

Abstract
Dopamine (DA) is a critical regulator of striatal network activity and is essential for motor activation and reward-associated behaviors. Previous work has shown that DA is influenced by the reward value of food, as well as by hormonal factors that reguate food intake and energy expenditure. Changes in striatal DA signaling also have been linked to aberrant eating patterns. Here we test the effect of leptin, an adipocyte-derived hormone involved in feeding and energy homeostasis regulation, on striatal DA release and uptake. Immunohistochemical evaluation identified leptin receptor (LepR) expression throughout mouse striatum, including on striatal cholinergic interneurons (ChIs) and their extensive processes. Using fast-scan cyclic voltammetry (FSCV), we found that leptin causes a concentration-dependent increase in evoked extra-cellular DA concentration ([DA]o) in dorsal striatum (dStr) and nucleus accumbens (NAc) core and shell in male mouse striatal slices, and also an increase in the rate of DA uptake. Further, we found that leptin increases ChI excitability, and that the enhancing effect of leptin on evoked [DA]o is lost when nicotinic acetylcholine (ACh) receptors are antagonized or when examined in striatal slices from mice lacking ACh synthesis. Evaluation of signaling pathways underlying leptin's action revealed a requirement for intracellular Ca2+, and the involvement of different downstream pathways in dStr and NAc core versus NAc shell. These results provide the first evidence for dynamic regulation of DA release and uptake by leptin within brain motor and reward pathways, and highlight the involvement of ChIs in this process.
SIGNIFICANCE STATEMENT Given the importance of striatal dopamine (DA) in reward, motivation, motor behavior and food intake, identifying the actions of metabolic hormones on DA release in striatal subregions should provide new insight into factors that influence DA-dependent motivated behaviors. We find that one of these hormones, leptin, boosts striatal DA release through a process involving striatal cholinergic interneurons (ChIs) and nicotinic acetylcholine (ACh) receptors. Moreover, we find that the intracellular cascades downstream from leptin receptor (LepR) activation that lead to enhanced DA release differ among striatal subregions. Thus, we not only show that leptin regulates DA release, but also identify characteristics of this process that could be harnessed to alter pathologic eating behaviors.
Keywords: acetylcholine, fast-scan cyclic voltammetry, glutamate, LepR, nAChR, NMDA receptor
Introduction
The striatal complex is the primary input site for the basal ganglia, and can be broadly subdivided into dorsal striatum (dStr) and the nucleus accumbens (NAc). These subdivisions differ in afferent and efferent circuitry, as well as in physiological roles. The dStr receives dopamine (DA) input from the substantia nigra pars compacta (SNc) and is involved in movement generation, motor learning and habit formation (Schultz, 2002; Pisani et al., 2005; Howe and Dombeck, 2016). The NAc receives afferents from DA neurons of the ventral tegmental area (VTA) and participates in reward processing and in goal-directed behaviors (Mogenson et al., 1980; Cardinal et al., 2002; Kelley, 2004; Mohebi et al., 2019).
Through this repertoire of roles, striatal DA transmission has been linked to the regulation of eating behavior and energy homeostasis, with both dStr and NAc involved in food intake and hedonic feeding (Ungerstedt, 1971; Szczypka et al., 2001; Roitman et al., 2004; Yun et al., 2004; Wise, 2006; de Araujo et al., 2008; Richard et al., 2013). Consistent with these roles, a decline in food intake and loss of body weight accompany the early stages of Parkinson's disease, characterized by the progressive loss of midbrain DA neurons and decreasing striatal DA (Beyer et al., 1995; Chen et al., 2003; Kistner et al., 2014). Interestingly, obesity is associated with decreased DA release, including in genetically obese mice (Fulton et al., 2006) and diet-induced obese rats (Geiger et al., 2009; Stouffer et al., 2015).
Circulating hormonal factors, which signal the long and short-term energy status of the body, modulate DA neuron activity and DA release in both nigrostriatal and mesolimbic DA pathways, fine-tuning adaptive responses to environmental and metabolic conditions (Abizaid et al., 2006; Quarta et al., 2009; Skibicka et al., 2011, 2013; Dickson et al., 2012; Labouèbe et al., 2013; Mietlicki-Baase et al., 2014, 2015; Stouffer et al., 2015). The metabolic hormone leptin, synthetized primarily in adipocytes, has attracted increasing attention since it was identified as a key player in the regulation of energy expenditure and appetite suppression (Halaas et al., 1995). Multiple leptin receptor (LepR) isoforms have been identified (Lee et al., 1996); however, leptin signaling is provided primarily by the long isoform (LepRb), especially in brain (Villanueva and Myers, 2008). The presence of LepRb on DA neurons in the VTA points to a role for leptin in the mesolimbic DA system (Fulton et al., 2000; Figlewicz et al., 2006; Hommel et al., 2006; Leinninger et al., 2009). Indeed, leptin has been shown to decrease the spontaneous activity of VTA DA neurons in ex vivo midbrain slices (Hommel et al., 2006; Murakami et al., 2018) and to decrease extracellular DA concentration ([DA]o) in the NAc in vivo after intracerebroventricular application (Krügel et al., 2003). Viral knock-down of LepRs in VTA DA neurons increases preference for sucrose and high fat food, implying a role in hedonic feeding (Hommel et al., 2006). Paradoxically, however, evoked [DA]o is decreased in ex vivo striatal slices from leptin-deficient ob/ob mice (Fulton et al., 2006).
Although these studies point to leptin-DA interactions, evidence that leptin might act within the striatum to modulate axonal DA release is lacking. Here, we assessed the distribution of LepRb in the striatum using immunohistochemistry and examined the influence of leptin on DA release using fast-scan cyclic voltammetry (FSCV) in mouse striatal slices. We identified LepRb on cholinergic interneurons (ChIs) and their processes, and observed that leptin enhances axonal DA release through an acetylcholine (ACh)-dependent microcircuit. Moreover, we evaluated molecular signaling pathways through which leptin acts and found regional differences among dStr, NAc core, and NAc shell. Our studies show that leptin, acting at discrete brain loci and using diverse molecular signaling pathways, influences striatal DA release, thus providing new insight into previously reported behavioral responses involving this metabolic hormone.
Materials and Methods
Animals
Mice were handled in accordance with the recommendations of the National Institutes of Health and with approvals granted by the New York University Grossman School of Medicine and the University of Michigan Animal Ethics Committees. Studies of DA release used primarily male C57BL/6J mice (5–12 weeks of age) from The Jackson Laboratory. Additionally, to examine LepRb expression in the striatum, we used male and female double homozygous LepRbEGFP mice, provided by Prof. Martin G. Myers Jr. from the University of Michigan, in which the detection of LepRb neurons was enhanced by EGFP expression; these were generated by breeding of Leprcre/cre mice with Gt(ROSA)26-Sortm2Sho mice (Leshan et al., 2010). Lastly, choline acetyltransferase (ChAT) knock-out (KO) animals were produced by crossing a conditional floxed allele of ChAT (ChATflox/flox) with a Nkx2.1Cre transgenic line to produce mutants (Nkx2.1Cre;ChATflox/flox) in which ACh synthesis is eliminated in the forebrain (Patel et al., 2012). These mice were genotyped using the Genotyping Core Laboratory of New York University Langone Health with transgenic littermates (ChATflox/flox) used as controls for studies involving ChAT KO mice. Animals were group-housed and maintained on a 12/12 h light/dark cycle (light on at 6:30 A.M.) with food and water ad libitum; all experiments were conducted in the morning.
Immunohistochemistry
LepRbEGFP reporter mice and ChAT KO mice were administered Euthasol (Virbac AH Inc.), then perfused transcardially with 0.1 m PBS (154 mm NaCl in 10 mm phosphate buffer, pH 7.3) followed by 4% paraformaldehyde in PBS (Sigma-Aldrich). Brains were removed, postfixed for 24 h, then sectioned in the coronal plane (50 µm) on a freezing microtome (Leica Cryocut 1800 cryostat, Belair Instrument Company). Free-floating sections were processed for immunohistochemistry, as described previously (Hikima et al., 2021). Briefly, sections were rinsed 3 × 15 min in PBS with 0.1% Triton X-100, blocked for 1 h in normal donkey serum in PBS with 0.3% Triton X-100, and incubated with the primary antibody at room temperature for 18–24 h. Then 3 × 15 min washes in PBS with 0.3% Triton X-100 were performed before incubation for 2 h with secondary antibodies diluted 1:200. Thereafter, sections were rinsed with PBS alone, mounted on glass slides, air-dried, dehydrated in graded alcohols, then Citrisolv and coverslipped in Krystalon (EMD Chemicals).
Immunoserum against EGFP (GFP-1020, dilution 1:500, Aves Labs) was used to identify LepRb-positive neurons. Immunoserum against ChAT (AB144P, dilution 1:200, Millipore) was used to label striatal ChIs. In sections from ChAT KO mice; neuronal profiles were visualized with an anti-smooth endoplasmic reticulum Ca2+ pump (SERCA2; Invitrogen MA3-910, dilution 1:400–1:1000). The absence of Chl immunostaining in ChAT KO mice established the specificity of the ChAT Ab (not illustrated). Secondary antibodies were: donkey anti-chicken Cy3 (catalog #703-165-155) and donkey anti-goat Cy2 (catalog #706-225-148), both from Jackson ImmunoResearch). Imaging was performed with a Nikon Eclipse C1 confocal microscope and data were processed and analyzed with Photoshop software (Adobe Systems Incorporated).
Slice preparation
Mice were anesthetized with isoflurane (Henry Schein Medical Inc.), the brains removed, and 300 µm thick coronal slices containing dStr and NAc core and shell cut using a Leica VT1200S vibrating microtome (Leica Microsystems). For FSCV experiments, slices were cut in ice-cold HEPES-based buffer containing (in mm): 120 NaCl, 20 NaHCO3, 10 glucose, 6.7 HEPES acid, 5 KCl, 3.3 HEPES sodium salt, 2 CaCl2, and 2 MgSO4, saturated with 95% O2/5% CO2. Slices were kept at room temperature in this HEPES-based buffer for 1 h before transfer to the recording chamber (Patel et al., 2012; Stouffer et al., 2015). For electrophysiology experiments, slices were prepared in ice-cold high-sucrose cutting solution containing (in mm): 225 sucrose, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 28 NaHCO3, 1.25 NaH2PO4, 7 glucose, 1 ascorbate, and 3 pyruvate, equilibrated with 95% O2/5% CO2. After cutting, slices were transferred to a holding chamber filled with oxygenated (95% O2/5% CO2) modified artificial CSF (aCSF) containing (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1 MgCl2, 2 CaCl2, 25 glucose, 1 ascorbate, 3 pyruvate, and 4 myo-inositol and bubbled with 95% O2/5% CO2. The initial temperature of this solution was 34°C, which then was allowed to cool to room temperature; slices were kept for at least 1 h under these conditions before transfer to the recording chamber. All FSCV and electrophysiology recordings were made in a submersion chamber that was perfused at 1.5 ml/min with aCSF at 32°C containing (in mm): 124 NaCl, 3.7 KCl, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 1.3 KH2PO4, 10 glucose, and 0.1 mg/ml bovine serum albumin (BSA) equilibrated with 95% O2/5% CO2. Recordings began after 30-min stabilization in the chamber.
Ex vivo slice FSCV
Local electrically evoked increases in [DA]o in ex vivo striatal slices were monitored using FSCV, as described previously (Patel et al., 2012; Patel and Rice, 2013; Stouffer et al., 2015; Bastioli et al., 2022). Peak oxidation currents, reflecting dynamic changes in DA concentration, were detected using a carbon fiber (7 µm in diameter) sealed into the tip of a pulled glass capillary with an exposed length of 30–70 µm (Patel and Rice, 2013). A triangular voltage waveform was applied to the microelectrode (from −0.7 to +1.3 V, then back to −0.7 V vs Ag/AgCl reference, at a rate of 800 V/s) every 100 ms using a Millar voltammeter (Dr. Julian Miller, Barts and the London School of Medicine and Dentistry, United Kingdom). The tip of the recording electrode was inserted 70–100 µm into the slice. Striatal DA release was evoked by local electrical stimulation using a concentric stimulating electrode positioned on the slice surface ∼100 away from the recording electrode. A single stimulus pulse (100-µs duration, 0.4-mA amplitude) sufficed to evoke DA release in lateral dStr and in NAc core, whereas a train of five pulses at 100 Hz was required to elicit a robust release response in NAc shell (Stouffer et al., 2015). The timing of the voltage sweeps and stimulation pulses was controlled by a Master-8 pulse generator (A.M.P.I.). Data were collected using a Molecular Devices Digidata 1550B controlled by AxoScope 10.7 software (Molecular Devices).
Experiments were performed using two different protocols. Initially, to determine the time required for the pharmacological effect of leptin, single-site recording was conducted in the dStr using single-pulse stimulation to elicit increases in [DA]o at 5-min intervals. Once a stable evoked [DA]o baseline was established (typically within three to five stimulations), leptin was applied and the response monitored until evoked [DA]o was again stable. For all the other experiments, we used multisite sampling with a single carbon-fiber microelectrode paired with a single stimulating electrode. For this approach, increases in [DA]o were evoked sequentially in four to five recording sites per striatal subregion in each of two slices from the same striatal level; total sampling time was ∼30 min. This approach is valuable because of site-to-site variability in evoked [DA]o (Stouffer et al., 2015; Patel et al., 2019; Longo et al., 2021; Bastioli et al., 2022). Based on the time at which leptin exerted its maximal effect, multisite sampling was initiated 50 min after application of leptin +/− other drugs, and in time-matched controls in aCSF alone or with a paired test drug. To evaluate the sensitivity of DA release to leptin, we conducted separate experiments in different concentrations of leptin using multisite sampling in each region. The concentration of leptin at which the increase in peak evoked [DA]o was half maximal (EC50) for each region was calculated by fitting sigmoidal concentration-response curves using nonlinear regression (GraphPad Prism 8; GraphPad Software Inc.). Drugs used to challenge leptin action were added at the desired concentration from stock solutions 10–15 min before leptin application with continual application throughout the experiment. In experiments with insulin or insulin plus leptin, sampling was initiated after 50-min superfusion and conducted in the continued presence of the hormone(s); in co-application experiments, the two hormones were applied simultaneously. Following each experiment, recording electrodes were calibrated in the recording chamber, using 1 µm DA in each experimental solution tested that day. Calibration solutions were prepared immediately before use from a stock solution of 2 mm DA in 0.1 m HClO4 stored at 4°C.
DA uptake analysis
Changes in DA uptake induced by leptin exposure were measured using a MATLAB script, provided by Charles Nicholson (New York University), as described previously (Li et al., 2010; Patel et al., 2019). The maximal uptake rate for DA (Vmax) was derived from analysis of single-pulse evoked [DA]o records from dStr and NAc core using the Michaelis-Menten equation with a Km fixed at 0.9 µm.
Ex vivo slice electrophysiology
Whole-cell patch clamp recordings were obtained from Chls in the dStr visualized at 40× using a water-immersion objective on an Olympus BX51WI microscope equipped with infrared differential interference contrast optics (Olympus America). Recording electrodes were made from fire polished 2.0 mm o.d. borosilicate capillary tubing (Sutter Instrument Company) using a P-97 Flaming/Brown micropipette puller (Sutter Instrument Company). Pipette resistance was ∼3–5 MΩ when filled with a solution containing (in mm): 129 K-gluconate, 11 KCl, 10 HEPES, 2 MgCl2, 1 EGTA, 2 Na2-ATP, and 0.3 Na3-GTP, adjusted to pH 7.3 with KOH.
ChIs were identified by their large somas and their electrophysiological properties, including a prominent hyperpoloarization-activated current (Ih) and broad action potentials (APs). The influence of leptin on ChI excitability was assessed in current-clamp mode by monitoring changes in the number of spike discharges in response to a series of depolarizing current injection pulses (100, 200, 300 pA), 3 s in duration and separated by 5 s. This series of depolarizing current pulses was applied every 2 min. Leptin superfusion began once responses were stable. Data were recorded with an Axopatch 200B amplifier, low pass filtered at 2 kHz, digitized (10 kHz, Digidata 1550B, Molecular Devices), collected using Clampex 10.7, and analyzed offline using Clampfit (all from Molecular Devices).
Drugs
Recombinant mouse leptin (carrier free; catalog number 498-OB), 2-aminoethoxydiphenyl borate (2-APB), cyclopiazonic acid (CPA), and dihydro-β-erythroidine (DHβE) were purchased from R&D Systems. Components of solutions used for voltammetry and electrophysiology were from Sigma-Aldrich, as were DA, EGTA-AM, BAPTA-AM, LY294002, BSA, and insulin. MK-2206 was from ApexBio Technology. A stock solution of leptin was prepared in 20 mm Tris-HCl buffer (pH 8.0) before addition to aCSF. Water soluble drugs were prepared as aqueous stock solutions; other drugs were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). Maximal final DMSO concentration was <0.1%. Drugs were diluted to their required concentrations in aCSF immediately before use; BSA was added as a carrier to the solutions used for voltammetry and electrophysiology to help maintain solubility of leptin and minimize adherence of leptin and other drugs to the tubing and recording chamber. Control data were obtained in the presence of the same volume of vehicle and did not differ from responses recorded in aCSF alone (data not illustrated).
Statistical analysis
Data analysis was performed using GraphPad Prism 8. All data are expressed as mean ± SEM. Significance of differences was assessed using Student's paired or unpaired t tests as appropriate, or ANOVA for multiple experimental groups. For voltammetry data, n indicates the number of recorded sites; two slices from each of three to four mice were used for each experimental group. For electrophysiology experiments, n indicates the number of paired stimulus responses in the absence and then presence of leptin. The confidence level of significance was set at 95%, such that a p ≤ 0.05 was considered to be statistically significant.
Results
Expression of functional LepRs in the striatum
We visualized LepRs in striatum by immunofluorescence in coronal brain sections from LepRbEGFP mice, in which EGFP (enhanced green fluorescent protein) is genetically co-expressed with LepRb (Leinninger et al., 2009; Leshan et al., 2010; Patterson et al., 2011). Particulate LepRb-immunoreactivity (ir) was observed throughout the striatum, appearing as grouped aggregates of various sizes (Fig. 1A, representative of immunostaining in three mice). In addition, a few large neurons expressed LepRb-ir throughout the perikaryon and initial portions of primary dendrites (Fig. 1A, arrow). Based on criteria of size and shape, we hypothesized that these large neurons were cholinergic and confirmed this with an antibody against ChAT. It is noteworthy that despite their low density, cholinergic neurons (Chls) have extensive axonal and dendritic processes (Fig. 1B). In a low magnification view, extensive overlap can be seen between ChAT-ir-positive perikarya and processes with LepRb-ir (Fig. 1C–E), with a portion of the Chl population showing intense LepRb-ir. At higher magnification, LepRb-ir can be seen in both the soma and processes of a single ChAT-ir neuron (Fig. 1F–H).
Figure 1.

LepRb expression in the striatum, including in ChI somata and processes. A, LepRb-EGFP-ir in the striatum of LepRbEGFP mice is generally distributed throughout the striatum and is aggregated in clusters of <1 µm to ∼5 µm. A few large neurons also show intense LepRb-EGFP-ir (arrow points to one of these). B, ChAT-ir identifies ChIs in mouse striatum; ChAT-ir is seen in large perikarya (arrow), and also in extensive processes that radiate throughout the striatum. C, Low-magnification (10×) section through dStr from a LepRbEGFP mouse showing widespread ChAT-ir in neuropil, as well as in sparse ChAT-ir ChI somata. D, The same section showing the distribution of LepRb-ir, including LepRb-ir neurons with varying fluorescence intensity. E, Merged image showing co-localization of LepRb-ir and ChAT-ir in somata and processes. C–E, Examples of ChIs expressing LepRb-ir are indicated by arrows. F, High magnification (100×) of a ChAT-ir neuron in dStr from a LepRbEGFP mouse. G, The same neuron showing LepRb-ir throughout the soma and processes. H, Merged image showing co-localization of LepRb-ir and ChAT-ir in the soma and its processes. Images from LepRbEGFP mice are representative of immunostained sections from three animals. Scale bars: 20 µm (A, B, D) and 10 µm (G).
To examine whether LepRb in the striatum is a regulator of DA release, we used FSCV to monitor evoked increases in [DA]o in striatal slices. In initial studies using single-site monitoring in dStr, we found that a physiologically relevant concentration of leptin (30 nm; Caro et al., 1996), caused an increase in evoked [DA]o that began 20 min after initiation of exposure, and reached a stable plateau after 40–50 min (data not shown). Thereafter, multisite sampling was used to quantify the effect of 30 nm leptin on evoked [DA]o in dStr, NAc core, and NAc shell. Leptin caused a significant increase in evoked [DA]o in each of these striatal subregions (dStr, 133 ± 11% of time-matched controls, p = 0.0051; NAc core, 130 ± 14% of controls, p = 0.0401; and NAc shell, 128 ± 10% of controls, p = 0.0376; n = 30 sites from three mice per condition; Fig. 2A). In each region, DA was identified by its characteristic voltammogram (Fig. 2A, insets). In ex vivo striatal slices, there is no ambiguity that DA is the electroactive substance detected throughout the striatum (Patel and Rice, 2013). Using multisite sampling, we then evaluated the concentration dependence of the response in 10–1000 nm leptin. Leptin produced concentration-dependent increases in evoked [DA]o throughout the striatum, with EC50 values in the 100 nm range for each region (Fig. 2B). We therefore applied 100 nm leptin in most subsequent pharmacological experiments.
Figure 2.
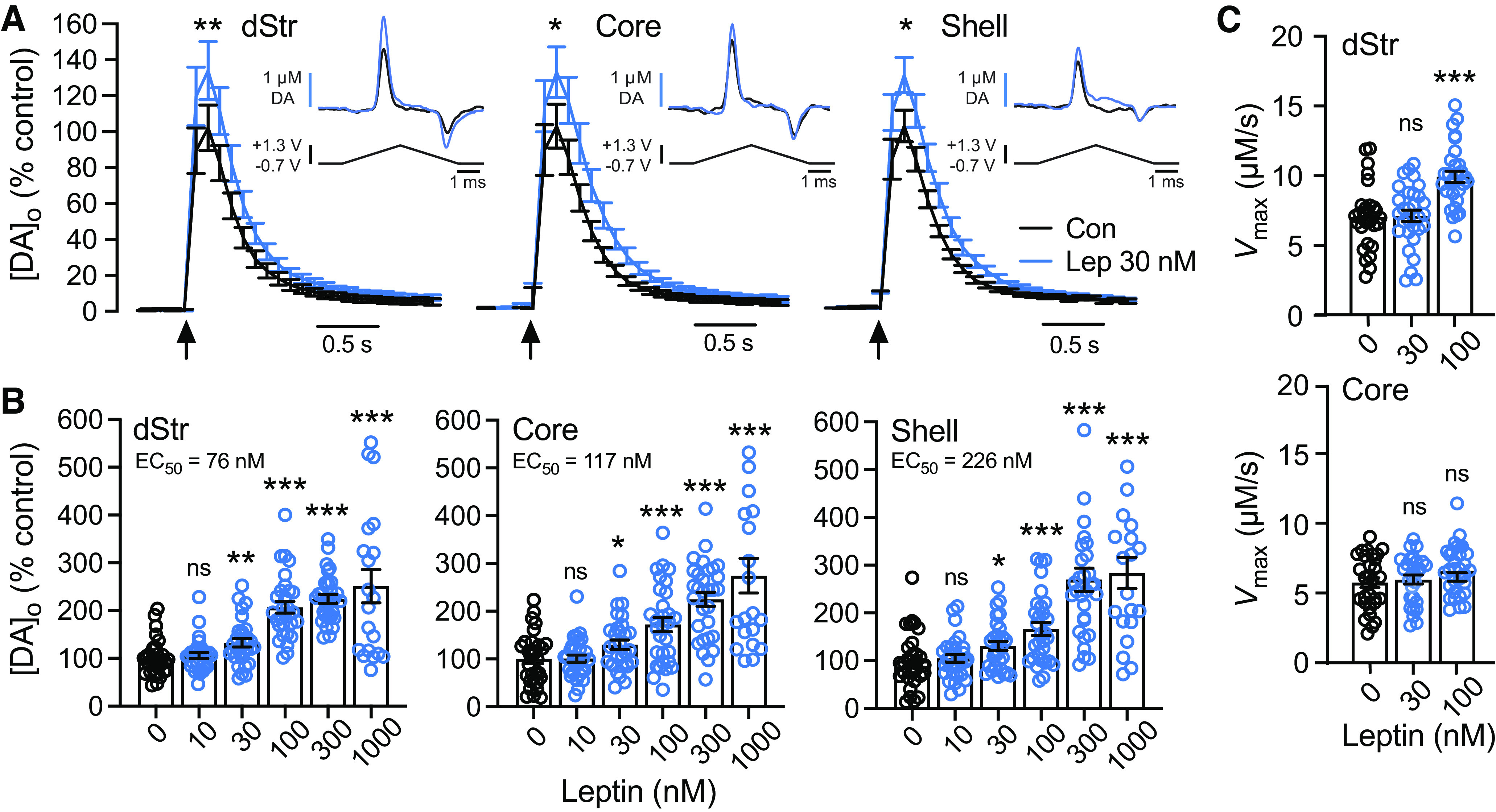
Leptin boosts axonal DA release and uptake. A, Averaged single-pulse-evoked [DA]o versus time in the absence or presence of leptin (30 nm) recorded in dStr, NAc core and shell. Arrows indicate stimulus onset. Evoked [DA]o was significantly higher in leptin than in time-matched controls in aCSF alone (dStr, 133 ± 11% of control; NAc core, 130 ± 14% of control; NAc shell, 128 ± 10% of control in NAc shell; n = 30 recording sites from 3 mice for each condition, data normalized to control; *p < 0.05, **p < 0.01; unpaired Student's t tests). Insets show representative DA voltammograms recorded in 30 nm leptin and in time-matched controls recorded in each striatal subregion. B, Concentration-dependence of the enhancing effect of leptin on evoked [DA]o in dStr, NAc core and shell. Leptin-enhanced increases in evoked [DA]o were concentration-dependent in all three subregions (dStr, n = 20–30, F(5,164) = 23.23, p < 0.001; NAc core, n = 20–30, F(5,164) = 18.66, p < 0.001; NAc shell, n = 20–30, F(5,163) = 21.78, p < 0.001; one-way ANOVA, Tukey's multiple comparisons tests). Calculated EC50 values are shown with the data set for each region. C, Vmax values determined from single-pulse evoked [DA]o records using a fixed Km of 0.9 μm in control conditions or in the presence of leptin. Data are mean ± SEM and were analyzed using a one-way ANOVA with Dunnett's post hoc test for each region; records with a goodness of fit of R2 < 0.95 were omitted (leptin vs control: ns, not significant; ***p < 0.0001; n = 28–30).
Previous studies of NAc synaptosomes showed that leptin increases Vmax for DA uptake via the DA transporter (DAT; Perry et al., 2010). We examined whether leptin increased DA uptake in our slice preparation by determining Vmax from evoked [DA]o recorded in the presence of 30 or 100 nm leptin versus time-matched controls. The method used distinguishes changes in release versus uptake by analysis of the falling phase of a single-pulse-evoked increase in [DA]o (Li et al., 2010; Stouffer et al., 2015; Patel et al., 2019). This allowed determination of Vmax from dStr and NAc core, in which increases in [DA]o were evoked using single-pulse stimulation (Li et al., 2010), but not from the NAc shell in which five-pulse trains were used to provide reliable DA release. Despite the significant increase in evoked [DA]o seen with 30 nm leptin (Fig. 2A,B), this concentration had no effect on Vmax in either dStr or NAc core (Fig. 2C). At 100 nm, however, leptin caused an increase in Vmax in both regions compared with time-matched controls in aCSF alone, which was significantly higher than control in the dStr analyzed using ANOVA (Fig. 2C), but also in NAc core when compared with control using a one-tailed unpaired t test (p = 0.0388; dStr: control, 7.05 ± 0.43 µm/s; 30 nm leptin, 7.13 ± 0.41 µm/s, p = 0.9848 vs control; 100 nm leptin, 9.93 ± 0.40 µm/s, p < 0.0001 vs control; NAc core: control, 5.74 ± 0.35 µm/s; 30 nm leptin, 5.97 ± 0.33 µm/s, p = 0.8523 vs control; 100 nm leptin, 6.59 ± 0.32 µm/s, p = 0.1269 vs control; ANOVA, n = 28–30 per group). These data support a direct effect of leptin on striatal DA axons. Given that evoked [DA]o reflects the sum of DA release and DA uptake (Sulzer et al., 2016), an increase in DA uptake alone would decrease evoked [DA]o. However, we observed an increase in evoked [DA]o, thereby revealing that leptin primarily enhances DA release, and does so at a lower concentration than that required to increase DA uptake.
Leptin amplifies striatal evoked DA release via ACh and nAChRs and increases excitability of striatal ChIs
Having established that leptin boosts striatal DA release, we next asked whether the effect was direct or indirect. Given the presence of LepRbs in ChIs and their striatal processes (Fig. 1E), we investigated the possible involvement of ACh, which is released concurrently with DA during local electrical stimulation, and which powerfully regulates striatal DA release via β2-subunit containing nicotinic ACh receptors (β2*-nAChRs) on DA axons (Rapier et al., 1990; Wonnacott et al., 2000; Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004; Cragg, 2006; Threlfell et al., 2010, 2012; Cachope et al., 2012; Exley et al., 2012; Patel et al., 2012). To test whether leptin-induced enhancement of DA release requires activation of nAChRs, we monitored [DA]o in the presence of a selective antagonist of β2*-nAChRs, DHβE. Application of DHβE (1 µm) alone decreased single-pulse evoked [DA]o (data not shown), as described previously (Zhou et at., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004). In the continued presence of this antagonist, the effect of leptin (100 nm) was lost in dStr, NAc core and NAc shell, pointing to a crucial role for nAChRs in the three striatal subregions examined (Fig. 3A; dStr, p = 0.2135; NAc core, p = 0.9223; NAc shell, p = 0.5510).
Figure 3.

Leptin-enhanced evoked [DA]o requires nAChR activation and ACh. A, Average peak evoked [DA]o in the absence or presence of leptin (100 nm) recorded in the presence of a nAChR antagonist, DHβE (1 μm). The usual enhancement in evoked [DA]o seen with leptin application (Fig. 2) was abolished in each striatal subregion by DHβE (n = 27–30 recording sites in each region in slices from 3 mice per condition; mean time-matched control in DHβE alone in each region is 100%; ns, not significant, unpaired t tests). B, Average peak evoked [DA]o in striatal slices from ChATflox/flox and ChAT KO mice following leptin (100 nm) application (ChATflox/flox-Lep, ChAT KO-Lep); mean time-matched control in each region for each genotype (ChATflox/flox Con; ChAT KO Con) is 100%. In striatal slices from ChATflox/flox mice, leptin caused an increase in evoked [DA]o to 193 ± 16% of time-matched controls in dStr, to 170 ± 18% of control in NAc core, and to 134 ± 11% of control in NAc shell, whereas no increase was seen in any region in KO slices (n = 26–30 recording sites, 3 mice for each experimental condition, data normalized to control; ***p < 0.001, *p < 0.05; ns, not significant; Student's unpaired t tests). C, Representative spiking pattern of striatal ChIs after a sweep of depolarizing current pulses (100, 200, and 300 pA; 3-s duration, 5-s interpulse interval) before (top) and following (bottom) application of leptin (30 nm). Leptin caused a decrease in spike frequency adaptation and an increase in the number of APs recorded during depolarizing current steps. D, Summary of the effect of leptin on ChI excitability, indicated by AP number during current injection before leptin application and at the peak of the response, with mean ± SEM (Con: 27 ± 2 APs, Lep: 44 ± 5 APs; n = 9 paired stimulations, 3 neurons, 3 mice; ***p < 0.001; paired Student's t test).
To confirm ACh involvement in leptin-enhanced DA release, we examined evoked [DA]o in ChAT KO mice bred to lack ACh synthesis in the forebrain (Patel et al., 2012; Stouffer et al., 2015). In slices from littermate control mice (ChATflox/flox), leptin (100 nm) caused a significant increase in evoked [DA]o in each striatal subregion (Fig. 3B; dStr, 193 ± 16% of time-matched recording in aCSF alone, p < 0.0001; NAc core, 170 ± 18%, p = 0.0008 of aCSF; NAc shell: 134 ± 11% increase, p = 0.0313; unpaired t tests, n = 26–30 recording sites in six slices from three mice per condition); however, in ChAT KO mice, the effect of leptin was absent in all subregions (Fig. 3B; dStr, p = 0.4665; NAc core, p = 0.1390; NAc shell, p = 0.4960 in leptin vs time-matched aCSF; n = 26–30 recording sites in six slices from three mice per condition). These data confirm that ACh intermediation is required for leptin-enhanced DA release.
Given previous work showing that insulin enhances DA release in a nAChR-dependent manner and also increases the excitability of ChIs in rat striatal slices (Stouffer et al., 2015), we tested whether leptin might have similar actions on ChIs in mouse striatal slices. Using current-clamp recording of ChIs in dStr, we found that depolarizing current steps (100–300 pA) induced reliable increases in AP generation that showed spike-frequency adaptation during prolonged current injection, as described previously (Stouffer et al., 2015; Tubert et al., 2016). Application of leptin (30 nm) attenuated the spike-frequency adaptation, with a maximal effect seen after 20–50 min of leptin exposure (Fig. 3C). The net result was an increase in AP number (Fig. 3C,D; baseline, 27 ± 2 APs; leptin 44 ± 5 APs; p = 0.0007, paired t tests; n = 9 stimulus pairs from three ChIs from 3 mice). In contrast, ChIs monitored over the same time period showed no increase in AP number during current injection in aCSF alone (p > 0.05, paired t test; data not shown). These data show that LepRbs on ChIs are functional, and provide an explanation for the nAChR-dependent amplification of evoked striatal DA release by leptin.
Leptin enhances evoked DA release with regional differences in signaling pathways
LepRbs are coupled to downstream signal transduction pathways (Fig. 4A) that use several of the same proteins required for insulin signaling (Frühbeck, 2006), including phosphoinositide 3-kinase (PI3K; Shepherd et al., 1998). Previous studies demonstrated that activation of PI3K by insulin is necessary for the nAChR-dependent enhancement of evoked striatal DA release (Stouffer et al., 2015). Based on the nAChR dependence of DA release enhancement by either leptin (Fig. 3A,B) or insulin, signaling pathways downstream from LepRbs are assumed to be primarily in ChIs. We first tested whether leptin, like insulin, acts via PI3K by monitoring the effect of leptin (100 nm) on evoked [DA]o in the presence of a PI3K inhibitor, LY294002 (1 µm; Stouffer et al., 2015), and in time-matched controls in LY294002 alone. In the continued presence of this PI3K inhibitor, no change in evoked [DA]o was seen with leptin (100 nm) in any striatal subregion (Fig. 4B; dStr, p = 0.7929; NAc core, p = 0.5398; NAc shell, p = 0.9979 in leptin + LY294002 vs time-matched controls in LY294002 alone, unpaired t tests; n = 30 recording sites per region and condition from three mice).
Figure 4.

Subregional differences in signaling pathways involved in DA release regulation by leptin. A, Schematic representation of key signaling pathways that can be activated by leptin acting at LepRb (based on Frühbeck, 2006). Proteins involved include: JAK2, Janus kinase 2; Grb2, growth factor receptor-bound protein 2; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-bisphosphate; PLC, phospholipase C; IP3, inositol 1,4,5-trisphosphate; DAG, diacylglycerol; PDK1, phosphoinositide-dependent kinase-1; PKC, protein kinase C; JNK, c-Jun N-terminal kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin. B, Evoked [DA]o in striatal slices in the absence or presence of leptin (Lep; 100 nm) in LY294002 (1 μm), a PI3K inhibitor. Leptin-induced enhancement of evoked [DA]o was abolished by LY284002 in all subregions. C, Evoked striatal [DA]o with or without leptin (100 nm) in the presence of a PKC inhibitor, GF-109203× (100 nm). Inhibition of PKC had no impact on the effect of leptin in dStr (166 ± 8% of time-matched control) or NAc core (137 ± 9% of control), but prevented the effect of leptin on evoked [DA]o in NAc shell. D, Mean evoked [DA]o in the absence or presence of leptin (100 nm) in an Akt inhibitor, MK-2206 (5 μm). Inhibition of Akt prevented the effect of leptin in NAc shell, but not in dStr (187 ± 20% of time-matched control) or NAc core (172 ± 19% of time-matched control). For B–D, mean time-matched control in each inhibitor alone in each region is 100%; ns, not significant; ***p < 0.001; unpaired Student's t test; n = 28–30 recording sites per region from 3 mice per condition. E, Average single-pulse-evoked [DA]o in the presence of leptin (100 nm), insulin (Ins, 30 nm), or 100 nm leptin plus 30 nm insulin (Lep + Ins). The presence of insulin did not significantly affect the leptin-induced enhancement in evoked [DA]o in dStr (199 ± 15% of time-matched control) or NAc core (204 ± 15% of time-matched control); however, in NAc shell, the usual enhancement with either insulin or leptin alone was lost when the hormones were applied together (dStr, F(3,116) = 24.23, p < 0.0001; NAc core, F(3,116) = 15.20, p < 0.0001; NAc shell, F(3,116) = 7.763, p < 0.0001; one-way ANOVA, Tukey's multiple comparisons tests; ns, not significant; *p < 0.05, **p < 0.01, ***p < 0.001; hormone vs time-matched control; +p < 0.05 leptin vs leptin + insulin; ##p < 0.001 insulin vs leptin + insulin; n = 28–30 recording sites per region).
Downstream products of PI3K typically stimulate protein kinases, including protein kinase C (PKC) and Akt (protein kinase B; Vanhaesebroeck and Waterfield, 1999). To probe the possible involvement of these molecules in mediating leptin effects on striatal DA release, we applied leptin (100 nm) in the presence of a PKC inhibitor, GF109203X (100 nm; Zhu et al., 2005), or in the presence of an Akt inhibitor, MK-2206 (5 µm; zur Nedden et al., 2018). Neither inhibitor altered the usual enhancement of evoked [DA]o by leptin in either dStr or NAc core (Fig. 4C,D; GF109203X: dStr, 166 ± 8% of control, p < 0.0001; NAc, 137 ± 9% of control, p = 0.0055; MK-2206: dStr, 187 ± 20% of control, p = 0.0003; NAc, 172 ± 19% of control in NAc core, p = 0.0020 unpaired t tests, n = 30 sites per region, three mice per condition). In contrast, inhibition of either PKC or Akt prevented the effect of leptin on evoked [DA]o in the NAc shell (Fig. 4C,D; p = 0.1252 for leptin in GF109203X and p = 0.9058 for leptin in MK-2206 vs time matched controls; n = 30 sites per region, three mice per condition). These findings suggest regional differences in intracellular signaling pathways activated by leptin, with greater reliance on PKC and Akt in NAc shell than in dStr or NAc core.
The finding that leptin, like insulin, activates PI3K suggests that these hormones might have either a synergistic or an occlusive effect on striatal DA release. We tested this by comparing the amplitude of evoked [DA]o after co-application of the two hormones with that in time-matched controls in aCSF or in leptin (100 nm) or insulin (30 nm) alone (Fig. 4E). Analysis by one-way ANOVA with Tukey's post hoc multiple comparisons test in each region showed that in dStr and NAc core, leptin, insulin, or the combination caused significant increases in evoked [DA]o compared with that in time-matched controls. The effect of leptin plus insulin was not additive, however, as evoked [DA]o in the presence of both hormones did not differ from that in leptin alone in either dStr or NAc core (dStr: F(3,116) = 24.23; p < 0.00001: leptin, 207 ± 14% of control, p < 0.00001; insulin, 145 ± 10% of control, p = 0.00003; leptin + insulin, 199 ± 15% of control, p < 0.00001; leptin + insulin vs leptin, p = 0.6403; leptin + insulin vs insulin, p = 0.0006; n = 28–30 sites from three mice per condition; NAc core: F(3,116) = 15.20; p < 0.00001: leptin, 172 ± 18% of control, p = 0.0002; insulin, 149 ± 13% of control, p = 0.0003; leptin + insulin, 204 ± 15% of control, p < 0.00001; leptin + insulin vs leptin, p = 0.0953; leptin + insulin vs insulin, p = 0.0001; n = 28–30 sites from three mice per condition; Fig. 4E). These data suggest competition between leptin and insulin for one or more components of their signaling cascades. The lack of an additive effect was even more striking in the NAc shell, where evoked [DA]o in the presence of leptin plus insulin did not differ from control in aCSF alone (NAc shell: F(3,116) = 7.763; p = 0.0001: leptin, 166 ± 17% of control, p = 0.0003; insulin, 158 ± 17% of control, p = 0.0006; leptin + insulin, 123 ± 13% of control, p = 0.0763; leptin + insulin vs leptin, p = 0.0070; leptin + insulin vs insulin, p = 0.0152; ANOVA, n = 28–30 sites from three mice per condition; Fig. 4E).
Regional differences in the dependence of leptin-enhanced striatal DA release on intracellular Ca2+ and Ca2+ stores
Given the relatively limited role of PKC and Akt in the influence of leptin on DA release, especially in dStr and NAc core, we turned our attention to another arm of leptin signaling pathways, which involves increased intracellular Ca2+. Previous studies found that leptin elicits a transient elevation of intracellular Ca2+ in neurons (Wang et al., 2008; Guimond et al., 2014; Mancini et al., 2014; Gavello et al., 2016). To examine the role of intracellular Ca2+ in the effect of leptin on DA release, we recorded evoked [DA]o in the presence of either of two membrane-permeable Ca2+ chelators, EGTA-AM or BAPTA-AM. Both agents provide effective global buffering of intracellular Ca2+; however, faster-acting BAPTA additionally buffers fast Ca2+ transients from Ca2+ entry at neuronal membranes (Adler et al., 1991; Eggermann et al., 2011; Cohen et al., 2016). At the low concentrations used, neither EGTA-AM (20 µm; Ermolyuk et al., 2013) nor BAPTA-AM (50 µm; Patel et al., 2009) altered evoked [DA]o significantly compared with time-matched control measurements in aCSF alone (data not shown). Each chelator was applied 15–20 min before leptin (100 nm) exposure; multisite sampling of DA release was initiated 50 min after the start of leptin application in the continued presence of EGTA-AM or BAPTA-AM or in time-matched controls in chelator alone. Both chelators prevented leptin-induced increases in evoked [DA]o in the dStr and in NAc core and shell (Fig. 5A–C); in the NAc shell, however, BAPTA-AM not only prevented the increase, but led to a decrease in evoked [DA]o with leptin (73 ± 7% of control; Fig. 5C; dStr, p = 0.0788; NAc core, p = 0.4489; NAc shell p = 0.9951 vs time-matched controls with EGTA-AM; dStr, p = 0.3068; NAc core, p = 0.4719; NAc shell, p = 0.0036 vs time-matched controls with BAPTA-AM; unpaired t tests; n = 30 sites per region from three mice per condition). The leptin-induced decrease with BAPTA-AM in NAc shell suggests a role for fast Ca2+ transients triggered by leptin.
Figure 5.

Leptin potentiation of DA release requires intracellular Ca2+. A–C, Evoked [DA]o recorded in the absence or presence of leptin (100 nm) in EGTA-AM (20 μm) or BAPTA-AM (50 μm) in dStr (A), NAc core (B), and NAc shell (C). The usual increase in evoked [DA]o was prevented in each striatal subregion by EGTA-AM or BAPTA-AM; in the NAc shell, the presence of BAPTA-AM reversed the effect of leptin, with a decrease in evoked [DA]o observed with leptin application (to 73 ± 7% of time-matched control in BAPTA-AM alone). For A–C, n = 30 recording sites per region from three sites per condition; mean time-matched control in each chelator alone in each region is 100% (***p < 0.001; ns = not significant; unpaired Student's t test). D–F, Evoked [DA]o in the dStr (D), NAc core (E), and NAc shell (F) recorded in the absence and presence of leptin (100 nm) in: CPA (30 μm), an endoplasmic reticulum Ca2+-ATPase inhibitor; 2-APB (30 μm), an IP3R antagonist; or dantrolene (DAN; 10 μm), a RyR antagonist. The presence of either CPA or 2-APB prevented the usual effect of leptin in all striatal subregions. In contrast, DAN had no effect on the effect of leptin in dStr (to 207 ± 11% of time-matched control in DAN alone) or NAc core (173 ± 13% of time-matched control), although it prevented the effect of leptin in NAc shell. For D–F, n = 30–40 recording sites per region from three to four mice per condition; mean time-matched control for each region with each drug is 100% (ns, not significant; ***p < 0.001; unpaired Student's t test).
We then investigated possible sources of Ca2+ recruitment by leptin, first by impairing Ca2+ storage using CPA (30 µm), an inhibitor of the endoplasmic reticulum Ca2+-ATPase (Patel et al., 2009). The increase in evoked [DA]o with leptin (100 nm) was completely prevented in the presence of CPA, implicating a role for intracellular Ca2+ stores in the action of leptin (Fig. 5D–F, dStr, p = 0.2421; NAc core, p = 0.3449; NAc shell, p = 0.2542 vs time-matched controls; unpaired t tests; n = 30–40 sites per region in slices from three to four mice per condition). We next tested involvement of receptors known to gate release of Ca2+ from stores using 2-APB (30 µm), an antagonist of inositol 1,4,5-trisphosphate receptors (IP3Rs) that mediate IP3-induced Ca2+ release and dantrolene (10 µm), an antagonist of ryanodine receptors (RyRs) that facilitate Ca2+-induced Ca2+ release (Patel et al., 2009). Leptin-induced increases in evoked [DA]o were prevented by 2-APB in all three striatal subregions (Fig. 5D–F, dStr, p = 0.8117; NAc core, p = 0.8124; NAc shell, p = 0.1001 vs time-matched controls; unpaired Student's t test; n = 29–30 sites per region from three mice per condition). By contrast, dantrolene was without effect in dStr and NAc core (Fig. 5D,E), but did prevent the usual enhancing effect of leptin in the NAc shell (Fig. 5F), highlighting the involvement of distinct regulatory pathways in this region (dStr, 207 ± 11% of time-matched controls, p < 0.00001; NAc core: 173 ± 13% of controls, p = 0.00001; NAc shell: 122 ± 6% of controls, p = 0.0598; unpaired t tests, n = 30 sites per region in slices from three mice per condition).
Leptin-induced enhancement of DA release in NAc shell requires NMDA receptors
The finding that BAPTA-AM not only prevented, but reversed the enhancing effect of leptin in the NAc shell suggested a role for transmembrane Ca2+ entry in ChIs in this region. Given previous evidence that leptin can potentiate NMDA receptor currents (Shanley et al., 2001; Neyens et al., 2020; Cochrane et al., 2020), we compared evoked [DA]o in leptin (100 nm) in the dStr, NAc core, and NAc shell in the absence or presence of a NMDA receptor antagonist, D-AP5 (50 µm; Kosillo et al., 2016). The effect of leptin on evoked [DA]o persisted in the presence of AP5 in both dStr and NAc core (Fig. 6A, dStr: 178 ± 10% of time-matched controls, p = 0.00000; NAc core: 141 ± 8% of controls, p = 0.00013; unpaired t tests, n = 30 sites per region, per condition, in slices from three mice). However, in NAc shell, the enhancing effect of leptin on DA release was absent in AP5 (Fig. 5A, p = 0.2512; n = 30 sites from three mice per condition). NMDA receptor activation requires depolarization, raising the possibility that differential involvement of NMDA receptors in NAc shell versus dStr or NAc core might be a consequence of the brief high frequency train (five pulses, 100 Hz) used in the NAc shell to provide reliable increases in [DA]o (Stouffer et al., 2015). We tested the possible influence of stimulation protocol by repeating leptin application in the presence of AP5 when increases in [DA]o were evoked in dStr and NAc core using five pulses at 100 Hz. Again, however, leptin-enhanced increases in evoked [DA]o in dStr and NAc core were unaffected by AP5 (Fig. 6B, dStr: 172 ± 14% of control in AP5 alone, p = 0.00001; NAc core: 162 ± 18% of control, p = 0.0020; n = 29–30 sites per region from three mice per condition). This result shows that the involvement of NMDA receptors in the NAc shell reflects differences in local microcircuitry, not differences in stimulation method.
Figure 6.

Activation of NMDA receptors in NAc shell contributes to leptin-enhanced evoked [DA]o. A, Mean evoked striatal [DA]o ± SEM in striatal slices exposed to leptin (100 nm) in the presence of D-AP5 (50 μm), an NMDA receptor antagonist; mean time-matched control in D-AP5 alone in each region is 100%. D-AP5 prevented the usual enhancement of evoked [DA]o by leptin in NAc shell, but not in dStr (178 ± 10% of time-matched control) or in NAc core (141 ± 8% of time-matched control). B, Evoked [DA]o in dStr and NAc core using the same brief pulse-train stimulus (5 pulses, 100 Hz) used to evoked DA release in NAc shell, recorded in the absence or presence of leptin in D-AP5 (50 μm; arrows indicate stimulus onset). As seen with single stimulation (A), NMDA receptor antagonism by D-AP5 had no effect on the enhancement of evoked DA]o by leptin in either dStr (172 ± 14% of time-matched control) or NAc core (162 ± 18% of time-matched control; n = 30 sites per region from 3 mice per condition; mean time-matched control in D-AP5 alone in each region is 100%; ns, not significant; ***p < 0.001; unpaired Student's t test).
Discussion
Leptin is a highly conserved peptide neuromodulator that contributes to energy homeostasis (Pan and Myers, 2018). Leptin actions in the arcuate nucleus and other nuclei within the hypothalamus are crucial for maintaining whole-body energy balance by helping to match food consumption with energy expenditure and motor output, with additional actions in the nucleus of the solitary tract helping to signal satiety (Dhillon et al., 2006; Hawke et al., 2009; Hayes et al., 2010; Ring and Zeltser, 2010; Neyens et al., 2020). Moreover, leptin has been shown to modulate feeding behavior by direct and indirect actions on midbrain DA neurons (Hommel et al., 2006; Leinninger et al., 2009). Expression of LepRb in VTA and SNc DA neurons (Scott et al., 2009; Leshan et al., 2010) implies that leptin might also act in brain regions that receive DA input from these neurons, including the dStr and NAc.
Indeed, we found a direct effect of leptin on DA uptake, with enhanced uptake in the dStr and NAc core, as suggested by previous work using NAc synaptosomes (Perry et al., 2010). If this were the only action of leptin, the net effect would be a decrease in evoked [DA]o. However, we found the opposite: a concentration-dependent increase in evoked [DA]o in the presence of leptin. This result is consistent with a previous in vivo microdialysis study showing an increase in [DA]o in the NAc after intranasal administration of leptin, an effect that was absent in LepR-deficient Zucker rats (Neto et al., 2016). Here, we observed that such increases are mediated indirectly by ChIs, which are key striatal targets of leptin. A leptin-induced increase in ChI excitability promotes ACh-dependent enhancement of DA release through activation of β2*-nAChRs on DA axons. Although leptin-enhanced DA release was seen throughout the striatum, we identified regional variation in the signaling pathways underlying these increases, indicating differences in the role and regulation of leptin's influence among striatal subregions.
Leptin acts via ACh throughout the striatum
ChIs are key modulators of striatal DA release (Rapier et al., 1990; Wonnacott et al., 2000; Zhou et al., 2001; Rice and Cragg, 2004; Zhang and Sulzer, 2004; Cragg, 2006; Threlfell et al., 2010, 2012; Cachope et al., 2012; Exley et al., 2012; Patel et al., 2012). Moreover, striatal ACh tone is affected by physiological state, including an increase in ACh release in the NAc in the later stages of feeding behavior (Mark et al., 1992; Avena et al., 2006). We show here that ChIs express LepRb, and respond directly to leptin by an increase in excitability. Compared with the abundance of LepRb-expressing neurons in other brain regions like the hypothalamus, the density of ChIs is low, constituting only 2% of striatal neurons. This low density presumably contributed to the previous conclusion that LepRb expression was absent in striatal regions in LepREGFP mice (Leshan et al., 2010; Patterson et al., 2011). A pivotal role for ChIs in leptin-enhanced DA release was supported further by the loss of leptin's effect in dStr and in the NAc core and shell in the presence of a β2*-nAChR selective antagonist in slices from wild-type mice, as well as in slices from ChAT KO mice lacking ACh synthesis.
Together, our findings highlight the ability of leptin to modulate the release of ACh and DA throughout the striatum, suggesting that leptin could influence both motor and motivated behaviors. Interestingly, previous studies have shown that peripherally or centrally administered leptin can dampen reward-related behaviors, and can even reverse the usual rewarding effects of sucrose or a high-fat diet (Domingos et al., 2011; Figlewicz et al., 2001, 2006; Bruijnzeel et al., 2011). Moreover, leptin administration can attenuate acute food deprivation-induced relapse to drug seeking (Morton et al., 2009). One explanation for these effects is that leptin inhibits the firing rate of VTA DA neurons (Hommel et al., 2006; Domingos et al., 2011; Trinko et al., 2011), which would be expected to decrease DA release in target regions in the striatum. However, our data showing that acute leptin elevation in the striatum increases axonal DA release through actions on ChIs suggest potential independence of DA release from changes in DA neuron activity. In support of such local regulation, baseline [DA]o and evoked DA release in the NAc are decreased in leptin-deficient ob/ob mice, and amphetamine-induced locomotion and sensitization are also lower in the absence of leptin compared to wild-type animals (Fulton et al., 2006). Thus, an alternative explanation for decreased attenuation of food reward following peripheral or central administration of leptin is that this exogenous leptin interferes with enhancement of DA release by endogenous leptin (and insulin) following food intake.
Regional difference in signaling pathways underlying the effect of leptin on DA release
The dependence of leptin-enhanced increases in striatal [DA]o on ACh and nAChRs, appears to reflect signaling pathways initiated by leptin primarily in striatal ChIs. Our results indicate that leptin acts via PI3K to mobilize Ca2+ from intracellular stores in an IP3R-dependent manner. Previous studies have shown that PI3K is also required for insulin-induced enhancement of striatal DA release (Stouffer et al., 2015). Consistent with the role of PI3K in the action of both hormones, the effect of leptin applied together with insulin on evoked [DA]o did not differ from the effect of leptin alone. This raises the possibility that high circulating levels of leptin, which are proportional to body weight, might be a contributing factor in the loss of the ability of insulin to enhance DA release in rats on a high-fat, high-sugar diet that promotes weight gain (Stouffer et al., 2015). Additionally, our data showing that leptin, like insulin, can catalyze interactions between ACh and DA (for review, see Cragg, 2006), point to the striatum as a locus of signal integration by these metabolic hormones.
Unexpectedly, we found that two additional signaling proteins, Akt and PKC, were involved in the increase of evoked [DA]o induced by leptin in the NAc shell, but not in dStr or NAc core. Both Akt and PKC act downstream from PI3K (Fig. 4A). Enhancement of [DA]o in the NAc shell also involved activation of RyRs, which are intracellular Ca2+ channels activated by Ca2+, including by Ca2+ entry through NMDA receptors (Schmitz et al., 2009). Based on previous work showing that leptin can enhance NMDA receptor signaling (Shanley et al., 2001; Cochrane et al., 2020; Neyens et al., 2020) and that Akt and PKC are implicated in NMDA receptor trafficking and gating (Lan et al., 2001; Lin et al., 2006; Yan et al., 2011; Chen and Roche, 2009), we hypothesized that similar processes might occur in striatum. The involvement of NMDA receptors in leptin-induced enhancement of evoked striatal [DA]o proved to be the case, but only in the NAc shell, not in dStr or NAc core. This implies amplification of glutamatergic signaling by leptin, in addition to the enhanced ACh-DA interactions already discussed. Although there is evidence for NMDA receptors on DA axons in the ventral striatum (Gracy and Pickel, 1996), our data are consistent with glutamatergic action on ChIs instead of DA axons, given that the effect of leptin on evoked [DA]o was lost in ChAT KO mice. Notably, our data also argue against a role for glutamate co-released by ChIs, as glutamate released by ChIs in NAc shell causes an inhibition of DA release via metabotropic glutamate receptor activation (Sakae et al., 2015). Future studies will be required to clarify whether an increased contribution of NMDA receptors in NAc DA release regulation is the result of an increase in receptor density or enhanced glutamate release, or both, as well as to identify the source(s) of regulatory glutamate.
In conclusion, our findings demonstrate that leptin conveys information to the striatal network by upregulating ACh and DA release, and may also enhance glutamate transmission. specifically in the NAc shell. By revealing local striatal microcircuits activated by leptin, our results point to unexpected sites of action in the basal ganglia. The data also contribute to the understanding of interactions among ACh, DA and glutamate in ingestive behavior and in obesity, which is a pathologic state involving perturbation of striatal function, as well as that of other brain regions (Ferrario et al., 2016). For example, glutamate input from cortex to the NAc contributes to DA-dependent reward-related behaviors (Kalivas et al., 2005; Berridge et al., 2009), which our findings suggest could be modulated by leptin. Similarly, DA-ACh interactions in the dStr and NAc contribute to the regulation of movement, with imbalance contributing to motor disruption in Parkinson's disease and in rodent models of Parkinson's (Hoebel et al., 2007; Quik and Wonnacott, 2011; McKinley et al., 2019; Ztaou and Amalric, 2019). Consequently, the findings reported here could point to new therapeutic strategies for disorders of motivated behavior characterized by dysregulated appetite and satiation (e.g., binge eating, bulimia, and anorexia), and potentially movement disorders (e.g., Parkinson's disease and Huntington's disease) in which circulating leptin levels are disrupted (Popovic et al., 2004; Lorefält et al., 2009).
Footnotes
This work was supported by the Marlene and Paolo Fresco Institute for Parkinson's Disease and Movement Disorders (M.E.R.). M.M. was a Fresco Postdoctoral Fellow. This work was also supported by National Institutes of Health Grants DA050165 (to M.E.R.) and DK122660 (to A.H.A.). We thank Professor Martin G. Myers Jr from the University of Michigan for providing LepRbEGFP mice and Professor Charles Nicholson from New York University Grossman School of Medicine for the software used to assess DA uptake. We also thank the Genotyping Core Laboratory of New York University Langone Health for genotyping services.
The authors declare no competing financial interests.
References
- Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao XB, Horvath TL (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116:3229–3239. 10.1172/JCI29867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler EM, Augustine GJ, Duffy SN, Charlton MP (1991) Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci 11:1496–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avena NM, Rada P, Moise N, Hoebel BG (2006) Sucrose sham feeding on a binge schedule releases accumbens dopamine repeatedly and eliminates the acetylcholine satiety response. Neuroscience 139:813–820. 10.1016/j.neuroscience.2005.12.037 [DOI] [PubMed] [Google Scholar]
- Bastioli G, Arnold JC, Mancini M, Mar AC, Gamallo-Lana B, Saadipour K, Chao MV, Rice ME (2022) Voluntary exercise boosts striatal dopamine release: evidence for the necessary and sufficient role of brain-derived neurotrophic factor. J Neurosci 42:4725–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer PL, Palarino MY, Michalek D, Busenbark K, Koller WC (1995) Weight change and body composition in patients with Parkinson's disease. J Am Diet Assoc 95:979–983. 10.1016/S0002-8223(95)00269-3 [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE, Aldridge JW (2009) Dissecting components of reward: 'liking', 'wanting', and learning. Curr Opin Pharmacol 9:65–73. 10.1016/j.coph.2008.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Corrie LW, Rogers JA, Yamada H (2011) Effects of insulin and leptin in the ventral tegmental area and arcuate hypothalamic nucleus on food intake and brain reward function in female rats. Behav Brain Res 219:254–264. 10.1016/j.bbr.2011.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF (2012) Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep 2:33–41. 10.1016/j.celrep.2012.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal RN, Parkinson JA, Hall J, Everitt BJ (2002) Emotion and motivation: the role of the amygdala, ventral striatum, and prefrontal cortex. Neurosci Biobehav Rev 26:321–352. 10.1016/s0149-7634(02)00007-6 [DOI] [PubMed] [Google Scholar]
- Caro JF, Sinha MK, Kolaczynski JW, Zhang PL, Considine RV (1996) Leptin: the tale of an obesity gene. Diabetes 45:1455–1462. 10.2337/diab.45.11.1455 [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW (2009) Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 62:471–478. 10.1016/j.neuron.2009.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhang SM, Hernán MA, Willett WC, Ascherio A (2003) Weight loss in Parkinson's disease. Ann Neurol 53:676–679. [DOI] [PubMed] [Google Scholar]
- Cochrane VA, Wu Y, Yang Z, ElSheikh A, Dunford J, Kievit P, Fortin DA, Shyng SL (2020) Leptin modulates pancreatic β-cell membrane potential through Src kinase-mediated phosphorylation of NMDA receptors. J Biol Chem 295:17281–17297. 10.1074/jbc.RA120.015489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Ma H, Kuchibhotla KV, Watson BO, Buzsáki G, Froemke RC, Tsien RW (2016) Excitation-transcription coupling in parvalbumin-positive interneurons employs a novel CaM kinase-dependent pathway distinct from excitatory neurons. Neuron 90:292–307. 10.1016/j.neuron.2016.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg SJ (2006) Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci 29:125–131. 10.1016/j.tins.2006.01.003 [DOI] [PubMed] [Google Scholar]
- De Araujo IE, Oliveira-Maia AJ, Sotnikova TD, Gainetdinov RR, Caron MG, Nicolelis MA, Simon SA (2008) Food reward in the absence of taste receptor signaling. Neuron 57:930–941. 10.1016/j.neuron.2008.01.032 [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S Jr, Elmquist JK, Lowell BB (2006) Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49:191–203. 10.1016/j.neuron.2005.12.021 [DOI] [PubMed] [Google Scholar]
- Dickson SL, Shirazi RH, Hansson C, Bergquist F, Nissbrandt H, Skibicka KP (2012) The glucagon-like peptide 1 (GLP-1) analogue, exendin-4, decreases the rewarding value of food: a new role for mesolimbic GLP-1 receptors. J Neurosci 32:4812–4820. 10.1523/JNEUROSCI.6326-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos AI, Vaynshteyn J, Voss HU, Ren X, Gradinaru V, Zang F, Deisseroth K, de Araujo IE, Friedman J (2011) Leptin regulates the reward value of nutrient. Nat Neurosci 14:1562–1569. 10.1038/nn.2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P (2011) Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci 13:7–21. 10.1038/nrn3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM, Volynski KE (2013) Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat Neurosci 16:1754–1763. 10.1038/nn.3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exley R, McIntosh JM, Marks MJ, Maskos U, Cragg SJ (2012) Striatal α5 nicotinic receptor subunit regulates dopamine transmission in dorsal striatum. J Neurosci 32:2352–2356. 10.1523/JNEUROSCI.4985-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrario CR, Labouèbe G, Liu S, Nieh EH, Routh VH, Xu S, O'Connor EC (2016) Homeostasis meets motivation in the battle to control food intake. J Neurosci 36:11469–11481. 10.1523/JNEUROSCI.2338-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ (2001) Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav 73:229–234. 10.1016/s0031-9384(01)00486-3 [DOI] [PubMed] [Google Scholar]
- Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW (2006) Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav 89:611–616. 10.1016/j.physbeh.2006.07.023 [DOI] [PubMed] [Google Scholar]
- Frühbeck G (2006) Intracellular signalling pathways activated by leptin. Biochem J 393:7–20. 10.1042/BJ20051578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton S, Woodside B, Shizgal P (2000) Modulation of brain reward circuitry by leptin. Science 287:125–128. 10.1126/science.287.5450.125 [DOI] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS (2006) Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 51:811–822. 10.1016/j.neuron.2006.09.006 [DOI] [PubMed] [Google Scholar]
- Gavello D, Carbone E, Carabelli V (2016) Leptin-mediated ion channel regulation: PI3K pathways, physiological role, and therapeutic potential. Channels (Austin) 10:282–296. 10.1080/19336950.2016.1164373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger BM, Haburcak M, Avena NM, Moyer MC, Hoebel BG, Pothos EN (2009) Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience 159:1193–1199. 10.1016/j.neuroscience.2009.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracy KN, Pickel VM (1996) Ultrastructural immunocytochemical localization of the N-methyl-D-aspartate receptor and tyrosine hydroxylase in the shell of the rat nucleus accumbens. Brain Res 739:169–181. 10.1016/s0006-8993(96)00822-0 [DOI] [PubMed] [Google Scholar]
- Guimond D, Diabir D, Porcher C, Bader F, Ferrand N, Zhu M, Appleyard SM, Wayman GA, Gaiarsa JL (2014) Leptin potentiates GABAergic synaptic transmission in the developing rodent hippocampus. Front Cell Neurosci 8:235. 10.3389/fncel.2014.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM (1995) Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269:543–546. 10.1126/science.7624777 [DOI] [PubMed] [Google Scholar]
- Hawke Z, Ivanov TR, Bechtold DA, Dhillon H, Lowell BB, Luckman SM (2009) PACAP neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J Neurosci 29:14828–14835. 10.1523/JNEUROSCI.1526-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, Grill HJ (2010) Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11:77–83. 10.1016/j.cmet.2009.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikima T, Lee CR, Witkovsky P, Chesler J, Ichtchenko K, Rice ME (2021) Activity-dependent somatodendritic dopamine release in the substantia nigra autoinhibits the releasing neuron. Cell Rep 35:108951. 10.1016/j.celrep.2021.108951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebel BG, Avena NM, Rada P (2007) Accumbens dopamine-acetylcholine balance in approach and avoidance. Curr Opin Pharmacol 7:617–627. 10.1016/j.coph.2007.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, DiLeone RJ (2006) Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51:801–810. 10.1016/j.neuron.2006.08.023 [DOI] [PubMed] [Google Scholar]
- Howe MW, Dombeck DA (2016) Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 535:505–510. 10.1038/nature18942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J (2005) Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45:647–650. 10.1016/j.neuron.2005.02.005 [DOI] [PubMed] [Google Scholar]
- Kelley AE (2004) Ventral striatal control of appetitive motivation: role in ingestive behavior and reward related learning. Neurosci Biobehav Rev 27:765–776. 10.1016/j.neubiorev.2003.11.015 [DOI] [PubMed] [Google Scholar]
- Kistner A, Lhommée E, Krack P (2014) Mechanisms of body weight fluctuations in Parkinson's disease. Front Neurol 5:84. 10.3389/fneur.2014.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosillo P, Zhang YF, Threlfell S, Cragg SJ (2016) Cortical control of striatal dopamine transmission via striatal cholinergic interneurons. Cereb Cortex 26:4160–4169. 10.1093/cercor/bhw252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krügel U, Schraft T, Kittner H, Kiess W, Illes P (2003) Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur J Pharmacol 482:185–187. 10.1016/j.ejphar.2003.09.047 [DOI] [PubMed] [Google Scholar]
- Labouèbe G, Liu S, Dias C, Zou H, Wong JC, Karunakaran S, Clee SM, Phillips AG, Boutrel B, Borgland SL (2013) Insulin induces long-term depression of ventral tegmental area dopamine neurons via endocannabinoids. Nat Neurosci 16:300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4:382–390. 10.1038/86028 [DOI] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM (1996) Abnormal splicing of the leptin receptor in diabetic mice. Nature 379:632–635. [DOI] [PubMed] [Google Scholar]
- Leinninger GM, Jo YH, Leshan RL, Louis GW, Yang H, Barrera JG, Wilson H, Opland DM, Faouzi MA, Gong Y, Jones JC, Rhodes CJ, Chua S Jr, Diano S, Horvath TL, Seeley RJ, Becker JB, Münzberg H, Myers MG Jr (2009) Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab 10:89–98. 10.1016/j.cmet.2009.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshan RL, Opland DM, Louis GW, Leinninger GM, Patterson CM, Rhodes CJ, Münzberg H, Myers MG Jr (2010) Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci 30:5713–5723. 10.1523/JNEUROSCI.1001-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. J Neurosci 30:1788–1797. 10.1523/JNEUROSCI.5604-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Jover-Mengual T, Wong J, Bennett MVL, Zukin RS (2006) PSD-95 and PKC converge in regulating NMDA receptor trafficking and gating. Proc Natl Acad Sci USA 103:19902–19907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo F, Mancini M, Ibraheem PL, Aryal S, Mesini C, Patel JC, Penhos E, Rahman N, Mamcarz M, Santini E, Rice ME, Klann E (2021) Cell-type-specific disruption of PERK-eIF2α signaling in dopaminergic neurons alters motor and cognitive function. Mol Psychiatry 26:6427–6450. 10.1038/s41380-021-01099-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorefält B, Toss G, Granérus AK (2009) Weight loss, body fat mass, and leptin in Parkinson's disease. Mov Disord 24:885–890. 10.1002/mds.22466 [DOI] [PubMed] [Google Scholar]
- Mancini M, Soldovieri MV, Gessner G, Wissuwa B, Barrese V, Boscia F, Secondo A, Miceli F, Franco C, Ambrosino P, Canzoniero LMT, Bauer M, Hoshi T, Heinemann SH, Taglialatela M (2014) Critical role of large-conductance calcium- and voltage-activated potassium channels in leptin-induced neuroprotection of N-methyl-d-aspartate-exposed cortical neurons. Pharmacol Res 87:80–86. 10.1016/j.phrs.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark GP, Rada P, Pothos E, Hoebel BG (1992) Effects of feeding and drinking on acetylcholine release in the nucleus accumbens, striatum, and hippocampus of freely behaving rats. J Neurochem 58:2269–2274. 10.1111/j.1471-4159.1992.tb10973.x [DOI] [PubMed] [Google Scholar]
- McKinley JW, Shi Z, Kawikova I, Hur M, Bamford IJ, Sudarsana Devi SP, Vahedipour A, Darvas M, Bamford NS (2019) Dopamine deficiency reduces striatal cholinergic interneuron function in models of Parkinson's disease. Neuron 103:1056–1072. 10.1016/j.neuron.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietlicki-Baase EG, Ortinski PI, Reiner DJ, Sinon CG, McCutcheon JE, Pierce RC, Roitman MF, Hayes MR (2014) Glucagon-like peptide-1 receptor activation in the nucleus accumbens core suppresses feeding by increasing glutamatergic AMPA/kainate signaling. J Neurosci 34:6985–6992. 10.1523/JNEUROSCI.0115-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mietlicki-Baase EG, Reiner DJ, Cone JJ, Olivos DR, McGrath LE, Zimmer DJ, Roitman MF, Hayes MR (2015) Amylin modulates the mesolimbic dopamine system to control energy balance. Neuropsychopharmacology 40:372–385. 10.1038/npp.2014.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohebi A, Pettibone JR, Hamid AA, Wong JT, Vinson LT, Patriarchi T, Tian L, Kennedy RT, Berke JD (2019) Dissociable dopamine dynamics for learning and motivation. Nature 570:65–70. 10.1038/s41586-019-1235-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY (1980) From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol 14:69–97. 10.1016/0301-0082(80)90018-0 [DOI] [PubMed] [Google Scholar]
- Morton GJ, Blevins JE, Kim F, Matsen M, Figlewicz DP (2009) The action of leptin in the ventral tegmental area to decrease food intake is dependent on Jak-2 signaling. Am J Physiol Endocrinol Metab 297:E202–210. 10.1152/ajpendo.90865.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Enjoji M, Koyama S (2018) Leptin attenuates D(2) receptor-mediated inhibition of putative ventral tegmental area dopaminergic neurons. Physiol Rep 6:e13631. 10.14814/phy2.13631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto S, Varatharajan R, Joseph K, Moser A (2016) Nasal administration of leptin dose-dependently increases dopamine and serotonin outflow in the rat nucleus accumbens. J Neural Transm (Vienna) 123:1247–1254. 10.1007/s00702-016-1591-9 [DOI] [PubMed] [Google Scholar]
- Neyens D, Zhao H, Huston NJ, Wayman GA, Ritter RC, Appleyard SM (2020) Leptin sensitizes NTS neurons to vagal input by increasing postsynaptic NMDA receptor currents. J Neurosci 40:7054–7064. 10.1523/JNEUROSCI.1865-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WW, Myers MG Jr (2018) Leptin and the maintenance of elevated body weight. Nat Rev Neurosci 19:95–105. 10.1038/nrn.2017.168 [DOI] [PubMed] [Google Scholar]
- Patel JC, Rice ME (2013) Monitoring axonal and somatodendritic dopamine release using fast-scan cyclic voltammetry in brain slices. Methods Mol Biol 96:243–273. [DOI] [PubMed] [Google Scholar]
- Patel JC, Witkovsky P, Avshalumov MV, Rice ME (2009) Mobilization of calcium from intracellular stores facilitates somatodendritic dopamine release. J Neurosci 29:6568–6579. 10.1523/JNEUROSCI.0181-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, Rossignol E, Rice ME, Machold RP (2012) Opposing regulation of dopaminergic activity and exploratory motor behavior by forebrain and brainstem cholinergic circuits. Nat Commun 3:1172. 10.1038/ncomms2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JC, Stouffer MA, Nicholson C, Mancini M, Carr KD, Rice ME (2019) Interactions between insulin and diet on striatal dopamine uptake kinetics in rodent brain slices. Eur J Neurosci 49:794–804. 10.1111/ejn.13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson CM, Leshan RL, Jones JC, Myers MG Jr (2011) Molecular mapping of mouse brain regions innervated by leptin receptor-expressing cells. Brain Res 1378:18–28. 10.1016/j.brainres.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry ML, Leinninger GM, Chen R, Luderman KD, Yang H, Gnegy ME, Myers MG Jr, Kennedy RT (2010) Leptin promotes dopamine transporter and tyrosine hydroxylase activity in the nucleus accumbens of Sprague-Dawley rats. J Neurochem 114:666–674. 10.1111/j.1471-4159.2010.06757.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Centonze D, Bernardi G, Calabresi P (2005) Striatal synaptic plasticity: implications for motor learning and Parkinson's disease. Mov Disord 20:395–402. 10.1002/mds.20394 [DOI] [PubMed] [Google Scholar]
- Popovic V, Svetel M, Djurovic M, Petrovic S, Doknic M, Pekic S, Miljic D, Milic N, Glodic J, Dieguez C, Casanueva FF, Kostic V (2004) Circulating and cerebrospinal fluid ghrelin and leptin: potential role in altered body weight in Huntington's disease. Eur J Endocrinol 151:451–455. 10.1530/eje.0.1510451 [DOI] [PubMed] [Google Scholar]
- Quarta D, Di Francesco C, Melotto S, Mangiarini L, Heidbreder C, Hedou G (2009) Systemic administration of ghrelin increases extracellular dopamine in the shell but not the core subdivision of the nucleus accumbens. Neurochem Int 54:89–94. 10.1016/j.neuint.2008.12.006 [DOI] [PubMed] [Google Scholar]
- Quik M, Wonnacott S (2011) α6β2* and α4β2* nicotinic acetylcholine receptors as drug targets for Parkinson's disease. Pharmacol Rev 63:938–966. 10.1124/pr.110.003269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapier C, Lunt GG, Wonnacott S (1990) Nicotinic modulation of [3H]dopamine release from striatal synaptosomes: pharmacological characterisation. J Neurochem 54:937–945. 10.1111/j.1471-4159.1990.tb02341.x [DOI] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci 7:583–584. 10.1038/nn1244 [DOI] [PubMed] [Google Scholar]
- Richard JM, Castro DC, Difeliceantonio AG, Robinson MJF, Berridge KC (2013) Mapping brain circuits of reward and motivation: in the footsteps of Ann Kelley. Neurosci Biobehav Rev 37:1919–1931. 10.1016/j.neubiorev.2012.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring LE, Zeltser LM (2010) Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J Clin Invest 120:2931–2934. 10.1172/JCI41985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24:1265–1271. 10.1523/JNEUROSCI.3823-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae DY, et al. (2015) The absence of VGLUT3 predisposes to cocaine abuse by increasing dopamine and glutamate signaling in the nucleus accumbens. Mol Psychiatry 20:1448–1459. 10.1038/mp.2015.104 [DOI] [PubMed] [Google Scholar]
- Schmitz Y, Luccarelli J, Kim M, Wang M, Sulzer D (2009) Glutamate controls growth rate and branching of dopaminergic axons. J Neurosci 29:11973–11981. 10.1523/JNEUROSCI.2927-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W (2002) Getting formal with dopamine and reward. Neuron 36:241–263. 10.1016/s0896-6273(02)00967-4 [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK (2009) Leptin targets in the mouse brain. J Comp Neurol 514:518–532. 10.1002/cne.22025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley LJ, Irving AJ, Harvey J (2001) Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J Neurosci 21:RC186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd PR, Withers DJ, Siddle K (1998) Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J 333:471–490. 10.1042/bj3330471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibicka KP, Hansson C, Alvarez-Crespo M, Friberg PA, Dickson SL (2011) Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience 180:129–137. 10.1016/j.neuroscience.2011.02.016 [DOI] [PubMed] [Google Scholar]
- Skibicka KP, Shirazi RH, Rabasa-Papio C, Alvarez-Crespo M, Neuber C, Vogel H, Dickson SL (2013) Divergent circuitry underlying food reward and intake effects of ghrelin: dopaminergic VTA-accumbens projection mediates ghrelin's effect on food reward but not food intake. Neuropharmacology 73:274–283. 10.1016/j.neuropharm.2013.06.004 [DOI] [PubMed] [Google Scholar]
- Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, Machold RP, Jones KT, Cabeza de Vaca S, Reith MEA, Carr KD, Rice ME (2015) Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun 6:8543. 10.1038/ncomms9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Cragg SJ, Rice ME (2016) Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6:123–148. 10.1016/j.baga.2016.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczypka MS, Kwok K, Brot MD, Marck BT, Matsumoto AM, Donahue BA, Palmiter RD (2001) Dopamine production in the caudate putamen restores feeding in dopamine-deficient mice. Neuron 30:819–828. 10.1016/s0896-6273(01)00319-1 [DOI] [PubMed] [Google Scholar]
- Threlfell S, Clements MA, Khodai T, Pienaar IS, Exley R, Wess J, Cragg SJ (2010) Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J Neurosci 30:3398–3408. 10.1523/JNEUROSCI.5620-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ (2012) Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75:58–64. 10.1016/j.neuron.2012.04.038 [DOI] [PubMed] [Google Scholar]
- Trinko R, Gan G, Gao XB, Sears RM, Guarnieri DJ, DiLeone RJ (2011) Erk1/2 mediates leptin receptor signaling in the ventral tegmental area. PLoS One 6:e27180. 10.1371/journal.pone.0027180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubert C, Taravini IRE, Flores-Barrera E, Sánchez GM, Prost MA, Avale ME, Tseng KY, Rela L, Murer MG (2016) Decrease of a current mediated by Kv1.3 channels causes striatal cholinergic interneuron hyperexcitability in experimental parkinsonism. Cell Rep 16:2749–2762. 10.1016/j.celrep.2016.08.016 [DOI] [PubMed] [Google Scholar]
- Ungerstedt U (1971) Adipsia and aphagia after 6-hydroxydopamine induced degeneration of the nigro-striatal dopamine system. Acta Physiol Scand Suppl 367:95–122. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Waterfield MD (1999) Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res 253:239–254. 10.1006/excr.1999.4701 [DOI] [PubMed] [Google Scholar]
- Villanueva EC, Myers MG Jr (2008) Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 32:S8–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Wang F, Yang MJ, Yu DF, Wu WN, Liu J, Ma LQ, Cai F, Chen JG (2008) Leptin regulated calcium channels of neuropeptide Y and proopiomelanocortin neurons by activation of different signal pathways. Neuroscience 156:89–98. 10.1016/j.neuroscience.2008.04.079 [DOI] [PubMed] [Google Scholar]
- Wise RA (2006) Role of brain dopamine in food reward and reinforcement. Philos Trans R Soc Lond B Biol Sci 361:1149–1158. 10.1098/rstb.2006.1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW (2000) Presynaptic nicotinic receptors modulating dopamine release in the rat striatum. Eur J Pharmacol 393:51–58. 10.1016/s0014-2999(00)00005-4 [DOI] [PubMed] [Google Scholar]
- Yan JZ, Xu Z, Ren SQ, Hu B, Yao W, Wang SH, Liu SY, Lu W (2011) Protein kinase C promotes N-Methyl-d-aspartate (NMDA) receptor trafficking by indirectly triggering calcium/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation. J Biol Chem 286:25187–25200. 10.1074/jbc.M110.192708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun IA, Wakabayashi KT, Fields HL, Nicola SM (2004) The ventral tegmental area is required for the behavioral and nucleus accumbens neuronal firing responses to incentive cues. J Neurosci 24:2923–2933. 10.1523/JNEUROSCI.5282-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sulzer D (2004) Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci 7:581–582. 10.1038/nn1243 [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4:1224–1229. 10.1038/nn769 [DOI] [PubMed] [Google Scholar]
- Zhu MH, Chae M, Kim HJ, Lee YM, Kim MJ, Jin NG, Yang DK, So I, Kim KW (2005) Desensitization of canonical transient receptor potential channel 5 by protein kinase C. Am J Physiol Cell Physiol 289:C591–C600. 10.1152/ajpcell.00440.2004 [DOI] [PubMed] [Google Scholar]
- Ztaou S, Amalric M (2019) Contribution of cholinergic interneurons to striatal pathophysiology in Parkinson's disease. Neurochem Int 126:1–10. 10.1016/j.neuint.2019.02.019 [DOI] [PubMed] [Google Scholar]
- zur Nedden S, Eith R, Schwarzer C, Zanetti L, Seitter H, Fresser F, Koschak A, Cameron AJM, Parker PJ, Baier G, Baier-Bitterlich G (2018) Protein kinase N1 critically regulates cerebellar development and long-term function. J Clin Invest 128:2076–2088. 10.1172/JCI96165 [DOI] [PMC free article] [PubMed] [Google Scholar]