

Abstract
RAD51 is a critical recombinase that functions in concert with auxiliary mediator proteins to direct the homologous recombination (HR) DNA repair pathway. We show that Cys319 RAD51 possesses nucleophilic characteristics and is important for irradiation-induced RAD51 foci formation and resistance to inhibitors of poly (ADP-ribose) polymerase (PARP). We have previously identified that cysteine (Cys) oxidation of proteins can be important for activity and modulated via binding to peroxiredoxin 1 (PRDX1). PRDX1 reduces peroxides and coordinates the signaling actions of protein binding partners. Loss of PRDX1 inhibits irradiation-induced RAD51 foci formation and represses HR DNA repair. PRDX1-deficient human breast cancer cells and mouse embryonic fibroblasts display disrupted RAD51 foci formation and decreased HR, resulting in increased DNA damage and sensitization of cells to irradiation. Following irradiation cells deficient in PRDX1 had increased incorporation of the sulfenylation probe DAz-2 in RAD51 Cys319, a functionally-significant, thiol that PRDX1 is critical for maintaining in a reduced state. Molecular dynamics (MD) simulations of dT-DNA bound to a non-oxidized RAD51 protein showed tight binding throughout the simulation, while dT-DNA dissociated from an oxidized Cys319 RAD51 filament. These novel data establish RAD51 Cys319 as a functionally-significant site for the redox regulation of HR and cellular responses to IR.
Keywords: Homologous recombination, Prdx1, RAD51, DNA repair, Sulfenylation
Graphical abstract
Highlights
-
•
A functionally-significant Cys319 was identified in RAD51 that possesses nucleophilic characteristics.
-
•
RAD51 Cys319 plays a central role in RAD51-mediated repair of DNA double strand breaks (DSB).
-
•
Loss of peroxiredoxin 1 (PRDX1) impairs DNA DSB repair by homologous recombination and results in DNA damage.
-
•
PRDX1 is critical for maintaining RAD51 Cys319 in a reduced state.
-
•
Molecular dynamic (MD) simulations suggest ssDNA to dissociate from sulfenylated and not reduced RAD51 Cys319.
1. Introduction
Oxidative damage to DNA occurs ∼104 times per cell on a daily basis [1]. The reactive species (RS) capable of DNA strand scission and base modification arise from basal metabolism, ultraviolet (UV) and ionizing radiation (IR), xenobiotic exposure and an assortment of primary and secondary products of tissue and inflammatory cell oxidases and oxygenases [2]. Maintenance of redox homeostasis is facilitated by low molecular weight oxidant scavengers, an array of enzymes that facilitate the reduction of free radicals, hydroperoxides and other oxidized cellular constituents and gene transcription mechanisms that sense cellular redox state [3]. Elevation of DNA-damaging radicals leads to mutations and genomic changes, with DSBs occurring upon replication fork collapse or damage on opposing strands [2].
The maintenance of genomic stability is important in preserving organismal homeostasis. More than 50,000 single-strand breaks (due to oxidative damage and base hydrolysis) and 50 DSBs are estimated to occur per cell per day [4,5]. Cells have developed an array of DNA repair pathways to ensure nucleotide insertion, deletion or mutational changes are minimized. DNA double-strand breaks (DSBs) are the most potent damaging events because they can lead to loss of genomic material. DSB are primarily repaired through the action of the nonhomologous end joining (NHEJ) and HR pathways or secondarily through the more deleterious single-strand annealing (SSA) and alternative end joining (Alt-EJ) pathways [6]. HR, SSA and Alt-EJ are downstream of DNA end resection pathways that produce 3′ ssDNA fragments, whereas the NHEJ doesn't utilize ssDNA substrates [7]. While individual DSBs are more frequently repaired through the more error-prone NHEJ, HR is a higher fidelity replication-associated pathway that does not cause loss of DNA and genomic instability when compared to NHEJ [6]. HR utilizes a homologous DNA template present during the S and G2 phases of the cell cycle to faithfully reconstitute lost genetic material surrounding the breakpoint. RAD51 is an essential recombinase for HR that works in concert with auxiliary mediator recombination proteins to assemble a pre-synaptic RAD51 filament that displaces RPA on ssDNA ends following end resection which then supports RAD51 filament growth to surround the ssDNA [[8], [9], [10], [11], [12], [13]]. The RAD51 filament then searches for a homologous sequence to perform strand invasion [14].
Endogenously and exogenously generated RS can both reversibly and irreversibly induce damage to proteins, lipids and DNA. Elevated peroxides are scavenged by the peroxiredoxin (PRDX) family, which consists of six members that enzymatically reduce peroxides via their active site peroxidatic cysteine (Cp), which is re-reduced predominantly by thioredoxin [15]. The PRDX proteins are subdivided into groups characterized by the presence of a “resolving” cysteine that enables a single (1-Cys; PRDX VI) or dual (2-Cys; PRDX I-IV and atypical 2-Cys; PRDX V) cysteine-dependent peroxide reduction [15]. The Cp of PRDXs is exceptionally reactive toward hydroperoxides. It has become evident that while peroxiredoxins were initially recognized as antioxidant proteins, they can influence various cell signaling pathways by direct protein interactions [16]. PRDX1 plays an important role in the prevention of malignancy through its peroxidase and protein chaperone signaling actions. Prdx1+/− and Prdx1−/− mice display decreased lifespan due to hemolytic anemia and the formation of lymphomas and various organ site sarcomas and carcinomas [17,18]. Prdx1−/− mouse embryonic fibroblasts (MEFs) display genomic instability with increased 8-oxoguanine DNA derivatives, compared with Prdx1+/+ MEFs [17]. Also, levels of intracellular reactive species are elevated in Prdx1−/− cells and display increased nuclear localization of c-Myc upon loss of PRDX1 activity [17,19]. The Prdx1 ortholog in yeast, Tsa1, also plays an important role in maintaining genomic stability and viability under aerobic conditions [20]. Specificity of the genome protective role of Prdx1 in comparison to Prdx2 has been shown in Tsa1 deletion mutants, which could be rescued with expression of human Prdx1, but not Prdx2 [21]. Tsa1 deletion mutants expressing Prdx1 compared to mutants expressing Prdx1 had decreased mutation rates in Canr (3-fold) and gross chromosomal rearrangements (GCR) (7-fold). Prdx1 was further able to rescue viability in cells challenged with H2O2 and 2.5 μg/ml antimycin A.
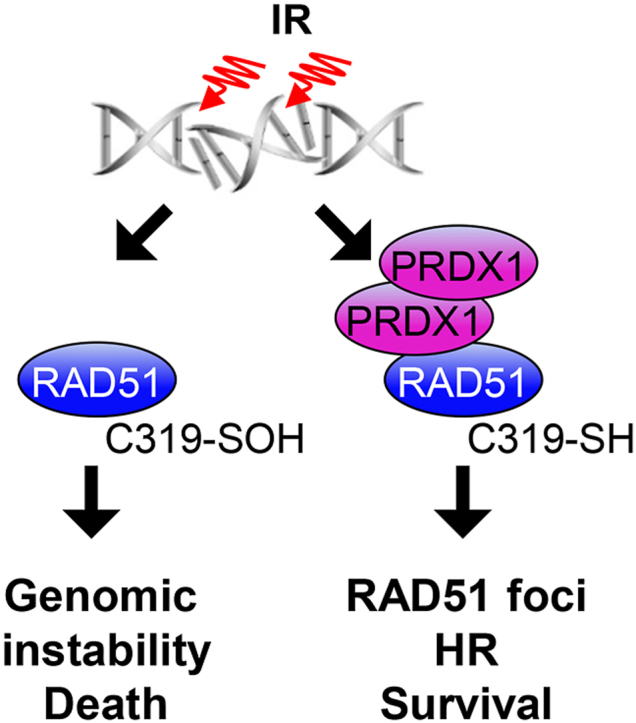
We report herein that PRDX1 protects RAD51 from oxidation and facilitates DNA repair reactions that promote cell survival. The critical role of a nucleophilic RAD51 Cys319 in promoting HR and cell survival in both non-malignant and breast cancer cells is reinforced by the observation that Cys319 mutant cells lose this adaptive phenotype. PRDX1-deficient breast cancer cells displayed heightened sensitivity to IR and decreased HR, as evidenced by impaired RAD51 foci formation. These results a) reinforce the concept that RS induce genomic perturbations via the disruption of HR, and b) demonstrate that HR can be regulated by modulating the redox state of RAD51 Cys319.
2. Results
2.1. RAD51 Cys319 has reactive nucleophilic characteristics
Our previous work demonstrated thiol alkylation of RAD51 Cys319 via a Michael Addition reaction to the electrophilic nitro-fatty acid OA-NO2 diminished HR [22]. Because the Cys319 residue displayed specific reactive properties to an endogenously detectable lipid signaling mediator and was able to alter RAD51 function, we further investigated the intrinsic nucleophilic properties of Cys residues and their function in RAD51 protein. Using the cryo EM structure (5NP7) and PROPKa 3.0, we calculated the pKa of Cys residues present within the three-dimensional RAD51 protein structure (Fig. 1A). A free Cys residue in solution exhibits a theoretical pKa of 8.6, which can be influenced by surrounding amino acids in the three-dimensional space of the folded protein. In monomeric RAD51, Cys31 had a calculated pKa of 9.0, while Cys137, 144 and 312 all had pKa values of 10 or higher. In contrast, PROPKa 3.0 calculated Cys319 RAD51 had a pKa value of 7.24, due to coulombic interactions with Arg96 that stabilize the negative charge of the deprotonated Cys thiolate. As RAD51 protein can form higher order homo-oligomeric structures to produce a pre-synaptic filament that surrounds ssDNA to promote recombination, we also investigated whether Cys reactivity was altered when RAD51 adopted this active recombination form. In the filament, Cys319 RAD51 on protomer B was present at the protomer-protomer interface and the opposing Arg167 RAD51 on protomer A was able to cause a two-unit decrease in the PROPKa calculated pKa value of Cys319 (Fig. 1B and C). These interactions at the boundary between RAD51 proteins in the filament led to a further shift in the theoretical calculated Cys319 pKa from 7.24 on the initial RAD51 protein, to between 5.3 and 5.6 in the subsequent Cys319 residues. The importance of changes to the Cys pKa value are exemplified by the proportion of reactive negatively charged Cys thiolates present as the pKa value approaches or drops below physiological pH as dictated by the Henderson-Hasselbalch equation for the deprotonation of Cys (Supplemental Fig. S1A equations). Equilibrium constant calculations show Cys thiolates in the RAD51 filament were primarily located on Cys319 residues, ranging from about 50% deprotonated on protomer A to 99% on protomers at the interface between RAD51 proteins in the filament (Fig. 1D). To observe the electrostatic effects of Arg167 on the pKa and proportion of thiolates to protonated thiols present in the RAD51 filament, Arg167Ala mutant calculations were made (Fig. 1E and Supplemental Fig. 1B). The pKa of Cys319 RAD51 in protomer A remained unchanged, as expected, but the pKa and thiolate character of the Cys319 in subsequent RAD51 protomers were increased to above 8 and below 5%, respectively. As thiol-based redox signaling through specific Cys residues can coordinate protein function, activity, abundance and binding partners, we specifically tested whether irradiation-induced RAD51 foci formation changes were present in Cys319Ser mutants compared to standard pKa Cys residues. Immunofluorescence microscopy image analysis showed that the average number of RAD51 foci/cell was significantly lower in cells harboring RAD51 Cys319Ser compared to RAD51 WT, Cys137 or Cys312 mutants, following IR exposure in DT40 cells (Fig. 1F and Supplemental S1C). Average γH2AX foci per cell was also significantly increased in RAD51 Cys319Ser mutant containing cells compared to RAD51 WT, Cys137 or Cys312 mutants (Fig. 1G).
Fig. 1.

RAD51 Cys319 has reactive nucleophilic characteristics. (A-C) The cryo EM structure of human RAD51 (5NP7) on single-stranded DNA and PROPKa 3.0, were used to calculate theoretical pKa values of Cys residues present within the three-dimensional RAD51 protein structure of monomeric RAD51 and the oligomerized RAD51 pre-synaptic filament (A-E). (B–C) RAD51 Cys319 is located at the interface between RAD51 protomers near intra- (Arg96) or intermolecular (Arg167) Arg residues that decrease thiol pKa. (D) Equilibrium constant calculations for the deprotonation of Cys were performed using the Henderson-Hasselbalch equation and the pKa values obtained in (C). Calculations of pKa were performed using PROPKa 3.0 on Arg167 RAD51. (F-G) RAD51 foci formation was decreased and γH2AX was increased in DT40 cells harboring RAD51C319S protein following IR with 5 Gy. DT40 cells were transduced with WT or cysteine mutant RAD51 constructs and dosed with 0 or 5 Gy and quantified by confocal fluorescence microscopy for average RAD51 foci per cell or γH2AX foci per cell in three independent experiments.
2.2. Cys319Ser RAD51 cells exhibit diminished IR-induced RAD51 foci formation and proliferation and increased sensitivity to PARP inhibitors
Previous studies have reported that recombinant RAD51 Cys319Ser proteins behave phenotypically like RAD51 WT proteins in D-loop and ATP hydrolysis assays in vitro [23], but cells harboring RAD51 Cys319 mutant proteins have not yet been reported in cell-based assays. Further testing of the effects of Cys319Ser RAD51 was performed in MDA-MB-231 cells mutated via CRISPR/Cas-9 to evaluate the functional consequences of the thiol at this site. Irradiation-induced RAD51 foci formation was diminished in cells with homozygous Cys319Ser mutation when compared to MDA-MB-231 cells that harbored WT RAD51 (Fig. 2A). Proliferation of cells with homozygous Cys319Ser RAD51 was also slower compared to WT RAD51 MDA-MB-231 cells (Fig. 2B). The functional importance of RAD51 Cys319 was further confirmed in DT40 cells with wild-type or mutant RAD51 expression under a Tet-repressible RAD51 transgene system with the endogenous RAD51 locus deleted [24]. Doxycycline (Dox) repression of RAD51 suppressed proliferation in control Tet-off DT40 cells and Tet-off DT40 cells that expressed human RAD51C319S, but not Tet-off DT40 cells that expressed RAD51 Cys137Ser or Cys312Ser mutants (Fig. 2C and Supplemental Figs. S2A and S2B). Cell cycle profiling of MDA-MB-231 cells showed a decreased number of cells in G1 phase in RAD51 Cys319Ser cells from 60% in control cells to 49% in Cys319Ser mutant cells; with an apparent G2 block, as an increase in G2 was also seen (23%–37%) (Fig. 2D). To determine if functional RAD51 was diminished and caused Cys319Ser mutant MDA-MB-231 cells to be more susceptible to single-strand DNA repair inhibitors, cells were treated with the poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) olaparib and talazoparib. Cys319Ser mutant MDA-MB-231 cells were sensitized to olaparib with EC50 values more than two-fold lower than cells harboring wild-type RAD51. Talazoparib treatment also exhibited that RAD51 Cys319Ser cells were sensitized, with EC50 values more than three-fold lower than controls (Fig. 2E). A soft agar colony formation assay was performed to measure whether anchorage-independent growth was altered and demonstrated that RAD51 mutation to Cys319Ser decreased colony formation more than two-fold (Supplemental Fig. S2C). Overall, these data suggest that the thiol moiety in Cys319 RAD51 is functionally important.
Fig. 2.

Cys319Ser RAD51 cells exhibit diminished IR-induced RAD51 foci formation and proliferation and increased sensitivity to PARP inhibitors.
(A) CRISPR/Cas-9 mutated homozygous Cys319Ser MM231 cells have decreased RAD51 foci formation (green) following IR with 5 Gy. DAPI stained nuclei (blue) merged images are shown. Cells plated on coverslips were dosed with 5 Gy then processed for IF after 5 h. Mean percentages of RAD51 WT (black) or RAD51 Cys319Ser (gray) cells with 5 or more foci from confocal z-stacked images dosed with 0 or 5 Gy + SEM from 3 independent experiments are shown.
(B) Proliferation of MM231 cells harboring RAD51 WT (black) or homozygous CRISPR/Cas-9 mutated Cys319Ser RAD51 (gray) was measured using the Incucyte ZOOM automated microscope to determine the percent confluence of phase contrast images over 144 h.
(C) Relative cell proliferation following doxycycline induced inhibition of conditionally Rad51 null DT40 cells. Doxycycline inhibits WT RAD51 (●) and leaves only transgenic expression of RAD51C319S (▼), C137S (■) or C312S (▲) mutants as indicated. Curve shows mean cell percentage difference ± SEM of the relative cell number in the presence and absence of the Tet-controlled transgene.
(D) Cell cycle analysis of propidium iodide stained RAD51 WT MM231 or CRISPR Cys319Ser RAD51 mutated cells was performed by flow cytometry. Mean ± SEM, n = 3 (E) Dose response assays (6-points, 0–50 μM) were performed with talazoparib or olaparib over 7-days and EC50 values are indicated as mean ± SEM, n = 3. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.3. PRDX1 binds RAD51 and inhibits sulfenylation
Thiolates are readily modified by post-translational modifications (PTMs) such as disulfide bonding, glutathionylation, or oxidation to sulfinic (SO2H) or sulfonic acid (SO3H) [25]. Our laboratory has previously identified Cys oxidation of proteins as an important repressor of activity in proteins such as the protein phosphatases PTEN [26] and MKP-5 [27], which can be controlled via binding to the peroxidase, PRDX1. We and others have shown that PRDX1 deficiency increases mutational frequency and genomic instability [[28], [29], [30]]. As this suggested that PRDX1 promotes genomic integrity, a role for PRDX1 in RAD51 control was explored. First, we tested if a direct PRDX1-RAD51 complex was observed in vitro by co-immunoprecipitation (IP) of purified RAD51 and PRDX1 proteins with PRDX1 antibody (Fig. 3A). We observed that PRDX1 antibody could precipitate RAD51 only when PRDX1 and RAD51 proteins were incubated together. To determine the KD of the interaction between the proteins, Alexa Fluor 546 labelled PRDX1 was incubated with RAD51 labelled with the QSY 35 quencher and the deceased signal was used to determine the KD. Two apparent KD values were observed at 140 and 610 nM (Fig. 3B). To observe if oxidative insults altered PRDX1-RAD51 complex formation in cells, FLAG-RAD51 was transfected into 293T cells and the ability of endogenous PRDX1 to bind FLAG-RAD51 was examined by co-immunoprecipitation (IP) analysis following irradiation (IR). There was an IR-dependent decrease in PRDX1-RAD51 complex formation for monomeric PRDX1, while dimeric PRDX1 levels remained consistent (Fig. 3C). Cells were lysed in buffer containing catalase and 100 mM N-ethylmaleimide to limit oxidation and thiol-disulfide exchange reactions to avoid the possibility of non-specific protein-protein interactions during sample processing. No disulfide linked PRDX1-RAD51 complexes were apparent in SDS-PAGE experiments when reducing and non-reducing conditions were compared (Supplemental Fig. S3A). To determine if loss of PRDX1 affected RAD51 protein cysteine oxidation, the relative concentration of Cys sulfenylation (Cys-OH) - the two-electron thiol oxidation product of Cys, was detected by measuring incorporation of an azide-tagged dimedone derivative (DAz-2) (Fig. 3D) [31]. Following IR (5 Gy), RAD51 sulfenylation was significantly increased in Prdx1−/− MEFs relative to Prdx1+/+ MEFs and Prdx1−/− MEFs expressing HA-PRDX1 (Fig. 3E). Under basal conditions in the absence of IR, Prdx1−/− MEFs displayed a 20% increase in DAz-2 incorporation when probing for cysteine sulfenylation in comparison to WT MEFs (Supplemental Fig. S3B). This result suggested that PRDX1 protects RAD51 cysteines from oxidation after genotoxic insult that produces DNA double-strand breaks. To identify site-specific cysteine sulfenylation of RAD51, RAD51 Cys mutants (solvent-exposed Cys312 and Cys319 as well as non-solvent-exposed Cys137 as a control) were engineered and tested in Prdx1−/− MEFs. RAD51 Cys319 mutant protein displayed a 2-fold decrease of oxidation as measured by DAz-2 incorporation, compared to wild-type RAD51 (Fig. 3F), suggesting that PRDX1 specifically prevents RAD51 Cys319 oxidation. Further studies to observe the relative levels of reversible Cys319 RAD51 oxidation products following IR in MCF10A non-transformed mammary epithelial cells were attempted, but failed due to issues related to Cys319 peptide detection. Mass spectrometry analysis of endogenous RAD51 immunoprecipitated from MCF10A lysates was able to detect co-precipitated PRDX1 peptides though, which corroborated the data seen in Fig. 3A, B and C.
Fig. 3.

PRDX1 binds RAD51 and loss of PRDX1 increases RAD51 sulfenylation.
(A) In vitro co-IP reactions were performed with purified RAD51 and PRDX1 proteins by precipitating PRDX1 with antibody and immunoblotting for RAD51.
(B) Alexa Fluor 546 labelled PRDX1 was incubated with increasing concentrations of RAD51 labelled with the QSY35 quencher (0–1600 nM) to produce a binding curve where the decease of signal was used for KD quantification.
(C) FLAG immunoprecipitation of 293T cells demonstrated that endogenous dimeric and monomeric PRDX1 could bind FLAG-RAD51 when probed under non-reducing conditions. 293T cells were transfected with FLAG-RAD51 and irradiated with 0–10 Gy.
(D) DAz-2 reaction scheme.
(E) Prdx1−/− MEFs transduced with FLAG-RAD51 and HA-Prdx1 exhibited decreased sulfenylation as measured by DAz-2 incorporation. MEFs were treated with DAz-2 (5 mM) for 1.5 h and DAz-2 incorporation was detected by chemiluminescence by utilizing ligation of p-biotin and binding of streptavidin-HRP. Mean ± SEM as indicated.
(F) Prdx1−/− MEFs transduced with WT or cysteine mutant RAD51 expression constructs exhibited decreased sulfenylation by DAz-2 incorporation in RAD51C319 mutant samples. MEFs transduced with a RAD51 expression construct were treated with DAz-2 (5 mM) for 1.5 h. DAz-2 incorporation was detected by chemiluminescence by utilizing ligation of p-biotin and binding of streptavidin-HRP. The mean and range of two experiments is indicated.
2.4. All atom MD simulations of a tetrameric RAD51-DNA filament: implications for oxidation of Cys319
To observe the implications of oxidation of Cys319 RAD51, computational studies on a RAD51 filament model were performed. Superimposing a previously published all atom homology model [32] onto a tetrameric cryo-EM structure of RAD51 (5NP7) [33], and poly-dT from (3CMU) [34] structure yielded a starting model for an MD-based refinement (Fig. 4A). The native system came to a global RMSD equilibrium after about 7 ns, then stabilized and the simulation was stopped at 44 ns. (Supplemental Fig. S4A). RMSD is a readout for the structural distance between coordinates. A parallel MD was performed for the oxidized Cys319, in which each Cys319 sulfur of the RAD51 filament was converted to the sulfenic acid form R-S-OH, starting from the energy minimized native form. This oxidized system took about 20 ns to come to a global RMSD equilibrium (Supplemental Fig. S4B). However, in the oxidized system, due to the large RMSF in the DNA and to some extent the protein itself, it was necessary to extend the simulation longer (∼149 ns) (Supplemental Figs. S4C and S4D). RMSF measures the average deviation of a protein residue over time from a reference point as the time-averaged position. Another notable feature of the oxidized system was the large reduction in the RMSF values for the all of the ATP residues compared to the native system (Supplemental Figs. S4E and S4F).
Fig. 4.

MD simulations of a tetrameric RAD51-DNA filament show sulfenylation of Cys319 destabilizes ssDNA binding.
(A) The model was designed by superposing our previously published all atom homology model [32] onto a tetrameric cryo-EM structure of RAD51 (5NP7), and subsequently superposing poly-dT from a RecA (3CMU) structure. This homology model was a starting point for an MD-based refinement, and subsequent MD simulations comparing native and Cys319 oxidized filaments. RAD51 protomers are labelled 1–4 with ssDNA.
(B) The last snap shot of a refined MD for native (upper) and oxidized (lower) RAD51 filaments with different protomers colored in purple, blue, green and yellow demonstrated ssDNA (poly-dT ball and stick) was destabilized in the oxidized RAD51 filament from binding residues in loops 232–237 and 278–288 present in the native RAD51 Cys319 filament.
(C) Superposed DNA snapshots without RAD51 filaments shown revealed that the native RAD51 filament yielded stable DNA positioning in an extended conformation throughout the simulation (left), while the RAD51 filament with oxidized Cys319 residues exhibited a complex behavior where the 5′ region of the ssDNA initially became unbound from monomer 1 and 2 (purple) and then migrates over the course of the simulation to bind the N-terminal domain of monomer 1 (green). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The oxidized simulations showed that the 5′ region of DNA became unbound from the first monomer's DNA-binding loops and eventually relocated to a new binding site on the N-terminal region of monomer 1. In order to summarize the large change in the 5′ region of the DNA ligand, we compared the last snapshots of the two simulations (native and oxidized) in Fig. 4B. The multi-colored molecular surface of the native RAD51 filament bound the ssDNA in the orthodox mode, to loops 232–237 and 278–288 throughout the simulation. However, there was a radical relocation of the 5′ end of the DNA in the oxidized system, in which it fully dissociated from the orthodox DNA binding loops in monomer 1, and largely from monomer 2 (Fig. 4B). As mentioned above, the DNA in the oxidized system had a much larger RMSF than the native system (Supplemental Figs. S4C and S4D). Plotting the superimposed snapshots where these transitions occurred revealed the DNA always remained bound in the orthodox manner in the native system (the protein portion is hidden for clarity). However, in the oxidized filament, radical changes in the location of the 5′ end of the DNA were observed during the course of the simulation (Fig. 4B). Initially, there was little to no contact with monomer 1, followed by a weak complexation, and finally, surprisingly binding to the N-terminal domain. This was also supported by plots comparing the DNA RMSD as a function of time (Supplemental Figs. S4G and S4H), which showed the oxidized system had periodic large spikes in RMSD as this transition was taking place). In order to capture all of these changes, Supplemental Fig. S4I to S4L) plot the per-residue contacts over the course of the simulation for the WT and oxidized systems, respectively. The highest quality contacts between the RAD51 and the DNA for the WT system were in monomers 1 and 2, which are depicted in Supplemental Figs. S4I and S4J, respectively. We observed that the WT system had varied and rich contacts with residues in both the loops 232–237 and 278–288. However, in the oxidized system contacts to the 232–237 loop were totally lost in monomer 1, and atrophied in the 278–288 loop, and eventually lost to binding of a loop in the N-terminal region (Supplemental Fig. S4K and S4L).
An important structural trend that occurred in the RAD51 filament was that the helix that flanked the 232–237 loop became more disordered in the oxidized system, and the loop pulled back from the DNA during the simulation. This was a key point in the unbinding of DNA. Dynamic Cross Correlation Matrices (DCCMs) from MD simulations (Supplemental Fig. S4M and S4N) showed that there were enhanced coupled motions between secondary structural elements in the oxidized system, particularly in intramolecular contacts between the beta-sheets and the alpha-helices in monomers 1 and 2, respectively. This may be contributing to the changes in the secondary structure that occur at the point of DNA unbinding.
2.5. PRDX1-deficiency increases DNA damage, but decreases RAD51 foci and HR
To determine the requirement for PRDX1 in IR-induced RAD51 nuclear foci formation, we examined RAD51 foci formation in MEFs following 5 Gy IR. The percentage of Prdx1−/− MEFs that contained more than 10 foci was significantly decreased 2 h following IR exposure compared with Prdx1+/+ MEFs, but not cells with 5–10 foci (Fig. 5A and Supplemental Fig. S5A). Elevated levels of DNA damage were present in irradiated Prdx1−/− MEFs compared to Prdx1+/+ MEFs as indicated by nuclear γH2AX levels following a dose of 0 or 5 Gy (Fig. 5B and Supplemental Fig. S5B). Experiments probing cytoplasmic and nuclear localization of RAD51 in irradiated Prdx1−/− and Prdx1+/+ MEFs showed that nuclear RAD51 protein levels were decreased in Prdx1−/− MEFs following 0 or 5 Gy IR (Fig. 5C). We next explored the role of PRDX1 in IR-dependent cell death by testing Prdx1−/− MEFs harboring HA-tagged wild-type or catalytically-inactive (CI) PRDX1 (C52S and C173S) [26]. A colony formation assay showed Prdx1−/− MEFs transduced with empty vector (EC50 = 4.6 ± 0.1 Gy) or HA-PRDX1 CI (EC50 = 5.2 ± 0.1 Gy) expression constructs were more sensitive to increasing doses of IR, compared with cells that expressed HA-wild-type PRDX1 (EC50 = 7.0 ± 0.1 Gy) (Fig. 5D). These changes were also evident when comparing Prdx1+/+ and Prdx1−/− MEFs (Supplemental S5C). Downstream RAD51 activity was studied by utilizing the direct repeat GFP assay (DR-GFP). In this assay, homologous-directed recombination of a modified tandem GFP reporter was measured, which contains two differently mutated GFP cDNA fragments. Transient I-SceI expression then initiates a DSB at the GFP-SceI site. Repair of the DSB by a non-crossover gene conversion downstream of the second GFP cDNA fragment results in reconstitution of a functional GFP coding sequence with loss of the I-SceI site and gain of a BcgI site (Supplemental Fig. S5D) [35]. The DR-GFP assay showed a more than 2-fold decrease of GFP recombination in Prdx1−/− MEFs after I-SceI overexpression, compared with Prdx1+/+ MEFs (Fig. 5E). The effect of Cys319 RAD51 mutation in HR-mediated repair was next tested by DR-GFP analysis in Prdx1−/− MEFs. HR-mediated DNA repair was decreased 2-fold in RAD51 Cys319Ser-expressing cells, compared to RAD51 WT cells (Fig. 5F). Similar levels of RAD51 WT and mutant protein expression were seen as detected by immunoblot when DSBs were induced by I-SceI in Prdx1−/− MEFs (Supplemental Fig. S5E). In summary, these data suggest that PRDX1 regulates HR-mediated DNA repair and thus genomic stability by affecting RAD51 nuclear foci formation.
Fig. 5.

PRDX1-deficiency sensitizes cells to IR and inhibits both HR and RAD51 foci formation.
(A) IF images of Prdx1+/+ or Prdx1−/− MEFs stained with RAD51 antibody post-IR with 5 Gy. Scale bar indicates 10 μm. PRDX1 deficiency decreased RAD51 foci formation when challenged with IR. Prdx1+/+ (black bars) or Prdx1−/− (white bars) MEFs underwent IR with 0 or 5 Gy and RAD51 foci were counted after 2 h. Values represent cells with more than 10 RAD51 foci per cell.
(B) PRDX1 deficiency increased ɣH2AX foci formation when challenged with IR. Prdx1+/+ (black bars) or Prdx1−/− (white bars) MEFs underwent IR with 0 or 5 Gy and ɣH2AX positive cells were measured after 2 h.
(C) Immunoblots of cell fractionation lysates of Prdx1+/+ or Prdx1−/− MEFs following 0 or 5 Gy with post-IR time points from 0 to 120 min.
(D) Loss of PRDX1 activity inhibited clonogenic growth with increasing ɣ-irradiation (IR). Prdx1−/− MEFs were reconstituted with empty vector (ㅇ) or HA tagged wild-type (■) or catalytically inactive PRDX1 ( ). EC50 values indicate mean + SEM, n = 3.
). EC50 values indicate mean + SEM, n = 3.
(E) Loss of PRDX1 activity decreased homologous recombination when DNA double strand breaks were induced in MEFs. Prdx1+/+ (black bars) or Prdx1−/− (white bars) MEFs were infected with a DR-GFP expression vector and the recombined GFP signal was measured in the presence or absence of I-SceI.
(F) DR-GFP assay shows the relative HR in Prdx1−/− MEFs transduced with WT or cysteine mutant RAD51 constructs transfected with I-SceI.
2.6. PRDX1-deficiency sensitizes breast cancer cells to IR and diminishes RAD51 foci intensity
To determine if PRDX1-deficient cancer cells are also sensitized to IR-induced cell death, PRDX1-proficient or deficient MDA-MB-231 triple-negative breast cancer cells were treated with 0 or 5 Gy IR. PRDX1-deficient MDA-MB-231 cells were more sensitive to IR (EC50 = 1.5 ± 0.1 Gy) than control cells (EC50 = 4.0 ± 0.2 Gy) (Fig. 6A). As observed in MEFs, the fold-change in IR-induced RAD51 foci formed was decreased in PRDX1-deficient MDA-MB-231 cells when compared to controls (Fig. 6B), although the percentage of irradiated shPRDX1 MDA-MB-231 cells that contained more than 5 foci was comparable to control cells (Supplemental Figs. S6A and S6B). Thus, RAD51 peak foci intensities were compared as both foci number and intensity can reflect the dynamics of RAD51 recruitment to DSB sites [36,37]. In three separate experiments, total foci quantified per group were MDA-MB-231 EV + 0 Gy (311), EV + 5 Gy (911), shPRDX1 + 0 Gy (277) and shPRDX1 + 5 Gy (1232) (Fig. 6C). RAD51 peak foci intensities were examined by comparing the proportion of foci over the intensity distributions using an empirical cumulative distribution function plot and comparing the populations by Mann-Whitney test (Fig. 6D) and Supplemental Table S1). It was found that the foci intensity of PRDX1-proficient and PRDX1-deficient MDA-MB-231 cells was increased significantly in both cell sample groups after IR exposure. Loss of PRDX1 did not significantly change peak foci intensity of non-irradiated MDA-MB-231 cells (p = 0.6), but irradiated shPRDX1 MDA-MB-231 cells did trend towards a decreased RAD51 foci intensity when compared to irradiated control MDA-MB-231 cells (p = 0.084). A box and whiskers plot indicates the spread of the RAD51 foci peak intensity values (Supplemental Fig. S6C). The changes in peak IR-induced foci intensity were significantly larger in EV MDA-MB-231 cells than shPRDX1 cells (p = 0.0006) (Fig. 6E). As RAD51 foci formation is highest during S-phase [38], cell cycle changes were also examined. No significant changes to the cell cycle were observed in propidium iodide stained PRDX1-deficient breast cancer cells when compared to control by flow cytometry (Supplemental Fig. S6D).
Fig. 6.

PRDX1-deficiency sensitizes breast cancer cells to IR and diminishes RAD51 foci intensity.
(A) PRDX1-deficient (□) MDA-MB-231 breast cancer cells were more sensitive to IR when compared to PRDX1-proficient EV (empty vector) cells (●). Clonogenic growth inhibition was compared in MDA-MB-231 cells infected with shPRDX1 or EV expression vectors dosed with 0–4 or 8 Gy. EC50 value indicate mean + SEM, n = 3. (B-E) PRDX1-deficient MDA-MB-231 cells had decreased RAD51 foci formation following IR with 5 Gy. Cells plated on 16-well coverslips were dosed with 5 Gy then processed for IF 6 h later.
(B) The mean fold-change of irradiated to non-irradiated cells with 5 or more RAD51 foci from confocal z-stacked images are indicated from PRDX1-proficient (black) ordeficient (white) samples + SEM, n = 3. (C-E) RAD51 peak foci intensities were measured with MATLAB from 3 independent experiments.
(C) Representative RAD51 foci (green) from raw pseudo-colored images. Scale bar indicates 500 nm. (D-E) IR enhanced RAD51 peak foci intensity more significantly in MDA-MB-231 EV cells compared to MDA-MB-231 shPRDX1 cells.
(D) The difference between EV and shPRDX1 ± IR are indicated. Mean ± SE
(E) An empirical cumulative distribution function plot of peak RAD51 foci intensity values of PRDX1-deficient (▬ or ▬) or proficient (▬ or ▬) MDA-MB-231 cells in the presence or absence of 5 Gy from more than 20 cells per group. Cell populations were normalized to 1 and the peak foci intensity percentages were compared. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Discussion
This study reveals the novel importance of Cys319 RAD51 in the intracellular environment. Previously, our lab has shown that Cys319 RAD51 is targeted by the electrophilic nitroalkene OA-NO2 [22]. Other reports have similarly demonstrated that adducts targeting Cys319 can inhibit RAD51 foci formation, namely RI-1 [39] and the maleimide linked fluorophore Alexa Fluor 594 [23]. Interestingly, recombinant RAD51 Cys319Ser proteins behave phenotypically like RAD51 WT proteins in D-loop and ATP hydrolysis assays in vitro [23], but our study shows inhibition of RAD51 foci formation and HR in cells harboring RAD51 Cys319Ser mutant proteins (Fig. 1, Fig. 2, Fig. 4). The cryo EM structure of RAD51 (5NP7) suggests that the side chain sulfur group of Cys319 in a population of monomeric RAD51 proteins exists with an equal proportion of protonated and de-protonated thiolates (Fig. 1). The thiolate character is stabilized by coulombic interactions with Arg96 that lower the Cys pKa. Oligomerization of RAD51 further enhances the proportion of thiolates present in the population due to the presence of Arg167 on the neighboring RAD51 protomer. RAD51 exists as monomeric and multimeric oligomers in the intracellular environment and this may have important redox implications for the temporal control of HR. Interestingly, the importance of Arg167 RAD51 has previously been noted to be involved in RAD51 filament oligomerization [40]. Mutation of Cys319 by site-directed mutagenesis or CRISPR/Cas-9 revealed that IR-induced RAD51 foci formation was diminished and cells were sensitized to PARPi (Fig. 1, Fig. 2). That under basal conditions we only see changes to RAD51 foci formation in the C319S RAD51 mutant cells without changes to γH2AX could reflect the ability of cells to bypass homologous recombination for other repair pathways. Probing RAD51 foci enables these homologous recombination DNA recombination repaired breaks to be visualized, but under basal conditions, cells may more easily bypass DNA double-strand break through RAD51 by choosing pathways such as NHEJ or secondarily through SSA and Alt-EJ to diminish DNA damage as seen via γH2AX with cells that harbor C319S RAD51 mutant protein. The absence of changes to basal DNA damage in C319S RAD51 may therefore reflect an elevation of other pathways that are more easily upregulated to meet the demands of DNA repair under basal conditions, but not when challenged with IR.
In the MD simulation (Fig. 4), it is surprising that a single heavy atom change in the Cys319 residue could create such a precipitous change in the basic DNA binding activity of RAD51. Nevertheless, it is not currently clear exactly how this subtle change is manifested in these large-scale dynamic changes. It is worth noting that whether or not the binding of the dislodged DNA to the N-terminal region in the oxidized simulation is an artifact of the simulation, the DNA is still unbinding from the orthodox DNA binding loops very early in the simulation, which strongly suggest an impairment of activity either way. Interestingly, there is evidence in the literature that the N-terminal region does possess a DNA-binding activity [41]. Finally, the Cys319 also is known to undergo reaction with covalent drugs as indicated above for OA-NO2 and RI-1, which obviously raises questions about whether the mode of action of these drugs might occur in a similar manner to the oxidative process described here.
The role of oxidation of proteins as a PTM to modulate activity is more frequently being recognized as significant and has been shown in proteins such as EGFR, PKC, Src, Akt2, APE1 and PTEN [42,43]. We and others have shown that PRDX1 can alter partner protein oxidation and that PRDX1 deficiency increases mutational frequency and genomic instability [17,[28], [29], [30]]. The importance of PRDX1 in maintaining genomic stability was shown in S. cerevisiae, where deletion of the PRDX1 ortholog TSA1 caused gross chromosomal rearrangements and increased mutation rates [29]. Loss of Tsa1 activity constitutively activated the DNA damage checkpoint and augmented intracellular deoxynucleotide concentrations [30]. Synthetic lethality could be achieved upon co-deletion of TSA1 and the DNA repair genes RAD51, RAD6 or MRE11 [44]. Genomic stability and synthetic lethality of TSA1 single and double mutants could be resolved when cells were grown under anaerobic conditions, which reinforced the significance of oxidant reactions in genome stability. We detected a direct PRDX1-RAD51 complex in vitro and in cell-based assays by co-IP using both purified or endogenous RAD51 and PRDX1 proteins (Fig. 3). Co-IP experiments under non-reducing conditions demonstrate monomeric and dimeric binding of PRDX1 on RAD51, which help explain two apparent KD values obtained in Fig. 3B. MS experiments found that endogenous RAD51 could bind Prdx1, but not peptides for Prdx2 were observed. It is possible that Prdx1 could influence RAD51 function through chaperone activity following Prdx1 oligomerization in addition to scavenging H2O2. Peroxide scavenging may not necessarily require RAD51 protein binding as well, which suggests other PRDX family members may also play a role. Based on the enzymatic activity of Prdx1/2 towards H2O2 (∼106−7 M−1s−1) in comparison with a general thiolate (∼10 M−1s−1), it is possible that RAD51 binding to Prdx1/2 may not necessarily be required for protection following IR as long as Prdx1/2 proximity is close enough to metabolize H2O2 before RAD51. Further investigations are necessary to define the role of other PRDX family members that don't bind RAD51.
Analysis of PRDX1 function in HR repair identifies the Cys319 of RAD51 as a functionally-significant target for redox regulation of HR. Specifically, the decrease of PRDX1 activity in both null MEFs and shRNA-treated malignant breast cancer cells sensitized them to the effects of IR-induced RAD51 foci formation, HR and colony formation. While antisense oligonucleotide-induced decreases in PRDX1 alter lung tumor IR sensitivity and NHEJ activity [45], the present study defines the mechanisms accounting for PRDX1 regulation of HR, specifically the protection of RAD51 protein from the oxidation of a functionally-significant Cys. The heightened oxidation of the RAD51 Cys thiol to sulfenic acid was decreased upon mutation of Cys319 (Fig. 3F).
Cancer cells are typically in a “pro-oxidative state” and use increased fluxes of hydrogen peroxide and other reactive species as mitotic signaling mechanisms [46]. Thus, elevated rates of production and increased steady state concentrations of reactive species stemming from metabolic stress, inflammation and xenobiotic exposure could cause defects in HR mediated DNA repair directly through oxidative PTMs to RAD51 protein, that in turn can promote genomic variations and possible carcinogenesis. As opposed to oxidative and free radical reactions that directly modify DNA bases, such as Fenton reactions, the inhibition of HR and an inability to cope with stalled replication forks would promote the accumulation of genomic damage over time. Similarly, once cells become transformed, decreases in various mechanisms that suppress oxidative stress would be expected to further destabilize cancer cell genomes during cell replication. Our results that Cys oxidation of RAD51 can influence HR would add to the previously seen HR oxidative stress-regulated proteins Ataxia–Telangiectasia mutated protein kinase (ATM) [47] and X-ray repair cross-complementing protein 3 (XRCC3) [48]. Interestingly, the DNA damage sensor protein ATM is activated by oxidation, while XRCC1 activity is inhibited. This may enable temporal control of the HR pathway to initiate the process of HR, but not proceed until homologous templates have been repaired from oxidative DNA damage to bases such as 8-oxoguanine. The base excision repair (BER) pathway has also been previously shown to be mediated by cysteine oxidation of proteins such as Alkyl-adenine DNA glycosylase (AAG), Apurinic/apyrimidinic endonuclease 1 (APE1), 8-oxoguanine glycosylase (OGG1), PARP1 and XRCC1 [49]. Recently, OGG1 was described to have distinct cysteine residues that are important for glycosylase activity, lyase activity and structural stability [50]. The role of redox regulation in DNA repair is therefore expanding with our current results and adds another layer of complexity to how and when the pathways work in a coordinated manner. Future studies are necessary to examine DNA repair pathway coordination and whether inactivation of Prdx1 through phosphorylation pathways can further control RAD51 as recently shown for LOX-dependent extracellular matrix remodeling in breast cancer [51].
The shRNA inhibition of PRDX1 expression in MDA-MB-231 cells decreased IR-dependent RAD51 foci formation when the fold-change of non-irradiated cells to irradiated cells was compared to controls (Fig. 6B). Total RAD51 foci numbers increased in shPRDX1 MDA-MB-231 cells and contrasted with results seen in MEF cells (Fig. 5A and Supplementary Fig. S5B). These differences could reflect the incomplete knock-down of PRDX1 or the heightened RAD51 expression in triple-negative breast cancer cells. However, quantification of RAD51 peak foci intensities in breast cancer cells following IR suggests that there is dysfunctional oligomerization of RAD51 in shPRDX1 MDA-MB-231 cells as IR affects PRDX1-proficient cells significantly more than PRDX1-deficient cells (Fig. 5D and E and Supplementary Fig. S5C).
In conclusion, this study highlights the importance of PRDX1 in maintaining proper HR by protecting the functionally significant Cys319 of RAD51 from oxidation. While the limitation of HR during oxidative stress may aid to ensure genome integrity and fidelity, a broader role for PRDX1 in maintaining genomic stability is likely. For example, alterations of RAD51 activity may impact the repair of stalled replication forks or DNA inter-strand crosslinks, thus further investigation of the role of PRDX1 in these areas is warranted [52,53]. The Cys319 residue in RAD51 is evolutionarily conserved throughout mammalian species and emphasizes the importance of PRDX1 in limiting RAD51 oxidation. The unique and high sensitivity of PRDX1 to reactive species thus enables an oxidative rheostat for the regulation of HR.
4. Materials and methods
4.1. Cell lines
Animals used for the isolation of Prdx1+/+ or Prdx1−/− murine embryonic fibroblasts (MEFs) were approved by and conducted according to the guidelines of the University of Pittsburgh IACUC as previously described [54]. MEFs were cultured at 37 °C with 5% CO2 in Dulbecco's modified Eagle's medium containing (DMEM) (Gibco) supplemented with 10% FBS (HyClone), 100 units/ml penicillin, 100 mg/ml streptomycin (Gibco), non-essential amino acids (Gibco) and 2 mM l-glutamine (Gibco). To generate MEFs with expression of wild-type or cysteine mutant RAD51, pLVX Neo-RAD51 vectors were utilized. Virus was produced in 150,000 293T cells plated in a 6-well plate and transfected with 6 μl Fugene 6 and the vectors pLVX Neo-RAD51 (WT or Cys mutant) (0.5 μg), psPAX2 (0.35 μg) and pMD2.G (0.1 μg) in OptiMEM (Gibco). Viral supernatant was used to transduce MEFs for 24 h in the presence of polybrene (0.8 μg/ml). Cells were selected with geneticin (500 μg/ml) (Gibco) for 1 week. HEK 293T, MDA-MB-231 and MDA-MB-468 cells (American Type Culture Collection) were cultured in DMEM growth media as indicated above. To generate PRDX1-deficient breast cancer cells and pLKO.1 empty vector (EV) control cells were produced as previously described [27].
DT40 lymphoblastoid cells Rad51−/−:tethRad51 (Clone 110, in which chicken RAD51 had been removed and replaced by a tet-controlled human wild-type RAD51) [24,55] were grown in RPMI 1640 medium with 10% FBS, 1% chicken serum, 100 units/ml penicillin and 100 μg/ml streptomycin at 39.5 °C under 5% CO2. Human full length RAD51 constructs containing the mutations C137S, C312S or C319S in the pApuro vector were digested overnight with ScaI and 25 μg DNA were used for stable transfections. Briefly, 10 × 106 Clone 110 cells at 106 cells/ml were spun at 320×g for 5 min and the medium was removed. Cells were resuspended into 500 μl sterile PBS and transferred to a 0.4 cm transfection cuvette (Biorad). After the addition of linearized DNA, cuvettes were incubated for 10 min on ice. Cells were electroporated using a Gene Pulser apparatus (BioRad) at 550 V and 25 μF, incubated on ice for a further 10 min, transferred to a 90 mm Petri dish with 20 ml pre-warmed media and incubated at 39 °C for 24 h.
Selection antibiotic puromycin (Sigma) was added to cells in 96-well plates and incubated at 39 °C for several days. Clones were tested for functional expression of human mutant RAD51 by surviving the addition of 0.1 μg/ml doxycycline. Un-transfected Clone 110 cells were used as wild-type hRad51 control.
4.2. Plasmids
HA-PRDX1 and HA-PRDX1 C52S, C173S were cloned into PCGN as previously described [26]. pCGN was a gift from Winship Herr (Addgene #53395). The direct repeat green fluorescent protein (DR-GFP) reporter and I-SceI pCAGGS plasmids were a kind gift from Prof. Maria Jasin [35,56]. pLKO.1-TRC cloning vector was a gift from David Root (Addgene #10878). pLKO1 puro EV and PRDX1 shRNA were previously described [26]. pMD2.G and psPAX2 were a gift from Didier Trono (Addgene #12259 and #12260. pLVX Neo-RAD51 was cloned by PCR amplification of RAD51 with PCR primers incorporating SpeI (5′) and BamHI (3’) restriction sites. The PCR product was then ligated into the corresponding restrictions sites of pLVX-Neo (Clontech) and transformed into DH5α Max efficiency cells (Invitrogen). pLVX Neo RAD51 cysteine to serine mutant plasmids (137, 312 or 319) were produced using the QuikChange II site-directed mutagenesis kit (Agilent) using pLVX Neo-RAD51 as a template.
4.3. γ-Irradiation
Experiments were conducted on cells dosed with 0–10 Gy utilizing a Gammacell 40 Exactor ɣ-Irradiator (Best Medical) with a dose rate of 69 R/min.
4.4. DT40 immunofluorescence and image analysis
Cells were either left at 39 °C (0 Gy group) or subjected to 5 Gy of ionizing radiation using a Millennium irradiator (Maintenance International Ltd), before being placed back at 39 °C to recover. After 4 h, cells were counted and 105 cells were centrifuged for 5 min at 72×g on to a poly-l–lysine-coated slide in a Shandon Cytospin. Slides were air-dried, cells were fixed with 4% paraformaldehyde for 10 min and rinsed in PBS (3x). Cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min, and washed in PBS. Slides were blocked in 1% BSA in PBS for 30 min at room temperature, before staining with rabbit anti-RAD51 antibody (PC-130, Merck Millipore) (1/500) for 1.5 h. After three 5 min PBS washes, slides were incubated with donkey anti-rabbit-AlexaFluor488-conjugated secondary antibody (1/200) for 45 min at 37 °C. After four 5 min washes in PBS in the dark, slides were mounted with DAPI in DABCO (1,4-diazadicyclol[2.2.2]octane). Slides were imaged using an Operetta High Content system (PerkinElmer), with a 40× Long Working Distance objective. Twenty-five fields of view per slide were acquired, with a Z-step of 1.2 μm over a depth of 6 μm, using 200 ms exposure time for DAPI (excitation filter 360–400 nm, emission filter 410–480 nm) and 300 ms for Alexa Fluor 488 (excitation filter 460–490 nm, emission filter 500–550 nm). Images were analyzed with Harmony High Content software (PerkinElmer). Stacks were processed as maximum intensity projections. The DAPI signal was used to segment the nucleus and the RAD51 signal was used to segment the cytoplasm. DAPI Intensity Contrast and Nucleus Roundness were used to select cells for analysis. RAD51 foci were identified using a Find Spots block, using Method C (Radius of 2.2 pixels, Contrast >0.10, Uncorrected Spot Intensity>1.3). Between 1300 and 8300 cells per slide were included in the analysis. Cells with more than 4 Rad51 foci were considered positive. Data shown are mean ± SEM of three independent experiments.
4.5. Immunostaining and imaging
To analyze RAD51 foci formation, cells were plated on coverslips (Fisher Scientific) in a 6-well plate at a density of 150,000 cells per well and were incubated overnight in 5% FBS media. Cells were irradiated with 5 Gy then incubated at 37 °C for 6 h before being processed for RAD51 foci formation or H2AX staining as previously described [22]. Antibodies used for overnight immunostaining at 4 °C were RAD51 (H-92) (Santa Cruz) at 1/100 dilution or phosphorylated γH2AX (JBW301) (EMD Biosciences) at 1/200 dilution. Z-stack images were acquired using NIS elements software on a Nikon A1R confocal microscope with a Nikon 60× oil objective NA 1.4.
4.6. HR repair assay
The HR repair assay was performed as previously described [35]. Briefly, MEFs were transfected with the DR-GFP plasmid and stably selected with (puromycin). Resistant cells were transfected with a plasmid containing I-SceI to induce DNA DSB. HR activity was measured by counting GFP-positive cells in the population by flow cytometry at the MWRI flow cytometry core utilizing a BD LSRII (BD Biosciences). Prdx1−/− MEFs were infected with (lentivirus/retrovirus) plasmid control, wild-type RAD51, or cysteine mutant RAD51 (C137S, C312S or C319S) and selected, then resistant Prdx1−/− MEFs were then transfected with the I-SceI and HR activity was measured as indicated above.
4.7. Western blotting
Cell lysates were prepared in 50 mM Tris, 150 mM NaCl, 1% Triton X-100, 0.5 mM EDTA, 0.5 mM EGTA and 10% glycerol supplemented with catalase (30 μg/ml), 100 mM N-ethylmaleimide (NEM) and protease (Halt protease inhibitors, Fisher Scientific) and phosphatase inhibitors (50 mM NaF, 1 mM NaVO4 and 40 mM β-glycerophosphate). Lysates were sonicated for 5 min (30 s pulse/30 s delay) at 4 °C, and then centrifuged to pellet insoluble material. Laemmli sample buffer (BioRad) supplemented with β-mercaptoethanol was added to 40 μg sample and incubated for 5 min at 95 °C. Protein lysates were separated on 10% tris-glycine gels and transferred onto nitrocellulose.
4.8. Immunoprecipitation
106 HEK 293T cells were transiently transfected with Fugene 6 (Promega) and 2 μg pQCXIP (EV) or FLAG-RAD51 pQCXIP plasmids. After 24 h cells were irradiated with between 0 and 10 Gy. Lysates were prepared as for Western blotting above. Protein concentrations were quantified by BCA assay kit (Fisher Scientific). 500 μg of cell lysate was incubated with 10 μL of acid treated Anti-FLAG M2 Affinity Gel and 400 μL lysis buffer at 25 °C for 3 h with rotation. Tubes containing precipitated proteins were centrifuged and washed four times in lysis buffer and once in 1x TBS. Laemmli sample buffer (BioRad) supplemented with β-mercaptoethanol was added to the sample and incubated for 10 min at 95 °C, then loaded onto a 10% tris-glycine gel. Prdx1+/+ MEFs were infected with HA-PRDX1 PCGN and/or FLAG-RAD51 pQCXIP, and selected with antibiotics. Cells were irradiated with 0–8 Gy and lysates were precipitated as above, but with monoclonal anti-HA-agarose for HA-PRDX1 precipitation.
4.9. Cell cycle analysis
Propidium iodide (PI) stained DNA content was measured as an indication of cell cycle phase in PRDX1-proficient and deficient MDA-MB-231 cells. 106 unsynchronized cells were harvested by trypsinization and following inactivation of trypsin, cells were pelleted and washed with ice cold 1xPBS (Gibco). Cells were fixed in 70% ethanol by adding ethanol dropwise with swirling and incubated for 20 min at 4 °C. Cells were then centrifuged at 2000 rpm and the supernatant removed. Pellets were washed with ice cold PBS then treated with 500 μl RNase A (50 ng RNAase/μl) for 15 min at 37 °C. Next, propidium iodide (Sigma) (50 ng PI/μl) was added and incubated for 30 min at 4 °C in a dark chamber. Samples were later analyzed at the Flow Cytometry Core at MWRI utilizing a BD LSRII (BD).
4.10. Mass spectrometry analysis and quantification
LC-MS/MS system consisted of an Orbitrap Eclipse Tribrid Mass Spectrometer (Thermo Scientific, Waltham, MA) and a Dionex Ultimate3000 nano-UPLC system (Thermo Scientific, Waltham, MA). An Acclaim PepMap 100 (C18, 5 μm, 100 Å, 100 μm x 2 cm) trap column and an Acclaim PepMap RSLC (C18, 2 μm, 100 Å, 75 μm x 50 cm) analytical column was be employed for peptide separation. MS spectra was acquired by parallel reaction monitoring consisting of MS/MS scans of the targeted precursor ions from the MS1 scan with dynamic exclusion option with 30 s of duration. Skyline (MacCoss Lab Software, University of Washington, Seattle, WA) was used for target peptides detection, peak feature extraction, and peak area calculation for quantitative data analysis. Peak areas were normalized using the total ion current (TIC) which is the sum of all peaks in the chromatogram acquired by complementary MS1 scan event. NIST mass spectral library was utilized to confirm peak selection for the analysis.
4.11. Protein purification
Recombinant His-tagged RAD51 in the pET21a vector were transformed into E. coli BL21(DE3)pLysS cells (EMD Millipore) and were purified using Ni-NTA beads. Protein was dialyzed against 20 mM Tris-HCl (pH 8.3) containing 200 mM NaCl and 1 mM DTT and stored at −80 °C as previously described. His-tagged ABL core protein in pVL1392 was purified from Sf9 insect cells upon co-expression with YopH, as described previously [57]. Protein was dialyzed against 20 mM Tris-HCl (pH 8.3) containing 100 mM NaCl and 3 mM DTT and stored at −80 °C as previously described [57].
4.12. Labeling of WT and mutant RAD51 sulfenylation in MEF cells
DAz-2, synthesized as previously described [58] was prepared in DMSO at a final concentration of 250 mM and stored at −20 °C. p-Biotin was prepared in DMSO at a final concentration of 5 mM and stored at −80 °C. Prdx1+/+ and Prdx1−/− MEFs expressing WT FLAG-RAD51 or Prdx1+/+ MEFs expressing C137S, C312S, or C391D FLAG-RAD51 were grown to ∼85% confluency. The cells were washed with sterile PBS ( × 3) and incubated in DMEM with 5 mM DAz-2 or 1% (v/v) DMSO for 1.5 h at 37 °C. Afterwards, the cells were washed with cold PBS (×3), scraped into RIPA buffer (250 μl) containing 1x mini complete protease inhibitors (Roche) with a rubber policeman, and lysed by incubating on ice for 15 min with swirling. Lysates were cleared by centrifugation at 4 °C for 15 min (14,000×g) and the protein concentration was determined by BCA assay (Pierce).
4.13. WT and mutant FLAG-RAD51 immunoprecipitation for sulfenylation assessment
For comparison of WT FLAG-RAD51 sulfenylation from Prdx1+/+ versus Prdx1−/− MEFs, Rad51 was immunoprecipitated from 400 μg of cell lysate by incubating with EZView Red Anti-FLAG M2 affinity gel (20 μl, Sigma) overnight at 4 °C with rocking. For assessment of sulfenylation of Rad51 mutants, WT and mutant Rad51 were immunoprecipitated from 750 μg of cell lysate by incubating with 30 μl resin as above. The resin was collected at 8200×g for 30 s, washed with RIPA buffer ( × 4, 1 ml) and enriched proteins were eluted by incubating with 20 resin volumes of elution buffer (50 mM Tris HCL, pH 7.5, 150 mM NaCl, 150 ng/ml 3x FLAG peptide) for 2 h at 4 °C with rocking. Staudinger ligation and western blotting were performed as described in supplemental methods [31].
4.14. DT40 growth curves
Cells were diluted to 5 × 104 cells/ml in pre-warmed media containing doxycycline and counted every 24 h for 72 h. Cell density was adjusted to the starting concentration when approaching confluency (106 cells/ml) and the dilution factor was taken into account when calculating cell numbers. Data shown are mean ± standard deviation of three independent experiments.
4.15. Computational studies on a RAD51 filament model
In previously published studies we reported a homology model of the RAD51 dimer, which was used in MD simulations, in order successfully predict BRC4 peptide binding energies and conformations [32]. This dimeric system lacked ssDNA (and dsDNA), but did contain ATP-Mg ligands. The recently published cryoEM structure of tetrameric RAD51 [33] was utilized in order to make an all atom model by propagating and superposing chain B of the RAD51 homology model from [32] using Coot. This crude all atom model was then superposed onto a RecA structure (3CMU) [34] that is complexed with DNA, to yield a starting RAD51-DNA complex for refinement. The filament model was refined with knowledge based forecefields using simulated annealing and molecular dynamics simulations specified below.
4.16. RAD51 filament model refinement with knowledge based forecefields using simulated annealing and molecular dynamics simulations
Our model filament structure, liganded with ssDNA (generated based on the RecA crystal structure and a cryoEM structure), as described above, was refined using a simulated annealing molecular dynamics procedure. The overall procedure for refinement was highly similar to our original work in generating the unliganded RAD51 dimer, as described previously [32]. Our general approach for utilizing MD refinement with knowledge based forcefields was described in detail previously [59,60]. The refined filament model yielded good overall Z-score of −0.57 (less than one standard deviation from ideal parameters for 1D and 3D packing). Our refined atomistic filament model of RAD51 allowed us to subject this complex to longer MD simulations with explicit solvent, under ambient conditions. The solvated filament system had ∼126,202 total atoms, and was conducted at 298K, pH 7.4, physiological NaCl concentration, using AMBER14 force field in YASARA Structure 20.12.24, with simulation box dimensions of 90 × 95 x 150 Å. The details of the simulation algorithms, are similar to those we employed in a system described in detail previously [60]. A parallel MD was performed for the oxidized Cys319, in which each Cys319 sulfur of the RAD51 filament was converted to the sulfenic acid form R-S-OH, starting from the energy minimized WT form. The oxidized system consisted of 126,220 atoms.
4.17. Statistical analysis
Data represents the mean ± SEM from 3 independent experiments unless otherwise noted. A p value < 0.05 was considered statistically significant. Non-linear curves were generated in GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA) for statistical analysis. EC50 values and standard error were calculated from three independent experiments utilizing a non-linear dose response variable slope model of groups challenged with IR. Significance was tested by one-way ANOVA for multiple groups with Tukey posttest for multiple comparisons between groups or by t-test when groups were less than three. RAD51 peak foci intensity was analyzed with MATLAB. For foci quantification, we segmented the nuclei and identified foci based on the local maxima with a peak intensity value of above 1000 using MATLAB. Then, each focus was fitted to a Gaussian function to obtain peak intensity values. Nuclear boundaries were individually identified in more than 20 cells per treatment group in three independent experiments. RAD51 peak intensity values were compared by sorting peak foci intensities from smallest to largest and normalizing the number of foci within experimental groups of more than 20 cells to 1. Intensity readings under the four conditions were evaluated (MDA-MB-231±IR and MDA-MB-231 shPRDX1±IR) from a total of 2739 values. The RAD51 peak intensity population values do display a Gaussian, or gamma distribution, so the nonparametric Mann-Whitney test was performed to compare groups.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
The authors would like to thank Dr. M. Jasin (Memorial Sloan Kettering Cancer Center, New York, NY) for providing HR plasmids, Dr. P. Grover and Dr. T. Smithgall (Univ. of Pittsburgh, Pittsburgh, PA) for providing purified RAD51 and ABL protein, Dr. S.Takeda (Kyoto Univ., Japan) for providing DT40 Rad51−/−::tethRad51 cells, Dr. D. Koes for computational biological analysis and Dr. B. Van Houten (Univ. of Pittsburgh, Pittsburgh, PA) and Dr. K. Bernstein (Univ. of Pittsburgh, Pittsburgh, PA) for helpful discussions and manuscript comments. This work was supported by National Institutes of Health (NIH) R01CA131350, P30CA047904 (UPCI, D.N.), Science Foundation Ireland (SFI) 10/IN.1/B2972 and 242361 BOOSTER, European Commission (EC) SEC-2009-4.3-02 (C.G.M.), NIH R56CA233817 (C.A.N.), Congressionally Directed Medical Research Programs Breast Cancer Research Program BC180467 (C.A.N.), the Univ. of Pittsburgh Dept. of Pharmacology and Chemical Biology predoctoral fellowship (A.A.), the William C. de Groat predoctoral fellowship (A.A.) and the Cotswold Foundation postdoctoral fellowship (J.J.S.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102443.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- 1.Ames B.N., Shigenaga M.K. Oxidants are a major contributor to aging. Ann. N. Y. Acad. Sci. 1992;663:85–96. doi: 10.1111/j.1749-6632.1992.tb38652.x. [DOI] [PubMed] [Google Scholar]
- 2.Kryston T.B., Georgiev A.B., Pissis P., Georgakilas A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011;711(1–2):193–201. doi: 10.1016/j.mrfmmm.2010.12.016. [DOI] [PubMed] [Google Scholar]
- 3.Jones D.P., Sies H. The redox code. Antioxidants Redox Signal. 2015;23(9):734–746. doi: 10.1089/ars.2015.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tubbs A., Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mehta A., Haber J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harbor Perspect. Biol. 2014;6(9) doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scully R., Panday A., Elango R., Willis N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20(11):698–714. doi: 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shinohara A., Ogawa H., Ogawa T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell. 1992;69(3):457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 9.Sung P., Robberson D.L. DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell. 1995;82(3):453–461. doi: 10.1016/0092-8674(95)90434-4. [DOI] [PubMed] [Google Scholar]
- 10.Gupta R.C., Bazemore L.R., Golub E.I., Radding C.M. Activities of human recombination protein Rad51. Proc. Natl. Acad. Sci. U. S. A. 1997;94(2):463–468. doi: 10.1073/pnas.94.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benson F.E., Stasiak A., West S.C. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J. 1994;13(23):5764–5771. doi: 10.1002/j.1460-2075.1994.tb06914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hilario J., Amitani I., Baskin R.J., Kowalczykowski S.C. Direct imaging of human Rad51 nucleoprotein dynamics on individual DNA molecules. Proc. Natl. Acad. Sci. U. S. A. 2009;106(2):361–368. doi: 10.1073/pnas.0811965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sung P., Krejci L., Van Komen S., Sehorn M.G. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003;278(44):42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 14.Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265(5176):1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- 15.Rhee S.G. Overview on peroxiredoxin. Mol. Cell. 2016;39(1):1–5. doi: 10.14348/molcells.2016.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hampton M.B., Vick K.A., Skoko J.J., Neumann C.A. Antioxid Redox Signal; 2018. Peroxiredoxin Involvement in the Initiation and Progression of Human Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann C.A., Krause D.S., Carman C.V., Das S., Dubey D.P., Abraham J.L., Bronson R.T., Fujiwara Y., Orkin S.H., Van Etten R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 18.Neumann C.A., Cao J., Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle. 2009;8(24):4072–4078. doi: 10.4161/cc.8.24.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egler R.A., Fernandes E., Rothermund K., Sereika S., de Souza-Pinto N., Jaruga P., Dizdaroglu M., Prochownik E.V. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24(54):8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 20.Iraqui I., Kienda G., Soeur J., Faye G., Baldacci G., Kolodner R.D., Huang M.E. Peroxiredoxin Tsa1 is the key peroxidase suppressing genome instability and protecting against cell death in Saccharomyces cerevisiae. PLoS Genet. 2009;5(6) doi: 10.1371/journal.pgen.1000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iraqui I., Faye G., Ragu S., Masurel-Heneman A., Kolodner R.D., Huang M.E. Human peroxiredoxin PrxI is an orthologue of yeast Tsa1, capable of suppressing genome instability in Saccharomyces cerevisiae. Cancer Res. 2008;68(4):1055–1063. doi: 10.1158/0008-5472.CAN-07-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asan A., Skoko J.J., Woodcock C.C., Wingert B.M., Woodcock S.R., Normolle D., Huang Y., Stark J.M., Camacho C.J., Freeman B.A., Neumann C.A. Electrophilic fatty acids impair RAD51 function and potentiate the effects of DNA-damaging agents on growth of triple-negative breast cells. J. Biol. Chem. 2019;294(2):397–404. doi: 10.1074/jbc.AC118.005899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Modesti M., Ristic D., van der Heijden T., Dekker C., van Mameren J., Peterman E.J., Wuite G.J., Kanaar R., Wyman C. Fluorescent human RAD51 reveals multiple nucleation sites and filament segments tightly associated along a single DNA molecule. Structure. 2007;15(5):599–609. doi: 10.1016/j.str.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Sonoda E., Sasaki M.S., Buerstedde J.M., Bezzubova O., Shinohara A., Ogawa H., Takata M., Yamaguchi-Iwai Y., Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17(2):598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies M.J. Protein oxidation and peroxidation. Biochem. J. 2016;473(7):805–825. doi: 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao J., Schulte J., Knight A., Leslie N.R., Zagozdzon A., Bronson R., Manevich Y., Beeson C., Neumann C.A. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009;28(10):1505–1517. doi: 10.1038/emboj.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner-Ivey B., Manevich Y., Schulte J., Kistner-Griffin E., Jezierska-Drutel A., Liu Y., Neumann C.A. Role for Prdx1 as a specific sensor in redox-regulated senescence in breast cancer. Oncogene. 2013;32(45):5302–5314. doi: 10.1038/onc.2012.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rani V., Neumann C.A., Shao C., Tischfield J.A. Prdx1 deficiency in mice promotes tissue specific loss of heterozygosity mediated by deficiency in DNA repair and increased oxidative stress. Mutat. Res. 2012;735(1–2):39–45. doi: 10.1016/j.mrfmmm.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang M.E., Kolodner R.D. A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol. Cell. 2005;17(5):709–720. doi: 10.1016/j.molcel.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 30.Tang H.M., Siu K.L., Wong C.M., Jin D.Y. Loss of yeast peroxiredoxin Tsa1p induces genome instability through activation of the DNA damage checkpoint and elevation of dNTP levels. PLoS Genet. 2009;5(10) doi: 10.1371/journal.pgen.1000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulsen C.E., Truong T.H., Garcia F.J., Homann A., Gupta V., Leonard S.E., Carroll K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011;8(1):57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanyam S., Jones W.T., Spies M., Spies M.A. Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res. 2013;41(19):9020–9032. doi: 10.1093/nar/gkt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Short J.M., Liu Y., Chen S., Soni N., Madhusudhan M.S., Shivji M.K., Venkitaraman A.R. High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic Acids Res. 2016;44(19):9017–9030. doi: 10.1093/nar/gkw783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z., Yang H., Pavletich N.P. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453(7194) doi: 10.1038/nature06971. 489-4. [DOI] [PubMed] [Google Scholar]
- 35.Moynahan M.E., Pierce A.J., Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 2001;7(2):263–272. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 36.Bee L., Fabris S., Cherubini R., Mognato M., Celotti L. The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression. PLoS One. 2013;8(7) doi: 10.1371/journal.pone.0069061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ochs F., Somyajit K., Altmeyer M., Rask M.B., Lukas J., Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 2016;23(8):714–721. doi: 10.1038/nsmb.3251. [DOI] [PubMed] [Google Scholar]
- 38.Rothkamm K., Kruger I., Thompson L.H., Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003;23(16):5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Budke B., Kalin J.H., Pawlowski M., Zelivianskaia A.S., Wu M., Kozikowski A.P., Connell P.P. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J. Med. Chem. 2013;56(1):254–263. doi: 10.1021/jm301565b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimizu H., Popova M., Fleury F., Kobayashi M., Hayashi N., Sakane I., Kurumizaka H., Venkitaraman A.R., Takahashi M., Yamamoto K. c-ABL tyrosine kinase stabilizes RAD51 chromatin association. Biochem. Biophys. Res. Commun. 2009;382(2):286–291. doi: 10.1016/j.bbrc.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 41.Aihara H., Ito Y., Kurumizaka H., Yokoyama S., Shibata T. The N-terminal domain of the human Rad51 protein binds DNA: structure and a DNA binding surface as revealed by NMR. J. Mol. Biol. 1999;290(2):495–504. doi: 10.1006/jmbi.1999.2904. [DOI] [PubMed] [Google Scholar]
- 42.Wani R., Nagata A., Murray B.W. Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 2014;5:224. doi: 10.3389/fphar.2014.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley M.R., Georgiadis M.M., Fishel M.L. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012;5(1):36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ragu S., Faye G., Iraqui I., Masurel-Heneman A., Kolodner R.D., Huang M.E. Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc. Natl. Acad. Sci. U. S. A. 2007;104(23):9747–9752. doi: 10.1073/pnas.0703192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen M.F., Keng P.C., Shau H., Wu C.T., Hu Y.C., Liao S.K., Chen W.C. Inhibition of lung tumor growth and augmentation of radiosensitivity by decreasing peroxiredoxin I expression. Int. J. Radiat. Oncol. Biol. Phys. 2006;64(2):581–591. doi: 10.1016/j.ijrobp.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 46.Liou G.Y., Storz P. Reactive oxygen species in cancer. Free Radic. Res. 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. ATM activation by oxidative stress. Science. 2010;330(6003):517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 48.Girard P.M., Graindorge D., Smirnova V., Rigolet P., Francesconi S., Scanlon S., Sage E. Oxidative stress in mammalian cells impinges on the cysteines redox state of human XRCC3 protein and on its cellular localization. PLoS One. 2013;8(10) doi: 10.1371/journal.pone.0075751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alnajjar K.S., Sweasy J.B. A new perspective on oxidation of DNA repair proteins and cancer. DNA Repair. 2019;76:60–69. doi: 10.1016/j.dnarep.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang K., Maayah M., Sweasy J.B., Alnajjar K.S. The role of cysteines in the structure and function of OGG1. J. Biol. Chem. 2021;296 doi: 10.1074/jbc.RA120.016126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Attaran S., Skoko J.J., Hopkins B.L., Wright M.K., Wood L.E., Asan A., Woo H.A., Feinberg A., Neumann C.A. Peroxiredoxin-1 Tyr194 phosphorylation regulates LOX-dependent extracellular matrix remodelling in breast cancer. Br. J. Cancer. 2021;125(8):1146–1157. doi: 10.1038/s41416-021-01510-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37(4):492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang A.T., Kim T., Wagner J.E., Conti B.A., Lach F.P., Huang A.L., Molina H., Sanborn E.M., Zierhut H., Cornes B.K., Abhyankar A., Sougnez C., Gabriel S.B., Auerbach A.D., Kowalczykowski S.C., Smogorzewska A. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol. Cell. 2015;59(3):478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neumann C.A., Krause D.S., Carman C.V., Das S., Dubey D.P., Abraham J.L., Bronson R.T., Fujiwara Y., Orkin S.H., Van Etten R.A. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 55.Takata M., Sasaki M.S., Sonoda E., Morrison C., Hashimoto M., Utsumi H., Yamaguchi-Iwai Y., Shinohara A., Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17(18):5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson C., Moynahan M.E., Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 1998;12(24):3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grover P., Shi H., Baumgartner M., Camacho C.J., Smithgall T.E. Fluorescence polarization screening assays for small molecule allosteric modulators of ABL kinase function. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0133590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leonard S.E., Reddie K.G., Carroll K.S. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 2009;4(9):783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 59.Hengel S.R., Malacaria E., Folly da Silva Constantino L., Bain F.E., Diaz A., Koch B.G., Yu L., Wu M., Pichierri P., Spies M.A., Spies M. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife. 2016;5 doi: 10.7554/eLife.14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dean S.F., Whalen K.L., Spies M.A. Biosynthesis of a novel glutamate racemase containing a site-specific 7-hydroxycoumarin amino acid: enzyme-ligand promiscuity revealed at the atomistic level. ACS Cent. Sci. 2015;1(7):364–373. doi: 10.1021/acscentsci.5b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.