

Abstract
Endometrial carcinomas (ECs) classified by The Cancer Genome Atlas (TCGA) as copy number-low (also referred to as “no specific molecular profile” [NSMP]) have a prognosis intermediate between POLE-mutated and copy number-high ECs. NSMP-ECs are a heterogeneous group, however, comprising both relatively indolent and aggressive ECs. We identified a total of 472 NSMP-ECs among 1,239 ECs that underwent clinical sequencing of 410–468 cancer-related genes. Somatic mutation and copy number alteration data were subjected to unsupervised hierarchical clustering, which identified three genomic clusters. Random sampling with stratification was used to choose ~80 endometrioid ECs from each cluster, resulting in a study size of 240 endometrioid ECs as well as an additional 44 non-endometrioid NSMP-ECs. Cluster 1 (C1, n=80) consisted primarily of NSMP-ECs with PTEN and PIK3R1 mutations, Cluster 2 (C2, n=81) of tumors with PTEN and PIK3CA mutations and Cluster 3 (C3, n=79) of NSMP-ECs with chromosome 1q high-level gain and lack of PTEN mutations. The majority (72.7%) of non-endometrioid NSMP-ECs mapped to C3. NSMP-ECs from C3 were more likely to be FIGO grade 3 (30%), estrogen receptor-negative/weak (54.5%) and FIGO stages III or IV. In multivariate analysis, molecular clusters were associated with worse overall survival outcomes with C3 tumors having the worst (hazard ratio: 4) and C1 tumors having the best outcome. In conclusion, NSMP-ECs are a heterogeneous group of tumors and comprise both aggressive and clinically low-risk ECs that can be identified based on mutation and copy number data.
INTRODUCTION
Molecular profiling of endometrioid and serous carcinomas of the endometrium by The Cancer Genome Atlas (TCGA) revealed four genomic groups, which include ultramutated tumors with POLE exonuclease domain mutations, hypermutated tumors with microsatellite instability (MSI)/ DNA mismatch repair (MMR) deficiency, copy number-high tumors with TP53 mutations, and copy number-low tumors, which lack all of the above-mentioned alterations [1]. The latter group was shown to be composed of tumors with predominately endometrioid morphology and considerable molecular heterogeneity, which has since been referred to as endometrial carcinoma (EC) of no specific molecular profile (NSMP) [2]. Follow-up studies suggested an exclusion surrogate approach (ProMisE) for identifying NSMP-EC, whereby ECs harboring any characteristic of the other TCGA groups are excluded, i.e. tumors lacking POLE exonuclease domain mutations, are MMR-proficient, and are p53/TP53 wild-type are designated as NSMP-ECs [3,4]. The designation of NSMP to this group of ECs derives from considerable molecular heterogeneity of this group and absence of defining molecular features. Additionally, the NSMP-EC have been shown to be a clinically and histologically diverse.
The molecular heterogeneity of NSMP-ECs has been shown to have clinical relevance [5]. For example, it has been reported that NSMP-ECs harboring 1q32.1 high level gain [6,7], CTNNB1 hotspot mutations [7] or expression of L1CAM [8] are associated with adverse outcomes.
Given the heterogeneity observed in NSMP-EC, one of the largest molecular subgroups of EC, in-depth studies are needed to further refine and understand the molecular underpinnings of these tumors [9]. In this study, we sought to define the clinicopathologic and molecular characteristics of a large group of NSMP-EC subjected to clinical sequencing of 410–468 cancer-related genes and attempt to subclassify these tumors based on characteristic molecular findings.
MATERIALS AND METHODS
Case selection and data extraction
This study, including review and analysis of data, was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center (MSK). ECs of all histologic types that underwent clinical tumor-normal targeted DNA next-generation sequencing (NGS) of 410–468 cancer-related genes using MSK-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT)[10,11] between 2014 and 2019 were evaluated (n=1,239). ECs of the NSMP-EC molecular subtype class in this MSK-IMPACT cohort were identified by employing a surrogate of the ProMisE model [12], as previously described [13]: tumors with hotspot POLE exonuclease domain mutations (n=60), with DNA MMR-deficiency defined either by immunohistochemistry and/or genomic determination (high MSIsensor score; n=357) [14], and with aberrant p53 immunohistochemical expression or TP53 genetic alterations including somatic mutations and homozygous deletions (n=297) were excluded. In addition, to ensure high quality data, sequenced tumors with very low tumor purity (<10% as estimated by the reviewing pathologist or average variant allele frequency <5%) were excluded from the analysis (n=53). The remaining cases were included in the downstream analysis (n=472; see CONSORT diagram Figure 1).
Figure 1. CONSORT diagram summarizing the selection process of NSMP endometrial carcinomas included in this study.
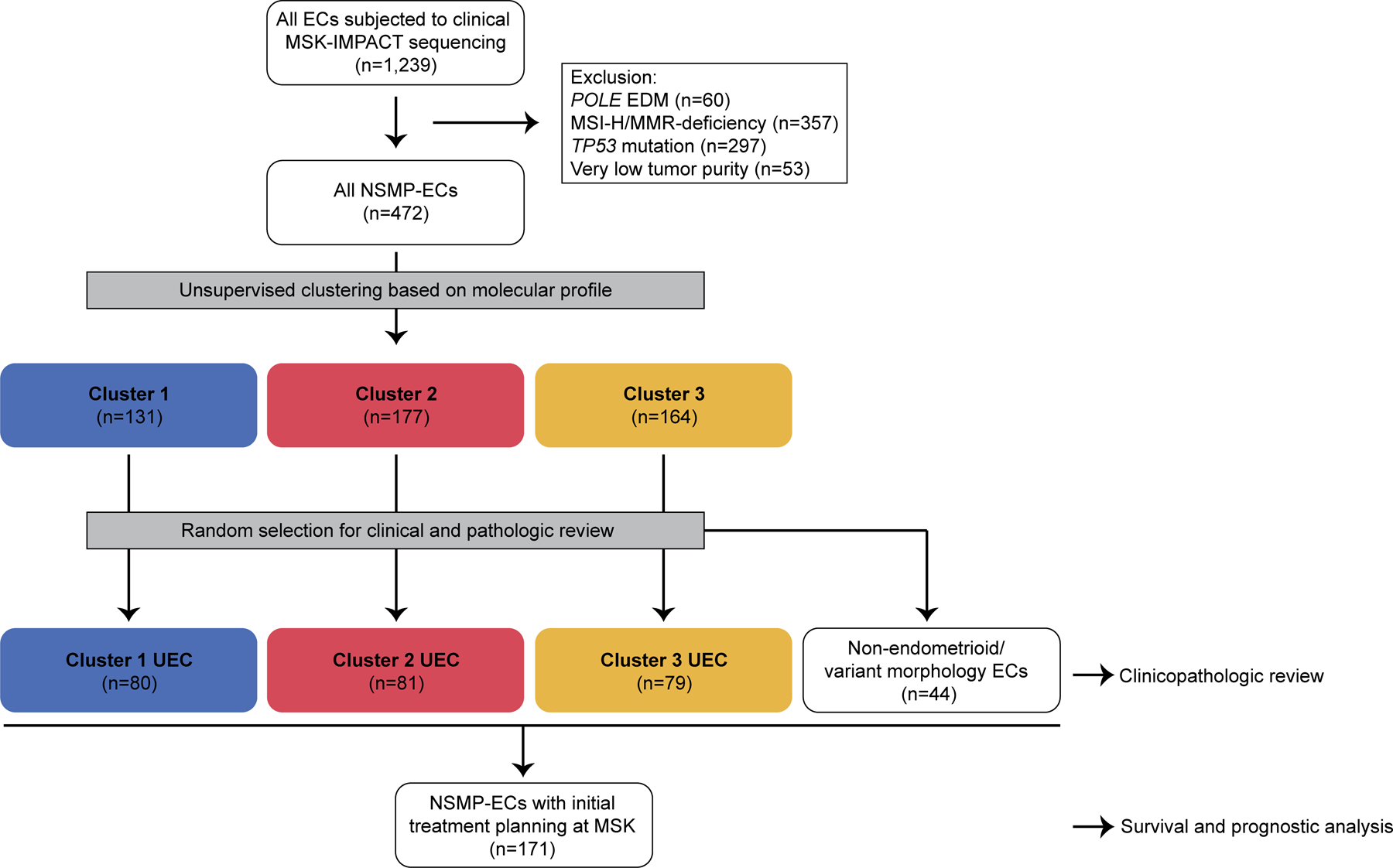
NSMP-EC, endometrial carcinomas of no special molecular profile; EDM, exonuclease domain mutant; MMR, DNA mismatch repair; MSI, microsatellite instability.
Genomic data
The MSK-IMPACT assay assesses somatic mutations, copy number (CN) alterations, structural variants, fraction of genome altered (FGA) and tumor mutation burden (TMB), as previously described [13]. GISTIC (version 2.0.23) was used to analyze the broad CN data [15]. The mutational data included chromosomal location, base-pair change, protein change, predicted functional impact of the mutation and the associated variant frequency. Data on allele specific CN alterations and ploidy were extracted using the ‘facets’ R package (version 0.5.14) [16]. Cancer cell fractions (i.e. clonality) of all somatic mutations were inferred using ABSOLUTE (v1.0.6), as described previously [17] using CN and tumor purity estimation information derived from FACETS [16]. Mutational signatures were determined using the MuSiCa R application (version 1.0) [18] in samples harboring ≥5 mutations (both synonymous and non-synonymous). Annotation of the genomic alterations for oncogenic properties was performed using the OncoKB database, as previously described [19]. Evaluation for mutual exclusivity/co-occurrence was performed using the ‘DISCOVER’ R package (v0.9) [20].
Pathology review
The histopathologic and morphologic data were extracted from the synoptic pathology report. The scanned pathology slides of the sequenced tumor were reviewed by a gynecologic pathologist (A.M.-B.) to confirm the findings of the pathology report. For histologic typing, to mitigate the effect of suboptimal interobserver concordance [21,22], we performed a single-institution study with a group of experienced gynecologic pathologists. Biweekly diagnostic consensus conferences encouraged a uniform diagnostic approach within the group, as did frequent review of each other’s cases for tumor board and quality assurance, as described [13].
Clustering and statistical analysis
A reduced segments matrix was calculated from the FACETS-derived CN segmentation file using the CNTools R package. The CN and the cancer cell fraction of somatic mutations were then used to create a combined data matrix which was subsequently normalized. Principal component (PC) analysis (PCA) was used to reduce the dimensionality of the matrix by employing the fast.prcomp function of the gmodels R package. Thirteen statistically significant PCs were identified using a permutation test. The output was visualized using t-distributed stochastic neighbor embedding (T-SNE; ‘rtsne’ R package; Supplementary Figure S1) [23]. To identify tumor clusters in our dataset, partitioning into separate clusters based on their scores along the thirteen significant PCs was performed using the HDBSCAN function of the DBSCAN R package [24].
Comparisons of quantitative data between the groups were performed using ANOVA with post-hoc Tukey test and comparison of qualitative data including associations between clinicopathologic features and molecular data were performed using chi-squared test with Fisher’s exact p-value calculation. All p-value were two tailed and p-values <0.05 were considered statistically significant. Disease-free survival (DFS) and overall survival (OS) were evaluated by calculating survival curves using the Kaplan–Meier method, using the Log-rank test to compare subgroups, with the start date set as the date of initial diagnostic biopsy. Univariate and multivariate Cox Proportional Hazards analysis was performed to determine the hazard ratio (HR).
Final case selection
Following genomic clustering, an equal number of endometrioid ECs were selected from each genomic cluster for further downstream analysis using random sampling with stratification (n= ~80 sample for each of 3 clusters; Figure 1). In addition, 44 non-endometrioid ECs or endometrioid variants meeting the same criteria used for NSMP-EC were selected and analyzed separately; these tumors consisted of 15 high-grade ECs with ambiguous morphology, 9 endometrial clear cell carcinomas, 7 mesonephric-like carcinomas, 5 carcinosarcomas, 2 corded and hyalinized endometrioid carcinomas, 2 dedifferentiated ECs, 2 undifferentiated ECs and 2 uterine serous carcinomas. The pathology reports and histopathology slides were reviewed for the 240 uterine endometrioid carcinomas and 44 non-endometrioid/variant endometrioid ECs, and clinical data relating to disease presentation and course were extracted from the electronic health records (Figure 1).
RESULTS
A large series of 472 NSMP-ECs subjected to clinical FDA-authorized tumor-normal MSK-IMPACT sequencing of 410–468 cancer-related genes, which included primarily uterine endometrioid carcinomas (UECs; n=367), but also non-endometrioid ECs and/or tumors with variant endometrioid histology (n=105), were clustered based on mutational and gene CN alteration data. Specifically, in this initial cohort of NSMP-EC (n=472), UEC was the most common tumor histologic type (367/472, 77.8%), followed by high-grade ECs with ambiguous morphology (46/472, 9.7%), uterine clear cell carcinoma (23/472, 4.9%), mesonephric-like carcinoma (12/472, 2.5%), uterine carcinosarcoma (9/472, 1.9%) with rare tumors categorized as uterine serous carcinoma (6/472, 1.3%), corded and hyalinized endometrioid carcinoma (4/472, 0.8%), dedifferentiated carcinoma (3/472, 0.6%), and undifferentiated carcinoma (2/472, 0.4%). The majority of UECs were FIGO grade 1 tumors (199/367, 54.2%), followed by FIGO grade 2 (97/367, 26.4%) and FIGO grade 3 (71/367, 19.3%) tumors.
The clustering revealed the presence of 3 distinct clusters: cluster 1 (C1), cluster 2 (C2) and cluster 3 (C3). 240 UECs in total were randomly selected with stratification for further clinical, pathologic and genomic review (C1, n=80; C2, n=81; C3, n=79; see Methods; Figure 1; Supplementary Figure S1).
Genomic landscape of endometrioid NSMP-ECs
Overall, in these 240 NSMP-UECs, the most commonly mutated genes were PTEN (n=154, 64%), ARID1A (n=108, 45%), PIK3CA (n=102, 42.5%), CTNNB1 (n=86, 36%), PIK3R1 (n=77, 32%), KRAS (n=70, 29%), CTCF (n=34, 14%) and BCOR (n=3, 14%). Other recurrent alterations included FBXW7, SOX17, FGFR2 (all n=21, 9%) and ESR1 (n=20, 8%; Figure 2A).
Figure 2. Recurrent somatic genetic alterations in NSMP endometrial carcinomas.
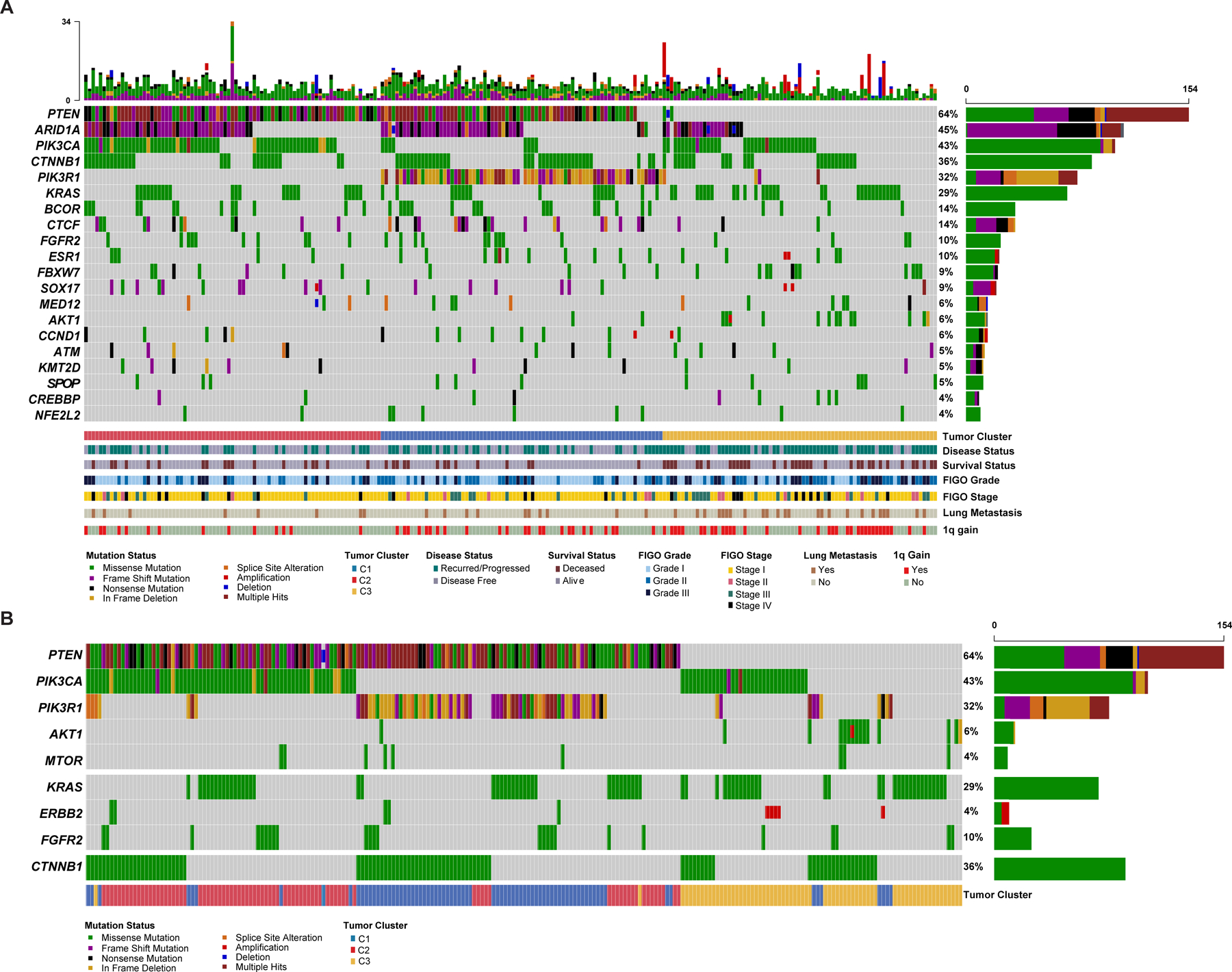
(A) Oncoprint depicting the most recurrent genomic alterations in uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC). Each column represents a tumor with the bar graph at the top depicting the number/distribution of alterations per sample, and the Oncoprint rows showing alterations for each gene. The bottom part of the graph shows the summary of histopathologic and clinical information for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene. Mutation types and clinicopathologic features are color-coded according to the legend. (B) Oncoprint depicting the most recurrent molecular alterations in NSMP-EEC focusing on alterations activating the PI3K/AKT/mTOR pathway in addition to CTNNB1 alterations. The bottom part of the graph shows the summary of molecular cluster information for each case. Note that majority of tumors harbor a combination of PIK3CA/PIK3R1 and PTEN mutations while AKT1 mutations occur in absence of upstream alterations. KRAS, ERBB2 and FGFR2 alterations are mutually exclusive. Mutation types and other features are color-coded according to the legend. C1, Cluster 1; C2, Cluster 2; C3, Cluster 3.
Of the 240 NSMP-UECs included in this study, 127 (52.9%) had alterations in combinations of PTEN/PIK3CA and PTEN/PIK3R1, other tumors had pathogenic mutations in AKT1 (6%) and MTOR (3%). KRAS, FGFR2 oncogenic mutations and ERBB2 amplification/activating mutations occurred in a mutually exclusive pattern in 29%, 10%, and 4% of NSMP-UECs, respectively. CTNNB1 hotspot mutations were present in 36% (n=87) of NSMP-UECs, and there was mutual exclusivity between KRAS/FGFR2/ERBB2 and CTNNB1 alterations (DISCOVER P<0.001; Figure 2B).
As a next step, the genomic landscape of NSMP-UECs according to the clusters was assessed and found to be distinct in terms of their genomic instability, mutational burden, CN and mutational landscapes (Figure 3). The fraction of genome altered, a measure of chromosomal instability, was significantly higher in the C3 NSMP-UECs compared to NSMP-UECs from the other two clusters (mean 0.17 versus 0.07, ANOVA P=0.0002; Figure 3B). Overall, amplifications and homozygous deletions were rare in the NSMP-UECs, with ERBB2 (n=5), AKT3 (n=4), and NTRK1 (n=4) amplification and CDKN2A deletions (n=4) being the most common. In terms of broad chromosomal arm level alterations, 1q gains were common in C3 (n=35, 44.9%) and C1 tumors (n=24, 30.4%), and less common in C2 tumors (n=14, 17.5%, X2 P=0.001; Figures 2A, 3A).
Figure 3. Global genomic landscape of NSMP uterine endometrioid carcinomas.
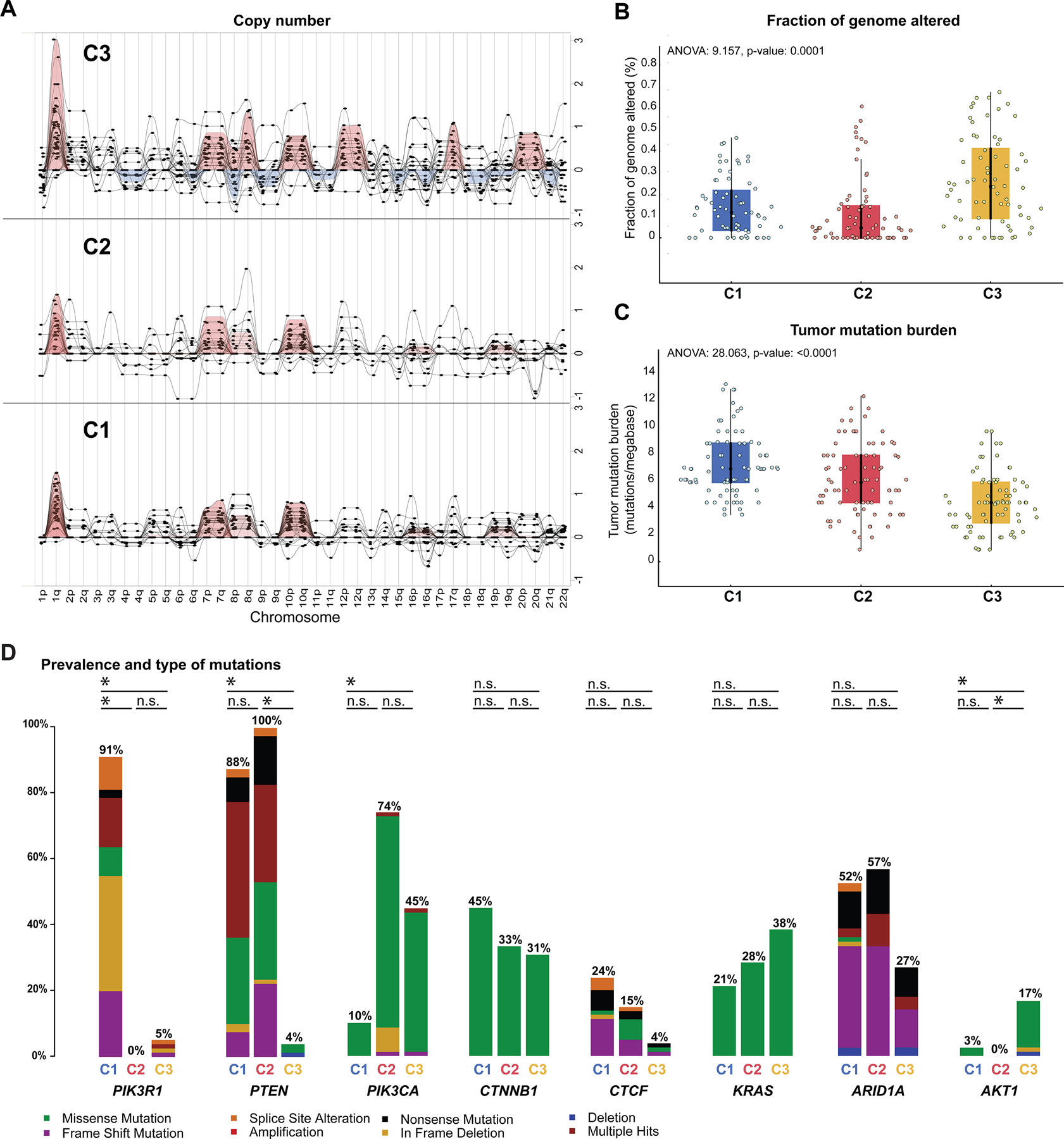
(A) Summary of broad copy number alterations in the three molecular clusters of uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC) based on GISTIC results. Each column indicates a chromosomal arm as shown in the X-axis labels. The Y-axis represents G-score which considers the amplitude of the aberration as well as the frequency of its occurrence across samples. (B) Comparison of fraction of genome altered values across the three NSMP-EC clusters. (C) Comparison of tumor mutational burden (#mutations/megabase) across the three NSMP-UEC clusters. (D) Bar-plots comparing the alteration frequency of 8 commonly altered genes among the three molecular clusters of NSMP-EC. The black horizontal lines marked with a * at the top of each bar-plot represent statistically significant difference in the frequency of the altered gene among the clusters; n.s., not significant. C1, Cluster 1; C2, Cluster 2; C3, Cluster 3.
The tumor mutational burden progressively decreased from C1 to C3 NSMP-UECs (median number of somatic mutations per megabase for C1, C2 and C3: 6.9, 5.9, 4.4, respectively ANOVA P: 0.0001; Table 1, Figure 3C). When assessing specific mutations, we found that while PTEN alterations were common in the C1 and C2 clusters (88% and 100% respectively), they were rare in C3 NSMP-UECs (5%, X2 P<0.0001; Figure 3D). PIK3R1 mutations, which were primarily truncating, were almost exclusively found in C1 tumors (91%; versus 0% in C2 and 5% in C3, X2 P<0.0001). Conversely, PIK3CA mutations, which preferentially affected the hotspot kinase domain codon H1047 (21.4%) or the helical domain hotspot codons E545 and E542 (10.7%, 4.5%), were uncommon in C1 (10%), but had higher frequencies in C2 and C3 NSMP-UECs (74% and 45%, X2 P<0.0001). AKT1 mutations, primarily the E17K hotspot mutation, were almost exclusively found in C3 NSMP-UECs (17% versus 3% in C1 and 0% in C2, X2 P<0.0001; Figure 3D), and they frequently co-occurred with CTNNB1 mutations (see below). No statistically significant difference in the frequency of alterations affecting CTNNB1, KRAS and CTCF was found between the clusters (X2 P>0.05 for all). We noted that the majority of non-synonymous somatic mutations identified in NSMP-UECs were oncogenic or likely/predicted oncogenic (72.9%; Supplementary Figures S2 and S3), including ESR1 mutations involving codons L536, Y537 and D538 which are associated with resistance to hormone therapy in breast cancer [25].
Table 1.
Summary of genomic findings in the three clusters of uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC).
| Cluster 1 (n=80) | Cluster 2 (n=81) | Cluster 3 (n=79) | All cases (n=240) | P-value | ||
|---|---|---|---|---|---|---|
| Tumor mutation burden (mutations/ megabase) | Median | 7.00 | 5.9 | 4.4 | 6.00 | <0.0001 |
| 95% IQR | 6–10 | 4.4–7.9 | 2.9–5.9 | 2–12 | ||
| Fraction of genome altered (%) | Median | 0.05 | 0.00 | 0.09 | 0.04 | <0.0001 |
| 95% IQR | 0–0.12 | 0–0.06 | 0–0.62 | 0.02–0.22 | ||
| DNA ploidy | Median | 2.16 | 2.03 | 2.19 | 2.14 | 0.498 |
| 95% IQR | 2.00–2.35 | 2.00–2.33 | 2.00–2.48 | 2.00–2.41 | ||
| Chromosome 1q total copy number | Median | 2.2 | 2.00 | 2.60 | 2.00 | <0.0001 |
| 95% IQR | 2–2.61 | 1.92–2 | 2–3.01 | 2–2.61 | ||
| PTEN alterations | n (%) | 70 (88%) | 81 (100%) | 3 (4%) | 154 (64%) | <0.0001 |
| PIK3R1 alterations | n (%) | 73 (91%) | 0 (0%) | 4 (5%) | 77 (32%) | <0.0001 |
| PIK3CA alterations | n (%) | 8 (10%) | 60 (74%) | 35 (45%) | 102 (42%) | <0.0001 |
| AKT1 alterations | n (%) | 2 (3%) | 0 (0%) | 13 (17%) | 15 (6%) | <0.0001 |
| KRAS alterations | n (%) | 18 (21%) | 22 (28%) | 30 (38%) | 70 (29%) | 0.106 |
| CTNNB1 alterations | n (%) | 35 (45%) | 28 (33%) | 23 (31%) | 86 (36%) | 0.0979 |
| ARID1A alterations | n (%) | 41 (52%) | 45 (57%) | 22 (27%) | 108 (45%) | 0.0004 |
IQR, interquartile range; n, number
Finally, we evaluated the cancer cell fractions/clonality of the mutations identified, meaning the bioinformatically inferred percentage of cancer cells harboring a given mutation in a tumor sample. This analysis revealed that in C1 NSMP-UECs PTEN mutations had the highest cancer cell fractions, and were primarily clonal, whereas PIK3R1, ARID1A and KRAS mutations occurred at lower cancer cell fractions. The same trend was observed in C2 NSMP-UECs with PTEN mutations having the highest cancer cell fractions, and PIK3CA, ARID1A and KRAS mutations with lower cancer cell fractions. In C3 NSMP-UECs, however, more heterogeneity was observed, with either AKT1, ARID1A or KRAS being clonal events in these tumors (Figure 4).
Figure 4. Clonality of somatic mutations in NSMP uterine endometrioid carcinomas and recurrent genomic alterations in non-endometrioid endometrial cancers of NSMP.
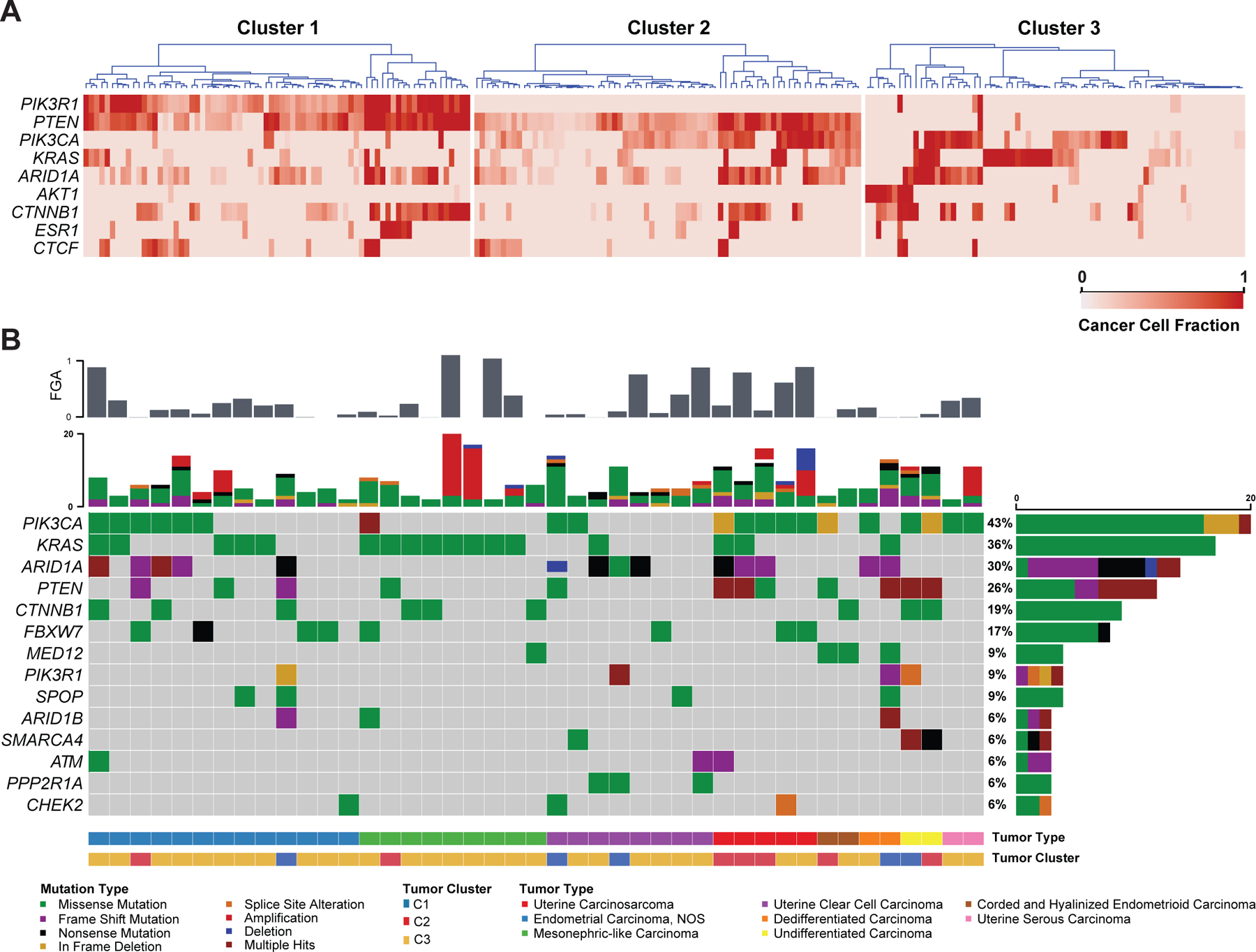
(A) Heatmaps showing the cancer cell fractions of the most commonly altered genes among the three clusters of uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC). Each column represents a tumor. The gradient of the heatmap is based on the calculated cancer cell fraction of the alteration in the sample, color-coded according to the legend. (B) Oncoprint depicting the most recurrent genomic alterations in non-endometrioid endometrial carcinomas of no special molecular profile (NSMP). Each column represents a tumor with the bar graph at the top depicting the number/distribution of alterations per sample, and the Oncoprint rows showing alterations for each gene. The bottom part of the graph shows the summary of histopathologic and clinical information for each case. The bar graph on the right of the panel shows the number and distribution of alterations for each gene. Mutation types and clinicopathologic features are color-coded according to the legend. C1, Cluster 1; C2, Cluster 2; C3, Cluster 3.
Taken together, NSMP-UECs are heterogeneous at the genetic level with C1 tumors being defined by recurrent PIK3R1 and PTEN alterations, C2 tumors by a combination of PTEN and PIK3CA mutations and C3 tumors by KRAS mutations along with 1q high level gain in the absence of PTEN alterations or alternatively AKT1 mutations along with CTNNB1 mutations.
Evaluation of the interaction matrix between the commonly altered genes in NSMP-UECs showed strong mutual exclusivity between PIK3CA and PIK3R1 mutations (DISCOVER P <0.0001), confirming previous reports [1,13]. In addition, CTNNB1 and KRAS as well as PTEN and AKT1 mutations were also found to be mutually exclusive (DISCOVER P<0.01; Supplementary Figure S4).
In terms of mutational signatures, 202 cases had sufficient mutations (≥5 mutations) to allow for investigation of mutational signatures. Consistent with previous results [26], signature 1 associated with aging was the most common mutational signature in NSMP-UECs; no statistically significant difference was observed between the 3 clusters (Supplementary Figure S5).
Genomic landscape of NSMPs of non-endometrioid and variant endometrioid histology
In addition to the 240 NSMPs of endometrioid subtype, 44 additional ECs subjected to clinical sequencing that fulfilled the criteria of NSMP were of non-endometrioid (n=19) or variant (n=21) histologic types. The majority of these non-endometrioid or variant endometrioid NSMP-ECs clustered with C3 UECs (72.7%, n=32); 15.9% clustered with C2 tumors (n=7) and 11.4% clustered with C1 UECs (n=5). One of the two cases of corded and hyalinized endometrioid carcinomas clustered with C2 tumors and the other with C3 tumors.
In terms of global genomic profile, these 44 non-UEC or variant UEC NSMP tumors showed a significantly higher fraction of genome altered (median: 0.0993, range: 0.024–0.76) compared to NSMP-UEC (median: 0.035, range: 0–0.67; Mann-Whitney U P<0.0001; Figure 4B). Furthermore, non-UEC or variant UEC NSMP tumors had a higher number of intrachromosomal breakpoints (median: 28, range: 23–74) compared to UEC tumors (median: 24, range: 21–72; Mann-Whitney U P<0.0001; data not shown) and had a lower overall number of somatic mutations (median: 5 versus 6 respectively; Mann-Whitney U P=0.009).
The 15 high-grade ECs with ambiguous morphology harbored alterations in PIK3CA (n=6), ARID1A and KRAS (n=5). The clear cell carcinomas had ARID1A (n=4) and PTEN alterations (n=1; Figure 4B). The seven mesonephric/mesonephric-like carcinomas all had activating KRAS mutations and chromosome 1q gain, with only two cases harboring concurrent PTEN or PIK3CA mutations [27]. The carcinosarcomas all had PIK3CA mutations (n=5) along with either PTEN (n=3) or FBXW7 (n=2) alterations. Both undifferentiated ECs showed truncating mutations in SMARCA4. Both variant UEC NSMPs of corded and hyalinized endometrioid carcinoma harbored MED12 mutations, with one also harboring PTEN and PIK3CA mutations and the other case a CTNNB1 mutation. Despite having a variant morphologic phenotype, the molecular phenotype of these tumors closely resembled that of other UECs; however, the limited number of cases prevents any firm conclusion to be drawn (Figure 4B).
Clinicopathologic features of NSMP ECs
Among the NSMP-UEC patients for whom in-depth clinicopathologic review was performed (n=240), the median age at diagnosis was 63 years (range: 28–91 years); there was no statistically significant difference between the clusters in terms of age distribution (Figure 5A). As expected, age had a strong association with the background endometrium with patients with background atrophic/inactive endometrium being older (median age: 65.5 years) compared to patients with either background endometrial hyperplasia (median age: 59 years) or proliferative endometrium (median age: 50.5 years; ANOVA P=0.0001). Of note, background endometrial hyperplasia was more likely to be observed in C2 (n=42) compared to C1 (n=29) and C3 (n=17) patients (X2 P=0.02; Figure 5B), which is in line with prior work showing frequent alteration of PTEN and PIK3CA in endometrial hyperplasia [28,29] while PIK3R1 alterations are reportedly rare in hyperplasia [30].
Figure 5. Clinicopathologic features of NSMP uterine endometrioid carcinomas.
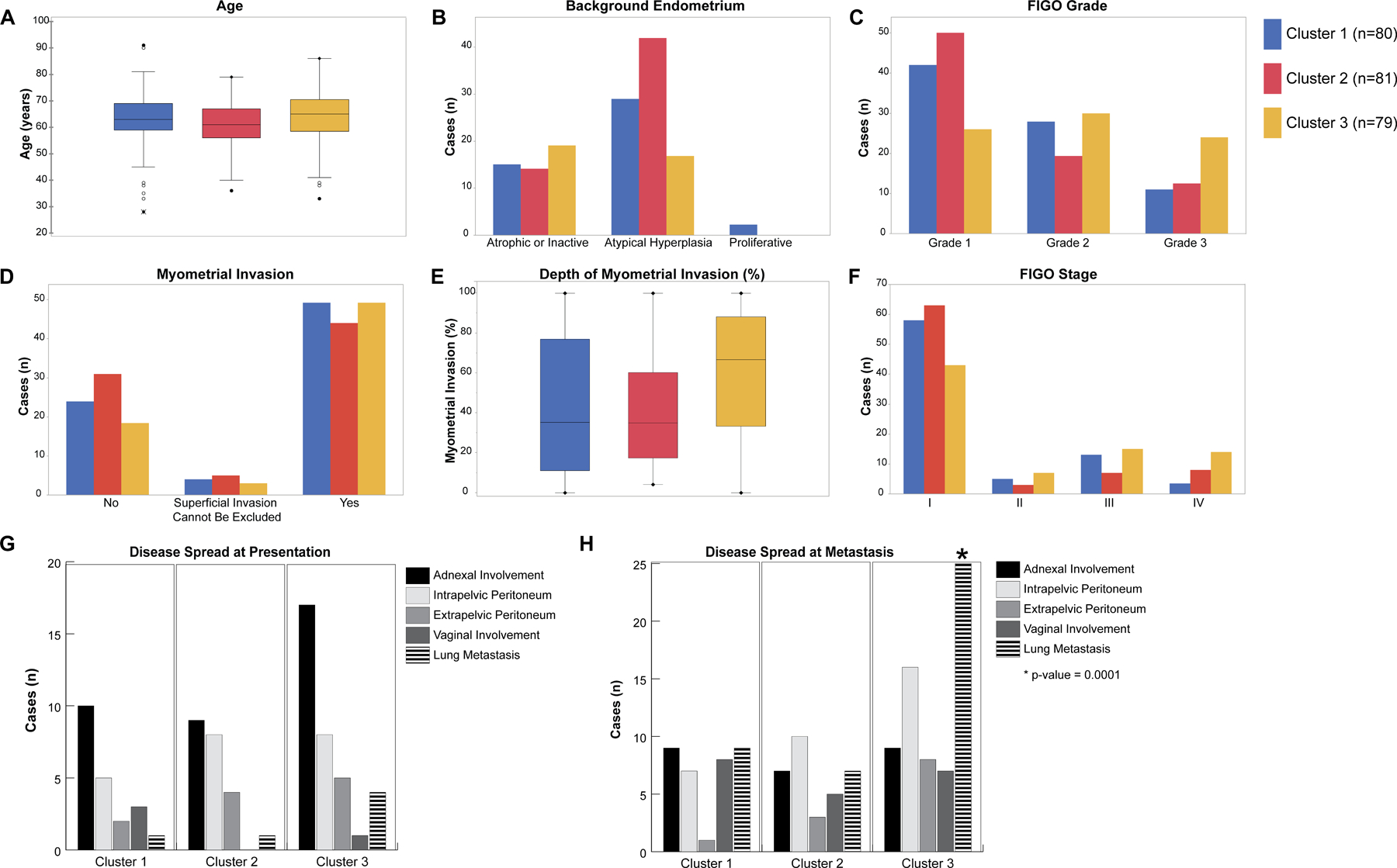
(A) Box and whisker plots comparing age distribution between the molecular clusters of uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC). (B) Bar-plot comparing the background endometrium findings among the molecular clusters. (C) Bar-plot comparing the tumor FIGO grade among the molecular clusters. (D) Bar-plot comparing the distribution of myometrial invasion among the molecular clusters. (E) Box and whisker plots comparing the depth of myometrial invasion among the molecular clusters. (F) Bar-plot comparing the distribution of tumor FIGO clinical stage among the molecular clusters. (G) Bar-plot comparing the tumor spread at the time of diagnosis among molecular clusters. (H) Bar-plot comparing the tumor spread at the time of recurrence among molecular clusters. Molecular clusters are color coded with blue representing Cluster 1, red representing Cluster 2 and yellow representing Cluster 3 tumors.
Most of the NSMP-UEC in the cohort were FIGO grade 1 tumors (n=117, 48.7%), followed by FIGO grade 2 (n=76, 31.7%) and FIGO grade 3 tumors (n=47, 19.6%). C3 tumors were more likely to have a high FIGO grade with 30% (24/79) having FIGO grade 3 morphology compared to 14.4% in the other clusters (23/161; X2 P: 0.0001; Figure 5C).
Estrogen receptor (ER) expression results by immunohistochemical analysis were available for 48 samples; C3 tumors were more likely to have negative or weak and focal ER expression (12/22, 54.5%) compared to C1/C2 tumors (23.1% (6/26); X2 P: 0.025; Supplementary Figure S6). Progesterone receptor (PR) expression as assessed by immunohistochemistry were available for 33 samples; C3 tumors were more likely to have negative or weak and focal PR expression (9/15, 60%) compared to C1/C2 tumors (16.7% (3/18); X2 P: 0.03; Supplementary Figure S7).
Patients with C3 NSMP-UECs were more likely to present with higher clinical stage disease with 36.7% (29/79) presenting at FIGO stages III and IV compared to 19.9% (32/161) for C1 and C2 NSMP-UECs (X2 P: 0.005; Table 2). The same was observed for pathologic T stage where 50.6% (40/79) of patients with C3 tumors presented at pathologic T2 stage or above compared to 23.6% (38/161) of C1/C2 tumors (X2 P: <0.001; Figure 5F). Follow-up data showed that patients with C3 tumors had a higher mortality rate (49.4%, 39/79) compared to C1 and C2 patients (17.5%, 14/80 and 22.2%, 18/81, respectively; X2 P: 0.0001). Lymphovascular invasion was identified in 78 tumors (32.5%). The C3 tumors were more likely to have lymphovascular invasion compared to the other clusters (41.8%, 33/79 versus 27.9%, 45/161 for C1/C2, X2 P: 0.032). On the other hand, C1/C2 tumors were more likely to have lymph node metastasis (62.7%, 101/161 versus 39.2%, 31/79; X2 P: 0.001) than C3 tumors (Table 2).
Table 2.
Summary of clinicopathologic findings in the three clusters of uterine endometrioid carcinomas of no special molecular profile (NSMP-UEC).
| Cluster 1 (n, %) | Cluster 2 (n, %) | Cluster 3 (n, %) | All Cases (n, %) | |
|---|---|---|---|---|
| Age median (n, min/max) | 63 (28–91) |
61 (36–79) |
65 (33–86) |
63 (28–91) |
| Tumor FIGO stage | ||||
| I | 58 (72.5%) | 63 (77.8%) | 43 (54.4%) | 164 (68.3%) |
| II | 5 (6.2%) | 3 (3.7%) | 7 (8.9%) | 15 (6.25%) |
| III | 13 (16.2%) | 7 (8.6%) | 15 (19%) | 35 (14.6%) |
| IV | 4 (5%) | 8 (9.9%) | 14 (17.7%) | 26 (10.8%) |
| Disease status | ||||
| Disease free | 42 (52.5%) | 51 (63%) | 18 (22.8%) | 111 (46.3%) |
| Persisted/ recurred/ progressed | 38 (47.5%) | 30 (37%) | 61 (77.2%) | 129 (53.7%) |
| Survival status | ||||
| Alive | 66 (82.5%) | 63 (77.8%) | 40 (50.6%) | 169 (70.4%) |
| Deceased | 14 (17.5%) | 18 (22.2%) | 39 (49.4%) | 71 (29.6%) |
| Tumor FIGO grade | ||||
| Grade I-II | 69 (86.2%) | 69 (85.2%) | 55 (69.6%) | 193 (80.4%) |
| Grade III | 11 (13.7%) | 12 (14.8%) | 24 (30.4%) | 47 (19.6%) |
| Tumor myometrial invasion | ||||
| Absent | 24 (30%) | 31 (38.3%) | 18 (22.8%) | 73 (30.4%) |
| Superficial invasion cannot be excluded | 4 (5%) | 5 (6.2%) | 3 (3.8%) | 12 (5%) |
| Present | 49 (61.2%) | 44 (54.3%) | 49 (62%) | 142 (59.2%) |
| N/A | 3 (3.7%) | 1 (1.2%) | 9 (11.4%) | 13 (5.4%) |
| Cervical stromal invasion | ||||
| Absent | 67 (83.7%) | 69 (85.2%) | 51 (64.6%) | 187 (77.9%) |
| Present | 10 (12.5%) | 9 (11.1%) | 19 (24%) | 38 (15.8%) |
| N/A | 3 (3.7%) | 3 (3.7%) | 9 (11.4%) | 15 (6.2%) |
| Lymphovascular invasion | ||||
| Absent | 52 (65%) | 58 (71.6%) | 36 (45.5%) | 146 (60.8%) |
| Present | 23 (28.7%) | 22 (27.2%) | 33 (41.8%) | 78 (32.5%) |
| N/A | 5 (6.2%) | 1 (1.2%) | 10 (12.7%) | 16 (6.7%) |
N/A, not available/missing data
Patients with C3 NSMP-UECs were more likely to have lung metastasis at recurrence (25/79 versus 16/161 for C1 and C2; X2 P: 0.0001), and to have involvement of extra-pelvic peritoneum and omentum on recurrence (16 and 8 out of 79 cases, respectively) compared to C1/C2 tumors (17 and 4 out of 161 cases, respectively; X2 P=0.04 and 0.011; Figures 5G and H).
Among genomic factors, 1q high level gain, absence of PTEN mutation and presence of AKT1 mutation were strongly associated with lung metastasis (X2 P=0.0001, 0.0001 and 0.005 respectively). Tumors with KRAS mutations in isolation were not associated with lung metastasis (X2 P=0.167), however in combination with an absence of PTEN mutations the association with lung metastasis was significant. Furthermore, 1q gain was associated with increased likelihood of myometrial invasion (91.3% (n=63/69) vs. 59.6% (n=102/171); X2 P=0.0001). Among the genomic alterations in our entire cohort, ESR1 mutations showed a strong correlation with age; patients with ESR1 mutations were strictly post-menopausal with a median age of 69 years (range 60–82 years) while the patients without ESR1 mutations were younger and included premenopausal patients (median age 62, range: 28–91 years; Mann-Whitney-U P=0.0001).
From the 284 patients with NSMP-EC, 171 patients were seen at the time of initial treatment planning with surgery performed at MSK, whereas the remainder of the patients (n=113) were seen at the time of recurrence. As an exploratory, hypothesis-generating analysis, we assessed whether the different clusters of NSMP-ECs identified would be associated with outcome (Figure 6). Mortality and disease recurrence were more likely in patients with C3 tumors with a 45.3% mortality rate (24/53) and a 66% recurrence rate (35/53). In contrast, patients with C1 and C2 tumors had lower mortality rates (5.3% (3/54) and 18.7% (13/64), respectively) and lower recurrence rates (31.9% (17/54) and 25% (16/64), respectively; Figure 6E). Log-rank survival analysis showed that the molecular clusters were associated with distinct overall survival (OS), with patients with C1 tumors having the best survival outcomes (Log-rank P: 0.0005; Figure 6A), while C2 and C3 tumors were statistically similar. In terms of disease-free survival (DFS), patients with C3 tumors had the worse outcomes, however, C1 and C2 tumors together showed similar DFS curves (Log-rank P: <0.0001; Figure 6B). Multivariate Cox-regression survival analysis showed that tumor clusters conferred OS disadvantage with C2 tumors having a hazard ratio (HR) of 3.7 and C3 tumors having a hazard ratio of 4.0 (P: 0.049 and 0.032 respectively). However, tumor clusters were not associated with disease-free survival on multivariate analysis. Interestingly, after controlling for stage, the most important prognostic factors for both OS and DFS were presence of FIGO grade 3 morphology (OS HR: 19.6 (P: 0.0003), DFS HR: 10.5 (P<0.0001)) and non-endometrioid morphology (OS HR: 30.6, P<0.0001; DFS HR: 12.9, P<0.0001; Figure 6C, D; Supplementary Table S1).
Figure 6. Association of molecular clusters and histologic grade with outcome in NSMP uterine endometrial carcinomas.
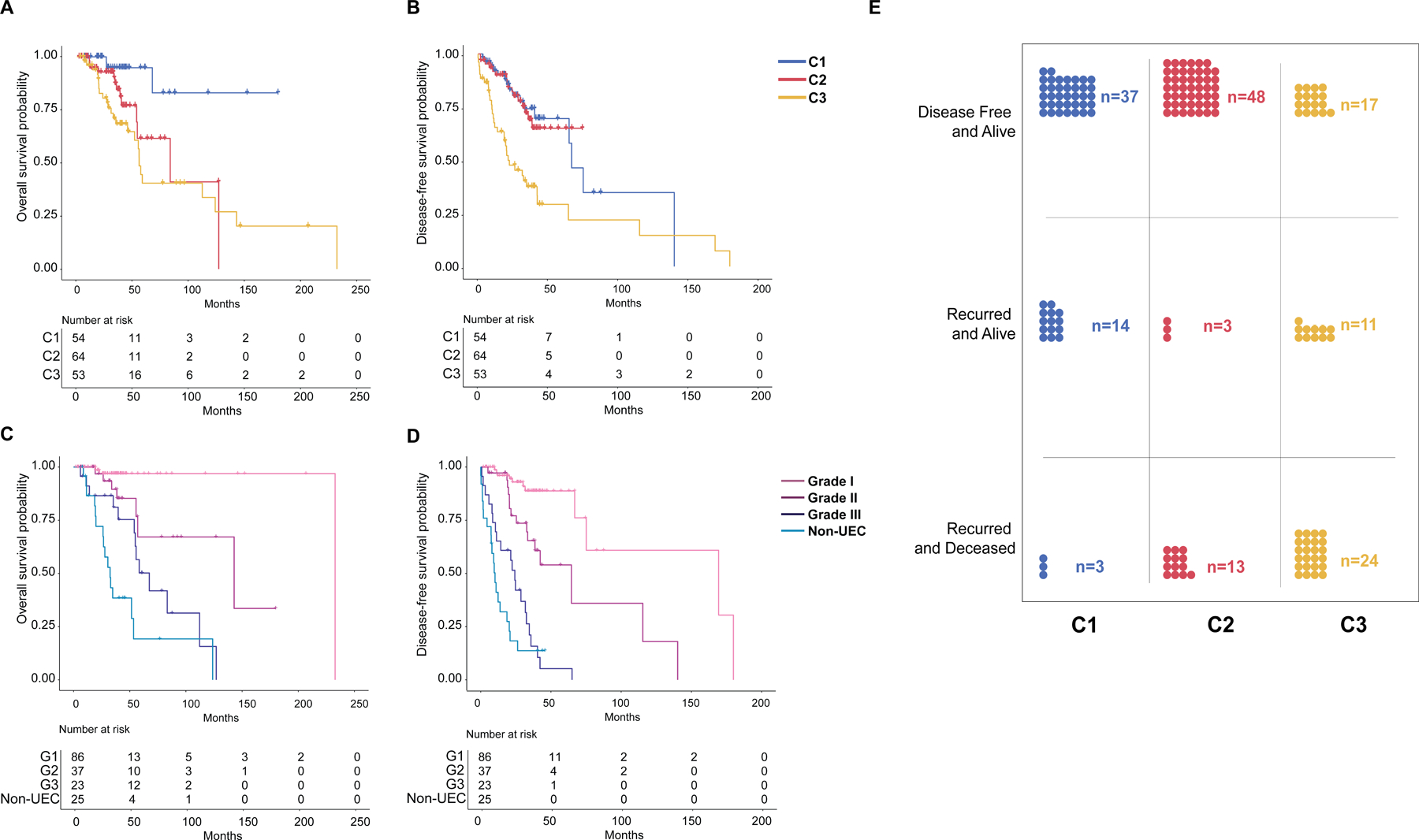
(A) and (B) Kaplan-Meier curves comparing overall survival and disease-free survival among molecular clusters of NSMP-EC. (C) and (D) Kaplan-Meier curves comparing overall survival and disease-free survival among histologic grades and type of NSMP-EC. (E) Distribution of outcome events among the three molecular clusters of NSMP-EC. C1, Cluster 1; C2, Cluster 2; C3, Cluster 3; G1-G3, Grades 1 through 3.
DISCUSSION
The NSMP-EC have been shown to have considerable morphologic and clinical diversity: In comparison with copy number-high or POLE ECs, patients with NSMP-EC have an intermediate prognosis comparable to that of women with MSI-H ECs [1]. However, considerable heterogeneity within the NSMP category exists in terms of clinical outcomes as some patients have an excellent prognosis, whereas others have a more aggressive disease course with associated morbidity and mortality [3,6]. While the majority of NSMP-EC are endometrioid, other morphologic variants including those with ambiguous morphology, endometrial clear cell carcinoma, mesonephric-like carcinoma, and other rare morphologic variants have been previously reported [31]. Current management of NSMP-EC is mainly driven by clinical stage and histopathologic features such as FIGO grade, and presence of lymphovascular invasion [32]. Recently, molecular features such as CTNNB1 alterations or L1CAM expression have been used in risk stratification of the NSMP tumors [33].
Our results demonstrate that the clinical and pathologic heterogeneity of the NSMP-ECs can be largely explained with the underlying molecular alterations. In fact, our results suggest that NSMP-ECs, which comprise the largest share of EC molecular subtypes, are composed of several distinct molecular subclades; based on clustering of mutations in cancer-related genes and copy number alterations, we have shown that NSMP-ECs are composed of at least 3 distinct molecular clusters: The first two clusters appear to be to be driven by activating alterations of the PI3K pathway, where mutations in PTEN are often paired with truncating alterations of PIK3R1 (C1) or with activating PIK3CA mutations (C2). Analysis of cancer cell fractions suggests that PTEN alterations are the initiating event for C1 and C2 tumors followed by alterations in either PIK3CA or PIK3R1, ARID1A, CTNNB1 and KRAS.
C3 tumors are markedly different from both C1 and C2 tumors. These tumors have a relative dearth of PTEN alterations. PI3K pathway alterations in C3 tumors were mostly single hits with either PIK3CA or AKT1 or KRAS activating alterations. A subset of C3 tumors harbored KRAS activating mutations along with chromosome 1q high level gain. The latter finding has been well-studied and believed to be associated with a mesonephric-like phenotype [34] with often adverse outcomes [35]. The mesonephric-like carcinomas are known to have a propensity for lung metastasis [36], a phenomenon which we also observed in our cohort. Unlike the KRAS mutated/1q gain tumors, which are well documented, characterization of an AKT1 mutated subgroup has only been observed anecdotally [37]. In this study, we report an even stronger propensity for lung metastasis compared to the KRAS mutated/1q gain tumors.
The genomic landscape of C1 and C2 NSMP-ECs supports the previous findings of the synergistic effect of PTEN and PIK3CA mutations [38–40], and in the absence of PIK3CA activating mutations, PIK3R1 truncating mutations may exert a similar synergistic effect [38,40,41]. The mutually exclusive nature of PIK3R1 and PIK3CA mutations has been previously shown in both endometrial and breast cancers [1,42]. AKT1 p.E17K mutation is another activating mechanism of the PI3K/AKT pathway [43,44] and we have shown that activating mutations of AKT1 gene occur in absence of alterations of upstream PTEN/PIK3CA/PIK3R1, thereby defining a novel molecular subcategory of NSMP-ECs.
Of interest CTNNB1 gene alterations were observed in relatively equal proportions in all three clusters (27%–49% of samples). While other reports suggest adverse clinical outcomes associated with CTNNB1 alterations [45–47], in our cohort, CTNNB1 hotspot mutations did not show any adverse survival effect. However, the previous studies did not follow the ProMisE classification and were limited to UECs, which may explain the different results observed in our cohort [45–47].
The molecular clustering of the NSMP-EC may portend clinical and prognostic significance. In our cohort, multivariate analysis showed a statistically significant OS hazard ratio associated with C2 and C3 tumors. Our findings suggest that NSMP-EC should be considered as separate molecular clades rather than a single group of clinically, pathologically, and molecularly heterogenous tumors. Previously, it was suggested that clustering of NSMP-EC based on 1q high level gain can also successfully separate the tumors into prognostic groups [6,9], however, in our cohort, 1q copy number status alone was not prognostically significant in multivariate analysis (see Supplementary Table S1).
In conclusion, we have shown here that the NSMP-EC molecular subset of endometrial carcinomas can be further subclassified based on their molecular landscape and that these molecular clusters are associated with meaningful clinical differences. We suggest that cluster 1 and 2 NSMP-EC can perhaps be defined as a PTEN and PI3K altered NSMP-EC group, and the cluster 3 tumors designated as either 1) PTEN wild-type AKT1 altered NSMP-EC or 2) PTEN wild-type KRAS altered NSMP-EC or 3) PTEN wild-type PIK3CA altered NSMP-EC. Further evaluation is needed to confirm the clinical or prognostic significance of these clusters.
Supplementary Material
FUNDING
BW was funded in part by Breast Cancer Research Foundation and Cycle for Survival grants. Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Footnotes
CONFLICTS OF INTEREST
C. Aghajanian reports membership of advisory boards/ personal fees from Tesaro, Eisai/Merck, Mersana Therapeutics, Roche/Genentech, Abbvie, AstraZeneca/Merck, Repare Therapeutics, and grants from Clovis, Genentech, AbbVie, Astra Zeneca, all outside the submitted work. N.R. Abu-Rustum reports Stryker/ Novadaq and GRAIL grants paid to the institution, outside the current study. B. Weigelt reports ad hoc membership of the scientific advisory board of REPARE Therapeutics, outside the submitted work. The remaining authors have no competing interests to disclose.
ETHICS APPROVAL/ CONSENT TO PARTICIPATE
This study was approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board and written informed consent was obtained from all patients.
DATA AVAILABILITY
The datasets used and analyzed in the current study are available from the corresponding authors upon reasonable request.
REFERENCES
- 1.Cancer Genome Atlas Network. Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McAlpine J, Leon-Castillo A, Bosse T. The rise of a novel classification system for endometrial carcinoma; integration of molecular subclasses. J. Pathol. 244, 538–549 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Talhouk A, McConechy MK, Leung S, Yang W, Lum A, Senz J, et al. Confirmation of ProMisE: a simple, genomics-based clinical classifier for endometrial cancer. Cancer 123, 802–813 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Murali R, Davidson B, Fadare O, Carlson JA, Crum CP, Gilks CB, et al. High-grade Endometrial Carcinomas: Morphologic and Immunohistochemical Features, Diagnostic Challenges and Recommendations. Int. J. Gynecol. Pathol. 38 Suppl 1, S40–S63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosse T, Nout RA, McAlpine JN, McConechy MK, Britton H, Hussein YR, et al. Molecular Classification of Grade 3 Endometrioid Endometrial Cancers Identifies Distinct Prognostic Subgroups. Am. J. Surg. Pathol. 42, 561–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Depreeuw J, Stelloo E, Osse EM, Creutzberg CL, Nout RA, Moisse M, et al. Amplification of 1q32.1 Refines the Molecular Classification of Endometrial Carcinoma. Clin. Cancer Res. 23, 7232–7241 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Stelloo E, Nout RA, Osse EM, Jurgenliemk-Schulz IJ, Jobsen JJ, Lutgens LC, et al. Improved Risk Assessment by Integrating Molecular and Clinicopathological Factors in Early-stage Endometrial Cancer-Combined Analysis of the PORTEC Cohorts. Clin. Cancer Res. 22, 4215–4224 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Kommoss FK, Karnezis AN, Kommoss F, Talhouk A, Taran F-A, Staebler A, et al. L1CAM further stratifies endometrial carcinoma patients with no specific molecular risk profile. Br. J. Cancer 119, 480–486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vermij L, Smit V, Nout R, Bosse T. Incorporation of molecular characteristics into endometrial cancer management. Histopathology 76, 52–63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 17, 251–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, et al. A clinically applicable molecular-based classification for endometrial cancers. Br. J. Cancer 113, 299–310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Momeni-Boroujeni A, Dahoud W, Vanderbilt CM, Chiang S, Murali R, Rios-Doria EV, et al. Clinicopathologic and Genomic Analysis of TP53-Mutated Endometrial Carcinomas. Clin. Cancer Res. 27, 2613–2623 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Middha S, Zhang L, Nafa K, Jayakumaran G, Wong D, Kim HR, et al. Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis. Oncol. 1, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids. Res. 44, e131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz-Gay M, Vila-Casadesus M, Franch-Exposito S, Hernandez-Illan E, Lozano JJ, Castellvi-Bel S. Mutational Signatures in Cancer (MuSiCa): a web application to implement mutational signatures analysis in cancer samples. BMC Bioinform. 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canisius S, Martens JW, Wessels LF. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol. 17, 1–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson J, McCluggage WG. Reclassifying endometrial carcinomas with a combined morphological and molecular approach. Curr. Opin. Oncol. 31, 411–419 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hoang LN, Kinloch MA, Leo JM, Grondin K, Lee CH, Ewanowich C, et al. Interobserver Agreement in Endometrial Carcinoma Histotype Diagnosis Varies Depending on The Cancer Genome Atlas (TCGA)-based Molecular Subgroup. Am. J. Surg. Pathol. 41, 245–252 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Van Der Maaten L Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Re.s 15, 3221–3245 (2014). [Google Scholar]
- 24.Hahsler M, Piekenbrock M, Doran D. dbscan: Fast density-based clustering with R. J. Stat. Softw. 91, 1–30 (2019). [Google Scholar]
- 25.Szostakowska M, Trebinska-Stryjewska A, Grzybowska EA, Fabisiewicz A. Resistance to endocrine therapy in breast cancer: molecular mechanisms and future goals. Breast Cancer Res. Treat. 173, 489–497 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashley CW, Da Cruz Paula A, Kumar R, Mandelker D, Pei X, Riaz N, et al. Analysis of mutational signatures in primary and metastatic endometrial cancer reveals distinct patterns of DNA repair defects and shifts during tumor progression. Gynecol. Oncol. 152, 11–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.da Silva EM, Fix DJ, Sebastiao APM, Selenica P, Ferrando L, Kim SH, et al. Mesonephric and mesonephric-like carcinomas of the female genital tract: molecular characterization including cases with mixed histology and matched metastases. Mod. Pathol. 34, 1570–1587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, et al. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin. Cancer Res. 12, 5932–5935 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Hsu AH, Lum MA, Shim K-S, Frederick PJ, Morrison CD, Chen B, et al. Crosstalk between PKCα and PI3K/AKT signaling is tumor suppressive in the endometrium. Cell Rep. 24, 655–669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russo M, Newell JM, Budurlean L, Houser KR, Sheldon K, Kesterson J, et al. Mutational profile of endometrial hyperplasia and risk of progression to endometrioid adenocarcinoma. Cancer 126, 2775–2783 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 28, 836–844 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Koh WJ, Abu-Rustum NR, Bean S, Bradley K, Campos SM, Cho KR, et al. Uterine Neoplasms, Version 1.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 16, 170–199 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Urick ME, Bell DW. Clinical actionability of molecular targets in endometrial cancer. Nat. Rev. Cancer 19, 510–521 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Na K, Kim HS. Clinicopathologic and Molecular Characteristics of Mesonephric Adenocarcinoma Arising From the Uterine Body. Am. J. Surg Pathol. 43, 12–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Euscher ED, Bassett R, Duose DY, Lan C, Wistuba I, Ramondetta L, et al. Mesonephric-like Carcinoma of the Endometrium: A Subset of Endometrial Carcinoma With an Aggressive Behavior. Am. J. Surg. Pathol. 44, 429–443 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Pors J, Segura S, Chiu DS, Almadani N, Ren H, Fix DJ, et al. Clinicopathologic Characteristics of Mesonephric Adenocarcinomas and Mesonephric-like Adenocarcinomas in the Gynecologic Tract: A Multi-institutional Study. Am. J. Surg. Pathol. 45, 498–506 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br. J. Cancer 101, 145–148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Yu M, Yang JX, Cao DY, Zhang Y, Zhou HM, et al. Genomic Comparison of Endometrioid Endometrial Carcinoma and Its Precancerous Lesions in Chinese Patients by High-Depth Next Generation Sequencing. Front. Oncol. 9, 123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nout RA, Bosse T, Creutzberg CL, Jurgenliemk-Schulz IM, Jobsen JJ, Lutgens LC, et al. Improved risk assessment of endometrial cancer by combined analysis of MSI, PI3K-AKT, Wnt/beta-catenin and P53 pathway activation. Gynecol.. Oncol 126, 466–473 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 1, 170–185 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thorpe LM, Spangle JM, Ohlson CE, Cheng H, Roberts TM, Cantley LC, et al. PI3K-p110alpha mediates the oncogenic activity induced by loss of the novel tumor suppressor PI3K-p85alpha. Proc. Natl. Acad. Sci. U S A 114, 7095–7100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Yang L, Yao L, Kuang XY, Zuo WJ, Li S, et al. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat. Commun. 9, 1357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen Y, Shalmon B, Korach J, Barshack I, Fridman E, Rechavi G. AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma. Gynecol. Oncol. 116, 88–91 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Wu W, Chen Y, Huang L, Li W, Tao C, Shen H. Effects of AKT1 E17K mutation hotspots on the biological behavior of breast cancer cells. Int. J. Clin. Exp. Pathol. 13, 332–346 (2020). [PMC free article] [PubMed] [Google Scholar]
- 45.Kurnit KC, Kim GN, Fellman BM, Urbauer DL, Mills GB, Zhang W, et al. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod. Pathol. 30, 1032–1041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Costigan DC, Dong F, Nucci MR, Howitt BE. Clinicopathologic and Immunohistochemical Correlates of CTNNB1 Mutated Endometrial Endometrioid Carcinoma. Int. J. Gynecol. Pathol. 39, 119–127 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding L, et al. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J. Natl. Cancer Inst. 106, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and analyzed in the current study are available from the corresponding authors upon reasonable request.