

Abstract
Arsenic, a widely existing environmental contaminant, is recognized to be toxic to multiple organs. Exposure to arsenic results in liver damage via excessive production of reactive oxidative species (ROS). PIN1 regulates the levels of ROS. N-acetyl-L-cysteine (NAC) is an ROS scavenger that protects the hepatic functions. Whether PIN1 plays a regulatory role in NAC-mediated antagonism against arsenic hepatotoxicity remains largely unknown. In our study, the protective effects of NAC against arsenic (NaAsO2)-induced hepatotoxicity were evaluated in vitro and in vivo. Arsenic exposure induced cytotoxicity by increasing the intracellular ROS production, impairing mitochondrial function and inducing apoptosis in L02 hepatocytes. Overexpression of PIN1 markedly protected against arsenic cytotoxicity, decreased ROS levels, and mitigated mitochondrial dysfunction and apoptosis in L02 cells. However, loss of PIN1 further aggravated arsenic-induced cytotoxicity and abolished the protective effects of NAC in L02 cells. An in vivo study showed that pretreatment with NAC rescued arsenic-induced liver injury by restoring liver function and suppressing hepatic oxidative stress. Overexpression of PIN1 in mice transfected with AAV-Pin1 relieved arsenic-induced liver dysfunction and hepatic oxidative stress. Taken together, our study identified PIN1 as a novel intervention target for antagonizing arsenic-induced hepatotoxicity, highlighting a new pharmacological mechanism of NAC targeting PIN1 in antagonism against arsenic toxicity.
Keywords: PIN1, arsenic, hepatotoxicity, ROS, mitochondrial dysfunction
1. Introduction
Arsenic is a ubiquitous metalloid element found in the environment and is classified as a group 1 carcinogen.1 Arsenic pollution from agricultural and industrial activities poses a worldwide threat to public health in modern society. Human exposure to arsenic through contaminated water, soil, air, and foods results in various adverse health effects.2,3,4,5 Epidemiological studies have revealed that arsenic exposure is closely associated with the occurrence of multiple diseases, such as neurological, immune, reproductive, and cardiovascular disorders, in addition to diabetes mellitus and various cancers.6,7,8,9,10 The liver is a major metabolic organ and plays a critical role in detoxification in the body. Previous studies have demonstrated that arsenic exposure causes liver damage and induces liver dysfunction. Furthermore, the toxic effects induced by arsenic promote the occurrence of liver fibrosis, cirrhosis, and cancer.11,12 Oxidative stress is considered to be one of the important biological mechanisms of arsenic-induced liver injury.13 Abnormally elevated production of ROS after arsenic exposure is mainly responsible for oxidative stress.14 Arsenic-induced ROS overproduction has been found to induce a series of pathological events, including DNA damage, abnormal gene expression, mitochondrial dysfunction, and even apoptosis.15 However, it remains elusive how arsenic exposure increases intracellular ROS production and disrupts mitochondrial functions at molecular level.
PIN1 is a peptidyl-prolyl cis/trans isomerase that specifically binds to phosphorylated Ser/Thr-pro motifs to catalytically regulate the post-phosphorylation conformation of its substrates.16 Conformational changes in substrate proteins regulated by PIN1 play an important role in diverse cellular processes, such as cell growth, the cell cycle, stress responses, cell survival, and apoptosis.17,18,19 Recent studies have found that PIN1 plays an important role in maintaining redox balance.17,20 Decreased PIN1 expression results in impairment of mitochondrial function through a significant increase in intracellular ROS production, which leads to apoptosis.20,21 In addition, suppression of PIN1 aggravates diabetic vascular disease by causing mitochondrial oxidative stress.17 These results indicate that PIN1 is functionally involved in regulating cellular oxidative stress. Previous studies on PIN1 mainly focused on its involvement in cancer development and neurodegenerative disorders.22,23 However, the role of PIN1 in oxidative stress in arsenic-induced hepatotoxicity is unclear and requires further experimental investigation.
Since arsenic exposure elicits oxidative stress, the application of antioxidants has been accepted as an effective approach for antagonizing arsenic toxicity. N-acetyl-L-cysteine (NAC) is an acetyl derivative of the amino acid cysteine and a precursor for the synthesis of glutathione.24 NAC, as a nonprescription drug without proven toxicity, is widely used as an antioxidant clinically.25,26 Multiple studies have suggested that NAC can alleviate arsenic-induced cytotoxicity by reducing oxidative stress and apoptosis in a variety of cell types, including hepatocytes,27 nerve cells,28 intestinal epithelial cells,29 and embryonic cells.30 In addition, pretreatment with NAC increases the expression level of PIN1.31 Moreover, administration of NAC to Alzheimer’s disease model mice slightly increases the levels of PIN1.32 However, the relationship between NAC, PIN1 and ROS has not been elucidated. The pharmacological mechanisms by which NAC counteracts arsenic-induced ROS production have not been fully clarified yet. Considering the critical role of PIN1 in regulating intracellular ROS production, whether PIN1 mediates the pharmacological actions of NAC in antagonizing arsenic hepatotoxicity needs to be elucidated.
In the present study, we aimed to investigate the underlying mechanisms by which NAC antagonizes arsenic hepatotoxicity. Our results demonstrated that NAC attenuated arsenic-induced hepatotoxicity both in vitro and in vivo, as indicated by the maintenance of cell viability, suppression on ROS production, preservation of mitochondrial functions, mitigation of oxidative stress, and inhibition of apoptosis. Mechanistically, we found that the expression of PIN1 was down-regulated by arsenic treatment in vitro and in vivo. However, overexpression of PIN1 relieved arsenic-induced hepatotoxicity, reduced ROS production, protected mitochondrial function, and suppressed apoptosis in vitro and in vivo. Silencing of PIN1 expression aggravated arsenic-induced hepatotoxicity and abolished the protective effect of NAC treatment. Our data demonstrated for the first time that PIN1 plays a suppressive role in arsenic-induced intracellular ROS production and serves as an intervention target for antagonizing arsenic hepatotoxicity.
2. Materials and methods
2.1 Materials and reagents
Human hepatocytes (L02 cells) were purchased from the Cell Bank of Shanghai Institute of Biological Sciences (Shanghai, China). Sodium arsenite (NaAsO2) of greater than 99% purity was purchased from Sigma–Aldrich (S7400, San Francisco, USA). N-Acetyl-L-cysteine (NAC) of greater than 99% purity was purchased from Beyotime (ST1546, Shanghai, China). The other reagents and assay kits used in the experiment are described in detail below.
2.2 Cell culture
The L02 cells were cultured and maintained in RPMI-1640 medium (Gibco, C11875500BT, USA) supplemented with 10% fetal bovine serum (Biological Industries, 04–001-1A, Israel) and 1% antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin; Gibco, 15140-122, USA). The cells were cultured in 75 cm2 flasks and incubated at 37 °C in a humidified atmosphere containing 5% CO2.
2.3 Cell viability assay
To assess arsenic cytotoxicity, L02 cells were seeded in 96-well flat-bottomed plates at a density of 8 × 103 cells per well and cultured overnight, and then arsenic was added to the culture medium at the concentration of 2.5, 5, 7.5, or 10 μM for 0 h, 12 h,, 24 h, or 48 h. To study the protective effect of NAC, the cells were pretreated with 2, 4, or 8 mM NAC for 4 h, and then exposed to arsenic for 24 h. Cell viability was measured using a CCK-8 assay kit (Dojindo Laboratories, CK04, Japan) according to the manufacturer's instructions. Briefly, CCK-8 reagent was mixed with fresh medium to produce a 1/10 (v/v) working solution, and 100 μL of the CCK-8 reagent working solution was added to each well. Then, the cells were cultured in an incubator at 37 °C for 1.5 h. The absorbance was read at 450 nm with a microplate reader (Infinite M200 PRO, TECAN, Switzerland).
2.4. Measurement of intracellular ROS levels
The levels of intracellular ROS were determined using a well-characterized probe, CM-H2DCFDA (Invitrogen, C6827, USA), as previously described.33 Briefly, L02 cells were pretreated with NAC (4 mM or 8 mM) for 4 h and coincubated with NAC and arsenic (7.5 μM) for 3 h. After treatment, the cells were carefully washed once with HBSS buffer. A working solution of 25 μM CM-H2DCFDA diluted in HBSS was added to the cells, and the cells were incubated for 30 min at 37 °C. Then, the probes were discarded, and the cells were washed twice with HBSS. The fluorescence value was determined at excitation and emission wavelengths of 485 and 538 nm, respectively, with a microplate reader (Infinite M200 PRO, TECAN, Switzerland) and quantified as a percentage of that in the untreated control group.
2.5 Mitochondrial membrane potential (ΔΨm) measurement
L02 cells were pretreated with NAC (4 mM or 8 mM) for 4 h and coincubated with NAC and arsenic (7.5 μM) for 3 h. Subsequently, the cells were mixed with 10 μg/mL JC-1 fluorescent probe (Beyotime, C2005, China), incubated in the dark at 37 °C for 20 min, washed twice with HBSS, and resuspended in HBSS. The fluorescence intensity was measured with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). For fluorescence quantitation, green (JC-1 monomers) and red (JC-1 aggregates) fluorescence intensity was measured at wavelengths of 488/525 nm (excitation/emission) and 560/590 nm (excitation/emission), respectively. The ratio of red/green represents the changes in the mitochondrial membrane potential.34
2.6 Measurement of intracellular adenosine triphosphate levels
L02 cells were pretreated with NAC (4 mM or 8 mM) for 4 h and coincubated with NAC and arsenic (7.5 μM) for 3 h. The intracellular adenosine triphosphate (ATP) levels were measured with an ATP Determination Kit (Molecular Probes, A22066, USA) following the manufacturer’s instructions. We prepared standard reaction solution by serially diluted 5 mM ATP standards and obtained a standard curve. ATP concentrations were calculated using the ATP standard curve.35
2.7 Mitochondrial morphology analysis
To assess the changes in mitochondrial morphology in L02 cells, cells were pretreated with NAC (4 mM) for 4 h and then coincubated with NAC and arsenic (7.5 μM) for 24 h. Then, the cells were incubated with 100 nM MitoTracker Red CMXRos probe (Beyotime, C1035, China) for 30 min at 37 °C according to the manufacturer’s instructions. After two washes with PBS, the L02 cells were visualized under a Leica confocal laser scanning microscope (Leica TCS SP5).36
2.8 Analysis of intracellular oxidative stress
To determine the status of intracellular oxidative stress, we measured intracellular antioxidant contents. L02 cells were pretreated with NAC (4 mM or 8 mM) for 4 h and then coincubated with NAC and arsenic (7.5 μM) for 24 h. After treatment, the cells were washed twice with cold PBS and lysed with lysis buffer on ice for 30 min. The supernatant was collected after centrifugation for 10 min (20,000 × g, 4 °C) and used to analyze the level of lipid peroxidation, reduced glutathione (GSH) content, and superoxide dismutase (SOD) and catalase (CAT) activity. Commercial assay kits for determining MDA (A003-1A), GSH (A006–2), SOD (A001–3) and CAT (A007–1) levels were purchased from Nanning Jiancheng Bioengineering Institute (Nanjing, China). All procedures were performed according to the manufacturer’s protocols. Briefly, the level of lipid peroxidation was measured by analyzing the content of MDA using the thiobarbituric acid (TBA) method.37 MDA reacts with TBA to form a red product. The absorbance value was measured at 532 nm with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). The GSH content was determined by using the 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) method.35 GSH reacts with DTNB to form a yellow product. The absorbance value was measured at 405 nm with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). SOD activity was estimated according to the WST-1 method.38 The reaction mixture, including enzyme supernatant solution, enzyme working solution, and substrate reaction solution, was incubated at 37 °C for 20 min. The absorbance value was measured at 450 nm with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). An enzyme activity unit was defined as the quantity of enzyme needed to cause 50% inhibition of the reaction solution. CAT activity was measured according to the ammonium molybdate method.38 The enzyme supernatant solution was reacted with the substrate solution for precisely 1 min at 37 °C. Stop solution was added to produce a yellow product. The absorbance value was measured at 405 nm with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). The protein concentrations of the samples were determined by the BCA method39 for normalization of the levels of MDA, GSH, SOD, and CAT.
2.9 Flow cytometry analysis
Apoptotic cells were detected by using an FITC Annexin V Apoptosis Detection Kit (BD Biosciences, 556,547, USA) according to the manufacturer’s instructions. After different treatments, the cells were harvested and washed twice with precooled Dulbecco's phosphate-buffered saline (D-PBS), resuspended in 200 μL of binding buffer containing 3 μL propidium iodide and 3 μL annexin V-FITC, and incubated for 15 min in the dark. All of the samples were analyzed immediately with a flow cytometer (BD AccuriC6, USA).40
2.10 Quantitative real-time PCR
Total cellular RNA was extracted using the RNAsimple Total RNA Kit (TIANGEN, DP419, China) according to the manufacturer’s instructions. cDNA was synthesized using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara, RR047A, Japan) and used for quantitative real-time reverse transcription polymerase chain reaction. To determine the expression of target genes, the expression levels determined by RT–PCR were normalized to the expression level of ACTB. The following PCR primer sequences were used. PIN1 forward: 5’-GGTGAAGCACATCCAGTCA-3′; PIN1 reverse: 5’-GGGCCTCCTCCTTGGTC-3′; ACTB forward: 5’-CCTGGCACCCAGCACAAT-3′; ACTB reverse: 5’-GGGCCGGACTCGTCATAC-3′. The relative mRNA expression levels of the target genes were quantified by using the 2-△△CT method.41
2.11 Pin1 plasmid and siRNA transfection
pcDNA3.1-Pin1 and pcDNA3.1-empty vector were constructed by Sangon Biotech (Shanghai, China). For Pin1 overexpression, pcDNA3.1-Pin1 and pcDNA3.1-empty vector were transfected into L02 cells by using Lipofectamine 2000 (Invitrogen, 11668027, USA) following the manufacturer’s instructions.41Pin1-specific siRNA and control siRNA were purchased from GenePharma (Shanghai, China). To silence Pin1 expression, Pin1 siRNA and control siRNA were transfected into L02 cells by using Lipofectamine 2000 (Invitrogen, 11668027, USA) following the manufacturer’s instructions.28 After transfection for 24 h, the cells were pretreated with NAC for 4 h and then exposed to arsenic alone or arsenic and NAC for 24 h.
2.12. AAV-Pin1 transfection in mice
To overexpress Pin1 in the mouse liver, the pAAV-CBh-MCS-EF1-mNeonGreen-WPRE empty vector was generated by OBio Technology (Shanghai, China). Pin1 cDNA was packaged into an empty vector (pAAV-CBh-PIN1-3xFLAGEF1-mNeonGreen-WPRE; titer: 8.53 × 1012 V.G/mL) by OBio Technology. The control was an empty vector (AAV-null; titer: 1.21 × 1013 V.G/mL). AAV-Pin1 transfection via tail vein injection was used to induce Pin 1 overexpression in mice.
2.13 Animal experiments
Male C57BL/6 J mice (20–24 g) aged 8 weeks were purchased from the animal center of Third Military Medical University. All animals were allowed to acclimatize to the environment for one week before being used in the experiments. The animals were housed in a room on a 12-h light/dark cycle and provided ad libitum access to standard laboratory food and fresh water during the entire experiment. The animal experiments were approved by the Third Military Medical University Animal Care and Use Committee.
To investigate whether NAC alleviates arsenic-induced liver injury in vivo, mice were divided randomly into 4 groups with 10 animals in each group: Group 1 (control group), which consisted of mice treated with normal saline once a day; Group 2, which consisted of mice administered NAC (100 mg/kg/day)42 by intraperitoneal injection once a day; Group 3, which consisted of mice treated with arsenic (10 mg/kg/day)43 by intragastric administration once a day; And Group 4, which consisted of mice treated with NAC (100 mg/kg/day, intraperitoneal injection) and arsenic (10 mg/kg/day, intragastric administration). NAC was given 2 h prior to arsenic administration. Arsenic exposure and NAC treatment continued for two weeks.
To investigate the protective effect of PIN1 against arsenic-induced liver injury in vivo, mice were randomly divided into the following 3 groups with 10 animals in each group: Group 1 (the AAV-null plus control group); Group 2 (the AAV-null plus arsenic group); And Group 3 (the AAV-Pin1 plus arsenic group). Mice in the AAV-Pin1 plus arsenic group were intravenously injected with 2 × 1011 AAV-Pin144 dissolved in saline. Mice in the AAV-null groups, received the same dose of AAV-null by injection. After 2 weeks, mice in the AAV-null plus arsenic and AAV-Pin1 plus arsenic group were treated with arsenic (10 mg/kg/day) by intragastric administration once a day, and mice in the AAV-null control group received the same volume of saline by injection.41 Arsenic exposure continued for two weeks.
Animals received the indicated treatments for two weeks and were then anesthetized with 20% ethyl carbamate and euthanized. Blood was immediately collected. Serum was isolated by centrifugation for 15 min at 1,000 × g and stored at −20 °C for biochemical analysis. The liver tissues were rapidly separated into two parts for further biochemical and histopathological examinations.
2.14 Serum biochemistry analysis
The activity of alanine transaminase (ALT) (C009–2-1) and aspartate transaminase (AST) (C010–2-1) in the serum was measured with commercial kits from Nanning Jiancheng Bioengineering Institute (Nanjing, China) according to the manufacturer’s protocol.45
2.15 Liver histopathology analysis
Histological examination of liver tissue was used to assess arsenic-induced hepatic damage and the protective effect of NAC. Liver specimens were fixed in 4% paraformaldehyde, embedded in paraffin, cut into 4-μm thick sections, and stained with hematoxylin and eosin (H&E). Morphological observation was conducted using a microscope (Eclipse Ci, Nikon, Japan). Representative images were obtained. Quantitative analysis of hepatic lesions was performed, and the injury score was determined according to Suzuki’s criteria.46
2.16 Measurement of liver oxidative stress
Liver tissues were prepared as 10% homogenates in .9% saline solution in an ice-water bath. Then, the homogenates were centrifuged at 1,000 × g for 10 min at 4 °C. The separated supernatants were used to analyze the level of lipid peroxidation, GSH content, and SOD and CAT activity. The detailed methods are described above.
2.17 Western blotting
Protein was extracted from cells and liver tissues using RIPA Lysis Buffer (Beyotime, P0013B, China) containing protease inhibitor cocktail (Roche, 04693116001, Switzerland) and centrifuged at 20,000 × g for 30 min at 4 °C. The protein concentration was determined with an Enhanced BCA Protein Assay Kit (Beyotime, P0010, China). Protein samples were mixed with 1× loading buffer and boiled for 15 min at 100 °C. Total protein (10 μg) was separated by SDS–PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Bio–Rad, 1,620,177, USA). The membranes were blocked in Quick-Block™ Blocking Buffer (Beyotime, P0252, China) for 1 h at room temperature and incubated with primary antibodies against PIN1 (Proteintech, 10,495–1-AP, 1:2,000) of rabbit source and ACTB (Beyotime, AF0003, 1:1,000) of mouse source overnight at 4 °C. After washing with TBST three times, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse (Beyotime, A0216, 1:1,000) or anti-rabbit (Beyotime, A0208, 1:1,000) IgG secondary antibody at room temperature for 1 h. The protein bands were visualized by using an enhanced chemiluminescence kit (Millipore, WBKLS0500, USA) and scanned with a ChemiDocTM XRS+ imaging densitometer (Bio–Rad, USA). The densities of the protein bands were quantified with Image Lab™ software. The expression level of PIN1 was normalized to the expression level of ACTB.41
2.18 Statistics
The experimental data are presented as the mean ± SEM. The data were analyzed with GraphPad Prism 5.0 software. Differences were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test and t test. Differences were considered statistically significant when P < .05.
3. Results
3.1 NAC relieves arsenic-induced cytotoxicity in L02 cells
Cytotoxicity induced by arsenic exposure resulted in a decrease in cell viability. Cells were exposed to different concentrations of arsenic (2.5, 5, 7.5, or 10 μM) for 12, 24 or 48 h. Cell viability was significantly decreased in a time- and dose-dependent manner (Fig. 1A, Fig. S1A and S1B). The IC50 value was approximately 30.4 μmol/L 24 h after arsenic exposure (Fig. S1C). NAC, a well-characterized antioxidant, was used to antagonize arsenic-induced hepatotoxicity. NAC treatment alone did not affect significantly cell viability, indicating that it had no cytotoxic effects (Fig. S1D). We found that compared with arsenic treatment alone, pretreatment with .5, 1, 2, 4, or 8 mM NAC for 4 h prior to exposure to 7.5 μM arsenic for 24 h markedly prevented the arsenic-induced decline in cell viability (Fig. S1E). The EC50 of NAC against arsenic toxicity was 4.81 mM (Fig. S1F). The data revealed that pretreatment with NAC for 4 h had the best protective effect, as indicated by the significant elevation of cell viability after pretreatment with 2, 4, and 8 mM NAC (Fig. 1B, Fig. S1G and S1H). Arsenic decreased cell viability and caused morphological damage. In the time-course study, arsenic treatment caused obvious morphological damage in a time-dependent manner. Pretreatment with 4 and 8 mM NAC considerably mitigated arsenic-induced morphological changes (Fig. 1C).
Fig. 1.

NAC relieves arsenic-induced cytotoxicity, intracellular ROS production and mitochondrial dysfunction in L02 cells. (A) Effects of different concentrations of arsenic on cell viability at 24 h after exposure. (B) Effects of NAC pretreatment for 4 h on the arsenic-induced decrease in cell viability at 24 h after exposure. (C) NAC pretreatment protects against arsenic-induced morphological damage, as observed under a light microscope at 10× magnification. (D) Changes in intracellular ROS levels at 3 h in the different treatment groups. (E) Changes in the mitochondrial membrane potential (ΔΨm) at 3 h in the different treatment groups. (F): Changes in intracellular ATP levels at 3 h in the different treatment groups. The data are presented as the mean ± SEM of four independent experiments. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group.
To explore the potential mechanisms of arsenic-induced cytotoxicity in L02 cells, intracellular ROS levels, mitochondrial function, oxidative stress, and apoptosis were measured. First, we found that the levels of intracellular ROS were markedly increased in the arsenic exposure group compared with the control group at 3 h after arsenic exposure. Arsenic treatment significantly decreased the mitochondrial membrane potential and ATP levels at 3 h after exposure (Fig. 1D-F). Second, arsenic treatment induced lipid peroxidation at 24 h, as indicated by the significant increase in MDA levels after treatment. Arsenic treatment resulted in a marked reduction in GSH content at 24 h after treatment. SOD and CAT activity was marked suppressed by arsenic at 24 h after treatment (Fig. S2A–S2D). Finally, arsenic resulted in severe apoptosis at 24 h after treatment (Fig. S2E and S2F). These results indicated that arsenic exposure could increase intracellular ROS production. This has been recognized as an early event leading to cell damage. Mitochondria are the main site of intracellular ROS production and important targets of oxidative damage.47 Overproduction of intracellular ROS disrupts the redox balance and causes oxidative stress, which ultimately leads to apoptosis.48 However, as the concentration of NAC increased, intracellular ROS levels, the mitochondrial membrane potential, ATP levels, MDA content, GSH content, SOD activity, CAT activity, and apoptosis of L02 cells were restored to control levels in the presence of 7.5 μM arsenic (Fig. 1D-F, Fig. S2A-S2F). These results suggested that arsenic exposure induced cytotoxicity in a dose- and time-dependent manner. NAC could attenuate arsenic-induced cytotoxicity by inhibiting intracellular ROS production, mitochondrial dysfunction, oxidative stress, and apoptosis in L02 cells.
3.2 NAC antagonizes the suppressive effect of arsenic on PIN1 expression in vitro and in vivo
Recent studies have revealed that PIN1 plays an important role in regulating intracellular ROS production. It is unclear whether arsenic exposure affects the expression of PIN1 and induces cellular oxidative stress. As shown in Fig. 2A-C, arsenic induced a decrease in PIN1 mRNA and protein expression in L02 cells after exposure for 24 h. Treatment with 4 and 8 mM NAC reversed the downregulation of PIN1 expression in a dose-dependent manner. Furthermore, administration of 100 mg/kg NAC markedly inhibited the arsenic-induced downregulation of PIN1 expression at the protein level in the liver (Fig. 2D and E). These in vitro and in vivo results indicated that arsenic exposure suppressed PIN1 expression and that NAC antagonized the decrease in the expression of PIN1 induced by arsenic. Therefore, we propose that PIN1 may play a critical role in arsenic-induced toxicity.
Fig. 2.

NAC antagonizes the decrease in the expression of PIN1 induced by arsenic in vitro and in vivo. (A) Changes in Pin1 mRNA levels in L02 cells in the different treatment groups. (B) Changes in PIN1 protein levels in L02 cells in the different treatment groups. (C) Semiquantification of PIN1 protein levels in L02 cells. (D) Changes in hepatic PIN1 protein levels in mice from the different treatment groups. (E) Semiquantification of hepatic PIN1 protein levels in mice from the different treatment groups. The data are presented as the mean ± SEM of four independent experiments or three mice. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group.
3.3 Pin1 overexpression attenuates arsenic-induced cytotoxicity in L02 cells
To further confirm the role of PIN1 in the antagonism of arsenic toxicity, we examined whether Pin1 overexpression could rescue arsenic-induced cytotoxicity by transfecting L02 with a Pin1-expressing plasmid. It was found that Pin1 plasmid transfection led to stable overexpression of PIN1 in L02 cells (Fig. 3A and B). Moreover, the arsenic-induced decrease in cell viability at 24 h was partially rescued by Pin1 overexpression (Fig. 3C and D). L02 cells were transfected with the Pin1 plasmid and treated with NAC, but no synergistic antagonist effect of Pin1 overexpression and NAC on arsenic-induced changes in PIN1 expression (Fig. 3A and B), morphological damage (Fig. 3C) or alterations in cell viability (Fig. 3D) were observed. Since Pin1 overexpression antagonized arsenic toxicity, we further evaluated whether Pin1 overexpression could rescue mitochondrial dysfunction, inhibit oxidative stress and suppress apoptosis in arsenic-treated L02 cells. Notably, compared with arsenic treatment alone, transfection of the Pin1 plasmid significantly suppressed the arsenic-induced elevation of ROS levels (Fig. 4A), obviously maintained the mitochondrial membrane potential (Fig. 4B), and markedly increased intracellular ATP levels (Fig. 4C). The combination of Pin1 transfection and NAC treatment did not significantly augment these protective effects. Moreover, mitochondrial damage, which was assessed with MitoTracker Red, was obviously mitigated by Pin1 plasmid transfection and NAC treatment (Fig. 4D). Pin1 plasmid transfection alone and NAC treatment alone significantly inhibited arsenic-induced apoptosis in L02 cells (Fig. 4E and F). Together, these data revealed that PIN1 is an important mediator of arsenic-induced cytotoxicity. PIN1 and NAC exerted the same protective effects against arsenic toxicity. Pin1 overexpression attenuated arsenic-induced cytotoxicity, intracellular ROS production, mitochondrial dysfunction, and apoptosis in L02 cells. However, the relationship between NAC and PIN1 in the antagonism of arsenic toxicity remains unclear.
Fig. 3.

Pin1 overexpression attenuates arsenic-induced cytotoxicity in L02 cells. (A) Representative images of PIN1 protein expression after Pin1 plasmid transfection in the different treatment groups. (B) Semiquantification of PIN1 protein levels after Pin1 plasmid transfection in the different treatment groups. (C) Pin1 plasmid transfection ameliorated the arsenic-induced morphological damage, as observed under a light microscope at 10× magnification. (D) Pin1 plasmid transfection inhibited the arsenic-induced decline in cell viability. The data are presented as the mean ± SEM of four independent experiments. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group.
Fig. 4.
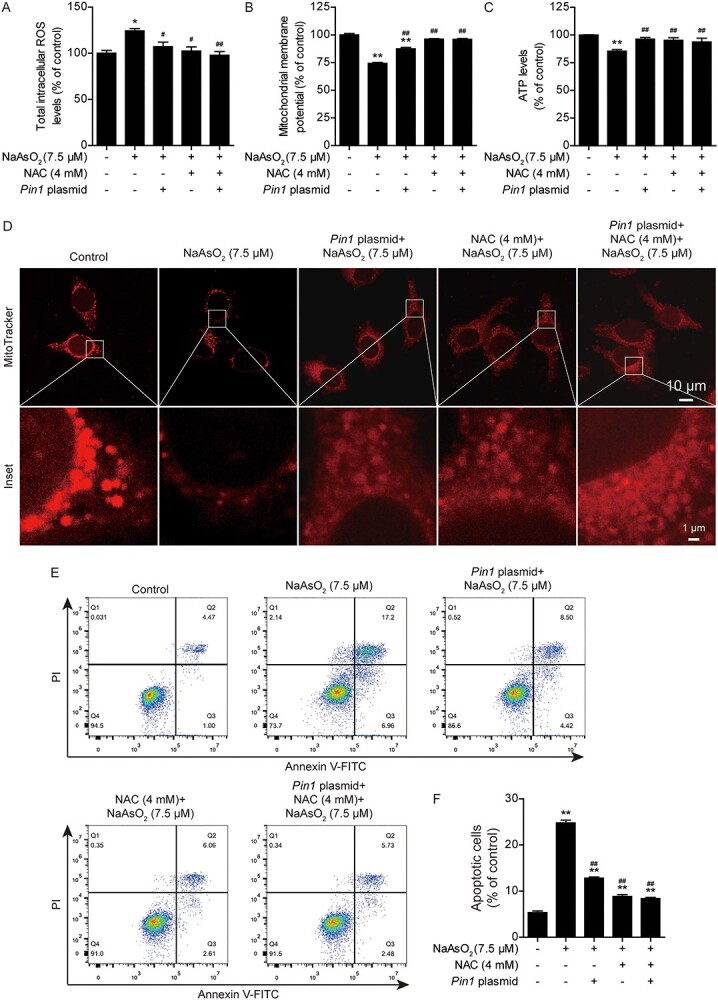
Pin1 overexpression attenuates arsenic-induced intracellular ROS production, mitochondrial dysfunction and apoptosis in L02 cells. (A) Changes in intracellular ROS levels in the different treatment groups. (B) Changes in the mitochondrial membrane potential (ΔΨm) in the different treatment groups. (C) Changes in intracellular ATP levels in the different treatment groups. (D) Mitochondrial damage, as indicated by the specific fluorescence probe MitoTracker red, was mitigated by Pin1 plasmid transfection. Scale bar: 10 or 1 μm. (E) Pin1 plasmid transfection suppressed arsenic-induced apoptosis, as analyzed by flow cytometry. (F) Quantification of apoptotic cells in the different treatment groups. The data are presented as the mean ± SEM of four independent experiments. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group.
3.4 Knockdown of Pin1 abolishes the protective effects of NAC against arsenic-induced cytotoxicity in L02 cells
Based on the above data, it is clear that PIN1 plays an important role in antagonizing arsenic toxicity. However, the relationship between PIN1 and NAC needs to be verified. Therefore, the expression of Pin1 was knocked down with specific siRNA (Fig. 5A and B). Downregulation of PIN1 expression markedly aggravated the arsenic-induced decline in cell viability. The protective effect of NAC on cell viability was almost completely abolished after Pin1 was silenced (Fig. 5C and D). Next, intracellular ROS levels and mitochondrial function were analyzed. The results revealed that loss of PIN1 expression increased intracellular ROS production and reduced the mitochondrial membrane potential and ATP levels in arsenic-treated cells (Fig. 6A-C). Furthermore, silencing of PIN1 expression increased cell apoptosis in arsenic-treated cells (Fig. 6D and E). Moreover, deficiency of PIN1 eliminated the protective effects of NAC against arsenic toxicity (Fig. 6A-E). Taken together, these data showed that PIN1 is a key regulator of the protective effects of NAC against arsenic-induced cytotoxicity, intracellular ROS production, mitochondrial dysfunction, and apoptosis in L02 cells.
Fig. 5.

Knockdown of Pin1 abolishes the protective effects of NAC against arsenic-induced cytotoxicity in L02 cells. (A) Representative images of PIN1 protein expression after Pin1 siRNA transfection in the different treatment groups. (B) Semiquantification of PIN1 protein levels after Pin1 siRNA transfection in the different treatment groups. (C) Pin1 siRNA transfection exacerbated arsenic-induced morphological damage and disrupted the protective effect of NAC against arsenic-induced morphological damage, as observed under a light microscope at 10× magnification. (D) Pin1 siRNA transfection aggravated the arsenic-induced decline in cell viability and eliminated the protective effect of NAC against the arsenic-induced decline in cell viability. The data are presented as the mean ± SEM of four independent experiments. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group. &P < .05, &&P < .01 vs. the NAC plus arsenic group.
Fig. 6.

Knockdown of Pin1 abolishes the protective effects of NAC against arsenic-induced intracellular ROS production, mitochondrial dysfunction and apoptosis in L02 cells. (A) Changes in intracellular ROS levels in the different treatment groups. (B) Changes in the mitochondrial membrane potential (ΔΨm) in the different treatment groups. (C) Changes in intracellular ATP levels in the different treatment groups. (D) Pin1 siRNA transfection aggravated arsenic-induced apoptosis and abolished the protective effect of NAC against arsenic-induced apoptosis, as analyzed by flow cytometry. (F) Quantification of apoptotic cells in the different treatment groups. The data are presented as the mean ± SEM of four independent experiments. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic group. &P < .05, &&P < .01 vs. the NAC plus arsenic group.
3.5 Pin1 overexpression mitigates arsenic-induced liver damage and oxidative stress in mice
We further investigated whether Pin1 overexpression plays a key role in arsenic-induced liver damage and oxidative stress in mice. An adeno-associated virus (AAV) vector expressing Pin1 was constructed and delivered to mice by tail vein injection. As shown in Fig. S3A and S3B, arsenic exposure significantly increased serum ALT and AST activity. Histopathological observation showed that arsenic exposure resulted in severe liver tissue damage. The affected hepatocytes were swollen, and their cytoplasms appeared cloudy and granular (Fig. S3C). The Suzuki injury score in the arsenic exposure group was significantly higher than that in the control group (Fig. S3D). Treatment with NAC reversed these liver function abnormalities and histopathological changes and lowered the Suzuki injury score (Fig. S3A-S3D). As expected, mice were successfully infected with AAV-Pin1, and PIN1 protein was overexpressed in these mice. Pin1 overexpression effectively restored the level of PIN1 in the arsenic exposure group (Fig. 7A and B). Similarly, compared with arsenic treatment alone, Pin1 overexpression obviously decreased ALT and AST activity and partially restored hydropic degeneration, necrosis and disordered arrangement of hepatocyte cords (Fig. 7C-E).
Fig. 7.

Pin1 overexpression mitigates arsenic-induced liver damage and oxidative stress in mice. (A) Representative images of PIN1 protein expression after AAV-Pin1 infection in mice from the different treatment groups. (B) Semiquantification of PIN1 protein levels after AAV-Pin1 infection in mice from the different treatment groups. (C) Changes in serum alanine transaminase (ALT) activity after AAV-Pin1 infection in mice from the different treatment groups. (D) Changes in serum aspartate transaminase (AST) activity after AAV-Pin1 infection in mice from the different treatment groups. (E) Representative images of hematoxylin and eosin showing liver damage after AAV-Pin1 infection in mice from the different treatment groups. Scale bar: 50 μm. (F-I) Changes in hepatic MDA levels (F), GSH levels (G), SOD activity (H) and CAT activity (I) after AAV-Pin1 infection in mice from the different treatment groups. The data are presented as the mean ± SEM of three or six mice. *P < .05, **P < .01 vs. the control group. #P < .05, ##P < .01 vs. the arsenic (AAV-null) group.
To assess whether the protective effect of PIN1 against arsenic-induced liver injury is associated with hepatic oxidative stress in mice, we further analyzed oxidative stress levels in Pin1-overexpressing mice. First, we found that the MDA level in the liver, which was shown to be increased by arsenic, was obviously decreased in NAC-treated mice (Fig. S3E). Moreover, the levels of GSH and activity of SOD and CAT, which are all antioxidants, were significantly higher in NAC-treated mice than in arsenic-treated mice (Fig. S3F-S3H). Interestingly, Pin1 overexpression also reduced MDA levels and increased GSH levels, SOD activity and CAT activity in the liver after arsenic exposure (Fig. 7F-I). Together, these findings indicated that Pin1 overexpression mitigated arsenic-induced liver damage and oxidative stress in mice.
4. Discussion
In this study, arsenic hepatotoxicity and antagonism by NAC were investigated in vitro and in vivo. Importantly, we identified PIN1 as a novel target for counteracting arsenic hepatotoxicity. Arsenic exposure reduced L02 cell viability, increased ROS production, caused mitochondrial dysfunction, stimulated oxidative stress, and led to apoptosis in L02 cells. Similarly, arsenic exposure caused liver damage by inducing oxidative stress in mice. More importantly, the toxic effects of arsenic exposure could be antagonized by NAC pretreatment. As reported previously, PIN1 exerts its functions by catalytically regulating conformational changes in substrate proteins after phosphorylation to further control the function of proteins involved in stress responses. We aimed to elucidate the potential mechanisms by which NAC relieves arsenic toxicity and found for the first time that PIN1 plays a pivotal role in protective effects of NAC treatment. These results provide new insight into arsenic cytotoxicity and hepatotoxicity, highlighting PIN1 as a novel therapeutic target of NAC in antagonism against arsenic hepatotoxicity.
It is well known that arsenic is a highly toxic metalloid.49 The toxicity of inorganic arsenic is higher than that of organic arsenic.50 Sodium arsenite (NaAsO2) has been widely used in the study of arsenic toxicity in vivo and in vitro.51 In our study, exposure to 7.5 μM arsenic induced obvious cytotoxicity in a time- and dose-dependent manner in L02 cells. Furthermore, treatment with 10 mg/kg/day arsenic for 14 days caused severe liver damage in mice. The World Health Organization (WHO) set a safety standard of 10 μg/L for arsenic in drinking water. The maximum arsenic concentration in well water tested in North Carolina was 806 μg/L.52 In the United States, arsenic concentrations greater than 3,000 μg/L have been found in wells.6 It has been reported that the groundwater concentration of arsenic varies greatly by region, from .5 to 5,000 μg/L.53 In the present study, 7.5 μM arsenic was equivalent to 1,000 μg/L. The arsenic concentration we selected was consistent with environmental exposure levels and concentrations used in previous studies.15,54 There is extensive epidemiological evidence for the multiple organ toxicity of arsenic exposure, and the data suggest that the liver is one of the major target organs of arsenic toxicity.12 Long-term arsenic exposure through drinking water impairs hepatic functions and increases the risk of liver cancer mortality.55 There are no antagonizing measures to control arsenic exposure at population level yet. Hence, it is important to evaluate the currently used drugs as antidotes for arsenic hepatotoxicity.
Oxidative stress is an important mechanism of arsenic-induced hepatotoxicity. It is well known that increased ROS production can impair mitochondrial function, elicit intracellular oxidative stress and induce apoptosis. These toxic effects have long been recognized as the most important molecular events in arsenic toxicity.56,57 The results of our in vitro and in vivo studies demonstrated that mitochondrial dysfunction and oxidative stress are crucial for arsenic hepatotoxicity. Increasing the cellular antioxidant capacity is one approach used to abolish arsenic toxicity. Antioxidants from natural and synthetic compounds may effectively reduce arsenic hepatotoxicity.58 NAC is an organosulfur antioxidant derived from Allium plants with a broad-spectrum antioxidant effect.59 It is reported to exert hepatoprotective effects.60 As a source of sulfhydryl groups, NAC can restore endogenous antioxidant potential, promote detoxification and act as a strong scavenger of toxic radicals such as OH• and H2O2.61 Additionally, NAC has a cytoprotective effect against inorganic arsenic toxicity.28 In the present study, we demonstrated the protective effects of NAC against liver injury induced by arsenic exposure both in vivo and in vitro. Specifically, biochemical indicators of arsenic-induced hepatocellular damage and ROS production were reversed by NAC treatment. Arsenic-induced histological and functional damage were significantly mitigated by NAC treatment. Mitochondria play critical roles in maintaining cell function by producing ATP and regulating cell metabolism.62 Our study suggested that arsenic-induced mitochondrial damage involves increased ROS production, ATP depletion and impaired mitochondrial integrity. However, the addition of the antioxidant NAC reversed this damage. To eliminate excessive intracellular ROS, the endogenous redox defense system needs to be activated. Cellular antioxidant enzymes (SOD and CAT) and GSH are the major components for scavenging free radicals.13,15 However, the generation of excess ROS disrupts the balance between oxidation and antioxidant homeostasis, induces intracellular oxidative stress and subsequently causes apoptosis.48 In our study, arsenic exposure triggered an increase in MDA levels and a decrease in GSH levels in vivo and in vitro. Furthermore, SOD and CAT activity was significantly decreased. These results suggested that arsenic exposure resulted in the production of excessive ROS and induced oxidative stress, emphasizing the importance of using antioxidants to alleviate arsenic toxicity. This notion was robustly supported by the fact that NAC treatment could effectively alleviate arsenic-induced oxidative stress and mitigate hepatotoxicity. Our results demonstrated that NAC could be an effective antioxidant for antagonizing arsenic hepatotoxicity. Although the production of ROS plays an important role in arsenic-induced hepatotoxicity, the mechanism through which arsenic induces ROS production is not fully understood, and the targets for NAC in alleviating arsenic-induced oxidative stress remain to be discovered.
Recently, PIN1 was recognized as an important regulator of intracellular ROS production.20 The biological functions of PIN1 have been widely investigated in various types of diseases. Previous studies on aberrant expression of PIN1 have focused on cancer progression and neurodegenerative disorders.22,23 However, whether PIN1 plays a role in arsenic toxicity and mediates the antioxidant effects of NAC is unknown. Research on the relationship between PIN1 and arsenic has mainly focused on cancer. Arsenic trioxide (ATO) induces PIN1 degradation and inhibits cell growth. Moreover, ATO, particularly in combination with all-trans retinoic acid (ATRA), blocks multiple cancer-driving pathways and eliminates tumor-initiating cells (TICs) in triple-negative breast cancer (TNBC) by targeting PIN1.63 However, the effect of PIN1 on arsenic-induced hepatotoxicity has not been studied. Herein, we identified PIN1 as an important regulator of arsenic-induced cytotoxicity. Arsenic exposure significantly suppresses PIN1 expression in vivo and in vitro. Moreover, cobalt, an environmental toxicant, dose-dependently decreases PIN1 expression and alterations in its substrates. Furthermore, cobalt-induced neurotoxicity is aggravated by genetic or chemical PIN1 inhibition but rescued by upregulation of PIN1 expression.64 These results and our study indicate the importance of PIN1 in heavy metal poisoning.
In the present study, downregulation of PIN1 expression decreased cell viability and promoted oxidative stress and apoptosis in L02 cells. Upregulation of PIN1 expression effectively antagonized arsenic-induced ROS production, alleviated mitochondrial dysfunction and protected against apoptosis in L02 cells. Furthermore, a decrease in PIN1 expression caused liver damage and hepatic oxidative stress after arsenic exposure, and overexpression of PIN1 by AAV-Pin1 infection relieved arsenic-induced hepatotoxicity. Our data showed that the mRNA and protein levels of PIN1 were decreased upon arsenic exposure in vitro and in vivo, indicating that the mechanism of arsenic-induced ROS production could be related to a reduction in PIN1 expression. Similar results were obtained in early studies, in which cobalt was found to cause cell damage, apoptosis and oxidative stress upon downregulation of PIN1 expression both in vitro and in vivo.64 PIN1 is a key effector involved in the Kras/ERK axis that synergistically mediates various cellular events in pancreatic ductal adenocarcinoma (PDAC). PIN1 expression is markedly upregulated in PDAC. Downregulation of PIN1 expression inhibits cell growth, causes mitochondrial dysfunction by obviously increasing intracellular ROS levels and induces apoptosis in PDAC cells. Inactivation of PIN1 can lead to the presence of aberrant dot-shaped mitochondria, a more interconnected mitochondrial network, and decreased perinuclear clustering, as indicated by MitoTracker Green staining.20 This is consistent with our results. In addition, PIN1 maintains redox balance through synergistic activation of c-Myc and NRF2 to upregulate the expression of antioxidant element-driven genes in PDAC cells. Silencing of PIN1 expression decreases the GSH/GSSG ratio and significantly downregulates the expression of antioxidant genes.20 In our present study, downregulation of PIN1 expression via arsenic decreased GSH content and reduced the activity of SOD and CAT in the mouse liver and caused lipid peroxidation. However, overexpression of PIN1 rescued the breakdown of the antioxidant defense system. Therefore, we further confirmed the importance of PIN1 in regulating arsenic-induced oxidative stress. In contrast, gene silencing of PIN1 suppresses p66Shc-dependent ROS production in human aortic endothelial cells,17 suggesting that PIN1 may have a dual role in the response to oxidative stress.
According to the above findings, there is an association between PIN1 and ROS. However, it is unknown whether PIN1 is also associated with NAC. (−)-Epigallocatechin-3-gallate (EGCG) induces differentiation by decreasing PIN1 expression and increasing ROS levels. Furthermore, treatment with the antioxidant NAC, together with EGCG, inhibits ROS production,65 but the relationship between PIN1 and NAC has not been elucidated. In our study, NAC rescued PIN1 protein expression in arsenic treatment in vitro and in vivo. Knockdown of PIN1 abolished the protective effects of NAC, relieving arsenic-induced cytotoxicity, ROS production, mitochondrial dysfunction and apoptosis, further confirming the important role of PIN1 in the protective effects of NAC. In addition, PIN1 plays key roles in aging-associated diseases. PIN1 protein expression is decreased in the sera of patients with age-related hearing loss (ARHL), in senescent HEI-OC1 cells, and in the cochlea of aged mice, and ROS levels are concomitantly increased. However, when cells are pretreated with NAC, the expression level of PIN1 is increased, suggesting that there may be a correlation between NAC and PIN1 levels.31 In early studies, PIN1 was shown to regulate oxidative stress through distinct pathways and substrate proteins, such as the PI3K/Akt/mTOR pathway,31 p53 protein,21 p66Shc protein19 and p38 MAPK pathway.66 Our study is the first to uncover the pivotal roles of PIN1 in arsenic toxicity and the pharmacological action of NAC. However, the exact molecular mechanism by which PIN1 mediates arsenic toxicity and regulates the effects of NAC needs to be further studied.
5. Conclusion
In conclusion, our study explicitly revealed that arsenic caused hepatotoxicity by increasing ROS production, disrupting mitochondrial function, eliciting oxidative stress, and inducing apoptosis in vitro and in vivo. Arsenic markedly suppressed PIN1 expression at the mRNA and protein levels. NAC pretreatment effectively antagonized arsenic toxicity and elevated PIN1 expression. Overexpression of PIN1 relieved arsenic-induced hepatotoxicity by inhibiting ROS production, restoring mitochondrial function, maintaining redox homeostasis, and decreasing apoptosis in vitro and in vivo. Conversely, silencing of PIN1 aggravated arsenic toxicity and abolished the protective effect of NAC. The major results of our study are summarized in Supplementary Fig. S4. Our study highlights PIN1 as a novel molecular target for antagonizing arsenic toxicity and deciphers the pharmacological mechanism by which NAC, as an effective antioxidant, can protect against arsenic toxicity.
Supplementary Material
Acknowledgments
Thanks to the technical supports from Department of Occupational Health of Third Military Medical University.
Contributor Information
Huijie Zhang, Medical College, Guangxi University, 100 University East Road, Xixiangtang District, Nanning, Guangxi, 530004, P. R. China.
Zhixin He, Department of Occupational Health, Third Military Medical University, 30 Gaotanyan Zhengjie, Shapingba District, Chongqing, 400038, P. R. China.
Ping Deng, Department of Occupational Health, Third Military Medical University, 30 Gaotanyan Zhengjie, Shapingba District, Chongqing, 400038, P. R. China.
Muxue Lu, Medical College, Guangxi University, 100 University East Road, Xixiangtang District, Nanning, Guangxi, 530004, P. R. China.
Chao Zhou, Department of Occupational Health, Third Military Medical University, 30 Gaotanyan Zhengjie, Shapingba District, Chongqing, 400038, P. R. China.
Lingling Yang, Department of Occupational Health, Third Military Medical University, 30 Gaotanyan Zhengjie, Shapingba District, Chongqing, 400038, P. R. China.
Zhengping Yu, Medical College, Guangxi University, 100 University East Road, Xixiangtang District, Nanning, Guangxi, 530004, P. R. China.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81872596) and Open Grants from Key Laboratory of Electromagnetic Radiation Protection, Ministry of Education, China (No. 2017DCKF005).
Conflict of interest statement: There are no conflicts of interest to declare.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Zhengping Yu designed the project. All experiments were performed by Huijie Zhang, Zhixin He, Muxue Lu, Chao Zhou, Lingling Yang and Ping Deng. Huijie Zhang and Muxue Lu contributed to the analysis and interpretation of the data. Huijie Zhang and Zhixin He completed the figures and wrote the manuscript. Zhengping Yu conducted critical revision of the manuscript and approval of the article. All authors read and approved the final manuscript.
Reference
- 1. Oberoi S, Barchowsky A, Wu F. The global burden of disease for skin, lung, and bladder cancer caused by arsenic in food. Cancer Epidemiology, Biomarkers & Prevention: A Publication Of The American Association for Cancer Research, Cosponsored by the American Society of Preventive Oncology. 2014:23(7):1187–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Singh AP, Goel RK, Kaur T. Mechanisms pertaining to arsenic toxicity. Toxicol Int. 2011:18(2):87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee MY, Bae ON, Chung SM, Kang KT, Lee JY, Chung JH. Enhancement of platelet aggregation and thrombus formation by arsenic in drinking water: a contributing factor to cardiovascular disease. Toxicol Appl Pharmacol. 2002:179(2):83–88. [DOI] [PubMed] [Google Scholar]
- 4. Tsuda T, Babazono A, Yamamoto E, Kurumatani N, Mino Y, Ogawa T, et al. . Ingested arsenic and internal cancer: a historical cohort study followed for 33 years. Am J Epidemiol. 1995:141(3):198–209. [DOI] [PubMed] [Google Scholar]
- 5. Brown KG, Chen CJ. Significance of exposure assessment to analysis of cancer risk from inorganic arsenic in drinking water in Taiwan. Risk Analysis: an Official Publication of the Society for Risk Analysis. 1995:15(4):475–484. [DOI] [PubMed] [Google Scholar]
- 6. Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, et al. . The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect. 2013:121(3):295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carlin DJ, Naujokas MF, Bradham KD, Cowden J, Heacock M, Henry HF, et al. . Arsenic and environmental health: state of the science and future research opportunities. Environ Health Perspect. 2016:124(7):890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu M, Xue J, Li Y, Zhang W, Ma D, Liu L, et al. . Resveratrol protects against arsenic trioxide-induced nephrotoxicity by facilitating arsenic metabolism and decreasing oxidative stress. Arch Toxicol. 2013:87(6):1025–1035. [DOI] [PubMed] [Google Scholar]
- 9. Yu S, Liao WT, Lee CH, Chai CY, Yu CL, Yu HS. Immunological dysfunction in chronic arsenic exposure: from subclinical condition to skin cancer. J Dermatol. 2018:45(11):1271–1277. [DOI] [PubMed] [Google Scholar]
- 10. Kuo CC, Moon KA, Wang SL, Silbergeld E, Navas-Acien A. The Association of Arsenic Metabolism with cancer, cardiovascular disease, and diabetes: a systematic review of the epidemiological evidence. Environ Health Perspect. 2017:125(8):087001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santra A, Das Gupta J, De BK, Roy B, Guha Mazumder DN. Hepatic manifestations in chronic arsenic toxicity. Indian Journal of Gastroenterology: official Journal of the Indian Society of Gastroenterology. 1999:18(4):152–155. [PubMed] [Google Scholar]
- 12. Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicological Sciences: An Official Journal of the Society of Toxicology. 2008:105(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu M, Niu Q, Hu Y, Feng G, Wang H, Li S. Proanthocyanidins antagonize arsenic-induced oxidative damage and promote arsenic methylation through activation of the Nrf2 Signaling pathway. Oxidative Med Cell Longev. 2019:2019:8549035. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14. Flora SJ. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011:51(2):257–281. [DOI] [PubMed] [Google Scholar]
- 15. Das S, Joardar S, Manna P, Dua TK, Bhattacharjee N, Khanra R, et al. . Carnosic acid, a natural diterpene, attenuates arsenic-induced hepatotoxicity via reducing oxidative stress, MAPK activation, and apoptotic cell death pathway. Oxidative Med Cell Longev. 2018:2018:1421438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007:8(11):904–916. [DOI] [PubMed] [Google Scholar]
- 17. Francesco P, Sarah C, Lorenzo C, Rodolfo B, Giuliana C, Sergio C, et al. . Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur Heart J. 2015:13:817–828. [DOI] [PubMed] [Google Scholar]
- 18. Lee TH, Pastorino L, Lu KP. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med. 2011:13:e21. [DOI] [PubMed] [Google Scholar]
- 19. Feng D, Yao J, Wang G, Li Z, Zu G, Li Y, et al. . Inhibition of p66Shc-mediated mitochondrial apoptosis via targeting prolyl-isomerase Pin1 attenuates intestinal ischemia/reperfusion injury in rats. Clinical Science (London, England: 1979). 2017:131(8):759–773. [DOI] [PubMed] [Google Scholar]
- 20. Liang C, Shi S, Liu M, Qin Y, Meng Q, Hua J, et al. . PIN1 maintains redox balance via the c-Myc/NRF2 Axis to counteract Kras-induced mitochondrial respiratory injury in pancreatic cancer cells. Cancer Res. 2019:79(1):133–145. [DOI] [PubMed] [Google Scholar]
- 21. Li L, Su Z, Zou Z, Tan H, Cai D, Su L, et al. . Ser46 phosphorylation of p53 is an essential event in prolyl-isomerase Pin1-mediated p53-independent apoptosis in response to heat stress. Cell Death Dis. 2019:10(2):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou XZ, Lu KP. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat Rev Cancer. 2016:16(7):463–478. [DOI] [PubMed] [Google Scholar]
- 23. Lanni C, Masi M, Racchi M, Govoni S. Cancer and Alzheimer's disease inverse relationship: an age-associated diverging derailment of shared pathways. Mol Psychiatry. 2020:26(1):280–295. [DOI] [PubMed] [Google Scholar]
- 24. Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014:141(2):150–159. [DOI] [PubMed] [Google Scholar]
- 25. Ezeriņa D, Takano Y, Hanaoka K, Urano Y, Dick TP. N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H(2)S and sulfane sulfur production. Cell Chem Biol. 2018:25(4):447–459.e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Raghu G, Berk M, Campochiaro PA, Jaeschke H, Marenzi G, Richeldi L, et al. . The multifaceted therapeutic role of N-acetylcysteine (NAC) in disorders characterized by oxidative stress. Curr Neuropharmacol. 2021:19(8):1202–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang C, Ning Z, Wan F, Huang R, Chao L, Kang Z, et al. . Characterization of the cellular effects and mechanism of arsenic trioxide-induced hepatotoxicity in broiler chickens. Toxicology In Vitro: An International Journal Published in Association with BIBRA. 2019:61:104629. [DOI] [PubMed] [Google Scholar]
- 28. He Z, Zhang Y, Zhang H, Zhou C, Ma Q, Deng P, et al. . NAC antagonizes arsenic-induced neurotoxicity through TMEM179 by inhibiting oxidative stress in Oli-neu cells. Ecotoxicol Environ Saf. 2021:223:112554. [DOI] [PubMed] [Google Scholar]
- 29. Jeong CH, Seok JS, Petriello MC, Han SG. Arsenic downregulates tight junction claudin proteins through p38 and NF-κB in intestinal epithelial cell line, HT-29. Toxicology. 2017:379:31–39. [DOI] [PubMed] [Google Scholar]
- 30. Zhang C, Liu C, Li D, Yao N, Yuan X, Yu A, et al. . Intracellular redox imbalance and extracellular amino acid metabolic abnormality contribute to arsenic-induced developmental retardation in mouse preimplantation embryos. J Cell Physiol. 2010:222(2):444–455. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Lv Z, Liu Y, Cao H, Yang J, Wang B. PIN1 protects hair cells and auditory HEI-OC1 cells against senescence by inhibiting the PI3K/Akt/mTOR pathway. Oxidative Med Cell Longev. 2021:2021:9980444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang Q, Aluise CD, Joshi G, Sultana R, St Clair DK, Markesbery WR, et al. . Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010:88(12):2618–2629. [DOI] [PubMed] [Google Scholar]
- 33. Oparka M, Walczak J, Malinska D, Oppen L, Szczepanowska J, Koopman WJH, et al. . Quantifying ROS levels using CM-HDCFDA and HyPer. Methods (San Diego, Calif). 2016:109:3–11. [DOI] [PubMed] [Google Scholar]
- 34. Hu J, Xu X, Zuo Y, Gao X, Wang Y, Xiong C, et al. . NPY impairs cell viability and mitochondrial membrane potential through Ca2+ and p38 Signaling pathways in neonatal rat cardiomyocytes. J Cardiovasc Pharmacol. 2017:70(1):52–59. [DOI] [PubMed] [Google Scholar]
- 35. Qian JY, Deng P, Liang YD, Pang L, Wu LC, Yang LL, et al. . 8-Formylophiopogonanone B antagonizes Paraquat-induced hepatotoxicity by suppressing oxidative stress. Front Pharmacol. 2019:10:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pi H, Xu S, Zhang L, Guo P, Li Y, Xie J, et al. . Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy. 2013:9(11):1780–1800. [DOI] [PubMed] [Google Scholar]
- 37. Liu L, Liu Y, Cui J, Liu H, Liu YB, Qiao WL, et al. . Oxidative stress induces gastric submucosal arteriolar dysfunction in the elderly. World J Gastroenterol. 2013:19(48):9439–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen Z, Xiao J, Liu H, Yao K, Hou X, Cao Y, et al. . Astaxanthin attenuates oxidative stress and immune impairment in D-galactose-induced aging in rats by activating the Nrf2/Keap1 pathway and suppressing the NF-κB pathway. Food Funct. 2020:11(9):8099–8111. [DOI] [PubMed] [Google Scholar]
- 39. Cao Z, Fang Y, Lu Y, Tan D, Du C, Li Y, et al. . Melatonin alleviates cadmium-induced liver injury by inhibiting the TXNIP-NLRP3 inflammasome. J Pineal Res. 2017:62(3):e12389. [DOI] [PubMed] [Google Scholar]
- 40. Li Y, Sun L, Cai T, Zhang Y, Lv S, Wang Y, et al. . Alpha-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res Bull. 2010:81(4–5):428–433. [DOI] [PubMed] [Google Scholar]
- 41. Liu M, Pi H, Xi Y, Wang L, Tian L, Chen M, et al. . KIF5A-dependent axonal transport deficiency disrupts autophagic flux in trimethyltin chloride-induced neurotoxicity. Autophagy. 2021:17(4):903–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turkmen R, Akosman MS, Demirel HH. Protective effect of N-acetylcysteine on MK-801-induced testicular oxidative stress in mice. Biomed Pharmacother. 2019:109:1988–1993. [DOI] [PubMed] [Google Scholar]
- 43. Akanda MR, Tae HJ, Kim IS, Ahn D, Tian W, Islam A, et al. . Hepatoprotective role of hydrangea macrophylla against sodium arsenite-induced mitochondrial-dependent oxidative stress via the inhibition of MAPK/Caspase-3 pathways. Int J Mol Sci. 2017:18(7):1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kiourtis C, Wilczynska A, Nixon C, Clark W, May S, Bird TG. Specificity and off-target effects of AAV8-TBG viral vectors for the manipulation of hepatocellular gene expression in mice. Biology Open. 2021:10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Modi M, Kaul RK, Kannan GM, Flora SJ. Co-administration of zinc and n-acetylcysteine prevents arsenic-induced tissue oxidative stress in male rats. Journal of Trace Elements in Medicine And Biology: Organ of the Society for Minerals and Trace Elements (GMS). 2006:20(3):197–204. [DOI] [PubMed] [Google Scholar]
- 46. Ge M, Yao W, Yuan D, Zhou S, Chen X, Zhang Y, et al. . Brg1-mediated Nrf2/HO-1 pathway activation alleviates hepatic ischemia-reperfusion injury. Cell Death Dis. 2017:8(6):e2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. . Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021:11(4):1845–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol. 2012:86(6):923–933. [DOI] [PubMed] [Google Scholar]
- 49. Garbinski LD, Rosen BP, Chen J. Pathways of arsenic uptake and efflux. Environ Int. 2019:126:585–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jomova K, Jenisova Z, Feszterova M, Baros S, Liska J, Hudecova D, et al. . Arsenic: toxicity, oxidative stress and human disease. JAT. 2011:31(2):95–107. [DOI] [PubMed] [Google Scholar]
- 51. Pace C, Dagda R, Angermann J. Antioxidants protect against arsenic induced mitochondrial cardio-toxicity. Toxics. 2017:5(4):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanders AP, Messier KP, Shehee M, Rudo K, Serre ML, Fry RC. Arsenic in North Carolina: public health implications. Environ Int. 2012:38(1):10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen QY, Costa M. Arsenic: a global environmental challenge. Annu Rev Pharmacol Toxicol. 2021:61:47–63. [DOI] [PubMed] [Google Scholar]
- 54. Tao Y, Qiu T, Yao X, Jiang L, Wang N, Jia X, et al. . Autophagic-CTSB-inflammasome axis modulates hepatic stellate cells activation in arsenic-induced liver fibrosis. Chemosphere. 2020:242:124959. [DOI] [PubMed] [Google Scholar]
- 55. Wang W, Cheng S, Zhang D. Association of inorganic arsenic exposure with liver cancer mortality: a meta-analysis. Environ Res. 2014:135:120–125. [DOI] [PubMed] [Google Scholar]
- 56. Dua TK, Dewanjee S, Khanra R. Prophylactic role of Enhydra fluctuans against arsenic-induced hepatotoxicity via anti-apoptotic and antioxidant mechanisms. Redox Report: Communications In Free Radical Research. 2016:21(4):147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Z, Pratheeshkumar P, Budhraja A, Son YO, Kim D, Shi X. Role of reactive oxygen species in arsenic-induced transformation of human lung bronchial epithelial (BEAS-2B) cells. Biochem Biophys Res Commun. 2015:456(2):643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Susan A, Rajendran K, Sathyasivam K, Krishnan UM. An overview of plant-based interventions to ameliorate arsenic toxicity. Biomed Pharmacother. 2019:109:838–852. [DOI] [PubMed] [Google Scholar]
- 59. Nissar AU, Farrukh MR, Kaiser PJ, Rafiq RA, Afnan Q, Bhushan S, et al. . Effect of N-acetyl cysteine (NAC), an organosulfur compound from allium plants, on experimentally induced hepatic prefibrogenic events in Wistar rat. Phytomedicine: International Journal of Phytotherapy and Phytopharmacology. 2013:20(10):828–833. [DOI] [PubMed] [Google Scholar]
- 60. Grisanti S, Cosentini D, Tovazzi V, Bianchi S, Lazzari B, Consoli F, et al. . Hepatoprotective effect of N-acetylcysteine in trabectedin-induced liver toxicity in patients with advanced soft tissue sarcoma. Supportive Care in Cancer: Official Journal of the Multinational Association of Supportive Care in Cancer. 2018:26(8):2929–2935. [DOI] [PubMed] [Google Scholar]
- 61. Pei Y, Liu H, Yang Y, Yang Y, Jiao Y, Tay FR, et al. . Biological activities and potential oral applications of N-acetylcysteine: progress and prospects. Oxidative Med Cell Longev. 2018:2018:2835787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wei S, Qiu T, Yao X, Wang N, Jiang L, Jia X, et al. . Arsenic induces pancreatic dysfunction and ferroptosis via mitochondrial ROS-autophagy-lysosomal pathway. J Hazard Mater. 2020:384:121390. [DOI] [PubMed] [Google Scholar]
- 63. Kozono S, Lin YM, Seo HS, Pinch B, Lian X, Qiu C, et al. . Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat Commun. 2018:9(1):3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zheng F, Li Y, Zhang F, Sun Y, Zheng C, Luo Z, et al. . Cobalt induces neurodegenerative damages through Pin1 inactivation in mice and human neuroglioma cells. J Hazard Mater. 2021:419:126378. [DOI] [PubMed] [Google Scholar]
- 65. Della Via FI, Shiraishi RN, Santos I, Ferro KP, Salazar-Terreros MJ, Franchi Junior GC, et al. . (−)-Epigallocatechin-3-gallate induces apoptosis and differentiation in leukaemia by targeting reactive oxygen species and PIN1. Sci Rep. 2021:11(1):9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao X, Wang D, Wan S, Liu X, Wang W, Wang L. The suppression of Pin1-alleviated oxidative stress through the p38 MAPK pathway in ischemia- and reperfusion-induced acute kidney injury. Oxidative Med Cell Longev. 2021:2021:1313847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.