

Abstract
MicroRNA (miRNA) loaded Argonaute (AGO) complexes regulate gene expression via direct base pairing with their mRNA targets. Previous works suggest that up to 60% of mammalian transcripts might be subject to miRNA‐mediated regulation, but it remains largely unknown which fraction of these interactions are functional in a specific cellular context. Here, we integrate transcriptome data from a set of miRNA‐depleted mouse embryonic stem cell (mESC) lines with published miRNA interaction predictions and AGO‐binding profiles. Using this integrative approach, combined with molecular validation data, we present evidence that < 10% of expressed genes are functionally and directly regulated by miRNAs in mESCs. In addition, analyses of the stem cell‐specific miR‐290‐295 cluster target genes identify TFAP4 as an important transcription factor for early development. The extensive datasets developed in this study will support the development of improved predictive models for miRNA‐mRNA functional interactions.
Keywords: integrative analysis, microRNA, miR‐290‐295 cluster, mouse embryonic stem cells, Tfap4
Subject Categories: Chromatin, Transcription & Genomics; RNA Biology; Stem Cells & Regenerative Medicine
Integrative analysis of transcriptome data from miRNA‐depleted mESC lines, miRNA interaction predictions and AGO‐binding profiles allows forthe identification of direct and functional miRNA targets. These data support the development of algorithms to predict functional miRNA‐mRNA interactions.

Introduction
The rapid rise in the discovery of microRNAs (miRNAs) as key regulators of gene expression in many biological processes has prompted an extensive search for their functional targets, as well as the development of tools for a reliable identification thereof. MiRNAs are endogenous small regulatory RNAs, approximately 22 nucleotides (nt) in length (Bartel, 2018), which are transcribed by RNA polymerase II to produce a capped and poly‐adenylated primary transcript (pri‐miRNA). They are further processed by the Microprocessor Complex consisting of one RNase III DROSHA and two double‐stranded RNA‐binding protein DGCR8 (Lee et al, 2002; Nguyen et al, 2015). The resulting miRNA precursor (pre‐miRNA) is exported to the cytoplasm and cleaved by DICER, another RNase III protein (Bernstein et al, 2001), leading to a double‐stranded miRNA duplex, subsequently loaded into an Argonaute (AGO) protein (Mourelatos et al, 2002; Song et al, 2004). The AGO‐miRNA complexes generally target the 3′ untranslated regions (UTRs) of their target mRNAs at miRNA response elements (MREs) (Lewis et al, 2005), although functional repression via binding to the coding sequence (CDS) has been reported (Reczko et al, 2012; Hausser et al, 2013). MREs are usually complementary to the seed sequence of the miRNA, which is at positions 2–7 of the mature miRNA's 5' end (Lewis et al, 2003). MiRNA binding can lead to the functional repression of its target by translation inhibition and decay of the mRNA (Guo et al, 2010).
Complementary computational and experimental approaches are commonly used to identify functional miRNA–mRNA interactions. AGO‐binding assays such as cross‐linking immunoprecipitation and sequencing (CLIP‐seq) precisely reveal binding sites of AGO to MREs allowing the identification of the regulatory miRNAs (Hafner et al, 2010). Of note, AGO‐binding does not necessarily elicit downstream repressive effects (Agarwal et al, 2015; Chu et al, 2020) and a diverse set of sequence‐associated features has been identified to influence the repression potential of a given interaction (Lewis et al, 2005; McGeary et al, 2019; Schäfer & Ciaudo, 2020). These features have been exploited by computational prediction models for the identification of miRNA interactions (Agarwal et al, 2015; Id et al, 2018; Schäfer & Ciaudo, 2020) and the integration of multiple approaches and datasets to obtain more accurate functional miRNA–mRNA interactions have been performed (Gosline et al, 2016; Oliveira et al, 2017; Chu et al, 2020). Notably, Gosline et al (2016) and Chu et al (2020) integrated AGO‐binding datasets with miRNA‐depletion and transcriptomics to identify miRNA targets in specific contexts (fibroblasts and HCT116 cells, respectively). Gosline et al (2016) further integrated epigenetics approaches, identifying miRNA‐regulated transcription factors (TFs), spanning broad regulatory networks, while Chu et al (2020) highlighted the difficulty to infer functional miRNA interactions directly from AGO‐binding data. However, it has been shown that computational models tend to predict a large number of nonfunctional interactions, where miRNAs do not exert any detectable repression to their respective targets (Pinzon et al, 2017; Chu et al, 2020).
In this study, we hypothesized that the inability of current miRNA target prediction models to provide an accurate view of functionally relevant miRNA interactions could be partially attributed to the lack of incorporating context‐specific factors. We performed an integrative analysis combining OMICs data from a unique series of miRNA‐deficient mouse Embryonic Stem Cell (mESC) lines generated in the same genetic background with other publicly available datasets (predictive models, AGO‐bound miRNAs) to determine the direct and functional miRNA interactions in mESCs. We further validated our findings by measuring the impact of the deletion of a stem‐cell‐specific miRNA cluster on gene expression. Altogether, we estimate that < 10% of expressed genes are subject to direct and functional miRNA‐regulation in mESCs. In addition, we identified the transcription factor TFAP4 as a miR‐290‐295 cluster target. Our data suggest an important role for TFAP4 in the regulation of the Wnt signaling pathway and gene regulation in mESCs.
Results and Discussion
Gene expression is not sufficient for accurate miRNA target prediction
To capture the extent of miRNA‐mediated gene regulation in mESCs, we established a unique set of miRNA knock‐out mESCs (miRNA_KOs) using a paired CRISPR/Cas9 approach (Wettstein et al, 2016) (Drosha_KO (Cirera‐Salinas et al, 2017), Dicer_KO (Bodak et al, 2017b) and Ago2&1_KO (this study) (Fig EV1A)). Principal component analysis (PCA) of RNA‐sequencing (RNA‐seq) from wildtype (WT) and miRNA_KO lines showed clustering of biological replicates and separation of samples (Fig EV1B and Dataset EV1). Differential expression (DE) analysis revealed a large number of upregulated genes in all miRNA_KOs (Fig EV1C). However, the overlap of upregulated genes across all three mutants was small relative to the total number of upregulated genes (Fig EV1D, 1,109 out of 6,408 upregulated genes, adjusted (adj.) P‐value < 0.2). All mutants exhibited a sizeable number of downregulated genes (Fig EV1C and D), indicating indirect effects.
Figure EV1. Gene expression is globally perturbed in miRNA_KO mESCs.
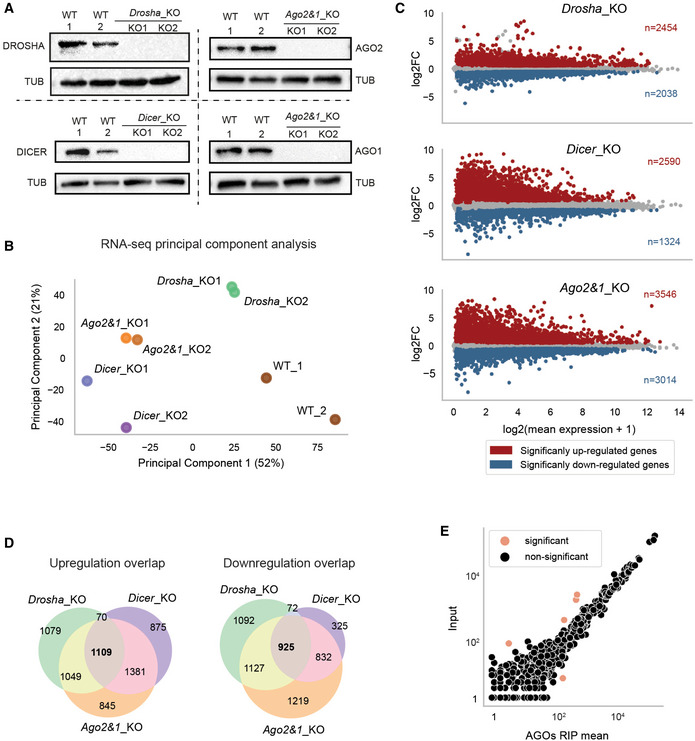
- Immunoblot analysis of DROSHA, DICER, AGO1, and AGO2 in WT and their respective KO mESC lines. TUBULIN was used as a loading control. Representative blot of three independent experiments is shown.
- PCA of gene expression as measured by RNA sequencing in miRNA_KO and WT samples. Biological replicates are indicated with corresponding colors.
- MA plots of the DGE analysis in miRNA_KO mutants versus WT. Significant up‐ and downregulated genes are colored in red and blue, respectively.
- Overlap of up‐ (left) and downregulated (right) genes in miRNA_KO mESCs.
- Comparison of miRNA loading for small RNA sequencing (Input) versus Argonaute2 & 1 (AGOs) RNA immunoprecipitation (RIP) and sequencing for all miRNAs. MiRNAs that show statistically significant difference between the two measured (adjusted P‐value < 0.1) are highlighted.
Data information: In (C and E), significance in differential analysis was determined using an adjusted P‐value threshold obtained from DESeq2 of 0.1. In (D), an adjusted P‐value threshold of 0.2 was applied.
Source data are available online for this figure.
In conclusion, the extended perturbation of gene expression observed in miRNA_KO mESC lines is not exclusively caused by the lack of miRNAs. Mutant‐specific effects and secondary regulation events strongly influence global gene expression profiles in stem cells, hindering direct inference of miRNA targets from these data.
Integrative transcriptomics data analysis predicts functional miRNA interactions in mESCs
To identify candidate genes directly and functionally targeted by miRNAs in mESCs, we integrated sequence‐based interaction prediction data from TargetScan (Agarwal et al, 2015), and published AGO2‐binding data (Li et al, 2020) with our AGO‐miRNA loading data from WT mESCs (Ngondo et al, 2018) and RNA‐seq from WT and miRNA_KO mESCs as visualized in Fig 1A. Potential interactions were established based on seed matching at AGO2 binding peaks and then filtered based on target mRNA upregulation in miRNA_KO mESCs, miRNA loading in AGOs and sequence‐based interaction predictions (Fig EV1E and Dataset EV2). The sum of all four normalized features led to an interaction score, which was used to rank interactions and candidate genes based on the evidence supporting their regulation by miRNAs, narrowing the number of detected miRNA candidate target genes down to 759 (Fig 1B and Dataset EV3).
Figure 1. Multi‐OMICs integration allows the identification of functional miRNA interactions in mESCs.
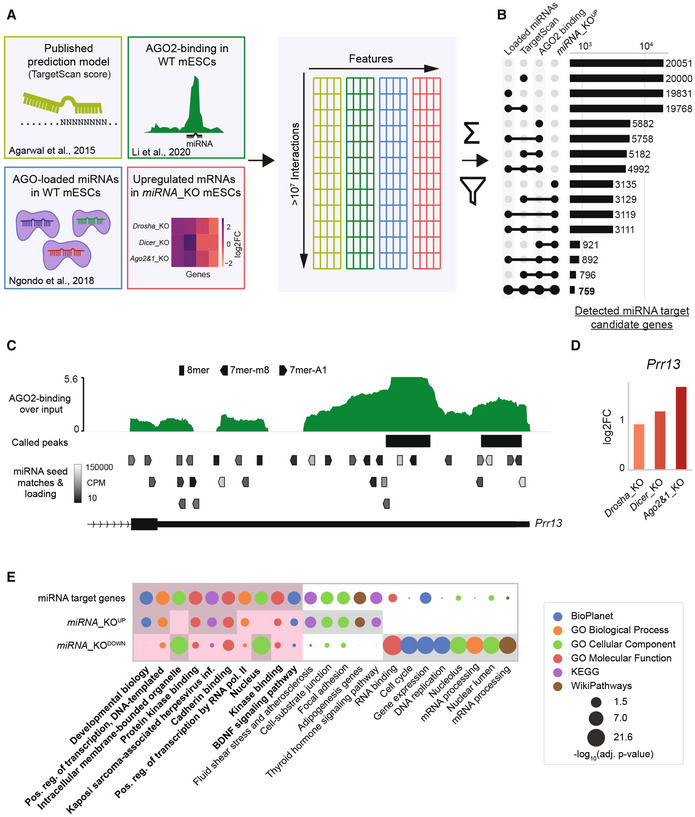
-
AGraphical overview of data sources and integration. Data are integrated on a per‐interaction basis, filtered by miRNA loading into Argonaute complexes (AGOs), AGO2‐binding, target upregulation and sequence‐based predictions and then scored on a per‐interaction and per‐gene basis (see Materials and Methods). Scoring allows for a confidence ranking of interactions and target genes.
-
BNumber of identified functional miRNA target genes for different integrative filtering approaches. The integration and filtering by mutant upregulation, AGO2‐binding, and TargetScan data leads to a restrictive selection of predicted target genes.
-
C, DExample of integrated data for the Prr13 gene with multiple lines of evidence for functional miRNA interactions. (C) shows AGO2‐binding profile and called peaks as obtained from (Li et al, 2020), along with predicted miRNA binding sites with the AGOs‐loading of the corresponding miRNA in WT mESCs (only binding sites for miRNAs with minimal expression of 10 CPM are shown). (D) shows the misregulation of Prr13 in miRNA_KO mutants (2 biological replicates each) in log2FoldChanges (log2FC) compared to WT.
-
EGO term analysis of gene sets representing miRNA target genes (759), commonly up‐ (3609), and downregulated (2956) genes. Top eight statistically most significant terms for each of the three gene groups are shown and highlighted with a gray background. Red background highlights the comparison of the top terms from miRNA target genes set.
Data Information: miRNA_KOUP and miRNA_KODOWN genes were determined by an adjusted P‐value threshold of 0.2 (DESeq2) in at least two miRNA_KO lines. Statistical significance for GO terms was determined by Enrichr and is indicated in the figure.
We identified miRNA–mRNA pairs, where the mRNA targets display high AGO2‐binding and significant upregulation in miRNA_KO mESCs, as well as loading of the targeting miRNAs in AGOs (Fig 1C and D). Our approach discards putative interactions with little evidence for the support of miRNA targeting, such as low AGO2‐binding, low targeting miRNA loading, and a lack of target upregulation in miRNA_KO mESCs (Fig EV2A and B). Consistent with other studies (Reczko et al, 2012; Hausser et al, 2013; Patel et al, 2020), we observed seemingly functional interactions in regions outside of the 3′UTR (Fig EV2C and D), which led us to include interactions in the 5′UTR and CDS despite the absence of a TargetScan score.
Figure EV2. Characterization of predicted miRNA interactions.
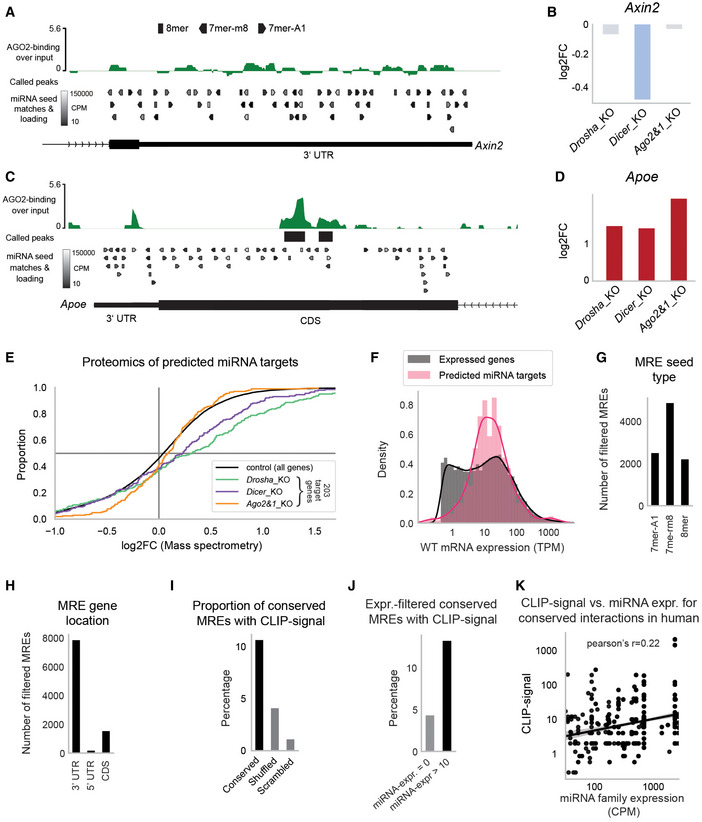
-
A, BExample of integrated data for the Axin2 gene with no evidence for functional interactions. For a more detailed subplot description, refer to the figure legend of Fig 1C and D.
-
C, DNo statistically significant AGO2‐binding in 3′UTR of the Apoe gene, yet statistically significant binding to the CDS at a strongly expressed 8mer binding site along with a consistent upregulation in all three mutants. For a more detailed subplot description, refer to the figure legend of Fig 1C and D.
-
ECumulative distribution function of differential protein abundance as detected by SWAT‐MS in miRNA_KO mESCs. Of 759 predicted miRNA target genes, 203 (27%) were detectable in the SWATH‐MS data and appear in the plot.
-
FExpression distribution of predicted miRNA targets in comparison to expression of all genes (filtered by TPM >0.5 to improve comparability).
-
GNumber of filtered (see Fig 1A and B) miRNA response elements (MREs) grouped by the three most effective seed types 7mer‐A1, 7mer‐m8, and 8mer.
-
HNumber of filtered (see Fig 1A and B) MREs grouped by their location within the messenger RNA (3′UTR, 5′UTR, and CDS).
-
IConservation analysis of predicted interactions in hESCs. An interaction is considered conserved if a miRNA seed matching MRE is found in the 3′UTR of the human ortholog gene. Shown is the proportion of mESC‐predicted interactions that have conserved MREs with associated cross‐linking immunoprecipitation (CLIP)‐seq reads in hESCs. As control, MRE search in hESCs was performed with shuffled and scrambled miRNA sequences.
-
JSimilar to (I), but only interactions with miRNA expression above 10 CPM (and = 0 CPM for control) were considered. miRNA expression was evaluated on a per‐family basis, corresponding to the seed‐match binding logic.
-
KCorrelation of hESC miRNA expression with the number of CLIP‐seq reads in hESCs for human‐conserved interactions (see Fig EV2I). Only interactions with at least one CLIP‐seq read and at least 10 CPM miRNA expression are considered. miRNA expression is shown on a family basis (expression of miRNAs with the same seed is summed).
Data information: In (E), Student's t‐test was applied to determine the significance of increased protein abundance for 203/759 miRNA targets (P < 0.0051 for every mutant). In (K), Pearson correlation was calculated, P < 1e−50.
Source data are available online for this figure.
A gene set enrichment analysis (GSEA) of the target candidates, compared to sets of commonly up‐ and downregulated genes, revealed a highly significant role for candidate genes in Developmental Biology and processes linked to the regulation of transcription (Fig 1E), in line with the known roles of miRNAs in early development and stem cells (DeVeale et al, 2021). Thus, multi‐OMICs integration facilitated the identification of 759 candidate miRNA targets corresponding to 7% of expressed genes. Interestingly, Tan et al (preprint: 2020) estimated that 6% of expressed genes were regulated by miRNAs in lymphoblastoid cell lines (preprint: Tan et al, 2020), suggesting that this percentage might reflect functionally relevant interaction number magnitudes in various contexts.
Ribosome profiling in miRNA_KO mESCs reinforces the identification of functional miRNA target genes
A substantial part of the data used in the integrative analysis was based on transcriptomics approaches, and we wondered whether the upregulation of miRNA targets in miRNA_KO mutants was also reflected at the translational level. We thus assessed the proteome of all miRNA mutant and WT mESCs using Sequential Window Acquisition of all THeoretical fragment ion spectra Mass Spectrometry (SWATH‐MS) (Gillet et al, 2012), and measured the differential protein abundance of candidate miRNA targets. This approach allowed us to capture protein abundances only for 27% of candidate miRNA targets (203 of 759, Fig EV2E). All miRNA_KO mutants showed significantly enriched positive log2‐fold‐changes (log2FCs) as compared to a control distribution of log2FCs, with at least 60% of genes exhibiting positive log2FCs (Fig EV2E and Dataset EV4). The low detection sensitivity of the approach was in line with previous mass spectrometry studies (Tacheny et al, 2012; Ding et al, 2013; preprint: Lai et al, 2021), which also described potentially high signal variance (preprint: Lai et al, 2021).
We, therefore, performed Ribosome profiling (Ribo‐seq) in miRNA_KO mESCs to measure the ribosome occupancy on mRNAs (Brar & Weissman, 2015). The high sensitivity of the approach allowed us to detect and compare ribosome occupancy for 96% of the candidates (728 of 759, Fig 2A). For every mutant, enrichments for positive log2FCs are statistically more significant and stronger than in the mass spectrometry experiment (P < 1e‐51 for every mutant, 76–79% of genes with positive log2FCs, Fig 2A and Dataset EV5). The Ribo‐seq approach thereby reinforces the validity of our integrative analysis and the list of candidates in mESCs.
Figure 2. Characterization of predicted miRNA interactions.
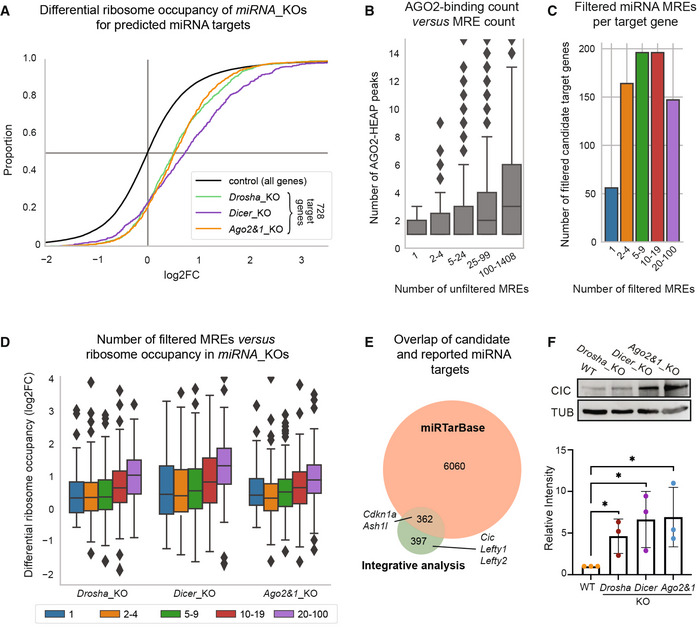
-
ACumulative distribution function of differential ribosome occupancy as detected by ribosome profiling in miRNA_KO mutants versus WT. Of 759 predicted miRNA target genes, 728 (96%) were detectable in the ribosome profiling data and appear in the plot.
-
BBoxplot of number of Argonaute2 (AGO2) HEAP peaks for genes grouped by the number of seed match‐based miRNA response elements (MRE) in their 3′ untranslated regions (UTRs). “Outliers” with more than 15 peaks are not shown.
-
CNumber of genes with different numbers of filtered MREs/interactions.
-
DDifferential ribosome occupancy in miRNA_KO mutants versus WT of genes grouped by MRE/interaction counts. Groups (colors) correspond to Fig 2C. Shown y‐axis range has been limited and some outliers are hidden.
-
EOverlap of candidate miRNA targets with previously reported miRNA targets as collected by the miRTarBase database. Selected genes described in the literature are labeled.
-
FTop: Immunoblot analysis of CIC in WT, Drosha_KO, Dicer_KO, and Ago2&1_KO mESCs. TUBULIN was used as a loading control. Blot is a representative image of three biological replicates. Bottom: Bar graph showing quantification of CIC intensity, normalized to TUBULIN and relative to the WT sample in three biological replicates.
Data information: In (A), Student's t‐test was applied to assess the significance for the increase in ribosome occupancy of the 728/759 miRNA targets (P‐value < 1e51 for every mutant). Box plots in (B and D) indicate median as the central band, 25–75%‐intervals as box, and 1.5 times the interquartile range as the whiskers. Group sizes in (B), are 27, 103, 773, 2,566, 3,293 (from left to right; genes with non‐detectable HEAP levels are excluded from this plot), group sizes for (D) are indicated in panel (C). Bar graph in Fig 2F shows the mean intensity of CIC signal ± SD normalized to TUBULIN. Values are relative to WT, which was set to 1. P‐values were calculated using a Student's t‐test comparing each value to the WT. *P‐value < 0.05.
Source data are available online for this figure.
Characterization of candidate miRNA target genes
Further characterization of our candidates showed that their WT expression resembled the expression distribution of all expressed genes with a slight enrichment of highly expressed genes (~ 1,000 TPM) (Fig EV2F). Of the roughly 10,000 interactions, about half were binding via a 7mer‐m8 seed match, while the other two quarters were comparably partitioned between the more potent 8mer and the less potent 7mer‐A1 MRE types (Fig EV2G, Bartel, 2018).
Most candidate interactions target the 3′UTR (>80%), the remaining of which mostly target the CDS and only very few shows binding to the 5′UTRs of target genes (Fig EV2H). High numbers of seed matching MREs in a given gene are associated with a high number of peaks identified in the AGO‐binding data (Fig 2B). Nevertheless, we did not observe a general correlation between AGO2‐binding signal and the number of MREs/interactions per gene (Appendix Fig S1). When grouping miRNA targets by number of interactions, we saw that around 50 genes were predicted to be targeted by only one miRNA, whereas the majority of candidates have at least two potential MREs, and almost 150 genes showed 20–100 predicted interactions (Fig 2C).
In order to test whether miRNAs can act in a cooperative manner to increase their repressive potential (Briskin et al, 2020), we compared the number of predicted interactions per gene with the gene's differential ribosome occupancy, which indeed revealed a positive correlation between the two metrics (Pearson's r = 0.2–0.24, P < 2.8e−8, Fig 2D).
Next, we scanned the 3′UTRs of human ortholog genes for seed matches from mESC‐predicted interactions and associated these conserved MREs with miRNA expression and AGO‐binding data in human ESCs (Lipchina et al, 2011). For 10% of interactions predicted in mESCs, we found conserved MREs with associated AGO‐binding, more than 2.5 times as many as for negative control seed sequences (Fig EV2I). Conserved MREs were three times more likely to show AGO‐binding when the binding site corresponded to an expressed miRNA as opposed to non‐expressed miRNAs (Fig EV2J). These interactions also showed a mild correlation between miRNA expression and the AGO‐binding signal (Pearson's r = 0.22, Fig EV2K). Taken together, a subset of miRNA interactions predicted in mESCs showed functional conservation in hESCs.
Finally, we compared our candidates with predicted miRNA targets in mice from the miRTarBase database, which collects miRNA interactions from the scientific literature and high‐throughput experimental data (Chou et al, 2018). Of the 6,422 miRNA targets described in miRTarBase and filtered for mESC‐loaded miRNAs, only half (362) were recapitulated by our approach, including Cdkn1a (Wang et al, 2008) and Ahs1l (Kanellopoulou et al, 2015) genes, but not other validated target genes like Lefty1&2 (Marson et al, 2008) and Cic (Choi et al, 2015) (Fig 2E). Since Cic was both absent from miRTarBase, and not previously described as a miRNA target in mESCs, we validated its upregulation in the miRNA_KO lines (Fig 2F). We conclude that our integrative analysis adds significantly to the set of previously described miRNA interactions, by providing a context‐specific window to functional miRNA interactions in mESCs.
Validation of candidate genes by miRNA cluster knockout in mESCs
MESCs have a specific miRNA expression pattern (Houbaviy et al, 2003; Ciaudo et al, 2009), with over 50% of all expressed miRNAs originating from five genomic clusters, including the miR‐290‐295 cluster (Fig EV3A) (Calabrese et al, 2007). This cluster is the most highly expressed (Appendix Fig S2) and its miRNA members are among the most abundantly AGO‐loaded miRNAs in mESCs (Fig EV3A and B). Additionally, half of our candidates are predicted to be targeted by at least one member of this cluster (360 out of 759 genes, Fig EV3A). The miR‐290‐295 cluster has been shown to be involved in multiple molecular processes in mESCs (Yuan et al, 2017). Its depletion results in partially penetrant embryonic lethality in mice (Medeiros et al, 2011), but is not sufficient to induce spontaneous differentiation in mESCs (Wang et al, 2017).
Figure EV3. Validation of predicted miRNA targets.
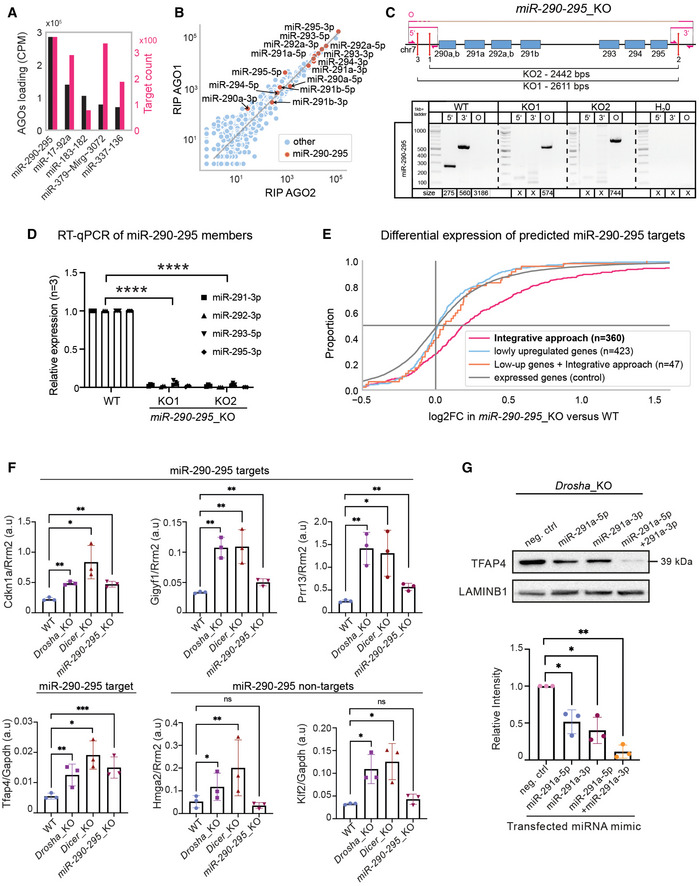
- MiRNA loading in Argonaute2 &1 (AGOs) and number of predicted target counts for top five loaded miRNA clusters.
- Loading of miRNAs in AGOs, highlighting miR‐290‐295 cluster members.
- MiR‐290‐295_KO strategy and genomic validation. Top: Schematic representation of the paired CRISPR/Cas9 KO approach for the generation of miR‐290‐295_KO cluster mESC lines. Genomic loci of miRNAs are indicated by blue boxes. Red lines mark sgRNA target sites. Primers for screening of the genomic deletion are indicated by pink half‐sided arrows and pink lines show PCR products. Bottom: Genomic PCR screening of miR‐290‐295_KO mESCs. Three screening primers, one around the 5′ cut site, one around the 3′ cut site, and one around the entire KO region (O) were used, as annotated at the top part of the subfigure. Expected amplicon sizes are denoted below the lanes. An X denotes no expected product.
- Quantitative real‐time PCR for representative miR‐290‐295 members in WT and miR‐290‐295_KO mESC lines. Expression levels were normalized to RNU6 in three biological replicates.
- Cumulative distribution function of differential expression in miR‐290‐295_KO for different gene groups, similar to Fig 3B. Colored log2FoldChange (log2FC) distributions correspond to different identification methods for miR‐290‐295 target genes based on different datasets (see Materials and Methods section). Integrative approach refers to the 360 miR‐290‐295‐targeted genes out of the 759 miRNA target genes that have been identified in the integrative analysis of this paper (Fig 1A). Lowly upregulated genes are those that show a log2FC between 0.1 and 0.5 in all three mutants. Low‐up genes + integrative filtering used the same set of genes but filtered for AGO2‐binding to one of three strongly expressed miR‐290‐295 members.
- Quantitative real‐time PCR showing relative expression levels of four predicted miR‐290‐295 targets and two miR‐290‐295 non‐targets normalized to the control genes Rrm2 or Gapdh in Drosha_KO Dicer_KO and miR‐290‐295_KO mESCs.
- Immunoblot analysis of TFAP4 after transfection of miRNA mimics (miR‐291a‐5p, miR‐291a‐3p and miR‐291a‐5p + miR‐291a‐3p combined) in Drosha_KO mESCs. LAMINB1 was used as a loading control. Blot is a representative image of three biological replicates. Bottom: Bar graph showing quantification of TFAP4 intensity, normalized to LAMINB1 and relative to the WT sample in three biological replicates.
Data information: In (D), statistical significance for RT‐qPCR of miR‐290‐295 members was assessed using a 2‐way ANOVA test. ****P < 0.0001. In (F), RT‐qPCR bar graphs show relative expression of miR‐290‐295 targets and non‐targets based on arbitrary units and normalized to a housekeeping gene (Rrm2 or Gapdh) for three biological replicates. P‐values were generated using a t‐test comparing each value to the WT. *P‐value < 0.05, **P‐value < 0.01, and ***P‐value < 0.001. In G, bar graphs show mean intensity of TFAP4 signal ± SD normalized to LAMINB1. Values are relative to the negative control mimic, which was set to 1. P‐values were calculated using a Student's t‐test comparing each value to the WT. *P‐value < 0.05, **P‐value < 0.01.
Source data are available online for this figure.
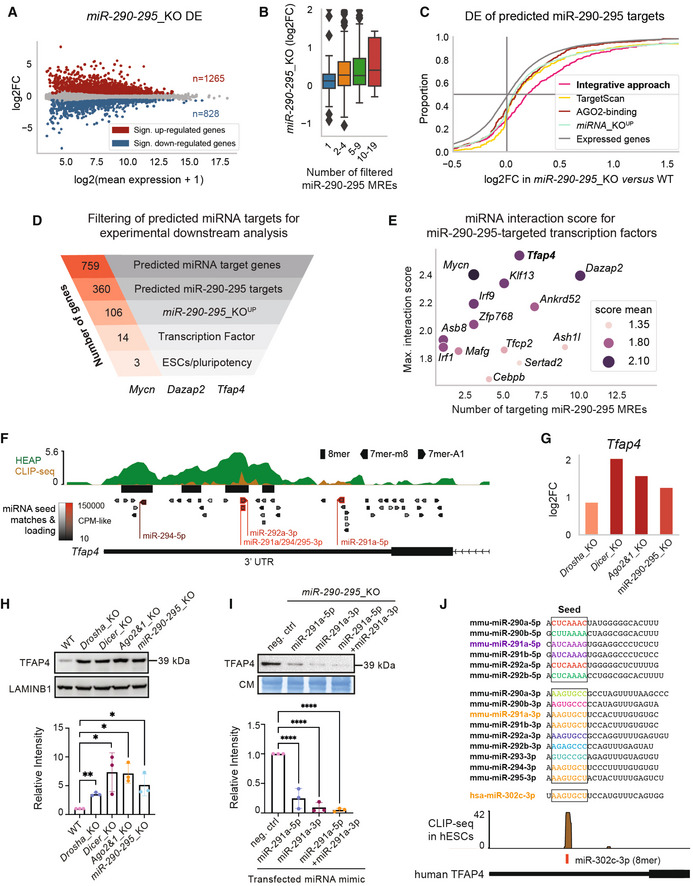
We, therefore, generated two independent miR‐290‐295_KO cell lines in the same genetic background as the miRNA_KO mESCs (Fig EV3C) and confirmed the integrity of the deletion at DNA and RNA levels (Fig EV3C and D). We next profiled the transcriptome of the miR‐290‐295_KO mESCs and performed a DE analysis (Dataset EV6). MiR‐290‐295_KO mESCs exhibited a strongly misregulated transcriptome with 1,265 up‐ and 828 downregulated genes (Fig 3A). In accordance with the differential ribosome occupancy (Fig 2D), the DE analysis also showed stronger upregulation for targets with larger numbers of predicted miR‐290‐295 interactions (Pearson's r = 0.22, P < 3.5e−5, Fig 3B). Candidates from the integrative analysis showed a strong enrichment for upregulation, with 72% of predicted genes (260 of 360) exhibiting a positive log2FC (pink curve in Fig 3C and Dataset EV6). Importantly, our integrative approach showed a significantly stronger enrichment for upregulated genes compared to when targets are predicted from each dataset individually (Fig 3C), indicating that integrating multiple datasets successfully led to a refined set of candidate miRNA target genes.
Figure 3. Validation of predicted miRNA targets.
-
AMA plot of the DGE analysis in miR‐290‐295_KO mESCs.
-
BDE of predicted miR‐290‐295 target genes in miR‐290‐295_KO versus WT for genes grouped by number of filtered miR‐290‐295 MREs/interactions (similar to Fig 2C).
-
CCumulative distribution function of DE in miR‐290‐295_KO for different gene groups. Colored log2FoldChange (log2FC) distributions correspond to different identification methods for miR‐290‐295 target genes based on different datasets (Fig 1A). Integrative approach refers to the 360 miR‐290‐295‐targeted genes out of the 759 candidate miRNA target genes that have been identified in the integrative analysis of this paper (Fig 1A). TargetScan targets were filtered for three strongly expressed members of the miR‐290‐295 cluster and from these the top 360 (in terms of TargetScan/context++ score) are shown. AGO2‐binding targets were first filtered by seed matches to the same three strongly expressed miR‐290‐295 members, then the 360 targets with the strongest binding signals were selected. miRNA_KO‐upregulated genes are the 3609 miRNA_KO UP genes used in the integrative analysis (see Materials and Methods section).
-
DFunnel analysis representing the number of genes identified by the integrative approach in each indicated category ending with miR‐290‐295 regulated transcription factors previously implicated in stem cell functions and pluripotency in the scientific literature.
-
EInteraction scores from our integrative analysis (Fig 1A) for all transcription factors predicted to be targeted by miR‐290‐295. The number of distinct miRNA binding sites for the miR‐290‐295 cluster, mean of interaction score, and maximum of interaction score is shown.
-
F, GIntegrated data for Tfap4 gene show multiple lines of evidence for its regulation by miR‐290‐295. In addition to the data shown and described in Fig 1C and D, a second AGO2‐binding dataset, based on cross‐linking immunoprecipitation and sequencing (CLIP‐seq), has been integrated. Response elements for miR‐290‐295 cluster members are denoted in red and labeled.
-
HTop: Immunoblot analysis of TFAP4 in WT, Drosha_KO, Dicer_KO, Ago2&1_KO, and miR‐290‐295_KO mESCs. LAMINB1 was used as a loading control. Blot is a representative image of three biological replicates. Bottom: Bar graph showing quantification of CIC intensity, normalized to LAMINB1 and relative to the WT sample in three biological replicates.
-
ITop: Immunoblot analysis of TFAP4 after transfection of miRNA mimics (miR‐291a‐5p, miR‐291a‐3p and miR‐291a‐5p + miR‐291a‐3p combined) in miR‐290‐295_KO mESCs. Immunoblots were stained with Coomassie blue dye as a loading control. Blot is a representative image of three biological replicates. Bottom: Bar graph showing quantification of TFAP4 intensity, normalized to Coomassie and relative to the WT sample in three biological replicates.
-
JTop: Mature sequences of 5p and 3p miRNAs of the miR‐290‐295 family and the mature sequence of the human miR‐302c‐3p. Colors indicate identical seed sequences. Bottom: AGO2 CLIP‐seq shows a peak at the miR‐302c‐3p binding site on human TFAP4.
Data information: In (A), significant genes in MA plot were determined using an adjusted P‐value of 0.1. In H and I, bar graphs show mean intensity of TFAP4 signal ± SD normalized to LAMINB1 or Coomassie. Values are relative to WT or the negative control mimic, which was set to 1. P‐values were calculated using a Student's t‐test comparing each value to the WT. *P‐value < 0.05, **P‐value < 0.01, ****P‐value < 0.0001. Box plot in panel (B) follows the same specification as described in Fig 2B and D. The sizes of the presented groups are 97, 164, 89, 10 (from left to right).
Source data are available online for this figure.
miRNAs have been described to often exert only low degrees of repression on their targets (Lai, 2015). We therefore, repeated our integrative approach based on lowly upregulated genes, which we defined as expressed (transcripts per million/TPM > 1) genes with a log2FC between 0.1 and 0.5 in all three mutants (low‐up genes n = 423). Filtering these genes for evidence of miRNA regulation (AGO2‐binding to miR‐290‐295 binding sites) indeed led to a minor enrichment of upregulation (mean log2FC = 0.10, Fig EV3E). Yet, only a fraction (< 40%) of the distribution showed increased log2FCs, which were minor. Given the small portion of verifiable low‐impact interactions, we continued to focus on interactions with more notable regulatory impact, as in our original integrative approach (Fig 1A).
In conclusion, all datasets contributed predictive power to the approach and their integration allowed for the most accurate prediction. Furthermore, generation of the miR‐290‐295_KO mutant cell lines allowed for the identification of a high‐confidence set of candidate miR‐290‐295 target genes.
Deletion of the miR‐290‐295 cluster combined with predicted functional interactions identifies novel key transcription factors regulated by miR‐290‐295
Out of the 360 predicted miR‐290‐295 targets, 106 showed a statistically significant upregulation in the miR‐290‐295_KO mutant (Dataset EV6 and Fig 3D). In order to validate these candidates, we measured their relative expression and observed an increase in the relative expression of all genes in the miRNA_KO mutants relative to WT, as well as an increase in the expression of predicted miR‐290‐295 targets in the miR‐290‐295_KO mutant (Fig EV3F).
Interestingly, 14 of the predicted miR‐290‐295 targets were annotated as transcription factors (TFs) by the mTFkb database (Sun et al, 2017) of which three, Tfap4, Dazap2 and Mycn had previously been implicated in stem cell functions, including pluripotency maintenance (Chappell & Dalton, 2013; Sugawara et al, 2020; Papathanasiou et al, 2021) (Fig 3D). TFs are proteins that primarily bind to promoter regions to modulate gene transcription (Spitz & Furlong, 2012), therefore potentially contributing to the observed differential gene expression (DGE) that cannot be explained by the depletion of miRNAs alone. Of all identified TFs, Tfap4 showed the highest interaction score from our analysis (Fig 3E). Its 3′UTR presents large AGO2‐binding peaks in both HEAP and CLIP‐seq data (Bosson et al, 2014; Li et al, 2020) with overlapping MREs for miR‐291a‐5p and miR‐291a‐3p (Fig 3F). This TF has recently been shown to be required for reprogramming mouse fibroblasts into pluripotent stem cells (Papathanasiou et al, 2021), but was never described as regulated by miRNAs. Tfap4 was upregulated in all miRNA_KO and miR‐290‐295_KO mESC lines at RNA and protein levels (Fig 3G and H).
To assess whether this upregulation was caused by the loss of miR‐290‐295‐mediated repression, we transfected miR‐290‐295_KO mESCs with miR‐291a‐5p and miR‐291a‐3p mimics individually or in combination and observed a significant downregulation of TFAP4 protein (Fig 3I). We repeated this experiment in the Drosha_KO mESCs and observed similar effects, suggesting a regulation of Tfap4 by specific miR‐290‐295 cluster miRNAs (Fig EV3G). Finally, mmu‐291a‐3p MRE is conserved in humans (hsa‐miR‐302c‐3p), and we observed AGO2 binding in the 3′UTR of human TFAP4 for this miRNA (Fig 3J), indicating a possible conservation for this miRNA regulation between human and mouse. Taken together, these data strongly suggest that Tfap4 is regulated by the miR‐290‐295 cluster in mESCs.
TFAP4 regulates important stem cell regulatory pathways
We hypothesized that a substantial number of misregulated genes observed in the miR‐290‐295_KO cell line (Fig 3A) may be in part a result of increased TF levels. To assess the transcriptional regulation of TFAP4 in mESCs, we attempted to rescue its expression level in miR‐290‐295_KO using a pool of small interfering RNAs (siPOOL) targeting Tfap4 mRNA (Fig EV4A). We monitored TFAP4 levels 36 h after transfection and observed that TFAP4 was indeed expressed at near WT levels in the miR‐290‐295_KO mESCs (Fig 4A). We then sequenced the transcriptome of the siPOOL‐transfected cells and assessed their DE (Dataset EV7), which also confirmed near‐WT Tfap4 RNA levels (Fig 4B). In addition, we observed a striking number of misregulated genes in the Tfap4 siPOOL samples with a large portion of them being inversely regulated (and therefore rescued back toward WT levels) compared to the initial DE in miR‐290‐295_KO mESC lines (Fig 4B). To discriminate and quantify rescued genes, we divided the log2FC observed in miR‐290‐295_KO vs. WT by the log2FC observed in the siPOOL‐transfected miR‐290‐295_KO mESCs and defined rescued genes to be within the range [−0.5, −2] (orange dots in Fig 4B). We observed 287 rescued genes for which TFAP4 acted in an activating and 210 for which it acted in a repressive manner. Thus, the misregulation of Tfap4 provoked by the loss of miR‐290‐295 might explain almost a quarter of the DEGs observed in the miR‐290‐295_KO mutant.
Figure EV4. Depletion of Tfap4 is not sufficient to rescue multiple Drosha_KO phenotypes.
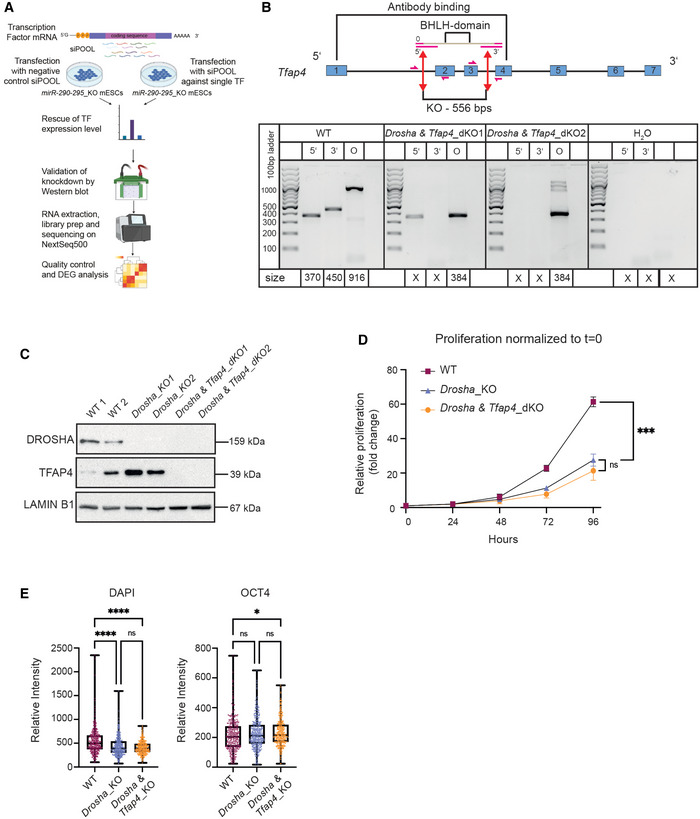
- Schematic representation describing the workflow to rescue TFAP4 expression by treating miR‐290‐295_KO mESCs with pool of siRNAs (siPOOL) targeting Tfap4. Transfected cells were harvested and analyzed on the transcriptomics level using RNA‐seq. Illustrations were extracted from BioRender.com.
- Top: Schematic representation of the paired CRISPR/Cas9 approach used to establish a functional Tfap4 knockout in Drosha_KO mESCs. Tfap4 exons are shown as blue boxes. Red double‐sided arrows indicate the sites of Cas9 sgRNA binding. Pink arrows show binding sites of the primers used for knockout validation by genomic DNA PCR. Bottom: Genomic PCR screening of Drosha & Tfap4_KO mESCs. Three screening primers pairs were used: one pair amplifies around the 5′ cut site, one around the 3′ cut site and one around the entire KO region (O) (see schematic representation above). Expected amplicon sizes are denoted below the lanes. An X denotes no expected product.
- Western blot validation of Drosha & Tfap4_KO clones. Immunoblot analysis of TFAP4 and DROSHA protein expression in WT, Drosha_KO and Drosha & Tfap4_KO. LAMINB1 was used as a loading control.
- Proliferation assay in WT, Drosha KO and Drosha & Tfap4_KO mESCs over 96 h. Relative change in proliferation is plotted for WT and KO cells as a mean of three biological replicates.
- Quantification of DAPI and OCT4 fluorescence signal intensity in WT, Drosha_KO and Drosha & Tfap4_KO mESCs. The central band represents the median relative intensity for each genotype. The boxes represent the interquartile range and the whiskers represent the maximum and minimum relative intensity values.
Data information: In (E), box plots show nuclear intensity of OCT4 and DAPI in ~ 300 cells per genotype. P‐values were generated using ordinary one‐way ANOVA. *P‐value < 0.05 and ****P‐value < 0.0001.
Source data are available online for this figure.
Figure 4. Tfap4 is a key regulator of gene expression in mESCs.
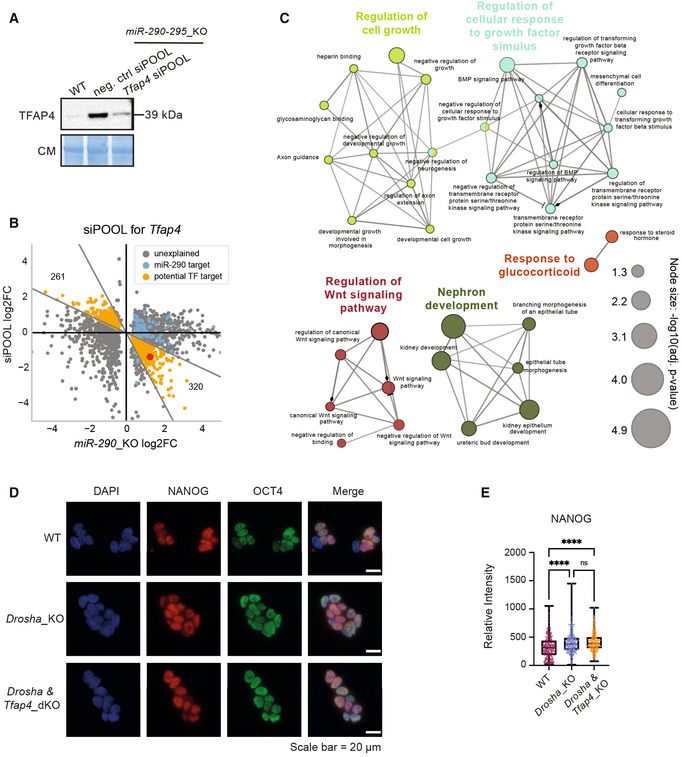
-
AImmunoblot validation of siPOOL‐mediated knock down of Tfap4. TFAP4 levels were compared between untreated WT versus miR‐290‐295_KO cells treated with a negative control and a Tfap4‐targeted siPOOL. Immunoblots were stained with Coomassie blue dye as a loading control. Blot is a representative image of two biological replicates.
-
BScatterplot of DEG in miR‐290‐295_KO control versus siPOOL‐Tfap4. SiPOOL experiments were performed in miR‐290‐295_KO cells and DE was assessed relative to a negative control siPOOL transfection. Only genes that are statistically significantly differentially expressed in miR‐290‐295_KO are shown. Genes predicted to be targeted by miR‐290‐295 are marked in blue. Genes are defined as rescued (orange) if the log2FoldChange‐ratio between miR‐290‐295_KO control and siPOOL‐Tfap4 is in the range [−2, −1/2]. Tfap4 is marked by a red dot.
-
CGene ontology analysis of 121 putative TFAP4 target genes using ClueGO. Only members of the top five terms (indicated by different colors) are shown. Colors and edges indicate associated terms.
-
DRepresentative images of NANOG (red) and OCT4 (green) immunofluorescence staining in WT, Drosha_KO and Drosha & Tfap4_KO mESCs. Scale bar = 20 μm.
-
EQuantification of NANOG fluorescence signal intensity in WT, Drosha_KO and Drosha & Tfap4_KO mESCs show no significant rescue of NANOG intensity in Drosha & Tfap4_KO.
Data information: In (C), statistical significance for GO terms was determined by ClueGO and is indicated in the figure. In (E), box plots show nuclear intensity of NANOG in ~ 300 cells per genotype. P‐values were generated using ordinary one‐way ANOVA. ****P‐value < 0.0001. In panel (E), the box plot's central band represents the median relative intensity for each genotype. The boxes represent the interquartile range and the whiskers represent the maximum and minimum relative intensity values.
Source data are available online for this figure.
The binding motif of TFAP4 has been previously described in humans (Jackstadt et al, 2013). To refine our set of rescue‐identified potential TFAP4‐targets, we scanned the genome for potential TFAP4‐binding sites, only keeping rescued genes with a binding site upstream of the promoter (< 1 kb distance), which resulted in 121 genes (Dataset EV7). We subjected this reduced set to a gene ontology analysis (Bindea et al, 2009). Most of the groups identified by the analysis (i.e., “regulation of cell growth,” “regulation of WNT signaling pathway”) (Fig 4C and Dataset EV7) are in line with previous reports of TFAP4 being an important regulator involved in developmental processes (Wong et al, 2021). TFAP4 has been shown to activate the Wnt/β‐catenin in human cancer and to drive cancer malignancy (Song et al, 2018; for review Wong et al, 2021). Furthermore, Tfap4 has been described to regulate stemness and proliferation (Jung et al, 2008; Jackstadt et al, 2013). Thus, we conclude that TFAP4 might be an essential regulator of stemness and development.
Restoration of TFAP4 levels is not sufficient to rescue miRNA_KO mutant phenotypes
In order to further assess the extent to which TFAP4 upregulation contributes to the observed multiple defects of miRNA_KO mESC lines (Bodak et al, 2017a), we deleted Tfap4 in Drosha_KO mESC line (Drosha &Tfap4_dKO) (Fig EV4B). The deletion of the BHLH domain of Tfap4 was validated at the DNA and protein levels (Fig EV4B and C). As previously observed in Dicer_KO mESCs (Bodak et al, 2017b), both Drosha_KO and Drosha & Tfap4_dKO mESC lines present a strong proliferation defect compared to WT mESCs (Fig EV4D), indicating that this defect is not caused by the upregulation of Tfap4.
We furthermore aimed to examine the pattern of expression of the pluripotency markers NANOG and OCT4 by immunofluorescence. WT mESCs have been shown to express NANOG heterogeneously, whereas miRNA_KO mESCs display homogenous expression of NANOG. In contrast, OCT4 expression remains homogenous (Cirera‐Salinas et al, 2017; Bakhtina et al, 2021). Both Drosha_KO and Drosha & Tfap4_dKO mESC lines present a homogenous expression of NANOG compared to the expected heterogeneity in WT mESCs (Fig 4D and E). OCT4 levels were similar in all cell lines (Figs 4D and EV4E).
Taken together, our data show that 24% of the perturbations in gene expression observed upon the loss of the miR‐290‐295 family are restored by the reduction of TFAP4 levels, suggesting an important role for TFAP4 in regulation of gene expression in mESCs. However, the deletion of Tfap4 in Drosha_KO is not sufficient to rescue mutant phenotypes. Our analysis revealed additional TFs as miRNA target candidates. Some of which, such as MYCN and DAZAP2, have been previously described with roles in pluripotency.
Limitations of the study
While our aim was to determine the functional miRNA targets in mESCs, a number of factors limits the accuracy of our results.
A substantial part of our integrative analysis was based on transcriptomics data, and while we showed that predicted targets are also affected at the protein level, we cannot exclude that some miRNA targets are only affected at the protein level and therefore not detected by our approach. Nevertheless, our datasets demonstrate a strong correlation between Ribo‐seq and RNA‐seq in mESCs as previously described in a different cellular context (Guo et al, 2010). The fact that a smaller subset of predicted miRNA targets does not show an increased ribosome occupancy in some of the mutants potentially reflects mutant‐specific misregulation events that overlay the effects caused by the loss of miRNAs.
Another aspect to take into account is that not all miRNAs require the full set of miRNA genes for their biogenesis (Bodak et al, 2017a, 2017b). Our approach is partially robust against this phenomenon, as we considered genes to be potential miRNA targets even if they are upregulated in only a subset of the miRNA_KO mutants (i.e., in at least two). Furthermore, based on miRNA_KOs' sRNA‐seq data, we identified only very few noncanonical miRNAs in mESCs and thus believe this manner is of low relevance in our context.
We consider limitations in the sensitivity of our approach as the most challenging to detect. Sensitivity limitations are likely to come with the filters for miRNA_KO‐upregulation and AGO2‐binding. Despite being functionally repressed by miRNAs, some genes might be downregulated in multiple mutants, for example, due to a miRNA‐regulated TF. Additionally, AGO2‐binding might remain undetected due to technical limitations. Here, we relied on data from a recent publication that employed a tagged version of AGO2 to circumvent the immunoprecipitation step, which can be a major source of variability. The sensitivity and specificity of thereby identified AGO2‐binding sites were reported to be drastically improved over previous methods (Bosson et al, 2014; Li et al, 2020). As the high specificity of our approach stems from the rigorous exclusion of interactions with limited evidence for functional miRNA interactions, it is challenging to improve upon the sensitivity without compromising the specificity.
One further limitation of our system lies in the use of mESCs to examine miRNA/mRNA interaction at the functional and molecular level. Due to the low transfection efficiency of mESCs, it was not feasible to perform the classical luciferase reporter assay as a means to validate the direct regulation of Tfap4 by members of the miR‐290‐295 cluster. Furthermore, performing these experiments in other cell types, such as the commonly used HEK293T cells, gave results that are contradictory to those observed in mESCs (Appendix Fig S3). This is potentially due to the fact that the miR‐290‐295 cluster is stem cell specific and may not be functional in other biological contexts.
Another challenge is that miRNAs are able to regulate their targets in a combinatorial manner, that is, several miRNAs can target the same mRNA at the same time (Cursons et al, 2018). This can lead to increased repression potential as shown in Fig 4B. Our works also show the importance of miRNAs as essential players in complex regulatory networks. Therefore, future works might improve our understanding of miRNA‐mediated regulation by extending miRNA interaction prediction to heterogeneous molecular network analysis. Here, machine learning models (Schäfer & Ciaudo, 2020), including graph‐aware deep learning models (Zhou et al, 2020) might help to overcome some of these barriers by generating a system's level understanding of regulation networks.
Our rigorous approach discards many interactions that would be falsely predicted by other methods, thus leading to a high‐confidence set of direct and functional miRNA interactions in mESCs. We expect these data will be useful to the scientific community and also trust that they will serve as a robust dataset on which to anchor future machine learning endeavors that can be applied to many different biological systems.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental models | ||
| WT Mouse ESCs (E14Tg2a) | ATCC | CRL‐1821 |
| Drosha_KO mESCs | Cirera‐Salinas et al (2017) | N/A |
| Dicer_KO mESCs | Bodak et al (2017a, 2017b) | N/A |
| Ago2&1_KO mESCs | This manuscript | N/A |
| miR‐290‐295_KO mESCs | This manuscript | N/A |
| Drosha & Tfap4_dKO mESCs | This manuscript | N/A |
| Recombinant DNA | ||
| pX458‐sgRNA_Ago1_1 | Addgene | #73533 |
| pX458‐sgRNA_Ago1_2 | Addgene | #73534 |
| pX458‐sgRNA_Ago1_3 | Addgene | #73535 |
| pX458‐sgRNA_Ago1_4 | Addgene | #73536 |
| pX458‐sgRNA_miR290‐295_1 | This manuscript | #172709 |
| pX458‐sgRNA_miR290‐295_2 | This manuscript | #172710 |
| pX458‐sgRNA_miR290‐295_3 | This manuscript | #172711 |
| pLentiCRISPR‐EGFP‐sgRNA_TFAP4_BHLH_5_1 | This manuscript | #185054 |
| pLentiCRISPR‐mCherry‐sgRNA_TFAP4_BHLH_3_2 | This manuscript | #185055 |
| Antibodies | ||
| Rabbit polyclonal anti‐CIC | ThermoFisher | Cat # PA1‐46018 |
| Mouse monoclonal anti‐LAMINB1 | Abcam | Cat#Ab16048 |
| Mouse monoclonal Anti‐alpha‐TUBULIN (clone DM1A) | Sigma‐Aldrich | Cat#T6199 |
| Rabbit monoclonal Anti‐Drosha (D28B1) | Cell Signaling Technology | Cat#D28B1 |
| Rabbit polyclonal Anti‐Dicer | Sigma‐Aldrich | SAB4200087 |
| Rabbit monoclonal Anti‐Ago2 (C34C6) | Cell Signaling Technology | Cat#2897S |
| Rabbit monoclonal Anti‐Ago1 (D84G10) | Cell Signaling Technology | Cat#5053 |
| Rabbit polyclonal Anti‐Tfap4 | Abcam | ab223771 |
| Rabbit monoclonal anti‐NANOG (D2A3) | Cell Signaling Technology | Cat#8822 |
| Mouse monoclonal anti‐OCT4 | BD Biosciences | Cat#611202 |
| anti‐mouse Alexa Fluor 488 | Invitrogen | Cat#A32731 |
| anti‐rabbit Alex Fluor 568 | Invitrogen | Cat#A11004 |
| Oligonucleotides and sequence‐based reagents | ||
| See Table EV1 | ||
| Chemicals, enzymes and other reagents | ||
| Trizol | Life Technologies | 15596018 |
| ESGRO recombinant mouse LIF protein | Millipore | ESG1107 |
| DMEM Media | Sigma‐Aldrich | D6429‐500ML |
| Opti‐MEM reduced serum media | Life Technologies | 31985070 |
| Lipofectamine RNAiMax Reagent | Life Technologies | 13778150 |
| Lipofectamine 2000 | Invitrogen | 52887 |
| Lipofectamine 3000 | Invitrogen | 100022052 |
| Penicillin/Streptomycin | Sigma‐Aldrich | P0781‐100ML |
| 0.05% Trypsin‐EDTA | Life Technologies | 25300054 |
| PBS1X | Life Technologies | 10010015 |
| 2‐ß‐mercaptoethanol | Life Technologies | 31350010 |
| FBS | Life Technologies | 10270‐106 |
| Urea | EuroBio | GEPURE00‐67 |
| NH4HCO3 | Sigma‐Aldrich | 9830 |
| Tris(carboxyethyl)phosphine | Sigma‐Aldrich | 68957 |
| Iodoacetamide | Sigma‐Aldrich | I1149 |
| Sequencing‐grade porcine trypsin | Promega | Cat#V5113 |
| Aqueous formic acid | Sigma‐Aldrich | F0507 |
| Acetonitrile | Sigma‐Aldrich | 271004 |
| Cycloheximide | Sigma‐Aldrich | 1810 |
| Phenol/Choloroform/Isoamyl Alcohol | Sigma‐Aldrich | P2069 |
| Tris‐HCl | AppliChem | A2937 |
| NaCl | Merck | 1.06404.1000 |
| IGEPAL CA‐630 | SIGMA | I3021‐50ML |
| Sodium deoxycholate | Sigma‐Aldrich | 30970 |
| Sodium dodecyl sulfate | Sigma‐Aldrich | L6026 |
| Protease inhibitor cocktail tablets | Roche | 5892791001 |
| Tween20 | Sigma‐Aldrich | P1379 |
| β‐galactosidase tryptic | Sigma‐Aldrich | Cat#4333606 |
| Coomassie | VWR | 443283M |
| Actinomycin D | Merck | A1410 |
| Dimethylsulfoxid | Sigma‐Aldrich | D2650 |
| Proteinase K | PanReac AppliChem | A3830 |
| Triton‐X | Roth | 9002‐93‐1 |
| EDTA | PanReac AppliChem | A2937 |
| Glycine | PanReac Applichem | A1067 |
| Ammonium persulfate (APS) | Sigma‐Aldrich | A3678 |
| TEMED | Sigma‐Aldrich | T9281 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma‐Aldrich | P8833 |
| Gelatin from porcine skin | Sigma‐Aldrich | G1890 |
| Acrylamide 4K Solution | PanReacAppliChem | A1672 |
| mirVana miRNA Mimic negative control | Thermo scientific | #4464058 |
| mirVana miRNA Mimic mmu‐miR‐291a‐3p | Thermo scientific | #4464066 |
| mirVana miRNA Mimic mmu‐miR‐291a‐5p | Thermo scientific | #4464066 |
| siPOOL Tfap4 + negative control | siTOOLs Biotech | 83383 |
| Software | ||
| Targetscan (v7.2) | Agarwal et al (2015) | http://www.targetscan.org/vert_72/ |
| Cytoscape (3.8.2) | Shannon et al (2003) | https://cytoscape.org/ |
| ClueGO (2.5.8) | Bindea et al (2009) | https://apps.cytoscape.org/apps/cluego |
| STAR (v2.4.2a) | Dobin et al (2013) | https://github.com/alexdobin/STAR/releases |
| featureCounts (1.5.0) | Liao et al (2014) | https://www.rdocumentation.org/packages/Rsubread |
| DESeq2 (1.18.1) | Love et al (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Cutadapt (1.8 and 1.13) | Martin (2011) | https://cutadapt.readthedocs.io/en/stable/ |
| Trim Galore | https://github.com/FelixKrueger/TrimGalore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Analyst TF (1.5.1) | AB Sciex | https://omictools.com/analyst‐tf‐tool |
| PeptideProphet | Keller et al (2002) | http://peptideprophet.sourceforge.net/ |
| iProphet | Shteynberg et al (2011) | http://tools.proteomecenter.org/wiki/index.php?title=Software:TPP |
| ProteoWizard (3.0.3316) | Chambers et al (2012) | http://proteowizard.sourceforge.net/ |
| OpenSWATH | Röst et al (2014) | http://openswath.org/en/latest/docs/openswath.html |
| SWATH2stats | Blattmann et al (2016) | http://bioconductor.org/packages/release/bioc/html/SWATH2stats.html |
| MSstats (MSstats.daily 2.3.5) | Choi et al (2014) | https://bioconductor.org/packages/release/bioc/html/MSstats.html |
| Bowtie2 (2.3.4.1) | Langmead & Salzberg (2012) | http://bowtie‐bio.sourceforge.net/bowtie2/index.shtml |
| Snakemake 5.26.1 | Mölder et al (2021) | https://github.com/snakemake/snakemake |
| snakePipes 2.3.1 | Bhardwaj et al (2019) | https://github.com/maxplanck‐ie/snakepipes |
| snakemake‐workflows/rna‐seq‐star‐deseq2 1.0.0 | Köster et al (2021) | https://zenodo.org/record/4741280 |
| NumPy | Harris et al (2020) | https://numpy.org/ |
| Pandas | McKinnery (2010) | https://pandas.pydata.org/ |
| SciPy | Virtanen et al (2020) | https://www.scipy.org/ |
| tqdm | https://tqdm.github.io/ | https://zenodo.org/record/4663456 |
| scikit‐learn | Pedregosa et al (2011) | https://scikit‐learn.org/ |
| scikit‐bio | http://scikit‐bio.org/ | https://github.com/biocore/scikit‐bio |
| BioPython | Cock et al (2009) | http://biopython.org/ |
| gffutils | https://github.com/daler/gffutils | https://github.com/daler/gffutils |
| pyBigWig | Ramírez et al (2016) | https://github.com/deeptools/pyBigWig |
| pyensembl | https://github.com/openvax/pyensembl | https://github.com/openvax/pyensembl |
| pybedtools | Dale et al (2009) | https://github.com/daler/pybedtools |
| bedtools | Quinlan and Hall (2010) | https://github.com/arq5x/bedtools2 |
| Matplotlib | Hunter (2007) | https://matplotlib.org/ |
| Matplotlib‐venn | https://github.com/konstantint/matplotlib‐venn | https://github.com/konstantint/matplotlib‐venn |
| seaborn | (Waskom) | https://seaborn.pydata.org/ |
| scikit‐plot | https://github.com/reiinakano/scikit‐plot | https://github.com/reiinakano/scikit‐plot |
| pyGenomeTracks | Lopez‐Delisle et al (2021) | https://github.com/deeptools/pyGenomeTracks |
| UpSetPlot | https://github.com/jnothman/UpSetPlot | https://github.com/jnothman/UpSetPlot |
| srna‐seq pipeline | This manuscript | https://github.com/moritzschaefer/srna‐seq |
| snakePipes runs | This manuscript | https://github.com/moritzschaefer/snakepipelines_pub |
| Auxiliary library | This manuscript | https://github.com/moritzschaefer/moritzsphd_pub |
| Main pipeline producing all data and figures | This manuscript | https://github.com/moritzschaefer/mesc‐regulation_pub |
| Omics datasets | ||
| Drosha_KO mESCs, RNA‐seq data | Cirera‐Salinas et al (2017) | GEO: GSE122627 |
| Dicer_KO mESCs, RNA‐seq data | Bodak et al (2017a, 2017b) | GEO: GSE78973 |
| Ago2&1_KO mESCs, RNA‐seq data | This manuscript | GEO: GSE110942 |
| WT Mouse ESCs (E14Tg2a), RNA‐seq data | Cirera‐Salinas et al (2017) | GEO: GSE78971 |
| mESC AGOs RIP‐seq data | Ngondo et al (2018) | GEO: GSE80454 |
| Full proteome data of mESC WT and miRNA_KO mutants | This manuscript | ProteomeXchange: PXD014484 |
| Ribosome Profiling data of mESC WT and miRNA_KO mutants | This manuscript | GEO: GSE135577 |
| QuantSeq data of mESC WT and miRNA‐290‐295_KO + sipools | This manuscript | GEO: GSE181393 |
| Halo‐enhanced AGO2 binding data in mESCs | Li et al (2020) | GSE139345 |
| CLIP‐seq data in hESCs | Lipchina et al (2011) | SRR359787 |
| iCLIP‐seq data in mESCs | Bosson et al (2014) | GSE61348 |
| Other | ||
| Roche Light Cycler 480 | Roche | |
| TruSeq stranded total RNA library prep | Illumina | 20020596 |
| TruSeq Small RNA Library Prep Kit | Illumina | RS‐200‐0012 |
| QuantSeq 3′ mRNA‐Seq Library Prep Kit FWD for Illumina | Lexogen | 15.24 |
| KAPA SYBR FAST for Roche LightCycler 480 | Sigma‐Aldrich | KK4611 |
| iRT‐Kit (RT‐kit WR) | Biognosys | Ki‐3002‐1 |
| TruSeq Ribo Profile Kit | Illumina | Cat.no. RPHMR12126 |
| Ribo‐Zero Gold rRNA Removal Kit | Illumina | Cat.no. MRZG12324 |
| RQ1 RNase‐Free DNase Kit | Promega | M6101 |
| GoScript Reverse Transcriptase | Promega | A5004 |
| miScript II RT Kit | Qiagen | Cat No./ID: 218161 |
| DC Protein Assay Reagent | Bio‐rad | 5000113‐5 |
| Clarity Western ECL Substrate | Bio‐rad | 1705061 |
| MicroSpin Column SilicaC18 (5‐60 μg capacity) | Nest Group Inc., Southborough, MA | CAT #SEMSS18V |
| MicroSpin S‐400 columns | GE Healthcare | GE27‐5140‐01 |
| PVDF membrane | Sigma‐Aldrich | GE10600023 |
| 1.5 ml Safe‐Lock Tubes | Eppendorf | 30120086 |
| 2 ml Safe‐Lock Tubes | Eppendorf | 30120094 |
| T25: 25 cm2 Filter Flask | TPP | 90026 |
| T75: 75 cm2 Filter Flask | TPP | 90076 |
| 96‐well plate | TPP | 92096 |
| 6‐well plate | TPP | 92006 |
| 15 ml CELLSTAR Tubes | Greiner | 188271 |
Methods and Protocols
Mouse ESC lines
WT E14, miRNA_KO (Drosha_KO, Dicer_KO, and Ago2&1_KO), miR‐290‐295_KO cluster, and Drosha & Tfap4_KO mESC lines (129/Ola background) were cultured in Dulbecco's Modified Eagle Media (DMEM) (Sigma‐Aldrich), containing 15% fetal bovine serum (FBS; Life Technologies) tested for optimal growth of mESCs, 100 U/ml LIF (Millipore), 0.1 mM 2‐ß‐mercaptoethanol (Life Technologies) and 1% Penicillin/Streptomycin (Sigma‐Aldrich), on 0.2% gelatin‐coated support in absence of feeder cells. The culture medium was changed daily. All cells were grown at 37°C in 8% CO2.
CRISPR/Cas9 mediated gene knockout
The generation of Drosha_KO and Dicer_KO mESC lines was previously described (Cirera‐Salinas et al, 2017; Bodak et al, 2017b). The Ago2&1_KO1 and KO2 cell lines as well as miR‐290‐295_KO1 and KO2, were generated using a paired CRISPR/Cas9 strategy on WT mESCs as described previously (Wettstein et al, 2016). We generated two independent clones for the Ago2&1_KO line (Ago2&1_KO1, Ago2&1_KO2) using two different pairs of gRNAs to delete one or more exons of the Ago1 gene in the previously described Ago2_KO1 mutant mESC line (Ngondo et al, 2018). Ago2_KO1 mESCs were transfected with pX458‐sgRNA_Ago1_1/2 (Addgene #73533 and #73534), and pX458‐sgRNA_Ago1_3/4 (Addgene #73535 and #73536) plasmids (Ngondo et al, 2018). We generated two independent miR‐290‐295_KO mESC lines by transfecting WT E14 mESCs with pX458‐sgRNA_miR290‐295_3/2 for KO1 (Addgene #172711, #172710) and pX458‐sgRNA_miR290‐295_1/2 for KO2 (Addgene #172709 and #172710). For the Drosha & Tfap4_KO lines, we generated one clone by transfecting Drosha_KO mESCs with pLentiCRISPR‐EGFP‐sgRNA_TFAP4_BHLH_5_1 and pLentiCRISPR‐mCherry‐sgRNA_TFAP4_BHLH_3_2 (Addgene #185054, #185055) to delete the BHLH domain of TFAP4. After 48 h, the GFP‐positive cells were single cell sorted in 96‐well plates. The deletion was genotyped by PCR using primers listed in Table EV1. All transfected plasmids are available in the Addgene repository. Positive clones were expanded and verified by genomic PCR and sequencing.
Extraction of total RNA from mESCs
Total RNA from 1 to 10 million cells was extracted using Trizol reagent (Life technologies) following the manufacturer's protocol (Bodak & Ciaudo, 2016). RNA was quantified using spectrophotometry on the Eppendorf Biophotometer. RNA integrity was visually controlled by running 1 μg of total RNA extract on a 1% agarose gel.
RNA‐seq
Tru‐seq
Prior to library preparation, the quality of isolated RNA was determined with a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). Up to 2 μg of polyA purified RNA was used for the library preparation using the TruSeq paired‐end stranded RNA Library Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer's recommendations. The library preparation and sequencing (Illumina HiSeq 2000) were performed by the FGCZ (Functional Genomic Center, Zurich). The paired‐end sequencing generated about 2 × 60 million reads per library.
QuantSeq
Total RNA of 500 ng was used for library preparation using the QuantSeq 3′ mRNA‐Seq Library Prep Kit FWD for Illumina (Lexogen) according to the manufacturer's recommendations. Sequencing was performed by the FGCZ (Functional Genomic Center, Zurich) on the Illumina NextSeq500 platform. Single‐end sequencing generated at least 20 million reads per library.
Small RNA‐seq
The Illumina TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, CA, USA) was used with 1 μg of total RNA for the construction of sequencing libraries by the Functional Genomic Center Zurich (Switzerland). Sequencing was performed on an Illumina Hiseq 2500 sequencer and generated between 20 and 30 million of single reads of 50 bp per library.
Genomic DNA extraction and PCR
Genomic DNA was extracted from 5 × 105 mESCs using Phenol/Chloroform/Isoamyl Alcohol (Sigma‐Aldrich). Each PCR reaction was performed using 50–100 ng of genomic DNA. Genotyping PCR primer sequences are listed in Table EV1.
Quantitative real‐time PCR analysis of miRNAs
For miRNA quantification, 1 μg total RNA was reverse transcribed using the miScript II Reverse Transcription kit (Qiagen) according to the manufacturer's instructions (Jay & Ciaudo, 2013). After reverse transcription, cDNA products were diluted in distilled water (1:5). Quantification of expression levels was performed on a Light Cycler 480 (Roche) using 2 μl of the diluted products, the KAPA SYBR FAST qPCR kit optimized for Light Cycler 480 (KAPA Biosystems), miScript Universal Primer (Qiagen) and a primer for the targeted miRNA. Differences between samples and controls were calculated based on the 2−ΔΔCT method using RNU6 control primer (Qiagen) as normalizer. Quantitative RT‐PCR assays were performed in triplicate (Jay & Ciaudo, 2013). All primers are listed in Table EV1.
Quantitative real‐time PCR analysis mRNAs
For mRNA expression evaluation, 1 μg total RNA was DNase treated using RQ1 DNase (Promega), according to the manufacturer's protocol. DNase‐treated RNA was then reverse transcribed using GoScript Reverse Transcriptase (Promega) after incubation with random primers. cDNA products were diluted in distilled water (1:5). Quantification of expression levels was performed on a Light Cycler 480 (Roche) using 2 μl of the diluted products, the KAPA SYBR FAST qPCR kit optimized for Light Cycler 480 (KAPA Biosystems) using gene‐specific primers. Relative expression levels for each gene were calculated based on the 2−ΔΔCT method using Gapdh or Rrm2 as a normalizer. Quantitative RT‐PCR assays were performed in triplicate (Jay & Ciaudo, 2013). All primers are listed in Table EV1.
Western blot analysis
Whole‐cell extracts were obtained by lysing the cells in RIPA buffer (50 mM Tris–HCl pH 8, 150 mM NaCl, 1% IGEPAL CA‐630 (w/v), 0.5% sodium deoxycholate (w/v), 0.1% sodium dodecyl sulfate (w/v) and protease inhibitors). Protein concentrations were determined by Bradford assay (Bio‐Rad Laboratories). The extracts were separated on SDS‐PAGE gels and transferred to polyvinylidene fluoride membranes (Sigma Aldrich). After blocking (5% milk in TBST: 50 mM Tris‐Cl, pH 7.5. 150 mM NaCl, 0.1% Tween20), membranes were incubated with primary antibodies diluted in blocking solution overnight at 4°C. Membranes were incubated with one of the following antibodies overnight: DROSHA antibody 1:2,000 (Cell Signaling; Cat#D28B1), DICER antibody 1:2,000 (Sigma Aldrich; SAB4200087), AGO1 antibody 1:1,500 (Cell Signaling; Cat#5053), AGO2 antibody 1:1,500 (Cell Signaling; Cat#2897S), TFAP4 antibody 1:1,000 (ab223771; Abcam), TUBULIN antibody 1:10,000 (Sigma Aldrich; Cat#T6119), CIC antibody 1:1,000 (Invitrogen: Cat#PA146018), LAMINB1 antibody 1:5,000 (Abcam: Cat#Ab16048). For secondary antibody incubation, the anti‐rabbit or anti‐mouse IgG HRP‐linked antibody (Cell Signaling Technology) was diluted to 1:10,000. Immunoblots were developed using the SuperSignal West Femto Maximum Sensitivity Substrate (Invitrogen) and imaged using the ChemiDoc MP imaging system (Bio‐Rad Laboratories). TUBULIN levels or the Coomassie brilliant blue staining of the membrane are used as loading controls.
ImageJ was used to quantify band intensity for each sample, which was then normalized to the Coomassie, TUBULIN, or LAMINB1 loading control. Band intensities in the mutants were represented as a fold‐change relative to the wild‐type sample. Three biological replicates were used to perform the quantification. Paired t‐tests between the control and each sample were used to assess statistical significance.
Transfection of mESCs with miRNA mimics
The 100,000 mESCs (miR‐290‐295_KO cell line or Drosha_KO) were seeded 24 h prior to transfection. Cells were transfected with miRNA mimics (Horizon, PerkinElmer) for miR‐291a‐5p (20 nM final concentration), miR‐291a‐3p (20 nM final concentration), or a combination of both (10 nM final concentration, each) using the Lipofectamine RNAiMax transfection reagent (Invitrogen), according to the manufacturer's protocol. A negative control miRNA mimic was also used (Table EV1). Media were changed 16 h after transfection and cells were harvested 36 h after transfection for protein extraction.
mESC transfection with siPOOLs
The 200,000 mESCs (miR‐290‐295_KO and Drosha_KO cell lines) were seeded 24 h prior to transfection. Cells were transfected with siPOOLs (siTOOLs Biotech) against Tfap4 or a negative control siPOOL at a final concentration of 5 nM using the Lipofectamine RNAiMax transfection reagent (Invitrogen), according to the manufacturer's protocol. Media were changed after 24 h of transfection and cells were harvested after 36 h of transfection using 0.05% Trypsin for further processing (RNA and protein extraction).
Proliferation assay
Proliferations assays were performed as previously described by (Cirera‐Salinas et al, 2017) in triplicate after 50,000 WT (E14), Drosha_KO, or Drosha & Tfap4_KO cells were seeded into gelatin‐coated 6‐well plates. The number of cells per ml was plotted using Prism. Two‐way ANOVA was used to assess significance between different genotypes.
Immunofluorescence
The 100,000 E14 mESCs per well were seeded on fibronectin (1 μg/ml in PBS) coated coverslips into a 6‐well plate on the day before the experiment. Washed cells were fixed with 3.7% formaldehyde in 1 x PBS for 10 min. After fixation, cells were washed two times with 1 x PBS at RT. Cells were permeabilized using CSK buffer (0.5% Triton X‐100, 10 mM HEPES, 100 mM NaCl, 3 mM MgCl2, and 300 mM Sucrose) for 4 min on ice followed by two washes with 1 x PBS at RT. Cells were blocked with 1% BSA in 1 x PBS‐T (1 × PBS + 0.1% Tween 20) for 20 min at RT before incubating with the primary antibody at RT for 1 h. The following primary antibody concentrations were used: NANOG (D2A3) antibody 1:500 (Cell Signaling; Cat#8822), OCT4 antibody 1:500 (BD Biosciences; Cat#611202). Afterward, coverslips were washed three times with 1 × PBS‐T and incubated with a fluorochrome‐conjugated secondary antibody for 1 h at RT in the dark. The following secondary antibodies were used: anti‐mouse Alexa Fluor 488 1:2,000 (Invitrogen; Cat#A32731), anti‐rabbit Alex Fluor 568 1:2,000 (Invitrogen, Cat#A11004). Prior to a counterstaining with DAPI (100 ng/ml) for 5 min at RT, the cells were washed two times with 1 × PBS‐T and once with 1 × PBS. Coverslips were mounted using Vectashield onto clean microscope slides, sealed with nail polish, and stored at −20°C until imaging with a DeltaVision Multiplex microscope at the ScopeM facility. The images were analyzed using Cell Profiler.
Proteome analysis by SWATH‐MS
The MS data acquisition (SWATH‐MS and DDA mode) was performed on TripleTOF 5600 mass spectrometer equipped with a NanoSpray III source and operated by Analyst TF 1.5.1 software (AB Sciex). The samples were injected onto a C18 nanocolumn packed in‐house directly in a fused silica PicoTip emitter (New Objective, Woburn, MA, USA) with 3‐μm 200 Å Magic C18 AQ resin (Michrom BioResources, Auburn, CA, USA) and reverse phase peptide separation was performed on a NanoLC‐Ultra 2D Plus system (Eksigent–AB Sciex, Dublin, CA, USA). The total acquired data were analyzed using a pipeline configured on the Euler‐Portal platform at ETH Zurich.
Sample preparation and protein digestion
The four distinct mESC lines (i.e., WT, Drosha_KO, Dicer_KO, and Ago2&1_KO) were prepared in biological duplicates (e.g., two independent CRISPR/Cas9 mutants), totaling 10 distinct samples for proteomic analysis. Corresponding cells from each 10 cm plate, were washed and scraped with ice‐cold phosphate‐buffered saline (PBS 1X). Then, their pellets (~ 5 × 106 cells) collected by centrifugation at 1000 rpm, were frozen in liquid nitrogen and left at −80°C. The cell pellets were lysed on ice using a lysis buffer containing 8 M urea (EuroBio), 50 mM NH4HCO3 (Sigma‐Aldrich), and complete protease inhibitor cocktail (Roche). The mixture was sonicated at 4°C for 5 min using a VialTweeter device (Hielscher‐Ultrasound Technology) at the highest setting and centrifuged at 800 x g at 4°C for 15 min to remove the insoluble material. An equal volume of 200 μl per sample was used for protein digestion, prior to which all samples were reduced by 5 mM tris(carboxyethyl)phosphine (Sigma‐Aldrich), and alkylated by 30 mM iodoacetamide (Sigma‐Aldrich). The samples, adjusted to 1.5 M UREA, were digested with sequencing‐grade porcine trypsin (Promega) at a 1:50 protease/protein ratio overnight at 37°C in 100 mM NH4HCO3 (Sigma‐Aldrich). The next day, the peptide digests were purified on MicroSpin Column SilicaC18 (5–60 μg capacity, Nest Group Inc., Southborough, MA), and solubilized in 50 μl of 0.1% aqueous formic acid (FA) with 2% acetonitrile (ACN). The final peptide amount was determined using Nanodrop ND‐1000 (Thermo Scientific), and the samples were adjusted to 1 μg/μl of peptide concentration. Prior to MS injection, an aliquot of retention time calibration peptides from an iRT‐Kit (RT‐kit WR, Biognosys) was spiked into each sample at a 1:20 (v/v) ratio to correct relative retention times between acquisitions, and each sample injected into the duplicates (i.e., technical replicates).
SWATH assay library generation
The samples were recorded in data‐dependent acquisition (DDA) mode to generate a mouse SWATH assay library, which is used for targeted data extraction from SWATH‐MS recorded data. Fifteen mESC samples recorded in DDA mode were combined with 65 available DDA files originating from fractionated mouse liver peptide digest to create a common mouse assay library. The nanoLC gradient used for all acquired DDA data was linear from 2 to 35% of buffer B (i.e., 0.1% formic acid in ACN) over 120 min at a 300 nl/min flow rate. Electrospray ionization was performed in positive polarity at 2.6 kV, and assisted pneumatically by nitrogen (20 psi). Mass spectra (MS) and tandem mass spectra (MS/MS) were recorded in “high‐sensitivity” mode over a mass/charge (m/z) range of 50 to 2,000 with a resolving power of 30,000 (full width at half maximum [FWHM]). DDA selection of the precursor ions in a survey scan of 250 ms was as follows: the 20 most intense ions (threshold of 50 counts) corresponding to 20 MS/MS‐dependent acquisitions of 50 ms each, charge state from 2 to 5, isotope exclusion of 4u, and precursor dynamic exclusion of 8 s leading to a maximum total MS duty cycle of 1.15 s. External mass calibration was performed by injecting a 100‐fmol solution of β‐galactosidase tryptic. Raw data files (.wiff) were centroided, and converted into mzXML as a final format using openMS.
The converted data files were searched in parallel using the search engines X! TANDEM Jackhammer TPP (2013.06.15.1 ‐ LabKey, Insilicos, ISB) and Comet (version “2016.01 rev. 3”) against the ex_sp 10090.fasta database (reviewed canonical Swiss‐Prot mouse proteome database, released 2017.12.01) appended with common contaminants and reversed sequence decoys (Elias & Gygi, 2007) and iRT peptide sequence. The search parameters were conducted using Trypsin digestion and allowing two missed cleavages. Included were ‘Carbamidomethyl (C)’ as static and ‘Oxidation (M)’ as variable modifications. The mass tolerances were set to 50 ppm for precursor ions and 0.1 Da for fragment ions. The identified peptides were processed and analyzed through the Trans‐Proteomic Pipeline (TPP v4.7 POLAR VORTEX rev 0, Build 201403121010) using PeptideProphet (Keller et al, 2002), iProphet (Shteynberg et al, 2011), and ProteinProphet scoring. Spectral counts and peptides for ProteinProphet were filtered at FDR of 0.009158 mayu‐protFDR (=0.998094 iprob). The raw spectral libraries were generated from all valid peptide spectra through an automated library generation workflow on the Euler‐Portal platform as described earlier (Schubert et al, 2015). The final generated spectral library contained high‐quality MS assays for 37,988 tryptic peptides from 4,107 mouse proteins.
SWATH‐MS measurement and data analysis
Reverse phase peptide separation during SWATH‐MS acquisition was performed with a linear nanoLC gradient from 2 to 35% of buffer B (0.1% formic acid in ACN) over 60 min at a 300 nl/min flow rate. Quadrupole settings in SWATH acquisition method were optimized for the selection of 64 variable‐wide precursor ion selection windows as described earlier (Röst et al, 2014). An accumulation time of 50 ms was used for 64 fragment‐ion scans operating in high‐sensitivity mode. At the beginning of each SWATH‐MS cycle, a TOF MS scan (precursor scan) was also acquired for 250 ms at high‐resolution mode, resulting in a total cycle time of 3.45 s. The swaths overlapped by 1 m/z, thus covering a range of 50–2,000 m/z. The collision energy for each window was determined according to the calculation for a charge 2+ ion centered upon the window with a spread of 15. Raw SWATH data files were converted into the mzXML format using ProteoWizard (version 3.0.3316) (Chambers et al, 2012), and data analysis was performed using the OpenSWATH tool (Röst et al, 2014) integrated in the Euler‐Portal workflow. The OpenSWATH workflow input files consisted of the mzXML files from the SWATH‐acquired data, the TraML assay library file created above, and the TraML file for iRT peptides. SWATH data were extracted with 50 ppm around the expected mass of the fragment ions and with an extraction window of +/−300 s around the expected retention time after performing iRT peptide alignment. The runs were subsequently aligned with a target FDR of 0.01 and a maximal FDR of 0.1 for aligned features. In the absence of a confidently identified feature, the peptide and protein intensities were obtained by integration of the respective background signal at the expected peptide retention time. The recorded feature intensities after OpenSWATH identification were filtered through R/Bioconductor package SWATH2stats (Blattmann et al, 2016) to reduce the size of the output data and remove low‐quality features. The filtered fragment intensities were introduced into the R/Bioconductor package MSstats (version MSstats.daily 2.3.5), and converted to a quantification matrix of relative protein abundances using functions of data pre‐processing, quality control of MS runs, and model‐based protein quantification (Choi et al, 2014). Quantification matrices were used as an input data template to perform further differential analysis by one‐way ANOVA test for multiple‐group comparison. A Tukey's HSD post hoc test revealed significant changes across control samples (WT) and three different cell line clones (i.e., Drosha_KO, Dicer_KO, and Ago2&1_KO). The raw counts and differential expression data are available as excel files (Dataset EV4).
Ribosome profiling and data analysis
Ribosome profiling sample/library preparation and sequencing
Ribosome profiling and parallel RNA‐seq were performed in duplicate for WT, Drosha_KO, Dicer_KO, and Ago2&1_KO mESC lines, following the TruSeq Ribo Profile Kit (RPHMR12126, Illumina) with minor modifications (see below), using one 15 cm dish of confluent mESCs per replicate. Cells were briefly pretreated with cycloheximide (0.1 mg/ml) for 2 min at 37°C and then immediately harvested by scraping down in ice‐cold PBS (supplemented with cycloheximide). The cell pellet was collected by brief centrifugation, snap‐frozen in liquid nitrogen, and stored at −80°C. From the cell pellets, lysates were prepared and ribosome‐protected mRNA fragments were generated by RNase I digestion as previously described using 5 units of RNase I per OD260 (Castelo‐Szekely et al, 2019). Of note, before RNase I digestion, mESC lysates were spiked‐in with Drosophila S2 cell lysates prepared using the same lysate buffers (spike‐in ratio 15mESC:1S2, based on OD260 measurements). After digestion, footprint‐containing monosomes were purified via MicroSpin S‐400 columns (GE Healthcare) and footprints were purified with miRNeasy Mini kit (217004 Qiagen). Fragmented RNA of 5 μg was used for ribosomal RNA removal using Ribo‐Zero Gold rRNA Removal Kit (MRZG12324 Illumina) according to Illumina's protocol for TruSeq Ribo Profile Kit (RPHMR12126, Illumina). Footprints were excised from 15% urea‐polyacrylamide gels (with single‐strand RNA oligonucleotides of 26 nt and 34 nt as size markers for excision). Sequencing libraries were generated essentially following the Illumina TruSeq Ribo Profile protocol. cDNA fragments were separated on a 10% urea‐polyacrylamide gel and gel slices between 70 and 80 nt were excised. The PCR‐amplified libraries were size‐selected on an 8% native polyacrylamide gel (footprint libraries were at ~ 150 bp). From the same initial extracts (containing the S2 lysate spike‐in), parallel RNA‐seq libraries were prepared essentially as described (PMID 30982898) and following the Illumina protocol. Briefly, after total RNA extraction using miRNeasy RNA Extraction kit (Qiagen), ribosomal RNA was depleted using Ribo‐Zero Gold rRNA (Illumina), and sequencing libraries were generated from the heat‐fragmented RNA as previously described (Castelo‐Szekely et al, 2019). All libraries were sequenced in‐house (Lausanne Genomic Technologies Facility) on a HiSeq2500 platform.
Ribosome profiling data analysis
Initial analysis, including mapping, and quantification of mRNA and footprint abundance, were performed as previously described (Castelo‐Szekely et al, 2017). Briefly, purity‐filtered reads were adapters and quality trimmed with Cutadapt v1.8 (Martin, 2011). Only reads with the expected read length (16 to 35 nt for the ribosome footprint and 35 to 60 nt for total RNA) were kept for further analysis. Reads were filtered out if they mapped to Mus musculus ribosomal RNA (rRNA) and transfer RNA (tRNA) databases (ENSEMBL v91, (Cunningham et al, 2019)) using bowtie2 v2.3.4.1 (Langmead & Salzberg, 2012). The filtered reads were aligned against Mus musculus transcripts database (ENSEMBL v91) using bowtie2 v2.3.4.1. Finally, the remaining reads were mapped against D. melanogaster transcript database (ENSEMBL v78). Reads mapping to transcripts belonging to multiple gene loci were filtered out. Reads were then summarized at a gene level using an in‐house script and mouse samples were then normalized by using the corresponding fly spike‐in read counts. Differential ribosome occupancy was assessed by DESeq2. The spike‐in normalized counts and differential expression analysis results are available in Dataset EV5.
RIP‐seq analysis
RIP‐seq data were obtained from (Ngondo et al, 2018). Reads were trimmed using Cutadapt 1.13 (Martin, 2011) with adapter TGGAATTCTCGGGTGCCAAGG and arguments “‐m 14 ‐M 40” and aligned to the mouse genome (GRCm38 primary assembly, annotation: GENCODE vM20) using STAR 2.4.2a (Dobin et al, 2013) with arguments “‐‐outFilterMismtachNoverLmax 0.05” to allow for 0 mismatches for reads < 20 bp. Next, reads were counted using subread‐featureCounts 1.5.0 (Liao et al, 2014) with arguments “‐f ‐O ‐s 1 ‐‐minOverlap 17” for miRbase v21 (Griffiths‐Jones et al, 2006). Differential loading (as compared to expression) was assessed using DESeq2 (v. 1.18.1) comparing AGO2 and AGO1 RIP‐seq versus Input (WT sRNA‐seq).
RNA‐seq analysis
Raw and normalized read counts for both genes and transcripts were computed by the RNA‐seq pipeline from snakePipes 2.3.1 (Bhardwaj et al, 2019) using the ENSEMBL GRCm38.98 primary assembly and annotation. The following command line arguments were passed “‐‐trim ‐‐trimmer trimgalore ‐‐trimmerOptions ‘‐‐illumina ‐‐paired’ ‐‐mode ‘alignment,alignment‐free,deepTools_qc ‐‐fastqc”. The RNA‐seq pipeline was further run for each miRNA_KO mESC line with an according sample sheet and the “‐‐sampleSheet” option to perform differential gene expression (DGE) analysis. Briefly, the pipeline employs TrimGalore/Cutadapt (Martin, 2011), STAR (Dobin et al, 2013), featureCounts (Liao et al, 2014), and DESeq2 (Love et al, 2014) to produce read counts and DGE data on a per‐gene basis. Salmon (Patro et al, 2017) was employed to derive per‐transcript expression.
QuantSeq analysis
In accordance with the QuantSeq manual, first, adapters were trimmed using TrimGalore with arguments “‐‐stringency 3 ‐‐illumina” then the polyA tail was trimmed using TrimGalore with argument “‐‐polyA.” Next, snakePipes (Bhardwaj et al, 2019) RNA‐seq pipeline was run with the arguments “‐‐libraryType 1 ‐‐featureCountOptions ‘‐‐primary’ ‐‐mode alignment, alignment‐free, deepTools_qc.” The RNA‐seq pipeline was further run for miR‐290‐295_KO mESC line with according to sample sheet and the additional “‐‐sampleSheet” option to perform DGE analysis.
Integration of multi‐OMICs miRNA interaction data
Retrieval and preparation of TargetScan score
Conserved and non‐conserved site context score tables were downloaded from TargetScan mouse 7.2 (Agarwal et al, 2015) and concatenated. The weighted context++ score from these data (explained in (Agarwal et al, 2015) and on the TargetScan website) is referred to as TargetScan score in this manuscript.
AGO2 binding data analysis
AGO2 binding peaks were downloaded from GEO (GSE139345) as provided by (Li et al, 2020) and miRNA seed matches (7merA1, 7merm8, and 8mer; 6mers were discarded) in peaks were identified for all mouse miRNAs (miRBase v21 (Griffiths‐Jones et al, 2006)). Seed matches were then mapped to gene regions to further associate them with a gene ID (if applicable) and the region type (5′UTR, CDS, or 3′UTR).
Integration of AGO2 binding, TargetScan, miRNA loading, and differential gene expression in miRNA_KO mESC lines
AGO2 binding data and TargetScan scores were preprocessed as explained above and reduced such that there was only a single entry for each (gene, region_type, miRNA, seed_match_type)‐tuple. In the rare cases of duplicates, the associated scores (peak size for AGO2 binding data and context++ score for TargetScan interactions) were computed as the exponentially decaying weighted sum (i.e., ). Next, prepared TargetScan scores were joined on the set of unique keys (gene, region_type, miRNA, seed_match_type) such that data entries without a match in one of the datasets were kept with missing data fields set to 0 (as in an outer‐join operation). The result was stored as integrated, unfiltered set of interactions.
Filtering and scoring of integrated interaction data
Filtering was performed in three steps. First, interactions with an AGO2‐binding value of 0 (which was the case for regions without detected peaks) were deleted. Next, interactions where the corresponding miRNA was loaded in Argonautes with less than 10 counts per million (CPM) in WT mESCs were deleted. Finally, interactions were deleted where the corresponding mRNA was not in the set of commonly upregulated genes (miRNA_KOUP ). This set was defined as those genes that passed a significance threshold (adj. P‐value < 0.2, which roughly corresponds to a P‐value of 0.05) with a positive log2FC (>0) in at least two miRNA_KO mutants. The loose significance threshold was chosen to avoid discarding functional miRNA targets with low levels of upregulation and we found it acceptable due to the increase in statistical power through the combination of multiple mutants. Filtering for negative TargetScan scores (< 0) was performed for interactions in the 3′UTR comparison, whereas interactions in the 5′UTR and CDS were discarded if the MRE showed the weak 7mer‐A1 seed type.
To allow for a confidence ranking of interactions, the mean of the four feature scores WT miRNA loading, AGO2 binding, TargetScan score, and mutant upregulation was used as the interaction score. The four scores were produced by scaling to [0, 1] after applying the log2 to the miRNA loading and AGO2 binding peak enrichment. The mutant upregulation was computed as count of mutants with statistically significant upregulation (adj. P‐value < 0.1, log2(fold‐change) > 0.5).
To allow for a ranking of genes, such that miRNA targets with the highest confidence are ranked highest, interaction scores were grouped and combined on a per‐gene basis in the following manner: the mutant upregulation score (which is logically the same for all interactions of the same gene) was added to the geometric mean of the maximum and of the sum of the three summed miRNA‐associated features. Here, the rationale was to rank genes higher if they were subject to larger numbers of interactions, however, to avoid high ranking of genes with a large number of interactions with low scores, the geometric mean dampened their score while favoring genes with large numbers of interactions and interactions with high scores.
Validation of predictions and integrative approach
The differential expression observed in the miR‐290‐295_KO was used as the basis to compare log2FC distributions for different sets of genes using CDF plots, distribution mean differences, and Kolmogorov–Smirnov tests. The control set of expressed genes was established using a TPM‐expression filter of 1. The gene set reflecting the integrative approach of this paper was defined by filtering the 360 miR‐290‐295‐targeted genes out of the 759 miRNA target genes that have been identified in the integrative analysis of this paper (Fig 1B). The other gene sets were generated and compared to demonstrate the relevance of the integration of multiple datasets for the high prediction accuracy. Where applicable, gene sets were purposefully sized to contain the same number of genes (360) as the miR‐290‐295‐filtered integrative approach gene set. Except from the integrative approach gene set, for the other gene sets miR‐290‐295‐filtering was performed more stringently such as to only include the strongly expressed members miR‐291‐3p, miR‐294‐3p, miR‐295‐3p, which all share the same seed sequence. The TargetScan targets were filtered for those three strongly expressed members of the miR‐290‐295 cluster and genes were ranked by TargetScan interaction scores (context++ score). AGO2‐binding targets were first filtered by seed matches to the three miR‐290‐295 members. Then the genes with the strongest AGO2‐binding signals were selected. The miRNA_KO‐upregulated genes were the commonly upregulated genes used in the integrative analysis and defined in the Methods section. Filtered‐up genes were the miRNA_KOUP genes filtered for the three miR‐290‐295 members. The Interaction score‐based set is based on target gene grouping of unfiltered, but scored (interaction score as previously defined) interactions, and ranking of the genes using the following aggregation: , where is the set of interaction scores for all interactions of a given gene. Lowly upregulated genes are defined as genes with a minimal expression of 1 TPM and a log2FC between 0.1 and 0.5 in all three mutants. Low‐up genes + integrative filtering used the same set of genes but filtered for AGO2‐binding to one of miR‐290‐295 members.
HEAP and CLIP coverage analyses in mESCs and hESCs
AGO2‐binding data in mESCs (Halo‐enhanced AGO2‐pulldown (HEAP) and iCLIP‐seq approaches) were obtained from GEO (GSE61348: Bosson et al, 2014; GSE139345: Li et al, 2020). For the iCLIP‐seq data, peaks were lifted from mm9 to mm10 annotation. AGO2 PAR‐CLIP‐seq data in hESCs were obtained from SRA (SRR359787: Lipchina et al, 2011) and processed using the snakePipes ChIP‐seq pipeline with default parameters and the GRCh38 Gencode 29 annotation. HESC miRNA expression was obtained from (Hinton et al, 2014). Human‐mouse ortholog genes were obtained from ENSEMBL BioMart (Smedley et al, 2015).
Conservation of miRNA interactions in human
The hESC PAR‐CLIP data (Lipchina et al, 2011) is of comparably low resolution and was likely not to lead to representative peak calling results. Instead, we used this data to test whether our mESC‐predicted interactions showed conservation through AGO2‐binding in hESCs. Potentially conserved interactions were identified by scanning for interaction‐specific seed matches in the 3′UTRs of the interaction's human gene orthologs. Such identified potentially conserved interactions were associated with the number of PAR‐CLIP read counts at the human MRE and with the hESC miRNA expression (Hinton et al, 2014) on a per‐miRNA family basis (given our seed‐based analysis). Three different negative control sets were used: (i) MREs/interactions that were lacking miRNA expression in hESCs, (ii) MREs in human orthologs matching randomly chosen seed sequences (from the set of seeds from mESC predicted interactions), (iii) MREs in human orthologs matching scrambled seed sequences. MREs identified in human that corresponded to the same seed sequence found in the mouse ortholog along with miRNA expression above 10 CPM and detected AGO2‐binding signal at the MRE site were considered as functionally conserved.
Analysis of cooperative miRNA effects
The number of MREs/interactions per gene was correlated with different metrics including the number of HEAP peaks, the mean HEAP peak intensity, and differential ribosome occupancy. These correlation analyses were performed for two groups of interactions. Filtered interactions as obtained from our integrative analysis and unfiltered interactions as derived from TargetScan interactions (but still filtered for mESC miRNA loading).
The number of MREs/interactions was also associated with the upregulation observed in miR‐290‐295_KO on a per‐gene basis, where interactions were filtered for miR‐290‐295 family members.
Gene ontology analysis of predicted miRNA targets
Predicted miRNA targets, commonly upregulated (described above) and commonly downregulated (defined same as their upregulation counterpart, but with negative log2FCs) genes were subjected to a gene set enrichment analysis using the gseapy python library on the gene set libraries BioPlanet 2019, WikiPathways 2019, KEGG 2019, GO Biological Processes 2021, GO Cellular Components 2021 and GO Molecular Function 2021. The visualization was performed customarily with seaborn and matplotlib.
Gene ontology analysis of Tfap4 targets
Potential Tfap4‐targets (as determined from their degree of rescue in the siPOOL‐treated cells) were further filtered for TFAP4‐binding sites in the genome. The PWMScan website (Ambrosini et al, 2018) was used with default parameters. The GRCm38/mm10 genome was selected and scanned for human CIS‐BP TFAP4 binding motifs. The resulting bed file was downloaded and used to select those genes that had a binding motif closely upstream to their transcription start site (< 1 kb distance). Gene ontology analysis was performed for these genes using ClueGO (Bindea et al, 2009) with the following options: Network specificity was set to medium‐1, GO term fusion was enabled, only pathways/terms with PV < 0.05 were shown and terms from WikiPathways, KEGG, GO Biological Processes, GO Cellular Components and GO Molecular Function from 2021/05/13 were used.
Custom data analyses, visualizations
Data analyses and visualizations were realized as described in the last sections using bash and python scripting, organized in a snakemake pipeline (Mölder et al, 2021). PCA analysis was performed using scikit‐learn (Pedregosa et al, 2011).
Author contributions
Moritz Schaefer: Conceptualization; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft. Amena Nabih: Conceptualization; formal analysis; supervision; validation; investigation; visualization; methodology; writing – original draft. Daniel Spies: Conceptualization; software; formal analysis; supervision; methodology. Victoria Hermes: Formal analysis; validation; investigation; visualization; methodology. Maxime Bodak: Formal analysis; supervision; validation; investigation; methodology. Harry Wischnewski: Formal analysis; validation; investigation; methodology. Patrick Stalder: Validation; investigation. Richard Patryk Ngondo: Supervision; validation; investigation; methodology. Luz Angelica Liechti: Formal analysis; investigation; methodology. Tatjana Sajic: Formal analysis; investigation; methodology. Ruedi Aebersold: Resources; supervision; methodology. David Gatfield: Resources; supervision; methodology. Constance Ciaudo: Conceptualization; resources; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
MS, DS, AN, and CC carried out conceptualization. MS, AN, VH, MB, HW, PS, and RPN were involved in laboratory experiments. TS carried out full proteome analysis. AL carried out ribo‐seq libraries preparation. MS and DS carried out computational analysis. MS, AN, and CC involved in writing—original draft preparation. RA and DG did expertise and editing. MS, AN, MB, and CC carried out visualization. CC carried out funding acquisition, supervision, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Source Data for Expanded View
Acknowledgements
We thank the members of the Ciaudo lab and Dr. Tobias Beyer for fruitful discussions and the critical reading of this manuscript. We are grateful to Bulak Arpat for help at the beginning of computational analyses of the Ribo‐seq datasets. This work was supported by the Swiss National Science Foundation (grants 31003A_173120 and 310030_196861) to C.C. C.C, H.W., and D.G. were supported by the NCCR RNA and Disease (grant 182880). We also thank the Functional Genomics Center Zurich (FGCZ) for their support with the preparation of RNA‐seq libraries and sequencing. Open access funding provided by Eidgenossische Technische Hochschule Zurich.
EMBO reports (2022) 23: e54762
Data availability
The datasets and computer code produced in this study are available in the following databases:
RNA‐seq data: Gene Expression Omnibus for Ago2&1_KO mESCs GSE110942 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110942)
QuantSeq data: Gene Expression Omnibus for mESC WT and miR‐290‐295_KO + siPools GSE181393 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181393)
Ribosome Profiling data: Gene Expression Omnibus GSE135577 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE135577)
Proteomic data: ProteomeXchange PXD014484 (http://www.ebi.ac.uk/pride/archive/projects/PXD014484)
Integrative analysis source code: GitHub (https://github.com/moritzschaefer/mesc‐regulation_pub)
RIP‐seq analysis pipeline: GitHub (https://github.com/moritzschaefer/srna‐seq)
RNA‐seq and QuantSeq analysis pipelines: GitHub (https://github.com/moritzschaefer/snakepipelines_pub)
Auxiliary scripts and functions: GitHub (https://github.com/moritzschaefer/moritzsphd_pub)
References
- Agarwal V, Bell GW, Nam J, Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: 1–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini G, Groux R, Bucher P (2018) PWMScan: a fast tool for scanning entire genomes with a position‐specific weight matrix. Bioinformatics 34: 2483–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtina NA, Müller M, Wischnewski H, Arora R, Ciaudo C (2021) 3D synthetic microstructures fabricated by two‐photon polymerization promote homogeneous expression of NANOG and ESRRB in mouse embryonic stem cells. Adv Mater Interfaces 8: 2001964 [Google Scholar]
- Bartel DP (2018) Metazoan microRNAs. Cell 173: 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363–366 [DOI] [PubMed] [Google Scholar]
- Bhardwaj V, Heyne S, Sikora K, Rabbani L, Rauer M, Kilpert F, Richter AS, Ryan DP, Manke T (2019) SnakePipes: facilitating flexible, scalable and integrative epigenomic analysis. Bioinformatics 35: 4757–4759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25: 1091–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattmann P, Heusel M, Aebersold R (2016) SWATH2stats: an R/bioconductor package to process and convert quantitative SWATH‐MS proteomics data for downstream analysis tools. PLoS One 11: e0153160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodak M, Ciaudo C (2016) Monitoring long interspersed nuclear element 1 expression during mouse embryonic stem cell differentiation. Methods Mol Biol 1400: 237–259 [DOI] [PubMed] [Google Scholar]
- Bodak M, Cirera‐Salinas D, Luitz J, Ciaudo C (2017a) The role of RNA interference in stem cell biology: beyond the mutant phenotypes. J Mol Biol 429: 1532–1543 [DOI] [PubMed] [Google Scholar]
- Bodak M, Cirera‐Salinas D, Yu J, Ngondo RP, Ciaudo C (2017b) Dicer, a new regulator of pluripotency exit and LINE‐1 elements in mouse embryonic stem cells. FEBS Open Bio 7: 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosson AD, Zamudio JR, Sharp PA (2014) Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56: 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Weissman JS (2015) Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16: 651–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briskin D, Wang PY, Bartel DP (2020) The biochemical basis for the cooperative action of microRNAs. Proc Natl Acad Sci U S A 117: 17764–17774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Seila AC, Yeo GW, Sharp PA (2007) RNA sequence analysis defines Dicer's role in mouse embryonic stem cells. Proc Natl Acad Sci U S A 104: 18097–18102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo‐Szekely V, Arpat AB, Janich P, Gatfield D (2017) Translational contributions to tissue specificity in rhythmic and constitutive gene expression. Genome Biol 18: 116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo‐Szekely V, De Matos M, Tusup M, Pascolo S, Ule J, Gatfield D (2019) Charting DENR‐dependent translation reinitiation uncovers predictive uORF features and links to circadian timekeeping via Clock. Nucleic Acids Res 47: 5193–5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J et al (2012) A cross‐platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 30: 918–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell J, Dalton S (2013) Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med 3: a014381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry‐based proteomic experiments. Bioinformatics 30: 2524–2526 [DOI] [PubMed] [Google Scholar]
- Choi N, Park J, Lee JS, Yoe J, Park GY, Kim E, Jeon H, Cho YM, Roh TY, Lee Y (2015) miR‐93/miR‐106b/miR‐375‐CIC‐CRABP1: a novel regulatory axis in prostate cancer progression. Oncotarget 6: 23533–23547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C‐H, Shrestha S, Yang C‐D, Chang N‐W, Lin Y‐L, Liao K‐W, Huang W‐C, Sun T‐H, Tu S‐J, Lee W‐H et al (2018) miRTarBase update 2018: a resource for experimentally validated microRNA‐target interactions. Nucleic Acids Res 46: D296–D302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Kilikevicius A, Liu J, Johnson KC, Yokota S, Corey DR (2020) Argonaute binding within 3′‐untranslated regions poorly predicts gene repression. Nucleic Acids Res 48: 7439–7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaudo C, Servant N, Cognat V, Sarazin A, Kieffer E, Viville S, Colot V, Barillot E, Heard E, Voinnet O (2009) Highly dynamic and sex‐specific expression of microRNAs during early ES cell differentiation. PLoS Genet 5: e1000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirera‐Salinas D, Yu J, Bodak M, Ngondo RP, Herbert KM, Ciaudo C (2017) Noncanonical function of DGCR8 controls mESC exit from pluripotency. J Cell Biol 216: 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B et al (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25: 1422–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, Bennett R, Bhai J, Billis K, Boddu S et al (2019) Ensembl 2019. Nucleic Acids Res 47: D745–D751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cursons J, Pillman KA, Scheer KG, Gregory PA, Foroutan M, Hediyeh‐Zadeh S, Toubia J, Crampin EJ, Goodall GJ, Bracken CP et al (2018) Combinatorial Targeting by MicroRNAs Co‐ordinates Post‐transcriptional Control of EMT. Cell Syst 7: 77–91.e7 [DOI] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, Quinlan AR (2011) Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27: 3423–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVeale B, Swindlehurst‐Chan J, Blelloch R (2021) The roles of microRNAs in mouse development. Nat Rev Genet 22: 307–323 [DOI] [PubMed] [Google Scholar]
- Ding C, Chan DW, Liu W, Liu M, Li D, Song L, Li C, Jin J, Malovannaya A, Jung SY et al (2013) Proteome‐wide profiling of activated transcription factors with a concatenated tandem array of transcription factor response elements. Proc Natl Acad Sci U S A 110: 6771–6776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP (2007) Target‐decoy search strategy for increased confidence in large‐scale protein identifications by mass spectrometry. Nat Methods 4: 207–214 [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R (2012) Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosline SJC, Gurtan AM, Jean‐Baptiste CK, Bosson A, Milani P, Dalin S, Matthews BJ, Yap YS, Sharp PA, Fraenkel E (2016) Elucidating microRNA regulatory networks using transcriptional, post‐transcriptional, and histone modification measurements. Cell Rep 14: 310–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths‐Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34: D140–D144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A‐C, Munschauer M et al (2010) Transcriptome‐wide identification of RNA‐binding protein and microRNA target sites by PAR‐CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ et al (2020) Array programming with NumPy. Nature 585: 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser J, Syed AP, Bilen B, Zavolan M (2013) Analysis of CDS‐located miRNA target sites suggests that they can effectively inhibit translation. Genome Res 23: 604–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton A, Hunter SE, Afrikanova I, Jones GA, Lopez AD, Fogel GB, Hayek A, King CC (2014) SRNA‐seq analysis of human embryonic stem cells and definitive endoderm reveals differentially expressed micrornas and novel isomirs with distinct targets. Stem Cells 32: 2360–2372 [DOI] [PubMed] [Google Scholar]
- Houbaviy HB, Murray MF, Sharp PA (2003) Embryonic stem cell‐specific MicroRNAs. Dev Cell 5: 351–358 [DOI] [PubMed] [Google Scholar]
- Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9: 90–95 [Google Scholar]
- Id CS, La TCD, Parveen A, Gretz N (2018) miRWalk: an online resource for prediction of microRNA binding sites. PLoS One 13: e0206239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, Röh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm G, Kirchner T, Menssen A et al (2013) AP4 is a mediator of epithelial‐mesenchymal transition and metastasis in colorectal cancer. J Exp Med 210: 1331–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay F, Ciaudo C (2013) An RNA tool kit to study the status of mouse ES cells: sex determination and stemness. Methods 63: 85–92 [DOI] [PubMed] [Google Scholar]
- Jung P, Menssen A, Mayr D, Hermeking H (2008) AP4 encodes a c‐MYC‐inducible repressor of p21. Proc Natl Acad Sci U S A 105: 15046–15051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C, Gilpatrick T, Kilaru G, Burr P, Nguyen CK, Morawski A, Lenardo MJ, Muljo SA (2015) Reprogramming of polycomb‐mediated gene silencing in embryonic stem cells by the miR‐290 family and the Methyltransferase Ash1l. Stem Cell Reports 5: 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Köster J, Forster J, Schmeier S & Salazar V (2021) snakemake‐workflows/rna‐seq‐star‐deseq2: Version 1.2.0, Zenodo. 10.5281/zenodo.5245549 [DOI]
- Lai EC (2015) Two decades of miRNA biology: lessons and challenges. RNA 21: 675–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MC, Ruiz‐Velasco M, Arnold C, Sigalova O, Gavin A‐C, Zaugg J (2021) Enhancer‐priming in ageing human bone marrow mesenchymal stromal cells contributes to immune traits. bioRxiv 10.1101/2021.09.03.458728 [PREPRINT] [DOI] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002) MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih I‐H, Jones‐Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20 [DOI] [PubMed] [Google Scholar]
- Li X, Pritykin Y, Concepcion CP, Lu Y, La Rocca G, Zhang M, King B, Cook PJ, Au YW, Popow O et al (2020) High‐resolution in vivo identification of miRNA targets by halo‐enhanced Ago2 pull‐down. Mol Cell 79: 167–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Lipchina I, Elkabetz Y, Hafner M, Sheridan R, Mihailovic A, Tuschl T, Sander C, Studer L, Betel D (2011) Genome‐wide identification of microRNA targets in human ES cells reveals a role for miR‐302 in modulating BMP response. Genes Dev 25: 2173–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Delisle L, Rabbani L, Wolff J, Bhardwaj V, Backofen R, Grüning B, Ramírez F, Manke T (2021) pyGenomeTracks: reproducible plots for multivariate genomic datasets. Bioinformatics 37: 422–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Anders S, Huber W (2014) Differential analysis of count data ‐ the DESeq2 package. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J et al (2008) Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134: 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet 17 10–12 [Google Scholar]
- McGeary SE, Lin KS, Shi CY, Pham TM, Bisaria N, Kelley GM, Bartel DP (2019) The biochemical basis of microRNA targeting efficacy. Science 366: eaav1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnery, W (2010) Data structures for statistical computing in python. In Proceedings of the 9th Python in Science Conference, Vol. 445, No. 1, pp 51–56 [Google Scholar]
- Medeiros LA, Dennis LM, Gill ME, Houbaviy H, Markoulaki S, Fu D, White AC, Kirak O, Sharp PA, Page DC et al (2011) Mir‐290‐295 deficiency in mice results in partially penetrant embryonic lethality and germ cell defects. Proc Natl Acad Sci U S A 108: 14163–14168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölder F, Jablonski KP, Letcher B, Hall MB, Tomkins‐Tinch CH, Sochat V, Forster J, Lee S, Twardziok SO, Kanitz A et al (2021) Sustainable data analysis with Snakemake. F1000Res 10: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G (2002) miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngondo RP, Cirera‐Salinas D, Yu J, Wischnewski H, Bodak M, Vandormael‐Pournin S, Geiselmann A, Wettstein R, Luitz J, Cohen‐Tannoudji M et al (2018) Argonaute 2 is required for extra‐embryonic endoderm differentiation of mouse embryonic stem cells. Stem Cell Reports 10: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Jo MH, Choi Y‐G, Park J, Kwon SC, Hohng S, Kim VN, Woo J‐S (2015) Functional anatomy of the human microprocessor. Cell 161: 1374–1387 [DOI] [PubMed] [Google Scholar]
- Oliveira AC, Bovolenta LA, Nachtigall PG, Herkenhoff ME (2017) Combining results from distinct microRNA target prediction tools enhances the performance of analyses. Front Genet 8: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papathanasiou M, Tsiftsoglou SA, Polyzos AP, Papadopoulou D, Valakos D, Klagkou E, Karagianni P, Pliatska M, Talianidis I, Agelopoulos M et al (2021) Identification of a dynamic gene regulatory network required for pluripotency factor‐induced reprogramming of mouse fibroblasts and hepatocytes. EMBO J 40: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, West JD, Jiang Y, Fogarty EA, Grimson A (2020) Robust partitioning of microRNA targets from downstream regulatory changes. Nucleic Acids Res 48: 9724–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 14: 417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B (2011) Scikit‐learn: machine learning in Python. F1000Res 10: 33 [Google Scholar]
- Pinzon N, Li B, Martinez L, Sergeeva A, Presumey J, Apparailly F, Seitz H (2017) MicroRNA target prediction programs predict many false positives. Genome Res 27: 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reczko M, Maragkakis M, Alexiou P, Grosse I, Hatzigeorgiou AG (2012) Functional microRNA targets in protein coding sequences. Bioinformatics 28: 771–776 [DOI] [PubMed] [Google Scholar]
- Röst HL, Rosenberger G, Navarro P, Gillet L, Miladinoviä SM, Schubert OT, Wolski W, Collins BC, Malmström J, Malmström L et al (2014) OpenSWATH enables automated, targeted analysis of data‐independent acquisition MS data. Nat Biotechnol 32: 219–223 [DOI] [PubMed] [Google Scholar]
- Schäfer M, Ciaudo C (2020) Prediction of the miRNA interactome ‐ established methods and upcoming perspectives. Comput Struct Biotechnol J 18: 548–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert O, Gillet L, Collins B, Navarro P, Rosenberger G, Wolski W, Lam H, Amodei D, Mallick PB, MacLean B et al (2015) Building high‐quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc 10: 426–441 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, Mendoza L, Moritz RL, Aebersold R, Nesvizhskii AI (2011) iProphet: multi‐level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics 10: M111.007690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G et al (2015) The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res 43: W589–W598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J‐J, Smith SK, Hannon GJ, Joshua‐Tor L (2004) Crystal structure of argonaute and its implications for RISC Slicer activity. Science 305: 1434–1437 [DOI] [PubMed] [Google Scholar]
- Song J, Xie C, Jiang L, Wu G, Zhu J, Zhang S, Tang M, Song L, Li J (2018) Transcription factor AP‐4 promotes tumorigenic capability and activates the Wnt/β‐catenin pathway in hepatocellular carcinoma. Theranostics 8: 3571–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM (2012) Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626 [DOI] [PubMed] [Google Scholar]
- Sugawara T, Miura T, Kawasaki T, Umezawa A, Akutsu H (2020) The hsa‐miR‐302 cluster controls ectodermal differentiation of human pluripotent stem cell via repression of DAZAP2. Regen Ther 15: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Wang H, Sun H (2017) MTFkb: a knowledgebase for fundamental annotation of mouse transcription factors. Sci Rep 7: 3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacheny A, Michel S, Dieu M, Payen L, Arnould T, Renard P (2012) Unbiased proteomic analysis of proteins interacting with the HIV‐1 5′LTR sequence: Role of the transcription factor Meis. Nucleic Acids Res 40: e168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JY, Abdulkarim B, Marques AC (2020) Noncanonical targeting contributes significantly to miRNA‐mediated regulation. bioRxiv 10.1101/2020.07.07.191023 [PREPRINT] [DOI] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J et al (2020) SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 17: 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R (2008) Embryonic stem cell–specific microRNAs regulate the G1‐S transition and promote rapid proliferation. Nat Genet 40: 1478–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X‐WW, Hao J, Guo W‐TT, Liao L‐QQ, Huang S‐YY, Guo X, Bao X, Esteban MA, Wang Y (2017) A DGCR8‐independent stable microrna expression strategy reveals important functions of miR‐290 and miR‐183–182 families in mouse embryonic stem cells. Stem Cell Reports 9: 1618–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein R, Bodak M, Ciaudo C (2016) Generation of a knockout mouse embryonic stem cell line using a paired CRISPR/Cas9 genome engineering tool. Methods Mol Biol 1341: 321–343 [DOI] [PubMed] [Google Scholar]
- Wong MMK, Joyson SM, Hermeking H, Chiu SK (2021) Transcription factor AP4 mediates cell fate decisions: to divide, age, or die. Cancer 13: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan K, Ai WB, Wan LY, Tan X, Wu JF (2017) The miR‐290‐295 cluster as multi‐faceted players in mouse embryonic stem cells. Cell Biosci 7: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Cui G, Hu S, Zhang Z, Yang C, Liu Z, Wang L, Li C, Sun M (2020) Graph neural networks: a review of methods and applications. AI Open 1: 57–81 [Google Scholar]