

Abstract

Male breast cancer (MBC) is a relatively rare disease, but emerging data recommend the development of novel therapeutics considering its alarming threats. Compared to female breast cancer (FBC), MBC is reportedly associated with inferior outcomes (poor survival) owing to their late diagnosis and lack of adequate treatment. Treatment typically correlates with FBC, involving surgical removal of the breast tissue along with chemo/hormonal/radiation therapy, the tamoxifen being a standard adjuvant. Considering the distinct immunophenotypic (implying different pathogenesis and progression) differences from FBC, the identification of diagnostics, prognostics, and therapeutics for MBC is highly desirable. In this context, we have analyzed the most deleterious nsSNPs of BRCA2, a human tumor suppressor gene constituting the potential biomarker for tumors including MBC, to predict the structural changes associated with the mutants hampering the normal protein–protein and protein–ligand interactions, resulting in MBC progression. Among 27 nsSNPs confined to 21 rsIDs pertaining to MBC, the 19 nsSNPs constituting 14 rsIDs have been predicted as highly deleterious. We believe that these nsSNPs could serve as potential biomarkers for diagnostic and prognostic purposes and could be the pivotal target for MBC drug discovery. Subsequently, the study highlights the exploration of the key nsSNPs (of BRCA2 associated with the MBC) and its applications toward the identification of therapeutic hit TIP006136 following the homology modeling, virtual screening of 5284 phytochemicals retrieved from the TIPdb (a database of phytochemicals from indigenous plants in Taiwan) database, molecular docking (against native and mutant BRCA2), dynamic simulations (against native and mutant BRCA2), density functional theory (DFT), and molecular electrostatic potential. To the best of our knowledge, this is the first report to use diverse computational modules to investigate the important nsSNPs of BRCA2 related to MBC, implying that TIP006136 could be a potential hit and must be studied further (in vitro and in vivo) to establish its anticancer property and efficacy against MBC.
Introduction
Breast cancer is most often found in women, but men can get breast cancer too. Statically, male breast cancer (MBC) accounts for <1% of all breast cancer cases, but emerging data recommend the early diagnosis and therapeutic development of MBC considering its alarming threats.1,2 As per the American cancer society projection, in the United States, approximately 2710 new cases of MBC will be diagnosed and 530 men will die from breast cancer in the year 2022.3 Epidemiologically, MBC has a similar geographic distribution to female breast cancer, with a higher prevalence in North America, Europe, and lower in Asia.4 Treatment typically correlating female breast cancer, involves surgery to remove the breast tissue along with radiation therapy/hormone therapy/chemotherapy based on the patient’s condition. Estrogen-blocking treatment is the gold standard of adjuvant hormonal therapies with tamoxifen as a standard adjuvant.5 The adverse effects of tamoxifen include but are not limited to headache, nausea, hot flashes, skin rash, fatigue (general), blood clots (deep vein thrombosis or pulmonary embolism), strokes, and increased risk of heart attacks (serious and life-threatening).6,7 Further, tamoxifen treatment leads to sexual dysfunction (decreased libido) in males resulting in poor patient compliance.
The pathogenesis of breast cancer in both men and women is influenced by common risk factors like genetic, hormonal, environmental factors, etc. However, it remains unclear whether the biological behavior and tumor progression associated with MBC parallels that of FBC. A recent study suggests that MBC presents distinct immunophenotypic differences from FBC, implying different pathogenesis and progression. Such differences may play key roles in therapeutic management, warranting different treatment strategies including the discovery of new drugs in comparison to FBCs.8
The genetic factor, particularly the loss-of-function mutations of BRCA1 and BRCA2 (tumor suppressor gene) carry cumulative lifetime MBC risk of 1–5 and 5%, respectively, with BRCA2 mutations occurring more frequently9,10 estimating about 3–40% of males as per population-based studies (see, Table S1, ESI-1). The BRCA2 gene encodes a large protein (3418 amino acids) and is involved in the repair of DNA double-strand breaks.11 Single base alterations in the amino acid sequence of such encoded protein (known as nonsynonymous single-nucleotide polymorphisms, nsSNPs) are linked to various diseases including cancers. This results in an extensive investigation of nsSNPs focusing on the impact of nsSNPs on specific proteins and protein–protein interactions, to provide more insight into the mechanisms by which nsSNPs might cause disease.12 However, due to a lack of genetic information from families carrying BRCA2 mutations, the importance of BRCA2 variants of uncertain significance (VUS) to male breast cancer has yet to be explored.13
In this context, in the present investigation, computational tooling of the deleterious and neutral nsSNPs of BRCA2 has been carried out to predict the structural and functional changes associated with the mutants hampering the normal protein–protein and protein–ligand interactions, resulting in MBC progression. Among 27 nsSNPs confined to 21 rsIDs pertaining to MBC, the 19 nsSNPs constituting 14 rsIDs were predicted as highly deleterious. We believe that these nsSNPs could serve as potential biomarkers for diagnostic and prognostic purposes and could be the pivotal target of MBC drug discovery. Subsequently, the study highlights the exploration of the key nsSNPs and their applications toward the identification of therapeutic novel hit TIP006136 among 5284 phytochemicals retrieved from the TIPdb database (a database of phytochemicals from indigenous plants in Taiwan) employing homology modeling, virtual screening, molecular docking (against native and mutant BRCA2), MD simulations (against native and mutant BRCA2), DFT, and molecular electrostatic potential. This is the first ever report that we are aware of using diverse computational modules to investigate the important nsSNPs of BRCA2 related to MBC, implying that TIP006136 could be a potential therapeutic hit for MBC.
Results
Retrieval of BRCA2 Gene Information and Its nsSNPs
The BRCA2 gene’s structure and functional information were obtained from the UniprotKB database using the query P51587 (BRCA2 HUMAN). Corresponding BRCA2 protein reported having the experimental 3D structures with less than 100 amino acids (AAs) chain length, which lack the mutation positions. The InterPro domain database reported three domains, BRCA2_OB_1 (IPR015187: 2670–2795), BRCA2_OB_3 (IPR015188: 3052–3185), and Tower domain (IPR015205: 2831–2872), in the region with amino acids sequence from position 2670–3185 which were associated with a BRCA2 mutation and corresponds to MBC. The natural variants with their corresponding nsSNPs were retrieved from the NCBI_dbSNP database, having a minor allele frequency (MAF) value <0.0001. 21 rsIDs associated with 27 nsSNPs were analyzed for the prediction of their neutral and deleterious effects on the structure and function of the BRCA2 gene.
Evaluation of the Functional Impact of Coding nsSNPs Using PredictSNP Web Server
Six of the most effective tools for predicting how a mutation would affect protein function are combined in the PredictSNP 1.0 web service to create a consensus classifier (see, Table S9, ESI-1). Among the analyzed 21 rsIDs and its corresponding 27 mutated positions, 14 rsIDs constituting 19 nsSNPs were identified to be deleterious with a better confidence level. The nsSNPs with rsIDs rs80359062, rs41293513, rs41293511, rs80359071, rs28897751, rs80359082, rs45580035, rs80359187, rs80359198, rs28897758, and rs28897759 were shown to be highly deleterious in all of the tools, whereas the other nsSNPs represented to be a combination of deleterious and neutral. These 19 deleterious mutations were used for converting the native BRCA2 into mutated BRCA2 protein at concerning amino acid positions.
Analysis of Protein Stability Change on Mutation in Native Hub Gene
The consensus results of mCSM, SDM, and DUET scores in kcal/mol represent the stability and de-stability of the protein structure upon mutation. From the DUET server, the values of 14 nsSNPs (V2728L, K2729N, G2793R, K2950N, A2951T, R3052W, G3076E, D3095E, L3101Q, L3101P, L3101R, I3103M, M3118T, and N3124S) could be able to predict the destabilization of the BRCA2 protein structure (see, Table S10, ESI-1).
Template-Based Homology Modeling and Model Evaluation of BRCA2
Since the corresponding BRCA2 protein experimental 3D structure has less than 100 amino acids (AAs) chain length and it lacks the mutation positions, the homology modeling was performed considering the AA sequence of length 2670–3185. The 3D structure of the Human BRCA2 was modeled using MODELLER 9.23 and the best model was selected based on the Discrete Optimized Protein Energy (DOPE) scores (Figure 1A). The obtained model was further validated using the PROCHECK server to get the Ramachandran plot. The plot reflected 86.8% residues found in most favored regions, 12.3% residues in additional allowed regions, 0.6% residues in generously allowed regions, and 0.2% residues in disallowed regions (Figure 1B). To get the overall quality of the modeled protein, the ProSA server has been used. The quality score (Z-score) was scaled for validating the BRCA2 from X-ray crystallography, structural NMR, and hypothetical predictions. The structure indicated a high-quality model in comparison to the known protein structures (Z-score −4.19) (Figure 1C).
Figure 1.

Validation of Human BRCA2. (A) The 3D structure of homology modeled BRCA2. (B) X-ray crystallography structure of BRCA2 by Ramachandran plot. (C) (Prosa Z-score plot of the BRCA2.
Prediction of Binding Site
The consensus results of the web servers depicted the residues SER2887; ARG2888, LEU2890, GLN2893, TYR2905, LYS2971, LEU2972, ARG2973, VAL2975, LYS2980, GLU2981, LYS2982, SER2984, ILE2986, SER2988, TRP2990, SER3016, LYS3017, SER3018, LYS3019, SER3020, GLU3021, ARG3022, ALA3023, ASN3024, GLN3026, ILE3048, TYR3049, GLN3050, GLN3066, PRO3067, SER3068, CYS3069, SER3070, GLU3071, LYS3104, TRP3106, ASN3124, LEU3125, GLN3126, TRP3127, ARG3128, PRO3129, THR3137, LEU3138, PHE3139, GLY3141, ASP3142, PHE3143, and SER3144 taking part in active site formation.
Virtual Screening of Phytochemicals (PCs) Enlisted in the TIPdb
Virtual screening is a computational tool routinely utilized to quest libraries of molecules having promising binding affinity to a drug target. In the present study, we carried out the virtual screening of phytochemicals (PCs) enlisted in the TIPdb, a database of phytochemicals from indigenous plants in Taiwan, using PyRx software.14−18 The screening reflected 14 (TIP001922, TIP002754, TIP003223, TIP003237, TIP003461, TIP005092, TIP006136, TIP008902, TIP008979, TIP009431, TIP010010, TIP011681, TIP012106, and TIP012114) out of 5284 PCs with better binding affinity (cutoff value: −10.0 kcal/mol) against modeled BRCA2.
These compounds were further analyzed with AutoDock 4.2 (Figure 2) (Table S11, ESI-1).
Figure 2.
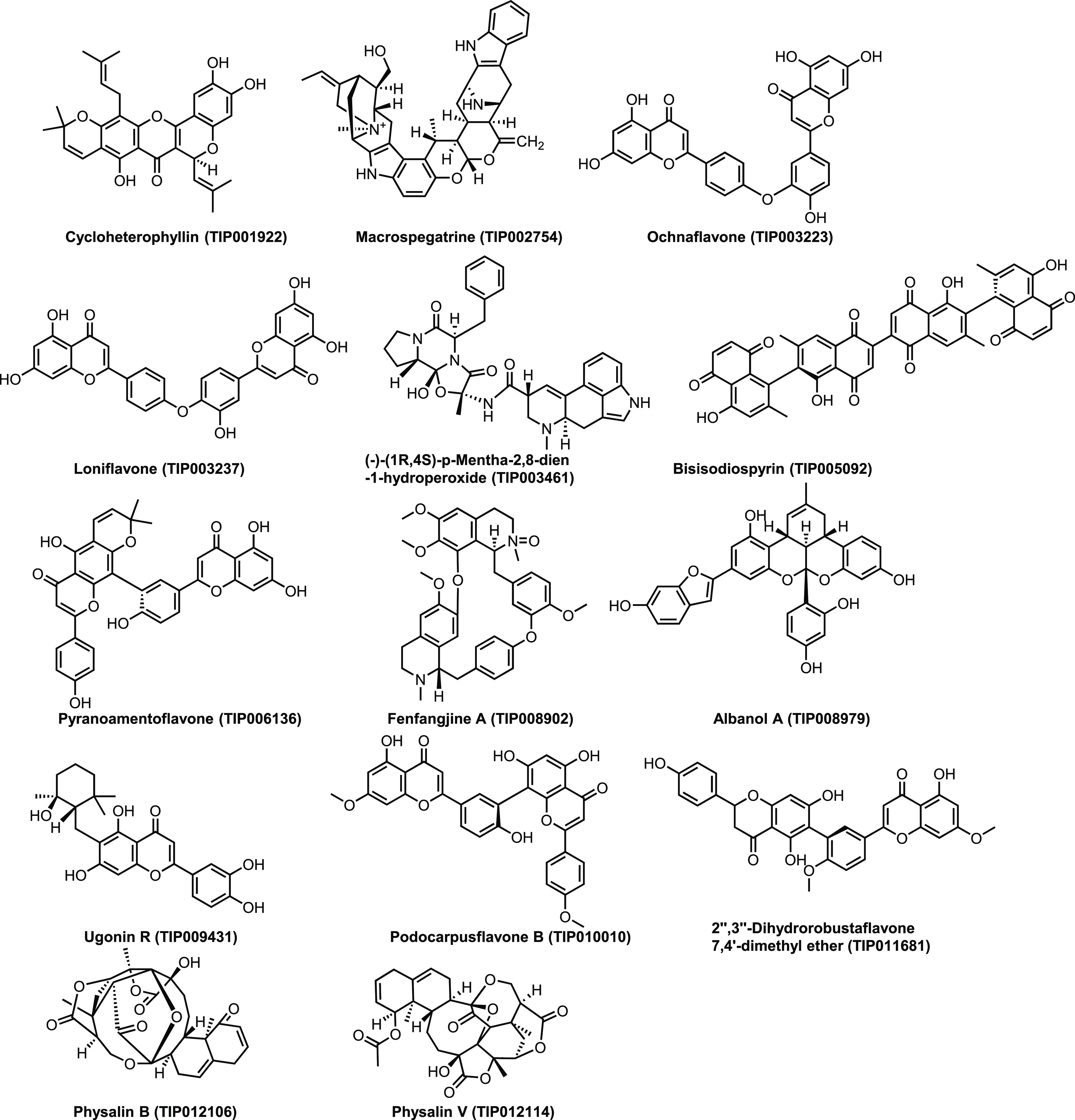
14 Phytochemicals selected from TIPdb based on virtual screening by PyRx.
Molecular Docking
The binding free energies of modeled native BRCA2 with all of the 14 selected PCs and tamoxifen (reference drug) interaction complexes are presented in Table S12, ESI-1. The best orientations with higher binding energies, H-bonds, and ligand efficiency were taken into consideration for intermolecular interactions analysis out of the 10 conformations obtained for each docking complex. BRCA2–TIP006136 docking complex (Figure 3B) represented the highest binding energy (−9.51 kcal/mol) followed by the BRCA2–TIP003461 complex (Figure 3C). The binding energy for the BRCA2–Tamoxifen complex was found to be −5.72 kcal/mol (Figure 3A). The TIP006136 and Tamoxifen were further docked against 19 mutant BRCA2 protein (built by modifying the amino acid of the native BRCA2 according to the positions of SNPs) (see Tables S13 and S14, ESI-1). R3052W–Tamoxifen and D2723H–TIP006136 complex depicted the highest binding affinity with −6.3 and −9.62 kcal/mol among all of the 19 complexes each.
Figure 3.
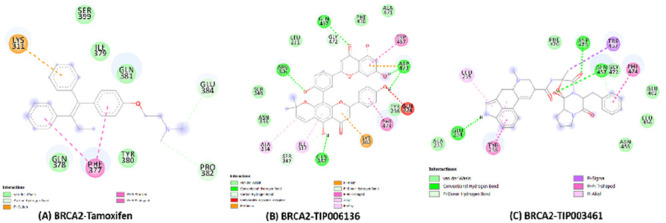
Intermolecular hydrogen bonding, electrostatic, and hydrophobic interactions formed between (A) BRCA2–Tamoxifen complex, (B) BRCA2–TIP006136 complex, and (C) BRCA2–TIP003461 complex. The images are drawn using BIOVIA Discovery Studio 20.1 Visualizer.
Quantum Chemical Calculation Using DFT
For TIP006136 and Tamoxifen, quantum computation was used to examine the molecular descriptors such as HOMO and LUMO, gap energy, and dipole moment (see, Table S15, ESI-1). Based on lowest band energy gap (ΔE = ELUMO – EHOMO, 9.675 kcal/mol), TIP006136 displayed higher reactivity (molecular interaction with protein) compared to Tamoxifen (ΔE = 11.416 kcal/mol) (Figure 4). Tamoxifen and TIP006136 were further suggested for MD simulation against both native and mutant BRCA2 using the combined results of molecular docking and DFT investigations.
Figure 4.

(A) LUMO and HOMO plots of TIP006136 with higher reactivity and low bandgap energy. (B) Tamoxifen’s LUMO and HOMO plots showed less reactivity and larger bandgap energy. Red color denotes positive electron density, whereas blue color denotes negative electron density.
Molecular Electrostatic Potential
(Tables S16–S23, ESI-1) show the results of optimized atomic coordinates, Zero Differential Overlap (ZDO) and Mulliken atomic charges, bond length, and bond angles of TIP006136 and tamoxifen, respectively. The geometric convergence curve of TIP006136 and tamoxifen was apparent in the energy form reduction; the lowest energies observed were −144344.6938 kcal/mol (Table S22, ESI-1) and −91813.0594 kcal/mol (Table S23, ESI-1) indicating that TIP006136 and tamoxifen could be stable at this point and able to interact with BRCA2 native and mutant proteins (Figures 5 and 6).
Figure 5.

Geometry convergence curve of (A) TIP006136 and (B)Tamoxifen.
Figure 6.

Electrostatic potential (ESP) mapped electron density surface of (A) TIP006136 and (B)Tamoxifen in opaque.
Molecular Dynamic (MD) Simulation: Trajectory Analysis
When it comes to predicting and evaluating the physical movements of atoms and molecules in the context of interactions between macromolecular structure and function, MD is a powerful computational tool of priority. For a predetermined period of time, the atoms and molecules are let to interact, reflecting the intricate “evolution” of the system.19 In the present study, GROMACS simulation package was used for nanosecond (ns)-scale MD simulations of all seven systems (BRCA2: Apo state and Holo states, native BRCA2–Tamoxifen complex: Holo1; native BRCA2–TIP006136 complex: Holo2; mutated R3052W–Tamoxifen complex: Holo3; mutated R3052W–TIP006136: Holo4; mutated D2723V–TIP006136: Holo5; mutated D2723H–TIP006136: Holo6). To evaluate the system’s stability and behavior in a dynamic environment, the backbone root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), intermolecular interactions, and principal component analysis (PCA) were aligned from the resultant MD trajectories.
The RMSD profile of the backbone atoms, which was plotted for 100 ns, was used to determine the dynamic stability of all seven systems (Apo, Holo1 to Holo6) (Figure 7A). The RMSD graph constituting of backbone atoms depicted a stable trajectory after 50 ns of simulation (Figure 7A) for all of the Holo states except Holo2 (∼0.9 to ∼1.75 nm) compared to the Apo state. A stable RMSD value between ∼0.75 to ∼1.0 nm (Holo1), ∼0.8 to ∼ 1.0 nm (Holo3), ∼0.75 to ∼0.8 nm (Holo4), ∼0.7 to ∼0.8 nm (Holo5), and ∼0.8 to ∼1.0 nm (Holo6) was observed during the simulation period of 50–100 ns. This depicts that the protein can be more stabilized by TIP006136 by changing its conformation. Moreover, the simulation of the mutated position R3052W reflected stronger interactions with TIP006136 as indicated by its restricted deviations. The graph also reflects higher deviations in Holo2 and restricted deviations in Holo4 in comparison to other Holo states, revealing native state offers higher deviations than mutated states.
Figure 7.

Conformational stability of the BRCA2 protein in the Apo and Holo states during the course of a 100 ns MD simulation. (A) BRCA2’s backbone-RMSD. (B) BRCA2’s C-RMSF profile. (C) BRCA2 radius of gyration (Rg) profile. (D) Total energy of the Holo1–Holo6 states and BRCA2 (Apo) throughout 100 ns of MD simulations. Black, red, green, blue, yellow, magenta, and cyan lines represent the Apo and Holo1–Holo6 states, respectively. RMSD, root mean square deviation; RMSF, root mean square fluctuation.
The variation of residues using RMSF was used to further validate the results of RMSD. The mobility of different residues was observed using RMSF plots (Figure 7B). Overall, Holo2 and Holo3 state showed higher fluctuations compared to other Holo states, which might be due to interaction with TIP006136 and tamoxifen during the course of the simulation. The amino acid residues between 3085–3100 and 2945–2960 of Holo2 and Holo3 states exhibited greater deviations in their Cα atoms in comparison to other regions, which may be due to the interaction of TIP006136 with BRCA2 protein. Holo4 represented restricted fluctuations compared with other states. The results of RMSF are well aligned with RMSD.
Rg vs time graphs were plotted to examine the compactness of all of the states. The Rg in the apo state varied from ∼2.77 to ∼3.45 nm, Holo2 ranged from ∼2.6 to ∼3.7 nm, whereas other Holo states represented close compactness with reference to Apo and Holo2 throughout the MD simulations particularly Holo4 and Holo5 with Rg ranging from ∼3.125 to ∼3.5 nm and ∼3.0 to ∼3.5 nm, respectively (Figure 7C). The mobility of residues in Holo states is lesser than in the Apo state, as seen by the energy map (Figure 7D), which is well backed up by the RMSF analysis.
Certain solvent exposure of amino acids is mediated by hydrophobic interactions. The exposed surface area directly correlates with the frequency of these interactions between the solvent and the core protein residues. In comparison to its Apo form, the available solvent surface in Holo states is smaller, as seen by the SASA graph (Figure 8A). The results of the SASA analysis revealed that the binding of TIP006136 and tamoxifen altered the hydrophilic and hydrophobic interaction regions. This alteration could potentially change the orientations of the protein surface due to the amino acid residue shift from the accessible area to the buried region. The Holo4 state’s SASA graphs depicted SASA with ∼285 to ∼310 nm2, which was less than Holo2 (∼270 to ∼308 nm2), and Holo3 state (∼275 to ∼312 nm2) during 50–100 ns of MD simulations. The SASA analysis reflected Holo4 to get less exposed to the solvent compared to other Holo states.
Figure 8.

(A) Analysis of the Apo and Holo states of the BRCA2 protein by solvent-accessible surface (SASA). (B) H-bond deviation played a role in the interaction. (C) The cloud is a representation of the trajectory eigenvector projection (EV1 and EV2). (D) BRCA2’s motion in its Apo and Holo modes projected along its first two main eigenvectors in phase space (EV1 and EV2) during 100 ns MD simulations. Black, red, green, blue, yellow, magenta, and cyan lines, respectively, show the Apo and Holo1–Holo6 states.
MD Simulation: Hydrogen-Bond Analysis
Intermolecular hydrogen bonds of all of the Holo states were tracked using the gmx_hbond tool of GROMACS (Figure 8B). A variable number of intermolecular hydrogen bonds were represented by the simulation of all holo states throughout the simulation. Holo5 and Holo6 represented the maximum no. of H-bonds in comparison to other states. In the case of Holo5 and Holo6, five and three H-bonds with an average number of H-bonds per time frame ∼1.851 out of 565100 possible with an atomic distance of 2.43 nm and ∼0.931 H-bonds per time frame out of 566605 possible with an atomic distance of 2.91 nm was noted, respectively. Tyr236, Asn455, Gln457, and Arg304 were the four H-bond-forming residues that broke during simulations of Holo5, but afterward, novel five H-bonds (Lys302, Asp473, and Phe474), van der Waals, and hydrophobic contacts were compensated for this (Figure 9).
Figure 9.

(A) Intermolecular hydrogen bonds, electrostatic connections, and hydrophobic contacts in the D2723V–TIP006136 complex were found in pre-MD simulations. (B) Various interactions (hydrogen bonds, electrostatic connections, and hydrophobic contacts) were still present in post-MD simulations. BIOVIA Discovery Studio 20.1 Visualizer was used to get these images.
Likewise, the residues Tyr236, Lys302, Glu402, Asn355, Ser347, and Cys400 contributed to hydrogen bonding (six H-bonds) with TIP006136 in Holo6 (Figure 10), which were broken during MD simulations and formed two novel H-bonds (Gln457, Gln357), but it did not compensate with H-bond forming residue Ser347. This reflects the potentiality of Ser347 as a crucial residue in boosting the BRCA2 binding. With an average of ∼0.955 H-bonds per time frame out of 565858 possible with an atomic distance of ∼2.90 nm, Holo2 represented two H-bonds, whereas in Holo4, ∼1.929 H-bonds per time frame out of 563608 possible with an atomic distance of ∼2.81 nm, represented one H-bond. The residues Arg304, Gln457, Asp473, and Gln357 were found to accomplish hydrogen bonding (Five H-bonds) with TIP006136 in Holo2, which were broken during the course of simulations and formed one novel H-bond (Asn355). The residue Asp473 (H-bond) was found to be intact through H-bond (Figure 11). The results suggest that the compound, TIP006136 exhibits its potentiality against targeted protein during post-MD simulation. Moreover, the residues Asp473, Glu402, Lys348 were found to contribute to hydrogen bonding (three H-bonds) with TIP006136 in Holo4 which were broken during the course of MD simulations and formed one novel H-bond (Gln224) (Figure 12).
Figure 10.

(A) Intermolecular hydrogen bonds, electrostatic connections, and hydrophobic contacts between the D2723H–TIP006136 complex were found in pre-MD simulations. (B) Various interactions (hydrogen bonds, electrostatic connections, and hydrophobic contacts) were still present in post-MD simulations. BIOVIA Discovery Studio 20.1 Visualizer was used to get these images.
Figure 11.

(A) Intermolecular hydrogen bonds, electrostatic connections, and hydrophobic contacts between the BRCA2–TIP006136 complex were found in pre-MD simulations. (B) Various interactions (hydrogen bonds, electrostatic connections, and hydrophobic contacts) were still present in post-MD simulations. BIOVIA Discovery Studio 20.1 Visualizer was used to get these images.
Figure 12.

(A) Intermolecular hydrogen bonds, electrostatic connections, and hydrophobic contacts between the R3052W–TIP006136 complex were found in pre-MD simulations. (B) Various interactions (hydrogen bonds, electrostatic connections, and hydrophobic contacts) were still present in post-MD simulations. BIOVIA Discovery Studio 20.1 Visualizer was used to get these images.
The BRCA2 protein’s atomic density maps for the Apo and Holo1 to Holo6 states are shown in Figure 13A–G. The Apo state of proteins had the highest atom density, measuring 16.6 nm–3, followed by the Holo states, which had densities of 20.7 nm–3 (Holo1), 22.8 nm–3 (Holo4), 23.9 nm–3 (Holo5), 28.1 nm–3 (Holo2), 30.9 nm–3 (Holo6), and 33.4 nm–3 (Holo3). The result was well aligned with large oscillation in Rg in Apo which displayed that the protein might be experiencing a significant structural transition. The consequences of these molecular changes were clearly observed in the atomic density distribution plot. Compared to other Holo and Apo states, the density distribution in the Holo3 and Holo6 states depicted a considerable alteration. Additionally, the Apo state has a higher atomic density distribution than Holo states (16.6 nm–3). It further indicates that Apo state has more flexibility than Holo states. The overall flexibility of various states of BRCA2 protein was further examined by the trace of the diagonalized covariance matrix of the Cα atomic positional fluctuations.
Figure 13.

Number density plot of BRCA2: (A) BRCA2: Apo, (B) BRCA2–Tamoxifen: Holo1, (C) BRCA2–TIP006136: Holo2, (D) R3052W–Tamoxifen: Holo3, (E) R3052W–TIP006136: Holo4, (F) D2723V–TIP006136: Holo5, and (G) D2723H–TIP006136: Holo6.
MD Simulation: Principal Component Analysis (PCA)
The trace values of the backbone atoms’ covariance matrix served as a restriction on and a determinant of Apo and Holo flexibility during each simulation protocol. The trajectory projections from PC1 and PC2 (Figure 8C), which were closely matched with RMSF, captured the movement of Apo and Holo states in phase space (Figure 7B). The trace values of Apo, Holo1, Holo2, Holo3, Holo4, Holo5, and Holo6 were 847.42, 463.289, 675.149, 412.762, 334.76, 276.326, and 392.822 nm2, respectively, confirming an overall increase in flexibility in the Apo and Holo2 state compared to other Holo states. The lower trace values supported the overall decrease in Holo5 and Holo4 states than other Holo states.
The vectorial representation of the individual components displayed the motion direction. The initial vectors display the majority of the internal motions, whereas EV1 and EV2 depict the majority of the overall movements. Eigenvalues showed steep slopes when they were plotted against eigenvectors (Figure 8D). The “cross-correlation matrix” of the Cα displacement revealed that the residues in the “BRCA2” protein experience both negatively (blue shades) and positively (red shades) linked motions (Figure 14A–G), which supports the “BRCA2” protein’s random movement.
Figure 14.

Comparative study of cross-correlation matrices of backbone atoms of (A) BRCA2: Apo, (B) BRCA2–Tamoxifen: Holo1, (C) BRCA2–TIP006136: Holo2, (D) R3052W–Tamoxifen: Holo3, (E) R3052W–TIP006136: Holo4, (F) D2723V–TIP006136: Holo5, and (G) D2723H–TIP006136: Holo6 during 100 ns. The movement’s range is depicted via different colors in the graph panel. Red shades indicate a positive correlation, whereas blue shades indicate an anticorrelation.
The investigation of the free energy landscape (FEL) revealed that the Gibbs free energy value ranges from 0 to 10 kJ/mol, 0 to 9.81 kJ/mol, 0 to 7.87 kJ/mol, 0 to 9.59 kJ/mol, 0 to 8.17 kJ/mol, 0 to 9.26 kJ/mol, and 0 to 8.88 kJ/mol for Apo (Figure 15A), Holo1 (Figure 15B), Holo2 (Figure 15C) Holo3 (Figure 15D), Holo4 (Figure 15E), Holo5 (Figure 15F), and Holo6 (Figure 15G), respectively. The Holo2 state and Holo4 have an energetically more pleasant transition from one conformation to another, which is why Holo2 showed lower energy followed by Holo4 than Apo and other Holo states. The MD simulations show that TIP006136’s binding to BRCA2 (Native/Holo2) and TIP006136’s binding to R3052W (Mutant/Holo4) raise the global minima (blue regions) of BRCA2, suggesting that these complexes may be more pleasant thermodynamically.
Figure 15.

Free energy landscape analysis of (A) BRCA2: Apo, (B) BRCA2–Tamoxifen: Holo1, (C) BRCA2–TIP006136: Holo2, (D) R3052W–Tamoxifen: Holo3, (E) R3052W–TIP006136: Holo4, (F) D2723V–TIP006136: Holo5, and (G) D2723H–TIP006136: Holo6. Higher Gibbs free energy (blue regions) presents an unfolded state of BRCA2, and lower Gibbs free energy (red regions) explains a folded state of BRCA2.
Discussion
MBC is an uncommon disease but possesses an alarming threat considering its elevated risk.1−3 Substantially, it is more common in families associated with BRCA2 germline mutations than BRCA1.5,20 The hub gene identification through STRING and WebGestalt protein–protein interaction network analysis revealed BRCA2 as hub gene which is well aligned with the experimental evidence.21,22 The drug association study using the WebGestalt server revealed mitomycin, hydroxyurea, and progesterone is effective against BRCA2-mediated MBC. However, based on the clinical evidence and practices, tamoxifen is a well-established adjuvant for the treatment of MBC.5,23 Thus, we chose tamoxifen as the reference drug for further study despite the reported adverse effects including life-threatening thromboembolism and its compliance in males (decreased libido). This in turn invites a worldwide quest to discover new hit/lead as a therapeutic candidate for the MBC.6,7
Missense (amino acid altering) mutations in cancer-risk genes make the clinical assessment of genetic testing results too difficult.24 Such missense mutations, intronic variants, in-frame deletions, and insertions, generally known as VUS or unclassified variants, in the BRCA2 gene, are yet to be discovered. Most of these mutations are rare; hence, there is a scarcity of genetic information from families that are susceptible to MBC risk.25 Thus, it has been challenging to determine the impact of numerous rare BRCA2 missense mutations on the MBC risk.25
The complete mechanism by which an SNP resulting a phenotypic/genotypic change is found to be mysterious to date. However, the advancement in the high-throughput computing tooling and in silico analysis facilitates the prediction of the genotypic and phenotypic effects of nsSNPs on physicochemical and structural-functional properties of the concerned BRCA2 protein and associated cancer biology. The current investigation was focused to explore the effect of rare variants on the function of BRCA2 gene that have been found to be major risk factors in MBC. The PredictSNP webserver explored 19 nsSNPs to be deleterious confined to 14 rsIDs. There are only a few experimental reports that show the association between deleterious nsSNPs (rs80359062, rs45580035, rs80359082, rs80359187, rs80359198, rs80359204) and MBC. In this investigation, these nsSNPs were validated through computational approach which are well collated with the existing reports.13,25 These variants could be pivotal in identifying disease-associated genes as potential biomarkers. Therefore, efforts were made to validate the nsSNPs that can modify the structure, function, and expression of the BRCA2 gene to complement this finding.
Four out of 27 nsSNPs, G2728D (rs80359071), D2723G (rs41293513), T2722R (rs80359062), and D3095E (rs80359198), were already analyzed in MBC patients of various ethnicities that were found to be favorable for causality.26 The missense VUS located in BRCA2 was counseled and tested genetically in southwest Germany and found that N3124I mutation was pathogenic. And, among seven families tested, it was noted that in one of the families this mutation corresponds to the MBC.27
The above-mentioned five mutations, investigated by Easton et al., and Surowy et al., were also found to be deleterious in our study which states that our in silico investigations are up to the mark in proving the consistency. Similarly, genetic analysis of D2723H (rs41293511), V2728I (rs28897749), and A2951T(rs11571769) in MBC patients in the USA revealed that D2723H was a harmful mutation, whereas V2728I and A2951T were neutral.28 Thus, the experimental evidence well coincided with the results obtained from our computational approach.
Furthermore, the SNPs such as L2792P (rs28897751), L3101R (rs28897758), N3124I (rs28897759), D2723H (rs41293511), D2723A (rs41293513), K2950N (rs28897754), and T3013I (rs28897755) were reported to be deleterious by Rajasekaran et al.29 Karchin et al. used the Protein Likelihood Ratio and Medical genetics method for computational validation and identification of the deleterious effect of the variants like D2723G (rs41293513), K2729N (rs80359065), G2748D (rs80359071), K2950N (rs28897754), D2723H (rs41293511), V2728I (rs28897749), and E2856A (rs11571747) in BRCA2.24 Among all, D2723G, G2748D, and D2723H were reported to be deleterious. Guidugli et al. had reported over 13 pathogenic mutations from BRCA2 through homology-directed DNA break repair (HDR) functional assay.13 These studies of Karchin et al. and Guidugli et al. were well aligned with our findings. Moreover, we analyzed the effect of these 19 deleterious nsSNPs through molecular docking studies of TIP006136 and Tamoxifen against mutated positions followed by MD simulations
The docking results of Tamoxifen against 19 mutated positions of BRCA2 represented R3052W with the highest binding energy (see, Table S14, ESI-1), whereas the docking of TIP006136 with the above-said positions represented D2723H with the highest binding energy (see, Table S13, ESI-1). Both these mutation positions are reported to be deleterious, which was at par with the existing reports.13,25,30 To evaluate the effect of nsSNP at the same position on the drug–target complex, Holo5 (D2723V-rs41293513) and Holo6 (D2723H-rs41293511), the simulation studies were carried out to explore the impact of amino acid properties during the course of MD simulation. Further validation of the docked complexes was carried out through MD simulations which represented Holo4 (R3052W–TIP006136 complex) and Holo6 (D2723H–TIP006136 complex) with stable conformations, revealing potential anticancer activity of TIP006136. The results were well aligned with the reported evidence that states these mutations favor cancer causality.13,25,30,31 The PC analysis and FEL also reflected a decrease in flexibility and lower Gibb’s free energy in Holo4 and Holo6 states. Taking all together the outcomes of various computational approaches and existing experimentally validated results, it is confident enough in stating that the above-said mutated positions may play a vital role in diagnostic, prognostic, and therapeutics for MBC. Further, TIP006136 could be a potential hit and must be studied further (in vitro and in vivo) to establish its anticancer property and efficacy against MBC.
Conclusions
In summary, we have analyzed the most deleterious nsSNPs of BRCA2 to predict the structural and functional changes associated with the mutants hampering the normal protein–protein and protein–ligand interactions, resulting in MBC progression. Among 27 nsSNPs confined to 21 rsIDs pertaining to MBC, the 19 nsSNPs constituting 14 rsIDs were predicted as highly deleterious. Among these, the current investigation explored the four novel mutations (G2793R-rs80359082, G3076E-rs80359187, I3103M-rs80359204) that are neither experimentally nor computationally reported to be deleterious. Further, for the first time, the study validates the experimental reported mutations (R3052W-rs45580035, D3095E-rs80359198, and T2722R-rs80359062) to be deleterious. We believe, these nsSNPs could serve as potential biomarkers for diagnostic and prognostic purposes and could be the pivotal target of MBC drug discovery. Further, the study highlights the exploration of the key nsSNPs (of BRCA2 associated with the MBC) and its applications toward the identification of therapeutic hit TIP006136 among 5284 phytochemicals retrieved from the TIPdb and recommends further in vitro and in vivo investigations to establish its anticancer property and efficacy against MBC.
Materials and Methods
Mining of Genes Associated with MBC from NHGRI-EBI GWAS Catalog
The NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/) offers a detailed, searchable, visualizable, and freely usable SNP–trait association database that can be conveniently combined with other tools. As of now, only two GWAS22,32 were conducted to identify the genomic risk variants in MBC of European ethnicity reporting about eight genes. The “Male Breast Carcinoma” disease search has been performed for the retrieval of the mapped gene using GWAS Catalog. This study resulted in One trait EFO_0006861 comprising 2 studies and 10 associations from discrete genomic locations of the human genome. The collected mapped genes were noted for establishing protein–protein network for the identification of hub gene responsible for male breast carcinoma.
Construction of Protein–Protein Interaction Network for Identification of Hub Gene
STRING (https://string-db.org/) is a database of known and anticipated protein–protein interactions. These interactions encompass both direct (physical) and indirect (functional) connections; they result from computational prediction, dissemination of information among species, and interactions gathered from the other (primary) database.33 The protein–protein interactions associated with MBC along with the hub gene identification were performed using the string database. The eight mapped genes retrieved from GWAS catalog and 71 genes reported in the literature were selected for the formation of the protein–protein network. In addition, the genes with significantly enriched biological processes were further selected for network formation for identifying hub gene.
Disease, Drug Association, and Gene Ontology Functional Analysis Using Webgestalt Server
″WebGestalt”34 (http://www.webgestalt.org/) is intended for practical genomic, proteomic, and large-scale genetic studies that continuously produce large numbers of gene lists (e.g., differentially expressed gene sets, co-expressed gene sets, etc.). Disease Association, Drug Association, Pathway common and GO analysis of hub gene was performed using WebGestalt. GO analysis was used to functionally annotate a total of 79 gene transcripts that comprise biological processes, molecular functions, and cellular component.
Retrieval of BRCA2 Gene Information and Its SNPs
The BRCA2 gene information was retrieved from the UniprotKB Database (https://www.uniprot.org/) with ID P51587 (BRCA2_HUMAN) for further studies. The PDB IDs reported in the UniprotKB database represented the protein structure with less than 100 amino acids that does not cover the mutated positions of BRCA2. The region with amino acids sequence from position 2670–3185 was reported to have the domains BRCA2_OB_1, BRCA2_OB_3, and Tower domain as per InterPro domain database which were involved in the progression of MBC. Natural variants consisting of nsSNPs with MAF values <0.0001 were chosen from the dbSNP and UniprotKB database. The mutation positions with respective rsIDs nsSNPs were analyzed in a PredictSNP for identifying the neutral and deleterious mutations.
Evaluation of the Functional Impact of Coding nsSNPs Using PredictSNP Web Server
PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp/) employs an unbiased evaluation of eight proven prediction tools: MAPP, nsSNP Analyzer, PANTHER, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, and SNAP, using a benchmark dataset including over 43,000 mutations.35 21 rsIDs constituting of 27 nsSNPs were analyzed by PredictSNP server, out of which 19 were found to be deleterious and 8 of them were neutral. Further analysis of the 19 deleterious SNPs was performed using various tools.
Analysis of Protein Stability Change on Mutation in Native Hub Gene
After implementing the 27 mutations (nsSNPs) in the identified hub gene, the DUET server (http://biosig.unimelb.edu.au/duet/) was utilized to analyze protein stability.36 The stability of mutant proteins was determined using the mCSM, SDM, and DUET scores in kcal/mol.
Template-Based Homology Modeling and Model Evaluation of BRCA2
The three-dimensional (3D) structure of BRCA2 (constituting amino acid sequence 2670–3185) was predicted by homology modeling using MODELLER 9.23.37 The UniprotKB database was used to acquire the BRCA2 protein sequence, and BLASTp38 was used to find appropriate templates for constructing 3D models. The consensus results retrieved from the above-mentioned tools were finally carried out for identifying the best templates with PDB IDs 1MIU_A, 1IYJ_B, and 1MJE_A for target template alignment and model building. Finally, the model with the lowest DOPE score was retained for more structural refinement. Using the Build action feature of BIOVIA DS 20.1 Visualizer, mutant BRCA2 protein structures were built by modifying the amino acid of the native type according to the positions of SNPs. Sidechain refinement was performed using WHATIF39 webserver. Using the PROCHECK40 tool, the resulting optimized model’s overall quality was assessed. The overall quality of the model was assessed using the Protein Structure Analysis (ProSA)41 tool; the resulting Z-score showed that this protein’s model was superior to all other proteins examined using X-ray crystallography and nuclear magnetic resonance imaging (NMR).
Prediction of Binding Site
BRCA2’s structure and active site were investigated using the Computed Atlas of Surface Topography of Proteins (CASTp),42 Grid-based HECOMi finder (GHECOM),43 and DEPTH44 tool. The consensus results were taken into consideration.
Retrieval of Drug-like Molecules from Taiwan Database
TIPdb14 is an accessible and systematic database containing antitubercular, anticancer, and antiplatelet PCs which are indigenous in Taiwan. The chemical structures in this database have been specially picked and may represent a valuable resource for QSAR and high-throughput screening of prospective anticancer candidates. All of these 5284 compounds have the unique property of adhering to the Lipinski rule of five. Virtual screening against modeled BRCA2 protein was performed using PCs from this database.
Virtual Screening and Molecular Docking
A total of 5284 compounds from TIPdb were virtually screened against BRCA2 using PyRx Python prescription 0.845 software. For the molecular docking study, AutoDock 4.246 was used, which is widely distributed public domain molecular docking software. The TIPdb compound as well as Tamoxifen were docked with the native type and 19 deleterious mutated positions (T2722R, D2723A, D2723G, D2723H, D2723V, G2748D, L2792P, G2793R, K2950N, T3013I, G3076E, D3095E, I3103M, L3101P, L3101Q, L3101R, N3124I, N3124S, and R3052W) of BRCA2. Space provided for docking was as follows: x-centering: 24.818, y-centering: 85.335, and z-centering: 11.816, based on grid dimensions. The resulting docked poses were chosen after giving due consideration to binding energy, ligand efficiency, and the intermolecular H-bond. The BIOVIA DS 20.1 Visualizer was used to evaluate ligand–protein interactions. Significant binding interactions with TIPdb compound against both native and mutant BRCA2 protein were explored for further analysis.
Quantum Chemical Calculation Using DFT
Using the concept from DFT, a quantum computational study was performed to know about the reactivity and efficacy of the screened PC and Tamoxifen. The Becke, three-parameter, LeeYang-Parr (B3LYP) correlation function of (DFT)47 was used to investigate the reactivity and efficiency of one TIPdb compound (TIP006136) and Tamoxifen with anticancerous activity against BRCA2 employing the lowest unoccupied molecular orbital (LUMO) energy and the highest occupied molecular orbital (HOMO) energy in a DFT-based study. To calculate, the energy ORCA Program version 5.0.248 was utilized, and inputs are generated using Avogadro 1.2.0n. For the TIPdb compound and Tamoxifen, the electronic energy, frontier HOMOs, LUMOs, gap energy, and dipole moment were measured.
Molecular Electrostatic Potential
Molecular electrostatic potential calculations were performed using the Argus Lab 4.0.1 software49 by performing semiempirical parameterized Model three (PM3) diminution. Electrostatic potential surfaces are useful in computer-assisted drug design because they help comprehend electrostatic interactions between macromolecules and drugs. Different inhibitors and substrates can be compared using these surfaces. Argus Lab 4.0.1 software was used to conduct an electrostatic potential-mapped electron density surface and conformational study of TIPdb compound and Tamoxifen. The sites of the molecule that would be vulnerable to nucleophilic and electrophilic attack were shown on an electrostatic potential-mapped electron density surface. The molecule’s minimal energy was anticipated by the geometry convergence curve.
Molecular Dynamics (MD) Simulations
GROMOS 54A7 force-field50 in the GROMACS suit (version 2020.3) MD simulation package was used to analyze the Apo (BRCA2: Apo; protein only) and Holo states (BRCA2–Tamoxifen complex: Holo1; BRCA2–TIPdb compound complex: Holo2; R3052W–Tamoxifen complex: Holo3; R3052W–TIPdb compound complex: Holo4; D2723V–TIPdb compound complex: Holo5; D2723H–TIPdb compound complex: Holo6) to understand the dynamic behavior, binding mode, and specificity of these inhibitors with BRCA2 and its mutated positions. According to the docking analysis, Holo1 had a lower binding affinity than Holo2 and Holo6 represented the highest binding affinity among all Holo states. To investigate the inhibitor specificity, dynamic behavior, and manner of binding activity of the aforementioned states, MD simulations were further processed. The application “pdb2gmx” from the GROMACS package was used to create the topology file. For energy minimization, the steepest descent approach with a tolerance of 1000 kJ/mol was utilized to release the competing interactions. For 1000 picoseconds (ps) in the first phase, a constant number of particles, volume, and temperature (NVT) ensemble was used to equilibrate the temperature by constraining the positions of the backbone atoms. In the second phase, a constant number of particles, pressure, and temperature (NPT) ensemble was used to equilibrate the pressure. Periodic boundary conditions (PBC) with constant temperature were used to set an MD time period of 100 ns for both the apo and holo states. To analyze the resulting trajectories that are built into GROMACS, Visual MD (VMD 1.9.1) was employed. Using the functions gmx_rmsd, gmx_rmsf, gmx gyrate, gmx_tenergy, and gmx_sasa, the RMSD, RMSF, Rg, total energy, and SASA were examined. Utilizing the gromacs package’s densmap tool, the density map was created to understand the atomic density, atomic orientation, and distribution of BRCA2 protein; we performed density distribution analysis of the molecular coordinates of each state during MD simulations.
Principal Component Analysis (PCA)
Using the gmx_covera and gmx_aneig tools in line with the software package of GROMACS protocol, PCA was performed to achieve the coordinated motions in the complex state of BRCA2 (Holo1, Holo2, Holo3, Holo4, Holo5, and Holo6) and Apo. A set of eigenvectors and eigenvalues were obtained after diagonalizing and computing the covariance matrix that represented the concerted motion of the molecules. FEL was performed to demonstrate the Gibbs free energy values.
For the data analysis of MD simulations, all 2D plots were graphed using GRaphing Advanced Computation and Exploration (GRACE 5.1.23) (https://www.its.hku.hk/services/research/hpc/software/grace).
Acknowledgments
D.K. acknowledges the different funding agencies (Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Gov. of India, DST-SERB, Gov. of India [Ramanujan Fellowship, File No. SB/S2/RJN-135/2017, Start-up Grant, SRG/2020/000658], and CSIR-HRDG, Gov. of India (File No. 02(0456)21/EMR-II).
Glossary
Abbreviations Used
- MBC
male breast cancer
- FBC
female breast cancer
- nsSNP
nonsynonymous single-nucleotide polymorphism
- VUS
variants of uncertain significance
- GWAS
genome-wide association studies
- GO
gene ontology
- AAs
amino acids
- MAF
minor allele frequency
- PCs
phytochemicals
- DFT
density functional theory
- ZDO
zero differential overlap
- MD
molecular dynamic
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- Rg
radius of gyration
- SASA
solvent-accessible surface area
- PCA
principal component analysis
- FEL
free energy landscape
- PM3
parameterized model three
- HOMO
highest occupied molecular orbital
- LUMO
lowest unoccupied molecular orbital
- ED
essential dynamics
- ns
nanoseconds
- ED
essential dynamics
- PBC
periodic boundary conditions
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c03851.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Gov. of India. DST-SERB, Gov. of India CSIR-HRDG, Gov. of India.
The authors declare no competing financial interest.
Supplementary Material
References
- Fox S.; Speirs V.; Shaaban A. M. Male Breast Cancer: An Update. Virchows Arch. 2022, 480, 85–93. 10.1007/s00428-021-03190-7. [DOI] [PubMed] [Google Scholar]
- Gaddam S.; Heller S. L.; Babb J. S.; Gao Y. Male Breast Cancer Risk Assessment and Screening Recommendations in High-Risk Men Who Undergo Genetic Counseling and Multigene Panel Testing. Clin. Breast Cancer 2021, 21, e74–e79. 10.1016/j.clbc.2020.07.014. [DOI] [PubMed] [Google Scholar]
- Key Statistics for Breast Cancer in Men. https://www.cancer.org/cancer/breast-cancer-in-men/about/key-statistics.html (accessed March 28, 2022).
- Weiss J. R.; Moysich K. B.; Swede H. Epidemiology of Male Breast Cancer. Cancer Epidemiol. Prev. Biomarkers 2005, 14, 20–26. 10.1158/1055-9965.20.14.1. [DOI] [PubMed] [Google Scholar]
- Fentiman I. S.; Fourquet A.; Hortobagyi G. N. Male Breast Cancer. Lancet 2006, 367, BMT32. 10.1016/S0140-6736(06)68226-3. [DOI] [PubMed] [Google Scholar]
- Fentiman I. S. Male Breast Cancer: A Neglected Disease. Breast Cancer Manag. 2019, 8, BMT32. 10.2217/bmt-2019-0020. [DOI] [Google Scholar]
- Wibowo E.; Pollock P. A.; Hollis N.; Wassersug R. J. Tamoxifen in Men: A Review of Adverse Events. Andrology 2016, 4, 776–788. 10.1111/andr.12197. [DOI] [PubMed] [Google Scholar]
- Muir D.; Kanthan R.; Kanthan S. C. Male Versus Female Breast Cancers. Arch. Pathol. Lab. Med. 2003, 127, 36–41. 10.5858/2003-127-36-MVFB. [DOI] [PubMed] [Google Scholar]
- Tai Y. C.; Domchek S.; Parmigiani G.; Chen S. Breast Cancer Risk Among Male BRCA1 and BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 2007, 99, 1811. 10.1093/jnci/djm203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breast Cancer Linkage Consortium T. Cancer Risks in BRCA2 Mutation Carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. 10.1093/jnci/91.15.1310. [DOI] [PubMed] [Google Scholar]
- Roy R.; Chun J.; Powell S. N. BRCA1 and BRCA2: Important Differences with Common Interests. Nat. Rev. Cancer 2012, 12, 372. 10.1038/nrc3181-c3. [DOI] [Google Scholar]
- Yates C. M.; Sternberg M. J. E. The Effects of Non-Synonymous Single Nucleotide Polymorphisms (NsSNPs) on Protein-Protein Interactions. J. Mol. Biol. 2013, 425, 3949–3963. 10.1016/j.jmb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Guidugli L.; Pankratz V. S.; Singh N.; Thompson J.; Erding C. A.; Engel C.; Schmutzler R.; Domchek S.; Nathanson K.; Radice P.; Singer C.; Tonin P. N.; Lindor N. M.; Goldgar D. E.; Couch F. J. A Classification Model for BRCA2 DNA Binding Domain Missense Variants Based on Homology-Directed Repair Activity. Cancer Res. 2013, 73, 265–275. 10.1158/0008-5472.CAN-12-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. C.; Wang C. C.; Chen I. S.; Jheng J. L.; Li J. H.; Tung C. W. TIPdb: A Database of Anticancer, Antiplatelet, and Antituberculosis Phytochemicals from Indigenous Plants in Taiwan. Scientific World J. 2013, 2013, 1–4. 10.1155/2013/736386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung C. W.; Lin Y. C.; Chang H. S.; Wang C. C.; Chen I. S.; Jheng J. L.; Li J. H. TIPdb-3D: The Three-Dimensional Structure Database of Phytochemicals from Taiwan Indigenous Plants. Database 2014, 2014, bau055. 10.1093/database/bau055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Garcia De Lomana M.; Friedrich N. O.; Kirchmair J. Characterization of the Chemical Space of Known and Readily Obtainable Natural Products. J. Chem. Inf. Model. 2018, 58, 1518–1532. 10.1021/acs.jcim.8b00302. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Sharma N.; Muddassir M.; Rahman Q. I.; Dwivedi U. N.; Akhtar S. Structure-Based Pharmacophore Modeling, Virtual Screening and Simulation Studies for the Identification of Potent Anticancerous Phytochemical Lead Targeting Cyclin-Dependent Kinase 2. J. Biomol. Struct. Dyn. 2021, 1–18. 10.1080/07391102.2021.1936178. [DOI] [PubMed] [Google Scholar]
- Gupta M. K.; Vadde R. In Silico Identification of Natural Product Inhibitors for γ-Secretase Activating Protein, a Therapeutic Target for Alzheimer’s Disease. J. Cell. Biochem. 2019, 120, 10323–10336. 10.1002/jcb.28316. [DOI] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorlacius S.; Olafsdottir G.; Tryggvadottir L.; Neuhausen S.; Jonasson J. G.; Tavtigian S. V.; Tulinius H.; Ögmundsdottir H. M.; Eyfjörd J. E. A Single BRCA2 Mutation in Male and Female Breast Cancer Families from Iceland with Varied Cancer Phenotypes. Nat. Genet. 1996, 13, 117–119. 10.1038/ng0596-117. [DOI] [PubMed] [Google Scholar]
- Couch F. J.; Farid L. M.; DeShano M. L.; Tavtigian S. V.; Calzone K.; Campeau L.; Peng Y.; Bogden B.; Chen Q.; Neuhausen S.; Shattuck-Eidens D.; Godwin A. K.; Daly M.; Radford D. M.; Sedlacek S.; Rommens J.; Simard J.; Garber J.; Merajver S.; Weber B. L. BRCA2 Germline Mutations in Male Breast Cancer Cases and Breast Cancer Families. Nat. Genet. 1996, 13, 123–125. 10.1038/ng0596-123. [DOI] [PubMed] [Google Scholar]
- Maguire S.; Perraki E.; Tomczyk K.; Jones M. E.; Fletcher O.; Pugh M.; Winter T.; Thompson K.; Cooke R.; Trainer A.; James P.; Bojesen S.; Flyger H.; Nevanlinna H.; Mattson J.; Friedman E.; Laitman Y.; Palli D.; Masala G.; Zanna I.; Ottini L.; Silvestri V.; Hollestelle A.; Hooning M. J.; Novaković S.; Krajc M.; Gago-Dominguez M.; Castelao J. E.; Olsson H.; Hedenfalk I.; Saloustros E.; Georgoulias V.; Easton D. F.; Pharoah P.; Dunning A. M.; Bishop D. T.; Neuhausen S. L.; Steele L.; Ashworth A.; Garcia Closas M.; Houlston R.; Swerdlow A.; Orr N. Common Susceptibility Loci for Male Breast Cancer. JNCI J. Natl. Cancer Inst. 2021, 113, 453. 10.1093/jnci/djaa101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S. H.; Buzdar A. U.; Hortobagyi G. N. Breast Cancer in Men. Ann. Intern. Med. 2002, 137, 678–687. 10.7326/0003-4819-137-8-200210150-00013. [DOI] [PubMed] [Google Scholar]
- Karchin R.; Agarwal M.; Sali A.; Couch F.; Beattie M. S. Classifying Variants of Undetermined Significance in BRCA2 with Protein Likelihood Ratios. Cancer Inform. 2008, 6, CIN.S618. 10.4137/CIN.S618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia D. J.; Agarwal M. K.; Pankratz V. S.; Deffenbaugh A. M.; Pruss D.; Frye C.; Wadum L.; Johnson K.; Mentlick J.; Tavtigian S. V.; Goldgar D. E.; Couch F. J. Functional Assays for Classification of BRCA2 Variants of Uncertain Significance. Cancer Res. 2008, 68, 3523–3531. 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton D. F.; Deffenbaugh A. M.; Pruss D.; Frye C.; Wenstrup R. J.; Allen-Brady K.; Tavtigian S. V.; Monteiro A. N. A.; Iversen E. S.; Couch F. J.; Goldgar D. E. A Systematic Genetic Assessment of 1,433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer–Predisposition Genes. Am. J. Hum. Genet. 2007, 81, 873–883. 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surowy H. M.; Sutter C.; Mittnacht M.; Klaes R.; Schaefer D.; Evers C.; Burgemeister A. L.; Goehringer C.; Dikow N.; Heil J.; Golatta M.; Schott S.; Schneeweiss A.; Bugert P.; Sohn C.; Bartram C. R.; Burwinkel B. Clinical and Molecular Characterization of the BRCA2 p.Asn3124Ile Variant Reveals Substantial Evidence for Pathogenic Significance. Breast Cancer Res. Treat. 2014, 145, 451–460. 10.1007/s10549-014-2943-5. [DOI] [PubMed] [Google Scholar]
- Ding Y. C.; Steele L.; Kuan C. J.; Greilac S.; Neuhausen S. L. Mutations in BRCA2 and PALB2 in Male Breast Cancer Cases from the United States. Breast Cancer Res. Treat. 2011, 126, 771–778. 10.1007/s10549-010-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran R.; George Priya Doss C.; Sudandiradoss C.; Ramanathan K.; Rituraj P.; Rao S. Computational and Structural Investigation of Deleterious Functional SNPs in Breast Cancer BRCA2 Gene. Chin. J. Biotechnol. 2008, 24, 851–856. 10.1016/S1872-2075(08)60042-4. [DOI] [PubMed] [Google Scholar]
- Lindor N. M.; Guidugli L.; Wang X.; Vallée M. P.; Monteiro A. N. A.; Tavtigian S.; Goldgar D. E.; Couch F. J. A Review of a Multifactorial Probability-Based Model for Classification of BRCA1 and BRCA2 Variants of Uncertain Significance (VUS). Hum. Mutat. 2012, 33, 8–21. 10.1002/humu.21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg Å.; Haile R. W.; Malone K. E.; Capanu M.; Diep A.; Törngren T.; Teraoka S.; Begg C. B.; Thomas D. C.; Concannon P.; Mellemkjaer L.; Bernstein L.; Tellhed L.; Xue S.; Olson E. R.; Liang X.; Dolle J.; Børresen-Dale A. L.; Bernstein J. L.; Reiner A. S.; Layne T. M.; Donnelly-Allen L.; Olsen J. H.; Andersson M.; Bertelsen L.; Guldberg P.; Epstein N.; Boice J. D.; Seminara D.; Shore R. E.; Jansen L.; Anton-Culver H.; Largent J.; Lynch C. F.; DeWall J.; Langholz B. M.; Zhou N.; Diep A. T.; Ter-Karapetova E.; Thompson W. D.; Stovall M.; Smith S.; Ramchurren N. Characterization of BRCA1 and BRCA2 Deleterious Mutations and Variants of Unknown Clinical Significance in Unilateral and Bilateral Breast Cancer: The WECARE Study. Hum. Mutat. 2010, 31, E1200–E1240. 10.1002/humu.21202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr N.; Lemnrau A.; Cooke R.; Fletcher O.; Tomczyk K.; Jones M.; Johnson N.; Lord C. J.; Mitsopoulos C.; Zvelebil M.; McDade S. S.; Buck G.; Blancher C.; Trainer A. H.; James P. A.; Bojesen S. E.; Bokmand S.; Nevanlinna H.; Mattson J.; Friedman E.; Laitman Y.; Palli D.; Masala G.; Zanna I.; Ottini L.; Giannini G.; Hollestelle A.; Van Den Ouweland A. M. W.; Novaković S.; Krajc M.; Gago-Dominguez M.; Castelao J. E.; Olsson H.; Hedenfalk I.; Easton D. F.; Pharoah P. D. P.; Dunning A. M.; Bishop D. T.; Neuhausen S. L.; Steele L.; Houlston R. S.; Garcia-Closas M.; Ashworth A.; Swerdlow A. J. Genome-Wide Association Study Identifies a Common Variant in RAD51B Associated with Male Breast Cancer Risk. Nat. Genet. 2012, 44, 1182–1184. 10.1038/ng.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D.; Gable A. L.; Lyon D.; Junge A.; Wyder S.; Huerta-Cepas J.; Simonovic M.; Doncheva N. T.; Morris J. H.; Bork P.; Jensen L. J.; Von Mering C. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.; Wang J.; Jaehnig E. J.; Shi Z.; Zhang B. WebGestalt 2019: Gene Set Analysis Toolkit with Revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. 10.1093/nar/gkz401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J.; Stourac J.; Salanda O.; Pavelka A.; Wieben E. D.; Zendulka J.; Brezovsky J.; Damborsky J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLOS Comput. Biol. 2014, 10, e1003440 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D. E. V.; Ascher D. B.; Blundell T. L. DUET: A Server for Predicting Effects of Mutations on Protein Stability Using an Integrated Computational Approach. Nucleic Acids Res. 2014, 42, W314. 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M.; Pieper U.; Sali A.. Comparative Protein Structure Modeling Using Modeller Curr. Protoc. Bioinforma. 2006, 15 , (1), 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Hekkelman M. L.; te Beek T. A. H.; Pettifer S. R.; Thorne D.; Attwood T. K.; Vriend G. WIWS: A Protein Structure Bioinformatics Web Service Collection. Nucleic Acids Res. 2010, 38, W719–W723. 10.1093/nar/gkq453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Wiederstein M.; Sippl M. J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkowski T. A.; Naghibzadeh S.; Liang J. CASTp: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2003, 31, 3352. 10.1093/nar/gkg512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata T. Detection of Cave Pockets in Large Molecules: Spaces into Which Internal Probes Can Enter, but External Probes from Outside Cannot. Biophys. Physicobiology 2019, 16, 391. 10.2142/biophysico.16.0_391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K. P.; Nguyen T. B.; Patel S.; Varadarajan R.; Madhusudhan M. S. Depth: A Web Server to Compute Depth, Cavity Sizes, Detect Potential Small-Molecule Ligand-Binding Cavities and Predict the PKa of Ionizable Residues in Proteins. Nucleic Acids Res. 2013, 41, W314–W321. 10.1093/nar/gkt503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S.; Olson A. J.. Small-Molecule Library Screening by Docking with PyRx. In Methods in Molecular Biology, 2015; Vol. 1263, pp 243–250. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; Ruth H.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill P. M. W.; Johnson B. G.; Pople J. A.; Frisch M. J. The Performance of the Becke—Lee—Yang—Parr (B—LYP) Density Functional Theory with Various Basis Sets. Chem. Phys. Lett. 1992, 197, 499–505. 10.1016/0009-2614(92)85807-M. [DOI] [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Thompson M. A. A QM/MM Molecular Dynamics Study of the Potential of Mean Force for the Association of K+ with Dimethyl Ether in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 11341–11344. 10.1021/ja00150a036. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindah E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.