

Abstract
Strained bicyclic substructures are increasingly relevant in medicinal chemistry discovery research because of their role as bioisosteres. Over the last decade, the successful use of bicyclo[1.1.1]pentane (BCP) as a para-disubstituted benzene replacement has made it a highly valuable pharmacophore. However, various challenges, including limited and lengthy access to useful BCP building blocks, are hampering early discovery research. Herein we report a single-step, transition metal-free, multi-component approach to synthetically versatile BCP boronates. Radicals derived from commonly available carboxylic acids and organohalides perform additions onto [1.1.1]propellane to afford BCP radicals, which then engage in polarity-matched borylation. A wide array of alkyl-, aryl-, and alkenyl-functionalized BCP boronates were easily prepared. Late-stage functionalization performed on natural products and approved drugs proceeded with good efficiency to generate the corresponding BCP conjugates. Various photoredox transformations forging C–C and C–N bonds were demonstrated by taking advantage of BCP trifluoroborate salts derived from the BCP boronates.
Editorial Summary:
Methods to access bicyclo[1.1.1]pentane building blocks are limited, with current routes requiring multiple steps. Now, a diverse array of bicyclo[1.1.1]pentane boronates can be accessed via a multi-component reaction in a single step. Alkyl-, aryl- and alkenyl substructures can be installed onto bicyclo[1.1.1]pentane boronates by the use of carboxylic acids and organohalides.
Successful drug discovery efforts rely on potent drugs presenting good activity/affinity toward the target of interest as well as satisfactory ADMET parameters.1 If unmet, these requirements spell the demise of clinical drug candidates.2 Modern medicinal chemistry has embraced the concept of “escaping from flatland” as a powerful tenet to circumvent toxicity and poor pharmacokinetics. To improve the physicochemical properties of drug candidates, practitioners are increasingly turning to bioisostere replacements to increase the proportion of sp3-hybridized carbon centers.3–7 Among the more prevalent pharmacophores, para-disubstituted aryl rings suffer from undesired metabolism to the corresponding quinones in vivo, and additionally such arenes are associated with low solubility in physiological media.8 The strategy of increasing three-dimensional space to alleviate these shortcomings has led to the incorporation of cubanes, adamantanes, and bicyclo[1.1.1]pentanes (BCPs), which, among others, have a demonstrated ability to fulfill all the requirements of good bioisosteres of arenes.9,10 Indeed, this strategy is becoming increasingly rewarding as demonstrated by a flow of reports, beginning with the seminal work from Pfizer.11 Since this pioneering effort, the number of patents published with BCP-containing drugs has skyrocketed, along with synthetic methods to incorporate this motif into scaffolds of interest using commercial feedstocks (Fig. 1a).
Figure 1. One-step, multi-component access to diverse BCP boronates.
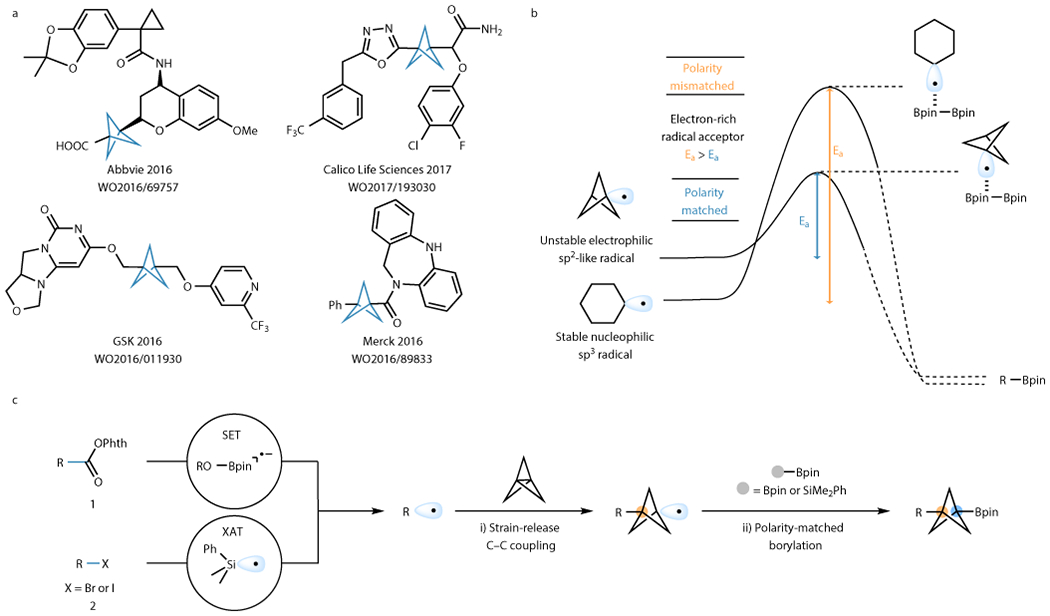
a. Representative BCP-containing drug candidates reported in recent years. b. Proposed kinetic barriers: polarity-matching requirement leads to favorable reactivity between strained sp2-like alkyl radicals with Bpin acceptors. c. This work: Leveraging lower kinetic barrier of radical addition to [1.1.1]propellane and favorable borylation reactivity between BCP radicals and Bpin acceptors to accomplish a simple transition metal-free multi-component reaction to afford the synthetically useful BCP boronates.
Over the decades since the first synthesis of [1.1.1]propellane by Wiberg,12 numerous efforts have been devoted to its functionalization by radical and anionic pathways.13–15 Although highly useful difunctionalizations of [1.1.1]propellane have been developed, especially in recent years by Knochel,16,17 Uchiyama,18,19 Anderson,20,21 Aggarwal,22 Leonori,23 MacMillan24 and others,25–27 the availability of versatile BCP building blocks that would allow late-stage functionalizations for medicinal chemistry studies remains limited. State-of-the-art appendages to elaborate BCPs include carboxylic acids, iodides, and boronates. Of the three, BCP boronates have proven particularly versatile for their ability to be functionalized in a wide range of bond-forming events.19,25,28–33 For example, the bridgehead hydroxy BCP synthesis can currently only be accomplished via BCP boronates. Though endowed with enormous and unique synthetic potential, the challenges of efficient access to BCP boronate derivatives are currently limiting their practicability in medicinal chemistry.
Decarboxylative borylation of BCP carboxylates, pioneered by Aggarwal, is currently the standard,34 but is limited by the lengthy synthesis of BCP carboxylates from [1.1.1]propellane. Furthermore, radical chain borylation using the Suginome borane reported by the Uchiyama group19 and the stepwise addition-borylation process reported by the Walsh group25 provide direct access to BCP boronic acid pinacol ester (Bpin) in one-pot, but these methods only address the synthesis of specific classes of BCP-Bpins, and the requirement for functional group-limiting organometallic reagents is also non-ideal. Finally, the Qin group came up with a modular access to BCP-Bpin by a cyclization approach,33 providing a versatile way to access bridge-substituted BCP boronates. However, a platform that allows rapid access to BCP-Bpin while incorporating diverse structural motifs simultaneously onto the BCP substructure is still elusive. Its development would greatly benefit medicinal chemists, facilitating therapeutic discoveries with BCP by adopting this versatile building block.
In recent years, the rapid development of transition metal-free radical borylation, especially using alkyl radicals, has allowed an expansive range of organoboron compounds to be prepared in a facile manner.35 A bifunctional boron reagent is usually used, with one handle functioning as activator of the radical precursor, while the other serves as the radical acceptor for borylation. Generally, trapping of nucleophilic alkyl radicals is accomplished with a (catecholato)boronate (Bcat) acceptor, which is more electron-poor than a Bpin acceptor. Similarly, trapping of the more electrophilic aryl radicals is usually achieved by using Bpin acceptors because they are relatively more electron-rich. We postulated that polarity matching between radicals and boronate acceptors could play an important role in three-component functionalizations of [1.1.1]propellane using organic radical and boronate partners, and that bridgehead BCP radicals, which display partial sp2 character,36 could be preferentially trapped by a more electron-rich Bpin-type acceptor instead of a Bcat-type acceptor (Fig. 1b).37–39 As evidence, computational studies have shown that the kinetic barrier of alkyl radical borylation with a Bpin acceptor is ~15 kcal/mol,38 which is higher than the barrier for addition onto [1.1.1]propellane (11-13 kcal/mol).20,21 Thus, we envisioned leveraging this difference in energy barriers to perform radical addition onto [1.1.1]propellane by strain-released C1-C3 bond cleavage to generate a BCP radical, which would then be trapped by a Bpin acceptor. In this manner, BCP-Bpins could be made in the simplest imaginable one-step manner. The generality of this approach was expected to depend largely on the diversity of radical progenitors, and because carboxylic acids (1) and organohalides (2) are among the most prevalent building blocks, we reasoned that they would allow access to the broadest range of chemical space. Herein, we report a general approach to substituted BCP-Bpins in a single step using [1.1.1]propellane and two different Bpin acceptors in which redox active esters (RAEs)40,41 derived from carboxylic acids or organohalides serve as radical precursors in conjunctive C–C/C–B couplings featuring easy setup, mild conditions, and broad scope (Fig. 1c).
Results and discussion
Visible-light mediated difunctionalization of [1.1.1]propellane with redox-active ester.
To realize the desired transformations, the reactivity of different alkyl radicals with a Bpin acceptor was first examined by competition experiments (Fig. 2). Five alkyl radicals derived from 1a-e were reacted competitively with the BCP radical from 1f together with bis(pinacolato)diboron (B2pin2) 3a.42 For alkyl radicals derived from sp3-hybridized precursors, only primary alkyl radical 1c shows competitive reactivity to form alkyl-Bpin product.43 We reasoned that the higher inherent reactivity of primary radicals, as well as their minimal steric footprint, make them more favorable partners in this borylation event in comparison with the more stabilized radicals from 1a and 1b. Interestingly, the degree of angle strain was found to correlate positively with the Bpin product formation. Cyclopropyl (1d) radical shows similar reactivity to BCP radicals, giving equal formation of alkyl-Bpin product, while the highly distorted cubyl radical (1e) outcompeted BCP radical, forming predominantly cubyl-Bpin. Additionally, the degree of s character was also found to correlate positively with the borylation yield when using B2pin2 3a (See Supplementary Section 8.1 for details). These results indicate that a polarity match between sp2-like alkyl radicals and Bpin acceptors allows efficient borylation. Moreover, this implies that B2pin2 3a can serve as a less expensive and more robust surrogate for borylation of sp2-like alkyl radicals in place of B2cat2.
Figure 2. Strain-induced increase in s character in alkyl radicals leads to favorable borylations in competition experiments.
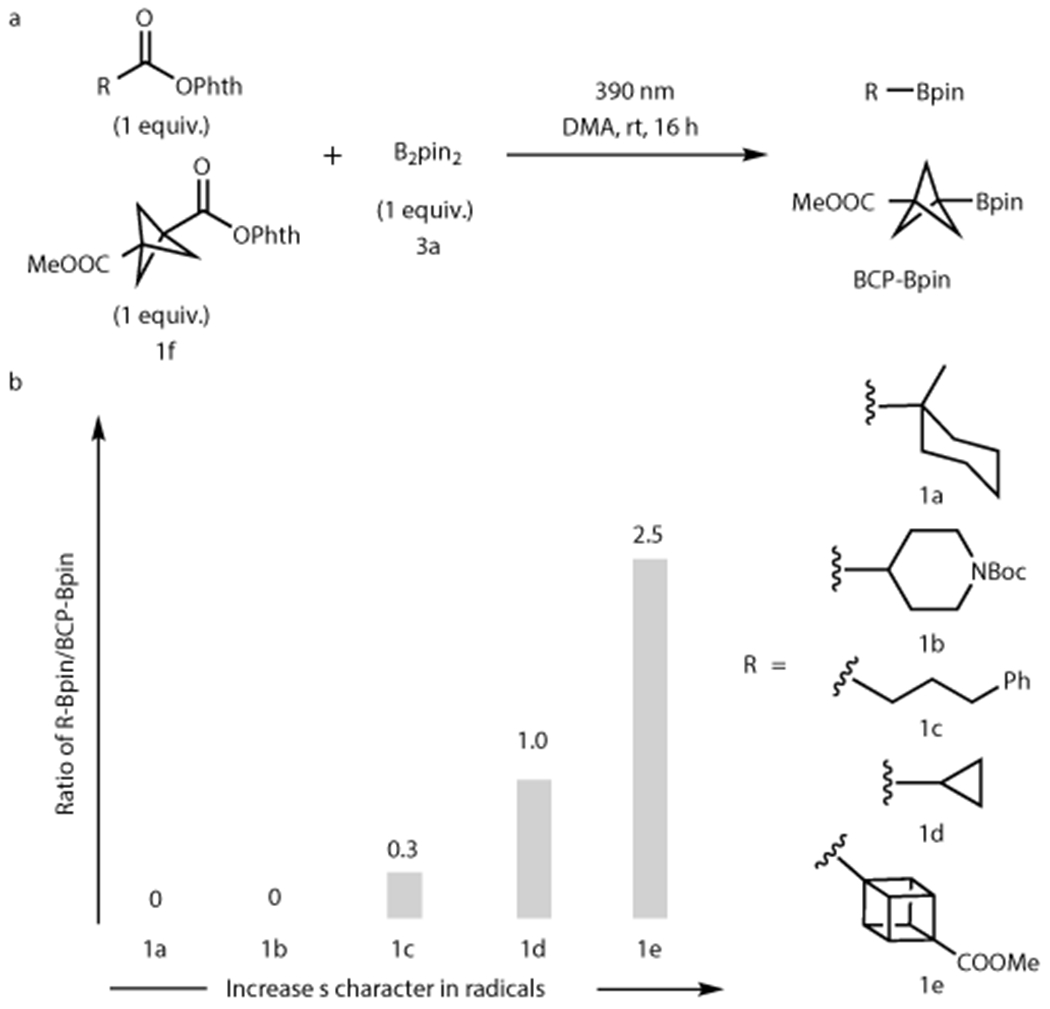
A positive correlation between the degree radical s character and borylation efficiency is demonstrated by competition experiments between radicals with varies s characters. See supplementary information section 8.1 for more details and discussion. a. Competition experiments were performed between BCP redox-active ester 1f and other alkyl bearing redox-active esters with B2pin2 in DMA under 390 nm light irradiation. b. Results indicate that higher s character in the radical leads to more favorable borylation with B2pin2.
Encouraged by this promising reactivity pattern, we explored the possibility of achieving a multi-component reaction forging a C–C and C–B bond in a single operation using [1.1.1]propellane as a linchpin (Table 1). Preliminary studies were carried out with RAE 1b as the radical precursor, [1.1.1]propellane as the BCP precursor, and B2pin2 3a as radical acceptor as well as the activator for the RAE. The multicomponent reaction performed with 1b (1 equiv.), [1.1.1]propellane (1.5 equiv.), and 3a (3 equiv.) in DMA (0.1 M) under irradiation of a 52 W Kessil 390 nm LED lamp provided the desired three-component product in 89% yield (entry 1). Furthermore, 3a is not the only acceptor capable of executing this transformation, as BCP boronic acid can be made in the same manner simply by using tetrahydroxydiboron [B2(OH)4] as the acceptor (entry 2). An attempt to improve the yield of the latter by increasing the loading of B2(OH)4 results in more direct alkyl-borylation product and a lower yield of the desired product (Supplementary Table S1). Not surprisingly, B2cat2, a reactive alkyl radical acceptor, exhibits undesired chemoselectivity, with the two-component alkyl-borylation being the dominant reaction (entry 3). Lowering the loading of 3a to 1.2 equivalents still allows good reactivity (entry 4). A non-amide type solvent was also suitable for the process, providing a slightly diminished yield, indicating the reaction is not mediated by DMA (entry 5).34 Moreover, the poor reactivity under 456 nm light irradiation (entry 6) and the fact that the reaction does not yield any product under thermal conditions in the absence of light (entry 7) indicates the importance of lower wavelength (390 nm) irradiation.
Table 1.
BCP Bpin and boronic acid synthesis by utilization of carboxylic acids as radical precursors
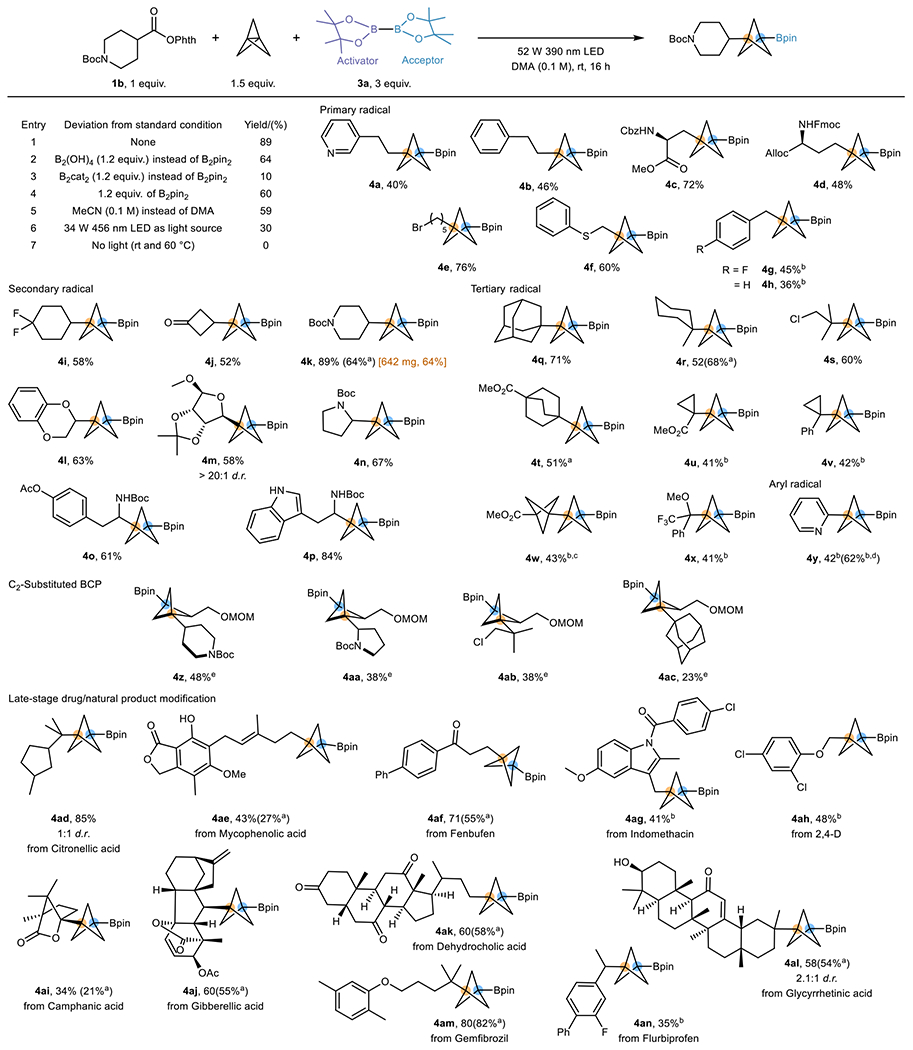
|
Optimization was performed on 0.3 mmol scale of 1b (See Supplementary Section 3.1 for details). Standard conditions for the scope study: RAE substrate (0.3 mmol, 1 equiv.), [1.1.1]propellane (0.45 mmol, 1.5 equiv.), B2pin2 (0.9 mmol, 3 equiv.), DMA (3 mL, 0.1 M), 16 h irradiation with 52 W 390 nm LEDs. All yields are isolated unless otherwise noted.
B2(OH)4 (1.2 equiv.) was used, and the yield was calculated by converting to the pinacolboronate by addition of pinacol (See Supplementary Section 4 for details).
3 equiv. of propellane were used.
1.2 equiv. of B2pin2 were used.
1H NMR yield using 1,3,5-trimethoxybenzene as internal standard
1.5 equiv. C2-substituted [1.1.1]propellane and 2 equiv. B2(OH)4 was used instead of [1.1.1]propellane and B2pin2. Products are racemic. (See Supplementary Section 4 for details).
After demonstrating the feasibility of the process, we next began to examine the scope of this transformation (Table 1). A wide variety of alkyl radicals derived from RAEs can be incorporated despite their dramatically different electrophilicities and steric encumbrances. Unactivated primary alkyl radicals bearing various functional groups afforded the desired BCP-Bpins in good to excellent yields (4a-e). Notably, 4c and 4d, derived from aspartic acid and glutamic acid, serve as unnatural amino acids by simple modifications. An alkyl bromide handle is also tolerated, thus enabling further modification by substitution (4e). Activated α-thioxy- (4f) and benzylic radicals (4g, 4h) also delivered BCP boronates in moderate to good yields. Secondary radicals proved to be highly efficient (4i-p). Notably, nucleophilic radicals, which are under-exploited in reactions with [1.1.1]propellane, were converted to the desired products in good yields (4l-p), demonstrating the generality of this method. Also of note, the reaction is scalable without significant loss of efficiency (4k). Somewhat surprisingly, the use of more sterically hindered tertiary radicals did not lead to a drop in yields (4q-x), even though the formation of vicinal quaternary centers is challenging. Radicals with significant sp2 character (4u, 4v) and electronically similar BCP radicals (4w) maintained good chemoselectivity. Notably, the radical precursor leading to 4x, which has low reactivity as a result of stabilization by a captodative effect, can be trapped by [1.1.1]propellane to deliver the corresponding product, showing the strong thermodynamic driving force accompanying the strain-release effect. Beyond alkyl radicals, an electron-deficient hetaryl radical was also found to be amenable as a result of fast decarboxylation (4x). To demonstrate the robustness of this method, various carboxylate-containing natural products and drug scaffolds were incorporated, and the results show no significant loss of efficiency (4ad-an) (Fig. 3c). Unsurprisingly, the radical derived from citronellic acid underwent 5-exo-trig cyclization to give product 4ad.21
Figure 3. One-pot synthesis of BCP-BF3K 6 and its diversification by photoredox-mediated processes.
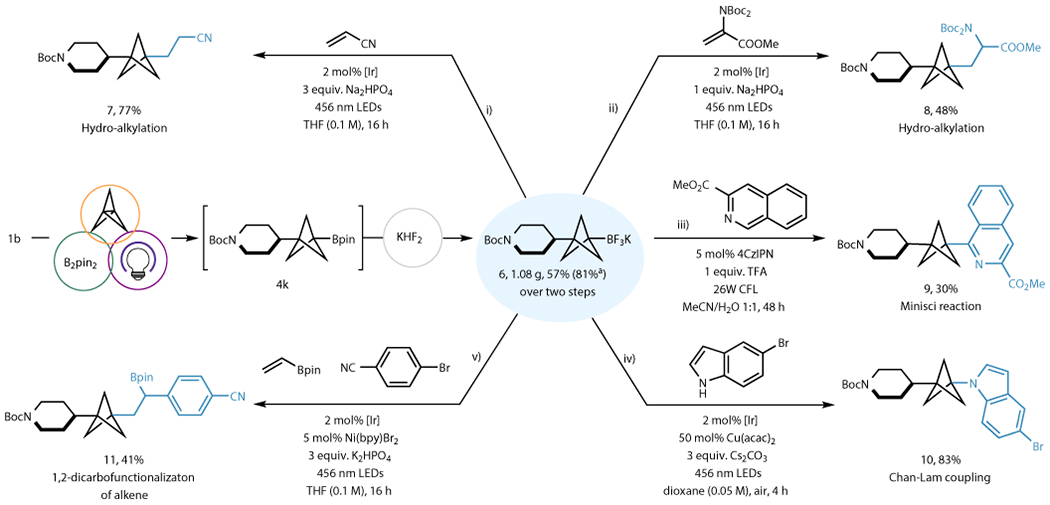
BCP trifluoroborate 6 can be synthesized in a one-pot manner from RAE without the need of column chromatography. The synthetic applications of 6 is demonstrated by several photoredox transformation: (i) hydroalkylation with acrylonitrile; (ii) hydroalkylation with dehydroalanine to yield an amino acid derivative; (iii) Minisci heteroarylation; (iv) single-electron mediated Chan-Lam C–N coupling; (v) 1,2-dicarbofunctionalization of vinyl boronic pinacol ester. See supplementary information section 7 for details on reaction conditions. [Ir]= Ir[dF(CF3)ppy]2(bpy)PF6. aYield by using isolated 4k.
Because boronic acids are more reactive than boronate esters in select transformations, thus providing beneficial advantages for further functionalization, several BCP boronic acids were accessed directly in the same manner using B2(OH)4 as the borylating agent (Table 1). Generally, the BCP boronic acids were synthesized in comparable yields to those of the boronate esters (4k, 4t, 4ae, 4af, 4ai-al), but tertiary radicals tended to perform better by using B2(OH)4 (4r, 4am). Moreover, ortho/meta mimics (4z-4ac) can also be synthesized using this method by using C2-substituted [1.1.1]propellane, albeit the less hindered B2(OH)4 is needed in place of B2pin2 because of steric effects exerted by the C2-substituent. Furthermore, as we realized the synthesis of RAE requires an extra step from the widely available carboxylic acid, we showcased a telescoped process by generating RAEs in situ, then demonstrated that the same reaction can be carried out with increased efficiency (4k, 4ao, 4ap) (Table 2). Remarkably, the in situ transformation is not only applicable in simple carboxylic acids, but more complex structures bearing multiple different functional groups as well (4aq, 4ar).
Table 2.
BCP Bpin synthesis by activating carboxylic acids in situ
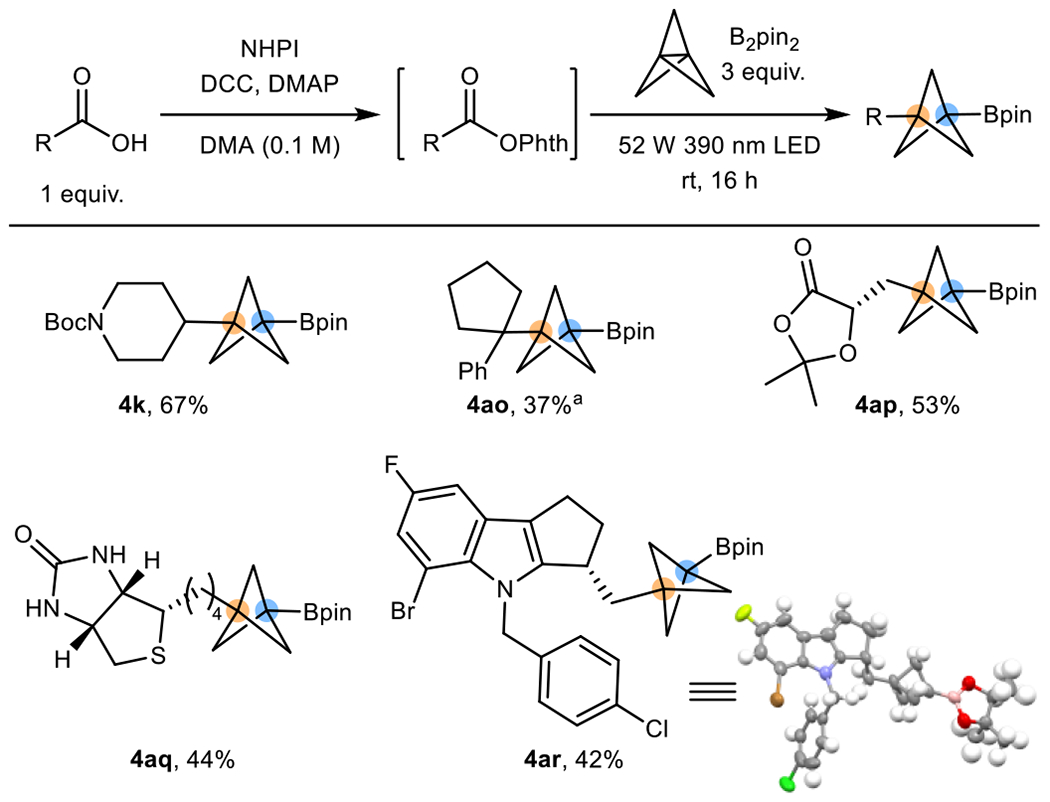
|
Standard conditions: alkyl carboxylic acid (0.3 mmol, 1 equiv.), NHPI (0.33 mmol, 1.1 equiv.), DCC (0.33 mmol, 1.1 equiv.), DMAP (0.015 mmol, 5 mol %), DMA (3 mL, 0.1 M), room temperature 4 h. Borylation; then [1.1.1]propellane (0.45 mmol, 1.5 equiv.), B2pin2 (0.9 mmol, 3 equiv.), 16 h irradiation with 52 W 390 nm LEDs.
3 equiv. of propellane were used.
Difunctionalization of [1.1.1]propellane with organohalides.
Having established a method utilizing B2pin2 3a and taking advantage of a boryl radical as a reductant, we sought to leverage complementary modes of activation that would engage other types of radical precursors to expand the BCP-Bpin library. Organohalides have been demonstrated to react with [1.1.1]propellane to afford BCP halides under thermal and light activation via an ATRA mechanism.20,21 Given the success realized with the RAE radical precursors, we hypothesized that we could also take advantage of abundant organohalides to accomplish BCP-Bpin synthesis. Suginome borane 3b (Table 3), an unsymmetrical reagent with silyl- and boryl handles, is an under-explored reagent in radical processes. The Uchiyama group has shown the possibility of using 3b to undergo Si–B bond homolytic cleavage to generate a silyl radical. This provided the inspiration to leverage the silyl radical as a halogen atom transfer (XAT) reagent to activate organohalides, while using its Bpin handle as a BCP radical acceptor.19 This unexplored mode of reactivity was expected to allow advantage to be taken of abundant organohalides as radical precursors to expand the chemical space of BCP-Bpin substructures (Table 3). Based on our established experience, we quickly discovered that the target product could be accessed in 66% yield using alkyl bromide 2a (1 equiv.) as a radical precursor, [1.1.1]propellane as a BCP linchpin, with Suginome borane 3b acting as an activator and BCP radical acceptor (entry 1). Similarly, alkyl iodide 2b is also compatible and gives a similar yield (entry 2). Control studies using B2pin2 3a give only a trace amount of product (entry 3), showing the necessity of having a strong silyl XAT reagent. The Bpin radical appears to be too sluggish to undergo XAT or single electron transfer (SET) with alkyl halides to generate radicals. The inclusion of a base (K3PO4) to neutralize the acidic byproducts is crucial for this reaction (entry 4). Irradiation at 390 nm works better than 456 nm because of the poor absorption of alkyl halides in the blue region (entry 5). Interestingly, the reaction carried out without light gives a detectable amount of product, which is likely induced by trace amounts of O2 in the reaction (entry 6).19
Table 3.
XAT mediated BCP boronate synthesis from organohalides
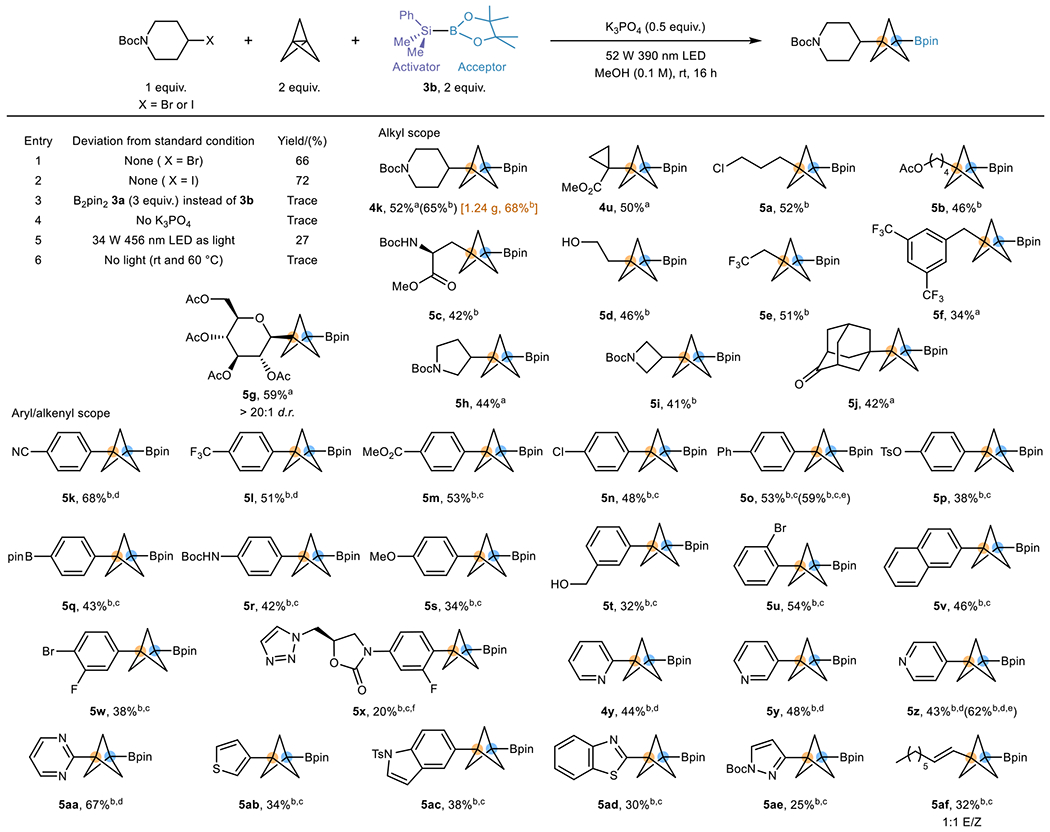
|
Optimization was performed using 0.1 mmol scale of 2a (See Supplementary Section 3.2 for details). Standard conditions for the scope study: organohalide substrate (0.3 mmol, 1 equiv.), [1.1.1]propellane (0.6 mmol, 2.0 equiv.), Me2PhSi-Bpin (0.6 mmol, 2 equiv.), K3PO4 (0.15 mmol, 0.5 equiv.), MeOH (3 mL, 0.1 M), 16 h irradiation with 52 W 390 nm LEDs. All yields are isolated unless otherwise noted.
The organobromide was used.
The organoiodide was used.
3 equiv. of propellane were used.
1.5 equiv. of propellane were used.
1H NMR yield using 1,3,5-trimethoxybenzene as an internal standard.
2:1 MeOH-acetone (0.1 M) mixed solvent was used.
With suitable conditions in hand, we then turned to investigate the scope of this transformation with various organohalides (Table 3). The first class of substrates we examined was alkyl halides, and overall, many types of alkyl halides were found to be compatible in this reaction. Notably, an electron-deficient alkyl radical with an electron-withdrawing group alpha to the radical gives higher yields by using an organobromide than using the carboxylic acid (4u), showing that the two methods are complementary. Also, a chloride handle (5a) and hydroxy group (5d) are tolerated, enabling the potential for post-functionalizations. The reaction was shown to be scalable by carrying out the gram-scale synthesis of 4k using an alkyl iodide.
The abundance of structurally diverse aryl- and heteroaryl halides provided another compelling rationale for using halides as radical precursors, allowing installation of (het)aryl substructures onto the BCP scaffold that were unsuccessful previously using (het)aryl RAEs owing to the challenging decarboxylation. Fortunately, various aryl iodides bearing electronically diverse para substituents were found to react under suitable conditions (5k-s). Moreover, aryl groups bearing many functionalizable handles can be used in the transformation (5p, 5q, 5u, 5w). The ability to incorporate multiple practical subunits on the various substrates in the reactions will facilitate customizable post-functionalizations on the BCP-Bpin platform. Furthermore, meta (5t), ortho (5u), and multi-substituted aryl units (5w, 5x) do not lead to drops in efficiency. Next, we moved on to evaluate the scope of heteroaryl iodides, which are of immense interest in medicinal chemistry. Both electron-deficient (4y, 5y-aa) as well as electron-rich heteroaryls (5ab-ae) delivered the corresponding BCP-Bpins. Notably, 1-benzothiazole and pyrazole have proven challenging to install onto BCP using cross-coupling strategies.44 Although 5ad and 5ae were obtained in moderate yields, access to such substructures demonstrates that the method provides a complementary pathway to high-value targets. Somewhat unusually, an alkenyl fragment could also be incorporated into the BCP substructure in modest yield (5af).
Photoredox functionalization of the boron handles.
The applicability of BCP boronates in subsequent functionalization has been demonstrated by many pioneering works. Numerous transformations have been shown to be successful on BCP boronates, including oxidation to alcohols, Suzuki-Miyaura coupling, and more.19,25,28,33 Thus, after exploiting the potential of the multicomponent reaction to access a wide array of BCP boronates, we sought to demonstrate the synthetic utility of the boron handle by taking advantage of BCP radicals generated from the boronate precursors. Our group is actively engaged in photoredox-mediated transformations using organotrifluoroborate salts, and the lack of reports on using BCP-containing boron compounds in photoredox transformations prompted us to pursue their applications using light-mediated reactions (Fig. 3). To showcase the feasibility of post-functionalizations, we first developed a straightforward process accessing BCP potassium trifluoroborate (BF3K) salt 6 in a one-pot procedure without the need of chromatography. The process can be run on gram-scale, demonstrating its practicality. Cyclic voltammetry was conducted on 6, revealing that even though a high s character is imparted by the strained BCP scaffold, 6 has a favorably low oxidation potential (E1/2 = +1.36 V vs SCE) that is similar to that of sp3 tertiary alkyl BF3Ks (E1/2 = +1.26 V vs SCE)45 and much lower than aryl BF3Ks (E1/2 = +1.84 V vs SCE),46 revealing the alkyl-like properties of the BCP substructure. The Anderson group recently reported a diverse array of BCP radical additions to vinylic acceptors by silyl radical-mediated XAT on BCP halides,47 and we envisioned BCP radicals generated under reductive quenching of a photocatalyst from 6 could also perform additions to electron-deficient alkenes (7, 8). The BCP radical was shown to undergo a smooth polarity-matched addition onto acrylonitrile (i) and dehydroalanine (ii). With the success of implementing these two net-neutral transformations with 6, the feasibility of a late-stage Minisci reaction, a net-oxidative transformation, was investigated.48 Importantly, the BCP radical was found to be modestly effective in this net-oxidative event, providing the Minisci product 9 in a 30% unoptimized yield (iii). Next, we moved on to a metal catalyzed transformation by investigating the feasibility of a photoredox Chan-Lam C–N coupling via a dual catalytic manifold. The Chan-Lam coupling is one of the more important transformations for organoboron compounds, and the C–N linkage is also a high-value bond connection in medicinal chemistry. However, engaging alkylboron compounds in Chan-Lam coupling has been challenging, with only sporadic reports and limited scope.30,49 We hypothesized that one of the challenges for alkylboron substrates in these reactions is the difficult two-electron transmetalation to Cu(II), which is often resolved by elevating the reaction temperature.50,51 Thus, we surmised we could leverage alkyl radical generation to perform a single-electron transmetalation to Cu(II), which would bypass the sluggish, more traditional two-electron transmetalation. Inspired by the work of the MacMillan group,52 we quickly identified suitable conditions for this photoredox-type Chan-Lam coupling, leading to a high yield of the desired product (10) (iv). Finally, the BCP trifluoroborate formed in a conjunctive, multicomponent transformation from readily available materials was subjected to another multicomponent reaction involving addition of the BCP radical to (vinyl)Bpin to form an α-boryl-stabilized radical. The displaced radical generated in this process was subsequently funneled into a Ni-catalyzed Csp2–Csp3 cross-coupling reaction with an aryl halide, providing the desired product 11 in 41% yield (v).53 The two-step process (1,3-difunctionalization of propellane followed by difunctionalization of the vinylboronate) thus dramatically increases molecular complexity by bringing together four very unique components in two simple transformations, with enormous flexibility for even further elaboration as a result of incorporating diverse functional groups from these subunits.
Mechanistic investigations of radical nature and origin of selectivity.
We next turned our attention to investigate the mechanism of this transformation, hoping to shed light on the radical intermediacy under light irradiation and the origin of the selectivity of this well-orchestrated, multi-component transformation. First, TEMPO trapping experiments were performed with both reactions (Fig. 4a). None of the reactions afforded the BCP-Bpin product, but rather the TEMPO adducts 12, which were derived from the radical precursors. Next, a RAE 1i-derived radical clock gives only ring-opened product 13, while 5-hexenyl radical from alkyl halide 2c undergoes 5-exo-trig cyclization to give ring-closing product 15 exclusively (Fig. 4b). Therefore, the radical nature of both reactions with RAE and organohalides were confirmed. Further, elaborations on different radical clocks showed that the time-scale for alkyl radical addition onto [1.1.1]propellane is significantly slower than addition onto electron-deficient alkenes, which is similar to what has been observed in alkyl-iodination of [1.1.1]propellane21 (Supplementary Section 8.3).
Figure 4. Mechanistic investigations into the radical intermediacy and origin of chemoselectivity.
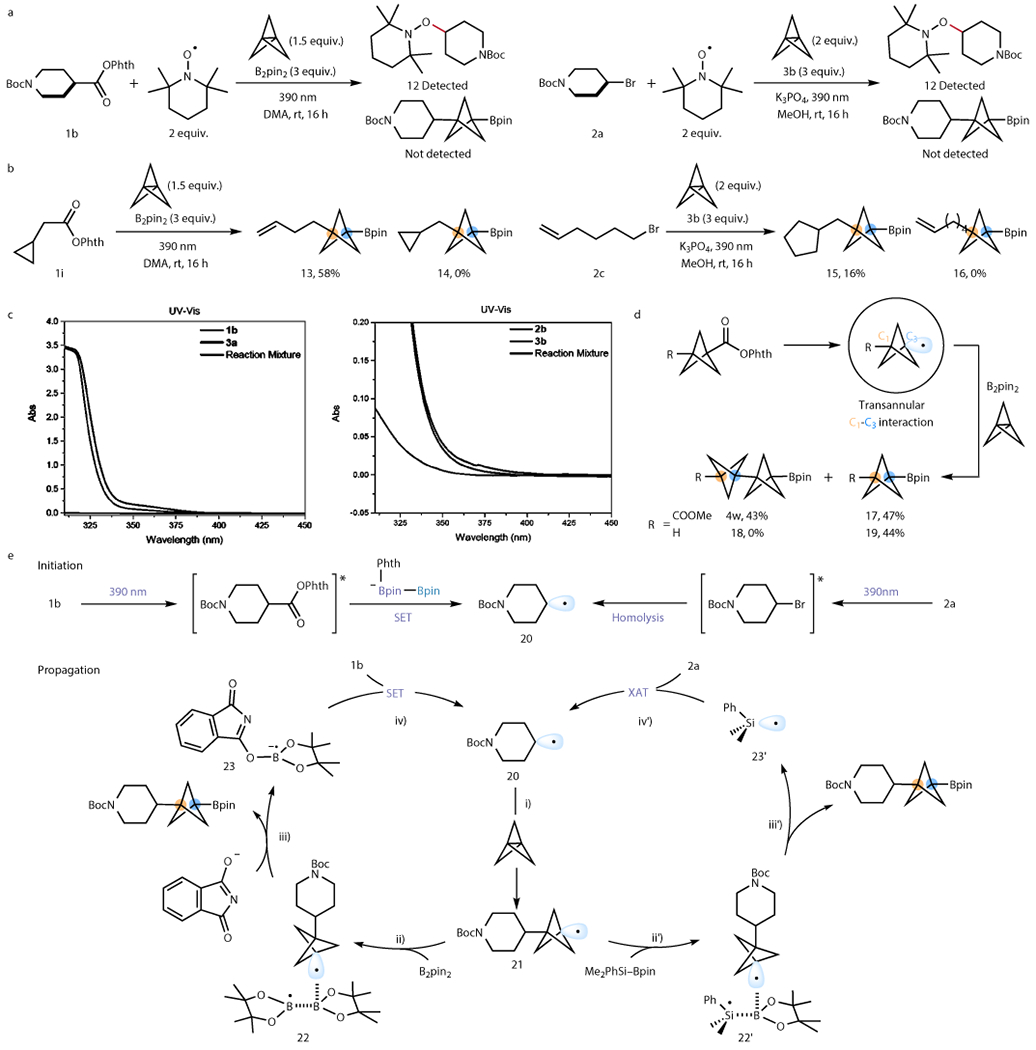
a. Addition of TEMPO completely shut down product formation, and TEMPO-adducts were formed. b. Radical clock experiments confirmed the radical nature of the two reactions and suggested slow addition between radical and [1.1.1]propellane. c. UV-vis studies indicate that at 390 nm, radical precursors are the only absorbing species, and there is no noticeable formation of an EDA complex. Thus, initiation is carried out by excitation of the RAE/organohalides. d. Electronic effect on BCP radical borylation vs. oligomerization: significant through-space effect on the BCP radical such that electron-rich BCP radical tends to undergo borylation whereas electron-poor BCP tends to oligomerize to form [2]staffane as the major product. e. Proposed mechanism based on literature and observations. The chain initiation involves light-mediated excitation of the radical precursor. Chain propagation involves four steps: i) radical addition to [1.1.1]propellane; ii) and ii’) the BCP radical formed coordinates to the Bpin acceptor; iii) and iii’) B–B/Si–B bond undergoes homolytic cleavage to generate BCP-Bpin; iv) and iv’) B/Si radical from the bond cleavage event initiates the next chain cycle.
With the radical nature of the reactions in mind, the genesis of the radicals was investigated. The involvement of O2 in radical generation was first ruled out by carefully degassing the reactions before subjecting the reaction to light irradiation – the results show no loss in reactivity by rigorous exclusion of O2. Thermal activation was also ruled out, because little to no product was observed in the absence of light (Supplementary Tables S1 & S3). UV-Vis studies suggest the only absorbing species are the radical precursors, and there is no bathochromic shift evident for electron donor-acceptor (EDA) complex formation, indicating that radical generation is accomplished by direct excitation of radical precursors (Fig. 4c). The above results support the formation of radicals via light-irradiation. Although numerous studies have demonstrated that the unsubstituted BCP radical is relatively slow to add to [1.1.1]propellane to form [2]staffanes,19,20,23 there is a lack of evidence on how the through-space interaction of substituted BCPs affects the staffane formation. Intriguingly, we observed that the C1-C3 field effect has a dramatic impact on [2]staffane formation (Fig. 4d). Two electronically different BCP radicals with ester (1f) and hydrogen (1h) substituents on C1 were generated from their corresponding RAEs. When the BCP radical bearing an electron-withdrawing group (ester) on C1 was subjected to the standard conditions, we observed a mixture of BCP monomer (17) and [2]staffane oligomer (4w), whereas BCP radicals with a more electron-neutral substituent only affords monomer 18. This explains the observation that multi-component reactions with alkyl radicals always afford BCP-Bpin exclusively, while engaging electrophilic aryl- as well as alkenyl radicals results in formation of a small amount (<20%) of [2]staffane byproducts. We reason that the origin of the phenomenon is variation of the electrophilicity and geometry in the BCP radicals. C1 substituents are known to have a significant impact on the BCP electrophilicity and geometry.23 The increase of electrophilicity favors the addition onto another [1.1.1]propellane instead of B2pin2 because of the electron-rich nature of [1.1.1]propellane, which causes the formation of [2]staffane product. Interestingly, kinetic and NMR studies suggest that phthalimide, a by-product from the RAE, serves as a Lewis base to stabilize the boryl radical, while control studies exclude the involvement of DMA-boryl radical in the chain propagation (See Supplementary Section 8.6 and 8.7 for details). Finally, we conducted quantum yield (Φ) studies with RAE and organohalide systems, and both quantum yields were measured to be larger than 1, indicating chain mechanisms in both cases (See Supplementary Section 8.8).
Based on the combined evidence in hand and previous literature, a mechanism that accounts for this multi-component borylation is proposed (Fig. 4e). In the initiation phase, the RAE is photoexcited by 390 nm irradiation, which then becomes a strong oxidant (E1/2 = +1.6 V)54 that can undergo SET with phthalimide-B2pin2 adduct, generating radical 20. Similarly, organohalide can be excited by 390 nm irradiation, leading to homolysis with generation of 20.39,55 Intermediate 20 is then funneled into the propagation phase by first being intercepted by [1.1.1]propellane (step i). The highly exothermic strain release leads to BCP radical 21, which then engages in borylation with boryl acceptor B2pin2/Me2PhSi-Bpin (step ii/ii′) to form a radical complex 22/22′. The B-B/Si-B bond is then cleaved to form BCP boronates (step iii/iii′) with the corresponding boryl/silyl radical 23/23′ activating another radical precursor by the corresponding SET/XAT event to sustain the chain propagation (step iv/iv′).
Conclusion
In summary, the transition metal-free, multi-component BCP-Bpin synthesis reported herein enables rapid access to a diverse array of BCP building blocks. The radical approaches allow incorporation of two widely available materials, carboxylic acids and organohalides, ensuring access to a broad expanse of novel chemical space. The excellent chemoselectivity enables various motifs, including α-heteroalkyls, strained alkyls, alkenyl- and (het)aryl groups, to be installed easily on the BCP scaffolds. The usefulness of this method is further boosted by direct access to stable BCP BF3K salts and the ability to engage them in photoredox-mediated transformations to build valuable C–C and C–N linkages. We anticipate more functional groups can be introduced into this new platform for BCP boron compound synthesis, and alternative radical acceptors could replace the boron acceptors to generate other BCP motifs, further leveraging the sp2 character of BCP radicals.
Methods
General Procedure for the preparation of [1.1.1]propellane-diethyl ether solution.
To an appropriately-sized round bottom flask is added 1,1-dibromo-2,2-bis(chloromethyl)cyclopropane (5.0 g, 16.8 mmol) and Et2O (10-12 mL) under an inert atmosphere. Once the reactants are dissolved, the reaction is cooled to −78 °C in a Dry Ice-acetone bath. The reaction turns into a slurry at −78 °C. To the light brown slurry is added PhLi (20 mL, 38.0 mmol, 2.3 equiv., 1.9 M solution in n-Bu2O) dropwise over 10 to 15 min. The reaction is then stirred at −78 °C for another 30 min and then is allowed to warm to 0 °C using an ice-water bath. After 2 h, the reaction turns into a dark-brown slurry, which indicates the reaction is finished. The product propellane is co-distilled with Et2O by house vacuum (ca. 4 Torr) as a clear, colorless solution. The receiving flask was submerged in a −78 °C bath or liquid nitrogen bath. The concentration of the [1.1.1]propellane-diethyl ether solution is determined by 1H NMR using 100μL of the solution and 0.100 mmol of 1,3,5-trimethoxybenzene as an internal standard in CDCl3.
General procedure for the preparation of BCP Bpin with redox-active esters and B2pin2.
To an 8 mL reaction vial equipped with a stirrer bar is added redox-active ester (1 equiv.) and dry B2pin2 (3 equiv.). The reaction vessel is sealed with a cap containing a TFE-lined silicone septum and then is evacuated and backfilled with argon three times. When done, degassed DMA (0.1 M) is added. Next, freshly prepared and titrated [1.1.1]propellane (1.5 - 3 equiv., 0.8 – 1.3 M solution in Et2O) is then added, and the vial is quickly sealed with Parafilm®. The reaction mixture is then irradiated under vigorous stirring at 390 nm irradiation using a 52 W Kessil® PR160L-390 nm lamp at 1 inch distance for 16 h. Room temperature is maintained by the use of two fans. After 16 h, the deep red reaction mixture is partitioned between Et2O and saturated aqueous NH4Cl. The organic layer is washed with brine twice, dried (MgSO4), filtered, and then concentrated under reduced pressure. The product is purified by flash column chromatography.
General procedure for the preparation of BCP Bpin with organohalides.
To an 8 mL reaction vial equipped with a stirrer bar is added organohalide (1 equiv.) and K3PO4 (0.5 equiv.). The vial is then transferred to a nitrogen-filled glovebox. Me2PhSi-Bpin (2 equiv.) is added, and then the vial is sealed with a cap containing a TFE-lined silicone septum and transferred out of the glovebox. MeOH (0.1 M) is added. Next, [1.1.1]propellane (1.5 – 3 equiv., 0.8 – 1.3 M solution in Et2O) is added, and the vial is quickly sealed with Parafilm®. The reaction mixture is then irradiated under vigorous stirring at 390 nm irradiation using a 52 W Kessil® PR160L-390 nm lamp at 1 inch distance for 16 h. Room temperature is maintained by the use of two fans. After 16 h, the crude material is passed through a pad of Celite® and eluted with another 10 mL of acetone. The filtrate is concentrated under reduced pressure and purified by flash column chromatography.
Data Availability
All data supporting the findings of this study are available in this article and its Supplementary Information. Crystallographic data for the structure 4ar reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under the deposition number CCDC 2105768. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
Supplementary Material
Supplementary Data 1 Crystallographic data for compound 4ar; CCDC reference 2105768
Acknowledgement
The authors thank financial support provided by NIGMS (R35 GM 131680 to G.M.). Financial support for this research was provided in part by AbbVie. AbbVie participated in the interpretation of data, review, and approval of the publication. The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplements awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr. Charles W. Ross, III (UPenn) for mass spectral data and Dr. Michael R. Gau (UPenn) for X-ray crystallography data. We thank Frontier for donations of B2pin2, BASF for donations of B2(OH)4, Kessil for donations of LED lamps, and Merck for donations of informer sets.
Footnotes
Supplementary Information General information, experimental procedures, extended optimizations, synthesis and characterization data, mechanistic studies, discussion, X-ray crystallographic data, references, NMR spectra, Supplementary Scheme S1, Figures S1–16 and Tables S1–13.
Competing Interests Statement
The authors declare no competing interest.
References
- 1.van de Waterbeemd H & Gifford E ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov 2, 192–204 (2003). [DOI] [PubMed] [Google Scholar]
- 2.M. Honorio K, L. Moda T & D. Andricopulo A Pharmacokinetic properties and in silico ADME modeling in drug discovery. Med. Chem 9, 163–176 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Lovering F, Bikker J & Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa M & Hashimoto Y Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 54, 1539–1554 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Meanwell NA Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem 54, 2529–2591 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Lovering F Escape from flatland 2: complexity and promiscuity. Med. Chem. Commun 4, 515–519 (2013). [Google Scholar]
- 7.Walker MA Novel tactics for designing water-soluble molecules in drug discovery. Expert Opin. Drug Discov 9, 1421–1433 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Ritchie TJ & Macdonald SJF The impact of aromatic ring count on compound developability – are too many aromatic rings a liability in drug design? Drug Discov. Today 14, 1011–1020 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Auberson YP et al. Improving nonspecific binding and solubility: bicycloalkyl groups and cubanes as para-phenyl bioisosteres. ChemMedChem 12, 590–598 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Mykhailiuk PK Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem 17, 2839–2849 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Stepan AF et al. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem 55, 3414–3424 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Wiberg KB & Walker FH [1.1.1]Propellane. J. Am. Chem. Soc 104, 5239–5240 (1982). [Google Scholar]
- 13.Wiberg KB Small ring propellanes. Chem. Rev 89, 975–983 (1989). [Google Scholar]
- 14.Kanazawa J & Uchiyama M Recent advances in the synthetic chemistry of bicyclo[1.1.1]pentane. Synlett 30, 1–11 (2019). [Google Scholar]
- 15.Pramanik MMD, Qian H, Xiao W-J & Chen J-R Photoinduced strategies towards strained molecules. Org. Chem. Front 7, 2531–2537 (2020). [Google Scholar]
- 16.Makarov IS, Brocklehurst CE, Karaghiosoff K, Koch G & Knochel P Synthesis of bicyclo[1.1.1]pentane bioisosteres of internal alkynes and para-disubstituted benzenes from [1.1.1]propellane. Angew. Chem. Int. Ed 56, 12774–12777 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Schwärzer K, Zipse H, Karaghiosoff K & Knochel P Highly regioselective addition of allylic zinc halides and various zinc enolates to [1.1.1]propellane. Angew. Chem. Int. Ed 59, 20235–20241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanazawa J, Maeda K & Uchiyama M Radical multicomponent carboamination of [1.1.1]propellane. J. Am. Chem. Soc 139, 17791–17794 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Kondo M et al. Silaboration of [1.1.1]propellane: a storable feedstock for bicyclo[1.1.1]pentane derivatives. Angew. Chem. Int. Ed 59, 1970–1974 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Caputo DFJ et al. Synthesis and applications of highly functionalized 1-halo-3-substituted bicyclo[1.1.1]pentanes. Chem. Sci 9, 5295–5300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nugent J et al. A general route to bicyclo[1.1.1]pentanes through photoredox catalysis. ACS Catal. 9, 9568–9574 (2019). [Google Scholar]
- 22.Yu S, Jing C, Noble A & Aggarwal VK 1,3-Difunctionalizations of [1.1.1]propellane via 1,2-metallate rearrangements of boronate complexes. Angew. Chem. Int. Ed 59, 3917–3921 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Kim JH, Ruffoni A, Al-Faiyz YSS, Sheikh NS & Leonori D Divergent strain-release amino-functionalization of [1.1.1]propellane with electrophilic nitrogen-radicals. Angew. Chem. Int. Ed 59, 8225–8231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shelp RA et al. Strain-release 2-azaallyl anion addition/borylation of [1.1.1]propellane: synthesis and functionalization of benzylamine bicyclo[1.1.1]pentyl boronates. Chem. Sci 12, 7066–7072 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin S, Lee S, Choi W, Kim N & Hong S Visible-light-induced 1,3-aminopyridylation of [1.1.1]propellane with N-aminopyridinium salts. Angew. Chem. Int. Ed 60, 7873–7879 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Wu Z, Xu Y, Zhang H, Wu X & Zhu C Radical-mediated sulfonyl alkynylation, allylation, and cyanation of propellane. Chem. Commun 57, 6066–6069 (2021). [DOI] [PubMed] [Google Scholar]
- 28.VanHeyst MD et al. Continuous flow-enabled synthesis of bench-stable bicyclo[1.1.1]pentane trifluoroborate salts and their utilization in metallaphotoredox cross-couplings. Org. Lett 22, 1648–1654 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Merchant RR & Lopez JA A general C(sp3)–C(sp3) cross-coupling of benzyl sulfonylhydrazones with alkyl boronic acids. Org. Lett 22, 2271–2275 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Zarate C et al. Development of scalable routes to 1-bicyclo[1.1.1]pentylpyrazoles. Org. Process Res. Dev 25, 642–647 (2021). [Google Scholar]
- 31.Yang Y et al. Practical and modular construction of C(sp3)-rich alkyl boron compounds. J. Am. Chem. Soc 143, 471–480 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Ripenko V, Vysochyn D, Klymov I, Zhersh S & Mykhailiuk PK Large-scale synthesis and modifications of bicyclo[1.1.1]pentane-1,3-dicarboxylic acid (BCP). J. Org. Chem (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y et al. An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nat. Chem 13, 950–955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fawcett A et al. Photoinduced decarboxylative borylation of carboxylic acids. Science 357, 283–286 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Friese FW & Studer A New avenues for C–B bond formation via radical intermediates. Chem. Sci 10, 8503–8518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levin MD, Kaszynski P & Michl J Bicyclo[1.1.1]pentanes, [n]staffanes, [1.1.1]propellanes, and tricyclo[2.1.0.02,5]pentanes. Chem. Rev 100, 169–234 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Jin S et al. Visible light-induced borylation of C–O, C–N, and C–X bonds. J. Am. Chem. Soc 142, 1603–1613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Q et al. Transition-metal-free borylation of alkyl iodides via a radical mechanism. Org. Lett 21, 6597–6602 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Cheng Y, Mück-Lichtenfeld C & Studer A Transition metal-free 1,2-carboboration of unactivated alkenes. J. Am. Chem. Soc 140, 6221–6225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toriyama F et al. Redox-active esters in Fe-catalyzed C–C coupling. J. Am. Chem. Soc 138, 11132–11135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin T et al. A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 352, 801–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fawcett A New developments in organoboron incorporation, homologation and functionalization Doctor of Philosophy thesis, The University of Bristol, (2018). [Google Scholar]
- 43.Hu D, Wang L & Li P Decarboxylative borylation of aliphatic esters under visible-light photoredox conditions. Org. Lett 19, 2770–2773 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Polites VC, Badir SO, Keess S, Jolit A & Molander GA Nickel-catalyzed decarboxylative cross-coupling of bicyclo[1.1.1]pentyl radicals enabled by electron donor–acceptor complex photoactivation. Org. Lett 23, 4828–4833 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Primer DN & Molander GA Enabling the cross-coupling of tertiary organoboron nucleophiles through radical-mediated alkyl transfer. J. Am. Chem. Soc 139, 9847–9850 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki J, Tanigawa M, Inagi S & Fuchigami T Electrochemical oxidation of organotrifluoroborate compounds. ChemElectroChem 3, 2078–2083 (2016). [Google Scholar]
- 47.Pickford HD et al. Twofold radical-based synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. J. Am. Chem. Soc 143, 9729–9736 (2021). [DOI] [PubMed] [Google Scholar]
- 48.Matsui JK, Primer DN & Molander GA Metal-free C–H alkylation of heteroarenes with alkyltrifluoroborates: a general protocol for 1°, 2° and 3° alkylation. Chem. Sci 8, 3512–3522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.West MJ, Fyfe JWB, Vantourout JC & Watson AJB Mechanistic development and recent applications of the Chan–Lam amination. Chem. Rev 119, 12491–12523 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Lam PYS in Synthetic Methods in Drug Discovery: Volume 1 Vol. 1 242–273 (The Royal Society of Chemistry, 2016). [Google Scholar]
- 51.Rossi SA, Shimkin KW, Xu Q, Mori-Quiroz LM & Watson DA Selective formation of secondary amides via the copper-catalyzed cross-coupling of alkylboronic acids with primary amides. Org. Lett 15, 2314–2317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lavagnino MN, Liang T & MacMillan DWC HARC as an open-shell strategy to bypass oxidative addition in Ullmann–Goldberg couplings. Proc. Natl. Acad. Sci. U. S. A 117, 21058–21064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Campbell MW, Compton JS, Kelly CB & Molander GA Three-component olefin dicarbofunctionalization enabled by nickel/photoredox dual catalysis. J. Am. Chem. Soc 141, 20069–20078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoon UC & Mariano PS The synthetic potential of phthalimide SET photochemistry. Acc. Chem. Res 34, 523–533 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Cheng Y, Mück-Lichtenfeld C & Studer A Metal-free radical borylation of alkyl and aryl iodides. Angew. Chem. Int. Ed 57, 16832–16836 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data 1 Crystallographic data for compound 4ar; CCDC reference 2105768
Data Availability Statement
All data supporting the findings of this study are available in this article and its Supplementary Information. Crystallographic data for the structure 4ar reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under the deposition number CCDC 2105768. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.