

SUMMARY
Using pre-treatment gene expression, protein/phosphoprotein and clinical data from the I-SPY2 neoadjuvant platform trial (NCT01042379), we create alternative breast cancer subtypes incorporating tumor biology beyond clinical hormone-receptor (HR) and Human Epidermal Growth Factor Receptor-2 (HER2) status to better predict drug responses. We assess predictive performance of mechanism-of-action biomarkers from ~990 patients treated with 10 regimens targeting diverse biology. We explore >11 subtyping schemas and identify treatment-subtype pairs maximizing pathologic complete response (pCR) rate over the population. The best performing schemas incorporate Immune, DNA-repair and HER2/Luminal phenotypes. Subsequent treatment allocation increases overall pCR rate to 63% from 51% using HR/HER2-based treatment selection. pCR gains from reclassification and improved patient selection are highest in HR+ subsets (>15%). As new treatments are introduced, the subtyping schema determines the minimum response needed to show efficacy. This data platform provides an unprecedented resource and supports the usage of response-based subtypes to guide future treatment prioritization.
Keywords: Breast cancer, clinical trial, multiple arms, platinum, immunotherapy, response prediction, subtyping, immune, DNA repair, Luminal
Graphical Abstract
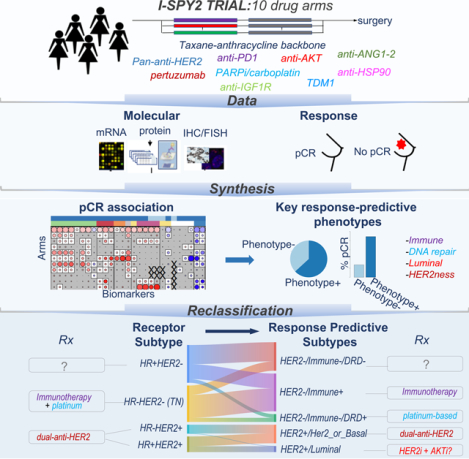
eTOC blurb
Wolf et. al. use gene expression, protein levels and response data from 10 drug-arms of the I-SPY2 neoadjuvant trial to create new breast cancer subtypes that incorporate tumor biology beyond clinical hormone-receptor (HR) and HER2 status. Use of these response predictive subtypes to guide treatment prioritization may improve patient outcomes.
INTRODUCTION
Though breast cancer treatment has improved over the past decades, over 40,000 women die annually in the US alone and worldwide, on average one in three patients will die of their disease (DeSantis et al., 2015). Patients who achieve pathologic complete response (pCR) after neoadjuvant therapy, defined by the absence of invasive disease in breast and lymph nodes, have excellent long-term outcomes (Spring et al., 2020; Yee et al., 2020). By improving pCR rates in the early disease setting, we can reduce the risk of subsequent metastatic disease and death from breast cancer. The I-SPY2 trial is an ongoing multicenter, Phase II neoadjuvant platform trial for high-risk, early-stage breast cancer designed to rapidly identify new treatments and treatment combinations with increased efficacy compared to standard-of-care (sequential weekly paclitaxel followed by doxorubicin/cyclophosphamide (T-AC) chemotherapy). In I-SPY2, multiple investigational treatment regimens are simultaneously and adaptively randomized against the shared control arm (Chien et al., 2019; Nanda et al., 2020; Park et al., 2016; Rugo et al., 2016). The primary efficacy endpoint is pCR (Yee et al., 2020).
The goal of the trial is to assess the activity of novel drugs, typically combined with weekly paclitaxel, in a priori defined biomarker subsets based on hormone receptor (HR), Human Epidermal Growth Factor Receptor-2 (HER2) expression, and MammaPrint (MP) status. Among HR+HER2− patients, only MammaPrint (MP) high cases are eligible for the trial. For all patients, tumor biology is further subdivided into high (MP1) or ultra-high (MP2) status (Chien et al., 2019; Nanda et al., 2020; Park et al., 2016; Rugo et al., 2016). An experimental arm “graduates” when it reaches ≥85% predictive probability of demonstrating superiority to control in a future 1:1 randomized 300-patient Phase III neoadjuvant trial in the most responsive subset (Chien et al., 2019; Clark et al., 2021; Nanda et al., 2020; Park et al., 2016; Rugo et al., 2016).
The value of a tumor subtyping schema is its utility in stratifying patients for efficacious treatment. It is well established that HR/HER2 subtyping is well suited for predicting response to endocrine and HER2-targeted agents (Waks and Winer, 2019). However, the landscape of targeted breast cancer therapeutics is expanding. Breast cancer treatment now includes platinum agents, PARP inhibitors, PIK3CA inhibitors, mTOR inhibitors, dual HER2-targeting regimens, and immunotherapy for specific HR/HER2-defined subtypes (Bergin and Loi, 2019; McAndrew and Finn, 2020; Wuerstlein and Harbeck, 2017). The aggregate mechanisms of action of the compendium of currently clinically available targeted therapeutics for breast cancer extends well beyond the biology that HR and HER2 expression captures. Therefore, we hypothesized that molecular subtyping categories incorporating biology beyond HR/HER2 could be created and that these categories will better inform novel agent selection for individual patients and maximize efficacy (i.e. pCR rate) over the entire treatment population.
The I-SPY2 trial and associated datasets present an opportunity to develop improved subtype classifications because of its comprehensive multi-omic molecular characterization of all tumors and the diverse array of drugs targeting different molecular pathways. As of September 2021, 1979 patients were randomized to I-SPY2, and 20 investigational agents were tested in the trial, of which 16 have completed evaluation. Experimental treatments include pan-HER2 inhibitors and anti-HER2 agents, PARP inhibitor/DNA damaging agent combinations, an AKT inhibitor, immunotherapy, and ANG1/2, IGF1R and HSP90 inhibitors added to standard of care chemotherapy. This manuscript includes analyses across 10 arms of I-SPY2: the first 9 experimental arms that completed evaluation and the control arm.
Within the I-SPY2 biomarker program, there are two primary biomarker platforms assayed at the pretreatment time-point – gene expression arrays and reverse phase protein arrays (RPPA). In the case of RPPA, upfront enrichment and purification of tumor epithelium, stromal, and intra-tumoral immune cell compartments via laser capture microdissection (LCM) is performed prior to separately assaying each population. Biomarkers are classified as standard, qualifying, or exploratory. Standard biomarkers are routinely used, US Food and Drug Administration cleared or approved, or have investigational device exemption (IDE) status (i.e. HR, HER2, MammaPrint, MRI functional tumor volume) and employed for clinical decision making. Qualifying biomarkers are pre-specified for analysis based on existing evidence suggesting a role in treatment response prediction and are tested in a CLIA setting; they may vary from drug to drug and are tested prospectively for their specific response-predictive value using a pre-specified statistical framework (Wolf et al., 2017, 2020a; Wulfkuhle et al., 2018). Exploratory biomarkers are hypothesis-generating and include discovery efforts using clinical data to identify predictive biomarkers (Sayaman et al., 2020).
In this paper, we summarize and further explore qualifying biomarker results across 10 arms of I-SPY2, combining information from standard and qualifying biomarkers to create biological treatment response-predicting subtypes (RPS) that represent better matches for our tested drugs than the standard HR/HER2-based subtypes (i.e., maximize pCR rate for a given drug, or class of agent, in a given subtype). We propose a RPS classification schema that will be prospectively used in the next phase of the I-SPY platform (I-SPY2.2). This manuscript is accompanied by the public release of the ISPY2–990 mRNA/RPPA Data Resource that includes gene expression and protein/phosphoprotein data for ~990 breast cancer patients, along with clinical annotation including treatment arm and response.
RESULTS
The I-SPY2–990 mRNA/RPPA Data Resource: patients and data
987 patients from 10 arms of I-SPY2 [210 Control (Ctr); 71 veliparib/carboplatin (VC); 114 neratinib (N); 93 MK2206; 106 ganitumab; 93 ganetespib; 134 trebananib; 52 TDM1/pertuzumab(P); 44 pertuzumab; 69 pembrolizumab (Pembro)] were included in this analysis (Figure 1a and 1b). 38% of tumors were HR+HER2−, 37% HR-HER2− (triple negative: TN), and 25% HER2+ (9% HR− and 16% HR+). Overall, 49% were classified MP2 class, and 51% MP1 class. 6 of these arms graduated within one or more receptor subtypes (purple bars) and 3 reached maximum accrual without graduation.
Figure 1. Trial design and data.
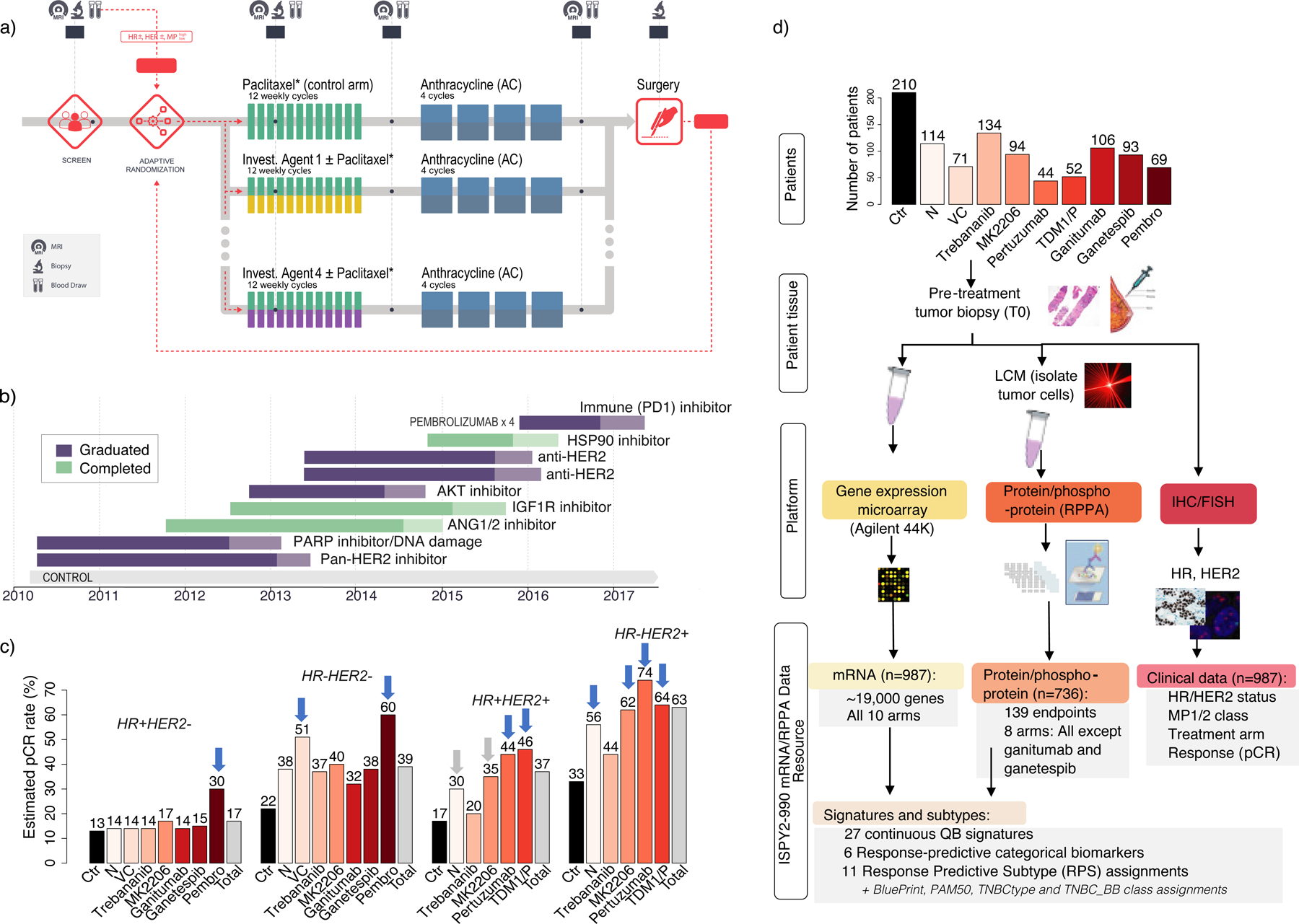
a) I-SPY2 trial schematic, b) Timeline of I-SPY2 investigational regimens, c) pCR rate across arms by receptor subtype (blue arrows=graduated; grey arrows=graduated in all HER2+, d) ISPY2–990 mRNA/RPPA Data Resource consort.
Estimated pCR rates by HR/HER2 receptor subtype for the 10 arms of the trial considered herein were previously reported and are summarized in Figure 1c (Chien et al., 2019; Clark et al., 2021; Nanda et al., 2020; Park et al., 2016; Pusztai et al., 2021; Rugo et al., 2016). Even in the highest-efficacy treatment arms, 70% of HR+HER2−, 40% of TN, 54% of HR+HER2+, and 26% of HR-HER2+ patients did not achieve pCR, further motivating the need for better biomarkers and subtyping schemas.
The I-SPY2–990 Data Resource contains gene expression, protein/phosphoprotein and clinical data for the patients included in this analysis (Figure 1d). All patients have pretreatment full transcriptome expression data on over ~19,000 genes assayed on Agilent 44K. 736 patients (all arms except ganitumab and ganetespib) have normalized LCM-RPPA data for 139 key signaling proteins/phosphoproteins in cancer (See STAR Methods). Clinical data includes HR, HER2 and MP status, response (pCR or no pCR), and treatment arm. The ISPY2–990 Data Resource is publicly available in NCBI’s Gene Expression Omnibus (GEO) (SuperSeries GSE196096, composed of SubSeries GSE194040 (mRNA) and GSE196093 (RPPA)) and through the I-SPY2 Google Cloud repository (http://www.ispytrials.org/results/data).
Predictive I-SPY2 ‘qualifying’ biomarkers across 10 arms of I-SPY2
Twenty-seven mechanism-of-action based gene expression signatures and proteins/phosphoproteins constituting our successful qualifying biomarkers reflect DNA repair deficiency (DRD; n=2), immune activation (n=8), estrogen receptor (ER) signaling (n=2), HER2 signaling (n=4), proliferation (n=3), (phospho) activation of AKT and mTOR (n=3), and ANG/TIE2 (n=1) pathways, among others (Table S1). Each pre-specified qualifying biomarker was originally found to predict response in a specific arm in one or more standard receptor subtypes, as previously reported (Lee et al., 2018; Wolf et al., 2018, 2017, 2020b, 2020a; Wulfkuhle et al., 2018; Yau et al., 2019). Table S1 also describes a newly developed VC-response biomarker for the TN subset (VCpred_TN) reflecting both DNA repair deficiency and Immune activation that was validated in BrighTNess (Loibl et al., 2018) and achieved qualifying status. In this analysis, we assessed whether they also predict response to different drugs included in other arms, with the goal of gaining biologic insight into which patients responded to what treatment and by what mechanism.
Figure 2 shows the unsupervised clustered heatmap of qualifying biomarker expression levels (Table S2). Biomarkers correlate by biologic pathway (Figure 2, side dendrogram). Although patient profiles largely cluster by receptor subtype (Figure 2), there is mixing between groups, highlighting the fact that for these patients, biological pathways other than HR/HER2 signaling are a stronger common denominator. Moreover, HR/HER2 sub-clusters appear to be characterized by immune-high (Figure 2; C4, C6, C7, top dendrogram) and immune-low (Figure 2; C1–3 and C5) signaling, though immune-high proportions differ by subtype (TN: 58%; HER2+: 41%; and HR+HER2−: 19%). Variability in ER/PGR, proliferation, and ECM signatures is visible as well.
Figure 2. Clustered heatmap of mechanism-of-action ‘qualifying’ biomarkers across 10 arms.
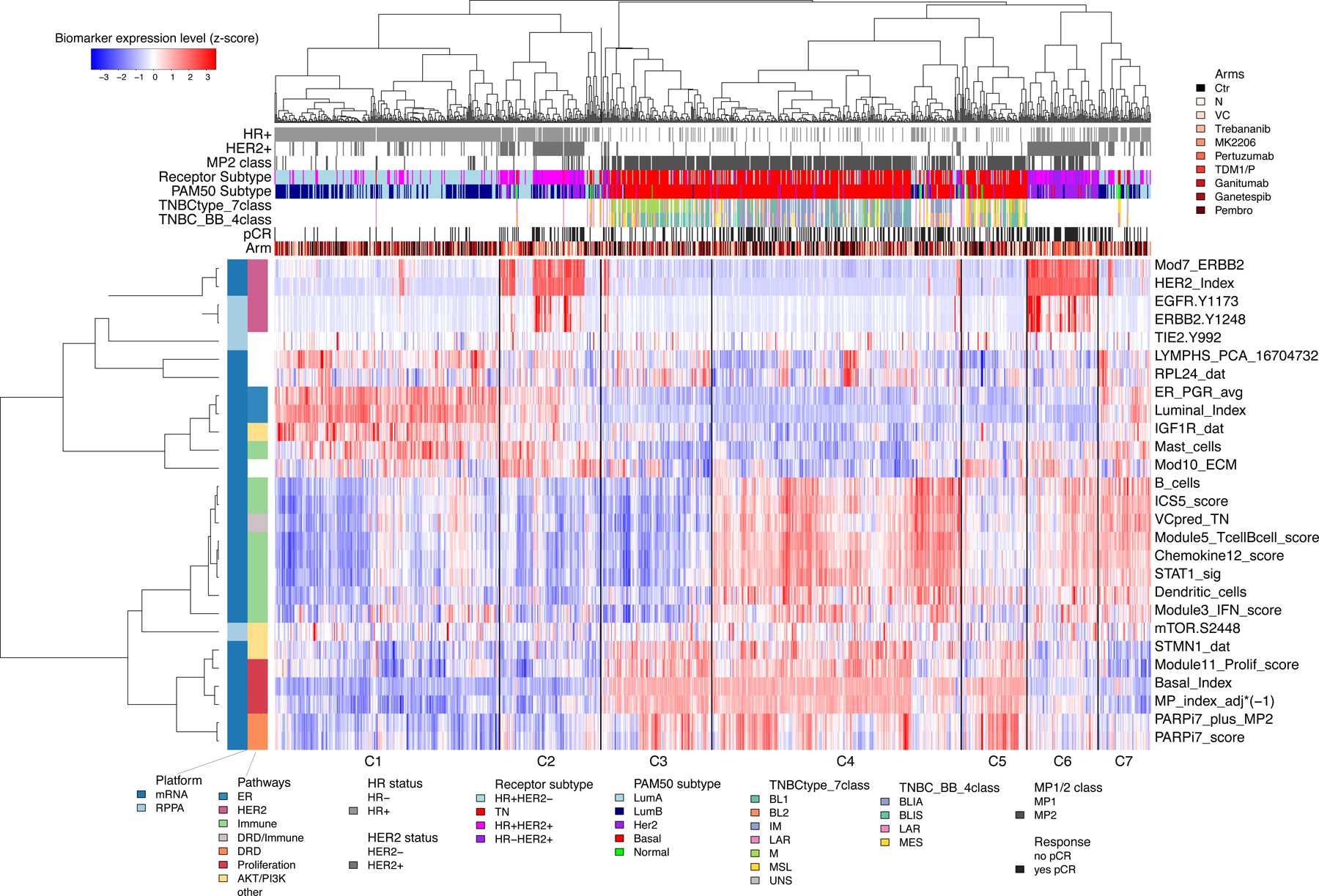
Unsupervised clustering of mechanism-of-action biomarkers (rows) and 987 patient samples (columns), with biomarkers annotated by platform and pathway; and samples annotated by HR/HER2, MP1/2 class, response, receptor subtype, PAM50, TN subtypes (7- and 4-classes), and arm. See also Table S1 and S2.
We used logistic regression to test the association of these 27 biomarkers with pCR in all 10 arms individually, in the population as a whole (adjusting for HR, HER2 and treatment arm), and within receptor subtypes (Figure 3 and Table S3). None of the 27 mechanism-of-action based biomarkers were associated with response exclusively in the arm where they were first proposed, indicating broader predictive function than anticipated.
Figure 3. pCR association analysis of continuous mechanism-of-action biomarkers across 10 arms.
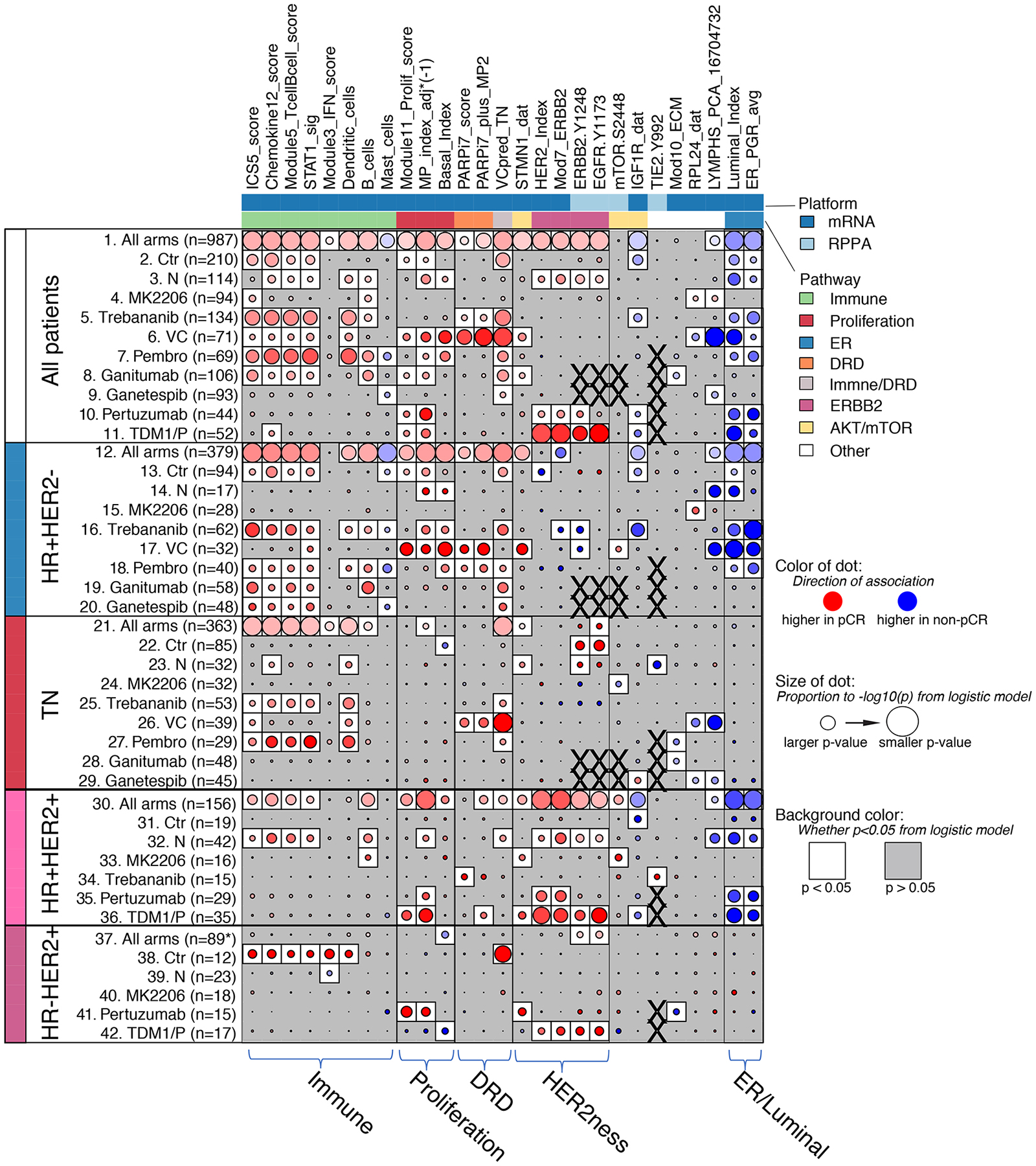
Dot-plot showing the level and direction of association between each signature (column) and pCR as labeled (rows): All patients (rows 1–11), HR+HER2− (rows 12–20), TN (rows 21–29), HR+HER2+ (rows 30–36) and HR-HER2+ (rows 37–42). Row labels denote treatment arm. Red/blue dot indicates higher/lower levels associate with pCR; darker intensity reflects larger effect size; size of dot reflects strength of association (1/p); white background indicates LR p<0.05; X denotes missing data. See also Table S3 and Figure S1.
The biomarkers with broadest predictive function across drug classes were from immune, proliferation and ER/luminal pathways (Figure 3 and Figure S1a). One or more immune signatures predicted response in 9 of the 10 arms in the overall population (Figure 3; rows 1–11, leftmost biomarker group-immune). However, different immune biomarkers were most predictive depending on receptor subtype and drug/drug class. For example, in the HER2+ subset, the B-cell gene signature predicts response to MK2206, neratinib and control chemotherapy, but was less predictive agents in the other arms (Figure 3, rows 30–42; and Figure S1b). In the TN subtype, the most predictive immune biomarkers are dendritic cells and STAT1_sig/chemokine12 gene signatures for pembrolizumab and the ANG1/2 inhibitor trebananib that affects macrophages and angiogenesis (Figure 3; rows 21–29). All immune biomarkers were higher in pCR than non-pCR cases. The exception to the rule was the mast cell signature, which was higher in cases with residual disease (RD) in the HR+HER2− subtype, mainly due to its negative association with pCR in the pembrolizumab arm.
Proliferation biomarkers (i.e., adjusted MP index and basal index (continuous scores), and module11 proliferation score) were also broadly predictive of higher pCR overall (in 7 of 10 arms; Figure 3 – rows 1–11, second biomarker group from left-proliferation) and also in HR+HER2− (5/8 arms) and HR+HER2+ (3/6 arms) subtypes (Figure 3; rows 12–20 and 30–36), but generally not in TN or HR-HER2+ cancers (Figure 3; rows 21–29 and 37–42).
Luminal/ER biomarkers (i.e. BluePrint_Luminal index, ER signature) predicted resistance to multiple therapies in the HR+HER2− subtype (5/8 arms: Pembro, Ctr, N, trebananib, and VC; Figure 3, rows 12–20, rightmost biomarker group-’ER/Luminal’). In HR+HER2+ and HER2+ subtypes they also associate with non-response in the HER2-only-targeted arms (control [trastuzumab+paclitaxel], N, THP and TDM1/P), but not in arms with agents that targeted other pathways (MK2206 or trebananib) added to trastuzumab (Figure 3, rows 30–36; Figure S1b). We also confirmed that HER2 biomarkers (i.e. HER2-EGFR co-activation, HER2index and Mod7_ERBB2 gene signatures) were predictive of pCR in multiple HER2-targeted arms (Figure 3, fourth biomarker group from the left-’HER2ness’). In the HR-HER2+ subtype, the BP-luminal and Her2ness did not generally predict response, other than HER2ness in TDM1/P (Figure 3, rows 37–42).
In different HR/HER2 subsets we also observed that the most specific biomarker (e.g., pMTOR for MK2206) may not be the most predictive (e.g. immune signals in the HER2+ subset in MK2206), and that phosphoproteins (e.g., pTIE2, pMTOR, pEGFR) may have greater predictive specificity than expression-based biomarkers (Figure 3). Moreover, it appeared that different biology may predict response to the same drugs in different receptor subtypes (e.g., trebananib: immune high in TN vs. pTIE2 in HER2+ (Figure 3 and (Wolf et al., 2018)); and MK2206: lower pMTOR in TN vs. higher pMTOR in HER2+ (Figure 3 and (Wolf et al., 2020a)). The number of significant biomarkers observed also differs by arm. Response to VC had the most significantly associated signatures and MK2206 the least (43% and 7% of biomarker-subtype pairs, respectively; Figure S1c). To assess whether this difference in the number of predictive biomarkers observed between agents is specific to the qualifying biomarker set selected, we performed whole-genome (n=19,000+ genes) analysis and observed similar results (Figure S1d).
A framework for identifying a response-predictive subtyping schema for prioritizing therapies
It is clear from our qualifying biomarker evaluation that within each HR/HER2 subtype, there is additional biology that further predicts response to I-SPY2 agents (Figure 3). Candidate biological phenotypes that may add value to HR/HER2 include proliferation, DRD, immune, luminal, basal, and HER2ness (Figure S2a). Of the 11+ response-predictive subtyping schemas that we explored (Figure S2b), our preferred schema incorporates biology that discriminates response to the treatments likely to be available in the clinic, such as platinum/PARP-inhibition and/or immunotherapy for HER2− patients, and dual-HER2 inhibition for HER2+ patients.
Our stepwise approach to developing this schema was as follows: since platinum-based and immunotherapy – separately and together – are becoming the standard of care for TN breast cancer, we first examined the overlap between DRD/platinum-response and immune biomarkers as the putative drug class-specific predictors and calculated response rates to VC and Pembro in TN patients positive for one, both, or neither biomarker (Figure 4a–c; see STAR Methods for biomarker implementation strategy). In TN, 67% were classified as DRD+, and 63% as Immune+ (Figure 4a–b). We note that though most patients classified immune-enriched by Brown & Burstein (Burstein et al., 2015) and Lehmann (Chen et al., 2012; Lehmann et al., 2011) schemas are also Immune+ in our implementation, many patients outside these (small) classes are predicted immune-responsive (Immune+) as well (Figure S2c–d). Immune+ TN patients had a high pCR rate to pembrolizumab (89%; Figure 4a) and the DRD+ TN patients had a high pCR rate to VC (75%; Figure 4b). There was considerable overlap between Immune and DRD biomarker status in this subset of patients: 56% of TN are high for both biomarkers, 7% are Immune+/DRD−, 11% Immune−/DRD+, and 26% are Immune−/DRD− (Figure 4c). The Immune+/DRD+ class had a very high pCR rate with either VC or pembrolizumab (pCR rates: VC: 74%, Pembro: 92%, control chemotherapy: 21%; Figure 4c, bottom right). In contrast, the Immune+/DRD− class, had the highest pCR rate to pembrolizumab (Pembro: 80%; Figure 4c, third down- right), whereas the Immune−/DRD+ class had the highest pCR to VC (VC: 80%, Pembro: 33%, control 38%; Figure 4c, second down-right). For the 26% of Immune−/DRD− TN patients, response rates were very low in all arms (<21%; Figure 4c, top right).
Figure 4. Clinically motivated response-based biomarker-subsets.
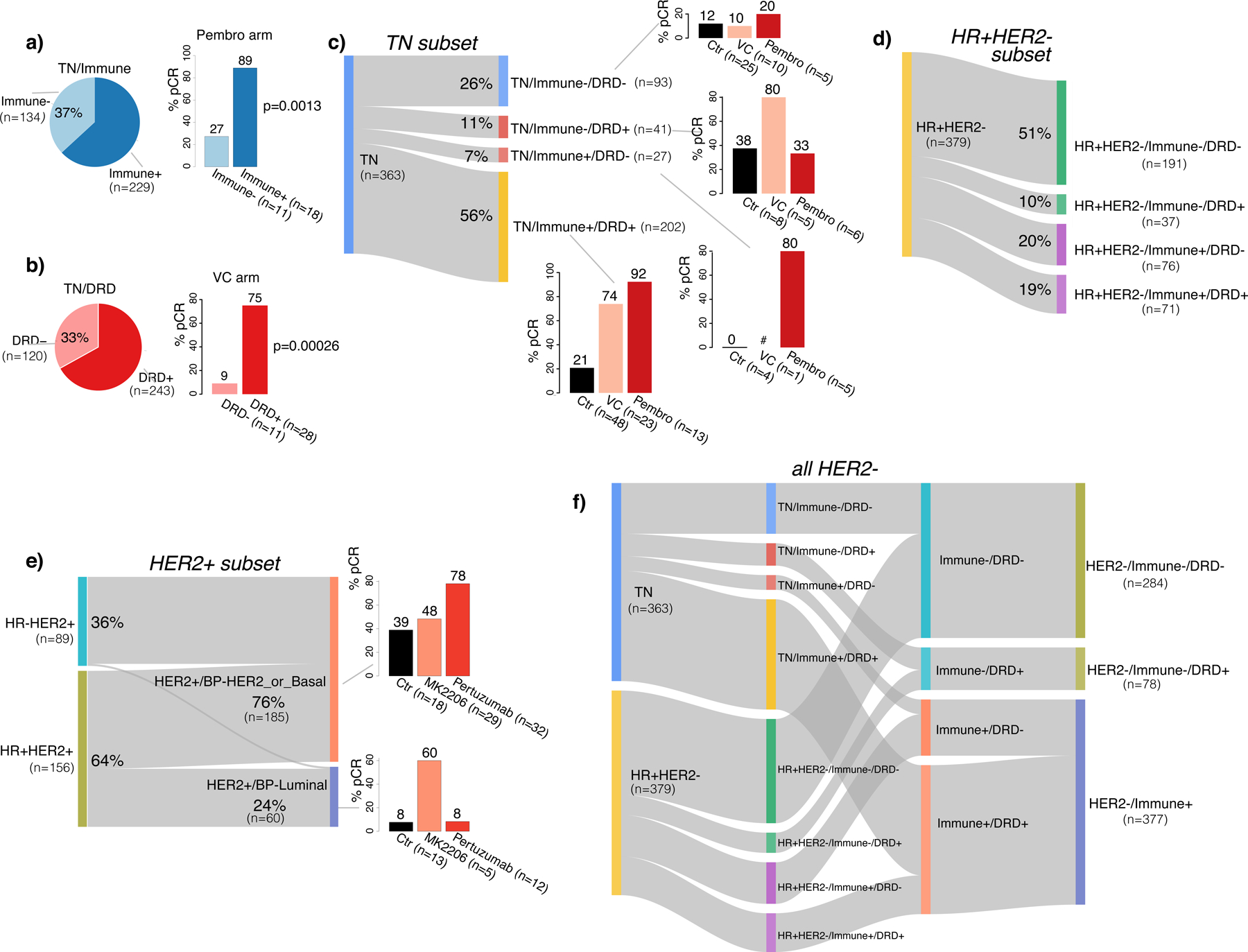
a) Overall prevalence and pCR rates in Pembro by immune subtype in TN. b) Overall prevalence and pCR rates in VC by DRD subtype in TN. p-values shown are from Fisher’s exact test. c) Sankey plot showing Immune/DRD subsets in TN, with barplots of pCR rates in VC, Pembro and control. d) Sankey plot showing Immune/DRD subsets in HR+HER2-. e) Sankey plot of HER2+/BP_Luminal and HER2+/BP_Her2_or_Basal in HER2+, with barplots of pCR rates in Ctr, TDM1/P and MK2206 arms. f) Sankey plot showing the collapse of Immune/DRD subtypes in HER2− from 8 to 3 classes. # denotes patient subset too small to be evaluable (<5). See also Figure S2.
Given that Pembro graduated in I-SPY2 for efficacy in HR+HER2− and that a DRD+ subset was found responsive to VC (Wolf et al., 2017), we applied the same strategy for HR+HER2− cancers as for TN and examined the overlap between DRD and Immune status. Nineteen percent of HR+HER2− are positive for both biomarkers, 20% are Immune+/DRD−, 10% Immune−/DRD+, and 51% are Immune−/DRD− (Figure 4d). While these proportions differ from those observed in TN, the pCR rates pattern is similar (Figure S2e–f). We note here that our example implementation of these response-predictive phenotypes is subtype specific (e.g. Dendritic-cell and STAT1/chemokine signatures define Immune+ in TN whereas B-cell and Mast-cell signatures define Immune+ in HR+HER2−; see STAR Methods).
In HER2+ cancers, motivated by the observation that high expression of the BP-Luminal index or an ER related gene signature associated with lack of pCR in the HER2-only-targeted arms (i.e., control [trastuzumab], N, THP and TDM1/P), but not in arms targeting an additional pathway (i.e., MK2206 or trebananib) (Figure 3), we defined a HER2+/Luminal phenotype and used the BluePrint subtypes to reclassify HER2+ patients by luminal signaling (Figure 4e). The HR+HER2+, triple positive, patients were assigned almost evenly into HER2+/BP-Luminal and HER2+/BP-HER2 or Basal classes, whereas nearly all HR-HER2+ cancers were HER2+/BP-HER2 or Basal, and hardly any BP-luminal. For HER2+/BP-HER2 or BP-Basal patients, the pCR rate in the pertuzumab arm is 78%, versus 48% in the MK2206 arm, and 39% in control. In the HER2+/BP-Luminal class, 60% of patients achieved pCR in the MK2206 arm versus 8% in the pertuzumab and control arms, although very few patients received MK2206 and this finding requires further validation.
Synthesis into a minimal set of response predictive subtypes: the RPS-5
Here, we combined the predictive biology described above to include all patients in one classification schema. If we added Immune, DRD, and BP-Luminal/Her2orBasal biomarkers to standard TN (Figure 4c), HR+/HER2− (Figure 4d), and HER2+ (Figure 4e) status per above, a 10-subtype schema would result. With 10 subtypes, some would include only a handful of patients and be difficult to statistically evaluate in a trial setting. Given this practical consideration, we combined all Immune+ patients in HR+HER2− and TN subsets into a single subtype HER2−/Immune+ (Figure 4f, right-bottom), as both subsets share pembrolizumab as the same best (highest pCR) agent (see Figures 4c and S2e–f). We also combined TN/Immune−/DRD+ and HR+HER2−/Immune−/DRD+ patients into the subtype HER2−/Immune−/DRD+ (Figure 4f, right-middle), as these subsets share VC as the highest-pCR arm (see Figures 4c and S2e–f). With this schema, we created the 5 subtypes that define the RPS-5 response-predictive subtyping schema (combined Figures 4f and 4e, respectively): HER2−/Immune−/DRD−, HER2−/Immune−/DRD+, HER2−/Immune+, HER2+/BP-HER2orBasal, and HER2+/BP-Luminal.
The Sankey diagram in Figure 5a shows the relationship between standard receptor subtypes and the RPS-5 subtyping schema in the I-SPY2 data. Receptor subtypes and their prevalence are shown on the left (starting with 38% HR+HER2−, 37% TN, 16% HR+HER2+, and 9% HR-HER2+) and the plot illustrates how receptor subtypes ‘flow’ into the RPS-5 subtypes on the right (stratifying into 29% HER2−/Immune−/DRD−, 38% HER2−/Immune+, 8% HER2−/Immune−/DRD+, 19% HER2+/BP-HER2orBasal, and 6% HER2+/BP-Luminal). pCR rates by drug arm within each subtype are shown in the bar plots to the left for the standard receptor subtypes and to the right for the RPS-5 subtypes.
Figure 5. Integrated treatment response-predictive subtyping 5 (RPS-5) schema combining Immune, DRD, HER2, and BP_subtype phenotypes.
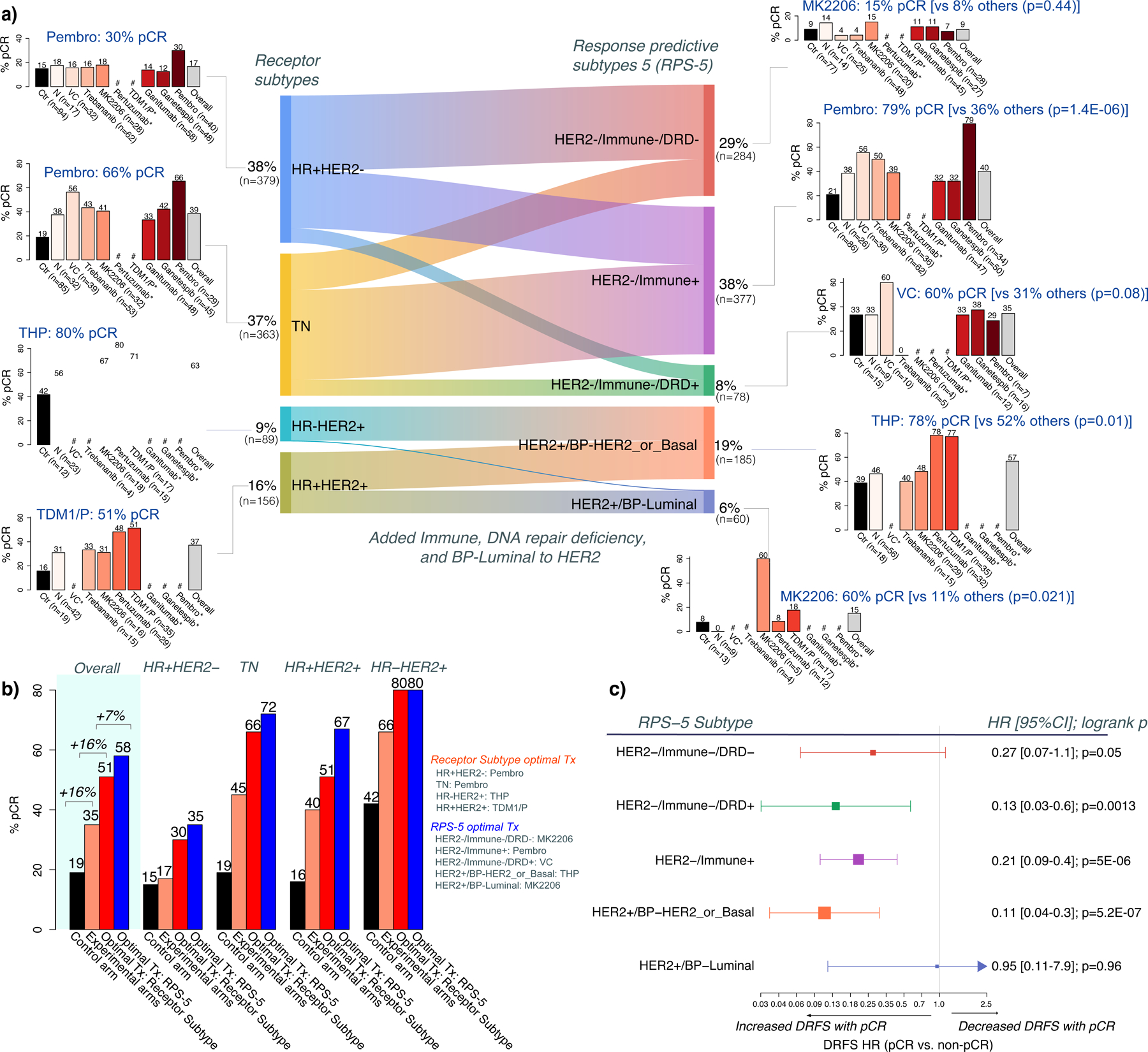
a) Sankey plot between receptor subtype and RPS-5 subtypes, with pCR rate barplots for each subtype (highest pCR rate labeled in blue). These pCR rates may differ from the reported estimated pCR in Figure 1c from Bayesian efficacy analyses. b) In silico experiment comparing pCR rates in I-SPY2’s control arm (black bar), experimental arms (orange bar); and estimated pCR rates if treatments had been ‘optimally’ assigned using receptor subtype (red bar;) or RPS-5 subtyping (blue bar). c) Hazard-ratio (HR) for Distant Recurrence-Free Survival (DRFS) for pCR versus non-pCR by RPS-5 subtype (box size=power; whiskers=95% CI). # denotes subsets with <5 patients, * denotes arm not open in subtype. p-values are from Fisher’s exact test. See also Figure S3.
Using the standard HR/HER2 receptor subtype to classify patients reveals that arms with the highest pCR rates include pembrolizumab for HR+HER2− and TN cancers with 30% and 66% pCR rates, respectively; pertuzumab for HR-HER2+ cancers with 80% pCR and TDM1/P for the HR+HER2+ subtype with 51% pCR. Using the RPS-5, the best drugs were pembrolizumab for HER2−/Immune+ with 79% pCR; VC for the HER2−/Immune-DRD+ cancers with 60% pCR; and MK2206 for HER2−/Immune−/DRD− cancers with 20% pCR though all arms performed similarly with low pCR in this subtype. In the HER2+ cancers, the best drug was pertuzumab for HER2+/BP-HER2orBasal cancers with 78% pCR; and MK2206 for HER2+/BP-Luminal cancers with 60% pCR, though numbers are small.
Impact of classification schema on trial population level pCR rates and maximization of patient benefit
A major goal of a response-predictive subtype schema is to increase the pCR rate in the population and to maximize the probability of pCR for an individual patient. To examine the impact of the RPS-5 schema, we performed an in silico experiment to calculate how the overall pCR rate would compare if treatments in the multi-arm adaptive randomization I-SPY2 trial (Figure 1A) had been assigned according to the RPS-5. The observed overall pCR rate in the standard of care control arm of I-SPY2 was 19% (black bar, Figure 5b, under “Overall”). In the 9 experimental arms of the trial taken together, the actual observed overall pCR rate was 35%, a 16% increase over the control arm (orange bar, Figure 5b). Had patients been assigned to the best experimental treatment arm (that became apparent only in hindsight) based on standard receptor subtypes, the estimated overall pCR rate in the experimental arms all together would have been 51%, a further 16% increase (red bar, Figure 5b). Finally, if we had assigned patients using the RPS-5 to their corresponding best treatment, the overall pCR rate in the combined experimental arms would be 58%, a further 7% improvement (blue bar, Figure 5b). Achieving a pCR results in excellent patient outcomes in all RPS-5 subtypes (Figure S3a). However, similar to differences observed among HR/HER2 subtypes (Spring et al., 2020; Yee et al., 2020), the relative survival benefit varies from RPS-5 subtype to subtype as well, with the highest hazard ratios observed in HER2−/Immune−/DRD+, HER2−/Immune+, and HER2+/BP-HER2_or_Basal (Figure 5c, Figure S3b).
The potential gain in pCR rate from RPS-5 reclassification was not evenly distributed across HR/HER2 subtypes. As illustrated to the right in Figure 5b, in the HR-HER2+ subtype there was no pCR increase by switching to the RPS-5 as they are all within the HER2+/HER2orBasal subtype, whereas in the HR+HER2+ receptor subtype switching to the RPS-5 could increase pCR rate by 16% (from 51% to 67%). In addition to boosting response rates over the population, a good subtyping schema should also discriminate between responders and non-responders over a wide range of treatment classes. We used bias-corrected mutual information, which quantifies the amount of uncertainty about pCR probability that is reduced by knowing subtype versus not knowing it, to compare the predictive power of different subtyping schemas. To visualize the pCR-predictive goodness of the RPS-5 schema vs. receptor subtype we plotted association p-value vs. bias-corrected mutual information for both classification schemas in each arm of the trial (Figure S3c). For most drug arms (7/10), the RPS-5 schema was more predictive of pCR than receptor subtype as can be seen by the higher concentration of points in the upper right quadrant with high BCMI and low p-values (Figure S3c).
Adapting response-predictive subtyping schemas to a rapidly evolving treatment landscape
Adding new drug classes to the trial in the future may call for incorporation of additional biomarkers and necessitate revisions to the classification schema. For example, an agent targeting HER2-low cancers, defined as HER2 IHC 2+ or 1+ and FISH-negative, is currently being evaluated in I-SPY2. If we transform HER2 status from the binary HER2+/− classes to 3 levels (HER2=0, HER2low, and HER2+) as shown in the Sankey diagram in Figure S4a, and integrate it with Immune, DRD, HR, HER2, and BP_Luminal, we arrived at a 7-subtype schema, the RPS-7, with subtypes S1: HER2+/BP-HER2orBasal, S2: HER2+/BP-Luminal, S3: HER2=0.or.low/Immune+, S4: HR-/HER2low/Immune−/DRD−, S5: HER2=0.or.low/Immune−/DRD+, S6: HER2=0/Immune−/DRD−, and S7: HR+HER2low/Immune-DRD− (Figure S4b). Agents yielding the highest pCR rates are THP [78%], MK2206 [60%], Pembro [79%], ganitumab [40%], VC [60%], N or MK2206 [20%], and MK2206 [20%] for S1–7, respectively. This schema added 11% pCR over optimal assignments using receptors only, even without a HER2 low targeted agent (pCR: 63% vs. 52%, Figure S4c).
The characteristics and relative pCR rates of RPS-5, RPS-7, and the nine other subtyping schemas defined in Figure S2b are summarized in Figure 6a–e. For example, the RPS-5 (third column from left) creates 5 classes (Figure 6a) defined by HER2, Immune, DRD, and Luminal status (Figure 6b), that if used to prioritize treatment arms by class would select Pembro, pertuzumab, MK2206, and VC (Figure 6c) and result in a pCR rate of 58% overall in the I-SPY2 population (Figure 6d), a 7% gain over the maximum possible for receptor status (Figure 6e). Similarly, the composition and performance of the RPS-7 (rightmost column) is summarized per above, including its selection of ganitumab and neratinib as the best agent within a subtype. Looking at these schemas together, we observed that different schemas select different ‘best’ treatments. Some agents were optimal for at least one subtype in nearly all schemas (e.g., Pembro and pertuzumab), while some were not selected in any schemas. Some agents are only selected when biological phenotypes in addition to HR/HER2 were incorporated (e.g. MK2206). All agents that graduated for efficacy appear as optimal in at least one schema, and two – ganetespib and ganitumab – that did not graduate for efficacy were selected as optimal in schemas incorporating the classes TN/Immune−/Basal or TN/HER2low/Immune−/DRD−, including the RPS-7, an illustration that conventional HR/HER2 subtyping may not be able to identify a responding subset. Estimated maximum pCR rates differed by subtyping schema as well, ranging from 49% to 63%, suggesting a cap of <65% pCR for the 10 treatments included in the ISPY2–990, irrespective of biomarker-based treatment assignment schema.
Figure 6. Response-predictive subtyping schema characteristics diagram for 11+ example schemas.
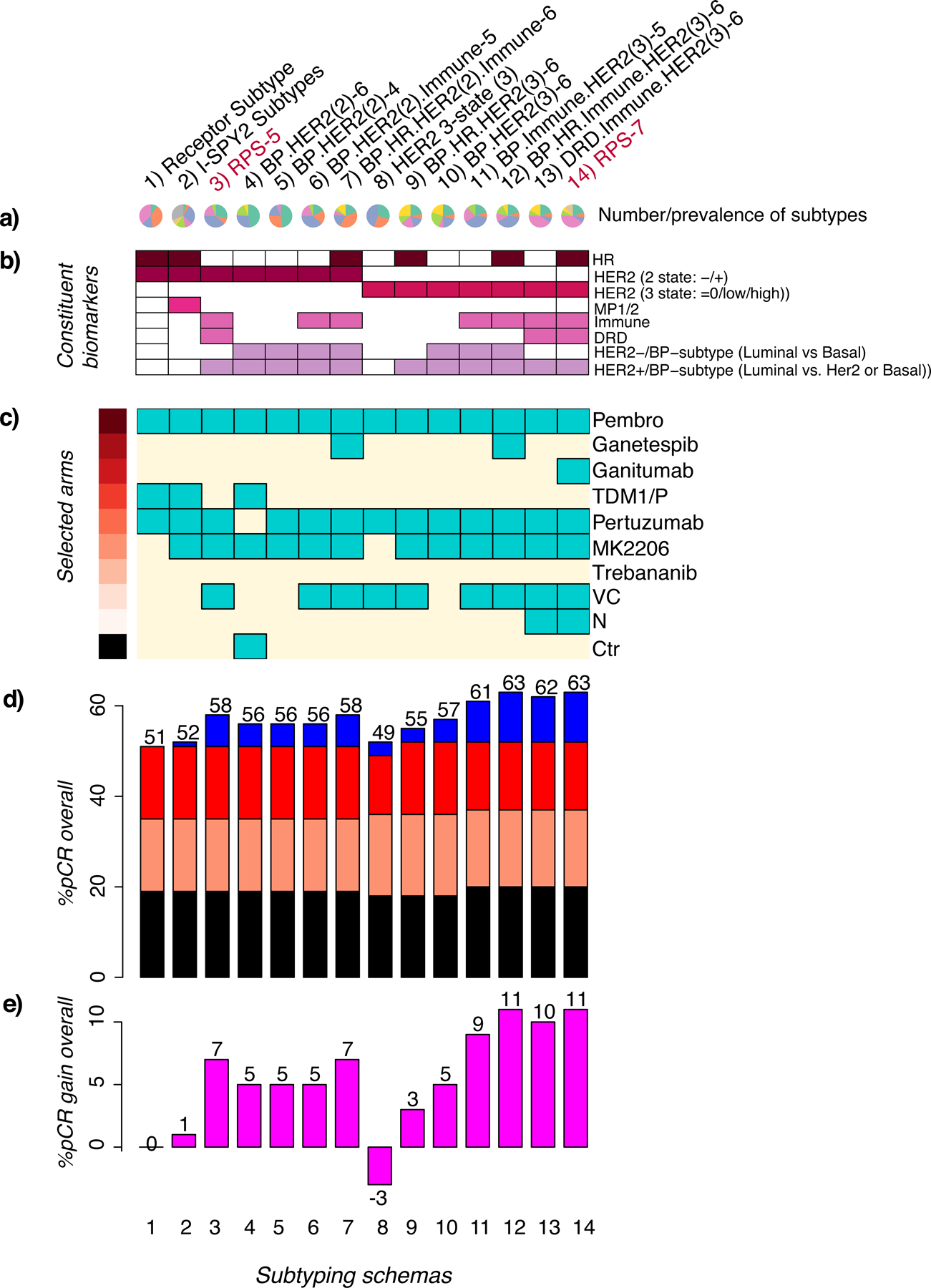
a) Pie charts showing the number (3–8) and prevalence of subtypes in each schema (column), b) Grid of constituent biomarkers (purple=present, white=absent), c) treatment arms with the highest pCR rate in one or more subtype (turquoise=selected, cream=not selected), and d) in silico experiment barplot showing pCR rates achieved in the control arm (black), experimental arms (orange); and estimated pCR rates if treatments had been optimally assigned using receptor subtype (red) or by the response-predictive schema in the column (blue). e) Barplot showing gain in pCR relative to receptor subtype. See also Figure S4.
The RPS-7 and other HER2 3-state-containing schemas also illustrated that when introducing a new class of agent such as a HER2low inhibitor, the minimum required efficacy to improve pCR rates depends strongly on the biomarker-subset in which it is tested. For example, in RPS-7 HER2low patients fall into four groups (RPS-7 classes S3-S5 and S7), with pCR rates to the most efficacious agent ranging from 20% to 70% with current I-SPY2 therapies (Figure S4b). In addition, other relevant HER2low subsets may include all HER2low or HR+HER2low, among others (Figure 7a). If tested in the HR+/HER2low/Immune−/DRD− group, a HER2low agent only has to reach a pCR rate of 20% to exceed the maximum response currently attainable from any agent tested so far in the trial (Figure 7b). This subset constitutes 20% of all HER2−, and 38% of HR+HER2− patients in the I-SPY2 trial. In contrast, if the developer were to test the agent in all HER2low patients, although the prevalence was higher (~65% of HER2−), the minimum efficacy for adding value to the I-SPY2 agent arsenal was considerably higher at 44% pCR (Figure 7b).
Figure 7. Impact of subtyping schema on minimum required efficacy of new agent (HER2low example).
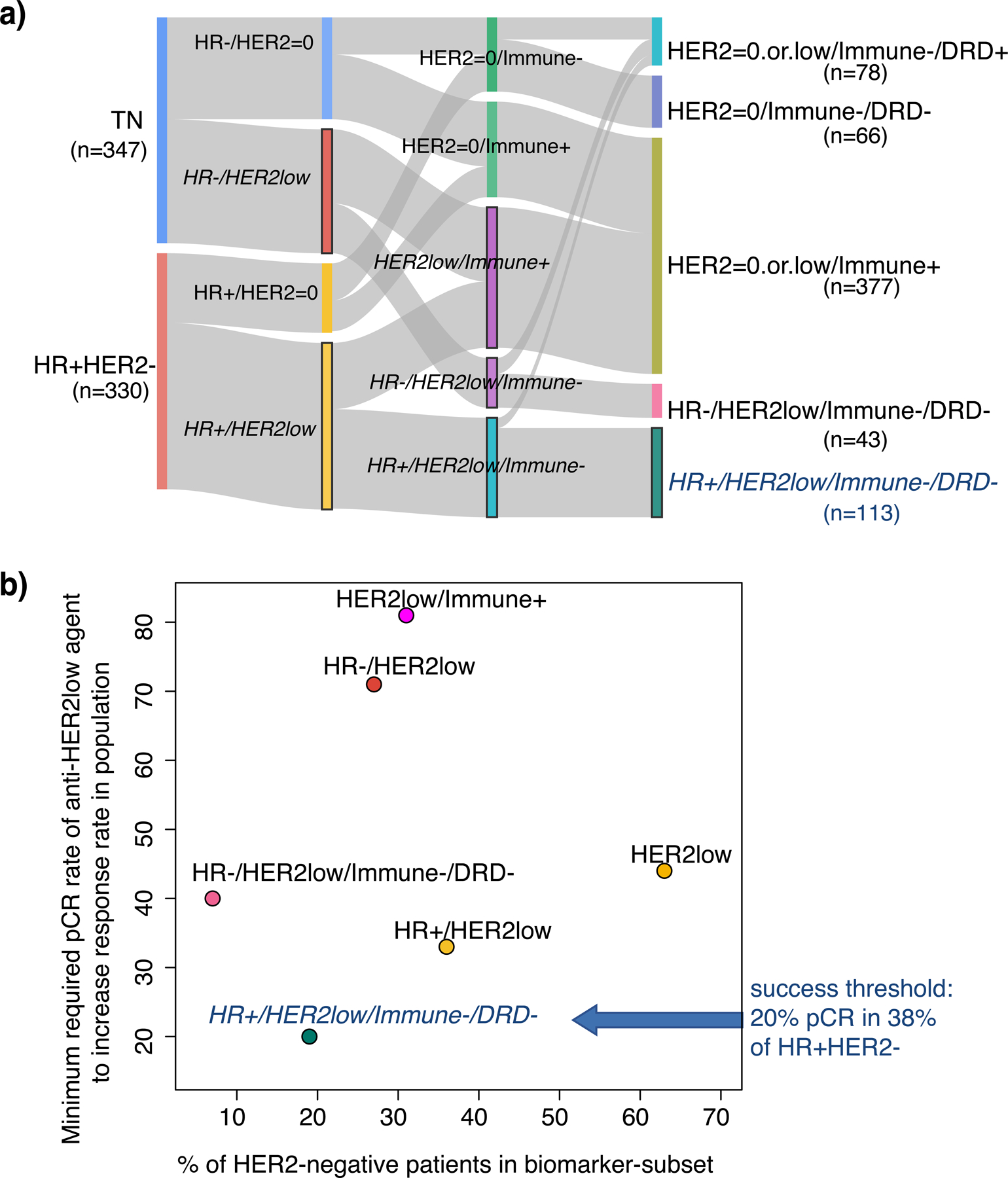
a) Sankey plot showing a variety of ways to combine HER2low status with HR and Immune/DRD. b) Scatter plot showing prevalence of HER2low subsets (x-axis) vs. the minimum pCR rate required for an anti-HER2low agent to equal that of the I-SPY2 agent with the highest response (minimum efficacy; y-axis).
DISCUSSION
With this manuscript we make public the ISPY2–990 mRNA/RPPA Data Resource, a data compendium containing pre-treatment gene expression data, tumor epithelium specific protein/phosphoprotein data and clinical/response information for ~990 breast cancer patients from the first 10 completed arms of the I-SPY2 neoadjuvant chemo-/targeted-therapy platform trial for high-risk, early-stage breast cancer. These high quality molecular data from common protocols and a centralized workflow constitute a unique resource containing patient-level response data to a wide variety of anti-cancer agents with very different mechanisms of action, including DNA damaging agents (platinum, anthracycline), PARP inhibitors, AKT inhibitors, angiogenesis inhibitors (Ang1/2; Tie2), immunotherapy (PD1), small molecule pan-HER2 inhibitors, and dual-HER2 targeting therapies.
To date, these data have been used to power our Qualifying (hypothesis testing) and Exploratory (discovery/hypothesis generating) Biomarker programs, where we have tested previously published mechanism-of-action biomarkers as predictors of response to platinum-based therapy (Wolf et al., 2017), neratinib (Wulfkuhle et al., 2018), AKT-inhibitor MK2206 (Wolf et al., 2020a), PD1 inhibitor pembrolizumab (Gonzalez-Ericsson et al., 2021), dual anti-HER2 therapies TDM1/P and pertuzumab (Clark et al., 2021; Wolf et al., 2020b) and anti-Ang1/2 therapy trebananib (Wolf et al., 2018), among others (Kim et al., 2021). In this manuscript, we extended our previous work by assessing the performance of successful biomarkers across arms and found that all examined biomarkers associated with response in at least one arm other than the one where they were proposed as predictors. Expression signatures from immune, proliferation and ER/luminal pathways are predictive of response to multiple regimens targeting diverse pathways in multiple subtypes, including HER2-targeted agents for HER2+ subtypes. In contrast, phosphoproteins from HER2, EGFR, AKT/mTOR and other pathways appear specific in predicting response to agents targeting related mechanisms of action. More generally, we found that the most specific biomarker may not be the most predictive, and that different receptor subtypes may have different predictive biomarkers to the same agents.
By viewing biomarker results in this larger 10-arm context, we here refine our understanding of who responds to which therapy and why. Responders to immunotherapy have high levels of immune signatures, but different receptor subtypes seem to have different predictive biology: high dendritic, chemokine, and STAT1 cells/signals best predict response for TN, whereas high B-cell combined with low mast cell best predict pCR in HR+HER2-. An exploratory cross-platform immune expression biomarker analysis further details immune subpopulations and their association with response (Yau et al., 2019). RPPA-based quantitative tumor epithelium MHCII levels and activation (phosphorylation) of STAT1 at pre-treatment were recently found to strongly associate with response to both pembrolizumab in I-SPY2 (Nanda et al., 2020) and durvalumab in the neo-adjuvant setting (NCT02489448)(Gonzalez-Ericsson et al., 2021). Platinum agent plus PARP inhibitor veliparib response is predicted by high DRD and STAT1-related immune signaling in TN and by both DRD and high proliferation in the HR+HER2− subset. HER2+ dual-HER2 targeted therapy responders tend to have higher HER2 signaling on expression, protein, phosphoprotein levels, with proliferation signals providing potential discrimination of response between TDM1/P and THP in the HR+HER2+ subset (Clark et al., 2021).
We then applied these insights and clinical considerations to develop response-predictive subtyping schemas that incorporate tumor biology beyond clinical HR/HER2 status that may better inform agent selection in a modern treatment landscape. Candidate ‘fit for purpose’ biological phenotypes to add to HR/HER2 included proliferation, DRD, Immune, luminal, basal, and HER2ness, selected because they predict response to newer agent classes likely to be found in the clinic today. However, when so many phenotypes are considered, there is a combinatorial explosion in the possible number of marker states, and many ways to collapse them into smaller useful response-predictive subtyping schemas. To help sort through the options, we reasoned that an ideal response-predictive subtyping schema should: 1) differentiate optimal treatments, meaning that different subtype classes should have different ‘best’ treatments yielding the highest pCR probability; 2) result in a higher pCR rate in the population if used to optimally assign/prioritize treatments; 3) differentiate between responders and non-responders over a wide range of treatments; and 4) be robust to platform and applicable across different drugs with the same mechanism of action and simple to implement clinically.
Of the 11+ potential mRNA expression-based response-predictive subtyping schemas we explored, we selected the treatment Response Predictive Subtype 5 (RPS-5) for prospective evaluation in I-SPY2. This schema was motivated by clinical considerations in TN and HER2+. Both immunotherapy and platinum-based therapy arms graduated in the TN subset in I-SPY2. These results were subsequently validated in the large randomized trials BrighTNess (Loibl et al., 2018) and KEYNOTE-522 (Schmid et al., 2020). These drugs are now increasingly used in clinical practice individually or together. We classified TN patients by Immune and DRD markers to determine whether the same, or different, populations are responding to each class of therapy and whether this information could be used to spare patients the toxicity of combined platinum-based and immunotherapy if both are not needed to achieve pCR. We applied the same stratification to HR+HER2− patients based on the efficacy of Pembro, the many immune markers associated with response in that arm and other immunotherapy arms in I-SPY2, and previous work showing that responders to VC can be identified by DRD biomarkers such as PARPi7 combined with MP2 class (Wolf et al., 2017), and also by the BluePrint(BP)-Basal subtype (Krijgsman et al., 2012). We used BP-Basal classification as our measure to assess the DRD phenotype in HR+HER2− because the assay is performed in a CLIA setting and is ready for clinical implementation with a pending IDE application submission to the US FDA, even though the research assay based PARPi7-high/MP2 performed somewhat better in this dataset. HER2+ patients were re-classified by luminal signaling to better identify subsets likely to respond to dual-anti-HER2 therapy vs. those that may need a different approach.
The resulting, simplified RPS-5 has five subtypes: HER2−/Immune−/DRD−, HER2−/Immune+, HER2−/Immune−/DRD+, HER2+/BP-HER2orBasal, and HER2+/BP-Luminal. Using this schema to maximize pCR rates, one would prioritize platinum-based therapy for HER2−/Immune−/DRD+, checkpoint inhibitor therapy for HER2−/Immune+, and dual-anti-HER2 therapy for HER2+ that are not luminal. HER2+/Luminal patients have very low response rates to dual-anti-HER2 therapy but may respond better to combination therapy including an AKT-inhibitor. HR-positivity, though very important in general for determining who should receive adjuvant endocrine therapy, is not used in this response-predictive schema, as further subdivisions based on HR-status would not impact agent prioritization. In our in silico experiment, treatment assignment based on matching HR/HER2 subsets to the most effective therapy improves trial level pCR from 19% to 51%; and assignment based on RPS-5 added a further 7% improvement to 58% pCR.
More generally, we showed that molecular subtyping categories incorporating biology outside HR/HER2 could be created and that these updated categories can better inform treatment assignment to new emerging therapies for breast cancer for individual patients and increase efficacy (i.e. pCR rate) over the entire treatment population. However, when comparing the relative contributions of improved biomarkers vs improved agents to response rate over the entire trial population, we observe that most of the pCR benefit appears to derive from the ‘right’ treatments (+30%) and an additional sizable pCR benefit comes from improved biomarker schemas (<=10–15%). With current agents, the highest pCR rate over the I-SPY2 population appears capped at ~65% in the best performing schemas incorporating Immune, Luminal and HER2-3state biomarkers. This limitation likely derives from a sizeable patient population with luminal biology who are Immune-negative and DRD-negative who did not respond to any of the treatments under study. Many of these patients are predicted endocrine responsive and may benefit from neoadjuvant endocrine therapy, an approach we are considering testing in the future.
We observe that different schemas have different sets of ‘best’ treatments, with some treatments (e.g., Pembro) chosen by all schemas, and others by a subset of schemas or not at all, although that is partially a consequence of the biological phenotypes included. As new agent classes that may help further improve response rate over the population become available, we will need to incorporate additional biological phenotypes into existing subtyping schemas that only classify cancers optimally for existing agents. Using HER2low-targeted agents as an example (an agent in this class is currently in I-SPY2), we developed a revised schema incorporating HER2 status as a 3-state variable (HER2-0, HER2-low, HER2+), and the resulting treatment Response Predictive Subtype 7 (RPS-7) classification further improved pCR rates in the overall population in our in silico experiments. This example also illustrates that the minimum efficacy required to demonstrate benefit (over best available agent) differs by biomarker subsets.
It is important to note that we make a distinction between predictive biological phenotypes like ‘Immune+’ and their implementation. For instance, in our study Immune+ is based on a variety of different subtype-specific signatures (e.g. B cell signature in HR+, STAT1/chemokine signature in TN). We acknowledge that other signatures reflecting similar biology may also be used to identify the same biological phenotype and may show similar or improved predictive performance, since biomarkers that capture the same biology are highly correlated and the underlying biological signals are robust. We acknowledge that the implementation we selected in this study would require translation to a straight-forward single-sample predictor for implementation in a clinical setting. CLIA compliant, clinically actionable versions of some of our selected biomarkers have been developed and an IDE submission is underway to enable prospective testing in the next-generation ‘I-SPY2.2’ trial. However, the idea is that as improved biomarkers are developed, the best available can be ‘swapped in’ to implement the phenotype in the clinic.
The ISPY2–990 Data Resource, and our analyses, have limitations. Though the overall resource represents an unparalleled cohort of clinically well-annotated neoadjuvant multi-arm targeted/chemo-therapy molecular data, each arm is relatively small (44–120 patients); further dividing these groups by receptor subtype or by one of the response-predictive subtyping schemas, the numbers become even smaller, and the cohort sizes are unequal. This limits the power of analysis. In addition, I-SPY2 uses adaptive randomization within HR/HER2/MP defined subtypes to enable efficient matching of novel regimens with their most responsive traditional clinical subtypes. This may result in the unbalanced prevalence of biomarker-positive subsets in experimental and control arms if a biomarker subset is correlated with a HR/HER2/MP subset that is preferentially enriched or depleted in an experimental arm by the randomization engine. For combination therapies (e.g. VC and TDM1/P) it is impossible to tease out relative contributions of each agent to response or to assess whether a biomarker is predictive of response to the individual agents within the combination. Altogether, these challenges limit our ability to draw definitive conclusions. Thus, our statistics are descriptive rather than inferential; and all individual predictors of response require further testing to assess their prediction characteristics within different treatment settings.
Another limitation to our underlying biomarker data is that while we utilized a multi-omic biomarker approach to generate multiplexed RNA-protein-phosphoprotein data as well as CLIA-based platforms, the study is limited to having only two biomarker platforms, and by the selection of the short list of continuous qualifying biomarkers as our focus. For instance, we cannot include some well-studied biomarkers, such as HRD and other DNA ‘scar’ assays for DNA repair deficiency, which requires DNA sequencing data, and we do not include exploratory whole-transcriptome or whole-RPPA array analyses, which are ongoing.
In conclusion, we expect the ISPY2–990 mRNA/RPPA Data Resource to be highly valuable to the breast cancer research and drug development community, and ultimately to patients. We found biomarkers predictive of response to a variety of agents with different mechanisms of action and proposed a framework for identifying a response-predictive subtyping schema for prioritizing therapies. Within this framework, we propose a clinically relevant breast cancer classification schema incorporating immune, DRD, and luminal-like biological phenotypes with HER2 status that may improve agent prioritization for individual patients and increase pCR rates over the population. We plan to prospectively test our response predictive subtyping schema in I-SPY2.2, an upcoming version of the I-SPY2 trial that incorporates a sequential multiple assignment randomize trial (SMART) scheme and adapts treatment within individual patients based on biology and response.
STAR * METHODS
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources or data should be directed to and will be fulfilled by Denise Wolf (Denise.Wolf@ucsf.edu)
Materials availability:
This study did not generate new unique reagents.
Data and code availability:
Transcriptomic, protein/phospho-protein and clinical data used in this study is available in NCBI’s Gene Expression Omnibus (GEO) SuperSeries GSE196096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196096) and its two SubSeries GSE194040 (mRNA: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194040) and GSE196093 (RPPA: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196093), and through the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data). Data on GEO represents the data as currently recorded in our database. Patient-level scores for the 27 qualifying biomarker scores and response data analyzed in this paper, and the RPS-5, RPS-7 and other subtype classifications and their constituent biomarkers presented herein are available in Table S2. Additional de-identified subject level data may be requested by qualified investigators. Details of the trial, data, contact information, proposal forms, and review and approval process are available at the following website: https://www.ispytrials.org/collaborate/proposal-submissions.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
I-SPY2 TRIAL Overview
I-SPY2 is an ongoing, open-label, adaptive, randomized phase II, multicenter trial of neoadjuvant therapy for early-stage breast cancer (NCT01042379; IND 105139). It is a platform trial evaluating multiple investigational arms in parallel against a common standard of care control arm. The primary endpoint is pCR (ypT0/is, ypN0), defined as the absence of invasive cancer in the breast and regional nodes at the time of surgery. As I-SPY2 is modified intent-to-treat, patients receiving any dose of study therapy are considered evaluable; those who switch to non-protocol therapy, progress, forgo surgery, or withdraw are deemed ‘non-pCR’. Secondary endpoints include residual cancer burden (RCB) and event-free and distant relapse-free survival (EFS and DRFS) (Symmans et al., 2007)
Trial Design
Assessments at screening establish eligibility and classify participants into subtypes defined by hormone receptor (HR) status, HER2, and 70-gene signature (MammaPrint®) status (Cardoso et al., 2016; Piccart et al., 2021). Adaptive randomization in I-SPY2 preferentially assigns patients to trial arms according to continuously updated Bayesian probabilities of pCR rates within each biomarker signature; 20% of patients are randomly assigned to the control arm (Berry, 2011). While accrual is ongoing, a statistical engine assesses the accumulating pathologic and MRI responses at weeks 3 and 12 and continuously re-estimates the probabilities of an experimental arm being superior to the control in each defined biomarker signature. An arm can be dropped for futility if the predicted probability of success in a future 300-patient, 1:1 randomized, phase 3 trial drops below 10%, or graduate for efficacy if the probability of success reaches 85% or greater in any biomarker signature. The clinical control arm for the efficacy analysis uses patients randomized throughout the entire trial. Experimental arms have variable sample sizes: highly effective therapies graduate with fewer patients in the experimental arm; arms that are equal to, or marginally better than, the control arm accrue slower and are stopped if they have not graduated, or terminated for lack of efficacy, before reaching a sample size of 75. During the design of each new experimental arm the investigators together with the pharmaceutical sponsor decide in which of the 10 a priori defined biomarker signatures the drug will be tested. Upon entry to the trial, participants are dichotomized into hormone receptor (HR) negative versus positive, HER2 positive versus negative, and MammaPrint High1 [MP1] versus High2 [MP2] status. From these 8 biomarker combinations (2×2×2) I-SPY has created 10 biomarker signatures that represent the disease subsets of interest (e.g. all patients, all HR+, all HER2+, HR+/HER2, etc., for complete list see reference Berry 2011) in which a drug can be tested for efficacy. Efficacy is monitored in each of these 10 biomarker signatures separately and an arm could graduate in any or all biomarker signature of interest. When graduation occurs, accrual to the arm stops, final efficacy results are updated when all pathology results are complete. The final estimated pCR results therefore may differ from the predicted pCR rate at the time of graduation. Additional details on the study design have been published elsewhere.(Park et al., 2016; Rugo et al., 2016)
Eligibility
Participants eligible for I-SPY2 are women >18 years of age with stage II or III breast cancer with a minimum tumor size of >2·5 cm by clinical exam, or >2·0 cm by imaging, and Eastern Cooperative Oncology Group performance status of 0 or 1 (Oken et al., 1982). HR-positive/HER2-negative cancers assessed as low risk by the 70-gene MammaPrint test are ineligible as they receive little benefit from systemic chemotherapy.
Treatment
This correlative study involved 987 women with high-risk stage II and III early breast cancer who were enrolled in 10 arms of I-SPY2: the first 9 experimental arms that completed evaluation and the control arm as shown in the schema of Fig 1A. During this same period (2010–2017), one arm was stopped due to toxicity with few patients enrolled and is not included in this evaluation. All patients received at least standard chemotherapy (paclitaxel alone followed by doxorubicin/cyclophosphamide (T->AC; or with trastuzumab (H) in HER2+, T+H->AC)) or in combination (taxane phase) with investigational agents: veliparib/carboplatin (VC; HER2− only: VC -> AC); neratinib (N; All patients: T+ N->AC); MK2206 (HER2−: T+MK2206->AC; HER2+: T+H+MK2206->AC); ganitumab (HER2− only: T+ganitumab->AC); ganetespib (HER2− only: T+ganetespib->AC); trebananib (HER2−: T+trebananib->AC; HER2+: T+H+trebananib->AC); TDM1/pertuzumab (P) (HER2+: TDM1/P->AC); pertuzumab (HER2+: T+H+pertuzumab->AC); and pembrolizumab (Pembro; HER2−: T+Pembro->AC). For HER2+ patients, N was administered instead of H, whereas MK2206 and trebananib were administered in addition to H. Dose reductions and toxicity management were specified in the protocol. Adverse events were collected according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. After completion of AC, patients underwent lumpectomy or mastectomy and nodal sampling, with choice of surgery at the discretion of the treating surgeon. Detailed descriptions of the design, eligibility, and efficacy of these 9 experimental arms of the I-SPY2 trial have been reported previously (Chien et al., 2019; Clark et al., 2021; Nanda et al., 2020; Park et al., 2016; Pusztai et al., 2021; Rugo et al., 2016).
Trial Oversight
I-SPY2 is conducted in accordance with the guidelines for Good Clinical Practice and the Declaration of Helsinki, with approval for the study protocol and associated amendments obtained from independent ethics committees at each site. Written, informed consent was obtained from each participant prior to screening and again prior to treatment. The I-SPY2 Data Safety Monitoring Board meets monthly to review patient safety.
METHOD DETAILS
Pretreatment Biopsy Processing and molecular profiling
Core needle biopsies of 16-gauge were taken from the primary breast tumor before treatment. Collected tissue samples are immediately frozen in Tissue-Tek® O.C.T.™ embedding media and then stored in −80°C until further processing. An 8μM section is stained with hematoxylin and eosin (H&E) and pathologic evaluation performed to confirm the tissue contains at least 30% tumor. A tissue sample meeting the 30% tumor requirement is further cryosectioned at 30 μM. Twenty to thirty sections are collected and emulsified in 0.5ml Qiazol solution and the tubes are sent on dry ice to Agendia, Inc., for RNA extraction and gene expression profiling on Agilent 44K (Agilent_human_DiscoverPrint_15746 with annotation GPL30493 (update of GPL16233); n=333) or 32K (Agendia32627_DPv1.14_SCFGplus with annotation GPL20078; n=654) expression arrays. For each array, the green channel mean signal was log2-tranformed and centered within array to its 75th quantile as per the manufacturer’s data processing recommendations. All values indicated for non-conformity are NA’d out; and a fixed value of 9.5 was added to avoid negative values. Probeset level data per array were mean-collapsed to the gene level, and genes common to the two platforms identified. Expression data from the first ~900 I-SPY2 patients distributed over the two platforms GPL30493 (n=333) and GPL20078 (n=545) were combined into a single gene-level dataset after batch-adjusting using ComBat (Johnson et al., 2007). Linear adjustment factors were derived from the larger ComBat operation, per platform, which can be used to batch correct raw files. The subsequent ~90 samples, assayed on GPL20078, were batch corrected using these factors and added to the original set, yielding a normalized expression dataset comprising 987 patients x 19,134 (common) genes. These transcriptomic data and the associated batch correction model coefficients are available in NCBI’s Gene Expression Omnibus (GEO), SubSeries GSE194040 (mRNA) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194040) and through the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data).
In addition, laser capture microdissection (LCM) was performed on pre-treatment biopsy specimens to isolate tumor epithelium for signaling protein and phospho-protein profiling by reverse phase protein arrays (RPPA) in the Petricoin Lab at George Mason University, as previously published (Wulfkuhle et al., 2018). Approximately 10,000 cells are captured per sample. RPPA samples were assayed on three arrays, each containing hundreds of samples from different arms of the trial quantifying up to 140 protein/phospho-protein endpoints (GPL28470). To remove batch effects we standardized each array prior to combining, by (1) sampling 5000 times, maintaining a receptor subtype balance equal to that of the first ~1000 patients (HR+HER2−: 0.384, TN:0.368, HR+HER2+:0.158, HR-HER2+:0.09); (2) calculating the mean(mean) and mean(sd) for each RPPA endpoint; (3) z-scoring each endpoint using the calculated mean/sd from (2). The consort diagram with the number of evaluable patients for each molecular profiling analysis is shown in Figure 1B. Details of the RPPA sample preparation and data processing are as previously described (Wulfkuhle et al., 2018). These RPPA data for 736 patients (all arms except ganitumab and ganetespib) are available in NCBI’s Gene Expression Omnibus (GEO), SubSeries GSE196093 (RPPA) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196093) and through the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data).
Continuous Gene Expression Biomarkers Assessed
Twenty-six prospectively defined, mechanism-of-action and pathway-based expression and protein/phospho-protein continuous signatures assayed from pre-treatment biopsies were previously found to be predictive in a particular agent/arm in pre-specified QBE analysis. We also include an exploratory VC-response signature for the TN subset reflecting both DNA repair deficiency and Immune expression that validated in BrighTNess and therefore achieved qualifying status, for a total of 27 continuous biomarkers considered in our analysis (see Table S1 for genes/proteins included per signature and scoring method; and Table S2 for patient-level biomarker scores).
VCpred_TN derivation:
VCpred_TN is a continuous gene expression signature that associates with response to VC in the TN subset. It differs from the other biomarkers in this study in that it was originally developed on I-SPY2 data, rather than previously published and in pre-specified analysis validated (qualified) in I-SPY2. We developed this signature in 2018, when the decision was made to switch I-SPY2 tumor biopsy tissue collection from fresh frozen (FF) as assayed for the I-SPY2–990 data compendium, to FFPE, and after performing expression studies of 72 matched FF:FFPE pairs from I-SPY2 that suggested that the previous DRD biomarker implementation frontrunner, PARPi7, may not translate well. In a quest to develop a more robust DRD biomarker that might better translate from FF to FFPE and between Agilent 44K platforms (GPL16233 and GPL20078) we developed VCpred_TN by: 1) collecting a large set of DNA repair related genes (Knijnenburg et al., 2018) including those in the PARPi7, and adding to them a subset of immune genes from module4 (Wolf et al., 2014) and IR7 (Teschendorff and Caldas, 2008), ESR1, and PGR, for a total of 162 genes; 2) filtering those 162 genes for presence on both Agilent 44K array types used in this study and for correlation between FF and FFPE samples using our 72-paired sample set (pearson correlation > 0.4), which yielded an 84 gene starting set for signature development; and 3) assessing association between expression levels of each of the 84 genes and pCR in the VC arm, in the TN subset using logistic modeling, after mean-centering the expression data. The resulting signature is the sum of -sign(coeff)*log(p) for the top 25 most correlated genes in the starting set, where sign(coeff) the sign of association between a gene and pCR (positive if higher levels associate with pCR, negative if higher levels associate with non-pCR), and p = the likelihood ratio test p–value. As also appears in the above Table S1, VCpred_TN = 13.60*CXCL13 −6.48*BRCA1 + 6.41*APEX1 + 5.32*FEN1 + 4.85*CD8A − 4.84*SEM1 + 4.78*APEX2 − 4.60*RNMT + 4.51*CCR7 + 3.99*H2AFX + 3.88*POLD3 − 3.49*PRKDC + 3.48*C1QA + 3.33*CLIC5 − 3.24*RAD51 + 3.10 *DDB2 − 2.83*SPP1 − 2.80 *POLD2 − 2.80*POLB + 2.72*LIG1 −2.67*GTF2H5 – 2.63*PMS2 + 2.60*LY9 −2.34*SHPRH + 6.27*ARAF; where the expression data is mean-centered by gene over all samples prior to evaluating this weighted sum, and the final signature is z-scored to have mean=0 and sd=1.
Biological response-predictive phenotypes: overview and implementation
Here we introduce the concept of and response-predictive biological phenotype, defined by considering promising treatments (e.g. Immunotherapy, dual-HER2, and platinum-based) and basic cancer biology (e.g. proliferation). Patients are considered Immune-positive (Immune+) if their immune-tumor state is such that they are likely to respond to immunotherapy, and DNA repair deficient/platinum-responsive (DRD+) if response to a platinum agent with or without PARP-inhibition is likely. As biomarkers representing the same biology are correlated and can be subtype-specific (Figure 2), multiple immune and DRD markers can be used to implement these biological phenotypes and perform similarly. Moreover, though we need to select example implementations for response predictive phenotypes like Immune, HER2ness, Luminal, DRD, and proliferation, we do so with the expectation that as improved biomarkers come available, they can be ‘swapped in’.
In general, we prefer to use categorical biomarkers, so as to not have to select thresholds using I-SPY2 trial data. Here we use BluePrint subtype (Agendia; BP-Luminal, BP-HER2, BP-Basal) to implement HER2ness, Luminal and Basal biological phenotypes, and MP2 class as a proliferation biomarker based on high levels of correlation to cell cycle/proliferation signatures. Where necessary, we also dichotomize continuous biomarkers using a subtype-specific cross-validation procedure to optimize performance as follows:
Biomarker dichotomization:
To identify optimal (exploratory) dichotomizing thresholds for select biomarkers in a particular patient subset, a cross-validation procedure was applied to selected endpoints associated with pCR in a selected treatment arm of the trial to identify potential cut points for biomarker positivity. Two-fold cross-validation was repeated 1000 times, with test and training sets balanced over pCR, using logistic models to assess association with response. A cutpoint was selected as ‘optimal’ if: (1) it was selected as optimal >100 times in the training set; (2) p<E-15 in the test sets (combined using the logit method (Dewey, 2018)); and (3) the prevalence is reasonably balanced.
Immune phenotype: example implementation:
Patients are considered Immune-positive (Immune+) if their immune-tumor state is such that they are likely to respond to immunotherapy. In general, immune signatures are correlated, therefore there are many possible implementations that may perform similarly. In this study we use a subtype-specific implementation. Based on our qualifying biomarker analysis, for TN patients we used the average of the dendritic cell and STAT1 signatures (Danaher et al., 2017; Rody et al., 2009; Yau et al., 2019). These biomarkers were the top two most predictive of TN response to pembrolizumab in this study (Figure 3) and the STAT1 signature has been further validated in the previously published durvalumab/olaparib arm of I-SPY2 (Pusztai et al., 2021) and in an independent Phase II trial (NCT02489448) (Blenman et al., 2020; Foldi et al., 2021; Pusztai et al., 2021). Specifically, we (1) z-scored their average ((STAT1_sig+Dendritic_sig)/2, denoted STAT1_Dendritic_ave), and (2) optimally dichotomized the averaged signatures per above using pCR data from the Pembro arm, yielding a cutpoint of 0 (TN/Immune-high: STAT1_Dendritic_ave>=0; and TN/Immune-low: STAT1_Dendritic_ave<0).
In the HR+HER2− subset, high B-cell and low mast-cell immune gene signatures were strong predictors of pCR to immunotherapy (Figure 3) and we use them in dichotomized form as an example implementation for our Immune+ phenotype in this subset. This choice was based on the observation that to achieve high predictive accuracy in the HR+HER2− subset, it is necessary to combine a ‘sensitivity’ immune biomarker (e.g. Bcell) with a second ‘resistance’ biomarker where high levels predict non-pCR (either Mast-cell or ESR1/PGR averaged). Applying the above dichotomization procedure yielded cutpoints 0.1495 for Bcell_score and 1.17 for MastCell_score (HR+HER2−/Immune-high: (B_cells>=0.1495) AND (Mast_cells<1.17); HR+HER2−/Immune-low: (B_cells<0.1495) OR (Mast_cells>=1.17)).
For HER2+ patients, we optimally dichotomized the B_cells signature in the combined MK2206, control and neratinib arms where immune signals associate with response, yielding a cutpoint of 0.58 (HER2+/Immune-high: B_cells>=0.58; HER2+/Immune-low: B_cells<0.58).
DRD phenotype: example implementation:
Our implementation of the DRD response-predictive phenotype is also subtype-specific. In the TN subset, we had intended to use the previously described PARPi7 gene signature (Figure 3; (Daemen et al., 2012; Wolf et al., 2017)) as an example implementation, but it did not validate in the BrighTNess trial (Filho et al., 2021; Loibl et al., 2018) (p>0.05). Instead we used the VCpred_TN signature developed in I-SPY2 (see above and Table S1), which validated in BrighTNess (p=5.08E-06) (Figure S5). We dichotomized the VCpred_TN using pCR data from the VC arm, using the above-described cross–validation optimization procedure and also taking into account our intention of using this biomarker in a multi-agent context with immunotherapy and an immune biomarker. Though the optimal cutpoint if only considering performance in VC is 0.35, this threshold results in a clinically important subset defined by Immune−/DRD+ that is too small (4%) to be clinically reasonable. Therefore we chose a ‘next best’ cutpoint of −0.31 (TN/DRD+: VCpred_TN>(−0.31); TN/DRD-: VCpred_TN<(−0.31)). With this cutpoint, the Immune−/DRD+ subset is a more clinically actionable size at 11%.
We used BP-Basal classification as our measure to assess the DRD phenotype in HR+HER2− (HR+HER2−/DRD+: BP_Basal; HR+HER2−/DRD−: BP_Luminal) because the assay is performed in a CLIA setting and is ready for clinical implementation with a pending IDE application submission to the US FDA, even though the research assay based PARPi7-high/MP2 performed somewhat better in this dataset (Daemen et al., 2012; Wolf et al., 2017).
Three-state clinical HER2 status: When considering a new HER2low-targeted agent, we used HER2 IHC levels (3+, 2+, 1+, 0) and HER2 FISH to define a 3-class clinical HER2 biomarker HER2-3state (HER2=0: IHC 0 and FISH-; HER2low: IHC 2+/1+ and FISH-; and HER2+: IHC 3+ or FISH+ as currently defined in the trial).
Combining response-predictive phenotypes and HR/HER2 status into response-predictive subtyping schemas
Once multiple response-predictive phenotypes are added to HR and HER2 status, there is a combinatorial explosion in the number of possible states, and many ways to collapse them into a practical number of subtypes (<8 or 9). To sort through the options, we reasoned that an ideal response-predictive subtyping schema should: R1) differentiate between treatments, meaning that different classes should have different best treatments yielding the highest pCR probability; R2) result in a higher pCR rate in the population if used to optimally assign/prioritize treatments; R3) differentiate between responders and non-responders over a wide range of treatment classes; and R4) be robust to platform and within-class treatments, simple to implement, and FDA approved or performed in a CLIA environment. For (R1) we generalize the ‘Carnaugh Map’ method used in circuit design to simplify digital logic (Brown, 1990). For example, if HR+HER2−/Immune−/DRD+ and TN/Immune−/DRD+ classes both have VC as the treatment yielding the highest pCR rate, we collapse them into a single class HER2−/Immune−/DRD+ as seen in Figure 5.
Implementation of previously published PAM50 and TNBC-4class and -7class subtyping schemas
In addition to standard clinical variables like HR, HER2, MP, pCR and Arm, several biomarker heatmaps (e.g., Figure 2) are annotated for PAM50 and two TNBC classification schemas as well, evaluated as previously described. PAM50 intrinsic subtyping was performed using Joel Parker’s centroid-based 50-gene classifier program (Parker et al., 2009) on a total of 1151 samples including 165 in the I-SPY low-risk registry (open to those who screen out of I-SPY2 due to assessment of low molecular risk by the 70-gene MammaPrint test). We included the low-risk registry patients in the dataset (mostly HR+HER2− Luminal A) prior to subtyping because I-SPY2 HR+HER2− patients are all MP high risk (mostly Luminal B) and we wanted the population to be more representative of the general breast cancer patient population as is required for sensible results. We also centered the genes on the mean value of repeated subsampling (500 times) of 1:1 ER+:ER− prior to running the code, as previously advised by Katie Hoadley (private communication) to obtain classifications most consistent with their original paper. Finally, we set to NA any call with a confidence level < 0.08, of which there were 14. TNBCtype classifications (7 classes: MSL, M, LAR, IM, BL2, BL1) were identified as published (Chen et al., 2012; Lehmann et al., 2011) by uploading (non-median centered) expression data from the TN subset (n=363) to the online calculator (https://cbc.app.vumc.org/tnbc/). The Burstein/Brown TN classifications (LAR, MES, BLIS, BLIA) were identified as published (Burstein et al., 2015), by: (1) quantile transforming over their predictor genes; (2) calculating Euclidean distance to the 4 published centroids; and (3) assigning class based on the closest (minimal distance) centroid. PAM50, TNBCtype and TNBC_BB subtype vectors are included in the Table S2 containing the biomarkers and response-predictive subtyping schemas explored in this manuscript.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis of Continuous Gene Expression Biomarkers
Unsupervised clustering was performed using Pearson correlation and complete linkage. We assess association between each continuous biomarker and response in the population as a whole and within each arm and HR/HER2 subtype using a logistic model. In whole-population analyses, models are adjusted for HR, HER2, and treatment arm (pCR~ biomarker + HR + HER2 + Tx). Within treatment arms, models are adjusted for HR and HER2 as appropriate. Markers are analyzed individually; likelihood ratio (LR) test p-values are descriptive. We also performed exploratory whole transcriptome analysis, per above, employing Benjamini-Hochberg multiple testing correction (Huang et al., 2009), with a significance threshold of BH LR p<0.05 (Figure S1). Analyses and visualizations were performed in the computing environment R (v.3.6.3) using R Packages ‘stats’ (v.3.6.3) and ‘lmtest’ (v.0.9–37) (Zeileis et al., 2002).
Response-predictive subtyping schema characterization
Sankey plots were used to visualize relationships between receptor subtype and alternative response predictive subtyping schemas using the R package GoogleVis (v.0.6.4) (Gesmann et al., 2011). For each subtype in each schema, we calculated pCR rates in each arm with sufficient patients and displayed the results (100*(number of patients with pCR)/total) in bar plots. A major goal of a response-predictive schema is to increase the pCR rate in the population and to maximize the probability of pCR for an individual patient (R2). To characterize the potential impact of the classification, we calculated the overall pCR rate in the I-SPY2 population had treatments been optimally assigned according to the response-predictive subtypes using the same 10 drugs. To this end, we: (1) calculated the prevalence of each subtype in the schema (prev_STi = (number of patients in STi)/(total number of patients), i=1:n, n=number of subtypes); (2) collected highest-pCR rates observed in an I-SPY2 arm for each subtype (pCR_max_STi); and (3) calculated a simple estimate of the pCR rate over the population as the weighted sum pCR_max_total = prev_ST1*pCR_max_ST1+ prev_ST2*pCR_max_ST2 +…prev_STn*pCR_max_STn. This calculation results in both an estimate of pCR over the population using the alternative subtyping schema, and identification of agents/combinations maximizing pCR for each subtype.
To characterize the pCR-predictive power of a subtyping schema within an arm (R3), we use bias corrected mutual information (BCMI; R package mpmi http://r-forge.r-project.org/projects/mpmi/) (Pardy et al., 2010), which quantifies the amount of uncertainty reduced about pCR by knowing subtype. These values are then visualized across arms in a scatter plot with BCMI and pCR-association p-values (LR p) on the axis, for both receptor subtype and a response-predictive subtyping schema to visualize differences.
In addition, we used Fisher’s exact test for associations with response, and Cox proportional hazards modeling to estimate DRFS hazard ratios for pCR within each RPS-5 subtype. The latter were performed using the coxph and Surv functions within the R package survival (Therneau et al., 2000).
ADDITIONAL RESOURCES
More information about the I-SPY 2 platform trial (NCT01042379) and associated resources can be found at https://clinicaltrials.gov/ct2/show/NCT01042379, https://www.ispytrials.org/i-spy-platform/i-spy2 and https://ispypatient.org.
Supplementary Material
Table S1. Continuous qualifying biomarkers: definition and implementation details, Related to Figure 2.
Table S2. Patient-level biomarker scores, subtype classes, and clinical/response data, Related to Figure 2.
Table S3. pCR association results for continuous qualifying biomarkers, Related to Figure 3.
KEY RESOURCES TABLE
| REAGENT or | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Tumor biopsy before treatment | I-SPY2 TRIAL | https://clinicaltrials.gov/ct2/show/NCT01042379 |
| Critical commercial assays | ||
| Custom Agilent 32K expression arrays (Agendia32627_DPv1 .14_SCFGplus) | Agendia, Inc | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL20078 |
| Custom Agilent 44K expression arrays (Agilent_human_Disc overPrint 15746) | Agendia, Inc | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL30493 |
| MammaPrint | Agendia, Inc | https://agendia.com/mammaprint/ |
| BluePrint | Agendia, Inc | https://agendia.com/blueprint/ |
| Reverse phase protein array (RPPA) | Petricoin Lab, George Mason University | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL28470 |
| Deposited data | ||
| Raw and processed transcriptomic data | This study | Gene Expression Omnibus (GEO) SubSeries GSE194040 (mRNA), (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194040, as part of the SuperSeries GSE196096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196096); and in the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data) |
| Raw and processed RPPA data | This study | Gene Expression Omnibus (GEO) SubSeries GSE196093 (RPPA) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196093, as part of the SuperSeries GSE196096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196096); and in the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data) |
| Patient-level expression signature and clinical data | This study | Gene Expression Omnibus (GEO) SuperSeries GSE196096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196096) and Table S2 and in the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data). |
| Software and algorithms | ||
| stats R package (v.3.6.3) | R Core Team (2020) | https://stat.ethz.ch/R-manual/R-devel/library/stats/html/stats-package.html |
| lmtest R package (v.0.9–37) | (Zeileis et al., 2002) | https://CRAN.R-proiect.org/package=lmtest |
| googleVis R package (v.0.6.4) | (Gesmann et al., 2011) | https://CRAN.R-proiect.org/package=googleVis |
| survival R package (v.3.1–12) | (Therneau et al., 2000) | https://CRAN.R-proiect.org/package=survival |
| mpmi R package (v.0.43) | (Pardy et al., 2010) | http://r-forge.r-proiect.org/proiects/mpmi/ |
Highlights.
The I-SPY2–990 Data Resource contains mRNA, protein, and response data over 10 drugs
Biomarkers are combined to create breast cancer subtypes to match modern treatments
Best subtyping schemas incorporate immune, DNA repair, Luminal, and HER2 phenotypes
Treatment assignment using these response predictive subtypes may improve outcome
ACKNOWLEDGEMENTS
With support from Quantum Leap Healthcare Collaborative, FNIH, NIH/NCI I-SPY2+ (Grant PO1-CA210961), NIH/NCI Imaging (Grant 28XS197 P-0518835), NIH/NCI CCMI (Grant U54CA209891), NIH/NCI CCSG (Grant P30- CA82103), NIH/NHGRI Big Data (Grant U54-HG007990), Safeway - an Albertsons Company, William K. Bowes Jr. Foundation, Breast Cancer Research Foundation (BCRF-20-165), UCSF, GMU, Gateway for Cancer Research (Grants G-16-900 and G-20-600), SideOut Foundation, the Biomarkers Consortium, Salesforce, OpenClinica, Formedix, Hologic Inc., TGen, CCS Associates, Berry Consultants, Breast Cancer Research - Atwater Trust, Stand up to Cancer, California Breast Cancer Research Program, and Give Breast Cancer the Boot; Angela and Shu Kai Chan Chair in Cancer Research (LvtV); IQVIA, Genentech, Amgen, Pfizer, Merck, Seattle Genetics, Daiichi Sankyo, AstraZeneca, Dynavax Technologies, Puma Biotechnology, AbbVie, Madrigal Pharmaceuticals (formerly Synta Pharmaceuticals), Plexxikon, Regeneron and Agendia. Sincere thanks to our DSMB, Independent Agent Selection Committee, Biomarker Working Group, our patients, advocates and investigators.
DECLARATION OF INTERESTS
Christina Yau consulted for NantOmics, LLC. JD Wulfkuhle reports honoraria from DAVA Oncology; consults for Baylor College of Medicine; has ownership in Theralink; and is co-inventor of RPPA technology, and phospho-HER2 and -EGFR response predictors w/filed patents. Minetta Liu reports support from Eisai, Genentech, GRAIL, Menarini Silicon Biosystems, Merck, Novartis, Seattle Genetics, and Tesaro. Paula R Pohlmann reports leadership and stock in Immunonet BioSciences; honoraria from ASCO, Dava Oncology, OncLive (Courses) and Frontiers (Editorship); consulting for Personalized Cancer Therapy, Immunonet BioSciences, Sirtex, CARIS Lifesciences, OncoPlex Diagnostics, Pfizer, Heron, Puma, AbbVie, BOLT, SEAGEN; and is occasional speaker for Genentech, Roche. Fraser Symmans is a co-founder of Delphi Diagnostics, co-inventor/patent holder for a (free) residual cancer burden calculator, holds shares in IONIS Pharmaceuticals,Eiger Biopharmaceuticals; is unpaid advisor/steering committee for Roche trials. Hope Rugo reports support from Pfizer, Merck, Novartis, Lilly, Genentech, Odonate, Daiichi, Seattle Genetics, Eisai, Macrogenics, Sermonix, Boehringer Ingelheim, Polyphor, Astra Zeneca and Immunomedics, and received honoraria from Puma Biotechnology, Mylan and Samsung. Claudine Isaacs reports consulting for Seattle Genetics, Genentech, AstraZeneca, Novartis, PUMA, Pfizer, and Esai. Angela DeMichele reports honoraria or consulting for Pfize, Context Therapeutics; and support from Novartis, Pfizer, Genentech, Calithera and Menarini. Doug Yee reports unrelated support from Boehringer Ingleheim. Donald Berry is co-owner of Berry Consultants, LLC, a company that designs adaptive clinical trials (including I-SPY2). Lajos Pusztai reports consulting fees and honoraria from Astra Zeneca, Merck, Novartis, Bristol-Myers Squibb Genentech, Eisai, Pieris, Immunomedics, Seattle Genetics, Clovis, Syndax, H3Bio and Daiichi. E.F. Petricoin reports leadership, stock/ownership, consulting/advisory and travel funds from Perthera and Ceres Nanosciences; stock and consulting/advisory for Avant Diagnostics; consulting/advisory for AZGen; support from Ceres Nanosciences, GlaxoSmithKline, Abbvie, Symphogen, and Genentech; patents/royalties from NIH, and filed patents for phospho-HER2 and -EGFR response predictors. Laura Esserman is an unpaid member of the board of directors of Quantum Leap Healthcare Collaborative (QLHC) and received grant support from QLHC for the I-SPY2 trial; is on the Blue Cross/Blue Shield Medical Advisory Panel and receives reimbursement for her time and travel; and received unrelated research support from Merck. Dr. van ‘t Veer is a co-inventor of the MammaPrint signature and part-time employee and stockholder of Agendia, NV.
INCLUSION AND DIVERSITY
We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Clinical Trial Registration: www.clinicaltrials.gov/ct2/show/NCT01042379
REFERENCES
- Bergin ART, and Loi S (2019). Triple-negative breast cancer: recent treatment advances. F1000research 8, F1000 Faculty Rev-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DA (2011). Adaptive clinical trials in oncology. Nature Reviews Clinical Oncology 9, 199–207. [DOI] [PubMed] [Google Scholar]
- Blenman KRM, Li X, Marczyk M, O’Meara T, Yaghoobi V, Gunasekharan V, Park T, Rimm D, and Pusztai L (2020). Abstract P3-09-05: Predictive markers of response to durvalumab concurrent with nab-paclitaxel and dose dense doxorubicin cyclophosphamide (ddAC) neoadjuvant therapy for triple negative breast cancer (TNBC). P3-09-05-P3-09-05.
- Brown FM (1990). Boolean reasoning : the logic of Boolean equations.
- Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, et al. (2015). Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso F, Veer L.J. van’t, Bogaerts J, Slaets L, Viale G, Delaloge S, Pierga J-Y, Brain E, Causeret S, DeLorenzi M, et al. (2016). 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. New England Journal of Medicine 375, 717–729. [DOI] [PubMed] [Google Scholar]
- Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, and Pietenpol JA (2012). TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Informatics 11, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien AJ, Tripathy D, Albain KS, Symmans WF, Rugo HS, Melisko ME, Wallace AM, Schwab R, Helsten T, Forero-Torres A, et al. (2019). MK-2206 and Standard Neoadjuvant Chemotherapy Improves Response in Patients With Human Epidermal Growth Factor Receptor 2– Positive and/or Hormone Receptor–Negative Breast Cancers in the I-SPY 2 Trial. J Clin Oncol 38, 1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AS, Yau C, Wolf DM, Petricoin EF, Veer L.J. van ‘t, Yee D, Moulder SL, Wallace AM, Chien AJ, Isaacs C, et al. (2021). Neoadjuvant T-DM1/pertuzumab and paclitaxel/trastuzumab/pertuzumab for HER2+ breast cancer in the adaptively randomized I-SPY2 trial. Nat Commun 12, 6428. (Pardy) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Wolf DM, Korkola JE, Griffith OL, Frankum JR, Brough R, Jakkula LR, Wang NJ, Natrajan R, Reis-Filho JS, et al. (2012). Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res Tr 135, 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, and Fling SP (2017). Gene expression markers of Tumor Infiltrating Leukocytes. J Immunother Cancer 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis CE, Bray F, Ferlay J, Lortet-Tieulent J, Anderson BO, and Jemal A (2015). International Variation in Female Breast Cancer Incidence and Mortality Rates. Cancer Epidemiol Biomarkers Prev 24, 1495–1506. [DOI] [PubMed] [Google Scholar]
- Dewey M (2018). metap: meta-analysis of significance values. R Package Version 1.0 [Google Scholar]
- Filho OM, Stover DG, Asad S, Ansell PJ, Watson M, Loibl S, G., C. E Jr, Bae J, Collier K, Cherian M, et al. (2021). Association of Immunophenotype With Pathologic Complete Response to Neoadjuvant Chemotherapy for Triple-Negative Breast Cancer: A Secondary Analysis of the BrighTNess Phase 3 Randomized Clinical Trial. JAMA Oncol 7, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldi J, Silber A, Reisenbichler E, Singh K, Fischbach N, Persico J, Adelson K, Katoch A, Horowitz N, Lannin D, et al. (2021). Neoadjuvant durvalumab plus weekly nab-paclitaxel and dose-dense doxorubicin/cyclophosphamide in triple-negative breast cancer. Npj Breast Cancer 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Ericsson PI, Wulfkhule JD, Gallagher RI, Sun X, Axelrod ML, Sheng Q, Luo N, Gomez H, Sanchez V, Sanders M, et al. (2021). Tumor-Specific Major Histocompatibility-II Expression Predicts Benefit to Anti–PD-1/L1 Therapy in Patients With HER2-Negative Primary Breast Cancer. Clin Cancer Res 27, 5299–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesmann M, de Castillo D (2011). googleVis: Interface between R and the Google Visualisation API. The R Journal, 3(2), 40–44 [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kim M, Park J, Bouhaddou M, Kim K, Rojc A, Modak M, Soucheray M, McGregor MJ, O’Leary P, Wolf D, et al. (2021). A protein interaction landscape of breast cancer. Science 374, eabf3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijgsman O, Roepman P, Zwart W, Carroll JS, Tian S, Snoo F.A. de, Bender RA, Bernards R, and Glas AM (2012). A diagnostic gene profile for molecular subtyping of breast cancer associated with treatment response. Breast Cancer Res Tr 133, 37–47. [DOI] [PubMed] [Google Scholar]
- Lee P, Zhu Z, Wolf D, Yau C, Audeh W, Glas A, Swigart L, Hirst G, DeMichele A, Investigators IS, et al. (2018). Abstract 2612: BluePrint Luminal subtype predicts non-response to HER2-targeted therapies in HR+/HER2+ I-SPY 2 breast cancer patients. Cancer Research 78, 2612–2612. [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, and Pietenpol JA (2011). Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loibl S, O’Shaughnessy J, Untch M, Sikov WM, Rugo HS, McKee MD, Huober J, Golshan M, Minckwitz G. von, Maag D, et al. (2018). Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, phase 3 trial. Lancet Oncol 19, 497–509. [DOI] [PubMed] [Google Scholar]
- McAndrew NP, and Finn RS (2020). Management of ER positive metastatic breast cancer. Semin Oncol 47, 270–277. [DOI] [PubMed] [Google Scholar]
- Nanda R, Liu MC, Yau C, Shatsky R, Pusztai L, Wallace A, Chien AJ, Forero-Torres A, Ellis E, Han H, et al. (2020). Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer. Jama Oncol 6, 676–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, and Carbone PP (1982). Toxicity and Response Criteria of the Eastern-Cooperative-Oncology-Group. American Journal of Clinical Oncology-Cancer Clinical Trials 5, 649–655. [PubMed] [Google Scholar]
- Pardy C, Wilson S (2010). A bioinformatic implementation of mutual information as a distance measure for identification of clusters of variables. ANZIAM Journal. 52. 10.21914/anziamj.v52i0.3959. [DOI] [Google Scholar]
- Park JW, Liu MC, Yee D, Yau C, Veer L.J. van t, Symmans WF, Paoloni M, Perlmutter J, Hylton NM, Hogarth M, et al. (2016). Adaptive Randomization of Neratinib in Early Breast Cancer. New England Journal of Medicine 375, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, et al. (2009). Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J Clin Oncol 27, 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccart M, Veer L.J. van ‘t, Poncet C, Cardozo JMNL, Delaloge S, Pierga J-Y, Vuylsteke P, Brain E, Vrijaldenhoven S, Neijenhuis PA, et al. (2021). 70-gene signature as an aid for treatment decisions in early breast cancer: updated results of the phase 3 randomised MINDACT trial with an exploratory analysis by age. Lancet Oncol. [DOI] [PubMed] [Google Scholar]
- Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, String-Reasor E, Boughey JC, Chien AJ, Elias AD, et al. (2021). Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell 39, 989–998.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A, Holtrich U, Pusztai L, Liedtke C, Gaetje R, Ruckhaeberle E, Solbach C, Hanker L, Ahr A, Metzler D, et al. (2009). T-cell metagene predicts a favorable prognosis in estrogen receptor-negative and HER2-positive breast cancers. Breast Cancer Res Bcr 11, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugo HS, Olopade OI, DeMichele A, Yau C, Veer L.J. van t, Buxton MB, Hogarth M, Hylton NM, Paoloni M, Perlmutter J, et al. (2016). Adaptive Randomization of Veliparib–Carboplatin Treatment in Breast Cancer. New England Journal of Medicine 375, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayaman RW, Wolf DM, Yau C, Wulfkuhle J, Petricoin E, Brown-Swigart L, Asare SM, Hirst GL, Sit L, O’Grady N, et al. (2020). Abstract P1-21-08: Application of machine learning to elucidate the biology predicting response in the I-SPY 2 neoadjuvant breast cancer trial. Cancer Res 80, P1-21-08-P1-21-08. [Google Scholar]
- Schmid P, Cortes J, Pusztai L, McArthur H, Kummel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N, et al. (2020). Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med 382, 810–821. [DOI] [PubMed] [Google Scholar]
- Spring LM, Fell G, Arfe A, Sharma C, Greenup R, Reynolds KL, Smith BL, Alexander B, Moy B, Isakoff SJ, et al. (2020). Pathologic Complete Response after Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive Meta-analysis. Clin Cancer Res 26, 2838–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmans WF, Peintinger F, Hatzis C, Rajan R, Kuerer H, Valero V, Assad L, Poniecka A, Hennessy B, Green M, et al. (2007). Measurement of Residual Breast Cancer Burden to Predict Survival After Neoadjuvant Chemotherapy. J Clin Oncol 25, 4414–4422. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, and Caldas C (2008). A robust classifier of high predictive value to identify good prognosis patients in ER-negative breast cancer. Breast Cancer Research 10, R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model. Springer, New York. ISBN 0-387-98784-3. [Google Scholar]
- Waks AG, and Winer EP (2019). Breast Cancer Treatment: A Review. JAMA 321, 288–300. [DOI] [PubMed] [Google Scholar]
- Wolf D, Yau C, Swigart L, Hirst G, Investigators IS, Asare S, Schwab R, Berry D, Esserman L, Albain K, et al. (2018). Abstract 2611: Evaluation of ANG/TIE/hypoxia pathway genes and signatures as predictors of response to trebananib (AMG 386) in the neoadjuvant I-SPY 2 TRIAL for Stage II-III high-risk breast cancer. Cancer Research 78, 2611–2611. [Google Scholar]
- Wolf DM, Lenburg ME, Yau C, Boudreau A, and Veer L.J. van ‘t (2014). Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS One 9, e88309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Yau C, Sanil A, Glas A, Petricoin E, Wulfkuhle J, Severson TM, Linn S, Brown-Swigart L, Hirst G, et al. (2017). DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. Npj Breast Cancer 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Yau C, Wulfkuhle J, Brown-Swigart L, Gallagher RI, Magbanua MJM, O’Grady N, Hirst G, I-SPY2 Trial Investigators, Asare S, et al. (2020a). Mechanism of action biomarkers predicting response to AKT inhibition in the I-SPY 2 breast cancer trial. Npj Breast Cancer 6, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DM, Yau C, Wulfkuhle J, Brown-Swigart L, Asare SM, Hirst GL, Sit L, Perlmutter J, Consortium, I.-S. 2 T., Liu M, et al. (2020b). Abstract P4-10-02: HER2 signaling, ER, and proliferation biomarkers predict response to multiple HER2-targeted agents/combinations plus standard neoadjuvant therapy in the I-SPY 2 trial. Cancer Res 80, P4-10-02-P4-10-02. [Google Scholar]
- Wuerstlein R, and Harbeck N (2017). Neoadjuvant Therapy for HER2-positive Breast Cancer. Rev Recent Clin Trials 12, 81–92. [DOI] [PubMed] [Google Scholar]
- Wulfkuhle JD, Yau C, Wolf DM, Vis DJ, Gallagher RI, Brown-Swigart L, Hirst G, Voest EE, DeMichele A, Hylton N, et al. (2018). Evaluation of the HER/PI3K/AKT Family Signaling Network as a Predictive Biomarker of Pathologic Complete Response for Patients With Breast Cancer Treated With Neratinib in the I-SPY 2 TRIAL. Jco Precis Oncol 2, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau C, Wolf D, Campbell M, Savas P, Lin S, Swigart L, Hirst G, Asare S, Zhu Z, Loi S, et al. (2019). Abstract P3-10-06: Expression-based immune signatures as predictors of neoadjuvant targeted-/chemo-therapy response: Experience from the I-SPY 2 TRIAL of ˜1000 patients across 10 therapies. Cancer Research 79, P3–10. [Google Scholar]
- Yee D, DeMichele AM, Yau C, Isaacs C, Symmans WF, Albain KS, Chen YY, Krings G, Wei S, Harada S, et al. (2020). Association of Event-Free and Distant Recurrence-Free Survival With Individual-Level Pathologic Complete Response in Neoadjuvant Treatment of Stages 2 and 3 Breast Cancer: Three-Year Follow-up Analysis for the I-SPY2 Adaptively Randomized Clinical Trial. JAMA Oncol 6, 1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeileis A, Hothorn T (2002). Diagnostic Checking in Regression Relationships. R News, 2(3), 7–10. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Continuous qualifying biomarkers: definition and implementation details, Related to Figure 2.
Table S2. Patient-level biomarker scores, subtype classes, and clinical/response data, Related to Figure 2.
Table S3. pCR association results for continuous qualifying biomarkers, Related to Figure 3.
Data Availability Statement
Transcriptomic, protein/phospho-protein and clinical data used in this study is available in NCBI’s Gene Expression Omnibus (GEO) SuperSeries GSE196096 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196096) and its two SubSeries GSE194040 (mRNA: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE194040) and GSE196093 (RPPA: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196093), and through the I-SPY2 Google Cloud repository (www.ispytrials.org/results/data). Data on GEO represents the data as currently recorded in our database. Patient-level scores for the 27 qualifying biomarker scores and response data analyzed in this paper, and the RPS-5, RPS-7 and other subtype classifications and their constituent biomarkers presented herein are available in Table S2. Additional de-identified subject level data may be requested by qualified investigators. Details of the trial, data, contact information, proposal forms, and review and approval process are available at the following website: https://www.ispytrials.org/collaborate/proposal-submissions.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.