

Abstract
The H3K4 demethylase KDM5B is overexpressed in multiple cancer types, and elevated expression levels of KDM5B is associated with decreased survival. However, the underlying mechanistic contribution of dysregulated expression of KDM5B and H3K4 demethylation in cancer is poorly understood. Our results show that loss of KDM5B in multiple types of cancer cells leads to increased proliferation and elevated expression of cancer stem cell markers. In addition, we observed enhanced tumor formation following KDM5B depletion in a subset of representative cancer cell lines. Our findings also support a role for KDM5B in regulating epigenetic plasticity, where loss of KDM5B in cancer cells with elevated KDM5B expression leads to alterations in activity of chromatin states, which facilitate activation or repression of alternative transcriptional programs. In addition, we define KDM5B-centric epigenetic and transcriptional patterns that support cancer cell plasticity, where KDM5B depleted cancer cells exhibit altered epigenetic and transcriptional profiles resembling a more primitive cellular state. This study also provides a resource for evaluating associations between alterations in epigenetic patterning upon depletion of KDM5B and gene expression in a diverse set of cancer cells.
INTRODUCTION
Histone 3 lysine 3 methylation (H3K4me) is important for regulating gene expression and chromatin architecture [1], and dysregulation of epigenetic processes is a contributing factor in tumor initiation and progression [2–7]. H3K4me3 is primarily localized at promoter regions of expressed genes [8, 9], where it functions as a recognition site for RNA polymerase II binding and target gene activation [10–12]. KDM5B, which is a member of the highly conserved family of H3K4 demethylases, is important for embryo development [13, 14], stem cell differentiation [15–19], mammary gland development [14], and is involved in pathogenic responses such as metastasis [14, 20–22].
KDM5B expression is moderate in adult tissues although it is overexpressed in human cancers. KDM5B expression is elevated in several cancers including ovarian [23], breast [20, 24, 25], prostate [22], bladder [26, 27], lung [26], melanoma [28], colon [29], gastric [30], gliomas [31], and liver cancer [32]. KDM5B has been shown to be amplified and overexpressed in ovarian epithelial cancer [23], and in luminal breast cancers [25], where it supports expression programs in differentiated luminal cells. Expression of KDM5B is also associated with drug resistance [33], including endocrine resistance in breast cancer, suggesting that KDM5B may regulate expression programs and cellular states in breast cancer [25]. KDM5B overexpression in melanoma cells is correlated with a slow-cycling state and resistance to anti-proliferative therapies [21, 34]. Dynamic expression of KDM5B in melanoma cells may allow melanoma cells to evade anti-proliferative therapies and facilitate acquired resistance to targeted therapies. KDM5B expression is also elevated in multi-drug resistant BRAFV600E-positive cells, suggesting that KDM5B may support a stem cell-like state in melanoma.
While these findings demonstrate that KDM5B is overexpressed in cancer, it is unclear whether depletion or inhibition of KDM5B as a therapeutic strategy is sufficient to reduce proliferation and collapse self-renewal transcriptional networks for KDM5B-overexpressing cancers. Moreover, the epigenetic mechanisms by which KDM5B contributes to tumor maintenance and tumor progression are largely unknown. While perturbation of gene expression resulting from stochastic changes in H3K4 methylation may promote altered expression profiles and thus lead to the formation of cancerous cells, it is not clear what triggers these events. Because KDM5 enzymes function in part to erase H3K4me3 epigenetic memory, it is possible that dysregulation of KDM5 family members, including KDM5B, in cancer cells may lead to epigenetic plasticity, or alterations in chromatin that facilitate a permissive state, and altered lineage fates. Misexpression of KDM5B may disrupt the homeostatic balance of chromatin to facilitate tumor evolution and adaptation. Downregulation of KDM5B may create an overly permissive chromatin structure that facilitates spurious gene activation and hinder differentiation by allowing cancer cells to sample various transcriptional profiles, some of which may allow cancer cells to adapt. Overexpression of KDM5B may create overly restrictive chromatin that is unable to activate apoptosis, DNA-repair or other programs. Thus, KDM5B-mediated deregulation of H3K4 methylation may support tumor potentiation, initiation, and progression.
RESULTS
Depletion of KDM5B in cancer cells leads to dedifferentiation
To investigate the mRNA expression level of KDM5B in a compendium of human cancers relative to normal samples we used Oncomine [35, 36]. These findings demonstrate that KDM5B is overexpressed in multiple types of cancer (bladder, breast, leukemia, lung, melanoma, ovarian, pancreas, prostate, renal, and uterus) (Fig. 1A). These findings are in alignment with results from previous studies which demonstrated that KDM5B is overexpressed in bladder [26, 27], breast [20, 24, 25], lung [26], melanoma [28], ovarian [23] and prostate [22] cancer. We further explored the expression level of KDM5B in a diverse panel of human cancer cell lines (NCI-60) representing nine types of cancer and tissue types including breast (BR), colon (CO), central nervous system (CNS), blood/leukemia (LE), lung (LU), ovary (OV), prostate (PR), skin/melanoma (ME) and kidney/renal (RE) [37]. This universally utilized NCI-60 cancer cell line panel is an ideal platform for studying cancer cells, and global RNA-Seq transcriptome analyses have been performed for these cells [38]. KDM5B is expressed at a relatively high level in numerous cancer cell lines (Fig. 1B), including cell lines representing breast, CNS, leukemia, lung, melanoma, ovarian, and renal cancer types. Moreover, Kaplan–Meier analyses revealed that breast, ovarian, renal, and lung cancer patients with high expression of KDM5B have a decreased rate of survival relative to patients with low expression of KDM5B (Fig. 1C) [35]. However, it is unclear whether depleting KDM5B in cancer cells that express elevated levels of KDM5B impacts cancer cell function and collapses essential transcriptional networks.
Fig. 1. KDM5B expression in cancer and establishment of cancer cell models.

A Expression analysis of KDM5B in normal and cancer samples. Oncomine was used to analyze public mRNA expression datasets from human cancers (bladder [79], breast-TCGA, leukemia [80], lung [81], ovarian-TCGA, melanoma-TCGA, pancreas [82], prostate [83], renal [84], uterine-TCGA). Boxplots indicate the 1st and 3rd quartiles (25th and 75th percentile, upper and lower bounds), 2nd quartile (center), and minima–maxima (1.5*interquartile range). B Z-score analysis of KDM5B expression in the NCI-60 panel of cancer cell lines. The top panel shows cancer cell lines with the highest expression of KDM5B, while the bottom panel shows cell lines with lower expression of KDM5B. BR breast, ME melanoma, LC lung cancer, CNS brain cancer, OV ovarian, LE leukemia, RE renal, PR prostate, CO colon. C Kaplan–Meier curve for the overall survival of cancer patients exhibiting high (red) or low (black) expression of KDM5B. D Schematic of experimental design. E qRT-PCR expression ±SEM (standard error of mean) of KDM5B in control (shLuc) and KDM5B depleted (shKDM5B) cancer cells (shLuc, Gray; shKdm5b, Black). F Western blot of KDM5B and H3K4me3 (rabbit monoclonal antibody H3K4me3; 17–614) in control and KDM5B depleted cancer cells. HSC70 (sc-7298) and total H3 (sc-517576) antibodies were used for total loading control and nuclear loading control, respectively.
To understand the contribution of KDM5B overexpression to dysregulation of epigenetic and transcriptional profiles across a diverse set of cancer cells, we generated KDM5B knockdown and control cancer cell lines representing a diverse set of 26 human cancer cell lines from the NCI-60 panel (breast, CNS, leukemia, lung, melanoma, ovarian, renal) (Fig. 1B, top). To study the effect of depleting KDM5B in cancer cells, we designed four shRNAs directed against KDM5B. To identify the shRNA that resulted in the greatest knockdown, MCF7 breast cancer cells were transduced with lentiviral particles encoding shRNAs directed against KDM5B or control luciferase (shLuc), and stably selected in the presence of 1 μg/ml puromycin. KDM5B-shRNA-2 resulted in the greatest knockdown of KDM5B mRNA in MCF7 cells and was used for subsequent studies (Fig. S1A). KDM5B-shRNA-1 was used as a second shRNA to confirm the knockdown phenotype of cancer cells.
Next, cancer cells with elevated KDM5B expression were transduced with lentiviral particles encoding shRNAs directed against KDM5B or control luciferase. KDM5B knockdown (shKDM5B; KDM5B-shRNA-2 or KDM5B-shRNA-1) and control (shLuc) cells were stably selected in the presence of 1–2 μg/ml puromycin (Fig. 1D). Q-Rt-PCR demonstrated that KDM5B mRNA levels decreased in shKDM5B (KDM5B-shRNA-2) cancer cells (Fig. 1E), which was confirmed using KDM5B-shRNA-1 (Fig. S1B). Western blotting confirmed knockdown of KDM5B protein levels, and increased levels of H3K4me3, in shKDM5B cancer cells (Fig. 1F).
KDM5B controls cancer type and cell-type-specific transcriptional networks
To interrogate the effect of depleting KDM5B on gene expression in cancer cells we performed RNA-Seq transcriptome analyses using control and shKDM5B cancer cells. K-means clustering was used to identify patterns of gene expression variability between cancer types and cell lines and between control and KDM5B-depleted cells (Fig. 2A and Table S1). This approach yielded 9 clusters of genes, including genes expressed lowly in leukemia cells (c1), leukemia/melanoma cells (c6), leukemia cells (c8, c9), and genes expressed highly in breast/CNS (c2), multiple cancer cells (c3), melanoma cells (c4), or leukemia cells (c7) (Fig. 2A). Gene ontology (GO) functional annotation analysis of the 9 clusters of genes revealed enrichment of multiple biological processes and disease terms. DAVID biological process GO analysis demonstrated that 23% of the statistically significant GO terms (p < 0.05) in c1, 31% in c2, 21% in c3, 24% in c4, 23% in c5, 27% in c6, 4% in c7, 24% in c8, and 17% in c9 were associated with development (Table S2). DAVID analysis revealed multiple diseases associated with genes in c1, c2, and enrichment of ovarian/lung/bladder cancer in c3, enrichment of melanoma and neuroblastoma in c4, ovarian/stomach/thyroid/prostate/lung/bladder cancer in c6, ovarian cancer/prostate neoplasm/cancer in c8, and ovarian/lung/bladder/breast/stomach/colorectal/oral/cervical/leukemia in c9 (Table S2). These findings also revealed diseases such as brain ischemia/stroke associated with genes in c7.
Fig. 2. KDM5B regulates the transcriptional repertoire of cancer cells.

A K-means clustering of RNA-Seq transcriptome data from control (shLuc) and shKDM5B cancer cells (>two-fold between any two groups, RPKM > 1, FDR < 0.001). Nine clusters (c1–c9) highlight cancer type-specific gene expression patterns. List and order of cell lines is shown in Table S1. B Bar plot and bubble plot representation of the number of differentially expressed genes (DE) between control and KDM5B depleted cancer cells. Bar plot shows the percentage of upregulated (red), downregulated (green), and genes with no expression change (gray), while the bubble plot shows the number of DE genes (upregulated, downregulated). The number of DE genes is represented by bubble size and color. C Principal component analysis (PCA) of RNA-Seq transcriptome data (RPKM) in 26 control and KDM5B depleted cancer cells. Seven cancer types are color coded (BR: breast, CNS: central nervous system, LC: lung cancer, LE: leukemia, ME: melanoma, OV: ovary, RE: renal). D Cancer type-specific PCA analysis of control and KDM5B depleted RNA-Seq expression profiles (X-axis, PC1; Y-axis, PC2). E Hierarchical clustering heatmap of DAVID biological process GO terms (−log10 p value) identified from differentially expressed genes in KDM5B depleted cancer cells. NCBI DAVID was used to calculate p values. F Scatter plot of REVIGO semantic similarity analysis of GO enrichment terms. G qRT-PCR expression of tumor progression and prognosis markers in control and KDM5B depleted cancer cells.
To further evaluate the functional effect of loss of KDM5B on gene expression programs of cancer cells we used edgeR [39] to identify differentially expressed (DE) genes between control and KDM5B-depleted cancer cells (see methods). Depletion of KDM5B resulted in a variable number and percentage of DE genes between control and KDM5B-depleted cells (Fig. 2B), where several cell lines such as MCF7, SNB-75, K562, MALME-3M, SK-MEL-28, OVCAR-3, OVCAR-4, A498, and CAKI-1 were more sensitive to gene expression changes following loss of KDM5B. A comparison of RNA-Seq data between control and KDM5B-depleted cells is shown in scatter plots in Fig. S2A. Principal component analysis (PCA) demonstrated heterogeneous expression profiles of 26 cancer cell lines and altered transcriptomes following loss of KDM5B (Fig. 2C). An evaluation of cancer type-specific transcriptional patterns using PCA revealed alterations in two-dimensional trajectories following depletion of KDM5B (Fig. 2D). Loss of KDM5B resulted in similar directional changes in the 2D space for breast cancer and CNS cells, while leukemia, lung, melanoma, ovarian, and renal cancer cells displayed scattered trajectories. These findings suggest that loss of KDM5B may result in divergent expression changes in a diverse set of cancer cells.
An investigation of GO term enrichment was performed using DAVID [40]. Hierarchical clustering of GO term p values (−log10) of genes that were DE in KDM5B-depleted cancer cells revealed several clusters of GO terms (Fig. 2E). These results demonstrated shared and distinct enrichment of GO terms in a cell and cancer type manner (Table S2). We also performed semantic analysis of GO terms enriched in DE genes across 26 cell lines using GoSemSim [41]. These findings enrichment patterns of GO terms for DE genes between control and KDM5B depleted cancer cells (Fig. S2B). Further GO term analysis using REVIGO [42] revealed common genes that are DE upon depletion of KDM5B across cancer types and cell lines (Figs. 2F and S3 and Table S3). These findings demonstrate that knockdown of KDM5B in cancer cells that exhibit elevated levels of KDM5B impacts expression of genes linked to multiple regulatory pathways.
Confirmation of dysregulated expression of proliferation genes and tumor progression genes was evaluated in multiple cancer cell lines following depletion of KDM5B using Q-RT-PCR. We observed upregulation of proliferation genes overexpressed in breast cancer such as Ki67, STK15, MYLB2, and MMP11 [43], and downregulation of the tumor suppressor SCUBE2 [44] (Fig. 2G). We also observed downregulation of ZAP70 and upregulation of LPL-1 in K562 leukemia cancer cells, downregulation of MCM4, OST, and HER3 in shKDM5B SK-MEL-28 melanoma cells, downregulation of Cyclin D1, Cyclin E1, and MMP2 and upregulation of EPHA and OPN in ovarian cancer cells, and upregulation of E-Cadherin (CDH1) and downregulation of AR and VEGF in CAKI-1 renal cancer cells (Fig. 2G).
To evaluate whether depletion of KDM5B impacts the expression state of “stemness” signature genes upregulated in human pluripotent stem cells (hESCs) [45], which are DE between control and KDM5B-depleted cancer cells, we used gene set enrichment analysis [46]. These results show that expression of stemness signature genes is enriched in 85% of KDM5B depleted cancer cell lines (22 of the 26) relative to control cells (Figs. 3A, B and S4), while expression of stemness signature genes was enriched in two control cancer cells lines (A498, SF-539), and two cell lines exhibited minor enrichment of stemness genes in control cancer cells relative to KDM5B-depleted cancer cells (BT-549, OVCAR-3). A further comparison of DE genes between control and KDM5B depleted cancer cells with a compendium of cancer expression datasets using Oncomine [35] revealed a correlation between genes that were upregulated upon loss of KDM5B and overexpressed in clear cell renal cell carcinoma, cutaneous melanoma, ovarian carcinoma, and leukemia versus normal tissues (Figs. 3C and S5 and S6). We also observed a correlation between genes that were underexpressed in KDM5B depleted ovarian cancer cells and underexpressed in human cancer cells (Fig. S7).
Fig. 3. Integrative transcriptome analysis reveals that loss of KDM5B leads to increased expression of stemness genes and an expression signature consistent with multiple cancers.
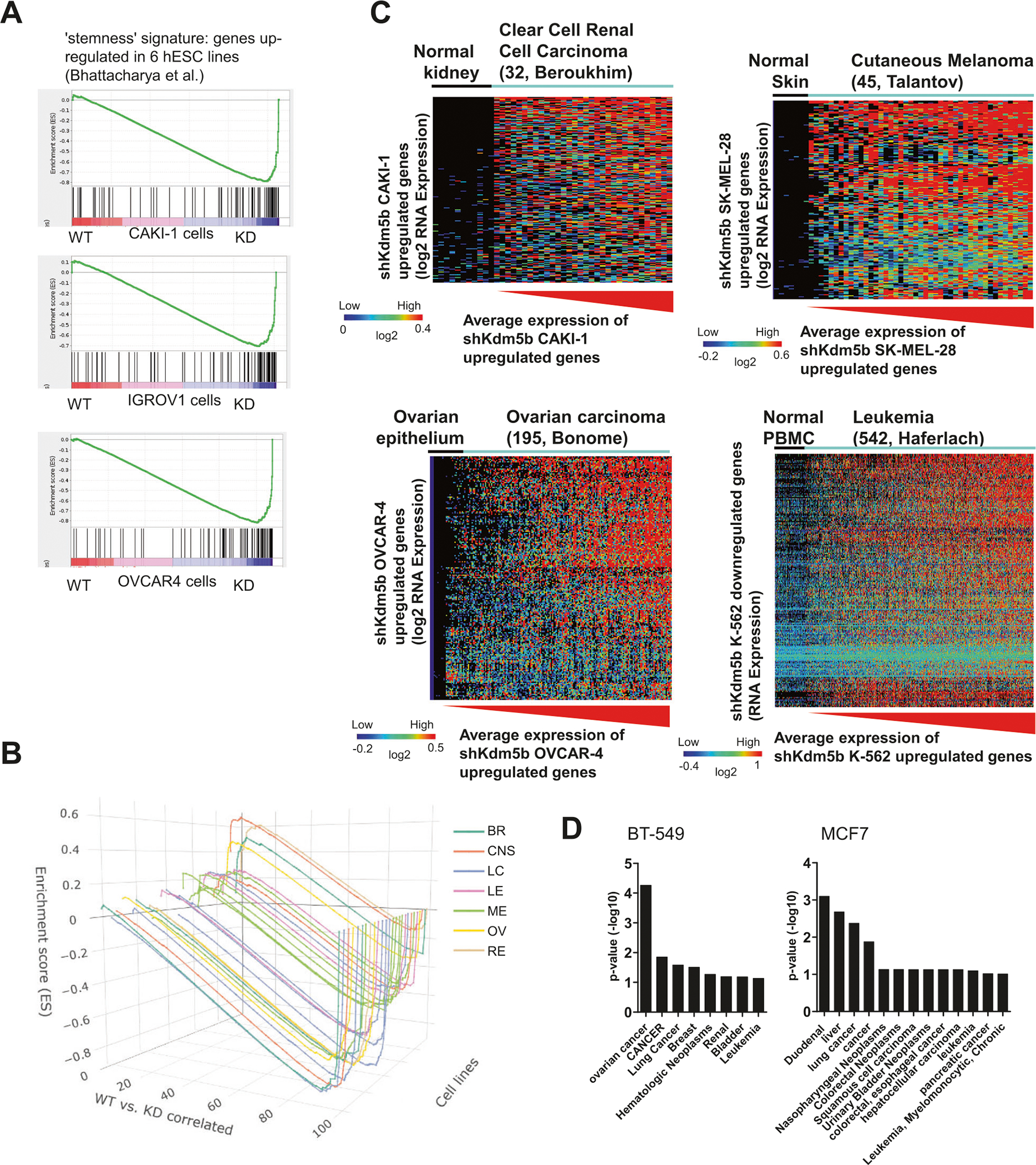
Gene set enrichment analysis (GSEA) plot of stemness genes (upregulated in hESCs) in control and KDM5B depleted (A) CAKI-1, IGROV1, and OVCAR cancer cells, and (B) 26 control and KDM5B depleted cancer cells. Loss of KDM5B leads to increased expression of stemness genes. C Meta-analysis of expression data from human renal, melanoma, ovarian, and leukemia tumors using Oncomine. Differentially expressed (DE) genes between control and KDM5B depleted cancer cells (>2 fold-change; FDR < 0.001) were analyzed using Oncomine (see “Methods”). Heatmap: Genes whose expression was upregulated in KDM5B depleted cancer cells were ordered by hierarchical clustering and tumors were sorted by average expression of genes upregulated in shKDM5B cancer cells from low to high (red). Loss of KDM5B leads to an expression signature consistent with multiple cancers. D DAVID gene ontology (GO) functional annotation of cancer disease terms in KDM5B depleted cancer cells relative to control cells in representative breast cancer cells.
Moreover, functional disease annotation of DE genes between control and KDM5B depleted cancer cells using DAVID revealed enrichment of cancer terms (cancer, neoplasm, carcinoma, leukemia/lymphoma) for 88% of the cancer cell lines (23 of 26) (Figs. 3D and S8 and S9 and Table S4). Loss of KDM5B resulted in differential expression of genes associated with multiple cancer types. While DE genes in KDM5B depleted triple negative breast cancer cells, BT-549, were associated with “breast cancer”, additional genes were associated with ovarian, lung, hematologic, renal, and bladder cancer. In addition, DE genes in other breast cancer cells MCF7 and T-47D were associated with various cancer subtypes, suggesting that loss of KDM5B impacts expression of cancer specific genes across multiple types of cancers.
KDM5B is a pan-cancer regulator of the H3K4me3 epigenome
To investigate the functional impact of depleting KDM5B on H3K4me3 distributions and levels across various cohorts of cancer cell lines we performed ChIP-Seq. A survey of H3K4me3 landscapes across 26 control and KDM5B depleted cancer epigenomes revealed varying numbers of peaks, where lung (HOP-92), melanoma (MALME-3M, SK-MEL-5, UACC-62), and ovarian cancer cells (OVCAR-3, OVCAR-4) displayed the greatest number of peaks, while a subset of breast (BT-549, MCF7), lung (EKVX, NCI-H322M, NCI-H522), and ovarian cancer cells (IGROV1) exhibited a smaller number of peaks (Fig. 4A). Intervene [47] pairwise intersections of H3Kme3 peaks (see methods) showed correlations between H3K4me3 occupancy across 26 control and KDM5B depleted cancer cell lines (Fig. 4B). While most pairs of control and KDM5B-depleted cancer cells clustered close to one another, control and KDM5B-depleted MCF7 breast, IGROV1 ovarian, and CAKI-1 renal cancer cells clustered further away from one another. These findings reveal increased sensitivity to depletion of KDM5B for a subset of cancer cells. These results also highlight relatively similar H3K4me3 patterning across a subset of breast, lung, melanoma, ovarian, and renal cancer cells, and between a subset of CNS and renal cancer cells. A comparison of H3K4me3 densities between control and KDM5B depleted cancer cells revealed variable sensitivity to depletion of KDM5B (Fig. 4C).
Fig. 4. Loss of KDM5B leads to epigenetic reprogramming of H3K4me3 in cancer cells.
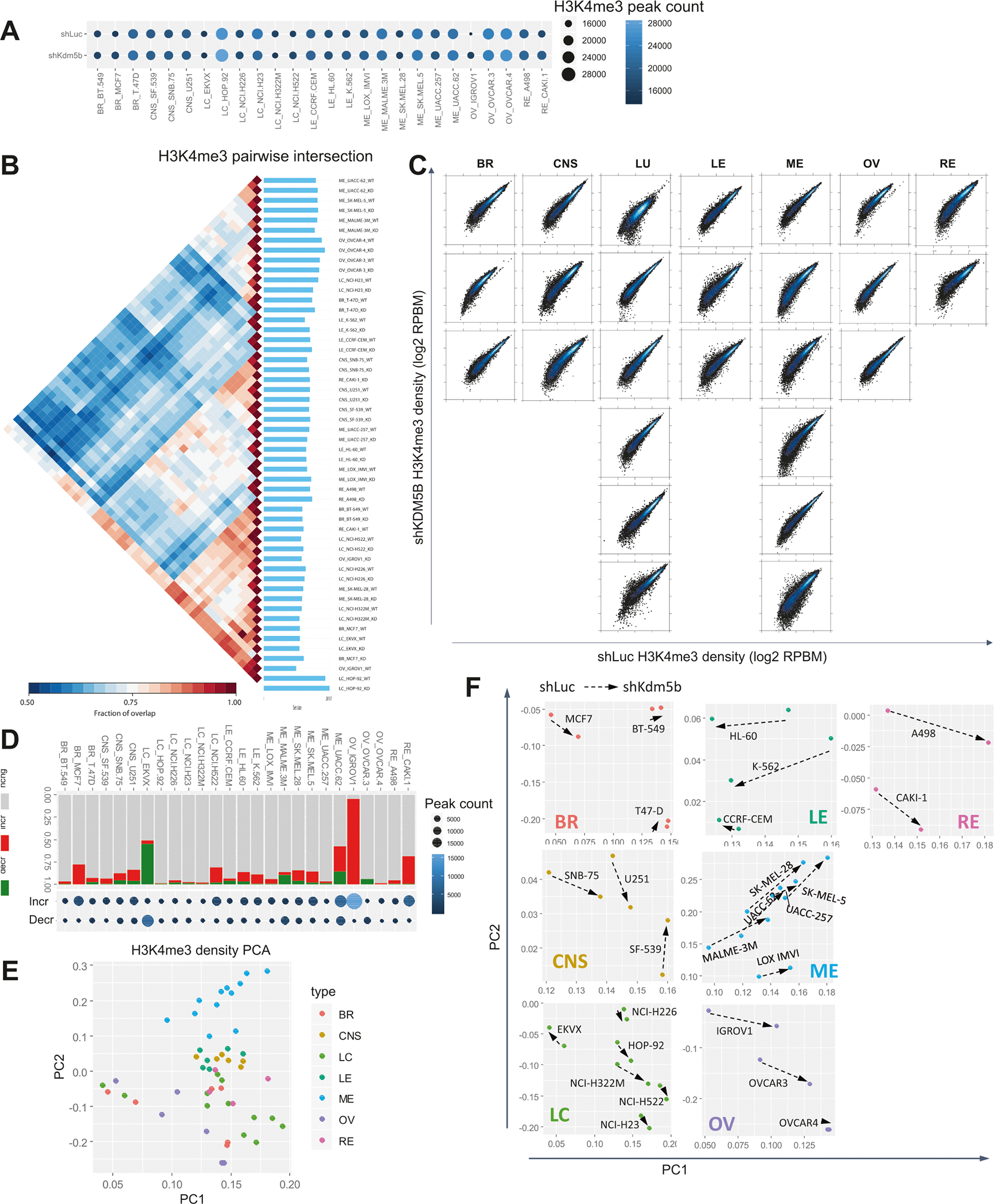
A Bubble plot showing the number of H3K4me3 peaks in control (shLuc) and KDM5B depleted (shKDM5B) cancer cells. B Intervene [47] pairwise intersection of SICER [49]-defined (FDR < 0.0001; see “Methods”) H3K4me3 enriched regions. Heatmap depicts pairwise intersection of genomic regions containing H3K4me3 peaks regions. C Scatter plots of H3K4me3 densities (log2 norm. tag density) across 26 cancer cell lines representing 7 cancer subtypes. D Bar plot and bubble plot showing SICER compare defined differential H3K4me3 peaks between control and KDM5B depleted cancer cells (see “Methods”). Bar plot shows the fraction of peaks with increased (red) or decreased (green) H3K4me3 density, or regions with no change (gray) in H3K4me3 density, while the bubble plot shows the number of differential H3K4me3 peaks (increased, decreased). The number of differential H3K4me3 peaks is represented by bubble size and color. E PCA analysis of H3K4me3 densities (norm. tag density) in 26 control and KDM5B depleted cancer cells. Seven types of cancer are color coded (BR: breast, CNS: central nervous system, LC: lung cancer, LE: leukemia, ME: melanoma, OV: ovary, RE: renal). F Cancer type-specific PCA analysis of control and KDM5B depleted H3K4me3 density profiles (X-axis, PC1; Y-axis, PC2).
An evaluation of genome coverage showed greater occupancy for several breast (T-47D), lung (HOP-92, NCI-H23), melanoma (MALME-3M, SK-MEL-5, UACC-62), and ovarian cancer cells (OVCAR-3, OVCAR-4), and lower occupancy for several breast (BT-549), lung (EKVX, NCI-H226, NCI-H322M, NCI-H522), melanoma (SK-MEL-28, UACC-257), and ovarian cancer cells (IGROV1) (Fig. S10A). We also observed increased coverage of H3K4me3 peaks in KDM5B depleted IGROV1 ovarian cancer cells. HOMER [48] annotation of H3K4me3 peaks demonstrated enrichment in promoter and intron regions, and to a lesser extent in intergenic regions (Fig. S10B). EKVX, MCF7, and IGROV1 exhibited a greater percentage of H3K4me3 promoter peaks while T-47D, HOP-92, NCI-H23, OVCAR-3, OVCAR-4, and SK-MEL-5 exhibited an increased percentage of intergenic H3K4me3 peaks. A comparison of H3K4me3 peaks with cytogenetic banding patterns showed that most genomic regions enriched with H3K4me3 are found in decondensed chromatin regions (Fig. S10C).
To investigate changes in H3K4me3 patterning upon loss of KDM5B we compared H3K4me3 distributions in control and KDM5B depleted cancer cells using Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) [49] islands (see “Methods”). Depletion of KDM5B resulted in increased H3K4me3 levels across most cancer cell lines (Fig. 4D), where KDM5B depleted MCF7 breast, NCI-H522 lung, UACC-62 melanoma, and IGROV1 ovarian, and CAKI-1 renal cancer cells exhibited the greatest increases in H3K4me3. We also observed decreased H3K4me3 levels in EKVX lung cancer cells following depletion of KDM5B. These findings reveal that loss of KDM5B leads to variable increases in H3K4me3 levels in most cancer cells. Our results also demonstrate variable sensitivity of H3K4me3 levels to loss of KDM5B across a diverse panel of cancer cells. These findings also demonstrate that cancer cell lines with higher expression of KDM5B exhibited greater alterations in H3K4me3 levels upon loss of KDM5B.
Next, we investigated whether loss of KDM5B impacts H3K4me3 patterning at promoters of stemness signature genes upregulated in hESCs [45] (described in Figs. 3 and S4). To this end, we evaluated SICER-defined alterations in H3K4me3 patterning at promoter regions of hESC stemness genes in KDM5B depleted cells. Depletion of KDM5B resulted in increased H3K4me3 levels at promoter regions of multiple stemness genes across several cancer cell lines (Fig. S11). These results link alterations in H3K4me3 due to depletion of KDM5B with increased expression of stemness genes. Elevated expression of stemness genes or natural variation in epigenetic or transcriptional response due to loss of KDM5B may contribute to the dedifferentiated phenotype of KDM5B depleted cancer cells.
PCA further demonstrated heterogeneous patterning of H3K4me3 densities across 26 cancer cell lines, where CNS, leukemia, and renal cancer cells were clustered close to each other while lung, melanoma, and ovarian cancer cells were more scattered throughout the 2D space (Fig. 4E). PCA analysis of cancer type-specific H3K4me3 densities demonstrated altered epigenomic trajectories following loss of KDM5B in multiple types of cancer cells (Fig. 4F). Depletion of KDM5B in MCF7 breast, SNB-75, SF, 549, and U251 CNS, EKVX and NCI-H322M lung, K-562 and HL-60 leukemia, MALM3–3M, SK-MEL-28, SK-MEL-5, and UACC-62 melanoma, IGROV1 and OVCAR-3 ovarian, and CAKI-1 and A498 renal cancer cells resulted in the greatest 2D shift. These findings highlight the variable impact of depleting KDM5B on sensitivity of cancer cells to H3K4me3 levels and distributions.
Mutations in regions enriched with H3K4me3
Next, we investigated whether mutations are enriched in genomic regions marked by H3K4me3, which are associated with DE genes in KDM5B depleted cancer cells. For this purpose, we calculated enrichment of genomic mutations in H3K4me3 regions across a panel of control and KDM5B depleted cancer cell lines using public whole-exome sequencing data [50–52]. These results revealed variable enrichment of mutations, including the greatest mutation burden in genomic regions associated with upregulated expression in BT-549 breast cancer cells, and downregulated expression in NCI-H522 lung, CCRF-CEM leukemia, SK-MEL-5 melanoma, and IGROV1 ovarian cancer cells (Fig. S12A). We also observed the elevated densities of deletion frameshift, insertion in frame, and substitution coding silent mutation subtypes in upregulated genes, while deletion in frame and complex insertion in frame mutation subtypes were enriched nearby genes whose expression was unchanged in KDM5B depleted cancer cells (Fig. S12B). These findings describe variation in mutation densities between chromatin regions enriched with H3K4me3, which are associated with genes that are DE in KDM5B depleted cancer cells.
Transcriptional dynamics of genes associated with altered H3K4me3 in KDM5B depleted cancer cells
Broad H3K4me3 domains are located at promoters of tumor suppressor genes (TSG) and genes that define cell identity in normal cells [53, 54]. Moreover, changes in the length of broad H3K4me3 regions near TSGs is correlated with altered gene expression, where decreased length of broad H3K4me3 regions is associated with repressed transcription. An evaluation of changes in the length of H3K4me3 domains revealed alterations in breadth of H3K4me3 domains in a panel of KDM5B depleted cancer cells (Fig. 5A). GO annotation of genes associated with differential H3K4me3 patterning revealed multiple cancer specific disease terms (Fig. 5B). Similar cancer terms were enriched in multiple cancer cells belonging to distinct cancer subtypes, suggesting that depletion of KDM5B may impact expression of genes involved in common pathways across distinct types of cancer cells.
Fig. 5. Breadth of H3K4me3 domains is linked to cancer cell identity.

A Scatter plots of H3K4me3 breadth (kb) in 26 control and KDM5B depleted cancer cell lines. B DAVID gene ontology (GO) functional annotation of genes whose promoters are associated with differential H3K4me3 between control and KDM5B depleted cancer cells. The p value (−log10) of cancer disease terms is represented by bubble size and color. Bubble plot showing the number of H3K4me3 peaks in control (shLuc) and KDM5B depleted (shKDM5B) cancer cells. C Scatter plot of H3K4me3 width (x-axis) and height (y-axis). Red and blue points represent broad and sharp peaks, respectively. D Enrichment p values of tumor suppressor (TSG), oncogenes (OG), and housekeeping genes (HKG) for genes associated with differential H3K4me3 peaks for all 26 cancer cell lines. E Percentage of all promoters with H3K4me3 or promoters with altered H3K4me3 in KDM5B depleted cancer cells enriched at TSG, OG, or HKG. F Change of H3K4me3 breadth in KDM5B depleted cancer cells. Boxplots of H3K4me3 peak width (kb) at differential broad (>4kb) and sharp (<4 kb) H3K4me3 peaks in representative control and KDM5B depleted cancer cells. H3K4me3 (G) widths (y-axis) or (H) heights (x-axis) and expression level (log2 RPKM; x-axis) for regions with differential H3K4me3 for a representative control and KDM5B depleted cancer cell line. Red and blue points show broad and sharp H3K4me3 peaks, respectively. I RNA-Seq expression level (log2 RPKM) of upregulated or downregulated DE genes associated with broad or sharp H3K4me3 peaks. p values were calculated using two-sided K–S tests.
We also observed a subset of lower density H3K4me3 regions, which exhibited altered levels in KDM5B depleted cells, that were wide and a distinct set of higher density H3K4me3 regions that were narrower (Figs. 5C and S13). To investigate whether altered H3K4me3 domains were associated with tumor suppressors, we evaluated enrichment of TSGs, oncogenes, and housekeeping genes at all promoters containing H3K4me3 and at promoters with altered H3K4me3 (SICER compare function; shKDM5B/shLuc; see “Methods”). Tumor suppressors and oncogenes were defined by somatic mutation profiles from >8000 paired tumor-normal samples [55] and housekeeping genes were also used as a control [56]. Our results show that while all broad H3K4me3 peaks and cancer type-specific broad H3K4me3 peaks are enriched with TSG relative to oncogenes (Figs. 5D and S14), promoters of tumor suppressor and housekeeping genes are less sensitive to alterations in breadth and level of H3K4me3 relative to oncogenes in KDM5B depleted cells (Fig. 5E). In addition, a cancer type-specific analysis revealed that while TSG and housekeeping genes exhibit lower sensitivity to alterations in H3K4me3 in KDM5B depleted cells, oncogenes exhibited variable sensitivity (Fig. S15), where breast, CNS, leukemia, melanoma, and ovarian displayed an increased percentage of promoters with altered H3K4me3, while lung cancer cells had a decreased percentage of promoters with altered H3K4me3. Also, while the length of broad H3K4me3 domains was mostly unchanged, we observed lengthening of sharp H3K4me3 domains in KDM5B depleted cancer cells (Figs. 5F and S16). Moreover, genes whose promoters exhibited broad H3K4me3 peaks (>4 kb) were expressed higher compared to genes with sharp H3K4me3 peaks (Figs. 5G–I and S17–S20). These findings also highlight cell line specific changes in expression of genes associated with sharp or broad H3K4me3 peaks. Alterations in breadth and expression of tumor suppressor, oncogenes, and housekeeping genes associated with broad H3K4me3 peaks at oncogenes may regulate tumor potentiation or progression.
Functional evaluation of KDM5B depleted cancer cells
Depletion of KDM5B in a subset of cell lines, SK-MEL-28 melanoma cells, CAKI-1 renal cancer, EKVX lung cancer, IGROV1 ovarian cancer, and MCF7 breast cancer cell lines, resulted in morphological changes indicative of dedifferentiation, and increased proliferation. Depletion of KDM5B in CAKI-1 renal cancer cells resulted in aggregation of cells and spontaneous 3D growth characteristics in a 2D cell culture dish (Figs. 6A and S21A, B), resembling a more primitive cellular state, while depletion of KDM5B in EKVX lung cancer cells results in decreased morphological heterogeneity (Figs. 6A and S21C). In addition, depletion of KDM5B in IGROV1 cells resulted in greater enrichment of shiny or circular IGROV1 cells (Fig. S21D). We also observed increased proliferation of KDM5B depleted SK-MEL-28, CAKI-1, EKVX, and IGROV1 cells in 3D Matrigel concentrically away from the original embedded cells protruding the matrix in all directions (Fig. 6B). In contrast, control SK-MEL-28, CAKI-1, EKVX, and IGROV1 cells formed 3D structures, but did not protrude as much into the matrix (Fig. 6B). We further evaluated the proliferation rate of a subset of KDM5B depleted cancer cells, which exhibited altered morphological characteristics. MTT assays demonstrated increased proliferation of KDM5B depleted MCF7, EKVX, SK-MEL-28, IGROV1, and CAKI-1 cells (Fig. 6C). We also investigated the in vivo consequence of depleting KDM5B in IGROV1 ovarian and CAKI-1 renal cancer cells, which exhibited the greatest increase in proliferation relative to control cancer cells. To this end, control or KDM5B-depleted IGROV1 or CAKI-1 cells were injected subcutaneously into SCID-beige mice. Mice injected with shKDM5B IGROV1 or CAKI-1 cells (Fig. 6D–G) exhibited increased tumor growth relative to tumors generated from shLuc IGROV1 or CAKI-1 cells. In vivo tumor growth was further evaluated using limiting dilution assays, which is a technique used to evaluate stemness of tumor cells in immunocompromised mice [57]. Bulk control (shLuc) or shKDM5B IGROV1 cells were injected subcutaneously into NOD scid gamma (NSG) mice. Results from these limiting dilution assays demonstrate that tumors appeared with decreased latency for KDM5B-depleted IGROV1 cells relative to control cells, and shKDM5B IGROV1 cells exhibited increased tumor growth (Fig. 6H). Combined, these results support a role for KDM5B in regulating cancer cell tumorigenicity.
Fig. 6. Functional and morphological characteristics of KDM5B depleted cancer cells.
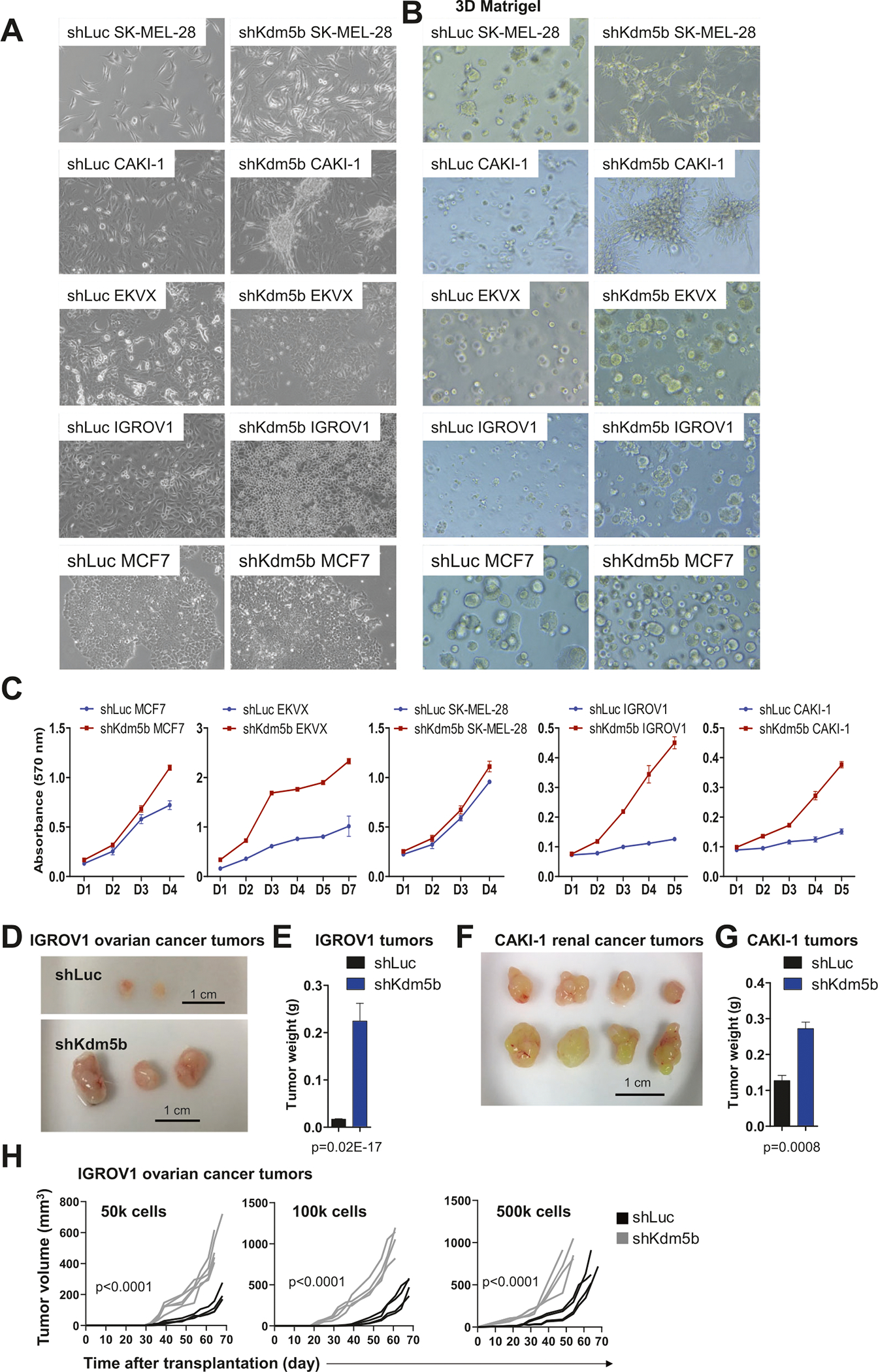
A Cancer cell lines transduced with shLuc or KDM5B-shRNA-2 (shKDM5B) lentiviral particles. shKDM5B CAKI-1, EKVX, and IGROV1 cancer cells exhibit morphological differences. KDM5B depleted CAKI-1 cells exhibit 3D growth characteristics, while KDM5B depleted EKVX are morphologically more homogenous, and “shiny” or circular IGROV1 cells became more prevalent upon knockdown of KDM5B. B shLuc and shKDM5B cancer cells cultured in 3D Matrigel. C MTT assays revealed that KDM5B-depleted cancer cells exhibit increased proliferation. Tumor growth assay. 106 shLuc or shKDM5B (D, E) IGROV1 or (F, G) CAKI-1 cells were subcutaneously injected into SCID-beige mice (n = 3 for IGROV1; n = 4 for CAKI1). E Weight of IGROV1 and G CAKI-1 tumors. Tumor weight plots are presented as mean ± SEM. H Limiting dilution tumor growth assay. 50k, 100k, or 500k shLuc or shKDM5B IGROV1 cells were subcutaneously injected into NSG mice (n = 4). Tumor volume was measured twice weekly. Statistical significance was determined using two-way ANOVA.
Cancer stem cells (CSC) have been identified in various solid and hematopoietic cancers including ovarian, leukemia, breast, colon, brain, lung, liver, skin, pancreatic, and prostate using a variety of cell markers such as CD133, CD15, CD44, and CD24 [58–60]. Profiling of CSC markers has also been evaluated for cells in the NCI60 panel [61]. Because we observed morphological changes in KDM5B depleted cancer cells indicative of dedifferentiation in a subset of cell lines, we evaluated the functional effect of depleting KDM5B on expression of CSC markers by performing flow cytometry analyses (see methods). Our results revealed increased levels of CD133 in OVCAR4 ovarian cancer cells, and minor increases in CD133 in K562 leukemia, CAKI1 and UACC-62 renal cancer cells (Fig. 7A). Depletion of KDM5B also resulted in greater enrichment of CD15+ and CD44+ cells in K562, UACC-62, IGROV1, OVCAR-4, CAKI-1, and LOX IMVI cancer cells. We also observed greater enrichment of CD15+ and CD24+ cells in OVCAR-3 ovarian cancer cells, and greater enrichment of CD24+ cells in CAKI-1 cells, and decreased enrichment of CD24+ in K562 cells. T-distributed stochastic neighbor embedding (t-SNE) analysis of KDM5B depleted ovarian cancer flow cytometry data further revealed altered distributions of CSC markers relative to control cancer cells (Fig. 7B). t-SNE projection of control and KDM5B depleted K-562 and IGROV1 cells revealed altered patterns of CD44, CD24, CD133, and CD15, while loss of KDM5B in UACC-62 revealed altered projections of CD24, CD133, and CD15. Combined, these results suggest that depletion of KDM5B leads to greater enrichment of CSC markers.
Fig. 7. Altered expression of cancer stem cell markers following loss of KDM5B.

A Expression analysis of CSC surface markers CD24, CD44, CD15, and CD133 was analyzed using flow cytometry and FlowJo software. CD24, CD44, CD15, and CD133 expression profiles are shown for control (shLuc; black) and shKDM5B (red) K562 leukemia, LOX IMVI and UACC-62 melanoma, IGROV1, OVCAR-3, and OVCAR-4 ovarian cancer cells, and CAKI-1 renal cancer cells. B t-SNE projection analysis defines CD24+, CD44+, CD15+, and CD133+ populations.
DISCUSSION
In this study, we evaluated the effect of loss of KDM5B on epigenetic patterning of H3K4me3, transcriptional repertoire, and function of multiple cancer cell lines representing a diverse set of cancer subtypes. Because KDM5B is overexpressed in a variety of cancer types, cancer biologists and clinicians have postulated that its targeted inhibition may represent a viable therapeutic strategy to decrease proliferation of cancer cells and decrease tumor growth [62, 63]. However, there are several considerations for the clinical application of KDM5B targeted inhibition as a cancer therapy [64]. It is unclear from previous studies whether elevated expression levels of KDM5B positively or negatively impacts the proliferation rate of cancer cells. It is possible that KDM5B expression may inhibit cancer progression in certain cancer subtypes or promote tumor progression in other cases. Results from this study show that depletion of KDM5B in cell lines representing multiple types of cancer, including breast, lung, melanoma, ovarian, and renal cancer cell lines, which express elevated levels of KDM5B, leads to increased proliferation (Fig. 6C). Our findings are in alignment with previous findings which showed that KDM5B plays a role in cell fate decisions in oral cancer [65]. Our results demonstrate that depletion of KDM5B in cancer cells which overexpress KDM5B leads to alterations in cell fate trajectories including a dedifferentiated phenotype as evaluated by flow cytometry profiling of CSC markers in a subset of KDM5B cancer cell lines and in vitro morphological changes, increased tumor growth in vivo, and epigenetic and transcriptional plasticity. Depletion of KDM5B in CAKI-1 renal cancer cells resulted in robust morphological changes resembling a dedifferentiated phenotype (Fig. 6A). Consistent with these findings, we observed altered profiles of putative cell surface markers of CSCs or tumor initiating cells (CD133, CD15, CD24, CD44) upon loss of KDM5B in multiple cancer cells (Fig. 7). While we observed variable expression of individual markers across multiple cancer cells lines, we observed systematic increases in CSC marker enrichment following depletion of KDM5B. In addition, we also observed changes in transcriptional patterning upon loss of KDM5B, where KDM5B depleted cancer cells exhibited increased expression of stemness signature genes (Figs. 3A and S4). Our previous findings demonstrate that KDM5B is a positive regulator of pluripotent ES cell differentiation [9, 17], where loss of KDM5B leads to sustained self-renewal in the absence of extrinsic or intrinsic signals. It is plausible that KDM5B broadly functions to regulate differentiation in normal and cancer cells, where loss of KDM5B delays differentiation or restricts differentiation capacity. In this case, loss of KDM5B may lead to a persistent population of self-renewing tumor initiating cells which may be resistant to anti-proliferative therapies.
Our findings reveal that depletion of KDM5B leads to increased H3K4me3 levels in most cancer cell lines. However, KDM5B depleted EKVX cells exhibit increased H3K4me3 levels at a subset of peaks (Fig. 4D). A possible explanation for increased H3K4me3 levels at a subset of peaks is enrichment of a more homogenous population of cells following depletion of KDM5B in EKVX cells, which may exhibit a distinct chromatin profile relative to control EKVX heterogenous bulk population of cells. In agreement with this hypothesis, KDM5B depleted EKVX cells exhibit decreased morphological heterogeneity (Figs. 6A and S21A, C), which may shift the population of cells from a heterogenous state to a more homogenous distribution.
CSCs have historically been associated with a slow cycling or quiescent, non-dividing cellular state as a response to drug treatment, where leukemic and solid tumors [66–68] have been shown to enter an arrested cell cycle after chemotherapy. Selective pressure shifts proliferative CSCs into a dormant, or quiescent, slow-cycling state that is associated with stress response, where cells can adapt as an integrated stress response[69]. However, the quiescent state is reversible: quiescent stem cells are poised for activation to a more highly proliferative state, where cell signals and epigenetic changes can accelerate CSC reawakening. Our findings, which show that depletion of KDM5B leads to a proliferative state, is in alignment with an activated CSC phenotype, where KDM5B depleted cancer cells exhibit increased proliferation. Also, in contrast to cells in a quiescent state, which exhibit chromatin enriched with repressive histone modifications [70], our findings show that loss of KDM5B leads to elevated H3K4me3, which is associated with open chromatin.
Epigenetic regulators such as histone demethylases are essential in establishing cell type-specific expression programs. While many studies focusing on intratumor heterogeneity have evaluated genetic mutations [5], epigenetic and transcriptional heterogeneity also likely contributes to tumor progression and drug resistance [71]. In support of this model, KDM5B activity has been shown to be a regulator of transcriptional heterogeneity in ER+ luminal breast cancers [33]. As inhibitors of KDM5B are currently being developed for their use in clinical trials for cancer patients, knowledge about the role for KDM5B in regulating cancer epigenomes will be instrumental as these therapies are utilized in a clinical setting. Future studies investigating the impact of depleting or targeted inhibition of KDM5B on patient survival with tumors exhibiting high or low expression levels of KDM5B will be impactful in understanding the role of this H3K4 demethylase in regulating tumor progression and response to therapies.
MATERIAL AND METHODS
Cell culture
The cancer cell lines were obtained from the NCI DTP Tumor Repository. The NCI DTP Tumor Repository performed Applied Biosystems AmpFLSTR Identifiler testing with PCR amplification to confirm consistency with the published Identifiler STR profile for each of the cancer cell lines. Cell culture was performed as described previously [72]. Briefly, cells were cultured in RPMI 1640/5% FBS media containing glutamine and pen/strep at 37 °C with 5% CO2. Cells were passaged by washing with PBS and dissociating with trypsin using serological pipettes (sc-200279 and sc-200281, Santa Cruz Biotechnology).
Lentiviral transduction
Human cancer cell lines were transduced with lentiviral particles encoding shRNAs as described previously [73]. Briefly, shRNAs were cloned into the pGreenPuro (pSIH1-H1-puro Vector; System Biosciences) according to the manufacture’s protocol. Next, HEK 293T cells were transfected with an envelope plasmid (pLP/VSVG), packaging vector (psPAX2), and shRNA expression vector using lipofectamine 2000. Twenty-four to 48 h post transfection, the medium containing lentiviral particles was filtered and used to transduce cancer cells. For cancer cells, the medium (RPMI 1640, glutamine, and 5% FBS) containing lentiviral particles was used to transduce human cancer cells overnight. Twenty-four hours post transduction cancer cells were stably selected in the presence of 1 μg/ml puromycin to generate a heteroclonal population.
ChIP-Seq analysis
Chromatin immunoprecipitation followed by next-generation sequencing (ChIP-Seq) was performed as previously described [2]. The rabbit monoclonal antibody H3K4me3 (17–614) antibody was obtained from Millipore [74, 75]. In brief, 15 million human cancer cells were trypsinized into a single-cell suspension and crosslinked with formaldehyde (1%) for 10 min at 37°C. Crosslinked cell pellets were flash frozen in liquid nitrogen and stored at −80 °C. Next, cell pellets were thawed and sonicated, and cell extracts (5 million cells) were used for ChIP experiments. DNA (ChIP-enriched and Input) was end-repaired using the End-It DNA End-Repair kit (Epicentre), followed by addition of a single A nucleotide, and ligation of Illumina adapters. PCR was performed using Phusion 2X High Fidelity PCR master mix. Libraries were sequenced on an Illumina HiSeq platform. Sequence reads were mapped to the human genome (hg19) using bowtie2 [76] with default settings. C++ programs to convert a SAM formatted file to a BED6 format from bowtie2 (Sam2Bed6_Bowtie2), to remove redundant reads from a BED6 file (RemoveRedundantReads), and to convert a BED6 file to a BEDGraph file (GenerateRPBMBasedSummary) were described previously [77].
ChIP-Seq peaks (H3K4me3 enriched regions) were identified relative to control Input using SICER [49] with a window size setting of 200 bps, a gap setting of 400 bps, and a false discovery rate (FDR) setting of 0.001. The SICER-compare function was used to compare multiple samples (FDR < 0.001, fold-change (FC) > 1.5). ChIP-Seq libraries were normalized by library size, where the measure read per base per million reads measure was used to calculate ChIP-Seq densities at genomic regions. Two biological replicates were performed for the ChIP-Seq analyses. The Kolmogorov–Smirnov test was used to obtain p value statistics for comparing density of ChIP-enrichment at genomic regions. The UCSC genome browser was used to visualize normalized ChIP data.
Broad H3K4me3 domains
We included a stringent definition of a broad H3K4me3 peak. H3K4me3 peaks that intersected TSS regions of hg19 refseq genes, and whose length exceeds 4 kb in length (≥4kb) were considered broad H3K4me3 domains, while H3K4me3 peaks whose length was less than 4 kb (<4kb) were considered sharp peaks.
RNA-Seq analysis
Poly-A mRNA was purified using the New England Biolabs NEBNext Ultra II RNA Library Prep Kit for Illumina. RNA-Seq libraries were sequenced at Novogene using an Illumina platform according to the manufacturer’s protocol. Sequence reads were mapped to the human genome (hg19) using bowtie2 [76] with default settings. The RPKM measure (read per kilobases of exon model per million reads) [78] was used to quantify the mRNA expression level of a gene from RNA-Seq data. DE genes were identified using edgeR (FDR < 0.001; FC > 1.5) [39]. Two biological replicates were performed for the RNA-Seq analyses.
Oncomine
DE genes between shKDM5B cancer and control (shLuc) cancer cells were evaluated using Oncomine [35].
Q-RT-PCR expression analysis
Total RNA was harvested from control and KDM5B depleted cancer cells using an RNeasy Mini Kit (Qiagen, Valencia, CA) and DNase treated. Reverse transcription was performed using a Superscript III kit (Invitrogen, Carlsbad, CA). Intron spanning primers used for Q-RT-PCR were designed using the Universal Probe Library Assay design Center (Roche) or Primer 3. Q-RT-PCR was performed using primers and SYBR green PCR Master Mix reagents (Applied Biosystems).
Xenograft tumor model
Human ovarian (IGROV1) and renal (CAKI-1) cancer cell lines were dissociated into single cells and 106 cells were injected subcutaneously into female SCID-beige mice (n = 3, IGROV1; n = 4 CAKI1), aged 6–8 weeks, in accordance with Institution Animal Care and Use Committee (IACUC) guidelines under current approved protocols at Wayne State University. After several weeks (4–6), when the tumors grew to ~1 cm in diameter, mice were euthanized and tumors were washed in PBS and weighed. For limiting dilution assays, shLuc or shKdm5b IGROV1 cells were suspended in PBS and different dilutions (50k, 100k or 500k) were injected subcutaneously into female NSG mice (n = 4), aged 6–8 weeks, in accordance with IACUC guidelines under approved protocols at WSU. Animals were monitored twice weekly for tumor growth. Tumor volume was calculated using the (L×W2)/2 formula. Statistical analysis of tumor growth was evaluated using a two-way ANOVA. For the experiments using immunocompromised mice (NSG or SCID-beige), the female animals with the same age were randomly grouped.
MTT assay
Cell viability was evaluated by MTT cell proliferation assays. Cancer cells were plated at a density of 5–10k cells per well in a 96-well culture plate and incubated overnight 37 °C with 5% CO2. Next, 10 μl MTT (0.5 mg/ml in cell culture media) was added to 100 μl medium for 3 h and subsequently replaced and dissolved by addition of 150 μl DMSO. 96-well plates were shaken for 10 min to dissolve crystals and absorbance at 570 nm wavelength was evaluated using a spectrophotometer. At least three biological replicates were performed.
Flow cytometry
Control (shLuc) or shKDM5B Cancer cells were washed with 1X PBS, trypsinized to a single-cell suspension, and re-suspended at a density of 106 cells/ml in FACS buffer (PBS + 1% FBS). Cells were incubated with 1 μl LIVE/DEAD™ Fixable Near-IR Dead Cell Stain (L34975) for 30 min at room temperature, and washed 1X with FACS buffer. Next, 100 μl of 106 cells/ml suspension was added to a FACS tube, and cells were incubated with anti-CD44, anti-CD24, anti-CD15, and anti-CD133 antibodies (3 μl per 100,000 cells) for 45 min on ice. After staining, cells were spun down at 1000 rpm for 5 min, and re-suspended in 250 μl FACS buffer. Antibodies were obtained from BD Bioscience (San Jose, CA). CD44 (BV510; BDB563029), CD24 (PE-Cy7; BDB561646), CD15 (BV786; BDB741013), CD133 (BV421; BDB56595). Compensation beads (BDB552843; BD Biosciences) were used to optimize compensation settings for colors and spectral overlap emitted by fluorochromes (within each laser and across lasers). Compensation beads were incubated with each antibody individually according to the manufacturer’s instructions. Unstained and stained beads were run separately. Gates were set to include GFP positive control and shKDM5B cancer cells. A BD LSR II instrument was used for flow cytometry analysis. Data were analyzed using FloJo (FlowJo, LLC). Three biological replicates were performed.
Mutation analysis
Whole-exome sequencing data for cancer cells [50] was downloaded from Cosmic [51]. An evaluation of mutations in regions enriched with differential H3K4me3 domains was performed using bedtools intersect. To compare enrichment of mutations across 26 cancer genomes, clustering was performed as shown in Fig. S12A. Annotation of mutations (e.g., deleterious or silent) and their frequencies across 26 cancer genomes is shown in Fig. S12B. Mutation density was calculated in SICER-defined regions enriched with differential H3K4me3 domains. Results from these analyses are shown in Fig. S12.
Gene ontology functional annotation
DAVID [40] was used to functionally annotate genes (p < 0.05 was considered significant). Enrichment of tumor suppressors, oncogenes, and housekeeping genes was evaluated using Fisher’s exact tests.
CONCLUSIONS
Our findings provide new insight into the role of the H3K4 demethylase, KDM5B, in regulating cancer cell epigenomes. Results presented in this study also provide a resource to evaluate genome-wide associations between alterations in H3K4me3 patterning and transcriptional changes upon depletion of KDM5B in a variety of cancer cell lines.
DATA AVAILABILITY
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE165959.
Supplementary Material
ACKNOWLEDGEMENTS
This work utilized the Wayne State University High Performance Computing Grid for computational resources (https://www.grid.wayne.edu/). Flow cytometry was performed in the Microscopy, Imaging, and Cytometry Resources (MICR) core at the Karmanos Cancer Institute and Wayne State University. We thank Lisa Polin for helpful discussions.
FUNDING
This work was supported by Wayne State University, Karmanos Cancer Institute, and a grant from the Elsa U. Pardee Foundation awarded to BLK.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
CONSENT FOR PUBLICATION
All authors have read and approved the final version of this manuscript.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-022-02311-z.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES
- 1.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. [DOI] [PubMed] [Google Scholar]
- 2.Gopi LK, Kidder BL. Integrative pan cancer analysis reveals epigenomic variation in cancer type and cell specific chromatin domains. Nat Commun. 2021;12:1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. [DOI] [PubMed] [Google Scholar]
- 5.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28. [DOI] [PubMed] [Google Scholar]
- 6.Muntean AG, Hess JL. Epigenetic dysregulation in cancer. Am J Pathol. 2009;175:1353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Timp W, Feinberg AP. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer. 2013;13:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. [DOI] [PubMed] [Google Scholar]
- 9.Kidder BL, Hu G, Zhao K. KDM5B focuses H3K4 methylation near promoters and enhancers during embryonic stem cell self-renewal and differentiation. Genome Biol. 2014;15:R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–11. [DOI] [PubMed] [Google Scholar]
- 11.Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. [DOI] [PubMed] [Google Scholar]
- 12.Sims RJ 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–39. [DOI] [PubMed] [Google Scholar]
- 13.Albert M, Schmitz SU, Kooistra SM, Malatesta M, Morales Torres C, Rekling JC, et al. The histone demethylase Jarid1b ensures faithful mouse development by protecting developmental genes from aberrant H3K4me3. PLoS Genet. 2013;9: e1003461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catchpole S, Spencer-Dene B, Hall D, Santangelo S, Rosewell I, Guenatri M, et al. PLU-1/JARID1B/KDM5B is required for embryonic survival and contributes to cell proliferation in the mammary gland and in ER+ breast cancer cells. Int J Oncol. 2011;38:1267–77. [DOI] [PubMed] [Google Scholar]
- 15.Dey BK, Stalker L, Schnerch A, Bhatia M, Taylor-Papidimitriou J, Wynder C. The histone demethylase KDM5b/JARID1b plays a role in cell fate decisions by blocking terminal differentiation. Mol Cell Biol. 2008;28:5312–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frankenberg S, Smith L, Greenfield A, Zernicka-Goetz M. Novel gene expression patterns along the proximo-distal axis of the mouse embryo before gastrulation. BMC Dev Biol. 2007;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kidder BL, Hu G, Yu ZX, Liu C, Zhao K. Extended self-renewal and accelerated reprogramming in the absence of Kdm5b. Mol Cell Biol. 2013;33:4793–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz SU, Albert M, Malatesta M, Morey L, Johansen JV, Bak M, et al. Jarid1b targets genes regulating development and is involved in neural differentiation. EMBO J. 2011;30:4586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie L, Pelz C, Wang W, Bashar A, Varlamova O, Shadle S, et al. KDM5B regulates embryonic stem cell self-renewal and represses cryptic intragenic transcription. EMBO J. 2011;30:1473–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S, et al. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–45. [DOI] [PubMed] [Google Scholar]
- 21.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA. 2007;104:19226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Mao Y, Du G, He C, Han S. Overexpression of JARID1B is associated with poor prognosis and chemotherapy resistance in epithelial ovarian cancer. Tumour Biol. 2015;36:2465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett A, Madsen B, Copier J, Lu PJ, Cooper L, Scibetta AG, et al. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: a new cancer/testis antigen? Int J Cancer. 2002;101:581–8. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto S, Wu Z, Russnes HG, Takagi S, Peluffo G, Vaske C, et al. JARID1B is a luminal lineage-driving oncogene in breast cancer. Cancer Cell. 2014;25:762–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T, et al. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer. 2010;9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Su Y, Pan J, Zhou Z, Song B, Xiong E, et al. Connexin 26 is down-regulated by KDM5B in the progression of bladder cancer. Int J Mol Sci. 2013;14:7866–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roesch A, Becker B, Schneider-Brachert W, Hagen I, Landthaler M, Vogt T. Re-expression of the retinoblastoma-binding protein 2-homolog 1 reveals tumor-suppressive functions in highly metastatic melanoma cells. J Invest Dermatol. 2006;126:1850–9. [DOI] [PubMed] [Google Scholar]
- 29.Ohta K, Haraguchi N, Kano Y, Kagawa Y, Konno M, Nishikawa S, et al. Depletion of JARID1B induces cellular senescence in human colorectal cancer. Int J Oncol. 2013;42:1212–8. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z, Tang F, Qi G, Yuan S, Zhang G, Tang B, et al. KDM5B is overexpressed in gastric cancer and is required for gastric cancer cell proliferation and metastasis. Am J Cancer Res. 2015;5:87–100. [PMC free article] [PubMed] [Google Scholar]
- 31.Dai B, Hu Z, Huang H, Zhu G, Xiao Z, Wan W, et al. Overexpressed KDM5B is associated with the progression of glioma and promotes glioma cell growth via downregulating p21. Biochem Biophys Res Commun. 2014;454:221–7. [DOI] [PubMed] [Google Scholar]
- 32.Shigekawa Y, Hayami S, Ueno M, Miyamoto A, Suzaki N, Kawai M, et al. Overexpression of KDM5B/JARID1B is associated with poor prognosis in hepatocellular carcinoma. Oncotarget. 2018;9:34320–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. 2018;34:939–53 e939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. 2013;23:811–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stinson SF, Alley MC, Kopp WC, Fiebig HH, Mullendore LA, Pittman AF, et al. Morphological and immunocytochemical characteristics of human tumor cell lines for use in a disease-oriented anticancer drug screen. Anticancer Res. 1992;12:1035–53. [PubMed] [Google Scholar]
- 38.Reinhold WC, Varma S, Sunshine M, Elloumi F, Ofori-Atta K, Lee S, et al. RNA sequencing of the NCI-60: integration into cellminer and cellminer CDB. Cancer Res. 2019;79:3514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 41.Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26:976–8. [DOI] [PubMed] [Google Scholar]
- 42.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–26. [DOI] [PubMed] [Google Scholar]
- 44.Cheng CJ, Lin YC, Tsai MT, Chen CS, Hsieh MC, Chen CL, et al. SCUBE2 suppresses breast tumor cell proliferation and confers a favorable prognosis in invasive breast cancer. Cancer Res. 2009;69:3634–41. [DOI] [PubMed] [Google Scholar]
- 45.Bhattacharya B, Miura T, Brandenberger R, Mejido J, Luo Y, Yang AX, et al. Gene expression in human embryonic stem cell lines: unique molecular signature. Blood. 2004;103:2956–64. [DOI] [PubMed] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khan A, Mathelier A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinforma. 2017;18:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, et al. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Res. 2012;72:3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, et al. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29:569–74. [DOI] [PubMed] [Google Scholar]
- 57.O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113–20. [DOI] [PubMed] [Google Scholar]
- 58.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–91. [DOI] [PubMed] [Google Scholar]
- 59.Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. 2013;15:338–44. [DOI] [PubMed] [Google Scholar]
- 60.Oskarsson T, Batlle E, Massague J. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 2014;14:306–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stuelten CH, Mertins SD, Busch JI, Gowens M, Scudiero DA, Burkett MW, et al. Complex display of putative tumor stem cell markers in the NCI60 tumor cell line panel. Stem Cells. 2010;28:649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor-Papadimitriou J, Burchell J. JARID1/KDM5 demethylases as cancer targets? Expert Opin Ther Targets. 2017;21:5–7. [DOI] [PubMed] [Google Scholar]
- 63.Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol. 2016;12:531–8. [DOI] [PubMed] [Google Scholar]
- 64.Xhabija B, Kidder BL. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin Cancer Biol. 2019;57:79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Facompre ND, Harmeyer KM, Sole X, Kabraji S, Belden Z, Sahu V, et al. JARID1B enables transit between distinct states of the stem-like cell population in oral cancers. Cancer Res. 2016;76:5538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AM, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.min M, Spencer SL. Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways. PLoS Biol. 2019;17: e3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA. H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell. 2013;24:3025–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. 2017;14:611–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu H, D’Andrade P, Fulmer-Smentek S, Lorenzi P, Kohn KW, Weinstein JN, et al. mRNA and microRNA expression profiles of the NCI-60 integrated with drug activities. Mol Cancer Ther. 2010;9:1080–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kidder BL, Hu G, Cui K, Zhao K. SMYD5 regulates H4K20me3-marked heterochromatin to safeguard ES cell self-renewal and prevent spurious differentiation. Epigenetics Chromatin. 2017;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.He R, Kidder BL. Culture of haploid blastocysts in FGF4 favors the derivation of epiblast stem cells with a primed epigenetic and transcriptional landscape. Sci Rep. 2018;8:10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu J, Kidder BL. KDM5B decommissions the H3K4 methylation landscape of self-renewal genes during trophoblast stem cell differentiation. Biol Open. 2018;7: bio031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu G, Zhao K. Correlating histone modification patterns with gene expression data during hematopoiesis. Methods Mol Biol. 2014;1150:175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. [DOI] [PubMed] [Google Scholar]
- 79.Dyrskjot L, Kruhoffer M, Thykjaer T, Marcussen N, Jensen JL, Moller K, et al. Gene expression in the urinary bladder: a common carcinoma in situ gene expression signature exists disregarding histopathological classification. Cancer Res. 2004;64:4040–8. [DOI] [PubMed] [Google Scholar]
- 80.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28:2529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci USA. 2001;98:13790–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology. 2008;55:2016–27. [PubMed] [Google Scholar]
- 83.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cutcliffe C, Kersey D, Huang CC, Zeng Y, Walterhouse D, Perlman EJ, et al. Clear cell sarcoma of the kidney: up-regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin Cancer Res. 2005;11:7986–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE165959.