

Abstract
Williams syndrome (WS) is a relatively rare microdeletion disorder that occurs in as many as 1:7,500 individuals. It arises due to mispairing of low-copy DNA repetitive elements at meiosis. Deletion size is similar across most individuals with WS and leads to loss of one copy of 25–27 genes on chromosome 7q11.23. The resulting unique disorder affects multiple systems, with cardinal features including, but not limited to, cardiovascular disease (characteristically stenosis of the great arteries and most notably supravalvar aortic stenosis), a distinctive craniofacial appearance and a specific cognitive and behavioural profile that includes intellectual disability and hypersociability. Genotype–phenotype evidence is strongest for the elastin (ELN) gene, which is responsible for the vascular and connective tissue features of WS, and for the transcription factor genes GTF2I and GTF2IRD1, which are known to affect intellectual ability, social functioning and anxiety. Mounting evidence also ascribes phenotypic consequences to deletion of BAZ1B, LIMK1, STX1A and MLXIPL, but more work is needed to understand the mechanism by which these deletions contribute to clinical outcomes. Age of diagnosis has fallen in regions of the world where technological advances, such as chromosomal microarray, enable clinicians to make the diagnosis of WS without formally suspecting it, allowing earlier intervention by medical and developmental specialists. Phenotypic variability is considerable for all cardinal features of WS, but the specific sources of this variability remain unknown. Further investigation to identify factors responsible for these differences may lead to mechanism-based rather than symptom-based therapies and, therefore, should be a high research priority.
Introduction
Williams syndrome (WS; also known as Williams–Beuren syndrome; OMIM 194050), is a distinctive multisystem disorder (Figure 1, Supplementary box 1). The most common areas of involvement include the cardiovascular, central nervous, gastrointestinal and endocrine systems, although any organ system could be affected. Recognition of WS as a distinct clinical syndrome dates back to the mid-20th century1–3, and knowledge of the phenotype has steadily expanded over the ensuing ~60 years.
Figure 1. Salient features of Williams syndrome.
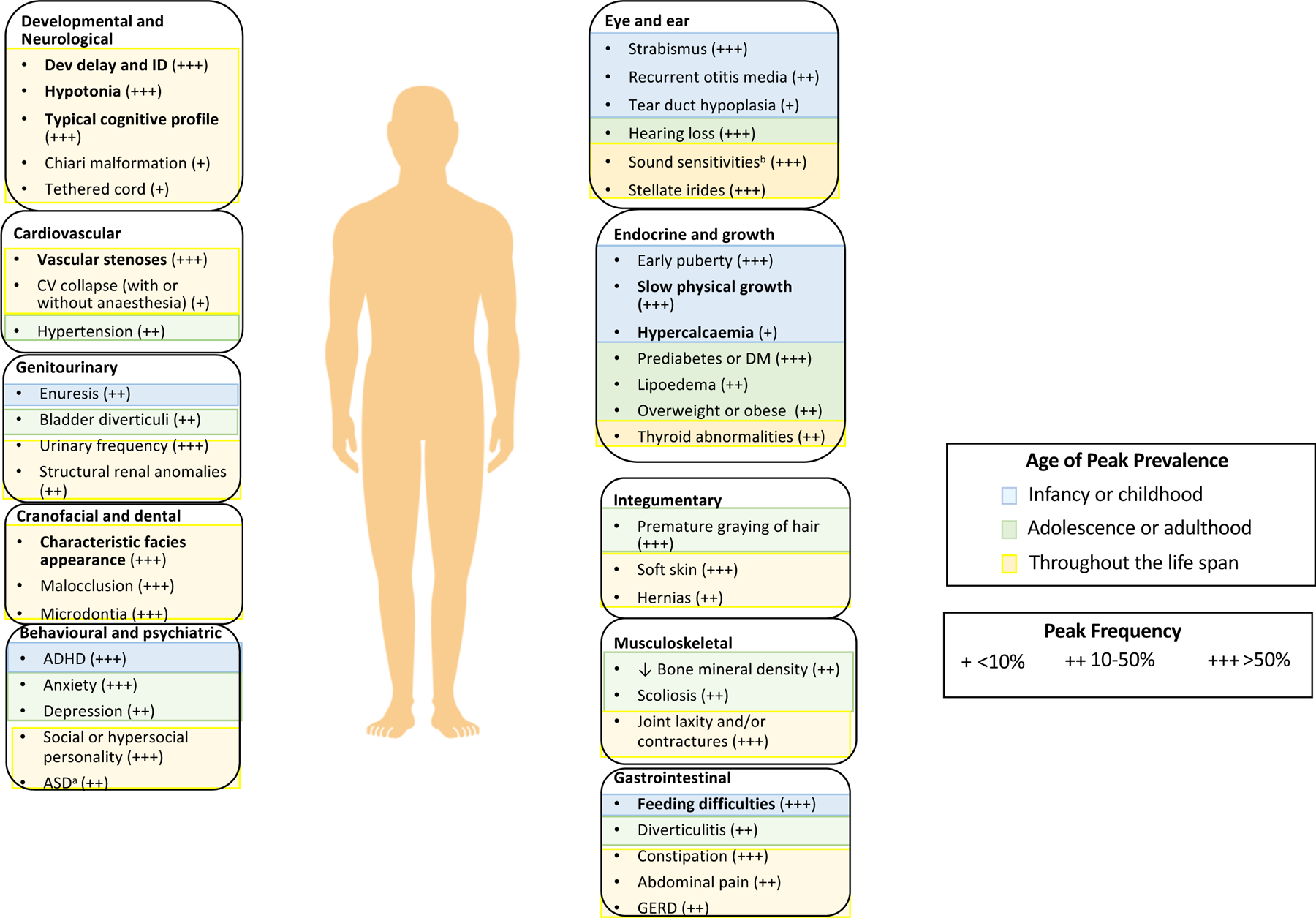
The age of peak prevalence and frequency of prominent signs or symptoms in organ systems affected in Williams syndrome are indicated. Features in bold are the common presenting features listed in the Clinical Diagnosis sectionin the main text. Further reading for features in plain text is included in Supplementary box 1.
aEstimates of co-occurring autism spectrum disorder (ASD) vary (12–20%). Most individuals with Williams syndrome who have ASD fit in Wing and Gould’s active-but-odd subtype of ASD292 rather than the aloof subtype of ASD232,292. As such, the diagnosis of, and interventions for, ASD in Williams syndrome are complex and ideally benefit from engagement of practitioners who are knowledgeable about both disorders. bSound sensitivities include one or more of the following: hyperacusis, odynacusis, auditory allodynia and auditory fascinations. ADHD, attention deficit hyperactivity disorder; CV, cardiovascular; DM, diabetes mellitus; GERD, gastro-oesophageal reflux disease; ID, intellectual disability.
Note: Figure 1 was re-drawn by the Nature Reviews Disease Primers art editor and permission for reproduction of the final figure was not granted by the publisher. Please see the published version of the paper for the final Fig. 1 at DOI: 10.1038/s41572-021-00276-z
We now know that cardiovascular disease is present in most individuals with WS4–6, with the most frequent abnormalities involving vascular stenoses of medium- and large-sized arteries (referred to as elastin arteriopathy)7. Depending on the location, severity and timing of onset, the management of the vasculopathy consists of noninvasive or surgical intervention complemented by life-long monitoring. Additional cardiovascular features include hypertension and a small but increased risk of cardiovascular sudden death8–12. The neurodevelopmental phenotype is unique and multifaceted. Mild to moderate intellectual disability is common but not universal and is seen in conjunction with a distinct cognitive profile of relative strengths and weaknesses13,14. In addition, there is a characteristic personality profile that includes overfriendliness, shortened attention span and/or distractibility, nonsocial specific phobias and anxiety15,16.
The genetic basis of WS was first identified in 1993, when fluorescence in situ hybridization (FISH) studies showed deletion of an elastin allele (ELN) on chromosome 7q17. We now know that WS is caused by a <2-million base pair (Mb) microdeletion on chromosome 7q11.23 and that the local genomic architecture predisposes to de novo occurrence of this deletion18. Individuals with WS are, therefore, hemizygous for the 25–27 genes that map to this interval, and reduction in the gene product of several key genes contributes to specific aspects of the WS phenotype.
Since WS was initially described, advances have been made in our understanding of the complexity and changing nature of the phenotype, the genetic basis of WS, the mechanisms that lead to selected phenotypes and the benefit of certain interventions. Yet, many more unanswered questions remain. Accordingly, our ability to optimize care and improve outcomes is modest.
In this Primer, we provide a snapshot of selected WS features across the lifespan, outline current and emerging diagnostic technologies, discuss the genetic basis of some of the most impactful WS features, and elaborate on pathophysiological mechanisms, where known. This information is key for formulating high-priority future research questions, the answers to which could accelerate WS-specific treatments.
Epidemiology
WS is a rare panethnic genetic condition. Although this syndrome has been described in different populations around the world, most reports address clinical and molecular findings, with little focus on epidemiological data. The most widely cited epidemiological study is from Norway19, which reports a prevalence of 1 in 7,500 live births, a prevalence which is higher than that often cited in many non-epidemiologic sources20,21. WS is sufficiently rare that it is unfamiliar to most doctors, scientists, and researchers.
The age of diagnosis of WS has trended towards younger ages over the past decades, most notably in high-income countries with greater availability of molecular diagnostic testing. In cohorts from the USA and Australia, the median age of diagnosis decreased by more than 2 years to around 1 year of age since the 1980s (B.A.K. and M.P., unpublished work). (Figure 2 and Supplementary box 2 were removed in post-acceptance processing). However, series from other countries indicate that diagnosis is often still established during childhood rather than infancy, even with access to molecular confirmation22,23. Several studies reported particular difficulty in diagnosing WS in African populations (or in those of African descent)24–26, owing to a variety of factors. Unfortunately, nearly all studies on WS, independent of topic, describe Caucasian individuals, and few studies whose participants represent diverse populations have been published27. Of note, the absence of clinically evident cardiovascular disease was associated with later diagnosis28.
Figure 2:
Note: Although Figure 2 (as cited in this text) was accepted after peer review, it was removed from the final manuscript at production. This figure does not exist in the final paper at DOI: 10.1038/s41572-021-00276-z
The prevalence of WS is comparable in males and females5,29. However, males are more likely to have severe cardiac disease30, especially supravalvar aortic stenosis (SVAS)29,31. There is no evidence that WS prevalence changes with parental age. Moreover, consistent differences in phenotype have been associated with the sex of the transmitting parent (that is, whether the deletion arose in the sperm or the egg)18,32.
Vascular anomalies, such as SVAS and stenosis of other large arteries including the pulmonary arteries, descending aorta, renals, mesenterics, and coronary arteries contribute to morbidity and mortality across the life span4. Other features, such as diverticular disease and aortic or mitral valve dysfunction, may also influence mortality in older age groups but are to-date insufficiently quantified in the literature. Sudden death incidence has been reported as 1:1000 patient years and is often associated with administration of sedation or anaesthesia for cardiac surgery; this rate is 25 to 100 times higher than in the age-matched general population12. Several adult cohorts have been described with participants as old as 86, but few studies included long-term adult follow up33,34, making it difficult to accurately estimate the life expectancy of individuals with WS.
Mechanisms/pathophysiology
General mechanisms underlying WS
In the 1990s, compelling evidence emerged indicating that WS is a genetic disorder with an autosomal dominant mode of inheritance35–38. Non-genetic risk factors are not known to contribute to the occurrence of WS.
Genomic structure and rearrangements.
WS is caused by the pathological loss of the Williams syndrome critical region (WSCR), a 1.55–1.83 Mb region which encompasses 25–27 unique protein-coding genes on chromosome 7q11.23. The WSCR frequently undergoes rearrangement due to the presence of large, complex segmental duplications termed low-copy repeats (LCRs), which are highly similar to one another and flank the WSCR39. The LCRs extend for several hundreds of kilobases, are comprised of genes and pseudogenes organized into distinct blocks (designated A, B and C), and contain extensive stretches of >99% nucleotide identity. They are thought to have emerged during primate evolution — first by duplication of smaller segments and later by transposable element (for example, Alu-mediated) shuffling, to produce the complex arrangement that exists in humans today40.
The LCRs mediate non-allelic homologous recombination (NAHR) events between the highly similar DNA sequences during meiosis, resulting in an increased rate of de novo copy number variation (CNV) events within the region39,41–43 (Figure 3). The WS deletion commonly occurs through NAHR between B block sequences in direct orientation with respect to each other, with the specific breakpoints depend on the precise site of NAHR39. The reciprocal event (that is, duplication of the same genomic area) produces a condition referred to as 7q11.23 duplication syndrome, resulting in 3 copies of each WSCR gene. Recombination between B blocks (LCRs with the highest nucleotide identity) in an inverted, rather than direct, orientation, results in an inversion of the intervening chromosome segment42 (Figure 3). This inversion, which is present in 6–7% of the general population44, does not cause symptoms45 but seems to increase the incidence of subsequent meiotic rearrangements42,44. Other LCR-specific rearrangements have also been observed at a higher frequency in the transmitting parents of children with WS41.
Figure 3. Genomic organization of the Williams syndrome critical region.
The vast majority of Williams syndrome (WS) deletions (~90%) span a 1.55-million base pair (Mb) region encompassing 25–27 protein-coding genes in the Williams syndrome critical region (WSCR) on chromosome 7q11.23, with the remainder being slightly larger (1.83 Mb)39. The larger, less common deletion occurs between low-copy repeats (LCRs) designated as ‘A blocks’, which are present in the great apes, whereas the smaller, more common deletion occurs between ‘B blocks’, which are only present in humans. B blocks originate from a more recent evolutionary duplication event and have a higher sequence identity than A blocks40. The WS deletion commonly occurs through non-allelic homologous recombination between B block sequences in direct orientation with respect to each other39; however, recombination between B blocks in an inverted orientation also occurs, resulting in inversion of the intervening chromosome segment42. Smaller, atypical deletions occur infrequently but can provide valuable insight into the genotype–phenotype relationship59,62,138–140. Several names exist for many of the genes mapping to WSCR (see 293and https://www.ncbi.nlm.nih.gov/gene/). ID, intellectual disability.
Note: Figure 3 was drawn by the Nature Reviews Disease Primers art editor and permission for reproduction of the final figure was not granted by the publisher. This material is labeled as Fig. 2 in the published version of the paper DOI: 10.1038/s41572-021-00276-z
Atypical deletions.
While most deletions span the typical 1.55–1.83 Mb interval at 7q11.23, there are individuals with rare deletions that encompass smaller or larger segments of the WSCR, often with one common and one unique breakpoint. Larger deletions that extend beyond the WSCR generally cause additional features. When these deletions extend toward the telomere and span the YWHAG and/or MAGI2 genes, seizures are common46,47, although there are reports of epilepsy in individuals with the typical deletion48. Inclusion of the AUTS2 gene on the centromeric side may result in smaller head size than is usually seen in WS49, and larger deletions can also alter the characteristic WS behavioural profile49,50. Deletion of HIP1 (formerly HSP27) in particular has been associated with more severe intellectual disability50,51.
Smaller deletions result in a subset of the phenotypic features seen in classic WS40,50,52–64, but clear-cut correlations between deletion size and specific phenotypic features are challenging, with the exception of ELN. This is likely due to the paucity and variety of small deletions, differing methods for phenotypic assessment, and high likelihood of the combinatorial effects of gene deletion. Several specific genotype–phenotype relationships are discussed in the next section.
Genomic analyses.
CNV events of the 7q11.23 region have been shown to affect both gene transcription and DNA methylation across the entire genome. Initial studies in WS lymphoblast cell lines identified dysregulation of genes involved in glycolysis and neuronal migration65, while subsequent studies of blood RNA highlighted upregulation of three gene expression modules linked to B cell activation, RNA processing and RNA transport66.
Transcriptome analysis of more relevant cell types has been made possible by the ability to reprogramme somatic cells into induced pluripotent stem cells (iPSCs) and direct them down specific cell lineages67. For example, iPSC-derived cortical neurons from individuals with WS show reduced expression of genes involved in neurotransmitter receptor activity, synaptic assembly and potassium channel complexes68. Comparison of gene expression in WS iPSCs with those from individuals with 7q11.23 duplication syndrome revealed that many of the differentially expressed genes have a symmetrically opposite pattern of expression69. A similar symmetrical gene–dose-dependent pattern was seen in DNA methylation analysis of blood DNA from individuals with WS (7q11.23 deletion) and those with the 7q11.23 duplication syndrome70, suggesting that CNV in this area affects epigenetic regulation of the genome.
Molecular mechanisms
The WSCR contains 25–27 genes and several noncoding RNAs. Knowledge about how each of these genes contributes to the WS phenotype is still growing (Figure 4). Several mouse models inform those efforts, including single gene knockouts, as well as deletion of the entire WSCR (CD)71–73 and two half-deletions (PD and DD)74. To refine genotype-phenotype correlations, we will focus only on selected single gene knockout models.
Figure 4. Phenotypic consequences of deleting key genes in the Williams syndrome critical region.
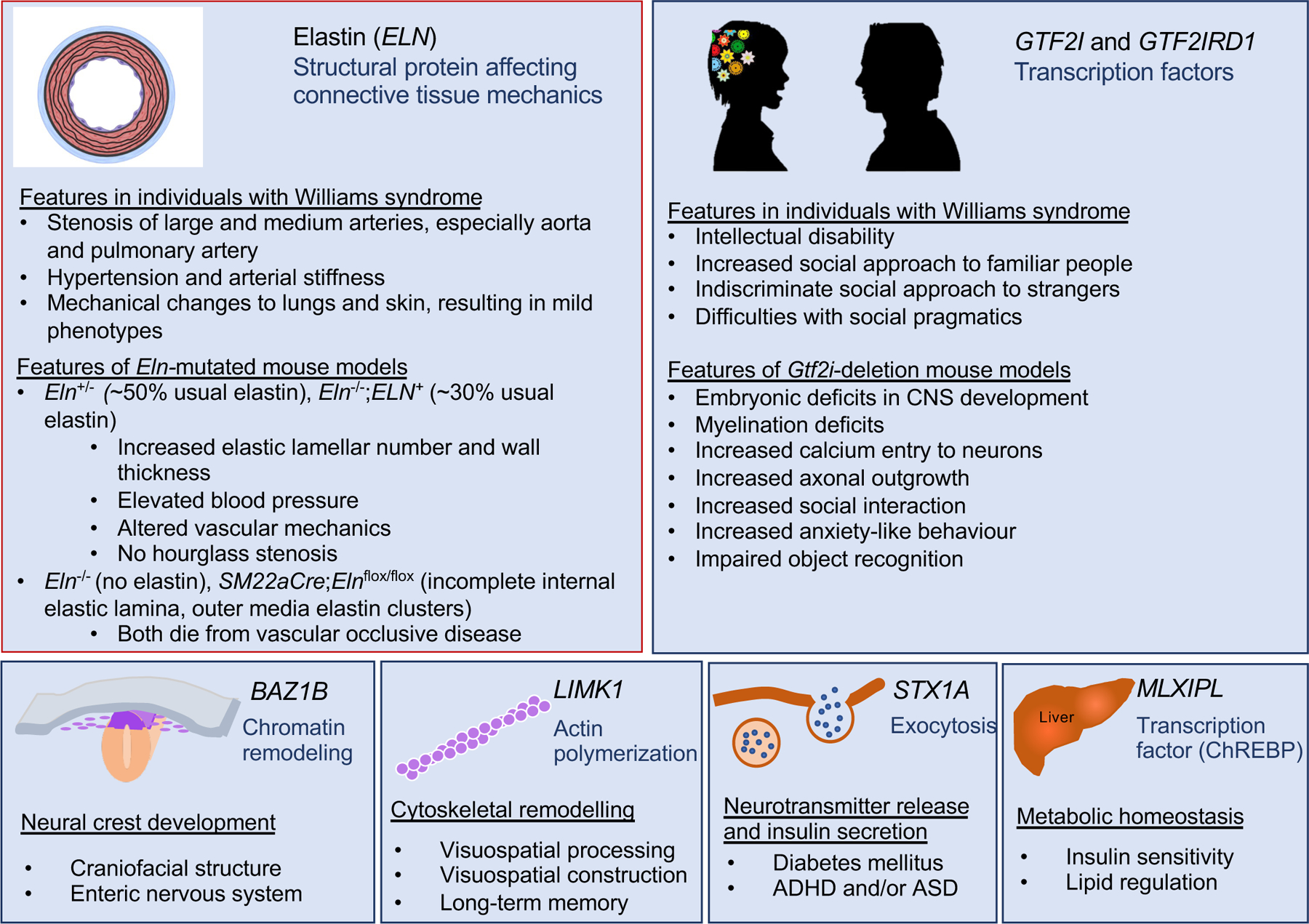
Putative or elucidated genotype–phenotype relationships for six genes in the Williams syndrome critical region (WSCR) are depicted, including elastin (ELN), general transcription factor II-I (GTF2I), BAZ1B, LIM domain kinase 1 (LIMK1), syntaxin 1A (STX1A) and carbohydrate-responsive element-binding protein (ChREBP). Phenotypes in mouse models and in individuals with Williams syndrome are indicated for ELN and the GTF2I genes because, presently, their mechanisms of action are best delineated and they offer the best targets for therapy.
Note: Figure 4 was redrawn by the Nature Reviews Disease Primers art editor and permission for reproduction of the final figure was not granted by the publisher. This version was designed using BioRender. This material is labeled as Fig. 3 in the published version of the paper DOI: 10.1038/s41572-021-00276-z
Seven genes (BAZ1B, VPS37D, STX1A, LIMK1, CLIP2, GTF2IRD1 and GTF2I) have a probability of loss-of-function intolerance (pLI) score of 0.9 or higher75, suggesting that only a subset of the genes in the WSCR directly contributes to phenotype. A pLI score is calculated by examining the frequency of loss of function variants in a population; fewer variants than expected is associated with a higher score and implies a greater likelihood of pathogenicity. It is important to point out that the gene with the greatest support for a role in the phenotypic consequences of WS is ELN, which has a pLI of 0. Similarly, considerable evidence implicates deletion of MLXIPL (pLI = 0.05) in metabolic aspects of WS. These two examples highlight the inadequacy of predictors such as pLI in identifying all pathogenetic genes. An overview of the best characterized genotype–phenotype correlations for WSCR genes is provided below.
Elastin.
The ELN gene is transcribed in tissues that stretch and recoil, such as the lungs, skin and elastic arteries (including the aorta, where elastin accounts for up to 50% of the vessel’s dry weight)76,77. The protein consists of repeating hydrophobic and crosslinking domains. The crosslinks allow monomers to be bound to one another in a highly interwoven polymer that allows for distribution of force, while the hydrophobic domains drive the recoil process through entropy when they are exposed to an aqueous environment with tissue expansion (stretch)78,79. The polymer is long-lived, with a short window for deposition and a calculated half-life of 74 years80. Interestingly, although robust elastogenesis occurs only during early growth and development, ELN is thought to be continually transcribed throughout the lifespan81. Outside of this tight developmental window, ELN transcripts are quickly turned over82,83. As such, this connection between transcription, translation and assembly is ripe for investigation.
Individuals with loss-of-function point mutations84–86 or intragenic deletions87 within ELN have ELN-associated familial SVAS, and develop cardiovascular manifestations that are indistinguishable from those found in WS. Common features include focal or long-segment stenosis (narrowing) of the large elastic arteries in the setting of a globally narrow and thick-walled vasculature4,6,7,30,88. Stenosis of the supravalvar aortic and supravalvar pulmonary arteries are the most common and show considerable variability in severity29,89. While pulmonary artery stenoses often improves with age, narrowing on the aortic side may stay the same, improve or worsen with time 5,88,90. Other vessels, such as the descending aorta, renal arteries, mesenteric arteries and coronaries may also exhibit stenosis, with symptomatology pointing to hypoperfusion of the associated end organ (hypertension, abdominal pain, and cardiac hypoperfusion with ST elevation or sudden death). Even in the absence of stenosis, individuals with either WS or familial SVAS have high rates of hypertension and vascular stiffness, which are detectable as early as infancy and childhood8,91. An increased sudden death relative risk with and without anaesthesia has been reported10,92–94; the precise mechanism by which this occurs is not currently known but it is expected to be multifactorial, reflecting the complex vascular pathophysiology95.
Heterozygous Eln knockout (Eln+/–) mice recapitulate relevant cardiovascular features of WS, including aortic wall thickening, hypertension and cardiac hypertrophy7,however, it has been difficult to replicate the hourglass-type supravalvar stenoses commonly seen in individuals with WS or familial SVAS. Neither Eln+/–nor transgenic Eln–/– mice expressing the human ELN gene (Eln–/–;ELN+ mice, which have 30% of normal elastin levels) develop frank stenosis, although Eln–/–; ELN+ mice exhibit more severe arterial wall thickening, luminal narrowing, hypertension and cardiac hypertrophy than Eln+/– mice96. Discrete stenosis or coarctation (congenital narrowing) of the aortic arch as well as the development of neointima (thickening of the intima characteristic of segmental aortic stenoses in WS) has been described in mice with a homozygous Eln deletion restricted to vascular smooth muscle cells97. Unfortunately, most of these mice do not survive past postnatal day 18.
Several hypotheses have been postulated for the mechanism by which elastin insufficiency causes large vessel arteriopathy. Segmental stenoses are thought to develop through increased proliferation and migration of vascular smooth muscle cells due to a reduction or lack of elastin98–100. Lineage tracing indicates that the excess smooth muscle cells responsible for inward remodeling of the arterial wall are not clonal but are derived from multiple existing smooth muscle cells in the media layer101. Inward remodeling is caused in part by excess integrin β3 signaling, as genetic or pharmacological inhibition of this pathway reduces vascular pathology and extends lifespan in Eln–/– mice101. Other work suggests that rather than an increase in proliferation, elastin insufficiency produces medial fibrosis, altered mobility of smooth muscle cells and abnormal circumferential growth, leading to smaller lumen size and thicker arterial walls102. Additional studies suggest that hypertension arises in Eln+/– mice as part of a developmental adaptation to normalize vessel wall stress, capitalizing on the increased pressures to prop open the narrow, stiff elastin-insufficient vessels103, although more recent studies indicate that reactive oxygen species (ROS) production may also have a role74. Several additional molecular and cellular mechanisms impact the pathogenesis of elastin arteriopathy, including mechanistic target of rapamycin (mTOR) perturbation of smooth muscle mechanosensing 104–106, and the adaptive immune system29.
In addition to vascular disease, patients (and mice) with elastin insufficiency have impaired pulmonary107–109 and skin110,111 elastic fibres, leading to impaired tissue mechanics. Other common connective tissue features of WS may also be linked to elastin insufficiency, such as periumbilical or inguinal hernias112, hoarse voice113, earlier-onset skin wrinkling111, atypical scar formation111 and genitourinary phenotypes112.
NCF1 modification of elastin-mediated hypertension.
Neutrophil cytosolic factor 1 (NCF1) resides at the telomeric end of the WSCR. Two NCF1 pseudogenes, NCF1B and NCF1C, are present in the LCR regions that flank the typical deletion. NCF1 is the regulatory subunit for several NADPH oxidase (NOX) complexes and generates ROS in multiple cell types, including endothelial cells, smooth muscle cells and leukocytes, following various stresses114,115. A deletion that removes NCF1 is found in ~50% of individuals with WS91,116. Loss of NCF1 has been associated with relative protection from hypertension and vascular stiffness in individuals with WS91,116 and in animal models74,117,118.
GTF2I and GTF2IRD1.
General transcription factor 2-I (GTF2I) and GTF2I repeat domain-containing 1 (GTF2IRD1) are paralogous genes located on adjacent loci at the telomeric end of the WSCR. They encode transcription factors and contribute to typical WS behaviour and development. On the molecular level, GTF2IRD1 and GTF2I encode BEN and GTFII-I, respectively, which are members of a versatile protein family with broad functional activities119–121.
GTFII-I is a highly conserved and ubiquitously expressed multifunctional transcription factor122–125 that regulates gene expression120,126,127 through interactions with tissue-specific transcription factors and complexes related to chromatin remodelling125. GTFII-I is activated in response to various extracellular signals and then translocates to the nucleus121,125,126,128. GTFII-I has been shown to be involved in multiple processes, which include regulating embryonic development122,129,130, the cell cycle125,127,131,132, actin cytoskeleton dynamics, axon guidance132 and epigenetic regulation133,134. Indeed, an iPSC-based study showed that GTF2I alterations are responsible for 10–20% of the transcription dysregulation in disease-relevant pathways in WS and in the 7q11.23 duplication syndrome, beginning in the pluripotent state and further amplified during development69.
From a phenotypic standpoint, individuals with classic deletions of the WSCR14 and those with shorter deletions that result in loss of GTF2IRD1 and GTF2I62 (Figure 3) typically show intellectual disability, high social approach to familiar persons and indiscriminate social approach to strangers (also referred to as social disinhibition or hypersociability), and difficulties in social communication (pragmatics). By contrast, individuals with deletions that spare these two genes typically exhibit neither intellectual disability40,54,56,59,135–137 nor these social characteristics59,138. Interestingly, individuals with shorter WS deletions that spare GTF2I but remove GTF2IRD1 also typically do not exhibit intellectual disability or hypersociability136,139, but they do show increased social approach to familiar people and difficulty with social pragmatics136. Further evidence for the role of GTF2I gene dose in intellectual ability comes from a family with a very short duplication affecting GTF2I but not GTF2IRD1; all family members with GTF2I duplication had intellectual disability, whereas the intellectual ability of those with the usual two copies of GTF2I was average140.
As in humans, partial hemizygous deletion of the mouse WSCR including Gtf2i results in increased sociability but also impairs motor coordination in mutant mice71. Homozygous deletion of Gtf2i in mice results in embryonic lethality owing to severe developmental abnormalities122,124,141, such as exencephaly and neural tube disclosure, whereas heterozygous mice show impaired social habituation to an unfamiliar mouse141,142. Selective deletion of Gtf2i in excitatory neurons leads to myelination alterations, motor deficits and hypersociability, which normalize with pharmacological rescue of myelination143,144. Intracisternal Gtf2i-gene therapy in the CD (full WS deletion) mouse model resulted in beneficial effects on behavioral deficits related to motor, social and anxiety-like behaviors145.
Taken together, these findings suggest that loss of a GTF2I allele is a major contributor to the intellectual disability and social disinhibition that are characteristics of individuals with WS. Deletion of GTF2IRD1, even without deletion of GTF2I, likely contributes to the social communication difficulties and generally increased social approach. Findings of dose-dependent Gtf2i-specific social and anxiety phenotypes in mouse models146,147 converge with those observed in the human hemideletion and duplication syndromes14,146,148,149. In a study of the effects of Gtf2i copy number on cortical neuron maturation and function, mice with a single copy of Gtf2i showed increased axonal outgrowth, whereas this outgrowth was decreased in mice with three Gtf2i copies150. The axonal growth effects of GTFII-I might occur through regulating the expression of the homeobox proteins DLX5 and DLX6151, thereby affecting the excitatory/inhibitory balance in the brain152. This balance has been suggested as a possible mechanism that causes autism spectrum disorder (ASD)153. In this regard, it is noteworthy that both deletion154,155 and duplication156 of the WSCR are associated with elevated rates of ASD.
Other candidate genes.
For several other genes within the WSCR, there is emerging evidence of an association with components of the Williams syndrome phenotype. Bromodomain adjacent to zinc finger domain 1B (BAZ1B), a member of the B-WICH chromatin remodelling complex, is essential for correct neural crest cell migration in vitro157 and in vivo158 and has been proposed as a master regulator of human craniofacial development157. As the enteric nervous system is also derived from the neural crest159, it is possible that abnormal innervation of the intestine contributes to WS gastrointestinal phenotypes, such as dysmotility and chronic constipation.
LIM domain kinase 1 (LIMK1) regulates actin cytoskeleton assembly and disassembly and has been linked with visual spatial cognitive ability in individuals with WS64 and in the general population160. Limk1–/– mice show visuospatial deficits, altered dendritic spine morphology and reduced synaptic plasticity, leading to reduced long-term memory. Limk1 expression can be upregulated by both brain-derived neurotrophic factor161 and cAMP response element-binding protein162, suggesting potential therapeutic avenues.
Syntaxin 1A (STX1A) is a key member of the protein complex that mediates exocytic vesicle fusion, thereby allowing the release of neurotransmitters into the synapse. Neuropsychiatric disorders found in individuals with WS have been associated with STX1A variants163,164. Insulin secretion from the pancreas is also dependent on exocytosis165, and STX1A levels are indeed reduced in members of the general population with type 2 diabetes mellitus166. Diabetes is common in adults with WS167, suggesting a possible physiological link with STX1A hemizygosity. As the MLXIPL gene, which encodes the ChREBP transcription factor that regulates both glucose168,169 and lipid metabolism170, is also located within the WSCR, both ChREBP and STX1A could contribute to the metabolic phenotypes in WS.
Bialleleic missense variation in DNAJC30, (DNAJ heat shock protein 40 family (Hsp40)) member 30 was recently associated with Leber’s hereditary optic neuropathy in humans171, a mitochondrial condition. Similar features have not, to date, been reported in WS. Bialleleic deletion of this gene in mice also produces mitochondrial dysfunction and behavioral changes172. More work is needed to investigate the impact of isolated hemideletion in humans, but potential impact of this gene on neurodevelopment should be considered.
Diagnosis, screening and prevention
Clinical diagnosis
WS is a multisystem disorder with a broad but characteristic pattern of organ involvement (Figure 1), including a distinctive facial appearance173–175 (Figure 5). As there is no newborn screening for WS, clinical consideration of the diagnosis is prompted by the presence of suggestive signs and/or symptoms. Below, we outline the most common presenting features prompting consideration of a WS diagnosis. Of note, the extent and exact distribution of system involvement can vary considerably from patient to patient.
Figure 5. Facial features of children and adults with Williams syndrome of different ethnic backgrounds.

Facial photos of individuals with molecularly-confirmed Williams syndrome of different racial and/or ethnic backgrounds aged 2 months to 52 years. Distinctive features in infants and children include broad forehead, peri-orbital fullness, flat bridge of the nose, full cheeks, long philtrum, and a small delicate chin. Many adolescents and adults continue to have micrognathia, and the face often elongates over time while the nasal bridge is no longer flat, and there is fullness of the lips with a wide mouth (especially appreciated when smiling).
Parents or caregivers for all pictured individuals signed consent for publication of their family member’s image. The presence of more male than female photos in adolescents and adults solely reflects availability of subjects.
Note: This material is labeled as Fig. 4 in the published version of the paper DOI: 10.1038/s41572-021-00276-z.
Craniofacial differences.
Individuals with WS often present with facial features that are not typical for their family. Prominent features in infants and young children include a broad forehead, peri-orbital fullness, flat bridge of the nose, full cheeks, long philtrum, and a small delicate chin. Adolescents and adults often continue to have micrognathia, but the face elongates over time, the nasal bridge is no longer flat, and there is fullness of the lips with a wide mouth (especially appreciated when smiling). Children and adults of different ethnic backgrounds from Brazil with molecularly confirmed WS are depicted in Figure 5.
Cardiovascular anomalies.
Cardiovascular disease in WS typically presents with a heart murmur. Evaluation in children most commonly reveals SVAS and/or stenosis of the main or branch pulmonary arteries. Pulmonary vascular disease is often less prominent in older individuals with WS. Other WS cardiovascular features, such as stenosis in other vessels, septal defects, hypertension, vascular stiffness or EKG abnormalities, are generally not the reason for referral but may be present at the time of diagnosis.
Hypercalcaemia.
Actionable hypercalcaemia (serum calcium >12.0 mg/dl) is seen in 5–10% of children with WS and when present usually occurs between 6 and 30 months of age176. While some children with hypercalcaemia are irritable and show poor oral intake, other cases are detected incidentally by laboratory testing.
Growth concerns.
On average, children and adults with WS are shorter than expected for age177. Once a diagnosis is made, WS-specific growth charts are available for plotting children’s expected growth178,179. In addition, many infants with WS exhibit prolonged colic may have difficulty feeding due to oral motor delays or sensitivities, and have difficulty gaining weight.
Developmental delay, intellectual disability and behavioural profile.
Developmental delay is almost universal and 75% of older children and adults with WS have intellectual disability (IQ <70)178, with most remaining individuals displaying borderline IQ (70–79) and/or more specific neuropsychological impairments13,14.
Many experienced parents almost immediately notice differences from typically developing infants, but other parents may not become concerned until they realize their child with WS is not meeting motor or language milestones. Features of ASD may also lead to referral.
Differential diagnosis
It is important to distinguish WS from other syndromes with overlapping features. Certain highly suggestive features (such as SVAS, hypercalcemia and characteristic facial features) exist that, when seen in combination by an experienced examiner, readily yield a correct clinical diagnosis of WS. And though a few other disorders are evocative of WS, in that they have a similar pattern of organ involvement (such as Fetal Alcohol syndrome, Rasopathies, FG syndrome), detailed examination of their specific features reveals distinct differences from WS. Some individuals come to medical attention, however, due to a single prominent feature. Depending on the specific presenting symptom, the differential diagnosis varies and Supplementary Table 1 is presented to aid practitioners in that setting. A clinical suspicion of WS should always be confirmed with genetic testing (see below).
Testing approaches
The most widely used laboratory methods available to detect the 7q11.23 microdeletion include FISH, polymorphic microsatellite markers, multiplex ligation-dependent probe amplification (MLPA) and chromosomal microarray analysis (CMA) (Table 1). CMA is the only current method that does not require the clinician to suspect a specific diagnosis of WS prior to testing. In addition to providing mapping for deletion boundaries and offering the ability to detect atypical deletions, CMA can also identify additional CNV events elsewhere in the genome. MLPA and polymorphic microsatellite markers are often used in low- and middle-income developing countries owing to their lower cost than FISH and CMA22,23,180,181.
Table 1.
Overview of methods for diagnosing Williams syndrome
| Method | Current Advantages | Current Disadvantages |
|---|---|---|
| Available methods | ||
| Microsatellite markers | Low cost | May be uninformative Need of trio sample |
| Multiplex ligation-dependent probe amplification | Low cost Highly effective Possibility of detecting other microdeletions/duplications (as determined by probe coverage) |
Requires ordering provider to suspect WS to order correct probe |
| Fluorescence in situ hybridization | High sensitivity May detect translocations (depending on availability of probe coverage) |
Higher cost False negative for smaller deletions Not possible to determinate deletion size Requires ordering provider to suspect WS to order correct probe |
| Chromosomal microarray | High positivity Able to determinate deletion size Able to determine CNVs elsewhere in genome. Ordering provider does not need to suspect WS to order this test |
Highest cost of currently available tests Cannot detect balanced translocation/inversion |
|
| ||
| Emerging methods | ||
| Noninvasive prenatal testing | Prenatal diagnosis of aneuploidies and large deletions or duplications | Low resolution (detects deletions >3 Mb) |
| Facial recognition software | Cost varies with some free software available online | Diagnosis is limited by number of photographs available in database May have different efficacy based on race or ethnicity of patient |
| Whole-exome sequencing | Deletion detection performed in research settings | Currently used clinically for single-nucleotide variants in most cases High cost Lowest accuracy for deletion detection |
| Whole-genome sequencing | Combined single-nucleotide variant and CNV or structural variant detection performed in research settings | High cost, slow turnaround in some settings |
WS, Williams syndrome; CNV, copy number variation; Mb, million base pairs
Newer technologies, such as facial recognition software182–184, may help focus the differential diagnosis and have been evaluated in individuals from various racial and ethnic backgrounds; the diagnostic precision of the software will likely continue to improve over time. Additional testing (gene sequencing or trinucleotide repeat expansion testing) may be indicated based on the differential diagnosis (Supplementary table 1). In the future, whole-genome sequencing may provide CNV and single-nucleotide polymorphism detection in a single test that will simultaneously assess for WS and other possible differential diagnoses.
Recurrence risk
If a parent has WS, the risk of WS in offspring is 50% for each pregnancy. However, owing to the complex medical and neurodevelopmental challenges associated with WS, few adults with WS have children.The recurrence risk for couples where neither parent has clinical findings of WS is extremely low185, since the 7q11.23 microdeletion arises de novo in the vast majority of cases. For these couples, parental testing for a 7q11.23 microdeletion is not indicated and neither is invasive prenatal testing of subsequent pregnancies (although many parents opt for the latter, especially in countries where molecular diagnostic testing is widely available). However, there are rare reports of recurrence in phenotypically normal parents. This recurrence is likely attributed to parental mosaicism186 or to one parent carrying an inversion of the WSCR185. The inversion has been associated with an approximately five-fold increase in risk of having a child with WS in each pregnancy; nevertheless, testing for the presence of inversion in phenotypically normal parents is not recommended, because their recurrence risk — despite being five times higher — remains well below 0.1%42,44,185.
Pregnancy and prenatal testing
Limited clinical information exists on pregnancies in women with WS187,188. Review of this literature suggests that mothers and fetuses (affected by WS or not) may require close monitoring, especially with regard to the maternal cardiovascular system.
In addition, there is no routine prenatal test that adequately screens for WS, although prenatal ultrasonography can sometimes detect relevant fetal anomalies. The most common prenatal finding is nonspecific, namely intrauterine growth retardation189. Various cardiovascular anomalies also can be seen and range from nonspecific (for example, ventricular septal defect) to nearly pathognomonic for elastin arteriopathy (for example, SVAS, although this finding is quite difficult to make by prenatal ultrasound)165. The prenatal detection of growth retardation combined with any cardiovascular defect may warrant performing high-resolution prenatal ultrasonography and genetic testing.
Noninvasive prenatal testing (NIPT) involves sequencing of fetal DNA circulating in the maternal circulation and can detect common fetal chromosomal aneuploidies in the first trimester. Currently, as even enhanced NIPT platforms can only detect deletions >3 Mb, they cannot be used to diagnose WS189. However, further technical advances in the NIPT technology are likely to enhance prenatal diagnosis and therefore affect the epidemiology of WS in the future. In addition, whole-genome sequencing may eventually be utilized to perform combined single-nucleotide polymorphism, copy number variant and structural variant detection in fetal samples. Prenatal diagnosis offers the opportunity to provide genetic counseling to families sooner, allowing them to avoid a diagnostic odyssey that can potentially last months and years after the child’s birth.
Management
The management of various aspects of WS has been extensively outlined in numerous research studies, reviews, and guidelines173,174,176–178,190–195.Here, we focus on the management of three key areas where improved therapeutics would have the greatest potential to improve health outcomes: elastin-associated vasculopathy, hypertension, and intellectual disability, social functioning and anxiety.
Elastin-associated vasculopathy
As mentioned previously, individuals with elastin insufficiency can develop focal stenosis and other vascular features. Vascular disease severity varies among persons with WS, with ~20% requiring intervention for SVAS, for example, while 30–40% have little to no stenosis in this location. At present, large vessel stenosis is predominantly managed surgically. To alleviate SVAS, patch aortoplasty is the most common approach196 and has undergone several technical improvements over time, evolving from the single-patch method197,198 to the pantaloon-shaped patch that enlarges the aorta and two aortic sinuses199. The most advanced method, which involves applying a patch to each of the three aortic sinuses200, exhibits notably lower residual pressure gradients and reoperation rates201 than the single-patch approach. Stenoses of pulmonary arteries can be treated with angioplasty, but catheter-based interventions on other arteries are often unsuccessful11,90,202. Surveillance of stenosis occurs through regular examinations by a cardiologist and associated imaging and should continue throughout an individual’s lifetime.
Individuals with WS experience increased rates of cardiovascular collapse with and without anaesthesia10,12, although the mechanism of this phenomenon is not completely understood. Young individuals and those with the most severe cardiovascular features (that is, biventricular outflow tract obstruction10) seem to have the highest risk, although some individuals with only minimal stenosis suffered sudden death in the setting of anaesthesia. This severe outcome may be influenced by various risk factors, including anatomical anomaly of the coronaries and reduction in perfusion pressure at induction and/or maintenance of anaesthesia. Therefore, careful preoperative screening should be performed to assess the anaesthesia-associated cardiovascular risk, and administration of intraoperative anaesthesia should ideally be provided by an anaesthesia team with knowledge of WS anaesthesia risks93–95,203,204. Care should be taken not to acutely lower blood pressure at induction of anaesthesia in order to maintain adequate perfusion of the coronaries during this sensitive time.
Studies suggest that novel pharmacological therapies may improve elastin-associated vasculopathy. Minoxidil, an ATP-dependent potassium channel opener, has received considerable attention based on evidence that it increased elastin production in cellular studies205 and in animal models206,207 of genetic elastin deficiency and that it ameliorated age-related degeneration of elastic fibres in mice208,209. However, a randomized, double-blind, placebo-controlled trial (NCT00876200) investigating the effect of a year-long minoxidil treatment in 17 individuals with WS failed to show improvement in the primary outcome measure (carotid intima-media thickness)210. This trial did show an increase in lumen size (a secondary finding, in line with previous mouse studies207) over the same time interval, along with an expected common adverse effect of hypertrichosis210.
Alternative therapeutic approaches may target various regulatory pathways that influence elastin expression. For example, both the coding region and the 3ʹ UTR of the ELN mRNA are enriched in binding sites for miR-29 and miR-15, two microRNAs that are upregulated in the late postnatal aorta, at the same time that the level of mature ELN mRNA decreases82. Antagonism of miR-29 increases elastin expression in haploinsufficient cells and bioengineered vessels211.
Hypertension
Elastin insufficiency is also associated with hypertension, but the risk of clinically significant blood pressure increases is modified by deletion size and NCF1 gene dosage91,116. Blood pressure should be measured in both arms and at least one leg due to possible right arm flow acceleration (the so-called Coanda effect) and/or coarctation of the aorta212. If consistent blood pressure elevations or pressure differentials (in the arms or as an arm-leg discrepancy, respectively) are detected, additional imaging of the heart and ascending aorta (by echocardiography) and the renal/abdominal vasculature (by Doppler ultrasonography, CT angiography or MRI/MR angiography) should be considered (when available), to assess the presence of focal or long-segment stenosis that affects renal perfusion and may benefit from surgical intervention5,9. Auscultation of the abdomen can also reveal a bruit (abnormal sound) in the region of the stenosis.
Blood pressure elevation often starts in childhood and increases in frequency with age8,9,116,213. Unlike stenosis, most cases of hypertension are treated pharmacologically5,8. Currently, there is no expert consensus on the best antihypertensive medication for individuals with WS214. Similarly, work in Eln+/– mice revealed no superior drug class for treating elastin-mediated hypertension215. However, because blood pressure influences lumen size, which in turn affects end-organ blood flow, care must be taken not to decrease pressure to the extent that lumen size, and therefore end-organ blood flow, is pathologically diminished.
For this reason, among others, diuretics may not be the ideal or initial choice for blood pressure management in individuals with WS. In addition, when reno-vascular causes of hypertension are suspected, angiotensin receptor blockers and angiotensin-converting enzyme (ACE) inhibitors should be used with caution. As hypertension risk has been linked to ROS production through the NOX signaling pathway, potential therapeutic strategies could include treatments that limit ROS production or affect signaling through ROS216.
Intellectual and social functioning
Deletion of multiple genes within the WSCR likely contribute to intellectual disability, altered social functioning and anxiety, although the roles of GTF2I and GTF2IRD1 (as outlined above) are the best described to date.
WS is associated with developmental delay (the expected developmental milestones are shown in Figure 6). The delays typically lead to mild to moderate intellectual disability, although a few individuals have severe intellectual disability or, at the other extreme, average intellectual ability14,217. This overall level of ability masks a phenotypic pattern of strengths and weaknesses relative to expectations for overall level of intellectual ability. In general, language and nonverbal reasoning abilities are stronger than expected for overall intellectual ability, whereas visuospatial construction (for example, handwriting and block construction) is considerably weaker than expected13,14,218. Visuospatial construction is facilitated by visual processing regions. Interestingly, research using resting-state functional MRI showed that for children in the general population, the intraparietal sulcus is functionally connected to more superior-anterior visual processing regions, whereas it is instead connected to social regions in children with WS160. The lack of this specific functional connection in individuals with WS contributes to their considerable weakness in visual spatial construction219. A more detailed description of the pattern of relative strengths and weaknesses within the language domain is provided in Table 2.
Figure 6. Developmental milestones for very young children with Williams syndrome.
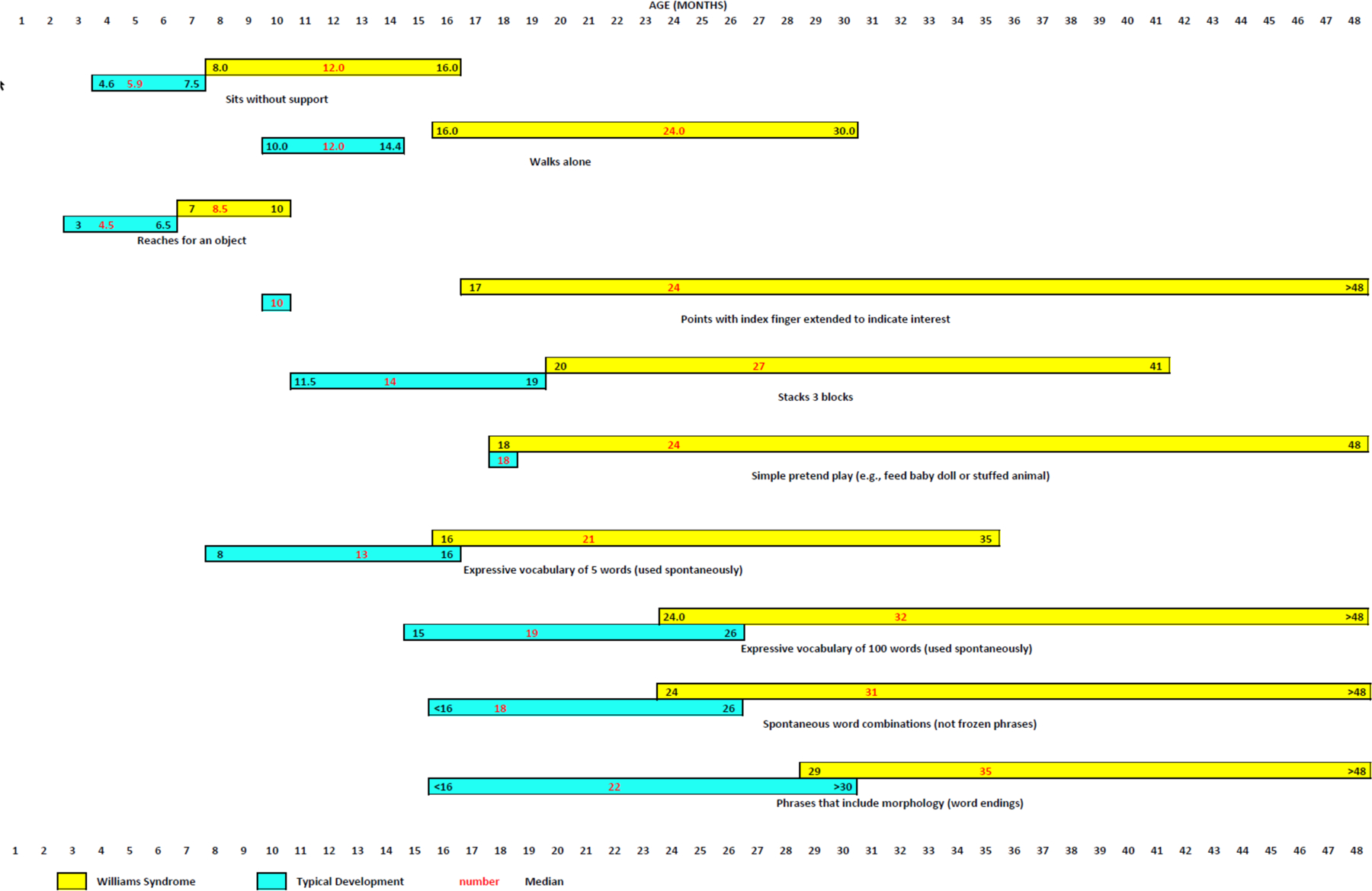
Median ages (in months) and 90th and 10th percentiles for attainment of various gross motor, fine motor, cognitive and language milestones by very young children with Williams syndrome (yellow bars) relative to typically developing children (aqua bars)154,294–302. Numbers in red typeface indicate median age; numbers in black typeface indicate 90th percentile (to the left of the median) and 10th percentile (to the right of the median). TD, typically developing.
Note: Figure 6 was redrawn by the Nature Reviews Disease Primers art editor and permission for reproduction of the final figure was not granted by the publisher. This material is labeled as Fig. 5 in the published version of the paper DOI: 10.1038/s41572-021-00276-z
Table 2.
Pattern of relative strengths and weaknesses in individuals with WSa.
| Category | Ability | Details | Level relative to overall intellectual ability |
|---|---|---|---|
| General pattern | Verbal | Vocabulary breadth and verbal analogies | Stronger than expected |
| Nonverbal reasoning | Matrix reasoning and pattern completion | Stronger than expected | |
| Visual-spatial construction | Drawing, writing and block construction | Considerably weaker than expected | |
| Language profile | Phonological processing | Knowledge of the sound structure of language and ability to manipulate that structure | Stronger than expected |
| Vocabulary breadth | Concrete vocabulary, picture identification and naming | Stronger than expected | |
| Verbal short-term memory | Ability to repeat back what was said verbatim | Stronger than expected | |
| Grammatical ability | Ability to speak grammatically and understand sentences someone else produced | At the expected level | |
| Verbal working memory | Ability to repeat back what was said in a different order (for example, in the reverse order) from the original | At the expected level | |
| Vocabulary depth | Ability to define words accurately and specifically | At the expected level | |
| Relational vocabulary | Understanding and use of spatial, temporal, and quantitative concepts and of complex conjunctions (for example, however) or disjunctions (for example, neither nor) | Considerably weaker than expected | |
| Nonliteral language | Understanding and use of metaphor and irony | Considerably weaker than expected | |
| Discourse skills | Event sequencing, asking and answering questions appropriately, maintaining the conversational topic | Considerably weaker than expected | |
| Comprehension monitoring | Evaluating whether one understood what was said or read and then taking appropriate action if one had not understood | Considerably weaker than expected |
Strengths and weaknesses relative to overall intellectual ability for broad categories of intellectual ability and within the language domain.
Current information indicates that cognitive ability remains stable, at least to mid-adulthood220–223. There is a possibility of IQ decline in older adults34, but the data sets are limited.
WS is also associated with a characteristic behavioural profile that is consistent across both Western and Eastern cultures224. At its most basic level, this profile includes gregariousness and hypersociability (or ‘overfriendliness’), accompanied by attention problems (often meeting diagnostic criteria for Attention Deficit Hyperactivity Disorder or ADHD), social problems, anxiety and emotional overreactivity15,16,225–229. Mastery motivation (that is, willingness to persist on a task one finds moderately difficult) is typically very limited14,227. The most common psychiatric problems associated with WS, and estimates of their prevalence based on meeting formal diagnostic criteria outlined in the Diagnostic and Statistical Manual of Mental Disorders IV (DSM), are delineated in Figure 7.
Figure 7. Prevalence of psychiatric diagnoses in individuals with Williams syndrome throughout the life span.
The genetic deletion in Williams syndrome (WS) has cascading effects on social, educational and vocational opportunities and on psychopathology. Estimated prevalence of psychiatric disorders in preschool-aged children, school-aged children and adolescents, and adults with Williams syndrome154,213,266,303,304 is depicted. Studies with participant over a wide age range that did not report results separately for children and adults were excluded.236,305–307 The term ‘anxiety disorders’ includes generalized anxiety disorder, specific phobia, separation anxiety or social anxiety, and panic disorder with agoraphobia.aMost individuals with WS who have autism spectrum disorder (ASD) fit in Wing and Gould’s292 active-but-odd subtype of ASD231 rather than the aloof subtype.
Note: Figure 7 was drawn by the Nature Reviews Disease Primers art editor and permission for reproduction of the final figure was not granted by the publisher. This material is labeled Fig. 6 in the published version of the paper for the final figure at DOI: 10.1038/s41572-021-00276-z
In most cases, the previously described learning and developmental differences are addressed by the school system through specialized education and therapy services, which are outlined below. In general, physical, occupational, and speech and language therapy are recommended for infants through school-aged children in the USA178,230 and in Australia192. By contrast, according to a parent survey in the UK, only 20–40% of school children with WS receive these services231.
The behavioural and psychiatric components of WS are managed with the help of structural and environmental supports at home and in school and vocational settings, including counselling and behavioural therapies14,156,178,232. For individuals whose symptoms greatly affect their daily functioning and quality of life (QOL), referral to a psychiatrist for consideration of pharmacological intervention may be indicated178. The anxiety associated with WS has been particularly difficult to treat. Trials are needed to identify appropriate therapeutics for anxiety, although, as has been shown for typically developing individuals, cognitive behaviour therapy seems promising as an effective treatment for anxiety in WS233,234. The same medication classes available for anxiolysis in the general population have been studied in small series of individuals with WS235,236 and are already widely used in clinical practice, but result in varying symptom relief. Furthermore, the risk:benefit ratio may be more unfavorable for some of these medications in those with WS, as reviewed by Thom et al237. Specifically, anxiety, mood, cardiac function and blood pressure all need to be monitored given the overlap between common side effects of ADHD medication in the general population and the psychological and physical difficulties associated with WS.
Educational and vocational provision
In high-income countries, a large proportion of primary school-aged children with WS are educated in mainstream schools and spend at least part of the day in classes with typically developing agemates; these proportions decrease considerably for secondary school231,238. Although there are no data specifically for children with WS, in low-income and middle-income countries, school attendance is much less common for children with disabilities than for typically developing children239, due not only to financial difficulties but also to discriminatory and negative views of persons with intellectual disability240.
Postsecondary education for individuals with WS is very limited worldwide, although opportunities are increasing in some high-income countries. In a US survey whose respondents were on average upper-middle class, 29% of individuals with WS had attended a postsecondary education programme241. These programmes typically focus on combinations of academics, vocational training (including job placement and coaching) and independent living skills.
After completing school, most adults with WS continue to live with their parent or another relative; <10% live independently241,242. Only 38–54% of adults are working at least part-time, generally in a special employment arrangement, either for pay or as a volunteer241,242.
Within the academic sphere, reading skills are considerably stronger than mathematics skills13,243 and range from an inability to read at all to reading comprehension at age or grade level243–246. About 30% of adolescents and adults with Williams syndrome have functional reading capability244. Children who were taught to read using systematic phonics instruction, which emphasizes letter–sound relations, read and comprehend significantly better than those taught using other instructional approaches246,247. There has been no research on written composition, and very little is known about the mathematical abilities of individuals with WS248.
Within the adaptive behaviour domain, socialization and communication skills are stronger than daily living and motor skills for children and adolescents with WS249. In longitudinal studies, adaptive behaviour standard scores declined significantly during childhood250 and in adulthood222, owing to stagnation or failure to increase adaptive skills at the rate needed to maintain a consistent standard score over time. For adolescents and adults with WS, adaptive behaviour is more limited than expected for their IQ251,252.
High but realistic parental expectations combined with better behavioural regulation234 and higher levels of mastery motivation are expected to positively affect both adaptive behaviour and academic achievement13. Even so, in a survey of teachers, most indicated they were not given adequate resources to teach children with WS238, and most parents surveyed thought their child’s teachers had little knowledge about WS238. Much more research is needed on effective instructional strategies for individuals with WS at all educational levels253.
Quality of life
The combination of intellectual disability, medical problems, and behavioural, psychological and adaptive impairments leads to considerable limitations on QOL in individuals with WS. Parents and teachers of children with WS report difficulties with peers, including problems establishing and maintaining friendships and increased social exclusion or isolation254. Although social vulnerability is high, self-awareness is limited255. Seventy-three percent of parents reported victimization of their child256, with the social interaction style of individuals with WS contributing significantly to their social vulnerability257. Even adolescents and adults with WS are typically not cognizant of stranger danger258,259. Intervention studies addressing these issues258,260 are both rare and crucial229.
Nearly all children with WS require multiple encounters with the healthcare system for management of medical or surgical problems261. These encounters place a financial and emotional burden on the individuals with WS and their families262 and often contribute to anticipatory anxiety about medical encounters and procedures263. Several studies of adults with WS ≥30 years of age demonstrated an increased frequency, variety and severity of medical morbidities33,34,213, and this trajectory accelerates among those >65 years of age (personal observation).
Prominent adult issues include cardiovascular disease, obesity (with/without lipoedema), diabetes mellitus, incontinence, hearing loss, consequences of poor oral health, gastrointestinal problems (including diverticulitis), decreased bone mineral density and sleep apnea174,213. Psychiatric concerns are often paramount. Health issues such as these may result in narrowed residential and vocational placements, restricted mobility and physical activity and, collectively, they promote further social isolation. Medical complications require focused management based on established guidelines, whereas general health could be improved by participation in programmes that promote healthy eating and increasing physical exercise activity to recommended adult levels264,265.
The presence of a child with WS may affect the QOL of other family members. Both generalized anxiety disorder266 and borderline or clinically significant levels of stress267,268 occur in a significantly higher proportion of mothers of children with WS than expected for same-aged women in the general population. Sensory modulation problems are very common among children with WS and are associated with more difficult temperament, more limited adaptive behaviour and emotion regulation difficulties and behavioural problems269. In turn, child behavioural problems (especially externalizing problems) contribute to increased maternal stress267,270 or challenges with raising the child (owing to, for example, the child’s difficulties with social skills or obsessions)271. The discrepancy between the general perception that children with WS are happy and have ‘easy’ temperaments and the reality that most children with WS have relatively difficult temperaments is in itself likely to increase maternal stress227. Worries regarding the child’s future are very common among family members and mothers in particular231,271,272. At the same time, most mothers also reported positive aspects of having a child with WS, including that the child brought joy and changed the mother’s outlook on life271 (Box 1).
Box 1. Patient perspective (family interview).
Molly (not her real name) is a 30-year-old woman with Williams syndrome who lives with her parents in a rural town in Australia. She has mild to moderate intellectual disability and the typical Williams syndrome personality. She completed school in a mainstream setting for the first six years, then moved to a special school. She currently works 3 days a week in sheltered employment after failed attempts to work in mainstream employment due to bullying. Molly enjoys music, softball and line dancing. Molly has struggled with many of the medical and psychological difficulties associated with Williams syndrome, but due to her determined nature and strong family support, she has made many wonderful life achievements. Please see Supplementary box 3 for the full interview.
Interviewer: Do you feel that others treat you any differently because you have Williams syndrome?
Molly: The way people talk to you is different, and they talk to mum instead of me… about my medical things …and I am like: “Hello, I am over here. Mum doesn’t have Williams syndrome. I do”.
Interviewer: What are the good things about having Williams syndrome?
Molly: I make people laugh and feel good. At home I am demanding. I love older people and their stories. I love to hear what they were up to back in the day.
Interviewer: What are the not so good/hard things about having Williams syndrome?
Molly: Lots of people don’t know about Williams syndrome. Most doctors don’t know about Williams syndrome. When I go to hospital, I always take an information sheet [about Williams syndrome].
I get bullied a lot. I think because of the way… oh this makes me feel sad… (now teary) …I think because of the way I act some people don’t realise it is hard to always be happy.
I know this might sound stupid, but I can’t tie my shoelaces. I wear shoes with elasticated shoelaces.
Interviewer: What is the biggest thing that having a child with Williams syndrome has taught you?
Parent: The biggest thing we have learnt is that a person with Williams syndrome is no different to you and me. We all have different personalities, have different needs and process information differently. We have a greater level of acceptance and empathy by having a person with Williams syndrome in our lives.
Interviewer: What have been the major challenges associated with having a daughter with Williams syndrome?
Parent: At first it is hectic. Appointments are on-going and seem to be never ending. You are always planning for the next stage. Learning how to feed and care for your child with Williams syndrome is the first step. Then moving on to therapy and the first stage of learning with early intervention and transition to school.
Also, stranger danger is a big concern as Molly accepts everyone as her best friend.
Interviewer: What is your take home message to new parents of a child with Williams syndrome?
Parent: Don’t be afraid to ask for support especially in the early years as there are times that you will be overwhelmed but as time goes on the positives can outweigh the negatives.
Interviewer: What are the three main points you want to make about Williams syndrome?
Parent: Williams syndrome is challenging but can be rewarding. The person with Williams syndrome is just like you and I but processes information slower. We just need to be more accepting, tolerant, non-judgmental. Treat a person with Williams syndrome with respect.
Outlook
Much has been learned about WS since its initial description, but important questions remain (Box 2). These questions centre on three major themes: molecular mechanisms of disease, interindividual variability and effective treatment strategies. A more complete understanding of the genes and pathways contributing to the phenotypes of individuals with WS would allow clinicians to move beyond symptom management and to instead use precisely targeted therapies to improve organ function and ultimately health outcomes.
Box 2. Key questions for future work in WS.
Long-term health in Williams syndrome (WS)
How do health needs change for individuals with WS across the lifespan? What recommendations can be made to optimize health of older individuals with WS?
Do different pathologies arise in WS at different developmental stages, and are any reversible? If treatments are designed targeting early processes, will this “normalize” future outcomes, or will it be necessary to target multiple stages and processes?
Phenotypic variability
What are the biological and environmental contributors to variability in outcomes in WS?
How do the ~20 WSCR genes without a specific phenotype designation affect health and development in people with WS? How should the combinatorial effects of multiple genes on disease variability be assessed?
How does variability in WSCR genes contribute to phenotypic variability in the general population?
Disease mechanism
What are the developmental neuropathological mechanisms underlying social and anxiety-related behaviour impairments in WS?
What factors impact transcription, translation, and deposition of elastin? Can those genes or pathways be harnessed to appropriately re-initiate elastin deposition outside of its normal developmental window?
What underlies metabolic differences in WS, such as glucose dysregulation, aberrant growth and aberrant body composition? How do changes in the genes contributing to these phenotypes affect health for people with and without WS?
Interventions and treatment
What interventions (medical, psychological or behavioural) would best improve quality of life for people with WS, especially pertaining to anxiety, mastery motivation and social vulnerability?
Should hypertension be aggressively treated in WS? What is the effect on short-term end organ perfusion and longer-term organ function?
What risk factors for adverse anaesthesia events need further refinement, such that management guidelines can be generated and widely shared?
Research priorities for WS
How can resources be developed to facilitate collaborative data collection and treatment trial coordination to optimize delivery of new treatments for people with WS?
Molecular mechanisms of disease
To date, only ELN, GTF2I and GTF2IRD1 have been definitively linked to key phenotypes that are identifiable in individuals with WS, but even for these genes, a significant knowledge gap between disease phenotype and gene function remains. For example, published literature has not yet clarified whether the global vascular narrowing seen in WS is driven by changes in cell proliferation99, radial growth of the vessel102 or alteration in cell–matrix interactions. In addition, the characteristic hourglass-type stenosis feature in WS seems to be driven by entirely different mechanisms than the more general narrow and stiff vessel pathology seen throughout the rest of the vasculature7,97. While for GTF2I and GTF2IRD1, clear effects on brain development and function are evident, the direct effect of reduced gene products on neural circuits, white matter properties273, developmental timing and brain activity is unknown.
To answer these questions, further studies on the roles of individual genes in human pathophysiology, at the cellular and molecular level, and in model systems are needed. In humans, a traditional approach has been to clinically characterize individuals with smaller WSCR deletions, in an effort to create more specificity for the traits associated with each gene (Figure 3). Currently, clinical exome and whole-genome sequencing are being used to identify rare single gene variation in individuals with phenotypes that overlap with WS (a phenotype to gene approach). In the future, the move toward big-data and genotype-first methods (a gene to phenotype approach) may offer new hope in the identification and further refinement of genotype–phenotype relationships that are part of the complex multisystem disorder of WS.
Further clarification of gene function can be obtained from studies in model systems. Cell systems offer ease of manipulation and provide precise ways to study protein–protein interactions and gene expression. However, they lack much of the complexity (for example, multiple cell types, tissue movement and endocrine signaling) needed to truly model human disease. Similarly, animal models (usually mice) may imperfectly match human disease outcomes; for example, the Eln+/– mouse does not have SVAS and, in the case of cognitive conditions, mouse behaviours may incompletely or inaccurately correspond to human behaviours. The advent of iPSCs, tissue engineering and ‘organs in a dish’ may offer the potential to bridge some of the gaps that are present in traditional cell culture systems68,69,274–276. As in all disease modeling, the biggest challenge lies in the ability of the in vitro system to truly mimic the complex in vivo environment. Brain organoids have shown promise for modeling developmental disorders such as autism and epilepsy277,278. Vascular organoids279 and numerous tissue engineering approaches 280 are being applied to the study of blood vessels.Progress is being made but it has been difficult to design complex vascular tissues that deposit mechanically competent and mature elastin281,282. Rapid improvement in these systems is expected in the coming years as more precise genetic approaches to adapting both cellular and animal systems facilitate the study of specific outcomes.
Interindividual variability
Phenotypic variation is readily apparent in WS, despite the vast majority of individuals carrying the typical 1.55–1.83 Mb WS deletion. Currently, the mechanisms underlying this variability are mostly unknown. It is likely that both environmental factors and genetic modifiers contribute to the overall penetrance of specific signs and symptoms in an individual with WS.
The study of polygenic risk is still in its relative infancy but offers essential insight into features of WS that overlap with common health conditions in the general population283,284. Variation in genes that globally influence hypertension or IQ, for example, likely act together with genes in the WSCR in additive and synergistic ways to produce much of the observed variation285. In this way, the study of genetic modifiers in individuals with microdeletion disorders may offer a shortcut to identify relevant disease pathways by acting as a sensitivity screen of sorts, with the notable downside being the difficulty of acquiring truly robust sample sizes.
Sets of polygenic risk genes for more unique WS features (such as SVAS or odynacusis286) are currently unavailable, but if identified, they may provide insight into targetable pathways that could impact these important outcomes. For example, ~20% of all individuals with WS require surgical correction for SVAS, but if it has been established that those who do need surgery have a variation in a particular pathway, treatment could be targeted to the modifier pathway rather that to elastin insufficiency itself. Some work has already been initiated in this area29 but larger studies covering a broader racial and ethnic distribution are needed.
Other, understudied areas of potential variation include epigenetic effects and somatic variation. In addition, little is known about how differences in the flanking LCR regions, which are difficult to study by current short-read sequencing methods, may affect disease outcomes. Another area requiring further study is environmental influences, prenatally and throughout life, on disease outcomes. The gut microbiome, which is potentially affected by feeding difficulties, early hospitalizations and increased medication use, also deserves consideration. Environment and gene x environment interactions, in particular, are likely to play a crucial role in educational achievement14,244,246,247 and adaptive functional outcomes 13,223,249.
Effective treatment strategies
Treatment strategies can be designed using a variety of approaches: targeting the genes and/or gene products themselves, targeting functional pathways and aiming to treat disease symptoms and signs. For the gene-based strategies, CRISPR- and viral vector-based gene therapy technologies287,288 are currently being studied in other rare conditions289,290. However, unlike single-gene diseases, microdeletion disorders such as WS offer particular challenges that relate to the large size of the material to be delivered and the sheer number of locations where the genes would need to be targeted to alter relevant phenotypes. It is possible that a subset of genes could be delivered using the methods previously described, which may improve efficiency of delivery and function. However, it is known that the 7q11.23 duplication syndrome149,291results from increased dosage of at least some of the genes in the WSCR, and therefore any therapeutic approach must involve carefully regulated gene expression or protein replacement. Although the ability to identify and modify target genes, either in utero or in early postnatal stages, offers the potential to greatly improve precise treatment in WS, much work is required before this strategy becomes a reality.
In the coming years, therapies are expected based on individual genes or pathways that are known to be important for influencing phenotype in WS. Studies in mouse models, for example, suggest that anti-miRNAs (such as anti-miR29a) may be useful for increasing elastin deposition211 and that influencing smooth muscle cell behaviour using inhibitors of mTOR106 or integrin β3101 could improve the vascular features of WS. Moreover, new pharmacological strategies targeting myelination have been proposed for improving neural outcomes143. However, each of these potential strategies comes with challenges in delivery, specificity or longevity. For example, miR29a regulates multiple transcripts, so although anti-miR29a administration may lead to increased elastin message stability (and therefore increased translation)211, other transcripts bound by miR29a may also be affected, potentially leading to other complications. Development of additional (non-rodent) WS models may be useful preclinical modalities prior to initiation of human trials. As people with WS retain one copy of each gene from the WSCR, future studies could be aimed at developing methods to target and upregulate expression of the remaining allele.
Additionally, patient-derived iPSCs may soon offer platforms through which to test novel therapeutics for impact on unique WS transcriptional pathways and phenotypes. Initial investigations incorporating this technology have been used to screen for medications that impact the increased smooth muscle cell proliferation seen in WS105. Rather than a goal of preventing stenosis, future studies that focus on resolving or reducing existing stenoses may be more relevant to treatment and lead to improved clinical outcomes, especially in young children who often come to medical attention with stenosis already present. When iPS cells or gene-edited versions thereof are being considered as deliverable therapeutics themselves, the timing and delivery mechanisms will be critically important.
In the interim, clinicians manage specific symptoms in individuals with WS (such as hypertension, anxiety, and hypercalcaemia) using medications and interventions that have been developed for these indications in the general population, without knowing if the mechanism of disease in WS is the same. For example, several classes of antihypertensive drugs are available, and monotherapy or polytherapy can reduce blood pressure in most individuals with WS. However, the targeting of these drugs is not mechanism-based, and formal studies on which medications in these classes are best for symptom control in the WS population are lacking214. The mechanistic and modifier studies previously defined could help narrow the options needed to pursue hypothesis-driven clinical trials. For the near term, longitudinal studies, as well as open label and double-blind randomized clinical trials, on common and high impact medical and behavioral problems are needed, the findings from which could provide valuable clinical algorithms to guide treatment.
Moving forward
To improve health outcomes for people with WS, a multifaceted approach is needed that brings together more investigators with expertise in the range of conditions and genes represented in WS. Newer topics, such as metabolic and immune differences29,66,190 in WS, are also in need of innovators. Funding is required to allow for the creation of a large international consortium that will collect prospective standardized data focused on the most relevant questions in WS and will incentivize the broad sharing of data acquired in diverse populations. Such a consortium will allow more rapid collection and dissemination of natural history data, which are needed to understand long-term outcomes and to choose appropriate endpoints for subsequent clinical trials. In addition, biospecimen collection can be optimized by the same mechanism. Ultimately, clinical trials focusing on therapies are critically needed to increase QOL for individuals with WS and their families.
Supplementary Material
Acknowledgements
The authors thank the anonymous individual with WS and her parents who provided the family experience interview, as well as those who provided facial photographs in Figure 5 and Supplementary Table 1.
B.A.K. was supported by the Department of Intramural Research the National Heart, Lung and Blood Institute of the National Institutes of Health. CAM was supported by the Williams Syndrome Association (grant number 0111). B.R.P. was supported by the Williams Syndrome Association, and the Williams Syndrome Charitable Foundation, USA. M.P. was funded by Williams Syndrome Australia Limited. B.B. was funded by the Fritz Thyssen Stiftung and the Israel Science Foundation (Grant No.2305/20). L.R.O. was supported by a Canada Research Chair in the Genetics of Neurodevelopmental Disorders. C.A.E.K. was supported by FAPESP 2019/21644–0 and CNPq 304897/2020–5.
The authors thank Z. Urban for discussion of elastin biology and management strategies for vascular disease in WS.
The authors thank the persons with WS and their families belonging to the Associação Brasileira de Síndrome de Williams, Canadian Association for Williams Syndrome, WIlliams Syndrome Australia Limited, and Williams Syndrome Association (USA) for continued participation in studies and support of research that allows knowledge of WS to grow.
Footnotes
Competing interests
The authors declare no conflicts of interest.
Informed consent
The authors affirm that human research participants provided informed consent for: publication of the photographs in Figure 5; publication of photographs in Supplementary Table 1; and taking part in the family experience interview.
Peer review information
Nature Reviews Disease Primers thanks A. Selicorni, D. Gothelf, P. Ortiz-Romero (V. Campuzano), J. Van Herwegen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
References
- 1.Beuren AJ, Apitz J & Harmjanz D Supravalvular aortic stenosis in association with mental retardation and a certain facial appearance. Circulation 26, 1235–1240, doi: 10.1161/01.cir.26.6.1235 (1962). [DOI] [PubMed] [Google Scholar]
- 2.Williams JC, Barratt-Boyes BG & Lowe JB Supravalvular aortic stenosis. Circulation 24, 1311–1318, doi: 10.1161/01.cir.24.6.1311 (1961). [DOI] [PubMed] [Google Scholar]
- 3.Fanconi G, Girardet P, Schlesinger B, Butler N & Black J Chronische Hypercalcaemie kombiniert mit Osteosklerose, Hyperazotaemie, Minderwuchs, und kongenitalen Missbildungen [Chronic hypercalcemia, combined with osteosclerosis, hyperazotemia, nanism, and congenital malformations]. Helvetica Paediatrica Acta, 314–349 (1952). [PubMed] [Google Scholar]
- 4.Cha SG et al. Long-term cardiovascular outcome of Williams syndrome. Congenit Heart Dis 14, 684–690, doi: 10.1111/chd.12810 (2019). [DOI] [PubMed] [Google Scholar]
- 5. Del Pasqua A et al. New findings concerning cardiovascular manifestations emerging from long-term follow-up of 150 patients with the Williams-Beuren-Beuren syndrome. Cardiol Young 19, 563–567, doi: 10.1017/S1047951109990837 (2009). This paper reports the cardiovascular outcomes from a large number of people with WS over a range of 0.5–25 years (average 6 years).
- 6.Collins RT 2nd. Cardiovascular disease in Williams syndrome. Curr Opin Pediatr 30, 609–615, doi: 10.1097/MOP.0000000000000664 (2018). [DOI] [PubMed] [Google Scholar]
- 7. Li DY et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest 102, 1783–1787, doi: 10.1172/JCI4487 (1998). This paper combines describes the impact of elastin insufficiency in humans and mice, cementing its role in the vasculopathy of WS.
- 8.Bouchireb K et al. Clinical features and management of arterial hypertension in children with Williams-Beuren syndrome. Nephrol Dial Transplant 25, 434–438, doi: 10.1093/ndt/gfp522 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Rose C, Wessel A, Pankau R, Partsch CJ & Bursch J Anomalies of the abdominal aorta in Williams-Beuren syndrome--another cause of arterial hypertension. Eur J Pediatr 160, 655–658, doi: 10.1007/s004310100835 (2001). [DOI] [PubMed] [Google Scholar]
- 10. Latham GJ et al. Perioperative morbidity in children with elastin arteriopathy. Paediatr Anaesth 26, 926–935, doi: 10.1111/pan.12967 (2016). This is the largest study that discusses risks associated with anestheisa in people with WS. N=48.
- 11.Furusawa EA et al. Diagnosis and management of systemic hypertension due to renovascular and aortic stenosis in patients with Williams-Beuren syndrome. Rev Assoc Med Bras (1992) 64, 723–728, doi: 10.1590/1806-9282.64.08.723 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Wessel A et al. Risk of sudden death in the Williams-Beuren syndrome. Am J Med Genet A 127A, 234–237, doi: 10.1002/ajmg.a.30012 (2004). This is the first study to demonstrate an elevated relative risk of sudden cardiovascular death in individuals with Williams syndrome.
- 13. Mervis CB & Greiner de Magalhães C in Pediatric neuropsychology: Research, theory, and practice (eds Beauchamp M et al. ) Ch. Williams syndrome, (2021). This article provides a thorough characterization of the Williams syndrome behavioral phenotype with special focus on intellectual disability, language and literacy development, memory, and executive function development along with suggested intervention approaches.
- 14.Mervis CB & John AE Cognitive and behavioral characteristics of children with Williams syndrome: implications for intervention approaches. Am J Med Genet C Semin Med Genet 154C, 229–248, doi: 10.1002/ajmg.c.30263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarvinen A, Korenberg JR & Bellugi U The social phenotype of Williams syndrome. Curr Opin Neurobiol 23, 414–422, doi: 10.1016/j.conb.2012.12.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein-Tasman BP & Mervis CB Distinctive personality characteristics of 8-, 9-, and 10-year-olds with Williams syndrome. Dev Neuropsychol 23, 269–290, doi: 10.1080/87565641.2003.9651895 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Ewart AK et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet 5, 11–16, doi: 10.1038/ng0993-11 (1993). [DOI] [PubMed] [Google Scholar]
- 18.Perez Jurado LA, Peoples R, Kaplan P, Hamel BC & Francke U Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am J Hum Genet 59, 781–792 (1996). [PMC free article] [PubMed] [Google Scholar]
- 19.Stromme P, Bjornstad PG & Ramstad K Prevalence estimation of Williams syndrome. J Child Neurol 17, 269–271, doi: 10.1177/088307380201700406 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Greenberg F Williams syndrome professional symposium. Am. J. Med. Genet 37, 85–88, doi: 10.1002/ajmg.1320370615 (1990). [DOI] [Google Scholar]
- 21.(NORD), N. O. f. R. D. Williams Syndrome, <https://rarediseases.org/rare-diseases/williams-syndrome/> (2006).
- 22.Honjo RS et al. Williams-Beuren Syndrome: A Clinical Study of 55 Brazilian Patients and the Diagnostic Use of MLPA. Biomed Res Int 2015, 903175, doi: 10.1155/2015/903175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P et al. Williams-Beuren Syndrome: Experience of 43 Patients and a Report of an Atypical Case from a Tertiary Care Center in India. Cytogenet Genome Res 146, 187–194, doi: 10.1159/000439205 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Gold NB et al. Delayed diagnosis of Williams-Beuren syndrome in an adolescent of Jamaican descent: examining racial disparities in genetics education. Clin Dysmorphol 30, 69–70, doi: 10.1097/MCD.0000000000000357 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Lumaka A et al. Williams-Beuren syndrome: pitfalls for diagnosis in limited resources setting. Clin Case Rep 4, 294–297, doi: 10.1002/ccr3.476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tekendo-Ngongang C et al. Challenges in clinical diagnosis of williams-beuren syndrome in sub-saharan africans: case reports from cameroon. Mol Syndromol 5, 287–292, doi: 10.1159/000369421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kruszka P et al. Williams-Beuren syndrome in diverse populations. Am J Med Genet A 176, 1128–1136, doi: 10.1002/ajmg.a.38672 (2018). This paper presents the largest series of facial photograph of persons with Williams syndrome from multiple regions throughout the world; this is crucial for increasing international awareness of the disorder.
- 28.Scheiber D et al. Echocardiographic findings in patients with Williams-Beuren syndrome. Wien Klin Wochenschr 118, 538–542, doi: 10.1007/s00508-006-0658-2 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Parrish PCR et al. Whole exome sequencing in patients with Williams-Beuren syndrome followed by disease modeling in mice points to four novel pathways that may modify stenosis risk. Hum Mol Genet 29, 2035–2050, doi: 10.1093/hmg/ddaa093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadler LS et al. Differences by sex in cardiovascular disease in Williams syndrome. J Pediatr 139, 849–853, doi: 10.1067/mpd.2001.118889 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Morris CA et al. Alpha 1 antitrypsin deficiency alleles are associated with joint dislocation and scoliosis in Williams syndrome. Am J Med Genet C Semin Med Genet 154C, 299–306, doi: 10.1002/ajmg.c.30265 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang MS et al. Molecular and clinical correlation study of Williams-Beuren syndrome: No evidence of molecular factors in the deletion region or imprinting affecting clinical outcome. Am J Med Genet 86, 34–43 (1999). [PubMed] [Google Scholar]
- 33.Elison S, Stinton C & Howlin P Health and social outcomes in adults with Williams syndrome: findings from cross-sectional and longitudinal cohorts. Res Dev Disabil 31, 587–599, doi: 10.1016/j.ridd.2009.12.013 (2010). [DOI] [PubMed] [Google Scholar]
- 34. Sauna-Aho O, Bjelogrlic-Laakso N, Siren A, Kangasmaki V & Arvio M Cognition in adults with Williams syndrome-A 20-year follow-up study. Mol Genet Genomic Med 7, e695, doi: 10.1002/mgg3.695 (2019). These authors provide long-term follow-up information on some of the oldest adults with Williams syndrome reported to date and document difficulties across numerous medical and functional domains.
- 35.Sadler LS, Robinson LK, Verdaasdonk KR & Gingell R The Williams syndrome: evidence for possible autosomal dominant inheritance. Am J Med Genet 47, 468–470, doi: 10.1002/ajmg.1320470406 (1993). [DOI] [PubMed] [Google Scholar]
- 36.Morris CA, Thomas IT & Greenberg F Williams syndrome: autosomal dominant inheritance. Am J Med Genet 47, 478–481, doi: 10.1002/ajmg.1320470409 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Metcalfe K, Simeonov E, Beckett W, Donnai D & Tassabehji M Autosomal dominant inheritance of Williams-Beuren syndrome in a father and son with haploinsufficiency for FKBP6. Clin Dysmorphol 14, 61–65, doi: 10.1097/00019605-200504000-00002 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Pankau R et al. Familial Williams-Beuren syndrome showing varying clinical expression. Am J Med Genet 98, 324–329, doi: (2001). [DOI] [PubMed] [Google Scholar]
- 39. Bayes M, Magano LF, Rivera N, Flores R & Perez Jurado LA Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet 73, 131–151, doi: 10.1086/376565 (2003). This paper dissects the complex make up of the low copy repeats that mediate the WBS deletion.
- 40.Antonell A et al. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet 47, 312–320, doi: 10.1136/jmg.2009.071712 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Cusco I et al. Copy number variation at the 7q11.23 segmental duplications is a susceptibility factor for the Williams-Beuren syndrome deletion. Genome Res 18, 683–694, doi: 10.1101/gr.073197.107 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osborne LR et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet 29, 321–325, doi: 10.1038/ng753 (2001). This paper identifies 7q11.23 inversion as a polymorphism that is a risk factor for the WBS deletion.
- 43.Somerville MJ et al. Severe expressive-language delay related to duplication of the Williams-Beuren locus. N Engl J Med 353, 1694–1701, doi: 10.1056/NEJMoa051962 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hobart HH et al. Inversion of the Williams syndrome region is a common polymorphism found more frequently in parents of children with Williams syndrome. Am J Med Genet C Semin Med Genet 154C, 220–228, doi: 10.1002/ajmg.c.30258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tam E et al. The common inversion of the Williams-Beuren syndrome region at 7q11.23 does not cause clinical symptoms. Am J Med Genet A 146A, 1797–1806, doi: 10.1002/ajmg.a.32360 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramocki MB et al. Recurrent distal 7q11.23 deletion including HIP1 and YWHAG identified in patients with intellectual disabilities, epilepsy, and neurobehavioral problems. Am J Hum Genet 87, 857–865, doi: 10.1016/j.ajhg.2010.10.019 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marshall CR et al. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23-q21.11. Am J Hum Genet 83, 106–111, doi: 10.1016/j.ajhg.2008.06.001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicita F et al. Epilepsy is a possible feature in Williams-Beuren syndrome patients harboring typical deletions of the 7q11.23 critical region. Am J Med Genet A 170A, 148–155, doi: 10.1002/ajmg.a.37410 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Lugo M et al. Social, neurodevelopmental, endocrine, and head size differences associated with atypical deletions in Williams-Beuren syndrome. Am J Med Genet A 182, 1008–1020, doi: 10.1002/ajmg.a.61522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fusco C et al. Smaller and larger deletions of the Williams Beuren syndrome region implicate genes involved in mild facial phenotype, epilepsy and autistic traits. Eur J Hum Genet 22, 64–70, doi: 10.1038/ejhg.2013.101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stock AD et al. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet A 120A, 320–325, doi: 10.1002/ajmg.a.20055 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Vandeweyer G, Van der Aa N, Reyniers E & Kooy RF The contribution of CLIP2 haploinsufficiency to the clinical manifestations of the Williams-Beuren syndrome. Am J Hum Genet 90, 1071–1078, doi: 10.1016/j.ajhg.2012.04.020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tassabehji M et al. Williams syndrome: use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes. Am J Hum Genet 64, 118–125, doi: 10.1086/302214 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tassabehji M et al. GTF2IRD1 in craniofacial development of humans and mice. Science 310, 1184–1187, doi: 10.1126/science.1116142 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Plaja A et al. A Novel Recurrent Breakpoint Responsible for Rearrangements in the Williams-Beuren Region. Cytogenet Genome Res 146, 181–186, doi: 10.1159/000439463 (2015). [DOI] [PubMed] [Google Scholar]
- 56. Morris CA et al. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A 123A, 45–59, doi: 10.1002/ajmg.a.20496 (2003). This paper demonstrates the roles of the deletion of centromeric and telomeric portions of the WSCR in the WS neurocognitive profile.
- 57.Hoeft F et al. Mapping genetically controlled neural circuits of social behavior and visuo-motor integration by a preliminary examination of atypical deletions with Williams syndrome. PLoS One 9, e104088, doi: 10.1371/journal.pone.0104088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirota H et al. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med 5, 311–321, doi: 10.1097/01.GIM.0000076975.10224.67 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Gagliardi C, Bonaglia MC, Selicorni A, Borgatti R & Giorda R Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J Med Genet 40, 526–530, doi: 10.1136/jmg.40.7.526 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrero GB et al. An atypical 7q11.23 deletion in a normal IQ Williams-Beuren syndrome patient. Eur J Hum Genet 18, 33–38, doi: 10.1038/ejhg.2009.108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delgado LM et al. A 1.3-mb 7q11.23 atypical deletion identified in a cohort of patients with williams-beuren syndrome. Mol Syndromol 4, 143–147, doi: 10.1159/000347167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Botta A et al. Detection of an atypical 7q11.23 deletion in Williams syndrome patients which does not include the STX1A and FZD3 genes. J Med Genet 36, 478–480 (1999). [PMC free article] [PubMed] [Google Scholar]
- 63.Alesi V et al. Atypical 7q11.23 deletions excluding ELN gene result in Williams-Beuren syndrome craniofacial features and neurocognitive profile. Am J Med Genet A 185, 242–249, doi: 10.1002/ajmg.a.61937 (2021). [DOI] [PubMed] [Google Scholar]
- 64.Frangiskakis JM et al. LIM-kinase1 hemizygosity implicated in impaired visuospatial constructive cognition. Cell 86, 59–69, doi: 10.1016/s0092-8674(00)80077-x (1996). [DOI] [PubMed] [Google Scholar]
- 65.Antonell A, Vilardell M & Perez Jurado LA Transcriptome profile in Williams-Beuren syndrome lymphoblast cells reveals gene pathways implicated in glucose intolerance and visuospatial construction deficits. Hum Genet 128, 27–37, doi: 10.1007/s00439-010-0817-4 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Kimura R et al. Integrated DNA methylation analysis reveals a potential role for ANKRD30B in Williams syndrome. Neuropsychopharmacology 45, 1627–1636, doi: 10.1038/s41386-020-0675-2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karagiannis P et al. Induced Pluripotent Stem Cells and Their Use in Human Models of Disease and Development. Physiol Rev 99, 79–114, doi: 10.1152/physrev.00039.2017 (2019). [DOI] [PubMed] [Google Scholar]
- 68.Khattak S et al. Human induced pluripotent stem cell derived neurons as a model for Williams-Beuren syndrome. Mol Brain 8, 77, doi: 10.1186/s13041-015-0168-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Adamo A et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat Genet 47, 132–141, doi: 10.1038/ng.3169 (2015). This paper uses iPSC models of 7q11.23 CNV to identify transcription changes associated with these genetic alterations.
- 70. Strong E et al. Symmetrical Dose-Dependent DNA-Methylation Profiles in Children with Deletion or Duplication of 7q11.23. Am J Hum Genet 97, 216–227, doi: 10.1016/j.ajhg.2015.05.019 (2015). This paper identifies extensive genome-wide changes in DNA methylation in WBS that could have significant impacts on gene regulation.
- 71.Segura-Puimedon M et al. Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum Mol Genet 23, 6481–6494, doi: 10.1093/hmg/ddu368 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Borralleras C et al. Synaptic plasticity and spatial working memory are impaired in the CD mouse model of Williams-Beuren syndrome. Mol Brain 9, 76, doi: 10.1186/s13041-016-0258-7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jimenez-Altayo F et al. Stenosis coexists with compromised alpha1-adrenergic contractions in the ascending aorta of a mouse model of Williams-Beuren syndrome. Sci Rep 10, 889, doi: 10.1038/s41598-020-57803-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Campuzano V et al. Reduction of NADPH-oxidase activity ameliorates the cardiovascular phenotype in a mouse model of Williams-Beuren Syndrome. PLoS Genet 8, e1002458, doi: 10.1371/journal.pgen.1002458 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291, doi: 10.1038/nature19057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harkness ML, Harkness RD & McDonald DA The collagen and elastin content of the arterial wall in the dog. Proc R Soc Lond B Biol Sci 146, 541–551, doi: 10.1098/rspb.1957.0029 (1957). [DOI] [PubMed] [Google Scholar]
- 77.Leung DY, Glagov S & Mathews MB Elastin and collagen accumulation in rabbit ascending aorta and pulmonary trunk during postnatal growth. Correlation of cellular synthetic response with medial tension. Circ Res 41, 316–323, doi: 10.1161/01.res.41.3.316 (1977). [DOI] [PubMed] [Google Scholar]
- 78.Li B & Daggett V Molecular basis for the extensibility of elastin. J Muscle Res Cell Motil 23, 561–573 (2002). [DOI] [PubMed] [Google Scholar]
- 79.Kozel BA & Mecham RP Elastic fiber ultrastructure and assembly. Matrix Biol 84, 31–40, doi: 10.1016/j.matbio.2019.10.002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shapiro SD, Endicott SK, Province MA, Pierce JA & Campbell EJ Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest 87, 1828–1834, doi: 10.1172/JCI115204 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parks WC Posttranscriptional regulation of lung elastin production. Am J Respir Cell Mol Biol 17, 1–2, doi: 10.1165/ajrcmb.17.1.f135 (1997). [DOI] [PubMed] [Google Scholar]
- 82.Ott CE et al. MicroRNAs differentially expressed in postnatal aortic development downregulate elastin via 3’ UTR and coding-sequence binding sites. PLoS One 6, e16250, doi: 10.1371/journal.pone.0016250 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang M & Parks WC Posttranscriptional regulation of lung elastin expression involves binding of a developmentally regulated cytosolic protein to an open-reading frame cis-element in the messenger RNA. Chest 116, 74S, doi: 10.1378/chest.116.suppl_1.74s (1999). [DOI] [PubMed] [Google Scholar]
- 84.Li DY et al. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet 6, 1021–1028, doi: 10.1093/hmg/6.7.1021 (1997). [DOI] [PubMed] [Google Scholar]
- 85.Tassabehji M et al. Elastin: genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum Mol Genet 6, 1029–1036, doi: 10.1093/hmg/6.7.1029 (1997). [DOI] [PubMed] [Google Scholar]
- 86.Urban Z et al. Isolated supravalvular aortic stenosis: functional haploinsufficiency of the elastin gene as a result of nonsense-mediated decay. Hum Genet 106, 577–588, doi: 10.1007/s004390000285 (2000). [DOI] [PubMed] [Google Scholar]
- 87.Olson TM et al. A 30 kb deletion within the elastin gene results in familial supravalvular aortic stenosis. Hum Mol Genet 4, 1677–1679, doi: 10.1093/hmg/4.9.1677 (1995). [DOI] [PubMed] [Google Scholar]
- 88.Pober BR, Johnson M & Urban Z Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Invest 118, 1606–1615, doi: 10.1172/JCI35309 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Collins RT 2nd, Kaplan P, Somes GW & Rome JJ Long-term outcomes of patients with cardiovascular abnormalities and williams syndrome. Am J Cardiol 105, 874–878, doi: 10.1016/j.amjcard.2009.10.069 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Collins RT 2nd. Cardiovascular disease in Williams syndrome. Circulation 127, 2125–2134, doi: 10.1161/CIRCULATIONAHA.112.000064 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Kozel BA et al. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 63, 74–79, doi: 10.1161/HYPERTENSIONAHA.113.02087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abu-Sultaneh S, Gondim MJ, Alexy RD & Mastropietro CW Sudden cardiac death associated with cardiac catheterization in Williams syndrome: a case report and review of literature. Cardiol Young, 1–5, doi: 10.1017/S1047951119000295 (2019). [DOI] [PubMed] [Google Scholar]
- 93.Matisoff AJ, Olivieri L, Schwartz JM & Deutsch N Risk assessment and anesthetic management of patients with Williams syndrome: a comprehensive review. Paediatr Anaesth 25, 1207–1215, doi: 10.1111/pan.12775 (2015). [DOI] [PubMed] [Google Scholar]
- 94. Staudt GE & Eagle SS Anesthetic Considerations for Patients With Williams Syndrome. J Cardiothorac Vasc Anesth 35, 176–186, doi: 10.1053/j.jvca.2020.01.022 (2021). This is a well-organized paper which reviews anesthesia risks and provides management guidelines for both children and adults with Williams syndrome.
- 95.Burch TM, McGowan FX Jr., Kussman BD, Powell AJ & DiNardo JA Congenital supravalvular aortic stenosis and sudden death associated with anesthesia: what’s the mystery? Anesth Analg 107, 1848–1854, doi: 10.1213/ane.0b013e3181875a4d (2008). [DOI] [PubMed] [Google Scholar]
- 96.Hirano E, Knutsen RH, Sugitani H, Ciliberto CH & Mecham RP Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res 101, 523–531, doi: 10.1161/CIRCRESAHA.107.153510 (2007). [DOI] [PubMed] [Google Scholar]
- 97. Lin CJ et al. Heterogeneous Cellular Contributions to Elastic Laminae Formation in Arterial Wall Development. Circ Res 125, 1006–1018, doi: 10.1161/CIRCRESAHA.119.315348 (2019). This paper dissects the role of endothelial vs smooth muscle cells on the elastin insufficiency phenotype and is the first mouse model to show neo-intima formation in an elastin mutant.
- 98.Karnik SK et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development 130, 411–423, doi: 10.1242/dev.00223 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Li DY et al. Elastin is an essential determinant of arterial morphogenesis. Nature 393, 276–280, doi: 10.1038/30522 (1998). [DOI] [PubMed] [Google Scholar]
- 100. Urban Z et al. Connection between elastin haploinsufficiency and increased cell proliferation in patients with supravalvular aortic stenosis and Williams-Beuren syndrome. Am J Hum Genet 71, 30–44, doi: 10.1086/341035 (2002). This paper links elastin insufficiency to changes in smooth muscle cell proliferation.
- 101.Misra A et al. Integrin beta3 inhibition is a therapeutic strategy for supravalvular aortic stenosis. J Exp Med 213, 451–463, doi: 10.1084/jem.20150688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jiao Y et al. Deficient Circumferential Growth Is the Primary Determinant of Aortic Obstruction Attributable to Partial Elastin Deficiency. Arterioscler Thromb Vasc Biol 37, 930–941, doi: 10.1161/ATVBAHA.117.309079 (2017). This paper posits that elastin insufficiency impacts circumferential growth of developing arteries.
- 103.Faury G et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest 112, 1419–1428, doi: 10.1172/JCI19028 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiao Y et al. mTOR (Mechanistic Target of Rapamycin) Inhibition Decreases Mechanosignaling, Collagen Accumulation, and Stiffening of the Thoracic Aorta in Elastin-Deficient Mice. Arterioscler Thromb Vasc Biol 37, 1657–1666, doi: 10.1161/ATVBAHA.117.309653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kinnear C et al. Everolimus Rescues the Phenotype of Elastin Insufficiency in Patient Induced Pluripotent Stem Cell-Derived Vascular Smooth Muscle Cells. Arterioscler Thromb Vasc Biol 40, 1325–1339, doi: 10.1161/ATVBAHA.119.313936 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li W et al. Rapamycin inhibits smooth muscle cell proliferation and obstructive arteriopathy attributable to elastin deficiency. Arterioscler Thromb Vasc Biol 33, 1028–1035, doi: 10.1161/ATVBAHA.112.300407 (2013). [DOI] [PubMed] [Google Scholar]
- 107.Wan ES, Pober BR, Washko GR, Raby BA & Silverman EK Pulmonary function and emphysema in Williams-Beuren syndrome. Am J Med Genet A 152A, 653–656, doi: 10.1002/ajmg.a.33300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shifren A, Durmowicz AG, Knutsen RH, Hirano E & Mecham RP Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema. Am J Physiol Lung Cell Mol Physiol 292, L778–787, doi: 10.1152/ajplung.00352.2006 (2007). [DOI] [PubMed] [Google Scholar]
- 109.Pangallo E et al. Pulmonary function in Williams-Beuren syndrome: Spirometric data of 22 Italian patients. Am J Med Genet A, doi: 10.1002/ajmg.a.61966 (2020). [DOI] [PubMed] [Google Scholar]
- 110.Urban Z et al. Elastin gene deletions in Williams syndrome patients result in altered deposition of elastic fibers in skin and a subclinical dermal phenotype. Pediatr Dermatol 17, 12–20, doi: 10.1046/j.1525-1470.2000.01703.x (2000). [DOI] [PubMed] [Google Scholar]
- 111.Kozel BA et al. Skin findings in Williams syndrome. Am J Med Genet A 164A, 2217–2225, doi: 10.1002/ajmg.a.36628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sammour ZM et al. Congenital genitourinary abnormalities in children with Williams-Beuren syndrome. J Pediatr Urol 10, 804–809, doi: 10.1016/j.jpurol.2014.01.013 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Vaux KK, Wojtczak H, Benirschke K & Jones KL Vocal cord abnormalities in Williams syndrome: a further manifestation of elastin deficiency. Am J Med Genet A 119A, 302–304, doi: 10.1002/ajmg.a.20169 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Drummond GR, Selemidis S, Griendling KK & Sobey CG Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10, 453–471, doi: 10.1038/nrd3403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lassegue B & Griendling KK NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30, 653–661, doi: 10.1161/ATVBAHA.108.181610 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Del Campo M et al. Hemizygosity at the NCF1 gene in patients with Williams-Beuren syndrome decreases their risk of hypertension. Am J Hum Genet 78, 533–542, doi: 10.1086/501073 (2006). This is the first paper to show a role for NCF1 gene dosage in modifying hypertension risk in WS.
- 117.Kozel BA et al. Genetic modifiers of cardiovascular phenotype caused by elastin haploinsufficiency act by extrinsic noncomplementation. J Biol Chem 286, 44926–44936, doi: 10.1074/jbc.M111.274779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Troia A et al. Inhibition of NOX1 mitigates blood pressure increases in elastin insufficiency. Function, In Revision (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caraveo G, van Rossum DB, Patterson RL, Snyder SH & Desiderio S Action of TFII-I outside the nucleus as an inhibitor of agonist-induced calcium entry. Science 314, 122–125, doi: 10.1126/science.1127815 (2006). [DOI] [PubMed] [Google Scholar]
- 120.Hakre S et al. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol Cell 24, 301–308, doi: 10.1016/j.molcel.2006.09.005 (2006). [DOI] [PubMed] [Google Scholar]
- 121.Roy AL Signal-induced functions of the transcription factor TFII-I. Biochim Biophys Acta 1769, 613–621, doi: 10.1016/j.bbaexp.2007.10.002 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Enkhmandakh B et al. Essential functions of the Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc Natl Acad Sci U S A 106, 181–186, doi: 10.1073/pnas.0811531106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hinsley TA, Cunliffe P, Tipney HJ, Brass A & Tassabehji M Comparison of TFII-I gene family members deleted in Williams-Beuren syndrome. Protein Sci 13, 2588–2599, doi: 10.1110/ps.04747604 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lucena J et al. Essential role of the N-terminal region of TFII-I in viability and behavior. BMC Med Genet 11, 61, doi: 10.1186/1471-2350-11-61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roy AL Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene 492, 32–41, doi: 10.1016/j.gene.2011.10.030 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheriyath V, Desgranges ZP & Roy AL c-Src-dependent transcriptional activation of TFII-I. J Biol Chem 277, 22798–22805, doi: 10.1074/jbc.M202956200 (2002). [DOI] [PubMed] [Google Scholar]
- 127.Desgranges ZP et al. Inhibition of TFII-I-dependent cell cycle regulation by p53. Mol Cell Biol 25, 10940–10952, doi: 10.1128/MCB.25.24.10940-10952.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roy AL Biochemistry and biology of the inducible multifunctional transcription factor TFII-I. Gene 274, 1–13, doi: 10.1016/s0378-1119(01)00625-4 (2001). [DOI] [PubMed] [Google Scholar]
- 129.Enkhmandakh B, Bitchevaia N, Ruddle F & Bayarsaihan D The early embryonic expression of TFII-I during mouse preimplantation development. Gene Expr Patterns 4, 25–28, doi: 10.1016/s1567-133x(03)00155-8 (2004). [DOI] [PubMed] [Google Scholar]
- 130.Makeyev AV & Bayarsaihan D New TFII-I family target genes involved in embryonic development. Biochem Biophys Res Commun 386, 554–558, doi: 10.1016/j.bbrc.2009.06.045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ashworth T & Roy AL Phase specific functions of the transcription factor TFII-I during cell cycle. Cell Cycle 8, 596–605, doi: 10.4161/cc.8.4.7728 (2009). [DOI] [PubMed] [Google Scholar]
- 132.Chimge NO, Makeyev AV, Ruddle FH & Bayarsaihan D Identification of the TFII-I family target genes in the vertebrate genome. Proc Natl Acad Sci U S A 105, 9006–9010, doi: 10.1073/pnas.0803051105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tussie-Luna MI, Bayarsaihan D, Seto E, Ruddle FH & Roy AL Physical and functional interactions of histone deacetylase 3 with TFII-I family proteins and PIASxbeta. Proc Natl Acad Sci U S A 99, 12807–12812, doi: 10.1073/pnas.192464499 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bayarsaihan D What role does TFII-I have to play in epigenetic modulation during embryogenesis? Epigenomics 5, 9–11, doi: 10.2217/epi.12.71 (2013). [DOI] [PubMed] [Google Scholar]
- 135.Howald C et al. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J Med Genet 43, 266–273, doi: 10.1136/jmg.2005.034009 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Karmiloff-Smith A et al. Social cognition in williams syndrome: genotype/phenotype insights from partial deletion patients. Front Psychol 3, 168, doi: 10.3389/fpsyg.2012.00168 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.van Hagen JM et al. Contribution of CYLN2 and GTF2IRD1 to neurological and cognitive symptoms in Williams Syndrome. Neurobiol Dis 26, 112–124, doi: 10.1016/j.nbd.2006.12.009 (2007). [DOI] [PubMed] [Google Scholar]
- 138.Karmiloff-Smith A et al. Using case study comparisons to explore genotype-phenotype correlations in Williams-Beuren syndrome. J Med Genet 40, 136–140, doi: 10.1136/jmg.40.2.136 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dai L et al. Is it Williams syndrome? GTF2IRD1 implicated in visual-spatial construction and GTF2I in sociability revealed by high resolution arrays. Am J Med Genet A 149A, 302–314, doi: 10.1002/ajmg.a.32652 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pinelli M et al. A small 7q11.23 microduplication involving GTF2I in a family with intellectual disability. Clin Genet 97, 940–942, doi: 10.1111/cge.13753 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sakurai T et al. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res 4, 28–39, doi: 10.1002/aur.169 (2011). [DOI] [PubMed] [Google Scholar]
- 142.Martin LA, Iceberg E & Allaf G Consistent hypersocial behavior in mice carrying a deletion of Gtf2i but no evidence of hyposocial behavior with Gtf2i duplication: Implications for Williams-Beuren syndrome and autism spectrum disorder. Brain Behav 8, e00895, doi: 10.1002/brb3.895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Barak B et al. Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat Neurosci 22, 700–708, doi: 10.1038/s41593-019-0380-9 (2019). This paper demonstrates the neuronal functions of Gtf2i in mediating myelination properties in the mouse brain, and that correcting myelination deficits rescues social and motor behavior deficits; light is also shed on molecular and cellular defects related to myelination deficits in the brain of individuals with WS.
- 144.Osso LA & Chan JR A surprising role for myelin in Williams syndrome. Nat Neurosci 22, 681–683, doi: 10.1038/s41593-019-0368-5 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Borralleras C, Sahun I, Perez-Jurado LA & Campuzano V Intracisternal Gtf2i Gene Therapy Ameliorates Deficits in Cognition and Synaptic Plasticity of a Mouse Model of Williams-Beuren Syndrome. Mol Ther 23, 1691–1699, doi: 10.1038/mt.2015.130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mervis CB et al. Duplication of GTF2I results in separation anxiety in mice and humans. Am J Hum Genet 90, 1064–1070, doi: 10.1016/j.ajhg.2012.04.012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Osborne LR Animal models of Williams syndrome. Am J Med Genet C Semin Med Genet 154C, 209–219, doi: 10.1002/ajmg.c.30257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mervis CB et al. Children with 7q11.23 duplication syndrome: psychological characteristics. Am J Med Genet A 167, 1436–1450, doi: 10.1002/ajmg.a.37071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morris CA et al. 7q11.23 Duplication syndrome: Physical characteristics and natural history. Am J Med Genet A 167A, 2916–2935, doi: 10.1002/ajmg.a.37340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Deurloo MHS et al. Transcription Factor 2I Regulates Neuronal Development via TRPC3 in 7q11.23 Disorder Models. Mol Neurobiol 56, 3313–3325, doi: 10.1007/s12035-018-1290-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang Y et al. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J Neurosci 30, 5334–5345, doi: 10.1523/JNEUROSCI.5963-09.2010 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Poitras L et al. An SNP in an ultraconserved regulatory element affects Dlx5/Dlx6 regulation in the forebrain. Development 137, 3089–3097, doi: 10.1242/dev.051052 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barak B & Feng G Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat Neurosci 19, 647–655, doi: 10.1038/nn.4276 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Becerra AM & Mervis CB Age at Onset of Declarative Gestures and 24-Month Expressive Vocabulary Predict Later Language and Intellectual Abilities in Young Children With Williams Syndrome. Front Psychol 10, 2648, doi: 10.3389/fpsyg.2019.02648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Richards C, Jones C, Groves L, Moss J & Oliver C Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry 2, 909–916, doi: 10.1016/S2215-0366(15)00376-4 (2015). [DOI] [PubMed] [Google Scholar]
- 156.Klein-Tasman BP & Mervis CB Autism Spectrum Symptomatology Among Children with Duplication 7q11.23 Syndrome. J Autism Dev Disord 48, 1982–1994, doi: 10.1007/s10803-017-3439-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zanella M et al. Dosage analysis of the 7q11.23 Williams region identifies BAZ1B as a major human gene patterning the modern human face and underlying self-domestication. Sci Adv 5, eaaw7908, doi: 10.1126/sciadv.aaw7908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Barnett C et al. Williams Syndrome Transcription Factor is critical for neural crest cell function in Xenopus laevis. Mech Dev 129, 324–338, doi: 10.1016/j.mod.2012.06.001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nagy N & Goldstein AM Enteric nervous system development: A crest cell’s journey from neural tube to colon. Semin Cell Dev Biol 66, 94–106, doi: 10.1016/j.semcdb.2017.01.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gregory MD et al. Williams syndrome hemideletion and LIMK1 variation both affect dorsal stream functional connectivity. Brain 142, 3963–3974, doi: 10.1093/brain/awz323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ravindran S, Nalavadi VC & Muddashetty RS BDNF Induced Translation of Limk1 in Developing Neurons Regulates Dendrite Growth by Fine-Tuning Cofilin1 Activity. Front Mol Neurosci 12, 64, doi: 10.3389/fnmol.2019.00064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Todorovski Z et al. LIMK1 regulates long-term memory and synaptic plasticity via the transcriptional factor CREB. Mol Cell Biol 35, 1316–1328, doi: 10.1128/MCB.01263-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Durdiakova J, Warrier V, Banerjee-Basu S, Baron-Cohen S & Chakrabarti B STX1A and Asperger syndrome: a replication study. Mol Autism 5, 14, doi: 10.1186/2040-2392-5-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sanchez-Mora C et al. Evaluation of common variants in 16 genes involved in the regulation of neurotransmitter release in ADHD. Eur Neuropsychopharmacol 23, 426–435, doi: 10.1016/j.euroneuro.2012.07.014 (2013). [DOI] [PubMed] [Google Scholar]
- 165.Aslamy A & Thurmond DC Exocytosis proteins as novel targets for diabetes prevention and/or remediation? Am J Physiol Regul Integr Comp Physiol 312, R739–R752, doi: 10.1152/ajpregu.00002.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Andersson SA et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol Cell Endocrinol 364, 36–45, doi: 10.1016/j.mce.2012.08.009 (2012). [DOI] [PubMed] [Google Scholar]
- 167.Pober BR et al. High prevalence of diabetes and pre-diabetes in adults with Williams syndrome. Am J Med Genet C Semin Med Genet 154C, 291–298, doi: 10.1002/ajmg.c.30261 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Morigny P et al. Interaction between hormone-sensitive lipase and ChREBP in fat cells controls insulin sensitivity. Nat Metab 1, 133–146, doi: 10.1038/s42255-018-0007-6 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Vijayakumar A et al. Absence of Carbohydrate Response Element Binding Protein in Adipocytes Causes Systemic Insulin Resistance and Impairs Glucose Transport. Cell Rep 21, 1021–1035, doi: 10.1016/j.celrep.2017.09.091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mejhert N et al. Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Mol Cell 77, 1251–1264 e1259, doi: 10.1016/j.molcel.2020.01.014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stenton SL et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J Clin Invest 131, doi: 10.1172/JCI138267 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tebbenkamp ATN et al. The 7q11.23 Protein DNAJC30 Interacts with ATP Synthase and Links Mitochondria to Brain Development. Cell 175, 1088–1104 e1023, doi: 10.1016/j.cell.2018.09.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pober BR Williams-Beuren syndrome. N Engl J Med 362, 239–252, doi: 10.1056/NEJMra0903074 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Pober BR & Morris CA Diagnosis and management of medical problems in adults with Williams-Beuren syndrome. Am J Med Genet C Semin Med Genet 145C, 280–290, doi: 10.1002/ajmg.c.30139 (2007). [DOI] [PubMed] [Google Scholar]
- 175.Morris CA in GeneReviews® [Internet] (University of Washington, Seattle, 1999. Apr 9 [Updated 2017 Mar 23]). [Google Scholar]
- 176. Sindhar S et al. Hypercalcemia in Patients with Williams-Beuren Syndrome. J Pediatr 178, 254–260 e254, doi: 10.1016/j.jpeds.2016.08.027 (2016). This is a large series showing relatively low frequency of “actionable” hypercalcemia in children with Williams syndrome, along with recommendations for medical management.
- 177.de Sousa Lima Strafacci A, Fernandes Camargo J, Bertapelli F & Guerra Junior G Growth assessment in children with Williams-Beuren syndrome: a systematic review. J Appl Genet 61, 205–212, doi: 10.1007/s13353-020-00551-x (2020). [DOI] [PubMed] [Google Scholar]
- 178. Morris CA, Braddock SR & Council On G Health Care Supervision for Children With Williams Syndrome. Pediatrics 145, doi: 10.1542/peds.2019-3761 (2020). These are the most recent USA general healthcare recommendations for children with WS.
- 179.Martin ND, Smith WR, Cole TJ & Preece MA New height, weight and head circumference charts for British children with Williams syndrome. Arch Dis Child 92, 598–601, doi: 10.1136/adc.2006.107946 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jehee FS et al. Using a combination of MLPA kits to detect chromosomal imbalances in patients with multiple congenital anomalies and mental retardation is a valuable choice for developing countries. Eur J Med Genet 54, e425–432, doi: 10.1016/j.ejmg.2011.03.007 (2011). [DOI] [PubMed] [Google Scholar]
- 181.Dutra RL et al. Copy number variation in Williams-Beuren syndrome: suitable diagnostic strategy for developing countries. BMC Res Notes 5, 13, doi: 10.1186/1756-0500-5-13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lumaka A et al. Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator. Clin Genet 92, 166–171, doi: 10.1111/cge.12948 (2017). [DOI] [PubMed] [Google Scholar]
- 183.Mishima H et al. Evaluation of Face2Gene using facial images of patients with congenital dysmorphic syndromes recruited in Japan. J Hum Genet 64, 789–794, doi: 10.1038/s10038-019-0619-z (2019). [DOI] [PubMed] [Google Scholar]
- 184.Elmas M & Gogus B Success of Face Analysis Technology in Rare Genetic Diseases Diagnosed by Whole-Exome Sequencing: A Single-Center Experience. Mol Syndromol 11, 4–14, doi: 10.1159/000505800 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Scherer SW et al. Observation of a parental inversion variant in a rare Williams-Beuren syndrome family with two affected children. Hum Genet 117, 383–388, doi: 10.1007/s00439-005-1325-9 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kara-Mostefa A et al. Recurrent Williams-Beuren syndrome in a sibship suggestive of maternal germ-line mosaicism. Am J Hum Genet 64, 1475–1478, doi: 10.1086/302362 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mulik VV, Temple KI & Howe DT Two pregnancies in a woman with Williams syndrome. BJOG 111, 511–512, doi: 10.1111/j.1471-0528.2004.00109.x (2004). [DOI] [PubMed] [Google Scholar]
- 188.van der Tuuk K, Drenthen W, Moons P & Budts W Three live-birth pregnancies in a woman with Williams syndrome. Congenit Heart Dis 2, 139–142, doi: 10.1111/j.1747-0803.2007.00088.x (2007). [DOI] [PubMed] [Google Scholar]
- 189.Yuan M, Deng L, Yang Y & Sun L Intrauterine phenotype features of fetuses with Williams-Beuren syndrome and literature review. Ann Hum Genet 84, 169–176, doi: 10.1111/ahg.12360 (2020). [DOI] [PubMed] [Google Scholar]
- 190.Stanley TL, Leong A & Pober BR Growth, body composition, and endocrine issues in Williams syndrome. Curr Opin Endocrinol Diabetes Obes 28, 64–74, doi: 10.1097/MED.0000000000000588 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Riby DM & Porter MA Williams syndrome. Adv Child Dev Behav 39, 163–209, doi: 10.1016/b978-0-12-374748-8.00005-6 (2010). [DOI] [PubMed] [Google Scholar]
- 192.Porter MA in Magill’s Medical Guide (eds Auday B, Buratovich M, Marrocco G, & Moglia P) 2385–2387 (Salem Press Inc, 2013). [Google Scholar]
- 193.Castro T, de Paula Martins Santos C, de Oliveira Lira Ortega A & Gallottini M Oral characteristics and medical considerations in the dental treatment of individuals with Williams syndrome. Spec Care Dentist 39, 108–113, doi: 10.1111/scd.12361 (2019). [DOI] [PubMed] [Google Scholar]
- 194.Hertzberg J, Nakisbendi L, Needleman HL & Pober B Williams syndrome--oral presentation of 45 cases. Pediatr Dent 16, 262–267 (1994). [PubMed] [Google Scholar]
- 195.Panel, W. S. F. P. A Vol. Revised (The Williams Syndrome Foundation UK, London, 2017). [Google Scholar]
- 196.Wu FY et al. Long-term Surgical Prognosis of Primary Supravalvular Aortic Stenosis Repair. Ann Thorac Surg 108, 1202–1209, doi: 10.1016/j.athoracsur.2019.04.094 (2019). [DOI] [PubMed] [Google Scholar]
- 197.Rastelli GC, McGoon DC, Ongley PA, Mankin HT & Kirklin JW Surgical treatment of supravalvular aortic stenosis. Report of 16 cases and review of literature. J Thorac Cardiovasc Surg 51, 873–882 (1966). [PubMed] [Google Scholar]
- 198.McGoon DC, Mankin HT, Vlad P & Kirklin JW The Surgical Treatment of Supravalvular Aortic Stenosis. The Journal of Thoracic and Cardiovascular Surgery 41, 123–133, doi: 10.1016/S0022-5223(20)31735-9 (1961). [DOI] [PubMed] [Google Scholar]
- 199.Doty DB, Polansky DB & Jenson CB Supravalvular aortic stenosis. Repair by extended aortoplasty. J Thorac Cardiovasc Surg 74, 362–371 (1977). [PubMed] [Google Scholar]
- 200.Brom AG in Cardiac surgery: safeguards and pitfalls in operative technique (ed Khonsari S) 276–280 (Aspen Publishers, 1988). [Google Scholar]
- 201.Kaushal S, Backer CL, Patel S, Gossett JG & Mavroudis C Midterm outcomes in supravalvular aortic stenosis demonstrate the superiority of multisinus aortoplasty. Ann Thorac Surg 89, 1371–1377, doi: 10.1016/j.athoracsur.2010.02.019 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Cohen JL, Glickstein JS & Crystal MA Drug-Coated Balloon Angioplasty: A Novel Treatment for Pulmonary Artery In-Stent Stenosis in a Patient with Williams Syndrome. Pediatr Cardiol 38, 1716–1721, doi: 10.1007/s00246-017-1646-1 (2017). [DOI] [PubMed] [Google Scholar]
- 203.Brown ML, Nasr VG, Toohey R & DiNardo JA Williams Syndrome and Anesthesia for Non-cardiac Surgery: High Risk Can Be Mitigated with Appropriate Planning. Pediatr Cardiol 39, 1123–1128, doi: 10.1007/s00246-018-1864-1 (2018). [DOI] [PubMed] [Google Scholar]
- 204.Collins Ii RT, Collins MG, Schmitz ML & Hamrick JT Peri-procedural risk stratification and management of patients with Williams syndrome. Congenit Heart Dis 12, 133–142, doi: 10.1111/chd.12447 (2017). [DOI] [PubMed] [Google Scholar]
- 205.Hayashi A et al. Minoxidil stimulates elastin expression in aortic smooth muscle cells. Arch Biochem Biophys 315, 137–141, doi: 10.1006/abbi.1994.1482 (1994). [DOI] [PubMed] [Google Scholar]
- 206.Slove S et al. Potassium channel openers increase aortic elastic fiber formation and reverse the genetically determined elastin deficit in the BN rat. Hypertension 62, 794–801, doi: 10.1161/HYPERTENSIONAHA.113.01379 (2013). [DOI] [PubMed] [Google Scholar]
- 207.Knutsen RH et al. Minoxidil improves vascular compliance, restores cerebral blood flow, and alters extracellular matrix gene expression in a model of chronic vascular stiffness. Am J Physiol Heart Circ Physiol 315, H18–H32, doi: 10.1152/ajpheart.00683.2017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Coquand-Gandit M et al. Chronic Treatment with Minoxidil Induces Elastic Fiber Neosynthesis and Functional Improvement in the Aorta of Aged Mice. Rejuvenation Res 20, 218–230, doi: 10.1089/rej.2016.1874 (2017). [DOI] [PubMed] [Google Scholar]
- 209.Fhayli W et al. Chronic administration of minoxidil protects elastic fibers and stimulates their neosynthesis with improvement of the aorta mechanics in mice. Cell Signal 62, 109333, doi: 10.1016/j.cellsig.2019.05.018 (2019). [DOI] [PubMed] [Google Scholar]
- 210.Kassai B et al. Minoxidil versus placebo in the treatment of arterial wall hypertrophy in children with Williams Beuren Syndrome: a randomized controlled trial. BMC Pediatr 19, 170, doi: 10.1186/s12887-019-1544-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhang P et al. Inhibition of microRNA-29 enhances elastin levels in cells haploinsufficient for elastin and in bioengineered vessels--brief report. Arterioscler Thromb Vasc Biol 32, 756–759, doi: 10.1161/ATVBAHA.111.238113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.French JW & Guntheroth WG An explanation of asymmetric upper extremity blood pressures in supravalvular aortic stenosis: the Coanda effect. Circulation 42, 31–36, doi: 10.1161/01.cir.42.1.31 (1970). [DOI] [PubMed] [Google Scholar]
- 213. Cherniske EM et al. Multisystem study of 20 older adults with Williams syndrome. Am J Med Genet A 131, 255–264, doi: 10.1002/ajmg.a.30400 (2004). This article presents multisystem assessments on adults with WS, emphasizing the major medical conditions in adults with WS, and provides recommendations for monitoring guidelines.
- 214.Walton JR, Martens MA & Pober BR The proceedings of the 15th professional conference on Williams Syndrome. Am J Med Genet A 173, 1159–1171, doi: 10.1002/ajmg.a.38156 (2017). [DOI] [PubMed] [Google Scholar]
- 215.Halabi CM et al. Chronic antihypertensive treatment improves pulse pressure but not large artery mechanics in a mouse model of congenital vascular stiffness. Am J Physiol Heart Circ Physiol 309, H1008–1016, doi: 10.1152/ajpheart.00288.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Troia A et al. Inhibition of NOX1 mitigates blood pressure increases in elastin insufficiency. Function, doi: 10.1093/function/zqab015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Martens MA, Wilson SJ & Reutens DC Research Review: Williams syndrome: a critical review of the cognitive, behavioral, and neuroanatomical phenotype. J Child Psychol Psychiatry 49, 576–608, doi: 10.1111/j.1469-7610.2008.01887.x (2008). [DOI] [PubMed] [Google Scholar]
- 218.Miezah D et al. Cognitive Profile of Young Children with Williams Syndrome. Journal of Intellectual Disability Research In press (2021). [DOI] [PubMed] [Google Scholar]
- 219.Meyer-Lindenberg A, Mervis CB & Berman KF Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci 7, 380–393, doi: 10.1038/nrn1906 (2006). [DOI] [PubMed] [Google Scholar]
- 220.Mervis CB & Pitts CH Children with Williams syndrome: Developmental trajectories for intellectual abilities, vocabulary abilities, and adaptive behavior. Am J Med Genet C Semin Med Genet 169, 158–171, doi: 10.1002/ajmg.c.31436 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Porter M & Dodd H A longitudinal study of cognitive abilities in Williams syndrome. Dev Neuropsychol 36, 255–272, doi: 10.1080/87565641.2010.549872 (2011). [DOI] [PubMed] [Google Scholar]
- 222.Fisher MH, Lense MD & Dykens EM Longitudinal trajectories of intellectual and adaptive functioning in adolescents and adults with Williams syndrome. J Intellect Disabil Res 60, 920–932, doi: 10.1111/jir.12303 (2016). [DOI] [PubMed] [Google Scholar]
- 223.Mervis CB, Kistler DJ, John AE & Morris CA Longitudinal assessment of intellectual abilities of children with Williams syndrome: multilevel modeling of performance on the Kaufman Brief Intelligence Test-Second Edition. Am J Intellect Dev Disabil 117, 134–155, doi: 10.1352/1944-7558-117.2.134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zitzer-Comfort C, Doyle T, Masataka N, Korenberg J & Bellugi U Nature and nurture: Williams syndrome across cultures. Dev Sci 10, 755–762, doi: 10.1111/j.1467-7687.2007.00626.x (2007). [DOI] [PubMed] [Google Scholar]
- 225. Leyfer OT, Woodruff-Borden J, Klein-Tasman BP, Fricke JS & Mervis CB Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome. Am J Med Genet B Neuropsychiatr Genet 141B, 615–622, doi: 10.1002/ajmg.b.30344 (2006). Clinical diagnostic interview of children and teens which demonstrated a high frequency of ADHD, and an increasing frequency of Generalized Anxiety Disorder with age.
- 226.Perez-Garcia D, Brun-Gasca C, Perez-Jurado LA & Mervis CB Behavioral Profiles of Children With Williams Syndrome From Spain and the United States: Cross-Cultural Similarities and Differences. Am J Intellect Dev Disabil 122, 156–172, doi: 10.1352/1944-7558-122.2.156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Tomc SA, Williamson NK & Pauli RM Temperament in Williams syndrome. Am J Med Genet 36, 345–352, doi: 10.1002/ajmg.1320360321 (1990). [DOI] [PubMed] [Google Scholar]
- 228.Vonarnim G & Engel P Mental Retardation Related to Hypercalcaemia. Dev Med Child Neurol 6, 366–377, doi: 10.1111/j.1469-8749.1964.tb08138.x (1964). [DOI] [PubMed] [Google Scholar]
- 229.Thurman AJ & Fisher MH The Williams syndrome social phenotypes: Disentangling the contributions of social interest and social difficulties. International Review of Research in Developmental Disabilities 49, 191–227 (2015). [Google Scholar]
- 230.Copes LE, Pober BR & Terilli CA Description of common musculoskeletal findings in Williams Syndrome and implications for therapies. Clin Anat 29, 578–589, doi: 10.1002/ca.22685 (2016). [DOI] [PubMed] [Google Scholar]
- 231.Van Herwegen J, Ashworth M & Palikara O Parental views on special educational needs provision: Cross-syndrome comparisons in Williams Syndrome, Down Syndrome, and Autism Spectrum Disorders. Res Dev Disabil 80, 102–111, doi: 10.1016/j.ridd.2018.06.014 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Klein-Tasman BP, van der Fluit F & Mervis CB Autism Spectrum Symptomatology in Children with Williams Syndrome Who Have Phrase Speech or Fluent Language. J Autism Dev Disord 48, 3037–3050, doi: 10.1007/s10803-018-3555-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Klein-Tasman BP & Albano AM Intensive, Short-Term Cognitive-Behavioral Treatment of OCD-Like Behavior With a Young Adult With Williams Syndrome. Clinical Case Studies 6, 483–492 (2017). [Google Scholar]
- 234.Phillips KD & Klein-Tasman BP Mental health concerns in Williams syndrome: Intervention considerations and illustrations from case examples. Journal of Mental Health Research in Intellectual Disabilities 2, 110–133 (2009). [Google Scholar]
- 235.Thom RP, Keary CJ, Waxler JL, Pober BR & McDougle CJ Buspirone for the Treatment of Generalized Anxiety Disorder in Williams Syndrome: A Case Series. J Autism Dev Disord 50, 676–682, doi: 10.1007/s10803-019-04301-9 (2020). [DOI] [PubMed] [Google Scholar]
- 236.Green T et al. Phenotypic psychiatric characterization of children with Williams syndrome and response of those with ADHD to methylphenidate treatment. Am J Med Genet B Neuropsychiatr Genet 159B, 13–20, doi: 10.1002/ajmg.b.31247 (2012). [DOI] [PubMed] [Google Scholar]
- 237.Thom RP, Pober BR & McDougle CJ Psychopharmacology of Williams syndrome: safety, tolerability, and effectiveness. Expert Opin Drug Saf, doi: 10.1080/14740338.2021.1867535 (2020). [DOI] [PubMed] [Google Scholar]
- 238.Reilly C, Senior J & Murtagh L A comparative study of educational provision for children with neurogenetic syndromes: parent and teacher survey. J Intellect Disabil Res 59, 1094–1107, doi: 10.1111/jir.12210 (2015). [DOI] [PubMed] [Google Scholar]
- 239.UNESCO. Education and disability: Analysis of data from 49 countries (Information Paper N. 49), 2018). [Google Scholar]
- 240.Karr V, Hayes A & Hayford S Inclusion of children with learning difficulties in literacy and numeracy in Ghana: A literature review. International Journal of Disability, Development and Education, doi: 10.1080/1034912X.2020.1792419 (2020). [DOI] [Google Scholar]
- 241.Fisher MH, Josol CK & Shivers CM An Examination of Social Skills, Friendship Quality, and Loneliness for Adults with Williams Syndrome. J Autism Dev Disord 50, 3649–3660, doi: 10.1007/s10803-020-04416-4 (2020). [DOI] [PubMed] [Google Scholar]
- 242.Howlin P & Udwin O Outcome in adult life for people with Williams syndrome-- results from a survey of 239 families. J Intellect Disabil Res 50, 151–160, doi: 10.1111/j.1365-2788.2006.00775.x (2006). [DOI] [PubMed] [Google Scholar]
- 243.Pagon RA, Bennett FC, LaVeck B, Stewart KB & Johnson J Williams syndrome: features in late childhood and adolescence. Pediatrics 80, 85–91 (1987). [PubMed] [Google Scholar]
- 244.Brawn G, Kohnen S, Tassabehji M & Porter M Functional basic reading skills in Williams syndrome. Dev Neuropsychol 43, 454–477, doi: 10.1080/87565641.2018.1455838 (2018). [DOI] [PubMed] [Google Scholar]
- 245.Levy Y & Antebi V Word reading and reading-related skills in Hebrew-speaking adolescents with Williams syndrome. Neurocase 10, 444–451, doi: 10.1080/13554790490894048 (2004). [DOI] [PubMed] [Google Scholar]
- 246.Mervis CB Language and Literacy Development of Children with Williams Syndrome. Top Lang Disord 29, 149–169, doi: 10.1097/TLD.0b013e3181a72044 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Mervis CB, Greiner de Magalhães C & Cardoso-Martins C Concurrent predictors of word reading and reading comprehension for 9-year-olds with Williams syndrome. Read Writ In press (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Van Herwegen J & Simms V Mathematical development in Williams syndrome: A systematic review. Res Dev Disabil 100, 103609, doi: 10.1016/j.ridd.2020.103609 (2020). [DOI] [PubMed] [Google Scholar]
- 249.Brawn G & Porter M Adaptive functioning in Williams syndrome: A systematic review. International Journal of Disability, Development and Education 65, 123–147 (2018). [Google Scholar]
- 250.Pitts CH, Klein-Tasman BP, Osborne JW & Mervis CB Predictors of specific phobia in children with Williams syndrome. J Intellect Disabil Res 60, 1031–1042, doi: 10.1111/jir.12327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Edgin JO, Pennington BF & Mervis CB Neuropsychological components of intellectual disability: the contributions of immediate, working, and associative memory. J Intellect Disabil Res 54, 406–417, doi: 10.1111/j.1365-2788.2010.01278.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fu TJ, Lincoln AJ, Bellugi U & Searcy YM The Association of Intelligence, Visual-Motor Functioning, and Personality Characteristics With Adaptive Behavior in Individuals With Williams Syndrome. Am J Intellect Dev Disabil 120, 273–288, doi: 10.1352/1944-7558-120.4.273 (2015). [DOI] [PubMed] [Google Scholar]
- 253.Palikara O, Ashworth M & Van Herwegen J Addressing the Educational Needs of Children with Williams Syndrome: A Rather Neglected Area of Research? J Autism Dev Disord 48, 3256–3259, doi: 10.1007/s10803-018-3578-x (2018). [DOI] [PubMed] [Google Scholar]
- 254.Gillooly AE, Riby DM, Durkin K & Rhodes SM Peer Relationships in Children with Williams Syndrome: Parent and Teacher Insights. J Autism Dev Disord, doi: 10.1007/s10803-020-04503-6 (2020). [DOI] [PubMed] [Google Scholar]
- 255.Lough E & Fisher MH Parent and Self-Report Ratings on the Perceived Levels of Social Vulnerability of Adults with Williams Syndrome. J Autism Dev Disord 46, 3424–3433, doi: 10.1007/s10803-016-2885-3 (2016). [DOI] [PubMed] [Google Scholar]
- 256.Fisher MH, Moskowitz AL & Hodapp RM Differences in Social Vulnerability among Individuals with Autism Spectrum Disorder, Williams Syndrome, and Down Syndrome. Res Autism Spectr Disord 7, 931–937, doi: 10.1016/j.rasd.2013.04.009 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ridley E, Riby DM & Leekam SR A cross-syndrome approach to the social phenotype of neurodevelopmental disorders: Focusing on social vulnerability and social interaction style. Res Dev Disabil 100, 103604, doi: 10.1016/j.ridd.2020.103604 (2020). [DOI] [PubMed] [Google Scholar]
- 258.Fisher MH Evaluation of a stranger safety training programme for adults with Williams syndrome. J Intellect Disabil Res 58, 903–914, doi: 10.1111/jir.12108 (2014). [DOI] [PubMed] [Google Scholar]
- 259.Riby DM, Kirk H, Hanley M & Riby LM Stranger danger awareness in Williams syndrome. J Intellect Disabil Res 58, 572–582, doi: 10.1111/jir.12055 (2014). [DOI] [PubMed] [Google Scholar]
- 260.Fisher MH & Morin L Addressing social skills deficits in adults with Williams syndrome. Res Dev Disabil 71, 77–87, doi: 10.1016/j.ridd.2017.10.008 (2017). [DOI] [PubMed] [Google Scholar]
- 261. Morris CA, Demsey SA, Leonard CO, Dilts C & Blackburn BL Natural history of Williams syndrome: physical characteristics. J Pediatr 113, 318–326, doi: 10.1016/s0022-3476(88)80272-5 (1988). A hallmark paper in delineating the natural history of Williams syndrome, in both children and adults.
- 262.Udwin O, Howlin P, Davies M & Mannion E Community care for adults with Williams syndrome: how families cope and the availability of support networks. J Intellect Disabil Res 42 ( Pt 3), 238–245, doi: 10.1046/j.1365-2788.1998.00122.x (1998). [DOI] [PubMed] [Google Scholar]
- 263.Pao M & Bosk A Anxiety in medically ill children/adolescents. Depress Anxiety 28, 40–49, doi: 10.1002/da.20727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Association, A. H. American Heart Association Recommendations for Physical Activity in Adults and Kids, <https://www.heart.org/en/healthy-living/fitness/fitness-basics/aha-recs-for-physical-activity-in-adults> (2018).
- 265.Disabilities, N. C. o. B. D. a. D. Physical Activity for People with Disability, <https://www.cdc.gov/ncbddd/disabilityandhealth/features/physical-activity-for-all.html#:~:text=For%20even%20greater%20health%20benefits,minutes%20a%20day%20every%20day.> (2020).
- 266.Leyfer O, Woodruff-Borden J & Mervis CB Anxiety disorders in children with williams syndrome, their mothers, and their siblings: implications for the etiology of anxiety disorders. J Neurodev Disord 1, 4–14, doi: 10.1007/s11689-009-9003-1 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Papaeliou C et al. Behavioural profile and maternal stress in Greek young children with Williams syndrome. Child Care Health Dev 38, 844–853, doi: 10.1111/j.1365-2214.2011.01306.x (2012). [DOI] [PubMed] [Google Scholar]
- 268.Sarimski K Behavioural phenotypes and family stress in three mental retardation syndromes. Eur Child Adolesc Psychiatry 6, 26–31, doi: 10.1007/BF00573637 (1997). [DOI] [PubMed] [Google Scholar]
- 269.John AE & Mervis CB Sensory modulation impairments in children with Williams syndrome. Am J Med Genet C Semin Med Genet 154C, 266–276, doi: 10.1002/ajmg.c.30260 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Fidler DJ, Hodapp RM & Dykens EM Stress in families of young children with Down syndrome, Williams syndrome, and Smith-Magenis syndrome. Early Education and Development 11, 395–406 (2000). [Google Scholar]
- 271.Reilly C, Murtagh L & Senior J The Impact on the Family of Four Neurogenetic Syndromes: A Comparative Study of Parental Views. J Genet Couns 24, 851–861, doi: 10.1007/s10897-015-9820-1 (2015). [DOI] [PubMed] [Google Scholar]
- 272.Ashworth M, Palikara O & Van Herwegen J Comparing parental stress of children with neurodevelopmental disorders: The case of Williams syndrome, Down syndrome and autism spectrum disorders. J Appl Res Intellect Disabil 32, 1047–1057, doi: 10.1111/jar.12594 (2019). [DOI] [PubMed] [Google Scholar]
- 273.Nir A & Barak B White matter alterations in Williams syndrome related to behavioral and motor impairments. Glia 69, 5–19, doi: 10.1002/glia.23868 (2021). [DOI] [PubMed] [Google Scholar]
- 274.Chailangkarn T et al. A human neurodevelopmental model for Williams syndrome. Nature 536, 338–343, doi: 10.1038/nature19067 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lalli MA et al. Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum Mol Genet 25, 1294–1306, doi: 10.1093/hmg/ddw010 (2016). [DOI] [PubMed] [Google Scholar]
- 276.Kinnear C et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med 2, 2–15, doi: 10.5966/sctm.2012-0054 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Amin ND & Pasca SP Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron 100, 389–405, doi: 10.1016/j.neuron.2018.10.007 (2018). [DOI] [PubMed] [Google Scholar]
- 278.Baldassari S et al. Brain Organoids as Model Systems for Genetic Neurodevelopmental Disorders. Frontiers in Cell and Developmental Biology 8, doi: 10.3389/fcell.2020.590119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Wimmer RA et al. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510, doi: 10.1038/s41586-018-0858-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Huang AH et al. Biaxial Stretch Improves Elastic Fiber Maturation, Collagen Arrangement, and Mechanical Properties in Engineered Arteries. Tissue Eng Part C Methods 22, 524–533, doi: 10.1089/ten.TEC.2015.0309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Liu C, Niu K & Xiao Q Updated Perspectives On Vascular Cell Specification And Pluripotent Stem Cell-Derived Vascular Organoids For Studying Vasculopathies. Cardiovasc Res, doi: 10.1093/cvr/cvaa313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ellis MW, Luo J & Qyang Y Modeling elastin-associated vasculopathy with patient induced pluripotent stem cells and tissue engineering. Cell Mol Life Sci 76, 893–901, doi: 10.1007/s00018-018-2969-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Kurki MI et al. Contribution of rare and common variants to intellectual disability in a sub-isolate of Northern Finland. Nat Commun 10, 410, doi: 10.1038/s41467-018-08262-y (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Magavern EF et al. An Academic Clinician’s Road Map to Hypertension Genomics: Recent Advances and Future Directions MMXX. Hypertension, HYPERTENSIONAHA12014535, doi: 10.1161/HYPERTENSIONAHA.120.14535 (2021). [DOI] [PubMed] [Google Scholar]
- 285.Girirajan S & Eichler EE Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 19, R176–187, doi: 10.1093/hmg/ddq366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Levitin DJ, Cole K, Lincoln A & Bellugi U Aversion, awareness, and attraction: investigating claims of hyperacusis in the Williams syndrome phenotype. J Child Psychol Psychiatry 46, 514–523, doi: 10.1111/j.1469-7610.2004.00376.x (2005). [DOI] [PubMed] [Google Scholar]
- 287.Levy G & Barak B Postnatal therapeutic approaches in genetic neurodevelopmental disorders. Neural Regen Res 16, 414–422, doi: 10.4103/1673-5374.293133 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Powell SK, Gregory J, Akbarian S & Brennand KJ Application of CRISPR/Cas9 to the study of brain development and neuropsychiatric disease. Mol Cell Neurosci 82, 157–166, doi: 10.1016/j.mcn.2017.05.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Ramdas S & Servais L New treatments in spinal muscular atrophy: an overview of currently available data. Expert Opin Pharmacother 21, 307–315, doi: 10.1080/14656566.2019.1704732 (2020). [DOI] [PubMed] [Google Scholar]
- 290.Oetjens MT, Kelly MA, Sturm AC, Martin CL & Ledbetter DH Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun 10, 4897, doi: 10.1038/s41467-019-12869-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Osborne LR & Mervis CB 7q11.23 deletion and duplication. Curr Opin Genet Dev 68, 41–48, doi: 10.1016/j.gde.2021.01.013 (2021). [DOI] [PubMed] [Google Scholar]
- 292.Wing L & Gould J Severe impairments of social interaction and associated abnormalities in children: epidemiology and classification. J Autism Dev Disord 9, 11–29, doi: 10.1007/BF01531288 (1979). [DOI] [PubMed] [Google Scholar]
- 293.Schubert C The genomic basis of the Williams-Beuren syndrome. Cell Mol Life Sci 66, 1178–1197, doi: 10.1007/s00018-008-8401-y (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Masataka N Why early developmental milestones are delayed in children with Williams syndrome: late onset of hand banging as a possible rate-limiting constraint on the emergence of canonical babbling. Developmental Science 4, 158–164, doi: 10.1111/1467-7687.00161 (2001). [DOI] [Google Scholar]
- 295.Plissart L & Fryns JP Early development (5 to 48 months) in Williams syndrome. A study of 14 children. Genet Couns 10, 151–156 (1999). [PubMed] [Google Scholar]
- 296.Sarimski K Early development of children with Williams syndrome. Genet Couns 10, 141–150 (1999). [PubMed] [Google Scholar]
- 297.Group, W. H. O. M. G. R. S. WHO Motor Development Study: windows of achievement for six gross motor development milestones. Acta Paediatr Suppl 450, 86–95, doi: 10.1111/j.1651-2227.2006.tb02379.x (2006). [DOI] [PubMed] [Google Scholar]
- 298.Bayley N Manual for the Bayley Scales of Infant Development 2nd edn, (Psychological Corp., 1993). [Google Scholar]
- 299.Fenson L et al. Variability in early communicative development. Monogr Soc Res Child Dev 59, 1–173; discussion 174–185 (1994). [PubMed] [Google Scholar]
- 300.Fenson L et al. MacArthur-Bates Communicative Development Inventories: User’s guide and technical manual 2nd edn, (Brookes, 2007). [Google Scholar]
- 301.Mervis CB, Becerra AM, Pitts CH & Marchman VA in Symposium on Research in Child Language Disorders (Madison, WI, 2019). [Google Scholar]
- 302.Weisberg DS Pretend play. Wiley Interdiscip Rev Cogn Sci 6, 249–261, doi: 10.1002/wcs.1341 (2015). [DOI] [PubMed] [Google Scholar]
- 303.Lincoln AJ, Searcy YM, Jones W & Lord C Social interaction behaviors discriminate young children with autism and Williams syndrome. J Am Acad Child Adolesc Psychiatry 46, 323–331, doi: 10.1097/chi.0b013e31802b9522 (2007). [DOI] [PubMed] [Google Scholar]
- 304. Dodd HF & Porter MA Psychopathology in Williams Syndrome: The Effect of Individual Differences Across the Life Span. Journal of Mental Health Research in Intellectual Disabilities 2, 89–109 (2009). Clinical diagnostic interview of psychopathology and behaviour (as opposed to just a symptom checklist). Looks at children and adults separately.
- 305.Dykens EM Anxiety, fears, and phobias in persons with Williams syndrome. Dev Neuropsychol 23, 291–316, doi: 10.1080/87565641.2003.9651896 (2003). [DOI] [PubMed] [Google Scholar]
- 306.Kennedy JC, Kaye DL & LS S. Psychiatric diagnoses in patients with Williams syndrome and their families. Jefferson J Psychiatry 20, 22–31 (2006). [Google Scholar]
- 307.Zarchi O et al. A comparative study of the neuropsychiatric and neurocognitive phenotype in two microdeletion syndromes: velocardiofacial (22q11.2 deletion) and Williams (7q11.23 deletion) syndromes. Eur Psychiatry 29, 203–210, doi: 10.1016/j.eurpsy.2013.07.001 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.