

Abstract
Cellular retinoic acid-binding protein 1 (CRABP1) binds retinoic acid (RA) specifically in the cytoplasm with unclear functions. CRABP1 is highly and specifically expressed in spinal motor neurons (MNs). Clinical and pre-clinical data reveal a potential link between CRABP1 and MN diseases, including the amyotrophic lateral sclerosis (ALS). We established a sequenced MN-muscle co-differentiation system to engineer an in vitro functional 3D NMJ model for molecular studies and demonstrated that CRABP1 in MNs contributes to NMJ formation and maintenance. Consistently, Crabp1 knockout (CKO) mice exhibited an adult-onset ALS-like phenotype with progressively deteriorated NMJs, characterized with behavioral, EchoMRI, electrophysiological, histological, and immunohistochemical studies at 2–20-months old. Mechanistically, CRABP1 suppresses CaMKII activation to regulate neural Agrn expression and downstream muscle LRP4-MuSK signaling, thereby maintaining NMJ. A proof-of-concept was provided by specific re-expression of CRABP1 to rescue Agrn expression and the phenotype. This study identifies CRABP1-CaMKII-Agrn signaling as a physiological pre-synaptic regulator in the NMJ. This study also highlights a potential protective role of CRABP1 in the progression of NMJ deficits in MN diseases.
Subject terms: Molecular biology, Neuroscience
Introduction
Cellular Retinoic Acid Binding Protein 1 (CRABP1) is a newly characterized cytosolic retinoic acid (RA) signaling molecule which has been suggested to regulate RA bioavailability and metabolism through its high-affinity binding for RA in the cytoplasm [1]. Recent studies have revealed non-canonical activities of CRABP1 to rapidly regulate cytosolic signaling pathways and modulate specific cellular processes in a context-dependent manner. For example, CRABP1 regulates ERK1/2 signal cascades in stem cell cycle control [2–4] and CaMKIIδ signaling in maintaining cardiomyocyte function [5]. Additionally, CRABP1 is highly and specially expressed in spinal motor neurons (MNs), but its function in MNs is still unclear.
The neuromuscular junction (NMJ) connects MNs and skeletal muscles by forming synapses, which controls and regulates all motor functions, and is essential for daily physical mobility and survival [6]. The formation and maintenance of NMJ are regulated by AGRIN-LRP4-MuSK signaling. Pre-synaptically, neural AGRIN (gene name: Agrn) binds to LRP4, which then binds and activates MuSk to initiate a series of phosphorylation on DOK7, CDK5, and RAPSYN in the post-synaptic compartment, muscle cells. Muscle LRP4 also contributes to the retrograde signaling, thereby regulating the pre-synaptic differentiation and functions including synaptic vesicles and active zone in the NMJ [7]. This signaling pathway induces AChR clustering and stabilizes the specialized NMJ structure [6]. Studies have found impairment in AGRIN-LRP4-MuSK signaling in MN diseases [8, 9]. However, the regulation of AGRIN-LRP4-MuSK signaling in deteriorated NMJs, as seen in diseased conditions or natural aging process, is still unknown.
Interestingly, Crabp1 expression is reduced in MN disease models, such as spinal muscular atrophy, Spinal-bulbar muscular atrophy, and amyotrophic lateral sclerosis (ALS) [10–13]. Importantly, Crabp1 gene expression is dramatically reduced in spinal MNs of sporadic ALS patients [14]. ALS is an adult-onset disorder, and the incidence and prevalence of ALS increase with age. An epigenetic study suggests that Crabp1 expression is decreased with age, mostly in people age ≥35 years old [15, 16]. The clinical and preclinical evidence suggest a potential relevance of reduced Crabp1 in disease progression and aging. The reduction/silencing of Crabp1 might contribute to MN diseases pathogenesis or disease progression.
In this study, we established a sequenced MN-muscle co-differentiation system to engineer a 3D-NMJ model for molecular studies. Further, Crabp1 knockout (CKO) mice indeed exhibited an adult-onset, progressive ALS-like phenotype with apparent defects in NMJ. Mechanistic studies revealed CaMKII as the direct target of CRABP1, which regulated neural Agrn expression. Finally, we provided a proof-of-concept for therapeutic application that neuron-specific re-expression of CRABP1 in young CKO mice recovered spinal MNs, restored NMJ, and rescued the deteriorated ALS phenotype at 10-months-old.
Results
Deleting Crabp1 causes a progressive ALS-like phenotype in mice
As shown in Fig. 1, CKO mice developed progressive ALS-like characteristics, evaluated with behavioral, EchoMRI, electrophysiological, and histological studies at 2–20-months-old, as outlined in Fig. 1A. CKO mice began to show defects in specific motor functions (grip strength and hanging tests), as early as 2-months-old (Fig. 1B and C), but not in locomotion or limb-clasping reflex (Fig. S1A and B). Grip strength is directly related to muscle contractility of limbs, which increasingly differed between WT and CKO with age (Fig. 1B; 7%, 16%, and 19% reduction in 2, 9, and 18-month-old, respectively), which is characteristic of adult-onset, progressively deteriorated motor defects. Most ALS symptoms start with limb weakness and eventual muscle atrophy. Body composition showed that body weight and lean muscle mass (Fig. 1D, E) but not fat or total water (Fig. S1C) were significantly reduced in 20-month-old CKO mice.
Fig. 1. Deleting Crabp1 causes a progressive ALS-like phenotype in adults.

A Experimental scheme for animal studies. Three independent groups of mice at ages 2, 9–11, or 18–20 months old were examined in open field, grip strength, and hanging tests. Following tests, mice were euthanatized, then spinal cord and lumbrical muscle were collected for analyses shown in Figs. 2 and S3. Key behavioral tests are shown in B grip strength and C hanging tests. (2 months old, WT: n = 18, CKO: n = 15; 9–11 months old, WT: n = 15, CKO: n = 14; 18–20 months old, n = 16/groups). EchoMRI body composition analyses are shown in D body weight, and E lean muscle mass. F Illustration of in vivo muscle contractility measurement. Contractility of the anterior crural muscle was assessed by stimulating the peroneal nerve and recorded as isometric tetanic torque at the ankle of 20 months old mice. Muscle contractility tests determine G peak isometric torque, H TA muscle weight, and I normalized peak isometric torque. J Representative H&E images of a TA muscle from 20 months old mice (Scale bar is 20 μm). K Quantification of the cross-sectional area (CSA) of TA muscle fibers. Results are presented as means ± SD, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, compared with WT group. #p ≤ 0.05, ##p ≤ 0.01, compared with same genotype group at 2 months old.
Electrophysiological measurement was performed to evaluate nerve and muscle function (Fig. 1F–I). The in vivo contractility of anterior crural muscles, including tibialis anterior (TA), extensor digitorum longus, and extensor hallucis longus; and TA muscle histology was examined. The peroneal nerve was stimulated via platinum subdermal needle electrodes attached to a stimulator and the isometric tetanic torque at the ankle were measured as a direct indicator of muscle contractility (Figs. 1F–I, S1D and E). At 20-months-old, the peak isometric torque, reflecting muscle strength of anterior leg, was 65% greater in WT than that of CKO (Fig. 1G). After normalizing the peak isometric torque to TA muscle mass, WT still showed ~36% greater strength as compared to CKO (Fig. 1H and I), indicating poorer intrinsic neuromuscular functional capacity in CKO.
Histology of TA muscle showed significantly smaller mean cross-sectional area (CSA) of TA muscle fibers in CKO (Fig. 1J and K), and left-shifted distribution of fiber CSA toward smaller fibers (Fig. S1F). Furthermore, central-located nuclei were observed in TA muscle (1J, black arrows) in ~1/3–1/4 of CKO samples. However, this phenomenon was rarely observed in WT samples. The data show that deleting Crabp1 leads to progressively weakened grip strength and reduced muscle contractility starting in relatively young adults (2-month-old), resulting in muscle atrophy later in life.
Deleting Crabp1 affects NMJ
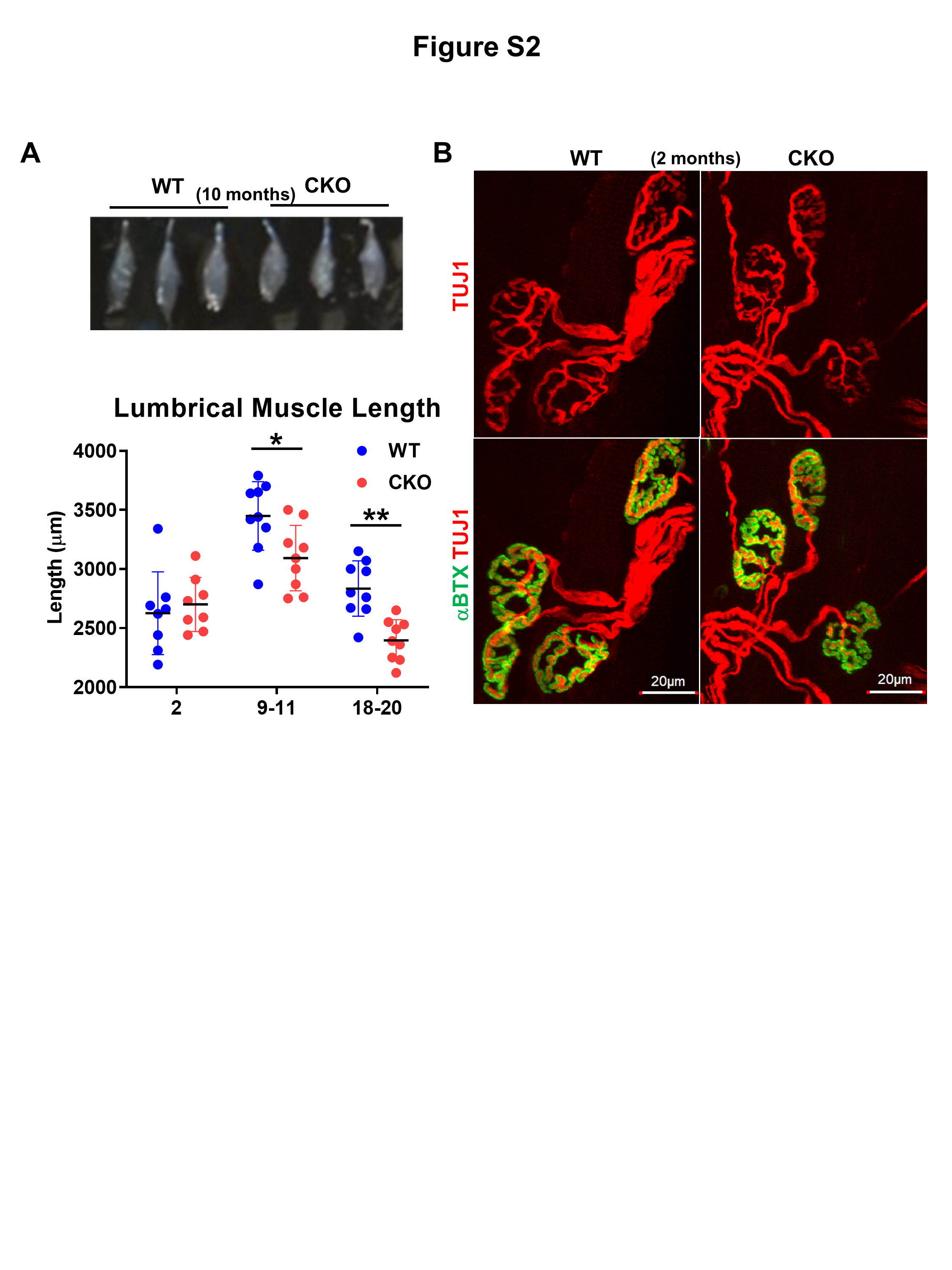
We examined the lumbrical muscle (which is directly responsible for grip strength, Fig. 2A), which was already significantly shortened in length in CKO mice at 10 months (Fig. S2A), indicating muscle atrophy in mid-adult stage and further deterioration with age. Lumbrical NMJs, marked with TUJ1 for pre-synaptic axon terminals and αBTX for post-synaptic acetylcholine receptors clusters (AChRs) in whole mount immunostaining were examined (Fig. 2B and C). In 10-months-old lumbrical muscle (Fig. 2B), WT NMJs had intact edges with a regular size (squares 1 and 2), but CKO had many irregular sized NMJs (very large, square 3; very small, square 4), indicating severely and progressively fragmented NMJs in CKO. Significantly fewer NMJs per lumbrical muscle were observed in CKO mice in all age groups (Fig. 2C), consistent with progressive decline of specific motor functions (Fig. 1B and C).
Fig. 2. Deleting Crabp1 causes defects in NMJs.

A Illustration of lumbrical muscle isolation and subsequent immunostaining experiments and confocal image analyses. B 3D representation of NMJs from WT and CKO lumbrical muscle. White squares 1 and 2 indicate regular-sized NMJs observed in WT mice. White squares 3 and 4 indicate irregular-sized NMJs observed in CKO mice. C NMJ quantification. CKO mice, from 2 to 20 months old, have significantly fewer NMJs as compared to WT mice for each age group (2 months old, n = 8/groups; 9–11 months old, n = 10/groups; 18–20 months old, n = 10/groups). Results are presented as means ± SEM, *p ≤ 0.05, **p ≤ 0.01, compare with WT group. D Immunostaining of NMJs showing axon bundles indicated with axon marker Tuj1 (Red) and acetylcholine receptor (AChR) labeled with αBTX (green). White squares mark axon bundles. Blue arrows indicate the loss AChR clusters at numerous nerve terminals in CKO, especially older (10 months and older), mice. E Enlarged images comparing details of normal and fragmented NMJs. Upper panels, stained with TUJ1 (red), show the morphology of axons and presynaptic terminals. Lower panels show αBTX (green) staining overlaid with TUJ1 staining. White squares mark discontinued AChR (green) clusters and abnormal (fragmented) morphology of NMJ in CKO mice.
Higher magnified NMJ images showed a clearly defined pretzel-like structure in WT and CKO mice at 2 months (Fig. S2B). But CKO NMJs appeared abnormal, starting at mid-adult (Fig. 2D and E). First, the number of NMJ on axon bundles (TUJ1, red) was reduced (Fig. 2D, white boxes). Secondly, some axon terminal branches lost specialized motor endplates (αBTX, green) (Fig. 2D; Blue arrows). Thirdly, compared to normal pre-synaptic terminals and post-synaptic AChR clusters in WT, CKO AChR clusters were clearly fragmented with poorly defined edges (Fig. 2E lower right). In pre-synaptic areas, axon terminals appeared degenerated (denervation, Fig. 2E upper white box; reinnervation, Fig. 2E lower white box). These results show that deleting Crabp1 impairs NMJ function, impacts the maintenance and/or stabilization of AChR clusters, and muscle contraction in adults.
Deleting Crabp1 progressively decreases spinal MN numbers and gray matter area
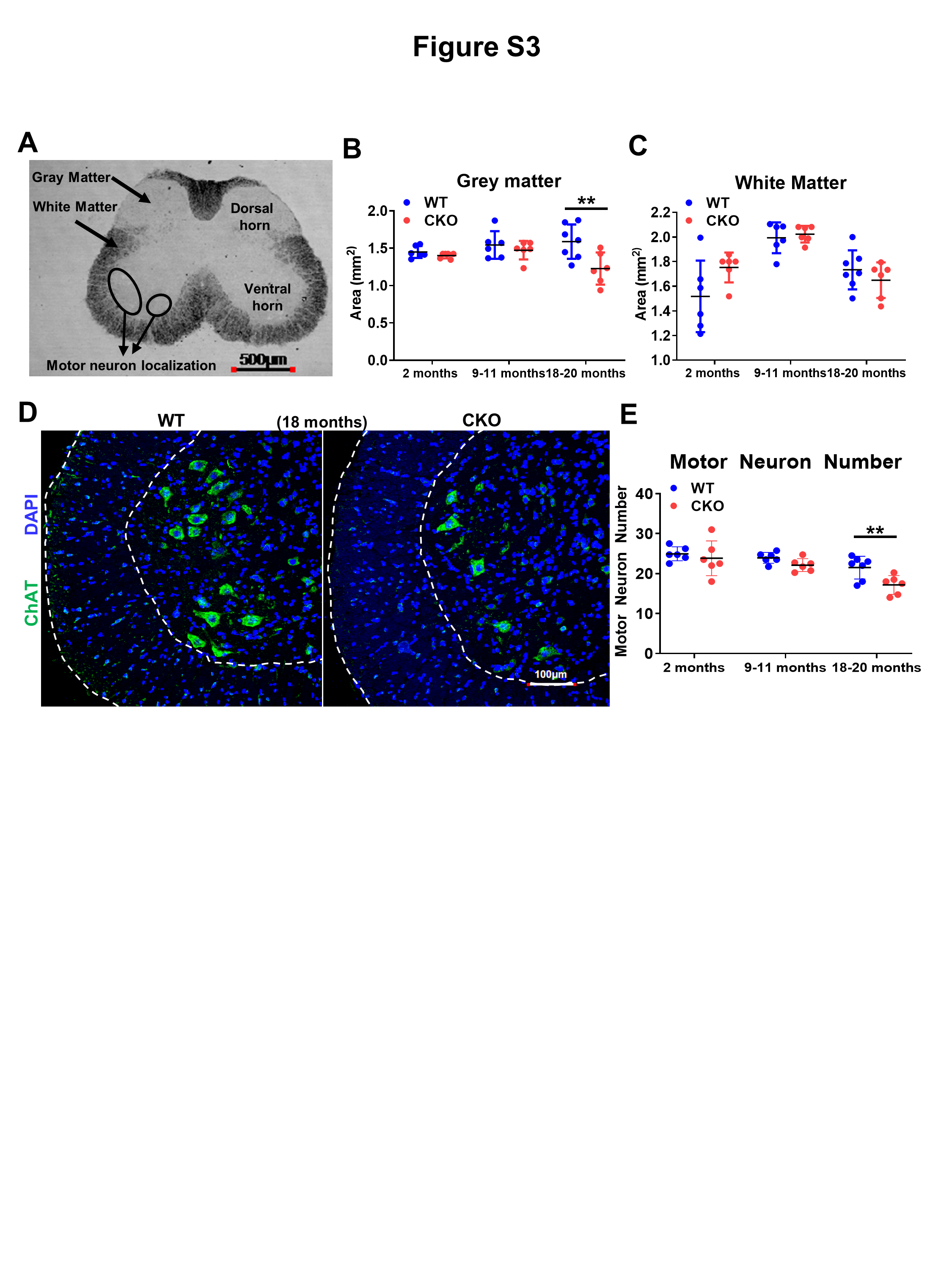
ALS is characterized by progressive loss of MNs. We examined spinal cord and spinal MNs, and compared gray matter and white matter of lumbar spinal cord. Figure S3A shows a bright-field image of white and gray matter, as well as MN localization. The gray matter area and number of MNs (Fig. S3D, indicated by ChAT+ signals) were significantly reduced in CKO mice only in the 20-months-old group (Fig. S3B and E). Thus, while NMJ abnormality develops relatively early in CKO mice (Figs. 1 and 2), MN loss is a late-onset event, suggesting a “dying-back” phenomenon where NMJ degeneration first occurs, followed by axonal degeneration and MN loss.
Expression data showed the highest CRABP1 protein level in spinal cord (Fig. 3A and B), but is not detected in skeletal muscle (TA muscle and diaphragm). Within the spinal cord, CRABP1 was detected in the ventral horn (marked by dotted lines) where MNs are localized (Figs. 3C, S3A, D). Co-staining with anti-CRABP1 and MN specific marker anti-ChAT showed clear overlap of ChAT with CRABP1 (Fig. 3D, top right panel). Furthermore, CRABP1 was not detected in glia (non-neuronal cells) marked with GFAP (Fig. 3D, bottom panels).
Fig. 3. CRABP1 is highly and specifically expressed in spinal MNs.

A, B Western blot analyses of various tissues and brain regions from 2-months old WT mice for the expression of CRABP1, NeuN. β-actin was used as a loading control. C Immunostaining of a spinal cord section (left) and bright field image (right) from 2-months-old WT mouse. CRABP1 is detected in the ventral horn, a region where motor neurons also localize. D Immunostaining of spinal cord section with CRABP1 (red), ChAT (motor neuron marker; green, upper panel), and GFAP (glial cell marker; green; bottom panel). E Immunostaining of lumbrical muscle from 2 months old mice to monitor pre-synaptic axon terminal marker Tuj1 (red; upper left), AChR cluster marker (αBTX; green upper right panel), and CRABP1 (White; bottom left). Overlay of Tuj1, αBTX, and CRABP1 images is shown in lower right.
Specific to NMJ (presynaptic axon marker TUJ1; post-synaptic AChR marker αBTX), CRABP1 was not detected in either pre- or post-synaptic region adjacent to the NMJ (Fig. 3E). Therefore, CRABP1 is specifically expressed in MN cell bodies.
CRABP1 in MNs enhances axon growth and myotube AChR clusters in 3D co-differentiation culture, forming organized and functional NMJ in vitro
We designed a sequenced, 3D co-differentiation culture system where MNs and muscle cells were sequentially co-differentiated to form a 3D structured NMJ (Fig. 4A) on an optimized micro-patterned gelatin hydrogel of 10 μm well width, 10 μm gap width, and 10 μm depth (see Methods). This bio-engineered hydrogel allowed differentiation, maturation, and alignment of C2C12-differentiated myotubes (Fig. 4B). On day 12, the length of axon and the size of AChR area were measured (Fig. 4C–F).
Fig. 4. Expressing Crabp1 in MNs enhances axon elongation, forming functional NMJs in 3D MN/muscle co-culture.

A Experimental scheme of engineering 3D NMJ in vitro using sequenced C2C12-MN1 co-differentiation culture, seeded on μmolded hydrogel. Seeded C2C12 was cultured in a myotube differentiation medium on day 2. On day 6, undifferentiated MN1 cells transfected with a control (GFP) or Crabp1-GFP vector were added onto differentiated C2C12 layer, and the co-culture was switched to a motor differentiation medium, allowing further co-differentiation. B Immunostaining of a sarcomere marker α-actinin (green) and AChR marker αBTX (red) on differentiated myotubes. The white arrow points to AChR puncta (αBTX, red) on Day 8 of differentiation. C A 3D image of the immunostaining of synaptic markers SV2 (white), axon marker Tuj1 (red), and αBTX (green) on a co-culture. Clustered AChRs (green, white arrows) near the SV2-positive synapses (white, blue arrows) indicate functional NMJs. D NMJ co-cultures (Ctrl vs. Crabp1-expression) stained for motor neuron (TUJ1) and myotube (αBTX). White arrows mark αBTX for AChR clusters. E Quantification data comparing axon length and F AChR signals in differentiated NMJs derived from Ctrl- vs. Crabp1-expressing group (Ctrl: n = 8; Crabp1: n = 7). Results are presented as means ± SD, *p ≤ 0.05, **p ≤ 0.01, compared with the control group.
While the sarcomere structure (α-actinin) readily formed by day 8 without MN1 neurons (Fig. 4B), only a few AChR clusters (αBTX) were present (Fig. 4B, white arrow). When MN1 cells were added for co-differentiation, MN1 neurons grew clear dendrites and axons (TUJ1), marked with synaptic marker SV2 (blue arrows) and increased AChR (αBTX; white arrows) adjacent to axon terminals (Fig. 4C).
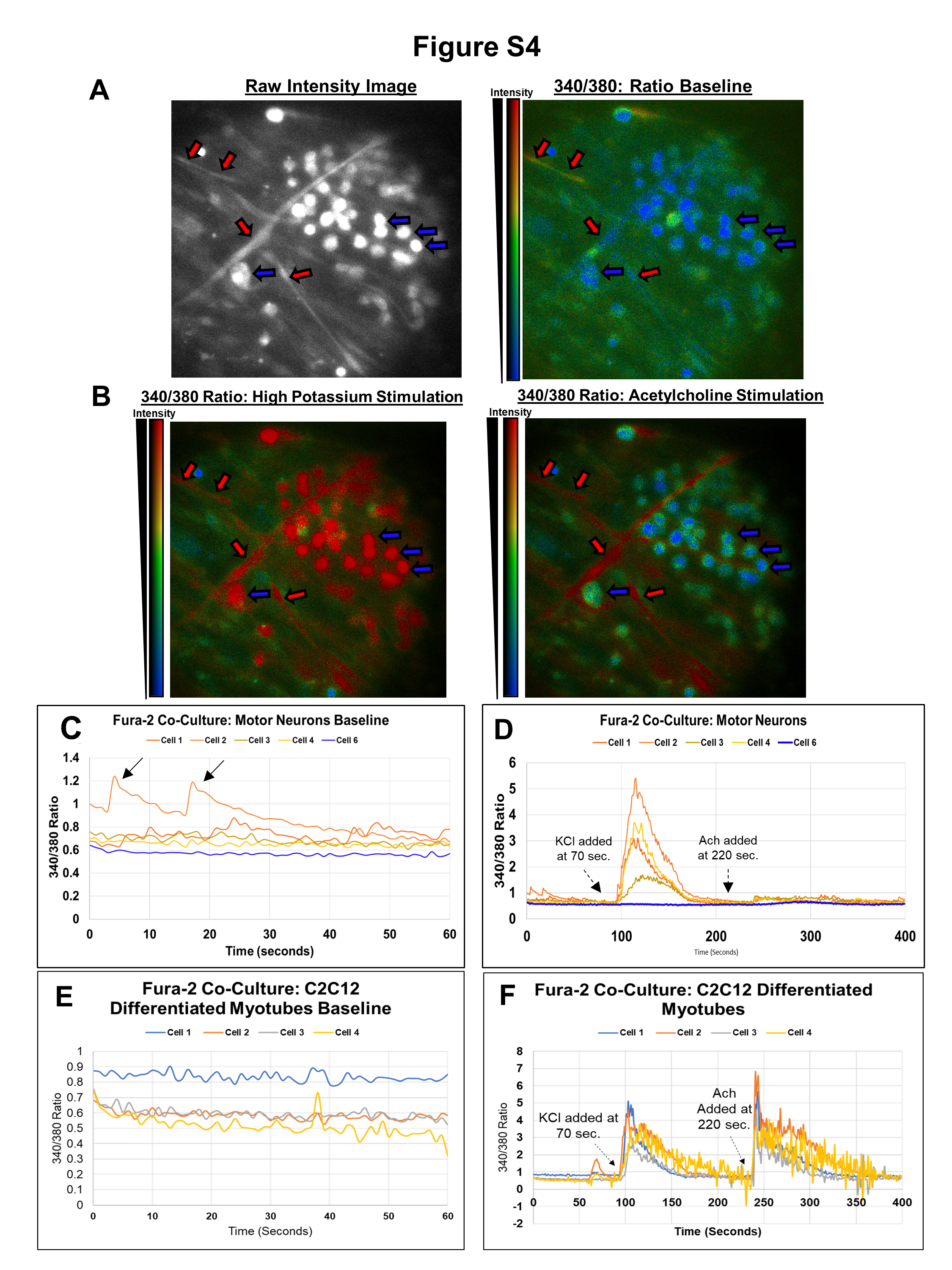
Moreover, the engineered NMJ exhibited contraction and calcium transients. Supplementary Videos showed myotube contractions in the presence (Videos 1 and 2) or absence (Video 3; fewer contracting myotubes) of MNs. Using Fura-2 AM, a fluorescent calcium indicator, we detected spontaneous and stimulated calcium transients in engineered NMJ (Fig. S4A and B; Video 4). MNs exhibited spontaneous calcium transients (Fig. S4C, black arrows), but myotubes did not (Fig. S4E). Stimulated with a high potassium concentration (50 mM; 1 min.) induce depolarization in both MN and myotubes (Fig. S4B left, representative image; Fig. S4D and F, calcium traces). We also determine if myotubes in the NMJ could respond to their physiologically relevant stimulus, acetylcholine (ACh). Indeed, under ACh stimulation (500 μM; 2 min), only the myotubes (not neurons) exhibited calcium transients (Fig. S4B right, representative image; Fig. S4F, calcium traces). These data validated the functionality, as well as physiological relevance, of the in vitro engineered NMJ.
In Fig. 4D, E, the MN1 neurons with elevated Crabp1 expression had ~50% longer axons and gave rise to >200% larger AChR areas in their post-synaptic compartments (Fig. 4D–F). The direct evidence indicated that the level of Crabp1 expression in MN is critical to axon growth and maintaining AChR clusters in the NMJ. Importantly, expressing Crabp1 in MNs improved the efficiency of forming and stabilizing structured NMJs.
CRABP1 in MNs affects neural Agrn expression, modulating LRP4-MuSK signaling in muscle
To determine pathways affected by CRABP1, we compared the expression of relevant genes in TA muscles and spinal cord MNs between WT and CKO mice at 2-months old (Fig. 5A). Agrn expression in MN, critical to NMJ formation and maintenance, was most robustly reduced (~60%) in CKO (Fig. 5B). Further, the two neuronal isoforms, Agrn 8 and Agrn 11, were most significantly reduced in CKO MNs (Fig. 5B). For pre-synaptic function, We examined the expression of SNARE (soluble N-ethylmaleimide–sensitive factor attachment protein receptor) complexes which mediated neurotransmitter release of synaptic vesicles in neurons [17]. The Target-SNARE (t-SNARE), associated with nerve terminal membranes, and Vesicle-SNARE (v-SNARE), associated with vesicle transportation and membrane fusion. The t-SNARE proteins, including Syt2, Snap25, and Snapin, were significantly altered in CKO (Fig. 5C), but v-SNARE proteins, including Vamp1, Vamp2, Vamp3, Unc13a, and Unc13b, were not affected (Fig. 5C). Neither were those genes for neurotransmitter acetylcholine metabolism/loading including ChAt, Cht, Vacht, and acetylcholinesterase (Ache) which hydrolyzes acetylcholine (Fig. 5C, D). These data showed deficits/changes in the release of pre-synaptic vehicles in the NMJ of CKO.
Fig. 5. Deleting Crabp1 causes deregulation in AGRIN-LRP4-MuSK signaling.

A Illustration of the experiment procedure. The spinal cord and TA muscle were collected from 2 months old mice (n = 4/group), and MNs were isolated from the spinal cord. B qPCR of total and neural Agrn expression in spinal MNs. CKO MNs express a significantly lowered level of Agrn as compared to WT MNs. C Expression of synaptic functional markers in spinal MNs. CKO MNs express a significantly lowered level of SYT2 and elevated expression of SNAP25 and SNAPIN as compared to WT. D Expression of genes for acetylcholine (ACh) metabolism in spinal MNs. E Expression of AGRIN-targeted LRP4-MuSK signaling components in TA muscle. CKO muscle expresses a significantly lowered LRP4 level, and elevated expression of MuSK and DOK7, as compared to WT. F Expression of AChE and AChRs in TA muscle. CKO muscle expresses a significantly lowered level of AChE, as compared to WT. G qPCR detect Agrn expression in WT and CKO muscle tissue. Results are presented as means ± SD, *p ≤ 0.05, **p ≤ 0.01, compared with WT group.
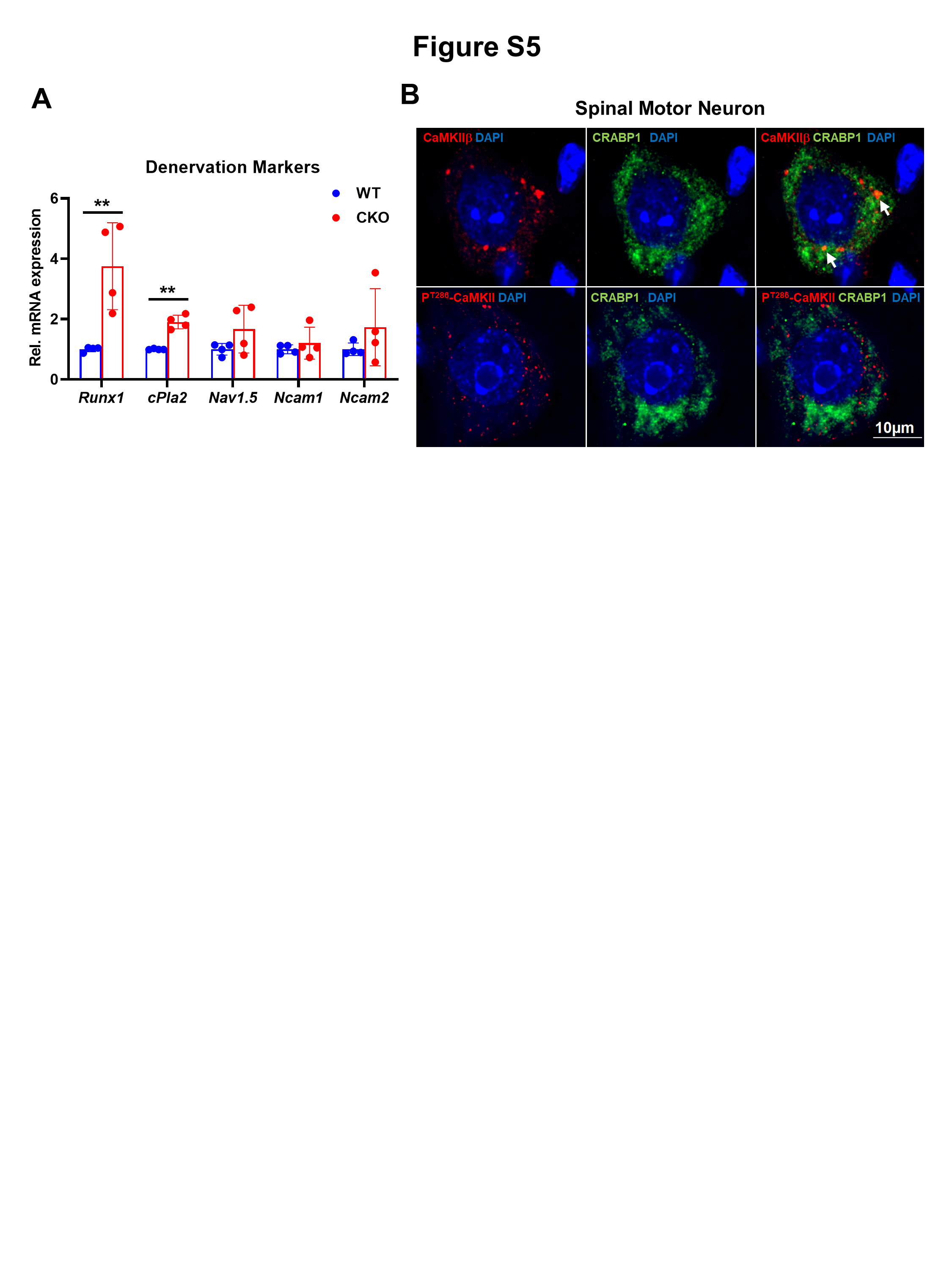
Neural AGRIN (in MN) regulates LRP4-MuSK signaling in skeletal muscle. Lrp4, AGRIN receptor, was lowered by ~80% in CKO muscle. Reduction in neural Agrn (Fig. 5B) and muscle Lrp4 (Fig. 5E) would suggest weakened AGRIN-LRP4-MuSK signaling in CKO. This might result in certain compensatory mechanism, as observed in the increase of Musk and Dok7 in CKO, although other further downstream associated components such as Rapsyn and Cdk5 showed no significant difference (Fig. 5E). Nevertheless, weakening in AGRIN-LRP4-MuSK signaling happened as early as 2-months-old in CKO mice, which could contribute to their defective motor function at a relatively young age. While Ache expression was significantly lower in CKO, AChRs components, including Achrα1, Achtγ, and Achrε, were not affected at 2-months-old (Fig. 5F). The gene expression patterns are consistent with the histological examinations of NMJ where AChR clusters remain intact in 2-months-old CKO (Fig. S2B). Muscle Agrn was not significantly affected (Fig. 5G), further supporting the specific pre-synaptic effect of CRABP1 in MNs. Given that the NMJ of CKO mice had decreased axon bundles and lost their specialized motor end plates (Fig. 2D, E), we thus examined whether markers for denervation were present. Indeed, in CKO muscle, the expression of several denervation markers including Runx1 and cPla2 was significantly increased, as compared to WT muscle. (Fig. S5A).
CRABP1 targets CaMKII to downregulate its activation, modulating neural AGRIN expression
Our previous studies have uncovered two kinase pathways as the potential targets of CRABP1, Mitogen-Activated Protein Kinase in proliferating stem/cancer cells and Ca2+/calmodulin-dependent protein kinase II (CaMKII) in non-proliferating cells [3, 18–20]. We suspected CaMKII as the target because it is most abundant in neurons. We compared the levels of PT286-CaMKIIα, PT286-CaMKIIβ, PT306-CaMKIIα, and PT306-CaMKIIβ in spinal cord between WT and CKO at 2-months-old (Fig. 6A, B). Interestingly, CaMKII activation (marked by PT286-CaMKIIα and PT286-CaMKIIβ) was significantly enhanced in CKO spinal cord. The data revealed elevated CaMKII signaling in CKO spinal cord, and suggested that CRABP1 might suppress CaMKII activation (see later).
Fig. 6. CRABP1 directly targets CaMKII to suppress its activation, thereby up-regulating AGRIN expression.

A Western blots detecting phospho-T286-CaMKII, phospho-T306-CaMKII, total CaMKII (α & β isoforms), CRABP1, AGRIN and β-actin in spinal cord of 2 months old WT and CKO mice. B Quantification of phospho-CaMKII (α & β isoforms) and AGRIN levels shown on panel A. C qPCR analyses for total Agrn mRNA in Crabp1-silenced MN1s treated with various CaMKII inhibitors. D Western blot analysis and quantification of AGRIN protein in Crabp1 silencing experiments in MN1 cells treated with CaMKII inhibitors mAIP and KN93. E Immunostaining shows co-localization of endogenous CRABP1 and phospho-CaMKII (PT286-CaMKII) in WT spinal MNs. F In vitro pull down showing direct interaction between CRABP1 and CaMKII. Left: Diagram showing flag-tagged CaMKII full-length (FL), kinase (K), regulatory (R), and association (A) domains. Right: In vitro pulldown assay using purified His-CRABP1 to pulldown flag-tagged CaMKII and its domains. The left panel shows input control of Flag-CaMKII in the mixture, the right panel shows Flag-CaMKII fragments pulled down by His-CRABP1. Input of CRABP1 in the mixture is shown at the bottom panel. CRABP1 interaction with R domain is negligible G Left: Western blot shows that expressing CRABP1 dampens basal (lane 4 & 5 vs. 1 & 2) and ionomycin (10 μM; 10 min.)-stimulated (lane 3 vs. 6) CaMKII activity in reconstituted HEK293T system. β-actin shows the loading control. Right: Quantification of data shown on the western blots. Results are presented as means ± SEM, *p ≤ 0.05, **p ≤ 0.01, compared with WT or the control group. ##p ≤ 0.01, compared with the KN92 group.
In CKO mice, AGRIN expression is significantly reduced in MN (Figs. 6A, B; 5B). To validate that CRABP1 and/or CaMKII regulate Agrn expression, we performed Crabp1-silencing experiments. As shown in Fig. 6 C, D, silencing of Crabp1 expression reduced Agrn mRNA and protein expression. Furthermore, CaMKII inhibitors KN93, KN62, and autocamtide-2-Related Inhibitory Peptide (myristoylated, cell-penetrant form; mAIP) all could enhance (rescued the inhibition) Agrn expression in MN1 cells (Fig. 6C, mRNA; Fig. 6D, protein). These data validated that CaMKII inhibited neuronal Agrn expression, which could be suppressed by CRABP1.
To examine potential CRABP1 and CaMKII interaction, we performed IHC staining of spinal cord sections. The results showed that endogenous CaMKIIβ and Crabp1 were partial co-localized in WT spinal MN (Fig. 6E; white arrows). Interestingly, CRABP1 rarely co-localized with PT286-CaMKII (Figs. 6E and S5B), implying that Crabp1 probably interacted more stably with the inactive CaMKII in comparison to the active enzyme, PT286-CaMKII. These data suggested an interesting mechanism for CRABP1-mediated regulation of CaMKII activation, that CRABP1 modulates (dampens) CaMKII activation probably by preferentially interacting with the inactive form of CaMKII. Next, rigorous molecular interaction (pull-down) tests with purified CaMKII sub-fragments, kinase (K), regulatory (R) and association (A) domains (Fig. 6F left) and Crabp1 showed that CRABP1 directly interacted with CaMKII through, mainly, the K and A domains (6F right, top western blot). Using a reconstituted HEK293T cell system, we validated that CRABP1 negatively regulated ionomycin-stimulated CaMKII activation [21], monitored with Phospho-threonine 286 (Fig. 6G). We concluded that through direct interaction with CaMKII, CRABP1 dampened basal and calcium-stimulated CaMKII activation, consistent with data obtained from CKO mice, that deleting Crabp1 resulted in an overall elevation of endogenous CaMKII activity.
Re-expressing CRABP1 in spinal MNs rescues CKO’s motor and NMJ defects
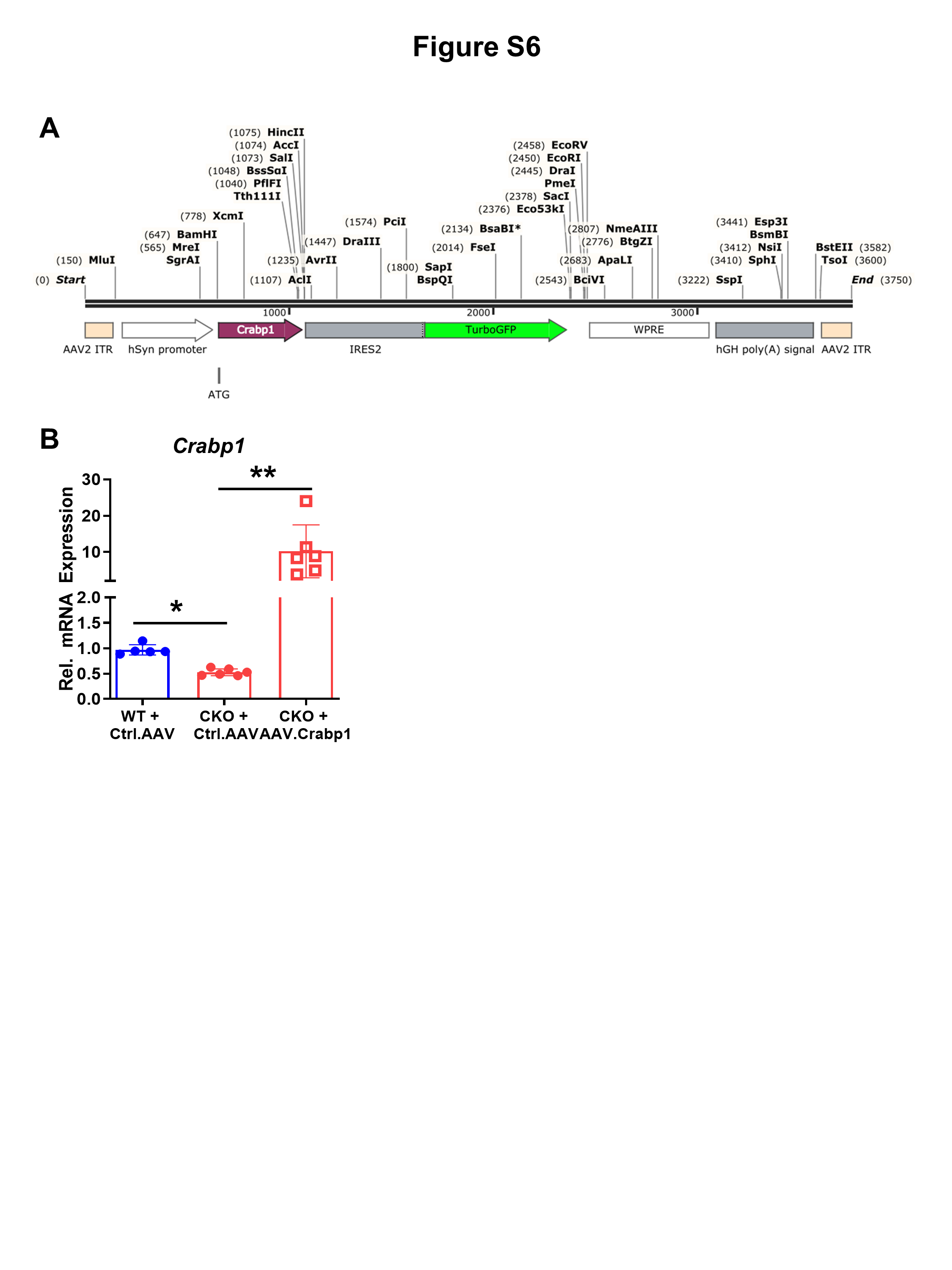
To confirm the functionality of CRABP1 in maintaining NMJ, we performed gene-rescuing experiments by post-natally re-expressing Crabp1 in MNs of CKO mice (3-weeks-old) with Crabp1-expressing AAVs (Figs. 7A, S6A). The engineered capsid, AAV.PHPeB, increased gene transfer efficiency in the CNS [22] and hSyn promoter driving Crabp1 expression specifically in neurons. Figure S6B shows efficient rescue/re-expression of CRABP1.
Fig. 7. Re-expressing Crabp1 in spinal MNs rescues motor defects, repairs NMJs and recovers neural Agrn expression.

A Experimental scheme for AAV-mediated rescue experiments. Control- or AAV.Crabp1-virus was injected into 3-weeks old mice. At 3-months old, spinal cord was collected for monitoring the expression rescue gene. At 10-months old, mice were subjected to behavioral tests, and then sacrificed. Spinal cord and lumbrical muscle were harvested for NMJ and gene expression analyses. B Western blots showing the efficiency and specificity of AAV virus-mediated expression of rescuing gene (GFP tagged Ctrl.AAV or GFP-tagged AAV.Crabp1), and the expression of AGRIN in spinal cord. Quantification of changes in CRABP1 and AGRIN expression in spinal cord is shown under the corresponding panel. N.S. indicates a non-specific signal above the band for CRABP1. C Left: Representative image of a spinal cord section from an AAV-infected mouse, detected with GFP (green) and MN marker, ChAT (red) (Scale bar is 50 um); Right: Quantification of GFP + (virus-infected cells) and ChAT+ MNs from Ctrl (GFP control) or AAV.Crabp1 injected mice. The results show greater than 60% MNs that harvest/express the injected Ctrl or AAV.Crabp1 (n = 2/group). D Grip strength. E Hanging tests (WT + Ctrl.AAV, n = 7; CKO + Ctrl.AAV, n = 9; CKO + AAV.Crabp1, n = 11). F Recovery in NMJ number of rescued CKO mice. G qPCR analysis showing recovered Agrn expression in spinal MN of rescued CKO mice (n = 6/group). Results are presented as means ± SD, *p ≤ 0.05, **p ≤ 0.01, compared with target group.
The specificity and duration of AAV-delivered Crabp1 expression in 3-month-old mice was monitored (Fig. 7B and C), showing control GFP and CRABP1 delivered to and strongly expressed in spinal cord, but not other peripheral organs (Fig. 7B), specifically in MNs (Fig. 7C). The quantified results showed effective infection/expression of the control group (~67% GFP-positive MNs) and CRABP1-expressing group (~63% Crabp1-positive MNs), which lasted for more than 2 months.
Motor function assessment showed that re-expressing CRABP1 in young CKO mice improved their performance in inverted screen test and grip strength without affecting their body mass later in life (10-month-old) (Fig. 7D, E). Histological and molecular examinations showed efficient recovery in the number of well-formed NMJs and Agrn expression in rescued CKO (Fig. 7F, G). These data validated specific functional role for Crabp1 gene in motor function, attributable to maintenance of NMJ through modulating AGRIN associated signaling pathways.
Discussion
In this study, we present the first evidence for a functional role of CRABP1 in NMJ formation and maintenance. We further determine the mechanism, that CRABP1-CaMKII-Agrn signal from MN modulates the post-synaptic LRP4-MuSK signaling in muscle. This raises two important questions. First, whether downregulation in Crabp1 expression in spinal MNs is common to various forms of MN diseases, such as ALS. Second, whether rescuing Crabp1 expression could be an effective therapy for most of these diseases. To this end, it has been documented that CRABP1 is up-regulated by GM602 which is a potential drug candidate of ALS [13].
It is interesting that CKO mice appear normal without significant motor deterioration or apparent NMJ abnormality until mid-adult stage (around 10-month-old). Thus, CRABP1-signalsome is not likely required for the initial NMJ formation or motor system in developmental stages. Rather, it most likely serves to maintain NMJ function, or plasticity, in adults, such as for repairing motor unit defects in diseases or ageing process.
The study determines CRABP1-signaling in spinal MNs as a potential, physiological regulator that maintains healthy NMJ. CRABP1 modulates neural Agrn expression in MNs, consequentially regulating its downstream, LRP4-MuSK, signaling in muscle. Muscle LRP4 also contributes to retrograde signaling, thereby regulating pre-synaptic differentiation and functions, including regulation of synaptic vesicles in the NMJ [7]. Disruption in the expression of t-SNARE, but not v-SNARE, in CKO mice would suggest specific deficits in synaptic vesicle (ACh) releasing, but not vesicle transportation in adult CKO mice. The deficits of nerve-muscle communication (AGRIN, LRP4 and ACh level) might contribute to the progressive ALS-like phenotype in CKO mice.
AGRIN is critical to MN function. Defect in AGRIN signaling has been observed in human MN diseases such as SMA and ALS [8, 23, 24]. A significant reduction in neural Agrn, as well as impaired AGRIN-LRP4-MuSK signaling, in CKO mice (Figs. 5, 6) would explain the defects in NMJ and retarded motor function. The key target of CRABP1, CaMKII, can regulate Agrn expression, which is supported by in vitro data that CaMKII activity modulates Agrin levels in MNs (Fig. 6). Further studies are needed to determine if CaMKII regulation of Agrn expression occurs through transcriptional and/or complicated post-transcriptional events.
CaMKII is a multimeric kinase extensively regulated by signal inputs affecting its intra- and/or inter-molecular interactions; but, calcium-calmodulin (Ca2+/CaM) binding is the best-understood mechanism of CaMKII activation [25]. CaMKII physically interacts with CRABP1 through at least two domains (Fig. 6F); but how this may lead to conformational change/enzyme activity remains to be determined in further biophysical and biochemical studies. It would also be important to determine whether and how RA regulates CRABP1-CaMKII interaction and its integration with other physiological processes/signals, and if the nutritional status of vitamin A affects this CRABP1 signalsome pathway in adults.
CKO NMJ/motor functional defects can be rescued by re-expressing Crabp1 in young animals, indicating that timely re-introducing Crabp1-signaling could effectively repair, or prevent NMJ/motor functional deterioration. This supports that the motor defect of CKO mice is not a developmental phenotype. Finally, supplementing Crabp1 may provide a potential preventive or therapeutic strategy in managing motor neuron diseases.
In summary (Fig. 8), consistent with reduced Crabp1 expression in spinal MNs of sALS patients, we found that CKO mice indeed exhibited an adult-onset progressive ALS-like phenotype (Fig. 8A). This phenotype was initiated from weakened grip strength and abnormal NMJs, then subsequent NMJ fragmentation, and, eventually, muscle denervation, weakness, wasting, and MN loss. Mechanistically (Fig. 8B), in MN, CRABP1 directly suppresses CaMKII activity, probably through its preferential interaction with the inactive enzyme, thereby regulating neural Agrn expression. Therefore, deleting Crabp1 downregulates AGRIN expression and leads to a disruption in nerve-muscle communication including AGRIN-LRP4-MuSK signaling and t-SNAREs expression in NMJ. Ultimately, these molecular signaling deficits in CKO mice cause progressive NMJ deterioration and the ALS-like phenotype later in life. This study also highlights a potential therapeutic strategy to manage NMJ degeneration in motor neuron diseases by correcting Crabp1 expression.
Fig. 8. A potential role of Crabp1 in protecting MN degeneration and maintaining healthy NMJ.

A The scheme illustrates that deleting Crabp1 in mice results in an adult-onset, progressive MN degenerative phenotype with specific defects in the NMJ. B The scheme illustrates the mechanism of CRABP1-CaMKII-AGRIN signal in spinal MNs, which modulates downstream events in NMJ. Dysregulated CRABP1-CaMKII-Agrn signaling in spinal MNs leads to a disrupted nerve-muscle communication (disrupted AGRIN-LRP4-MuSK signaling and t-SNARE expression) in adult NMJ, and, subsequently, progressive NMJ deficits and a MN degenerative phenotype.
Materials and methods
Animal experiments
Crabp1 knockout mice were generated as described in Lin et al. [4]. WT and Crabp1 knockout (CKO) mice, 2–22 weeks old were used in these studies. The mice were housed and bred in the University of Minnesota Research Animal Resources facilities. Animals were housed in temperature-controlled (22 ± 1 °C) housing on a 14/10 light/dark cycle (lights on/off at 0600/2000) with ad libitum food and water. The experimental procedures were conducted according to National Institutes of Health guidelines and were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Locomotion activity
Locomotion test was performed as described [4]. Briefly, mice were placed in an open field comprised of a Plexiglass box with 40 × 25 × 22 cm dimensions in a dimly lit environment. Each mouse was allowed 30 min of free movement. Data was collected via a ceiling-mounted camera and analyzed using Any-mazeTM software (Stoelting Co, USA).
Grip strength
Forelimb grip strength was measured with a semi-automated system (Animal Grip Strength System; San Diego Instruments, CA, USA). The forelimb of mouse is placed on a plate containing a bar, the animal is then slowly and steadily pulled across the plate by the base of the tail, followed by four successive trials measuring the combined forelimb grip strength.
Hanging test
Hanging test was measured using a custom wired mesh (15 × 15 cm). The mice were placed on the wired mesh at 45 cm high. Then the mesh was inverted. Latency to fall was measured by recording the time at which the mice fell onto a soft surface.
Body composition analysis
Body composition analysis of live mice was performed by the University of Minnesota Physiology Core. EchoMRITM was used to acquire measurements of fat, lean, free water, and total water masses.
Ankle joint mechanics
Ankle joint mechanics of each mouse were measured as previously described [26–28]. Briefly, each mouse was anesthetized via isoflurane inhalation (1.5%) with an O2 flow rate of 125 mL/min. Passive torque about the left ankle joint was assessed by stabilizing the knee and placing the foot in a plate attached to a servomotor (Aurora Scientific, Aurora, Ontario, Canada). The servomotor rotated the ankle to four angles of dorsiflexion (5°, 10°, 15°, and 20°) and four angles of plantarflexion (5°, 10°, 15°, and 20°) and torque was measured at each angle.
In vivo anterior crural muscles contractile function measurement
Immediately following testing of joint mechanics and while still anesthetized under isoflurane, mice underwent contractile testing of the left leg. Maximal isometric torque produced by the anterior crural muscles was measured in vivo in anesthetized mice as previously described [26]. Briefly, while mice were anesthetized under isoflurane, in vivo contractility of the anterior crural muscles (tibialis anterior, extensor digitorum longus, and extensor hallucis longus) was assessed by stimulating the peroneal nerve. With the left knee stabilized by a clamp and the foot secured in a plate attached to the shaft of the servomotor, the peroneal nerve was stimulated via platinum subdermal needle electrodes attached to a stimulator and stimulus isolation unit (Grass Telefactor, Warwick, RI). Parameters on the stimulator were set for a 200-ms contraction duration consisting of 0.5-ms square wave pulses at 200 Hz to produce maximal isometric tetanic torque at the ankle. The voltage on the stimulator started at 3.0 V and was adjusted between 1.0 and 8.0 V until torque no longer increased.
Euthanasia and tibialis anterior muscle excision
For harvesting, mice were euthanized via intraperitoneal injection with a 200 mg/kg body weight overdose of pentobarbital sodium (Diamondback Drugs; Scottsdale, AZ) and left side TA muscles (in vivo testing side) were excised and weighted. TA muscles for histology were placed in Tissue Tek® Optimal Cutting Temperature (O.C.T.) compound (Sakura Finetek; Torrance, CA) and frozen in isopentane. Muscle samples were snap frozen in liquid nitrogen and then stored in −80 °C until further processed.
Muscle fiber cross-sectional area (CSA)
Muscle fiber cross-sectional area of each TA muscle was measured as previously described [26]. All histological evaluations were conducted on 6–7 mice per group. Briefly, using a microtome cryostat at –20 °C, ten cross-sections (10 µm thick) were cut at each of 6 levels equally spaced along the length of the TA muscle. Cross-sections of TA muscles were stained with hematoxylin-eosin (H&E) for blinded histological assessment. Images were taken at 5×, 10×, or 20× magnification using a Leica DM5500B upright microscope (Leica Microsystems; Buffalo Grove, IL).
CSA analysis was performed by a blinded individual. Using FIJI software, the border of each H&E-stained muscle fiber was traced by hand using a stylus on a touch-screen device. The area of each muscle fiber was then quantified. Two-sided t-test with Welch’s correction was used to determine if there was a significant difference between the mean CSA of WT compared to CKO mice.
Spinal cord isolation
Mice were euthanized by CO2. Immediately mice were processed for spinal cord isolation by removing the spinal column then using a syringe to perfuse 1XPBS to expel the spinal cord. The lumbar region of the spinal cord was used in the following experiments.
MN isolation
MNs from mouse spinal cord were isolated as described [29] with modifications. Briefly the spinal cord was isolated from 2-month-old male mice. Then the spinal cord tissue was minced and incubated in 0.25% trypsin-EDTA solution (Thermo) at 37 °C for 20 min. Every 5 min the mixture was triturated throughout the incubation. Then DMEM supplemented with 10% FBS was added, and the mixture was centrifuged at 300 × g. After removing the supernatant, the pellet was resuspended in 4 ml of NABG (97% Neuralbasal medium, 2% B27, 0.5% glutamine, 0.5% penicillin–streptomycin). Then 4 ml of separation solution (87.6% DMEM/F12 medium, 12.4% OptiPrep) was added. The mixture was centrifuged at 800 × g at 4 °C for 15 min. The mixture is now separated into three layers: F1, F2, F3. MNs were isolated from F2 layer. Then 4 ml of HBSS was added to the isolated F2 layer and centrifuged at 500 × g at 4 °C for 5 min. The pellet containing MNs were then processed for RNA isolation and RT-qPCR.
Cryosection of spinal cord and TA muscle
The isolated lumbar spinal cords or TA muscle were embedded in OCT compound and snap-frozen in liquid nitrogen-chilled isopentane. The tissue sections were obtained in 20-μm-thick slices by Cryostat.
Immunostaining and image analysis for NMJ
Lumbrical muscle was isolated as described [30]. Lumbrical muscle was fixed in 4% PFA (paraformaldehyde) for 10 min. Tissue was blocked and permeabilized by incubating in 2% BSA and 0.2% triton-X100 for 1 h at room temperature. Primary antibody, Tuj1 (1:1000; Thermofisher, MA) in 1XPBS was incubated overnight. Then after washing with 1XPBS three times, fluorescent secondary antibody (1:1000; Thermofisher) and αBTX conjugated to a fluorescent dye (1:400) in 1XPBS were incubated overnight. Images were acquired on an Olympus Fluoview FV1000 BX2 Upright Confocal.
Image acquisition and NMJ quantification were performed by two separate, blinded individuals. Using FIJI software and the 3D objects counter plug-in [31] each image was individually thresholded and all images were quantified using the size-cut offs of 40 voxels as the minimum and 25165824 voxels as the maximum.
For spinal cord sections
Spinal cord sections were processed for IF as described above. Primary anti-body ChAT (1:400; Cell Signaling Technology, MA), CaMKII (1:400; Cell Signaling Technology), and Crabp1 (1:100; Sigma-Aldrich, MO) was used. Fluorescent secondary antibody was used at 1:1000. Images were acquired on an Olympus Fluoview FV1000 BX2 Upright Confocal (Olympus, Japan). The area of white matter, gray matter, and the number of MNs were quantified using FIJI software (NIH, USA).
Western blotting
Spinal cord tissue was sonicated in RIPA buffer with phosphatase inhibitors (Thermofisher). 50 μg of protein was loaded for SDS-PAGE. Primary antibodies, p-CaMKII T286 (1:1000; Cell Signaling), total CaMKII α and β (1:1000; Santa Cruz, TX), p-CaMKII T306 (1:1000; PhosphoSolutions, CO), Crabp1 (1:1000; Sigma), Beta actin (1:2000; Santa Cruz). Crabp1 AAV rescue western blot HRP signal was analyzed via ImageJ [32]. Mean gray values were measured and background subtracted. Then results of anti- Crabp1 or anti-AGRIN were normalized to actin. Fold changes of Crabp1 and AGRIN shown in Fig. 7B were quantified by normalizing to the Ctrl AAV group. All the full length original western blots are shown in Supplementary Fig S8.
In vitro pull-down assay
In vitro pull-down assay for CRABP1-CaMKII detected was performed as described ([18]). Briefly purified His-tagged CRABP1 was captured on Ni-NTA beads (Qiagen, Germany) and incubated with Flag-tagged CaMKII full-length or domains for 2 h to overnight in reaction buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.2% (v/v) NP-40, 10% (v/v) glycerol and 10 mM Imidazole). Flag-tagged domains were generated using in vitro transcription and translation (Promega). Beads were washed three times with reaction buffer and then western blot performed. Interaction was probed for using anti-Flag antibody 1:1000 (Sigma) and horseradish peroxidase-conjugated mouse secondary antibody 1:2000 (GeneTex, CA).
HEK293T enzyme assay
HEK293T cells were seeded into a 10 cm dish. 5 μg GFP-CaMKII (Addgene #21227) and 5ug of empty flag vector or flag- CRABP1 for a total of 10ug of DNA was transiently transfected using the calcium phosphate method [33]. After transfection, cells were trypsinized and 0.8–1 million cells were seeded into six well plates. Medium was replaced with dextran-coated charcoal DMEM (Gibco) for 18 h. Cells were then treated with 10 μM ionomycin or DMSO (Sigma) for 10 min.
Cells were harvested in lysis buffer (50 mm HEPES, 0.1 mm EGTA, 0.1 mm EDTA, 120 mm NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM NaF, 1 mg/ml NaVO4, 1 mM PMSF and 1× Protease & Phosphatase Inhibitor Cocktail (Cell Signaling) on ice. 40–50 μg of protein lysate was loaded proceeded by SDS-PAGE and western blot. The PVDF membrane was blocked with 5% non-fat milk in 1× TBST for 1 h. After three 10-min washes with 1× TBST (0.1% Tween-20 in TBS), the membranes were incubated in primary antibodies, pCaMKII-T286 (Cell Signaling; 1:1000), anti-Flag (Sigma;1:2000) or beta actin (Santa Cruz; 1:2000) with antibody buffer (0.1% Tween 20 and 5% BSA in TBS) overnight. Blots were then incubated in secondary horseradish peroxidase-conjugated antibody (1:2000; GeneTex). The HRP Signals were detected with Immobilon Western Chemiluminescent HRP substrate (Advansta). Signals were detected with MyECL imager (ThermoFisher) and the relative level of protein was analyzed by ImageJ.
Fabrication of PDMS stamp and microcontact printing (Micro CP)
A silicon master mold was fabricated by the University of Minnesota Nano Center (MNC). Briefly a silicon wafer was coated with SU-8 photoresist. Then the SU-8 was treated with a light exposure through a chrome mask. Unexposed SU-8 was washed away resulting in the desired pattern ready for PDMS stamp fabrication.
PDMS stamps were fabricated as described elsewhere [34]. Briefly, the silicon wafer mold was treated with trichloromethyl silane (Millipore Sigma) vapor for 30 min as a releasing agent. PDMS mixture (PDMS: curing agent = 10:1, Sylgard 184, Dow Corning, MI) was poured onto a master mold and cured for 2 h at 60 °C. The cured PDMS was cut and peeled from the master mold and trimmed to provide the stamp. The PDMS stamp was cleaned with oxygen plasma (irradiation intensity: 200 W) for 10 min.
C2C12 and MN1 Co-differentiation Culture System, Immunostaining and Data Analysis
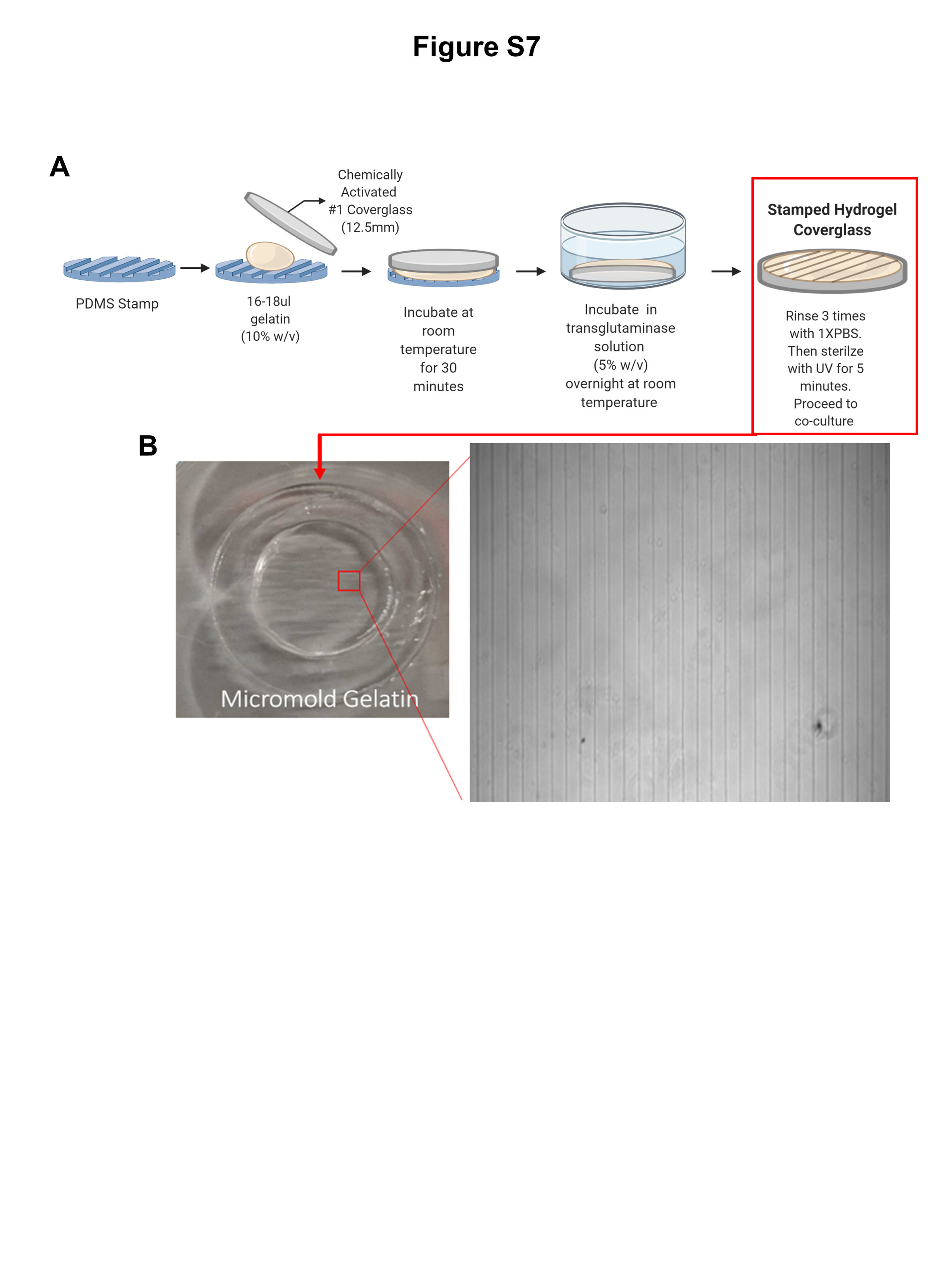
Hydrogels were fabricated as described in [35]. Briefly, 10% Type A gelatin solution (Sigma) dissolved in sterile ddH20 at 65 °C. 20 μl of gelatin solution was added onto 12.5 mm circular glass cover slips (Fisher Scientific). The PDMS stamp was added on top of the cover slips and then incubated at room temperature for 30 min. The PDMS stamp was carefully removed and the cover slips with the microprinted hydrogels were transferred to a 12-well plate and then incubated in a 5% microbial transglutaminase enzyme (AJINOMOTO) overnight at room temperature. The hydrogels were rinsed with 1XPBS three times and UV sterilized for using in co-culture studies (Fig. S7A, B).
C2C12 and MN1 cells were maintained in growth media (high glucose DMEM medium containing 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mM L-glutamine, and 10% FBS). For the co-culture (Fig. 4A), 2 × 104 C2C12 cells/cm2 were seeded on hydrogels in 12-well plates and cultured in growth media. Once confluent (about 2 day), myotube differentiation medium (DMEM with 2% horse serum containing 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mM L-glutamine) was added. C2C12 were differentiated for 4 days and the myotube differentiation media was replenished every 2 days. Then 1 × 104 MN1 cells/cm2 were added to each well of 12- well, on top of the differentiated C2C12 cells. MN1 cells were previously transfected with either empty vector control or GFP- CRABP1 prior to being seeded onto C2C12 cells. The co-culture was maintained in MN differentiation media (DMEM/F12 media with 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mM L-glutamine, 1% MEM non-essential amino acids for 6 days. The MN differentiation medium was replenished every 2 days.
For IF staining, cells were fixed in 4% PFA for 15 min on ice. After three 5-min 1XPBS washes, 0.2% of tritonX-100 was added for 15 min for permeabilization. For blocking, 2% BSA in 1XPBS for 1 h at room temperature was performed. Primary antibodies, α-actinin (1:200; Abcam), Tuj1 (1:1000; ThermoFischer) and SV2 (1:20; DSHB) in blocking buffer were incubated overnight at 4 °C. After three 5-min 1XPBS washes, fluorescent secondary antibodies (1:400), αBTX conjugated dye (1:400) were incubated for 1 h, and then DAPI (1:1000) for 10 min. Images were acquired on Olympus Fluoview FV1000 BX2 Upright Confocal.
Data were analyzed by a blinded individual. Axon length and acetylcholine receptor (AChR) clusters were analyzed using FIJI software. Axon length of GFP positive MN1 cells visible in the field was measured. NMJ clusters were analyzed by quantifying the total fluorescent intensity of αBTX signal in the field [36].
Quantitative RT-PCR
RNA from isolated spinal motor and TA muscle was extracted by TRIzol Reagent (ThermoFisher). RNA concentration was measured with NanoDrop and cDNA was synthesized by High-Capacity cDNA Reverse Transcription Kit (ThermoFisher). Quantitative RT-PCR was performed using the SYBR™ Green PCR Master Mix (ThermoFisher), and following primers were listed below. GAPDH was used for normalization. Real-time RT-PCR was conducted on an StepOne™ Real-Time PCR System (Applied Biosystems, MO) or. Mx3000P QPCR Systems (Agilent, CA). Primers are listed in Tables S1 and S2.
Statistical analysis
Data were analyzed using the Student’s t test (WT V.S. CKO), one-way ANONA with Bonferroni post hoc test (CaMKII inhibitors test), and two-way ANONA with Turkey post hoc test in mice phenotype (Genotype × Age). Statistical analyses were performed using Prism 8.0 (GrapgPad, CA). All tests were performed at a significance level of p ≤ 0.05, and data are presented as mean ± standard error or mean ± SD of the mean as indicated.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Video 1 co-culture with motor neuron 10X
Video 2 Co-Culture With Motor Neuron 20X-1_mp4_CORRECTED.mp4
Video 3 C2c12 Myotube Only (Co-Culture Without Motor Neuron) 10X-1_MP4_CORRECTED.mp4
Video 4 Co-Culture Fura2-1_MP4_CORRECTED.mP4
Acknowledgements
We like to acknowledge University of Minnesota Viral Vector Core (VVC), Physiology Core, University Imaging Center (UIC) and staff support from Dr. Guillermo Marques and Dr. Mary Brown, and the Minnesota Nano Center (MNC) supported by the National Science Foundation through the National Nanotechnology Coordinated Infrastructure (NNCI) under Award Number ECCS-2025124. We would also like to thank Dr. Stanley Thayer for imaging equipment and analysis software. Figures depicting experimental procedures were Created with BioRender.com. This study was supported by DK54733 and DK60521, and University of Minnesota Medical School Dean’s Commitment to LNW, and F31DK123999 to JN. DJL and S-HO acknowledge support by the Minnesota Environment and Natural Resources Trust Fund as recommended by the Legislative-Citizen Commission on Minnesota Resources (LCCMR) and the Sanford P. Bordeau Chair in Electrical Engineering.
Author contributions
YL designed and performed experiments and analyzed data; JN performed experiments and analyzed data; PL performed experiments and analyzed data; GL performed experiments and analyzed data; DJL generated reagents and materials; CW performed experiments. YWL performed experiments. S-HO provided reagents and materials and technical input DL provided reagents and materials and technical input. L-NW designed the experiments, analyzed the data, coordinated the study, and provided financial support. L-NW is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Data availability
The data generated during this study are available upon reasonable request from the corresponding author.
Competing interests
The authors declare no competing interests.
Ethics statement
All experimental procedures were conducted according to National Institutes of Health guidelines and were approved by the University of Minnesota Institutional Animal Care and Use Committee.
Footnotes
Edited by L Greene.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-022-00959-4.
References
- 1.Napoli JL. Retinoic acid: its biosynthesis and metabolism. Prog Nucleic Acid Res Mol Biol. 1999;63:139–88. doi: 10.1016/S0079-6603(08)60722-9. [DOI] [PubMed] [Google Scholar]
- 2.Persaud SD, Lin Y-W, Wu C-Y, Kagechika H, Wei L-N. Cellular retinoic acid binding protein I mediates rapid non-canonical activation of ERK1/2 by all-trans retinoic acid. Cell Signal. 2013;25:19–25. doi: 10.1016/j.cellsig.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Persaud SD, Park SW, Ishigami-Yuasa M, Koyano-Nakagawa N, Kagechika H, Wei LN. All trans-retinoic acid analogs promote cancer cell apoptosis through non-genomic Crabp1 mediating ERK1/2 phosphorylation. Sci Rep. 2016;6:22396. doi: 10.1038/srep22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin YL, Persaud SD, Nhieu J, Wei LN. Cellular retinoic acid-binding protein 1 modulates stem cell proliferation to affect learning and memory in male mice. Endocrinology. 2017;158:3004–14. doi: 10.1210/en.2017-00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park SW, Persaud SD, Ogokeh S, Meyers TA, Townsend DW, Wei LN. CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J Endocrinol. 2018;236:151–65. doi: 10.1530/JOE-17-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–33. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012;489:438–42. doi: 10.1038/nature11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, Xiong WC, Mei L. Neuromuscular junction formation, aging, and disorders. Annu Rev Physiol. 2018;80:159–88. doi: 10.1146/annurev-physiol-022516-034255. [DOI] [PubMed] [Google Scholar]
- 9.Taetzsch T, Valdez G. NMJ maintenance and repair in aging. Curr Opin Physiol. 2018;4:57–64. doi: 10.1016/j.cophys.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizzo F, Nizzardo M, Vashisht S, Molteni E, Melzi V, Taiana M, et al. Key role of SMN/SYNCRIP and RNA-Motif 7 in spinal muscular atrophy: RNA-Seq and motif analysis of human motor neurons. Brain. 2019;142:276–94. doi: 10.1093/brain/awy330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maeda M, Harris AW, Kingham BF, Lumpkin CJ, Opdenaker LM, Mccahan SM, et al. Transcriptome profiling of spinal muscular atrophy motor neurons derived from mouse embryonic stem cells. PLoS One. 2014;9:e106818. doi: 10.1371/journal.pone.0106818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin YL, Lin YW, Nhieu J, Zhang X, Wei LN. Sonic hedgehog-gli1 signaling and cellular retinoic acid binding protein 1 gene regulation in motor neuron differentiation and diseases. Int J Mol Sci. 2020. 10.3390/ijms21114125. [DOI] [PMC free article] [PubMed]
- 13.Swindell WR, Bojanowski K, Kindy MS, Chau RMW, Ko D. GM604 regulates developmental neurogenesis pathways and the expression of genes associated with amyotrophic lateral sclerosis. Transl Neurodegener. 2018;7:1–24. doi: 10.1186/s40035-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005;57:236–51. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- 15.Arellano-Ortiz AL, Salcedo-Vargas M, Vargas-Requena CL, López-Díaz JA, De la Mora-Covarrubias A, Silva-Espinoza JC, et al. DNA methylation of cellular retinoic acid-binding proteins in cervical cancer. Genet Epigenet. 2016;8:53–7. doi: 10.4137/GEG.S40847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka K, Imoto I, Inoue J, Kozaki K, Tsuda H, Shimada Y, et al. Frequent methylation-associated silencing of a candidate tumor-suppressor, CRABP1, in esophageal squamous-cell carcinoma. Oncogene. 2007;26:6456–68. doi: 10.1038/sj.onc.1210459. [DOI] [PubMed] [Google Scholar]
- 17.Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013. 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed]
- 18.Nhieu J, Lin YL, Wei LN. Noncanonical retinoic acid signaling. In: Methods in Enzymology. Academic Press Inc., 2020, pp 261–81. [DOI] [PubMed]
- 19.Wook Park S, Nhieu J, Persaud SD, Miller MC, Xia Y, Lin YW, et al. A new regulatory mechanism for Raf kinase activation, retinoic acid-bound Crabp1. Sci Rep. 2019;9:10929. doi: 10.1038/s41598-019-47354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park SW, Nhieu J, Lin YW, Wei LN. All-trans retinoic acid attenuates isoproterenol-induced cardiac dysfunction through Crabp1 to dampen CaMKII activation. Eur J Pharm. 2019;858:172485. doi: 10.1016/j.ejphar.2019.172485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Hermann TE. Characterization of ionomycin as a calcium ionophore. J Biol Chem. 1978;253:5892–4. doi: 10.1016/S0021-9258(17)34550-7. [DOI] [PubMed] [Google Scholar]
- 22.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20:1172–9. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold AS, Gueye M, Guettier-Sigrist S, Courdier-Fruh I, Coupin G, Poindron P, et al. Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab Investig. 2004;84:1271–8. doi: 10.1038/labinvest.3700163. [DOI] [PubMed] [Google Scholar]
- 24.Boido M, de Amicis E, Valsecchi V, Trevisan M, Ala U, Ruegg MA, et al. Increasing agrin function antagonizes muscle atrophy and motor impairment in spinal muscular atrophy. Front Cell Neurosci. 2018;12:17. doi: 10.3389/fncel.2018.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharyya M, Karandur D, Kuriyan J. Structural insights into the regulation of Ca2+ /calmodulin-dependent protein kinase II (Camkii) Cold Spring Harb Perspect Biol. 2020;12:1–20.. doi: 10.1101/cshperspect.a035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baltgalvis KA, Call JA, Nikas JB, Lowe DA. Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve. 2009;40:443–54. doi: 10.1002/mus.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greising SM, Call JA, Lund TC, Blazar BR, Tolar J, Lowe DA. Skeletal muscle contractile function and neuromuscular performance in Zmpste24-/-mice, a murine model of human progeria. Age. 2012;34:805–19. doi: 10.1007/s11357-011-9281-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garlich MW, Baltgalvis KA, Call JA, Dorsey LL, Lowe DA. Plantarflexion contracture in the mdx mouse. Am J Phys Med Rehabil. 2010;89:976–85. doi: 10.1097/PHM.0b013e3181fc7c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang W, Qi B, Lv H, Wu F, Liu L, Wang W, et al. A new method of isolating spinal motor neurons from fetal mouse. J Neurosci Methods. 2017;288:57–61. doi: 10.1016/j.jneumeth.2017.06.014. [DOI] [PubMed] [Google Scholar]
- 30.Sleigh JN, Burgess RW, Gillingwater TH, Cader MZ. Morphological analysis of neuromuscular junction development and degeneration in rodent lumbrical muscles. J Neurosci Methods. 2014;227:159–65. doi: 10.1016/j.jneumeth.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bolte S, Cordelières FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 32.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–67. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 34.Théry M, Piel M. Adhesive micropatterns for cells: a microcontact printing protocol. Cold Spring Harb Protoc. 2009;4. 10.1101/pdb.prot5255. [DOI] [PubMed]
- 35.Bettadapur A, Suh GC, Geisse NA, Wang ER, Hua C, Huber HA. et al. Prolonged culture of aligned skeletal myotubes on micromolded gelatin hydrogels. Sci Rep. 2016. 10.1038/srep28855. [DOI] [PMC free article] [PubMed]
- 36.Cammer .Measure intensity of a Z stack. http://microscopynotes.com/imagej/measure_Zstack_intensity/index.html. Accessed 15 Feb 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1 co-culture with motor neuron 10X
Video 2 Co-Culture With Motor Neuron 20X-1_mp4_CORRECTED.mp4
Video 3 C2c12 Myotube Only (Co-Culture Without Motor Neuron) 10X-1_MP4_CORRECTED.mp4
Video 4 Co-Culture Fura2-1_MP4_CORRECTED.mP4
Data Availability Statement
The data generated during this study are available upon reasonable request from the corresponding author.