO-1 expression |
[203] |
|
| ||||
| Geniposidic acid (GPA) | APP/PS1 transgenic C57BL/6J mice | 25, 50, and 75 mg/kg every day for 90 days | (a) Improving spatial learning and memory abilities and reducing brain Aβ deposition in mice (b) Inhibiting the activation of astrocytes and microglia, downregulating the expression of proinflammatory cytokines and iNOS, and upregulating the expression of anti-inflammatory cytokines and Arg-1 (c) Downregulating the expression of TLR2, TLR4, RAGE, MyD88, and NF-κB p65 |
[204] |
|
| ||||
| Chotosan (CTS) | Aβ1-42-induced AD model in ICR male mice | 375, 750 mg/kg/day for 3 weeks | (a) Improving memory impairment in mice (b) Decreasing TLR-4 and NF-κB p65 expression and reducing the release of proinflammatory cytokines (including TNF-α and IL-1β) in the hippocampus (c) Increasing the Bcl-2/Bax ratio and decreasing caspase-3 activity, thereby inhibiting neuronal apoptosis |
[213] |
5.1. Flavonoids
Flavonoids are most helpful for treating neurodegenerative diseases due to their anti-inflammatory, antioxidant, and antiapoptotic properties [118, 119]. Sinensetin (SIN), a polymethoxyflavonoid, is a significant active compound predominant in citrus plants and indicated to have antitumor, antioxidant, and anti-inflammatory pharmacological activities [120–123]. Zhi et al. found that SIN preconditioning inhibited Aβ25-35-regulated upregulation of TLR4 and nuclear translocation of NF-κB p65. SIN protected SH-SY5Y cells from Aβ25-35-regulated neurotoxicity by inhibiting oxidative stress, inflammation, and apoptosis via suppressing the TLR4/NF-κB pathway [124]. Moreover, overexpression of TLR4 eliminated the protective role of SIN. Hesperidin is a bioactive flavonoid produced by hesperidin and found in citrus fruits [125]. Reports showed that hesperidin exerted neuroprotective effects in diverse models [126, 127]. Hesperidin is a powerful free-radical scavenger that facilitates cellular anti-inflammatory properties [128]. Muhammad et al. found that hesperidin significantly inhibited the expression of TLR4, P-NF-κB, TNF-α, and IL-1β induced by Aβ1-42, showing that hesperidin had the same effect as specific inhibitors [129]. This finding demonstrated that hesperidin effectively mitigated Aβ-induced pathological changes in mice and cells by modulating TLR4/NF-κB to inhibit neuroinflammation and was a promising and reasonable neuroprotective agent. Soybean isoflavone (SIF) is a type of soybean phytochemical with anti-inflammatory, antioxidant, and antiosteoporosis properties. Youn et al. demonstrated that plant polyphenols could suppress LPS-induced TLR4 dimerization and thus regulate TLR-regulated inflammation [130]. As a polyphenol, SIF may also act on TLR4 and suppress the toxicity of Aβ, further attenuating downstream inflammatory pathways. Ding et al. demonstrated that SIF treatment significantly reversed Aβ1-42-induced elevated expression of TLR4 and NF-κB p65 subunit, decreased expression of TNF-α and IL-1β, improved Aβ-induced brain injury, and ameliorated learning and memory in rats [131]. These results suggested that SIF could play a neuroprotective effect in AD through the TLR4/NF-κB signaling pathway, which was beneficial to AD treatment. In summary, these flavonoids can inhibit the expression of TLR4 and play a significant role in AD.
5.2. Medications
5.2.1. Circumdatin D
Acetylcholine (ACh) is a crucial neurotransmitter in memory and learning processes. The functional decline of cognition in AD patients is related to a lack of the neurotransmitter Ach in the brain. Acetylcholinesterase (AChE) is a hydrolytic enzyme that hydrolyses acetylcholine to choline and acetic acid [132, 133]. The study found a 20% increase in plasma levels of AChE in AD patients compared to age- and sex-matched controls [134]. Suppression of AChE averted the breakdown of ACh and subsequently added its concentration and duration of action, thought to be of clinical benefit for patients with AD. AChE inhibitors currently account for four of the five treatments prescribed for AD. These cholinergic drugs are less effective in curing the disease and can only relieve some AD symptoms by facilitating cholinergic signaling [135, 136]. Therefore, it is essential to develop novel acetylcholinesterase inhibitors for therapeutic intervention in AD. Natural products (particularly alkaloids) are potential sources for novel AChE inhibitors [137]. Circumdatins are a group of alkaloids of marine origin with dual inhibitory activities of AChE and proinflammatory reactions. Among them, circumdatin D showed a promising neuroprotective effect through the multitarget strategy. Zhang et al. found that cyclostatin D regulated TLR4-mediated NF-κB, MAPKs, and JAK/STAT signaling pathways associated with inflammation in LPS-stimulated BV-2 cells and protected primary neurons against LPS-regulated neurotoxicity [138]. Therefore, circumdatin D may be a potential drug to exert the neuroprotective effect of AD via TLR4.
5.2.2. GX-50
Sichuan pepper is one of the main spices used in Chinese food and has anti-inflammatory, antifungal, and analgesic activities [139]. GX-50, a natural ingredient extracted from Sichuan pepper, is a promising agent for AD therapy [140–142]. The overactivated microglial cells, proinflammatory factors, and continued deposition of Aβ plaques led to a positive feedback loop of persistent and irreversible inflammation, ultimately leading to the progression of AD. Studies have suggested that GX-50 may be involved in this inflammatory circuit. Studies showed that GX-50 could directly decrease the aggregation of Aβ oligomer, reduce neuroinflammation, and improve the cognitive function of APP transgenic mice. Shi et al. found that GX-50 exerted an anti-inflammatory role on Aβ-induced microglial overactivation through a mechanism involving the TLR4-mediated NF-κB/MAPK signaling pathway. GX-50 inhibited Aβ-stimulated activation of TLR4, subsequently inhibited MyD88 and TRAF6 recruitment, resulting in inhibition of NF-κB and MAPK, and inhibited Aβ-induced inflammatory reaction [73]. These results suggest that TLR4/NF-κB/MAPK may be an important pathway in GX-50 treatment of AD.
5.2.3. Resveratrol
Several epidemiological data suggest that red wine consumption in moderation is related to a lower incidence of dementia and AD [143–145]. Studies in mouse AD models have shown that red wine consumption reduced brain amyloid deposition and Aβ-related cognitive impairment [146, 147]. Resveratrol is a natural polyphenol found in red wine. Recent studies have shown that this polyphenol is related to antiamyloidosis and neuroprotective activities of cell lines in vitro and mice in vivo. The relative abundance of resveratrol found in red wine is related to possible neuroprotective effects, thus interpreting the advantageous roles of wine consumption on AD. Resveratrol may have anti-inflammatory activities in multiple systems, involving activated microglia [148]. Evidence showed that resveratrol significantly reduced Aβ-induced microglial inflammation in vitro and in vivo. Resveratrol effectively inhibited LPS-regulated inflammation in microglia BV-2 cells and RAW 264.7 macrophages. Resveratrol preferentially antagonized the IKK/IκBα/NF-κB signaling pathway under LPS stimulation, repressed NF-κB transcription and the expression of multiple NF-κB target genes, such as TNF-α and IL-6. Moreover, oral resveratrol reduced the number of activated microglia related to cortical amyloid plaque formation in an in vivo study of a mouse model of amyloid deposition in the brain [149]. In conclusion, resveratrol negatively controls Aβ-induced microglial inflammation in vitro and in vivo, which may be associated with TLR4-related signaling pathways and is a potential treatment for AD.
5.3. Bacteria Associated with the Gut
Probiotics offer a potential preventive effect on AD progression [150, 151]. Multiple probiotics reduce the Mini-Mental State Examination (MMSE) score and some metabolic characteristics in patients with AD. A probiotic mixture was identified to regulate the gut microbiome and ameliorate memory dysfunction and oxidative stress in rats injected with β-amyloid protein (1e42)[152]. Probiotic-4 is a probiotic preparation made of Lactobacillus casei, Bifidobacterium lactate, Lactobacillus acidophilus, and Bifidobacterium bifidum. Studies have shown that probiotic-4 regulates age-related gut microbiome dysregulation improving microbiome axis defects and cognitive dysfunction. The mechanism of probiotic-4 neuroprotective effects was related to inhibition of TLR4 and RIG-I mediated NF-κB pathway and inflammatory reactions [153]. Glucagon-like peptide-1 (GLP-1) is a type of endogenous hormone secreted by endocrine cells in the ileum that facilitates insulin secretion and suppresses glucagon secretion, lowering blood glucose in a glucose-dependent manner [154]. Studies have shown that GLP-1 plays a neuroprotective role in affecting the proliferation and apoptosis of neural cells; ameliorating learning, memory, and motor deficits; reducing the deposition of Aβ plaques in the brain; reducing the loss of dopaminergic neurons; and promoting neural regeneration [155]. GLP-1 also reduced hyperphosphorylated tau and neurofilament proteins in rodents to improve AD-like neurodegeneration, which was related to memory amelioration and learning dysfunction [156]. GLP-1 has demonstrated efficacy in clinical trials in patients with AD [157, 158]; however, GLP-1 undergoes easy degradation, and therefore, continuous intravenous infusions or continuous subcutaneous injections are necessary to perform therapeutic roles, thus limiting its clinical application [154]. The engineered strain may be a novel intervention for treating AD through alleviating neuroinflammation. Fang et al. constructed an engineered Lactococcus lactis strain MG1363-PMG36E-GLP-1 capable of continuously expressing GLP-1. They found that MG1363-PMG36E -GLP-1 reduced neuroinflammation by downregulating the TLR4/NF-κB signaling, upregulated the AKT/GSK3β pathway, and restored the disturbed microbiome to normality, remarkably alleviating spatial learning and memory impairment in AD mice [159]. These results provide theoretical support for applying GLP-1 engineered strain in treating AD through TLR4.
5.4. Molecular Inhibitors and Agonists
5.4.1. TAK-242
TAK-242 is a selective inhibitor of TLR4 with a low molecular weight that destroys the interaction of TLR4 with the cohesive molecules and suppresses its downstream pathway. Due to its high lipid solubility and low molecular weight, TAK-242 has the ability to cross the BBB [160]. Cui et al. found that TAK-242 could protect APP/PS1 transgenic AD mice from injury. They proved that TLR4 expression increased in mice with AD, and inhibition of TLR4 triggered microglia from an inflammatory M1 phenotype to a protective M2 phenotype and protected neurons from cytotoxicity of activated BV2 microglia by downregulating MyD88/NF-κB and NLRP3 signaling pathways in AD [33].
5.4.2. Monophosphoryl Lipid A (MPL)
Monophosphoryl lipid A (MPL), a chemically detoxified part of the lipid A, is an LPS-derived TLR4 agonist that displays distinct immunoregulatory activities at nonpyrogenic doses [161, 162]. Moreover, MPL is secure in humans, and millions of patients have received it as part of several vaccine formulations, including Cervarix [163]. Studies have shown that chronic systemic administration of MPL can stimulate the phagocytosis of innate immune cells, induce a moderate inflammatory response, reduce the brain Aβ load, and improve the cognitive function of AD mice. These results have shown that MPL stimulates p38 and facilitates Aβ uptake selectively while preventing the production of potentially harmful proinflammatory factors triggered by other pathways [162]. Studies showed that p38, ERK, JNK, and NF-κB induced TLR-mediated factor production, but p38 could upregulate scavenger receptor expression and induce phagocytic activity [164, 165]. Although MPL did not stimulate ERK or JNK and induced NF-κB to a lower extent than LPS, it strongly activated p38 and drove SR-A expression in microglia. These findings showed that MPL stimulated p38, facilitated Aβ uptake selectively, and avoided the production of potentially harmful proinflammatory factors triggered by other signal pathways. TLR4 stimulates the detoxification ligand MPL to significantly mitigate AD-related pathology, and its application in treating AD remains to be explored. Taken together, molecular inhibitors and agonists focusing on the regulation of the TLR4 may be potential treatment strategies for AD.
5.5. Clinical Drugs
5.5.1. Alogliptin
Type 2 diabetes mellitus (T2DM) is the main risk factor for progression to AD. Therefore, antidiabetic agents may be effective treatments for AD. Dipeptidyl peptidase-4 (DPP-4) suppressors are widely known antidiabetic agents for managing T2DM via attenuating endogenous GLP-1 degradation, which leads to inhibition of glucagon release and glucose-dependent rise in insulin secretion [166]. In addition to the glycemic effect, DPP-4 suppressors have neuroprotective effects. Among them, sitagliptin, vildagliptin, and saxagliptin inhibited the accumulation of Aβ and abnormal phosphorylation of tau to alleviate inflammation and reverse behavioral defects in AD rats [167, 168]. Studies showed that alolliptin, a highly selective and effectual DPP-4 suppressor, could ameliorate cognitive dysfunction and depressive symptoms in obese ApoE-/- mice. Ayman et al. found that alolliptin reversed the LPS-activated TLR4/MyD88/NF-κB signaling pathway and reduced TNF-α and IL-6 levels, thus playing a neuroprotective role [169]. This finding suggests that alolliptin may improve amyloidation-related cognitive decline by inhibiting TLR4 activation-related neuroinflammation and may be a potential treatment for AD.
5.5.2. Cattle Encephalon Glycoside and Ignotin (CEGI)
Cattle encephalon glycoside and ignotin (CEGI) involves monosialotetrahexosyl ganglioside (GM1), hypoxanthine, free amino acids, and polypeptides [170]. In China, CEGI injections are commonly used for treating central and peripheral nerve injury disorders, such as acute ischemic stroke and diabetic peripheral neuropathy [171, 172]. Gao et al. found that CEGI inhibited activated microglia in APP/PS1 mice, suggesting that CEGI may be a prospective agent for treating AD. In addition, they showed that CEGI inhibited elevated TLR4 levels in APP/PS1 mice and Aβ-induced BV2 cells and inhibited NF-κB p65 phosphorylation in vitro and in vivo [173]. Based on these results, the authors indicated the role of CEGI in antineuroinflammation by inhibiting the TLR4/NF-κB signaling pathway. This result provides new theoretical support for applying CEGI in managing AD.
5.5.3. Fasudil
Several studies have shown that the Rho/Rho kinase (ROCK) signaling pathway participates in many pathological processes, such as oxidative stress, inflammatory cell migration, and immune cell activation, which may influence the occurrence and progression of various neurodegenerative diseases, such as AD. Studies showed that ROCK inhibitors could prevent neuronal injury, neuroinflammation, and demyelination. Thus, ROCK has become a possible target for new therapies for AD [174, 175]. Fasudil is an effective ROCK inhibitor shown to have several neuroprotective effects in the CNS: promoting axonal regeneration, inhibiting inflammatory responses, and protecting against Aβ-induced neurodegeneration [176–178]. Guo et al. found that microglia-astrocyte crosstalk through the TLR4/MyD88/NF-κB signaling pathway played an important role in the neuroinflammation of AD. They observed that fasudil inhibited the expression of TLR4, MyD88, and NF-κB in APP/PS1 Tg mice, thus suppressing microglia activation and inducing their transformation to an anti-inflammatory phenotype and further transforming astrocytes from an A1 phenotype to an A2 phenotype; the cognitive defects of APP/PS1 Tg mice showed improvement [179]. These results suggest that fasudil may play a therapeutic role in AD through the TLR4 signaling pathway.
5.5.4. Atorvastatin
Statins are 3-hydroxy-3-methylglutaryl-coA (HMGCoA) reductase suppressors and the most efficacious cholesterol-lowering drugs of all lipid-lowering medicines [180]. Clinical data showed a decrease in the incidence of AD among statin users compared to nonusers [181]. Studies have shown that the main effect of statins on AD patients is to reduce the inflammatory reaction caused by microglial activation rather than the accumulation of Aβ [182]. Atorvastatin, a lipophilic member of the statin family, has shown a promising therapeutic anti-inflammatory role [183]. Research showed that after 3 weeks of atorvastatin pretreatment, the levels of the anti-inflammatory factor IL-4 enhanced in the hippocampus and the intracellular expression of Aβ-induced proinflammatory factors TNF-α, IL-6, and IL-1β decreased [184, 185]. We found that chronic treatment with high-dose atorvastatin significantly reduced the levels of TLR4, TRAF6, and NF-κB, inhibited the activation of microglia and astrocytes, alleviated hippocampal pathologic alterations and neuronal apoptosis, and improved spatial learning ability and memory impairment in AD rats. They also found a tendency for continuous low-dose atorvastatin administration to ameliorate Aβ-induced cognitive dysfunction; however, the difference was not prominent, suggesting that the neuroprotective role of atorvastatin in AD appears to depend on the dose and treatment regimen used [37]. In conclusion, appropriate modification of the TLR4-mediated signal pathway may be one of the potent targets for atorvastatin treatment of Aβ-regulated neurotoxicity in AD.
5.6. Traditional Chinese Therapy
5.6.1. Chinese Herbal Medicine
(1) Tetrandrine. Tetrandrine is a natural product isolated from the Chinese herbal medicine Stephania tetrandra, which has many biological activities [186]. Previous studies have demonstrated that tetrandrine has anti-inflammatory properties in various cell types, such as astrocytes, T cells, and monocytes.[187–190]. Yang et al. reported that tetrandrine inhibited inflammatory activation of LPS-induced microglia in culture [191]. He et al. demonstrated that tetrandrine ameliorates cognitive disorder by suppressing NF-κB activity and inflammation in a rat model of AD induced by Aβ1-42 [192]. They found that in 5XFAD mice, tetrandrine administration dose-dependently reduced amyloid plaque deposition in the brain, diminished apoptosis in the hippocampus, and reduced cognitive performance. Tetrandrine repressed the expression of TLR4 and p65 in BV2 cells induced by Aβ 1-42 and effectively suppressed the inflammatory activation of BV2 cells [193]. These results suggests that tetrandrine may promote the clearance of amyloid plaques by regulating the functional transformation of microglia. Together, these findings indicate that tetrandrine may be a promising agent for treating AD through TLR4-mediated neuroinflammation.
(2) Pinoresinol diglucoside (PDG). Eucommia ulmoides, a traditional Chinese herbal medicine, is widely used in China. Pinoresinol diglucoside (PDG), extracted form of Eucommia ulmoides, is one of the primary lignans with anti-inflammatory, antioxidant, antivascular injury, anticancer, antihypertensive actions, and prevention of osteoporosis [194–200]. Studies have reported that PDG inhibits NF-κB and induces Nrf2-dependent HO-1 activation by regulating the MAPK, PI3K/Akt, and GSK-3β pathways to suppress the proinflammatory reaction of BV-2 microglia [201]. Water extract of Eumoides ulmoides bark ameliorates Aβ25-35-induced learning and memory dysfunction in mice and plays a neuroprotective role mediated by cholinergic protection and enhancement [202]. Lei et al. demonstrated an improved influence of PDG on Aβ1-42 neurotoxicity in mice. They found that PDG treatment remarkably inhibited the expression of TLR4 and NF-κB upregulated by Aβ1-42, thereby suppressing the expression of TNF-α and IL-1β, and relieving inflammatory reactions, suggesting that PDG is involved in the regulation of the TLR4/NF-κB pathway [203]. This finding highlights the neuroprotective activity of PDG, which decreases neuroinflammation by modulating the TLR4/NF-κB pathway, thus reducing cognitive dysfunction in AD mice, suggesting that PDG is a potential drug for AD treatment.
(3) Geniposidic acid (GPA). Geniposidic acid (GPA) is an iridoid glycoside isolated from Eucommia ulmoides. Several studies have shown that GPA exhibits anti-inflammatory, antioxidant, antiaging, and neurotrophic roles. Zhou et al. found that the distinct contribution of GPA to the APP/PS1 transgenic AD mouse model was partly regulated by its anti-inflammatory effect. They confirmed that GPA could ameliorate Aβ plaque load and learning and memory function in APP/PS1 mice by blocking the TLR4/2–MyD88 signaling pathway in reactive microglia when HMGB-1 protein expression was reduced [204]. This finding provides theoretical support for the application of GPA in the treatment of AD.
(4) Curcumin. Curcumin is the yellow component of the spice turmeric isolated from the root of turmeric. Studies have shown that curcumin has powerful anti-inflammatory, antioxidant, and antiamyloidosis activities and may play neuroprotective roles in AD [205–208]. Curcumin could reduce the expression levels of HMGB1, TLR4, or TLR2 in lipopolysaccharide-treated human endothelial cells [209]. He et al. found that curcumin pretreatment significantly inhibited the expression of TLR4 and RAGE in Aβ25-35-induced microglia. Curcumin also significantly reduced the expression of TNF-α and IL1β. These results suggest that curcumin may inhibit HMGB1-mediated inflammation in microglia stimulated by Aβ25-35, partly by suppressing the expression of TLR4 and RAGE [210]. In conclusion, TLR4 suppression is a critical target of curcumin in treating AD.
(5) Chotosan (CTS). Chotosan (CTS) is a traditional compound herbal formula consisting of 11 raw herbs often used to improve chronic headaches and high blood pressure symptoms and treat related neurological diseases [211]. Various animal models showed that CTS could effectively alleviate cognitive dysfunction and memory deficits associated with disorders that involve more or less abnormal microglial activation and inflammation of the CNS, such as chronic cerebral hypoperfusion, ischaemic stroke, diabetes, and aging. Rhynchophylline and isorhynchophylline, derived from Uncaria sinensis, are representative herbal components of CTS. They had preservative effects against Aβ25–35-induced cellular neurotoxicity; moreover, they inhibited LPS-induced proinflammatory factors, such as TNF-α and IL-1β and NO produced by mouse N9 microglia [212]. Although clinical practice showed that CTS could treat cognitive impairment and behavioral and psychological symptoms in patients with AD, the specific pharmacological mechanisms needed to be clarified. Chen et al. found that CTS administration decreased the level of TLR4 induced by Aβ1–42 and inhibited TLR-4-mediated inflammatory reaction. The production of NF-κB p65, TNF-α, and IL-1β was reduced in the hippocampus. Aβ1–42 enhanced the activity of caspase-3 and reduced the Bcl-2/Bax ratio, leading to obvious apoptotic reactions in the hippocampus. However, CTS treatment showed strong antiapoptotic effects by reversing the upregulation of proapoptotic proteins [213]. These results demonstrates that CTS therapy can ameliorate Aβ1–42-induced learning and memory disorder through neuroinflammation and apoptosis mediated by the TLR-4/NF-κB signal pathway. This finding provides an idea for the treatment of CTS in AD.
5.6.2. Electroacupuncture
Acupuncture originates from ancient China and is one of the oldest treatments worldwide. Acupuncture, a part of traditional Chinese medicine (TCM), has been an alternative and complementary therapy for over millennia. It belongs a complement of traditional medicine in various eastern and western countries recently [214]. Electroacupuncture (EA) combines acupuncture and electrical stimulation to treat multiple diseases effectively. Acupuncture therapy has a beneficial effect in mitigating cognitive impairment in AD [215, 216]. Recent research has shown that EA treatment improves neuroinflammation and thus cognitive dysfunction in mice with AD [217, 218]. EA therapy inhibited microglial activation, which showed that electroacupuncture at GV 20 and ST 36 points could relieve neuroinflammation effectively and thus reduce cognitive impairment. Previous research has also reported that EA at ST36 represses TLR4/NF-κB signaling, thereby reducing LPS-regulated inflammation in rat models [219]. Moreover, acupuncture could regulate the gut microbiome [220]. He et al. demonstrated that inflammatory substances produced by dysbiosis of intestinal flora could trigger subsequent cerebral inflammatory reactions. They also observed that preventive EA therapy during ageing could rescue learning and memory dysfunction in ageing rats via modulating the gut microbiome and impaired microbiome-gut-brain axis. Furthermore, this protective effect of EA might be related to the downregulation of the TLR4/NF-κB signaling pathway [221]. In conclusion, the neuroprotective effect of EA in suppressing the TLR4 pathway activation may be an effective treatment for AD.
6. Conclusion
Although therapeutic targeting of TLR4 in AD models has yielded promising results, further research is needed to translate these approaches into clinical practice. The design and optimization of agonists that selectively target the TLR4-MyD88 pathway may be a promising future agent that can preferentially enhance the phagocytic activity of glial cells while moderately upregulating glial cytokines and cytotoxins. Since the activation of TLR4 may modulate neuroinflammation and promote glial scarring, further preclinical and clinical safety research is needed before recommending these agents for clinical trials.
Moreover, the key adverse activities of TLR4 antagonists may be associated with their inhibition of abnormal protein and cell fragment clearance and interference with myelination. One challenge facing the clinical development of some TLR4-specific agonists and antagonists is their large molecular structure, which may prevent them from crossing the BBB. Besides, the effectiveness of therapeutic strategies targeting TLR4 may rely on the stage of the disease. For instance, prevention of Aβ accumulation with TLR4 agonists may be beneficial only in early AD. Therefore, early AD diagnosis is the key to effective treatment of TLR4. Thus, the specific involvement of TLR4 in disease staging in AD pathology is an important goal. The imbalance of the immune system in AD is complex, with interactions and interacting factors affecting neuroinflammation. Therefore, a single inflammatory mediator may not be entirely harmful or beneficial. In the future, it will be significant to explore the mechanisms between TLR4 and other receptors to determine effective therapeutic strategies for AD.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (Nos. 82072531 and 81971228) and Natural Science Foundation of Hebei Province (Nos. H2021206160 and H2021206021).
Contributor Information
Min Zhang, Email: hebmuzhangmin@163.com.
Feng Zhang, Email: zjk20019@126.com.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Linyu Wu, Xiaohui Xian, and Guangyu Xu contributed equally to this article.
References
- 1.Jia J., Zhang X., Xu G., Zeng X., Li L. Thioredoxin-1 inhibits amyloid-β-induced activation of NLRP1/caspase-1/GSDMD pyroptotic pathway in PC12 cells. Molecular Biology Reports . 2022;49(5):3445–3452. doi: 10.1007/s11033-022-07177-8. [DOI] [PubMed] [Google Scholar]
- 2.Andreone B., Przybyla L., Llapashtica C., et al. Alzheimer’s-associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nature Neuroscience . 2020;23(8):927–938. doi: 10.1038/s41593-020-0650-6. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y., Chen Y., Xu C., Zhang H., Lin C. TLR4 targeting as a promising therapeutic strategy for Alzheimer disease treatment. Frontiers in Neuroscience . 2020;14:p. 602508. doi: 10.3389/fnins.2020.602508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eratne D., Loi S., Farrand S., Kelso W., Velakoulis D., Looi J. Alzheimer’s disease: clinical update on epidemiology, pathophysiology and diagnosis. Australasian psychiatry : bulletin of Royal Australian and New Zealand College of Psychiatrists . 2018;26(4):347–357. doi: 10.1177/1039856218762308. [DOI] [PubMed] [Google Scholar]
- 5.Chen R., Zhang Q., Yan Y., Zhang Y., Zhang T. Legumain knockout protects against Aβ-induced AD-like cognitive deficits and synaptic plasticity dysfunction via inhibiting neuroinflammation without cleaving APP. Molecular Neurobiology . 2021;58(4):1607–1620. doi: 10.1007/s12035-020-02219-3. [DOI] [PubMed] [Google Scholar]
- 6.Lu J., Zhang C., Lv J., et al. Antiallergic drug desloratadine as a selective antagonist of 5HT receptor ameliorates pathology of Alzheimer's disease model mice by improving microglial dysfunction. Aging Cell . 2021;20(1):p. e13286. doi: 10.1111/acel.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia J., Zeng X., Song X., Zhang P., Chen L. Diabetes mellitus and Alzheimer's disease: the protection of epigallocatechin-3-gallate in streptozotocin injection-induced models. Frontiers in Pharmacology . 2017;8:p. 834. doi: 10.3389/fphar.2017.00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryu J., Zimmer E., Rosa-Neto P., Yoon S. Consequences of metabolic disruption in Alzheimer's disease pathology. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics . 2019;16(3):600–610. doi: 10.1007/s13311-019-00755-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galvin J., Howard D., Denny S., Dickinson S., Tatton N. The social and economic burden of frontotemporal degeneration. Neurology . 2017;89(20):2049–2056. doi: 10.1212/wnl.0000000000004614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saido T. Alzheimer's disease as proteolytic disorders: anabolism and catabolism of beta-amyloid. Neurobiology of aging . 1998;19:S69–S75. doi: 10.1016/s0197-4580(98)00033-5. [DOI] [PubMed] [Google Scholar]
- 11.Bertram L., Lill C., Tanzi R. The genetics of Alzheimer disease: back to the future. Neuron . 2010;68(2):270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 12.Lattanzi S., Brigo F., Vernieri F., Silvestrini M. Visit-to-visit variability in blood pressure and Alzheimer's disease. Journal of clinical hypertension (Greenwich, Conn.) . 2018;20(5):918–924. doi: 10.1111/jch.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lattanzi S., Vernieri F., Silvestrini M. Blood pressure variability and neurocognitive functioning. Journal of clinical hypertension (Greenwich, Conn.) . 2018;20(4):645–647. doi: 10.1111/jch.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia J., Xu G., Zhu D., Liu H., Zeng X., Li L. Advances in the functions of thioredoxin system in central nervous system diseases. Antioxidants & Redox Signaling . 2022 doi: 10.1089/ars.2022.0079. [DOI] [PubMed] [Google Scholar]
- 15.Fülöp T., Itzhaki R., Balin B., Miklossy J., Barron A. Role of microbes in the development of Alzheimer's disease: state of the art - an International Symposium presented at the 2017 IAGG Congress in San Francisco. Frontiers in Genetics . 2018;9:p. 362. doi: 10.3389/fgene.2018.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Querfurth H., LaFerla F. Alzheimer’s Disease. The New England Journal of Medicine . 2010;362(4):329–344. doi: 10.1056/nejmra0909142. [DOI] [PubMed] [Google Scholar]
- 17.Yang J., Wise L., Fukuchi K. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer's disease. Frontiers in Immunology . 2020;11:p. 724. doi: 10.3389/fimmu.2020.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Z., Zhong C. Oxidative stress in Alzheimer's disease. Neuroscience Bulletin . 2014;30(2):271–281. doi: 10.1007/s12264-013-1423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adlard P., Tran B., Finkelstein D., et al. A review of β-amyloid neuroimaging in Alzheimer's disease. Frontiers in Neuroscience . 2014;8:p. 327. doi: 10.3389/fnins.2014.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castellani R., Perry G. Pathogenesis and disease-modifying therapy in Alzheimer's disease: the flat line of progress. Archives of medical research . 2012;43(8):694–698. doi: 10.1016/j.arcmed.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 21.Gao L., Yu X., Chen Q., Zhou D. Alzheimer’s disease therapeutics: current and future therapies. Minerva Medica . 2016;107(2):108–113. [PubMed] [Google Scholar]
- 22.Calvo-Rodriguez M., García-Rodríguez C., Villalobos C., Núñez L. Role of Toll like receptor 4 in Alzheimer's disease. Frontiers in Immunology . 2020;11:p. 1588. doi: 10.3389/fimmu.2020.01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takeuchi O., Akira S. Pattern recognition receptors and inflammation. Cell . 2010;140(6):805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 24.Kawasaki T., Kawai T. Toll-like receptor signaling pathways. Frontiers in Immunology . 2014;5:p. 461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bsibsi M., Ravid R., Gveric D., van Noort J. Broad expression of Toll-like receptors in the human central nervous system. Journal of Neuropathology and Experimental Neurology . 2002;61(11):1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 26.Okun E., Griffioen K., Mattson M. Toll-like receptor signaling in neural plasticity and disease. Trends in Neurosciences . 2011;34(5):269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carty M., Bowie A. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochemical pharmacology . 2011;81(7):825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Pineau I., Lacroix S. Endogenous signals initiating inflammation in the injured nervous system. Glia . 2009;57(4):351–361. doi: 10.1002/glia.20763. [DOI] [PubMed] [Google Scholar]
- 29.Kluwe J., Mencin A., Schwabe R. Toll-like receptors, wound healing, and carcinogenesis. Journal of Molecular Medicine (Berlin, Germany) . 2009;87(2):125–138. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rakoff-Nahoum S., Medzhitov R. Toll-like receptors and cancer. Nature Reviews Cancer . 2009;9(1):57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 31.Frangogiannis N. The immune system and cardiac repair. Pharmacological Research . 2008;58(2):88–111. doi: 10.1016/j.phrs.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moresco E., LaVine D., Beutler B. Toll-like receptors. Current Biology : CB . 2011;21(13):R488–R493. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 33.Cui W., Sun C., Ma Y., Wang S., Wang X., Zhang Y. Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLRP3 inflammasome in Alzheimer’s disease. Frontiers in Neuroscience . 2020;14:p. 444. doi: 10.3389/fnins.2020.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muhammad T., Ikram M., Ullah R., Rehman S., Kim M. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients . 2019;11(3):p. 648. doi: 10.3390/nu11030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao L., Zhang L., Gao R., Jiang F., Cao J., Liu H. Andrographolide alleviates acute brain injury in a rat model of traumatic brain injury: possible involvement of inflammatory signaling. Frontiers in Neuroscience . 2018;12:p. 657. doi: 10.3389/fnins.2018.00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He Y., Ruganzu J., Jin H., et al. LRP1 knockdown aggravates Aβ-stimulated microglial and astrocytic neuroinflammatory responses by modulating TLR4/NF-κB/MAPKs signaling pathways. Experimental Cell Research . 2020;394(2):p. 112166. doi: 10.1016/j.yexcr.2020.112166. [DOI] [PubMed] [Google Scholar]
- 37.Wang S., Zhang X., Zhai L., et al. Atorvastatin attenuates cognitive deficits and neuroinflammation induced by Aβ involving modulation of TLR4/TRAF6/NF-κB pathway. Journal of Molecular Neuroscience : MN . 2018;64(3):363–373. doi: 10.1007/s12031-018-1032-3. [DOI] [PubMed] [Google Scholar]
- 38.Tahara K., Kim H., Jin J., Maxwell J., Li L., Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain : a Journal of Neurology . 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin Y., Liu Y., Hao W., et al. Stimulation of TLR4 attenuates Alzheimer's disease-related symptoms and pathology in tau-transgenic mice. Journal of Immunology . 2016;197(8):3281–3292. doi: 10.4049/jimmunol.1600873. [DOI] [PubMed] [Google Scholar]
- 40.Buchanan M., Hutchinson M., Watkins L., Yin H. Toll-like receptor 4 in CNS pathologies. Journal of Neurochemistry . 2010;114(1):13–27. doi: 10.1111/j.1471-4159.2010.06736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimazu R., Akashi S., Ogata H., et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. The Journal of Experimental Medicine . 1999;189(11):1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagai Y., Akashi S., Nagafuku M., et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nature Immunology . 2002;3(7):667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 43.Gangloff M., Weber A., Gibbard R., Gay N. Evolutionary relationships, but functional differences, between the Drosophila and human Toll-like receptor families. Biochemical Society Transactions . 2003;31:659–663. doi: 10.1042/bst0310659. [DOI] [PubMed] [Google Scholar]
- 44.Rittirsch D., Flierl M., Day D., et al. Cross-talk between TLR4 and FcgammaReceptorIII (CD16) pathways. PLoS Pathogens . 2009;5(6):p. e1000464. doi: 10.1371/journal.ppat.1000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawai T., Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends In Molecular Medicine . 2007;13(11):460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 46.Ferrao R., Zhou H., Shan Y., et al. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly, Molecular Cell . 2014;55(6):891–903. doi: 10.1016/j.molcel.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton G. M., Medzhitov R. Toll-like receptor signaling pathways. ScienceNew York . 2003;300(5625):1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 48.Batool M., Anwar M. A., Choi S. Toll-like receptors targeting technology for the treatment of lymphoma. Expert Opinion On Drug Discovery . 2016;11(11):1047–1059. doi: 10.1080/17460441.2016.1233964. [DOI] [PubMed] [Google Scholar]
- 49.Xia Z.-P., Sun L., Chen X., et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature . 2009;461(7260):114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Medzhitov R. Toll-like receptors and innate immunity. Nature Reviews. Immunology . 2001;1(2):135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 51.Guijarro C., Egido J. Transcription factor-kappa B (NF-kappa B) and renal disease. Kidney International . 2001;59(2):415–424. doi: 10.1046/j.1523-1755.2001.059002415.x. [DOI] [PubMed] [Google Scholar]
- 52.Tajalli-Nezhad S., Karimian M., Beyer C., Atlasi M. A., Tameh A. A. The regulatory role of Toll-like receptors after ischemic stroke: neurosteroids as TLR modulators with the focus on TLR2/4. Cellular and Molecular Life Sciences: CMLS . 2019;76(3):523–537. doi: 10.1007/s00018-018-2953-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nozaki K., Nishimura M., Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Molecular Neurobiology . 2001;23(1) doi: 10.1385/mn:23:1:01. [DOI] [PubMed] [Google Scholar]
- 54.Nito C., Kamada H., Endo H., Narasimhan P., Lee Y.-S., Chan P. H. Involvement of mitogen-activated protein kinase pathways in expression of the water channel protein aquaporin-4 after ischemia in rat cortical astrocytes. Journal of Neurotrauma . 2012;29(14):2404–2412. doi: 10.1089/neu.2012.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yasukawa H., Sasaki A., Yoshimura A. Negative regulation of cytokine signaling pathways. Annual Review of Immunology . 2000;18:143–164. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]
- 56.Nakagawa R., Naka T., Tsutsui H., et al. SOCS-1 participates in negative regulation of LPS responses. Immunity . 2002;17(5):677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 57.Kawai T., Akira S. TLR signaling. Seminars in Immunology . 2007;19(1):24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 58.Ostuni R., Zanoni I., Granucci F. Deciphering the complexity of Toll-like receptor signaling. Cellular and Molecular Life Sciences: CMLS . 2010;67(24):4109–4134. doi: 10.1007/s00018-010-0464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kawai T., Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity . 2011;34(5):637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 60.Yamamoto M., Sato S., Hemmi H., et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science . 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 61.Miron J., Picard C., Frappier J., Dea D., Théroux L., Poirier J. TLR4 gene expression and pro-inflammatory cytokines in Alzheimer’s disease and in response to hippocampal deafferentation in rodents. Journal of Alzheimer’s Disease: JAD . 2018;63(4):1547–1556. doi: 10.3233/jad-171160. [DOI] [PubMed] [Google Scholar]
- 62.Wang X., Yin Z., Cao P., et al. NaoXinTong capsule ameliorates memory deficit in APP/PS1 mice by regulating inflammatory cytokines. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie . 2021;133:p. 110964. doi: 10.1016/j.biopha.2020.110964. [DOI] [PubMed] [Google Scholar]
- 63.McLarnon J. G., Ryu J. K. Relevance of abeta1-42 intrahippocampal injection as an animal model of inflamed Alzheimer's disease brain. Current Alzheimer Research . 2008;5(5):475–480. doi: 10.2174/156720508785908874. [DOI] [PubMed] [Google Scholar]
- 64.Balducci C., Frasca A., Zotti M., et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer's disease. Brain, Behavior, and Immunity . 2017;60:188–197. doi: 10.1016/j.bbi.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 65.Balistreri C. R., Grimaldi M. P., Chiappelli M., et al. Association between the polymorphisms of TLR4 and CD14 genes and Alzheimer’s disease. Current Pharmaceutical Design . 2008;14(26):2672–2677. doi: 10.2174/138161208786264089. [DOI] [PubMed] [Google Scholar]
- 66.Balistreri C. R., Colonna-Romano G., Lio D., Candore G., Caruso C. TLR4 polymorphisms and ageing: implications for the pathophysiology of age-related diseases. Journal of Clinical Immunology . 2009;29(4):406–415. doi: 10.1007/s10875-009-9297-5. [DOI] [PubMed] [Google Scholar]
- 67.Minoretti P., Gazzaruso C., Vito C. D., et al. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer's disease. Neuroscience Letters . 2006;391(3):147–149. doi: 10.1016/j.neulet.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 68.Miron J., Picard C., Lafaille-Magnan M.-É., et al. Association of TLR4 with Alzheimer’s disease risk and presymptomatic biomarkers of inflammation. Alzheimer’s & Dementia: the Journal of the Alzheimer's Association . 2019;15(7):951–960. doi: 10.1016/j.jalz.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 69.Ahmad M. H., Fatima M., Mondal A. C. Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: rational insights for the therapeutic approaches. Journal of Clinical Neuroscience . 2019;59:6–11. doi: 10.1016/j.jocn.2018.10.034. [DOI] [PubMed] [Google Scholar]
- 70.Regen F., Hellmann-Regen J., Costantini E., Reale M. Neuroinflammation and Alzheimer’s disease: implications for microglial activation. Current Alzheimer Research . 2017;14(11):1140–1148. doi: 10.2174/1567205014666170203141717. [DOI] [PubMed] [Google Scholar]
- 71.Lee C. Y. D., Daggett A., Gu X., et al. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron . 2018;97(5):1032–1048.e5. doi: 10.1016/j.neuron.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Currais A., Prior M., Dargusch R., et al. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer's disease transgenic mice. Aging Cell . 2014;13(2):379–390. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi S., Liang D., Chen Y., et al. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer’s disease. European Journal of Immunology . 2016;46(3):665–676. doi: 10.1002/eji.201545855. [DOI] [PubMed] [Google Scholar]
- 74.Kirkley K. S., Popichak K. A., Afzali M. F., Legare M. E., Tjalkens R. B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. Journal of Neuroinflammation . 2017;14(1):p. 99. doi: 10.1186/s12974-017-0871-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hickman S., Izzy S., Sen P., Morsett L., El Khoury J. Microglia in neurodegeneration. Nature Neuroscience . 2018;21(10):1359–1369. doi: 10.1038/s41593-018-0242-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dansokho C., Heneka M. T. Neuroinflammatory responses in Alzheimer’s disease. Journal of Neural Transmission . 2018;125(5):771–779. doi: 10.1007/s00702-017-1831-7. [DOI] [PubMed] [Google Scholar]
- 77.Salter M. W., Stevens B. Microglia emerge as central players in brain disease. Nature Medicine . 2017;23(9):1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- 78.Hansen D. V., Hanson J. E., Sheng M. Microglia in Alzheimer's disease. The Journal of Cell Biology . 2018;217(2):459–472. doi: 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Salminen A., Ojala J., Suuronen T., Kaarniranta K., Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. Journal of Cellular and Molecular Medicine . 2008;12(6A):2255–2262. doi: 10.1111/j.1582-4934.2008.00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blasko I., Stampfer-Kountchev M., Robatscher P., Veerhuis R., Eikelenboom P., Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer's disease in old age: the role of microglia and astrocytes. Aging Cell . 2004;3(4):169–176. doi: 10.1111/j.1474-9728.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 81.Dilshara M. G., Lee K.-T., Jayasooriya R. G. P. T., et al. Downregulation of NO and PGE2 in LPS-stimulated BV2 microglial cells by trans-isoferulic acid via suppression of PI3K/Akt-dependent NF-κB and activation of Nrf2-mediated HO-1. International Immunopharmacology . 2014;18(1):203–211. doi: 10.1016/j.intimp.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 82.Park H. Y., Kim T. H., Kim C. G., et al. Purpurogallin exerts anti-inflammatory effects in lipopolysaccharide-stimulated BV2 microglial cells through the inactivation of the NF-κB and MAPK signaling pathways. International Journal of Molecular Medicine . 2013;32(5):1171–1178. doi: 10.3892/ijmm.2013.1478. [DOI] [PubMed] [Google Scholar]
- 83.De Vita T., Albani C., Realini N., et al. Inhibition of serine palmitoyltransferase by a small organic molecule promotes neuronal survival after astrocyte amyloid beta 1-42 injury. ACS Chemical Neuroscience . 2019;10(3):1627–1635. doi: 10.1021/acschemneuro.8b00556. [DOI] [PubMed] [Google Scholar]
- 84.Walter S., Letiembre M., Liu Y., et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cellular Physiology and Biochemistry . 2007;20(6):947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 85.Song M., Jin J., Lim J.-E., et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of Neuroinflammation . 2011;8:p. 92. doi: 10.1186/1742-2094-8-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Go M., Kou J., Lim J.-E., Yang J., Fukuchi K.-I. Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer's mouse model: implication of TLR4 signaling in disease progression. Biochemical and Biophysical Research Communications . 2016;479(2):331–337. doi: 10.1016/j.bbrc.2016.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nan K., Han Y., Fang Q., et al. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-κB signaling in primary hippocampal neurons induced by Aβ. International Immunopharmacology . 2019;67:294–301. doi: 10.1016/j.intimp.2018.12.027. [DOI] [PubMed] [Google Scholar]
- 88.Paudel Y. N., Angelopoulou E., Piperi C., Balasubramaniam V. R. M. T., Othman I., Shaikh M. F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: updates on receptor signalling. European Journal of Pharmacology . 2019;858:p. 172487. doi: 10.1016/j.ejphar.2019.172487. [DOI] [PubMed] [Google Scholar]
- 89.Takata K., Takada T., Ito A., et al. Microglial amyloid-β1-40 phagocytosis dysfunction is caused by high-mobility group box protein-1: implications for the pathological progression of Alzheimer's disease. International Journal of Alzheimer's Disease . 2012;2012, article 685739:11. doi: 10.1155/2012/685739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lue L. F., Walker D. G., Brachova L., et al. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Experimental Neurology . 2001;171(1):29–45. doi: 10.1006/exnr.2001.7732. [DOI] [PubMed] [Google Scholar]
- 91.Fujita K., Motoki K., Tagawa K., et al. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer's disease. Scientific Reports . 2016;6:p. 31895. doi: 10.1038/srep31895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mazarati A., Maroso M., Iori V., Vezzani A., Carli M. High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and receptor for advanced glycation end products, Experimental Neurology . 2011;232(2):143–148. doi: 10.1016/j.expneurol.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paudel Y. N., Angelopoulou E., Piperi C., Othman I., Aamir K., Shaikh M. F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer's disease (AD): from risk factors to therapeutic targeting. Cells . 2020;9(2):p. 383. doi: 10.3390/cells9020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhong L., Xu Y., Zhuo R., et al. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer’s disease model. Nature Communications . 2019;10(1):p. 1365. doi: 10.1038/s41467-019-10950-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lue L.-F., Schmitz C. T., Serrano G., Sue L. I., Beach T. G., Walker D. G. TREM2 protein expression changes correlate with Alzheimer's disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathology (Zurich, Switzerland) . 2015;25(4):469–480. doi: 10.1111/bpa.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guerreiro R., Wojtas A., Bras J., et al. TREM2 variants in Alzheimer's disease. The New England Journal of Medicine . 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ewers M., Franzmeier N., Suárez-Calvet M., et al. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Science Translational Medicine . 2019;11(507) doi: 10.1126/scitranslmed.aav6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tanaka M., Saito S., Inoue T., Satoh-Asahara N., Ihara M. Potential therapeutic approaches for cerebral amyloid angiopathy and Alzheimer’s disease. International Journal of Molecular Sciences . 2020;21(6) doi: 10.3390/ijms21061992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ito H., Hamerman J. A. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. European Journal of Immunology . 2012;42(1):176–185. doi: 10.1002/eji.201141679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhou J., Yu W., Zhang M., Tian X., Li Y., Lü Y. Imbalance of microglial TLR4/TREM2 in LPS-treated APP/PS1 transgenic mice: a potential link between Alzheimer's disease and systemic inflammation. Neurochemical Research . 2019;44(5):1138–1151. doi: 10.1007/s11064-019-02748-x. [DOI] [PubMed] [Google Scholar]
- 101.Long H., Zhong G., Wang C., et al. TREM2 attenuates Aβ1-42-mediated neuroinflammation in BV-2 cells by downregulating TLR signaling. Neurochemical Research . 2019;44(8):1830–1839. doi: 10.1007/s11064-019-02817-1. [DOI] [PubMed] [Google Scholar]
- 102.Sims R., Van Der Lee S. J., Naj A. C., et al. Rare coding variants in PLCG2_ , ABI3 , and TREM2 implicate microglial- mediated innate immunity in Alzheimer's disease. Nature Genetics . 2017;49(9):1373–1384. doi: 10.1038/ng.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakanishi A., Kaneko N., Takeda H., et al. Amyloid β directly interacts with NLRP3 to initiate inflammasome activation: identification of an intrinsic NLRP3 ligand in a cell-free system. Inflammation and Regeneration . 2018;38:p. 27. doi: 10.1186/s41232-018-0085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Halle A., Hornung V., Petzold G. C., et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nature Immunology . 2008;9(8):857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lim J.-E., Song M., Jin J., et al. The effects of MyD88 deficiency on exploratory activity, anxiety, motor coordination, and spatial learning in C57BL/6 and APPswe/PS1dE9 mice. Behavioural Brain Research . 2012;227(1):36–42. doi: 10.101S6/j.bbr.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lim J.-E., Kou J., Song M., et al. MyD88 deficiency ameliorates β-amyloidosis in an animal model of Alzheimer’s disease. The American Journal of Pathology . 2011;179(3):1095–1103. doi: 10.1016/j.ajpath.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Calvo-Rodríguez M., de la Fuente C., García-Durillo M., García-Rodríguez C., Villalobos C., Núñez L. Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca responses, and neuron cell death in cultured rat hippocampal neurons. Journal of Neuroinflammation . 2017;14(1):p. 24. doi: 10.1186/s12974-017-0802-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lipinski M. M., Zheng B., Lu T., et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America . 2010;107(32):14164–14169. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Peric A., Annaert W. Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction. Acta Neuropathologica . 2015;129(3):363–381. doi: 10.1007/s00401-014-1379-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nixon R. A., Wegiel J., Kumar A., et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of Neuropathology and Experimental Neurology . 2005;64(2):113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 111.Yang D.-S., Stavrides P., Mohan P. S., et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain: a Journal of Neurology . 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boland B., Kumar A., Lee S., et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. Journal of Neuroscience . 2008;28(27):6926–6937. doi: 10.1523/jneurosci.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.He C., Klionsky D. J. Regulation mechanisms and signaling pathways of autophagy. Annual Review of Genetics . 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spielman L. J., Gibson D. L., Klegeris A. Unhealthy gut, unhealthy brain: the role of the intestinal microbiota in neurodegenerative diseases. Neurochemistry International . 2018;120:149–163. doi: 10.1016/j.neuint.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 115.Clemente J. C., Ursell L. K., Parfrey L. W., Knight R. The impact of the gut microbiota on human health: an integrative view. Cell . 2012;148(6):1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dutta G., Zhang P., Liu B. The lipopolysaccharide Parkinson's disease animal model: mechanistic studies and drug discovery. Fundamental & Clinical Pharmacology . 2008;22(5):453–464. doi: 10.1111/j.1472-8206.2008.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ghosh A., Roy A., Liu X., et al. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America . 2007;104(47):18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lan X., Han X., Li Q., Wang J. (-)-Epicatechin, a natural flavonoid compound, protects astrocytes against hemoglobin toxicity via Nrf2 and AP-1 signaling pathways. Molecular Neurobiology . 2017;54(10):7898–7907. doi: 10.1007/s12035-016-0271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jung K.-Y., Park J., Han Y.-S., Lee Y. H., Shin S. Y., Lim Y. Synthesis and biological evaluation of hesperetin derivatives as agents inducing apoptosis. Bioorganic & Medicinal Chemistry . 2017;25(1):397–407. doi: 10.1016/j.bmc.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 120.Lyckander I. M., Malterud K. E. Lipophilic flavonoids from Orthosiphon spicatus prevent oxidative inactivation of 15-lipoxygenase. Prostaglandins, Leukotrienes, and Essential Fatty Acids . 1996;54(4):239–246. doi: 10.1016/s0952-3278(96)90054-x. [DOI] [PubMed] [Google Scholar]
- 121.Laavola M., Nieminen R., Yam M. F., et al. Flavonoids eupatorin and sinensetin present in Orthosiphon stamineus leaves inhibit inflammatory gene expression and STAT1 activation. Planta Medica . 2012;78(8):779–786. doi: 10.1055/s-0031-1298458. [DOI] [PubMed] [Google Scholar]
- 122.Shin H.-S., Kang S.-I., Yoon S.-A., Ko H.-C., Kim S.-J. Sinensetin attenuates LPS-induced inflammation by regulating the protein level of IκB-α. Bioscience, Biotechnology, and Biochemistry . 2012;76(4):847–849. doi: 10.1271/bbb.110908. [DOI] [PubMed] [Google Scholar]
- 123.Tan K.-T., Lin M.-X., Lin S.-C., Tung Y.-T., Lin S.-H., Lin C.-C. Sinensetin induces apoptosis and autophagy in the treatment of human T-cell lymphoma. Anti-Cancer Drugs . 2019;30(5):485–494. doi: 10.1097/cad.0000000000000756. [DOI] [PubMed] [Google Scholar]
- 124.Zhi Z., Tang X., Wang Y., Chen R., Ji H. Sinensetin Attenuates amyloid beta-induced oxidative stress, inflammation, and apoptosis in SH-SY5Y cells through the TLR4/NF-κB signaling pathway. Neurochemical Research . 2021;46(11):3012–3024. doi: 10.1007/s11064-021-03406-x. [DOI] [PubMed] [Google Scholar]
- 125.Garg A., Garg S., Zaneveld L. J., Singla A. K. Chemistry and pharmacology of the Citrus bioflavonoid hesperidin. Phytotherapy Research: PTR . 2001;15(8):655–669. doi: 10.1002/ptr.1074. [DOI] [PubMed] [Google Scholar]
- 126.Rainey-Smith S., Schroetke L.-W., Bahia P., et al. Neuroprotective effects of hesperetin in mouse primary neurones are independent of CREB activation. Neuroscience Letters . 2008;438(1):29–33. doi: 10.1016/j.neulet.2008.04.056. [DOI] [PubMed] [Google Scholar]
- 127.Choi E. J., Ahn W. S. Neuroprotective effects of chronic hesperetin administration in mice. Archives of Pharmacal Research . 2008;31(11):1457–1462. doi: 10.1007/s12272-001-2130-1. [DOI] [PubMed] [Google Scholar]
- 128.Pollard S. E., Whiteman M., Spencer J. P. E. Modulation of peroxynitrite-induced fibroblast injury by hesperetin: a role for intracellular scavenging and modulation of ERK signalling. Biochemical and Biophysical Research Communications . 2006;347(4):916–923. doi: 10.1016/j.bbrc.2006.06.153. [DOI] [PubMed] [Google Scholar]
- 129.Ikram M., Muhammad T., Rehman S. U., et al. Hesperetin Confers neuroprotection by regulating Nrf2/TLR4/NF-κB signaling in an Aβ mouse model. Molecular Neurobiology . 2019;56(9):6293–6309. doi: 10.1007/s12035-019-1512-7. [DOI] [PubMed] [Google Scholar]
- 130.Youn H. S., Saitoh S. I., Miyake K., Hwang D. H. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochemical Pharmacology . 2006;72(1):62–69. doi: 10.1016/j.bcp.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 131.Ding B. J., Ma W. W., He L. L., et al. Soybean isoflavone alleviates β-amyloid 1-42 induced inflammatory response to improve learning and memory ability by down regulation of Toll-like receptor 4 expression and nuclear factor-κB activity in rats. International Journal of Developmental Neuroscience . 2011;29(5):537–542. doi: 10.1016/j.ijdevneu.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 132.Anand P., Singh B. A review on cholinesterase inhibitors for Alzheimer's disease. Archives of Pharmacal Research . 2013;36(4):375–399. doi: 10.1007/s12272-013-0036-3. [DOI] [PubMed] [Google Scholar]
- 133.Galimberti D., Scarpini E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opinion On Investigational Drugs . 2016;25(10):1181–1187. doi: 10.1080/13543784.2016.1216972. [DOI] [PubMed] [Google Scholar]
- 134.Sun W., Chen L., Zheng W., et al. Study of acetylcholinesterase activity and apoptosis in SH-SY5Y cells and mice exposed to ethanol. Toxicology . 2017;384:33–39. doi: 10.1016/j.tox.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 135.Ritchie C. W., Ames D., Clayton T., Lai R. Metaanalysis of randomized trials of the efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer disease. The American Journal of Geriatric Psychiatry . 2004;12(4):358–369. doi: 10.1097/00019442-200407000-00003. [DOI] [PubMed] [Google Scholar]
- 136.Li X., Wang H., Lu Z., et al. Development of multifunctional pyrimidinylthiourea derivatives as potential anti-Alzheimer agents. Journal of Medicinal Chemistry . 2016;59(18):8326–8344. doi: 10.1021/acs.jmedchem.6b00636. [DOI] [PubMed] [Google Scholar]
- 137.Ng Y. P., Or T. C. T., Ip N. Y. Plant alkaloids as drug leads for Alzheimer’s disease. Neurochemistry International . 2015;89:260–270. doi: 10.1016/j.neuint.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 138.Zhang C., Hu L., Liu D., Huang J., Lin W. Circumdatin D exerts neuroprotective effects by attenuating LPS-induced pro-inflammatory responses and downregulating acetylcholinesterase activity In Vitro and In Vivo. Frontiers in Pharmacology . 2020;11:p. 760. doi: 10.3389/fphar.2020.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Q., Luyten W., Pellens K., et al. Antifungal activity in plants from Chinese traditional and folk medicine. Journal of Ethnopharmacology . 2012;143(3):772–778. doi: 10.1016/j.jep.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 140.Gu R.-X., Gu H., Xie Z.-Y., et al. Possible drug candidates for Alzheimer's disease deduced from studying their binding interactions with alpha7 nicotinic acetylcholine receptor. Medicinal Chemistry (Shariqah (United Arab Emirates)) . 2009;5(3):250–262. doi: 10.2174/157340609788185909. [DOI] [PubMed] [Google Scholar]
- 141.Tang M., Wang Z., Zhou Y., et al. A novel drug candidate for Alzheimer's disease treatment: Gx-50 derived from Zanthoxylum bungeanum. Journal of Alzheimer’s Disease: JAD . 2013;34(1):203–213. doi: 10.3233/jad-121831. [DOI] [PubMed] [Google Scholar]
- 142.Hurst R. S., Hajós M., Raggenbass M., et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: in vitro and in vivo characterization, Journal of Neuroscience . 2005;25(17):4396–4405. doi: 10.1523/jneurosci.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Orgogozo J. M., Dartigues J. F., Lafont S., et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Revue Neurologique . 1997;153(3):185–192. [PubMed] [Google Scholar]
- 144.Lindsay J., Laurin D., Verreault R., et al. Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology . 2002;156(5):445–453. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]
- 145.Truelsen T., Thudium D., Grønbaek M. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology . 2002;59(9):1313–1319. doi: 10.1212/01.wnl.0000031421.50369.e7. [DOI] [PubMed] [Google Scholar]
- 146.Wang J., Ho L., Zhao Z., et al. Moderate consumption of Cabernet Sauvignon attenuates Abeta neuropathology in a mouse model of Alzheimer’s disease. FASEB Journal . 2006;20(13):2313–2320. doi: 10.1096/fj.06-6281com. [DOI] [PubMed] [Google Scholar]
- 147.Ho L., Chen L. H., Wang J., et al. Heterogeneity in red wine polyphenolic contents differentially influences Alzheimer's disease-type neuropathology and cognitive deterioration. Journal of Alzheimer’s Disease: JAD . 2009;16(1):59–72. doi: 10.3233/jad-2009-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang F., Liu J., Shi J.-S. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in microglial activation. European Journal of Pharmacology . 2010;636(1-3):1–7. doi: 10.1016/j.ejphar.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 149.Capiralla H., Vingtdeux V., Zhao H., et al. Resveratrol mitigates lipopolysaccharide- and Aβ-mediated microglial inflammation by inhibiting the TLR4/NF-κB/STAT signaling cascade. Journal of Neurochemistry . 2012;120(3):461–472. doi: 10.1111/j.1471-4159.2011.07594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mancuso C., Santangelo R. Alzheimer’s disease and gut microbiota modifications: the long way between preclinical studies and clinical evidence. Pharmacological Research . 2018;129:329–336. doi: 10.1016/j.phrs.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 151.Shen L., Liu L., Ji H.-F. Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. Journal of Alzheimer’s Disease: JAD . 2017;56(1):385–390. doi: 10.3233/jad-160884. [DOI] [PubMed] [Google Scholar]
- 152.Azm S. A. N., Djazayeri A., Safa M., et al. Lactobacilli and bifidobacteria ameliorate memory and learning deficits and oxidative stress in β-amyloid (1-42) injected rats. Applied Physiology, Nutrition, and Metabolism = Physiologie Appliquee, Nutrition Et Metabolisme . 2018;43(7):718–726. doi: 10.1139/apnm-2017-0648. [DOI] [PubMed] [Google Scholar]
- 153.Yang X., Yu D., Xue L., Li H., Du J. Probiotics modulate the microbiota-gut-brain axis and improve memory deficits in aged SAMP8 mice. Acta Pharmaceutica Sinica. B . 2020;10(3):475–487. doi: 10.1016/j.apsb.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Athauda D., Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discovery Today . 2016;21(5):802–818. doi: 10.1016/j.drudis.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 155.Kim D. S., Choi H.-I., Wang Y., Luo Y., Hoffer B. J., Greig N. H. A new treatment strategy for Parkinson's disease through the gut-brain axis: the glucagon-like peptide-1 receptor pathway. Cell Transplantation . 2017;26(9):1560–1571. doi: 10.1177/0963689717721234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen S., Zhou M., Sun J., et al. DPP-4 inhibitor improves learning and memory deficits and AD-like neurodegeneration by modulating the GLP-1 signaling. Neuropharmacology . 2019;157:p. 107668. doi: 10.1016/j.neuropharm.2019.107668. [DOI] [PubMed] [Google Scholar]
- 157.Aviles-Olmos I., Dickson J., Kefalopoulou Z., et al. Exenatide and the treatment of patients with Parkinson's disease. The Journal of Clinical Investigation . 2013;123(6):2730–2736. doi: 10.1172/JCI68295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gejl M., Gjedde A., Egefjord L., et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Frontiers in Aging Neuroscience . 2016;8:p. 108. doi: 10.3389/fnagi.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fang X., Zhou X., Miao Y., Han Y., Wei J., Chen T. Therapeutic effect of GLP-1 engineered strain on mice model of Alzheimer's disease and Parkinson's disease. AMB Express . 2020;10(1):p. 80. doi: 10.1186/s13568-020-01014-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hua F., Tang H., Wang J., et al. TAK-242, an antagonist for Toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. Journal of Cerebral Blood Flow and Metabolism . 2015;35(4):536–542. doi: 10.1038/jcbfm.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Casella C. R., Mitchell T. C. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cellular and Molecular Life Sciences: CMLS . 2008;65(20):3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Michaud J.-P., Hallé M., Lampron A., et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proceedings of the National Academy of Sciences of the United States of America . 2013;110(5):1941–1946. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Garçon N., Van Mechelen M. Recent clinical experience with vaccines using MPL- and QS-21-containing adjuvant systems. Expert Review of Vaccines . 2011;10(4):471–486. doi: 10.1586/erv.11.29. [DOI] [PubMed] [Google Scholar]
- 164.Doyle S. E., O'Connell R. M., Miranda G. A., et al. Toll-like receptors induce a phagocytic gene program through p38. The Journal of Experimental Medicine . 2004;199(1):81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kong L., Ge B.-X. MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Research . 2008;18(7):745–755. doi: 10.1038/cr.2008.65. [DOI] [PubMed] [Google Scholar]
- 166.Gallwitz B. Clinical use of DPP-4 inhibitors. Frontiers in Endocrinology . 2019;10:p. 389. doi: 10.3389/fendo.2019.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kosaraju J., Gali C. C., Khatwal R. B., et al. Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology . 2013;72:291–300. doi: 10.1016/j.neuropharm.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 168.Kosaraju J., Murthy V., Khatwal R. B., et al. Vildagliptin: an anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. The Journal of Pharmacy and Pharmacology . 2013;65(12):1773–1784. doi: 10.1111/jphp.12148. [DOI] [PubMed] [Google Scholar]
- 169.El-Sahar A. E., Shiha N. A., El Sayed N. S., Ahmed L. A. Alogliptin attenuates lipopolysaccharide-induced neuroinflammation in mice through modulation of TLR4/MYD88/NF-κB and miRNA-155/SOCS-1 signaling pathways. The International Journal of Neuropsychopharmacology . 2021;24(2):158–169. doi: 10.1093/ijnp/pyaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ma K., Li R., Zhao H., et al. Cattle encephalon glycoside and ignotin reduce early brain injury and cognitive dysfunction after subarachnoid hemorrhage in rats. Neuroscience . 2018;388:181–190. doi: 10.1016/j.neuroscience.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 171.Zhang H., Li C.-L., Wan F., et al. Efficacy of cattle encephalon glycoside and ignotin in patients with acute cerebral infarction: a randomized, double-blind, parallel-group, placebo-controlled study. Neural Regeneration Research . 2020;15(7):1266–1273. doi: 10.4103/1673-5374.272616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sun J., Zheng H., Qin X., Qi L. Effects of immunocytokine combined with cattle encephalon glycoside and ignotin on CTGF, HO-1 and NT-3 in patients with type 2 diabetic peripheral neuropathy. Iranian Journal of Public Health . 2017;46(12):1632–1638. [PMC free article] [PubMed] [Google Scholar]
- 173.Gao Y., Zhang J., Li S., et al. Cattle encephalon glycoside and ignotin protects neurons against microglia-induced neuroinflammation via elevating BDNF expression and inhibiting TLR4/NF-κB pathway. Neurochemical Research . 2021;46(2):326–336. doi: 10.1007/s11064-020-03168-y. [DOI] [PubMed] [Google Scholar]
- 174.LoGrasso P. V., Feng Y. Rho kinase (ROCK) inhibitors and their application to inflammatory disorders. Current Topics in Medicinal Chemistry . 2009;9(8):704–723. doi: 10.2174/156802609789044452. [DOI] [PubMed] [Google Scholar]
- 175.Yan Y., Yu J., Gao Y., et al. Therapeutic potentials of the Rho kinase inhibitor Fasudil in experimental autoimmune encephalomyelitis and the related mechanisms. Metabolic Brain Disease . 2019;34(2):377–384. doi: 10.1007/s11011-018-0355-7. [DOI] [PubMed] [Google Scholar]
- 176.Hou S.-W., Liu C.-Y., Li Y.-H., et al. Fasudil ameliorates disease progression in experimental autoimmune encephalomyelitis, acting possibly through antiinflammatory effect. CNS Neuroscience & Therapeutics . 2012;18(11):909–917. doi: 10.1111/cns.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Song Y., Chen X., Wang L.-Y., Gao W., Zhu M.-J. Rho kinase inhibitor fasudil protects against β-amyloid-induced hippocampal neurodegeneration in rats. CNS Neuroscience & Therapeutics . 2013;19(8):603–610. doi: 10.1111/cns.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yu J.-W., Li Y.-H., Song G.-B., et al. Synergistic and superimposed effect of bone marrow-derived mesenchymal stem cells combined with fasudil in experimental autoimmune encephalomyelitis. Journal of Molecular Neuroscience: MN . 2016;60(4):486–497. doi: 10.1007/s12031-016-0819-3. [DOI] [PubMed] [Google Scholar]
- 179.Guo M.-F., Zhang H.-Y., Li Y.-H., et al. Fasudil inhibits the activation of microglia and astrocytes of transgenic Alzheimer's disease mice via the downregulation of TLR4/Myd88/NF-κB pathway. Journal of Neuroimmunology . 2020;346:p. 577284. doi: 10.1016/j.jneuroim.2020.577284. [DOI] [PubMed] [Google Scholar]
- 180.Geifman N., Brinton R. D., Kennedy R. E., Schneider L. S., Butte A. J. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimer’s Research & Therapy . 2017;9(1):p. 10. doi: 10.1186/s13195-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chou C.-Y., Chou Y.-C., Chou Y.-J., Yang Y.-F., Huang N. Statin use and incident dementia: a nationwide cohort study of Taiwan. International Journal of Cardiology . 2014;173(2):305–310. doi: 10.1016/j.ijcard.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 182.Wolozin B. Cholesterol, statins and dementia. Current Opinion in Lipidology . 2004;15(6):667–672. doi: 10.1097/00041433-200412000-00007. [DOI] [PubMed] [Google Scholar]
- 183.Zhao L., Zhao Q., Zhou Y., Zhao Y., Wan Q. Atorvastatin may correct dyslipidemia in adult patients at risk for Alzheimer’s disease through an anti-inflammatory pathway. CNS & Neurological Disorders Drug Targets . 2016;15(1):80–85. doi: 10.2174/1871527315666151215095742. [DOI] [PubMed] [Google Scholar]
- 184.Clarke R. M., O'Connell F., Lyons A., Lynch M. A. The HMG-CoA reductase inhibitor, atorvastatin, attenuates the effects of acute administration of amyloid-beta1-42 in the rat hippocampus in vivo. Neuropharmacology . 2007;52(1):136–145. doi: 10.1016/j.neuropharm.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 185.Zhang Y.-Y., Fan Y.-C., Wang M., Wang D., Li X.-H. Atorvastatin attenuates the production of IL-1β, IL-6, and TNF-α in the hippocampus of an amyloid β1-42-induced rat model of Alzheimer’s disease. Clinical Interventions in Aging . 2013;8:103–110. doi: 10.2147/CIA.S40405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bhagya N., Chandrashekar K. R. Tetrandrine--a molecule of wide bioactivity. Phytochemistry . 2016;125:5–13. doi: 10.1016/j.phytochem.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 187.Ho L.-J., Juan T.-Y., Chao P., et al. Plant alkaloid tetrandrine downregulates IkappaBalpha kinases-IkappaBalpha-NF-kappaB signaling pathway in human peripheral blood T cell. British Journal of Pharmacology . 2004;143(7):919–927. doi: 10.1038/sj.bjp.0706000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wu S.-J., Ng L.-T. Tetrandrine inhibits proinflammatory cytokines, iNOS and COX-2 expression in human monocytic cells. Biological & Pharmaceutical Bulletin . 2007;30(1):59–62. doi: 10.1248/bpb.30.59. [DOI] [PubMed] [Google Scholar]
- 189.Zhang H., Li Y.-Y., Wu X.-Z. Effect of Tetrandrine on LPS-induced NF-kappaB activation in isolated pancreatic acinar cells of rat. World Journal of Gastroenterology . 2006;12(26):4232–4236. doi: 10.3748/wjg.v12.i26.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lin S.-T., Wang Y., Xue Y., Feng D.-C., Xu Y., Xu L.-Y. Tetrandrine suppresses LPS-induced astrocyte activation via modulating IKKs-IkappaBalpha-NF-kappaB signaling pathway. Molecular and Cellular Biochemistry . 2008;315(1-2):41–49. doi: 10.1007/s11010-008-9787-4. [DOI] [PubMed] [Google Scholar]
- 191.Xue Y., Wang Y., Feng D.-C., Xiao B.-G., Xu L.-Y. Tetrandrine suppresses lipopolysaccharide-induced microglial activation by inhibiting NF-kappaB pathway. Acta Pharmacologica Sinica . 2008;29(2):245–251. doi: 10.1111/j.1745-7254.2008.00734.x. [DOI] [PubMed] [Google Scholar]
- 192.He F.-Q., Qiu B.-Y., Zhang X.-H., et al. Tetrandrine attenuates spatial memory impairment and hippocampal neuroinflammation via inhibiting NF-κB activation in a rat model of Alzheimer's disease induced by amyloid-β(1-42) Brain Research . 2011;1384:89–96. doi: 10.1016/j.brainres.2011.01.103. [DOI] [PubMed] [Google Scholar]
- 193.Ren D., Fu Y., Wang L., et al. Tetrandrine ameliorated Alzheimer's disease through suppressing microglial inflammatory activation and neurotoxicity in the 5XFAD mouse. Phytomedicine: International Journal of Phytotherapy and Phytopharmacology . 2021;90:p. 153627. doi: 10.1016/j.phymed.2021.153627. [DOI] [PubMed] [Google Scholar]
- 194.During A., Debouche C., Raas T., Larondelle Y. Among plant lignans, pinoresinol has the strongest antiinflammatory properties in human intestinal Caco-2 cells. The Journal of Nutrition . 2012;142(10):1798–1805. doi: 10.3945/jn.112.162453. [DOI] [PubMed] [Google Scholar]
- 195.Ding Z.-J., Liang C., Wang X., et al. Antihypertensive activity of Oliv: male flower extract in spontaneously hypertensive rats. Evidence-based Complementary and Alternative Medicine: ECAM . 2020;2020, article 6432173:6. doi: 10.1155/2020/6432173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Saleem M., Kim H. J., Ali M. S., Lee Y. S. An update on bioactive plant lignans. Natural Product Reports . 2005;22(6):696–716. doi: 10.1039/b514045p. [DOI] [PubMed] [Google Scholar]
- 197.Li Q., Zhang Y., Shi J.-L., et al. Mechanism and anticancer activity of the metabolites of an endophytic fungi from Eucommia ulmoides Oliv. Anti-cancer Agents in Medicinal Chemistry . 2017;17(7):982–989. doi: 10.2174/1871520616666160923094814. [DOI] [PubMed] [Google Scholar]
- 198.Horn-Ross P. L., John E. M., Lee M., et al. Phytoestrogen consumption and breast cancer risk in a multiethnic population: the Bay Area Breast Cancer Study. American Journal of Epidemiology . 2001;154(5):434–441. doi: 10.1093/aje/154.5.434. [DOI] [PubMed] [Google Scholar]
- 199.Horn-Ross P. L., John E. M., Canchola A. J., Stewart S. L., Lee M. M. Phytoestrogen intake and endometrial cancer risk. Journal of the National Cancer Institute . 2003;95(15):1158–1164. doi: 10.1093/jnci/djg015. [DOI] [PubMed] [Google Scholar]
- 200.Yao J., Zou Z., Wang X., Ji X., Yang J. Pinoresinol diglucoside alleviates oxLDL-induced dysfunction in human umbilical vein endothelial cells. Evidence-based Complementary and Alternative Medicine: ECAM . 2016;2016, article 3124519:10. doi: 10.1155/2016/3124519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kwon S.-H., Ma S.-X., Hwang J.-Y., et al. The anti-inflammatory activity of Eucommia ulmoides Oliv. Bark. involves NF-κB suppression and Nrf2-dependent HO-1 induction in BV-2 microglial cells. Biomolecules & Therapeutics . 2016;24(3):268–282. doi: 10.4062/biomolther.2015.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kwon S.-H., Ma S.-X., Joo H.-J., Lee S.-Y., Jang C.-G. Inhibitory Effects of Eucommia ulmoides Oliv. Bark on scopolamine-induced learning and memory deficits in mice. Biomolecules & Therapeutics . 2013;21(6):462–469. doi: 10.4062/biomolther.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lei S., Wu S., Wang G., Li B., Liu B., Lei X. Pinoresinol diglucoside attenuates neuroinflammation, apoptosis and oxidative stress in a mice model with Alzheimer’s disease. Neuroreport . 2021;32(3):259–267. doi: 10.1097/wnr.0000000000001583. [DOI] [PubMed] [Google Scholar]
- 204.Zhou Z., Hou J., Mo Y., et al. Geniposidic acid ameliorates spatial learning and memory deficits and alleviates neuroinflammation via inhibiting HMGB-1 and downregulating TLR4/2 signaling pathway in APP/PS1 mice. European Journal of Pharmacology . 2020;869:p. 172857. doi: 10.1016/j.ejphar.2019.172857. [DOI] [PubMed] [Google Scholar]
- 205.Goozee K. G., Shah T. M., Sohrabi H. R., et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer's disease. The British Journal of Nutrition . 2016;115(3):449–465. doi: 10.1017/s0007114515004687. [DOI] [PubMed] [Google Scholar]
- 206.Massa F., Meli R., Morbelli S., Nobili F., Pardini M. Serum neurofilament light chain rate of change in Alzheimer’s disease: potentials applications and notes of caution. Annals of Translational Medicine . 2019;7(Suppl 3):p. S133. doi: 10.21037/atm.2019.05.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lim G. P., Chu T., Yang F., Beech W., Frautschy S. A., Cole G. M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. The Journal of Neuroscience . 2001;21(21):8370–8377. doi: 10.1523/jneurosci.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ye J., Zhang Y. Curcumin protects against intracellular amyloid toxicity in rat primary neurons. International Journal of Clinical and Experimental Medicine . 2012;5(1):44–49. [PMC free article] [PubMed] [Google Scholar]
- 209.Kim D.-C., Lee W., Bae J.-S. Vascular anti-inflammatory effects of curcumin on HMGB1-mediated responses in vitro. Inflammation Research . 2011;60(12):1161–1168. doi: 10.1007/s00011-011-0381-y. [DOI] [PubMed] [Google Scholar]
- 210.He W., Yuan K., Ji B., Han Y., Li J. Protective effects of curcumin against neuroinflammation induced by Aβ25-35 in primary rat microglia: modulation of high-mobility group box 1, toll-like receptor 4 and receptor for advanced glycation end products expression. Annals of Translational Medicine . 2020;8(4):p. 88. doi: 10.21037/atm.2019.12.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Murakami Y., Zhao Q., Harada K., Tohda M., Watanabe H., Matsumoto K. Choto-san, a Kampo formula, improves chronic cerebral hypoperfusion-induced spatial learning deficit via stimulation of muscarinic M1 receptor. Pharmacology, Biochemistry, and Behavior . 2005;81(3):616–625. doi: 10.1016/j.pbb.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 212.Yuan D., Ma B., Yang J.-Y., et al. Anti-inflammatory effects of rhynchophylline and isorhynchophylline in mouse N9 microglial cells and the molecular mechanism. International Immunopharmacology . 2009;9(13-14):1549–1554. doi: 10.1016/j.intimp.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 213.Chen L., Hu L., Zhao J., et al. Chotosan improves Aβ1-42-induced cognitive impairment and neuroinflammatory and apoptotic responses through the inhibition of TLR-4/NF-κB signaling in mice. Journal of Ethnopharmacology . 2016;191:398–407. doi: 10.1016/j.jep.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 214.Kim S. K., Bae H. Acupuncture and immune modulation. Autonomic Neuroscience: Basic & Clinical . 2010;157(1-2):38–41. doi: 10.1016/j.autneu.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 215.Jia Y., Zhang X., Yu J., et al. Acupuncture for patients with mild to moderate Alzheimer's disease: a randomized controlled trial. BMC Complementary and Alternative Medicine . 2017;17(1):p. 556. doi: 10.1186/s12906-017-2064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kan B.-H., Yu J.-C., Zhao L., et al. Acupuncture improves dendritic structure and spatial learning and memory ability of Alzheimer's disease mice. Neural Regeneration Research . 2018;13(8):1390–1395. doi: 10.4103/1673-5374.235292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cai M., Lee J.-H., Yang E. J. Electroacupuncture attenuates cognition impairment via anti-neuroinflammation in an Alzheimer's disease animal model. Journal of Neuroinflammation . 2019;16(1):p. 264. doi: 10.1186/s12974-019-1665-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang Y., Wang Q., Ren B., et al. Olfactory three-needle enhances spatial learning and memory ability in SAMP8 mice. Behavioural Neurology . 2020;2020:2893211. doi: 10.1155/2020/2893289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chen T., Xiong Y., Long M., et al. Electro-acupuncture pretreatment at Zusanli (ST36) acupoint attenuates lipopolysaccharide-induced inflammation in rats by inhibiting Ca2+ influx associated with cannabinoid CB2 receptors. Inflammation . 2019;42(1):211–220. doi: 10.1007/s10753-018-0885-5. [DOI] [PubMed] [Google Scholar]
- 220.Wang H., Wang Q., Liang C., et al. Acupuncture regulating gut microbiota in abdominal obese rats induced by high-fat diet. Evidence-based Complementary and Alternative Medicine: ECAM . 2019;2019, article 4958294:12. doi: 10.1155/2019/4958294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.He C., Huang Z.-S., Yu C.-C., et al. Preventive electroacupuncture ameliorates D-galactose-induced Alzheimer's disease-like inflammation and memory deficits, probably via modulating the microbiota-gut-brain axis. Iranian Journal of Basic Medical Sciences . 2021;24(3):341–348. doi: 10.22038/ijbms.2021.49147.11256. [DOI] [PMC free article] [PubMed] [Google Scholar]