

Abstract
Ermin is an actin‐binding protein found almost exclusively in the central nervous system (CNS) as a component of myelin sheaths. Although Ermin has been predicted to play a role in the formation and stability of myelin sheaths, this has not been directly examined in vivo. Here, we show that Ermin is essential for myelin sheath integrity and normal saltatory conduction. Loss of Ermin in mice caused de‐compacted and fragmented myelin sheaths and led to slower conduction along with progressive neurological deficits. RNA sequencing of the corpus callosum, the largest white matter structure in the CNS, pointed to inflammatory activation in aged Ermin‐deficient mice, which was corroborated by increased levels of microgliosis and astrogliosis. The inflammatory milieu and myelin abnormalities were further associated with increased susceptibility to immune‐mediated demyelination insult in Ermin knockout mice. Supporting a possible role of Ermin deficiency in inflammatory white matter disorders, a rare inactivating mutation in the ERMN gene was identified in multiple sclerosis patients. Our findings demonstrate a critical role for Ermin in maintaining myelin integrity. Given its near‐exclusive expression in myelinating oligodendrocytes, Ermin deficiency represents a compelling “inside‐out” model of inflammatory dysmyelination and may offer a new paradigm for the development of myelin stability‐targeted therapies.
Keywords: Ermin, inflammatory demyelination, myelin sheath, white matter pathology
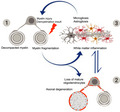
A working model for the sequence of pathological events in Ermin‐deficient mice (1) Loss of Ermin leads to compromised myelin that is de‐compacted, out folded, and fragmented; (2) As Ermin‐deficient mice age, augmentation of myelin fragmentation and myelin sheath breakdown lead to loss of myelinating oligodendrocytes. Due to compromised myelin sheaths and oligodendrocyte function, loss of trophic support to axons also leads to axonal damage and degeneration; (3) Excessive myelin debris along with oligodendrocyte loss and axonal damage/degeneration triggers white matter inflammation. Activated microglia and astrocytes further exacerbate cell loss. This ensuing inflammation may act as a feed‐forward demyelinating insult causing further damage to unhealthy myelin sheaths in Ermin deficient mice.
1. INTRODUCTION
Myelin is a multilayered, compact membrane structure that insulates and supports axons in the central (CNS) and peripheral (PNS) nervous systems [1]. The myelin sheath is exceptionally stable and is one of the most complex biological membranes, with unique composition and architecture. Despite its importance, much of the molecular and cellular choreography underlying the generation, assembly, maintenance, and remodeling of myelin sheaths and how it differs between the CNS and PNS remains poorly understood.
Although CNS and PNS myelin have similar functions, each is unique in several respects, including their molecular composition [1]. For example, whereas proteolipid protein (PLP) is the most abundant protein constituent of myelin in the mammalian CNS, it represents less than 1% of PNS myelin. Conversely, the single membrane P0 glycoprotein, the most abundant protein in PNS myelin, is nearly absent from mammalian CNS myelin. The shift from P0 to PLP as a major constituent of CNS myelin is a key evolutionary event in the transition from fish to higher vertebrates [2] and is paralleled by the emergence of a protein named Ermin expressed exclusively in the CNS and which localizes to myelin sheaths of mature, myelinating oligodendrocytes [3, 4, 5].
Unlike classical myelin proteins such as PLP, MBP, CNP, MOG, and MAG, Ermin represents less than 1% of the myelin protein, making it a quantitatively minor myelin protein [6]. Ermin was first described as an oligodendrocyte‐specific gene and a new member of the ERM (Ezrin, Radixin, and Moesin) family of proteins [4, 5]. ERM proteins cross‐link actin filaments with plasma membranes in diverse cell types, including Schwann cells [7]. Structurally, Ermin exhibits key similarities and differences with other ERM proteins: it shares a highly conserved C‐terminus actin‐binding domain but lacks an N‐terminus FERM regulatory sequence. In oligodendrocytes, Ermin was shown to promote 2′,3′‐cyclic nucleotide 3′‐phospho‐diesterase (CNPase) trafficking [4]. Because of the putative interaction of CNPase with microtubules and the role of actin rearrangement in myelinogenesis [8], Ermin was suggested to (1) provide links between the actin‐based microfilaments and tubulin‐based microtubules and (2) play a role as an interaction hub in the rearrangement of cytoskeletal proteins during myelin wrapping and compaction. Furthermore, in vitro crystallography studies suggest that Ermin has a disordered structure, a feature it shares with other cytoskeletal and myelin‐specific proteins such as myelin basic protein (MBP) [9]. Notably, myelin compaction processes, including MBP polymerization, are thought to begin at the outer layer of myelin sheaths where Ermin is localized [4, 10], supporting a possible role for Ermin in myelin compaction.
Ermin's expression follows the progression of myelination, appearing in a caudal‐to‐rostral and ventral‐to‐dorsal manner, and is found to lag behind MBP by 2–3 days in the postnatal period [4]. Based on its spatial and temporal expression patterns as well as its role in actin‐binding and remodeling, Ermin has been suggested to play a role in the maintenance of the myelin sheath [5], although this has not been directly examined in vivo. Furthermore, changes in Ermin expression have been linked to several disorders such as epilepsy [11], schizophrenia [12], and autism [13], although no disease‐causing mutations in ERMN nor a pathological role for it have been reported to date.
Here, we wondered whether the loss of Ermin compromised the structural and functional integrity of myelin sheaths. We found that Ermin deficiency led to structural myelin abnormalities and impaired nerve conduction. These deficits were coupled with progressive neurological phenotypes, white matter inflammation, and increased susceptibility to an immune‐mediated demyelinating insult. Our findings demonstrate an essential role for Ermin in maintaining the integrity of CNS myelin and the pathological consequences of its loss. Furthermore, we have identified a germline inactivating mutation in ERMN in a multi‐incidence multiple sclerosis family. The etiology of inflammatory white matter disorders such as multiple sclerosis is multi‐factorial and associated with significant clinical and pathological heterogeneity [14, 15]. While ample evidence supports “outside‐in” etiologies in which myelin sheath pathology and degeneration are caused by processes that originate outside oligodendrocytes, an “inside‐out” pathogenic process has been more challenging to prove. Given its near‐exclusive expression in myelinating oligodendrocytes, Ermin deficiency represents a compelling model of “inside‐out” inflammatory dysmyelination.
2. MATERIALS AND METHODS
2.1. Generation of Ermin knockout mice using CRISPR/Cas9
Ermin knockout (KO) mice were generated on a C57BL/6 background using CRISPR/Cas9. The following guide RNAs that target exon 1 of Ermn were used: gRNA_1, “GACGAGTTGGTTTCGAACTGT” and gRNA_2, “GCATACTACAAGGTTGAAC”. Ermin heterozygous (HTZ) mice were bred together to generate Ermin KO mice. Mice were housed with littermates of mixed genotype (2–5 per cage) in individually ventilated cages and kept in a room with a 12‐h light/dark cycle (lights on at 09:00). Water and food (Altromin 1324 irradiated modified 18% Protein and 6% Fat) were available in cages throughout the study. Mice were maintained under standard conditions, and all animal procedures were performed with the approval of the Institutional Animal Care and Use Committee (IACUC # 161186) at Biological Resource Centre (BRC), A*STAR, and by their approved guidelines.
2.2. Genotyping
Genomic DNA was extracted from the tail tissue of mice using the DNeasy Tissue kit (Qiagen). To visualize the successful deletion of 201 nucleotides of Ermn gene in KO mice, the PCR products were run on a 3% agarose gel with SYBR Safe DNA gel stain (Invitrogen). The genotyping primers are: 5′ CCGGGCTGGTTACCAAACT 3′ (forward) and 5′ GATCTTCTCATCTTCAGGCCCTT 3′ (reverse).
2.3. Transmission electron microscopy (TEM)
Mice (n = 3 per group) were transcardially perfused with freshly prepared 2.5% glutaraldehyde, and 2.5% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS). Whole brains were post‐fixed overnight at 4°C in the same fixative solution, washed in PBS, and stored in 5% sucrose and 0.1% sodium azide at 4°C. Coronal slices at the level of Bregma −1 mm were obtained consistently from one hemisphere using a stainless‐steel mouse brain slicer matrix (Agnthos), followed by micro‐dissection of the anterior region of the corpus callosum (~Bregma 1.10–0.50 mm, according to the Mouse Brain Atlas [16]) and washed in PBS. Tissues were then post‐fixed in 1% Osmium tetroxide for 1 h at room temperature (RT), dehydrated through an ascending ethanol series (25%–100%) and 100% acetone wash, and infiltrated with 1:1 acetone: resin for 30 min at RT and 1:6 acetone: resin overnight at RT. Sections were then transferred to 100% resin for embedding and polymerization at (30 min at 40°C, 1 h at 45 and 50°C, respectively, and 60°C for 24 h). Ultrathin sections (90 nm) were obtained using Ultracut E with a diamond knife (Diatome, Ultra45, 3mm length) attachment and embedded on a copper grid. TEM imaging was performed on an FEI TECNAI Spirit G2 for g‐ratios and a JEM1010 Microscope equipped with a bottom‐mount SIA model 12C high‐resolution full‐frame CCD camera (16bit, 4K) for periodicity. Over 300 axons and myelin fiber diameters per animal were measured using ImageJ. G‐ratios were obtained by calculating the ratio between axon diameter and axon diameter plus the outermost layer of surrounding myelin. G‐ratios were calculated within equal axonal distributions. Periodicity was calculated by dividing myelin thickness by the number of myelin layers.
2.4. Protein analysis
RIPA buffer (Sigma‐Aldrich) with 1mM PMSF (Sigma‐Aldrich), 5mM Z‐VAD (Promega), 1mM NaVan (Sigma‐Aldrich), and 1x Complete Protease Inhibitor Cocktail tablets (Roche) was used to prepare Protein lysate of half brain and CD11b+ cells lysis from mice. For western blotting, 30 µg of protein lysate was separated on 12% Bis‐Tris protein gel (Novex) with 20X MES running buffer (Novex) and transferred on nitrocellulose membranes. Primary antibodies (listed in Table S5) were incubated at 4°C overnight. Secondary antibodies (1:10,000) were used: Alexa‐Fluor goat anti‐rabbit 800, Alexa‐Fluor goat anti‐mouse 680, Alexa‐Fluor goat anti‐rabbit 680, and Alexa‐Fluor goat anti‐rat 800 (all from Life Technologies). The membrane was imaged using the LiCor Imaging System and Odyssey V3.0 software (LiCor), followed by intensity analysis with ImageJ.
2.5. Corpora callosa slice preparation and electrophysiology
Three months old male mice were used to measure compound action potentials (CAPs) in CC. After cervical dislocation, brains were dissected and placed in oxygenated (95% O2 + 5% CO2) ice‐cold sucrose based artificial cerebrospinal fluid (aCSF) solution (206 mM Sucrose, 2 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM Glucose, 2 mM MgCl2, 2 mM MgSO4, 1 mM CaCl2; 305–315 mOsm, pH 7.3–7.4). Brains were sliced coronally in sucrose‐based ACSF using a vibratome (Leica VT1200s) with 450 µm thickness, and slices were collected approximately from Bregma 1.10 to −0.22 (3 slices per mouse) in Mouse Brain Atlas (Franklin and Paxinos, 3rd ed.) [16]. Slices were immediately transferred to a holding chamber filled with pre‐warmed (37°C) oxygenated (95% O2 + 5% CO2) standard aCSF solution (124 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 24 mM NaHCO3, 5 mM HEPES, 12.5 mM Glucose, 2 mM MgSO4, 2 mM CaCl2), and the slices were recovered for 30 min at 37°C, afterward kept at room temperature till recording.
Compound action potentials (CAPs) evoked by electrical stimulation were recorded in the corpora callosa at three different distances between stimulation and recording electrodes: 1.5 mm, 1.0 mm, and 0.5 mm distance. The recording electrode assembled glass pipette (1–3 M) filled with 3 M NaCl was placed 1 mm away from the midline of the brain slice, and the stimulation electrode was placed 0.5 mm away from the middle line on the opposite side of the recording pipette. The recording was conducted with an order from 1.5 mm to 0.5 mm by moving in the recording electrode. The intensity of the stimulus was increased from 0.5 mA to 5 mA using an external stimulator (model 2100; A‐M System), and responses were recorded at each step‐in triplicate. The Multiclamp 700B, Digidata 1550B and pClamp10 software (Molecular Devices) were used for recordings. From acquired data, amplitudes, area of N1 (myelinated) and N2 (non‐myelinated) were calculated using the Population spike analysis tool in AxoGraph X (AxoGraph Scientific). The area and duration of peaks on 5 mA stimulation at 1 mm distance were used to compare compared between groups.
2.6. Study design of behavioral analysis
All behavioral tests were performed during the dark phase of the reversed light/dark‐ cycle. Two cohorts of mice were behaviorally tested at 3, 5, and 7 months of age. Mice were evaluated for motor and psychiatric‐like behavior. For all behavioral tests, mice were allowed to acclimatize to the testing room for at least 30 min prior to the commencement of the behavioral test. Investigator was blinded for all test sessions.
2.7. Spontaneous locomotor activity test
The spontaneous locomotor activity test is used to measure gross and fine motor movements in mice [17]. The aspects of activity measured are horizontal activity such as distance traveled and vertical activity such as vertical and jump counts. In order to assess motor function, mice were monitored for 30 min using the Med Associates spontaneous activity chambers (27.3 [L] × 27.3 [W] × 20.3 [H] cm) with 16 beams (Med Associate Inc).
2.8. Rotarod test of motor coordination
Motor coordination and balance were evaluated using the accelerating rotarod task performed on a UGO Basile 47600 Rotarod with a rotating rod diameter of 3 cm [18]. The training was performed at three months of age and consisted of three trials (120 s) per day at a fixed speed of 18 rpm for three consecutive days. Testing was performed at 3, 5, and 7 months of age and consisted of three trials, spaced 2 h apart, with a rod acceleration from 5 to 40 rpm within 5 min. Rotarod scores are the average of three trials.
2.9. Climbing test
The climbing test is used to evaluate motor function in mice [19]. Testing was performed at 3, 5, and 7 months of age. Mice were placed at the bottom end of a closed‐top metal wire mesh cylinder (10.5 cm of diameter and 15.5 cm high) and were video recorded for 5 min. The number of the climbing event was counted manually and blinded. Climbing was determined when all four paws of the mouse were on the walls of the cylinder.
2.10. Elevated plus‐maze test of anxiety‐like behavior
The Elevated plus maze (EPM) test is classically used to assess anxiety‐like in rodents [20]. The EPM apparatus is a cross‐like shape with two open arms perpendicular to two closed arms of equal dimensions. The closed arms are surrounded by three 10‐cm high walls. Because mice have an innate fear of high open spaces, they prefer to spend less time in the open arms. Time spent in the open arms is taken as a measure of anxiety‐like behavior. Test sessions lasted 5 min and time spent in the open arms were recorded using an automated video‐based tracking system (Noldus EthoVision 9, Netherlands).
2.11. Porsolt forced swim test (FST) of depression‐like behavior
The Porsolt FST is used to evaluate depressive‐like behavior in mice. The test was performed at seven months of age as previously described [19]. Mice were put in individual cylinders (25 [H] × 19 [W] cm) filled with room temperature water (23–25°C) to a depth of 15 cm for a period of 6 min. The test sessions were recorded and examined blinded. The last 4 min of the test session was scored using a time‐sampling technique to rate the predominant behavior, including swimming and immobility, over 5‐s intervals. Time spent immobile is considered as a measure of depressive‐like behavior.
2.12. Bar test of catatonia‐like behavior
This test was performed as previously described [21, 22, 23]. Briefly, the mouse was carried by the tail to a horizontal bar made of stainless steel (12 [L] × 4–5 [H] cm, and 2.5 cm of diameter) and allowed to grasp the bar with both forepaws. Upon grasping the bar with both forepaws, the mouse was moved downwards so that its hind paws had contact with the floor before its tail was released. The time a mouse stood nonmoving with at least one forepaw on the bar, and both hind paws on the ground was measured. The test was performed three times for each mouse, and the catatonia‐like score for each mouse was the average of the three trials.
2.13. Brain tissue collection for molecular and sequencing analysis
Corpora callosa from 8‐month‐old animals were dissected, snap‐frozen in liquid nitrogen and stored at −80°C until further use.
2.14. RNA‐Seq analysis
RNA was extracted from mouse corpora callosa (CC) using Trizol (Life Technologies) and subsequently an RNeasy plus mini kit (Qiagen) according to the manufacturer's instructions. Subsequent library preparation and paired‐end 150 bp sequencing and 15 M reads/per sample using HiSeq were performed by Novogene (Hong Kong).
RNA‐Seq data analysis was done on the AIR platform (www.transcriptomics.cloud). The trimming is performed with BBduk to remove low‐quality bases (minimum quality 25, minimum length 35 bp). High‐quality reads were then aligned to the mm10 genome (Ensembl version 89) using STAR 020201 (https://github.com/alexdobin/STAR) and quantified using featureCounts (http://bioinf.wehi.edu.au/featureCounts/), genes expressed at a low level were removed with the package HTSFilter (http://www.bioconductor.org/packages/release/bioc/html/HTSFilter.html), Differential expression of the filtered genes was analyzed by the NOIseq method, pairwise. Genes were considered statistically differentially expressed if the corrected P‐value (false discovery rate, FDR) was <10%. Enrichment analysis was performed using Hypergeometric Optimization of Motif Enrichment (HOMER) [24] on the proportion of GO categories between the differentially expressed genes (DEGs) and the background of expressed genes; GO categories were considered enriched if the FDR of the test was <0.05. An expression heat map was constructed for a pre‐selection of genes of interest using the web‐based tool Heatmapper (https://software.broadinstitute.org/morpheus/), transforming transcripts per million (TPM) values using a Z‐score scaling.
2.15. Immunohistochemistry and stereological measurements
For immunohistochemistry, one mouse cohort was used with N = 6–8 per genotype. Half brains were post‐fixed overnight in 4% PFA and switched to 30% sucrose in 1X phosphate‐buffered saline (PBS). Half brains were cut via cryostat (Leica CM3050S) into a series of 25μm sagittal sections free‐floating in 1X PBS with 0.01% sodium azide. For all stainings, sections were washed three times with 1X PBS and then blocked in 5% normal goat serum (NGS) in PBS containing 0.1% Triton X‐100 (TX) for 120 min at room temperature (RT). Sections were then incubated with primary antibodies in 1% NGS in PBS‐TX overnight at 4ºC. The following antibody concentrations were used: rabbit anti‐GST‐pi at 1:3000 (MBL, #311), rabbit anti‐Iba1 at 1:500 (Wako, #01919741) and rabbit anti‐GFAP at 1:1000 (Millipore, #OBT0030). Sections were washed three times with PBS and stained using a biotinylated anti‐rabbit secondary antibody (1:1000, Vector Laboratories) for 90 min at RT. Sections were then incubated using ABC Elite Kit (Vector Laboratories) to amplify the signal, and then in 3,3'‐diaminobenzidine (DAB, Thermo Scientific) for signal detection.
For stereological measurements, corpus callosum (CC), fimbria, hippocampus and caudate regions from one hemisphere were traced using the Stereo Investigator software (MBF Bioscience). Iba1 positive cells were counted in all regions, while GFAP and GST‐pi positive cells were counted in the CC using the Optical Director probe. The following parameters were used: Iba1 – 100 µm × 100 µm counting frame, 450 µm × 450 µm sampling grid; GFAP – 150 µm × 150 µm counting frame, 300 µm × 300 µm sampling grid; GST‐pi – 50 µm × 50 µm counting frame, 300 µm × 300 µm sampling grid. The volume of counted areas was determined using the Cavalieri Estimator probe. Activated microglia and astrocytes were visually identified by increased cell body size and more intense IBA1 or GFAP staining [25, 26]. Another significant morphology of activated microglia/astrocytes is contracted ramifications that appear thicker than in their resting state.
2.16. Experimental autoimmune encephalomyelitis (EAE) induction and evaluation
10 WT and 8 Ermin KO mice were immunized by Hooke kits (Hooke laboratories, EK‐0115, Lawrence, MA, USA) according to the manufacturer's instructions. Briefly, 0.1 ml MOG35‐55/CFA emulsion was injected subcutaneously into both flanks of each mouse (0.2 ml/animal) after anesthesia. Then, pertussis toxin (0.1 ml/animal/day, i.p.) was injected intraperitoneally (IP) on the same day and 24 h later.
Two researchers scored the clinical signs of mice daily from day 6 to day 25 post‐immunization using the scale 0 to 5 point EAE scoring system was based on Hooke lab protocol briefly as follows: 0, no clinical sign; 0.5, Tip of the tail is limp; 1.0, complete tail paralysis; 1.5, Limp tail and hind leg inhibition; 2.0, Limp tail and weakness of hind legs.; 2.5, unilateral hind limb paralysis; 3, complete hind limb paralysis; 3.5, hind limb paralysis and forelimb weakness; 4.0, complete paralysis (tetraplegia), and 5.0, moribund or dead. The successful induction of EAE in all WT and KO mice was confirmed by this scoring scale. These clinical scoring results were analyzed to compare the course of EAE between the two groups.
2.17. Microglia and macrophages enrichment protocol
Brain or spinal cord were harvested and put in 3 ml or 1 ml solution, respectively, comprised of RPMI, 10%FCS, Collagenase (0.2 mg/ml), and DNAse (30 µg/ml) using 12 wells plates. Cut in small pieces and incubated at 37°C for 30 min. Using 3 ml syringes with the blunt needle, the tissues were dissociated by aspiring and flushing 10 times. The cell suspension was filtered on a cell strainer 70 µM and falcon tube 50 ml. The cells were washed with 14 ml of FACS buffer (PBS +5% BSA +2 mM EDTA) and centrifuge 4 min at 1600 rpm 4°C. The supernatant was removed, and cells were resuspended in 1 ml FACS buffer +100 µl of microbeads anti‐mouse CD11b for the brain, and the spinal cord was resuspended in 200 µl + 20 µl of microbeads. Cells were incubated for 15 min at 4°C. 5 ml of FACS buffer was added after incubation, and cells were centrifuged at 1600 rpm 4 min 4°C. The supernatant was removed, and 2 ml of FACS buffer added. Using the automatics possel function, CD11b+ cells were retrieved. CD11b+ cells were centrifuged at 1600 rpm 4min 4°C.
2.18. Microglia and macrophage flow cytometry (FACS) staining
Antibodies were diluted in blocking buffer containing FACS buffer +1% rat serum +1% mouse serum. All antibodies were used at 1/200 dilution. 100 µl of staining mixture was used per brain, and 50uL was used per spinal cord. Cells were incubated for 15min with antibodies mixed at 4°C and then washed with 1 ml of FACS buffer and resuspended in 200 µl of PBS + DAPI (1 µg/ ml). The list of FACS antibodies used is listed in Table S6.
2.19. Discovery of novel mutations in ERMN in a multi‐incident MS family
Board‐certified clinical neurologists evaluated all patients clinically. The entire family was recruited under an Isfahan University of Medical Sciences ethics‐approved research protocol with informed consent.
We applied whole‐exome sequencing (WES) analysis to a multi‐incident MS family consisting of 4 individuals over two generations, two of whom were diagnosed with MS for WES, 1 µg of gDNA of healthy father, affected mother and affected daughter (1, 2 and 3) used for exome library preparation on an Ion OneTouch System. Amplified samples were sequenced on an Ion Proton instrument (Life Technologies, Carlsbad, CA, USA) using one ION PI chip. Sequence reads were aligned to the human GRCh37/hg19 assembly (UCSC Genome browser). The variants were filtered based on functional prediction scores, including SIFT, PolyPhen2, Grantham and M‐CAP (Mendelian Clinically Applicable Pathogenicity), as well as PhyloP conservation scores. Variants were further filtered for common single nucleotide polymorphisms (SNPs) using the “common and no known medical impacts” database (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/vcf_GRCh37/) and the Exome Aggregation Consortium (ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.2/). Variants were next compared to an in‐house (IMCB) database of 332 previously sequenced samples, and those that were present in more than 1% of the previously sequenced samples were removed. WES of family members 1,2 and 3 gDNA generated a total of 14.5 Gb with an average read length of 189 bp. Average coverage of 181.9X was achieved across the exome, with 96.89% of the targeted sequences covered at 20X. A total of 67,763 variants were identified across protein‐coding exons, untranslated region (UTRs), splice sites and flanking introns. Sanger sequencing was used to confirm that the identified mutation segregates in the family. The primers used for Sanger sequencing were: 5′‐ggacaattgtggctcctgtt‐3′ (forward) and 5′‐ggtgtccagctcatttcgat‐3′ (reverse).
2.20. Accession numbers
The RNA‐Seq data reported in this paper have been deposited into the Sequence Read Archive under accession number: PRJNA612161. [Reviewers’ link: https://dataview.ncbi.nlm.nih.gov/object/PRJNA612161?reviewer=9pp52uvff3e2uq8fm1v7vvpul0].
2.21. Statistical analysis
Prism 6 (GraphPad Software) was used for all statistical analyses. The normal distribution of data was checked by D'Agostino & Pearson normality test. If the data were normally distributed, Student's t‐test was used to calculate statistical significance when only two experimental groups were compared. For multiple comparisons, a one‐way or two‐way ANOVA was performed, followed by a post‐hoc Tukey's multiple comparisons test. If the data were not normally distributed, nonparametric tests such as the Kruskal‐Wallis test were used. Differences were considered statistically significant when p < 0.05.
3. RESULTS
3.1. Generation of Ermin knockout mice
To investigate the physiological functions of Ermin, we created an Ermin knockout (KO) mouse model using the CRISPR/Cas9 technology [27]. Briefly, Cas9 mRNA and two different guide RNAs (gRNAs) targeting the promoter and exon 1 region of Ermn were co‐injected into zygotes with a C57BL/6 background (Figure 1A). As a result, the promoter region, 5′ UTR and 122 nucleotides of exon 1 were deleted. The deletion was confirmed by genomic PCR (Figure 1B). Immunoblot analysis of brain lysates confirmed ~50% reduction in heterozygotes (HTZ) and complete loss of expression in Ermin knockout (KO) mice (Figure 1C,D). Knockout mice were born at Mendelian ratios and were viable. Immunoblotting the antibody that specifically binds to the C‐terminus of Ermin, confirmed lack of truncated protein (Figure S1).
FIGURE 1.
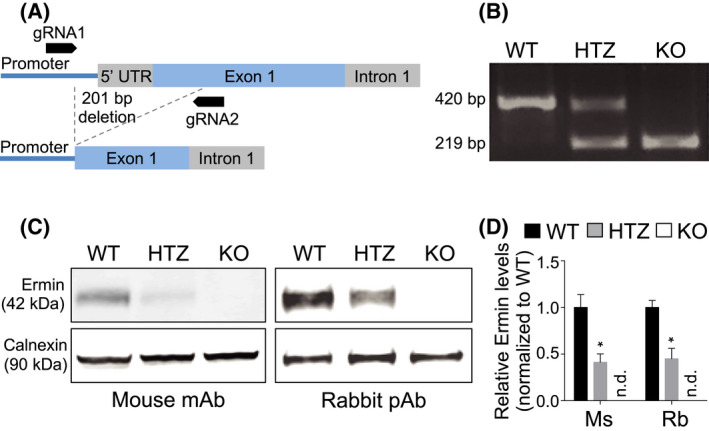
Generation of Ermin knockout mice. (A) Schematic depicting the location of CRISPR/Cas9 gRNA1 and gRNA2 used to generate a 201bp deletion in the Ermn gene. (B) Genomic PCR on wild‐type (WT), heterozygous (HTZ) and homozygous (KO) mice confirming germline transmission of a 201bp deletion in Ermn. (C) Immunoblotting using two different antibodies on whole brain lysates of 3 months old mice confirmed reduction of Ermin protein in HTZ mice and complete absence in KO mice. mAb, monoclonal antibody; pAb, polyclonal antibody. (D) Ermin protein levels are reduced by ~50% in HTZ and absent in KO mice. Ermin levels relative to respective Calnexin loading controls were normalized to WT mice. Values shown as mean ± SEM, n = 6 per genotype, *p < 0.05 compared to WT was determined by one‐way ANOVA with Tukey's post‐hoc test (adjusted p value). Ms, mouse antibody; n.d., not detected; Rb, rabbit antibody
3.2. Loss of Ermin leads to compromised myelin and axonal damage
To assess the impact of the loss of Ermin on myelin microstructures, we used transmission electron microscopy to visualize myelinated fibers in the corpus callosum (CC), the largest white matter structure in the brain, in 5 months old mice (Figure 2A). We evaluated the g‐ratio, calculated as the ratio of axon diameter (axon caliber) to myelinated fiber diameter, as a measure of myelin sheath thickness. Plotting g‐ratios against axonal diameters demonstrated that g‐ratios of larger caliber axons were lower in Ermin KO mice compared with WT littermates (Figure 2B). In line with these findings, the mean g‐ratio of myelinated axons was lower for Ermin KO mice compared to WT, and the cumulative frequency distributions of g‐ratios demonstrated a shift to the left in the population suggesting a thickening of myelin sheaths in Ermin KO mice (Figure 2B). Furthermore, periodicity, a measure of myelin compaction calculated as the mean distance between two major dense lines, was significantly increased in Ermin KO mice (Figure 2C,D), suggesting that loss of Ermin leads to de‐compacted myelin sheaths in the CC of Ermin KO mice compared with WT mice (Figure 2A–D). Thus, the increase in thickness of myelin sheaths in Ermin‐deficient mice is likely related to its de‐compaction in these mice.
FIGURE 2.
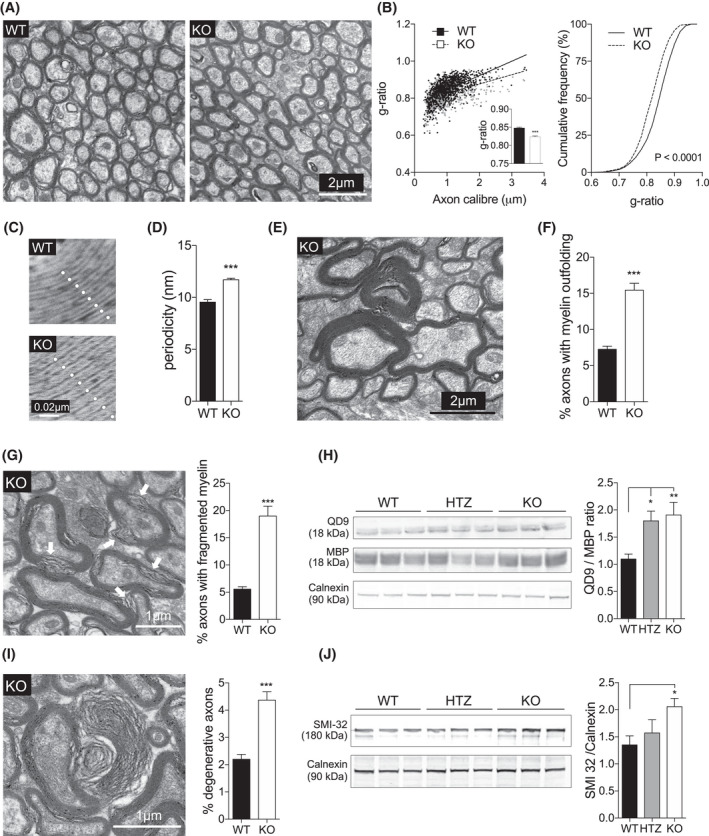
Loss of Ermin results in de‐compacted myelin sheath, myelin out folding and fragmentation, and axonal pathology. Ultrastructural electron microscopy (EM) analysis of myelin was performed on 5 months old mice. (A) Representative EM images of myelinated axons in the corpus callosum in WT and KO mice. (B–D) Ermin KO mice exhibit lower g‐ratios (B) and higher periodicity (C and D), suggesting less compact myelin sheath. n = 3 per genotype; ~300 axons quantified per animal for g‐ratio calculation. Data show means ± SEM; F 2,2 = 3.35, **P < 0.01; two‐tailed unpaired Student's test. (E) Representative EM image of myelin out folding in the corpus callosum of KO mice. (F) Quantification reveals increased myelin out folding in Ermin KO mice. Data shown as mean ± SEM; F 34,33 = 5.54, ***p < 0.001 by two‐tailed unpaired Student's test. (G) Representative EM image and quantification of axons with fragmented myelin in the corpus callosum of KO mice. Data show means ± SEM; F 34,33 = 20.39, ***p < 0.001; two‐tailed unpaired Student's test. (H) Immunoblot showing QD9 and corresponding MBP band, and Calnexin as loading control. Quantification of MBP/QD9 ratio, a measure of myelin damage and fragmentation, at 3 months of age: n = 9/genotype. Data represent means ± SEM, one‐way ANOVA with Tukey's post‐hoc test (adjusted p value). *p < 0.05, **p < 0.001. (I) Representative EM image and quantification of degenerating/ed axons in the corpus callosum of KO mice. Data show means ± SEM; F 34,33 = 3.32, ***p < 0.001 by two‐tailed unpaired Student's test. (J) Immunoblot of SMI‐32, a marker of axonal damage, and Calnexin as loading control. Quantification of normalized SMI‐32 level at 3 months of age: n = 9/genotype. Data represent means ± SEM; one‐way ANOVA followed by Tukey's test (adjusted p value). WT vs. KO, *p < 0.05
We next analyzed the number of myelinated axons with morphological abnormalities in the CC of Ermin KO mice. Comparing Ermin KO mice to their WT littermates, we observed a significant increase in the percentage of axons with myelin out folding, a phenotype seen in several myelin gene mutant mice [28] (Figure 2E,F). Similarly, the percentage of axons with fragmented myelin (Figure 2G) and degenerative phenotype (Figure 2I) was increased in Ermin KO mice compared with WT mice.
We further investigated whether QD9/MBP ratio was altered. QD9 is an antibody shown to detect an epitope of MBP primarily accessible under conditions of myelin fragmentation or damage [29, 30]. We found a significant increase in the QD9/MBP ratio in the brain of 3 months old Ermin KO mice compared with WT (Figure 2H), indicating compromised myelin sheath integrity in Ermin‐deficient mice.
Given the increase in axons with degenerating/ed phenotype, we assessed the levels of SMI32, a measure of the dephosphorylated form of neurofilament proteins and a marker of axonal damage [30]. SMI32 levels were significantly increased in the brain of 3 months old Ermin KO mice, supporting the presence of increased axonal pathology (Figure 2J). Altogether, our results suggest that loss of Ermin leads to compromised myelin and axonal damage. The MBP/QD9 ratios and levels of SMI32 are comparable between young Ermin deficient and WT mice (Figure S2).
3.3. Ermin deficiency leads to slower conduction velocity
Facilitation of conduction is one of the primary functions of myelin sheaths. In order to evaluate possible consequences of the compromised myelin sheaths in Ermin KO mice, we recorded the compound action potential (CAP) across the CC of Ermin KO and WT mice at 3 months of age by electrophysiological analysis (Figure 3A). Quantification of the average stimulus‐response time (Figure 3B) revealed that the conduction velocity was delayed in myelinated fibers of the N1 component (Figure 3C) but not in unmyelinated fibers (N2) (Figure 3D), in the Ermin KO mice compared with WT mice. Interestingly, myelinated fibers of Ermin Heterozygote (HTZ) mice behaved similarly to those of Ermin KO mice (Figure 3C). There were no alterations in the amplitudes, area, and duration of peaks in CAPs across the CC in Ermin KO mice (Figure S3). Altogether, these results suggest that loss of Ermin compromises the conductivity of myelinated, but not unmyelinated, fibers. In addition, Latency of the CAP responses from the corpus callosum N1 and N2 fibers are unaltered in 1.5 months old Ermin KO mice (Figure S4).
FIGURE 3.
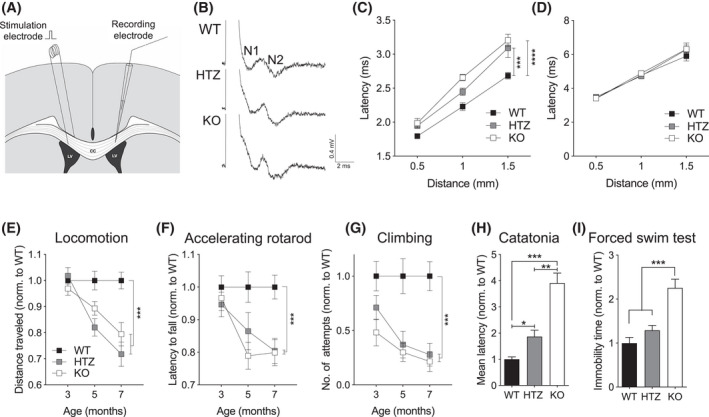
Reduced conduction velocity and progressive neurological deficit in Ermin KO mice. (A) Schematic showing the placement of stimulation and recording electrodes for measurement of compound action potentials (CAPs) in the corpus callosum, (B) representative CAP traces evoked with 5 mA stimuli. (C and D) Conduction was delayed in (C) myelinated fibers (N1), but not (D) unmyelinated fibers (N2) for KO and HTZ mice compared to WT mice. Each plot represents mean ± SEM, n = 12 slices from 4 mice per genotypes, ***p < 0.001 by two‐way ANOVA with Tukey's post‐hoc test (adjusted p value). (E–G) Ermin‐deficient mice exhibited progressive motor deficits based on (E) spontaneous activity, (F) accelerating rotarod performance, and (G) climbing attempts. (H) Ermin KO mice showed catatonic phenotype in the bar test at 7 months of age and (I) increased immobility in the Porsolt FST of depressive‐like behavior at 7 months of age. Data represent means ± SEM; N = 17–25 per genotype; *p < 0.05, **p < 0.01 ***p < 0.001 by (H) Kruskal‐Wallis test, and (I) one‐way ANOVA or (E–G) two‐way ANOVA followed by Tukey's post‐hoc test (adjusted p value)
3.4. Progressive neurological deficits develop in Ermin‐deficient mice
In light of the myelin structural and functional (conduction) abnormalities, we hypothesized that Ermin deficiency may result in neurological phenotypes. To test this hypothesis, we evaluated mice at 3, 5 and 7 months of age using a battery of behavioral tests. Ermin KO mice exhibited deficits in motor function such as deficits in spontaneous activity (decrease in distance traveled) (Figure 3E), accelerating rotarod performance (Figure 3F), climbing (Figure 3G). Ermin KO mice also displayed catatonia (Figure 3H), a phenomenon observed in a number of myelinopathies [31], as well as depressive‐like behavior in the Porsolt forced swim test (FST) (Figure 3I). Other behavioral results are summarized in (Figure S5).
3.5. The transcriptional analysis points to white matter inflammation in Ermin‐deficient mice
To gain insights into the processes impacted by the loss of Ermin, we performed RNA‐seq analysis on corpora callosal (CC) isolated from 8 months old WT, Ermin HTZ, and KO mice (deposited in SRA under accession number PRJNA612161). Principle component analysis reveals general separation of WT samples from the Ermin HTZ and KO samples which showed relatively tighter clustering (Figure 4A). We identified 579 differentially expressed genes (DEGs; false discovery rate (FDR) <10%) between WT and HTZ, of which 331 genes were upregulated and 248 were downregulated in HTZ. Furthermore, there were 828 differentially expressed genes identified between WT and KO, of which 556 genes were upregulated and 272 were downregulated in KO. Comparison of the differentially expressed genes (DEGs, relative to WT) showed more DEGs in Ermin KO mice compared with HTZ mice (Figure 4B; Tables S1 and S2), suggesting a greater impact of Ermin deficiency in KO mice compared with the HTZ mice. Venn analysis of DEGs showed the transcriptional changes in HTZ mice overlap with those seen in KO mice (Figure 4C). Functional annotation of the DEGs for Ermin KO mice revealed enrichment for axonal genes as well as genes associated with myelin sheath and myelination amongst the downregulated DEGs (Figure 4D; Table S3). Amongst upregulated genes, there was an enrichment of genes associated with immune and inflammatory responses (Figure 4D; Table S4). A heatmap of representative inflammation‐related genes such as Nfkb1, Itgax, and Lcn2 upregulated in Ermin KO mice is shown in Figure 4E. Overall, the differential gene expression analysis points to inflammatory activation in the corpora callosa of Ermin‐deficient mice, amongst other changes related to axonal and myelination processes.
FIGURE 4.
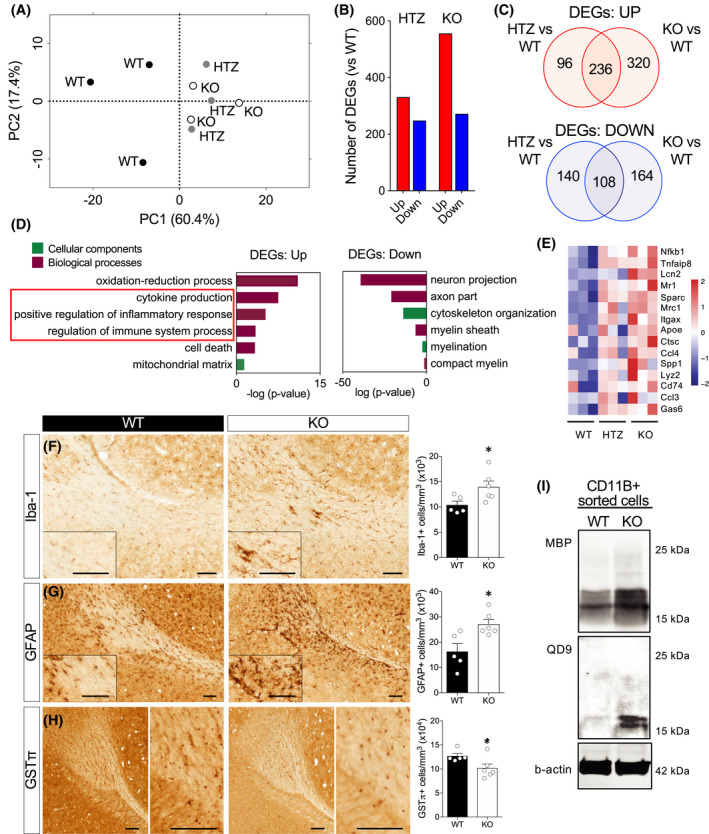
Loss of Ermin leads to inflammatory activation and microgliosis. (A–E) Transcriptional analysis of corpora callosa of Ermin‐deficient mice at 8 months of age. (A) PCA plot showing clustering of WTs (n = 3), HTZ (n = 3) and KO (n = 3) mice, (B) The number of deferentially expressed genes (DEGs), both upregulated (UP) and downregulated (DOWN), in HTZ and KO mice compared with WT (FDR<5%). (C) Venn analysis showed a substantial overlap of the UP and DOWN genes between HTZ and KO mice. (D) GO analysis of DEGs between KO and WT mice showed enrichment for terms related to inflammatory processes amongst the upregulated DEGs and myelin‐related processes in the downregulated DEGs. (E) Heat map of mean gene expression for selected immune‐ and myelination‐related in WT, HTZ and KO mice. (F–H) Immunohistological analysis shows microgliosis (Iba‐1+ counts), F 5,4 = 2.97 (F), astrogliosis (GFAP+ counts), F 4,5 = 2.40 (G), and loss of mature oligodendrocytes (GSTπ+ counts), F 5,4 = 4.05 in the corpus callosum Ermin KO mice at 8 months of age. Data show means ± SEM; *p < 0.05; two‐tailed unpaired Student's test. (I) Immunoblot analysis of CD11b+ (microglia/macrophages) isolated from brains of 12 months old male mice shows increased levels of phagocytosed MBP and fragmented MBP (QD9)
3.6. Ermin deficiency triggers microgliosis, astrogliosis, and loss of mature oligodendrocytes
To investigate inflammatory and oligodendrocyte changes suggested by the transcriptional analysis, we performed stereological analysis of microglia (IBA‐1+), astrocytes (GFAP+) and mature oligodendrocytes (GSTπ +) in CC of 8 months old Ermin KO mice. We observed a significant increase in the total number of microglia (Figure 4F) and astrocytes (Figure 4G), and a significant decrease in the total number of mature oligodendrocytes (Figure 4H) in KO mice compared with WT. These results indicate that loss of Ermin causes increased white matter inflammation and loss of mature myelinating oligodendrocytes.
Clearance of myelin debris is a key function of microglia [32]. To investigate whether the microgliosis observed may be due, at least in part, to an increased demand for clearance of myelin debris in Ermin KO mice, we isolated brain CD11b+ cells (microglia/macrophages), same protein lysate amount 30 µg and assessed the levels of MBP and fragmented MBP (QD9) they contained. Increased levels of MBP and fragmented MBP were detected in CD11b+ cells isolated from Ermin KO cells (Figure 4I), confirming a higher level of phagocytosed total and damaged myelin in Ermin KO mice.
We have also analyzed the number of microglia in different brain regions, including the caudate nucleus and hippocampus, and observed that microgliosis in KO mice is more pronounced in white matter rich regions such as the CC and Fimbria (Figure S6). In addition, we performed stereological analysis of microglia (IBA‐1+), astrocytes (GFAP+), and mature oligodendrocytes (GSTπ+) in the CC of Ermin KO mice at one month age. We observed no difference in the total number of microglia (Figure S7A) astrocytes (Figure S7B), or mature oligodendrocytes (Figure S7C) in KO mice compared with WT. These results indicate that white matter inflammation and loss of mature myelinating oligodendrocytes are not observed in young Ermin deficient mice.
3.7. Increased susceptibility to inflammatory demyelinating insult in Ermin knockout mice
Immune and inflammatory activation is a feature of several demyelinating white matter disorders. Given the compromise in myelin sheaths and the heightened inflammatory milieu in brains of Ermin KO mice, we sought to examine the impact of Ermin deficiency on the susceptibility to demyelination insult using the widely used experimental autoimmune encephalomyelitis (EAE) model (Figure 5A) [33]. EAE‐induced pathology is characterized by inflammatory demyelination and neurological phenotypes. In 9 weeks old Ermin KO mice, we observed significantly earlier onset of phenotypic (neurological) deficits compared with WT mice (Figure 5B,C). Furthermore, a smaller proportion of Ermin KO mice were symptom‐free post‐induction compared with WT mice (Figure 5D). Mixed effect model analysis of clinical score of EAE indicated indicates significant time and genotype effects (p < 0.0001 and p = 0.048, respectively). However, the maximum disease score in both WT and KO groups was similar between the genotypes (KO; 2 ± 0.93, WT; 2 ± 1.15, F 3,4 = 1.52, p = 0.9). Similarly, the cumulative disease score during day 6 to 24 was comparable (KO; 16.88 ± 7.9, WT; 13.30 ± 7.9, F 4,3 = 1.01, p = 0.5).
FIGURE 5.
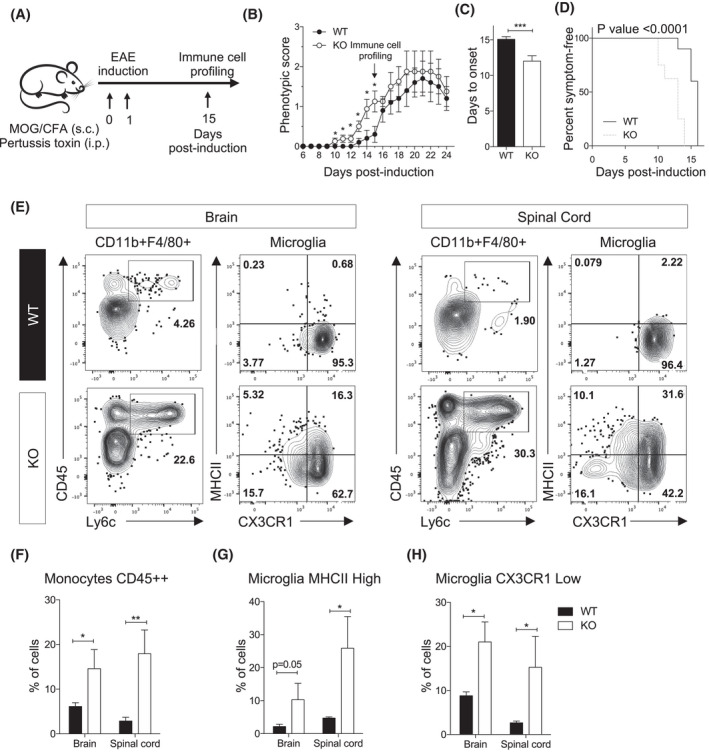
Loss of Ermin increases susceptibility to demyelination insult in the EAE model. (A) Schematic representation of the EAE experiment performed on 9‐week‐old female mice. (B) Daily scoring shows earlier onset of phenotypic deficits in Ermin KO mice compared to WT. (C) Onset of phenotypic deficits is significantly earlier in KO mice compared to WT. (D) Analysis of symptom‐free mice post‐induction shows enhanced susceptibility to EAE in the KO mice (p < 0.001; Mann‐Whitney test). Graph represents a Kaplan‐Meier curve with significance calculated using the log‐rank test. (E) Flow cytometry analysis of CD11b+ enriched cells isolated from the brains and spinal cords of WT and Ermin KO mice at day 15 of post‐induction. The graphs show representative dot plots and related quantification of (F) Monocytes CD45++, and (G and H) Microglia based on (G) MHCII High and (H) CX3CR1 Low levels. Data show means ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001; Mann‐Whitney test
Given such difference in phenotypic onset, we examined the cellular immune response in Ermin KO and WT mice. Flow cytometry analysis demonstrated a massive infiltration of monocytes (CD45 High Ly6C+) as well as non‐conventional monocytes (CD45 High Ly6C−) in brain and spinal cord of Ermin KO mice compared with WT (Figure 5E,F). Microglia constitute the major central nervous system macrophage population and has been shown to play a crucial role in the clearing of myelin in the brain [32]. In fact, we also observed higher levels of fragmented myelin inside microglia in Ermin KO compared to WT, as well (Figure 4I). In Ermin KO mice, microglia acquire higher expression levels of MHCII (MHCII High ) (Figure 5G). MHCII has been described to be associated to microglia activation status [34]. In Ermin KO mice, microglia also down regulate more the fractalkine receptor, CX3CR1 (CX3CR1 Low ) compared with WT mice (Figure 5H). Down regulation of CX3CR1 has been shown to be associated with the emergence of disease‐associated microglia (DAMs) [35]. These data suggest that Ermin KO mice have an increased susceptibility to inflammatory demyelination associated with higher monocyte infiltration, microglia activation, and DAM microglia (CX3CR1 Low ) in brain and spinal cord.
3.8. Discovery of mutations in ERMN in a multi‐incident multiple sclerosis family
Having established the pathological and functional consequences of Ermin loss in mice, we next sought to investigate whether Ermin deficiency could have similar effects in humans. Using GeneMatcher [36], an online tool that matches researchers with interest in a common gene, we identified an Iranian family segregating a rare inactivating germline mutation in ERMN in which multiple members have been diagnosed with multiple sclerosis (MS). This multi‐incident MS family consists of 4 individuals over two generations, two of whom were diagnosed with MS based on the McDonald criteria [37] (Figure 6A,B). Whole exome sequencing (WES) was performed on the affected mother, affected daughter and healthy father. After applying several filters on the analyzed trio, 367 HTZ and 1 HMZ variants remained. The identified variants were further filtered as depicted in (Figure 6C). Of those, 9 HTZ variants passed filtering criteria that include (a) minor allele frequency (MAF) below 1% in public and proprietary databases of variants, (b) exonic or highly conserved intronic variants (PhyloP score), (c) pathogenic score based on SIFT, PolyPhen and M‐CAP (Mendelian Clinically Applicable Pathogenicity) [38] (Figure 6C).
FIGURE 6.
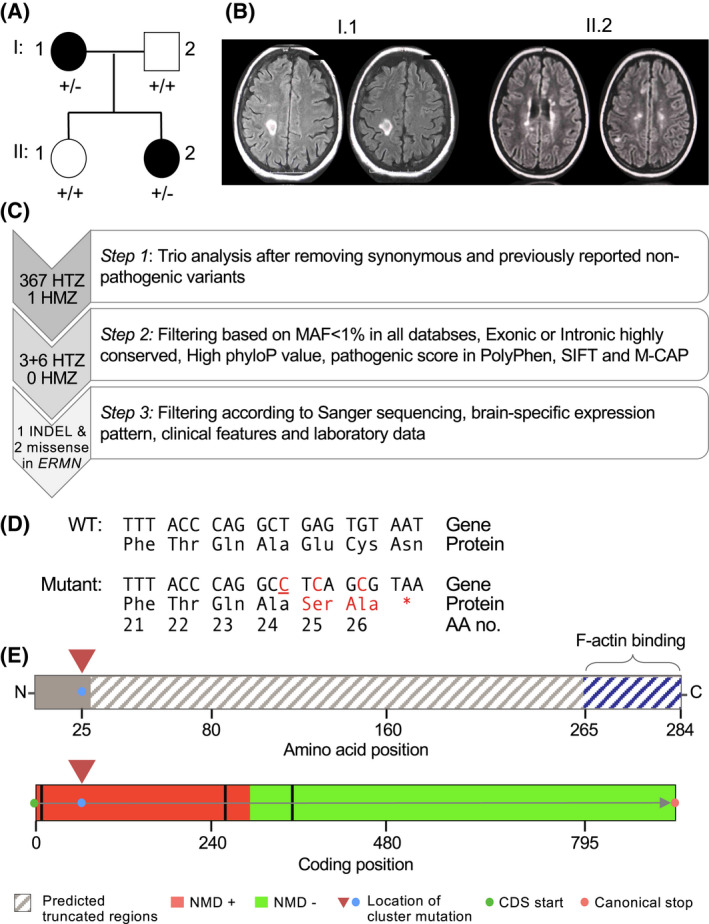
Discovery of novel cluster ERMN mutations in multi‐incident multiple sclerosis family. (A) Pedigree of the multi‐incidence MS family. Patients diagnosed with MS have black‐filled symbols. (B) T2‐FLAIR MRI images of the affected mother (I.1) and daughter (II.2) with demyelinating plaques. The mother (I.1) had plaques in the subcortical and periventricular white matter; images show tumefactive plaque in the right centrum semiovale. The affected daughter (II.2) had plaques in the pons, basal ganglia, corpus callosum; images show paraventricular and centrum semiovale plaques. (C) Criteria used to filter the variants identified by whole exome sequencing. The filtering approach included trio analysis, in silico prediction of conservation and pathogenicity, as well as confirmation by Sanger sequencing. Following filtering, a cluster of three mutations (an insertion and two missense variants) in the ERMN gene remained. (D) Predicted amino acid sequence of the mutant ERMN allele. Underlined nucleotide represents an insertion. (E) Premature stop codon results in the truncation of ~92% of the ERMIN protein 260/284, including the highly conserved C‐terminus actin‐binding domain (top). Based on the position of the cluster mutation, in silico analysis using the NMDEscPredictor algorithm predicts it to cause nonsense‐mediated decay (bottom)
Three adjacent mutations were all found in ERMN: two missense mutations and one frame‐shift mutation were clustered within 5 nucleotides of each other (Figure 6D). It is of note that such clustering has been reported as a feature of de novo mutations [39]. The cluster mutation is predicted to result in a truncated Ermin fragment that is devoid of Ermin's highly conserved C‐terminus actin‐binding protein (Figure 6D). The mutation is also predicted to trigger nonsense‐mediated decay based on in silico analysis using the NMDEscPredictor algorithm (Figure 6E) [40]. We cross‐referenced our cluster of SNPs with all publicly available databases such as ExAC, ESP6500, UK10K, 1000G, and dbSNP and none were detected (MAF = 0). This finding of an inactivating mutation in ERMN in patients with MS‐like presentation suggests that similar to our observations in Ermin KO mice, Ermin‐deficient may be associated with inflammatory white matter disorders in humans.
4. DISCUSSION
Ermin, a CNS‐specific, actin‐binding myelin protein expressed almost exclusively in mature oligodendrocytes, represents less than 1% of the myelin proteome [6]. Despite its minor abundance, we present here several lines of evidence, including structural, functional, transcriptional, and genetic analyses that demonstrate an essential role for Ermin in the maintenance and stability of myelin sheaths and the marked pathological consequences of its loss (summarized in Figure 7).
FIGURE 7.
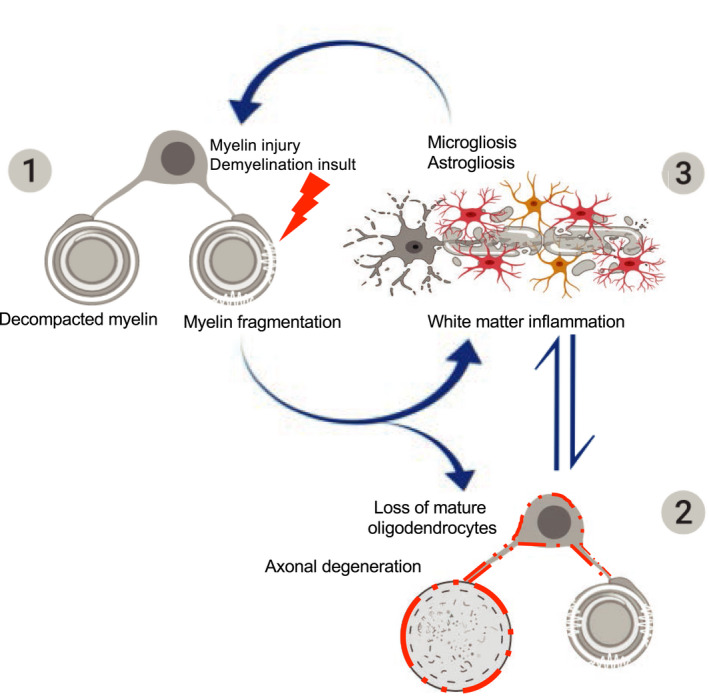
A working model for the sequence of pathological events in Ermin‐deficient mice. The pathology in Ermin‐deficient mice originates from the myelin sheaths: (1) Loss of Ermin leads to compromised myelin that is de‐compacted, out folded and fragmented; (2) As Ermin‐deficient mice age, augmentation of myelin fragmentation and myelin sheath breakdown leads to loss of myelinating oligodendrocytes. Due to compromised myelin sheaths and oligodendrocyte function, loss of trophic support to axons also leads to axonal damage and degeneration; (3) Excessive myelin debris along with oligodendrocyte loss and axonal damage/degeneration triggers white matter inflammation. Activated microglia and astrocytes further exacerbate cell loss. This ensuing inflammation may act as a feed‐forward demyelinating insult causing further damage to unhealthy myelin sheaths in Ermin deficient mice
On the level of myelin ultrastructure, myelinated callosal axons of Ermin‐deficient mice exhibited signs of myelin de‐compaction, fragmentation, and out folding. Consistent with a compromised myelin sheath, the ratio of QD9/MBP, a measure of myelin damage and fragmentation [30], was elevated in Ermin‐deficient mice. That this elevation in the ratio of QD9/MBP was observed at 3 months, but not 1 month, of age indicates that the myelin damage in Ermin‐deficient mice is progressive in nature. These structural myelin abnormalities were coupled with impaired saltatory conduction as early as three months of age. The reduced velocity of nerve conduction along myelinated callosal fibers in Ermin‐deficient mice is likely caused by myelin sheath de‐compaction and fragmentation, which are known to increase axonal capacitance [41].
In addition to abnormal myelin ultrastructure, signs of axonal pathology were also evident in Ermin‐deficient mice. The morphological changes were corroborated by elevated levels of SMI‐32, a marker of axonal damage [42, 43]. Since oligodendrocytes and their myelin sheaths play an important role in providing axons with trophic support and neuroprotection [44, 45], the observed manifestations of axonal damage provide further evidence of compromised myelin sheath integrity in Ermin‐deficient mice. Our observations of earlier clinical onset but no significant impact on the peak of disease severity in the EAE model is similar to what has been reported in other knockout mouse models with increased susceptibility to EAE [46]. We propose that compromised myelin sheath in the Ermin KO mice makes them vulnerable to mild demyelination insults, thus accelerating onset of clinical features in the EAE model without altering the nature or severity of the immune response trigger.
Ermin deficiency also resulted in a number of neurological deficits, including impaired motor performance, affective phenotypes, and catatonic behavior. The motor phenotypes were age‐dependent, with worsening performance over time suggestive of progressive myelin and axonal pathology in Ermin‐deficient mice. Interestingly, catatonia has been shown to be a common neurological manifestation in a number of myelinopathies [21, 22, 23].
Transcriptionally, genes downregulated in the corpus callosum of Ermin‐deficient mice showed enrichment for several myelin‐related cellular components and biological processes such as “myelin sheath”, “myelination,” and “compact myelin”. A number of genes downregulated in Ermin‐deficient mice are key components of compacted myelin such as Mbp, Pllp, and Sept8 [46, 47, 48], changes that may reflect and/or contribute to the de‐compaction of myelin sheaths observed in the Ermin‐deficient mice. Similarly, and consistent with the signs of axonal pathology observed, gene ontology terms related to axons and neuron death were enriched amongst the genes downregulated in Ermin‐deficient mice. Analysis of genes upregulated in the corpus callosum of Ermin‐deficient mice revealed an increase in inflammation and immune‐related processes. These include “cytokine production”,” positive regulation of inflammatory response”, and “regulation of immune system process”. Among these are Nfkb1, a master regulator of inflammation [49], as well as Mrc1, Itgax and Lcn2, all genes linked to inflammation and innate responses [50, 51, 52]. These signs of inflammatory and immune activation were corroborated by histological analyses showing marked microgliosis and astrogliosis in the corpus callosum of aged Ermin‐deficient mice. In line with the progressive nature of the myelin abnormalities, the microgliosis and astrogliosis were observed in aged, but not young, in Ermin‐deficient mice.
Microgliosis and astrogliosis are cardinal features of neurodegenerative conditions, including those that involve demyelination and oligodendrocyte loss such as multiple sclerosis [53, 54]. The etiology and sequence of events leading to these characteristic pathologies and the associated axonal atrophy remain subjects of debate. In the context of multiple sclerosis for example, a dominating view posits that an aberrantly activated immune system initiates a pathological cascade that culminates in oligodendrocyte death and demyelination, paralleled by axonal damage and degeneration. This view represents the initiating trigger as an “outside‐in” event [15]. An alternative “inside‐out” hypothesis that has been more challenging to prove proposes that the pathology originates within the oligodendrocytes or the myelin sheaths, with the immune and inflammatory activation being a secondary event that propagates the pathogenic cycle [55, 56]. Given Ermin's almost exclusive expression in mature, myelinating oligodendrocytes of the CNS, our study presents compelling evidence in support of the “inside‐out” hypothesis. The clinical and pathological heterogeneity of such inflammatory white matter disorders suggests that multiple etiologies, entailing both “outside‐in” and “inside‐out” pathogenic processes, are likely involved.
How might microgliosis and inflammatory activation arise in the context of Ermin deficiency? Since microglia play a key role in the removal of myelin debris [32], one initiating trigger may be the increased demand for clearance of myelin debris as a result of the instability of myelin sheaths in Ermin‐deficient mice. This possibility is supported by the elevated myelin content we detected in brain microglia/macrophages (CD11b+) cells from Ermin‐deficient mice. Alternatively, the initiating event may be the degeneration of oligodendrocytes as suggested by the decrease in GSTπ‐positive cells we observed in the corpus callosum of Ermin‐deficient mice. Furthermore, oligodendrocytes are known to express a wide range of immunomodulatory molecules and are thought to be capable of immunomodulation in CNS especially during the initiation of inflammatory responses [57]. Thus, loss or malfunction of oligodendrocytes in Ermin‐deficient mice may potentially contribute to the immune activation and microgliosis observed. We observed microgliosis and inflammatory milieu in the major white matter‐rich region of the brain corpus callosum in the aged Ermin KO mice. We did not observe a significant difference in other brain regions such as gray matter‐rich caudate and hippocampus and only detected a trend in fimbriae. We posit that inflammation is white matter specific and more pronounced in the white matter rich areas where the myelin sheath is compromised in Ermin deficient mice. Although the lack of phenotypic deficits in a subset of the tests for HTZ mice appears inconsistent with a haploinsufficiency mechanism of action, additional testing in older HTZ mice is warranted to fully test this possibility. Indeed, on the whole, our results indicate a slower rate of progression of abnormalities in HTZ compared with KO mice, suggesting the need for a more extensive time‐course analysis of phenotypic deficits in older Ermin‐deficient mice in future studies.
Another key feature of a number of demyelinating neurodegenerative disorders such as multiple sclerosis is an increase in the susceptibility to autoimmune‐mediated insults. Indeed, many of the therapies currently available for multiple sclerosis act largely through modulation of inflammatory processes [15]. Our findings show that the inflammatory milieu that develops in Ermin‐deficient mice is sufficient to increase susceptibility to autoimmune‐mediated demyelination. Two further key points can be deduced from these results. First, conditions that compromise the integrity of the myelin sheath are likely to increase susceptibility to immune‐mediate demyelination. This is consistent with recent studies in which pharmacologically‐ induced subtle biochemical changes in myelin sheaths were found to trigger inflammatory demyelination [58]. Second, treatments based solely on the modulation of inflammatory and immune activation in conditions where compromised myelin integrity is the initial trigger are not likely to be effective in the long term. In this respect, strategies that can improve myelin sheath stability and integrity may be important for effective combinatorial therapeutic interventions.
The possible contribution of impaired Ermin function to human white matter disorders is suggested by our findings of an inactivating mutation in ERMN in patients with MS‐like presentation. A recent analysis also reported down‐regulation of ERMN expression in blood samples from patients with multiple sclerosis [59]. While this limited evidence points to dysregulation of ERMN in multiple sclerosis, further targeted analyses of Ermin in multiple sclerosis and related conditions is warranted to validate its possible role in susceptibility to demyelinating insults and white matter disorders in humans.
Overall, our findings demonstrate that deficits in factors essential for myelin integrity may contribute to the pathogenesis of inflammatory white matter disorders. More specifically, our study broadly suggests that factors that, similar to Ermin deficiency, result in increasing myelin pathology are likely to similarly trigger a cascade of microglial and astrocytic activation with age, and may as a result increase the susceptibility to autoimmune insults. Our results are further corroborated by a recent study demonstrating compromised myelin stability, motor deficits, and increased susceptibility to cuprizone‐induced demyelination in Ermn knockout mice [60]. Our study is consistent with the notion that low‐abundance myelin sheath components such as Ermin may not necessarily be functionally minor [61]. It further lends support for the “inside‐out” theory of inflammatory white matter degenerative disorders, the notion that compromised myelin integrity can trigger inflammation and increase susceptibility to demyelinating insults. Finally, given its near‐exclusive expression in myelinating oligodendrocytes, the Ermin deficiency model we describe may offer a new paradigm for the development of myelin stability‐targeted therapies.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Amin Ziaei and Mahmoud A. Pouladi designed research; Amin Ziaei, Marta Garcia‐Miralles, Carola I. Radulescu, Aymeric Silvin, Han‐Gyu Bae, Harwin Sidik, Nur Amirah Binte Mohammad Yusof, Costanza Ferrari Bardile, Liang Juin Tan, Sumanty Tohari, Lily Henry, Xin Yi Yeo, Sejin Lee, and Leila Dehghani performed research; Amin Ziaei, Aymeric Silvin, Han‐Gyu Bae, Carine Bonnard, Alvin Yu Jin Ng, Sarah R. Langley, and Mahmoud A. Pouladi analyzed data; Byrappa Venkatesh, Vahid Shaygannejad, Bruno Reversade, Sangyong Jung, Florent Ginhoux provided intellectual input; Amin Ziaei and Mahmoud A. Pouladi wrote the paper with feedback from the other authors; and Mahmoud A. Pouladi conceptualized the study.
Supporting information
FIGURE S1 Immunoblotting analysis of Ermin expression on whole brain lysates of 3 months old mice using a rabbit polyclonal antibody (Abcam #ab243730) raised against amino acids encoded by exons 2/3 of Ermin. No truncated fragments of Ermin are detected in the heterozygous or homozygous knockout mice
FIGURE S2 The MBP/QD9 ratios and levels of SMI32 are comparable between young Ermin deficient and WT mice. (A) Quantification of the MBP/QD9 ratio, a measure of myelin damage and fragmentation, at 1 month of age: n = 9/genotype. Data represent means ± SEM, (B) Immunoblot of SMI‐32 and Calnexin as loading control. Quantification of normalized SMI‐32 level at 1 months of age: n = 9/genotype. Data represent means ± SEM
FIGURE S3 No alteration in amplitudes of CAPs across CC in Ermin KO mice. The amplitude was recorded in 1 mm distance between stimulation and recording electrodes with various stimulation strength (from 0.1 to 5 mA), and the averaged amplitudes of N1 (A) and N2 (B) were plotted against simulations. The areas (N1, C; N2, D) and duration (N1, E; N2, F) were calculated from the traces responded to 5 mA stimulation at 1 mm distance. The traces show the way to measure the area (C and D) and duration (E and F). Data were represented with means ± SEM; 12 brain slices (n) from 4 mice (N) per genotype.
FIGURE S4 Latency of the CAP responses from the corpus callosum N1 and N2 fibers are unaltered in 1.5 months old Ermin KO mice. (A) Representative CAP traces from the corpus callosum of control (WT), heterozygote (HTZ), and Ermin KO mice. There is no significant difference in the N1 (B) and N2 (C) fiber CAP latency between WT, HTZ, and KO samples with 0.5 mm, 1.0 mm or 1.5 mm between the stimulation and recording electrode. Data presented as mean ± SEM, statistical analysis conducted with two‐way ANOVA. WT n = 15, HTZ n = 15, KO n = 11.
FIGURE S5 Additional behavioral assessments of the Ermin knockout mice. (A and B) Mice were assessed for motor symptoms. progressive locomotion deficits in the jump time and vertical count in 30 min (C). Mice were assessed for anxiety‐like symptoms by elevated plus maze. (D and E) Rotarod training test in three consecutive days as a cognitive assessing test. N = 17–25 per genotype, littermates, mixed gender. Data show means ± SEM; ***p < 0.001; Two‐way ANOVA followed by Tukey's multiple comparisons test were applied for all behavioral studies
FIGURE S6 Immunohistochemistry and stereological analysis of IBA‐1 markers in Fimbria, Hippocampus and Caudate nucleolus regions of 8months, male mice, n = 5–7 per genotypes. Data show means ± SEM
FIGURE S7 Microglia (IBA‐1+), astrocytes (GFAP+), and mature oligodendrocytes (GSTπ+) counts in the corpus callosum of 1 month old Ermin KO and WT mice, male mice, n = 5 per groups. Data show means ± SEM
TABLE S1 Up‐regulated differential expression gene in KOs compare to WTs
TABLE S2 Up‐regulated differential expression gene in HTZs compare to WTs
TABLE S3 Functional enrichment terms for down‐regulated genes in KOs compare to WTs
TABLE S4 Functional enrichment terms for up‐regulated genes in KOs compare to WTs
TABLE S5 Antibodies used for immunoblotting and immunohistochemistry
TABLE S6 FACS antibodies used
ACKNOWLEDGEMENTS
We thank members of the Pouladi and Reversade labs for helpful discussions and comments. We thank Dr Fengyi Liang for the gift of the polyclonal anti‐Ermin antibody and helpful discussions. We thank Dr Ehsan Ziaei, Dr Giti Sadeghian, Dr Sahar Dehghan Kelishadi, Shaun Tan and Eri Aung for technical assistance and the A*STAR Microscopy Platform for assistance in sample imaging. The work was partly funded by a Strategic Positioning Fund for Genetic Orphan Diseases (SPF2012/005) and SUREKids (IAF311019) from the Agency for Science Technology and Research (A*STAR, Singapore) to MAP and BR, grants from the National Multiple Sclerosis Society USA (RG‐1906‐34251) and the BC Children’s Hospital Research Institute Investigator Grant Award Program (IGAP) to MAP, and a Joint Council Office grant (BMSI/15‐ 800003‐SBIC‐00E) from A*STAR to SJ. MAP is a recipient of a Scholar Award from the Michael Smith Foundation for Health Research. CFB is supported by a fellowship from the Hereditary Disease Foundation.
Ziaei A, Garcia‐Miralles M, Radulescu CI, Sidik H, Silvin A, Bae H‐G, et al. Ermin deficiency leads to compromised myelin, inflammatory milieu, and susceptibility to demyelinating insult. Brain Pathol. 2022;32:e13064. 10.1111/bpa.13064
DATA AVAILABILITY STATEMENT
The RNA‐Seq data reported in this paper have been deposited in the SRA.
REFERENCES
- 1. Nave K‐A, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30(1):503–33. [DOI] [PubMed] [Google Scholar]
- 2. Yoshida M, Colman DR. Parallel evolution and coexpression of the proteolipid proteins and protein zero in vertebrate myelin. Neuron. 1996;16(6):1115–26. [DOI] [PubMed] [Google Scholar]
- 3. Kriventseva EV, Tegenfeldt F, Petty TJ, Waterhouse RM, Simão FA, Pozdnyakov IA, et al. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res [Internet]. 2015. Jan;43(D1):D250–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25428351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang B, Cao Q, Guo A, Chu H, Chan YG, Buschdorf JP, et al. Juxtanodin: an oligodendroglial protein that promotes cellular arborization and 2’,3’‐cyclic nucleotide‐3’‐phosphodiesterase trafficking. Proc Natl Acad Sci U S A [Internet]. 2005;102(32):11527–32. Available from http://www.ncbi.nlm.nih.gov/pubmed/16051705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brockschnieder D. Ermin, A myelinating oligodendrocyte‐specific protein that regulates cell morphology. J Neurosci. 2006;26(3):757–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40(1):55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scherer SS, Xu T, Crino P, Arroyo EJ, Gutmann DH. Ezrin, radixin, and moesin are components of Schwann cell microvilli. J Neurosci Res [Internet]. 2001. Jul;65(2):150–64. Available from http://www.ncbi.nlm.nih.gov/pubmed/11438984 [DOI] [PubMed] [Google Scholar]
- 8. Brown TL, Macklin WB. The actin cytoskeleton in myelinating cells. Neurochem Res. 2020;45:684–93. 10.1007/s11064-019-02753-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruskamo S, Chukhlieb M, Vahokoski J, Bhargav SP, Liang F, Kursula I, et al. Juxtanodin is an intrinsically disordered F‐actin‐binding protein. Sci Rep. 2012;2:899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snaidero N, Velte C, Myllykoski M, Raasakka A, Ignatev A, Werner HB, et al. Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell Rep [Internet]. 2017;18(2):314–23. Available from http://www.ncbi.nlm.nih.gov/pubmed/28076777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang T, Jia L, Lv B, Liu B, Wang W, Wang F, et al. Human Ermin (hErmin), a new oligodendrocyte‐specific cytoskeletal protein related to epileptic seizure. Brain Res. 2011;1367:77–84. 10.1016/j.brainres.2010.10.003 [DOI] [PubMed] [Google Scholar]
- 12. Martins‐de‐Souza D, Gattaz WF, Schmitt A, Rewerts C, Maccarrone G, Dias‐Neto E, et al. Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2009. Apr 22;259(3):151–63. 10.1007/s00406-008-0847-2 [DOI] [PubMed] [Google Scholar]
- 13. Homs A, Codina‐Solà M, Rodríguez‐Santiago B, Villanueva CM, Monk D, Cuscó I, et al. Genetic and epigenetic methylation defects and implication of the ERMN gene in autism spectrum disorders. Transl Psychiat. 2016;6(7):e855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple sclerosis. Lancet. 2018. Apr;391(10130):1622–36. [DOI] [PubMed] [Google Scholar]
- 15. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med. 2018. Jan;378(2):169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Franklin KBJ, Paxinos G. Paxinos and Franklin’s the mouse brain in stereotaxic coordinates. Elsevier. [Google Scholar]
- 17. Teske JA, Perez‐Leighton CE, Billington CJ, Kotz CM. Methodological considerations for measuring spontaneous physical activity in rodents. Am J Physiol Regul Integr Comp Physiol [Internet]. 2014. May;306(10):R714–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24598463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pouladi MA, Stanek LM, Xie Y, Franciosi S, Southwell AL, Deng Y, et al. Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full‐length BACHD and YAC128 mice. Hum Mol Genet. 2012. May;21(10):2219–32. 10.1093/hmg/dds037 [DOI] [PubMed] [Google Scholar]
- 19. Garcia‐Miralles M, Geva M, Tan JY, Yusof NABM, Cha Y, Kusko R, et al. Early pridopidine treatment improves behavioral and transcriptional deficits in YAC128 Huntington disease mice. JCI Insight. 2017;2(23):e95665. 10.1172/jci.insight.95665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety‐related behavior in rodents. Nat Protoc. 2007;2(2):322–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagemeyer N, Goebbels S, Papiol S, Kästner A, Hofer S, Begemann M, et al. A myelin gene causative of a catatonia‐depression syndrome upon aging. EMBO Mol Med. 2012;4(6):528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wieser GL, Gerwig UC, Adamcio B, Barrette B, Nave K‐A, Ehrenreich H, et al. Neuroinflammation in white matter tracts of Cnp1 mutant mice amplified by a minor brain injury. Glia. 2013. Jun;61(6):869–80. [DOI] [PubMed] [Google Scholar]
- 23. Poggi G, Boretius S, Möbius W, Moschny N, Baudewig J, Ruhwedel T, et al. Cortical network dysfunction caused by a subtle defect of myelination. Glia. 2016;64(11):2025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010. May;38(4):576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996. Aug;19(8):312–8. [DOI] [PubMed] [Google Scholar]
- 26. Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005. Jun;50(4):427–34. [DOI] [PubMed] [Google Scholar]
- 27. Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas‐mediated genome engineering. Nat Protoc [Internet]. 2014. Aug;9(8):1956–68. Available from: http://www.nature.com/articles/nprot.2014.134 [DOI] [PubMed] [Google Scholar]
- 28. Patzig J, Erwig MS, Tenzer S, Kusch K, Dibaj P, Möbius W, et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 2016. Aug;5:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG, et al. Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol. 1997. Apr;150(4):1253–66. [PMC free article] [PubMed] [Google Scholar]
- 30. Weil M‐T, Möbius W, Winkler A, Ruhwedel T, Wrzos C, Romanelli E, et al. Loss of myelin basic protein function triggers myelin breakdown in models of demyelinating diseases. Cell Rep. 2016;16(2):314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arinrad S, Balmuth E, Pan H, Nave K‐A, Homuth G, Goebbels S, et al. Microglia ablation alleviates myelin‐associated catatonic signs in mice. J Clin Invest [Internet]. 2017;128(2):734–45. Available from: https://dm5migu4zj3pb.cloudfront.net/manuscripts/97000/97032/cache/97032.2‐20180123102350‐covered‐253bed37ca4c1ab43d105aefdf7b5536.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age‐related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci [Internet]. 2016. Aug;19(8):995–8. Available from: http://www.nature.com/articles/nn.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sheng W, Yang F, Zhou YI, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM‐CSF‐producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res [Internet]. 2014. Dec;24(12):1387–402. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25412660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scheffel J, Regen T, Van Rossum D, Seifert S, Ribes S, Nau R, et al. Toll‐like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia. 2012. Dec;60(12):1930–43. 10.1002/glia.22409 [DOI] [PubMed] [Google Scholar]
- 35. Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell [Internet]. 2017. Jun;169(7):1276–1290.e17. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0092867417305780 [DOI] [PubMed] [Google Scholar]
- 36. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat [Internet]. 2015. Oct;36(10):928–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26220891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–73. 10.1016/S1474-4422(17)30470-2 [DOI] [PubMed] [Google Scholar]
- 38. Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet [Internet]. 2016;48(12):1581–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27776117 [DOI] [PubMed] [Google Scholar]
- 39. Chan K, Gordenin DA. Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet [Internet]. 2015;49:243–67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26631512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coban‐Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain‐of‐function alleles. Am J Hum Genet. 2018. Aug;103(2):171–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suminaite D, Lyons DA, Livesey MR. Myelinated axon physiology and regulation of neural circuit function. Glia. 2019. Jun;67(11):2050–62. 10.1002/glia.23665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Su JH, Cummings BJ, Cotman CW. Plaque biogenesis in brain aging and Alzheimer’s disease. I. Progressive changes in phosphorylation states of paired helical filaments and neurofilaments. Brain Res. 1996. Nov;739(1–2):79–87. [DOI] [PubMed] [Google Scholar]
- 43. Hoehn HJ, Kress Y, Sohn A, Brosnan CF, Bourdon S, Shafit‐Zagardo B. Axl‐/‐ mice have delayed recovery and prolonged axonal damage following cuprizone toxicity. Brain Res. 2008. Nov;1240:1–11. [DOI] [PubMed] [Google Scholar]
- 44. Morrison BM, Lee Y, Rothstein JD. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol. 2013. Dec;23(12):644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saab AS, Nave K‐A. Myelin dynamics: protecting and shaping neuronal functions. Curr Opin Neurobiol. 2017. Dec;47:104–12. [DOI] [PubMed] [Google Scholar]
- 46. Patzig J, Erwig MS, Tenzer S, Kusch K, Dibaj P, Möbius W, et al. Septin/anillin filaments scaffold central nervous system myelin to accelerate nerve conduction. eLife. 2016;5:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Snaidero N, Velte C, Myllykoski M, Raasakka A, Ignatev A, Werner HB, et al. Antagonistic functions of MBP and CNP establish cytosolic channels in CNS myelin. Cell Rep [Internet]. 2017. [cited 2019 May 20];18(2):314–23. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28076777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bosse F, Hasse B, Pippirs U, Greiner‐Petter R, Müller HW. Proteolipid plasmolipin: localization in polarized cells, regulated expression and lipid raft association in CNS and PNS myelin. J Neurochem. 2003;86(2):508–18. [DOI] [PubMed] [Google Scholar]
- 49. Liu T, Zhang L, Joo D, Sun S‐C. NF‐κB signaling in inflammation. Signal Transduct Target Ther [Internet]. 2017;2:17023. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29158945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hendrickx DAE, van Scheppingen J, van der Poel M, Bossers K, Schuurman KG, van Eden CG, et al. Gene expression profiling of multiple sclerosis pathology identifies early patterns of demyelination surrounding chronic active lesions. Front Immunol [Internet]. 2017. Dec;8:1810. Available from: http://journal.frontiersin.org/article/ 10.3389/fimmu.2017.01810/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lehmann ML, Weigel TK, Elkahloun AG, Herkenham M. Chronic social defeat reduces myelination in the mouse medial prefrontal cortex. Sci Rep [Internet]. 2017. Jun;7(1):46548. Available from: http://www.nature.com/articles/srep46548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jordão MJC, Sankowski R, Brendecke SM, Sagar LG, Tai Y‐H, et al. Single‐cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science [Internet]. 2019. Jan;363(6425):eaat7554. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30679343 [DOI] [PubMed] [Google Scholar]
- 53. Luo C, Jian C, Liao Y, Huang Q, Wu Y, Liu X, et al. The role of microglia in multiple sclerosis. Neuropsychiatr Dis Treat [Internet]. 2017. [cited 2019 Apr 28];13:1661–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28721047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med [Internet]. 2000. Sep;343(13):938–52. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11006371 [DOI] [PubMed] [Google Scholar]
- 55. Tsunoda I, Fujinami RS. Inside‐Out versus Outside‐In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol. 2002;24(2):105–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Geurts JJG, Kooi E‐J, Witte ME, van der Valk P. Multiple sclerosis as an “Inside‐out” disease. Ann Neurol. 2010;68(5):767–8. [DOI] [PubMed] [Google Scholar]
- 57. Zeis T, Enz L, Schaeren‐Wiemers N. The immunomodulatory oligodendrocyte. Brain Res. 2016. Jun;1641(Pt A):139–48. [DOI] [PubMed] [Google Scholar]
- 58. Caprariello AV, Stys PK. Turned inside out: will myelin‐protective therapies become the next‐generation anti‐inflammatories? DNA Cell Biol. 2018;38(3):219–22. [DOI] [PubMed] [Google Scholar]
- 59. Salek Esfahani B, Gharesouran J, Ghafouri‐Fard S, Talebian S, Arsang‐Jang S, Omrani MD, et al. Down‐regulation of ERMN expression in relapsing remitting multiple sclerosis. Metab Brain Dis [Internet]. 2019;34(5):1261–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31123898 [DOI] [PubMed] [Google Scholar]
- 60. Wang S, Wang T, Liu T, Xie RG, Zhao XH, Wang L, et al. Ermin is a p116RIP‐interacting protein promoting oligodendroglial differentiation and myelin maintenance. Glia. 2020. Jun 12;68(11):2264–76. 10.1002/glia.23838 [DOI] [PubMed] [Google Scholar]
- 61. Ishii A, Dutta R, Wark GM, Hwang S‐I, Han DK, Trapp BD, et al. Human myelin proteome and comparative analysis with mouse myelin. Proc Natl Acad Sci USA [Internet]. 2009. Aug;106(34):14605–10. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19706548 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Immunoblotting analysis of Ermin expression on whole brain lysates of 3 months old mice using a rabbit polyclonal antibody (Abcam #ab243730) raised against amino acids encoded by exons 2/3 of Ermin. No truncated fragments of Ermin are detected in the heterozygous or homozygous knockout mice
FIGURE S2 The MBP/QD9 ratios and levels of SMI32 are comparable between young Ermin deficient and WT mice. (A) Quantification of the MBP/QD9 ratio, a measure of myelin damage and fragmentation, at 1 month of age: n = 9/genotype. Data represent means ± SEM, (B) Immunoblot of SMI‐32 and Calnexin as loading control. Quantification of normalized SMI‐32 level at 1 months of age: n = 9/genotype. Data represent means ± SEM
FIGURE S3 No alteration in amplitudes of CAPs across CC in Ermin KO mice. The amplitude was recorded in 1 mm distance between stimulation and recording electrodes with various stimulation strength (from 0.1 to 5 mA), and the averaged amplitudes of N1 (A) and N2 (B) were plotted against simulations. The areas (N1, C; N2, D) and duration (N1, E; N2, F) were calculated from the traces responded to 5 mA stimulation at 1 mm distance. The traces show the way to measure the area (C and D) and duration (E and F). Data were represented with means ± SEM; 12 brain slices (n) from 4 mice (N) per genotype.
FIGURE S4 Latency of the CAP responses from the corpus callosum N1 and N2 fibers are unaltered in 1.5 months old Ermin KO mice. (A) Representative CAP traces from the corpus callosum of control (WT), heterozygote (HTZ), and Ermin KO mice. There is no significant difference in the N1 (B) and N2 (C) fiber CAP latency between WT, HTZ, and KO samples with 0.5 mm, 1.0 mm or 1.5 mm between the stimulation and recording electrode. Data presented as mean ± SEM, statistical analysis conducted with two‐way ANOVA. WT n = 15, HTZ n = 15, KO n = 11.
FIGURE S5 Additional behavioral assessments of the Ermin knockout mice. (A and B) Mice were assessed for motor symptoms. progressive locomotion deficits in the jump time and vertical count in 30 min (C). Mice were assessed for anxiety‐like symptoms by elevated plus maze. (D and E) Rotarod training test in three consecutive days as a cognitive assessing test. N = 17–25 per genotype, littermates, mixed gender. Data show means ± SEM; ***p < 0.001; Two‐way ANOVA followed by Tukey's multiple comparisons test were applied for all behavioral studies
FIGURE S6 Immunohistochemistry and stereological analysis of IBA‐1 markers in Fimbria, Hippocampus and Caudate nucleolus regions of 8months, male mice, n = 5–7 per genotypes. Data show means ± SEM
FIGURE S7 Microglia (IBA‐1+), astrocytes (GFAP+), and mature oligodendrocytes (GSTπ+) counts in the corpus callosum of 1 month old Ermin KO and WT mice, male mice, n = 5 per groups. Data show means ± SEM
TABLE S1 Up‐regulated differential expression gene in KOs compare to WTs
TABLE S2 Up‐regulated differential expression gene in HTZs compare to WTs
TABLE S3 Functional enrichment terms for down‐regulated genes in KOs compare to WTs
TABLE S4 Functional enrichment terms for up‐regulated genes in KOs compare to WTs
TABLE S5 Antibodies used for immunoblotting and immunohistochemistry
TABLE S6 FACS antibodies used
Data Availability Statement
The RNA‐Seq data reported in this paper have been deposited in the SRA.