

SUMMARY
Great progress has been made in understanding gut microbiome’s products and their effects on health and disease. Less attention, however, has been given to the inputs that gut bacteria consume. Here we quantitatively examine inputs and outputs of the mouse gut microbiome, using isotope tracing. The main input to microbial carbohydrate fermentation is dietary fiber, and to branched-chain fatty acids and aromatic metabolites is dietary protein. In addition, circulating host lactate, 3-hydroxybutyrate and urea (but not glucose or amino acids) feed the gut microbiome. To determine nutrient preferences across bacteria, we traced into genus-specific bacterial protein sequences. We find systematic differences in nutrient use: Most genera in the phylum Firmicutes prefer dietary protein, Bacteroides dietary fiber, and Akkermansia circulating host lactate. Such preferences correlate with microbiome composition changes in response to dietary modifications. Thus, diet shapes the microbiome by promoting the growth of bacteria that preferentially use the ingested nutrients.
Graphical Absctract
In Brief:
Isotope tracing into bacterial-specific protein sequences allows for a determination of nutrient preferences across gut microbes in vivo, and reveals how diet alters microbiome composition.
INTRODUCTION
The gut microbiome possesses an enormous diversity of enzymes, exceeding the number in mammals’ genomes by more than 100-fold (Qin et al., 2010). This enzymatic capacity enables the processing of incoming dietary nutrients into a broad spectrum of microbial metabolites. Some of these reach the host circulation at substantial concentrations (Lai et al., 2021; Quinn et al., 2020). Microbial metabolites can play important roles in host pathophysiology. For example, short-chain fatty acids (SCFAs; acetate, propionate, butyrate) (Dalile et al., 2019; Koh et al., 2016), trimethylamine N-oxide (Tang et al., 2013), secondary bile acids (Arab et al., 2017; Funabashi et al., 2020), indole-3-propionate (Wikoff et al., 2009), and imidazole propionate (Koh et al., 2018) affect immune maturation (Campbell et al., 2020; Hang et al., 2019), insulin sensitivity (Koh et al., 2018), cancer growth (Garrett, 2015; Yoshimoto et al., 2013), and cardiovascular disease (Nemet et al., 2020; Wang et al., 2011).
Both to replicate themselves and to release metabolic products, gut bacteria require nutrient inputs. These come in forms including ingested food, host-synthesized gut mucus (Desai et al., 2016; Sicard et al., 2017), and host circulating metabolites (Scheiman et al., 2019). The availability of dietary nutrients to gut microbiota depends on the extent of host absorption: nutrients that are absorbed in the small intestine, like starch, are not available to the colonic microbiome. In contrast, nutrients that are poorly digested in the upper gastrointestinal tract, like fiber, can be key microbiome feedstocks (Lund et al., 2021; Wong and Jenkins, 2007).
Isotope tracing enables quantitative measurement of the inputs to metabolites and biomass. Studies employing radioactive tracers defined the basics of mammalian metabolism (Wolfe, 1984). Recent work has increasingly relied on stable isotope tracers coupled to mass spectrometry detection, which enables the measurement of labeling in specific downstream products (Fernández-García et al., 2020; McCabe and Previs, 2004). This approach has revealed fundamental features of host metabolism, such as circulating lactate being a major TCA fuel (Faubert et al., 2017; Hui et al., 2017). In addition, it has provided important insights into host-microbiome metabolic interplay. For example, it revealed that dietary fructose is processed by the microbiome into acetate, which fuels hepatic lipogenesis (Jang et al., 2018; Zhao et al., 2020).
In principle, stable isotope tracing coupled to mass spectrometry can also be applied to determine the metabolic inputs to specific microbes, based on measuring labeling in bacteria-specific peptide sequences (Berry et al., 2015; Holmes et al., 2017; Oberbach et al., 2017; Reese et al., 2018; Zhang et al., 2016a, 2016b). By infusing nitrogen-labeled threonine to label host mucus, investigators were able to compare the contribution of dietary versus mucus protein to the gut microbiome and observed a shift towards more mucus contribution in mice fed a low-protein diet (Holmes et al., 2017).
Here, we perform large-scale, quantitative assessment of the metabolic inputs to the gut microbiome and its products. We examine the contributions from dietary starch, fiber, and protein, and from host mucus. We also examine most major circulating host nutrients, finding that lactate, 3-hydroxybutyrate, and urea stand out for passing from the host to the gut microbiome. Based on the measurement of bacteria-specific peptide sequences, we assess the nutrient preferences of different bacterial genera and show that these preferences align with microbiome composition changes in response to altered diet.
RESULTS
Microbiome consumes less digestible dietary components
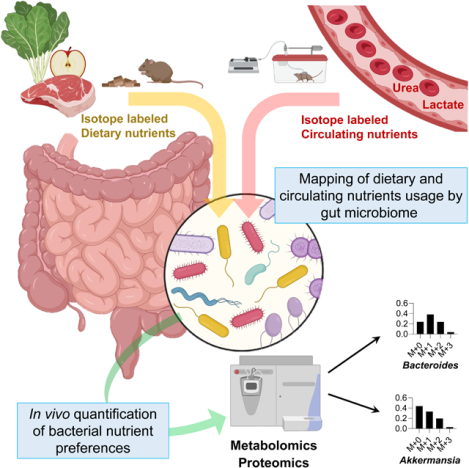
A major mechanism by which the microbiome may impact host physiology is via secreted metabolic products. We measured, in the portal, systemic circulation and the cecal contents, the absolute concentrations of more than 50 metabolites characterized in the literature as microbiome-derived (Campbell et al., 2020; De Vadder et al., 2014; Han et al., 2021; Hang et al., 2019; Koh et al., 2018; Mager et al., 2020; Ridlon et al., 2014; Wikoff et al., 2009) (Figure S1A-B, Table 1, S1). Most were elevated in the portal circulation relative to systemic blood, and all but two (inosine and N-acetyl-tryptophan, which are apparently mainly host derived) were depleted by antibiotics treatment.
Table 1. Absolute concentrations and sources of microbiota-associated metabolites. See alsoTable S1,Figure S1,S4.
Data are from ad lib fed state (ZT0); for ad lib fasted state (ZT12), see Supplementary Table S1. Absolute concentration is mean, N = 5 mice. Portal/systemic = fold-change in concentration between the portal vein and tail vein (median, N = 5 mice). Abx/Conv refers to fold-change in portal blood concentration between mice treated with antibiotics cocktail versus not (median, N = 5 mice/group). Source bar indicates the relative contribution to the indicated metabolite from dietary inulin, algal protein and circulating lactate (based on isotope tracing). Percentages indicate quantitative relative contributions from those nutrients (median, N = 4). Numbers typically add up to less than 100%, as other sources (e.g., mucins) contribute.
|
The dominant excreted products on a molar basis (0.4 – 2 mM in the portal blood) are SCFAs. Other relatively abundant microbiome products (10 – 30 uM) are aromatic amino acid fermentation products (phenol, indoxyl sulfate, and 3-phenylpropionate) and branched-chain fatty acids (valerate, isovalerate, 4-methylvalerate, isobutyrate, 2-methylbutyrate). Primary bile acids, while present in the portal circulation at up to ~ 10 uM concentration, are produced by the host and accordingly were not included in Table 1. Secondary bile acids, which are produced from primary bile acids by the microbiome, were lower in absolute concentration, the most abundant being tauroursodeoxycholic acid (3 uM in portal vein).
To probe the dietary inputs to gut microbial products, we began by feeding mice via oral gavage, starch (readily digestible glucose polymer) and inulin (slowly digestible fructose polymer, i.e., soluble fiber) (Figure S1C). Following 13C-starch gavage, labeled glucose, lactate, and alanine quickly appeared in the portal circulation and accounted for most starch carbons (~75%) (Figure S1D–F, S1G) (Jang et al., 2018). In contrast, after 13C-inulin gavage, substantial labeled fructose, glucose, lactate, and alanine were not observed, and instead labeled portal metabolites slowly appeared in the form of SCFAs, with ~ 40% of inulin carbons becoming SCFAs and the remainder being undigested and excreted in the feces (Figure S1E–F, S1H–I). Moreover, dietary inulin, but not starch, extensively labeled glycolytic and TCA intermediates and amino acids in the cecal content (Figure S1G).
We next carried out similar experiments, comparing the gavage of a free amino acid mixture to algal protein, both uniformly 13C-labeled (Figure S1C). The free amino acids resulted in the rapid appearance of labeled amino acids in portal circulation (Figure S1J–L), while the algal protein substantially labeled amino acids within the cecal contents (Figure S1M). Moreover, the algal protein copiously labeled microbiome-derived portal vein metabolites: SCFAs, branched-chain fatty acids, and aromatics (indole, indole-3-propionate, 3-phenylpropionate) (Figure S1K–L). Thus, poorly digestible carbohydrates and protein feed the microbiome directly, and the host indirectly via microbiome-derived products.
Few circulating metabolites reach the microbiome
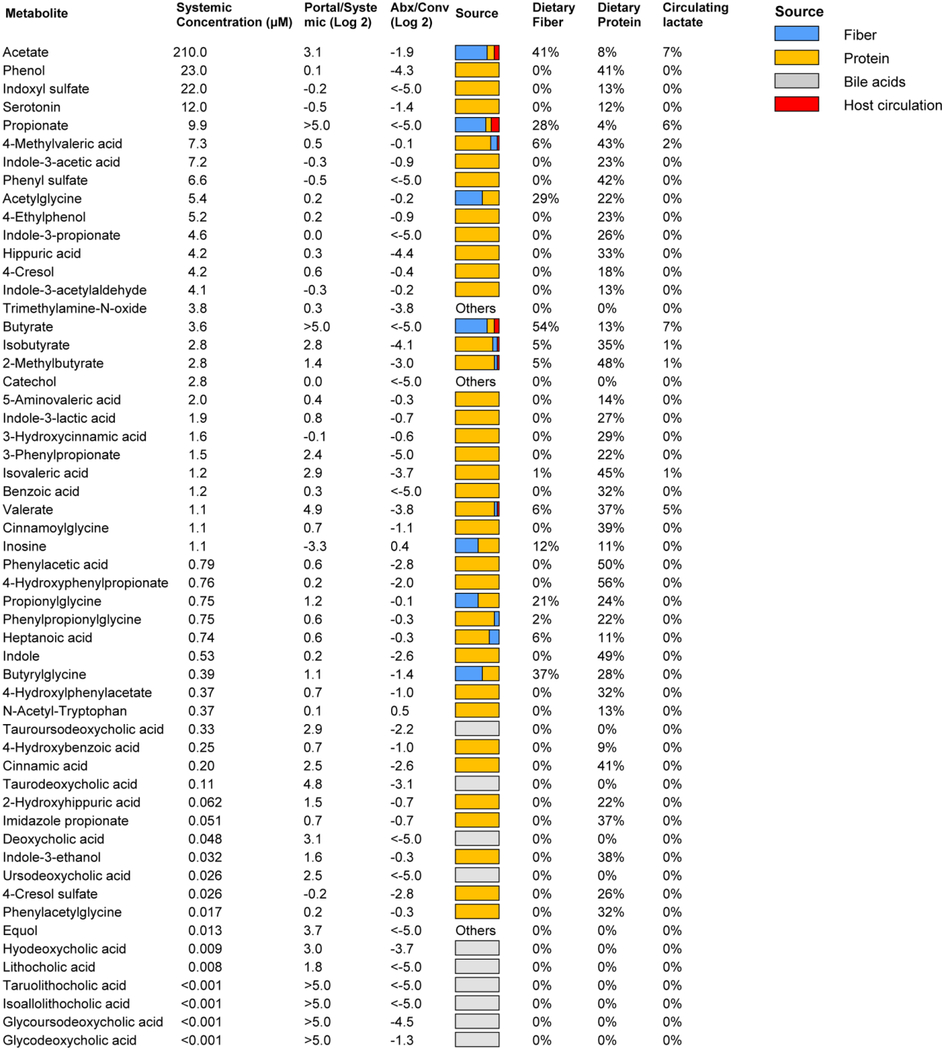
Next, we examined the possibility that nutrients in host circulation feed the gut microbiota. We infused deuterated water and eighteen major circulating nutrients (13C-labeled) into the systemic circulation of pre-catheterized mice (Figure 1A). The infusion rates were selected to achieve modest but readily measurable labeling without substantially perturbing circulating concentrations. Circulating labeling reached a steady state by 2.5 h, at which time we collected serum and feces to quantitate the carbon contributions of each circulating nutrient to the corresponding fecal metabolites. Upon intravenous infusion of 13C-lactate, fecal lactate labeled rapidly (Figure 1B). Most infused circulating nutrients, however, did not penetrate the feces (Figure 1C–D). Indeed, while water fully exchanged with the feces, among abundant circulating carbon carriers, only lactate and 3-hydroxybutyrate penetrated. Glucose, amino acids, TCA intermediates and fatty acids did not. Both lactate and 3-hydroxybutyrate are substrates of monocarboxylate transporters (MCTs), which are highly expressed in the colonic epithelium (Halestrap and Price, 1999, p. 1). Pharmacological MCT inhibition prevented lactate from penetrating the feces (Figure 1E). Thus, in contrast to most host circulating metabolites, which do not reach the colonic microbiome, monocarboxylic transporters render circulating lactate and 3-hydroxybutyrate accessible to gut microbes.
Figure 1. Circulating lactate and 3-hydroxybutyrate feed the gut microbiome. See alsoFigure S2.
(A) Schematic of intravenous infusion of isotope-labeled nutrients to identify circulating metabolites that feed gut microbiome.
(B) Circulating lactate rapidly enters the feces. Mice were infused with 13C-lactate and serum and fresh feces enrichment were compared. Mean±s.e., N = 3.
(C) Circulating citrate does not enter the feces. As in (B), for 13C-citrate. Mean±s.e., N = 3.
(D) Passage of circulating 13C-labeled nutrients into the feces. Mice were infused with labeled nutrients for 2.5 h, and labeling fraction in feces was normalized to labeling fraction in serum. Mean±s.e., N = 3 except for lactate (N = 8) and 3-hydroxybutyrate (N = 7).
(E) Pharmacological inhibition of MCT1 transporter decreases the passage of circulating lactate to feces. Mice were injected i.p. with saline or 100 mg/kg AZD3965, and fresh feces lactate enrichment measured. Mean±s.e. N = 6 for saline and N = 5 for AZD3965. ***P<0.001 by two-sided Student’s t-test.
(F) Passage of circulating 15N-labeled nutrients into the feces. As in (D), for 15N-lableing. Mean±s.e. N = 3 except for urea (N = 4) and ammonia (N = 5).
Circulating urea is a microbiome nitrogen source
In addition to carbon, nitrogen is a fundamental constituent of all living cells. To assess nitrogen sources of the gut microbiome, we infused twelve abundant circulating nutrients in 15N-labeled form. Nitrogen from circulating urea and ammonia, but not amino acids, penetrates the feces and contributes to microbiome amino acids and ammonia (Figure 1F, S2A–B). Urea usage by the microbiome involves its re-conversion to ammonia via the enzyme urease, which is expressed by a subset of gut microbes (Mora and Arioli, 2014; Ni et al., 2017), and gnotobiotic mice colonized intentionally with only urease-negative bacteria showed no labeling from urea (Figure S2C-D).
Urea, which is made from ammonia in the liver, was a quantitatively greater source of microbiome nitrogen than ammonia. Moreover, urea but not ammonia was more abundant in the host circulation than cecal lumen, consistent with only urea being able to passively flow into the gut lumen (Figure S2E–F). We hypothesized that circulating host ammonia might be feeding the microbiome mainly indirectly, after being converted by the host liver into circulating urea (Figure S2G) (Bartman et al., 2021). This indirect contribution was calculated by multiplying circulating urea’s contribution to fecal amino acids (LAAs←urea) by the circulating urea fraction that comes from circulating ammonia (). It fully explained the observed microbiome labeling from circulating ammonia (Figure S2H). Further supporting the indirect pathway, antibiotics treatment blocked both circulating urea and ammonia from becoming cecal ammonia (Figure S2I–J), which makes sense if flux of ammonia into the cecal contents goes through host urea and microbial urease (Figure S2K).
Microbiota synthesize amino acids from fiber and urea
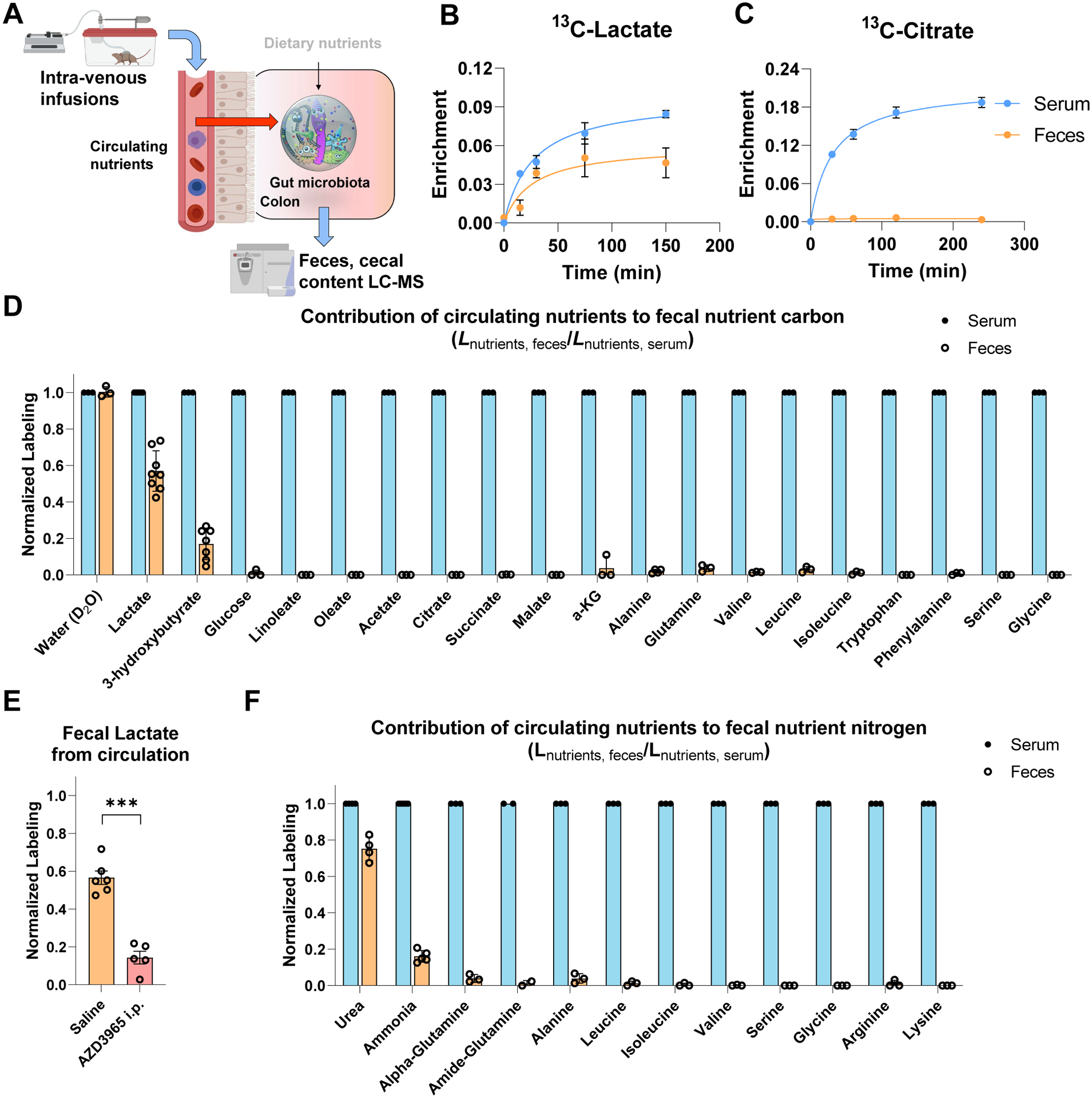
To determine quantitatively the sources of microbiome metabolites, we measured their labeling after ad libitum feeding of isotopically enriched food. To this end, we fed mice standard chow with a portion of the fiber, fat, or protein 13C-labeled, with cecal labeling reaching steady-state within 12 h (Figure S3A). To account for circulating nutrient inputs, we also infused 13C-lactate or 3-hydroxybutyrate (Figure 2A). These studies identified a majority of the carbon feeding into most microbiome central metabolites, with glycolytic and pentose phosphate metabolites labeling almost exclusively coming from dietary fiber (inulin), while pyruvate and TCA metabolites are also labeled from dietary protein and circulating lactate (Figure 2B and S4A).
Figure 2. Quantitative analysis of dietary and circulating nutrient contributions to gut microbiome. See alsoFigure S3,S4.
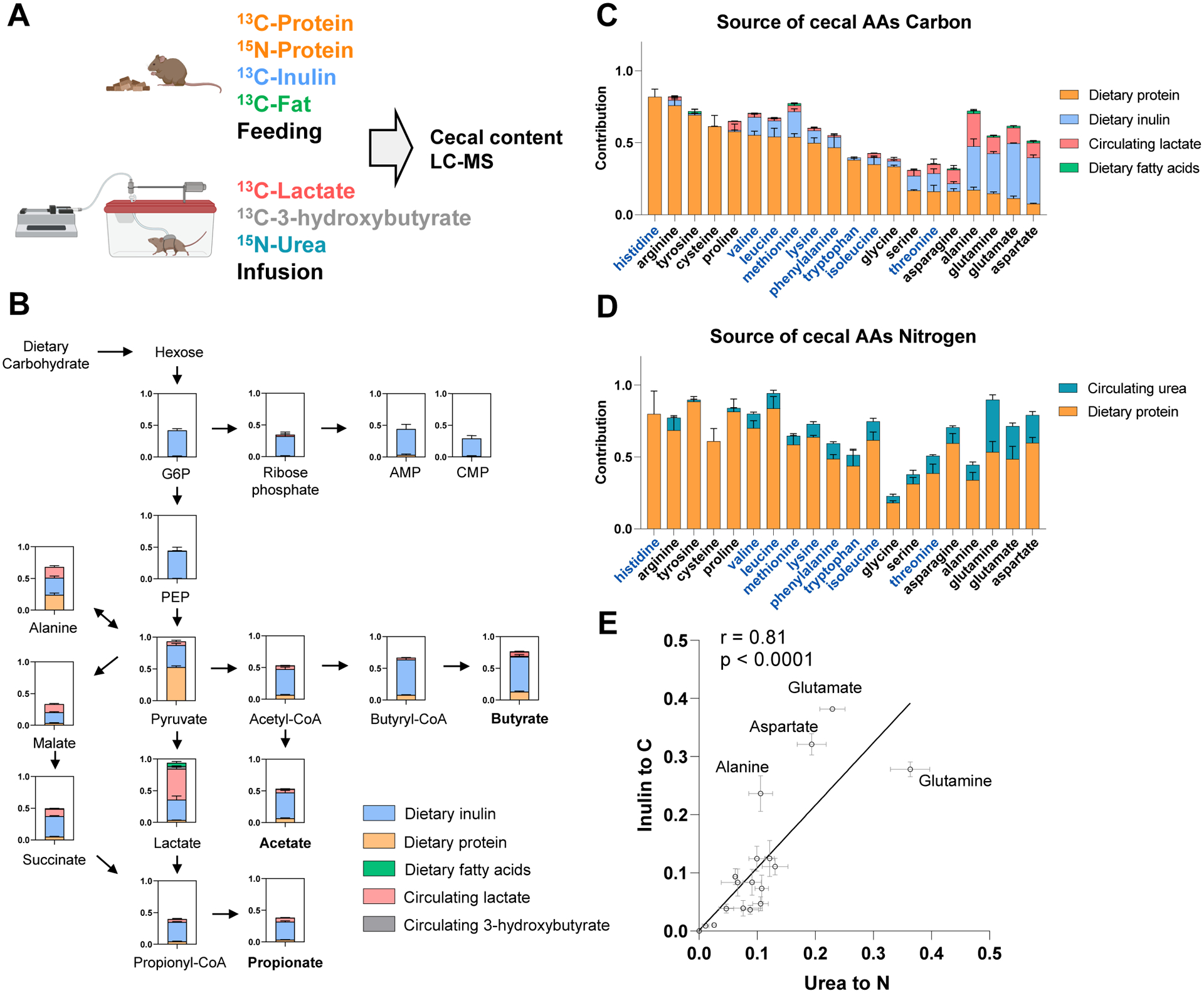
(A) Experimental design. Mice were fed chow containing 13C-protein, 13C-inulin, 13C-fatty acids, or 15N-protein for 24 h. Alternatively, mice were intravenously infused with 13C-lactate, 13C-3-hydroxybutyrate or 15N-urea for 24 h. The labeling of cecal content metabolites was analyzed by LC-MS.
(B) Contribution of dietary and circulating nutrients to carbohydrate fermentation pathways in gut microbiome. Mean ± s.e. N = 4.
(C) Contribution of dietary and circulating nutrients to cecal amino acid carbon. The names of essential amino acids (EAA) are written in blue and non-essential amino acids (NEAA) in black. Mean ± s.e. N = 4.
(D) Contribution of dietary and circulating nutrients to cecal amino acid nitrogen. As in (C), for nitrogen.
(E) Positive correlation, across amino acids in the cecal contents, of carbon contribution from dietary inulin and nitrogen contribution from circulating urea. Mean ± s.e. N = 4.
We next examined inputs to microbiome free amino acids, tracing also with 15N-labeled dietary protein and infused urea. Unlike mammals, most gut bacteria have the biosynthetic capacity to make all 20 proteogenic amino acids. Nevertheless, we observed that “essential amino acids,” which cannot be made by mammals and require the expression of extensive biosynthetic pathways in bacteria, are derived mainly from dietary proteins (Figure 2C). In contrast, “non-essential amino acids” are primarily synthesized within the gut microbiome, using dietary inulin and circulating lactate as carbon sources. Microbiota depletion with antibiotics or in germ-free mice favored cecal accumulation of those amino acids coming (based on our isotope tracing studies) largely from dietary protein, and depletion of those being synthesized by the microbes (Figure S3B–G).
Dietary protein was the main nitrogen source for both essential and non-essential amino acids, with host urea also contributing substantially to the non-essential amino acids (Figure 2D). Dietary protein provides nitrogen to cecal amino acids mainly directly, not through circulating urea (Figure S3H–I). Consistent with the gut microbiome synthesizing amino acids from fiber carbon and urea nitrogen, across amino acids, urea’s nitrogen contribution correlated with inulin’s carbon contribution (Figure 2E).
Amino acid labeling from inulin was typically partial (i.e., one or a few of the amino acid’s carbons atoms were labeled), reflecting inulin’s carbons being scrambled with other inputs into central metabolism (Figure S3J). In contrast, labeling from 13C, 15N-proteins were typically complete (or complete except for the nitrogen label, Figure S3K), indicating direct usage of intact amino acids after proteolysis (sometimes after a cycle of deamination and re-amination). Consistent with such re-amination, the combination of 15N-urea infusion and 13C-protein feeding produced some double-labeled (13C,15N-labeled) amino acids (Figure S3L).
Lastly, the amino acids synthesized by the microbiome, stay in the microbiome: We do not observe discernible labeling of these amino acids in the host (Figure S3M). Taken together, we found that: (i) essential amino acids, although capable of being synthesized by the microbiome, come mainly from the diet and do not go through any carbon rearrangements, (ii) the most closely TCA-linked non-essential amino acids are substantially synthesized by the microbiome using carbon from fiber scrambled with other carbon via central metabolic reactions, and (iii) transamination reactions partially mix nitrogen from diet-derived amino acids with nitrogen from host urea.
Diverse microbiome products come from dietary protein
We next examined the carbon inputs to the other major microbiome products, especially the ones excreted into the portal circulation (Table 1). As expected, SCFAs, the most abundant microbial metabolites, come mainly from dietary fiber. Many less abundant ones, however, are mainly derived from dietary protein (Table 1).
In addition to classical microbiome products, we also observed metabolites that are made in a collaborative manner, with the host carrying out the final synthesis using microbiome-derived inputs. For example, a wide range of microbiome-derived carboxylic acids are conjugated to glycine in the liver and the kidneys to make different acyl-glycines (Figure S4B–E) (Wikoff et al., 2009).
We also examined the host clearance mechanisms of microbiome metabolites, based on arterial-venous gradients across the liver and kidney and levels in the urine. SCFAs and branched-chain fatty acids were avidly consumed by the liver. Most microbiome-derived metabolites were excreted by the kidney into the urine, with the notable exception of SCFAs, which are actively reabsorbed (Table S1A) (Jang et al., 2019; Ullrich et al., 1982). Thus, we establish dietary protein as a major precursor to many microbiome metabolites and identify host-microbiome interplay in the metabolism of SCFAs, including their renal reabsorption and use by liver and kidney for the synthesis of acyl-glycines.
Circulating levels of microbiota metabolites are controlled by protein digestibility
We found that many microbiome-derived metabolites are derived from unabsorbed dietary protein that reaches the colon. We hypothesized that the circulating levels of such metabolites would depend on the extent of dietary protein reaching the colon microbiome. To manipulate this, we fed mice diets in which a portion of the protein (casein, which in part reaches the colonic microbiome) was replaced with free amino acids (which are essentially fully absorbed in the small intestine) (Figure 3A). After 2 weeks, we performed metabolomics on the systemic blood. As expected, diets with less intact protein and more free amino acids tended to increase circulating amino acid levels (Figure 3B). Importantly, protein-derived circulating microbial metabolites (phenols, indoles, and acyl-glycines) fell in tandem (Figure 3C–I). Thus, knowledge of the nutrient sources of microbiome metabolites can be applied to manipulate their systemic levels.
Figure 3. Circulating levels of microbiota metabolites depend on protein reaching the microbiome.
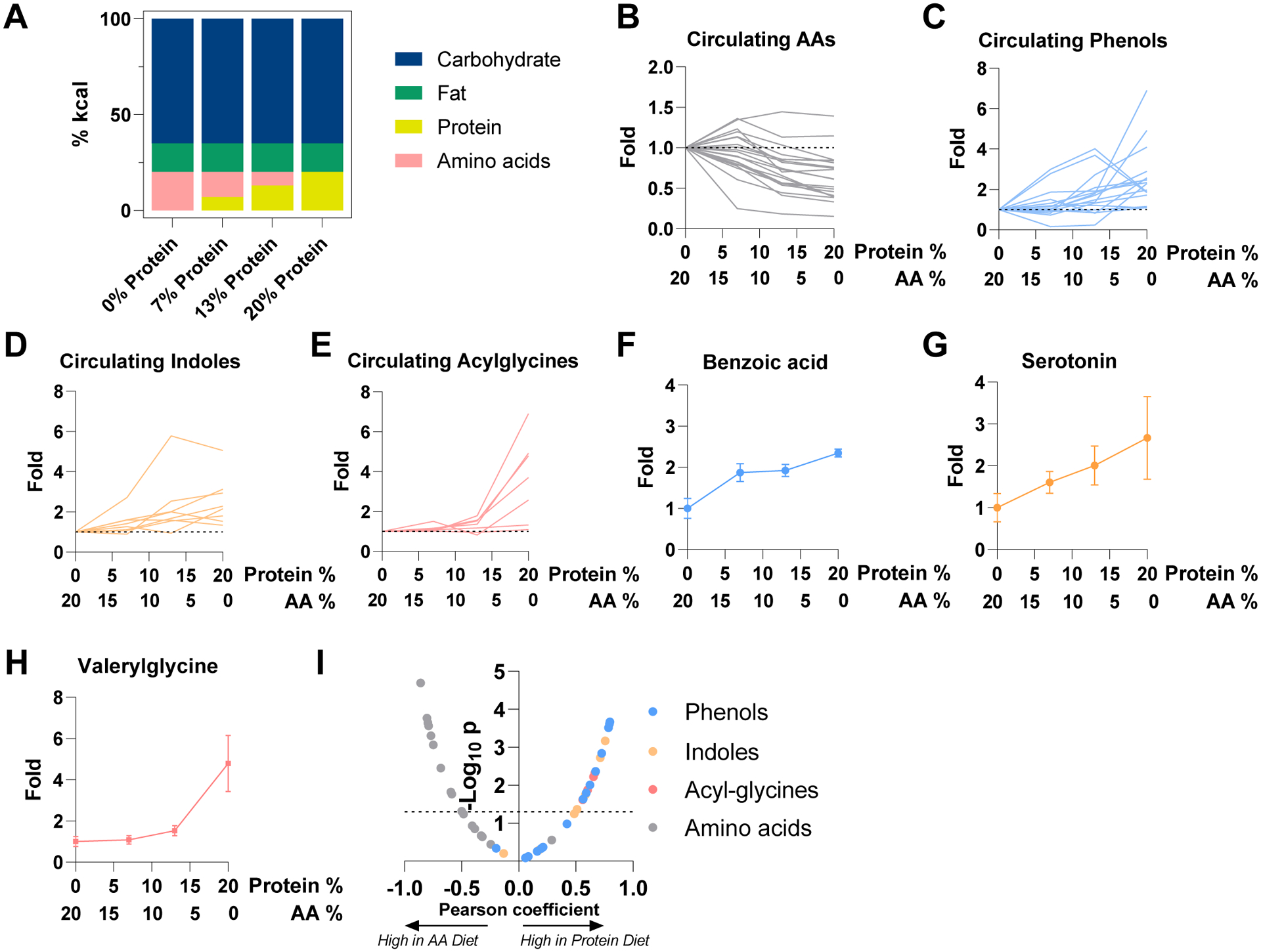
(A) Compositions of diets used in the figure. “Protein” is casein. “Amino acids” are composition-matched free amino acids.
(B) Concentration of circulating amino acids in systemic circulation after two weeks test diet relative to free amino acids diet. Serum was taken at ad lib fed state. Each metabolite is a line. Mean, N = 4 mice.
(C) As in (B), for phenols. Mean, N = 4 mice.
(D) As in (B), for indoles. Mean, N = 4 mice.
(E) As in (B), for acylglycines. Mean, N = 4 mice.
(F) As in (B) for benzoic acid. Mean ± s.e., N = 4 mice.
(G) As in (F), for serotonin. Mean ± s.e., N = 4 mice.
(H) As in (F), for valerylglycine. Mean ± s.e., N = 4 mice.
(I) Correlation between dietary protein (as opposed to free amino acid) fraction in diet and metabolite abundances (relative to amino acid diet). The volcano plot shows Pearson coefficient and P value of correlation between metabolite levels to casein abundance in diet.
Gut bacterial growth is synchronized with host feeding
Thus far, we have reported inputs and outputs of the gut microbiome as a whole. We now shift to examining the growth and metabolism of specific bacterial genera. To this end, we deployed proteomics to measure gut microbial peptides and their labeling, focusing on peptide sequences specific to a single bacterial genus (Figure 4A).
Figure 4. Growth rate of different gut bacterial genera quantified by isotope tracing. See alsoFigure S5.
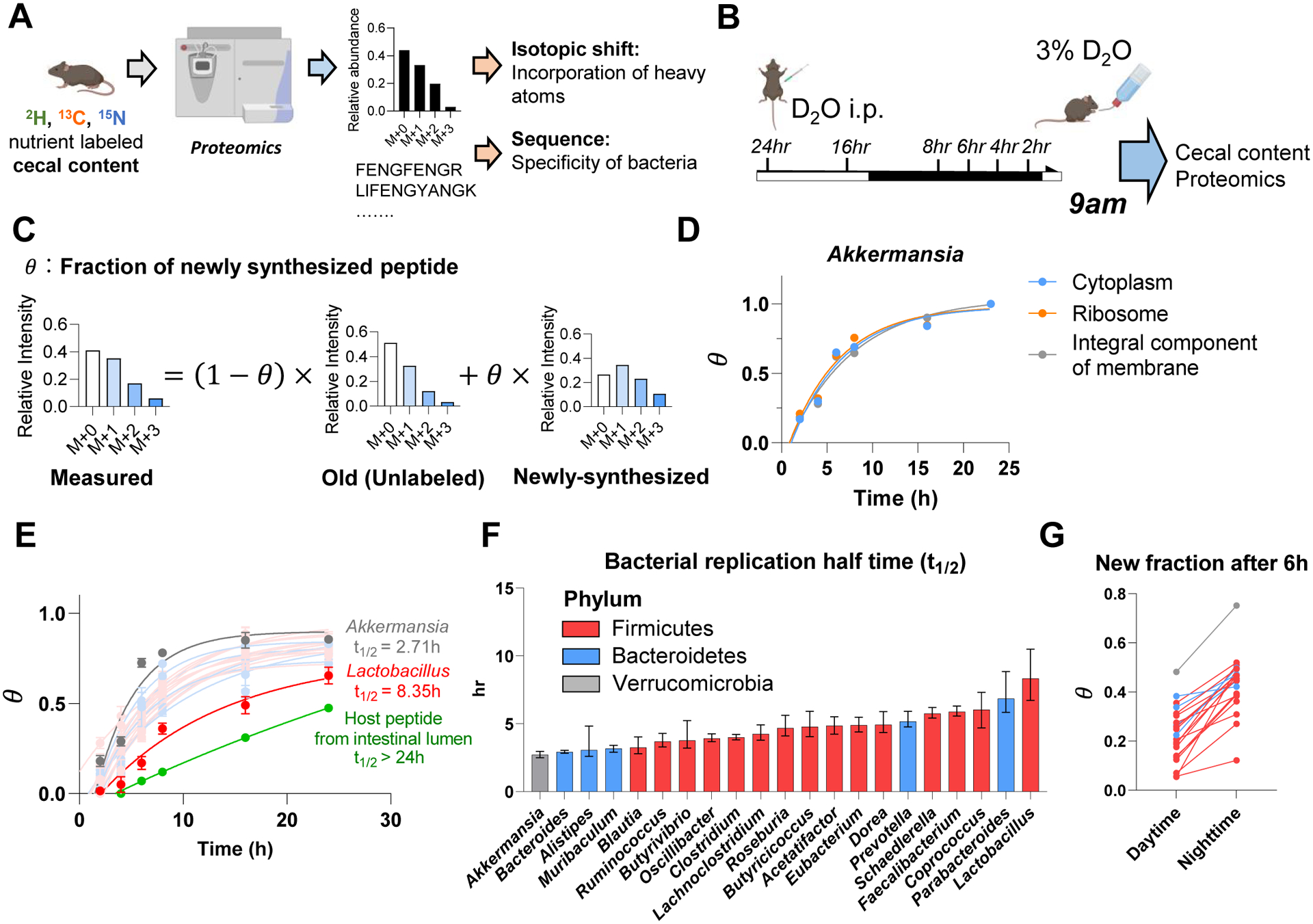
(A) Experimental approach for isotope tracing into specific gut bacteria. Only peptides that are specific to a particular bacterial genus were examined.
(B) Growth rate quantification using D2O. Mice received D2O by i.p. injection followed by D2O drinking water and cecal content labeling was measured over time by proteomics and metabolomics. Mice were fed ad lib; tissues were harvested at 9am.
(C) Calculation of newly synthesized peptide fraction (θ). The experimentally observed peptide mass isotope distribution was fit to a linear combination of unlabeled peptide (“old,” heavy forms from natural isotope abundance) and newly synthesized peptide (“new,” heavy forms from isotope labeling pattern of free cecal amino acids and from natural isotope abundance).
(D) Different cellular compartments from the same bacterial genus show similar labeling rate. Mean, N = 5 mice for each time point.
(E) Genus-specific growth rates were determined by a single exponential fitting, as a function of time, of θ (mean across both different peptides measured from that genus and replicate mice). Mean±s.e., N = 5 mice for each time point.
(F) Bacterial replication half time of different gut bacteria. Data are exponential fits ±s.e.
(G) The gut bacteria synthesize protein in sync with the physiological feeding patterns of the host. The figure shows the average newly synthesized peptide fraction (θ) for different gut bacterial genera after D2O labeling during daytime vs nighttime. Each line connects the daytime and nighttime measurements for one genus. Mean, N = 5 mice for daytime and for nighttime.
To quantify protein synthesis in different gut microbial genera, we used deuterated water (D2O) tracing (Holmes et al., 2015; O’Brien et al., 2020). To achieve steady-state labeling of body water, we gave mice D2O by bolus injection followed by mixing it into drinking water. Peptide labeling in the cecal contents was then measured by proteomics (Figure 4B).
A key technical challenge in using proteomics to read out metabolic activity is the complexity, arising from natural isotope abundances, of peptide mass spectra. We used liquid chromatography-high resolution mass spectrometry to obtain the full scan (MS1) mass isotope distribution for each peptide of interest, with MS/MS analysis of the unlabeled form used to determine the peptide’s identity. We then calculated, based on the mass isotope distribution, the fraction of peptide that was newly synthesized (θ). To this end, first, we calculated the mass isotope distribution of unlabeled peptides based on natural isotope abundances (“old”). Second, we calculated the expected mass isotope distribution of a newly-synthesized peptide generated from cecal free amino acids, whose labeling we experimentally measured by metabolomics. Then, we determined the fraction of newly synthesized (θ) by linear interpolation between the “old” and “newly synthesized” spectra (Figure 4C). To verify this approach in vitro, we cultured Clostridium sporogenes and Bacteroides dorei in media enriched with D2O and measured growth rate as is typically done (based on OD600) and as above (using media in place of cecal amino acid labeling), finding good agreement (Figure S5A–C).
We then measured the newly synthesized fraction (θ) for a minimum of 5 peptides for each bacterial genus in vivo, with abundant gut bacteria yielding θ for over 100 characteristic peptides. Irrespective of their intracellular location, different peptides from the same bacterial genus tended to label at a similar rate (Figure 4D, S5D–E). Labeling rate varied across bacterial genera, with a half doubling time ranging from 2.5 h for Akkermansia to 8 h for Lactobacillus, which still markedly exceeded the labeling rate of host intestinal proteins (> 24 h half doubling time) (Figure 4E–F, Figure S5F).
Our prior analyses revealed that the microbiome is fed substantially by dietary components. Accordingly, we hypothesized that microbial growth synchronizes with physiological feeding, which in mice occurs mainly during the nighttime. To assess the diurnal rhythm of gut bacterial protein synthesis, mice were given D2O for 6 h intervals throughout the diurnal cycle, followed by proteomic analysis of their cecal contents. Every measured bacterial genus showed greater protein synthesis during nighttime than daytime (Figure 4G). Thus, gut bacteria grow in sync with the physiological feeding patterns of the host.
Preferred carbon sources differ across gut bacteria
Next, we quantitated the carbon feedstocks of different microbes, by combining 13C-nutrient labeling and proteomics. Each 13C-labeled nutrient (dietary inulin, dietary algal protein, or circulating lactate) was provided for 24 hours, which is sufficient to achieve steady-state labeling in the gut bacteria. Our analysis strategy involved two steps: first, we calculated, based on each genus-specific peptide’s observed mass isotope distribution, its relative 13C-enrichment (γ) compared to that of cecal free amino acids (Figure 5A). Mathematically, this calculation is identical to the calculation of θ in the D2O case, except here, the tracer is a particular 13C-labeled nutrient, which unlike D2O is used preferentially by certain bacterial genera. The observed peptide’s relative 13C-enrichment multiplied by the average contribution of that 13C-tracer to the gut microbial amino acids pool (LAA_avg←nutrient) gives a quantitative measure of the tracer’s contribution to the observed genus-specific peptide. Averaging across such peptides gives a fractional contribution of the 13C-labeled nutrient to protein synthesis in a bacterial genus.
Figure 5. Preferred carbon sources differ across gut bacteria. See alsoFigure S6.
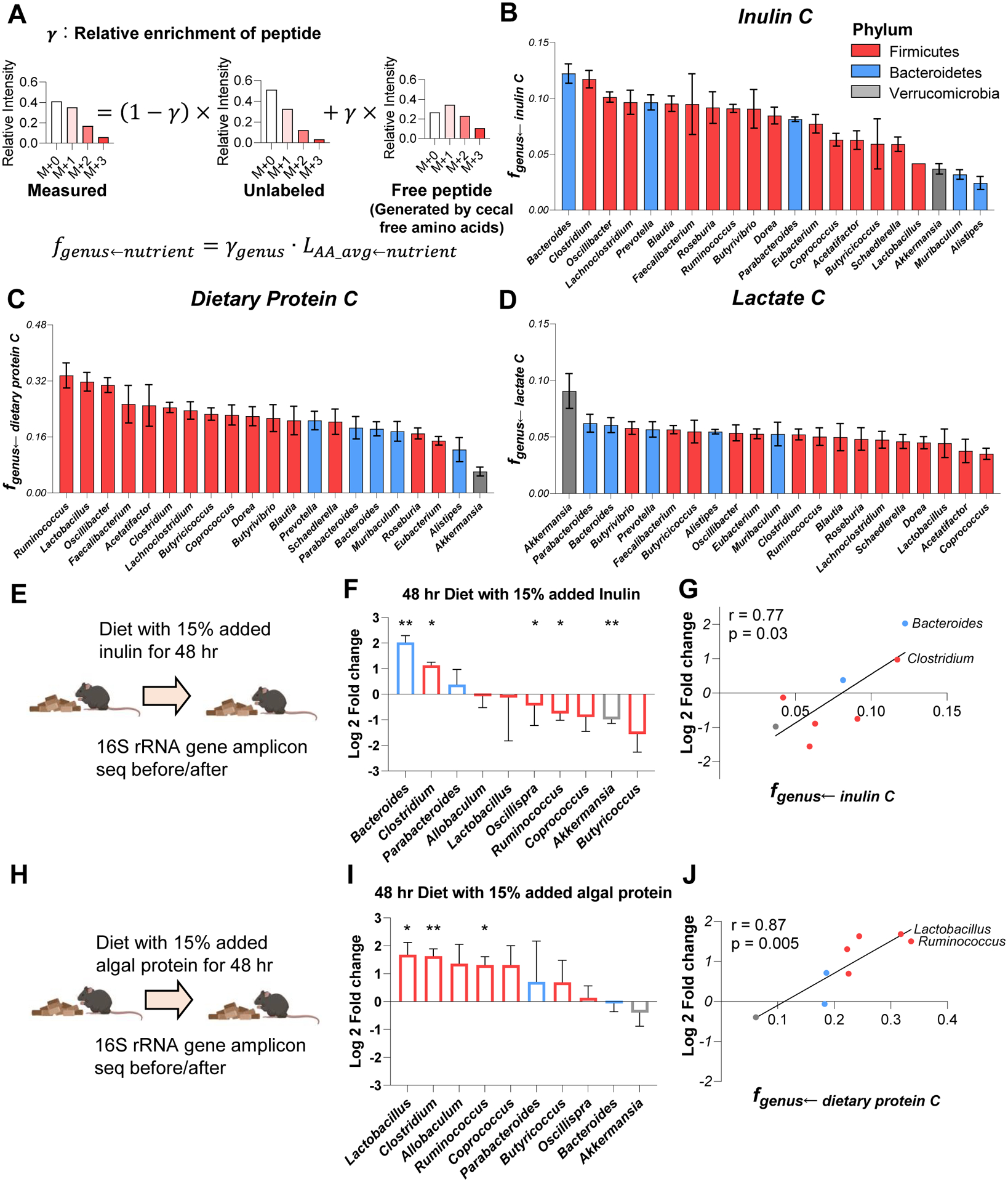
(A) Calculation of peptide relative 13C-enrichment (γ) and carbon contribution from the tracer to a bacterial genus (fgenus←nutrient). First, the experimentally observed peptide mass isotope distribution was fit to a linear combination of unlabeled peptide (heavy forms from natural isotope abundance) and a peptide made from free cecal amino acids (heavy forms from isotope labeling pattern of free cecal amino acids and from natural isotope abundance), yielding. Then, fgenus←nutrient was determined by correcting for the fractional contribution of that tracer to the cecal free amino acid pools.
(B) Carbon contribution of dietary inulin across bacterial genera. Mean±s.e., N = 4 mice.
(C) Carbon contribution of dietary algal protein across bacterial genera. Mean±s.e., N = 6 mice.
(D) Carbon contribution of circulating lactate across bacterial genera. Mean±s.e., N = 7 mice.
(E) Experimental scheme of high-inulin diet feeding followed by 16S rRNA gene amplicon sequencing.
(F) Genus-level microbiota composition changes after high-inulin diet. The genera increased after high-inulin diet prefer inulin in (B). Mean±s.e., N=3 mice. *P<0.05 and **P<0.01 by two-sided Student’s t-test.
(G) Correlation between genera abundance changes and carbon-source preference.
(H- J) As in (E - G), for algal protein-supplemented diet.
Using this method, we measured feedstocks of the bacterial genera that were detected in every proteomics experiment. We were also able to make species-specific measurements in some cases (Figure S6A–F). We observed marked differences in nutrient preferences across microbiota. For example, Bacteroides and Clostridium use over four-fold more inulin than Akkermansia, Muribaculum, or Alistipes (Figure 5B, S6A). Overall, bacteria from the phylum Firmicutes, used more dietary protein than did Bacteroidetes (Firmicutes 0.237 ± 0.052; Bacteroidetes 0.175 ± 0.031, p = 0.02). Akkermansia, which is generally considered a health-promoting gut microbe, used among the least dietary inulin and protein (Figure 5B–C, S6A–B). In contrast, it used by far the most circulating lactate from the host (Figure 5D, S6C).
We were curious whether these bacterial nutrient preferences predict microbiome composition changes upon dietary changes. To explore this possibility, we fed mice an inulin-enriched or algal protein-enriched diet for two days and measured microbiome composition by 16S rRNA gene amplicon sequencing. Bacteroides, the top consumer of 13C-inulin, increased by 4-fold after high inulin diet (Figure 5E–G). Clostridium, another high inulin consumer, also increased by 2-fold. Other genera that use less inulin carbon were either unchanged or slightly decreased. Similar consistency between microbes’ nutrient preference and abundance changes was observed in mice fed the algal protein-enriched diet (Figure 5H–J). Carbon-source preference measured by proteomics (fgenus←nutrient) correlates with abundance change following a diet shift measured by 16S rRNA gene amplicon sequencing, for both the inulin and algal protein conditions (Figure 5G, J). Thus, the nutrient preferences of different gut bacteria help explain microbiome compositional changes following dietary manipulations (David et al., 2014).
Firmicutes consume dietary protein while Bacteroidetes consume secreted host protein
Lastly, we turned to the nitrogen source preferences of different gut bacteria, comparing 15N-labeled dietary protein feeding to 15N-urea infusion. The analytical approach was identical to that employed above for carbon source preferences. Bacterial genera that highly use carbon from dietary protein also highly use nitrogen from dietary protein, consistent with amino acids from dietary protein being assimilated intact in bacterial proteomes (Figure 6A, S6D, S6G).
Figure 6. Firmicutes favor dietary protein while Bacteroidetes prefer secreted host protein. See alsoFigure S6,S7.
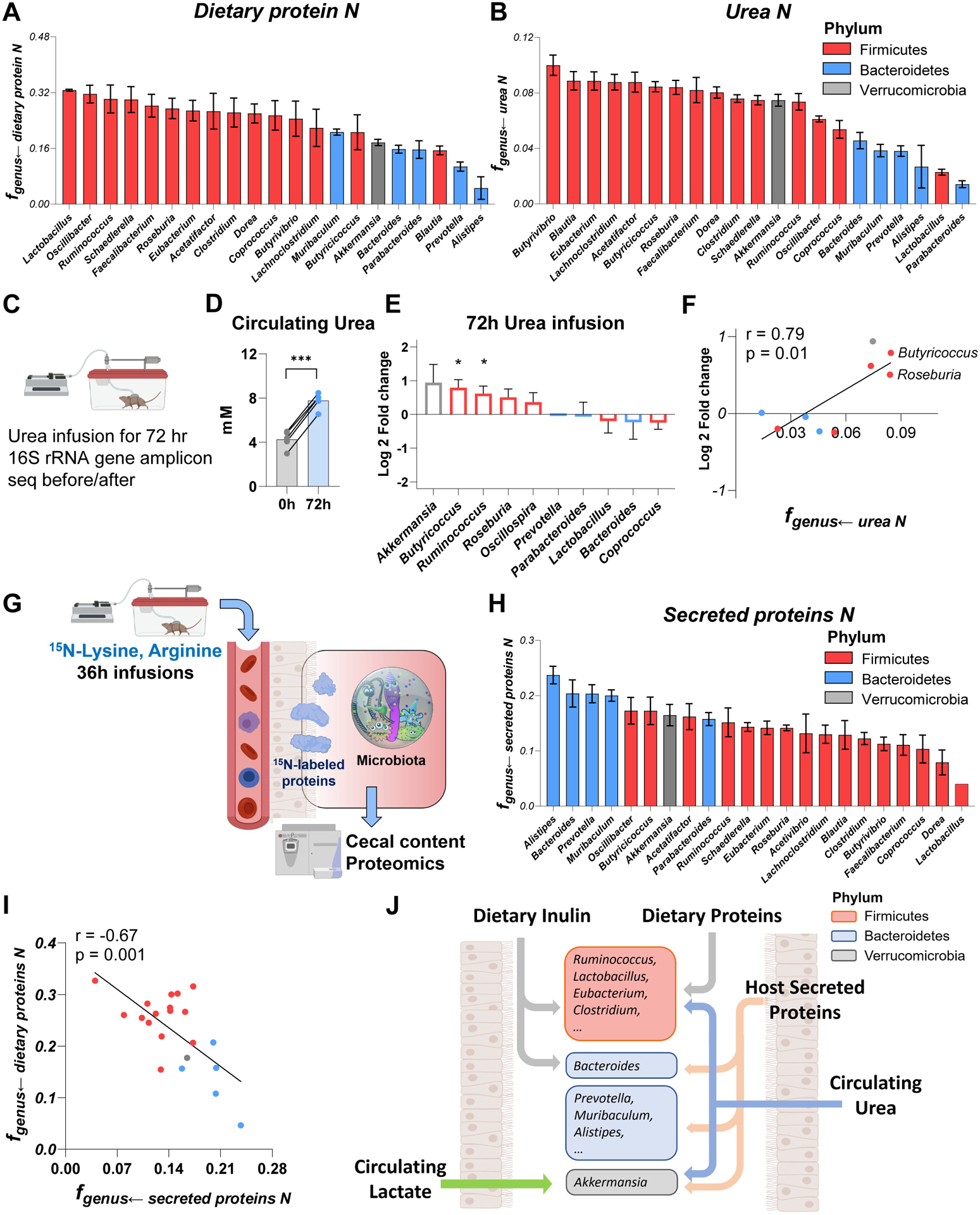
(A) Nitrogen contribution of dietary algal protein across bacterial genera. Mean±s.e., N = 6 mice.
(B) Nitrogen contribution of circulating urea across bacterial genera. Mean±s.e., N = 6 mice.
(C) Experimental scheme of 72 hr urea infusion followed by 16S rRNA gene amplicon sequencing.
(D) Urea infusions increased urea concentration in systemic circulation. N = 5 mice. ***P<0.001 by two-sided Student’s t-test.
(E) Genus-level microbiota composition changes after urea infusion. The genera increased after urea infusion prefer urea in (B). Mean±s.e., N=5 mice. *P<0.05 by two-sided Student’s t-test.
(F) Correlation between genera abundance changes and nitrogen-source preference.
(G) Experimental schematic of long-term 15N-lysine and 15N-arginine infusion to probe the contribution of secreted host proteins to different bacterial genera.
(H) Nitrogen contribution of secreted host proteins across bacterial genera. Mean±s.e., N = 5 mice.
(I) Negative correlation between fgenus←dietary proteins N and fgenus←secreted proteins N.
(J) Summary of carbon and nitrogen inputs to different gut bacteria. Firmicutes prefer dietary carbon sources (fiber and protein) and nitrogen from host circulating urea. Bacteroidetes heavily use dietary fiber, while using on host secreted proteins for nitrogen. Verrucomicrobia prefers host secreted nutrients, both protein and circulating small molecules (lactate, urea).
Conversely, among members of the phylum Firmicutes, genera preferring urea nitrogen tended to be avid inulin users, i.e., to synthesize their own amino acids using inulin and urea (Figure 6B, S6E, S6H). This includes some urease-negative genera, which presumably acquire urea nitrogen via cross-feeding. Moreover, again among Firmicutes, we also saw the expected trade-off where some genera prefer nitrogen from dietary protein, and others from circulating urea (Figure S6I). Following intravenous urea infusions to raise circulating urea concentrations, abundance of those Firmicutes preferring urea, along with Akkermansia, increased substantially (Figure 6C–F).
Compared to Firmicutes, the lower use of both dietary protein and circulating urea nitrogen by Bacteroidetes raised a key question: How do Bacteroidetes get nitrogen? Some members of gut microbiome (e.g. Bacteroides and Akkermansia) are capable of digesting host secreted proteins such as mucins (Berry et al., 2013; Reese et al., 2018). We hypothesized that host secreted proteins are a key source of Bacteroidetes nitrogen. To probe this possibility, we performed long-term 15N-labeled lysine and arginine infusions (12, 18, 36 h) to label host proteins in the colon (Figure 6G and Figure S7A–E). Despite not directly feeding the microbiome (Figure 1F, S7E), lysine and arginine did contribute after 36-h infusion, consistent with the labeling occurring via host proteins. Such labeling occurred preferentially in Bacteroidetes and Akkermansia (Figure 6H, S6F). The nitrogen contributions from dietary and secreted host proteins were anti-correlated, consistent with some gut bacteria preferentially consuming dietary protein, and others host protein (Figure 6I). Bacterial genera with a greater preference for dietary protein, whose availability depends on host feeding, grow more differently between daytime and nighttime (Figure S6J–K). Thus, dietary proteins and circulating urea are the major nitrogen feedstock of Firmicutes, while secreted host proteins provide nitrogen to Bacteroidetes.
DISCUSSION
As for most microbial communities, the composition of the gut microbiome is shaped by nutrient availability. Here we developed quantitative isotope tracing approaches to measure the nutrient preferences of gut bacteria. In addition to dietary fiber and secreted host proteins, we establish dietary protein and circulating host lactate, 3-hydroxybutyrate, and urea as important nutrients feeding gut bacteria. Importantly, we rule out direct contributions from other circulating host nutrients, like glucose and amino acids, to the colonic microbiome.
A key technical achievement is enabling tracing from different carbon and nitrogen sources into bacteria-specific peptides, thereby revealing the nutrient preferences of different bacteria within the complex and competitive gut lumen environment. We find that Firmicutes and Bacteroidetes differ systematically in their utilization of host secreted protein versus dietary protein: Firmicutes tend to acquire amino acids from dietary protein, while Bacteroidetes rely more on secreted host protein (Figure 6J). This may relate to different localization of bacteria within the colon, either in terms of central versus peripheral (closer to host mucus) or distal versus proximal (closer to incoming food remnants) (Albenberg et al., 2014; Li et al., 2015; Yasuda et al., 2015).
Within these two major families of gut bacteria, we found marked disparities in the use of dietary fiber as a carbon source. The most abundant Bacteroidetes’ genus is Bacteroides, and it was the most avid assimilator of fiber (inulin). In contrast, other types of bacteria in the same phylum hardly consumed inulin. Likewise, some Firmicutes like Clostridium avidly used fiber, while others did not. Strikingly, feeding a fiber-enriched diet led to an increased abundance of Bacteroides and Clostridium, the precise genera that most actively assimilate fiber based on isotope tracing.
A similar trend was observed in the case of dietary supplementation with algal protein: Firmicutes, which actively use such protein, tended to increase in abundance. Algal protein (the only type commercially available in bulk in 13C-labeled form) may be particularly hard for mammals to digest. This is reflected in the limited appearance of 13C-labeled amino acids from algal protein in the portal circulation, and instead extensive passage from the intestine into the colon. This influx of dietary protein to the microbiome was a major contributor to secreted microbiome metabolites: As shown by replacing intact dietary protein with more absorbable (and thus less microbiome-accessible) free amino acids, the production and hence systemic concentration of these products depends on dietary protein reaching the colonic microbiome. An important future question is whether the nature of dietary protein (e.g. plant or animal-based) impacts passage through the small intestine to the colonic microbiome and thereby shapes microbiome composition or metabolite secretion (Madsen et al., 2017; Wali et al., 2021).
Host circulating metabolite levels may also impact microbiome nutrient access and ultimately composition. Here we show such effects are likely limited to the few host metabolites that meaningfully penetrate the microbiome: urea, 3-hydroxybutyrate, and lactate. Among them, lactate was recently shown to feed the gut microbiome in human marathon runners (Scheiman et al., 2019). Among gut bacteria, Akkermansia most avidly use circulating lactate. Akkermansia are mucin degraders, and their proximity to the gut epithelial wall may augment their access to lactate from the host circulation. Akkermansia are more abundant in athletes, and exercise increases their levels in mice and human (Liu et al., 2017; Munukka et al., 2018). A possible mechanism involves increased circulating lactate levels following exercise directly feeding Akkermansia. Whether lactate induced Akkermansia growth in part mediates beneficial effects of exercise is an important open question. Consistent with their urea preferences measured by isotope tracing, Akkermansia and certain genera within Firmicutes (e.g., Roseburia, Butyricoccus, Ruminococcus) also increase in abundance upon experimental elevation of circulating host urea.
Ultimately, manipulating the microbiome requires understanding which nutrients different bacteria consume, and how such consumption impacts microbiome composition and product secretion. Through isotope tracing, including proteomic measurements that offer bacterial genus specificity, we provide foundational knowledge about which nutrients feed the gut microbiome, and which bacteria prefer which nutrients. The methodologies developed here are poised for broader application, to eventually contribute to the holistic and quantitative understanding of the diet-microbiome-health connection.
LIMITATIONS OF THE STUDY
Our investigation focuses solely on healthy mice fed standard chow (in some cases with specific fiber or protein supplements). Measurements of microbiome feedstocks are limited to isotope tracing and mass spectrometry. Feedstocks of different bacteria are determined based on the isotopic signatures of bacteria-specific peptides. Peptide identification involves a 2% false discovery rate. Taxonomic assignment is based on bacterial proteome sequences available on Uniprot (Gurdeep Singh et al., 2019). Orthogonal approaches, which could provide measurement validation or complementary information, such as fluorescent-activated cell sorting of bacteria, were not explored (Batani et al., 2019). In most cases, taxonomic assignment was limited to the genus level, due to lack of sufficient specificity of the detected peptide sequences. In the future, improved sensitivity may enable species- or strain-specific peptide sequence measurements.
STAR*METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Professor Joshua D. Rabinowitz (joshr@princeton.edu).
Materials Availability
This study did not generate new unique regents or new mouse lines.
Data and Code Availability
The proteomics datasets generated during this study are deposited in PRIDE: PXD031015. The isotope tracing data are included in Table S2. The taxonomic assignment of the detected tryptic peptides in the study are included in Table S3. Composition of the diet used in the study are included in Table S4. The 16S rRNA gene amplicon sequencing datasets generated during this study are available in Table S5. The code for peptide enrichment calculations generated during this study is available at GitHub: (https://github.com/xxing9703/pepMID_simul). Any additional information required to re-analyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse studies.
Mouse studies followed protocols approved by the Princeton University Animal Care and Use Committee. Unless otherwise indicated, 7–9-week-old male C57BL/6NCrl mice (strain 027; Charles River Laboratories) were group-housed on a normal light-dark cycle (8:00–20:00) with free access to water and chow.
Bacterial culture studies.
B. dorei, C. sporogenes, E. coli, S. aureus and L. reuteri glycerol stocks were brought into an anerobic chamber (70% N2, 25% CO2, 5% H2) and grown in liquid media: L. reuteri was grown on MRS (MRS Broth, Sigma); E. coli was grown on LB (Luria Broth, Sigma); S.aureus was grown in TSB (Tryptic Soy Broth, Bacto) and C. sporogenes and B. dorei were grown in GAM (GAM Broth Modified, HyServe).
METHOD DETAILS
Mouse gavage and nutrient feeding.
For the 13C-nutrient gavage experiments, mice were fasted at 9 am and received a 1:2:4 mixture of inulin, protein/amino acids, and starch (0.5 g kg−1 inulin, 1 g kg−1 protein/amino acids 2g kg−1 starch dissolved in water) at 3 pm via oral gavage with a plastic feeding tube (Instech Laboratories). Food was given back at 8 pm.
For the mouse experiments involving labeled nutrient feeding, the labeled diet was prepared by adding 13C/15N-nutrients to a diet mixture premix (modified from normal diet with reduced protein, inulin, and starch content, Research diets Inc, D20030303). The final enrichment for each labeled dietary nutrient was 10% - 25% (with observed labeling corrected by dividing by the fraction dietary nutrient labeled). The contribution of each dietary nutrient to metabolites is calculated by the metabolite labeling enrichment normalized to the final enrichment of each labeled dietary nutrient. All diets shared the same final macronutrient composition (40% starch, 20% protein or amino acids, 7.5% inulin and 2.5% cellulose). Mice were first adapted to a non-labeled diet (of identical composition to the subsequent labeled diet) for 10 days, and then fed labeled diet for 24 h prior to sacrifice.
For the deuterium water drinking experiment, mice were administered a bolus intraperitoneal injected of D2O (1.26 % w/w relative to body weight), followed by having ad lib access to 3% D2O drinking water.
For the protein and amino acids diet feeding experiment, mice were fed on casein or compositional matched amino acids diet (20% casein, 13% casein + 7% amino acids, 7% casein +13% amino acids, and 20% amino acids as protein/amino acids sources, Table S4) for 2 weeks. Serum was sampled by tail-bleed at 9 am ad lib.
Intravenous infusions.
To quantify contribution of circulating nutrients to microbiota metabolism, 9–11-week-old C57BL/6 mice were catheterized in house in the right jugular vein. The mice were infused with carbon or nitrogen-labeled tracer starting at 3:30 pm without any fasting. Infusion rate was 0.1 ul/min/g. Infusion solutions are described in Table S2A. Overnight (24 h) infusions both started and finished around 9 am. The contribution of circulating nutrient to each metabolite is calculated by the metabolite labeling enrichment normalized to the average tracer serum enrichment throughout 24 hr.
Antibiotics treatment.
To deplete the mouse resident microbiome, an antibiotic drinking water protocol was used. In brief, mice were treated with a cocktail of antibiotics (1 g/L ampicillin, 1 g/L neomycin, 1 g/L metronidazole, and 1 g/L vancomycin) in both their drinking water 14 days. To make the drinking water more palatable, 5% aspartame was added. The effectiveness of antibiotics treatments was verified by observing much lower SCFAs in the feces by LC-MS.
Sample collection.
Systemic blood samples (~6 μl) were collected by tail bleeding. For sampling from tissue-specific draining veins, a mouse was put under anesthesia and different tissue veins were exposed, and blood samples were pulled with an insulin syringe (BD insulin syringes, # SY8290328291) insertion into the vein. Successful isolation of portal vein was confirmed by much higher (> 10x) concentrations of SCFAs and secondary bile acids (deoxycholic acid and lithocholic acid) than systemic vein; hepatic vein was confirmed by much lower secondary bile acids, SCFAs and higher glucose, 3-hydroxybutyrate levels compared to portal vein. Mouse urine was collected from the urinary bladder using a syringe. All serum samples were placed on ice without anticoagulant for 15 min, and centrifuged at 16,000 × g for 15 min at 4 C.
Tissues were harvested by quick dissection and snap freezing (<5 sec) in liquid nitrogen with a pre-cooled Wollenberger clamp; intestinal contents were removed before clamping. For cecal content sampling, the mouse cecum was first removed and cut on the surface, then the cecal content was squeezed out using a tweezer followed by freeze clamping. Whole liver, intestine, and intestinal contents were collected and grounded to homogenous powder. To sample fresh feces, the mouse belly was gently massaged to induce defecation and fresh feces were freeze clamped. For long-term feces collection, a mouse was transferred to a new cage and mouse fecal pellets on the bedding were collected every 1~2 h and freeze clamped. Serum, tissue, and feces samples were kept at −80 °C until further analysis.
16S rRNA gene amplicon sequencing and analysis.
Extraction of Bacterial DNA from cecal or fecal samples was performed using the Power Soil DNA Isolation kit (QIAGEN). A section of the 16S rRNA gene (~250 bp, V4 region) was amplified, and Illumina sequencing libraries were prepared from these amplicons according to a previously published protocol and primers (Caporaso et al., 2012). Libraries were further pooled together at equal molar ratios and sequenced on an Illumina HiSeq 2500 Rapid Flowcell or MiSeq as paired-end reads. These reads were 2×150 bp with an average depth of ~20,000 reads. Also included were 8 bp index reads, following the manufacturer’s protocol (Illumina, USA). Pass-Filter reads were generated from raw sequencing reads using Illumina HiSeq Control Software. Samples were de-multiplexed using the index reads. The DADA2 plugin within QIIME2 version 2018.6 was used to inferred Amplicon sequencing variants (ASVs) from the unmerged paired-end sequences (Bolyen et al., 2019; Callahan et al., 2016). The forward reads were trimmed at 150 bp and the reverse reads trimmed at 140 bp, with all other DADA2 as default. Taxonomy was assigned to the resulting ASVs with a naïve Bayes classifier trained on the Greengenes database version 13.8, with only the target region of the 16S rRNA gene used to train the classifier (Bokulich et al., 2018; McDonald et al., 2012). Downstream analyses were performed MATLAB (Hunter, 2007; McKinney, 2010).
Bacterial culture studies.
For the D2O experiment, 250 – 1000 μl D2O was added into the media (5–10 mL, to reach a final enrichment of 5–10%) with either B. dorei or C. sporogenes, and OD600 was recorded at the addition. After every 25–30 min, OD600 was recorded and 100–200 μl bacterial solution was taken for metabolomics and proteomics analysis. The newly synthesized fraction of bacteria was calculated by (OD600 – OD600, 0min)/OD600.
Bacterial colonization in mice.
Mice were treated with antibiotics in drinking water for 10 days. On day 11, no antibiotics were administered, and mice were gavaged with 250 μl of bacterial consortia consisting of urease-negative bacteria (B. dorei, C. sporogenes and E. coli) or a combination of urease-negative and urease-positive bacteria (B. dorei, C. sporogenes, E. coli, S. aureus and L. reuteri).
Metabolite extraction.
For serum samples, 3 ul serum was added to 90 ul methanol and incubated on ice for 10 min, followed by centrifugation at 17,000 × g for 10 min at 4°C. The supernatant was transferred to an MS vial until further analysis. For tissues and feces samples, frozen samples were first ground at liquid nitrogen temperature with a cryomill (Restch, Newtown, PA). The resulting tissue powder was extracted with 40:40:20 methanol: acetonitrile: water (40 ul extraction solvent per 1 mg tissue) for 10 min on ice, followed by centrifugation at 17,000 × g for 10 min, the supernatant was transferred to a MS vial until further analysis.
Measurements of metabolites, protein, and polysaccharides.
To measure metabolites in serum, tissue and feces samples, a quadrupole orbitrap mass spectrometer (Q Exactive; Thermo Fisher Scientific) was coupled to a Vanquish UHPLC system (Thermo Fisher Scientific) with electrospray ionization and scan range m/z from 60 to 1000 at 1 Hz, with a 140,000 resolution. LC separation was performed on an XBridge BEH Amide column (2.1×150 mm, 2.5 μm particle size, 130 Å pore size; Waters Corporation) using a gradient of solvent A (95:5 water: acetonitrile with 20 mM of ammonium acetate and 20 mM of ammonium hydroxide, pH 9.45) and solvent B (acetonitrile). Flow rate was 150 μl/min. The LC gradient was: 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; and 30 min, 85% B. Injection volume was 5–10 μl and autosampler temperature was set at 4°C. For cysteine measurement, samples were derivatized before measurement as follows: Serum, cecal content or feces samples were extracted and centrifuged. To the supernatant, 2 mM N-ethylmaleimide was added and incubated at room temperature for 20 min. The resulting mixture was transferred to a MS vial. Derivatized cysteine has a m/z at 245.06015 in negative mode.
To quantify the metabolite concentration in serum and tissue samples, either isotope spike-in or standard spike-in was performed. For isotope spike-in, known concentrations of isotope-labeled standard were added to the serum or tissues extraction solution, then the concentration was calculated by the ratio of labeled and unlabeled metabolites. When isotope standard is not available, a serially diluted non-labeled standard was added, and a linear fitting between measured total ion count and added concentration of standard was generated. Then, the concentration of endogenous metabolite was determined by the x intercept of the fitting line.
Starch and inulin were measured by acid hydrolysis and LC-MS. In brief, 5–10 mg sample was mixed with 10 μl 2 M hydrochloric acid, and samples were incubated at 80°C for 2 h. After cooling down, the resulting mixture was neutralized with 12 μl saturated sodium bicarbonate, followed with 88 μl 1:1 acetonitrile: methanol solution. After centrifugation at 17,000 × g for 10 min at 4°C, the supernatant was transferred to a MS vial. Inulin and starch concentration in samples was inferred from total ion count of fructose and glucose, respectively.
SCFAs and BCFAs were derivatized and measured by LC-MS. Serum (5 μl) or tissue samples (~10 mg) were added to 100 μl derivatizing reagents containing 12 mM 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide, 21 mM 3-Nitrophenylhydrazine hydrochloride acid and pyridine (2.4% v/v) in methanol. The reaction was incubated at 4°C for 1 h. Then, the reaction mixture was centrifuged at 17,000 g for 10 min. 20 μl supernatant was quenched with 200 μl 0.5 mM beta-mercaptoethanol in 0.1% formic acid water. After centrifugation at 17,000 g for 10 min, the supernatant was transferred to MS vials until further analysis. The measurement of SCFAs and BCFAs are performed using the same Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer with different column and LC setup. LC separation was on Acquity UPLC BEH C18 column (2.1 mm × 100 mm, 1.7 5 μm particle size, 130 Å pore size, Waters, Milford, MA) using a gradient of solvent A (water) and solvent B (methanol). Flow rate was 200 μL/min. The LC gradient was : 0 min, 10% B; 1 min, 10% B; 5 min, 30% B; 11 min 100% B; 14 min, 100% B; 14.5 min 10% B; 22 min 10 % B. Autosampler temperature was 5 °C, column temperature was 60 °C and injection volume was 10 μl. Ion masses for derivatized acetate, propionate, butyrate, iso-butyrate, valeric acid, isovaleric acid, 2-methylbutyrate, 4-methylvaleric acid were 194.0571, 208.0728, 222.0884, 222.0884, 236.1041, 236.1041, 236.1041, 250.1197 in negative mode, respectively.
The ammonia derivatization method was modified from the previous reported Berthelot reaction assay (Spinelli et al., 2017b, 2017a). In brief, 20 mg tissue or 10 μl serum was extracted by using 200 μl 80% methanol. 100 μl of the metabolite extract was mixed with 100 μl. Solution #1 (100 mM Phenol, 50 mg/L sodium nitroprusside) and 100 μl. Solution #2 (0.38 M dibasic sodium phosphate, 125 mM NaOH, 1% sodium hypochlorite, available chlorine 10–15%). The mixture was incubated at 40°C for 30 min. Then, 100 μl reaction solution was mixed with 200 μl methanol to oversaturate the inorganic salt to quench the reaction. The final solution was centrifuged for 30 min. Then the supernatant was loaded to LC-MS for analysis. Ion mass for derivatized ammonia is 198.05605 in negative ion mode.
Protein amino acid composition was measured by acid hydrolysis. Approximately 10 mg of protein was extracted with 400 μl methanol, 200 μl chloroform and 300 μl water, followed by centrifugation at 20,000 × g for 10 min at 4 °C. The upper layer was removed. The resulting mixture was further extracted with 600 μl methanol twice and supernatant was discarded. The resulting precipitate was dried under nitrogen gas and then hydrolyzed with 250 μl 6 M hydrochloric acid incubated overnight at 115°C. After incubation, the samples were dried under nitrogen gas and reconstituted in 1 mL methanol, and the supernatant was transferred to a MS vial for analysis. Amino acid composition of the proteins used in the study are shown in Figure S4F, and such differences in protein amino acid composition do not correlate with the quantified dietary protein contribution to cecal amino acids (Figure S4G).
Proteomics sample preparation.
Proteomics samples were prepared mostly as previously described (Gupta et al., 2018; Wühr et al., 2014). Mouse cecal samples (10 mg each) were dissolved in 400 μl lysis buffer (6M guanidium chloride, 2% cetrimonium bromide, 5 mM dithiothreitol, 50 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), pH 7.2). Then the sample mixture was put on ice and sonicated for 10 cycles (30 s on and 30 s off cycle, amplitude 50%) by a sonicator (Qsonica), followed by centrifugation at 20,000 × g for 20 min at 4 °C. The supernatant was taken and alkylated with 20 mM N-ethylmaleimide for 20 min at room temperature, 5 mM dithiothreitol was added to quench the excessive alkylating reagents. Proteins were purified by methanol-chloroform precipitation. The dried protein pellet was resuspended in 10 mM EPPS (N-(2-Hydroxyethyl) piperazine-N’-(3-propanesulfonic acid)) at pH 8.5 with 6 M guanidine hydrochloride. Samples were heated at 60°C for 15 min and protein concentration was determined by BCA assay (Pierce BCA Protein Assay Kit, Thermo Scientific). The protein mixture (30~50 μg) was diluted with 10 mM EPPS pH 8.5 to 2 M GuaCl and digested with 10 ng/μL LysC (Wako) at room temperature overnight. Samples were further diluted to 0.5 M GuaCl with 10 M EPPS pH 8.5 and digested with an additional 10 ng/μL LysC and 20 ng/μL sequencing grade Trypsin (Promega) at 37°C for 16 h. Samples were desalted using a SepPak cartridges (Waters) and then vacuum-dried and resuspended in 1% formic acid before mass spectrometry analysis.
Proteomics peptide measurement.
Samples were analyzed on an EASY-nLC 1200 (Thermo Fisher Scientific) HPLC coupled to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) with Tune version 3.3. Peptides were separated on an Aurora Series emitter column (25 cm × 75 μm ID, 1.6 μm C18) (Ionopticks, Australia) and held at 60°C during separation using an in-house built column oven over 180 min, applying nonlinear acetonitrile gradients at a constant flow rate of 350 nL/min. The Fusion Lumos was operated in data dependent mode. The survey scan was performed at a resolution setting of 120k in orbitrap, followed by MS2 duty cycle of 1.5 s. The normalized collision energy for CID MS2 experiments was set to 30%.
Solvent A consisted of 2% DMSO (LC-MS-grade, Life Technologies), 0.125% formic acid (98%+, TCI America) in water (LC-MS-grade, OmniSolv, VWR), solvent B of 80% acetonitrile (LC-MS-grade, OmniSolv, Millipore Sigma), 2% DMSO and 0.125% formic acid in water. The following 120 min-gradient with percentage of solvent B were applied at a constant flow rate of 350 nL/min after thorough equilibration of the column to 0% B: 0% – 6% in 5 min; 6 – 25% in 160 min; 25% –100% in 10 min; 100% for 5 min. For electrospray ionization, 2.6 kV were applied between minutes 1 and 113 (or minutes 1 and 83 for fractionated samples) of the gradient through the column. To avoid carry-over of peptides, 2,2,2-trifluoroethanol (>99% Reagent plus, Millipore Sigma) was injected in a 30 min wash between each sample.
Proteomics data analysis.
The data was analyzed using GFY software licensed from Harvard (Nusinow et al., 2020). Thermo Fisher Scientific. raw files were converted to mzXML using ReAdW.exe. MS2 spectra assignment was performed using the SEQUEST algorithm v.28 (rev. 12) by searching the data against the combined reference proteomes for Mus Musculus, Bos Taurus, and all the abundant bacterial families detected in 16S rRNA gene amplicon sequencing (Bacteroidaceae, Porphyromonadaceae, Prevotellaceae, Rikenellaceae, Muribaculaceae, Lachnospiraceae, Ruminococcaceae, Erysipelotrichaceae, Oscillospiraceae, Clostridiaceae, Eubacteriaceae, Lactobacillaceae and Verrucomicrobiaceae) acquired from Uniprot on Jan 2021 (SwissProt + Trembl) along with common contaminants such as human keratins and trypsin. The target-decoy strategy was used to construct a second database of reverse sequences that were used to estimate the peptide false discovery rate (Elias and Gygi, 2007). A 20-ppm precursor ion tolerance with the requirement that both N- and C- terminal peptide ends are consistent with the protease specificities of LysC and Trypsin was used for SEQUEST searches, two missed cleavage was allowed. NEM was set as a static modification of cysteine residues (+125.047679 Da). An MS2 spectral assignment false discovery rate of 0.5% was achieved by applying the target decoy database search strategy. Linear Discriminant analysis was used for filtering with the following features: SEQUEST parameters XCorr and unique ΔXCorr, absolute peptide ion mass accuracy, peptide length and charge state. Forward peptides within three standard deviations of the theoretical m/z of the precursor were used as positive training set. All reverse peptides were used as negative training set. Linear Discriminant scores were used to sort peptides with at least seven residues and to filter with the desired cutoff. Furthermore, we performed a filtering step on the protein level by the “picked” protein FDR approach (Savitski et al., 2015). Protein redundancy was removed by assigning peptides to the minimal number of proteins which can explain all observed peptide, with above-described filtering criteria.
To quantify the intensities of all the isotopic peaks of the peptides, we used raw intensity. Missed cleavage peptides (more than one K or R in the peptide) and low signal to FT-noise peptides (M0 S/N < 20) were removed. Peptide phylogenetic assignment was performed using Unipept 4.0 (Gurdeep Singh et al., 2019), ‘Equate I and L’ and ‘Advanced missed cleavage handling’ were not selected. Only peptides that are specific at a genus level were used for further analysis.
Quantification of newly-synthesized fraction of peptide.
To determine the newly synthesized fraction of a bacterial peptide in D2O drinking water experiment, we first measured the cecal content free amino acids deuterium labeling pattern using metabolomics. Then, for each peptide, we simulated the expected isotope envelope pattern if the peptide were old, i.e., unlabeled with deuterium (Iold), versus if it were newly synthesized by taking up free amino acids from the cecal content (Inew). Iold was calculated based on the peptide’s molecular formula and 13C, 15N, 2H, 17O, 18O, 32S, 33S and 36S natural abundance. Inew was calculated based on the peptide’s sequence and experimentally observed labeling of the corresponding cecal free amino acids (after natural isotope correction), and the natural isotope abundance of the unlabeled atoms in the peptide’s formula. The simulation of expected peptide isotope distribution and fitting was performed using a MATLAB code: https://github.com/xxing9703/pepMID_simul. Exact mass isotopic peaks with appreciable abundances were bundled by nominal mass into fraction M+0, M+1, …M+n, constituting the final simulated spectrum. A least square fit was used to find the scalar θ that best fit the measured peptide isotopic distribution (Imeasured) to a linear combination of Iold and Inew:
The root mean square error was determined for each peptide fitting, and any fitting with a root mean square error > 1% was removed. For genus-level turnover quantification, only genera with more than two measurements were kept in the analysis, with the median value across peptides reported.
Quantification of contribution of labeled nutrient to peptide.
To determine the contribution of a 13C- or 15N-labeled nutrient to a bacterial peptide, similar to the above approach, we first measured the cecal content free amino acids 13C- or 15N-labeling using metabolomics. Then, for each peptide, we simulated the expected isotope envelope pattern if the peptide were unlabeled (Iunlabeled) versus if it were synthesized from free cecal amino acids (Ifree). A scalar γ (analogous to θ above) can then be determined by fitting the measured peptide isotope distribution (Imeasured) to a linear combination of Iunlabeled and Ifree. Note that γ will exceed 1 when a bacterial genus uses a particular nutrient in excess of that nutrient contribution’s to cecal free amino acids. Because the 13C- and 15N-labeling patterns are simpler than the D2O labeling patterns, in lieu of carrying out this fitting, we instead determined γ (with the same conceptual and mathematical meaning) using simple algebraic equations.
Specifically, we measured γ for each peptide as follows:
where (with the exception of 13C-protein feeding data, discussed immediately below) φ is the average number of extra neutrons in a given peptide (or simulated peptide), relative to the M+0 form. This was calculated based on the experimentally observed (or simulated, as above) fraction of M+0, M+1, M+2, and M+3, which account for > 90% of the isotopes for each peptide (with more heavily labeled forms too low abundance and noisy to contribute productively to the measurements):
For the 13C-protein feeding experiments, the most readily detected labeled forms involve incorporation of a single midsized U-13C-amino acid, which manifests as M+5 or M+6 peptide labeling. Other isotopic forms were sufficiently noisier, as to render their inclusion unhelpful. Accordingly, we calculated γ based on φ′:
The above equations give nearly identical values for γ as fitting (as done to determine θ).
For genus-level measurements of feedstock contributions, only genera with more than 3 peptides measured per mouse was kept in the analysis, with the median value across peptides reported as γgenus. Only genera that were consistently detected in proteomics, and the family of that genera detected (> 0.5%) in 16S rRNA gene amplicon sequencing were analyzed. The product of γgenus and the contribution of each nutrient to cecal free amino acids (LAA_avg←nutrient) was used to determine the contribution of each nutrient to bacterial genus (fgenus←nutrient):
where the contribution of each nutrient to bacterial protein pool (LAA_avg←Nutrient) was calculated as the average labeling across amino acids, weighted based on their abundance in that genus’ protein and corrected for fraction of the nutrient interest labeled (T):
with w%AA,bacteria taken from literature (Purser and Buechler, 1966).
Quantification and statistical analysis.
A two-tailed, unpaired student’s t-test was used to calculate P values, with P<0.05 used to determine statistical significance.
Supplementary Material
Figure S1. Microbiome consumes dietary fiber and protein. Related to Table 1.
(A) Composition of the measured portal microbial metabolome. The pie charts show the relative molar abundance of different gut microbiota-associated metabolites in mice (N = 6 mice).
(B) As in (A), for cecal content microbial metabolome. (N = 6 mice)
(C) Experimental scheme. Mice received an oral gavage of 4:2:1 starch: protein (or free amino acids): inulin by weight. In each dietary condition, one component was 13C-labeled. After gavage of the labeled meal, tissue and serum metabolite labeling were measured over time by LC-MS.
(D) Dietary starch feeds the host, while dietary inulin feeds the microbiome. The data shows concentrations of labeled carbons in hexose in portal circulation (mean ± s.e., N = 3 mice).
(E) As in (D), for acetate (mean ± s.e., N = 3 mice).
(F) Heatmap showing the percentage of labeled carbon atoms in the indicated metabolites in portal circulation, following gavage of 4:2:1 starch: protein: inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(G) Heatmap showing the molarity of total labeled carbon atoms in the indicated metabolites in cecal content, following gavage of 4:2:1 starch: protein: inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(H) Metabolic fate of inulin and starch. Stacked bars show the fraction of gavaged inulin and starch that is converted into each of the indicated metabolic products, with the undigested fraction being excreted in the feces (mean ± s.e., N = 3 mice).
(I) Undigested carbohydrate in the feces. Graph shows the fraction of labeled hexose after cecal content hydrolysis, 12 h following gavage as above (mean ± s.e., N = 3 mice).
(J) Dietary amino acids feed the host while dietary algal protein feeds the microbiome. The data shows concentrations of labeled carbons in valine in portal circulation (mean ± s.e., N = 3 mice).
(K) As in (J), for acetate (mean ± s.e., N = 3 mice).
(L) Heatmap showing the percentage of labeled carbon atoms in the indicated amino acids in portal circulation, following gavage of 4:2:1 starch: protein (or free amino acids): inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(M) Heatmap showing the molarity of total labeled carbon atoms in the indicated amino acids in cecal content, following gavage of 4:2:1 starch: protein (or free amino acids): inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
Figure S2. Circulating ammonia contributes to microbiota metabolism via circulating urea. Related to Figure 1.
(A) Normalized labeling of serum urea, ammonia and fecal amino acids after 15N-urea infusion (mean ± s.e., N = 4 mice). Feces labeling fraction is normalized to serum infused tracer (urea) labeling fraction.
(B) As in (A), for 15N-ammonia infusion (mean ± s.e., N = 5 mice).
(C) Experimental design. Gnotobiotic mice colonized with urease-negative bacteria (C.sporogenes, B.dorei and E.coli) or a combination of urease-negative and positive bacteria (C.sporogenes, B.dorei, E.coli, L.reuteri and S.aureus) were intravenously infused with 15N-urea for 24 hr. Cecal content amino acids labeling were measured by LC-MS.
(D) Normalized labeling of cecal ammonia and amino acids after 15N-urea infusion in urease-negative bacteria colonized gnotobiotic mice and urease-positive and negative bacteria colonized gnotobiotic mice (mean ± s.e., N = 3 mice). *P<0.05, **P<0.01 and ***P<0.001 by two-sided Student’s t-test.
(E) Ammonia concentration in systemic circulation and cecal content (mean ± s.e., N = 5 mice).
(F) As in (E), for urea (mean ± s.e., N = 5 mice).
(G) Contribution calculation from circulating ammonia to circulating urea, then to fecal amino acids.
(H) Contribution of circulating ammonia to fecal amino acids are quantitatively explained by circulating urea (mean ± s.e., N = 4 mice).
(I) Normalized labeling of fecal ammonia and amino acids after 15N-urea infusion in control and antibiotics treated mice (mean ± s.e., N = 3 mice). *P<0.05, **P<0.01 and ***P<0.001 by two-sided Student’s t-test.
(J) Normalized labeling of fecal ammonia after 15N-ammonia infusion in control and antibiotics treated mice (mean ± s.e., N = 5 mice for control mice, N = 4 for antibiotics-treated mice). **P<0.01 by two-sided Student’s t-test.
(K) Schematic of the pathway from circulating ammonia to luminal ammonia.
Figure S3. Quantitative analysis of microbiome amino acid metabolism. Related to Figure 2.
(A) Labeling fraction of cecal amino acids from 12 h or 24 h 13C-algal protein feeding. Mean ± s.e. N = 4 mice.
(B) Contribution of dietary and circulating nutrients to cecal amino acid carbon in antibiotics treated mice. Mean ± s.e. N = 3 mice.
(C) As in (B), for nitrogen.
(D) Cecal amino acids concentration fold-change in antibiotics-treated mice relative to mock treated mice. Mean ± s.e. N = 3. TCA-related amino acids are shown in blue bars and the others are shown in grey bars.
(E) Cecal amino acids coming directly from diet tend to go up with antibiotics treatment. Plot shows the correlation, across amino acids (each point is an individual amino acid), between concentration fold change in antibiotics-treated mice and carbon contribution from dietary proteins based on isotope tracing (without any antibiotic treatment).
(F) As in (D), for germ-free mice relative to conventional mice. Mean ± s.e. N = 3. TCA-related amino acids are shown in blue bars and the others are shown in grey bars.
(G) As in (E), for germ-free mice.
(H) Schematic of the possibility for dietary protein nitrogen to contribute to cecal amino acid nitrogen via circulating urea.
(I) Contribution from dietary proteins to cecal amino acid nitrogen are mostly independent of circulating urea. Mean ± s.e. N = 3.
(J) Cecal amino acid isotopic labeling forms from 13C,15N-Algal protein feeding. Mean ± s.e. N = 3.
(K) Cecal amino acid isotopic labeling forms from 13C-Inulin feeding. Mean ± s.e. N = 3.
(L) Total ion chromatogram of 13C5, 15N1-glutamate and 13C4, 15N1-aspartate from simultaneous 15N-urea infusion and 13C-protein feeding (relative to the 15N-urea infusion or 13C-protein feeding separately).
(M) Amino acids synthesized in the gut microbiome, stay in the microbiome, as urea contributes to microbiome amino acids but not host circulating amino acids. Mean ± s.e. N = 4 mice.
Figure S4. Sources of cecal metabolites and acyl-glycines. Related to Figure 2, Table 1.
(A) Heat maps showing the contribution of dietary or circulating nutrients to cecal metabolites. For experimental design, see Figure 3. N = 4 mice.
(B) Experimental design. Mice were intravenously infused with [U-13C] glycine for 2.5 h and tissue and serum glycine and acyl-glycine labeling were measured.
(C) Circulating acyl-glycines are made from circulating glycine. Mean ± s.e. N = 4 mice.
(D) Tissue phenylpropionyl-glycine labeling (normalized to circulating glycine labeling). Mean ± s.e. N = 4 mice.
(E) Tissue butyryl-glycine labeling (normalized to circulating glycine labeling). Mean ± s.e. N = 4 mice.
(F) Amino acid composition of algal protein and casein measured by acid hydrolysis. Mean ± s.e. N = 3 for algal protein and N = 1 for casein.
(G) Contribution of dietary algal protein versus casein to cecal amino acid carbon correlates poorly with relative amino acids abundances in algal protein versus casein.
Figure S5. Single exponential fit of newly synthesized fraction of microbial peptides over time. Related to Figure 4.
(A) Growth rate quantification using D2O in vitro. Clostridium sporogenes or Bacteroides dorei were cultured in media with 5% D2O, and bacteria growth over time was measured by either OD600, or proteomics.
(B) Proteomics measured growth rate mirrors the growth rate measured by OD600. Mean±s.e. N > 200 characteristic peptides for each time point.
(C) As in (B), for Bacteroides dorei.
(D) Different cellular compartments from the genus Bacteroides show similar labeling rate.
(E) As in (D), for genus Ruminococcus.
(F) Single exponential fit was applied to determine genus-level microbial turnover. Data are mean ± s.e. N = 5 mice.
Figure S6. Species level and correlation analysis of nutrient preferences across different gut bacterial genera. Related to Figure 5, 6.
(A) Carbon contribution of dietary inulin across bacterial species. Mean±s.e. N=4 mice.
(B) Carbon contribution of dietary algal protein across bacterial species. Mean±s.e. N=6 mice.
(C) Carbon contribution of circulating lactate across bacterial species. Mean±s.e. N=7 mice.
(D) Nitrogen contribution of circulatory urea across bacterial species. Mean±s.e. N=6 mice.
(E) Nitrogen contribution of dietary algal protein across bacterial species. Mean±s.e. N=6 mice.
(F) Carbon contribution of secreted host proteins across bacterial species. Mean±s.e. N=5 mice.
(G) Positive correlation between fgenus←dietary proteins C and fgenus←dietary proteins N.
(H) Positive correlation between fgenus←inulin C and fgenus←urea N in Firmicutes.
(I) Negative correlation between fgenus←urea N and fgenus←dietary proteins N in Firmicutes.
(J) Negative correlation between and fgenus←dietary proteins N.
(K) Positive correlation between and fgenus←secreted proteins N.
Figure S7. Host mucins are synthesized using circulating host amino acids and eventually contribute to fecal amino acids. Related to Figure 6.
(A) Experimental design. Mice were infused with 15N-lysine and 15N-arginine and mouse colon proteins were analyzed by proteomics.
(B) MUC2 labeling (multiple different MUC2 peptides). N = 2 mice for 12 h, 18 h, N = 3 mice for 36 h.
(C) As in (B), for MUC3.
(D) As in (B), for MUC13.
(E) Passage of circulating amino acids from serum into host colonic protein, and from there into host proteins and finally fecal free amino acids. Figure shows the enrichment of serum AA, colonic MUC2 proteins, host proteins in intestinal lumen and fecal AAs normalized to the serum AA enrichment at 36 h following 15N-lysine and 15N-arginine infusions. Mean±s.e., N = 2 mice for 12 h, 18 h, N = 3 mice for 36 h for serum and fecal AAs. N = 14 characteristic MUC2 peptides for 12 h, 18 h, N = 28 characteristic MUC2 peptides for 36 h. N > 50 characteristic peptides for host proteins in intestinal lumen for 12 h, 18 h and 36 h.
Table S1A. Microbiota metabolites concentration in portal and systemic circulation. Related to Table 1.
Mean, 95% confidence intervals, N = 5 mice. Renal, portal, hepatic refer to concentrations in those veins. Systemic = tail vein.
Table S1B. Microbiota-associated metabolite concentrations in tissues (ad lib fed state, ZT1). Related to Table 1.
Mean, 95% confidence interval, N = 5 mice. Red indicates metabolite is enriched in cecal content and colon, blue indicates the metabolite is enriched in kidney and yellow indicates enriched in liver.
Table S2A. Infusion parameters. Related to STAR Methods
Table S2B. Labeling data generated in the study. Related to Figure 1–2, STAR Methods
Table S2C. Proteomics data information. Related to Figure 4–6, STAR Methods
Table S2D. Cecal content amino acids labeling data. Related to Figure 4–6, STAR Methods
Table S3. Taxonomic assignment of the detected peptides in the study. Related to STAR Methods
Table S4. Diet composition. Related to STAR Methods
Table S5. Tabular results of the 16S rRNA gene amplicon sequencing of diet manipulation experiment. Related to Figure 5, 6, STAR Methods
Key Resources Table
| Chemicals, Peptides, and Recombinant Proteins | ||
| DEUTERIUM OXIDE (D, 99.9%) | Cambridge Isotope Laboratories | Cat# DLM-4-PK |
| D-GLUCOSE (U-13C6, 99%) | Cambridge Isotope Laboratories | Cat# CLM-1396-PK |
| SODIUM L-LACTATE (13C3, 98%) 20% W/W in H2O | Cambridge Isotope Laboratories | Cat# CLM-1579-PK |
| L-GLUTAMINE (13C5, 99%) | Cambridge Isotope Laboratories | Cat# CLM-1822-H-PK |
| L-GLUTAMINE (ALPHA-15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-1016-PK |
| L-GLUTAMINE (AMIDE-15N, 98%+) | Cambridge Isotope Laboratories | Cat# NLM-557-PK |
| SODIUM D-3-HYDROXYBUTYRATE (13C4, 99%) 97% CHEMICAL PURITY | Cambridge Isotope Laboratories | Cat# CLM-3853-PK |
| LINOLEIC ACID, POTASSIUM SALT (U-13C18, 98%) | Cambridge Isotope Laboratories | Cat# CLM-8835-PK |
| OLEIC ACID, SODIUM SALT (U-13C18, 98%) | Cambridge Isotope Laboratories | Cat# CLM-8763-PK |
| SODIUM PALMITATE (U-13C16, 98%+) | Cambridge Isotope Laboratories | Cat# CLM-6059-PK |
| SODIUM ACETATE (1,2–13C2, 99%) | Cambridge Isotope Laboratories | Cat# CLM-440-PK |
| CITRIC ACID (13C6, 99%) | Cambridge Isotope Laboratories | Cat# CLM-9021-PK |
| SUCCINIC ACID (13C4, 99%) | Cambridge Isotope Laboratories | Cat# CLM-1571-PK |
| L-MALIC ACID (13C4, 99%) | Cambridge Isotope Laboratories | Cat# CLM-8065-PK |
| ALPHA-KETOGLUTARIC ACID, DISODIUM SALT (1,2,3,4–13C4, 99%) | Cambridge Isotope Laboratories | Cat# CLM-4442-PK |
| L-ALANINE (13C3, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2184-H-PK |
| L-ALANINE (15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-454-PK |
| L-VALINE (13C5, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2249-H-PK |
| L-VALINE (15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-316-PK |
| L-LEUCINE (13C6, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2262-H-PK |
| L-LEUCINE (15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-142-PK |
| L-ISOLEUCINE (13C6, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2248-H-PK |
| L-ISOLEUCINE (15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-292-PK |
| L-SERINE (13C3, 99%; 15N, 99%) | Cambridge Isotope Laboratories | Cat# CNLM-474-H-PK |
| GLYCINE (13C2, 97–99%) | Cambridge Isotope Laboratories | Cat# CLM-1017-PK |
| GLYCINE (13C2, 99%; 15N, 99%) | Cambridge Isotope Laboratories | Cat# CNLM-1673-H-PK |
| L-TYROSINE (13C9, 99%) | Cambridge Isotope Laboratories | Cat# CLM-2263-H-PK |
| UREA (15N2, 98%+) | Cambridge Isotope Laboratories | Cat# NLM-233-PK |
| AMMONIUM CHLORIDE (15N, 99%) | Cambridge Isotope Laboratories | Cat# NLM-467-PK |
| L-LYSINE:2HCL (ALPHA-15N, 98%) | Cambridge Isotope Laboratories | Cat# NLM-143-PK |
| L-ARGININE:HCL (ALPHA-15N, 98%+) | Cambridge Isotope Laboratories | Cat# NLM-1267-PK |
| ALGAL STARCH (U-13C, 98%+) | Cambridge Isotope Laboratories | Cat# CLM-1699-PK |
| INULIN (FROM CHICORY) (U-13C, 97%+) 97% CHEMICAL PURITY | Cambridge Isotope Laboratories | Cat# CLM-9181-PK |
| Algal crude protein extract-13C | Sigma-Aldrich | Cat# 642878 |
| Algal crude protein extract-15N | Sigma-Aldrich | Cat# 586773 |
| Algal crude protein extract-13C,15N | Sigma-Aldrich | Cat# 608254 |
| Algal amino acid mixture-13C | Sigma-Aldrich | Cat# 426199 |
| Algal fatty acid mixture-13C | Sigma-Aldrich | Cat# 487937 |
| AZD 3965 | AstraZeneca | Cat# AZD3965 |
| Ampicillin | Sigma-Aldrich | Cat# A0166 |
| Neomycin trisulfate salt hydrate | Sigma-Aldrich | Cat# N6386 |
| Metronidazole | Sigma-Aldrich | Cat# M1547 |
| Vancomycin hydrochloride from Streptomyces orientalis | Sigma-Aldrich | Cat# V1130 |
| Trypsin | Promega | Cat# V5113 |
| Lysyl endopeptidase R | Wako Chemicals USA | Cat# 12902541 |
| Aspartame | Sigma-Aldrich | Cat# 47135 |
| GAM Broth Modified | HyServe | Cat# 5433 |
| LB Broth (Miller, Luria Broth) | Sigma | Cat# L3522 |
| MRS Broth | Sigma | Cat# 69966 |
| TSB (Tryptic Soy Broth) | Bacto | Cat# 211825 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Charles River Laboratories | Cat #027 |
| Strain: Bacteroides dorei (CL02T00C15) | BEI | #HM-717 |
| Strain: Clostridium sporogenes (ATCC 15579) | ATCC | 15579 |
| Strain: Escherichia coli (ATCC 25922) | ATCC | 25922 |
| Strain: Lactobacillus reuteri (CF-48–34A) | BEI | #HM-102 |
| Strain: Staphylococcus aureus subsp. Aureus Rosenbach | ATCC | 29213 |
| Software and Algorithms | ||
| El-MAVEN software | Elucidata | https://resources.elucidata.io/elmaven |
| AccuCor | GitHub | https://github.com/XiaoyangSu/AccuCor |
| PepMID | GitHub | https://github.com/xxing9703/pepMID_simul |
| MATLAB R2021b | MathWorks | N/A |
| Others | ||
| PicoLab Rodent Diet 20 | LabDiet | Cat# 5053 |
| 20% Diet premix | Research Diets | Cat# D11112201Npx2i |
| 20% Amino acids diet | Research Diets | Cat# A11112201Bi |
Gut microbiome feedstocks mapped by isotope tracing into bacteria-specific peptides
Major contributors are dietary fiber and protein, and host lactate, urea, and mucins
Microbiome composition shifts towards bacteria fed their preferred nutrients
Microbial metabolites’ systemic levels reflect dietary precursors reaching microbiome
ACKNOWLEDGEMENTS.
This work is supported by the NIH Pioneer award IDP1DK113643 (J.D.R.), Stand Up to Cancer Convergence Award 3.14.16 probing the cancer-microbiome connection (J.D.R.), Ludwig Cancer Research (J.D.R.), NIH grant R35GM128813 (M.W.), Princeton Catalysis Initiative (M.W.), Eric and Wendy Schmidt Transformative Technology Fund (M.W), and the Pew Biomedical Scholars Program (M.S.D). M.G. is funded by Harold W. Dodds Fellowship. F.C.K is funded by EMBO ALTF 601-2018. J.G.L. is funded by a National Science Foundation Graduate Research Fellowship (2017249408). Y.-C.J.L. is funded by the High Meadows Environmental Institute at Princeton University through the generous support of the William Clay Ford, Jr. ‘79 and Lisa Vanderzee Ford ‘82 Graduate Fellowship fund and by a training grant from the National Institute of General Medicine Sciences (T32GM007388). M.D.N. is supported by NIH 5T32CA257957. We thank Sheng Hui, Michel Nofal, Yihui Shen, Lingfan Liang, Won Dong Lee and other Rabinowitz lab members for the input and advice. We thank Seema Chatterjee for the assistance with DNA extraction. We thank Wei Wang and the Lewis Sigler Institute sequencing core facility for assistance with HT sequencing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARIATION OF INTERESTS. J.D.R. is a member of the Rutgers Cancer Institute of New Jersey and the University of Pennsylvania Diabetes Research Center; a co-founder and stockholder in Empress Therapeutics and Serien Therapeutics; and advisor and stockholder in Agios Pharmaceuticals, Bantam Pharmaceuticals, Colorado Research Partners, Rafael Pharmaceuticals, Barer Institute and L.E.A.F. Pharmaceuticals. M.S.D. is a member of the scientific advisory board of DeepBiome Therapeutics and VastBiome.
References
- Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, Thom SR, Bushman FD, Vinogradov SA, Wu GD, 2014. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology 147, 1055–1063.e8. 10.1053/j.gastro.2014.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab JP, Karpen SJ, Dawson PA, Arrese M, Trauner M, 2017. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatol. Baltim. Md 65, 350–362. 10.1002/hep.28709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartman CR, TeSlaa T, Rabinowitz JD, 2021. Quantitative flux analysis in mammals. Nat. Metab 3, 896–908. 10.1038/s42255-021-00419-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batani G, Bayer K, Böge J, Hentschel U, Thomas T, 2019. Fluorescence in situ hybridization (FISH) and cell sorting of living bacteria. Sci. Rep 9, 18618. 10.1038/s41598-019-55049-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Mader E, Lee TK, Woebken D, Wang Y, Zhu D, Palatinszky M, Schintlmeister A, Schmid MC, Hanson BT, Shterzer N, Mizrahi I, Rauch I, Decker T, Bocklitz T, Popp J, Gibson CM, Fowler PW, Huang WE, Wagner M, 2015. Tracking heavy water (D2O) incorporation for identifying and sorting active microbial cells. Proc. Natl. Acad. Sci 112, E194–E203. 10.1073/pnas.1420406112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry D, Stecher B, Schintlmeister A, Reichert J, Brugiroux S, Wild B, Wanek W, Richter A, Rauch I, Decker T, Loy A, Wagner M, 2013. Host-compound foraging by intestinal microbiota revealed by single-cell stable isotope probing. Proc. Natl. Acad. Sci 110, 4720–4725. 10.1073/pnas.1219247110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J, 2018. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. 10.1186/s40168-018-0470-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG, 2019. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP, 2016. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin W-B, Guo C-J, Violante S, Ramos RJ, Cross JR, Kadaveru K, Hambor J, Rudensky AY, 2020. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 1–5. 10.1038/s41586-020-2193-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R, 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. 10.1038/ismej.2012.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalile B, Oudenhove LV, Vervliet B, Verbeke K, 2019. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol 1. 10.1038/s41575-019-0157-3 [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ, 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Bäckhed F, Mithieux G, 2014. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156, 84–96. 10.1016/j.cell.2013.12.016 [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, Young VB, Henrissat B, Wilmes P, Stappenbeck TS, Núñez G, Martens EC, 2016. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353.e21. 10.1016/j.cell.2016.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP, 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. 10.1038/nmeth1019 [DOI] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, Li H, Huet G, Yuan Q, Wigal T, Butt Y, Ni M, Torrealba J, Oliver D, Lenkinski RE, Malloy CR, Wachsmann JW, Young JD, Kernstine K, DeBerardinis RJ, 2017. Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e9. 10.1016/j.cell.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-García J, Altea-Manzano P, Pranzini E, Fendt S-M, 2020. Stable Isotopes for Tracing Mammalian-Cell Metabolism In Vivo. Trends Biochem. Sci 45, 185–201. 10.1016/j.tibs.2019.12.002 [DOI] [PubMed] [Google Scholar]
- Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, Guo C, Higginbottom S, Almo SC, Fischbach MA, 2020. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570. 10.1038/s41586-020-2396-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, 2015. Cancer and the microbiota. Science 348, 80–86. 10.1126/science.aaa4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Sonnett M, Ryazanova L, Presler M, Wühr M, 2018. Quantitative Proteomics of Xenopus Embryos I, Sample Preparation, in: Vleminckx K. (Ed.), Xenopus: Methods and Protocols, Methods in Molecular Biology. Springer, New York, NY, pp. 175–194. 10.1007/978-1-4939-8784-9_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdeep Singh R, Tanca A, Palomba A, Van der Jeugt F, Verschaffelt P, Uzzau S, Martens L, Dawyndt P, Mesuere B, 2019. Unipept 4.0: Functional Analysis of Metaproteome Data. J. Proteome Res 18, 606–615. 10.1021/acs.jproteome.8b00716 [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Price NT, 1999. The proton-linked monocarboxylate transporter (MCT) family: structure, function and regulation. Biochem. J 343, 281–299. [PMC free article] [PubMed] [Google Scholar]
- Han S, Van Treuren W, Fischer CR, Merrill BD, DeFelice BC, Sanchez JM, Higginbottom SK, Guthrie L, Fall LA, Dodd D, Fischbach MA, Sonnenburg JL, 2021. A metabolomics pipeline for the mechanistic interrogation of the gut microbiome. Nature 595, 415–420. 10.1038/s41586-021-03707-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, Zheng Y, Longman RS, Rastinejad F, Devlin AS, Krout MR, Fischbach MA, Littman DR, Huh JR, 2019. Bile acid metabolites control T H 17 and T reg cell differentiation. Nature 576, 143–148. 10.1038/s41586-019-1785-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AJ, Chew YV, Colakoglu F, Cliff JB, Klaassens E, Read MN, Solon-Biet SM, McMahon AC, Cogger VC, Ruohonen K, Raubenheimer D, Le Couteur DG, Simpson SJ, 2017. Diet-Microbiome Interactions in Health Are Controlled by Intestinal Nitrogen Source Constraints. Cell Metab. 25, 140–151. 10.1016/j.cmet.2016.10.021 [DOI] [PubMed] [Google Scholar]
- Holmes WE, Angel TE, Li KW, Hellerstein MK, 2015. Chapter Seven - Dynamic Proteomics: In Vivo Proteome-Wide Measurement of Protein Kinetics Using Metabolic Labeling, in: Metallo CM (Ed.), Methods in Enzymology, Metabolic Analysis Using Stable Isotopes. Academic Press, pp. 219–276. 10.1016/bs.mie.2015.05.018 [DOI] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Zhan L, Guo JY, White E, Rabinowitz JD, 2017. Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. 10.1038/nature24057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD, 2007. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 9, 90–95. 10.1109/MCSE.2007.55 [DOI] [Google Scholar]
- Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, Liu W, Tesz GJ, Birnbaum MJ, Rabinowitz JD, 2018. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 27, 351–361.e3. 10.1016/j.cmet.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, Morscher RJ, Reyes J, Frezza C, Hwang HY, Imai A, Saito Y, Okamoto K, Vaspoli C, Kasprenski L, Zsido GA, Gorman JH, Gorman RC, Rabinowitz JD, 2019. Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab. 30, 594–606.e3. 10.1016/j.cmet.2019.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, Bergh P-O, Gerdes V, Hartstra A, Brauw M. de, Perkins R, Nieuwdorp M, Bergström G, Bäckhed F, 2018. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 175, 947–961.e17. 10.1016/j.cell.2018.09.055 [DOI] [PubMed] [Google Scholar]
- Koh A, Vadder FD, Kovatcheva-Datchary P, Bäckhed F, 2016. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 165, 1332–1345. 10.1016/j.cell.2016.05.041 [DOI] [PubMed] [Google Scholar]
- Lai Y, Liu C-W, Yang Y, Hsiao Y-C, Ru H, Lu K, 2021. High-coverage metabolomics uncovers microbiota-driven biochemical landscape of interorgan transport and gut-brain communication in mice. Nat. Commun 12, 6000. 10.1038/s41467-021-26209-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Limenitakis JP, Fuhrer T, Geuking MB, Lawson MA, Wyss M, Brugiroux S, Keller I, Macpherson JA, Rupp S, Stolp B, Stein JV, Stecher B, Sauer U, McCoy KD, Macpherson AJ, 2015. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun 6, 8292. 10.1038/ncomms9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Liu H-Y, Zhou H, Zhan Q, Lai W, Zeng Q, Ren H, Xu D, 2017. Moderate-Intensity Exercise Affects Gut Microbiome Composition and Influences Cardiac Function in Myocardial Infarction Mice. Front. Microbiol 8. 10.3389/fmicb.2017.01687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund PJ, Gates LA, Leboeuf M, Smith SA, Chau L, Friedman ES, Lopes M, Saiman Y, Kim MS, Petucci C, Allis CD, Wu GD, Garcia BA, 2021. Stable Isotope Tracing in vivo Reveals A Metabolic Bridge Linking the Microbiota to Host Histone Acetylation. bioRxiv 2021.07.05.450926. 10.1101/2021.07.05.450926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen L, Myrmel LS, Fjære E, Liaset B, Kristiansen K, 2017. Links between Dietary Protein Sources, the Gut Microbiota, and Obesity. Front. Physiol 8, 1047. 10.3389/fphys.2017.01047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M, Lewis IA, Geuking MB, McCoy KD, 2020. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 10.1126/science.abc3421 [DOI] [PubMed] [Google Scholar]
- McCabe BJ, Previs SF, 2004. Using isotope tracers to study metabolism: application in mouse models. Metab. Eng 6, 25–35. 10.1016/j.ymben.2003.09.003 [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P, 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney W, 2010. Data Structures for Statistical Computing in Python. Proc. 9th Python Sci. Conf 56–61. 10.25080/Majora-92bf1922-00a [DOI] [Google Scholar]
- Mora D, Arioli S, 2014. Microbial Urease in Health and Disease. PLOS Pathog. 10, e1004472. 10.1371/journal.ppat.1004472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munukka E, Ahtiainen JP, Puigbó P, Jalkanen S, Pahkala K, Keskitalo A, Kujala UM, Pietilä S, Hollmén M, Elo L, Huovinen P, D’Auria G, Pekkala S, 2018. Six-Week Endurance Exercise Alters Gut Metagenome That Is not Reflected in Systemic Metabolism in Over-weight Women. Front. Microbiol 9. 10.3389/fmicb.2018.02323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemet I, Saha PP, Gupta N, Zhu W, Romano KA, Skye SM, Cajka T, Mohan ML, Li L, Wu Y, Funabashi M, Ramer-Tait AE, Prasad SVN, Fiehn O, Rey FE, Tang WHW, Fischbach MA, DiDonato JA, Hazen SL, 2020. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 180, 862–877.e22. 10.1016/j.cell.2020.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Shen T-CD, Chen EZ, Bittinger K, Bailey A, Roggiani M, Sirota-Madi A, Friedman ES, Chau L, Lin A, Nissim I, Scott J, Lauder A, Hoffmann C, Rivas G, Albenberg L, Baldassano RN, Braun J, Xavier RJ, Clish CB, Yudkoff M, Li H, Goulian M, Bushman FD, Lewis JD, Wu GD, 2017. A role for bacterial urease in gut dysbiosis and Crohn’s disease. Sci. Transl. Med 9. 10.1126/scitranslmed.aah6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER, Kalocsay M, Jané-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, Golji J, Porter DA, Rejtar T, Wang YK, Kryukov GV, Stegmeier F, Erickson BK, Garraway LA, Sellers WR, Gygi SP, 2020. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402.e16. 10.1016/j.cell.2019.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberbach A, Haange S-B, Schlichting N, Heinrich M, Lehmann S, Till H, Hugenholtz F, Kullnick Y, Smidt H, Frank K, Seifert J, Jehmlich N, von Bergen M, 2017. Metabolic in Vivo Labeling Highlights Differences of Metabolically Active Microbes from the Mucosal Gastrointestinal Microbiome between High-Fat and Normal Chow Diet. J. Proteome Res 16, 1593–1604. 10.1021/acs.jproteome.6b00973 [DOI] [PubMed] [Google Scholar]
- O’Brien JJ, Narayan V, Wong Y, Seitzer P, Sandoval CM, Haste N, Smith M, Rad R, Gaun A, Baker A, Kukurugya M, Martin-McNulty B, Zhang C, Kolumam G, Sidrauski C, Jojic V, McAllister F, Bennett B, Buffenstein R, 2020. Precise Estimation of In Vivo Protein Turnover Rates. bioRxiv 2020.11.10.377440. 10.1101/2020.11.10.377440 [DOI] [Google Scholar]
- Purser DB, Buechler SM, 1966. Amino Acid Composition of Rumen Organisms. J. Dairy Sci 49, 81–84. 10.3168/jds.S0022-0302(66)87791-3 [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li Shaochuan Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li Shengting, Jian M, Zhou Y, Li Y, Zhang X, Li Songgang, Qin N, Yang H, Wang Jian, Brunak S, Doré J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang, Jun, 2010. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn RA, Melnik AV, Vrbanac A, Fu T, Patras KA, Christy MP, Bodai Z, Belda-Ferre P, Tripathi A, Chung LK, Downes M, Welch RD, Quinn M, Humphrey G, Panitchpakdi M, Weldon KC, Aksenov A, da Silva R, Avila-Pacheco J, Clish C, Bae S, Mallick H, Franzosa EA, Lloyd-Price J, Bussell R, Thron T, Nelson AT, Wang M, Leszczynski E, Vargas F, Gauglitz JM, Meehan MJ, Gentry E, Arthur TD, Komor AC, Poulsen O, Boland BS, Chang JT, Sandborn WJ, Lim M, Garg N, Lumeng JC, Xavier RJ, Kazmierczak BI, Jain R, Egan M, Rhee KE, Ferguson D, Raffatellu M, Vlamakis H, Haddad GG, Siegel D, Huttenhower C, Mazmanian SK, Evans RM, Nizet V, Knight R, Dorrestein PC, 2020. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129. 10.1038/s41586-020-2047-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AT, Pereira FC, Schintlmeister A, Berry D, Wagner M, Hale LP, Wu A, Jiang S, Durand HK, Zhou X, Premont RT, Diehl AM, O’Connell TM, Alberts SC, Kartzinel TR, Pringle RM, Dunn RR, Wright JP, David LA, 2018. Microbial nitrogen limitation in the mammalian large intestine. Nat. Microbiol 3, 1441–1450. 10.1038/s41564-018-0267-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS, 2014. Bile Acids and the Gut Microbiome. Curr. Opin. Gastroenterol 30, 332–338. 10.1097/MOG.0000000000000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Wilhelm M, Hahne H, Kuster B, Bantscheff M, 2015. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteomics MCP 14, 2394–2404. 10.1074/mcp.M114.046995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiman J, Luber JM, Chavkin TA, MacDonald T, Tung A, Pham L-D, Wibowo MC, Wurth RC, Punthambaker S, Tierney BT, Yang Z, Hattab MW, Avila-Pacheco J, Clish CB, Lessard S, Church GM, Kostic AD, 2019. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat. Med 1. 10.1038/s41591-019-0485-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard J-F, Le Bihan G, Vogeleer P, Jacques M, Harel J, 2017. Interactions of Intestinal Bacteria with Components of the Intestinal Mucus. Front. Cell. Infect. Microbiol 7. 10.3389/fcimb.2017.00387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Kelley LP, Haigis MC, 2017a. An LC-MS Approach to Quantitative Measurement of Ammonia Isotopologues. Sci. Rep 7, 10304. 10.1038/s41598-017-09993-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC, 2017b. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946. 10.1126/science.aam9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WHW, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL, 2013. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk [WWW Document]. http://dx.doi.org/10.1056/NEJMoa1109400. 10.1056/NEJMoa1109400 [DOI] [PMC free article] [PubMed]
- Ullrich KJ, Rumrich G, Klöss S, 1982. Reabsorption of monocarboxylic acids in the proximal tubule of the rat kidney. II. Specificity for aliphatic compounds. Pflüg. Arch. Eur. J. Physiol 395, 220–226. 10.1007/BF00584813 [DOI] [PubMed] [Google Scholar]
- Wali JA, Milner AJ, Luk AWS, Pulpitel TJ, Dodgson T, Facey HJW, Wahl D, Kebede MA, Senior AM, Sullivan MA, Brandon AE, Yau B, Lockwood GP, Koay YC, Ribeiro R, Solon-Biet SM, Bell-Anderson KS, O’Sullivan JF, Macia L, Forbes JM, Cooney GJ, Cogger VC, Holmes A, Raubenheimer D, Le Couteur DG, Simpson SJ, 2021. Impact of dietary carbohydrate type and protein–carbohydrate interaction on metabolic health. Nat. Metab 1–19. 10.1038/s42255-021-00393-9 [DOI] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, DuGar B, Feldstein AE, Britt EB, Fu X, Chung Y-M, Wu Y, Schauer P, Smith JD, Allayee H, Tang WHW, DiDonato JA, Lusis AJ, Hazen SL, 2011. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. 10.1038/nature09922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G, 2009. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci 106, 3698–3703. 10.1073/pnas.0812874106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe RR, 1984. Tracers in metabolic research: radioisotope and stable isotope/mass spectrometry methods. Lab. Res. Methods Biol. Med 9, 1–287. [PubMed] [Google Scholar]
- Wong JMW, Jenkins DJA, 2007. Carbohydrate Digestibility and Metabolic Effects. J. Nutr 137, 2539S–2546S. 10.1093/jn/137.11.2539S [DOI] [PubMed] [Google Scholar]
- Wühr M, Freeman RM, Presler M, Horb ME, Peshkin L, Gygi SP, Kirschner MW, 2014. Deep Proteomics of the Xenopus laevis Egg using an mRNA-Derived Reference Database. Curr. Biol 24, 1467–1475. 10.1016/j.cub.2014.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Oh K, Ren B, Tickle TL, Franzosa EA, Wachtman LM, Miller AD, Westmoreland SV, Mansfield KG, Vallender EJ, Miller GM, Rowlett JK, Gevers D, Huttenhower C, Morgan XC, 2015. Biogeography of the Intestinal Mucosal and Lumenal Microbiome in the Rhesus Macaque. Cell Host Microbe 17, 385–391. 10.1016/j.chom.2015.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N, 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- Zhang X, Ning Z, Mayne J, Deeke SA, Li J, Starr AE, Chen R, Singleton R, Butcher J, Mack DR, Stintzi A, Figeys D, 2016a. In Vitro Metabolic Labeling of Intestinal Microbiota for Quantitative Metaproteomics. Anal. Chem 88, 6120–6125. 10.1021/acs.analchem.6b01412 [DOI] [PubMed] [Google Scholar]
- Zhang X, Ning Z, Mayne J, Moore JI, Li J, Butcher J, Deeke SA, Chen R, Chiang C-K, Wen M, Mack D, Stintzi A, Figeys D, 2016b. MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome 4, 31. 10.1186/s40168-016-0176-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, Trefely S, Fernandez S, Carrer A, Miller KD, Schug ZT, Snyder NW, Gade TP, Titchenell PM, Rabinowitz JD, Wellen KE, 2020. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591. 10.1038/s41586-020-2101-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Microbiome consumes dietary fiber and protein. Related to Table 1.
(A) Composition of the measured portal microbial metabolome. The pie charts show the relative molar abundance of different gut microbiota-associated metabolites in mice (N = 6 mice).
(B) As in (A), for cecal content microbial metabolome. (N = 6 mice)
(C) Experimental scheme. Mice received an oral gavage of 4:2:1 starch: protein (or free amino acids): inulin by weight. In each dietary condition, one component was 13C-labeled. After gavage of the labeled meal, tissue and serum metabolite labeling were measured over time by LC-MS.
(D) Dietary starch feeds the host, while dietary inulin feeds the microbiome. The data shows concentrations of labeled carbons in hexose in portal circulation (mean ± s.e., N = 3 mice).
(E) As in (D), for acetate (mean ± s.e., N = 3 mice).
(F) Heatmap showing the percentage of labeled carbon atoms in the indicated metabolites in portal circulation, following gavage of 4:2:1 starch: protein: inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(G) Heatmap showing the molarity of total labeled carbon atoms in the indicated metabolites in cecal content, following gavage of 4:2:1 starch: protein: inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(H) Metabolic fate of inulin and starch. Stacked bars show the fraction of gavaged inulin and starch that is converted into each of the indicated metabolic products, with the undigested fraction being excreted in the feces (mean ± s.e., N = 3 mice).
(I) Undigested carbohydrate in the feces. Graph shows the fraction of labeled hexose after cecal content hydrolysis, 12 h following gavage as above (mean ± s.e., N = 3 mice).
(J) Dietary amino acids feed the host while dietary algal protein feeds the microbiome. The data shows concentrations of labeled carbons in valine in portal circulation (mean ± s.e., N = 3 mice).
(K) As in (J), for acetate (mean ± s.e., N = 3 mice).
(L) Heatmap showing the percentage of labeled carbon atoms in the indicated amino acids in portal circulation, following gavage of 4:2:1 starch: protein (or free amino acids): inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
(M) Heatmap showing the molarity of total labeled carbon atoms in the indicated amino acids in cecal content, following gavage of 4:2:1 starch: protein (or free amino acids): inulin, with the indicated nutrient labeled. Each data point is median of N = 3 mice.
Figure S2. Circulating ammonia contributes to microbiota metabolism via circulating urea. Related to Figure 1.
(A) Normalized labeling of serum urea, ammonia and fecal amino acids after 15N-urea infusion (mean ± s.e., N = 4 mice). Feces labeling fraction is normalized to serum infused tracer (urea) labeling fraction.
(B) As in (A), for 15N-ammonia infusion (mean ± s.e., N = 5 mice).
(C) Experimental design. Gnotobiotic mice colonized with urease-negative bacteria (C.sporogenes, B.dorei and E.coli) or a combination of urease-negative and positive bacteria (C.sporogenes, B.dorei, E.coli, L.reuteri and S.aureus) were intravenously infused with 15N-urea for 24 hr. Cecal content amino acids labeling were measured by LC-MS.
(D) Normalized labeling of cecal ammonia and amino acids after 15N-urea infusion in urease-negative bacteria colonized gnotobiotic mice and urease-positive and negative bacteria colonized gnotobiotic mice (mean ± s.e., N = 3 mice). *P<0.05, **P<0.01 and ***P<0.001 by two-sided Student’s t-test.
(E) Ammonia concentration in systemic circulation and cecal content (mean ± s.e., N = 5 mice).
(F) As in (E), for urea (mean ± s.e., N = 5 mice).
(G) Contribution calculation from circulating ammonia to circulating urea, then to fecal amino acids.
(H) Contribution of circulating ammonia to fecal amino acids are quantitatively explained by circulating urea (mean ± s.e., N = 4 mice).
(I) Normalized labeling of fecal ammonia and amino acids after 15N-urea infusion in control and antibiotics treated mice (mean ± s.e., N = 3 mice). *P<0.05, **P<0.01 and ***P<0.001 by two-sided Student’s t-test.
(J) Normalized labeling of fecal ammonia after 15N-ammonia infusion in control and antibiotics treated mice (mean ± s.e., N = 5 mice for control mice, N = 4 for antibiotics-treated mice). **P<0.01 by two-sided Student’s t-test.
(K) Schematic of the pathway from circulating ammonia to luminal ammonia.
Figure S3. Quantitative analysis of microbiome amino acid metabolism. Related to Figure 2.
(A) Labeling fraction of cecal amino acids from 12 h or 24 h 13C-algal protein feeding. Mean ± s.e. N = 4 mice.
(B) Contribution of dietary and circulating nutrients to cecal amino acid carbon in antibiotics treated mice. Mean ± s.e. N = 3 mice.
(C) As in (B), for nitrogen.
(D) Cecal amino acids concentration fold-change in antibiotics-treated mice relative to mock treated mice. Mean ± s.e. N = 3. TCA-related amino acids are shown in blue bars and the others are shown in grey bars.
(E) Cecal amino acids coming directly from diet tend to go up with antibiotics treatment. Plot shows the correlation, across amino acids (each point is an individual amino acid), between concentration fold change in antibiotics-treated mice and carbon contribution from dietary proteins based on isotope tracing (without any antibiotic treatment).
(F) As in (D), for germ-free mice relative to conventional mice. Mean ± s.e. N = 3. TCA-related amino acids are shown in blue bars and the others are shown in grey bars.
(G) As in (E), for germ-free mice.
(H) Schematic of the possibility for dietary protein nitrogen to contribute to cecal amino acid nitrogen via circulating urea.
(I) Contribution from dietary proteins to cecal amino acid nitrogen are mostly independent of circulating urea. Mean ± s.e. N = 3.
(J) Cecal amino acid isotopic labeling forms from 13C,15N-Algal protein feeding. Mean ± s.e. N = 3.
(K) Cecal amino acid isotopic labeling forms from 13C-Inulin feeding. Mean ± s.e. N = 3.
(L) Total ion chromatogram of 13C5, 15N1-glutamate and 13C4, 15N1-aspartate from simultaneous 15N-urea infusion and 13C-protein feeding (relative to the 15N-urea infusion or 13C-protein feeding separately).
(M) Amino acids synthesized in the gut microbiome, stay in the microbiome, as urea contributes to microbiome amino acids but not host circulating amino acids. Mean ± s.e. N = 4 mice.
Figure S4. Sources of cecal metabolites and acyl-glycines. Related to Figure 2, Table 1.
(A) Heat maps showing the contribution of dietary or circulating nutrients to cecal metabolites. For experimental design, see Figure 3. N = 4 mice.
(B) Experimental design. Mice were intravenously infused with [U-13C] glycine for 2.5 h and tissue and serum glycine and acyl-glycine labeling were measured.
(C) Circulating acyl-glycines are made from circulating glycine. Mean ± s.e. N = 4 mice.
(D) Tissue phenylpropionyl-glycine labeling (normalized to circulating glycine labeling). Mean ± s.e. N = 4 mice.
(E) Tissue butyryl-glycine labeling (normalized to circulating glycine labeling). Mean ± s.e. N = 4 mice.
(F) Amino acid composition of algal protein and casein measured by acid hydrolysis. Mean ± s.e. N = 3 for algal protein and N = 1 for casein.
(G) Contribution of dietary algal protein versus casein to cecal amino acid carbon correlates poorly with relative amino acids abundances in algal protein versus casein.
Figure S5. Single exponential fit of newly synthesized fraction of microbial peptides over time. Related to Figure 4.
(A) Growth rate quantification using D2O in vitro. Clostridium sporogenes or Bacteroides dorei were cultured in media with 5% D2O, and bacteria growth over time was measured by either OD600, or proteomics.
(B) Proteomics measured growth rate mirrors the growth rate measured by OD600. Mean±s.e. N > 200 characteristic peptides for each time point.
(C) As in (B), for Bacteroides dorei.
(D) Different cellular compartments from the genus Bacteroides show similar labeling rate.
(E) As in (D), for genus Ruminococcus.
(F) Single exponential fit was applied to determine genus-level microbial turnover. Data are mean ± s.e. N = 5 mice.
Figure S6. Species level and correlation analysis of nutrient preferences across different gut bacterial genera. Related to Figure 5, 6.
(A) Carbon contribution of dietary inulin across bacterial species. Mean±s.e. N=4 mice.
(B) Carbon contribution of dietary algal protein across bacterial species. Mean±s.e. N=6 mice.
(C) Carbon contribution of circulating lactate across bacterial species. Mean±s.e. N=7 mice.
(D) Nitrogen contribution of circulatory urea across bacterial species. Mean±s.e. N=6 mice.
(E) Nitrogen contribution of dietary algal protein across bacterial species. Mean±s.e. N=6 mice.
(F) Carbon contribution of secreted host proteins across bacterial species. Mean±s.e. N=5 mice.
(G) Positive correlation between fgenus←dietary proteins C and fgenus←dietary proteins N.
(H) Positive correlation between fgenus←inulin C and fgenus←urea N in Firmicutes.
(I) Negative correlation between fgenus←urea N and fgenus←dietary proteins N in Firmicutes.
(J) Negative correlation between and fgenus←dietary proteins N.
(K) Positive correlation between and fgenus←secreted proteins N.
Figure S7. Host mucins are synthesized using circulating host amino acids and eventually contribute to fecal amino acids. Related to Figure 6.
(A) Experimental design. Mice were infused with 15N-lysine and 15N-arginine and mouse colon proteins were analyzed by proteomics.
(B) MUC2 labeling (multiple different MUC2 peptides). N = 2 mice for 12 h, 18 h, N = 3 mice for 36 h.
(C) As in (B), for MUC3.
(D) As in (B), for MUC13.
(E) Passage of circulating amino acids from serum into host colonic protein, and from there into host proteins and finally fecal free amino acids. Figure shows the enrichment of serum AA, colonic MUC2 proteins, host proteins in intestinal lumen and fecal AAs normalized to the serum AA enrichment at 36 h following 15N-lysine and 15N-arginine infusions. Mean±s.e., N = 2 mice for 12 h, 18 h, N = 3 mice for 36 h for serum and fecal AAs. N = 14 characteristic MUC2 peptides for 12 h, 18 h, N = 28 characteristic MUC2 peptides for 36 h. N > 50 characteristic peptides for host proteins in intestinal lumen for 12 h, 18 h and 36 h.
Table S1A. Microbiota metabolites concentration in portal and systemic circulation. Related to Table 1.
Mean, 95% confidence intervals, N = 5 mice. Renal, portal, hepatic refer to concentrations in those veins. Systemic = tail vein.
Table S1B. Microbiota-associated metabolite concentrations in tissues (ad lib fed state, ZT1). Related to Table 1.
Mean, 95% confidence interval, N = 5 mice. Red indicates metabolite is enriched in cecal content and colon, blue indicates the metabolite is enriched in kidney and yellow indicates enriched in liver.
Table S2A. Infusion parameters. Related to STAR Methods
Table S2B. Labeling data generated in the study. Related to Figure 1–2, STAR Methods
Table S2C. Proteomics data information. Related to Figure 4–6, STAR Methods
Table S2D. Cecal content amino acids labeling data. Related to Figure 4–6, STAR Methods
Table S3. Taxonomic assignment of the detected peptides in the study. Related to STAR Methods
Table S4. Diet composition. Related to STAR Methods
Table S5. Tabular results of the 16S rRNA gene amplicon sequencing of diet manipulation experiment. Related to Figure 5, 6, STAR Methods
Data Availability Statement
The proteomics datasets generated during this study are deposited in PRIDE: PXD031015. The isotope tracing data are included in Table S2. The taxonomic assignment of the detected tryptic peptides in the study are included in Table S3. Composition of the diet used in the study are included in Table S4. The 16S rRNA gene amplicon sequencing datasets generated during this study are available in Table S5. The code for peptide enrichment calculations generated during this study is available at GitHub: (https://github.com/xxing9703/pepMID_simul). Any additional information required to re-analyze the data reported in this work is available from the lead contact upon request.