

Abstract
Schwann cell precursors (SCPs) are nerve‐associated progenitors that can generate myelinating and non‐myelinating Schwann cells but also are multipotent like the neural crest cells from which they originate. SCPs are omnipresent along outgrowing peripheral nerves throughout the body of vertebrate embryos. By using single‐cell transcriptomics to generate a gene expression atlas of the entire neural crest lineage, we show that early SCPs and late migratory crest cells have similar transcriptional profiles characterised by a multipotent “hub” state containing cells biased towards traditional neural crest fates. SCPs keep diverging from the neural crest after being primed towards terminal Schwann cells and other fates, with different subtypes residing in distinct anatomical locations. Functional experiments using CRISPR‐Cas9 loss‐of‐function further show that knockout of the common “hub” gene Sox8 causes defects in neural crest‐derived cells along peripheral nerves by facilitating differentiation of SCPs towards sympathoadrenal fates. Finally, specific tumour populations found in melanoma, neurofibroma and neuroblastoma map to different stages of SCP/Schwann cell development. Overall, SCPs resemble migrating neural crest cells that maintain multipotency and become transcriptionally primed towards distinct lineages.
Keywords: multipotency, neural crest, regulons, Schwann cell precursors, Schwann cell lineage
Subject Categories: Development, Neuroscience
A transcriptional, temporal and spatial single‐cell profiling resource uncovers Schwann cell progenitors as a multipotent source of neural crest‐like cells in the developing mouse embryo.

Introduction
Schwann cell precursors (SCPs) represent a nerve‐associated embryonic cell type that spreads throughout the body using peripheral nerves as navigational scaffolds during vertebrate embryonic development (Furlan & Adameyko, 2018). SCPs give rise to melanocytes, peripheral neurons, Schwann, neuroendocrine and mesenchymal cells (Joseph et al, 2004; Adameyko et al, 2009; Nitzan et al, 2013; Dyachuk et al, 2014; Kaukua et al, 2014; Uesaka et al, 2015; Espinosa‐Medina et al, 2017; Furlan et al, 2017; Kastriti et al, 2019). While initially regarded as progenitors of Schwann cells (SCs) prior to fate restriction towards myelination (Jessen & Mirsky, 1992), both in vitro and ex vivo studies revealed that these nerve‐associated cells can give rise to many downstream fates (Ciment et al, 1986; Morrison et al, 1999; Dupin et al, 2000). Thus, SCPs are multipotent in vivo and associated with essential developmental functions beyond myelination and support of axonal homeostasis within the peripheral nervous system.
Schwann cell precursors arise from neural crest (NC) cells that settle on outgrowing peripheral nerves (Weston, 1963). They spread throughout the developing body via branching innervation, detach from the nerves in specific locations and produce large quantities of pigment cells, autonomic and enteric neurons, chromaffin cells of the adrenal medulla and specific mesenchymal populations within nerves and cranial locations (Joseph et al, 2004; Adameyko et al, 2009; Nitzan et al, 2013; Dyachuk et al, 2014; Uesaka et al, 2015; Espinosa‐Medina et al, 2017; Furlan et al, 2017; Kastriti et al, 2019). Based on this wide array of SCP‐derived fates, SCPs can be viewed as the nerve‐associated state of neural crest‐like cells that persists into later developmental stages. In line with this concept, we suggested that the evolutionary ancient NC‐like cells navigated via the nerves in early chordates and may have resembled SCPs (Ivashkin & Adameyko, 2013). Experimental support for this hypothesis comes from lamprey and zebrafish, where enteric neurons are generated from the NC‐derived nerve‐associated SCPs navigating via the trunk nerves to the gut of developing larvae (Green et al, 2017; El‐Nachef & Bronner, 2020).
In adults, SCPs and nerve‐associated peripheral glial cells may represent a source of the adult NC‐like stem cells residing in numerous tissues. For example, the skin contains multipotent skin‐derived precursor cells (SKP cells; Toma et al, 2001; Fernandes et al, 2004; Wong et al, 2006). Similarly, peripheral glia associated with innervation are known to release signals that stimulate the formation of the regenerating limb blastema (Lehoczky et al, 2011; Johnston et al, 2016; Carr et al, 2019; Storer et al, 2020). Thus, identifying adult cells capable of reverting to the NC‐like multipotency has potentially important implications for regenerative medicine.
The fact that SCPs generate diverse cell types in particular locations raises questions about what determines their cell fate choice. For instance, SCPs covering preganglionic autonomic motor nerves generate chromaffin cells near the dorsal aorta, whereas SCPs associated with sensory fibres innervating the skin generate melanocytes and specialised sensory skin end organs (Adameyko et al, 2009; Furlan et al, 2017; Harty & Monk, 2017; Lumb et al, 2018; Abdo et al, 2019; Jessen & Mirsky, 2019). This suggests that SCPs might be intrinsically heterogeneous and become fate‐restricted or biased by unknown early cues generated by their local environment. This is consistent with the mechanisms that drive sympathoadrenal differentiation via CXCL12 and BMP signals secreted by dorsal aorta (Saito et al, 2012) and by signalling factors inducing melanocyte formation (WNTs, EDN3, KIT ligand) from the NC lineage (Baynash et al, 1994; Hosoda et al, 1994; Reid et al, 1996; Dorsky et al, 1998; Dunn et al, 2000; Hou et al, 2000; Lewis et al, 2004). Understanding the spatiotemporal heterogeneity of SCPs is key to answering these mechanistic questions.
In the present study, we employ single‐cell transcriptomic analysis to generate the atlas of the neural crest lineage and elucidate SCP identity, heterogeneity and similarity to the NC and NC‐derived cell types in cancer. Our results show that early SCPs and late neural crest cells (NCCs) share a multipotent “hub” state containing differentially biased cells. We further find that loss‐of‐function of the “hub” gene Sox8 in the trunk neural crest affects SCPs along the nerve and sympathoadrenal cell fate choice. Once SCPs are driven towards the SC lineage, they diverge from the NC‐like state via orchestrated transcriptional activation, with spatial heterogeneity and sets of genes biasing them to myelinating and non‐myelinating SCs; terminal SCs of neuromuscular junctions and endoneurial fibroblasts. Finally, the comparisons of SCP transcriptional profiles with cells from melanoma, neuroblastoma and neurofibromatosis type 1 predict the reactivation of embryonic‐like cell states.
Results
Migratory neural crest cells transit into Schwann cell precursors via a joint multipotent “hub” transcriptional state
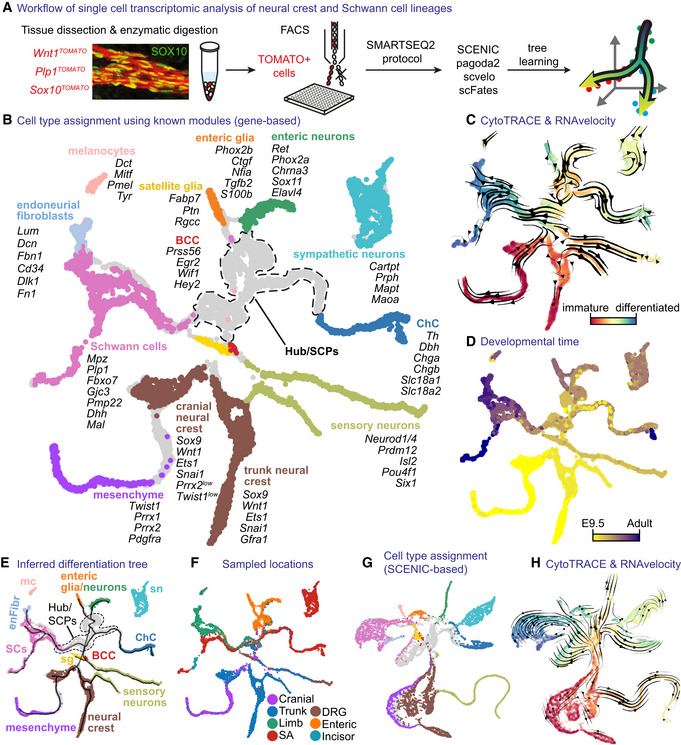
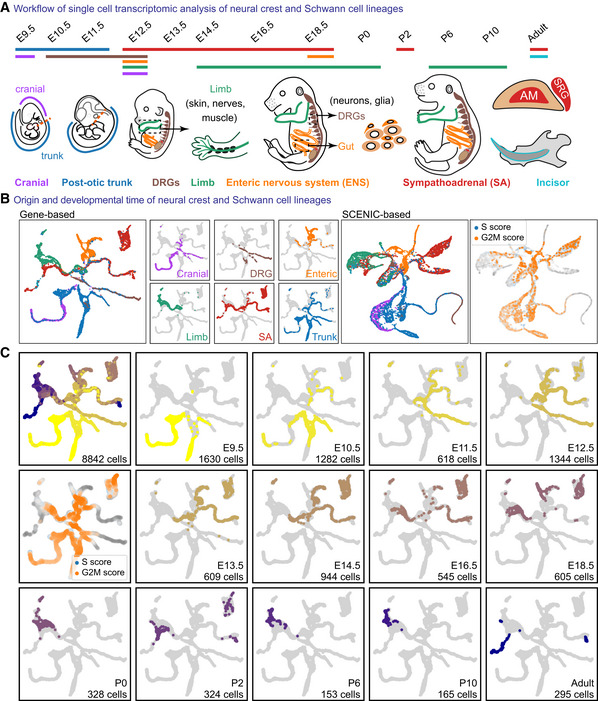
To examine the spatiotemporal heterogeneity of the developing neural crest and SC lineage, we used the Smartseq2 single‐cell transcriptomics approach, which allows identifying the expression of 7,000–8,000 genes per cell on average (Fig 1A and Appendix Fig S1). We combined our newly generated data corresponding to a range of developmental and postnatal stages (from E9.5 to adult) and multiple body locations (Fig EV1) with previously generated data sets of mouse NC populations (Soldatov et al, 2019). Neural crest‐derived cells were obtained via Cre‐based labelling with the constitutively active Wnt1‐Cre;R26 TOMATO and tamoxifen‐inducible Plp1 CreERT2 ;R26 TOMATO and Sox10 CreERT2 ;R26 TOMATO transgenes, with recombination induced 48 h prior to tissue collection (Fig 1A). Additionally, enrichment of sensory neurons was achieved using Isl1 Cre ;R26 TOMATO from newly generated and already published data sets (Faure et al, 2020). Starting from E12.5 onwards, we separately examined anatomical regions of the developing embryos to discern regional differences between sympathoadrenal, sensory, SCPs, melanocytes and enteric NC lineages (Fig EV1B). After computational clean‐up of contaminating populations using published markers (including predelaminating neural crest based on neural tube signature defined as Olig3 +/Sox2 +/Msx1 + cells; Simoes‐Costa & Bronner, 2015; Tabula Muris et al, 2018) and low‐quality transcriptomes (Appendix Figs S1 and S2), we recovered the transcriptomes of 8,842 cells covering embryonic and postnatal stages, different locations and neural crest‐derived fates (Fig EV1A–C). The resulting atlas is available for data mining online via internet browser (please see “Data availability” section).
Figure 1. Transcriptomic analysis of neural crest, Schwann cell precursors and their downstream lineages.
-
AOverview of single‐cell sampling, transcriptomic analysis and developmental tree assembly.
-
BGene expression‐based UMAP embedding. Cells assigned to defined cell types using shown markers.
-
CUMAP with CytoTRACE values and overlaid RNA velocity‐derived vector streams.
-
DUMAP with developmental stage.
-
EUMAP embedding as in (B) with overlaid learned tree.
-
FUMAP embedding with sampled locations.
-
GUMAP embedding based on SCENIC activity scores (same colouring as B).
-
HSCENIC‐based UMAP with the same information as (C).
Data information: BCC, boundary cap cells; ChC, chromaffin cells; DRG, dorsal root ganglia; Mc, melanocytes; SA, sympathoadrenal; SCs, Schwann cells; sn, sympathetic neurons.
Figure EV1. Overview and composition of neural crest and Schwann cell data set.
-
AOverview of the sampled locations and time points in mice. AM, adrenal medulla; SRG, suprarenal ganglion.
-
BColour‐coded projections of the tissue of origin, including SCENIC regulon‐based UMAP embedding, with cell cycle shown (right).
-
CUMAP embeddings colour‐coded according to the developmental stage, with cell cycle shown.
Next, we processed the data set via pagoda2 and SCENIC pipelines with joint UMAP embedding based on: (i) multiscale diffusion space derived from PCA space (generated by gene‐based pagoda2); (ii) multiscale diffusion space derived from regulon activity scores (AUC, generated by SCENIC) showing striking similarities in their structure (Fig 1B–H). This observation suggested that the main differentiation paths of the NC lineage could be abstracted at the level of transcription factor (TF) activity alone. In other words, the detected transactivation activity of identified TFs (i.e. regulons) sufficiently defined the trajectory and the structure of the data set. Therefore, we decided to base our main trajectory analysis by fitting a principal tree on the multiscale diffusion space of the SCENIC AUC regulon activity scores. Visualisations of gene expression were displayed on the UMAP embedding generated from the same diffusion space. Using the combined expression of known markers for NCCs and their derivatives (Appendix Fig S3), we first mapped the cells that clearly define the main populations on the SCENIC‐based UMAP embedding. This resulted in a pool of multipotent NCCs derived from E9.5 and E10.5 (expressing Wnt1, Ets1, Sox9; Wu et al, 2003; Yanfeng et al, 2003; Bronner & Simoes‐Costa, 2016) projecting through cellular streams towards multiple definitive fates (Fig 1B and C).
The structure of the NC pool revealed that cells within this population were divided into two subgroups, separated by the presence of mesenchymal bias in cranial neural crest cells (Figs 1B and EV1B). Intuitively, two parallel paths of NC differentiation converge in terms of nonmesenchymal fates at later time points. The examination of directionality of the NC and downstream nonmesenchymal populations with CytoTRACE and RNA velocity further produced a continuum of transitions and cell states towards the most differentiated transcriptional states (Fig 1C and H). This indicates that the molecular changes that govern the switch from migrating NC to nerve‐bound SCPs take place as a smooth transition of transcriptional states.
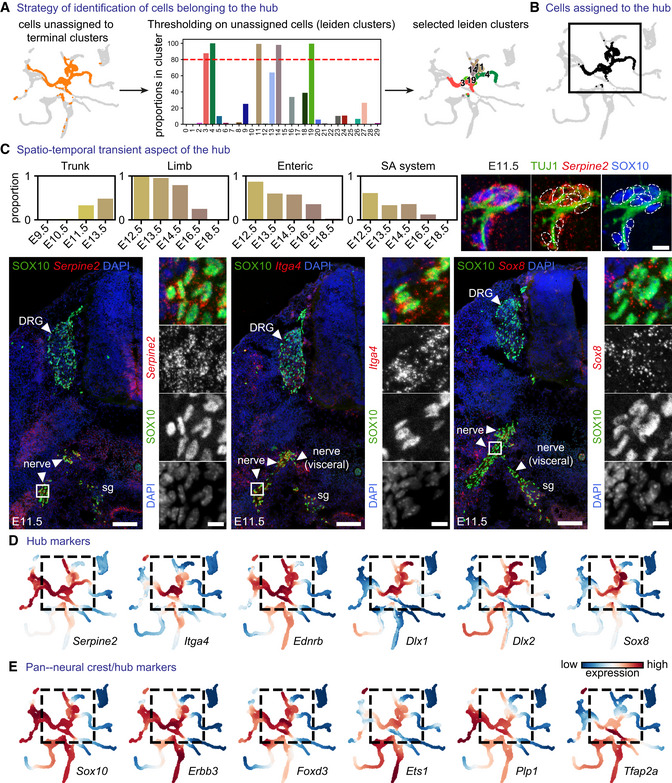
When focusing on the root of the resulting tree of transcriptional events, we observed that one of the directions of differentiation emerging from the NC included a cell population extending towards sensory neurons (Fig 1B). This population appeared to be proliferative, consisting of the late NCCs expressing Neurod1/4, Prdm12, Isl2 Pou4f1 and Six1 (Fig EV1B and Appendix Fig S3). In contrast to cells differentiating towards this sensory neurogenesis branch, we detected a separating pool of cycling progenitors appearing as a “hub” for downstream fates (Figs 1B and E, 2A and B, and EV1B). The “hub” cells (represented as grey in Fig 1B) mainly originated from E10.5, E11.5 and E12.5 stages (Figs 2C and EV1B) with few admixed E9.5 NCCs. The “hub” cells expressed both pan‐neural crest and SCP markers (Fig 2D and E) and spanned all peripheral locations, including mixed nerves and developing autonomic and enteric ganglia as validated experimentally with combined SOX10 immunodetection and RNAscope® for the “hub” genes Itga4, Serpine2 and Sox8 (Appendix Figs S5–S7). Even though SOX10+ cells representing neural crest migrate freely at E9.5, very few of them are found near the emerging axons. At E10.5, most SOX10+ cells become nerve (or peripheral neuron)‐associated. Starting from E11.5, all SOX10+ cells are associated with the outgrowing peripheral nerves or neurons consistent with previous studies (Adameyko et al, 2012; Dyachuk et al, 2014; Furlan et al, 2017). Thus, the late non‐sensory‐biased NCCs and nerve‐associated SCPs (at E9.5, E10.5 and E11.5) from cranial and trunk regions converge on a similar multipotent transcriptional state we refer to as “hub,” from which other subtrajectories emerge towards multiple definitive fates according to RNA velocity and trajectory analysis (Fig 1B, C and F). The dynamics of nerve association combined with the onset of detectable “hub” signature (Itga4, Serpine2, Sox8) by RNAscope® in neural crest cells at postdelamination stages at E9.5 (Appendix Figs S5–S7) and progressive upregulation with an observed peak at E12.5 (Fig 2C) proposes that the onset of the “hub” state initiates in late neural crest but consolidates and reaches a peak upon cell attachment onto peripheral nerves. The cells of the “hub” state differentiate towards SCs as detected via the onset of expression of Gjc3, Plp1, Pmp22, Mpz; (Altevogt et al, 2002; Jessen & Mirsky, 2005), melanocytes expressing Mitf, Dct, Tyr, Pmel (Steingrimsson et al, 2004), enteric neurons (Hoxb5, Hoxa5, Chrnb4, Chrna3, Chodl, Elavl4), enteric glia (S100b, Phox2b, Tgfb2, Nfia, Ctgf; Steingrimsson et al, 2004), autonomic neurons (Prph, Cartpt, Mapt, Maoa) and chromaffin cells (Chga, Chgb, Th, Dbh, Slc18a1, Slc18a2; Furlan et al, 2017; Kastriti et al, 2019; Kameneva et al, 2021).
Figure 2. The “hub” state is a transient cell pool occupying peripheral locations with unique markers.
-
AStrategy of “hub” identification: Leiden clusters containing ≥ 80% of unassigned cells (red dashed lined demarcating the cutoff for clusters characterised by ≥ 80% of unassigned cells that were considered as “hub”).
-
BCells assigned to the “hub.”
-
CProportion of “hub” cells across sampling locations and developmental stage and (Cox et al, 2004) validation of the “hub” gene signature using SOX10 as a pan‐”hub” marker and RNAscope® in situ hybridization for Serpine2, Itga4 and Sox8. Upper right: SOX10+ cells expressing “hub” markers associate with peripheral nerves at E11.5. Scale bar in overview pictures is 100 μm and 10 μm in insets. Scale bar for upper right subpanel = 10 μm.
-
DMAGIC‐imputed expression of selected markers among the top 25 differentially expressed in “hub” cells.
-
EMAGIC‐imputed expression of known neural crest markers. SA system: sympathoadrenal system.
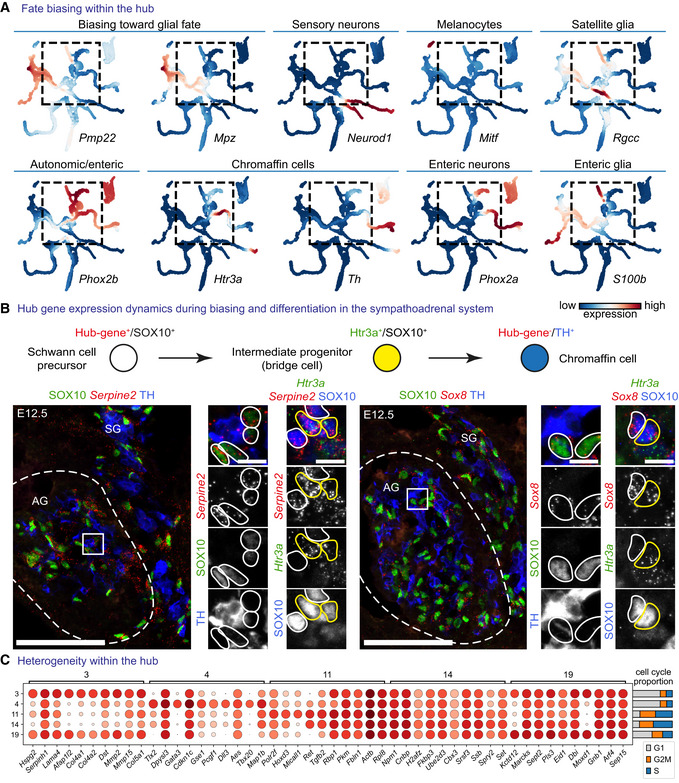
Despite the expression of core neural crest transcription factors (Sox10, FoxD3, Tfap2a, Ets1; Bronner & Simoes‐Costa, 2016) and signalling genes reported to be upregulated in the neural crest (Erbb3, Ednrb, Ngfr; Bernd, 1985; Nataf et al, 1996; Britsch et al, 1998), the “hub” cell population expressed high levels of specific genes in a unique combination, including Sox8, Dlx1, Itga4 and Serpine2, rendering the “hub” different from the majority of delaminating and early migrating neural crest cells (Fig 2D and E). These “hub”‐specific genes were downregulated when the cells entered differentiation into terminal neural crest fates (Figs 2C–E, and 3A and B). This transient nature of the “hub” was validated experimentally in the developing adrenal gland using immunostaining against SOX10 (SCP marker) and TH (chromaffin cell marker) and RNAscope® to visualise the intermediate Htr3a‐expressing progenitors or “hub” genes Serpine2 and Sox8. Namely, we observed the high expression of Serpine2 and Sox8 in SOX10+ SCPs and Htr3a‐expressing bridge cells but no expression upon differentiation towards TH+ chromaffin cells (Fig 3B).
Figure 3. Cells in the “hub” state are subject to fate bias and a high degree of heterogeneity.
-
AMAGIC‐imputed markers of fate‐biasing within the “hub.”
-
BRNAscope® in situ hybridization for “hub” genes Serpine2 and Sox8 in the developing adrenal gland combined with immunohistochemistry for SOX10 (progenitors—Schwann cell precursors (SCPs)), TH (differentiated chromaffin cells) or RNAscope® for committed SCPs towards chromaffin fate—Htr3a. Scale bar in overview pictures (left) is 100 and 10 μm in insets (right).
-
CDot plot of top eight differentially expressed genes between Leiden clusters within the “hub.”
On the other hand, the “hub” cells appeared heterogeneous by showing mixed expression of terminal fate‐related genes, which suggests a number of fate biasing programs operating within the “hub” (preceding particular commitment and further differentiation; Fig 3C). This is supported by the expression of fate‐specific genes Pmp22 and Mpz (biasing towards SCs) or Phox2a/b and S100b (biasing towards autonomic neurons and glia), in the “hub” population (Fig 3A).
When it comes to the developmental origin of melanocytes, the corresponding branch in the gene‐based embedding (Fig 1B and E) does not attach to the rest of the tree likely because of the multiple convergent origins of pigment cells, which are derived from the migratory neural crest and from different populations of cranial and trunk SCPs. At the same time, the SCENIC‐based embedding (Fig 1G) connects the melanocyte branch to the rest of the tree, which suggests common mechanisms in melanocyte fate biasing, specification and differentiation. Similarly, the sympathetic neuronal branch stays disconnected in the gene‐based embedding, as sympathoblasts can originate directly from the ventral pathway of neural crest migration and via SCPs along the ventral nerves (Weston, 1970; Le Douarin & M‐AM, 1974; Kastriti et al, 2019). In the SCENIC‐based embedding (Fig 1G), the sympathetic neuron branch connects perfectly due to the common regulation of the differentiation process.
Finally, we identified and connected the satellite glial cells found in association with sensory neurons within dorsal root ganglia, and boundary cap stem cells (BCCs) associated with motor exit points from the neural tube. Using previously known markers (Coulpier et al, 2009; Mapps et al, 2022), we found the satellite glia and BCCs in the position where the neural crest populations transit into the “hub” (Fig 1B and Appendix Fig S4). The expression of “hub” markers in these cell populations suggests that they represent highly specialised subtypes of multipotent neuron‐ and nerve‐associated cells.
Loss of “hub” gene Sox8 results in SCPs defects and biased cell differentiation
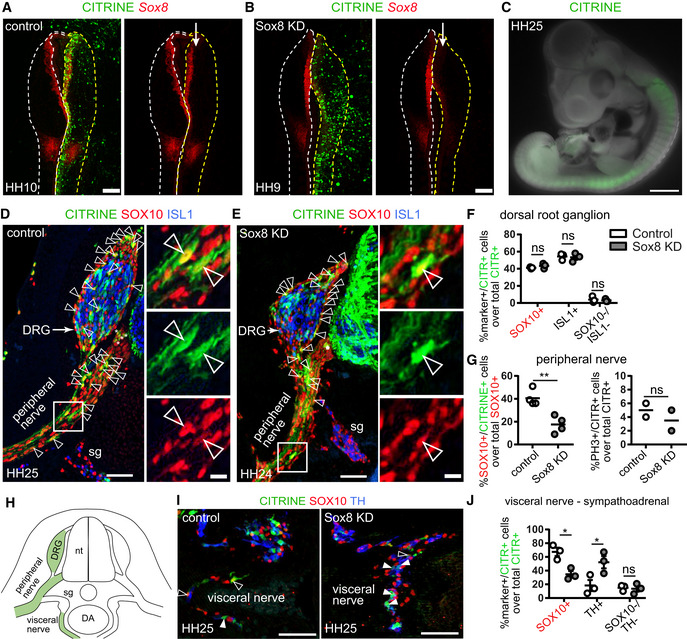
To test the function of the “hub”‐specific transcription factor Sox8, we electroporated the trunk neural tube of HH11 chick embryos with a single plasmid containing gRNA, Cas9 and a citrine reporter (Gandhi et al, 2021) to knockout Sox8 in neural crest‐derived cells (Fig 4A and B). Embryos were then allowed to develop until HH24‐25 (Fig 4C), by which time the dorsal root ganglia (DRG) and visceral nerves had developed (Fig 4H). Quantification revealed a markedly reduced proportion of electroporated CITRINE+/SOX10+ cells along the ventral nerve after the loss of Sox8 as compared to controls (Fig 4D, E and G). No significant differences were noted in the numbers of cells undergoing mitosis between experimental and control embryos, suggesting that Sox8 might be involved in regulating migration or other processes rather than the proliferation of neural crest‐derived cells along the ventral nerve. This is consistent with a previous study suggesting a role for Sox8 in neural crest migration (O'Donnell et al, 2006). Despite the phenotype in the ventral nerves, the loss of Sox8 did not alter the number of glial (SOX10+) or neuronal (ISL1+) cells in the DRGs (Fig 4D–F), which supports that loss of SOX8 does not affect cell survival. By contrast, in the sympathoadrenal domain, the proportion of CITRINE+/SOX10+ nerve‐associated cells appeared reduced, whereas the proportion of CITRINE+/TH+ cells significantly increased (Fig 4I and J). This result suggests elevated rates of conversion of sympathoadrenal SCPs into adrenergic fates. Taken together, high levels of Sox8 might specifically stabilise the SCP/“hub” phenotype and assist the migration of neural crest‐derived SOX10+ cells along the peripheral nerves.
Figure 4. CRISP‐Cas9‐mediated knock down of Sox8 in developing chicken late neural crest affects migration and differentiation of “hub” cells.
-
AElectroporation of the control CRISPR‐Cas9 plasmid (CITRINE+ cells) does not affect Sox8 expression as seen by HCR against Sox8. The arrow points to the unilaterally electroporated side of the embryo. Scale bar = 200 μm.
-
BValidation of Sox8 knock down (KD) using the CRISPR‐Cas9 plasmid containing a Sox8 guide RNA by HCR. The arrow points to the unilaterally electroporated side of the embryo. Scale bar = 200 μm.
-
CCITRINE+ (electroporated) cells found migrating away from the neural tube after 3 days of culture following unilateral electroporation. Scale bar = 1 mm.
-
D, EImmunofluorescence against CITRINE (electroporated cells), SOX10 (Schwann cell precursors and Schwann cells) and ISL1 (sensory and sympathetic neurons) on embryos electroporated with either control CRISPR plasmid (D) or a CRISPR plasmid containing a guide RNA against Sox8 (E). CITRINE+ cells populate the developing dorsal root ganglia (DRG) and peripheral nerves. Examples of CITRINE+/SOX10+ cells shown by arrowheads. Asterisks show ventral boundary cap glia. Scale bar is 50 μm in overviews and 10 μm in insets.
-
FQuantification of the fate distribution of CITRINE+ cells as a % between glial (SOX10+) cells, sensory neurons (ISL1+) or neither (SOX10−/ISL1−) in the DRG of control and Sox8KD chick embryos and Sox8KD chick embryos. Biological replicates – N = 3 embryos per condition (wild‐type versus Sox8 KD). Data represented as mean ± SEM. Statistical significance determined using the Holm–Sidak method (α = 0.05; multiple t‐tests, unpaired). SOX10+ cells: P = 0.7901, ISL1+ cells: P = 0.9206, SOX10−/ISL1− cells: P = 0.9206. For statistical significance: nonsignificant P‐value ≥ 0.05.
-
GQuantification of (left) the % of SOX10+ cells in the peripheral nerves that are CITRINE+ in control and Sox8KD chick embryos and (right) the % of PH3+ CITRINE+ cells corresponding to proliferative cells. Biological replicates – N = 4 embryos per condition for SOX10+ distribution and N = 2 for PH3 quantification (wild‐type versus Sox8 KD). Data represented as mean ± SEM. Statistical significance determined unpaired t‐test with two‐tailed P‐value. SOX10+ cells: P = 0.0044, PH3+ cells: P = 0.4929. For statistical significance: nonsignificant P‐value ≥ 0.05, **P‐value < 0.01.
-
HSchematic representation of analysed anatomical locations.
-
IImmunofluorescence against CITRINE (electroporated cells), SOX10 (Schwann cell precursors and Schwann cells) and TH (sympathetic neurons and chromaffin cells) of the sympathoadrenal domain on control and Sox8KD embryos. CITRINE+/SOX10+ cells shown by empty arrowheads while CITRINE+/TH+ cells are shown by filled arrowheads. Scale bar = 50 μm.
-
JQuantification of the fate distribution of CITRINE+ cells as a % between glial (SOX10+) cells, chromaffin cells (TH+) or neither (SOX10−/TH−) in the proximity of the dorsal aorta and visceral nerve of wild‐type and Sox8 KD chick embryos. Biological replicates – N = 4 embryos per condition. Data represented as mean ± SEM. Statistical significance determined using the Holm–Sidak method (α = 0.05; multiple t‐tests, unpaired). SOX10+ cells: P = 0.0067, TH+ cells: P = 0.0067, SOX10−/TH− cells: P = 0.8819. For statistical significance: nonsignificant P‐value ≥ 0.05, *P‐value < 0.05.
Data information: In total, six embryos were analysed for the control electroporation and seven embryos were analysed for the Sox8 knock down electroporation, and data depicted in graphs correspond to mean ± SEM per embryo (corresponding to biological replicates): 3–4 electroporated embryos were analysed per condition (control and SOX8 knock down) with 4–5 sections stained and analysed per embryo per region of interest (DRG, ventral nerve, sympathoadrenal domain). The only exception is the analysis of pH3 staining where two electroporated embryos were analysed per condition with five sections stained and analysed per embryo. DA, dorsal aorta; DRG, dorsal root ganglia; nt, neural tube; sg, sympathetic ganglion.
Coordinated metaregulons maintain the neural crest‐like state in “hub” cells
The “hub” population represents a transcriptional state common to some late NCCs and SCPs before commitment to other fates (Fig 1B). This favours the interpretation of “hub” cells as a nerve‐associated, late‐NCCs. In cases, when migratory NCCs do not immediately give rise to sensory and autonomic neurons, ectomesenchyme or melanocytes (Soldatov et al, 2019), NCCs reach the nerve‐associated “hub” state before differentiation towards “nerve‐derived” autonomic neurons, chromaffin cells and melanocytes. BCCs and satellite glial cells can also give rise to sensory neurons of the second wave and, consistently, they occupy the intermediate position on the embedding expressing “hub”‐enriched markers, such as high levels of Serpine2 and Ednrb, lower levels of Itga4 and Sox8, with the absence of Dlx1/2 as opposed to the rest of the “hub” (Fig 2D).
To investigate the in‐depth regulatory principles in early migratory NCCs and “hub”/SCPs on the portion of the SC trajectory, we employed pseudotime analysis to reconstruct the finest events of the correlated transcriptional activation. To achieve this, we fitted a principal curve spanning from the cluster of the trunk neural crest proximal to the “hub” state towards the tip of the differentiation of SCs or the emergence point of immature Schwann cells (iSCs; Fig 5A). We then tested and fitted significant AUC scores to the inferred differentiation trajectory and superimposed the biological developmental time transferred from the individual cell transcriptomes (Fig 5B). This allowed us to delineate changes in the transcriptional activation along developmental time. We then relied on the concept of regulons, which correspond to a cluster of coherently co‐activated genes driven by a specific transcription factor. Observations of such coordinated transcriptional activation highlight the key regulatory events driving developmental transitions and help to predict possible causal molecular interactions related to multipotency and cell fate choice.
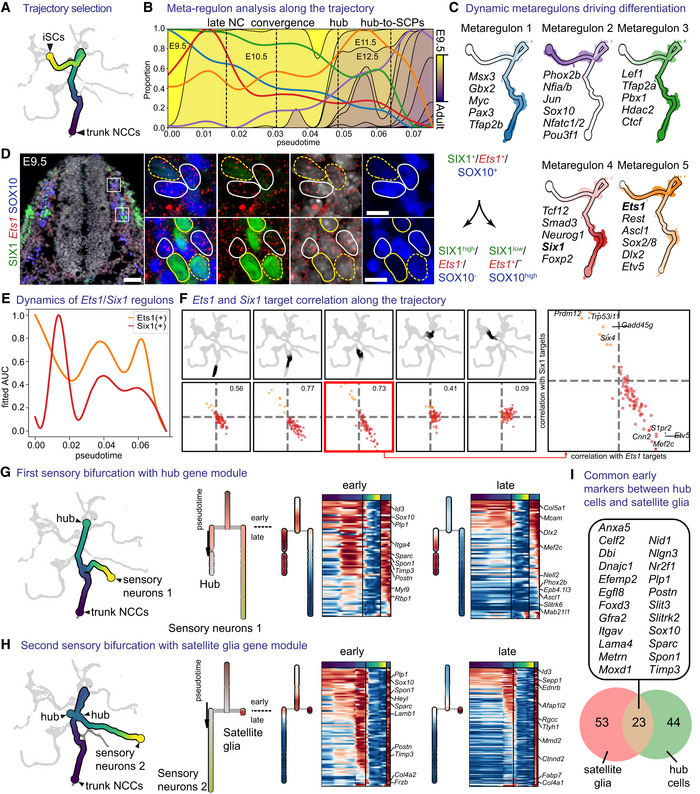
Figure 5. Dynamic changes of metaregulons shape biologically distinct stages of neural crest and Schwann cell precursor development and differentiation.
-
ASubset of the tree containing the trajectory of trunk neural crest to immature Schwann cells.
-
BProgression of the metaregulons over the trajectory selected in (A), combined with developmental time. A metaregulon is the mean expression of all regulons composing a cluster over pseudotime.
-
CTrajectory coloured by metaregulons, with a selection of regulons composing them.
-
DValidation of SIX1 and Ets1 as markers for biasing towards sensory and glia fates, respectively. Scale bar is 50 μm in the overview picture and 10 μm in the insets. Stainings were repeated on two separate occasions on two embryos from the same litter. Cells surrounded by solid white lines correspond to SIX1-/Ets1+/SOX10+ SCPs or glial fate-biased progenitors, while cells surrounded by dashed yellow lines SIX1+/Ets1- or low/SOX10- or low sensory fate-biased progenitors.
-
ENormalised regulon activity scores of Ets1 and Six1 over the trajectory.
-
FInter‐/intra‐correlation analysis of the target genes of Ets1 and Six1 regulons on nonintersecting windows of cells over the trajectory.
-
G, HBifurcation analysis over two successive sensory branches, highlighting the early and late genes biasing towards “hub” cells (G) and satellite glia (H).
-
IVenn diagram of early genes expressed in the bifurcation between “hub” and satellite glia. Gene list shows the common genes between the two.
Data information: iSCs, immature Schwann cells; NCCs, neural crest cells.
Clustering of the scaled fitted transcriptional activity scores resulted in identifying five groups of coordinated regulons, which we termed metaregulons (Figs 5C and EV2). Of these, two metaregulons were characterised by high activity during the early migratory NC‐stage (E9.5 and to a lesser degree E10.5) that subsequently decreased as the trajectory proceeds towards the “hub”/SCP state and further to SCs (metaregulons 1 and 3). Metaregulon 1 and to a lesser degree metaregulon 3 were composed of classical neural crest specification genes (Msx3, Gbx2, Pax3, Tfap2a/b; Basch et al, 2006; Khudyakov & Bronner‐Fraser, 2009) or regulators of WNT1 signalling (Myc, Lef1; Hao et al, 2019). Metaregulon 3 included genes involved in chromatin remodelling and accessibility, such as Ctcf and Hdac2, with key roles in Schwann cell specification and myelination (Jacob et al, 2011; Lee et al, 2022). The dynamics of metaregulon 3 also show multiple peaks reflecting critical periods during which chromatin remodelling and biasing to promyelinating fate might occur. Comparison of the activity of regulons governing early migratory crest and multipotent “hub”/SCPs leaning towards SC fate uncovered a remarkable similarity of these progenitor types (Figs 5B and EV2). For instance, a significant part of this similarity could be attributed to the joint activity of the Ets1(+) (metaregulon 5), Pax3(+) and Tfap2b (metaregulon 1) and Tfap2a (metaregulon 3) regulons (Fig 5C). Concomitantly, we detected smooth and incrementally increasing differences in overall gene expression starting from the NC towards SCPs at the transition between late E10.5 to E11.5 and E12.5, including the onset of activity of Jun, Sox10, Nfatc1/2 and Pou3f1, all genes with described roles in myelination (metaregulon 2; Fig 5C; Mirsky et al, 2008). Notably, Jun represses myelination (Parkinson et al, 2008), whereas Pou3f1 and Sox10 synergistically promote myelination (Britsch et al, 2001; Ghislain & Charnay, 2006; Schreiner et al, 2007). The simultaneous presence of active repressors and facilitators of myelination within the same group of coordinated regulons reflects opposing cell biasing tendencies, which eventually result in separation of SC fates towards myelinating and non‐myelinating, as seen by the decrease in Jun activity and increase in Pou3f1 activity (Fig EV2). Interestingly, Nfatc1/2 factors have not been studied so far in SCs, although they are indispensable for myelination by oligodendrocytes, where they synergize with Sox10 (Weider et al, 2018).
Figure EV2. Metaregulon composition.
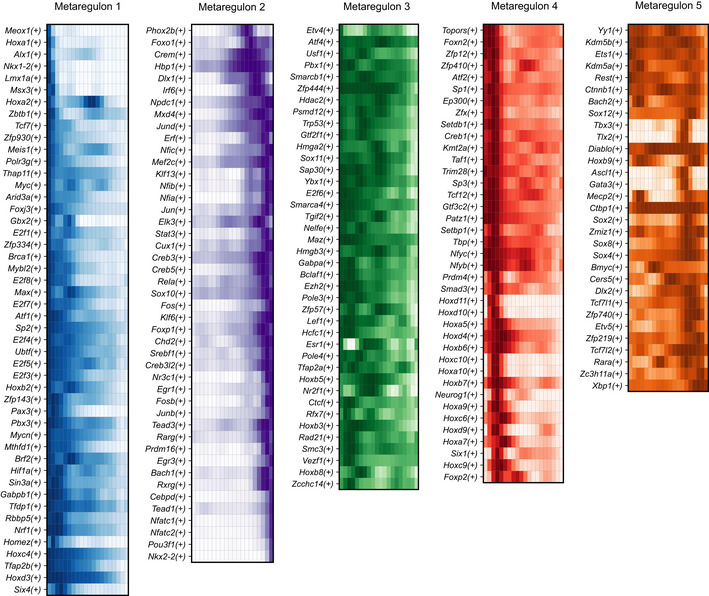
Heatmaps of regulon activity (AUC scores) of individual regulons making up metaregulons 1–5, over the neural crest to immature Schwann cell trajectory, summarised into 20 pseudotime bins.
The active regulons belonging to metaregulon 4 corresponded to neurogenic sensory bias activated in NC and early “hub” cells at E9.5‐E10.5 and included Six1 and Neurog1 regulons (Fig EV2; Sommer et al, 1996; Zou et al, 2004). Surprisingly, metaregulon 5 showed two peaks of activity, interrupted by the peak of the pro‐neurogenic metaregulon 4, which resolved upon neuronal differentiation as the two lineages split, with Ets1 expression retained in SOX10+ SCPs but downregulated in SIX1+ sensory progenitors and neurons, as confirmed by experimental validations using immunofluorescence and RNAscope® (Fig 5D–F). Consistent with this, overexpression of neurogenic factor Six1 suppresses the expression of Ets1 in vitro (Hosseinipour, 2017). The temporary downregulation of Ets1 metaregulon in the middle of the differentiation trajectory is due to mosaic compositional effects that result from a significant proportion of cells upregulating the activity of the neurogenic Six1 regulon and downregulating the activity of Ets1 regulon; other cells in the same location on the trajectory maintained high levels of Ets1 regulon without switching on Six1 regulon (and were intermixed with the first population; Fig 5F). Consistently, the sensory neurogenic subtrajectory started from the Ets1 + NCCs and generated the first branch of Six1‐activating sensory neurons, and also gave rise to the “hub” cells (Fig 5G) and sensory satellite glia (Fig 5H). Sensory satellite glia converged with the “hub” and generated the second sensory neurogenesis branch via activation of Six1 regulon (Zou et al, 2004). The comparison between satellite glia and neighbouring “hub” cells uncovered a set of commonly expressed genes (including classical neural crest “core” of transcription factors), even though the two cellular populations were additionally characterised by separate gene expression programs (Fig 5I).
Metaregulon 5 included the Ets1 and Sox8 regulons (part of the essential NC gene expression programs; O'Donnell et al, 2006; Bronner & Simoes‐Costa, 2016) and the regulon of the transcriptional repressor Rest (Chong et al, 1995), which initially showed high activity during the early NC phase. Additionally, Ascl1, Sox2 and Etv5 regulons, all expressed in embryonic multipotent Schwann cell precursors, belong to metaregulon 5. Accordingly, members of metaregulon 5 were reactivated in the rest of the “hub” and SCPs before commitment to terminal fates in cells from E11.5 to E14.5. TFs Ets1 and Sox8 are indispensable for the NC development (Tahtakran & Selleck, 2003; O'Donnell et al, 2006; Nie & Bronner, 2015), and their activity may convey multipotency and control the spectrum of available fates in “hub”/SCP populations, which is consistent with our functional experiments with targeting Sox8 (Fig 4). Furthermore, Ascl1 correlates with fate‐biasing of SCPs towards autonomic neurons and chromaffin cells (Memic et al, 2016; Furlan et al, 2017); Sox2 inhibits melanocyte fate (Adameyko et al, 2012) and Schwann cell differentiation/myelination (Hagedorn et al, 2000; Le et al, 2005; Balakrishnan et al, 2020), whereas Etv5 has been shown to have a dynamic expression in the SCP lineage. Thus, metaregulon 5 reflects transcription factor activity facilitating multipotency at discrete stages of the neural crest and SCP lineage fate restriction.
Next, we questioned the differences in regulon activity and transition into the “hub” in the cranial and trunk neural crest cells, which are both multipotent populations distinguished by the ability of the cranial neural crest to give rise to ectomesenchyme, a property lacking in the trunk neural crest. Additionally, a significant portion of the cranial neural crest cells delaminates from the anterior Hox‐negative neuroepithelium. During a comparative regulon analysis between the trajectories of cranial versus trunk neural crest progressing towards the iSCs stage (Appendix Fig S8), we observed a significant overlap between regulons detected in the cranial and trunk trajectory (175 regulons in common, with 28 trunk‐specific and 14 cranial‐specific). While cranial‐specific regulons included Tfap2c, Msx1/2, Lbx2, trunk‐specific regulons included, as expected, Hox genes and Six1/4, Neurog1, which correspond to sensory neurogenesis prominent in the trunk region. However, both cranial and trunk neural crest cells converge later towards the “hub” state, as shown by RNAvelocity and Cytotrace (Fig 1C and H) and contribute equally to the generation of multipotent Schwann cell precursors found on cranial and trunk peripheral nerves. This is supported by the experimental evidence showing that SCPs covering cranial nerves are multipotent and generate the majority of cranial melanocytes and chromaffin‐like cells of the carotid oxygen‐sensing organ (Adameyko et al, 2012; Hockman et al, 2018).
Finally, our analysis suggested that although transcriptional changes along the differentiation trajectory and developmental time are smooth, they can be broken down into overlapping patterns of multiple transcriptional activation events. The analysis of regulons provides a discrete picture compared with total gene expression, which allows for defining, isolating and discriminating critical transitions and developmental states of the NC lineage. For instance, the peaks of activity of metaregulon 5 overlap with the developmental stages of experimentally validated SCP and NC multipotency (at E9.5–E10.5 and E11.5–E13.5) and, thus, can be used to define the “hub”/early SCP state versus committed glial progenitor state characterised by the activity of metaregulon 2. Similarly, the activity of NC‐specific metaregulon 1 and cell reprogramming‐related metaregulon 3 helps to set a border between the early migratory NC and later phases connected to the nerve association and “hub” state with what we anatomically term as SCP state (SOX10+ cells associated with nerves). Therefore, the “hub”/SCPs might be defined as a cell state with reactivation of metaregulon 5 coinciding with gradually reducing the activity of metaregulons 1 and 3 (linked to neural crest properties and cell reprogramming) before the activation of metaregulon 2 (linked to the terminal differentiation of Schwann cells; Fig 5B and C). Thus, the “hub” state is distinct in terms of regulation from the majority of the neural crest cells where metaregulons 1 and 3 are dominating.
The “hub”/SCP state is a mixture of differentially biased progenitors that cross‐consolidate multiple fates
The “hub”/SCP state appears to be a point of transcriptional regulation, where fate decisions take place before commitment to definitive cell types. The depth and the quality of the obtained data allowed us to probe the molecular mechanisms of fate selection at this state.
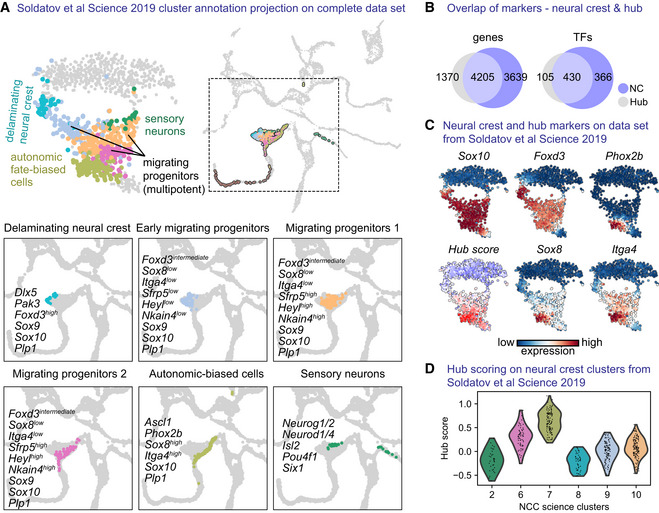
Recently, we reported that NCCs differentiate towards downstream fates through a system of consecutive bifurcations mediated by mutually repressive transcriptional programs leading to noncompatible fates (Soldatov et al, 2019). With the help of our “hub”‐specific gene expression signature (Sox8, Itga4 etc.), we mapped the “hub”‐state onto the neural crest Soldatov et al data set to detect the emergence of the early “hub” program (Fig EV3). For this, we relied on the described states of neural crest development including delaminating, multipotent migrating progenitors, autonomic‐biased cells and cells differentiating towards neurons (Fig EV3A). Briefly, delaminating neural crest cells are defined by Dlx5, Pak3, Sox9, Sox10 genes and migrating groups are characterised by Sfrp5, Heyl and onset of Nkain4 expression, while sensory neurons expressed Neurog1/2, Pou4f1, Isl1, Six1 and Neurod1/4. Joined analysis of the genes expressed by the neural crest and “hub” cells uncovered different degrees of similarity in gene expression (Fig EV3B and Dataset EV1). Some cells previously considered as multipotent migrating progenitors in Soldatov et al data set appeared more similar to the “hub” state as compared to others with the highest “hub” signature identified in the NCCs biased to the autonomic fate before neuro‐glial separation (Fig EV3A, C and D). This observation reinforces the idea that the emerging “hub” state is seen at late neural crest stages, prior to nerve association but is reinforced and upregulated following the attachment of cells to the peripheral nerves. Taking into account these similarities, we analysed the fate choice mechanisms mediating transitions from “hub”/SCPs towards immature Schwann cells (iSCs), enteric populations and sympathoadrenal progenitors in order to compare them with those reported for the NC by Soldatov et al.
Figure EV3. Mapping and comparison of the “hub” state on previously published single‐cell data set of the neural crest.
-
AtSNE embedding and annotated clusters from Soldatov et al (2019), overlayed onto the UMAP embedding containing our own data set.
-
BVenn diagram showing genes and transcription factors (TFs) positively regulated when comparing, respectively, neural crest cells and “hub” cells to the rest of the cells of the data set (Wilcoxon rank‐sum test).
-
C“hub” scoring on previous data set using gene scoring from the top 25 differentially expressed genes specific to the “hub.”
-
DViolin plot of “hub” score over the published annotated clusters.
First, we focused on a developmental paradigm specific to the sympathoadrenal domain. During development, spinal preganglionic sympathetic axons innervate the developing adrenal gland, being covered with SCPs (Furlan et al, 2017; Lumb et al, 2018). In mice, between E11.5 and E13.5, SCPs detach from the preganglionic axons and differentiate towards chromaffin cells that will populate the medulla of the adrenal gland. However, not all SCPs detach from the nerves; some remain associated with axons and generate iSCs (Jessen & Mirsky, 2019). We examined progenitors giving rise to these two cell types starting from NC cells (Fig 6A). Similarly to the mechanisms reported by Soldatov et al, this analysis revealed the initial co‐activation of lineage‐specific gene modules composed of “early” genes prior to the bifurcation, then subsequent repression of these modules (detected as the negatively correlated expression of genes belonging to the opposing modules) and, finally, activation of “late” genes around the bifurcation point (Fig 6B–E). In agreement with the previous studies, the onset of Ascl1 and Phox2b (Huber et al, 2002, 2005) genes biased SPCs towards sympathoadrenal fate, while important drivers of iSC fate were Ednrb, Timp3, Moxd1, Postn, Fabp7 and others (Fig 6C and E).
Figure 6. Biasing of bipotent progenitors through the “hub” state towards sympathoadrenal progenitors and immature Schwann cells.
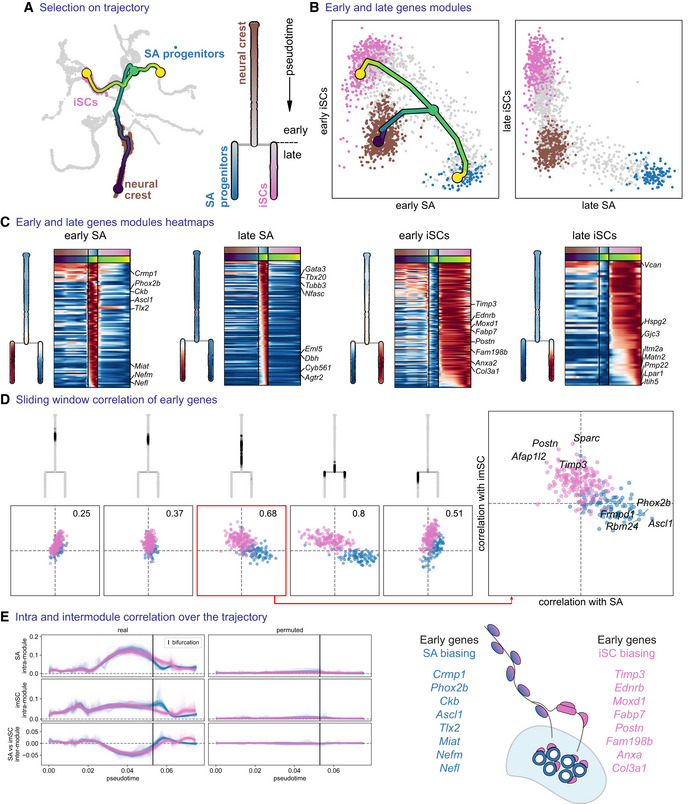
-
ASubset tree selection of the trajectory from trunk NCCs to immature Schwann cells versus sympathoadrenal progenitors. An abstract dendrogram is displayed on the right.
-
BMean expression of early and late gene modules for each branch and overlayed tree on the early gene modules.
-
CHeatmap of early and late genes for each branch, with mean expression shown on the dendrogram representations.
-
DInter‐/intra‐correlation analysis of early gene modules of both branches, on nonintersecting windows of cells over the trajectory.
-
EInter‐ and intra‐module correlation performed on a sliding window of cells (over 100 probabilistic mappings of the trajectory) along the pseudotime axis. iSCs, immature Schwann cells; SA progenitors, sympathoadrenal progenitors.
Next, we focused on SCPs that share gene signatures leading towards chromaffin cells and enteric glia (Appendix Fig S10A–C). Even though the two lineages share a common core of genes (i.e. Ascl1, Phox2b, Hand2), we could not see a real bifurcation and concomitant fate choice taking place, as we did not identify co‐activation of the two competing prebiasing modules and found that these modules were expressed in different cells (Appendix Fig S10D and E). This is not surprising given that most of the enteric glia come from vagal neural crest cells, whereas sympathoadrenal progenitors arise from the trunk being spatially segregated from the vagal. This result supports the robustness of our approach, which can identify real fate choices from the artificial situations resulting from the transcriptional similarity.
Lastly, we focused on the development of enteric glia (found in the intrinsic enteric ganglia) versus the iSCs found in the extrinsic innervation that reaches the gut (Appendix Fig S11A–C). During development, the enteric nervous system (enteric neurons and glia) is derived from vagal NCCs that migrate to the posterior trunk and populate the developing intestine. However, some SCPs delivered by the external innervation can also differentiate between enteric glia and neurons (Uesaka et al, 2015; El‐Nachef & Bronner, 2020). Our bifurcation analysis confirmed the initial co‐expression and then mutual repulsion of “early” enteric genes Ret, Ascl1, Phox2b and Hand2, and the opposing Igfbp5, Prss12, Moxd1, Egfl8 program biasing the cells towards remaining on the extrinsic nerves and becoming iSCs (Appendix Fig S11C–E).
Overall, we found that the “hub”/SCP state is comprised of a mixture of differentially biased progenitors expressing early opposing programs, thus highlighting the local heterogeneity of cells in this developmental phase. The basic mechanisms of cell fate choice based on the co‐activation and subsequent mutual repulsion of transcriptional programs leading to definitive fates also appeared consistent with the earlier findings reported for the NC as described by (Soldatov et al, 2019).
Immature Schwann cells are biased towards myelinating, non‐myelinating Schwann cells and other fates
Previous studies established that SCPs along the nerves transit to iSCs before differentiating towards two alternative fates: myelinating SCs or non‐myelinating SCs (also known as Remak cells; Jessen & Mirsky, 2005). Additionally, during perinatal stages in rodents, a Dhh‐expressing subpopulation of iSCs has been described to generate endoneurial fibroblasts (Parmantier et al, 1999; Joseph et al, 2004; Sharghi‐Namini et al, 2006).
Even though genes driving myelination in SCs are well known (Sox10, Egr2, Pou3f1; Topilko et al, 1994; Bermingham Jr. et al, 1996; Jaegle et al, 1996, 2003; Le et al, 2005; Schreiner et al, 2007; Finzsch et al, 2010), the exact mechanisms underlying fate choice between the three lineages (myelinating, Remak or fibroblastic) are not fully elucidated. Studies point to the crucial role of NRG1‐type III levels supplied by the nerve and Sox10 expression, while SOX2 has been shown to suppress myelination (Britsch et al, 2001; Taveggia et al, 2005; Schreiner et al, 2007; Finzsch et al, 2010; Roberts et al, 2017). To address this, we focused on cells sampled from E11.5 onwards (a time point where all SOX10+ cells are considered to be SCPs and no longer NCCs due to complete association with nerves or peripheral neurons), in order to understand fate separations towards myelinating, non‐myelinating SCs, neuromuscular junction‐associated terminal glial cells and endoneurial fibroblasts (Fig 7B).
Figure 7. Heterogeneity of Schwann cells and fate biasing in maturing peripheral nerves.
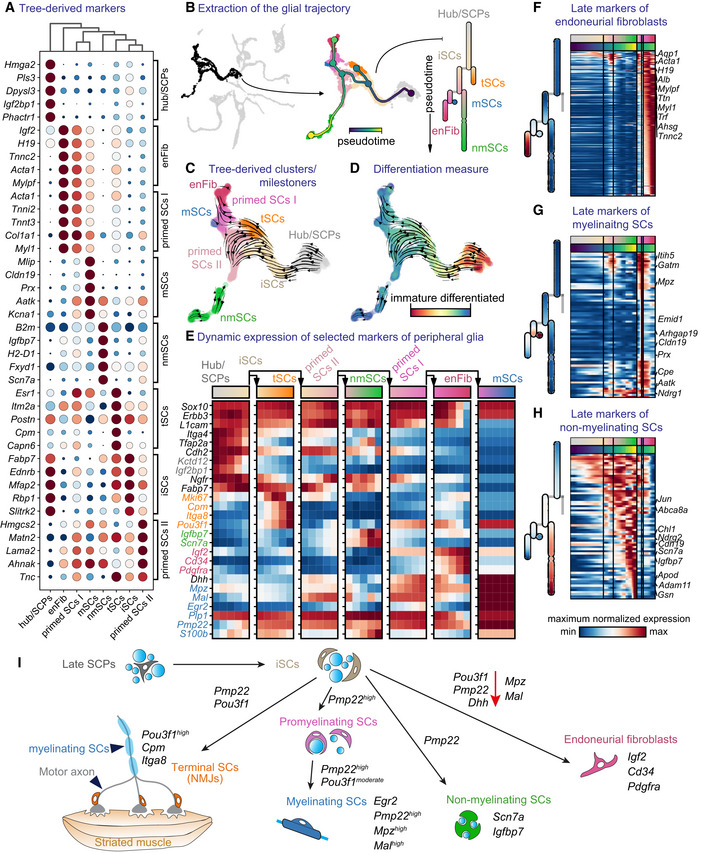
-
ADot plot of the top five differentially expressed genes from the milestones of the glial trajectory.
-
BLeft: selected cells (Foster et al, 2020) of the glial trajectory, middle: constructed tree overlayed on the UMAP, right: abstract dendrogram.
-
CRNA velocity on tree‐derived clusters (milestones).
-
DRNA velocity with CytoTRACE overlayed on the UMAP.
-
EDynamics of expression of known markers over the trajectory summarised over five bins per segment of the tree.
-
F–HHeatmap of late markers of the three endpoints of differentiation from immature Schwann cells: endoneurial fibroblasts (F) and myelinating (G) or nonmyelinating Schwann cells (H).
-
ISchematic representation of transitions from late “hub” cells biased to immature Schwann cells to all end fates along with mixed motor and sensory nerves.
Data information: enFibr, endoneurial fibroblasts; iSCs, immature Schwann cells; mSCs, myelinating Schwann cells; NMJs, neuromuscular junctions; nmSCs, non‐myelinating Schwann cells; SCPs, Schwann cell precursors; tSCs, terminal Schwann cells.
Firstly, RNA velocity and CytoTRACE analysis both agreed on the directionality of the transition from Sox10 +/Erbb3 + /Ngfr + /L1cam + /Itga4 high /Cdh2 + /Tfap2a + /Fabp7 + /Plp1 + SCPs/“hub” cells towards iSCs, identified by the decrease in Itga4 levels and progressive S100b upregulation (Jessen & Mirsky, 2005; Fig 7A and C–E). Differentially expressed markers suggested an early splitting cell type derived from iSCs, which corresponded to terminal SCs of neuromuscular junction SCs (tSCs; Figs 7C and E, and EV4), based on previously reported markers such as Cspg4, Itga8, Slitrk3, Cpm, Pou3f1 (Castro et al, 2020). Biological validations showed the early origin of Itga8 + /Cpm + /Pou3f1 + terminal SCs as seen by RNAscope® or immunofluorescence against OCT6 (encoded by Pou3f1) on E18.5 Plp1 YFP ‐traced embryos when recombination was induced at E13.5 (Figs 8E and EV4E).
Figure EV4. Late embryonic and early postnatal terminal glia representation in the data set.
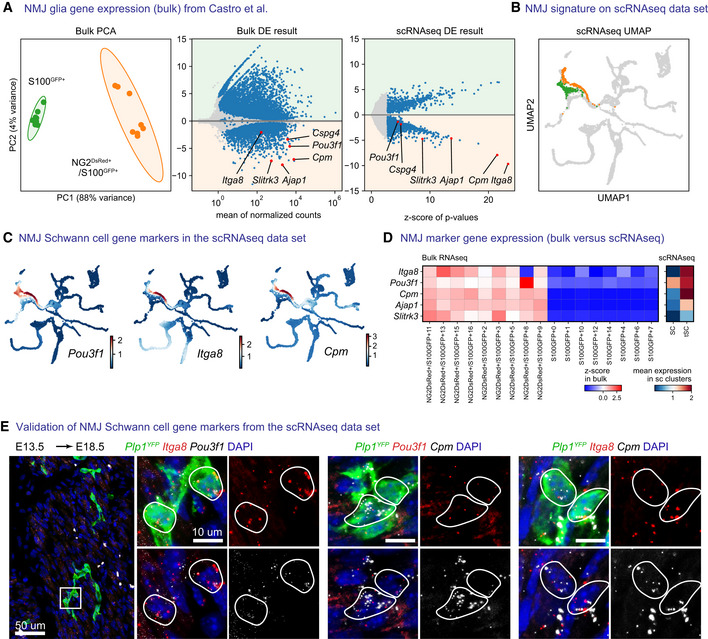
-
ARe‐analysis of bulk RNA sequencing data of S100GFP+ cells and NG2dsRED+/S100GFP+ cells isolated from adult neuromuscular junctions from Castro et al (2020) with PCA plot (left) or DESeq2 differential gene expression analysis (center) and comparison of differential expression results (right) from Leiden clusters of terminal Schwann cells and fate‐biased Schwann cells in our own data set.
-
BLeiden clusters defining terminal Schwann cells (orange) and fate‐biased Schwann cells (Green et al, 2017).
-
CMAGIC‐imputed expression of Pou3f1, Itga8 and Cpm on UMAP embeddings.
-
DComparison of five terminal Schwann cells markers, as z‐scores on bulk data per sample (left) and as mean expression in the Leiden clusters shown in (A and B) in our own data set (right).
-
ERNAscope in situ hybridization validation of Pou3f1, Itga8 and Cpm as markers of terminal Schwann cells at E18.5 combined with immunofluorescence against Plp1 YFP on hindlimbs of an embryo injected with tamoxifen at E13.5. Stainings were repeated on two separate occasions on multiple embryos from the same litter.
Figure 8. Positional code of heterogeneity of Schwann cell precursors and Schwann cells.
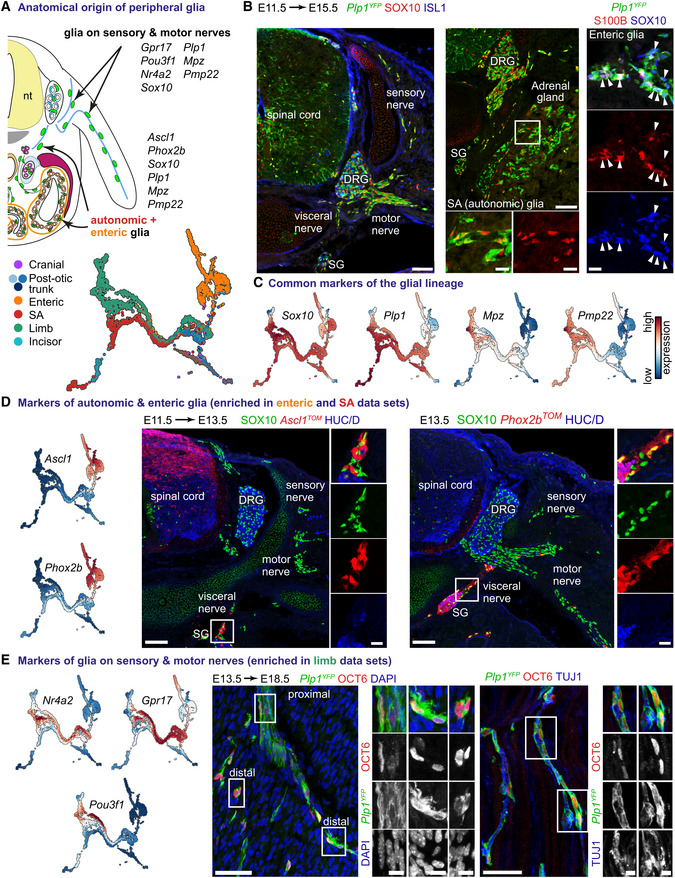
-
ATranscriptional code of peripheral glial cells of various anatomical locations.
-
BImmunofluorescent staining against SOX10 (a marker of all peripheral glia) and ISL1 (neuronal marker; left panel) or S100B (a marker of a subset of peripheral glia) and SOX10 (right panel) on E11.5 to E15.5 Plp1 CreERT2 ;R26 YFP trunk in various anatomical locations. Scale bar in the overview pictures is 100 and 10 μm in insets. Arrowheads point to SOX10+/S100B+ enteric glia.
-
CUMAPs of classical genes shared in peripheral glia.
-
DLeft: UMAPs of Ascl1 and Phox2b expression. Right: Immunofluorescent staining against SOX10, Ascl1 TOM or Phox2b TOM (lineage tracing of autonomic and enteric glia) and HUC/D (neuronal marker) on E13.5 trunk in various anatomical locations. Scale bar in overview pictures is 100 and 10 μm in insets.
-
ELeft: UMAPs of genes enriched in peripheral glia sampled from the developing limbs. Middle: Immunofluorescent staining against OCT6 (encoded by Pou3f1) and TUJ1 (axonal marker; left panel) on hindlimbs of E13.5 to E18.5 Plp1 CreERT2 ;R26 YFP trunk showing the various levels of OCT6 immunoreactivity and variable morphology of Plp1 YFP+/OCT6+ cells along the motor nerves. Scale bar in overview pictures is 50 and 10 μm in insets. Stainings were repeated on two separate occasions on multiple embryos from the same litter.
Data information: DRG, dorsal root ganglion; SA, sympathoadrenal; SG, sympathetic ganglion.
Next, the analysis of the differentiation downstream of iSCs and the inference of a tree on the multiscale diffusion space generated from SCENIC AUC scores revealed a bifurcation between two primed states of SCs (primed SCs I and II). Primed SCs I show progressive downregulation of Ngfr/Cdh2 with Pou3f1/Dhh upregulation differentiating towards Igf2 + /Cd34 + /Pdgfra endoneurial fibroblasts (Fig 7C–F; Richard et al, 2014; Carr et al, 2019). Primed SCs II expressed lower levels of Dhh, retained Cdh2 expression, whereas Fabp7 downregulation coincided with Ngfr increase and eventually gave rise to either Mpz + /Prx + /Pmp22 + myelinating or Scn7a + /Jun + non‐myelinating SCs (Fig 7C–E, G and H).
Interestingly, biased early SCs (downstream of iSCs), display an overlap of expression of multiple lineage‐specific markers, such as Mpz, Mal, Dhh, Pmp22 and Pou3f1 albeit at variable levels (Fig 7E). This suggests that these SCs are experiencing conflicting gene programs for selecting different fates. These observations are compatible with the classical model of myelination onset, as cells of these three paths are characterised by varying expression of Pou3f1 before the onset of Egr2 expression, which defines the myelinating terminal state. Surprisingly, the path with the highest experimentally validated expression of Pou3f1 led to the terminal neuromuscular SCs, side‐tracking early SCs away from the myelinating fate via an unknown mechanism (Figs 6E and EV4). This is consistent with the reasoning that cells along the adjacent fibroblast‐biased path have less chance of acquiring myelinating or non‐myelinating SC fate, which opens up new interesting domains of research in regards to unveiling the factors determining this fate restriction.
Overall, our analysis revealed a complex system of transcriptional control and gene expression characterising differentiation programs that likely play a role in biasing or priming SC populations. For instance, even though Pou3f1 (coding for OCT6) has been previously reported to play a crucial role in myelination, it additionally emerged as a candidate biasing factor towards terminal neuromuscular junction SCs, suggesting a novel dual role in nerve development.
Schwann cell lineage heterogeneity reflects positional codes
Given the fate bias‐related heterogeneity of a “hub”/SCP population identified in our single‐cell analysis, we hypothesised that one of the critical aspects driving heterogeneity might be the spatial distribution of SCPs in the embryo (Fig 8A–C). If this hypothesis is correct, the differences in downstream fate acquisition might correlate with differences in location‐specific transcriptional programs in “hub”/SCPs.
For example, our bifurcation analysis revealed gene expression programs biasing “hub” cells/SCPs towards sympathoadrenal progenitors (Ascl1 and Phox2b expression) or future mature glia (Ednrb, Moxd1, Postn expression; Fig 6). Additionally, we observed that peripheral glial cells derived from the sympathoadrenal system, gut and developing limbs intermingle in the computed SCP branch towards non‐myelinating SCs (Remak cells) and primed SCs II (Fig 8A). However, glia of sympathoadrenal/enteric locations separated from the branches towards myelinating SC, endoneurial fibroblast and primed SCs I (Fig 8A). This suggests that glia along the visceral nerves (preganglionic axons sprouting from visceral motor neurons and postganglionic axons of the autonomic nervous system) might carry a different positional code as compared to the glia found on sensory and motor nerves that reach the skin or axial muscles. Such positional code could restrict nerve‐associated cells to autonomic neurogenesis and non‐myelinating SCs. In line with this hypothesis, lineage tracing in E13.5 Ascl1 CreERT2 ;R26 TOMATO embryos (traced at E11.5) and E13.5 Phox2b Cre ;R26 TOMATO embryos, revealed two lineages restricted to the sympathetic chain, adrenal gland and paraganglia, and, correspondingly, enteric nervous system (Fig 8D and Appendix Fig S12).
Further examination of cells from E11.5 and later stages revealed correlations of gene expression with the anatomical origin of cells (Appendix Fig S13). For example, Ascl1 and Phox2b expression in glia was limited exclusively to the autonomic and enteric nervous system at E12.5. On the other hand, E12.5 glia sampled from the developing limb exclusively expressed high levels of Nr4a2 (validated using RNAscope®) and increasing levels of Gjc3 (coding for CX29), which was also observed at E14.5, E16.5, E18.5 and perinatal stages (Fig 8E and Appendix Fig S12). This regionalized expression of Nr4a2 coincided with that of Gpr17, Pou3f1 and other Schwann cell markers (Cpm, Itga8; Fig 8E and Appendix Fig S13). Accordingly, we hypothesised that NURR1 (coded by Nr4a2) might be a novel gene in SC fate consolidation or myelination. However, analysis of Nr4a2 −/− newborn mice through immunofluorescence showed no apparent defect in SC distribution on the axons innervating the limbs when compared to Nr4a2 +/+ littermates (Appendix Fig S12D).
Next, OCT6 high cells (encoded by Pou3f1) were predominantly found in developing and postnatal limbs, with an increasing gradient of OCT6 levels from the proximal‐to‐distal part of the peripheral nerves (Fig 8E). These cells were characterised by the signature of terminal neuromuscular SCs as seen by the expression of Cpm, Itga8, Slitrk3, Cspg4 and other markers previously shown enriched in this SC subpopulation (Fig EV4A, B and D, and Appendix Fig S13). As compared to this subgroup of limb‐dwelling glial cells, sympathoadrenal or enteric glia had much lower Gjc3 expression during development, which caught up with expression levels of Gjc3 found in embryonic limbs only postnatally (Appendix Fig S13).
Many genes associated with the early SC lineage are expressed transiently (for instance, Nrp1, Nrp2, Ednra/b and Ngfr with a peak of expression during E11.5–E16.5 followed by progressive downregulation; Appendix Fig S13C). Our analysis revealed several novel transient iSC‐specific genes (Kctd12, Prss12, Slitrk2, Moxd1) that appeared downregulated predominantly in sympathoadrenal and limb‐derived SCs (Appendix Fig S13C and F).
All these observations point to specific spatial and temporal codes in SCPs and SCs welcoming more focused future research lines.
Melanoma, neurofibromatosis and neuroblastoma reactivate the transcriptional signatures of “hub”/SCPs and NC
Plasticity and intra‐tumoral heterogeneity are prominent features of NC‐derived tumours, including neuroblastoma, melanoma and neurofibromatosis (Thomas et al, 2012; Boeva et al, 2017; Grzywa et al, 2017; Monroe et al, 2017). Such heterogeneity and transitions between cell types within tumours may be related to reactivated NC/SCP multipotency. This is consistent with the fact that SCPs give rise to pigment, sympathoadrenal and mesenchymal cells during normal development. To check if neuroblastoma, melanoma and neurofibromatosis tumour cells reactivate developmental cell states, we searched for SCP‐ and the NC‐specific signatures in different types of NC‐derived tumours.
First, we examined whether we could pinpoint the SCP‐like populations in already published single‐cell transcriptomic analyses of human melanoma (Durante et al, 2020). Using CONOS (Barkas et al, 2019). For this, we mapped single‐cell transcriptomic data from human uveal melanoma onto our data set (Appendix Fig S14A and C). We extracted single cells forming a trajectory from a single patient and applied CytoTRACE to this subset to correlate the progression of the tumour cells (Appendix Fig S15A). As expected, we found that the majority of the tumour cells along the entire progression identified by CytoTRACE mapped to the population of embryonic melanocytes (Appendix Fig S15A). At the same time, a significant portion of melanoma cells appeared transcriptionally similar to SCPs and the NC (Appendix Fig S14C).
Next, we performed the same analysis using available bulk transcriptomic data derived from cell lines from type 1 neurofibromatosis patients (Appendix Fig S14B). We found that all samples mapped to the mesenchymal NC branch, showing the high bias of such tumours to this fate.
Finally, we used the same approach to associate single‐cell transcriptomic data from neuroblastoma patients (Dong et al, 2020) with our transcriptional map of the NC lineage. The single‐cell data derived from eight different patients were analysed separately via pagoda2 and scvelo pipeline (Appendix Fig S15B). For each patient, CONOS was employed for developmental milestone and pseudotime values propagation onto the tumour data. While neuroblastoma cells are mostly similar to adrenergic cells from our developmental trajectory (Appendix Fig S14D and E), some tumour cells also mapped onto embryonic SC lineage (Appendix Fig S15C). This might suggest that some malignant cells reactivate the developmental gene expression signatures becoming similar to SCPs and the NC.
Discussion
The term “Schwann cell precursor” (SCP) was coined by Jessen and Mirsky. depicted an NC‐derived cell type associated with the outgrowing nerves at the end of the NC migration (Jessen & Mirsky, 1991). In recent years, the NC‐like multipotency of SCPs has become apparent and attracted significant attention (Joseph et al, 2004; Adameyko et al, 2009; Nitzan et al, 2013; Dyachuk et al, 2014; Kaukua et al, 2014; Uesaka et al, 2015; Espinosa‐Medina et al, 2017; Furlan et al, 2017; Lumb et al, 2018; Kastriti et al, 2019). However, SCPs differ from the migratory NC in several aspects such as nerve association and specific morphology (Jessen & Mirsky, 1991, 1992, 2005), raising the intriguing challenge of identifying the molecular identity and cell state of SCPs. Indeed, SCPs could potentially represent nerve‐associated NCCs or, alternatively, might be a distinct cell type. The shared transcription factor code and common downstream genes have made it difficult to distinguish between these alternative scenarios (Furlan & Adameyko, 2018). For example, SCPs express transcription factors in common with migratory NC, including Sox9, Sox10, FoxD3, Tfap2a/b and Ets1 (Britsch et al, 2001; Paratore et al, 2001; Parkinson et al, 2002; Nitzan et al, 2013; Balakrishnan et al, 2016).
To resolve the conundrum of SCP identity and similarity to the neural crest, we performed a single‐cell transcriptomic analysis of the entire NC lineage starting from the NC initiation until postnatal stages using Smartseq2 protocol, which produced individual transcriptomes of much higher quality as compared to droplet‐based methods. This atlas is available online for in‐depth exploration by the research community (see “Data availability” section). Our results revealed the existence of a “hub” state of transcriptional convergence, where late NCCs and SCPs mix while becoming nerve‐associated and biased towards final fates. Notably, most cells contributing to the “hub” state are SCPs from E10.5 and later stages and express a set of specific markers including Sox8, Itga4 and other genes downregulated in the early migratory crest or differentiating Schwann cells. Consistently, targeting Sox8 in the NC lineage resulted in a deficiency in the nerve‐associated SCPs with a concomitant proportional increase in sympathoadrenal cells. This is consistent with computational predictions, where the “hub” state transited towards SCs and other NC fates, including sympathoadrenal, enteric and melanocytes. Thus, the “hub” cells represent a continuation of the multipotent neural crest in postmigratory and nerve‐associated phases before commitment and differentiation towards definitive cell types. This fits the evolutionary scenario in which proto‐NCCs migrated from the neural tube following sensory and motor nerves in a nerve‐associated state (Ivashkin & Adameyko, 2013).
While visualisation and construction of trees of transcriptional events from individual transcriptomes help to formulate testable hypotheses regarding actual lineage transitions towards terminal states, these may not accurately reflect the actual clonal lineage tree. Based on the generated tree of transcriptional states, there are three possible scenarios of actual NCC lineage transition towards terminal fates. In the first scenario, some NCCs slide through the “hub” state without nerve association to produce differentiated cell types. The second scenario suggests that NCCs do not switch on the “hub” state gene program and instead proceed to the terminal fates while bypassing the “hub” or tunnelling through the “hub.” In the third option, numerous NCCs become nerve‐associated SCPs expressing “hub” genes, followed by the generation of definitive cell types in a nerve‐dependent fashion. The real clonal lineage portrait can be the sum of all three options mixed in different proportions. In any case, the intercalation of the transcriptomes of cells of the “hub”/SCP state between those of NCCs and downstream terminal fates suggests that SCPs are indeed a cellular state functioning as an extension of NCCs in terms of multipotency and expressed genes. The cells of this “extension state” differ from the NCCs in that they use peripheral nerves as their navigation routes and express additional gene expression programs. According to experimental evidence, these additional programs gradually increase in the neural crest‐derived “hub” cells after nerve association and are likely driven by strengthening interactions between the cells and the nerve fibres.
Based on the convergence of SCPs and late NC in the “hub” state, we expected to find similarities in the general logic of cell fate decisions reported previously for the NC. Accordingly, we analysed the fate splits (called “bifurcations”) and found the conserved three‐step model of a fate choice (co‐activation of competing modules, repulsion of competing modules and commitment), as described previously for the murine NCCs at E9.5 (Soldatov et al, 2019).
Recent advancements in single‐cell transcriptomics established incremental fluidity of evolving transcriptional profiles during cell lineage development (Marques et al, 2018; Nowotschin et al, 2019; Mu et al, 2020). While observing the fluidity of gene expression programs, we also attempted to detect rather discrete steps in cell differentiation and developmental dynamics based on the molecular transitions within the expanded NC tree. Indeed, it is useful to define cell types and their developmental progression via understanding multigenic modules (regulons) controlled by a specific gene regulatory network (GRN) subcircuit and corresponding to specific functions. The multigenic modules switch on and off rather discretely, driven by specific transcriptional activators. However, since this is happening in the background of other many programs with overlapping dynamic behaviour, the sum of all transcriptional up‐ and downregulation event results is a smooth incremental change of the overall gene expression during differentiation.
To disentangle this fluidity into a set of overlapping discrete programs that help to separate the “hub” from the early migratory crest and differentiating SCs at the level of regulation, we utilised the approach of elucidating orchestrated gene expression modules‐regulons (Soldatov et al, 2019; Van de Sande et al, 2020). Metaregulons are groups of regulons with co‐regulated or correlated expression profiles. We took advantage of the regulon concept to assist in defining the transient NC, “hub”/SCP and iSC states towards the terminal stages of SC development. Our analysis of regulons revealed how the uninterrupted and smooth NC and SCP differentiation trajectory is divided into discrete steps, which assisted the regulation‐based definition of the “hub.” Furthermore, we discovered that Ets1 metaregulon is specifically active in both emerging NCCs and SCPs before their transition towards mature SCs or commitment to neuronal fates, suggesting its role in multipotency maintenance. Indeed, the time window of this metaregulon coincides with the actual multipotency of the NC and SCP populations, which is evident from reported lineage‐tracing experiments (Dyachuk et al, 2014; Furlan et al, 2017; Kastriti et al, 2019).
After defining the converging transcriptional states of the NC and SCPs based on the discrete activity of identified regulons, we set out to investigate the transition from SCPs towards a diversity of mature Schwann cell types. Our results showed transcriptional and regulatory paths towards myelinating, non‐myelinating, neuromuscular junction terminal SCs and endoneurial fibroblasts. For instance, endoneurial fibroblasts arise from the nerve‐associated Dhh‐expressing cells that lose contact with axons during late embryonic and early postnatal stages of development (Parmantier et al, 1999; Joseph et al, 2004; Sharghi‐Namini et al, 2006). According to our results, the profile of Dhh expression reconciles the current and previous findings and provides the precise molecular identity of progenitors biased towards endoneurial fibroblasts.
SC differentiation is guided by the type and diameter of the nerve fibres (key molecules in this process are NEUREGULIN‐1 type III and endothelin) and possibly other currently unknown spatial signals (Brennan et al, 2000; Taveggia et al, 2005). Following this idea, we attempted to reveal spatial aspects of heterogeneity of SCPs and more advanced glial progenitors. Our results clearly showed that nearly all SCPs belonging to the autonomic nervous system express markers biasing them towards neuronal or neuroendocrine chromaffin fates, such as Phox2b and Ascl1. On the other hand, SCPs and more advanced glial progenitors that cover sensory nerves in the skin, specifically express Nr4a2 nuclear receptor gene. In line with these findings, some degree of spatial heterogeneity of SCPs has been reported previously, as evident from the proximal‐to‐distal gradient of SOX2 in SCPs, which affects the SCP‐to‐melanocyte conversion (Adameyko et al, 2009, 2012; Nitzan et al, 2013).
Finally, the heterogeneity of SCPs might be related to transcriptional states leading to a spectrum of embryonic and adult tumours, including melanoma, neuroblastoma and neurofibromatosis. Furthermore, examples of extra‐cutaneous melanomas and melanoma cells with NC‐like phenotype favour the potential SCP‐dependent origin of some tumour populations (Schatton et al, 2008; Boiko et al, 2010; Civenni et al, 2011; Kaufman et al, 2016; Diener & Sommer, 2021). In the case of neurofibromatosis type 1, the tumour‐initiating cells are located within embryonic nerves and may reflect specific SCP populations or other cells of the NC or SC lineage, as reviewed by Li et al (2020). Since SCPs generate chromaffin and sympathetic cells (Furlan et al, 2017; Kastriti et al, 2019), their transitory states might be essential for understanding the origin and plasticity of malignant cells in neuroblastoma (Dong et al, 2020; Hanemaaijer et al, 2021; Kameneva et al, 2021). To address this, we compared the transcriptional states within the NC lineage with type 1 neurofibromatosis, uveal melanoma and neuroblastoma cell populations. Our results revealed subsets of cells with a “reverted” developmental‐like phenotype, which suggests that heterogeneous tumours might contain NC‐like or SCP‐like malignant cells capable of migration via the nerves and potentially metastasizing to the other parts of the body. These results fit the current trend of discovering the role of developmental genetic modules in tumour evolution, spread and survival, including the dynamic transitions between malignant cell populations under treatment (Chakrabarti et al, 2012; Boeva et al, 2017; Kameneva et al, 2021).
In summary, our results revealed the transcriptional identity and heterogeneity of SCPs and other cells of the NC lineage presented here as an open and accessible high‐resolution atlas (7,000–8,000 genes revealed per cell on average). Based on these computational atlas, experimental validations and functional experiments, we defined “hub”/SCPs as NC‐like cells in a multipotent state with multiple detectable fate biases and additional gene expression programs. These programs provide multipotency and control recruitment of nerve‐associated “hub”/SCP cells to other fates. At last, the comparisons of the single‐cell atlas of the entire embryonic NC and “hub” lineage with pathological states provided methodological examples that can be relevant to questioning the cell of origin for specific NC tumour subtypes or for predicting the mechanisms mediating intra‐tumoral plasticity.
Materials and Methods
Mouse husbandry, strains and genetics
All mouse work was performed under a permit approved by the Ethical Committee on Animal Experiments (Stockholm North committee) of Sweden or the Ethical Committee for Advice and Assessment of Research Projects on Animals of the Medical University of Vienna (Ethik‐Kommission der MedUni Wien zur Beratung und Begutachtung von Forschungsprojekten am Tier) of Austria adhering to Swedish, Austrian and European regulations and guidelines for animal experimentation.
Ascl1::CreERT2 mice were received from The Jackson Laboratory, stock number 012882 (full strain name: Ascl1tm1.1(Cre/ERT2)Jejo/J). Sox10::CreERT2 mice are available from the laboratory of Vassilis Pachnis (The Francis Crick Institute, UK) under a material transfer agreement with the institution. Phox2b‐Cre;R26 TOMATO embryos were received from the Jean‐François Brunet laboratory (D'Autreaux et al, 2011). Ret::CreERT2 mice were received from D. Ginty laboratory (Harvard University, USA; http://www.informatics.jax.org/allele/MGI:4437245). Plp1::CreERT2 were received from U. Suter laboratory (ETH Zurich, Switzerland; http://www.informatics.jax.org/allele/MGI:2663093). R26R YFP and R26R TOMATO mice were received from The Jackson Laboratory (stock number 006148 and 007914, respectively). Wnt1‐Cre mice were received from The Jackson Laboratory, stock number 009107 (full strain name B6.Cg‐Tg(Wnt1‐cre)11Rth Tg(Wnt1‐GAL4)11Rth/J). Isl1‐Cre mice were received from The Jackson Laboratory (stock number 024242). Nr4a2 knockout embryos and postnatal tissue were received from the laboratory of Thomas Perlmann (available through The Jackson Laboratory, stock number 017859).
Tamoxifen‐induced lineage tracing
For all experiments using embryonic tissue, following time‐mating, the day of vaginal plug detection was considered E0.5. In the case of postnatal collections, the day of birth was considered as P0. In the case of inducible lineage tracings (Plp1::CreERT2, Sox10::CreERT2 and Ascl1::CreERT2 strains), tamoxifen (Sigma, Cat No T5648) was dissolved in corn oil (Sigma, Cat No 8267) and delivered via intraperitoneal (i.p.) injection to pregnant females (0.1 mg/g of body weight). For inducible lineage‐tracing experiments intended for tissue to be subjected to single‐cell transcriptomics, tamoxifen was administered 48 h prior to tissue harvesting.
Tissue collection from mice and preparation for downstream analysis
Pregnant females or young pups were sacrificed following isoflurane overdose with cervical dislocation. Embryonic or postnatal tissue was dissected within 15 min of the collection and placed on ice. Dissected tissue was fixed with 4% PFA, 4°C, with mild agitation for 2–16 h depending on the developmental stage. Following washes with 1× PBS, tissue was submerged into 30% sucrose in 1× PBS for cryoprotection for 24–48 h, 4°C before embedding in O.C.T. and snap freezing. Frozen tissue blocks were stored at −20 to −80°C until cryosectioning took place. Cryosections of 14–16 μm were collected onto SuperFrost Plus slides, dried at room temperature and stored at −20°C until further use.
Avian embryos and Sox8 knock down
Embryos
Wild‐type chicken embryos were obtained from Rhode Island Red hens (Sunstate Ranch). Eggs were incubated in humidified incubators at 38°C to the desired stage. Embryos were staged according to Hamburger & Hamilton (1992).
Making Crispr constructs
Crispr constructs were designed and made according to the protocol by Gandhi et al (2021). The plasmid contains a sequence for Cas9, citrine and a guide RNA. The sequence for the guide RNA used in this study to knock down SOX8: 5′ ‐ ATCCACCTTAGCGCCCAGCG – 3′. The control plasmid used contained a synthetic DNA construct not found in the chicken genome in place of the guide RNA: 5′ ‐ GCACTGCTACGATCTACACC – 3′. Cripr plasmids for all experiments were used at a final concentration of 2.5 μg/μl, diluted in EB buffer. To enable visualisation of the injection solution, 0.5 μl of 2% blue food dye was added per 10 μl injection mix.
Verifying Cripr construct
HH4 stage embryos were electroporated in vitro and cultured overnight. The Cripr construct was injected between the epiblast and the membrane on the right half of the area pellucida. Electroporation was achieved using a flat electrode placed above the injected area with the following settings: 5.7 volts, 50 ms pulses, 5 pulses, off for 100 ms between pulses. Embryos were then cultured overnight until HH9‐10 using the EC method (Chapman et al, 2001). The expression pattern of SOX8 was visualised by HCR. Electroporated cells were identified by HCR against citrine. The “Molecular Technologies” protocol for HCR was used.
Embryology
HH10‐11 stage embryos were injected and cultured in ovo. The Crispr plasmid was injected into the neural tube via a capillary needle. The needle was used to pierce the dorsal part of the neural tube at the rostral end of the trunk. The solution was injected until the trunk was filled to the posterior neural pore. Unilateral electroporation of the trunk was achieved using a two‐pronged electrode, with the electrodes placed 4 mm apart, either side of the neural tube with the following settings: 15 volts, 50 ms pulses, 5 pulses, off for 100 ms between pulses. Embryos were then cultured for 3 days in ovo at 38°C until they reached HH24‐25.
Histology
Embryos were fixed in 4% PFA in PB overnight at 4°C, rinsed three times in PBS with 0.3% triton, then washed overnight at 4°C. Embryos were equilibrated in 5% sucrose in PBS for 2–4 h at 4°C then in 15% sucrose overnight at 4°C. Embryos were transferred to melted gelatin and allowed to equilibrate overnight at 38°C. Embryos were then snap frozen using liquid nitrogen and stored at −80°C for at least overnight before sectioning using a cryostat into 16 μm sections. Sections were kept at room temperature overnight prior to immunohistochemistry staining.
Immunohistochemistry
Slides were left in PBS at 42°C for 7 min to remove gelatin, washed in PBS with 0.3% triton‐X100 twice for 15 min at room temperature and blocked (10% donkey serum) for 1 h at room temperature. The following primary antibodies were diluted in block at the following concentrations and kept on slides for 2 days at 4°C: rabbit anti‐Tyrosine Hydroxylase (# AB152, Millipore) 1:500; mouse anti‐Islet (39.4Ds, DSHB) 1:100; rabbit anti‐SOX10 (# HPA068898‐100UL, Sigma) 1:500; mouse anti‐SOX10 (# SC365692, Santa‐Cruz Biotechnology) 1:100; mouse anti‐pH3 (# ab14955, Abcam) 1:500. Slides were then washed three times for 15 min in PBS with 0.3% triton‐X100. The following secondary antibodies were diluted in 10% donkey serum block at the following concentrations and kept on slides overnight at 4°C: Alexa Fluor 647 donkey anti‐rabbit IgG (# A31573, Invitrogen) 1:1,000; Alexa Fluor 647 donkey anti‐mouse IgG (# A31571, Invitrogen) 1:1,000; Alexa Fluor 568 donkey anti‐rabbit IgG (# A10042, Invitrogen) 1:1,000; Alexa Fluor 568 donkey anti‐mouse IgG (# A11037, Invitrogen) 1:1,000; Alexa Fluor 488 donkey anti‐goat IgG (# A11055, Invitrogen) 1:1,000. Slides were then washed two times for 15 min in PBS with 0.3% triton‐X100, one time in PBS for 15 min and for 7 min in PBS with DAPI (1 μg/ml). Slides were rinsed in PBS and mounted using Fluoromount‐G (# 0100–01, Southern Biotech).
Imaging and image processing
Immunostained sections were imaged using a Zeiss Imager MZ with an ApoTome module. Images were processed using the “Fiji” software, using the “Cell Counter” plugin for counting cells. Four to five sections were quantified per embryo from the forelimb axial level, and 2–4 embryos were quantified per condition.
Immunofluorescent staining on mouse tissue
Slides were equilibrated to room temperature, and antigen retrieval was performed using a steam cooker by submerging the slides in 1× Target Retrieval Solution (Dako, S1699) for 20 min. Sections were washed three times in PBS with 0.1% Tween‐20 (1× PBST) and incubated at 4°C, overnight with primary antibodies diluted in 1× PBST. Next, sections were washed in 1× PBST and incubated with secondary antibodies diluted in 1× PBST at room temperature for 1 h, washed three times in 1× PBST and mounted using Mowiöl mounting medium.
Primary antibodies used: Goat anti‐GFP (1:500, Abcam, #ab6662), chicken anti‐GFP (1:500, Aves Labs Inc., #GFP‐1020), chicken anti‐mCherry (1:1,000, EnCor Biotech, #CPCA‐mCherry), goat anti‐SOX10 (1:500, Santa‐Cruz, #sc‐17,342), goat anti‐SOX10 (1:1,000, R&D systems, #AF2864), chicken anti‐NF200 (1:1,000, Abcam, #ab4680), chicken anti‐P0 (1:1,000, Abcam, ab134439), mouse anti‐HUC/D (1:100, ThermoFisher Scientific, #A‐21271), mouse anti‐ISL1 (1:250, DSHB, clone 39.4D5‐s), rabbit anti‐S100B (1:500, Synaptic Systems, #287003), mouse anti‐NEUROFILAMENTS (1:100, DSHB, clone 2H3), mouse anti‐βIII Tubulin (1:1,000, Promega, clone G712A), rabbit anti‐OCT6 (1:500, Abcam, ab272925).
When DAPI (Sigma, D9542) was used, it was applied on the sections simultaneously with the secondary antibodies at a concentration of 0.5 mg/ml. For detection of the primary antibodies, secondary antibodies raised in donkeys and conjugated with Alexa‐488, ‐555 and ‐647 fluorophores were used (1:1,000, Molecular Probes, ThermoFisher Scientific).
RNAscope® in situ hybridization
In situ hybridization using the RNAscope® Fluorescent Multiplex Assay kit and a commercially available probe against mouse Nr4a2 (Cat No. 423351), Itga4 (Cat No. 542901), Serpine2 (Cat No. 435241), Sox8 (Cat No. 454781), Htr3a (Cat No. 411141‐C3), Itga8 (Cat No. 417941), Pou3f1 (Cat No. 436421‐C2), Cpm (Cat No. 1121871‐C3) was performed using cryosections of fixed frozen tissue according to the manufacturer's instructions.
Microscopy
Images were acquired using LSM 780, LSM 880 Zeiss confocal microscopes equipped with 20×, 40× and 63× objectives or a Zeiss Imager MZ with an ApoTome module. Images were acquired in the lsm format and processed with ImageJ for export as tiff files. Figures were compiled using Adobe Photoshop and Illustrator.
Quantification of RNAscope® in situ hybridization
Following export as RGB tiff files, images were cropped by annotated region and loaded onto cellpose for segmentation, using cytoplasm model, SOX10 signal and a diameter parameter of 20 pixels (Stringer et al, 2021). From each segmented cell, we calculated (i) the mean intensity of SOX10 and (ii) the background‐corrected mean intensity of RNAscope®‐detected marker. To filter out nonglial cells, two different approaches were used depending on the collected data in a defined region. When the distribution of the mean intensity of SOX10 per cell presented two clear distributions, the SOX10low population was removed (corresponding to cells biased to sensory fate) using a SOX10 mean intensity threshold. When the region contained a specific population of SOX10low and very high intensity of the RNAscope®‐detected marker, with a high correlation between the two values, these cells were considered as blood cells. These contaminants were removed either by applying k‐means with two clusters on the scaled mean intensities of SOX10 and RNAscope®‐detected marker and discarding the blood cell cluster when successfully captured. A SOX10 mean intensity threshold was employed for unsuccessful clustering results. Mean intensities results were then combined and annotated by location and developmental day.
Statistical analysis and sample size
Sample size was decided according to accepted practices in the field and initial preliminary experiments to estimate variability within the same litter while adhering to the use of the minimum number of animals. Analysis of statistical significance (Fig 4) was performed using GraphPad Prism 7 and the Holm‐Sidak method (α = 0.05; multiple t‐tests, unpaired) or unpaired t‐test with a two‐tailed P‐value. Blinding was not applied, as most validations were performed on wild‐type tissue. In the case of the analysis of Sox8 KD (Fig 4), quantification was performed the same way on all chick embryos.
Single‐cell suspension preparation, fluorescence‐activated cell sorting (FACS) and single‐cell transcriptomic sequencing
Collection and dissection of embryonic and postnatal traced tissue were carried out using a stereotactic microscope equipped with a fluorescent light source to enrich the tissue in relevant cells. Dissected tissue was collected in 1× PBS on ice, and either 0.05% Trypsin/0.02% EDTA or 2 mg/ml collagenase P was used for enzymatic digestion of embryonic or postnatal tissue, respectively, incubated at 37°C for 5–20 min depending on the developmental stage. Following enzymatic digestion, the tissue was triturated with a P‐1000 and then P‐200 pipette until dissociation, and cold 10% FBS was added to quench the enzymes. The cell suspension was spun down, 300 × g, 5 min, 4°C and washed three times with 1× PBS. Finally, the cellular suspension was filtered through a 40 μm‐pore size cell strainer and collected into a FACS tube on ice.
Single TOMATO+ cells were sorted into 384‐well plates prefilled with lysis buffer according to the previously published SmartSeq2 protocol (Picelli et al, 2014) using a BD FACSAria Fusion Cell Sorter B5/R3/V3 system with a three‐laser configuration (488, 633 and 405 nm) and 16 fluorescence detectors. Single‐cell transcriptomic sequencing was performed as previously described (Picelli et al, 2014).
Generation of count matrices, QC and filtering
The single‐cell transcriptome data were generated at the Eukaryotic Single‐cell Genomics facility at Science for Life Laboratory in Stockholm, Sweden. The samples were analysed by first demultiplexing the fastq files using deindexer (https://github.com/ws6/deindexer) using the nextera index adapters and the 384‐well plate layout. Individual fastq files were then mapped to mm10_ERCC genome using the STAR aligner using 2‐pass alignment (Dobin et al, 2013). Reads were filtered for only uniquely mapped and were saved in BAM file format; count matrices were subsequently produced. Estimated count matrices were gathered and converted to anndata python object prior to scanpy processing. Cells with 5 × 104 ≤ transcripts ≥ 6 × 104, 1,000 ≤ genes ≥ 10,000 or ≤ 15% of ERCC reads were kept.
Overview and selection of developing glial trajectories
The analysis of the count matrix was performed using the scanpy and scFates python packages with scFates used for a function detecting overdispersed genes following the approach proposed by pagoda2 R package (Fan et al, 2016). PCA was performed on the scaled count matrix subset by overdispersed genes. A nearest neighbour graph was generated with 30 neighbours and on 30 PC components. 2D embedding and Clusters were then identified from NN graph using Leiden algorithm (Traag et al, 2019) and UMAP, respectively (preprint: McInnes et al, 2020). Cells in clusters expressing Sox10 were kept, as well as linked Isl1 + neurogenic clusters. The subset count matrix was further processed in a similar way at the difference of using 15 neighbours for the NN graph. At this stage, scrublet was run on the filtered count raw matrix, and two Leiden clusters enriched with high doublet scores were removed from the data set. As a final step, Palantir (Setty et al, 2019) was applied to generate diffusion maps with a NN graph with neighbours on the first 50 PCs. The first nine diffusion components were used for generating a new NN graph, with 40 neighbours, followed by PAGA, which was then used as the basis for a final UMAP embedding (Wolf et al, 2019). Using these diffusion components, the count matrix was then imputed using Palantir's function MAGIC. The subset count matrix was used as input for the CytoTRACE pipeline (Gulati et al, 2020; using default parameters).
Cell type assignment and identification of cells assigned to the “hub”
Cell types were assigned by performing gene scoring using the MAGIC‐imputed gene expression known markers (see Appendix Fig S3). Cells were annotated by setting a specific threshold for each cell type. Any cells assigned to two or more cell types were discarded from annotation. “hub” cells were identified as being a member of a Leiden cluster containing more than 80% of unannotated cells. “hub” markers were identified by performing differential gene expression via Wilcoxon rank‐sum test, comparing the “hub” cells to the rest.
RNA velocity
BAM files from each plate were processed using python command‐line velocity tool (La Manno et al, 2018) using run‐smartseq2 command with GENCODE M21 genome and repeat masker annotation files, leading to a loom file for each plate containing spliced and unspliced transcript counts. The loom file was combined and subset to the previously filtered out cells. Using scvelo tool on python (Van de Sande et al, 2020), genes with less than 20 spliced counts or unspliced counts were excluded, and the 2,000 top highly variable genes were kept, from which spliced and unspliced counts were then knn smoothed with 30 neighbours. The smoothed values were used to calculate velocity values. The data set was then used as input to BRIE2 pipeline (Huang & Sanguinetti, 2021) to identify differential momentum genes, using the previously measured CytoTRACE value as a continuous covariate. BRIE2 detected 120 genes displaying differential momentum (FDR < 1 × 10−40), which were used to generate the velocity graph and were computed using default parameters. Extrapolated states were then projected on the UMAP embedding produced during the initial analysis.
SCENIC analysis
The SCENIC pipeline was applied using the python package pySCENIC (Aibar et al, 2017; Van de Sande et al, 2020). Firstly, the normalised count matrix from the pagoda2 pipeline was used as input, combined with a list of known transcription factors (TFs), to generate regulons based on correlation with putative target genes. Next, using the generated adjacency matrix combined with cisTraget databases (mm10 500bpUp100Dw and TSS+/−10kbp), the regulons are refined by pruning targets that do not present an enrichment for a corresponding motif of the TF. Lastly, cells were scored for each regulon with a measure of recovery of target genes from a given regulon. These AUC scores are then used to generate diffusion maps using Palantir (run_diffusion_maps, knn = 40), with the first seven diffusion components used to generate a UMAP embedding. In downstream analyses, the AUC scores of the regulons were weighted according to the log10 fpm expression of the related TF. In such cases, the weight is equal to the log10 fpm value between 0 and 1 and then set to 1 if log10(fpm) > 1.
Pseudotime tree inference
These steps were performed using scFates v0.4.1, a python package built‐in continuity of the crestree R package (Soldatov et al, 2019). The trajectory inference was performed in two stages. A 3,000‐nodes tree was fitted, using SimplePPT approach (Mao et al, 2015) on the diffusion space using tl.tree function (scFates, method = “ppt,” sigma = 0.0005, lambda = 200, metric = “euclidean”). Using the developmental time and CytoTRACE score two roots were selected, indicating the vagal and posterior trunk NC. The tree is composed of a soft assignment matrix R, where each node of the tree contains a value for all cells, each cell has an assignment strength to a node between 0 and 1, with 1 being the closest to the node. Pseudotime value was then generated as a distance on the tree from the selected roots and projected to cells using R assignment matrix.
For the glial trajectory, the first tree only captured nmSC and endoneurial fibroblasts. To capture and justify the existence of the mSC fate, the cells were the first subset and processed via cell rank, which used RNA velocity information weighted with cell connectivities. Detected macrostates using Leiden clustering identified nmSC, mSC and endoneurial Fibroblast states, which were selected as terminal states and for absorption probabilities computation. The results were then converted into a 300 nodes principal tree via scFates, which was used as input the absorption probabilities simplex from cell rank and CytoTRACE values. Finally, in order to capture the tSC population and have a more biologically meaningful tree, these cells were further re‐analysed by fitting a tree on the UMAP embedding, allowing to capture all four cell types.
To facilitate the representation of the lineages, highlight bifurcations and roots and endpoints, we employed a concept of milestones, inspired by Saelens et al (2019). Specifically, milestones are either a tip or a fork of the fitted tree and cells are assigned to the closest milestone in pseudotime distance.
Testing for features associated with the tree
Feature expression was modelled as a function of pseudotime in a branch‐specific manner, using cubic spline regression for each branch independently. This tree‐dependent model is then compared with the unconstrained model using F‐test. P‐values were then corrected for multiple testing, features were considered significant if FDR < 0.0001.
log10(fpm) count matrix was used to test with GAM to model which genes are significantly changing along the whole tree (scFates, tl.test_association, default parameters), with significant genes being fitted using GAM to obtain smoothed trends (scFates, tl.fit, default parameters). Whole tree was also used in combination with SCENIC‐derived AUC score, to detect significantly changing regulon activities. To do so, AUC scores were tested (scFates, test_association, A_cut = 0.025) and fitted (scFates, tl.fit, default parameters) via GAM.
Bifurcation analysis
Branch‐specific genes were first detected via amplitude testing using the following GAM model:
From s(pseudotime): Branch interaction term, P‐values were extracted and then corrected for multiple testing (scFates, tl.test_fork, fdr_cut = 0.1).
Then, each significant gene was tested for its upregulation along the path from progenitor to terminal state, using the linear model . Differentially expressed genes were then assigned between two postbifurcation branches with fdr < 0.05 and defined differences in expression cutoffs, using tl.branch_specific (scFates, cutoffs were specifically set for each bifurcation). Finally, the pseudotime of activation was estimated by separating the trajectory into 10 bins and by calculating the relative expression rate at a specific bin:
Where f(b) is the mean fitted expression at a specific bin, if the rate was higher than a defined threshold, the gene was considered to activate at the pseudotime value of the related bin.
To analyse molecular mechanisms of cell fate biasing, cell composition was approximated by a sliding window of cells along the pseudotime axis, cells were manually selected in order to represent the different steps of differentiation. The local gene–gene correlation reflecting the coordination of genes around a given pseudotime t was defined as a gene–gene Pearson correlation within each window of cells. The local correlation of a gene g with a module was assessed as a mean local correlation of that gene with the other genes comprising the module. Similarly, intra‐module and inter‐module correlations were taken to be the mean local gene–gene correlations of all possible gene pairs inside one module, or between the two modules, respectively (scFates, tl.slide_cors, default parameters).
Neural crest to Schwann cell trajectory analysis and glial heterogeneity
The linear trajectory from posterior neural crest cells to immature Schwann cells was a subset from the main tree using the tree milestones. Pseudotime was robustly calculated by running 100 probabilistic mappings where in each run a cell was assigned to a node by using their assignment values as probabilities (see section “Pseudotime tree inference”). Regulons associated with the trajectory were detected over these 100 mappings using tl.test_association (scFates, A_cut = 0.02) on AUC score matrix from SCENIC pipeline. Next, associated regulons were fitted using tl.fit (scFates, default parameters), and fitted regulons were clustered using tl.cluster using cosine distance metric and 40 neighbours.
Similar to bifurcation analysis, two groups of target genes from Six1(+) and Ets1(+) regulons, respectively, were used for correlation analysis, where cell composition was approximated by a sliding window of cells along the pseudotime axis between iSCs and NCC. The local gene–gene correlation reflecting the coordination of genes around a given pseudotime t was defined as a gene–gene Pearson correlation within each window of cells.
To focus the analysis on the SCPs and Schwann cell population, the tree generated previously (see section “Pseudotime tree inference”) was subset to keep only SCPs, SCs, tSCs and endoneurial fibroblasts. Differential gene expression analysis was performed on the tree milestones using Wilcoxon rank‐sum test from scanpy package via tl.rank_genes_groups (scanpy, method = “Wilcoxon”).
Neural crest and Schwann cell lineage comparison with transcriptomic data from tumours
Comparison with neurofibromatosis type 1 data
Bulk RNAseq data from neurofibromatosis type 1 plexiform neurofibroma‐derived Schwann cells were obtained as a tpm matrix and were log‐transformed from a published data set (Ferrer et al, 2018). Mouse gene names from the SS2 data were converted to human symbols to allow comparison. Both data sets were subsets to keep only common gene names. Pairwise correlation between cells from both data sets was computed using either all common genes or genes presenting a low correlation with cell cycle aspect score identified via pagoda2 pipeline (< 0.1). Bulk data points were projected onto the UMAP embedding of the mouse‐derived SS2 data set by building a kNN graph from the pairwise correlations with five neighbours and by computing a weighted average of both dimension coordinates with the weights of the connectivity between neighbours.
Comparison with uveal melanoma data
Using the raw data from a published study on scRNAseq analysis of human uveal melanoma (Durante et al, 2020), count matrices from all patients were combined and preprocessed with scanpy python package. Filtering was performed as follows: cells having between 200 and 8,000 detected genes, less than 10% of mitochondrial genes and more than 400 UMIs were kept. Filtered data were normalised to a target sum of UMIs of 10e6 and log normalised (base 10). Highly variable genes were detected using cellRanger approach and were used to compute PCA on scaled data. kNN graph was generated with 50 neighbours on the 10 first PCs, followed by UMAP embedding. Upon inspection of Sox10 and Mitf expression, the data were subset into a single trajectory. Following the same strategy as the original paper, we identified tumour cells with MLANA, MITF and DCT expression. We focused on PRAME+ class 2 tumour cells from a single patient and data kept originating from a single patient to avoid batch effect‐related aspects, and cycling cells were removed. Lastly, human gene names were converted to mouse symbols to allow integration. PCA was performed on this subset, and kNN graph was generated with 50 neighbours on the 5 first PCs. Palantir was also applied to the PCA space, and tSNE embedding was generated on the multiscale diffusion space. CytoTRACE was applied to the raw count matrix with default parameters. The melanoma trajectory was reprocessed with pagoda2 pipeline (basicP2proc function, default parameters) and CONOS (Barkas et al, 2019) was performed (k = 15, k.self = 5, space = “PCA,” ncomps = 30) between the melanoma trajectory and the complete mouse‐derived NCC and Schwann cell trajectory. Cell‐type assignment labels from our developmental data set were transferred to the melanoma trajectory. Projection of the cells onto developmental embedding was performed by computing the weighted average of the developmental UMAP coordinates, using the CONOS‐derived connectivities as weights.
Comparison with neuroblastoma
Using the raw data from a published study on scRNAseq analysis of neuroblastoma (Dong et al, 2020), count matrices from all patients were combined and preprocessed with scanpy python package. Filtering was performed as follows: cells having between 200 and 10,000 detected genes, less than 10% of mitochondrial genes and more than 400 UMIs were kept. Filtered data were normalised to a target sum of UMIs of 10e6 and log normalised (base 10). Highly variable genes were detected using the cellRanger approach and were used to compute PCA on scaled data. kNN graph was generated with 30 neighbours on the 30 first PCs, followed by UMAP embedding. Data were then subsetted to the existing published cell‐type annotation and separated according to patients to avoid the batch effect. Lastly, human gene names were converted to mouse symbols to allow integration. PCA was performed on this subset, and kNN graph was generated with 50 neighbours on the 5 first PCs. Palantir was also applied to the PCA space, and tSNE embedding was generated on the multiscale diffusion space. CytoTRACE was applied to the raw count matrix with default parameters. CONOS (Barkas et al, 2019) was performed (k = 15, k.self = 5, space = “PCA,” ncomps = 30) between the neuroblastoma data sets and the complete mouse‐derived NCC and Schwann cell trajectory. Cell‐type assignment labels from our developmental (SmartSeq2) data set were transferred to the melanoma trajectory. Projection of the cells onto developmental embedding was performed by computing the weighted average of the developmental UMAP coordinates, using the CONOS‐derived connectivities as weights.
Comparative validation of terminal Schwann cells and satellite glia populations
Validation of tSC population using published NMJ bulk RNAseq data
Published bulk RNA seq data containing nonmyelinating, synaptic glia of the neuromuscular junction (NMJ; Mapps et al, 2022) was downloaded and processed as follows: fastq files were aligned with STAR tool and count matrices generate via featureCounts using mouse genome annotation gencode M27. Count matrix was then analysed via DESeq2 pipeline, where the covariate to compare was FACS sorting of cells being S100β‐GFP+ or S100β‐GFP+/NG2‐dsRed. Differential gene expression was generated, and on the top of the results were highlighted five main top markers defining tSC in our data set.
Validation of satellite glia using published scRNAseq data set
Published scRNAseq data containing cells from mouse DRG and SCG were downloaded and processed via the kallisto bus pipeline (Bray et al, 2016; Melsted et al, 2021) using mouse genome GRCm38 release 97. Cells with less than 200 genes and more than 7.5% of mitochondrial proportion were filtered out. Highly variable genes were detected via scFates wrapper for pagoda2 over dispersion detection approach. PCA was generated from the scaled count matrix and used for the NN graph with 20 PCs and 30 neighbours. Cluster‐defining satellite glia (Plp1+/Fabp7+) was selected and only cells from DRG were used for subsequent analysis. The same process was applied to that subset, and Leiden clustering generated five clusters that could be easily annotated. Mapping was applied by processing that subset and our developmental data set to CONOS pipeline, which consisted in reprocessing with pagoda2, alignment in PCA space (20 PCs) using 15 neighbours and five self‐neighbours, and propagation of cell‐type assignment labels generated from our developmental data (see section Cell‐type assignment and “hub” cell identification). Furthermore, projection of the cells onto developmental embedding was performed by computing the weighted average of the developmental UMAP coordinates, using the CONOS‐derived connectivities as weights.
Author contributions
Maria Eleni Kastriti: Conceptualization; data curation; formal analysis; supervision; validation; investigation; visualization; writing – original draft; project administration; writing – review and editing. Louis Faure: Resources; data curation; software; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Dorothea Von Ahsen: Formal analysis; validation; investigation; visualization; methodology. Johan Boström: Resources; investigation; writing – review and editing. Thibault Gerald Bouderlique: Validation; investigation; visualization; writing – review and editing. Tatiana Solovieva: Data curation; formal analysis; investigation; visualization; methodology; writing – review and editing. Cameron Jackson: Investigation. Marianne Bronner: Resources; supervision; funding acquisition; methodology; writing – review and editing. Dies Meijer: Writing – review and editing. Saida Hadjab: Resources; funding acquisition; writing – review and editing. Francois Lallemend: Resources; funding acquisition; writing – review and editing. Alek Erickson: Resources; writing – original draft. Marketa Kaucka: Resources; writing – review and editing. Viacheslav Dyachuk: Resources; writing – review and editing. Thomas Perlmann: Resources; writing – review and editing. Laura Lahti: Resources; writing – review and editing. Jan Krivanek: Resources; investigation; writing – review and editing. Jean‐Francois Brunet: Resources; writing – review and editing. Kaj Fried: Writing – review and editing. Igor Adameyko: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Acknowledgements
The authors want to thank the Eukaryotic Single‐Cell Genomics facility (Stockholm, Sweden) and in particular Henrik Gezelius and Anastasios Glaros for excellent sequencing services and customer support. MEK was supported by the Novo Nordisk Foundation (Postdoc fellowship in Endocrinology and Metabolism at International Elite Environments, NNF17OC0026874) and Stiftelsen Riksbankens Jubileumsfond (Erik Rönnbergs fond stipend). LF was supported by Austrian Science Fund DOC 33‐B27. T.G.B. was supported by a Lise Meitner grant from the Austrian Science Fund (M2688‐B28). TS was supported by NIH grant DE027568 to MB. SH is supported by Swedish Research Council, Brain Foundation and StratNeuro. VD was supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075‐15‐2022‐301). TP was supported by the Swedish Research Council (2020‐00884); Knut and Alice Wallenberg's Foundation and Söderberg's Foundation. IA was supported by Paradifference Foundation, The Swedish Cancer Society, Bertil Hållsten Research Foundation, Swedish Research Council, ERC Consolidator and EMBO Young Investigator Grants.
The EMBO Journal (2022) 41: e108780
See also: S Stierli & L Sommer (September 2022)
Data availability
Newly generated single‐cell transcriptomic data and previously published data sets have been uploaded under GSE201257 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201257). Two pagoda2 web applications are available on the following link: https://adameykolab.srv.meduniwien.ac.at/glia_gene_umap/ with the (gene‐based embedding, and https://adameykolab.srv.meduniwien.ac.at/glia_scenic_umap/ with (SCENIC‐based UMAP embedding).
Cropped images used for quantification of the “hub” markers Itga4, Serpine2 and Sox8 following RNAscope® in situ hybridization can be found at DOI 10.6084/m9.figshare.19620102.
Code and data for downstream analysis are found on the following repository: https://github.com/LouisFaure/glialfates_paper.
References
- Abdo H, Calvo‐Enrique L, Lopez JM, Song J, Zhang MD, Usoskin D, El Manira A, Adameyko I, Hjerling‐Leffler J, Ernfors P (2019) Specialized cutaneous Schwann cells initiate pain sensation. Science 365: 695–699 [DOI] [PubMed] [Google Scholar]
- Adameyko I, Lallemend F, Aquino JB, Pereira JA, Topilko P, Muller T, Fritz N, Beljajeva A, Mochii M, Liste I et al (2009) Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 139: 366–379 [DOI] [PubMed] [Google Scholar]
- Adameyko I, Lallemend F, Furlan A, Zinin N, Aranda S, Kitambi SS, Blanchart A, Favaro R, Nicolis S, Lubke M et al (2012) Sox2 and Mitf cross‐regulatory interactions consolidate progenitor and melanocyte lineages in the cranial neural crest. Development 139: 397–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aibar S, Gonzalez‐Blas CB, Moerman T, Huynh‐Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J et al (2017) SCENIC: single‐cell regulatory network inference and clustering. Nat Methods 14: 1083–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002) Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22: 6458–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Belfiore L, Chu TH, Fleming T, Midha R, Biernaskie J, Schuurmans C (2020) Insights into the role and potential of Schwann cells for peripheral nerve repair from studies of development and injury. Front Mol Neurosci 13: 608442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Stykel MG, Touahri Y, Stratton JA, Biernaskie J, Schuurmans C (2016) Temporal analysis of gene expression in the murine Schwann cell lineage and the acutely injured postnatal nerve. PLoS ONE 11: e0153256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkas N, Petukhov V, Nikolaeva D, Lozinsky Y, Demharter S, Khodosevich K, Kharchenko PV (2019) Joint analysis of heterogeneous single‐cell RNA‐seq dataset collections. Nat Methods 16: 695–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch ML, Bronner‐Fraser M, Garcia‐Castro MI (2006) Specification of the neural crest occurs during gastrulation and requires Pax7. Nature 441: 218–222 [DOI] [PubMed] [Google Scholar]
- Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M (1994) Interaction of Endothelin‐3 with endothelin‐B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 79: 1277–1285 [DOI] [PubMed] [Google Scholar]
- Bermingham JR Jr, Scherer SS, O'Connell S, Arroyo E, Kalla KA, Powell FL, Rosenfeld MG (1996) Tst‐1/Oct‐6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev 10: 1751–1762 [DOI] [PubMed] [Google Scholar]
- Bernd P (1985) Appearance of nerve growth factor receptors on cultured neural crest cells. Dev Biol 112: 145–156 [DOI] [PubMed] [Google Scholar]
- Boeva V, Louis‐Brennetot C, Peltier A, Durand S, Pierre‐Eugene C, Raynal V, Etchevers HC, Thomas S, Lermine A, Daudigeos‐Dubus E et al (2017) Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 49: 1408–1413 [DOI] [PubMed] [Google Scholar]
- Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ et al (2010) Human melanoma‐initiating cells express neural crest nerve growth factor receptor CD271. Nature 466: 133–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L (2016) Erratum: near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34: 888 [DOI] [PubMed] [Google Scholar]
- Brennan A, Dean CH, Zhang AL, Cass DT, Mirsky R, Jessen KR (2000) Endothelins control the timing of Schwann cell generation in vitro and in vivo . Dev Biol 227: 545–557 [DOI] [PubMed] [Google Scholar]
- Britsch S, Goerich DE, Riethmacher D, Peirano RI, Rossner M, Nave KA, Birchmeier C, Wegner M (2001) The transcription factor Sox10 is a key regulator of peripheral glial development. Genes Dev 15: 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britsch S, Li L, Kirchhoff S, Theuring F, Brinkmann V, Birchmeier C, Riethmacher D (1998) The ErbB2 and ErbB3 receptors and their ligand, neuregulin‐1, are essential for development of the sympathetic nervous system. Genes Dev 12: 1825–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, Simoes‐Costa M (2016) The neural crest migrating into the twenty‐first century. Curr Top Dev Biol 116: 115–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr MJ, Toma JS, Johnston APW, Steadman PE, Yuzwa SA, Mahmud N, Frankland PW, Kaplan DR, Miller FD (2019) Mesenchymal precursor cells in adult nerves contribute to mammalian tissue repair and regeneration. Cell Stem Cell 24: 240–256 [DOI] [PubMed] [Google Scholar]
- Castro R, Taetzsch T, Vaughan SK, Godbe K, Chappell J, Settlage RE, Valdez G (2020) Specific labeling of synaptic schwann cells reveals unique cellular and molecular features. eLife 9: e56935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Abou‐Antoun T, Vukmanovic S, Sandler AD (2012) Reversible adaptive plasticity: a mechanism for neuroblastoma cell heterogeneity and chemo‐resistance. Front Oncol 2: 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman SC, Collignon J, Schoenwolf GC, Lumsden A (2001) Improved method for chick whole‐embryo culture using a filter paper carrier. Dev Dyn 220: 284–289 [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia‐Ramirez J, Kim S, Toledo‐Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G (1995) REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80: 949–957 [DOI] [PubMed] [Google Scholar]
- Ciment G, Glimelius B, Nelson DM, Weston JA (1986) Reversal of a developmental restriction in neural crest‐derived cells of avian embryos by a phorbol ester drug. Dev Biol 118: 392–398 [DOI] [PubMed] [Google Scholar]
- Civenni G, Walter A, Kobert N, Mihic‐Probst D, Zipser M, Belloni B, Seifert B, Moch H, Dummer R, van den Broek M et al (2011) Human CD271‐positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long‐term growth. Cancer Res 71: 3098–3109 [DOI] [PubMed] [Google Scholar]
- Coulpier F, Le Crom S, Maro GS, Manent J, Giovannini M, Maciorowski Z, Fischer A, Gessler M, Charnay P, Topilko P (2009) Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia 57: 1450–1457 [DOI] [PubMed] [Google Scholar]
- Cox TC, Sadlon TJ, Schwarz QP, Matthews CS, Wise PD, Cox LL, Bottomley SS, May BK (2004) The major splice variant of human 5‐aminolevulinate synthase‐2 contributes significantly to erythroid heme biosynthesis. Int J Biochem Cell Biol 36: 281–295 [DOI] [PubMed] [Google Scholar]
- D'Autreaux F, Coppola E, Hirsch MR, Birchmeier C, Brunet JF (2011) Homeoprotein Phox2b commands a somatic‐to‐visceral switch in cranial sensory pathways. Proc Natl Acad Sci USA 108: 20018–20023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener J, Sommer L (2021) Reemergence of neural crest stem cell‐like states in melanoma during disease progression and treatment. Stem Cells Transl Med 10: 522–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R, Yang R, Zhan Y, Lai HD, Ye CJ, Yao XY, Luo WQ, Cheng XM, Miao JJ, Wang JF et al (2020) Single‐cell characterization of malignant phenotypes and developmental trajectories of adrenal Neuroblastoma. Cancer Cell 38: 716–733 [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Moon RT, Raible DW (1998) Control of neural crest cell fate by the Wnt signalling pathway. Nature 396: 370–373 [DOI] [PubMed] [Google Scholar]
- Dunn KJ, Williams BO, Li Y, Pavan WJ (2000) Neural crest‐directed gene transfer demonstrates Wnt1 role in melanocyte expansion and differentiation during mouse development. Proc Natl Acad Sci USA 97: 10050–10055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin E, Glavieux C, Vaigot P, Le Douarin NM (2000) Endothelin 3 induces the reversion of melanocytes to glia through a neural crest‐derived glial‐melanocytic progenitor. Proc Natl Acad Sci USA 97: 7882–7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante MA, Rodriguez DA, Kurtenbach S, Kuznetsov JN, Sanchez MI, Decatur CL, Snyder H, Feun LG, Livingstone AS, Harbour JW (2020) Single‐cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Commun 11: 496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyachuk V, Furlan A, Shahidi MK, Giovenco M, Kaukua N, Konstantinidou C, Pachnis V, Memic F, Marklund U, Muller T et al (2014) Neurodevelopment. Parasympathetic neurons originate from nerve‐associated peripheral glial progenitors. Science 345: 82–87 [DOI] [PubMed] [Google Scholar]
- El‐Nachef WN, Bronner ME (2020) De novo enteric neurogenesis in post‐embryonic zebrafish from Schwann cell precursors rather than resident cell types. Development 147: dev186619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa‐Medina I, Jevans B, Boismoreau F, Chettouh Z, Enomoto H, Muller T, Birchmeier C, Burns AJ, Brunet JF (2017) Dual origin of enteric neurons in vagal Schwann cell precursors and the sympathetic neural crest. Proc Natl Acad Sci USA 114: 11980–11985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Salathia N, Liu R, Kaeser GE, Yung YC, Herman JL, Kaper F, Fan JB, Zhang K, Chun J et al (2016) Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat Methods 13: 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure L, Wang Y, Kastriti ME, Fontanet P, Cheung KKY, Petitpre C, Wu H, Sun LL, Runge K, Croci L et al (2020) Single cell RNA sequencing identifies early diversity of sensory neurons forming via bi‐potential intermediates. Nat Commun 11: 4175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KJ, McKenzie IA, Mill P, Smith KM, Akhavan M, Barnabe‐Heider F, Biernaskie J, Junek A, Kobayashi NR, Toma JG et al (2004) A dermal niche for multipotent adult skin‐derived precursor cells. Nat Cell Biol 6: 1082–1093 [DOI] [PubMed] [Google Scholar]
- Ferrer M, Gosline SJC, Stathis M, Zhang X, Guo X, Guha R, Ryman DA, Wallace MR, Kasch‐Semenza L, Hao H et al (2018) Pharmacological and genomic profiling of neurofibromatosis type 1 plexiform neurofibroma‐derived schwann cells. Sci Data 5: 180106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzsch M, Schreiner S, Kichko T, Reeh P, Tamm ER, Bosl MR, Meijer D, Wegner M (2010) Sox10 is required for Schwann cell identity and progression beyond the immature Schwann cell stage. J Cell Biol 189: 701–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Marshall CD, Gulati GS, Chinta MS, Nguyen A, Salhotra A, Jones RE, Burcham A, Lerbs T, Cui L et al (2020) Elucidating the fundamental fibrotic processes driving abdominal adhesion formation. Nat Commun 11: 4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlan A, Adameyko I (2018) Schwann cell precursor: a neural crest cell in disguise? Dev Biol 444 (Suppl 1): S25‐S35 [DOI] [PubMed] [Google Scholar]
- Furlan A, Dyachuk V, Kastriti ME, Calvo‐Enrique L, Abdo H, Hadjab S, Chontorotzea T, Akkuratova N, Usoskin D, Kamenev D et al (2017) Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 357: eaal3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Li Y, Tang W, Christensen JB, Urrutia HA, Vieceli FM, Piacentino ML, Bronner ME (2021) A single‐plasmid approach for genome editing coupled with long‐term lineage analysis in chick embryos. Development 148: dev193565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain J, Charnay P (2006) Control of myelination in Schwann cells: a Krox20 cis‐regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep 7: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SA, Uy BR, Bronner ME (2017) Ancient evolutionary origin of vertebrate enteric neurons from trunk‐derived neural crest. Nature 544: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzywa TM, Paskal W, Wlodarski PK (2017) Intratumor and Intertumor heterogeneity in melanoma. Transl Oncol 10: 956–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S et al (2020) Single‐cell transcriptional diversity is a hallmark of developmental potential. Science 367: 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn L, Paratore C, Brugnoli G, Baert JL, Mercader N, Suter U, Sommer L (2000) The Ets domain transcription factor Erm distinguishes rat satellite glia from Schwann cells and is regulated in satellite cells by neuregulin signaling. Dev Biol 219: 44–58 [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL (1992) A series of normal stages in the development of the chick embryo. 1951. Dev Dyn 195: 231–272 [DOI] [PubMed] [Google Scholar]
- Hanemaaijer ES, Margaritis T, Sanders K, Bos FL, Candelli T, Al‐Saati H, van Noesel MM, Meyer‐Wentrup FAG, van de Wetering M, Holstege FCP et al (2021) Single‐cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc Natl Acad Sci USA 118: e2022350118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao YH, Lafita‐Navarro MC, Zacharias L, Borenstein‐Auerbach N, Kim M, Barnes S, Kim J, Shay J, DeBerardinis RJ, Conacci‐Sorrell M (2019) Induction of LEF1 by MYC activates the WNT pathway and maintains cell proliferation. Cell Commun Signal 17: 129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty BL, Monk KR (2017) Unwrapping the unappreciated: recent progress in Remak Schwann cell biology. Curr Opin Neurobiol 47: 131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockman D, Adameyko I, Kaucka M, Barraud P, Otani T, Hunt A, Hartwig AC, Sock E, Waithe D, Franck MCM, Ernfors P, Ehinger S, Howard MJ, Brown N, Reese J, Baker CVH (2018) Striking parallels between carotid body glomus cell and adrenal chromaffin cell development. Dev Biol 444 Suppl 1: S308‐S324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, Yanagisawa M (1994) Targeted and natural (piebald‐lethal) mutations of endothelin‐B receptor gene produce Megacolon associated with spotted coat color in mice. Cell 79: 1267–1276 [DOI] [PubMed] [Google Scholar]
- Hosseinipour M (2017) Functional role of the homeobox transcription factor Six1 in neoplastic transformation of human keratinocytes. PhD Thesis
- Hou L, Panthier JJ, Arnheiter H (2000) Signaling and transcriptional regulation in the neural crest‐derived melanocyte lineage: interactions between KIT and MITF. Development 127: 5379–5389 [DOI] [PubMed] [Google Scholar]
- Huang Y, Sanguinetti G (2021) BRIE2: computational identification of splicing phenotypes from single‐cell transcriptomic experiments. Genome Biol 22: 251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K, Bruhl B, Guillemot F, Olson EN, Ernsberger U, Unsicker K (2002) Development of chromaffin cells depends on MASH1 function. Development 129: 4729–4738 [DOI] [PubMed] [Google Scholar]
- Huber K, Karch N, Ernsberger U, Goridis C, Unsicker K (2005) The role of Phox2B in chromaffin cell development. Dev Biol 279: 501–508 [DOI] [PubMed] [Google Scholar]
- Ivashkin E, Adameyko I (2013) Progenitors of the protochordate ocellus as an evolutionary origin of the neural crest. Evodevo 4: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C, Christen CN, Pereira JA, Somandin C, Baggiolini A, Lotscher P, Ozcelik M, Tricaud N, Meijer D, Yamaguchi T et al (2011) HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat Neurosci 14: 429–436 [DOI] [PubMed] [Google Scholar]
- Jaegle M, Ghazvini M, Mandemakers W, Piirsoo M, Driegen S, Levavasseur F, Raghoenath S, Grosveld F, Meijer D (2003) The POU proteins Brn‐2 and Oct‐6 share important functions in Schwann cell development. Genes Dev 17: 1380–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaegle M, Mandemakers W, Broos L, Zwart R, Karis A, Visser P, Grosveld F, Meijer D (1996) The POU factor Oct‐6 and Schwann cell differentiation. Science 273: 507–510 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (1991) Schwann cell precursors and their development. Glia 4: 185–194 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (1992) Schwann cells: early lineage, regulation of proliferation and control of myelin formation. Curr Opin Neurobiol 2: 575–581 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2005) The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci 6: 671–682 [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (2019) Schwann cell precursors; multipotent glial cells in embryonic nerves. Front Mol Neurosci 12: 69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston AP, Yuzwa SA, Carr MJ, Mahmud N, Storer MA, Krause MP, Jones K, Paul S, Kaplan DR, Miller FD (2016) Dedifferentiated Schwann cell precursors secreting paracrine factors are required for regeneration of the mammalian digit tip. Cell Stem Cell 19: 433–448 [DOI] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer D, Anderson DJ, Morrison SJ (2004) Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 131: 5599–5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, Erickson A, Semsch B, Andersson ER, Ratz M et al (2021) Single‐cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet 53: 694–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastriti ME, Kameneva P, Kamenev D, Dyachuk V, Furlan A, Hampl M, Memic F, Marklund U, Lallemend F, Hadjab S et al (2019) Schwann cell precursors generate the majority of Chromaffin cells in Zuckerkandl organ and some sympathetic neurons in Paraganglia. Front Mol Neurosci 12: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ et al (2016) A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 351: aad2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukua N, Shahidi MK, Konstantinidou C, Dyachuk V, Kaucka M, Furlan A, An Z, Wang L, Hultman I, Ahrlund‐Richter L et al (2014) Glial origin of mesenchymal stem cells in a tooth model system. Nature 513: 551–554 [DOI] [PubMed] [Google Scholar]
- Khudyakov J, Bronner‐Fraser M (2009) Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev Dyn 238: 716–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lonnerberg P, Furlan A et al (2018) RNA velocity of single cells. Nature 560: 494–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin NM, M‐AM T (1974) Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Dev Biol 41: 162–184 [DOI] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J (2005) Analysis of congenital hypomyelinating Egr2Lo/lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci USA 102: 2596–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R, Kang MK, Kim YJ, Yang B, Shim H, Kim S, Kim K, Yang CM, Min BG, Jung WJ et al (2022) CTCF‐mediated chromatin looping provides a topological framework for the formation of phase‐separated transcriptional condensates. Nucleic Acids Res 50: 207–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoczky JA, Robert B, Tabin CJ (2011) Mouse digit tip regeneration is mediated by fate‐restricted progenitor cells. Proc Natl Acad Sci USA 108: 20609–20614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JL, Bonner J, Modrell M, Ragland JW, Moon RT, Dorsky RI, Raible DW (2004) Reiterated Wnt signaling during zebrafish neural crest development. Development 131: 1299–1308 [DOI] [PubMed] [Google Scholar]
- Li S, Chen Z, Le LQ (2020) New insights into the neurofibroma tumor cells of origin. Neurooncol Adv 2: i13–i22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb R, Tata M, Xu X, Joyce A, Marchant C, Harvey N, Ruhrberg C, Schwarz Q (2018) Neuropilins guide preganglionic sympathetic axons and chromaffin cell precursors to establish the adrenal medulla. Development 145: dev162552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Q, Yang L, Goodison S, Sun Y (2015) SimplePPT: a simple principal tree algorithm. Proceedings of the 2015 SIAM International Conference on Data Mining (SDM), 792–800. [Google Scholar]
- Mapps AA, Thomsen MB, Boehm E, Zhao H, Hattar S, Kuruvilla R (2022) Diversity of satellite glia in sympathetic and sensory ganglia. Cell Rep 38: 110328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Väremo L, Giacomello S, Falcão AM, Meijer M, Björklund ÅK et al (2018) Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev Cell 46: 504–517.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy, J , Melville J (2020) UMAP: uniform manifold approximation and projection for dimension reduction. arXiv 10.48550/arXiv.1802.03426 [PREPRINT] [DOI] [Google Scholar]
- Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min KHJ, da Veiga BE, Hjorleifsson KE, Gehring J, Pachter L (2021) Modular, efficient and constant‐memory single‐cell RNA‐seq preprocessing. Nat Biotechnol 39: 813–818 [DOI] [PubMed] [Google Scholar]
- Memic F, Knoflach V, Sadler R, Tegerstedt G, Sundstrom E, Guillemot F, Pachnis V, Marklund U (2016) Ascl1 is required for the development of specific neuronal subtypes in the enteric nervous system. J Neurosci 36: 4339–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsky R, Woodhoo A, Parkinson DB, Arthur‐Farraj P, Bhaskaran A, Jessen KR (2008) Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J Peripher Nerv Syst 13: 122–135 [DOI] [PubMed] [Google Scholar]
- Monroe CL, Dahiya S, Gutmann DH (2017) Dissecting clinical heterogeneity in Neurofibromatosis type 1. Annu Rev Pathol 12: 53–74 [DOI] [PubMed] [Google Scholar]
- Morrison SJ, White PM, Zock C, Anderson DJ (1999) Prospective identification, isolation by flow cytometry, and in vivo self‐renewal of multipotent mammalian neural crest stem cells. Cell 96: 737–749 [DOI] [PubMed] [Google Scholar]
- Mu T, Xu L, Zhong Y, Liu X, Zhao Z, Huang C, Lan X, Lufei C, Zhou Y, Su Y et al (2020) Embryonic liver developmental trajectory revealed by single‐cell RNA sequencing in the Foxa2(eGFP) mouse. Commun Biol 3: 642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataf V, Lecoin L, Eichmann A, Le Douarin NM (1996) Endothelin‐B receptor is expressed by neural crest cells in the avian embryo. Proc Natl Acad Sci USA 93: 9645–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie S, Bronner ME (2015) Dual developmental role of transcriptional regulator Ets1 in Xenopus cardiac neural crest vs. heart mesoderm. Cardiovasc Res 106: 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzan E, Pfaltzgraff ER, Labosky PA, Kalcheim C (2013) Neural crest and Schwann cell progenitor‐derived melanocytes are two spatially segregated populations similarly regulated by Foxd3. Proc Natl Acad Sci USA 110: 12709–12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotschin S, Setty M, Kuo YY, Liu V, Garg V, Sharma R, Simon CS, Saiz N, Gardner R, Boutet SC et al (2019) The emergent landscape of the mouse gut endoderm at single‐cell resolution. Nature 569: 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell M, Hong CS, Huang X, Delnicki RJ, Saint‐Jeannet JP (2006) Functional analysis of Sox8 during neural crest development in Xenopus . Development 133: 3817–3826 [DOI] [PubMed] [Google Scholar]
- Paratore C, Goerich DE, Suter U, Wegner M, Sommer L (2001) Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development 128: 3949–3961 [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur‐Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R et al (2008) C‐Jun is a negative regulator of myelination. J Cell Biol 181: 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Langner K, Namini SS, Jessen KR, Mirsky R (2002) Beta‐Neuregulin and autocrine mediated survival of Schwann cells requires activity of Ets family transcription factors. Mol Cell Neurosci 20: 154–167 [DOI] [PubMed] [Google Scholar]
- Parmantier E, Lynn B, Lawson D, Turmaine M, Namini SS, Chakrabarti L, McMahon AP, Jessen KR, Mirsky R (1999) Schwann cell‐derived desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23: 713–724 [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R (2014) Full‐length RNA‐seq from single cells using smart‐seq2. Nat Protoc 9: 171–181 [DOI] [PubMed] [Google Scholar]
- Reid K, Turnley AM, Maxwell GD, Kurihara Y, Kurihara H, Bartlett PF, Murphy M (1996) Multiple roles for endothelin in melanocyte development: regulation of progenitor number and stimulation of differentiation. Development 122: 3911–3919 [DOI] [PubMed] [Google Scholar]
- Richard L, Vedrenne N, Vallat JM, Funalot B (2014) Characterization of Endoneurial fibroblast‐like cells from human and rat peripheral nerves. J Histochem Cytochem 62: 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SL, Dun XP, Doddrell RDS, Mindos T, Drake LK, Onaitis MW, Florio F, Quattrini A, Lloyd AC, D'Antonio M et al (2017) Sox2 expression in Schwann cells inhibits myelination in vivo and induces influx of macrophages to the nerve. Development 144: 3114–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, Saeys Y (2019) A comparison of single‐cell trajectory inference methods. Nat Biotechnol 37: 547–554 [DOI] [PubMed] [Google Scholar]
- Saito D, Takase Y, Murai H, Takahashi Y (2012) The dorsal aorta initiates a molecular cascade that instructs sympatho‐adrenal specification. Science 336: 1578–1581 [DOI] [PubMed] [Google Scholar]
- Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga‐Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C et al (2008) Identification of cells initiating human melanomas. Nature 451: 345–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner S, Cossais F, Fischer K, Scholz S, Bosl MR, Holtmann B, Sendtner M, Wegner M (2007) Hypomorphic Sox10 alleles reveal novel protein functions and unravel developmental differences in glial lineages. Development 134: 3271–3281 [DOI] [PubMed] [Google Scholar]
- Setty M, Kiseliovas V, Levine J, Gayoso A, Mazutis L, Pe'er D (2019) Characterization of cell fate probabilities in single‐cell data with Palantir. Nat Biotechnol 37: 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharghi‐Namini S, Turmaine M, Meier C, Sahni V, Umehara F, Jessen KR, Mirsky R (2006) The structural and functional integrity of peripheral nerves depends on the glial‐derived signal desert hedgehog. J Neurosci 26: 6364–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes‐Costa M, Bronner ME (2015) Establishing neural crest identity: a gene regulatory recipe. Development 142: 242–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov R, Kaucka M, Kastriti ME, Petersen J, Chontorotzea T, Englmaier L, Akkuratova N, Yang Y, Haring M, Dyachuk V et al (2019) Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364: eaas9536 [DOI] [PubMed] [Google Scholar]
- Sommer L, Ma Q, Anderson DJ (1996) Neurogenins, a novel family of atonal‐related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Mol Cell Neurosci 8: 221–241 [DOI] [PubMed] [Google Scholar]
- Steingrimsson E, Copeland NG, Jenkins NA (2004) Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 38: 365–411 [DOI] [PubMed] [Google Scholar]
- Storer MA, Mahmud N, Karamboulas K, Borrett MJ, Yuzwa SA, Gont A, Androschuk A, Sefton MV, Kaplan DR, Miller FD (2020) Acquisition of a unique mesenchymal precursor‐like blastema state underlies successful adult mammalian digit tip regeneration. Dev Cell 52: 509–524 [DOI] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, Pachitariu M (2021) Cellpose: a generalist algorithm for cellular segmentation. Nat Methods 18: 100–106 [DOI] [PubMed] [Google Scholar]
- Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; PRINCIPAL Investigators (2018) Single‐cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562: 367–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahtakran SA, Selleck MA (2003) Ets‐1 expression is associated with cranial neural crest migration and vasculogenesis in the chick embryo. Gene Expr Patterns 3: 455–458 [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P et al (2005) Neuregulin‐1 type III determines the ensheathment fate of axons. Neuron 47: 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas L, Mautner VF, Cooper DN, Upadhyaya M (2012) Molecular heterogeneity in malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Hum Genomics 6: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma JG, Akhavan M, Fernandes KJ, Barnabe‐Heider F, Sadikot A, Kaplan DR, Miller FD (2001) Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol 3: 778–784 [DOI] [PubMed] [Google Scholar]
- Topilko P, Schneider‐Maunoury S, Levi G, Baron‐Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P (1994) Krox‐20 controls myelination in the peripheral nervous system. Nature 371: 796–799 [DOI] [PubMed] [Google Scholar]
- Traag VA, Waltman L, van Eck NJ (2019) From Louvain to Leiden: guaranteeing well‐connected communities. Sci Rep 9: 5233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uesaka T, Nagashimada M, Enomoto H (2015) Neuronal differentiation in Schwann cell lineage underlies postnatal neurogenesis in the enteric nervous system. J Neurosci 35: 9879–9888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q et al (2020) A scalable SCENIC workflow for single‐cell gene regulatory network analysis. Nat Protoc 15: 2247–2276 [DOI] [PubMed] [Google Scholar]
- Weider M, Starost LJ, Groll K, Kuspert M, Sock E, Wedel M, Frob F, Schmitt C, Baroti T, Hartwig AC et al (2018) Nfat/calcineurin signaling promotes oligodendrocyte differentiation and myelination by transcription factor network tuning. Nat Commun 9: 899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston JA (1963) A radioautographic analysis of the migration and localization of trunk neural crest cells in the chick. Dev Biol 6: 279–310 [DOI] [PubMed] [Google Scholar]
- Weston JA (1970) The migration and differentiation of neural crest cells. Adv Morphog 8: 41–114 [DOI] [PubMed] [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Gottgens B, Rajewsky N, Simon L, Theis FJ (2019) PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 20: 59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CE, Paratore C, Dours‐Zimmermann MT, Rochat A, Pietri T, Suter U, Zimmermann DR, Dufour S, Thiery JP, Meijer D et al (2006) Neural crest‐derived cells with stem cell features can be traced back to multiple lineages in the adult skin. J Cell Biol 175: 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Saint‐Jeannet JP, Klein PS (2003) Wnt‐frizzled signaling in neural crest formation. Trends Neurosci 26: 40–45 [DOI] [PubMed] [Google Scholar]
- Yanfeng W, Saint‐Jeannet JP, Klein PS (2003) Wnt‐frizzled signaling in the induction and differentiation of the neural crest. Bioessays 25: 317–325 [DOI] [PubMed] [Google Scholar]
- Zou D, Silvius D, Fritzsch B, Xu PX (2004) Eya1 and Six1 are essential for early steps of sensory neurogenesis in mammalian cranial placodes. Development 131: 5561–5572 [DOI] [PMC free article] [PubMed] [Google Scholar]