

SUMMARY
Metastatic colonization is the primary cause of death from colorectal cancer (CRC). We employed genome-scale in vivo short hairpin RNA (shRNA) screening and validation to identify 26 promoters of CRC liver colonization. Amongst these genes, we identified a cluster containing multiple targetable genes including ITPR3 that promoted liver metastatic colonization and elicited similar downstream gene expression programs. ITPR3 is a caffeine-sensitive inositol 1,4,5-triphosphate (IP3) receptor that releases calcium from the endoplasmic reticulum and enhanced metastatic colonization by inducing expression of RELB, a transcription factor associated with non-canonical NF-κB signaling. Genetic, cell biological, pharmacologic, and clinical association studies revealed that ITPR3 and RELB drive CRC colony formation by promoting cell survival upon substratum detachment or hypoxic exposure. RELB was sufficient to drive colonization downstream of ITPR3. Our findings implicate the ITPR3/calcium/RELB axis in CRC metastatic colony formation and uncover multiple clinico-pathologically associated targetable proteins as drivers of CRC metastatic colonization.
Keywords: Colorectal cancer, metastasis, ITPR3, calcium, RELB, non-canonical NF-κB, hypoxia
eTOC Blurb
Moy et al. define a clinically relevant pathway that promotes colorectal cancer cell survival and liver metastasis via the type 3 inositol 1,4,5-triphosphate receptor ITPR3, a calcium channel that controls calcium release from the endoplasmic reticulum, and RELB, a transcription factor associated with non-canonical NF-κB signaling.
Graphical Abstract
INTRODUCTION
Colorectal cancer (CRC) is the second leading cause of cancer mortality worldwide (Sung et al., 2021), and as is the case for most solid tumors, metastatic progression is the overwhelming cause of CRC death (Lambert et al., 2017). While modern treatment regimens have modestly improved outcomes, long-term survival in metastatic CRC remains poor, with less than 15 percent of patients alive at five years (Siegel et al., 2020). Therefore, a better understanding of the cellular and molecular mechanisms driving CRC metastasis is warranted.
The liver is the most common site of metastasis in CRC, and over 50% of patients will harbor liver metastases at diagnosis or during their treatment course (Engstrand et al., 2018; Zarour et al., 2017). The establishment of metastases depends on a series of complex events termed the invasion-metastasis cascade (Lambert et al., 2017; Talmadge and Fidler, 2010). CRC cells transit to the liver via the portal circulation and encounter permeable fenestrated sinusoids, suggesting that extravasation is not a major barrier to dissemination (Braet and Wisse, 2002). Rather, physiologic barriers posed by the metastatic microenvironment dictate the ability of CRC cells to colonize the liver and other tissues. Prior studies revealed that colonization represents a major bottleneck to metastatic progression, with less than 0.01% of tumor cells having the capacity to form metastatic colonies in the liver (Chambers et al., 2002; Luzzi et al., 1998). However, the fundamental genes and pathways that regulate metastatic initiation within the liver microenvironment are poorly defined.
To discover key regulators of CRC metastasis, we performed a systematic large-scale short hairpin (shRNA) screen using an in vivo mouse model of CRC liver colonization. A preliminary focused analysis identified liver and red blood cell pyruvate kinase (PKLR) as a metastasis promoter that promotes metastatic colonization through induction of the antioxidant metabolite glutathione (Nguyen et al., 2016). Here, we present full results from this screen, which yielded a list of 26 candidate genes that promote CRC liver colonization. While some of these genes have been linked to tumorigenesis in certain contexts, their roles in metastatic colonization are poorly characterized. By individually silencing these 26 genes and performing mRNA sequencing (RNA-seq), we found that transcriptional changes elicited by the depletion of type 3 inositol 1,4,5-triphosphate (IP3) receptor (ITPR3), a calcium channel and receptor that mediates release of calcium from the endoplasmic reticulum (ER) (Mangla et al., 2020), are correlated with clinical outcomes of CRC patients. We validated that loss of ITPR3 in multiple CRC models substantially reduces liver metastatic burden in vivo, defining ITPR3 as a critical driver of metastatic colonization. We thus focused our efforts on elucidating the mechanisms by which ITPR3 promotes liver metastatic progression and evaluating its potential as a therapeutic target.
RESULTS
Genome-scale in vivo shRNA screen identifies top drivers of CRC metastatic colonization
To elucidate the molecular programs that CRC utilizes during metastatic colonization, we performed a genome-scale in vivo shRNA screen using three different human CRC cell lines (LS174T, SW620, WiDr) (Nguyen et al., 2016). Cells were transduced with shRNA-encoding lentiviruses containing 54,591 hairpins targeting 14,095 genes, and in parallel, cultured in vitro or directly injected into mouse livers to select for cells that are efficient in colonization. ShRNA inserts were sequenced from liver tumors or cultured cells, with loss of a particular shRNA suggesting that silencing of that gene had impaired liver colonization or cell growth. This primary screen identified 556 gene hits that putatively promote liver colonization to varying degrees (Figure 1A, Table S1). To obviate the concern for RNA interference off-target effects and increase confidence in these genes, we performed a secondary screen targeting the top 209 genes (Table S2) from our primary screen with two new independent shRNAs in highly metastatic LS174T-LvM3b cells. From this list, we focused on the top 26 genes that exhibited the greatest magnitude of shRNA depletion in vivo (Figure 1A-B). To verify that the candidate genes regulate metastatic colonization, we individually silenced several of these genes in luciferase-expressing CRC cells by shRNAs and monitored metastatic liver burden after intrasplenic injection, which models the delivery of CRC cells from the portal circulation. Depletion of several of these putative liver colonization genes including RELB, DDR2, UBR4, PLG, and GPR56 suppressed metastatic colonization in SW480 (Figure 1C) and LS174T-LvM3b cells (Figure 1D), validating the screen’s identification of bona fide metastasis promoters.
Figure 1. Genome-scale in vivo shRNA screen identifies top drivers of CRC metastatic colonization.

The top 26 liver colonization genes as defined by strongest shRNA depletion in the secondary screen are shown. (A) Human CRC lines (LS174T, WiDr, SW620) were transduced with a genome-scale shRNA library and cultured in vitro or directly injected into the liver. ShRNAs were sequenced from cells or liver tumors, and their abundance was compared to the initial population. (B) The top 209 genes which dropped out from the in vivo primary screen were selected for a secondary screen including two new shRNAs in a highly metastatic CRC cell line, LS174T-LvM3b. Heatmap indicates z-score depletion (A) or log-ratio relative to original representation (B). Yellow number indicates the number of hairpins that were absent after selective pressure (A) or depleted to less than 2-logs (B). (C,D) 5×105 SW480 cells (C) or LS174T-LvM3b cells (D) expressing individual shRNAs targeting the indicated gene were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence after 21 days (n=5). Values were normalized to median of control bioluminescence; *p<0.05, **p<0.01 ***p<0.001, Student’s t-test. (E) SW480 cells were transfected with control siRNA or siRNA targeting each of the 26 liver colonization genes in duplicate, cultured for 72 hours and processed for RNA-seq. UMAP plot showing distances for transcriptomes of each gene knockdown. (F,G) Kaplan-Meier analysis of progression-free survival from TCGA data comparing patients with primary tumor transcriptional profile positively correlating (gene-low) or negatively correlating (gene-high) with the gene signature from the indicated knockdown for ITPR3 (F) or PLK4 (G). See also Table S1 and S2.
To systematically define the downstream transcriptional programs regulated by the 26 liver colonization genes, we performed mRNA sequencing (RNA-seq) on SW480 cells individually transfected with siRNAs targeting each gene. Interestingly, uniform manifold approximation and projection (UMAP) analysis depicting the distances separating the transcriptome of each gene knockdown (KD) revealed that subsets of genes clustered together, suggesting that they may function in similar pathways (Figure 1E). We focused our attention on a cluster containing several genes including PLK4, GPR56, ITPR3, RELB, and DDR2, as these genes exhibited distinct transcriptomic profiles relative to control cells and several representatives were individually validated as metastasis promoters (Figure 1C-D).
We leveraged comprehensive mRNA expression analysis of CRC patients from The Cancer Genome Atlas (TCGA) to determine if transcriptional alterations caused by these genes associate with clinical outcomes (Cancer Genome Atlas Network, 2012). Of this set, we identified ITPR3 and PLK4 as genes whose depletion was sufficient to elicit genes expression changes that correlate with progression-free survival (Figure 1F-G). Overall, these data reveal that genes identified as liver colonization promoters enhance metastatic colonization and identified ITPR3 and PKL4 as genes that regulate downstream transcriptional programs associated with patient survival.
ITPR3 promotes metastatic liver colonization in CRC
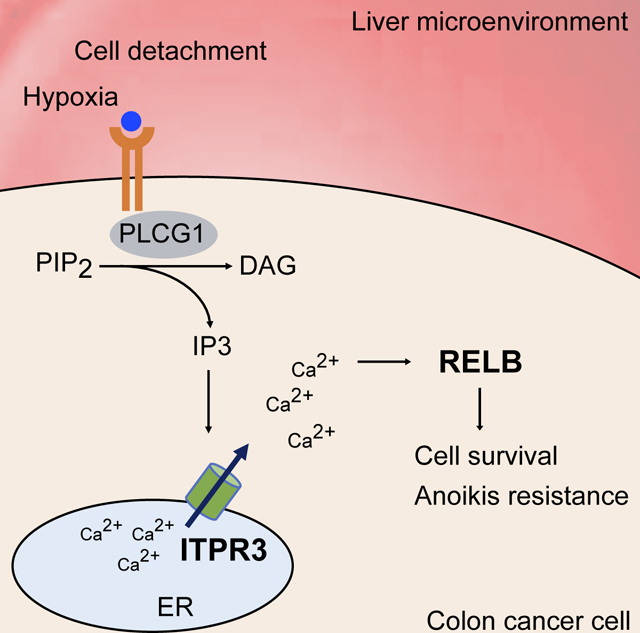
Given correlations with clinical outcome in patients with CRC, we focused further studies on ITPR3, which mobilizes calcium from the ER (Mangla et al., 2020). ITPR3 channel opening is activated by IP3, which is generated by phospholipase C (PLC) enzymes that hydrolyze phosphatidylinositol 4,5-bisphosphonate (PIP2) to IP3 and diacylglycerol in response to G protein-coupled receptor (GPCR) or receptor tyrosine kinase (RTK) signaling. Interestingly, compared to the two other ITPR subtypes, ITPR3 is selectively expressed in human primary CRC tumors, and high ITPR3 expression at the invasive margin is clinically associated with increased liver metastasis and poor overall survival (Shibao et al., 2010). However, the mechanism by which ITPR3 promotes CRC metastasis is not defined.
To assess ITPR3 expression in CRC liver metastases, we analyzed publicly available microarray or RNA-seq datasets which also included samples from normal mucosa and primary tumors (Kim et al., 2014; Sheffer et al., 2009). We observed higher ITPR3 expression in primary tumors compared to normal colon, with further increased ITPR3 expression in liver metastases (Figure 2A-B). In contrast, transcript levels of ITPR1 and ITPR2 were decreased in primary tumors and liver metastases (Figure 2C-D). Thus, among the known IP3 receptors, ITPR3 may play a selective role in regulating CRC progression.
Figure 2. ITPR3 promotes metastatic liver colonization in CRC.

(A) ITPR3 expression as measured by microarray from normal colon, CRC primary tumors or liver metastases (GSE41258). (B) ITPR3 expression as measured by RNA-seq (GSE50760). (C,D) ITPR1 (C) or ITPR2 (D) expression as measured by microarray (GSE41258). (E) ITPR3 expression by immunoblot in CRISPR-edited sgCTRL or sgITPR3 SW480 cells. (F) 5 × 105 SW480 cells were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence at the indicated timepoints (n=8 mice). Representative liver bioluminescence and gross pathology are shown. (G) Immunoblot for ITPR3 protein in LS174T cells expressing the indicated shRNAs. (H) 5 × 105 LS174T cells were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=5 mice). (I) Itpr3 protein expression by immunoblot in CRISPR-edited sgCTRL or sgItpr3 MC38 cells. (J) 5 × 105 MC38 cells were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=10–11 mice). Median (A-D) or mean ± SEM (F,H,J); *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, Mann-Whitney test. See also Figure S1.
To define the function of ITPR3 in metastatic colonization in vivo and further validate our shRNA screen results, we used CRISPR/Cas9 to delete ITPR3 in SW480 CRC cells and injected these cells into the portal circulation of mice (Figure 2E and S1A). ITPR3 knockout (KO) cells exhibited significantly reduced capacity to form liver metastases relative to control cells (Figure 2F and S1B). We observed a significant decrease in both the number and size of metastatic tumor nodules with loss of ITPR3 (Figure S1C-E). In addition, we knocked down ITPR3 in a second human CRC cell line (LS174T) (Figure 2G) and similarly found that ITPR3 depletion decreased metastatic liver colonization (Figure 2H and S1F). Consistent with our results in human cells, ITPR3 deletion in mouse CRC cells (MC38) also significantly reduced liver metastasis in an immunocompetent model, including both the number and size of metastases (Figure 2I-J and S1G-I). In contrast to liver metastasis, ITPR3 loss did not affect lung metastasis after tail vein injection, suggesting a selective role for ITPR3 in metastatic liver colonization (Figure S1J). To evaluate the role of ITPR3 in primary tumor growth, we implanted control or ITPR3 KO cells into the flanks of mice. Loss of ITPR3 reduced subcutaneous xenograft growth in the SW480 (Figure S1K), albeit to a lesser extent than metastatic liver tumor burden, while we observed no change in MC38 subcutaneous xenograft growth (Figure S1L). Taken together, these studies implicate ITPR3 as a key promoter of CRC liver colonization.
Because ITPR3 mediates calcium release from ER stores into the cytoplasm, we hypothesized that cytoplasmic calcium accumulation promotes metastatic colonization. To test this, we inhibited intracellular calcium by pretreating CRC cells with the cell-permeable calcium chelator BAPTA-AM or vehicle control prior to portal circulation injection. Transient calcium chelation significantly reduced liver metastasis (Figure S1M), revealing that cytosolic calcium facilitates an early stage of liver colonization. These findings support a model whereby increased metabolic accumulation of IP3 and its activation of ITPR3 on the ER enhances cytosolic calcium, thereby promoting the initiation of CRC metastatic liver colonization.
Genes implicated in IP3 signaling and metabolism regulate metastatic CRC liver colonization
To further confirm whether ITPR3 signaling regulates metastatic colonization, we interrogated genes involved in IP3 metabolism (Figure 3A). PLC enzymes generate IP3 from PIP2 (Kadamur and Ross, 2013), and interestingly we observed increased expression of one of these genes (PLCG1) in liver metastases relative to primary tumors and normal colonic tissue (Figure 3B and S2A). ShRNA-mediated depletion of PLCG1 in SW480 cells (Figure S2B) markedly inhibited metastatic liver colonization (Figure 3C and S2C). IP3 can also be converted to other products by inositol 5-phosphatases such as INPP5A (Pirruccello et al., 2014), which exhibited reduced expression in CRC liver metastases (Figure 3B and S2A). We hypothesized that depleting INPP5A may inhibit IP3 breakdown and enhance ITPR3-mediated signaling, thereby augmenting metastasis. Indeed, depletion of INPP5A in SW480 cells was sufficient to enhance liver metastatic colonization after portal circulation injection (Figure 3D and S2D). We found similar effects on CRC liver metastasis by silencing PLCG1 and INPP5A in LS174T cells (Figure 3E-F and S2E-F). These findings support a role for IP3 signaling in CRC liver colonization by identifying an IP3 generating enzyme (PLCG1) as a metastasis promoter and an IP3 metabolizing enzyme (INPP5A) as a metastasis suppressor.
Figure 3. Genes implicated in IP3 signaling and metabolism regulate metastatic CRC liver colonization.

(A) Schematic of IP3 signaling and metabolism. (B) PLCG1 and INPP5A expression as measured by microarray from normal colon, CRC primary tumors or liver metastases (GSE41258). (C) 5×105 SW480 cells expressing control or PLCG1 shRNA were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=5 mice). (D) 5×105 SW480 cells expressing control or INPP5A shRNA were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=5 mice). (E,F) 5×105 LS174T cells expressing control, PLCG1 or INPP5A shRNA were inoculated by portal circulation injection, and metastatic colonization was assessed by liver mass (E) or metastatic tumor nodule count on liver histology (F). Median is denoted for B. All other graphs show mean ± SEM. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, Mann-Whitney test. See also Figure S2.
ITPR3 regulates initiation of metastatic colonization in vivo
We speculated that ITPR3 enables CRC cells to overcome physiological barriers inherent to the hepatic microenvironment. To assess the impact of ITPR3 deletion on the fate of liver-metastatic CRC cells as a function of time in vivo, we labeled ITPR3 KO and control CRC cells with CellTracker Red and Green dyes, respectively, and injected a 1:1 mixture into the portal circulation (Figure 4A). Livers were harvested at two hours or 48 hours after injection, and ITPR3 KO and control cells were visualized by immunofluorescence (Figure 4B-C). Strikingly, while control and ITPR3 KO cells were observed at equal representation two hours after injection (Figure 4D), there was a marked reduction (~10-fold) in ITPR3 KO cells compared to control cells by 48 hours (Figure 4E). It is well established that cancer cells arriving within the metastatic microenvironment undergo considerable attrition. Consistent with this, we observed a ~30% reduction in the number of control (red) cells. ITPR3 deleted cells (green) exhibited a ~90% decrease in cell number, consistent with a substantial reduction in survival of ITPR3 deleted cells within 48 hours of arrival in the hepatic microenvironment. These findings reveal that ITPR3 is critical in promoting the survival of CRC cells at the initial stage of metastatic liver colonization.
Figure 4. ITPR3 regulates initiation of metastatic colonization in vivo.

(A) SW480 cells labeled with CellTracker Red (sgCTRL) or Green (sgITPR3) were mixed 1:1, and 1× 106 cells were introduced into the portal circulation. Livers were harvested at the indicated timepoints (n=5 mice). (B,C) Representative liver surface (B) or liver section images by confocal microscopy (C) from livers harvested at 2H or 48H after injection. (D,E) Number of sgCTRL or sgITPR3 cells per field of view at 2H (D) or 48H (E) after portal circulation inoculation. Mean ± SEM; ** p < 0.01, Student’s t-test.
RELB promotes CRC metastatic liver colonization downstream of ITPR3
To define genes or pathways that promote liver colonization downstream of ITPR3, we extracted control or ITPR3 KO CRC cells from established liver metastases and performed RNA-seq. Interestingly, pathways related to immune response and non-canonical NF-κB signaling were enriched among the genes that were down-regulated in ITPR3 KO cells (Figure S3). Using qRT-PCR, we observed decreased expression of the transcription factor RELB as well as TNF in ITPR3 KO cells relative to control cells isolated from metastases, validating the RNA-seq results (Figure 5A-B). The non-canonical NF-κB pathway differs from the canonical pathway in its components and primarily functions through the transcription factors RELB and p52 (Sun, 2017). Under basal conditions, RELB is typically associated with p100 and sequestered in the cytoplasm as a RELB-p100 dimer; activation leads to ubiquitination and processing of p100, association of RELB with p52 and nuclear translocation of the RELB-p52 dimer. While RELB expression and non-canonical NF-κB signaling have been associated with clinical outcomes in CRC (Tao et al., 2018), their exact roles in metastatic progression are unclear.
Figure 5. RELB promotes CRC metastatic liver colonization downstream of ITPR3.

(A,B) RNA expression levels of RELB (A) or TNF (B) from ex vivo SW480 sgCTRL or sgITPR3 cells harvested from liver metastases as measured by qRT-PCR (n=3). (C) RELB expression in LS174T cells expressing control or RELB shRNA by qRT-PCR. (D) 5×105 LS174T cells were inoculated by portal circulation injection, and metastatic colonization was assessed by liver mass. (E) Immunoblot for RELB protein levels from CRISPR-edited sgCTRL or sgRELB cells (two independent guide RNAs). (F) 5 × 105 SW480 cells were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=5 mice). (G) Immunoblot of sgCTRL or gITPR3 cells stably overexpressing RELB or empty vector (EV) control. (H,I) 5×105 sgCTRL (H) or sgITPR3 (I) cells expressing RELB or EV were inoculated by portal circulation injection, and metastatic colonization was measured by liver bioluminescence (n=5–6 mice). Mean ± SEM; *p < 0.05, ** p < 0.01, *** p < 0.001, Mann-Whitney test. See also Figure S3 and S4.
Interestingly, RELB was also one of the top liver colonization genes identified in our large-scale shRNA screen (Figure 1B), and therefore we further validated the role of RELB in liver colonization in vivo. RELB depletion in LS174T cells by shRNA (Figure 5C) substantially reduced metastatic burden after portal circulation injection (Figure 5D and Figure S4A). Additionally, we deleted RELB in SW480 cells using CRISPR/Cas9 (Figure 5E) and similarly found that RELB KO cells exhibited dramatically reduced liver metastatic colonization capacity up to 72-fold (Figure 5F). Like ITPR3 deficiency, loss of RELB decreased both the number and size of metastatic tumors (Figure S4B-C). RELB deficiency also significantly inhibited CRC liver metastatic colonization in an immunocompetent model (Figure S4D-E). To determine if RELB acts downstream of ITPR3, we ectopically expressed RELB in control or ITPR3 KO cells and performed metastatic liver colonization assays (Figure 5G). While we observed no further increases liver metastatic colonization upon RELB overexpression in control cells (Figure 5H), RELB overexpression rescued the colonization defect of ITPR3-deleted cells (Figure 5I). Collectively, these data reveal that ITPR3-mediated activation of RELB is a critical driver of metastatic colonization.
ITPR3 and RELB regulate CRC survival under substratum detachment and hypoxia in cells and patient-derived xenograft organoids
We next assessed the cellular mechanisms by which ITPR3 and RELB facilitate metastatic colonization. ITPR3 loss did not affect cell growth under anchorage-dependent normal tissue culture conditions (Figure 6A). In addition, ITPR3 deletion did not impair cell migration in vitro (Figure S5A). CRC cells arriving in the liver parenchyma through the portal circulation undergo profound cellular stress, likely exacerbated by the lack of attachment to the extracellular matrix (ECM) of the primary epithelial site, which normally provides important survival cues (Buchheit et al., 2014). We sought to recapitulate these stresses in vitro by culturing control and ITPR3 KO SW480 cells in ultra-low attachment (ULA) plates, which contain a covalently bound hydrogel layer that prevents cell attachment. Under these conditions, we observed a significantly lower number of ITPR3 KO cells relative to control cells (Figure 6B). Because early liver-metastatic CRC cells must proliferate from single cells to form colonies, we also assessed CRC cell growth when plated at clonal density (one cell per well) in ULA plates. ITPR3 KO cells exhibited reduced capacity to form colonies (Figure 6C), consistent with a role for ITPR3 in enabling colony formation in the absence of normal substratum attachment. Lastly, using an ATP-based cell viability assay, we found that ITPR3 loss reduced viability in CRC cells grown under detached conditions (Figure 6D and Figure S5B).
Figure 6. ITPR3 and RELB regulate CRC survival under substratum detachment and hypoxia in cells and patient-derived xenograft organoids.

(A) 1×105 SW480 cells were grown under normoxia and attached cell culture conditions. (B) 50,000 SW480 cells were seeded in triplicate in ultra-low attachment (ULA) plates and counted on day 5. (C) SW480 cells were sorted at single-cell clonal density into ULA plates and the number of colonies per plate was counted at day 21 (n=3 independent experiments). (D) Viability of SW480 cells cultured in ULA plates for 5 days as assessed by ATP-based luminescent cell viability assays (n=4 independent experiments). (E) Apoptosis levels of SW480 cells cultured in ULA plates for 2 days as assessed by caspase 3/7 activity (n=3 independent experiments). (F) Immunoblot for cleaved caspase-3 levels in sgCTRL or sgITPR3 cells cultured under attached or detached conditions (48H) (n=3 independent experiments). (G) Viability of control or ITPR3 KO SW480 cells overexpressing RELB or empty vector (EV) cultured in ULA plates (n=3 independent experiments). (H, I) SW480 (H) and LS174T (I) cells were counted after 5 days in culture under hypoxia. (J) CRC PDXs organoids (PDXOs) were generated form the CLR28 PDX. Control, ITPR3 or RELB KO PDXOs were cultured for 5 days under 0.5% O2 and assessed for viability using the MTT assay. (K) Venn diagram showing number of overlapping differentially expressed up-regulated and down-regulated genes under normoxia or hypoxia (0.5% O2 for 72 hours) in ITPR3 and RELB KO SW480 cells relative to control cells. (L) Volcano plot of gene sets enriched in shared differentially expressed genes that are down-regulated or up-regulated in hypoxia-exposed ITPR3 and RELB KO cells. Mean ± SEM; *p < 0.05, ** p < 0.01, *** p < 0.001, Student’s t-test. See also Figure S5, Figure S6, Table S3, and Table S4.
As ECM detachment can activate several downstream pathways leading to cell death (Buchheit et al., 2014; Simpson et al., 2008), we next characterized whether ITPR3 deficiency predisposes CRC cells to apoptosis. In the absence of substratum attachment, ITPR3 KO cells exhibited increased caspase-3 and caspase-7 activity (Figure 6E), as well as higher cleaved caspase-3 levels relative to control cells (Figure 6F). RELB overexpression in detached ITPR3 KO cells rescued cell viability equivalent to that of control cells (Figure 6G). These findings reveal that ITPR3 enhances CRC survival and inhibits apoptosis after ECM detachment in a RELB-dependent manner, thereby providing an adaptive mechanism for outgrowth within the metastatic microenvironment.
Another major hallmark of the liver parenchyma is prominent hypoxia (Jungermann and Kietzmann, 2000). Previous studies have demonstrated that hypoxia is a major barrier for CRC liver colonization (Loo et al., 2015; Nguyen et al., 2016; Yamaguchi et al., 2019); we thus hypothesized that hypoxia may render CRC dependent on ITPR3 and RELB for survival. Consistent with this, loss of ITPR3 or RELB significantly reduced the growth of SW480 and LS174T CRC cells cultured in 0.5% O2 (Figure 6H-I) and promoted apoptosis upon hypoxia exposure as revealed by cleaved caspase-3 expression (Figure S5C-D).
We next interrogated the role of ITPR3 and RELB in a clinically relevant patient-derived model. Using human CRC tissues from primary or metastatic tumor sites, we previously developed highly metastatic PDX models of CRC liver metastasis that recapitulate the histology and architecture of human tumors (Yamaguchi et al., 2019). Another model that has gained traction is patient-derived organoids, three-dimensional cultures of cancer cells which are more amenable to genome editing (Drost et al., 2015, 2017; Weeber et al., 2017). Organoids have been successfully generated from CRC and accurately model treatment responses in patients (Vlachogiannis et al., 2018; van de Wetering et al., 2015). We therefore generated PDX-derived organoids (PDXOs) from a highly metastatic CRC PDX (CLR28) and deleted ITPR3 and RELB using CRISPR/Cas9. Consistent with our results in CRC cell lines, ITPR3 and RELB KO PDXOs exhibited significantly reduced viability when cultured under hypoxia (Figure 6J). Taken together, these data suggest that ITPR3 and RELB additionally impact metastatic colonization by promoting CRC survival within hypoxic microenvironments like the tumor or liver metastatic microenvironment.
To determine if similar mechanisms controlling CRC survival are active in vivo, we interrogated markers of proliferation and cell death in liver tissue obtained from animals bearing hepatic metastases. We observed no difference in Ki67 positivity between control and ITPR3 KO cells at early and late timepoints, suggesting that ITPR3 was not enhancing cell proliferation (Figure S5E-F). In contrast, using an in vivo caspase-3/7-dependent reporter, we observed increased apoptotic cell burden in ITPR3 KO cells (Figure S6G). Taken together, these data support a role for ITPR3 in mediating resistance to apoptosis.
We next evaluated effector pathways and target genes downstream of ITPR3 and RELB in the context of cell detachment and hypoxia exposure. ECM detachment has been shown to activate autophagy, which can promote cell survival and mediate anoikis resistance (Avivar-Valderas et al., 2013; Fung et al., 2008). However, we observed robust lipidated microtubule-associated protein light chain 3 (LC3-II) accumulation in ITPR3- and RELB-deficient cells upon detachment, suggesting that these genes are not required for detachment-induced autophagy (Figure S5H). RELB has been shown to transcriptionally upregulate BCL3 to promote CRC tumorigenesis (Tao et al., 2018); while BCL3 levels were lower in detached RELB KO cells by qRT-PCR relative to control cells, BCL3 expression was higher in ITPR3 KO cells, suggesting that BCL3 is not a main target gene downstream of this axis (Figure S5I). In addition, we examined the expression of anti-apoptotic and pro-apoptotic genes, whose transcriptional control has been more strongly linked to the canonical NF-κB pathway. We did not observe significant changes in BCL2 expression in ITPR3 KO cells compared to control cells (Figure S5J). In contrast, expression of BCL2 modifying factor (BMF) was significantly increased basally and upon cell detachment in both ITPR3 and RELB KO cells (Figure S5K). Notably, BMF is a critical regulator of anoikis in intestinal epithelial cells (Hausmann et al., 2011) and has been shown to be transcriptionally repressed by RELB (Vallabhapurapu et al., 2015). Thus, BMF may be a potential target gene controlled by the ITPR3-RELB axis that could contribute to enhanced survival upon cell detachment.
We observed that BMF was not increased in ITPR3 and RELB KO cells cultured for 72 hours under hypoxic conditions (Figure S5L), suggesting that other downstream pathways may be involved in the hypoxia response. To define gene programs regulated by ITPR3 and RELB upon hypoxia, we performed mRNA-sequencing of control, ITPR3 KO and RELB KO cells. Using a systematic information-theoretic pathway analysis (iPAGE) (Goodarzi et al., 2009), we identified gene sets that were individually modulated by ITPR3 and RELB. In ITPR3 KO cells exposed to hypoxia, genes related to endoplasmic reticulum unfolded protein response, ubiquitination, and intracellular receptor signaling pathways were underrepresented, whereas genes related to ribosomal structural components, respiratory electron transport chain, and carboxypeptidase activity were overrepresented (Figure S6A). Loss of RELB was associated with lower expression of genes related to translation initiation and unfold protein response, while genes related to carboxypeptidase activity and lipid catabolic process showed higher expression (Figure S6B). Next, we compared up-regulated and down-regulated genes in both ITPR3 and RELB KO cells under normoxia and hypoxia to define overlapping gene signatures (Figure S6C-D and Table S3). We observed significant overlap of differentially expressed genes in ITPR3 and RELB KO cells under normoxia (p<10−74) (Figure 6K). Notably, the majority of genes that were down-regulated or up-regulated in ITPR3 KO cells relative to control cells under hypoxia showed similar expression patterns in RELB KO cells (p<10−74) (Figure 6K). To elucidate possible pathways controlled by the ITPR3-RELB axis in response to hypoxia, we performed gene set enrichment analysis of the overlapping differentially expressed genes (Figure 6L and Table S4). Interestingly, the common down-regulated genes were enriched for genes related to growth factor activity, chemical homeostasis, and blood vessel development, including VEGFA, a key promoter of CRC progression and validated target in CRC treatment (Bhattacharya et al., 2016), as well as GDF15, which has been associated with CRC metastasis (Li et al., 2016). The most significantly up- regulated common genes were related to endopeptidase inhibitor activity, including genes with tumor suppressive activity such as SERPINB5 (Lockett et al., 2006). These data reveal putative gene pathways that are regulated by the ITPR3-RELB axis.
Treatment with the ITPR3 inhibitor caffeine reduces CRC metastatic capacity
Finally, we examined whether inhibiting ITPR3 pharmacologically could reduce CRC liver metastasis. Previous studies had demonstrated that caffeine is an inhibitor of ITPR3-mediated calcium flux and inhibits invasion in a xenograft model of glioblastoma (Brown et al., 1992; Kang et al., 2010). Intriguingly, while data on caffeine intake in CRC risk are mixed, prospective studies have shown that increased caffeinated coffee consumption in patients with stage III CRC is associated with reduced metastatic recurrence and improved survival (Guercio et al., 2015). However, the molecular basis underlying a possible protective effect of caffeine is unknown.
Caffeine treatment in vitro reduced CRC cell growth after cell detachment (Figure S7). To determine the effect of caffeine treatment in vivo, we first pretreated LS174T CRC cells for 24 hours with caffeine or vehicle control prior to injection into the portal circulation. Caffeine pretreatment significantly reduced metastatic colonization, suggesting that transient caffeine exposure prior to arrival at the metastatic environment can inhibit metastatic colonization (Figure 7A-B). To assess whether the effect of caffeine is ITPR3-dependent, we exposed sgCTRL or sgITPR3 cells to caffeine or vehicle control prior to portal circulation injection. Notably, caffeine treatment reduced the liver metastatic capacity of sgCTRL and sgITPR3 cells (Figure 7C-D), suggesting that caffeine may exert its metastasis suppressor effects on additional protein targets, although the magnitude of decrease in sgITPR3 cells was approximately half that in control cells. Finally, RELB overexpression curtailed the effect of caffeine pretreatment on liver metastasis, consistent with RELB being sufficient to promote metastasis downstream of ITPR3 (Figure 7E). These data reveal that caffeine inhibits CRC metastatic colonization and that these effects are mediated in part through ITPR3, though other ITPR3-independent pathways are likely active.
Figure 7. Treatment with the ITPR3 inhibitor caffeine reduces CRC metastatic capacity.

(A,B) LS174T cells were cultured in the presence of 10 mM caffeine or vehicle control for 24H prior to portal circulation injection. Liver metastatic burden was quantified by bioluminescence (A) or liver mass (B). (C,D) Control or ITPR3 KO SW480 cells were cultured in the presence of 10 mM caffeine or vehicle control for 24H prior to portal circulation injection. Liver metastatic burden was quantified by bioluminescence by bioluminescence (C) or liver mass (D). (E) Cells overexpressing RELB were cultured in the presence of 10 mM caffeine or vehicle control for 24H prior to portal circulation injection. Liver metastatic burden was quantified by bioluminescence by bioluminescence. (F) Proposed model showing hypoxia and lack of normal cellular attachment within the liver microenvironment activating caffeine-sensitive IP3 and ITPR3-mediated signaling leading to RELB activation and CRC cell survival. Mean ± SEM; *p < 0.05, ** p < 0.01, Mann-Whitney test. See also Figure S7.
DISCUSSION
Metastatic colonization is the primary driver of mortality in CRC. Here, we report a genome-scale loss-of-function screen that identified 26 promoters of CRC liver colonization, validating several candidates using in vivo assays and identifying genes that function in a common critical molecular pathway that drives metastasis (Figure 7F).
One of the key metastasis promoters that we identified is ITPR3, which we found regulates a transcriptional program that is associated with CRC patient survival. Recent studies have revealed emerging context-dependent roles for ITPR3 in tumorigenesis, but its role in metastatic colonization is poorly defined. For example, one study found that high ITPR3 expression in resected CRC primary tumors correlates with poor clinical outcomes including liver metastasis and overall survival (Shibao et al., 2010). Loss of ITPR3 in CRC cell lines enhanced sensitivity to apoptosis inducers (Shibao et al., 2010). ITPR3 has also been implicated in promoting the growth of other tumor types, such as hepatocellular carcinoma and cholangiocarcinoma (Guerra et al., 2019; Ueasilamongkol et al., 2020). In contrast, some studies have demonstrated a tumor suppressive role for ITPR3 through its involvement in transferring calcium from the ER to mitochondria, a key step in apoptosis (Bononi et al., 2017; Katona et al., 2019; Kuchay et al., 2017). BAP1 has been shown to deubiquitinate ITPR3, whereas PTEN competes with the ubiquitin ligase FBXL2 for binding to ITPR3–both mechanisms leading to higher ITPR3 expression and increased ITPR3-and calcium-mediated apoptosis (Bononi et al., 2017; Kuchay et al., 2017). Oncogenic KRASG13D has also been demonstrated to repress ITPR3 expression, thereby rendering cells resistant to apoptosis and promoting oncogenic transformation (Pierro et al., 2014). While ITPR3 may have both pro- and anti-tumorigenic roles at the primary tumor site, our data suggest that ITPR3 contributes to calcium remodeling in CRC and is a critical driver of metastasis.
Focused studies revealed that ITPR3 and IP3 signals are essential for metastasis initiation during liver colonization. This effect may be due to ITPR3’s role in supporting the survival of cancer cells in response to physiological stressors in the metastatic microenvironment, such as loss of normal cell attachment and exposure to hypoxia. Indeed, multiple cell types respond to hypoxia by increasing intracellular calcium levels (Seta et al., 2004). Moreover, matrix deprivation has been shown to induce a surge of ITPR3-dependent ER calcium release, leading to oxidant signaling and activation of AMP-activated protein kinase, which is involved in anoikis resistance (Sundararaman et al., 2016). A previous report found that loss of all IP3 receptor activity and calcium transfer to the mitochondria in tumor cells leads to mitotic catastrophe and necrosis due to altered metabolism (Cárdenas et al., 2016). Metabolite profiling of IP3 receptor-deficient DT40 cells demonstrated alterations in energy charge and reactive oxygen species homeostasis, although the effect of IP3 receptors and specifically ITPR3 on CRC metabolism is not known (Wen et al., 2015). Taken together, our data and others suggest that ITPR3 may be a key player in coordinating stress responses as encountered by disseminating CRC cells.
By interrogating transcriptional programs controlled by ITPR3, we identified non-canonical NF-κB signaling as a possible downstream mediator in metastatic liver colonization. Interestingly, the key transcription factor associated with the non-canonical NF-κB pathway, RELB, was also discovered independently as a top hit in our screen. While the canonical NF-κB pathway has been widely studied in cancer, roles for the non-canonical NF-κB pathway are less clear. Interestingly, high RELB expression in CRC has been associated with poor survival outcomes (Tao et al., 2018; Zhou et al., 2018). We found that loss of RELB in CRC cells diminishes metastatic efficiency in vivo and that RELB overexpression rescues the metastatic defect of ITPR3 KO cells, suggesting that RELB acts downstream of ITPR3 within the same pathway. We moreover found that both ITPR3 and RELB cooperate to promote survival of CRC cells under hypoxia and matrix deprivation. Canonical NF-kB has been shown to be activated by hypoxia in a calcium and calcium/calmodulin-dependent protein kinase II manner (Culver et al., 2010), and there is extensive crosstalk between canonical NF-kB and hypoxia inducible factor (HIF) signaling (D’Ignazio et al., 2017); however, the interplay between hypoxia, calcium and non-canonical NF-κB activation is not defined.
We found that ITPR3 and RELB are important mediators of resistance to apoptosis in response to ECM detachment and hypoxia. Through transcriptomic gene expression analyses, we observed that ITPR3 and RELB KO cells exhibited higher expression of the pro-apoptotic gene BMF, which was induced upon cell detachment and was previously implicated in promoting anoikis in intestinal epithelial cells. This function may be context-dependent, as transcriptional profiling of cells exposed to hypoxia demonstrated that ITPR3 and RELB loss is associated with reduced growth factor expression, including VEGFA and GDF15. Additional studies are needed to further dissect the molecular mechanism by which ITPR3 controls RELB activity and characterize the mechanisms of downstream gene’s actions in this process.
Finally, we investigated the therapeutic potential of targeting this ITPR3/RELB axis. While there are no existing specific inhibitors of ITPR3, previous data suggest that caffeine inhibits ITPR3 as well as additional target genes (Brown et al., 1992; Kang et al., 2010). Epidemiological data for caffeine on CRC incidence have been mixed, with meta-analyses of prospective cohort studies suggesting no significant associations between coffee intake and CRC incidence (Vieira et al., 2017; Zhang et al., 2010). However, coffee or caffeine may have a more potent effect on suppressing CRC metastasis. A recent analysis of the Nurses’ Health Study and Health Professionals Follow-up Study demonstrated an association between higher coffee intake and reduced CRC mortality (including both caffeinated and decaffeinated coffee), although this study was retrospective and lacking recurrence data (Hu et al., 2018). Notably, a prospective study suggested that high caffeinated coffee intake but not decaffeinated coffee intake significantly associates with improved recurrence-free and overall survival in stage III CRC patients receiving adjuvant chemotherapy (Guercio et al., 2015). We found that exposure of CRC cells to caffeine significantly reduced metastatic burden in a manner that is partially dependent on ITPR3. Given the pleiotropic targets of caffeine, its effect on CRC progression is likely complex, but our data raises the potential promise of more specific inhibitors of ITPR3 as therapeutic suppressors of metastasis in the adjuvant setting.
In summary, our genome-scale screen has identified several therapeutically amenable candidate genes that interact in cellular and molecular pathways such as the ITPR3/calcium/RELB axis to drive CRC liver colonization and metastatic progression.
LIMITATIONS OF THE STUDY
This study identified an important link between ITPR3, intracellular calcium, and RELB in promoting CRC liver metastasis, but the exact mechanism by which ITPR3-dependent calcium signaling regulates RELB remains unclear. We did not define whether ITPR3 and calcium predominantly control RELB expression or activation (such as nuclear translocation or transcriptional output). The mechanisms by which tumor hypoxia controls ITPR3 and RELB expression or activity were also not fully addressed. Finally, caffeine is a low affinity inhibitor of ITPR3, necessitating high intracellular concentrations, and it has complex physiological effects that may impact the colonization process. Future investigations to define molecular interactions underlying this ITPR3/calcium/RELB axis and understand its therapeutic potential are warranted.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sohail Tavazoie (sohail.tavazoie@rockefeller.edu).
Materials availability
Unique materials and reagents generated in this study are available upon request from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
RNA-seq data have been deposited in GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All data reported in this paper will be shared by the lead contact upon request.
The paper does not report original code.
Any additional information to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ITPR3 | BD Biosciences | Cat# 610313; RRID: AB_397705 |
| Rabbit polyclonal anti-RELB | Cell Signaling Technology | Cat# 4954; RRID: AB_330626 |
| Mouse monoclonal anti-Tubulin | Cell Signaling Technology | Cat# 3873; RRID: AB_1904178 |
| Mouse monoclonal anti-NFkB p52 | Millipore | Cat# 05-361; RRID: AB_309692 |
| Rabbit polyclonal anti-PLCG1 | Cell Signaling Technology | Cat# 2822; RRID: AB_2163702 |
| Rabbit polyclonal anti-INPP5A | Proteintech | Cat# 21723-1-AP; RRID: AB_10734438 |
| Rabbit polyclonal anti-Cleaved caspase-3 | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Mouse monoclonal anti-GAPDH | Proteintech | Cat# 60004-1-Ig; RRID: AB_2107436 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Rabbit polyclonal anti-LC3B | Cell Signaling Technology | Cat# 2775S; RRID: AB_915950 |
| HRP goat anti-mouse secondary antibody | Thermo Fisher Scientific | Cat# A16078; RRID: AB_2534751 |
| HRP goat anti-rabbit secondary antibody | Thermo Fisher Scientific | Cat# 65-6120; RRID: AB_2533967 |
| Bacterial and virus strains | ||
| Chemically competent One shot Stbl3 | Invitrogen | Cat# C737303 |
| Biological samples | ||
| Highly metastatic human CRC PDX | Tavazoie Lab (Yamaguchi et al., 2019) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Polybrene | Santa Cruz | Cat# sc-134220 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| G418 sulfate | Thermo Fisher Scientific | Cat# 10131027 |
| D-Luciferin | GoldBio | Cat# LUCK-100 |
| Caffeine | Sigma-Aldrich | Cat# C0750 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Lipofectamine RNAiMax | Thermo Fisher Scientific | Cat# 13778150 |
| Turbofect | Thermo Fisher Scientific | Cat# FERR0531 |
| cOmplete mini protease inhibitor | Roche | Cat# 11836153001 |
| RIPA lysis and extraction buffer | G-Biosciences | Cat# 786-490 |
| ECL Western blotting substrate | Pierce | Cat# 32106 |
| Fast SYBR Green Master Mix | Thermo Fisher Scientific | Cat# 43-856-18 |
| ACK lysing buffer | VWR | Cat# 12002-070 |
| Optiprep density gradient medium | Sigma-Aldrich | Cat# D1556 |
| Collagenase, type IV | Worthington | Cat# LS004188 |
| Matrigel | Corning | Cat# 354262 |
| Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | Cat# 31985062 |
| Fetal bovine serum | Sigma-Aldrich | Cat# F2442 |
| CellTracker Red CMPTX | Thermo Fisher Scientific | Cat# C34552 |
| CellTracker Green CMFDA | Thermo Fisher Scientific | Cat# C7025 |
| Tween-20 | Sigma-Aldrich | Cat# P9416 |
| DNAse1 | Sigma-Aldrich | Cat# D5025 |
| Y-27632 | Sigma-Aldrich | Cat# Y0503 |
| Collagenase, type XI | Sigma-Aldrich | Cat# C9407 |
| R-spondin | R&D | Cat# 4645-RS |
| Wnt 3a | R&D | Cat# 5036-WN |
| Cell recovery solution | Corning | Cat# 354253 |
| TrypLE Express Enzyme | Thermo Fisher Scientific | Cat# 12604013 |
| Lenti-X concentrator | Takara | Cat# 631231 |
| Tissue-Tek O.C.T Compound | VWR | Cat# 25608-930 |
| DAPI | Roche | Cat# 10236276001 |
| N-acetylcysteine | Sigma-Aldrich | Cat# A9165 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 |
| Recombinant murine Noggin | Peprotech | Cat# 250-38 |
| Recombinant human EGF | Peprotech | Cat# AF-100-15 |
| Recombinant human FFG-10 | Peprotech | Cat# 100-26 |
| Gastrin I (human) | TOCRIS | Cat# 3006 |
| A 83-01 | TOCRIS | Cat# 2939 |
| B27 supplement | Thermo Fisher Scientific | Cat# 17504044 |
| Prostaglandin E2 (PGE2) | TOCRIS | Cat# 2296 |
| Critical commercial assays | ||
| CellTiterGlo 2.0 Assay | VWR | Cat# PAG9241 |
| MTS cell proliferation assay kit | Abcam | Cat# ab197010 |
| Total RNA purification kit | Norgen | Cat# 37500 |
| CaspaseGlo 3/7 Assay | Promega | Cat# G8091 |
| VivoGlo Caspase 3/7 Substrate | Promega | Cat# P1782 |
| Mouse cell depletion kit | Miltenyi Biotec | Cat# 130-104-694 |
| Truseq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2002 |
| Quantseq 3’ mRNA-seq Library Prep Kit FWD | Lexogen | Cat# 015.96 |
| SuperScript III First-Strand Synthesis System for RT-PCR | Thermo Fisher Scientific | Cat# 18080051 |
| Deposited data | ||
| Microarray expression data from CRC patients | (Sheffer et al., 2009) | GSE41258 |
| RNA-seq expression data from CRC patients | (Kim et al., 2014) | GSE50760 |
| RNA-seq of ITPR3 KO in SW480 CRC metastases | This paper | GSE182257 |
| RNA-seq of SW480 CRC cells transfected with siRNAs targeting different putative metastasis promoting genes | This paper | GSE182386 |
| RNA-seq of SW480 CRC cells with loss of ITPR3 or RELB cultured under normoxic or hypoxic conditions | This paper | GSE197576 |
| Human refence genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Broad Institute TCGA data | (Broad Institute TCGA Genome Data Analysis Center, 2016) | https://doi.org/10.7908/C11G0KM9 |
| Experimental models: Cell lines | ||
| Human: SW480 cells | ATCC | Cat# CCL-228; RRID: CVCL_0546 |
| Human: LS174T cells | ATCC | Cat# CL-188; RRID: CVCL_1384 |
| Human: SW620 cells | ATCC | Cat# CCL-227; RRID: CVCL_0547 |
| Human: WiDr cells | ATCC | Cat# CCL-218; RRID: CVCL_2760 |
| Human: 293LTV cells | Cell Biolabs | Cat# LTV100; RRID: CVCL_JZ09 |
| Human: LS14T-LV2 cells | Tavazoie Lab | Derived from in vivo selection of LS174T cells |
| Mouse: MC38 cells | Kerafast | Cat# ENH204-FP; RRID: CVCL_B288 |
| Experimental models: Organisms/strains | ||
| Mouse: NSG | The Jackson Laboratory | Strain: 005557 |
| Mouse: C57Bl/6 | The Jackson Laboratory | Strain: 000664 |
| Oligonucleotides | ||
| shRNA sequences, see Table S5 | This paper | N/A |
| sgRNA sequences, see Table S5 | This paper | N/A |
| siRNAs, see Table S6 | Dharmacon | N/A |
| Primers for qRT-PCR, see Table S7 | This paper | N/A |
| Recombinant DNA | ||
| pLKO.1-puro | Addgene | Cat# 8453; RRID: Addgene_8453 |
| pCMV-VSG-G | Cell Biolabs | Cat# VPK-206 |
| pCgpV | Cell Biolabs | Cat# VPK-206 |
| pRSV-Rev | Cell Biolabs | Cat# VPK-206 |
| pSpCas(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat# 62988; RRID: Addgene_62988 |
| lentiCRISPR V2.0 | Addgene | Cat# 59261; RRID: Addgene_52961 |
| pCMV6-Entry | Origene | Cat# PS100001 |
| pCMV6-RELB | Origene | Cat# SC122747 |
| Software and algorithms | ||
| Prism 9 | GraphPad | https://www.graphpad.com; RRID:SCR_002798 |
| R v3.5.1. | R Core Team | https://www.r-project.org/; RRID: SCR_001905 |
| Fiji | (Schindelin et al., 2012) | https://imagej.net/software/fiji/ |
| STAR aligner (v2.6.0a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| featureCounts (v1.6.3) | (Liao et al., 2014) | https://bio.tools/featurecounts |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ReactomePA package for R | (Yu and He, 2016) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Package for survival analysis in R | (Therneau, 2020) | https://cran.r-project.org/web/packages/survival/index.html |
| Survminer package for R | (Kassambara et al., 2017) | https://cran.r-project.org/web/packages/survminer/index.html |
| QuPath (v0.3.2) | (Bankhead et al., 2017) | https://qupath.github.io |
| iPAGE | (Goodarzi et al., 2009) | https://tavazoielab.c2b2.columbia.edu/iPAGE |
| topGO package for R | (Alexa and Rahnenfuhrer, 2021) | https://bioconductor.org/packages/release/bioc/html/topGO.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture:
SW480, LS174T and WiDr cell lines were obtained from ATCC, MC38 cells were obtained from Kerafast, and HEK-293LTV cells were obtained from Cell Biolabs. The cells were labeled with a luciferase reporter as described previously (Loo et al., 2015; Ponomarev et al., 2004). The in vivo-selected LS174T-LvM3b line was derived in the laboratory as previously described (Loo et al., 2015). Cell lines were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum. All cell lines were maintained at 37°C and 5% CO2 and regularly checked for mycoplasma contamination.
Animal studies:
All animal work was conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at The Rockefeller University. NOD/SCID gamma male mice (The Jackson Laboratory) or C57BL/6 male mice (The Jackson Laboratory) aged 6 to 10 weeks were used for xenograft experiments, respectively. For direct liver injections, cells were mixed with Matrigel (Corning) at a 1:1 ratio, and the indicated number of cells was injected into the liver. For portal circulation injections, 500,000 cells resuspended in 50 μL PBS were injected into the spleen followed by splenectomy. For caffeine pretreatment experiments, cells were cultured in the presence of 10 mM caffeine (Sigma) or vehicle (water) for 24H prior to injection. For BAPTA-AM pretreatment experiments, cells were incubated in the presence of 20 μM BAPTA-AM (Sigma) or vehicle (DMSO) for 2H prior to injection. For lung colonization assays, 200,000 cells resuspended in 100 μL PBS were injected intravenously into the lateral tail vein. Animals were excluded from studies if the inoculated cells did not enter the liver as assessed by bioluminescence. To monitor tumor burden non-invasively in vivo, mice were injected with 50 μL of 7.5 mg/ml d-luciferin substrate (GoldBio) by retro-orbital injection and imaged using an IVIS Lumina II (Caliper Life Science) at the indicated timepoints, and the liver bioluminescence signal was normalized to the baseline bioluminescence on day 0. To monitor caspase 3/7 activity in vivo, the liver bioluminescence signal using VivoGlo Caspase 3/7 Substrate (Promega) was normalized to the bioluminescence signal from d-luciferin. Mice were imaged at the indicated timepoints, and experiments were terminated when the luciferase signal was saturated or the mice were too ill, whichever occurred first. For primary tumor growth assays, 500,000 cells mixed 1:1 with Matrigel (Corning) were subcutaneous injected into the flanks of mice in a total volume of 100 μL. Tumor size was measure on the indicated days using digital calipers and tumor volume was calculated using the formula (small diameter)2 x (large diameter) x π/6.
Generation of knockdown and knockout cells:
Cells were depleted of the indicated gene using lentivirus-mediated delivery of shRNA as described previously. Sequences for shRNAs are provided in Table S5 and were cloned into the pLKO.1-puro vector (Addgene). For lentivirus generation, 293LTV cells (Cell Biolabs) seeded in 10cm plates at ~50% confluency were incubated with 9 μg of pLKO.1-puro shRNA vector, 3 μg pRSV-Rev (Cell Biolabs), 3 μg pCgpV (Cell Biolabs), 3 μg pCMV-VSV-G (Cell Biolabs) and Lipofectamine 2000 (Invitrogen) diluted in OptiMEM media (Gibco) per the manufacturer’s instructions. Media was changed to antibiotic-free media after five hours, and lentivirus was harvested by filtering the supernatant through a 0.45 μM filter (Pall). Cells plated one day prior were transduced with lentivirus by incubating with lentivirus media supplemented with polybrene (8 μg/mL) for 16 hours. At 48 hours after transduction, shRNA-containing cells were selected with puromycin-supplemented media (2 μg/mL). For generation of CRISPR knockout cells, single guide RNA sequences (Table S5) were cloned into the pSpCas(BB)-2A-Puro (PX459) V2.0 vector (Addgene) or lentiCRISPR V2.0 vector (Addgene). Cells were either transduced with lentivirus as above or plated one day prior to transfection and transfected with 3 μg plasmid and Turbofect (Thermo Scientific) diluted in serum-free media per the manufacturer’s instructions. At 24 hours, transfected cells were selected by changing to puromycin-supplemented media (2 μg/mL). Single clones were selected by limiting dilution by plating cells at 0.5 cells/well of a 96 well plate and isolating single colonies. Knockout was verified by western blot and 6–10 confirmed control or knockout clones were pooled.
Generation of stable RELB-overexpressing cells:
Cells were transfected with 3 μg with empty vector control or pCMV6-RELB (Origene) and Turbofect (Thermo Scientific) diluted in serum-free media per the manufacturer’s instructions. At 24 hours, transfected cells were selected by changing to G418-containing media (1 mg/mL) for seven days. RELB expression was confirmed by immunoblot.
siRNA knockdown:
For siRNA knockdown experiments, SMARTpool siRNAs (Dharmacon) were mixed with Lipofectamine RNAiMAX (Thermo Fisher). siRNAs are provided in Table S6. Cells were plated at 400,000 per well of 6 well plate and the Lipofectamine/siRNA mixture was added to the well for reverse transfection. Cells were collected at 72 hours for RNA analysis.
shRNA drop-out screening:
Genome-wide screen identifying 556 liver colonization gene hits, selected by number of hairpins absent after the selection pressure in vivo, was performed as described previously (Nguyen et al., 2016). The top 209 genes that had an effect in vivo but not in vitro (z-score < −2 in vivo and z-score > −2 in cell culture) were selected for additional hairpin validation screening. The secondary screen was generated by cloning a top-scoring shRNA used in the genome-wide screen and an additional 2 hairpins not previously used, respectively, into the pLKO.1 vector and producing a viral library that was subjected to the experimental procedures described previously (Nguyen et al., 2016). The additional hairpin validation screen was performed with two biological replicates for each condition. Data processing was performed by taking the log-ratios, and the average of the top 2 scoring shRNAs was used. From this secondary screen, the top 26 colonization genes were selected based on the greatest magnitude of shRNA dropout in vivo, minimal shRNA dropout in cell culture and adequate shRNA representation (at least two shRNAs that could be analyzed from the secondary screen).
In vitro cell growth assays:
Cells were seeded in six-well plates in triplicate at 100,000 cells per well or in 24-well plates at 50,000 cells per well and allowed to grow for three to five days. Cells were lifted using trypsin 0.25% and quantified by cell counting and trypan blue dye exclusion. For hypoxia experiments, cells were seeded one day prior to placing them at 0.5% oxygen in a hypoxia chamber (INVIVO). For proliferation assays in the absence of substratum attachment, cells were seeded into 24-well ultra-low attachment plates (Corning) at 50,000 cells per well and counted on day five. For cell viability assays, cells were processed using Cell-Titer Glo 2.0 (Promega) and luminescence was measured using a luminometer (Molecular Devices). For single cell counting assays, cells were sorted using a FACSAria II (BD) at a clonal density of one cell per well into 96-well ultra-low attachment plates (Corning) containing EGM-2 media (Lonza). The presence of single cells was confirmed using bright-field microscopy, and the number of wells per plate with colonies was counted on day 21. For each independent experiment (n = 3), two plates were seeded per cell type.
Western blotting:
Cells were lysed in ice cold RIPA buffer (G-Biosciences) supplemented with cOmplete protease inhibitor (Roche). Samples were denatured, separated by SDS-PAGE using either 4–12% Bis-tris or 3–8% Tris-acetate gels, and transferred to Immobilon-P PVDF membrane (Millipore). Membranes were blocked with 5% bovine serum albumin in PBST (1X PBS, 0.1% Tween-20 (Sigma)) and probed with primary antibody overnight at 4°C. Membranes were washed with PBST, incubated with secondary antibodies conjugated to horseradish peroxidase (1:10,000), and developed using ECL Western Blotting Substrate (Pierce) and the SRX-101A (Konica Minolta) developer according to the manufacturer’s instructions.
Quantitative RT-PCR:
Total RNA Purification Kit (Norgen Biotek) was used to extract RNA from cells grown on tissue culture dishes per the manufacturer’s instructions. cDNA was generated using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative real-time PCR was performed in 384-well PCR microplates (Axygen) using a 7900HT Fast Real-Time PCR System (Thermo Fisher). cDNA samples were analyzed in quadruplicates. Each well contained a total volume of 10 μL including 0.5 μL cDNA, 2.0 μL primers (2.5 mM), 5.0 μL Fast SYBR Green Master Mix (Thermo Fisher) and 2.5 μL molecular grade water. Data were quantified using the comparative Ct (ΔΔCt) method. Primers for qPCR are listed in Table S7.
Isolation of ex vivo cells from liver metastases:
For ex vivo sequencing, livers were harvested from tumor-bearing mice at the indicated time points, chopped using a scalpel and incubated with 300 U/ml collagenase IV (Worthington) in DMEM on a shaker at 37°C for one hour. The suspension was centrifuged, and the supernatant removed. Red blood cells were lysed using 5 mL of ACK lysing buffer (Lonza) for five minutes at room temperature. The tumor suspension was pelleted, and the supernatant discarded. Live tumor cells were isolated by density gradient using a 40% and 20% solution of OptiPrep Density Gradient Medium (Sigma-Aldrich) and centrifuging at 800g for 20 minutes. Tumor cells were passed through a 100 μM and 70 μM cell strainer (VWR), and human tumor cells were isolated using a mouse cell depletion kit (Miltenyi Biotec) per the manufacturer’s instructions.
RNA sequencing library:
RNA was extracted using the Total RNA Purification Kit (Norgen Biotek) and DNAse treated using the RNase-Free Dnase I kit (Norgen Biotek). The QuantSeq 3’ mRNA-Seq Library Prep Kit (Lexogen) or Truseq RNA Library Prep Kit V2 (Illumina) was used for generation of RNAseq libraries. RNAseq libraries were quantified using a Bioanalyzer (Agilent) and pooled samples were sequenced on an Illumina NextSeq 500.
RNA-seq analysis:
Libraries were sequenced on an Illumina NextSeq sequencer. For RNA-seq of sgCTRL versus sgITPR3 liver metastases, polyA and adapter sequences were trimmed using the BBDuk utility (v38.31, sourceforge.net/projects/bbmap/; options k=13, ktrim=r, forcetrimleft=11, useshortkmers=t, mink=5, qtrim=t, trimq=10, minlength=20). Reads were aligned to the human genome (assembly GRCh38) using STAR aligner (v2.6.0a) (Dobin et al., 2013) using the ENCODE settings, apart from “--outFilterMismatchNoverLmax 0.1” as recommended by Lexogen (personal communication) for RNA-seq for sgCTRL versus sgITPR3 liver metastases and using default settings except for “--outFilterMultimapNmax 1” for RNA-seq of siRNA transfected cells. Reads mapping to genes were counted with featureCounts (v1.6.3) (Liao et al., 2014). Further analysis was performed using R (v3.5.1). Differential gene expression was calculated using DESeq2 (Love et al., 2014). For ranking of genes for visualizations and as input for gene set enrichment analysis (GSEA) (Subramanian et al., 2005), logfold-changes were shrunken using the apeglm method (Zhu et al., 2019). For calculating enriched pathways of the Reactome database, the ReactomePA package for R (v1.28) was used to perform GSEA (Yu and He, 2016). We carried out Gene Ontology enrichment analyses to identify processes underlying differentially expressed genes in ITPR3 and RELB KO cells cultured under normoxia or hypoxia. (R package ‘topGO’ 2.22.0). Given the hierarchical nature of these ontological definitions, we sought to reduce redundancy in the enriched terms (for visualization) by grouping GO terms with similar gene memberships together. We achieved this by first calculating the Jaccard similarity between every pair of enriched GO terms and then using this as input to clustering by Affinity Propagation (Frey and Dueck, 2007). For each exemplar GO term, we calculated the log-ratios between the mean expression of its associated genes in the perturbed (sgITPR3 and sgRELB) and in the control cells. This log-ratio represents if the set of genes associated with the enriched GO term are up- (> 0) or down-regulated (< 0) in the perturbed cells. Volcano plots were generated by plotting the log-ratios of the exemplar GO terms and their p-values from the GO enrichment analysis.
Survival analysis:
For every knockdown, we defined a differential expression (DE) signature from the DESeq2 analyses (described above). Clinical and RNA-seq data for colon cancer patients were obtained from Liu et al. and Broad Institute’s Genome Data Analysis Center (Broad Institute TCGA Genome Data Analysis Center, 2016; Liu et al., 2018). Gene-wise standardized expression (z-score) was calculated by subtracting the mean expression of the gene divided by its standard deviation. For each gene knockdown, we correlated its DE signature with the standardized gene expression patterns of colon cancer patients and grouped patients into positively and negatively correlated groups. Thus, patient transcriptome patterns that were concordant (discordant) with gene expression changes resulting from the knock-down, would be positively (negatively) correlated. Progression-free interval survival differences were quantified between the two groups (Therneau, 2020) and were visualized using the R package “survminer” (version 0.4.8) (Kassambara et al., 2017).
UMAP analysis:
Count data that were regularized and log-transformed (rlog function in DESeq2) was used as input to umap (R function umap) with default parameters. For each knockdown, two biological replicates and the average of the two replicates were included and their relative distances in 2-dimensions were visualized as scatter plots.
iPAGE analysis:
We used iPAGE to discover perturbed pathways in genes differentially expressed in cells harboring perturbations in ITPR3 (or RELB) and controls (https://tavazoielab.c2b2.columbia.edu/iPAGE/). iPAGE utilizes an information theoretic framework to systematically discover pathways that are significantly informative of gene expression profiles without any explicit thresholding requirements. Input to iPAGE included gene symbols and the log2-ratio between sgITPR3 (or sgRELB) and controls from the differential expression analysis (described earlier). iPAGE was run in continuous mode (“exptype”) with 3, 5, 7 bins (“ebins”) to minimize redundancy in the set of pathways identified (“ind=1”).
Establishment of CRC PDXO:
CLR28 PDX (Yamaguchi et al., 2019) was harvested at a size of 100 mm3 from NSG mice. Grossly necrotic tissue was trimmed out and the remaining tumor was washed in ice-cold PBS and minced with a #10 scalpel. The tumor fragments were incubated with 5 ml Collagenase type XI (Sigma) in DMEM at 5 mg/ml supplemented with 10 μg/ml DNAse1 (Sigma) and Y-27632 (Sigma) 10μM at 37°C for 1H. The digested tumors were triturated three times and allowed to sink by gravity, and 3 ml of supernatant was removed (fraction 1). Fresh DMEM (3 mL) was added and triturated 3 times to repeat the process for fractions 2–4. The fractions were examined under a bright-field microscope and the fraction most enriched with colonic epithelium was chosen for downstream processing. Tubes were centrifuged at 200G for 5 minutes at 4°C, the supernatant was discarded, the pellet was resuspended in Matrigel (Corning), 30 μl of the matrigel-colonic epithelium mix was plated in a well of a 24-well plate on a heating pad and allowed to solidify, and then 500 μL of organoid growth media (Advanced DMEM/F-12, HEPES 10 mM, GlutaMAX supplement, BSA 0.1%, Wnt3a, R-spondin, B27, nicotinamide 1.25 mM, N-acetylcysteine 1.25mM, primocin 100 μg/mL, mNoggin 100 ng/mL, hEGF 50 ng/mL, hFGF 100 ng/mL, hGastrin I 10nM, A 83–01 500 nM, Y-27632 10.5 μM, PGE2 1μM) was added.
Generation of CRISPR KO PDXOs:
Fully grown organoids were disrupted with Cell Recovery Solution (Corning), incubated on ice for 1H, and centrifuged for 5 minutes at 200G at 4°C. The supernatant was discarded and 1 ml of TrypLE solution (Thermo-Fisher) was added, and the sample was incubated at 37°C degree for 10 minutes. Cell clumps were disrupted with a micropipette, and the pellet was resuspended with organoid was media to 5 ml. 200,000 cells were taken per group, and 1 ml of concentrated CRISPR lentivirus concentrated with Lenti-X concentrator (Takara) with 8 μg/mL polybrene was added. The virus-organoid mix was plated in a 24-well plated, and spinfection at 2000 RPM was performed for 1H at 25°C. Organoid cells were resuspended with Matrigel and plated into 3–4 wells of a 24-well plate with organoid growth media. Puromycin selection at 2 μg/mL was started 48H after the transduction, and the media was changed to normal growth media after 48H.
Organoid viability assay:
Fully grown organoids were disrupted with Cell Recovery Solution (Corning), incubated on ice for 1H, and centrifuged for 5 minutes at 200G at 4°C. The supernatant was discarded and 1 ml of TrypLE solution (Thermo-Fisher) was added, and the sample was incubated at 37°C degree for 10 minutes. Cell clumps were disrupted with a micropipette, and the pellet was resuspended with organoid was media to 5 ml. Cells were counted, and 5000 cells were plated in 5 μL of Matrigel per well of a 96-well plate with 100 μL of growth media. Organoids were cultured overnight under normoxia and then transferred to a hypoxia chamber at 0.5% O2 for 96H. The media was changed to fresh growth media and 10 μL of MTS assay reagent (Abcam, ab197010) was added. The plate was shaken and incubated for 2H at 37°C under normoxia, and the absorbance was measured at 490 nM using a plate reader (Molecular Devices). Average absorbance of a non-MTS reagent control was subtracted, and cohorts were normalized to the average of the control to obtain the relative viability.
In vivo competition assay:
Cells were labeled with CellTracker Red CMPTX (Invitrogen) or CellTracker Green CMFDA (Invitrogen) per the manufacturer’s instructions. Cells were washed with PBS, lifted with 0.25% trypsin, pooled at 1:1 ratio and resuspended in PBS. For portal circulation injections, 1 × 106 cells were injected into the spleen followed by splenectomy. At the indicated timepoints, livers were harvested and visualized on a Zeiss Axiovert fluorescent microscope prior to being fixed in 4% paraformaldehyde for 24H at 4°C. Livers were washed, incubated overnight in 25% sucrose, and embedded in OCT cryosectioning compound (Sakura Finetek). Frozen livers were sectioned using a cryotome, slides were counterstained with DAPI and imaged on a confocal microscope.
Histology:
Tumor-bearing livers were harvested from mice at the indicated day and fixed in 4% paraformaldehyde for 24H at 4°C. The samples were then dehydrated in 70% ethanol for 24H at room temperature. The samples were embedded in paraffin and section in 5 μM slices, sectioned and stained with hematoxylin and eosin (Histoserv). Tumor nodule size was quantified using QuPath v0.3.2 (Bankhead et al., 2017).
Clinical analysis:
The GSE41258 and GSE50760 datasets were used for RNA-seq and microarray analysis of gene expression in CRC.
Statistical analysis:
Results are presented as mean ± SEM unless otherwise indicated. The number of samples for each group was chosen based on the expected levels of variation and consistency. Non-parametric tests were used when normality could not be assumed. Mann-Whitney test and Student’s t-test were used when comparing groups. One-tailed tests were used when a difference was predicted to be in one direction; otherwise, a two-tailed test was used. A p-value less than or equal to 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism version 9.0.2. or R version 4.0.0.
Wound closure assay:
500,000 cells were seeded into 6 wells in triplicate and the media was changed to DMEM/0.2% FBS for 12 hours to prevent cell division. A 200 ul pipette tip was used to draw a scratch through each well, the cells were washed with PBS, and the media was replaced with DMEM/0.2% FBS. The scratch was imaged on a Zeiss Axiovert microscope at time 0H and 24H with 3 images per well, and the gap area was quantified using Fiji.
Supplementary Material
Highlights.
Loss of ITPR3 in colorectal cancer cells inhibits metastatic liver colonization
RELB acts downstream of ITPR3 to promote colorectal cancer liver metastasis
ITPR3 and RELB regulate survival in response to hypoxia and substratum detachment
Exposure of CRC cells to ITPR3 inhibitor caffeine reduces liver metastasis
ACKNOWLEDGMENTS
We thank V. Padmanaban, D. Hsu and other members of the Tavazoie laboratory for thoughtful discussions on the manuscript. We thank Rockefeller University resource centers including the Genomics Resource Center and the veterinary staff from the Comparative Bioscience Center for animal husbandry and care. R.H.M was a Robert Black Fellow of the Damon Runyon Cancer Research Foundation (DRG 121-20) and was supported by grants from the NIH (T32CA00927 and K08CA263304). C.M.K. is a member of the German Academic Scholarship Foundation (Studienstiftung des deutschen Volkes). A.N. was supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the NIH (T32GM07739) to the Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program. N.Y. was a Meyer Clinical Scholar (Meyer Foundation) and is supported by the National Center for Advancing Translational Sciences (UL1TR001866) and the Black Family Foundation. This study was supported by the Black Family Foundation. S.T was supported by grants from the NIH (R01-HG009065 and R01-CA257153). S.F.T. was supported by a U54 grant from the National Cancer Institute of the National Institute of Health under Award number U54CA261701, the Black Family Foundation, a Howard Hughes Medical Institute Faculty Scholar Award, a Breast Cancer Research Foundation Award, and the Reem-Kayden award.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, and Aguirre-Ghiso JA (2013). Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 32, 4932–4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci Rep 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Ye X-C, Wang R, Ling X, McManus M, Fan F, Boulbes D, and Ellis LM (2016). Intracrine VEGF Signaling Mediates the Activity of Prosurvival Pathways in Human Colorectal Cancer Cells. Cancer Res 76, 3014–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, Pellegrini L, Signorato V, Olivetto F, Pastorino S, et al. (2017). BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braet F, and Wisse E (2002). Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center (2016). Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run.
- Brown GR, Sayers LG, Kirk CJ, Michell RH, and Michelangeli F (1992). The opening of the inositol 1,4,5-trisphosphate-sensitive Ca2+ channel in rat cerebellum is inhibited by caffeine. Biochem. J. 282 ( Pt 2), 309–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchheit CL, Weigel KJ, and Schafer ZT (2014). Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat. Rev. Cancer 14, 632–641. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, Urra F, Smith N, Molgó J, Diehl JA, et al. (2016). Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep 14, 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, and MacDonald IC (2002). Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2, 563–572. [DOI] [PubMed] [Google Scholar]
- Culver C, Sundqvist A, Mudie S, Melvin A, Xirodimas D, and Rocha S (2010). Mechanism of hypoxia-induced NF-kappaB. Mol Cell Biol 30, 4901–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ignazio L, Batie M, and Rocha S (2017). Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus GJ, Begthel H, et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature 521, 43–47. [DOI] [PubMed] [Google Scholar]
- Drost J, van Boxtel R, Blokzijl F, Mizutani T, Sasaki N, Sasselli V, de Ligt J, Behjati S, Grolleman JE, van Wezel T, et al. (2017). Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrand J, Nilsson H, Strömberg C, Jonas E, and Freedman J (2018). Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer 18, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey BJ, and Dueck D (2007). Clustering by passing messages between data points. Science 315, 972–976. [DOI] [PubMed] [Google Scholar]
- Fung C, Lock R, Gao S, Salas E, and Debnath J (2008). Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 19, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Elemento O, and Tavazoie S (2009). Revealing global regulatory perturbations across human cancers. Mol Cell 36, 900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guercio BJ, Sato K, Niedzwiecki D, Ye X, Saltz LB, Mayer RJ, Mowat RB, Whittom R, Hantel A, Benson A, et al. (2015). Coffee Intake, Recurrence, and Mortality in Stage III Colon Cancer: Results From CALGB 89803 (Alliance). J. Clin.Oncol. 33, 3598–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra MT, Florentino RM, Franca A, Lima Filho AC, Dos Santos ML, Fonseca RC, Lemos FO, Fonseca MC, Kruglov E, Mennone A, et al. (2019). Expression of the type 3 InsP3 receptor is a final common event in the development of hepatocellular carcinoma. Gut 68, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann M, Leucht K, Ploner C, Kiessling S, Villunger A, Becker H, Hofmann C, Falk W, Krebs M, Kellermeier S, et al. (2011). BCL-2 modifying factor (BMF) is a central regulator of anoikis in human intestinal epithelial cells. J Biol Chem 286, 26533–26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Ding M, Yuan C, Wu K, Smith-Warner SA, Hu FB, Chan AT, Meyerhardt JA, Ogino S, Fuchs CS, et al. (2018). Association Between Coffee Intake After Diagnosis of Colorectal Cancer and Reduced Mortality. Gastroenterology 154, 916–926.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungermann K, and Kietzmann T (2000). Oxygen: modulator of metabolic zonation and disease of the liver. Hepatology 31, 255–260. [DOI] [PubMed] [Google Scholar]
- Kadamur G, and Ross EM (2013). Mammalian phospholipase C. Annu. Rev. Physiol.75, 127–154. [DOI] [PubMed] [Google Scholar]
- Kang SS, Han K-S, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, Woo DH, Brat DJ, Hwang EM, et al. (2010). Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 70, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassambara A, Kosinski M, Biecek P, and others (2017). survminer: Drawing Survival Curves using’ggplot2’. R Package Version 0.3 1.
- Katona BW, Glynn RA, Paulosky KE, Feng Z, Davis CI, Ma J, Berry CT, Szigety KM, Matkar S, Liu Y, et al. (2019). Combined Menin and EGFR Inhibitors Synergize to Suppress Colorectal Cancer via EGFR-Independent and Calcium-Mediated Repression of SKP2 Transcription Cancer Res. 79, 2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-K, Kim S-Y, Kim J-H, Roh SA, Cho D-H, Kim YS, and Kim JC (2014). A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol Oncol 8, 1653–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, Washburn MP, Collazo-Lorduy A, Castillo-Martin M, et al. (2017). PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Wang J, Kong J, Tang J, Wu Y, Xu E, Zhang H, and Lai M (2016). GDF15 promotes EMT and metastasis in colorectal cancer. Oncotarget 7, 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockett J, Yin S, Li X, Meng Y, and Sheng S (2006). Tumor suppressive maspin and epithelial homeostasis. J Cell Biochem 97, 651–660. [DOI] [PubMed] [Google Scholar]
- Loo JM, Scherl A, Nguyen A, Man FY, Weinberg E, Zeng Z, Saltz L, Paty PB, and Tavazoie SF (2015). Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, and Groom AC (1998). Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 153, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangla A, Guerra MT, and Nathanson MH (2020). Type 3 inositol 1,4,5-trisphosphate receptor: A calcium channel for all seasons. Cell Calcium 85, 102132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A, Loo JM, Mital R, Weinberg EM, Man FY, Zeng Z, Paty PB, Saltz L, Janjigian YY, de Stanchina E, et al. (2016). PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. J. Clin. Invest. 126, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierro C, Cook SJ, Foets TCF, Bootman MD, and Roderick HL (2014). Oncogenic K-Ras suppresses IP₃-dependent Ca2+ release through remodelling of the isoform composition of IP₃Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci 127, 1607–1619. [DOI] [PubMed] [Google Scholar]
- Pirruccello M, Nandez R, Idevall-Hagren O, Alcazar-Roman A, Abriola L, Berwick SA, Lucast L, Morel D, and De Camilli P (2014). Identification of inhibitors of inositol 5-phosphatases through multiple screening strategies. ACS Chem. Biol. 9, 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev V, Doubrovin M, Serganova I, Vider J, Shavrin A, Beresten T, Ivanova A, Ageyeva L, Tourkova V, Balatoni J, et al. (2004). A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur. J. Nucl. Med. Mol. Imaging 31, 740–751. [DOI] [PubMed] [Google Scholar]
- Seta KA, Yuan Y, Spicer Z, Lu G, Bedard J, Ferguson TK, Pathrose P, Cole-Strauss A, Kaufhold A, and Millhorn DE (2004). The role of calcium in hypoxia-induced signal transduction and gene expression. Cell Calcium 36, 331–340. [DOI] [PubMed] [Google Scholar]
- Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, and Domany E (2009). Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 106, 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, Iwakiri Y, Nathanson MH, and Yamaguchi K (2010). The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 48, 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, and Jemal A (2020). Colorectal cancer statistics, 2020. CA Cancer J Clin. [DOI] [PubMed]
- Simpson CD, Anyiwe K, and Schimmer AD (2008). Anoikis resistance and tumor metastasis. Cancer Lett 272, 177–185. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S-C (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaman A, Amirtham U, and Rangarajan A (2016). Calcium-Oxidant Signaling Network Regulates AMP-activated Protein Kinase (AMPK) Activation upon Matrix Deprivation. J Biol Chem 291, 14410–14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, and Bray F (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. [DOI] [PubMed]
- Talmadge JE, and Fidler IJ (2010). AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 70, 5649–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Liu Z, Hou Y, Wang S, Liu S, Jiang Y, Tan D, Ge Q, Li C, Hu Y, et al. (2018). Alternative NF-κB signaling promotes colorectal tumorigenesis through transcriptionally upregulating Bcl-3. Oncogene 37, 5887–5900. [DOI] [PubMed] [Google Scholar]
- Therneau TM (2020). A Package for Survival Analysis in R.
- Ueasilamongkol P, Khamphaya T, Guerra MT, Rodrigues MA, Gomes DA, Kong Y, Wei W, Jain D, Trampert DC, Ananthanarayanan M, et al. (2020). Type 3 Inositol 1,4,5-Trisphosphate Receptor Is Increased and Enhances Malignant Properties in Cholangiocarcinoma. Hepatology 71, 583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu SD, Noothi SK, Pullum DA, Lawrie CH, Pallapati R, Potluri V, Kuntzen C, Khan S, Plas DR, Orlowski RZ, et al. (2015). Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat Commun 6, 8428. [DOI] [PubMed] [Google Scholar]
- Vieira AR, Abar L, Chan DSM, Vingeliene S, Polemiti E, Stevens C, Greenwood D, and Norat T (2017). Foods and beverages and colorectal cancer risk: a systematic review and meta-analysis of cohort studies, an update of the evidence of the WCRF-AICR Continuous Update Project. Ann Oncol 28, 1788–1802. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernández-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber F, Ooft SN, Dijkstra KK, and Voest EE (2017). Tumor Organoids as a Pre-clinical Cancer Model for Drug Discovery. Cell Chem Biol 24, 1092–1100. [DOI] [PubMed] [Google Scholar]
- Wen H, Xu WJ, Jin X, Oh S, Phan CHD, Song J, Lee SK, and Park S(2015). The roles of IP3 receptor in energy metabolic pathways and reactive oxygen species homeostasis revealed by metabolomic and biochemical studies. Biochim Biophys Acta 1853, 2937–2944. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161, 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi N, Weinberg EM, Nguyen A, Liberti MV, Goodarzi H, Janjigian YY, Paty PB, Saltz LB, Kingham TP, Loo JM, et al. (2019). PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, and He Q-Y (2016). ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst 12, 477–479. [DOI] [PubMed] [Google Scholar]
- Zarour LR, Anand S, Billingsley KG, Bisson WH, Cercek A, Clarke MF, Coussens LM, Gast CE, Geltzeiler CB, Hansen L, et al. (2017). Colorectal Cancer Liver Metastasis: Evolving Paradigms and Future Directions. Cell Mol Gastroenterol Hepatol 3, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Albanes D, Beeson WL, van den Brandt PA, Buring JE, Flood A, Freudenheim JL, Giovannucci EL, Goldbohm RA, Jaceldo-Siegl K, et al. (2010). Risk of colon cancer and coffee, tea, and sugar-sweetened soft drink intake: pooled analysis of prospective cohort studies. J Natl Cancer Inst 102, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Shan Z, Yang H, Xu J, Li W, and Guo F (2018). RelB plays an oncogenic role and conveys chemo-resistance to DLD-1 colon cancer cells. Cancer Cell Int. 18, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences.Bioinformatics 35, 2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited in GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. All data reported in this paper will be shared by the lead contact upon request.
The paper does not report original code.
Any additional information to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ITPR3 | BD Biosciences | Cat# 610313; RRID: AB_397705 |
| Rabbit polyclonal anti-RELB | Cell Signaling Technology | Cat# 4954; RRID: AB_330626 |
| Mouse monoclonal anti-Tubulin | Cell Signaling Technology | Cat# 3873; RRID: AB_1904178 |
| Mouse monoclonal anti-NFkB p52 | Millipore | Cat# 05-361; RRID: AB_309692 |
| Rabbit polyclonal anti-PLCG1 | Cell Signaling Technology | Cat# 2822; RRID: AB_2163702 |
| Rabbit polyclonal anti-INPP5A | Proteintech | Cat# 21723-1-AP; RRID: AB_10734438 |
| Rabbit polyclonal anti-Cleaved caspase-3 | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Mouse monoclonal anti-GAPDH | Proteintech | Cat# 60004-1-Ig; RRID: AB_2107436 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Rabbit polyclonal anti-LC3B | Cell Signaling Technology | Cat# 2775S; RRID: AB_915950 |
| HRP goat anti-mouse secondary antibody | Thermo Fisher Scientific | Cat# A16078; RRID: AB_2534751 |
| HRP goat anti-rabbit secondary antibody | Thermo Fisher Scientific | Cat# 65-6120; RRID: AB_2533967 |
| Bacterial and virus strains | ||
| Chemically competent One shot Stbl3 | Invitrogen | Cat# C737303 |
| Biological samples | ||
| Highly metastatic human CRC PDX | Tavazoie Lab (Yamaguchi et al., 2019) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Polybrene | Santa Cruz | Cat# sc-134220 |
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| G418 sulfate | Thermo Fisher Scientific | Cat# 10131027 |
| D-Luciferin | GoldBio | Cat# LUCK-100 |
| Caffeine | Sigma-Aldrich | Cat# C0750 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Lipofectamine RNAiMax | Thermo Fisher Scientific | Cat# 13778150 |
| Turbofect | Thermo Fisher Scientific | Cat# FERR0531 |
| cOmplete mini protease inhibitor | Roche | Cat# 11836153001 |
| RIPA lysis and extraction buffer | G-Biosciences | Cat# 786-490 |
| ECL Western blotting substrate | Pierce | Cat# 32106 |
| Fast SYBR Green Master Mix | Thermo Fisher Scientific | Cat# 43-856-18 |
| ACK lysing buffer | VWR | Cat# 12002-070 |
| Optiprep density gradient medium | Sigma-Aldrich | Cat# D1556 |
| Collagenase, type IV | Worthington | Cat# LS004188 |
| Matrigel | Corning | Cat# 354262 |
| Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | Cat# 31985062 |
| Fetal bovine serum | Sigma-Aldrich | Cat# F2442 |
| CellTracker Red CMPTX | Thermo Fisher Scientific | Cat# C34552 |
| CellTracker Green CMFDA | Thermo Fisher Scientific | Cat# C7025 |
| Tween-20 | Sigma-Aldrich | Cat# P9416 |
| DNAse1 | Sigma-Aldrich | Cat# D5025 |
| Y-27632 | Sigma-Aldrich | Cat# Y0503 |
| Collagenase, type XI | Sigma-Aldrich | Cat# C9407 |
| R-spondin | R&D | Cat# 4645-RS |
| Wnt 3a | R&D | Cat# 5036-WN |
| Cell recovery solution | Corning | Cat# 354253 |
| TrypLE Express Enzyme | Thermo Fisher Scientific | Cat# 12604013 |
| Lenti-X concentrator | Takara | Cat# 631231 |
| Tissue-Tek O.C.T Compound | VWR | Cat# 25608-930 |
| DAPI | Roche | Cat# 10236276001 |
| N-acetylcysteine | Sigma-Aldrich | Cat# A9165 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 |
| Recombinant murine Noggin | Peprotech | Cat# 250-38 |
| Recombinant human EGF | Peprotech | Cat# AF-100-15 |
| Recombinant human FFG-10 | Peprotech | Cat# 100-26 |
| Gastrin I (human) | TOCRIS | Cat# 3006 |
| A 83-01 | TOCRIS | Cat# 2939 |
| B27 supplement | Thermo Fisher Scientific | Cat# 17504044 |
| Prostaglandin E2 (PGE2) | TOCRIS | Cat# 2296 |
| Critical commercial assays | ||
| CellTiterGlo 2.0 Assay | VWR | Cat# PAG9241 |
| MTS cell proliferation assay kit | Abcam | Cat# ab197010 |
| Total RNA purification kit | Norgen | Cat# 37500 |
| CaspaseGlo 3/7 Assay | Promega | Cat# G8091 |
| VivoGlo Caspase 3/7 Substrate | Promega | Cat# P1782 |
| Mouse cell depletion kit | Miltenyi Biotec | Cat# 130-104-694 |
| Truseq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2002 |
| Quantseq 3’ mRNA-seq Library Prep Kit FWD | Lexogen | Cat# 015.96 |
| SuperScript III First-Strand Synthesis System for RT-PCR | Thermo Fisher Scientific | Cat# 18080051 |
| Deposited data | ||
| Microarray expression data from CRC patients | (Sheffer et al., 2009) | GSE41258 |
| RNA-seq expression data from CRC patients | (Kim et al., 2014) | GSE50760 |
| RNA-seq of ITPR3 KO in SW480 CRC metastases | This paper | GSE182257 |
| RNA-seq of SW480 CRC cells transfected with siRNAs targeting different putative metastasis promoting genes | This paper | GSE182386 |
| RNA-seq of SW480 CRC cells with loss of ITPR3 or RELB cultured under normoxic or hypoxic conditions | This paper | GSE197576 |
| Human refence genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Broad Institute TCGA data | (Broad Institute TCGA Genome Data Analysis Center, 2016) | https://doi.org/10.7908/C11G0KM9 |
| Experimental models: Cell lines | ||
| Human: SW480 cells | ATCC | Cat# CCL-228; RRID: CVCL_0546 |
| Human: LS174T cells | ATCC | Cat# CL-188; RRID: CVCL_1384 |
| Human: SW620 cells | ATCC | Cat# CCL-227; RRID: CVCL_0547 |
| Human: WiDr cells | ATCC | Cat# CCL-218; RRID: CVCL_2760 |
| Human: 293LTV cells | Cell Biolabs | Cat# LTV100; RRID: CVCL_JZ09 |
| Human: LS14T-LV2 cells | Tavazoie Lab | Derived from in vivo selection of LS174T cells |
| Mouse: MC38 cells | Kerafast | Cat# ENH204-FP; RRID: CVCL_B288 |
| Experimental models: Organisms/strains | ||
| Mouse: NSG | The Jackson Laboratory | Strain: 005557 |
| Mouse: C57Bl/6 | The Jackson Laboratory | Strain: 000664 |
| Oligonucleotides | ||
| shRNA sequences, see Table S5 | This paper | N/A |
| sgRNA sequences, see Table S5 | This paper | N/A |
| siRNAs, see Table S6 | Dharmacon | N/A |
| Primers for qRT-PCR, see Table S7 | This paper | N/A |
| Recombinant DNA | ||
| pLKO.1-puro | Addgene | Cat# 8453; RRID: Addgene_8453 |
| pCMV-VSG-G | Cell Biolabs | Cat# VPK-206 |
| pCgpV | Cell Biolabs | Cat# VPK-206 |
| pRSV-Rev | Cell Biolabs | Cat# VPK-206 |
| pSpCas(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat# 62988; RRID: Addgene_62988 |
| lentiCRISPR V2.0 | Addgene | Cat# 59261; RRID: Addgene_52961 |
| pCMV6-Entry | Origene | Cat# PS100001 |
| pCMV6-RELB | Origene | Cat# SC122747 |
| Software and algorithms | ||
| Prism 9 | GraphPad | https://www.graphpad.com; RRID:SCR_002798 |
| R v3.5.1. | R Core Team | https://www.r-project.org/; RRID: SCR_001905 |
| Fiji | (Schindelin et al., 2012) | https://imagej.net/software/fiji/ |
| STAR aligner (v2.6.0a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| featureCounts (v1.6.3) | (Liao et al., 2014) | https://bio.tools/featurecounts |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ReactomePA package for R | (Yu and He, 2016) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Package for survival analysis in R | (Therneau, 2020) | https://cran.r-project.org/web/packages/survival/index.html |
| Survminer package for R | (Kassambara et al., 2017) | https://cran.r-project.org/web/packages/survminer/index.html |
| QuPath (v0.3.2) | (Bankhead et al., 2017) | https://qupath.github.io |
| iPAGE | (Goodarzi et al., 2009) | https://tavazoielab.c2b2.columbia.edu/iPAGE |
| topGO package for R | (Alexa and Rahnenfuhrer, 2021) | https://bioconductor.org/packages/release/bioc/html/topGO.html |