

Abstract
The activating interplay of thrombosis and inflammation (thromboinflammation) has been established as a major underlying pathway, driving not only cardiovascular disease but also autoimmune disease, and most recently, Coronavirus disease 2019 (COVID-19). Throughout the years, innate immune cells have emerged as important modulators of this process. As the most abundant white blood cell in humans, neutrophils are well-positioned to propel thromboinflammation. This includes their ability to trigger an organized cell death pathway with release of decondensed chromatin structures called neutrophil extracellular traps (NETs). Decorated with histones and cytoplasmic as well as granular proteins, NETs exert cytotoxic, immunogenic and prothrombotic effects accelerating disease progression. Distinct steps leading to extracellular DNA release (NETosis) require the activities of protein arginine deiminase 4 (PAD4) catalyzing citrullination of histones and are supported by neutrophil inflammasome. By linking the immunological function of neutrophils with the pro-coagulant and pro-inflammatory activities of monocytes and platelets, PAD4 activity holds important implications for understanding the processes that fuel thromboinflammation. We will also discuss mechanisms whereby vascular occlusion in thromboinflammation depends on the interaction of NETs with ultra-large von Willebrand Factor (ULVWF), and speculate on the importance of PAD4 in neutrophil inflammasome assembly and NETs in thromboinflammatory diseases including atherosclerosis and COVID-19. This article, primarily focused on work from the Wagner lab, is based on the 2021 Russell Ross Memorial Lecture in Vascular Biology. Professor Ross was a mentor to Dr Denisa Wagner and influenced her lab’s investigations in the field of atherosclerosis.
Since first leads about an inflammatory cause of atherosclerosis were given by the pioneering work of Russell Ross, our knowledge of the role of inflammation in atherosclerosis and other chronic diseases has flourished. Soon it became clear that inflammation and innate immune cells promote thrombotic events, such as thrombosis after plaque rupture leading to myocardial infarction. We proposed early on, that the procoagulant state, promoted by platelet activation, propels the vicious circle of inflammation and thrombosis (1). In the last ten years the concept that innate immune cells drive thrombosis was developed (2). First understood as being a protective mechanism from pathogen invasion through localized fibrin deposition, it now is clear that innate immune cells play a profound role in sterile pathological thrombosis as well (3). Today, thromboinflammation is widely accepted as an important target for therapy in a broad range of human disorders. Central to the concept of thromboinflammation is the activation of platelets and immune cells as well as endothelial activation/dysfunction (Figure 1) building up to microvascular thrombosis, ultimately compromising organ function.
Fig.1. Key mechanisms driving thromboinflammation.
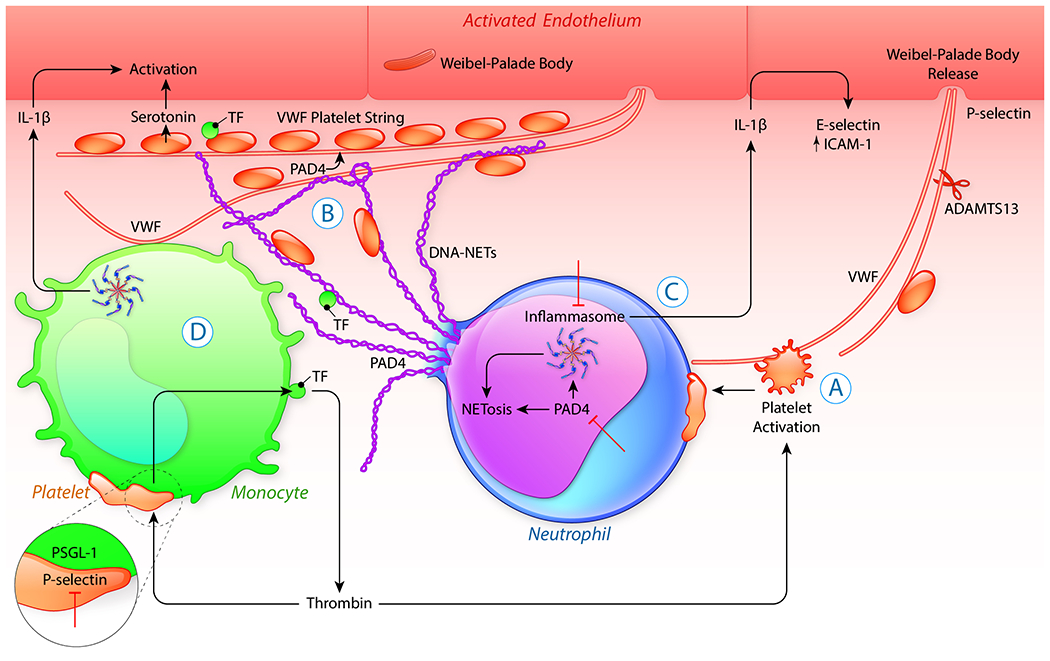
Once activated, platelets (A) form heterotypic, activating complexes with neutrophils and monocytes. Translocated from platelet granules to the membrane, the adhesion molecule P-selectin pairs with PSGL-1 to reprogram cellular functions. The platelet-neutrophil interaction promotes extrusion of neutrophil extracellular traps (NETosis) (B). NET production, in many cases, is preceded by neutrophil inflammasome assembly (C). Both the activation of the inflammasome in the cytoplasm as well as the citrullination of histones in the nucleus are promoted by PAD4, and required for NETosis. Thrombin generation, localized on the surface of activated platelets or other stimuli produced by the inflammatory process itself, lead to the activation of endothelial cells and rapid secretion of VWF from Weibel-Palade bodies. Once released VWF self-associates into long strands and exerts multiple prothrombotic effects forming a scaffold attaching NETs to the vessel wall (B). PAD4 released with NETs (B) inhibits ADAMTS13 activity to cleave VWF and platelet strings persist at the vessel wall. The VWF-DNA scaffold recruits and activates more platelets and innate immune cells. Histones, as well as cytoplasmic and granular proteins entangled in NETs, have pleiotropic effects fueling pathological responses. Platelet-monocytes binding also induces inflammasome activation in monocytes (D). The inflammasome, through generation of active caspase 1, causes the release of the important vascular effector cytokine, IL-1β. IL-1β in turn supports the endothelial expression of adhesion molecules such as E-selectin, propelling leukocyte recruitment. The interplay of platelets with immune cells can, partly through IL-1β production, present an autocrine feedback loop, which can snowball into a cytokine storm. Activated platelets also stimulate tissue factor synthesis and its release in vesicles from monocytes (D). This facilitates thrombin generation, fueling a systemic procoagulant state and with further platelet activation, systemically releasing VWF, cumulates in the vicious cycle of thromboinflammation. Inhibition (⊥) of PAD4, P-selectin/PSGL-1, inflammasome or providing additional ADAMTS13 could potentially disrupt this process.
Platelets and P-selectin: Key players in Thromboinflammation
Platelets have emerged as key regulators in thromboinflammation (Figure 1A) (4). Once activated, they form heterotypic, activating complexes with monocytes and neutrophils. This interaction is through binding of the adhesion molecule P-selectin (CD62P), translocated from the alpha granules, with its constitutively expressed counter receptor on leukocytes, P-selectin glycoprotein ligand 1 (PSGL-1). P-Selectin, being stored and released by platelets as well as Weibel-Palade bodies (WPBs) of endothelial cells (ECs), has a pivotal role in leukocyte recruitment and activation (5, 6). Interaction of P-selectin with PSGL-1 also stimulates P-selectin shedding (7, 8). This soluble P-selectin (sP-selectin), retains many of its procoagulant and stimulatory properties and elevated levels of sP-selectin in plasma are associated with increased risk of myocardial infarction, stroke and cardiovascular disease in man and mice (9, 10). In monocytes, binding to P-selectin triggers rapid surface exposure of tissue factor (TF), the instigator of coagulation (11, 12). Over time, an increased TF gene expression causes shedding of TF in extracellular vesicles from the monocyte surface and the activated monocyte becomes the primary source of blood-borne TF (13) (Figure 1). A complex of TF-with Factor VIIa facilitates the assembly of the prothrombinase complex on the surface of activated platelets, resulting in the generation of large amounts of thrombin. Interestingly, we showed in a preclinical model of atherosclerosis, guided by the discoveries of Russell Ross, that a specific P-selectin-deficiency in platelets alters initiation of atherosclerosis, perhaps by reducing the migration of smooth muscle cells thus dramatically affecting the lipid content and cellularity of the developing plaque (14, 15). However, studies of acute coronary syndrome patients treated with inclacumab, an anti-P-selectin antibody, reduced troponin release but did not alter adverse events in the SELECT-ACS trial (16). This might be due to expression of yet another selectin, the adhesion molecule E-Selectin through endothelial activation by Platelet factor 4 or other cytokines (17, 18). Maybe a dual approach, including the inhibition of both P- and E-selectin, is needed in atherothrombosis.
Activated platelets release a plethora of activating agents among which we think serotonin (5-HT) to be of special interest. 5-HT not only induces vasoconstriction but amplifies platelet and endothelial activation. That is made clear by 5-HT facilitating WPB exocytosis (8, 19). Our group has shown, that in sterile inflammation, platelets also trigger neutrophils to release Neutrophil Extracellular Traps (NETs) through P-selectin and PSGL-1 signaling (described below) (20).
All of these events complemented by platelet aggregation and fibrin deposition result in pathological vessel occlusion occuring in the absence of a physical vessel injury (21).
Nevertheless, it is important to remember that the role of platelets in inflammation is complex. Platelets also have a protective function for the blood vessel in that they prevent hemorrhaging of the inflamed venules caused by endothelial junction disruption by transmigrating leukocytes (22, 23).
Vascular occlusion in thromboinflammation depends on the interaction of ULVWF with NETs.
von Willebrand-Factor and ADAMTS13
The multimeric glycoprotein von Willebrand factor (VWF) recruits and activates platelets through binding to platelet glycoprotein Ibα. VWF not only can be found in the same location as P-selectin (WPBs of ECs and α-granules) but supports platelet tethering and leukocyte adhesion in a similar way. If left uncleaved, ultra-large von Willebrand factor (ULVWF) stored in WPBs forms long strings anchored transiently to the endothelial surface (24). In synthetic endothelialized microvessels, VWF secreted from stimulated endothelial cells self-associates into long strands, which can easily span the vascular lumen (25, 26). Such ULVWF shears red blood cells (RBCs), producing schistocytes as occurs in thrombotic thrombocytopenic purpura (TTP) a disease, where ADAMTS13 activity is impaired (27, 28). RBC fracture leaks heme, which activates NETosis, amplifying the thromboinflammatory process (29, 30). With its binding sites stretched/activated, the uncleaved VWF initiates the formation of platelet strings (Figure 1) and micro-thrombosis (31, 32). Elevated VWF levels correlate with disease severity in cardiovascular disease and stroke (33–35). In mice ADAMTS13 deficiency is both pro-thrombotic and pro-inflammatory (36, 37). We have shown a beneficial anti-inflammatory effect of an administered recombinant ADAMTS13 in both stroke and myocardial ischemia/reperfusion injury in mice (38, 39).
Histological analysis of thrombi from animals and humans exhibits localized intertwined fibrin, NETs and VWF in the solid matrix of thrombi (3). VWF exerts multiple thrombus-stabilizing effects forming a bridge between vessel wall and NETs while histones released with NETs promote VWF release from endothelial cells and platelet activation (24, 40). There is a direct interaction between VWF and NETs (Figure 1B) (24). Both DNA and histones bind VWF, thus retaining NETs at the site of inflammation. Indeed, before NETs were discovered, a specific interaction of A1 domain of VWF with histones was observed in Michael Berndt’s lab (41). Now we finally understand the purpose of this binding site on VWF. Recombinant ADAMTS13 treatment removes VWF from endothelial surfaces and it has been observed by our group to consequently also remove NETs (42).
Neutrophil extracellular traps
In response to a variety of pathological stimuli, including ischemia, a subset of neutrophils undergoes an organized cell death pathway called NETosis (43). We have recently reviewed the cell biology of the process (44). The master of ceremony in NETosis is the enzyme protein arginine deiminase 4 (PAD4) (45). PAD4 is the only PAD with a nuclear localization signal. Once in the nucleus, PAD4 drives chromatin decondensation by catalyzing the citrullination of positively charged arginyl residues abundant in histones to citrulline, an amino acid that lacks charge. Loss of the positive charge loosens histones interaction within nucleosomes and with DNA, ultimately allowing histone proteolysis and unwinding of chomatin into NETs. The citrullinated histones H3 [H3Cit] or H4 [H4Cit]) are useful biomarkers for the production of NETs in animals or humans, and can be evaluated in plasma samples and tissue sections.
PAD4 likely also has some cytoplasmic substrates that influence the cell biology of NETosis and neutrophil inflammasome assembly (44, 46). Neutrophils lacking PAD4 activity, either genetically or through inhibition, exhibit a serious defect in NETosis. Using high-resolution time-lapse microscopy of stimulated mouse neutrophils and human neutrophil-like cells to induce NETosis, we were able to show that distinct steps such as nuclear envelope rupture, as well as extracellular DNA release, required the enzymatic and nuclear localization activities of PAD4. Correspondingly, our lab has provided evidence that in patients with type 1 diabetes, mellitus increased NETosis is paralleled by elevated neutrophil PAD4 protein expression, explaining their pro-NETotic phenotype (47, 48). Another important step necessary for neutrophils to undergo cell death with release of chromatin is gasdermin D dependent membrane pore assembly (49, 50). However, it appears that not all pathways of NET formation lead to neutrophil death and well-functioning cytoplasts (enucleated cells) were detected during an infection, capable of phagocytosis (51, 52).
Decorated with histones and cytoplasmic as well as granular proteins, NETs provide a large scaffold that is harmful to surrounding tissue and presents neoantigens thus inducing autoimmune disease. Recently we have shown that induction of NETs by G-CSF together with collagen injection initiated arthritis and joint erosion in a strain of mice, normally resistant to the disease (53). Within the blood vessel NETs, similar to VWF, offer a platform for platelet adhesion and coagulation initiation (Figure 1B). Active PAD4, released together with NETs, also mediates the citrullination of ADAMTS13, which inhibits VWF cleavage, thus permitting platelet strings to persist near the vessel wall in the presence of PAD4 (54) (Figure 1B).
Some neutrophils release fragments of inflammasome containing ASC (Apoptosis-associated speck-like protein containing a CARD) entangled in NETs (46). Ingestion of inflammasome-debris is known to propagate inflammasome assembly in those cells, analogous to prion protein, thus enhancing IL-1β production and accelerating systemic inflammatory responses (55).
Fitting with the procoagulant role of NETs, studies have found TF-containing microparticles (MPs) deposited in NETs. These can be from multiple origins. Monocytes are the primary source of procoagulant MPs but MPs are also derived from other cells, including malignant cells (56). NETosis as well as NET-associated TF increase has recently been related to levels of systemic inflammation and IL-1β indicating a common regulating pathway (57). Finally, recent studies highlight that the activation of canonical and non-canonical inflammasomes triggers the release of TF from activated macrophages and monocytes (58).
Over the years, a large body of clinical and experimental evidence has implicated NETs in a variety of ischemia-induced and other thromboinflammatory diseases (3, 52, 59, 60). The inhibition of NET formation or their cleavage by DNases were proposed as new therapies similar to the cleavage of VWF by ADAMTS13 (49).
The Inflammasome: One ring to rule them all.
Cellular interactions aside, the process of thrombinflammation is orchestrated by a complex interplay between the coagulation cascade, the complement system and cytokines primarily of the interleukin 1 (IL-1) family. Many of the involved regulatory proteins are synthesized as inactive precursors and require proteolytic processing to attain biological function. First described in 1991, caspase 1, the major intracellular protease implicated in processing of pro-IL-1β, was later found to be the work horse in the inflammasome (61, 62). Inflammasomes are a family of macromolecular ring-like multi-component protein structures that assemble upon activation in innate immune cells (63). Having been mainly studied in monocytes/macrophages, recent reports now also provide evidence for inflammasome assembly in neutrophils (Figure 1C and D). While neutrophils respond to the same stimuli as monocytes, they have been shown, in vitro, to not need LPS priming as opposed to monocytes, suggesting a faster responsiveness (46). This is consistent whith their role as the first line of host immune response.
Major stimulus of inflammasome assembly in sterile thromboinflammation are activated platelets (64) (Figure 1). However, there is a plethora of other intracellular and extracellular inducers acting on different activation pathways for which we refer the reader elsewhere (65). Nevertheless, the importance of the inflammasome for pro-IL-1β processing puts it in the center in the development of thromboinflammation promoting it as a good target to alter inflammatory pathways. The activated receptor NLR family pyrin domain containing 3 (NLRP3) is the most studied inflammasome in IL-1β activation (66).
Once activated and released, IL-1β is the major pro-inflammatory cytokine driving the synthesis of endothelial adhesion molecules that recruit leukocytes such as E-selectin, intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 (67). Certain rare genetic diseases highlight the importance of the inflammasome in controlling the overwhelming proinflammatory potential of the released IL-1β. Cryopyrin-associated periodic syndromes (CAPS) for example arise from gain-of-function mutations in the NLRP3 inflammasome. A specific anti-IL-1β antibody, canakinumab, has received approval as an orphan drug for treating these diseases. We are very excited about a recent report showing that in mice, a mutation only expressed in neutrophils, prediposing for inflammasome assembly, is sufficient to promote CAPS, prompting a prominent role of neutrophils in CAPS (68).
Our lab was recently able to detect ASC speck formation, which is accepted as readout for inflammasome activity, occurring before chromatin decondensation in murine neutrophils (48). Correspondingly, we have shown that NLRP3 deficiency drastically reduces NETosis by neutrophils stimulated in vitro and also causes significantly lower density of NETs in thrombi produced by a stenosis-induced mouse model of deep vein thrombosis (46). This is very exciting since it puts inflammasome upstream of yet another thromboinflammatory process: NETosis. To make things even more entangled, citrullination supports inflammasome assembly (69) and in neutrophils the citullinating enzyme is PAD4 (46) (Figure 1C).
We are convinced that the process of PAD4 mediated inflammasome assembly in neutrophils and subsequent NETosis are at the very center of pathological thromboinflammation. It links the immunological function of neutrophils with the activation of platelets and monocytes, thus producing a superior-platform for both thrombosis and inflammation. PAD4 is a promising target (45) to inhibit the thromboinflammatory process at a proximal point in a complex series of interconnected multi-cellular processes.
Thromboinflammation in Atherosclerosis and COVID-19
It stands to reason that the activating interplay of platelets with VWF, NETs and inflammasomes in thromboinflammation fuels disease pathology. Here we choose to discuss two different pathological processes in which thromboinflammation is increasingly thought to be implicated.
Atherosclerosis
Targeting inflammation in atherosclerosis, as proposed by Russell Ross (70), has become clinical reality in 2019 with the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS), providing proof, that the inflammatory response is driving atherosclerosis development and progression (66, 71). CANTOS showed that administration of an anti-IL-1β antibody to patients who were cardiovascularly stable after a myocardial infarction and treated according to current guidelines, reduced recurrent major adverse cardiovascular events. Unfortunately those results were dearly bought, as they were accompanied by a statistically significant increase in infections including some with fatal outcome (63). In the following, we wish to focus on how several key players of thromboinflammation already discussed in this review are linked to the process of atherogenesis and offer possible targets for therapy. Please refer elsewhere for general review on the condition (71, 72).
Similar to other thromboinflamatory diseases, in atherosclerosis, activated endothelium and platelets are key in the early stages of leukocyte recruitment (73). Platelets secrete chemokines (e.g. CCL5) and cytokines (IL-1β) that when deposited on the endothelium promote adhesion of monocytes and neutrophils (74). Both platelet and endothelial P-selectin facilitate leukocyte adhesion and atherosclerotic lesion development (14). Soluble E-selectin also increases the risk for cardiovascular disease (75). A combined deficiency of both P- and E-selectin had the strongest negative impacts on lesion progression in mice (76).
Ultrasound molecular imaging has shown that an increase in VWF-mediated platelet adhesion to the endothelium occurs even prior to atherosclerotic plaque development (77) indicating that VWF may take part in the lesion initiation function of platelets. This observation may explain our surprising early observation in 2001, that in VWF-deficient mice compared to wild type controls, lesions not only formed later after start of the high fat diet, but formed outside of the usual atherosclerosis-susceptible locations (78). Perhaps under turbulent flow, where lesions preferably initiate, VWF is required to support the initial platelet adhesion that marks the site for monocyte recruitment. Also, the fatty streaks that finally formed in the VWF-deficient mice were smaller and contained fewer monocytes. This observation fits with the finding that endothelial and not platelet-derived VWF is important for atherosclerosis development in mice (79). In agreement with all animal disease models in which VWF release plays a pathological role, ADAMTS13 deficiency has the opposite effect of VWF deficiency, for example, dramatically accelerating atherosclerotic lesion development (80, 81).
Similar to VWF in humans, high levels of sP-selectin in plasma correlate with cardiovascular disease severity (9). We produced knock-in mice with a deletion of the cytoplasmic domain that is necessary for P-selectin storage. The resulting mice had a high procoagulant state caused by excessive thrombin generation and were prone to develop atherosclerosis (10).
While monocytes, in this lipid-driven disease, are clearly most important in atherosclerosis, recent work provided genetic evidence for the involvement of neutrophil- derived NETs during a process resembling endothelial erosion in mice (71). PAD4 deficiency, which inhibits NETosis as well as the destruction of NETs by DNAse administration, reduced endothelial discontinuity and endothelial cell apoptosis in a mouse model (82). This treatment not only targeted the prothrombotic effects driven by NETs but also NET-associated cytotoxicity. Amongst other implications, histone H4 in NETs has been recently shown to cause the death of intimal smooth muscle cells, accelerating atherosclerotic plaque destabilization (83). This could provide the answer regarding how acute infections, resulting in excessive NETosis, augment a pre-existing cardiovascular risk (71, 84, 85). Targeting NETs could be of value in secondary prevention therapy for plaque stability, for example in carotid disease and stable coronary artery disease (71).
There is a loop from PAD4 mediated inflammasome assembly and NETosis all the way to IL-1β activation, which further enhances NETosis and atherosclerosis (86) (Figure 1C). Experimental evidence shows that cholesterol crystals activate NLRP3 inflammasome putting it directly onto the pathway of atherogenesis. Consequently, inhibition of NLRP3, or its genetic deletion, reduces atherosclerosis in mice (71, 87, 88). Since NLRP3-deficiency also reduces NETosis and the deposition of NETs in thrombi (46), PAD4 and/or NLRP3 inhibition could be new targets to prevent atherothrombosis.
Thromboinflammation in COVID-19
In the wake of the COVID-19 pandemic and the serious coagulopathy often present in affected patients, thromboinflammation also gained momentum. In reminiscence of the very definition of thromboinflammation, COVID-19 patients have been reported to frequently thrombose dialysis and ECMO circuits despite proper anticoagulation (89, 90). Endothelial dysfunction (91, 92) and thromboinflammation (60) have emerged early on as the key pathogenic mechanisms driving COVID-19 pathology. Indeed, patients present with increased platelet activation and platelet-monocyte aggregate formation with a marked increase in monocyte TF expression (93). Nevertheless, antiplatelet therapy has proven ineffective in improving outcome in hospitalized patients (94, 95). This indicates that not platelet aggregation alone, but the interplay of platelets with different factors, including NETs, drives severe COVID-19. A COVID-19-specific coagulopathy is epitomized by elevated levels of fibrinogen, fibrin degradation product D-dimer and highly increased levels of VWF in the blood of patients (96, 97). Evidence suggests that increased endothelial cell activation, with the consecutive release of large VWF multimers, paired with COVID-19 related relative insufficient VWF cleavage either due to ADAMTS13 consumption or disease pathophysiology, may account for increased platelet–vessel wall VWF interactions underlying thrombotic microangiopathy (98). Hyperactivation of the complement and coagulation systems have been reported in COVID-19 (99, 100).
Due to its procoagulant activity, it is not surprising that sP-selectin has been found associated with disease severity in COVID-19 patients (101). However, in a placebo-controlled, randomized trial of mild COVID-19 patients, testing the effect of a single dose of crizanlizumab, a P-selectin inhibitor, in COVID-19 showed no significant differences from placebo for clinical endpoints despite evidence of a decrease in thrombin activation as well as levels of sP-selectin (102). A larger trial with a higher number of doses is being planned.
In a similar way, NETs are also likely to be involved in thromboinflammation driving COVID-19 and the severe lung injury observed in this infection (103). Already in 2012 our group and that of Mark Looney showed a pathological role for NETs in acute lung injury with NETs being deposited in the lungs of transfusion-related acute lung injury (TRALI) associated acute respiratory distress syndrome (ARDS) in mice and humans (104, 105). Correspondingly, NETs have also been identified in the lung parenchyma and alveolar space in autopsy case reports of COVID-19 patients (106). As part of a clinical trial on DNase 1 inhalation (NCT04402944) we studied freshly isolated neutrophils from severe COVID-19 patients. We found not only that they are highly poised for NETosis (40 percent of nuclei were positive for citrullinated histone H3) but also 2% of neutrophils from blood or tracheal aspirate (i.e. from the lung) were forming inflammasome as detected by ASC speck assembly. The specks were seen adjacent to multilobulated neutrophil nuclei, before the typical nuclear rounding of NETosis (107). Thus, inflammasome assembly precedes NETosis in affected COVID-19 patients, labelling it a promising target for therapy. Although, in our study neutrophils and monocytes showed a similar rate of inflammasome positivity (2%), considering that there are 10 times as many neutrophils as monocytes in the bloodstream, the importance of IL-1β production by neutrophils could be grossly underestimated. The same autocrine IL-1β-feed-forward loop unleashed by inflammasome activation must contribute to the development of acute adult respiratory distress syndrome, cytokine storm, and microvascular thrombosis and can culminate in failure of multiple organs in severe COVID-19 cases.
Summary and future directions
It became evident that in disease, neither thrombosis nor inflammation occur in isolation. With more knowledge, vascular processes are revealing their complexity. Clot formation is no longer understood to contain solely cleaved fibrinogen with plasma proteins and the clot DNA content now has to be considered for clot lysis. Similarly, platelet plugs are far from being only platelets crosslinked by fibrinogen. Times change: many of us were surprised when mice lacking both fibrinogen and VWF were still able to produce thrombi after injury (108). Likewise, deep vein thrombosis has recently been discovered to require not only platelets and red blood cells, but also monocytes and neutrophils (109). In inflammation, the role of platelets and platelet derived factors was recognised early on, amongst others by Russell Ross, and platelet impact on inflammation continues to be supported by new studies. The classical cellular players of inflammation fully intermingle with those of thrombosis, justifying the term thromboinflammation.
In this review, we chose to place neutrophils with their new-found ability for inflammasome assembly and NET-production in the center of thromboinflammation (Figure 1). With NETs being equally involved in thrombosis, as well as perpetuating the immune response and tissue injury, they certainly are in the middle of it all. NETosis activates platelets, releases toxic histones (110) and active enzymes, modifying thrombus stability (111). In a similar way, the assembly of the inflammasome paralleled by increased PAD4 activity leads to proinflammatory and prothrombotic cytokine release and activation of endothelium, thus further boosting leukocyte recruitment and inflammasome assembly in bystander cells. It is easy to imagine that PAD4 and inflammasome are promising points to target in anti-thromboinflammatory therapy. However, many mechanistic unanswered questions remain. For example, what are the protein target(s) of PAD4 citrullination that are necessary for inflammasome assembly? Reciprocally, which are the intracellular substrates of caspase 1, the enzyme produced by inflammasome, whose cleavage is necessary for NETosis to proceed? Understanding all of it will give us a better handle on how to tame the beast in the neutrophil.
Highlights.
Platelets and P-selectin are Key players in Thromboinflammation
Vascular occlusion in thromboinflammation depends on the interaction of ULVWF with NETs.
The neutrophil, with its inflammasome and NETs, takes centre stage in pathologies involving thromboinflammation.
Acknowledgments
We thank Dr. Bruce Ewenstein, MD/Ph.D for his helpful suggestions and critical reading of the manuscript, Ms. Kristen Douthit for careful editing and manuscript preparation, and Mr. Ben Smith for the artwork. We thank the anonymous reviewers for their helpful comments and suggestions.
Sources of Funding
DDW is funded by a grant from the National Heart Lung and Blood Institute (NIH R35OIA HL135765). LAH received funding from the German Research Foundation (DFG; HE 8679/1-1:1);
Abbreviations
- PSGL-1
P-selectin glycoprotein ligand 1
- WPB
Weibel-Palade body
- EC
Endothelial cell
- sP-selectin
Soluble P-selectin
- TF
Tissue factor
- 5-HT
Serotonin
- NET
Neutrophil Extracellular Trap
- ULVWF
Ultra-large von Willebrand factor
- PAD4
Protein arginine deiminase 4
- MPs
Microparticles
- COVID-19
Coronavirus disease 2019
Footnotes
Disclosures
None.
References
- 1.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111(11):5271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34–45. [DOI] [PubMed] [Google Scholar]
- 3.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003;23(12):2131–7. [DOI] [PubMed] [Google Scholar]
- 5.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993;74(3):541–54. [DOI] [PubMed] [Google Scholar]
- 6.Polgar J, Matuskova J, Wagner DD. The P-selectin, tissue factor, coagulation triad. J Thromb Haemost. 2005;3(8):1590–6. [DOI] [PubMed] [Google Scholar]
- 7.Dole VS, Bergmeier W, Patten IS, Hirahashi J, Mayadas TN, Wagner DD. PSGL-1 regulates platelet P-selectin-mediated endothelial activation and shedding of P-selectin from activated platelets. Thromb Haemost. 2007;98(4):806–12. [PubMed] [Google Scholar]
- 8.Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106(7):2334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ridker PM, Buring JE, Rifai N. Soluble P-selectin and the risk of future cardiovascular events. Circulation. 2001;103(4):491–5. [DOI] [PubMed] [Google Scholar]
- 10.Kisucka J, Chauhan AK, Zhao BQ, Patten IS, Yesilaltay A, Krieger M, et al. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood. 2009;113(23):6015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grover SP, Mackman N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709–25. [DOI] [PubMed] [Google Scholar]
- 12.Ivanov II, Apta BHR, Bonna AM, Harper MT. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci Rep. 2019;9(1):13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hrachovinova I, Cambien B, Hafezi-Moghadam A, Kappelmayer J, Camphausen RT, Widom A, et al. Interaction of P-selectin and PSGL-1 generates microparticles that correct hemostasis in a mouse model of hemophilia A. Nat Med. 2003;9(8):1020–5. [DOI] [PubMed] [Google Scholar]
- 14.Burger PC, Wagner DD. Platelet P-selectin facilitates atherosclerotic lesion development. Blood. 2003;101(7):2661–6. [DOI] [PubMed] [Google Scholar]
- 15.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(5):812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libby P, Everett BM. Novel Antiatherosclerotic Therapies. Arterioscler Thromb Vasc Biol. 2019;39(4):538–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu G, Rux AH, Ma P, Bdeir K, Sachais BS. Endothelial expression of E-selectin is induced by the platelet-specific chemokine platelet factor 4 through LRP in an NF-kappaB-dependent manner. Blood. 2005;105(9):3545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur J Immunol. 2011;41(7):2074–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. 2013;121(6):1008–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yun SH, Sim EH, Goh RY, Park JI, Han JY. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed Res Int. 2016;2016:9060143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O’Donnell E, Zhao BQ, et al. Inflammation induces hemorrhage in thrombocytopenia. Blood. 2008;111(10):4958–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008;68(16):6851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang J, Wu Z, Long Q, Huang J, Hong T, Liu W, et al. Insights Into Immunothrombosis: The Interplay Among Neutrophil Extracellular Trap, von Willebrand Factor, and ADAMTS13. Front Immunol. 2020;11:610696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Y, Chen J, Lopez JA. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015;6:7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Chung DW. Inflammation, von Willebrand factor, and ADAMTS13. Blood. 2018;132(2):141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauhan AK, Walsh MT, Zhu G, Ginsburg D, Wagner DD, Motto DG. The combined roles of ADAMTS13 and VWF in murine models of TTP, endotoxemia, and thrombosis. Blood. 2008;111(7):3452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadler JE. Pathophysiology of thrombotic thrombocytopenic purpura. Blood. 2017;130(10):1181–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117(4):850–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123(24):3818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(9):1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, et al. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002;99(11):3971–7. [DOI] [PubMed] [Google Scholar]
- 33.Peetermans M, Meyers S, Liesenborghs L, Vanhoorelbeke K, De Meyer SF, Vandenbriele C, et al. Von Willebrand factor and ADAMTS13 impact on the outcome of Staphylococcus aureus sepsis. J Thromb Haemost. 2020;18(3):722–31. [DOI] [PubMed] [Google Scholar]
- 34.Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006;4(6):1186–93. [DOI] [PubMed] [Google Scholar]
- 35.Hanson E, Jood K, Karlsson S, Nilsson S, Blomstrand C, Jern C. Plasma levels of von Willebrand factor in the etiologic subtypes of ischemic stroke. J Thromb Haemost. 2011;9(2):275–81. [DOI] [PubMed] [Google Scholar]
- 36.Chauhan AK, Kisucka J, Brill A, Walsh MT, Scheiflinger F, Wagner DD. ADAMTS13: a new link between thrombosis and inflammation. J Exp Med. 2008;205(9):2065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gandhi C, Motto DG, Jensen M, Lentz SR, Chauhan AK. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Meyer SF, Savchenko AS, Haas MS, Schatzberg D, Carroll MC, Schiviz A, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood. 2012;120(26):5217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009;114(15):3329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michels A, Albanez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, et al. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost. 2016;14(11):2274–86. [DOI] [PubMed] [Google Scholar]
- 41.Ward CM, Tetaz TJ, Andrews RK, Berndt MC. Binding of the von Willebrand factor A1 domain to histone. Thromb Res. 1997;86(6):469–77. [DOI] [PubMed] [Google Scholar]
- 42.Wong SL, Goverman J, Staudinger C, Wagner DD. Recombinant human ADAMTS13 treatment and anti-NET strategies enhance skin allograft survival in mice. Am J Transplant. 2020;20(4):1162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular Mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong SL, Wagner DD. Peptidylarginine deiminase 4: a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018:fj201800691R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front Immunol. 2021;12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiam HR, Wong SL, Qiu R, Kittisopikul M, Vahabikashi A, Goldman AE, et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci U S A. 2020;117(13):7326–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorvillo N, Cherpokova D, Martinod K, Wagner DD. Extracellular DNA NET-Works With Dire Consequences for Health. Circ Res. 2019;125(4):470–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). [DOI] [PubMed] [Google Scholar]
- 51.Krishnamoorthy N, Douda DN, Bruggemann TR, Ricklefs I, Duvall MG, Abdulnour RE, et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci Immunol. 2018;3(26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–94. [DOI] [PubMed] [Google Scholar]
- 53.Fukui S, Gutch S, Fukui S, Cherpokova D, Aymonnier K, Sheehy CE, et al. The prominent role of hematopoietic peptidyl arginine deiminase 4 in arthritis: collagen and G-CSF induced arthritis model in C57BL/6 mice. Arthritis Rheumatol. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorvillo N, Mizurini DM, Coxon C, Martinod K, Tilvawala R, Cherpokova D, et al. Plasma Peptidylarginine Deiminase IV Promotes VWF-Platelet String Formation and Accelerates Thrombosis After Vessel Injury. Circ Res. 2019;125(5):507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and ’prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15(8):727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liman TG, Bachelier-Walenta K, Neeb L, Rosinski J, Reuter U, Bohm M, et al. Circulating endothelial microparticles in female migraineurs with aura. Cephalalgia. 2015;35(2):88–94. [DOI] [PubMed] [Google Scholar]
- 57.Liberale L, Holy EW, Akhmedov A, Bonetti NR, Nietlispach F, Matter CM, et al. Interleukin-1beta Mediates Arterial Thrombus Formation via NET-Associated Tissue Factor. J Clin Med. 2019;8(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu R, Wang N, Comish PB, Tang D, Kang R. Inflammasome-Dependent Coagulation Activation in Sepsis. Front Immunol. 2021;12:641750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doring Y, Libby P, Soehnlein O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ Res. 2020;126(9):1228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21(5):319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chan AH, Schroder K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med. 2020;217(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Libby P. Targeting Inflammatory Pathways in Cardiovascular Disease: The Inflammasome, Interleukin-1, Interleukin-6 and Beyond. Cells. 2021;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rolfes V, Ribeiro LS, Hawwari I, Bottcher L, Rosero N, Maasewerd S, et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep. 2020;31(6):107615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grebe A, Hoss F, Latz E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ Res. 2018;122(12):1722–40. [DOI] [PubMed] [Google Scholar]
- 67.Wang X, Feuerstein GZ, Gu JL, Lysko PG, Yue TL. Interleukin-1 beta induces expression of adhesion molecules in human vascular smooth muscle cells and enhances adhesion of leukocytes to smooth muscle cells. Atherosclerosis. 1995;115(1):89–98. [DOI] [PubMed] [Google Scholar]
- 68.Stackowicz J, Gaudenzio N, Serhan N, Conde E, Godon O, Marichal T, et al. Neutrophil-specific gain-of-function mutations in Nlrp3 promote development of cryopyrin-associated periodic syndrome. J Exp Med. 2021;218(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mishra N, Schwerdtner L, Sams K, Mondal S, Ahmad F, Schmidt RE, et al. Cutting Edge: Protein Arginine Deiminase 2 and 4 Regulate NLRP3 Inflammasome-Dependent IL-1beta Maturation and ASC Speck Formation in Macrophages. J Immunol. 2019;203(4):795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340(2):115–26. [DOI] [PubMed] [Google Scholar]
- 71.Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20(8):589–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524–33. [DOI] [PubMed] [Google Scholar]
- 73.Gawaz M, Langer H, May AE. Platelets in inflammation and atherogenesis. J Clin Invest. 2005;115(12):3378–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res. 2007;100(1):27–40. [DOI] [PubMed] [Google Scholar]
- 75.Roldan V, Marin F, Lip GY, Blann AD. Soluble E-selectin in cardiovascular disease and its risk factors. A review of the literature. Thromb Haemost. 2003;90(6):1007–20. [DOI] [PubMed] [Google Scholar]
- 76.Dong ZM, Brown AA, Wagner DD. Prominent role of P-selectin in the development of advanced atherosclerosis in ApoE-deficient mice. Circulation. 2000;101(19):2290–5. [DOI] [PubMed] [Google Scholar]
- 77.Wu MD, Atkinson TM, Lindner JR. Platelets and von Willebrand factor in atherogenesis. Blood. 2017;129(11):1415–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Methia N, Andre P, Denis CV, Economopoulos M, Wagner DD. Localized reduction of atherosclerosis in von Willebrand factor-deficient mice. Blood. 2001;98(5):1424–8. [DOI] [PubMed] [Google Scholar]
- 79.Doddapattar P, Dhanesha N, Chorawala MR, Tinsman C, Jain M, Nayak MK, et al. Endothelial Cell-Derived Von Willebrand Factor, But Not Platelet-Derived, Promotes Atherosclerosis in Apolipoprotein E-Deficient Mice. Arterioscler Thromb Vasc Biol. 2018;38(3):520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gandhi C, Khan MM, Lentz SR, Chauhan AK. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood. 2012;119(10):2385–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin SY, Tohyama J, Bauer RC, Cao NN, Rader DJ, Zheng XL. Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler Thromb Vasc Biol. 2012;32(8):1817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ Res. 2018;123(1):33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569(7755):236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang CC, et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA. 2015;313(3):264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dalager-Pedersen M, Sogaard M, Schonheyder HC, Nielsen H, Thomsen RW. Risk for myocardial infarction and stroke after community-acquired bacteremia: a 20-year population-based cohort study. Circulation. 2014;129(13):1387–96. [DOI] [PubMed] [Google Scholar]
- 86.Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, et al. Novel Role of IL (Interleukin)-1beta in Neutrophil Extracellular Trap Formation and Abdominal Aortic Aneurysms. Arterioscler Thromb Vasc Biol. 2018;38(4):843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, et al. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler Thromb Vasc Biol. 2017;37(8):1457–61. [DOI] [PubMed] [Google Scholar]
- 88.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bemtgen X, Zotzmann V, Benk C, Rilinger J, Steiner K, Asmussen A, et al. Thrombotic circuit complications during venovenous extracorporeal membrane oxygenation in COVID-19. J Thromb Thrombolysis. 2021;51(2):301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grenon E, Canet E. High Incidence of Circuit Clotting in Critically-ill COVID-19 Patients Treated With Renal Replacement Therapy. J Am Soc Nephrol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Libby P, Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Levy JH, Iba T, Gardiner EE. Endothelial Injury in COVID-19 and Acute Infections: Putting the Pieces of the Puzzle Together. Arterioscler Thromb Vasc Biol. 2021;41(5):1774–6. [DOI] [PubMed] [Google Scholar]
- 93.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136(11):1330–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Group RC. Aspirin in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet. 2022;399(10320):143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Investigators R-CWCftR-C, Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z, et al. Effect of Antiplatelet Therapy on Survival and Organ Support-Free Days in Critically Ill Patients With COVID-19: A Randomized Clinical Trial. JAMA. 2022;327(13):1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gauchel N, Rieder M, Krauel K, Goller I, Jeserich M, Salzer U, et al. Complement system component dysregulation is a distinctive feature of COVID-19 disease: a prospective and comparative analysis of patients admitted to the emergency department for suspected COVID-19 disease. J Thromb Thrombolysis. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kander T. Coagulation disorder in COVID-19. Lancet Haematol. 2020;7(9):e630–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pascreau T, Zia-Chahabi S, Zuber B, Tcherakian C, Farfour E, Vasse M. ADAMTS 13 deficiency is associated with abnormal distribution of von Willebrand factor multimers in patients with COVID-19. Thromb Res. 2021;204:138–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Afzali B, Noris M, Lambrecht BN, Kemper C. The state of complement in COVID-19. Nat Rev Immunol. 2022;22(2):77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levi M, Thachil J, Iba T, Levy JH. Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. 2020;7(6):e438–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yatim N, Boussier J, Chocron R, Hadjadj J, Philippe A, Gendron N, et al. Platelet activation in critically ill COVID-19 patients. Ann Intensive Care. 2021;11(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leucker TM, Osburn WO, Reventun P, Smith K, Claggett B, Kirwan BA, et al. Effect of Crizanlizumab, a P-Selectin Inhibitor, in COVID-19: A Placebo-Controlled, Randomized Trial. JACC Basic Transl Sci. 2021;6(12):935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122(7):2661–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood. 2012;119(26):6335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d’Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. 2020;217(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aymonnier K, Ng J, Fredenburgh LE, Zambrano-Vera K, Munzer P, Gutch S, et al. Inflammasome activation in neutrophils of patients with severe COVID-19. Blood Adv. 2022;6(7):2001–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, et al. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. J Clin Invest. 2000;106(3):385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187(5):2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Varju I, Sorvillo N, Cherpokova D, Farkas AZ, Farkas VJ, Komorowicz E, et al. Citrullinated Fibrinogen Renders Clots Mechanically Less Stable, but Lysis-Resistant. Circ Res. 2021;129(2):342–4. [DOI] [PMC free article] [PubMed] [Google Scholar]