

Abstract
The W215A/E217A mutant thrombin is called “anticoagulant thrombin” because its activity towards its procoagulant substrate, fibrinogen, is reduced more than 500-fold whereas in the presence of thrombomodulin (TM) its activity towards its anticoagulant substrate, protein C, is reduced less than 10-fold. To understand how these mutations so dramatically alter one activity over the other, we compared the backbone dynamics of wild type thrombin to those of the W215A/E217A mutant thrombin by hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS). Our results show that the mutations cause the 170s, 180s and 220s C-terminal β-barrel loops near the sites of mutation to exchange more, suggesting that the structure of this region is disrupted. Far from the mutation sites, residues at the N-terminus of the heavy chain, which need to be buried in the Ile pocket for correct structuring of the catalytic triad, also exchange much more than in wild type thrombin. TM binding causes reduced H/D exchange in these regions and also alters the dynamics of the β-strand that links the TM binding site to the catalytic Asp 102 in both wild type thrombin and in the W215A/E217A mutant thrombin.. In contrast, whereas TM binding reduces the dynamics the 170s, 180s and 220s C-terminal β-barrel loops in WT thrombin, this region remains disordered in the W215A/E217A mutant thrombin. Thus, TM partially restores the catalytic activity of W215A/E217A mutant thrombin by allosterically altering its dynamics in a manner similar to that of wild type thrombin.
Keywords: serine protease, protein dynamics, hydrogen-deuterium exchange, relaxation dispersion, allostery
Introduction
The serine protease, thrombin, circulates in the blood as the zymogen prothrombin. Vascular tissue damage triggers the clotting cascade, which ultimately converts prothrombin to α-thrombin via proteolytic cleavage near the site of the compromised tissue (1). Once cleaved, the newly formed N-terminal residue of the thrombin heavy chain, Ile 16CT (37seq), inserts into the Ile cleft, and is known to play an important role in establishing the structure of the serine protease catalytic triad (2, 3). Thrombin is often described as a “dual-action” protease as this enzyme is also capable of stimulating the anticoagulative pathway by cleaving -and thereby activating-protein C (4). The binding of the protein cofactor, thrombomodulin (TM), to thrombin is the key regulatory switch that toggles thrombin substrate specificity away from procoagulative substrates and towards cleavage and activation of protein C. Using amide exchange mass spectrometry (HDX-MS) we discovered that in solution, the N-terminus of the heavy chain remains solvent accessible until either a substrate analog or thrombomodulin (TM) binds (5). This result suggested that a key role of TM binding is to establish the catalytically active structure of thrombin for cleavage of protein C.
The W215A/E217A double mutant thrombin has been shown to act as a safe and efficacious anticoagulant intervention in vivo (6–11). The W215A/E217A thrombin is called an anticoagulant thrombin because shifts the balance of these two thrombin activities away from procoagulative and toward anticoagulative substrates (12, 13). The loss of ability to cleave fibrinogen is much more significant than the loss of activity towards protein C, which still requires TM (14, 15). A crystallographic study suggested that the W215A/E217A mutation causes the primary substrate binding pocket of thrombin to collapse (16) providing an explanation for its strongly decreased activity towards procoagulative substrates. However, it is not clear how TM renders this mutant with a closed substrate pocket capable of cleaving protein C. We hypothesized that the collapsed substrate binding pocket and/or the gain of function upon TM binding may indicate that the W215A/E217A thrombin is dynamic, and perhaps TM binding shifts the dynamic ensemble of thrombin states allowing protein C cleavage.
Numerous studies over the years using a variety techniques such as X-ray crystallography (17), HDX-MS (5), and isothermal titration calorimetry (ITC) (18), surface plasmon resonance (SPR) (19), accelerated molecular dynamics simulations (20) have been conducted on the complex formed between thrombin and TM456 -the minimal fragment of TM necessary for triggering the anticoagulative activity of thrombin. However, only the recent publication of nuclear magnetic resonance (NMR) Carr-Purcell-Meiboom-Gill (CPMG) experiments measuring the dynamics of thrombin-TM456 has been able to reveal the residue-level changes in thrombin that result when TM456 binds (21). These experiments showed that TM456 binding reduces µs-ms motions in the 170sCT, 180sCT, and 220sCT loops, which are implicated in thrombin substrate recognition and binding, and residues Trp 215CT (263seq) and Glu 217 (265seq) were highlighted as residues that link the 170sCT and 220sCT loops through sidechain interactions. HDX-MS and accelerated MD simulations showed that mutation of residue 215CT induces disorder and increased dynamics in the 170sCT and the 220sCT loop (22, 23), raising questions regarding the effect of the combined W215A and E217A mutations on thrombin dynamics and on the allosteric influence of TM.
In the experiments presented here, we utilized HDX-MS to evaluate the changes in thrombin dynamics induced by the W215A/E217A double mutations in the absence and presence of TM456. We found that these mutations cause changes in dynamics that could be traced from the 170sCT, 180sCT, and 220sCT loops of thrombin all the way to the anion binding exosite 1 (ABE1) where TM binds. The increased dynamics in the 170sCT, 180sCT, and 220sCT loops induced by the W215A/E217A mutations are likely responsible for the collapsed structure observed by X-ray crystallography, but it may not prevent substrate binding. TM binding did not alter the dynamics of the 170sCT, 180sCT, and 220sCT loops. TM binding did, however, stabilize the allosteric pathway linking ABE1 to the mutant thrombin active site resulting in correct positioning of the N-terminus of the heavy chain as was also observed in wild type thrombin (24). Thus, the critical dynamic motions necessary for establishing the catalytically competent conformation of thrombin could still be induced by TM456 when the W215A/E217A mutations were present.
Methods
Thrombin expression and purification.
The W215A/E217A mutant thrombin plasmid was created through site-directed mutagenesis using primers from Integrated DNA Technologies. W215A/E217A and WT thrombin (P00734) were expressed and refolded from E. coli as previously described (24, 25). To correctly refold thrombin from E. coli, 18 amino acids of the pro-thrombin sequence are required (referred to here as pre-thrombin). Once the protein has been refolded, this segment must be removed (25). is the refolded pre-thrombin was purified on a HiLoadS column with an NaCl gradient. Correctly refolded thrombin eluted last off the MonoS column. WT thrombin was activated by the addition of E. carinatus venom (Sigma-Aldrich Car.#V8250) for 2–10 hr at room temperature after diluting with 50 mM Tris/HCl pH 7.4, 20 mM CaCl2, 1 mg/mL PEG8000, and 5% (v/v) glycerol. To activate W215A/E217A thrombin, the pooled HiLoadS fractions corresponding to correctly-folded W215A/E217A were diluted in the same buffer and E. carinatus venom was added as described for the WT protein. After 3 hr, previously purified α-thrombin was added to a molar ratio of 1:30 WT:W215A/E217A and incubation continued for 12–16 hr at 30 °C. The addition of WT thrombin is necessary to remove the 18 amino acids from the N-terminus of the protein. WT α-thrombin eluted off the MonoS at ~340 mM NaCl, and W215A/E217A α-thrombin eluted at ~310 mM NaCl. Fractions containing the target α-thrombin species were pooled and stored at −80oC for no longer than 1 month before use. This method of thrombin purification has been shown to result in >95% α-thrombin, and previous NMR analysis of isotopically labeled thrombin prepared this way demonstrated that the species present was α-thrombin (19, 25).
Activity of the W215A/E217A mutant thrombin.
We previously reported activity towards the chromogenic substrate, S-2238 of a number of thrombin mutants including the single W215A mutant (22). Our activities generally agree with those reported previously by Di Cera (26). The W215A mutant had s/sWT(that is the ratio of kcat/KM for mutant/WT) of 0.053 (22). The W215A/E217A mutant gave s/sWT = 0.00013, which is somewhat higher than that reported by Di Cera, but on the same order of magnitude. We also measured the protein C activation activity of the W215A/E217A mutant thrombin in the presence of TM as previously described (22, 27) The protein C activity (kcat/KM) of W215A/E217A was 20-fold lower than WT thrombin, again on the same order of magnitude as reported previously (15).
Hydrogen-Deuterium Exchange Mass Spectrometry.
Purified WT and W215A/E217A α-thrombin samples were concentrated to 5 µM using pre-rinsed 6 mL 10K MWCO Vivaspin concentrators, spinning at 1000 x g in 5 min intervals at 4 °C. These apo-thrombin samples were passed through a 0.2 micron filter, and 130 µL of each was saved for the HDX-MS experiment. The remaining apo-thrombin samples were then concentrated to 10 µM, and 50 µL was used for peptide identification. To make the α-thrombin-TM456m complex for HDX-MS, purified TM456m was added to purified WT and W215A/E217A α-thrombin samples at a molar ratio of 1:10 thrombin:TM456m, which ensured 99% of thrombin would be bound to TM456m at the concentrations used in these experiments. These samples were left at 4 °C overnight before being concentrated to 5 µM and 10 µM for the HDX-MS and ID runs respectively using 6 mL 3K MWCO Vivaspin concentrators, spinning at 3000 rpm in 15 min intervals at 4 °C.
HDX-MS was performed using a Waters Synapt G2Si system with HDX technology (Waters Corporation) (28). Deuterium exchange reactions were prepared using a Leap HDX PAL autosampler (Leap technologies, Carrboro, NC). D2O buffer was prepared by lyophilizing 1 mL of 250 mM phosphate pH 6.5 along with 850 mM NaCl for the apo-thrombin experiments and 1000 mM NaCl for the thrombin-TM456m experiments, before being resuspended in 10 mL 99.96% D2O immediately before use. Each deuterium exchange time point (0 min, 30 sec, 1 min, 2 min, and 5 min) was measured in triplicate. For each deuteration time point, 5 µL of protein was held at 25 °C for 5 min before being mixed with 55 µL of D2O buffer, which ensured a concentration of 100 mM NaCl for both the apo and TM456-bound samples at the time of the HDX experiments. The NaCl concentration of 100 mM was selected because we found that HDX was more sensitive to thrombin dynamics at this concentration. The deuterium exchange was quenched for 1 min at 1 °C by combining 50 µL of the deuteration reaction with 50 µL of 250 mM TCEP pH 2.5. The quenched sample was then injected into a 50 µL sample loop, followed by digestion and an in-line pepsin column (immobilized pepsin, Pierce, Inc.) at 15°C and a flow rate of 25µL/min. The resulting peptides were captured on a BEH C18 Vanguard pre-column, separated by analytical chromatography (Acquity UPLC BEH C18, 1.7 µM, 1.0 × 50 mm, Waters Corporation) using a 7–85% acetonitrile in 0.1% formic acid over 7.5 min at a flow rate of 40 µL/min, and electrosprayed into the Waters Synapt G2Si quadrupole time-of-flight mass spectrometer. The mass spectrometer was set to collect data in the Mobility, ESI+ mode; mass acquisition range 200–2,000 (m/z); scan time 0.4 s. Continuous lock mass correction was accomplished with infusion of leu-enkephalin every 30 s (mass accuracy of 1 ppm for calibration standard). For peptide identification, the mass spectrometer was set to collect data in MSE, mobility ESI+ mode instead. Peptides masses were identified from triplicate analyses of 10 μM α-thrombin, and data were analyzed using PLGS 2.5 (Waters Corporation). Peptides masses were identified using a minimum number of 250 ion counts for low energy peptides and 50 ion counts for their fragment ions.
The peptides identified in PLGS were then analyzed in DynamX 3.0 (Waters Corporation). Additional filters in DynamX 3.0 included a cut-off score of 7, minimum products per amino acid of 0.2, maximum MH+ error tolerance of 5 ppm, retention time standard deviation of 5%, and requiring that the peptide be present in at least 2 of the 3 peptide identification runs. The final identified peptide data was then used to analyze the deuteration data. The deuterium uptake for each peptide was calculated by comparing the centroids of the mass envelopes of the deuterated samples vs. the undeuterated controls. For all HDX-MS data, at least 2 biological replicates were analyzed, each with 3 technical replicates. Data are represented as mean values +/− SEM of 3 technical replicates due to processing software limitations, however biological replicates were highly reproducible due to use of the LEAP robot for all experiments. The deuterium uptake was corrected for back-exchange using a global back exchange correction factor (typically 25%) determined from the average percent exchange measured in disordered termini of various proteins. ANOVA analyses and t tests with a p value cutoff of 0.05 implemented in the program, DECA (github.com/komiveslab/DECA), were used to determine the significance of differences between HDX data points (29). The peptides reported on the coverage maps are actually those from which deuterium uptake data were obtained. The HDX-MS data for WT apo-thrombin and thrombin-TM456 reported here are the same as reported in another study (21).
Results
Amide exchange at the TM binding site differs between WT and the W215A/E217A mutant when TM is bound.
HDX-MS experiments were conducted on WT and on W215A/E217A thrombin in the absence or presence of TM. Deuterium uptake was determined for 99% of the α-thrombin sequence for both WT and the W215A/E217A mutant thrombin in all experiments (Figure S1). Consistent with the role of the thrombin 70sCT loop as part of the TM-binding site of thrombin, peptides spanning the 70sCT loop- residues 66–84CT (residues 97–116seq; MH+ 2473.357)- of both WT and W215A/E217A thrombin showed decreased exchange in the presence of TM (Figure 1).
Figure 1.
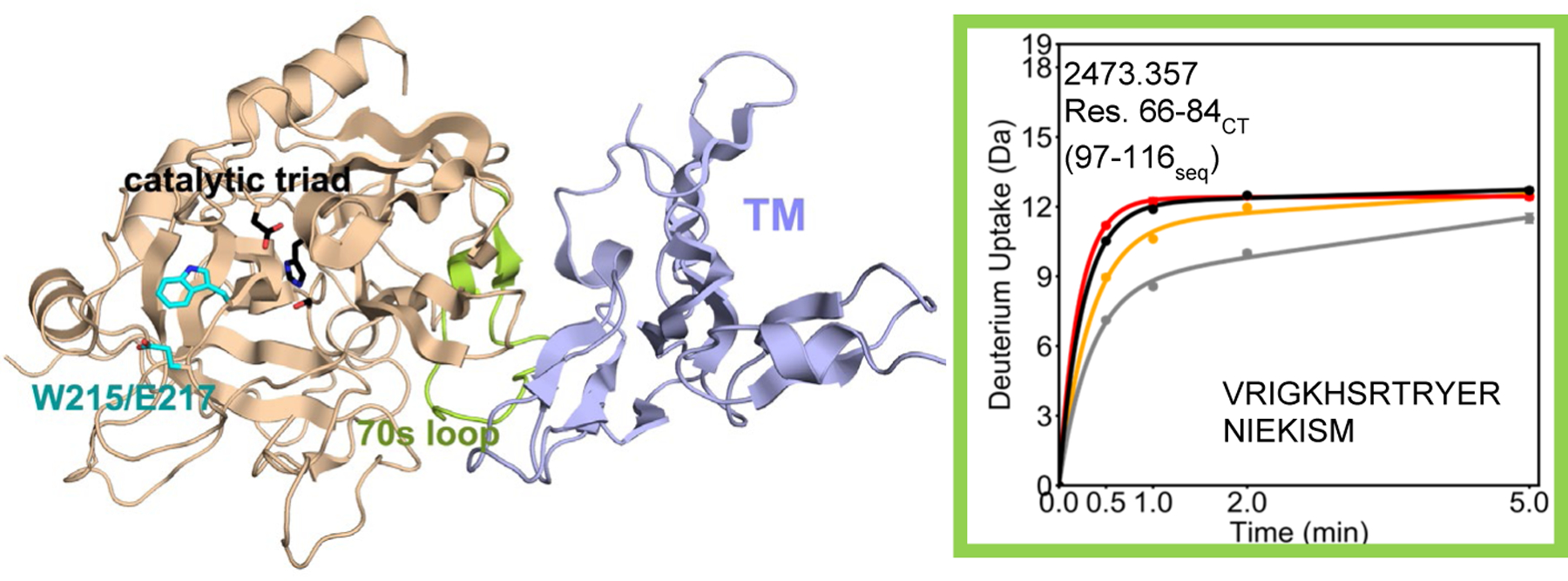
Crystal structure of thrombin (wheat) bound to TM456 (light blue) [PDB ID: 1DX5]. Note that crystal structures of serine proteases are numbered such that the catalytic triad is residues 57, 102, and 195 regardless of the sequential numbering. As a result all residue numbers in this manuscript are given in both numbering conventions. The HDXMS deuterium uptake plot for residues 66–84CT (97–116seq; MH+ 2473.357) is shown and this region is colored lime green on the structure. Sidechains are shown for the catalytic triad (black), and for Trp 215 and Glu 217 (cyan). The black and grey curves correspond to WT thrombin and WT thrombin-TM456, and the red and orange curved correspond to W215A/E217A thrombin and W215A/E217A-TM456 respectively. HDXMS error bars are shown but often lie within the symbols.
The presence of the W215A/E217A mutations did not affect the degree of exchange of the 70s loop (residues 66–84CT 97–116seq) which was ~12 deuterons for both WT and the W215A/E217A mutant in the absence of TM. However, the degree to which TM provided protection from the exchange of deuterons at this binding interface region was impacted by the W215A/E217A mutations. In the presence of TM, WT thrombin exchanged ~ 4 fewer deuterons at 30 sec whereas the W215A/E217A mutant thrombin exchanged only ~2 fewer deuterons. We do not think this difference is due to TM not binding as well to the mutant protein because the structurally adjacent residues 106–114CT (139–147seq; MH+ 1061.654), which link ABE1 to the catalytic Asp 102CT (135seq), exchanged the same in both WT and the W215A/E217A mutant, and showed similar reduction of exchange of ~1 deuteron upon addition of TM (Figure 2).
Figure 2.
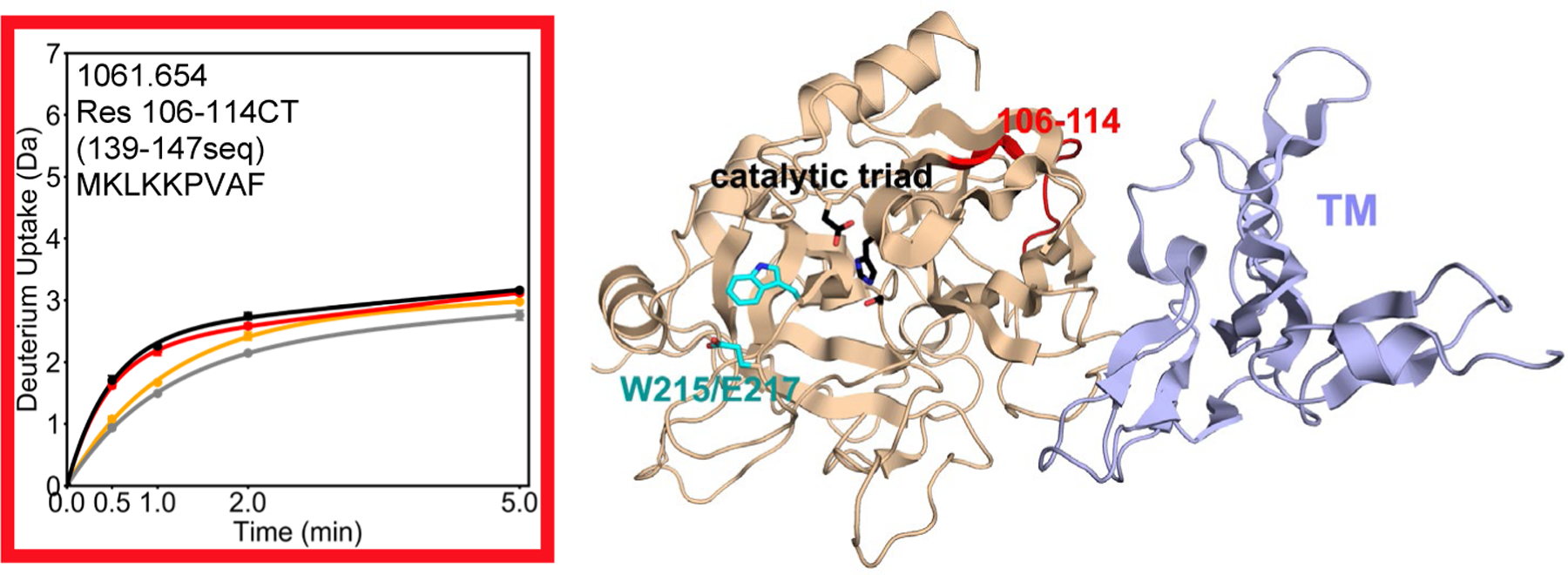
Crystal structure of thrombin (wheat) bound to TM456 (light blue) [PDB ID: 1DX5], and the HDXMS deuterium uptake plot for residues 106–114CT (139–147seq) colored red on the structure. Sidechains are shown for the catalytic triad (black), and for Trp 215 and Glu 217 (cyan). The black and grey curves correspond to WT thrombin and WT thrombin-TM456, and the red and orange curved correspond to W215A/E217A thrombin and W215A/E217A-TM456 respectively. HDXMS error bars are shown but often lie within the symbols.
The W215A/E217A mutations destabilize the 170sCT, 180sCT and 220sCT loops of thrombin.
HDX-MS revealed dramatically increased exchange, ~5 deuterons more, for residues 161–180CT (residues 202–221seq; MH+ 2343.227), which span the 170sCT loop, in the W215A/E217A mutant thrombin as compared to WT thrombin (Figure 3). Similarly, deuterium uptake into residues 182–207CT (residues 223–255seq; MH+ 3575.589) was ~2 deuterons higher in the W215A/E217A mutant compared to WT. Subtraction of the deuterium uptake into residues 198–207 (residues 244–255seq; MH+ 1522.762), from that into residues 182–207CT (residues 223–255seq; MH+ 3575.589) allowed localization of the difference in uptake to residues 182–198CT, which encompasses the 180sCT loop of thrombin. The presence of TM causes a decrease in uptake of ~1 deuteron in residues 161–180CT (residues 202–221seq; MH+ 2343.227), and a decrease in uptake of ~2 deuterons in residues 198–207CT (residues 244–255seq; MH+ 1522.762) (i.e. residues 182–198CT) in WT thrombin. In contrast, TM had no effect on the deuterium uptake in either the 170sCT or the 180sCT loop of on the W215A/E217 mutant thrombin. A comparison of the peptides that cover the 220sCT loop, residues 212–227CT (residues 260–275seq; MH+ 1615.753 for W215A/E217A; MH+ 1788.801 for WT) showed that the W215A/E217A mutant exchanged ~3 deuterons more as compared to WT thrombin (Figure 4).
Figure 3.
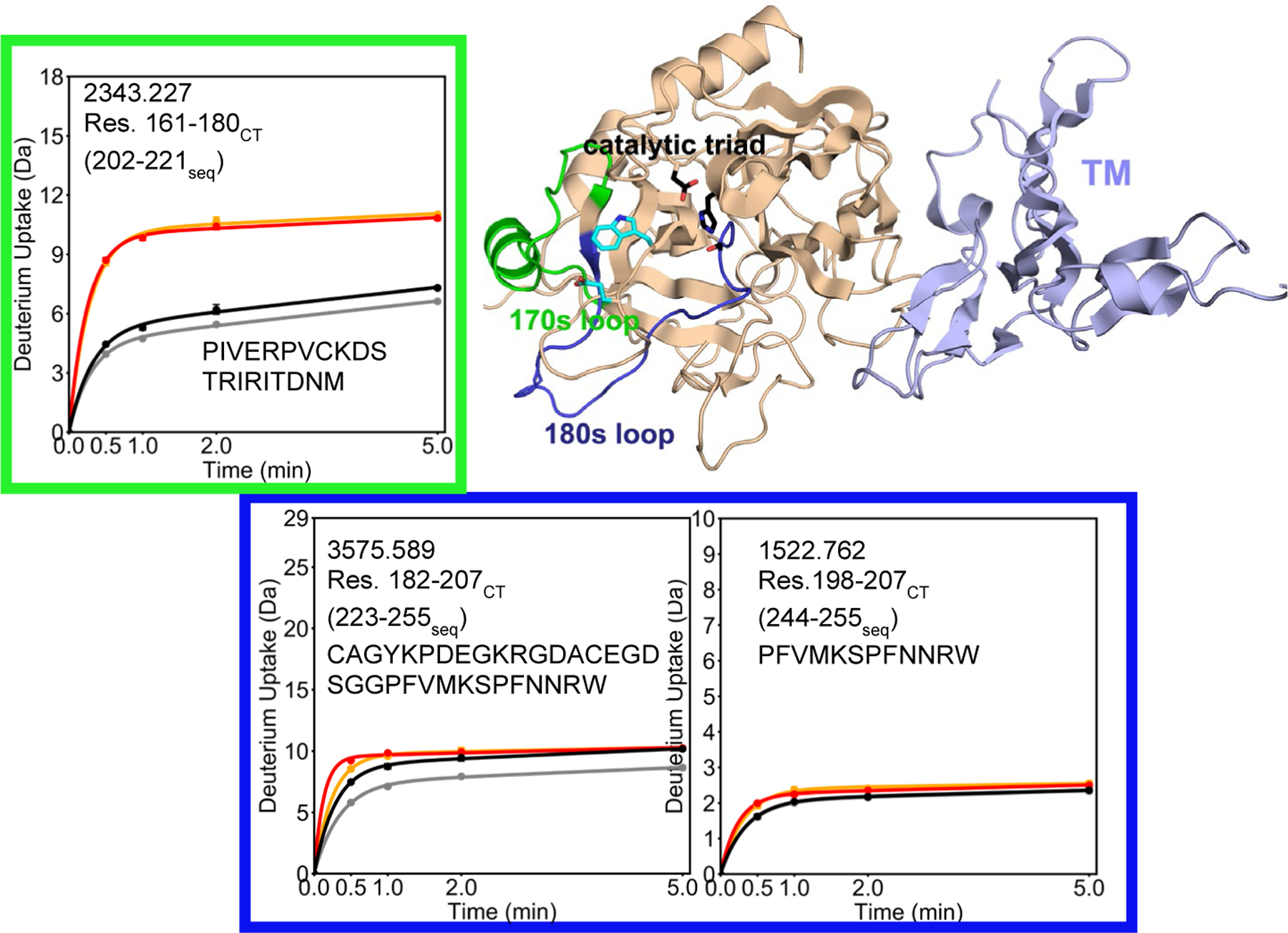
Crystal structure of thrombin (wheat) bound to TM456 (light blue) [PDB ID: 1DX5], and the HDXMS deuterium uptake plot for residues 161–180CT (202–221seq; MH+ 2343.227) colored green and residues 182–197 (223–243seq) are colored dark blue on the structure. Residues 182–197 (223–243seq) correspond to the region of thrombin that showed an uptake difference between experiments when peptide subtraction is applied to residues 182–207CT (223–255seq; MH+ 3575.589) and 198–207CT (244–255seq; MH+ 1522.762). Sidechains are shown for the catalytic triad (black), and for Trp 215 and Glu 217 (cyan). The black and grey curves correspond to WT thrombin and WT thrombin-TM456, and the red and orange curved correspond to W215A/E217A thrombin and W215A/E217A-TM456 respectively. HDXMS error bars are shown but often lie within the symbols.
Figure 4.
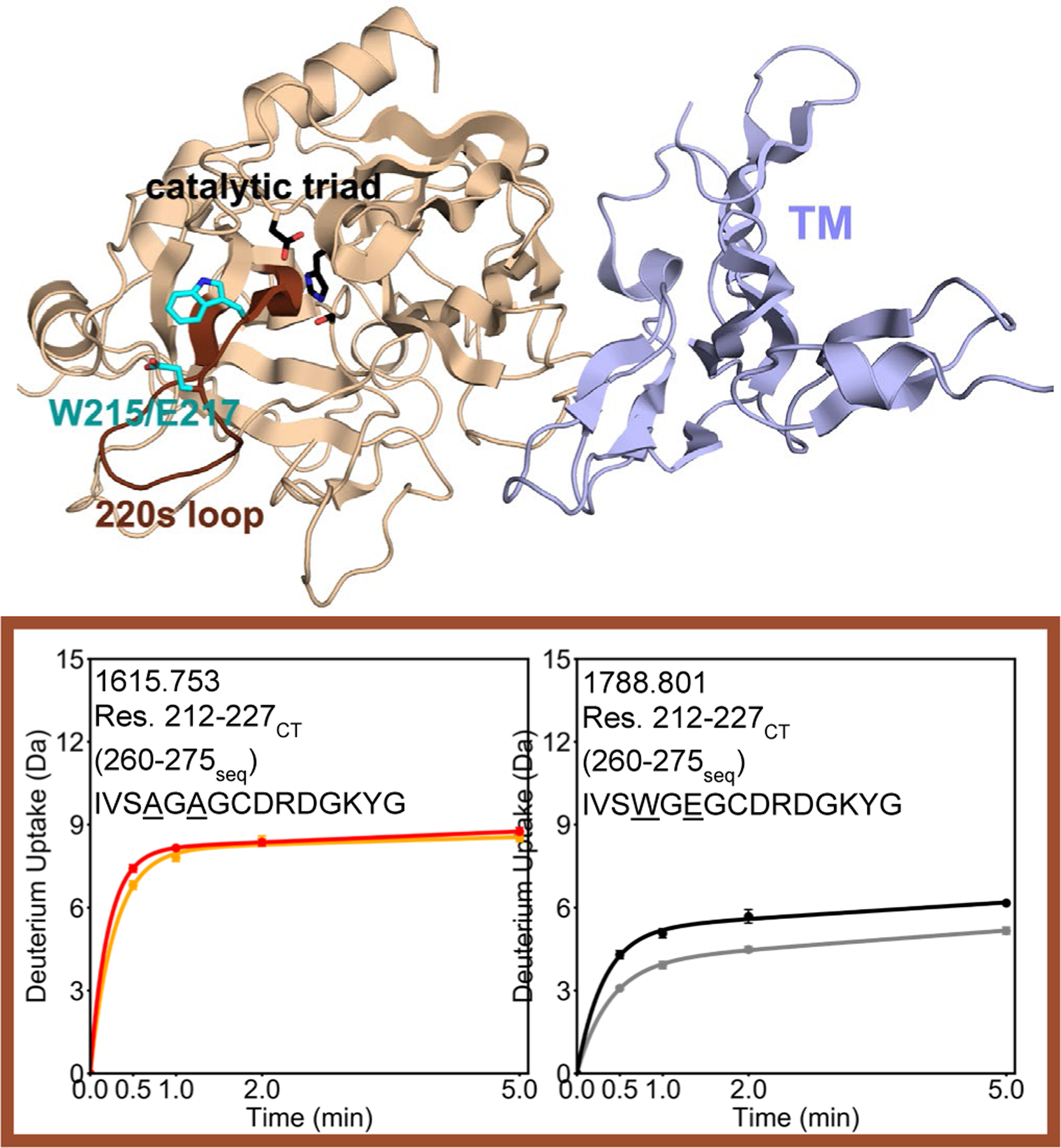
Crystal structure of thrombin (wheat) bound to TM456 (light blue) [PDB ID: 1DX5], and the HDXMS deuterium uptake plot for residues 212–227CT (260–275seq) are colored brown on the structure. Because this region of thrombin contains residues 215 and 217, the uptake plots for WT (MH+ 1788.801) and W215A/E217A (MH+ 1615.553) are shown separately. Sidechains are shown for the catalytic triad (black), and for Trp 215 and Glu 217 (cyan). The black and grey curves correspond to WT thrombin and WT thrombin-TM456, and the red and orange curved correspond to W215A/E217A thrombin and W215A/E217A-TM456 respectively. HDXMS error bars are shown but often lie within the symbols.
Similar to the peptides covering the 170sCT and 180sCT loops, the 220sCT loop, TM binding caused a decrease in exchange in residues 212–227 (residues 260–275seq; MH+ 1615.753 for W215A/E217A; MH+ 1788.801 for WT) only for WT thrombin. TM did not appear to affect this region in the W215A/E217A mutant. Thus, the peptides spanning the 170sCT, 180sCT, and 220sCT loops all exchanged more in the W215A/E217A mutant as compared to WT thrombin, and the mutations make these regions unable to respond allosterically to TM-binding.
Allosteric effects at the N-terminus of the heavy chain when TM is bound.
We previously showed that the N-terminus of the heavy chain exchanges substantially in WT thrombin, but that TM binding allosterically decreases exchange of this region likely stabilizing the catalytically active form of thrombin, which must have the new N-terminus of the heavy chain embedded in the Ile pocket (5). We were able to analyze this same region in the W215A/E217A mutant thrombin. The peptide spanning residues 16–23CT (residues 37–44seq; MH+ 819.373), which contains the N-terminus of the heavy chain, exchanged significantly more in the W215A/E217A mutant thrombin than in WT (4 deuterons in the W215A/E217A mutant vs. 3 deuterons in WT thrombin). Upon TM binding, this region showed a substantial decrease in exchange of ~1.5 deuterons for WT thrombin within 1 min of exchange. The decrease in exchange observed for the W215A/E217A mutant with TM present was ~0.5 deuterons bringing it to the same level of exchange for WT thrombin in the absence of TM (Figure 5). This region is remote, over 20 Å away from the 70sCT loop where TM binds, and ~12 Å away from the W215A/E217A mutations. CPMG NMR experiments showed the dynamics of the N-terminus of the heavy chain to be linked to dynamics of the 140sCT loop (21). However, the 140sCT is highly disordered, so peptides spanning this region completely exchanged their amide hydrogens in all HDX-MS experiments performed (Figure S2). As a result, the effect of the W215A/E217A mutations on the 140s loop could not be evaluated by HDX-MS.
Figure 5.
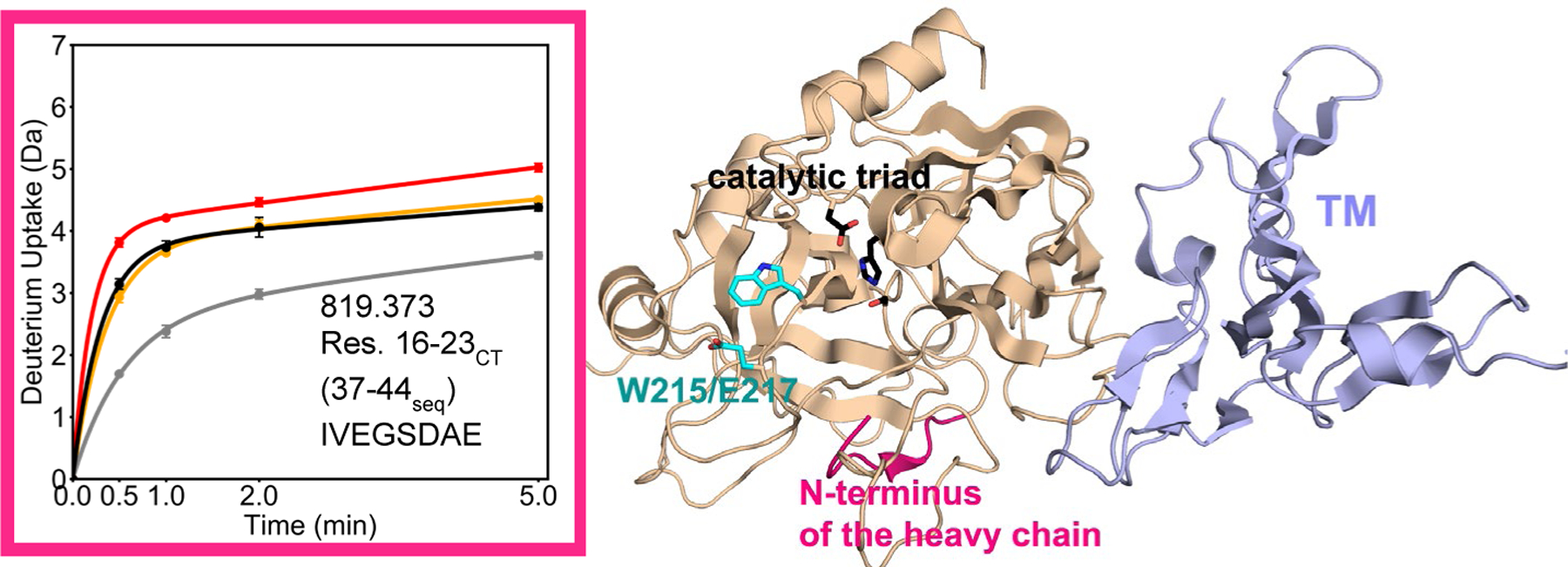
Crystal structure of thrombin (wheat) bound to TM456 (light blue) [PDB ID: 1DX5], and the HDXMS deuterium uptake plot for residues 16–23CT (37–44seq; MH+ 819.373) colored pink on the structure. Sidechains are shown for the catalytic triad (black), and for Trp 215 and Glu 217 (cyan). The black and grey curves correspond to WT thrombin and WT thrombin-TM456, and the red and orange curved correspond to W215A/E217A thrombin and W215A/E217A-TM456 respectively. HDXMS error bars are shown but often lie within the symbols.
Discussion
Both the Trp 215CT (263seq) and the E217A (265seq) sidechains are key for the recognition of procoagulative substrates. The W215A mutation causes a 500-fold loss in activity of thrombin towards fibrinogen. The mutation of E217A alone causes a 60-fold loss and together the loss of procoagulant activity is over 2000-fold. In contrast, the double W215A/E217A mutations only cause less than a 20-fold loss of activity towards PC (15). An explanation of how the W215A/E217A mutations dramatically reduce thrombin’s activity towards procoagulative substrates was provided by the crystal structure of W215A/E217A thrombin, which showed a collapsed S1 pocket (16). Our H/D exchange results are not consistent with a collapsed S1 pocket because collapse is expected to reduce the HD exchange in the S1 pocket of the W215A/E217A thrombin as compared to WT thrombin. In fact, the W215A and E217A mutations caused a dramatic increase in exchange in the 170sCT, 180aCT, and 220sCT loops that all participate in the formation of the primary substrate binding pocket of thrombin (30). These changes are in similar locations but slightly larger than for the W215A mutant alone (22). It is important to note that the differences in intrinsic H/D exchange rates of the two substituted residues Trp and Glu to Ala (31) are not large enough to explain the ~3 deuteron difference in exchange observed between the mutant and WT thrombin.
In addition, the increased exchange is not localized to residues 212–227CT (260–275seq) of the thrombin 220sCT loop which contains the mutations, but it extends to the 170sCT and 180sCT loops. The side chains of Trp 215CT and Glu 217CT make contacts with Ile 172CT and Thr 174CT of the 170sCT loop in the crystal structures of WT thrombin both with and without TM456 (17, 30), explaining how disruption of these contacts may propagate disorder from the 220sCT loop to the 170sCT loop. Multiple studies demonstrated that the mutation of residues in the 170sCT, 180sCT, and 220sCT loops can significantly impact the structural integrity of this region of thrombin. Crystal structures of thrombin containing Y225P, D221A/D222K, and W215A/E217A mutations led to decreased Na+ coordination between the 180sCT and 220sCT loops (16, 32, 33) and loss of electron density for some residues in the 180sCT loop or in the 220sCT loop. A monomeric crystal structure of human W215A/E217A (PDB ID: 3EE0) is also missing electron density within the 220sCT loop. The H/D exchange results presented here are consistent with the idea that the W215A/E217A mutations cause a loss of structure and/or an increase in dynamics of both the 180sCT and 220sCT loops, providing an explanation for the missing residues in the aforementioned crystal structures. Our results suggest that the dramatic decrease in activity of the W215A/E217A thrombin towards pro-coagulant substrates is most likely due to the loss of an important interaction between the substrate and W215 in the W215A mutant and the subsequent disordering of the substrate binding pocket.
As has been shown for WT thrombin, the W215A/E217A thrombin mutant still requires TM to exhibit anticoagulative activity (14, 15, 34). TM binding to ABE1 allosterically decreases the dynamics of the 170sCT, 180sCT, and 220sCT loops, which are 20 – 30 Å from ABE1 (21). In fact, these loops showed decreased H/D exchange in WT thrombin upon TM binding in the work presented here. However, the H/D exchange in the 170sCT, 180sCT, and 220sCT loops of the W215A/E217A mutant was essentially unaffected by TM, suggesting that TM can no longer allosterically structure the substrate binding pocket in the W215A/E217A mutant. It is possible that the disordered substrate binding pocket may still support PC binding while it is less able to support binding of procoagulative substrates that depend more strongly on hydrophobic interactions being correctly formed.
We previously showed that TM binding to the 70sCT loop allosterically changes the dynamics of residues 106–114CT (139–147seq) which lead to the catalytic Asp 102CT (135seq), and the residues surrounding the N-terminus of Ile 16CT (37seq) which interacts with the backbone of Asp 189CT (235seq) and helps coordinate the oxyanion hole through interactions with the sidechain of Asp 194CT (240seq) (3, 21, 35, 36). It is likely that the TM-induced 200-fold increase in the catalytic rate of thrombin towards PC can be attributed to these TM-induced dynamics changes directly at the active site (37). TM binding caused similar dynamics changes in these regions of the W215A/E217A mutant. Although the W215A/E217A mutations have not been reported to influence the TM binding affinity, we observed somewhat less protection of the 70sCT loop upon TM binding in the W215A/E217A mutant as compared to WT thrombin. TM-induced dynamics changes were fully recapitulated in residues 106–114CT (139–147seq) of the W215A/E217A mutant as compared to WT thrombin strongly supporting that TM was fully bound under the experimental conditions and supporting that TM could still remodel the thrombin active site in the mutant protein.
Finally, the residues at the N-terminus of the heavy chain of W215A/E217A thrombin exchanged more compared to WT thrombin. The new N-terminus was completely exchanging in the W215A/E217A mutant indicating that it was not well packed into the Ile pocket. This observation may provide additional explanation for the lack of activity of the W215A/E217A thrombin toward pro-coagulative substrates. We previously showed that TM binding causes a dramatic decrease in exposure of the new N-terminus in WT thrombin, and here we show that TM also causes protection of this region in the W215A/E217A mutant. Thus, TM stabilizes the insertion of the N-terminus of the heavy chain in both WT and the W215A/E217A thrombin, although the mutant has its N-terminus more exposed than it is in the WT thrombin. This disparity helps explain why the specific activity of the W215A/E217A mutant towards PC is somewhat lower than that measured for WT thrombin in the presence of TM (15). Thus, the W215A/E217A thrombin mutations do not abolish the function of TM as an allosteric activator of the thrombin N-terminal β-barrel. TM binding likely enhances the activity of W215A/E217A thrombin towards PC by allosterically communicating with the N-terminus of the thrombin heavy chain and with the catalytic Asp 102CT. However, TM cannot promote the cleavage procoagulative substrates in these ways, because procoagualtive substrates compete with TM for binding to the thrombin ABE1 (38).
In conclusion, the W215A/E217A mutations impede TM-induced allosteric remodeling of the C-terminal β-barrel loops. These loops are much more disordered in the mutant than in WT thrombin. They become even more structured upon TM binding to WT thrombin but remain disordered upon TM binding to the W215A/E217A mutant thrombin. It is likely that this difference contributes to the much more dramatic defect in activity of the W215A/E217A mutant thrombin toward procoagulant substrates as compared to anticoagulant substrates. The W215A/E217A mutant thrombin does respond to TM-induced allosteric remodeling of the N-terminal β-barrel. Thus, TM allosterically structures the active site of both the WT and the W215A/E217A mutant thrombin, which promotes activity only towards Protein C.
Supplementary Material
Acknowledgements
This work was supported by NIH R01HL127041.
Footnotes
Supporting information
Figure S1 – coverage map of thrombin and mutants
Figure S2 – HDX Uptake plots for all peptides
Table S1 – HDX Summary table.
Protein Accession Codes
Human thrombin: Uniprot P00734 (https://www.uniprot.org/uniprot/P00734)
Human thrombomodulin Uniprot P07204 (https://www.uniprot.org/uniprot/07204)
Data Availability
HDX-MS data is available at massive.ucsd.edu data set MSV000088425, password WE The data will be made public without a password after the manuscript is accepted.
References
- 1.Fenton JW (1986) Thrombin, Annals New York Acad Sci 485, 5–15. [DOI] [PubMed] [Google Scholar]
- 2.Bode W (2006) The structure of thrombin: a janus-headed proteinase., Semin Thromb Hemost 32 16–31. [DOI] [PubMed] [Google Scholar]
- 3.Huber R, and Bode W (1978) Structural basis of the activation and action of trypsin, Acc Chem Res 11, 114–122. [Google Scholar]
- 4.Esmon CT (2000) Regulation of blood coagulation., Biochim Biophys Acta 1477, 349–360. [DOI] [PubMed] [Google Scholar]
- 5.Handley LD, Treuheit NA, Venkatesh VJ, and Komives EA (2015) Thrombomodulin binding selects the catalytically active form of thrombin, Biochemistry 54, 6650–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruber A, Cantwell AM, Di Cera E, and Hanson SR (2002) The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo., J Biol Chem 277, 27581–27584.. [DOI] [PubMed] [Google Scholar]
- 7.Gruber A, Fernández JA, Bush L, Marzec U, Griffin JH, Hanson SR, and E., D. C. (2006) Limited generation of activated protein C during infusion of the protein C activator thrombin analog W215A/E217A in primates., J Thromb Haemost 4, 392–397. [DOI] [PubMed] [Google Scholar]
- 8.Berny MA, White TC, Tucker EI, Bush-Pelc LA, Di Cera E, Gruber A, and McCarty OJ (2008) Thrombin mutant W215A/E217A acts as a platelet GPIb antagonist., Arterioscler Thromb Vasc Biol 28, 329–334. [DOI] [PubMed] [Google Scholar]
- 9.Vicente CP, Weiler H, Di Cera E, and Tollefsen DM (2012) Thrombomodulin is required for the antithrombotic activity of thrombin mutant W215A/E217A in a mouse model of arterial thrombosis, Thrombosis Res 130, 646–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker EI, Verbout NG, Markway BD, Wallisch M, Lorentz CU, Hinds MT, Shatzel JJ, Pelc LA, Wood DC, McCarty OJT, Di Cera E, and Gruber A (2020) The protein C activator AB002 rapidly interrupts thrombus development in baboons, Blood 135, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berny-Lang MA, Hurst S, Tucker EI, Pelc LA, Wang RK, Hurn PD, Di Cera E, McCarty OJ, and Gruber A (2011) Thrombin mutant W215A/E217A treatment improves neurological outcome and reduces cerebral infarct size in a mouse model of ischemic stroke., Stroke 42, 1736–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbs CS, Coutré SE, Tsiang M, Li WX, Jain AK, Dunn KE, Law VS, Mao CT, Matsumura SY, Mejza SJ, Paborsky LR, and Leung LLK (1995) Conversion of thrombin into an anticoagulant by protein engineering., Nature 378, 413–416. [DOI] [PubMed] [Google Scholar]
- 13.Arosio D, Ayala YM, and Di Cera E (2000) Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C, Biochemistry 39, 8095–8101. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka KA, Gruber A, Szlam F, Bush LA, Hanson SR, and Di Cera E (2008) Interaction between thrombin mutant W215A/E217A and direct thrombin inhibitor., Blood Coagul Fibrinolysis 19, 465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cantwell AM, and Di Cera E (2000) Rational design of a potent anticoagulant thrombin, J Biol Chem 275, 39827–39830. [DOI] [PubMed] [Google Scholar]
- 16.Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, and Di Cer, a. E. (2004) The anticoagulant thrombin mutant W215A/E217A has a collapsed primary specificity pocket., J Biol Chem 279, 39824–39828. [DOI] [PubMed] [Google Scholar]
- 17.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, and Bode W (2000) Structural basis for the anticoagulant activity of the thrombin-thrombomodulin complex, Nature 404, 518–525. [DOI] [PubMed] [Google Scholar]
- 18.Treuheit NA, Beach MA, and Komives EA (2011) Thermodynamic compensation upon binding to exosite 1 and the active site of thrombin, Biochemistry 50, 4590–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baerga-Ortiz A, Rezaie AR, and Komives EA (2000) Electrostatic dependence of the thrombin-thrombomodulin interaction., J Mol Biol 296, 651–658. [DOI] [PubMed] [Google Scholar]
- 20.Gasper PM, Fuglestad B, Komives EA, Markwick PR, and McCammon JA (2012) Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities., Proc Natl Acad Sci U S A 109, 21216–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peacock RB, McGrann T, Tonelli M, and Komives EA (2021) Serine protease dynamics revealed by NMR analysis of the thrombin-thrombomodulin complex., Sci Rep 11, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peacock RB, Davis JR, Markwick PRL, and Komives EA (2018) Dynamic Consequences of Mutation of Tryptophan 215 in Thrombin., Biochemistry 57, 2694–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Markwick PRL, Peacock RB, and Komives EA (2019) Accurate Prediction of Amide Exchange in the Fast Limit Reveals Thrombin Allostery., Biophys J 116, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Handley LD, Fuglestad B, Stearns K, Tonelli M, Fenwick RB, Markwick PR, and Komives EA (2017) NMR reveals a dynamic allosteric pathway in thrombin., Sci Rep 7, 39575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuglestad B, Gasper PM, Tonelli M, McCammon JA, Markwick PRL, and Komives EA (2012) The dynamic structure of thrombin in solution, Biophys J 103, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marino F, Pelc LA, Vogt A, Gandhi PS, and Di Cera E (2010) Engineering thrombin for selective specificity toward protein C and PAR1, J Biol Chem 285, 19145–19152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White CE, Hunter MJ, Meininger DP, White LR, and Komives EA (1995) Large-scale expression, purification and characterization of small fragments of thrombomodulin: the roles of the sixth domain and of methionine 388, Protein Engineering, Design and Selection 8, 1177–1187. [DOI] [PubMed] [Google Scholar]
- 28.Wales TE, Fadgen KE, Gerhardt GC, and Engen JR (2008) High-speed and high-resolution UPLC separation at zero degrees Celsius, Anal Chem 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lumpkin RJ, and Komives EA (2019) DECA, A Comprehensive, Automatic Post-processing Program for HDX-MS Data., Mol Cell Proteomics 18, 2516–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bode W, Mayr I, Baumann U, Huber R, Stone SR, and Hofsteenge J (1989) The refined 1.9 A crystal structure of human alpha-thrombin: interaction with D-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment, The EMBO Journal 8, 3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai Y, Milne JS, Mayne L, and Englander SW (1993) Primary structure effects on peptide group hydrogen exchange., Proteins 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu W, Chen Z, Gandhi PS, Vogt AD, Pozzi N, Pelc LA, Zapata F, and Di Cera E (2011) Crystallographic and kinetic evidence of allostery in a trypsin-like protease., Biochemistry 50, 6301–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang QD, Sabetta M, and Di Cera E (1997) Selective loss of fibrinogen clotting in a loop-less thrombin., J Biol Chem 272, 19649–19651. [DOI] [PubMed] [Google Scholar]
- 34.Vicente CP, Weiler H, Di Cera E, and Tollefsen DM (2021) Thrombomodulin is required for the antithrombotic activity of thrombin mutant W215A/E217A in a mouse model of arterial thrombosis, Thromb Res 130, 646–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bode W, Schwager P, and Huber R (1978) The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution, J Mol Biol 118, 99–112. [DOI] [PubMed] [Google Scholar]
- 36.Stojanovski BM, Chen Z, Koester SK, Pelc LA, and Di Cera E (2019) Role of the I16-D194 ionic interaction in the trypsin fold., Sci Rep 9, 18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esmon CT (1989) The roles of protein C and thrombomodulin in the regulation of blood coagulation., J Biol Chem 264, 4743–4746. [PubMed] [Google Scholar]
- 38.Kurosawa S, Stearns DJ, Jackson KW, and Esmon CT (1988) A 10-kDa cyanogen bromide fragment from the epidermal growth factor homology domain of rabbit thrombomodulin contains the primary thrombin binding site., J Biol Chem 263, 5993–5996. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HDX-MS data is available at massive.ucsd.edu data set MSV000088425, password WE The data will be made public without a password after the manuscript is accepted.