

Abstract
Background: Prostatic inflammation is closely linked to the development and progression of benign prostatic hyperplasia (BPH). Clinical studies of non-steroidal anti-inflammatory drugs, which inhibit cyclooxygenase-2 (COX-2), targeting prostate inflammation patients with symptomatic BPH have demonstrated conflicting results, with some studies demonstrating symptom improvement and others showing no impact. Thus, understanding the role of the cyclooxygenases in BPH and prostatic inflammation is important. Methods: The expression of COX-1 was analyzed in a cohort of donors and BPH patients by immunohistochemistry and compared to previously determined characteristics for this same cohort. The impact of mitochondrial dysfunction on COX-1 and COX-2 was determined in experiments treating human benign prostate epithelial cell lines BPH-1 and RWPE-1 with rotenone and MitoQ. RWPE-1 cells were transfected with small interfering RNA specific to complex 1 gene NDUFS3. Results: COX-1 expression was increased in the epithelial cells of BPH specimens compared to young healthy organ donor and normal prostate adjacent to BPH and frequently co-occurred with COX-2 alteration in BPH patients. COX-1 immunostaining was associated with the presence of CD8+ cytotoxic T-cells, but was not associated with age, prostate size, COX-2 or the presence of CD4+, CD20+ or CD68+ inflammatory cells. In cell line studies, COX protein levels were elevated following treatment with inhibitors of mitochondrial function. MitoQ significantly decreased mitochondrial membrane potential in RWPE-1 cells. Knockdown of NDUFS3 stimulated COX-1 expression. Conclusion: Our findings suggest COX-1 is elevated in BPH epithelial cells and is associated with increased presence of CD8+ cytotoxic T-cells. COX-1 can be induced in benign prostate epithelial cells in response to mitochondrial complex I inhibition, and knockdown of the complex 1 protein NDUFS3. COX-1 and mitochondrial dysfunction may play more of a role than previously recognized in the development of age-related benign prostatic disease.
Keywords: COX-1, COX-2, prostate inflammation, BPH, aging
Introduction
Benign prostatic hyperplasia (BPH) is an age-related disease characterized by benign prostate enlargement due to periurethral stromal and/or glandular expansion [1]. The etiology of BPH is not fully understood. BPH does not develop in the absence of androgens; and estrogens, stromal expansion and inflammatory cytokines have also been implicated as factors involved in BPH development [2-6]. Globally, the population of men > 65 years of age is expected to rise to ~17% over the next three decades [7], thus both the overall incidence of BPH and the number of years living with this condition is expected to increase and elucidating the underlying mechanisms contributing to BPH is of critical importance for the development of therapeutic strategies for this highly prevalent condition.
The cyclooxygenase (COX) enzyme family consists of two isoforms, COX-1 and COX-2. Both enzymes catalyze the conversion of arachidonic acid to prostaglandin H2 (PGH2), an intermediate in the biosynthetic pathway that generates many inflammatory mediators including other prostaglandins and thromboxanes. In almost all tissues, COX-1 is constitutively expressed while COX-2 is rapidly induced in response to growth factors and pro-inflammatory cytokines (Reviewed in [8]). Inhibition of the COX isoforms by non-steroidal anti-inflammatory drugs (NSAIDS) acutely reduces inflammation, pain and fever [9]. Upregulation of COX-2, and to a lesser degree COX-1 is observed in symptomatic BPH and is associated with resistance to treatment with 5α-reductase inhibitors (5ARI) or an α-adrenergic receptor antagonist [10]. Prior immunohistochemical (IHC) analyses of prostate tissue revealed that COX-1 and COX-2 expression are predominantly localized within the basal epithelial layer and in luminal epithelial cells associated with prostatic inflammation as well as in the stromal compartment [10,11]. Patients with prostatic inflammation are more likely to have symptomatic BPH and lower urinary tract symptoms (LUTS) [12,13], suggesting that targeting prostatic inflammation could provide a therapeutic benefit. However, studies of patients taking NSAIDS have not demonstrated consistent results, with some studies finding an improvement in symptoms [14] and others showing no significant reductions in BPH incidence or symptoms [15,16]. Thus, the role of COXs in driving the production of prostatic inflammatory mediators that impact the development of LUTS in patients with BPH remains obscure.
In this report we sought to provide additional insights into the expression of distinct COXs in the aging prostate and uncover potential molecular mechanisms contributing to alternative regulation of COX-1 and COX-2 in prostate epithelial cells. Our results demonstrate significant increases in epithelial COX-1 expression in the prostates of BPH patients. Furthermore, cell culture studies reveal the potential impact of selective alterations in mitochondrial function in dysregulation of COX-1 and COX-2 expression. Activation of the COX pathway by aberrant mitochondrial function may result in increased prostatic inflammation, contributing to BPH development or progression.
Methods
Patient cohort
Human prostate tissue sections were from a previously described cohort of 14 patients who received transurethral resection of the prostate or simple prostatectomy for symptomatic BPH, and six young organ donors [17,18]. All prostate tissues were from the transition zone or central zone and not from the peripheral zone of donor or BPH patients without any prior history of chemo-, radio, or hormone therapy. BPH specimens were composed of mixed hyperplastic nodules with both glandular and stromal expansion. Two patients displayed prostatitis, one donor and one BPH, and were therefore not included in the analyses. The criteria used for defining prostatitis was the presence of inflammatory cells in the prostate stroma infiltrating into prostate glands with or without the presence of crypt abscess formation. The “deidentified” specimens were retrieved from the clinical files of UPMC by the University of Pittsburgh Biospecimen Core (PBC) with approval from the University of Pittsburgh Institutional Review Board (IRB) for this research project under protocol #17010177. PBC also provided deidentified pathology reports for the patients constituting the study cohort through their IRB-approved Honest Broker System, HB015, to ensure research integrity. All participating patients or their next of kin provided informed consent for the banking protocol.
Inclusion criteria: 1) for BPH tissues, evidence of BPH by transrectal ultrasound and/or digital rectal exam, and prostate glands must be > 30 grams to qualify; for donor tissues, patients must be < 35 years of age to qualify and tissues must appear histologically normal; 2) no prior use of finasteride or dustateride; 3) no prior chronic NSAID use.
Exclusion criteria: 1) prior use of finasteride or chronic NSAIDs; 2) peptic ulcer disease and/or asthma; 3) previous prostate surgery for BPH (TURP or simple open prostatectomy); 4) previous minimally invasive procedures for BPH (TUNA, laser treatments, thermotherapies such as microwave treatment).
Serial sections for histopathologic and immunohistochemistry (IHC) analyses
Serial sections of prostate tissues were cut from each patient sample for histopathologic and IHC studies. Sections on glass slides were deparaffinized and stained for hematoxylin and eosin (H&E) and IHC using a standard histology protocol. Antigen retrieval was performed using a citrate buffer (Dako, Carpinteria, CA) in a BioCare Decloaking chamber (BioCare Medical, Pacheco, CA) at 123°C followed by 5 minutes rinsing in Tris-buffered saline (TBS) buffer. The slides were stained using an Autostainer Plus (Dako) platform with TBS containing Tween 20 (TBST) rinse buffer (Dako) using anti-rabbit polyclonal COX-1 antibody (#4841, Cell Signaling Technology, Danvers, MA). The substrate used was 3,3, diaminobenzidine (Dako) and slides were counterstained with hematoxylin (Cell Signaling). Immunostained sections were imaged with a Leica DM LB microscope (Leica Microsystems Inc, Bannockburn, IL) equipped with an Imaging Source NII 770 camera (The Imaging Source Europe GmbH, Bremen, Germany) and NIS-Elements Documentation v 4.6 software (Nikon Instruments, Inc., Mellville, NY). All tissues were examined by a board-certified genitourinary pathologist (RD) and a pathology resident (PS). Tissues exhibiting BPH and normal morphology (normal adjacent to BPH) from BPH patients were identified by the pathologists using light microscopy.
COX-1 immunostaining intensity in the prostate tissue sections was determined semi-quantitatively. The percentage of prostate epithelial cells of a specific histological phenotype (donor, normal adjacent to BPH and BPH) that expressed the antigen was estimated in at least three randomly selected fields at a magnification of 40×. Staining intensity was evaluated by two parameters (staining intensity and percentage of cells exhibiting each level of intensity). Intensity of reaction product was based on a 4-point scale-none (0), faint/equivocal (1), moderate (2) and intense (3). An H-Score was calculated for each immunostain by cell type using the following formula:
H-Score = 0 (% no stain) + 1 (% faint/equivocal) + 2 (% moderate) + 3 (% intense).
Cell culture and treatment
BPH-1 cells [19] were a gift from Dr. Simon Hayward (Northshore University Health System, USA) and were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS; Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin (Corning, Manassas, VA). RWPE-1 cells (ATCC, Manassas, VA) were maintained in Gibco Keratinocyte Serum Free Medium supplemented with bovine pituitary extract and human recombinant epidermal growth factor (Life Technologies, Rockville, MD, USA). For experimental treatments, cells were seeded at 2.5-3×105 cells per well in 6-well plates. After 24 h (for BPH-1 cells) or 48 h (for RWPE-1 cells), cells were treated with a dose range of 0-500 nM rotenone or 0-3 μM mitoquinol (MitoQ) (both from Cayman Chemical, Ann Arbor, MI, USA) for 48 h, then briefly rinsed with ice-cold phosphate-buffered saline (PBS). Cells were lysed in Cell Extraction Buffer containing complete protease inhibitor cocktail tablet (Roche, Penzberg, Germany) and freshly made phenylmethylsulfonyl fluoride according to the manufacturer’s instructions. Cellular debris in samples was removed by centrifugation at 10,000× g for 10 min at 4°C. Protein samples were stored at -20°C.
Western blotting
Protein concentration was determined using a BCA protein assay kit (Pierce, Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of total protein from cell extracts were incubated with 4× Laemmli reducing sample buffer (Bio-Rad Laboratories, Hercules, CA), then heated at 100°C for 5 min before loading onto 10% gels. Proteins were separated via sodium dodecyl sulphate-polyacrylamide gel electrophoresis using the Mini Protein 3 System (Bio-Rad), then transferred onto 0.2 µm nitrocellulose membranes (Bio-Rad). Membranes were blocked with 1% bovine serum albumin in PBS containing 0.05% Tween 20 at room temperature for 1 h, then incubated with the appropriate primary and secondary antibodies. Primary antibodies used in this study were: anti-rabbit polyclonal COX-1 antibody (#4841, Cell Signaling Technology Danvers, MA), anti-rabbit polyclonal COX-2 antibody (ab15191, abcam, Cambridge, UK), mouse monoclonal anti-β-actin antibody (Clone AC-15, A5441, Sigma-Aldrich, St. Louis, MO), and rabbit polyclonal anti-NDUFS3 (15066-1-AP, Proteintech, Rosemont, IL, USA). Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE) was used to detect specific proteins. Secondary antibody was labeled with either IRDye 680LT (for 680 nm) or 800CW (for 780 nm). Different fluorescent signals from specific proteins were scanned and discriminated using two independent infrared detection channels at 685 and 785 nm excitation wavelengths, which were quantified by the NIH ImageJ software [20]. Full length western blot images were provided as Figures S1, S2, S3, S4.
Mitochondrial membrane potential (ΔΨm)
Changes in the mitochondrial membrane potential (ΔΨm) in live RWPE-1 cells were assessed using the JC-1 Mitochondrial Membrane Potential Assay Kit from Abcam (Cambridge, MA). RWPE-1 cells were seeded at 1.5×104 cells per well in a 96-well black tissue culture plate with a clear bottom (VWR, Radnor, PA). After 48 h, cells were treated with either rotenone or MitoQ (both from Cayman Chemical, Ann Arbor, MI) for 2 h and 24 h. After treatment, media was replaced with fresh culture medium containing 5 µM JC-1 for 15 min at 37°C and fluorescence was assayed using a fluorescent plate reader (Spectra Max M2, Molecular Devices, San Jose, CA, USA). The red to green ratio was calculated and compared to untreated control to determine loss of ΔΨm. Data were collected and averaged for at least three independent experiments.
RNA isolation and quantitative real-time PCR (qPCR)
RNA was isolated from RWPE-1 and BPH-1 cells after 24 h treatment using a RNeasy Mini Kit from Qiagen (Germantown, MD). cDNA synthesis was performed using the iScript cDNA Synthesis Kit (Bio-Rad). qPCR was performed using the CFX96 Touch Real Time Detection System with products generated using iTaq Universal SYBR Green Supermix (Bio-Rad). Gene-specific primers (COX-1, forward: 5’-TGCCCAGCTCCTGGCCCGCCGCTT-3’ and reverse: 5’-GTGCATCAACACAGGCGCCTCTTC-3’; COX-2, forward: 5’-ATCACAGGCTTCCATTGACC-3’ and reverse: 5’-CAGGATACAGCTCCACAGCA-3’; and HPRT1, forward: 5’-TGGCGTCGTGATTAGTGATG-3’ and reverse: 5’-ACCCTTTCCAAATCCTCAGC-3’) were used to validate gene expression levels using the comparative CT method; HPRT1 served as a reference gene.
RNA interference of NDUFS3
siRNA knockdown of NDUFS3 protein was performed in RWPE-1 cells using ON-TARGETplus human NDUFS3 siRNA or a scrambled siRNA control (Horizon Discovery/Dharmacon, Lafayette, CO). RWPE-1 cells were plated onto 6-well plates overnight as described above. Cells were then transfected with siRNAs for 48 h according to the manufacturer’s procedure. The final concentration of transfecting siRNA was 25 nM and the efficiency of siRNA interference for NDUFS3 was monitored by western blot.
Statistical analyses
An analysis of variance (ANOVA) used to compare mean H scores between donor, normal adjacent and BPH tissue. Alteration frequency was determined by quantification of the number of patients with increased (i.e., greater than the mean for each cohort) immunostaining intensity for COX-1. Quantification of COX-2 immunostaining intensity was determined previously [18]. A Fisher’s exact test and a two-tailed p-value was used to compare alteration frequency between groups. The Pearson correlation coefficient was used to determine the correlation between COX-1 immunostaining with previously reported age, prostate size, COX-2 immunostaining and infiltration of inflammatory cells data from this cohort of donor and BPH patients [18]. Comparisons between groups were calculated using the Student t test for data from cell line experiments. A p value of P < 0.05 was considered significant. Data are expressed as the mean ± S.D. or mean ± S.E.M. GraphPad Prism version 9 was used for graphics (GraphPad Software, San Diego, CA, USA).
Results
Expression of COX-1 in human prostate
We sought to more fully characterize the expression of both isoforms of COX enzymes in the prostate epithelium by examining COX-1 expression in this same cohort of patients. Similar to COX-2, COX-1 immunostaining intensity was low in donor and normal glands from BPH patients and not altered with age. However, elevation of COX-1 immunostaining intensity was observed in the luminal cytoplasm of several BPH nodules and infrequently in the nuclei of isolated basal epithelial cells of BPH glands (Figure 1A, 1B), suggesting that in contrast to COX-2, COX-1 may be up-regulated in BPH epithelium. Elevated COX-1 immunostaining was rarely observed in donor or normal adjacent prostate tissues but was frequently observed in BPH tissues (Figure 1C). The frequency of elevated COX-1 and COX-2 immunostaining was higher in BPH compared with donor [66.7% (6 of 9) vs 0% (0 of 5), respectively, P = 0.03] or normal adjacent to BPH tissues [66.7% (6 of 9) vs 0% (0 of 6), respectively, P = 0.03] (Figure 1C).
Figure 1.
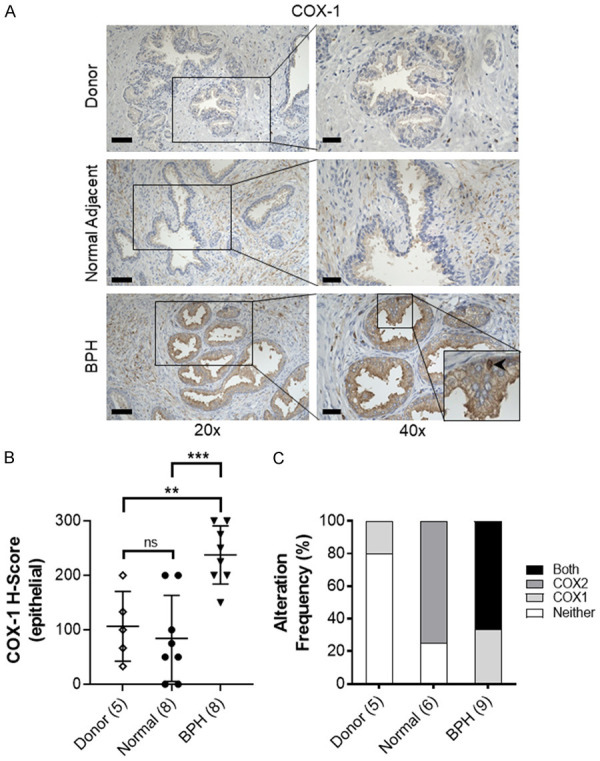
Expression of COX-1 in the prostate. (A) Representative immunostaining of COX-1 (brown) expression in young healthy donor (Donor), normal adjacent to BPH prostate (Normal Adjacent) and BPH specimens. Scale bars indicate 100 µm in 20×, 50 µm in 40×. BPH inset, bottom right, showing nuclear COX-1 staining (black arrow). (B) Quantification of mean COX-1 staining intensity H-score in prostate epithelial cells. (C) Alteration frequency of specimens with no alteration in COX-1 or COX-2 expression (Neither), upregulated COX-1 (COX1), upregulated COX-2 (COX2), or concurrent upregulation of COX-1 and COX-2 in Donor, Normal Adjacent and BPH specimens. Scoring was quantified for patients with both COX-1 and COX-2 immunostaining scores; specimens missing either COX-1 or COX-2 data were not included. Number of patients in parentheses. Data represent mean ± S.D.; ns, not significant, **P < 0.01; ***P < 0.001.
There was no significant correlation between COX-1 and previously reported COX-2 [18] staining in prostate epithelial cells (Pearson = -0.01, P = 0.95) (Table 1). COX-1 expression was moderately correlated with the presence of CD8 positive cytotoxic T-cells (Pearson = 0.40, P = 0.04). COX-1 immunostaining was not significantly correlated with age, prostate size, or the presence of CD68 macrophages, CD20 B-cells, or CD4 T-cells.
Table 1.
Pearson correlation of COX-1 immunostaining with age and inflammatory cells in prostate
| COX-1 vs. | Age | Prostate mass | Prostate volume | COX-2 (epi) | CD4 | CD8 | CD20 | CD68 |
|---|---|---|---|---|---|---|---|---|
| r | 0.28 | 0.23 | 0.16 | 0.01 | 0.36 | 0.40 | 0.11 | 0.24 |
| 95% CI | -0.12 to 0.60 | -0.21 to 0.58 | -0.27 to 0.54 | -0.38 to 0.40 | -0.035 to 0.65 | 0.017 to 0.68 | -0.29 to 0.48 | -0.16 to 0.58 |
| R squared | 0.08 | 0.05 | 0.03 | 0.00 | 0.13 | 0.16 | 0.01 | 0.06 |
| p (two-tailed) | 0.17 | 0.30 | 0.46 | 0.95 | 0.07 | 0.04 | 0.59 | 0.23 |
CI: confidence interval; Age, prostate size and inflammatory cell data were analyzed in silico from previously published data [18].
Regulation of COX-1 and COX-2 expression in human prostate epithelial cell lines
To test whether ROS activation through the mitochondrial electron transport pathway (established regulators of COX enzymes) could influence COX-1 and COX-2 expression in benign prostate epithelial cells, we utilized rotenone, a mitochondrial complex I inhibitor. COX-1 protein expression was induced by rotenone in RWPE-1 (Figure 2A) but not BPH-1 (Figure 2B) cells. Similar to the impact on HL-60 cells [21], rotenone induced a dose-dependent increase in COX-2 protein in both RWPE-1 and BPH-1 cells (Figure 2C, 2D). Rotenone generated distinct effects on COX mRNA expression in RWPE-1 cells inducing an increase in COX-1 but not COX-2 mRNA (Figure 2E).
Figure 2.
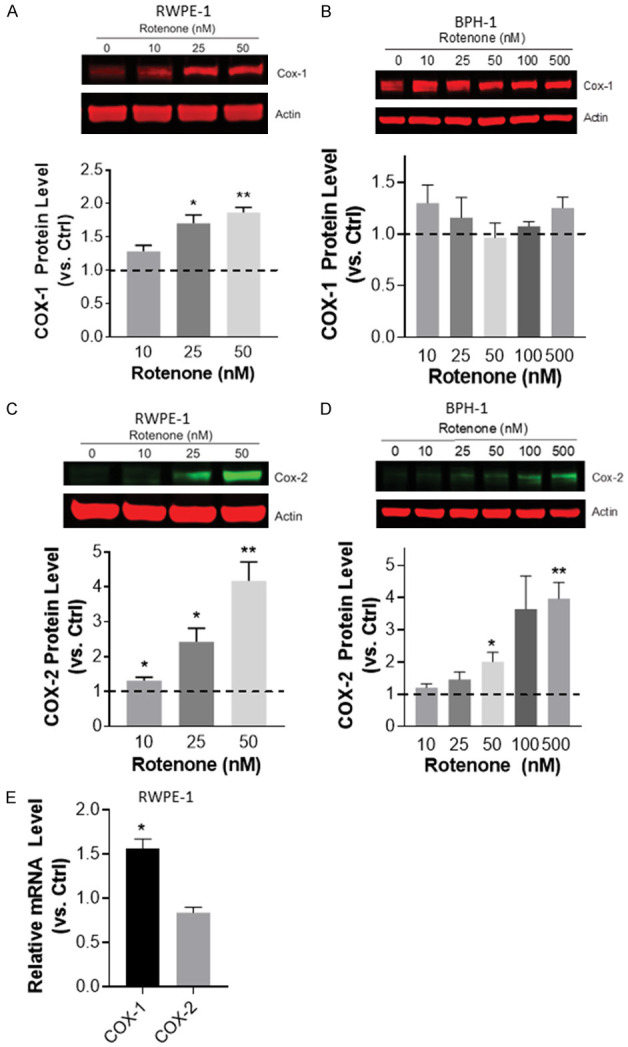
Induction of COX expression by rotenone in benign prostate epithelial cells in vitro. (A) Western blot analysis of COX-2 protein expression levels following rotenone treatment of (A) RWPE-1 and (B) BPH-1 cells for 48 h. (C) COX-1 expression following rotenone treatment for 48 h in RWPE-1 and (D) BPH-1 cells. Band intensities were quantified and the ratios of COX protein relative to actin were normalized to untreated control (ctrl, dashed line). Data were calculated from at least 3 independent experiments. *P < 0.05 and **P < 0.01. (E) qPCR analysis of COX-1 and COX-2 mRNA expression in RWPE-1 cells following 24 h of treatment with 25 nM rotenone. Expression in treated cells shown as relative to untreated control cells (Ctrl, dashed line). Data represent mean ± S.E.M. *p < 0.05 and **P < 0.01.
MitoQ also induced an increase in COX-1 and COX-2 protein expression in RWPE-1 cells (Figure 3). Rotenone did not decrease mitochondrial membrane potential in RWPE-1 cells at 2 h, and generated only a minor but significant decrease following 24 h of treatment (Figure 4A, 4B). In contrast, MitoQ treatment of RWPE-1 cells induced an acute (i.e. 2 h) dose-dependent decrease in the red-green JC-1 fluorescence ratio relative to untreated control cells (Figure 4C). This decrease in mitochondrial membrane potential was long lasting and was more pronounced at 24 h (Figure 4D).
Figure 3.
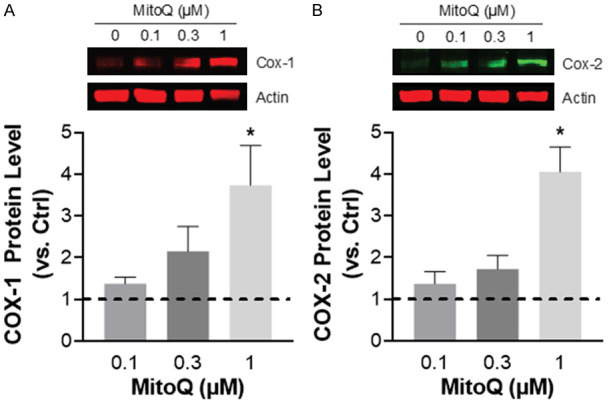
Induction of COX expression by MitoQ in the RWPE-1 benign prostate epithelial cell line. (A) Western blot analysis of COX-1 and (B) COX-2 protein expression levels following MitoQ treatment (1 µM) for 48 hours. Results are from 4-5 independent experiments. (Ctrl, dashed line). Data represent mean ± S.E.M. *P < 0.05.
Figure 4.
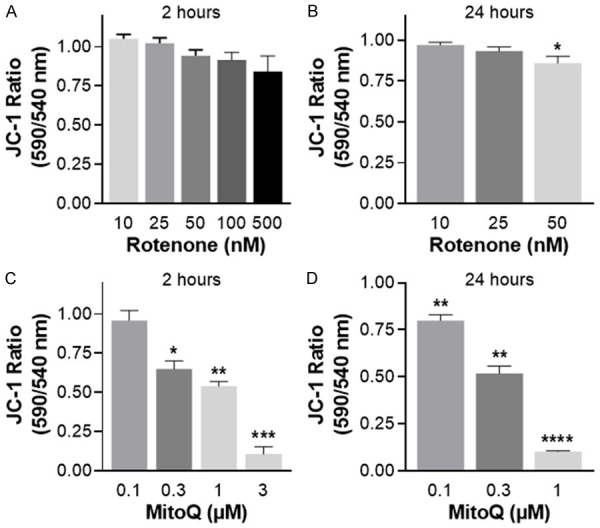
Loss of mitochondrial membrane potential (ΔΨm) induced by rotenone or MitoQ in RWPE-1 cells, measured by quantitative analysis of JC-1 fluorescence. (A) Impact of rotenone treatment after 2 h, or (B) 24 h. (C) Impact of MitoQ treatment after 2 h, or (D) 24 h. Data are expressed relative to untreated control cells 3-4 independent experiments. Data represent mean ± S.E.M. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
To corroborate the impact of complex I dysfunction on COX-1 and COX-2, RWPE-1 cells were siRNA ablated of NDUFS3, a protein subunit that is essential for the assembly of mitochondrial respiratory chain complex I. COX-1 protein expression was induced in RWPE-1 cells following NDUFS3 knockdown (Figure 5). However, NDUFS3 knockdown did not induce COX-2 protein expression (data not shown).
Figure 5.
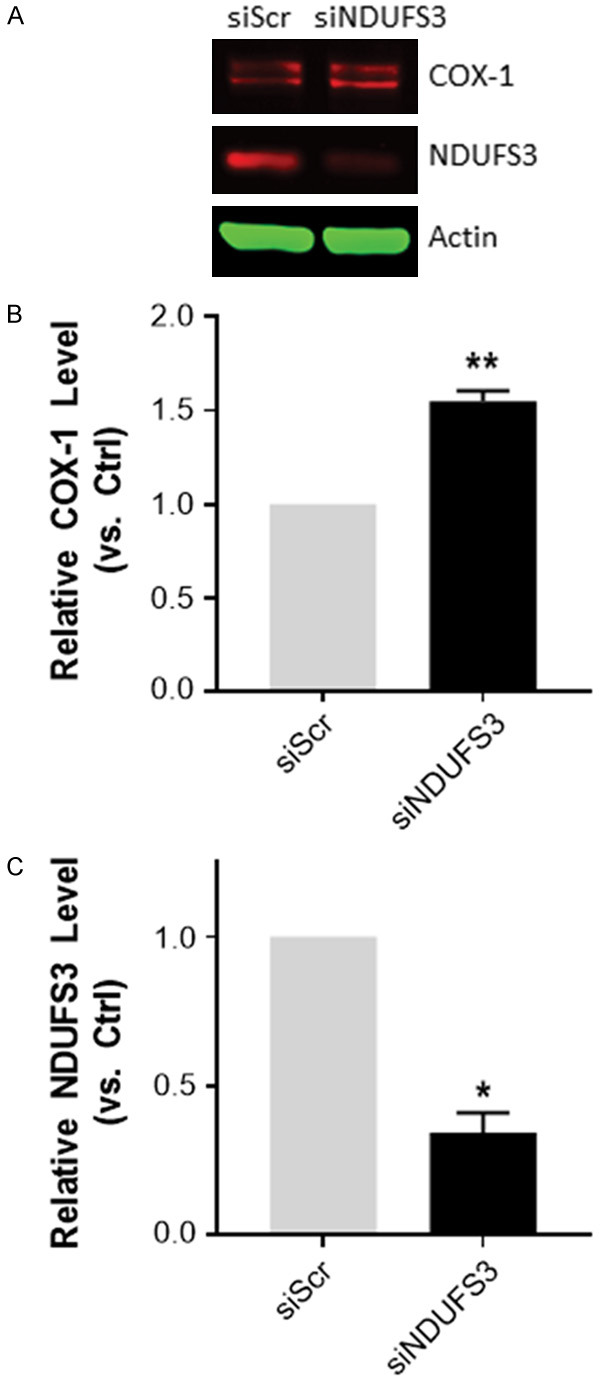
Induction of COX-1 protein expression in response to NDUFS3 siRNA knock-down in RWPE-1 cells. (A) Western blot analysis of COX-1 and NDUFS3 expression in RWPE-1 cells following 48 h NDUFS3 siRNA-transfection cells (ND3), control cells were treated with a scramble control (Scr). (B) Quantification of band intensities and the ratios of COX-1, and (C) NDUFS3 protein relative to actin were normalized to siRNA scramble control. Final siRNA concentration was 25 nM. Data were calculated from at least 3 independent experiments. *P < 0.05 and **P < 0.01. Data represent mean ± S.E.M. *P < 0.05 and **P < 0.01.
Discussion
The effectiveness of anti-inflammatory therapeutics has been inconclusive for BPH/LUTS patients. While some studies have demonstrated no effect on BPH prevention or symptom improvement [15,16], others suggest that NSAIDS can improve BPH symptoms [14]. Treatment of BPH patients with COX-2 inhibitors and NSAIDs has been shown to significantly reduce IPSS scores, however COX-2 blockade has not been shown to prevent BPH symptomatology once it has occurred [12]. Targeting prostatic inflammation directly/alone may be insufficient for the prevention or treatment of symptomatic BPH and additional mechanisms may be involved in BPH development and progression. COX-2 is the major COX enzyme involved in inflammatory responses but a few experimental paradigms that can trigger inflammation have also detected increases in COX-1 (Reviewed in [22,23]). Furthermore, COX-1 and COX-2 serve non-overlapping roles in the generation of downstream inflammatory mediators [24,25]. We recently showed COX-2 was increased with aging and inflammation in the human prostate stroma while epithelial COX-2 was not altered [18]. Stromal COX-1 and COX-2 are increased in BPH tissues, specifically in patients resistant to 5ARI treatment [10]. The differential expression of the COX enzymes in the aging prostate and in BPH suggests that they have distinct, cell-type specific roles in the maintenance of prostate homeostasis.
Here, we show that COX-1 immunostaining is rare in the prostate epithelium of young donor tissues and normal adjacent prostate of BPH patients but is significantly increased in BPH epithelial cells and is associated with an increased presence of CD8+ cytotoxic T-cells. Interestingly, COX-1 expression was not correlated with an increase in CD4+ T-cells, macrophages or B-cells, or with COX-2 expression. Thus, distinct pathophysiological changes that accompanied with the development of BPH might be responsible for the elevation of COX-1 in prostatic epithelial cells. In order to identify potential mechanisms responsible for elevated COX-1 in BPH, we used human benign prostatic epithelial cell lines to disrupt one metabolic system that declines with age, mitochondrial bioenergetics. Both pharmacological and molecular approaches were used to disrupt a specific component of mitochondria, electron transport chain complex I, which alters mitochondrial bioenergetics and can trigger elevation of reactive oxygen species (ROS). We consider this relevant since COX expression can be regulated by intracellular ROS [21], and since oxidative stress has been hypothesized to contribute to prostatic inflammation and urinary dysfunction [26]. In cell line experiments, COX-2 and COX-1 expression levels were increased in response to pharmacological disruption of mitochondrial function, particularly respiratory complex I. More selective inhibition of complex I upon siRNA knockdown of one of its essential components (i.e., NDUFS3), suggest that COX-1 may be specifically sensitive in prostate epithelial cells to disruption in the mitochondrial respiratory complex. Knockdown of NDUFS3 has been reported previously to impair complex I activity in different cell types including HEK [27] and HeLa cells [28-30]. Whether the enhancement of COX-1 in the prostate epithelium of BPH patients is reflective of mitochondrial disruption awaits more detailed comparisons of mitochondrial function and integrity in BPH. The difference of COX-2 response to pharmacological disruption of complex I versus NDUFS3 knockdown suggest that either NDUFS3 knockdown does not trigger sufficient alterations in complex I function to induce COX-2, or that off target effects of rotenone are responsible for its induction.
The differential impact of COX-1 and COX-2 on the signaling mediators derived from arachidonic acid metabolism has not been thoroughly investigated in many organ systems but given our results, could provide insights into BPH pathogenesis. Thus, although COX-1 and COX-2 catalyze the production of a common precursor (i.e., PGH2) of various prostaglandins, prostacyclins, and thromboxanes, their physical association with distinct enzymes in the metabolic pathway of these inflammatory mediators can influence the products derived from PGH2 and ultimately cellular response [31,32]. Mice with conventional deletion of COX-1 displayed reduced platelet aggregation and a decreased inflammatory response to arachidonic acid suggesting that COX-1 is an important inflammatory mediator, and that COX-2 cannot fully compensate for COX-1 loss [33]. In a genetically engineered mouse model where the elements regulating COX-1 and COX-2 (Ptgs1 and Ptgs2 respectively) transcription were swapped, exposure to lipopolysaccharide differentially altered the activity of prostanoid synthases and thromboxane synthase [34]. The change in synthases led to significant differences in thromboxane production in mice that were unable to constitutively express COX-1. Prostaglandin E2 and 6-keto-PGF1 alpha levels were significantly lower in the genetically altered mice compared to wild type mice as well [35]. These data support the physiological relevance of distinct, if not sometimes synergistic, activities of COX-1 and COX-2 enzymes.
Our in vitro studies utilizing a mitochondrial electron transport chain (ETC) complex I inhibitor, rotenone, and a disrupter of mitochondrial potential, MitoQ, revealed robust induction of COX-1 and COX-2 in response to disruption of mitochondrial function within the benign prostate epithelial cell line RWPE-1. BPH has long been recognized as a disease entwined with the aging process and much groundbreaking work has been performed linking a cumulative burden of mitochondrial disfunction due to age with inflammation [36]. We previously showed that stromal, but not epithelial expression of COX-2 was associated with aging in the human prostate [18]. Here, COX-1 expression was also not different in young donor compared to aged BPH patients. However, COX-1 immunostaining was significantly increased in BPH glands compared to both normal adjacent tissue and the epithelium of donor tissues and was associated with the increased presence of cytotoxic T-cells. Our findings provide evidence that the elevation of COX-1 immunostaining observed in BPH epithelial cells could be triggered by mitochondrial dysfunction, potentially contributing to an increase in prostatic inflammation. NSAIDS targeting both COX-1 and COX-2 may provide greater therapeutic impact on prostate inflammation than those targeting COX-2 alone.
Mitochondrial dysfunction provoked with oligomycin has been shown to induce not only COX-2 but also prostaglandin E2 within normal human synoviocytes indicating a possible analogous role within the human prostate epithelium [37]. Our results in a prostate epithelial cell line suggest that mitochondrial dysfunction may be partly responsible for altering the balance of arachidonic acid metabolites driving tissue homeostasis through selective effects of COX-1. Inhibition of ETC complex I leads to the generation of ROS [38,39], which are well established inducers of COX-2 [21,40,41]. Here, we show that complex I inhibition also induces COX-1 in benign prostate epithelial cells. The mechanisms responsible for regulation of COX-1 transcription remain poorly understood although some studies have identified GC-rich transcriptional response elements in the COX-1 proximal promoter that could be responsive via the SP1 or KLF10 transcription factors to complex I dysfunction [42-44].
This study was limited in that the number of prostate specimens examined was relatively small. Future IHC studies focusing on essential components of the mitochondrial ETC (e.g. NDUFS3) with larger patient populations will provide additional insights into how mitochondrial dysfunction and COX-1 contribute to prostate inflammation associated with BPH and LUTS. Furthermore, preclinical studies with genetic models allowing for conditional ablation of select mitochondrial components in prostate [45] will provide an opportunity to test the progression of benign prostatic alterations as mitochondrial function is compromised.
In conclusion, COX-1 is elevated in BPH epithelium and was associated with the presence of CD8+ cytotoxic T-cells in the prostate but is not altered with aging and is not associated with prostate size or COX-2 expression. COX-1 and COX-2 can be induced in benign prostate epithelial cells in response to mitochondrial complex I inhibition. Thus, the aging dependent decline in mitochondrial function may be one, but not an exclusive contributor to BPH progression, acting to alter the fine tuning of extracellular mediators derived from arachidonic acid (i.e., COX enzymes) even in the absence of blatant inflammation.
Acknowledgements
We are grateful to Anthony Green and Paul Knizner for technical support. This was funded in part by NIH grants U54 from NIDDK, DK112079 (ZW), R56 DK107492 (ZW), and a CAIRIBU Convergence Award (LEP). This project used the Hillman Cancer Center Tissue and Research Pathology Services (TARPS) and the Pitt Biospecimen Core and was supported in part by award P30CA047904 with additional support from the University of Pittsburgh Institute for Precision Medicine.
Disclosure of conflict of interest
None.
Abbreviations
- BPH
Benign prostatic hyperplasia
- COX-1
Cyclooxygenase-1
- COX-2
Cyclooxygenase-2
- PGH2
Prostaglandin H2
- NSAIDS
Non-steroidal anti-inflammatory
- 5ARAI
5α-reductase inhibitors
- IHC
Immunohistochemistry
- LUTS
Lower urinary tract symptoms
- PBC
Pitt Biospecimen Core
- H&E
Hematoxylin and eosin
- TBS
Tris-buffered saline
- MitoQ
Mitoquinol
- PBS
Phosphate buffered saline
- siRNA
Silencing ribonucleic acid
- ANOVA
Analysis of variance
- ROS
Reactive oxygen species
- ETC
Electron transport chain
- ΔΨm
Mitochondrial membrane potential
Supporting Information
References
- 1.Vuichoud C, Loughlin KR. Benign prostatic hyperplasia: epidemiology, economics and evaluation. Can J Urol. 2015;22(Suppl 1):1–6. [PubMed] [Google Scholar]
- 2.Barrack ER, Berry SJ. DNA synthesis in the canine prostate: effects of androgen and estrogen treatment. Prostate. 1987;10:45–56. doi: 10.1002/pros.2990100108. [DOI] [PubMed] [Google Scholar]
- 3.Nickel JC. Inflammation and benign prostatic hyperplasia. Urol Clin North Am. 2008;35:109–115. doi: 10.1016/j.ucl.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schauer IG, Rowley DR. The functional role of reactive stroma in benign prostatic hyperplasia. Differentiation. 2011;82:200–210. doi: 10.1016/j.diff.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schauer IG, Ressler SJ, Tuxhorn JA, Dang TD, Rowley DR. Elevated epithelial expression of interleukin-8 correlates with myofibroblast reactive stroma in benign prostatic hyperplasia. Urology. 2008;72:205–213. doi: 10.1016/j.urology.2007.11.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Pascal LE, Wang K, Dhir R, Sims AM, Campbell R, Gasper G, DeFranco DB, Yoshimura N, Wang Z. Differential impact of paired patient-derived BPH and normal adjacent stromal cells on benign prostatic epithelial cell growth in 3D culture. Prostate. 2020;80:1177–1187. doi: 10.1002/pros.24044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Launer BM, McVary KT, Ricke WA, Lloyd GL. The rising worldwide impact of benign prostatic hyperplasia. BJU Int. 2021;127:722–728. doi: 10.1111/bju.15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faki Y, Er A. Different chemical structures and physiological/pathological roles of cyclooxygenases. Rambam Maimonides Med J. 2021;12:e0003. doi: 10.5041/RMMJ.10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 10.Jin R, Strand DW, Forbes CM, Case T, Cates JMM, Liu Q, Ramirez-Solano M, Milne GL, Sanchez S, Wang ZY, Bjorling DE, Miller NL, Matusik RJ. The prostaglandin pathway is activated in patients who fail medical therapy for benign prostatic hyperplasia with lower urinary tract symptoms. Prostate. 2021;81:944–955. doi: 10.1002/pros.24190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirschenbaum A, Klausner AP, Lee R, Unger P, Yao S, Liu XH, Levine AC. Expression of cyclooxygenase-1 and cyclooxygenase-2 in the human prostate. Urology. 2000;56:671–676. doi: 10.1016/s0090-4295(00)00674-9. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd GL, Marks JM, Ricke WA. Benign prostatic hyperplasia and lower urinary tract symptoms: what is the role and significance of inflammation? Curr Urol Rep. 2019;20:54. doi: 10.1007/s11934-019-0917-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torkko KC, Wilson RS, Smith EE, Kusek JW, van Bokhoven A, Lucia MS. Prostate biopsy markers of inflammation are associated with risk of clinical progression of benign prostatic hyperplasia: findings from the MTOPS study. J Urol. 2015;194:454–461. doi: 10.1016/j.juro.2015.03.103. [DOI] [PubMed] [Google Scholar]
- 14.Kahokehr A, Vather R, Nixon A, Hill AG. Non-steroidal anti-inflammatory drugs for lower urinary tract symptoms in benign prostatic hyperplasia: systematic review and meta-analysis of randomized controlled trials. BJU Int. 2013;111:304–311. doi: 10.1111/j.1464-410X.2012.11559.x. [DOI] [PubMed] [Google Scholar]
- 15.Sutcliffe S, Grubb Iii RL, Platz EA, Ragard LR, Riley TL, Kazin SS, Hayes RB, Hsing AW, Andriole GL Urologic Diseases in America Project. Non-steroidal anti-inflammatory drug use and the risk of benign prostatic hyperplasia-related outcomes and nocturia in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. BJU Int. 2012;110:1050–1059. doi: 10.1111/j.1464-410X.2011.10867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falahatkar S, Mokhtari G, Pourreza F, Asgari SA, Kamran AN. Celecoxib for treatment of nocturia caused by benign prostatic hyperplasia: a prospective, randomized, double-blind, placebo-controlled study. Urology. 2008;72:813–816. doi: 10.1016/j.urology.2008.04.069. [DOI] [PubMed] [Google Scholar]
- 17.Pascal LE, Dhir R, Balasubramani GK, Chen W, Hudson CN, Srivastava P, Green A, DeFranco DB, Yoshimura N, Wang Z. Claudin-1 down-regulation in the prostate is associated with aging and increased infiltration of inflammatory cells in BPH. Am J Clin Exp Urol. 2021;9:53–64. [PMC free article] [PubMed] [Google Scholar]
- 18.Pascal LE, Dhir R, Balasubramani GK, Chen W, Hudson CN, Srivastava P, Green A, DeFranco DB, Yoshimura N, Wang Z. E-cadherin expression is inversely correlated with aging and inflammation in the prostate. Am J Clin Exp Urol. 2021;9:140–149. [PMC free article] [PubMed] [Google Scholar]
- 19.Hayward SW, Dahiya R, Cunha GR, Bartek J, Deshpande N, Narayan P. Establishment and characterization of an immortalized but non-transformed human prostate epithelial cell line: BPH-1. In Vitro Cell Dev Biol Anim. 1995;31:14–24. doi: 10.1007/BF02631333. [DOI] [PubMed] [Google Scholar]
- 20.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbieri SS, Eligini S, Brambilla M, Tremoli E, Colli S. Reactive oxygen species mediate cyclooxygenase-2 induction during monocyte to macrophage differentiation: critical role of NADPH oxidase. Cardiovasc Res. 2003;60:187–197. doi: 10.1016/s0008-6363(03)00365-1. [DOI] [PubMed] [Google Scholar]
- 22.Ghazanfari N, van Waarde A, Dierckx R, Doorduin J, de Vries EFJ. Is cyclooxygenase-1 involved in neuroinflammation? J Neurosci Res. 2021;99:2976–2998. doi: 10.1002/jnr.24934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueda N, Yamashita R, Yamamoto S, Ishimura K. Induction of cyclooxygenase-1 in a human megakaryoblastic cell line (CMK) differentiated by phorbol ester. Biochim Biophys Acta. 1997;1344:103–110. doi: 10.1016/s0005-2760(96)00131-2. [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto C. Roles of COX-1 and COX-2 in gastrointestinal pathophysiology. J Gastroenterol. 1998;33:618–624. doi: 10.1007/s005350050147. [DOI] [PubMed] [Google Scholar]
- 25.Suchiva P, Takai T, Kamijo S, Maruyama N, Yokomizo T, Sugimoto Y, Okumura K, Ikeda S, Ogawa H. Inhibition of both cyclooxygenase-1 and -2 promotes epicutaneous Th2 and Th17 sensitization and allergic airway inflammation on subsequent airway exposure to protease allergen in mice. Int Arch Allergy Immunol. 2021;182:788–799. doi: 10.1159/000514975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas S, Hao L, DeLaney K, McLean D, Steinke L, Marker PC, Vezina CM, Li L, Ricke WA. Spatiotemporal proteomics reveals the molecular consequences of hormone treatment in a mouse model of lower urinary tract dysfunction. J Proteome Res. 2020;19:1375–1382. doi: 10.1021/acs.jproteome.9b00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramanujan VK. Metabolic imaging in multiple time scales. Methods. 2014;66:222–229. doi: 10.1016/j.ymeth.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. Suppression of mitochondrial complex I influences cell metastatic properties. PLoS One. 2013;8:e61677. doi: 10.1371/journal.pone.0061677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y, Yuen WH, Fu J, Huang G, Melendez AJ, Ibrahim FB, Lu H, Cao X. The mitochondrial respiratory chain controls intracellular calcium signaling and NFAT activity essential for heart formation in Xenopus laevis. Mol Cell Biol. 2007;27:6420–6432. doi: 10.1128/MCB.01946-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang G, Chen Y, Lu H, Cao X. Coupling mitochondrial respiratory chain to cell death: an essential role of mitochondrial complex I in the interferon-beta and retinoic acid-induced cancer cell death. Cell Death Differ. 2007;14:327–337. doi: 10.1038/sj.cdd.4402004. [DOI] [PubMed] [Google Scholar]
- 31.Brock TG, McNish RW, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem. 1999;274:11660–11666. doi: 10.1074/jbc.274.17.11660. [DOI] [PubMed] [Google Scholar]
- 32.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Mazaleuskaya LL, Yuan C, Ballantyne LL, Meng H, Smith WL, FitzGerald GA, Funk CD. Flipping the cyclooxygenase (Ptgs) genes reveals isoform-specific compensatory functions. J Lipid Res. 2018;59:89–101. doi: 10.1194/jlr.M079996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Mazaleuskaya LL, Ballantyne LL, Meng H, FitzGerald GA, Funk CD. Genomic and lipidomic analyses differentiate the compensatory roles of two COX isoforms during systemic inflammation in mice. J Lipid Res. 2018;59:102–112. doi: 10.1194/jlr.M080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valcárcel-Ares MN, Riveiro-Naveira RR, Vaamonde-García C, Loureiro J, Hermida-Carballo L, Blanco FJ, López-Armada MJ. Mitochondrial dysfunction promotes and aggravates the inflammatory response in normal human synoviocytes. Rheumatology (Oxford) 2014;53:1332–1343. doi: 10.1093/rheumatology/keu016. [DOI] [PubMed] [Google Scholar]
- 38.Fato R, Bergamini C, Bortolus M, Maniero AL, Leoni S, Ohnishi T, Lenaz G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim Biophys Acta. 2009;1787:384–392. doi: 10.1016/j.bbabio.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao RZ, Jiang S, Zhang L, Yu ZB. Mitochondrial electron transport chain, ROS generation and uncoupling (review) Int J Mol Med. 2019;44:3–15. doi: 10.3892/ijmm.2019.4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdel-Aziz AM, Gamal El-Tahawy NF, Salah Abdel Haleem MA, Mohammed MM, Ali AI, Ibrahim YF. Amelioration of testosterone-induced benign prostatic hyperplasia using febuxostat in rats: the role of VEGF/TGFβ and iNOS/COX-2. Eur J Pharmacol. 2020;889:173631. doi: 10.1016/j.ejphar.2020.173631. [DOI] [PubMed] [Google Scholar]
- 41.Onodera Y, Teramura T, Takehara T, Shigi K, Fukuda K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio. 2015;5:492–501. doi: 10.1016/j.fob.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taniura S, Kamitani H, Watanabe T, Eling TE. Transcriptional regulation of cyclooxygenase-1 by histone deacetylase inhibitors in normal human astrocyte cells. J Biol Chem. 2002;277:16823–16830. doi: 10.1074/jbc.M200527200. [DOI] [PubMed] [Google Scholar]
- 43.Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002;68-69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 44.Yang DH, Hsu CF, Lin CY, Guo JY, Yu WC, Chang VH. Kruppel-like factor 10 upregulates the expression of cyclooxygenase 1 and further modulates angiogenesis in endothelial cell and platelet aggregation in gene-deficient mice. Int J Biochem Cell Biol. 2013;45:419–428. doi: 10.1016/j.biocel.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Pereira CV, Peralta S, Arguello T, Bacman SR, Diaz F, Moraes CT. Myopathy reversion in mice after restauration of mitochondrial complex I. EMBO Mol Med. 2020;12:e10674. doi: 10.15252/emmm.201910674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.