

Abstract
Childhood obesity is one of the most serious global public-health challenges of the twenty-first century. Over the past four decades, the number of children and adolescents with obesity has risen more than tenfold. Worldwide, an increasing number of youth are facing greater exposure to obesity throughout their lives, and this increase will contribute to the early development of type 2 diabetes, fatty liver and cardiovascular complications. Herein, we provide a brief overview of trends in the global shifts in, and environmental and genetic determinants of, childhood obesity. We then discuss recent progress in the elucidation of the central role of insulin resistance, the key element linking obesity and cardiovascular-risk-factor clustering, and the potential mechanisms through which ectopic lipid accumulation leads to insulin resistance and its associated cardiometabolic complications in obese adolescents. In the absence of effective prevention and intervention programs, childhood obesity will have severe public-health consequences for decades to come.
Paediatric obesity, in both childhood and adolescence, constitutes a major global public-health crisis of our time1–5. Although understanding of its pathogenesis and dynamics has become more nuanced, prevention and treatment remain elusive. Paediatric obesity, particularly in adolescents, is a pervasive disorder with a high risk of continuing into adulthood6. The body mass index (BMI) is the accepted standard measure of overweight and obesity for children 2 years of age and older7–11. Consensus committees have recommended that children and adolescents be considered overweight or obese if their BMI exceeds the 85th or 95th percentile in curves generated from the 1963–1965 and 1966–1970 US National Health and Nutrition Examination Survey (NHANES)5,12.
Epidemiology
Global prevalence and trends of childhood obesity and underweight from 1975 to 2016: the double burden of malnutrition.
The Non-Communicable Disease Risk Factor Collaboration (NCD-RisC) study, led by Ezzati et al.11, using a comprehensive global database of BMI from 200 countries, has indicated that the prevalence of paediatric obesity rose dramatically from 4% in 1975 to 18% in 2016. From 1975 to 2016 (Fig. 1), obesity increased from 5 million to 50 million girls and from 6 million to 74 million boys11. The largest increases have been in East Asia, the Middle East and North Africa, South Asia and high-income English-speaking regions11. Notably, in high-income countries, the rise in childhood obesity has recently plateaued, whereas it continues to rise in low-income and middle-income countries11.
Fig. 1 |. Trends in the number of children and adolescents with obesity and with moderate and severe underweight by region.
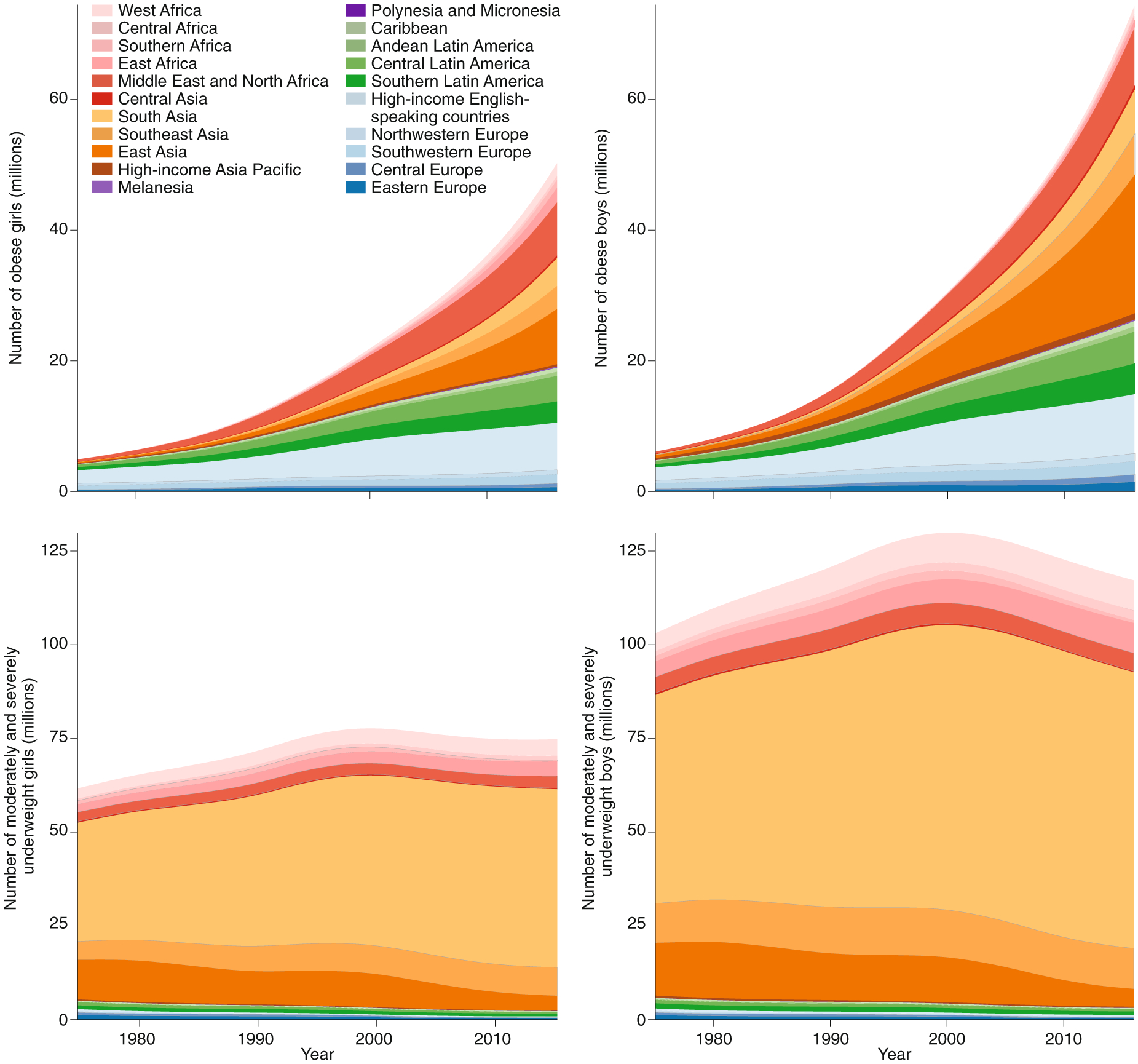
Image reprinted with permission from ref.11 under a Creative Commons license CC BY 4.0.
Undernutrition and obesity commonly coexist side by side within the same country, community or household. The experiences of East Asia, Latin America and the Caribbean have shown that the transition from underweight to overweight and obesity can be rapid, thus creating obstacles to a nation’s ability to implement a transition to healthful nutrition. An unhealthful nutritional transition—that is, an increase in nutrient-poor, energy-dense foods—can lead to stunted growth along with weight gain in youth, thus resulting in higher BMI and poorer health outcomes throughout the life course11,13.
Trends in childhood obesity and its increasing severity in the United States.
NHANES, using calculated BMI from measured weights and heights, has described the history of childhood obesity from 1963–1965 through 2015–2016 in both children and adolescents14 (Fig. 2).
Fig. 2 |. Trends in the prevalence of childhood obesity in the United States from 1963 to 2016.
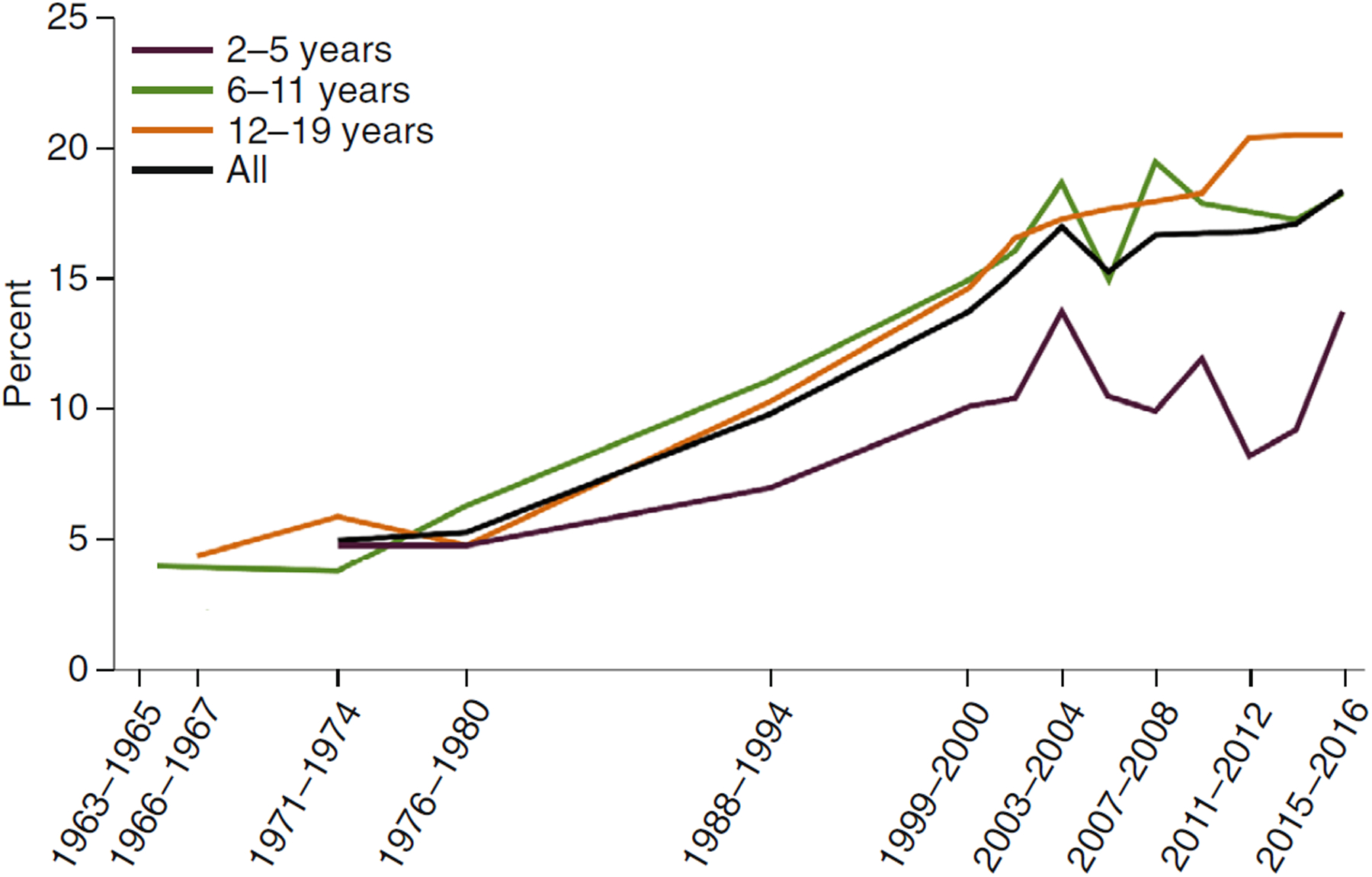
Image reprinted with permission from ref.14, National Center for Health Statistics.
Substantial ancestral disparities exist in the prevalence of obesity among US children and adolescents: the recent prevalence rates of obesity are 25.1% in non-Hispanic black girls, 23.6% in Hispanic girls, 13.5% in non-Hispanic white girls and 10.1% in non-Hispanic Asian girls15.
The severity of obesity in adults is subcategorized into class I (BMI 30–35), class II (BMI 35–40) and class III (BMI ≥40). Skinner et al.15–17 have used a definition of severe obesity applying to only children and adolescents, in which BMI ≥95th percentile is class I obesity, BMI ≥120% of the 95th percentile is class II obesity, and BMI ≥140% of the 95th percentile is class III obesity15–17. Under this definition, the prevalence of severe obesity in adolescents was ~10% in non-Hispanic white girls, 20% in non-Hispanic black girls and 16% in Mexican American girls15. The rightward shift in the BMI, as classified into class II and class III obesity, is particularly notable in adolescents and in non-Hispanic black individuals12,16.
Despite evidence that the severity of childhood obesity negatively affects health in youth, current guidelines for screening do not differentiate among degrees of obesity18.
Risk factors for childhood and adolescent obesity
Environmental determinants.
The root causes of obesity development in childhood and adolescence reflect complex interactions among environmental, socioeconomic, behavioural and genetic factors. The dramatic environmental changes worldwide in recent decades have fuelled the rise in the prevalence of childhood obesity19–21. Countless environmental changes that foster eating more frequently have occurred, such as the availability of cheap foods with higher energy content; the growth of soda and fast-food industries generating ultraprocessed foods; and the increased number and marketing of snacks, which continue to play a critical role in the weight and health of youth6,22. In the United States and other Western countries, and more recently in underdeveloped countries, in parallel with the rise in obesity, the consumption of added sugars has increased significantly17,19. Adolescents are the highest consumers of added sugar20,21 and are particularly vulnerable to sugar’s central reward effects and effects on obesity development. Using functional magnetic resonance imaging, Jastreboff et al.23,24 have assessed brain perfusion responses to drinking two common monosaccharides, glucose and fructose, in obese and lean adolescents. Obese adolescents show impaired prefrontal executive-control responses to drinking glucose and fructose, whereas their homeostatic and hedonic responses appear to be heightened. Thus, obesity-related brain adaptations to glucose and fructose consumption in obese adolescents may contribute to excessive consumption of both sugars, thereby promoting further weight gain.
Sedentary behaviours are key contributors to the development of obesity. Opportunities in daily life to increase energy expenditure have diminished: children have many hours of ‘media time’ per day, physical education has markedly decreased in schools, many neighbourhoods lack sidewalks for safe walking, and bicycles have been replaced by motorized bicycles and scooters6.
Genetics of childhood obesity: from twin studies to the exome era to GWAS.
Since 1970, several studies have tested the hypothesis that human obesity is a heritable trait25–27. Seminal studies by Stunkard et al.26,27 have estimated BMI heritability to be 0.77 by age 20 and 0.84 by age 25 (refs.26,27). As stated by the authors: “the concept of ‘heritability’ does not refer to a constant genetic effect, rather it describes the genetic effect under certain environmental exposures, thus for the same individuals under different environmental conditions, different estimates of heritability may be obtained”27.
One of the milestones that propelled the field of genetics of obesity came in 1994 with the discovery of the leptin gene (LEP) and its product, the leptin hormone, which is synthetized mainly by adipose tissue and acts in the hypothalamus by decreasing appetite and increasing satiety28.
Soon after the cloning of the LEP gene in humans, O’Rahilly et al. described two severely obese children homozygous for a frame-shift mutation in codon 133 of the LEP gene29. One of the children (a 9-year-old girl) was successfully treated with recombinant leptin for 12 months (ref.30). After leptin’s discovery, studies of individuals with severe early-onset obesity indicated the importance of the melanocortigenic pathway in modulating leptin’s action. Subsequently, α-melanocyte-stimulating hormone, a compound derived from proopiomelanocortin (POMC), was clearly implicated in the control of appetite through its receptor, melanocorin-4-receptor (MC4R)31. Furthermore, mutations in the MC4R gene are the most common monogenic cause of severe obesity32. The prevalence of mutations in the MC4R gene ranges from 0.5% to 5%, with an average of 3%, among obese individuals32–35.
The discovery of the melanocortinergic system has been extremely important, because it has led to the development of drugs targeting this system that are effective in people with MC4R mutations and leptin deficiency36,37.
The second pivotal landmark in the study of the genetics of obesity was the availability of genome-wide association studies (GWAS). In 2007, a key large GWAS study discovered a locus within the FTO gene that is strongly associated with BMI and obesity risk38. GWAS have used anthropometric measures as outcomes, such as BMI, waist-to-hip ratio adjusted for BMI (WHRadjBMI) and percentage body mass39–41. More than 500 genetic loci have been associated with obesity-related traits in a recent GWAS performed in nearly 700,000 individuals41. In a recent study, 346 loci and 463 signals associated with WHRadjBMI were identified41, but the combined variants explained only 3.9% of the outcome variance41. The investigators also observed a sex-dependent effect, in which the heritability of WHRadjBMI was found to be stronger in women than in men41. Of note, loci associated with WHRadjBMI appear to be enriched in genes involved in adipose tissue biology40, whereas loci associated with BMI primarily affect brain-expressed genes involved in appetite regulation39.
Although most GWAS discoveries have been made in adults, studies in children have been performed in recent years42–45. The main difference between studies in adults and children is the smaller sample size in the latter. GWAS in paediatric groups have shown that most of the loci discovered in adults can be replicated in children, thus suggesting an overlap between the genetic architecture of obese children and obese adults42,43. Some paediatric studies have shown that the FTO locus is strongly associated with obesity, although the strongest association was not consistently observed in all the studies42, contrary to the results from studies in adults. Paediatric GWAS have also served as a discovery tool for new signals not identified in adult populations (such as OLFM4 at chromosome 13q14 and HOXB5 at chromosome 17q21)42,43. Very recently, a large paediatric GWAS meta-analysis in groups of children with different ancestries has confirmed that adult and paediatric obesity may share the same genetic underpinnings and that not all the loci found in adults can be replicated in children, primarily because of the smaller sample size of the latter group46. Overall, in children, as in adults, the relevant loci together explain a small portion of heritability46.
Despite the lack of functional studies for most the genes or loci associated with obesity, in 2019, a polygenic risk score, using gene variants associated with obesity through GWAS, was derived and shown to be associated with the degree of obesity as well as the development of obesity over time, starting from the age of 12 years (ref.47).
The possibility of selectively sequencing all coding regions of the genome (exome) enables examination of whether rare variants (with a minor allele frequency <1%) rather than common variants (explored through GWAS) may explain the so-called ‘missing heritability’. A recent exome-wide search for low-frequency and rare genetic variants associated with BMI has been performed by using exome-targeted genotyping arrays in 718,734 individuals48. Interestingly, the investigators discovered 14 new variants in 13 genes and found that the rare variants had an effect size ten times larger than the common variants previously identified by GWAS48. This finding suggests that rare and low-frequency variants rather than common variants may play major roles in the genetics of obesity.
The effects of the FTO genotype on food intake in children: a potential contributor to obesity.
GWAS studies have reliably established that single-nucleotide polymorphisms in the first intron of FTO are strongly associated with greater BMI across different ages and populations38,49,50. People homozygous for the A allele of FTO rs9939609 have an obesity risk 1.7-fold greater than that of people homozygous for the low-risk T allele22. The association between FTO and BMI is predominantly driven by an increased drive to eat, probably because of impaired satiety51,52. FTO is highly expressed in brain regions controlling feeding, such as the hypothalamus, particularly the arcuate nucleus, which is critical for the appetite-stimulating effect of ghrelin53–56. A preference for energy-dense foods among children has been found to be associated with the FTO genotype independently of body weight22,57,58.
Highlighting the mechanistic link between FTO and ghrelin in energy homeostasis, Karra et al.59, in a group of lean men, have shown that people homozygous for the FTO A allele, compared with people homozygous for the major allele, have attenuated suppression of ghrelin levels and hunger as well as diminished hypothalamic neural responses after consuming a meal. Rosenbaum et al.60 have suggested that even before the development of obesity, children carrying the rs9939609 A risk allele exhibit greater food intake, and each copy of the A allele is associated with approximately 65 additional calories consumed, thereby explaining 3% of variance in intake60. The influence of genotype dose on energy intake adds support to the hypothesis that FTO genotypic associations with body weight may be mediated by effects on food intake rather than on energy expenditure. Despite these observations, Claussnitzer et al. have proposed that the FTO effect may be mediated by a different gene variant that decreases energy expenditure61. This intriguing observation contrasts with findings from another study indicating that the FTO rs9939609 variant is not associated with any phenotype related to energy expenditure62.
Syndromic forms of obesity: what they can teach us about bodyweight regulation.
Although the above-mentioned studies tested the effects of common and rare variants on the risk of developing essential non-syndromic obesity early in life, some complex genetic syndromes are characterized by early-onset obesity. Among these, Prader–Willi syndrome (PWS) is the most common syndrome associated with severe obesity63. PWS is a congenital multisystem disorder characterized by neonatal hypotonia and feeding problems, developmental delay, short stature, hypogonadotropic hypogonadism and diminished resting energy expenditure. PWS results from a defect in the expression of genes in the paternally inherited chromosomal region 15q11.2–q13. Although the genetic defects underlying PWS are known, their relationships with the PWS phenotype remain unclear. Recent findings suggest that the PCSK1 gene might be responsible for early-onset obesity64. Indeed, Burnett et al., using induced pluripotent stem cells from people with PWS, have shown that the PCSK1 gene, which encodes the enzyme prohormone convertase 1 (PC1), is weakly expressed in PWS64. This finding is extremely interesting because a defect in the PCSK1 gene was originally found to cause early-onset obesity by affecting the melanocortinergic pathway65.
Further insights into the pathophysiology of paediatric obesity have come from Alström syndrome (AS) and Bardet–Biedl syndrome (BBS), two ciliopathies characterized by early-onset obesity. AS is an autosomal recessive disease caused by mutations in the ALMS1 gene66 characterized by hearing and visual impairment, early-onset obesity, high plasma triglycerides, severe insulin resistance (IR), diabetes and fatty liver66, along with developmental delays66. Knockout of Alms1 in mice suggests that Alms1 deficiency leads to insulin hypersecretion, owing to a β-cell glucose-sensing defect. Consequent hyperinsulinaemia leads to β-cell exhaustion, thus resulting in diabetes66. This model suggests that body-fat accumulation in these patients may be caused by hyperinsulinaemia, and studies comparing people with AS and BMI-matched controls have highlighted the extreme insulin-resistance characteristic of AS67.
BBS is characterized by ciliary dysfunction along with obesity, early-onset diabetes, retinitis and kidney disease68. Mutations causing BBS result in obesity through a dysregulation of food-seeking activity69. People with BBS experience severe leptin resistance70, probably because the underlying ciliopathy affects leptin signaling70. Variable BBS gene mutations are associated with different degrees and distribution of adiposity, thus suggesting that fat accumulation has a different pathogenesis in BBS and nonsyndromic obesity70.
Overall, these observations imply that insights from syndromic obesity must be taken into account in the study of non-syndromic obesity, because they can provide essential clues regarding the complex pathophysiology of weight regulation.
Cardiometabolic complications and their burden in childhood
Paediatric obesity is increasingly associated with type 2 diabetes, fatty liver and cardiovascular disease71–76. The clustering of cardiovascular risk factors (CVRFs) in early childhood is concerning, given that ~80% of obese youth remain obese in adulthood77–79. Below, we describe the role of IR, the key element linking obesity and CVRF clustering, and potential mechanisms leading to its development in obese children.
Ectopic fat storage in obese youth: a potential cause of IR.
The subcutaneous adipose tissue (SAT) has been proposed to act as a ‘sink’ accommodating excess energy as triglycerides and thus preventing the flow of lipids to other organs80–82. Of note, not all obese youth develop metabolic complications; in fact, many obese youth have favourable metabolic profiles83. One hypothesis explaining this paradox is that total body fat is not the culprit of an unfavourable metabolic profile; instead, the relative proportion of lipids in various fat depots is the determinant of metabolic risk. In other words, inadequate SAT expansion results in lipid overflow into visceral adipose tissue (VAT) and non-adipose tissues84,85. The ‘adipose expandability hypothesis’86,87 suggests that after AT storage capacity is exceeded, the net lipid flux to non-adipose tissues increases, thus causing lipotoxicity and leading to IR87. Our studies in obese adolescents have indicated that IR is related to a particular abdominal fat distribution and ectopic fat accumulation88,89. To unravel the mechanisms responsible for the inefficient storage of fat in the abdominal SAT, fat-cell size and the transcription of genes regulating lipogenesis and adipogenesis have been measured in two groups of obese adolescents with similar degrees of obesity but profound differences in abdominal fat distribution (low VAT/SAT ratio versus high VAT/SAT ratio)83. Compared with obese adolescents with low VAT/SAT, obese adolescents with high VAT/SAT showed coexistence of large and small adipocytes, downregulation of key lipogenic and adipogenic genes, and an inflammatory profile characterized by macrophage infiltration and decreased SIRT1 expression90.
To gain further insights into the potential causes of inefficient storage of triglycerides in the SAT and gluteal depots, we measured in vivo dynamic fluxes of SAT triglycerides, de novo lipogenesis and adipocyte turnover in obese girls with distinct differences in abdominal fat distribution but similar degrees of obesity91. We found that obese girls with a high VAT/(VAT + SAT) display increased in vivo rates of lipolysis and unaltered de novo lipogenesis in both abdominal and gluteal SAT; higher adipocyte turnover, with no difference in preadipocyte proliferation; and a strong relationship between the increased lipolytic rates and intrahepatic lipid accumulation. These findings suggest that increased turnover of triglycerides and mature adipocytes in SAT, rather than decreased triglyceride deposition capacity or proliferation of new adipocytes, contributes to the development of fatty liver and related metabolic impairment.
The deposition of lipids within insulin-responsive tissues, such as the skeletal muscle, liver and adipose tissue, is associated with localized IR in molecular pathways related to glucose metabolism, thus leading to hyprinsulinaemia92. In insulin-responsive tissues, a selective resistance to the effects of insulin is observed in pathways related to glucose metabolism, whereas other insulin-mediated pathways respond normally to the resultant hyperinsulinaemia (such as hepatic lipogenesis or renal sodium reabsorption)93. This selective IR is largely caused by intracellular fatty acid derivates such as fatty acyl-CoA, diacylglycerol and ceramides94. In skeletal muscle, the result of such IR is decreased trafficking of the glucose transporter GLUT-4 to the cellular membrane and thus lower uptake of systemic glucose. In the liver, the result is increased hepatic glucose production mediated by a decreased suppression of gluconeogenesis85. Similarly, adipose tissue IR is manifested increased lipolysis and decreased glucose uptake, thereby resulting in increased systemic flux of free fatty acids (‘lipotoxicity’) into the skeletal muscle and liver95. These cellular and molecular defects of IR are manifested early in life in obese youth88,91,95–97. First, obese adolescents who develop impaired glucose tolerance have profound IR and are characterized by increased intramuscular and intra-abdominal lipid deposition despite being as obese (according to anthropometric and percentage-body-fat indices) as obese adolescents with normal glucose tolerance95. Second, obese youth with greater degrees of intrahepatic steatosis (despite comparable degrees of obesity to their peers) have been found to have higher 2-hour glucose levels and profound whole-body IR, and most have substantial CVRF clustering98. Adipose IR in this context is tightly related to increased 2-hour glucose levels and decreased postprandial suppression of free fatty acid levels99. Importantly, in the presence of IR, the β-cells in obese youth are challenged by an increased allostatic load100. The presence of baseline insulin secretion defects, together with continuous weight gain, further aggravates β-cell failure and ultimately leads to further deterioration of glucose metabolism101.
The systemic response to IR related to altered lipid partitioning is thus manifested as altered glucose metabolism and, in parallel, as the appearance of adverse cardiovascular biomarkers such as elevated concentrations of triglycerides, small particles containing oxidized-low-density-lipoprotein cholesterol102 and markers of subclinical inflammation103, along with diminished levels of high-density lipoprotein (HDL) cholesterol and adiponectin104. Together, the cluster of CVRFs such as altered glucose metabolism, dyslipidaemia, elevated blood pressure and subclinical inflammation has been named insulin-resistance syndrome and is highly prevalent in obese youth75. The clinical correlates of obesity-related CVRF clustering are increased intima–media thickness and left-ventricular hypertrophy105 in childhood106, which continue into adulthood107, thus suggesting the presence of accelerated atherogenesis.
Non-alcoholic fatty liver disease in obese youth: pathogenesis and genetic underpinnings.
Non-alcoholic fatty liver disease (NAFLD) defines a spectrum of disease including intrahepatic fat accumulation, steatohepatitis, fibrosis and cirrhosis108. Its prevalence in children increases with BMI and is approximately 38% in obese adolescents109; however, the prevalence differs according to ancestry and is highest in Hispanic (45%) and in non-Hispanic white people (~30%) and lowest in non-Hispanic black people (~13%)110. The natural history of paediatric NAFLD is poorly understood, given the paucity of longitudinal data. A retrospective study has shown that adolescents with NAFLD at the age of 13.9 years have a survival free of end-stage liver disease lower than that expected in the general US population of the same age and sex111. Obese youth with NAFLD are more insulin resistant than those without NAFLD112, and have a higher prevalence of prediabetes, type 2 diabetes113 and dyslipidaemia114. NAFLD and IR are intrinsically linked, although which precedes the other is unknown. A recent longitudinal study has shown that youth who develop NAFLD within 2 years tend to have higher levels of insulin and c-peptide at baseline than those who do not develop NAFLD110, thus suggesting that IR may precede the development of NAFLD.
Although a certain degree of IR is present in all obese youth, not all of them develop NAFLD, and the heritability of NAFLD ranges between 35% and 50% (refs.115–117). The first gene variant associated with NAFLD through a GWAS was the rs738409 variant in the PNPLA3 gene118, which encodes a substitution of isoleucine with methionine in a highly conserved codon (I148M)118. The PNPLA3 gene encodes the protein adiponutrin, which promotes the transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets119. Although the mechanism through which this variant predisposes people to NAFLD has not yet been elucidated, this is the strongest genetic signal associated with NAFLD120,121. Along with this variant in PNPLA3, other variants in genes expressed in the liver have been consistently associated with NAFLD in youth122–124. Notably, the effects of these gene variants on the development of NAFLD are amplified by the degree of adiposity, thus indicating that the effect of the genotype on the phenotype increases with increasing BMI125. In Fig. 3, we propose potential mechanisms linking NAFLD to IR and cardiac dysfunction in obese adolescents.
Fig. 3 |. Proposed pathophysiological mechanisms linking NAFLD to IR and cardiac dysfunction in obese adolescents.
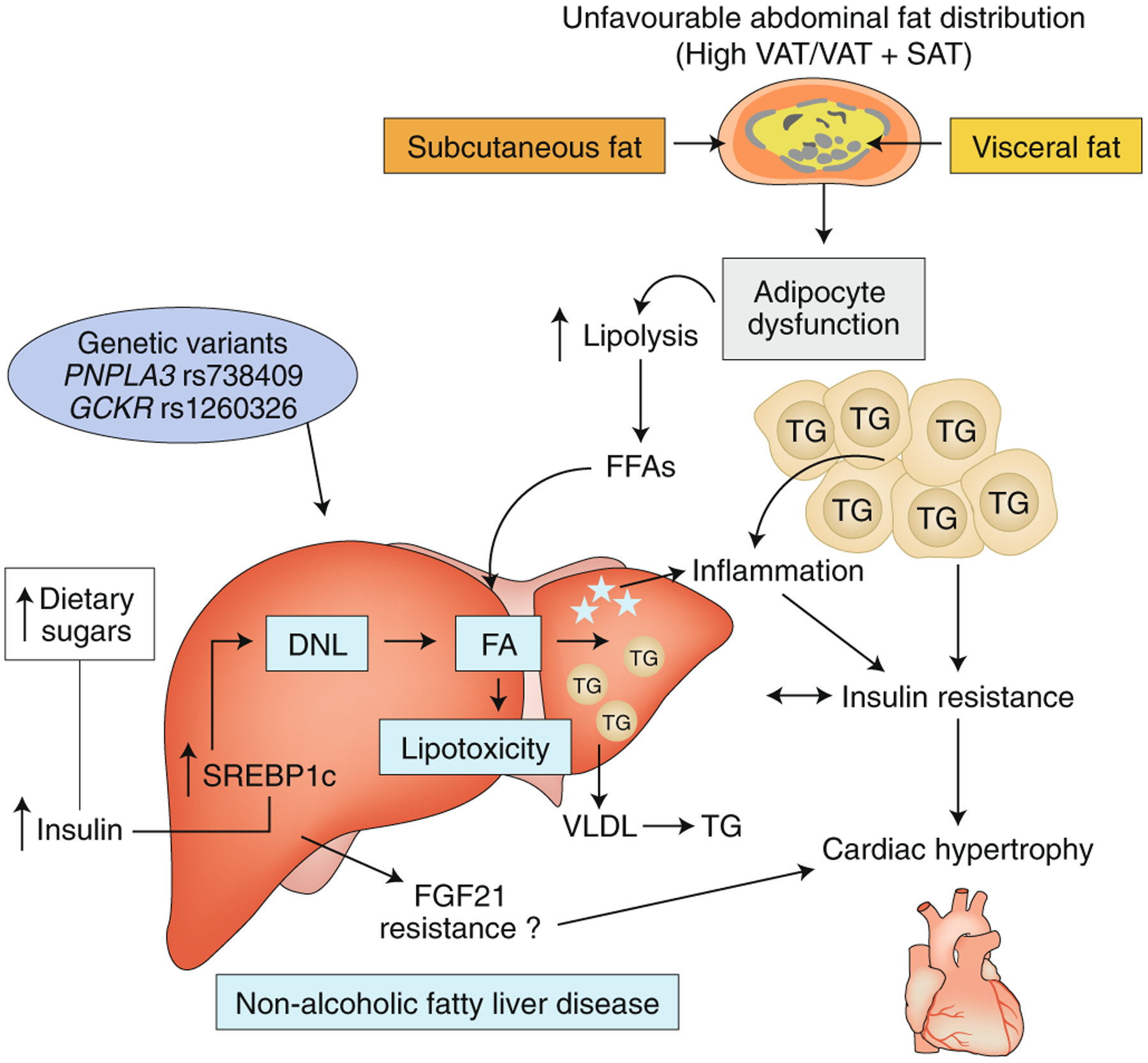
NAFLD is a result of genetics and environmental factors (dietary habits, IR, increased de novo lipogenesis and adipose tissue lipolysis). FFAs, free fatty acids; FA, fatty acid, VLDL, very low-density lipoprotein; TG, triglyceride; DNL, de novo lipogenesis; FGF21, fibroblast growth factor 21; SREBP1c, sterol regulatory element–binding protein 1c. Image adapted with permission from ref.172, American Association for the Study of Liver Diseases.
Obesity dynamics and CVRF stability in obese adolescents.
CVRF clustering (CVRFs, altered glucose metabolism, elevated blood pressure and triglycerides, low HDL-cholesterol levels and elevated biomarkers of inflammation) is tightly associated with IR and is common amongst obese youth75. As described above, altered lipid partitioning is the main mechanistic driver linking obesity and the development of IR in obese children. Adolescents with BMI greater than the 99th percentile for age and sex have a significantly greater risk of having CVRF clustering than those with lower degrees of obesity126. Using conservative thresholds for prediabetes, dyslipidaemia and elevated blood pressure, a large multi-ancestral cohort has demonstrated that the prevalence of the CVRF is directly and independently linked to the degree of both obesity and IR75. Across a spectrum of BMI, each half unit of the BMI z score increases the risk of having CVRF clustering by 55% (hazard ratio, 1.55; 95% confidence interval, 1.16–2.08). Independently of the degree of obesity, increasing IR (an additional unit of homeostatic model assessment of IR) increases the risk of CVRF clustering by 12% (hazard ratio, 1.12; 95% confidence interval, 1.07–1.18). Skinner et al., using NHANES data16, have shown that values for some but not all CVRFs are higher in people with increasing severity of obesity, while also demonstrating that after adjustment for age, ancestry and sex, a greater degree of obesity increases the risk of lower HDL-cholesterol levels, high systolic and diastolic blood pressure and elevated plasma triglycerides. In an obesity-clinic-derived cohort127, greater adiposity has been shown to be associated with greater risk of CVRF clustering, yet this risk tends to plateau and not increase further in people with BMIs greater than 40 kilograms per square metre.
Weight loss, resulting in decreased ectopic lipid deposition, is expected to reverse the presence of such risk factors. Multiple clinical trials combining dietary modifications, physical activity and family-oriented therapy have demonstrated that modest weight loss, or even cessation of weight gain, may significantly improve metabolic phenotypes128,129. Linking the clinical-marker improvement associated with weight loss has revealed that a decrease of 0.30 BMI s.d. in obese adolescents is associated with a significant decrease in intrahepatic and intramuscular lipid deposition130, thus resulting in significantly increased insulin sensitivity131. Moreover, an exercise program leads to weight loss, better insulin sensitivity and improvements in clinical biomarkers of atherogenesis such as intima–media thickness132.
The studies described above led to a critical question: what magnitude of BMI s.d. score (or percentage body weight) decrease is needed in obese adolescents to improve insulin sensitivity sufficiently to enable recovery from obesity-related metabolic morbidity? The amount of weight loss needed to induce substantial changes in insulin sensitivity that translate to normalization of clinical risk markers has been postulated to be approximately 0.25 BMI s.d., and achieving weight loss >0.50 BMI s.d. has the greatest benefit133,134. Importantly, such improvements may be sustained months after the completion of an intervention program135.
However, longitudinal changes in insulin sensitivity are strongly related specifically to fat-mass accrual over time136, thus highlighting the mechanistic role of adipose tissue excess in the development of IR. Specifically, weight gain is tightly associated with decreased insulin sensitivity and is the best predictor of deteriorating glucose tolerance137. The relationship between weight dynamics and insulin sensitivity is best appreciated when interventions are evaluated upon completion, and their sustainability is determined over time. In such studies, weight loss immediately after the completion of a successful intervention program is paralleled by improvements in proxies for insulin sensitivity, yet after subsequent weight regain, insulin sensitivity returns to its low baseline level138.
Interventions for reversing the obese state in youth
Recently published guidelines recommend that therapeutic approaches to paediatric obesity be limited to counselling about physical activity and lifestyle changes7. Yet, noncompliance and high attrition rates are highly frequent among obese youth139,140. Therefore, the addition of pharmacological treatments to lifestyle-intervention programmes may be an effective therapeutic strategy in paediatrics.
Pharmacological approaches.
In children, unlike adults, few US Food and Drug Administration (FDA)-approved obesity drugs are available, owing to the difficulty in demonstrating the safety profiles of centrally acting drugs in to the context of growth and development. Among the few pharmacological options in paediatric medicine, orlistat, an inhibitor of pancreatic lipase (limiting fat absorption from the gut)) is currently the only FDA-approved medication for treatment of paediatric obesity for children from 12 years of age141. However, its effect on weight loss is very modest142.
Recently, in a clinical trial in obese adolescents with type 2 diabetes, liraglutide, a GLP-1 agonist, has shown similar safety, tolerability and pharmacokinetic profiles to those in adults143. The investigators have demonstrated the superiority of liraglutide over placebo in decreasing haemoglobin A1c and plasma glucose, although they did not show the superiority of liraglutide in decreasing the BMI z score after 26 weeks. Testing of the effect of liraglutide on the BMI z score in 24 prepubertal obese children (7–11 years of age) has demonstrated that liraglutide is superior to placebo in decreasing the BMI z score (−0.28; P = 0.0062)144. Therefore, GLP-1 agonists may be a promising medication for treating obesity in children. The mechanism through which GLP-1 causes weight loss is not entirely clear yet seems to be related to an effect on the central nervous system. GLP-1 crosses the blood–brain barrier and reaches regions critical for the control of appetite, such as the hypothalamus. In particular, animal studies have suggested that GLP-1 acts in the arcuate nucleus of the hypothalamus, where it stimulates POMC/CART neurons and inhibits AgRP neurons, thereby determining satiety145.
The discovery of the melanocortinergic system as a key pathway in appetite control has led to the development of drugs targeting MC4R. In particular, recent trials using setmelanotide, an MC4R agonist, have been performed in obese individuals carrying rare variants in the MC4R, POMC and leptin-receptor genes36,37,146. Specifically, the first trial was performed in two people with a POMC deficit and showed a loss of 51.0 kilograms after 42 weeks in one patient and 20.5 kilograms after 12 weeks in the other146. In another phase 1b clinical trial, 49 obese adults were enrolled and randomized to treatment with either setmelanotide or placebo for 14 or 28 days (ref.37). Significant weight loss was observed in obese people receiving ≥0.01 milligrams of setmelanotide per kilogram in 24 hours, compared with placebo37. Setmelanotide has also effectively led to weight loss in three adults with leptin-receptor mutations36 and is well tolerated36,37,146.
Importantly, monogenic causes of obesity are rare, and thus agents such as setmelanotide do not provide a solution for most obese children.
To address the metabolic effects of altered lipid partitioning in obese youth, several trials have used rosiglitazone. This agent induces preadipocyte differentiation and thus expands subcutaneous fat while diverting lipid from the liver and skeletal muscle. In a 4-month randomized intervention in obese youth with prediabetes, rosiglitazone indeed improved insulin sensitivity in association with increased subcutaneous fat, decreased intrahepatic fat and normalized glucose metabolism in 58% of those who received it147.
Bariatric surgery in paediatric obesity.
The important long-term morbidity associated with severe obesity in children and adolescents and the modest effects of conservative interventions in this age group have led to more aggressive interventions such as bariatric surgical procedures. Bariatric procedures differ in their degree of anatomical modification, thus resulting in variable relative combinations of mechanical and hormonal effects. Nonetheless, they will be discussed herein as a group regarding their effects on weight and insulin sensitivity in morbidly obese adolescents. Patient selection for bariatric surgery in adolescents requires candidates to have reached >95% of their projected height and to have severe obesity-related complications (BMI ≥35 kilograms per square metre or 120% of the 95th percentile with clinically relevant comorbid conditions such as obstructive sleep apnoea (apnoea hypopnea index >5), type 2 diabetes, idiopathic intracranial hypertension, steatohepatitis, Blount’s disease or hypertension; or BMI either ≥40 kilograms per square metre or 140% of the 95th percentile, whichever is lower). Surgery candidates must be assessed by a multidisciplinary team to decide whether the patients and families have the ability and motivation to adhere to preoperative and postoperative regimens148. Bariatric surgery is contraindicated in adolescents with medical or psychiatric problems that may hamper adherence to postoperative dietary and pharmacological regimens, recent substance abuse, planned pregnancy or a medically correctable cause of obesity. In the Teen-Longitudinal Assessment of Bariatric Surgery (Teen-LABS) consortium, a multicentre observational study, data from 242 morbidly obese adolescents showed a significant weight loss (26–28%) postsurgery (for both Roux-en-Y gastric bypass and sleeve gastrectomy). Weight loss was associated with type 2 diabetes remission (95% of patients), and recovery from dyslipidaemia (66% of patients) and hypertension (74% of patients)149. The main conclusion from Teen-LABS was that the 5-year outcomes of bariatric surgery in obese adolescents are similar to those demonstrated in adults regarding weight and are slightly better regarding remission from obesity-related comorbidities150. Similarly, The Adolescent Morbid Obesity Surgery (AMOS) study has reported the superiority of 5-year outcomes in adolescents receiving Roux-en-Y gastric bypass compared with conservative treatment151. AMOS demonstrated a significant weight loss, which was maintained for 5 years and was associated with a 74–100% resolution of altered glucose metabolism, hypertension, dyslipidaemia and inflammatory markers. Despite the clear and impressive short-term effects of bariatric procedures on weight and related comorbidities, these clinical benefits should be balanced with the potential comorbidities associated with such surgical interventions, including the need for additional surgical procedures (25% of people in the AMOS study) and the development of nutritional deficiencies (72%) that might affect future bone health if they occur during a critical period of bonemass development. Inge et al. have reported nutritional deficits after 2 years of follow-up in ~50% of adolescents and 26% of adults, whereas the rate of a second surgery was higher in adolescents than in adults151. A 5-year follow-up study has highlighted a high prevalence of gastroesophageal reflux symptoms, especially in adolescents receiving vertical sleeve gastrectomy152. A tendency towards a higher prevalence of alcohol abuse and self-harm after surgery has also been described153.
Importantly, substantial ethical issues exist regarding these procedures: they are irreversible (with the exception of gastric banding), the long-term effects on morbidity and mortality (specifically in people without major obesity-related morbidity in adolescence) are unknown, and not all adolescents seem to benefit from them154. One of the main concerns relates to future bone health, because such procedures have been shown to be associated with substantial early bone loss in adolescents and adults155 as well as with an overall increased risk of fractures156. Most patient-selection guidelines for such procedures in adolescence highlight the importance of prior participation in a lifestyle intervention (to demonstrate adherence), thus ruling out major psychiatric conditions and demonstrating a viable family support system. Because adherence to obesity-management programs in this age group is notoriously low157, and given the high prevalence of psychiatric morbidity in obese youth158, strict adherence to such guidelines results in very few suitable surgical candidates159.
In cases of syndromic or hypothalamic obesity, the results of bariatric procedures are less promising. In patients with PWS compared with those with non-syndromic obesity, the results of surgery are poorer, specifically in weight regain160. Similarly, in patients with hypothalamic obesity after resection of craniopharyngioma, surgery outcomes are quite variable161. Therefore, in cases of syndromic or hypothalamic obesity, given the lower success rates, bariatric surgery should be considered only as a last resort in patients with substantial comorbidity.
Halting the epidemic of childhood obesity: reasons for patchy progress.
To understand the beginning of the epidemic of obesity in the United States, using NHANES data from the early 1970s, Rodgers et al.162 have noted that in most obese adults and children in the United States, the excess weight gain began at approximately the same time (1970s)162. Further, they have argued that the precipitous global spread of obesity could not have been driven by changes in genetic predisposition, which do not occur over short periods of time162. Hence, to determine the potential drivers of the rise in obesity, they focused on the changes in US farm bills in 1970, which caused an increase in food production, thus increasing the availability of cheap food and fast food, and allowing restaurants to serve larger portions163. Another important role has been played by the expanded use of inexpensive sweeteners such as high-fructose corn syrup164.
Given the complex nature of obesity, it has become increasingly clear that confronting and reversing obesity trends should be addressed through comprehensive multisectoral action165. The World Health Organization has been, and still is, at the forefront of the problem in developing global strategies for the prevention of childhood obesity and non-communicable diseases166, and improving maternal and child health167. As a result of its global efforts to prevent childhood obesity, the World Health Organization established the Commission on Ending Childhood Obesity (ECHO). Although not entirely novel, the plan and recommendations of the ECHO program are well developed, described and illustrated in the Commission’s report166, which proposes a range of governmental recommendations aimed at reversing the rising trend in children becoming obese. As a result of these global efforts, high-level policies, strategies and targets for addressing childhood obesity have been agreed upon, yet translating these recommendations into specific policies at the national level has been challenging168. Roberto et al. have described the implementation of these strategies as being “patchy” at best168. The reasons for the poor translation of global recommendations into national policies lie mainly in the imbalance between the private sector and government/society. The prioritization of free-market goals has influenced the deregulation of markets in many countries, and privileged commercial interests are affecting policy-making169. Indeed, fast-food and soft-drink conglomerates have had critical roles in halting efforts to introduce public-health regulation of these industries170.
Future outlook.
A major concern related to childhood obesity is that obese children tend to become obese adults with all the associated risks and comorbidities (including diabetes, fatty liver disease and cardiovascular disease, among many others). A recent study171 has estimated that among children between the ages of 2 and 19 years in 2016, more than half (57.3%) will be obese by the age of 35 years. Early development of obesity predicts obesity in adulthood, especially for children who were severely obese. Thus, these findings highlight the importance of promoting healthy weight throughout childhood and adulthood. A narrow focus solely on preventing childhood obesity will not avert potential future damage to health that may be induced by the ongoing obesity epidemic171.
Acknowledgements
The authors are grateful to B. Pierpont and M. Savoye for their support and dedication to this work. S.C. is supported by US National Institutes of Health (NIH) grants R01-DK111038 and R01-HD028016. N.S. is supported by NIH grant R01-DK114504. The work of S.C. and N.S. at Yale is also made possible by NIH grant P30DK045735. This publication was also made possible by CTSA grant UL1 TR000142 from the National Center for Advancing Translational Science (NCATS), a component of the NIH. The contents herein are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Consideration of the Evidence on Childhood Obesity for the Commission on Ending Childhood Obesity: Report of the Ad Hoc Working Group on Science and Evidence for Ending Childhood Obesity, Geneva, Switzerland (World Health Organization, 2016). [Google Scholar]
- 2.Lobstein T, Baur L & Uauy R Obesity in children and young people: a crisis in public health. Obes. Rev 5 (Suppl. 1), 4–104 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Prevalence of obesity. World Obesity Federation https://www.worldobesity.org/about/about-obesity/prevalence-of-obesity (2015).
- 4.Wang Y & Lobstein T Worldwide trends in childhood overweight and obesity. Int. J. Pediatr. Obes 1, 11–25 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Ogden CL, Carroll MD, Kit BK & Flegal KM Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 311, 806–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koplan JP & Dietz WH Caloric imbalance and public health policy. JAMA 282, 1579–1581 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Styne DM et al. Pediatric obesity—assessment, treatment, and prevention: an Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab 102, 709–757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freedman DS & Sherry B The validity of BMI as an indicator of body fatness and risk among children. Pediatrics 124 (Suppl. 1), S23–S34 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Freedman DS et al. Classification of body fatness by body mass index-for-age categories among children. Arch. Pediatr. Adolesc. Med 163, 805–811 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole TJ & Lobstein T Extended international (IOTF) body mass index cut-offs for thinness, overweight and obesity. Pediatr. Obes 7, 284–294 (2012). [DOI] [PubMed] [Google Scholar]
- 11.NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 390, 2627–2642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.National Center for Health Statistics. National Health Examination Surveys II (ages 6–11) and III (ages 12–17), and National Health and Nutrition Examination Surveys I, II and III, and 1999–2006 Centers for Disease Control and Prevention https://www.cdc.gov/nchs/nhanes/index.htm (2020).
- 13.Yanovski JA Trends in underweight and obesity—scale of the problem. Nat. Rev. Endocrinol 14, 5–6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fryar CD, Carroll MD & Ogden CL Prevalence of Overweight, Obesity, and Severe Obesity Among Children and Adolescents Aged 2–19 Years: United States, 1963–1965 Through 2015–2016 (National Center for Health Statistics, 2018); https://www.cdc.gov/nchs/data/hestat/obesity_child_15_16/obesity_child_15_16.pdf [Google Scholar]
- 15.Skinner AC & Skelton JA Prevalence and trends in obesity and severe obesity among children in the United States, 1999–2012. JAMA Pediatr. 168, 561–566 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Skinner AC, Perrin EM, Moss LA & Skelton JA Cardiometabolic risks and severity of obesity in children and young adults. N. Engl. J. Med 373, 1307–1317 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Skinner AC, Ravanbakht SN, Skelton JA, Perrin EM & Armstrong SC Prevalence of obesity and severe obesity in US children, 1999–2016. Pediatrics 141, e20173459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grossman DC et al. Screening for obesity in children and adolescents: US Preventive Services Task Force recommendation statement. JAMA 317, 2417–2426 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Jeffery RW & Utter J The changing environment and population obesity in the United States. Obes. Res 11 (Suppl.), 12S–22S (2003). [DOI] [PubMed] [Google Scholar]
- 20.Ebbeling CB, Pawlak DB & Ludwig DS Childhood obesity: public-health crisis, common sense cure. Lancet 360, 473–482 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Campbell KJ et al. Associations between the home food environment and obesity-promoting eating behaviors in adolescence. Obesity (Silver Spring) 15, 719–730 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Gluckman P, Nishtar S & Armstrong T Ending childhood obesity: a multidimensional challenge. Lancet 385, 1048–1050 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Jastreboff AM et al. Altered brain response to drinking glucose and fructose in obese adolescents. Diabetes 65, 1929–1939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jastreboff AM et al. Leptin is associated with exaggerated brain reward and emotion responses to food images in adolescent obesity. Diabetes Care 37, 3061–3068 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brook CG, Huntley RM & Slack J Influence of heredity and environment in determination of skinfold thickness in children. Br. Med. J 2, 719–721 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stunkard AJ, Foch TT & Hrubec Z A twin study of human obesity. JAMA 256, 51–54 (1986). [PubMed] [Google Scholar]
- 27.Stunkard AJ, Harris JR, Pedersen NL & McClearn GE The body-mass index of twins who have been reared apart. N. Engl. J. Med 322, 1483–1487 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y et al. Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- 29.Montague CT et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature 387, 903–908 (1997). [DOI] [PubMed] [Google Scholar]
- 30.Farooqi IS et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N. Engl. J. Med 341, 879–884 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Huszar D et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88, 131–141 (1997). [DOI] [PubMed] [Google Scholar]
- 32.Farooqi IS et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Invest 106, 271–279 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farooqi IS et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N. Engl. J. Med 348, 1085–1095 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Miraglia Del Giudice E et al. Low frequency of melanocortin-4 receptor (MC4R) mutations in a Mediterranean population with early-onset obesity. Int. J. Obes. Relat. Metab. Disord 26, 647–651 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Santoro N et al. Prevalence of pathogenetic MC4R mutations in Italian children with early onset obesity, tall stature and familial history of obesity. BMC Med. Genet 10, 25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clément K et al. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat. Med 24, 551–555 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Collet TH et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab 6, 1321–1329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frayling TM et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Locke AE et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 518, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shungin D et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature 518, 187–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pulit SL et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet 28, 166–174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felix JF et al. Genome-wide association analysis identifies three new susceptibility loci for childhood body mass index. Hum. Mol. Genet 25, 389–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradfield JP et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat. Genet 44, 526–531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao J et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity (Silver Spring) 17, 2254–2257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J et al. Role of BMI-associated loci identified in GWAS meta-analyses in the context of common childhood obesity in European Americans. Obesity (Silver Spring) 19, 2436–2439 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Bradfield JP et al. A trans-ancestral meta-analysis of genome-wide association studies reveals loci associated with childhood obesity. Hum. Mol. Genet 28, 3327–3338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khera AV et al. Polygenic prediction of weight and obesity trajectories from birth to adulthood. Cell 177, 587–596.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turcot V et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat. Genet 50, 26–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dina C et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet 39, 724–726 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Lauria F et al. Prospective analysis of the association of a common variant of FTO (rs9939609) with adiposity in children: results of the IDEFICS study. PLoS One 7, e48876 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wardle J et al. Obesity associated genetic variation in FTO is associated with diminished satiety. J. Clin. Endocrinol. Metab 93, 3640–3643 (2008). [DOI] [PubMed] [Google Scholar]
- 52.den Hoed M, Westerterp-Plantenga MS, Bouwman FG, Mariman EC & Westerterp KR Postprandial responses in hunger and satiety are associated with the rs9939609 single nucleotide polymorphism in FTO. Am. J. Clin. Nutr 90, 1426–1432 (2009). [DOI] [PubMed] [Google Scholar]
- 53.McTaggart JS et al. FTO is expressed in neurones throughout the brain and its expression is unaltered by fasting. PLoS One 6, e27968 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olszewski PK et al. Hypothalamic FTO is associated with the regulation of energy intake not feeding reward. BMC Neurosci. 10, 129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fredriksson R et al. The obesity gene, FTO, is of ancient origin, up-regulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology 149, 2062–2071 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Cowley MA et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37, 649–661 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Cecil JE, Tavendale R, Watt P, Hetherington MM & Palmer CN An obesity-associated FTO gene variant and increased energy intake in children. N. Engl. J. Med 359, 2558–2566 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Qi Q et al. Fried food consumption, genetic risk, and body mass index: gene-diet interaction analysis in three US cohort studies. BMJ 348, g1610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karra E et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J. Clin. Invest 123, 3539–3551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ranzenhofer LM et al. The FTO gene and measured food intake in 5-to 10-year-old children without obesity. Obesity (Silver Spring) 27, 1023–1029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Claussnitzer M et al. FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med 373, 895–907 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Melhorn SJ et al. FTO genotype impacts food intake and corticolimbic activation. Am. J. Clin. Nutr 107, 145–154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Angulo MA, Butler MG & Cataletto ME Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest 38, 1249–1263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burnett LC et al. Deficiency in prohormone convertase PC1 impairs prohormone processing in Prader-Willi syndrome. J. Clin. Invest 127, 293–305 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackson RS et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet 16, 303–306 (1997). [DOI] [PubMed] [Google Scholar]
- 66.Paisey RB et al. in GeneReviews (eds Adam MP et al. ) (University of Washington, 1993). [Google Scholar]
- 67.Han JC et al. Comprehensive endocrine-metabolic evaluation of patients with Alström syndrome compared with BMI-matched controls. J. Clin. Endocrinol. Metab 103, 2707–2719 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forsythe E, Kenny J, Bacchelli C & Beales PL Managing Bardet-Biedl Syndrome: now and in the future. Front Pediatr. 6, 23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sherafat-Kazemzadeh R et al. Hyperphagia among patients with Bardet-Biedl syndrome. Pediatr. Obes 8, e64–e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feuillan PP et al. Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J. Clin. Endocrinol. Metab 96, E528–E535 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daniels SR et al. Overweight in children and adolescents: pathophysiology, consequences, prevention, and treatment. Circulation 111, 1999–2012 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Morrison JA, Barton BA, Biro FM, Daniels SR & Sprecher DL Overweight, fat patterning, and cardiovascular disease risk factors in black and white boys. J. Pediatr 135, 451–457 (1999). [DOI] [PubMed] [Google Scholar]
- 73.Morrison JA, Sprecher DL, Barton BA, Waclawiw MA & Daniels SR Overweight, fat patterning, and cardiovascular disease risk factors in black and white girls: The National Heart, Lung, and Blood Institute Growth and Health Study. J. Pediatr 135, 458–464 (1999). [DOI] [PubMed] [Google Scholar]
- 74.Pinhas-Hamiel O et al. Increased incidence of non-insulin-dependent diabetes mellitus among adolescents. J. Pediatr 128, 608–615 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Weiss R et al. Obesity and the metabolic syndrome in children and adolescents. N. Engl. J. Med 350, 2362–2374 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Berenson GS et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults: the Bogalusa Heart Study. N. Engl. J. Med 338, 1650–1656 (1998). [DOI] [PubMed] [Google Scholar]
- 77.Freedman DS et al. The relation of childhood BMI to adult adiposity: the Bogalusa Heart Study. Pediatrics 115, 22–27 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Juonala M et al. Childhood adiposity, adult adiposity, and cardiovascular risk factors. N. Engl. J. Med 365, 1876–1885 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Ludwig DS Childhood obesity: the shape of things to come. N. Engl. J. Med 357, 2325–2327 (2007). [DOI] [PubMed] [Google Scholar]
- 80.Wajchenberg BL Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr. Rev 21, 697–738 (2000). [DOI] [PubMed] [Google Scholar]
- 81.Frayn KN Adipose tissue as a buffer for daily lipid flux. Diabetologia 45, 1201–1210 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Smith SR et al. Contributions of total body fat, abdominal subcutaneous adipose tissue compartments, and visceral adipose tissue to the metabolic complications of obesity. Metabolism 50, 425–435 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Taksali SE et al. High visceral and low abdominal subcutaneous fat stores in the obese adolescent: a determinant of an adverse metabolic phenotype. Diabetes 57, 367–371 (2008). [DOI] [PubMed] [Google Scholar]
- 84.Shulman GI Cellular mechanisms of insulin resistance. J. Clin. Invest 106, 171–176 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shulman GI Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N. Engl. J. Med 371, 1131–1141 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Gray SL & Vidal-Puig AJ Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr. Rev 65, S7–S12 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Virtue S & Vidal-Puig A Adipose tissue expandability, lipotoxicity and the metabolic syndrome: an allostatic perspective. Biochim. Biophys. Acta 1801, 338–349 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Kursawe R et al. Cellularity and adipogenic profile of the abdominal subcutaneous adipose tissue from obese adolescents: association with insulin resistance and hepatic steatosis. Diabetes 59, 2288–2296 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kursawe R et al. Decreased transcription of ChREBP-α/β isoforms in abdominal subcutaneous adipose tissue of obese adolescents with prediabetes or early type 2 diabetes: associations with insulin resistance and hyperglycemia. Diabetes 62, 837–844 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gillum MP et al. SirT1 regulates adipose tissue inflammation. Diabetes 60, 3235–3245 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nouws J et al. Altered in vivo lipid fluxes and cell dynamics in subcutaneous adipose tissues are associated with the unfavourable pattern of fat distribution in obese adolescent girls. Diabetes 68, 1168–1177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petersen MC & Shulman GI Mechanisms of insulin action and insulin resistance. Physiol. Rev 98, 2133–2223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reaven G Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation 106, 286–288 (2002). [DOI] [PubMed] [Google Scholar]
- 94.Roden M et al. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest 97, 2859–2865 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weiss R et al. Prediabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet 362, 951–957 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perry RJ et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 160, 745–758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagarajan A et al. MARCH1 regulates insulin sensitivity by controlling cell surface insulin receptor levels. Nat. Commun 7, 12639 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cali AM et al. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology 49, 1896–1903 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hershkop K et al. Adipose insulin resistance in obese adolescents across the spectrum of glucose tolerance. J. Clin. Endocrinol. Metab 101, 2423–2431 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Weiss R et al. Degree of obesity and glucose allostasis are major effectors of glucose tolerance dynamics in obese youth. Diabetes Care 30, 1845–1850 (2007). [DOI] [PubMed] [Google Scholar]
- 101.Giannini C et al. Evidence for early defects in insulin sensitivity and secretion before the onset of glucose dysregulation in obese youths: a longitudinal study. Diabetes 61, 606–614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cali AM et al. Intrahepatic fat accumulation and alterations in lipoprotein composition in obese adolescents: a perfect proatherogenic state. Diabetes Care 30, 3093–3098 (2007). [DOI] [PubMed] [Google Scholar]
- 103.Caprio S, Perry R & Kursawe R Adolescent obesity and insulin resistance: roles of ectopic fat accumulation and adipose inflammation. Gastroenterology 152, 1638–1646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Winer JC et al. Adiponectin in childhood and adolescent obesity and its association with inflammatory markers and components of the metabolic syndrome. J. Clin. Endocrinol. Metab 91, 4415–4423 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Brady TM The role of obesity in the development of left ventricular hypertrophy among children and adolescents. Curr. Hypertens. Rep 18, 3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reinehr T, Kiess W, de Sousa G, Stoffel-Wagner B & Wunsch R Intima media thickness in childhood obesity: relations to inflammatory marker, glucose metabolism, and blood pressure. Metabolism 55, 113–118 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Yajnik CS et al. Higher glucose, insulin and insulin resistance (HOMA-IR) in childhood predict adverse cardiovascular risk in early adulthood: the Pune Children’s Study. Diabetologia 58, 1626–1636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Angulo P Nonalcoholic fatty liver disease. N. Engl. J. Med 346, 1221–1231 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Schwimmer JB et al. Prevalence of fatty liver in children and adolescents. Pediatrics 118, 1388–1393 (2006). [DOI] [PubMed] [Google Scholar]
- 110.Tricò D et al. Metabolic features of non-alcoholic fatty liver (NAFL) in obese adolescents: findings from a multi-ethnic cohort. Hepatology 68, 1376–1390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feldstein AE et al. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 58, 1538–1544 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.D’Adamo E et al. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 33, 1817–1822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Newton KP et al. Prevalence of prediabetes and type 2 diabetes in children with non-alcoholic fatty liver disease. JAMA Pediatr. 170, e161971 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schwimmer JB, Pardee PE, Lavine JE, Blumkin AK & Cook S Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation 118, 277–283 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wagenknecht LE et al. Correlates and heritability of nonalcoholic fatty liver disease in a minority cohort. Obesity (Silver Spring) 17, 1240–1246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwimmer JB et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 136, 1585–1592 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Loomba R et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 149, 1784–1793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Romeo S et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet 40, 1461–1465 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitsche MA, Hobbs HH & Cohen JC Patatin-like phospholipase domain–containing protein 3 promotes transfer of essential fatty acids from triglycerides to phospholipids in hepatic lipid droplets. J. Biol. Chem 293, 6958–6968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian C, Stokowski RP, Kershenobich D, Ballinger DG & Hinds DA Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet 42, 21–23 (2010). [DOI] [PubMed] [Google Scholar]
- 121.Núñez-Torres R et al. The PNPLA3 genetic variant rs738409 influences the progression to cirrhosis in HIV/hepatitis C virus coinfected patients. PLoS One 11, e0168265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Santoro N et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 55, 781–789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Goffredo M et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: a multiethnic study. Hepatology 63, 117–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Santoro N et al. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J. Clin. Endocrinol. Metab 100, E1125–E1132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Stender S et al. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat. Genet 49, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Freedman DS, Mei Z, Srinivasan SR, Berenson GS & Dietz WH Cardiovascular risk factors and excess adiposity among overweight children and adolescents: the Bogalusa Heart Study. J. Pediatr 150, 12–17.e2 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Zabarsky G et al. Impact of severe obesity on cardiovascular risk factors in youth. J. Pediatr 192, 105–114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Al-Khudairy L et al. Diet, physical activity and behavioural interventions for the treatment of overweight or obese adolescents aged 12 to 17 years. Cochrane Database Syst. Rev 6, CD012691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rajjo T et al. The association of weight loss and cardiometabolic outcomes in obese children: systematic review and meta-regression. J. Clin. Endocrinol. Metab 102, 758–762 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Fonvig CE et al. Multidisciplinary care of obese children and adolescents for one year reduces ectopic fat content in liver and skeletal muscle. BMC Pediatr. 15, 196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kloppenborg JT et al. The effect of impaired glucose metabolism on weight loss in multidisciplinary childhood obesity treatment. Pediatr. Diabetes 19, 366–374 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Farpour-Lambert NJ et al. Physical activity reduces systemic blood pressure and improves early markers of atherosclerosis in pre-pubertal obese children. J. Am. Coll. Cardiol 54, 2396–2406 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Reinehr T et al. Which amount of BMI-SDS reduction is necessary to improve cardiovascular risk factors in overweight children? J. Clin. Endocrinol. Metab 101, 3171–3179 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Reinehr T, Kleber M & Toschke AM Lifestyle intervention in obese children is associated with a decrease of the metabolic syndrome prevalence. Atherosclerosis 207, 174–180 (2009). [DOI] [PubMed] [Google Scholar]
- 135.Savoye M et al. Long-term results of an obesity program in an ethnically diverse pediatric population. Pediatrics 127, 402–410 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Adam TC et al. Insulin sensitivity as an independent predictor of fat mass gain in Hispanic adolescents. Diabetes Care 32, 2114–2115 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Weiss R et al. Predictors of changes in glucose tolerance status in obese youth. Diabetes Care 28, 902–909 (2005). [DOI] [PubMed] [Google Scholar]
- 138.van der Baan-Slootweg O et al. Inpatient treatment of children and adolescents with severe obesity in the Netherlands: a randomized clinical trial. JAMA Pediatr. 168, 807–814 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Zeller M et al. Predictors of attrition from a pediatric weight management program. J. Pediatr 144, 466–470 (2004). [DOI] [PubMed] [Google Scholar]
- 140.Hampl S, Paves H, Laubscher K & Eneli I Patient engagement and attrition in pediatric obesity clinics and programs: results and recommendations. Pediatrics 128 (Suppl. 2), S59–S64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Guerciolini R Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord 21 (Suppl. 3), S12–S23 (1997). [PubMed] [Google Scholar]
- 142.Chanoine JP, Hampl S, Jensen C, Boldrin M & Hauptman J Effect of orlistat on weight and body composition in obese adolescents: a randomized controlled trial. JAMA 293, 2873–2883 (2005). [DOI] [PubMed] [Google Scholar]
- 143.Klein DJ et al. Liraglutide’s safety, tolerability, pharmacokinetics, and pharmacodynamics in pediatric type 2 diabetes: a randomized, double-blind, placebo-controlled trial. Diabetes Technol. Ther 16, 679–687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mastrandrea LD et al. Liraglutide effects in a paediatric (7–11 y) population with obesity: a randomized, double-blind, placebo-controlled, short-term trial to assess safety, tolerability, pharmacokinetics, and pharmacodynamics. Pediatr. Obes 14, e12495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Secher A et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J. Clin. Invest 124, 4473–4488 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kühnen P et al. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N. Engl. J. Med 375, 240–246 (2016). [DOI] [PubMed] [Google Scholar]
- 147.Cali AM et al. Rosiglitazone improves glucose metabolism in obese adolescents with impaired glucose tolerance: a pilot study. Obesity (Silver Spring) 19, 94–99 (2011). [DOI] [PubMed] [Google Scholar]
- 148.Pratt JSA et al. ASMBS pediatric metabolic and bariatric surgery guidelines, 2018. Surg. Obes. Relat. Dis 14, 882–890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Inge TH et al. Weight loss and health status 3 years after bariatric surgery in adolescents. N. Engl. J. Med 374, 113–123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Inge TH et al. Five-year outcomes of gastric bypass in adolescents as compared with adults. N. Engl. J. Med 380, 2136–2145 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Olbers T et al. Laparoscopic Roux-en-Y gastric bypass in adolescents with severe obesity (AMOS): a prospective, 5-year, Swedish nationwide study. Lancet Diabetes Endocrinol. 5, 174–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dewberry LC et al. Change in gastrointestinal symptoms over the first 5 years after bariatric surgery in a multicenter cohort of adolescents. J. Pediatr. Surg 54, 1220–1225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zeller MH et al. Severe obesity and comorbid condition impact on the weight-related quality of life of the adolescent patient. J. Pediatr 166, 651–659.e4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ells LJ et al. Surgery for the treatment of obesity in children and adolescents. Cochrane Database Syst. Rev 10.1002/14651858. CD011740 (2015). [DOI] [PubMed] [Google Scholar]
- 155.Yu EW et al. Two-year changes in bone density after Roux-en-Y gastric bypass surgery. J. Clin. Endocrinol. Metab 100, 1452–1459 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lu CW et al. Fracture risk after bariatric surgery: a 12-year nationwide cohort study. Medicine (Baltimore) 94, e2087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Denzer C, Reithofer E, Wabitsch M & Widhalm K The outcome of childhood obesity management depends highly upon patient compliance. Eur. J. Pediatr 163, 99–104 (2004). [DOI] [PubMed] [Google Scholar]
- 158.Rankin J et al. Psychological consequences of childhood obesity: psychiatric comorbidity and prevention. Adolesc. Health Med. Ther 7, 125–146 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Beamish AJ & Reinehr T Should bariatric surgery be performed in adolescents? Eur. J. Endocrinol 176, D1–D15 (2017). [DOI] [PubMed] [Google Scholar]
- 160.Scheimann AO, Butler MG, Gourash L, Cuffari C & Klish W Critical analysis of bariatric procedures in Prader-Willi syndrome. J. Pediatr. Gastroenterol. Nutr 46, 80–83 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bretault M et al. Clinical review: bariatric surgery following treatment for craniopharyngioma: a systematic review and individual-level data meta-analysis. J. Clin. Endocrinol. Metab 98, 2239–2246 (2013). [DOI] [PubMed] [Google Scholar]
- 162.Rodgers A, Woodward A, Swinburn B & Dietz WH Prevalence trends tell us what did not precipitate the US obesity epidemic. Lancet Public Health 3, e162–e163 (2018). [DOI] [PubMed] [Google Scholar]
- 163.Young LR & Nestle M Expanding portion sizes in the US marketplace: implications for nutrition counseling. J. Am. Diet. Assoc 103, 231–234 (2003). [DOI] [PubMed] [Google Scholar]
- 164.Bray GA, Nielsen SJ & Popkin BM Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr 79, 537–543 (2004). [DOI] [PubMed] [Google Scholar]
- 165.Population-based Approaches to Childhood Obesity Prevention (World Health Organization, 2012). [Google Scholar]
- 166.Report of the Commission on Ending Childhood Obesity (World Health Organization, 2016); https://www.who.int/end-childhood-obesity/publications/echo-report/en/. [Google Scholar]
- 167.Institute of Medicine. Early Childhood Obesity Prevention Policies (National Academy of Sciences, 2011); http://www.nationalacademies.org/hmd/Reports/2011/Early-Childhood-Obesity-Prevention-Policies.aspx [Google Scholar]
- 168.Roberto CA et al. Patchy progress on obesity prevention: emerging examples, entrenched barriers, and new thinking. Lancet 385, 2400–2409 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Swinburn BA et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet 378, 804–814 (2011). [DOI] [PubMed] [Google Scholar]
- 170.Stuckler D & Nestle M Big food, food systems, and global health. PLoS Med. 9, e1001242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ward ZJ et al. Simulation of growth trajectories of childhood obesity into adulthood. N. Engl. J. Med 377, 2145–2153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Santoro N & Caprio S Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis in obese adolescents: a looming marker of cardiac dysfunction. Hepatology 59, 372–374 (2014). [DOI] [PubMed] [Google Scholar]