

ABSTRACT
Alphaherpesvirus infection results in severe health consequences in a wide range of hosts. USPs are the largest subfamily of deubiquitinating enzymes that play critical roles in immunity and other cellular functions. To investigate the role of USPs in alphaherpesvirus replication, we assessed 13 USP inhibitors for PRV replication. Our data showed that all the tested compounds inhibited PRV replication, with the USP14 inhibitor b-AP15 exhibiting the most dramatic effect. Ablation of USP14 also influenced PRV replication, whereas replenishment of USP14 in USP14 null cells restored viral replication. Although inhibition of USP14 induced the K63-linked ubiquitination of PRV VP16 protein, its degradation was not dependent on the proteasome. USP14 directly bound to ubiquitin chains on VP16 through its UBL domain during the early stage of viral infection. Moreover, USP14 inactivation stimulated EIF2AK3/PERK- and ERN1/IRE1-mediated signaling pathways, which were responsible for VP16 degradation through SQSTM1/p62-mediated selective macroautophagy/autophagy. Ectopic expression of non-ubiquitinated VP16 fully rescued PRV replication. Challenge of mice with b-AP15 activated ER stress and autophagy and inhibited PRV infection in vivo. Our results suggested that USP14 was a potential therapeutic target to treat alphaherpesvirus-induced infectious diseases.
Abbreviations ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; ATG5: autophagy related 5; ATG12: autophagy related 12; CCK-8: cell counting kit-8; Co-IP: co-immunoprecipitation; CRISPR: clustered regulatory interspaced short palindromic repeat; Cas9: CRISPR associated system 9; DDIT3/CHOP: DNA-damage inducible transcript 3; DNAJB9/ERdj4: DnaJ heat shock protein family (Hsp40) member B9; DUBs: deubiquitinases; EIF2A/eIF2α: eukaryotic translation initiation factor 2A; EIF2AK3/PERK: eukaryotic translation initiation factor 2 alpha kinase 3; EP0: ubiquitin E3 ligase ICP0; ER: endoplasmic reticulum; ERN1/IRE1: endoplasmic reticulum (ER) to nucleus signaling 1; FOXO1: forkhead box O1; FRET: Förster resonance energy transfer; HSPA5/BiP: heat shock protein 5; HSV: herpes simplex virus; IE180: transcriptional regulator ICP4; MAP1LC3/LC3: microtube-associated protein 1 light chain 3; MOI: multiplicity of infection; MTOR: mechanistic target of rapamycin kinase; PPP1R15A/GADD34: protein phosphatase 1, regulatory subunit 15A; PRV: pseudorabies virus; PRV gB: PRV glycoprotein B; PRV gE: PRV glycoprotein E; qRT-PCR: quantitative real-time polymerase chain reaction; sgRNA: single guide RNA; siRNA: small interfering RNA; SQSTM1/p62: sequestosome 1; TCID50: tissue culture infective dose; UB: ubiquitin; UBA: ubiquitin-associated domain; UBL: ubiquitin-like domain; UL9: DNA replication origin-binding helicase; UPR: unfolded protein response; USPs: ubiquitin-specific proteases; VHS: virion host shutoff; VP16: viral protein 16; XBP1: X-box binding protein 1; XBP1s: small XBP1; XBP1(t): XBP1-total.
KEYWORDS: Alphaherpesvirus, ER stress, PRV VP16, selective autophagy, USP14
Introduction
Herpesviruses are enveloped, double-stranded DNA viruses that encompass over 200 species and undergo latent and lytic replication [1]. They are divided into three subfamilies, alpha-, beta- and gammaherpesviruses, according to their distinct biological properties. The alphaherpesviruses include HSV-1, HSV-2, PRV, varicella zoster virus, bovine herpesvirus, and duck enteritis virus [2]. HSV-1 is ubiquitous and highly neurotropic, with serious consequences in affected patients. Currently, more than 3.7 billion people (67%) under the age of 50 years are infected with HSV-1 and 417 million people between the age of 17 and 49 years suffer from HSV-2 infection [3]. Several clinical drugs are used to treat HSV infection, such as valacyclovir, famciclovir, phosphonoformate, iododeoxyuridine, and cidofovir [4]. PRV is the causative agent of pseudorabies, a disease of great economic and welfare importance in swine [5]. The tegument protein VP16 is conserved between Alphaherpesvirinae and it induces transcription of viral immediate-early genes that are critical for the viral life cycle [6].
Post-translational modification of proteins by ubiquitin is critical for protein quality control [7]. Ubiquitin can be covalently attached to proteins in a process termed ubiquitination that utilizes ubiquitin-activating, ubiquitin-conjugating, and ubiquitin ligase enzymes [8]. DUBs hydrolyze ubiquitin adducts and can be classified into five families based on their sequence and structural homology [9]. USPs represent the largest family of DUBs, and several studies have reported that DUBs are involved in immunity and infectious diseases [10]. Zebrafish USP5 activates interferon resistance to spring viremia caused by carp virus by increasing the expression of Ddx58/retinoic acid-inducible gene I [11]. USP13 deconjugates polyubiquitin chains from the STING1 (stimulator of interferon response cGAMP interactor 1) and prevents the recruitment of TBK1 (TANK-binding kinase 1) to the signaling complex, thereby negatively regulating cellular antiviral responses [12]. USP15 participates in hepatitis C virus propagation through the regulation of viral RNA translation and lipid droplet formation [13]. USP29 maintains the stability of cGAS (cyclic GMP-AMP synthase) and promotes cellular antiviral responses and autoimmunity [14].
Macroautophagy/autophagy is an essential cellular mechanism that controls intracellular homeostatic pathways [15]. ER stress is a common cellular stress response and is closely related to the activation of autophagy. Activation of ER stress triggers the activation of EIF2AK3/PERK, ERN1/IRE1 and ATF6 to buffer ER stress and orchestrate the recovery of ER function [16]. The activation of EIF2AK3 inhibits general protein translation by EIF2A/eIF2α phosphorylation that subsequently upregulates transcription of the stress-inducible transcription factor ATF4 [17]. ER stress stimulates ERN1 oligomerization in ER membranes and auto-phosphorylation of ERN1’s cytosolic domain is required for Xbp1 mRNA processing [18]. ATF6 translocates to the Golgi compartment upon ER stress and is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum [19]. ER stress signaling activates autophagy through inhibition of MTOR (mechanistic target of rapamycin kinase) complex 1, activation of adenosine 5′-monophosphate-activated protein kinase, and induction of Becn1/beclin-1 and Atg expression [20].
In this study, we investigated the effect of USP inhibitors on PRV proliferation. We demonstrated that a USP14 inhibitor, as well as USP14 deficiency, activated ER stress, and EIF2AK3- and ERN1-mediated autophagy induction. Inhibition of USP14 promoted the conjugation of K63-linked polyubiquitin chains on K168 in PRV VP16 and subsequent degradation of VP16 through SQSTM1/p62-mediated selective autophagy. These findings provided insight into the mechanism by which inhibition of USP14 prevented alphaherpesviruses infection.
Results
B-AP15 prevents PRV proliferation
The post-translational modification of proteins by ubiquitination is involved in a number of infectious diseases [21]. To explore the roles of ubiquitination in alphaherpesvirus proliferation, we assessed the effect of 13 USPs inhibitors on PRV proliferation, which was measured using our previously established GFP reporter assay [22]. These inhibitors target USP1 (SJB3-019A, SJB2-043 and ML-323), USP2 (PR-619), USP5 (Degrasyn), USP7 (USP7, USP47 inhibitor, USP7-IN-1, P22077 and P005091), USP8 (DUBs-IN-1, DUBs-IN-2 and DUBs-IN-3) and USP14 (b-AP15) (Fig. S1A). Interestingly, we found that all of the compounds could inhibit PRV proliferation in a dose-dependent manner (Fig. S1B–N). We focused our attention on the inhibitor of USP14, because among all the inhibitors, b-AP15 showed the most dramatic effect on the inhibition of PRV-GFP proliferation (Fig. S1N).
We next verified whether b-AP15 indeed inhibited PRV proliferation in vitro. We treated PK-15 and 3D4/21 cells with b-AP15 (0–1 μM) for 24–48 h. Cell viability measurements, assessed by the CCK-8 cell counting assay, indicated that b-AP15 was harmless to both cells (Figure 1A). Fluorescence microscopy analysis found that b-AP15 could limit PRV-GFP proliferation in PK-15 and 3D4/21 cells (Figure 1B). Quantification of GFP-positive cells by flow cytometry proved that PRV-GFP replication was reduced upon b-AP15 treatment (Figure 1C a nd D). PRV gB (viral late protein) plays a central role in the induction of immunity and is involved in virus entry and virulence [23]. Treatment of PK-15 and 3D4/21 cells with b-AP15 inhibited PRV gB expression in a dose dependent manner (Figure 1E). We also detected the multiplication of PRV progeny virus in response to b-AP15 treatment using a viral titer assay. b-AP15 significantly inhibited the production of PRV progeny virus (Figure 1F a nd G). To gain further insight into the effect of b-AP15 on PRV infection, we monitored the growth kinetics of PRV. b-AP15 resulted in the decrease of viral titer starting at 2 h post infection (Figure 1H a nd I). The IC50 value for b-AP15 was 4.081 μM on the basis of the viral titer in 3D4/21 cells measured by the TCID50 assay (Figure 1J). These data indicated that b-AP15 inhibited PRV proliferation in vitro.
Figure 1.
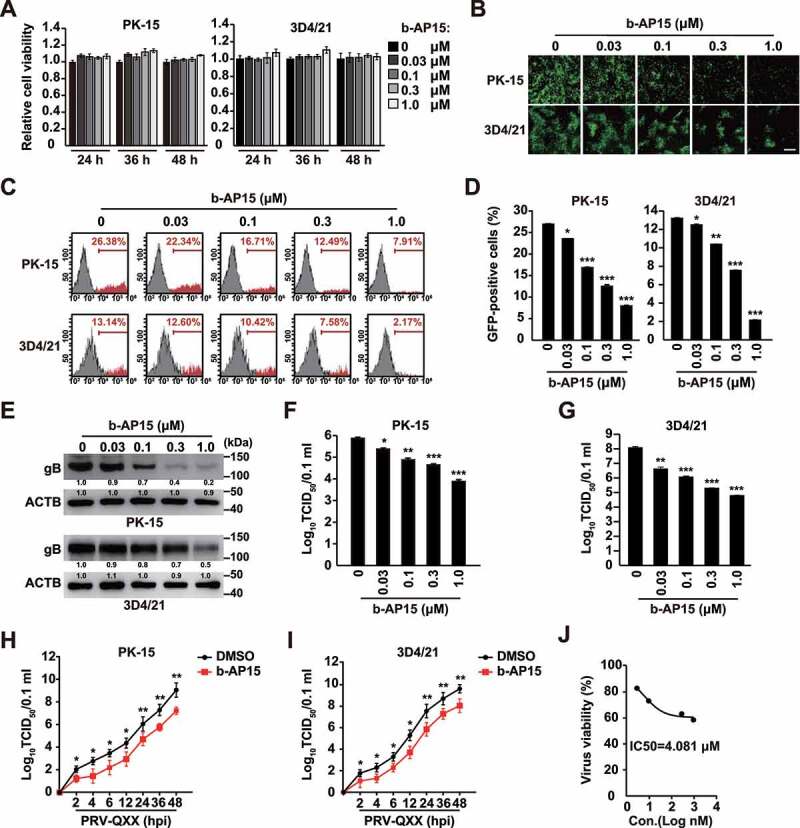
B-AP15 inhibits PRV infection in vitro. (A) PK-15 and 3D4/21 cells were treated with b-AP15 (0–1 μM) for 24–48 h. Cell viability was assessed using the CCK-8 cell counting assay. (B) PK-15 and 3D4/21 cells were infected with PRV-GFP (MOI = 0.01) and simultaneously treated with b-AP15 (0–1 μM) for 36 h. The fluorescence of GFP was detected by fluorescent microscopy. Scale bar: 200 μm. (C and D) Quantification of the percentage of GFP-positive cells from B by flow cytometry. (E) PK-15 and 3D4/21 cells were infected with PRV-QXX (MOI = 0.1) and simultaneously treated with b-AP15 (0–1 μM) for 24 h. PRV gB was assessed by immunoblot analysis. (F and G) PK-15 (F) and 3D4/21 (G) cells were infected with PRV-QXX (MOI = 0.1) and simultaneously treated with b-AP15 (0–1 μM) for 24 h. Viral titers were assessed by a TCID50 assay. (H and I) PK-15 (H) and 3D4/21 (I) cells were infected with PRV-QXX (MOI = 0.01) and simultaneously treated with DMSO or b-AP15 (1 μM) for 0–48 h. One-step growth curves of PRV-QXX were assessed using a TCID50 assay of viral titers. hpi, hour post infection. (J) Determination of IC50 value of b-AP15 from G. Data were shown as mean ± SD based on three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test.
USP14 deficiency restricts PRV proliferation
Because b-AP15 is a selective inhibitor of USP14 [24], we ablated USP14 to identify whether it was involved in PRV proliferation. CRISPR-Cas9 technology was employed to generate USP14 knockout cell lines (Figure 2A). The CCK-8 cell counting assay indicated that USP14 deficiency did not result in a limitation of cell proliferation for 72 h (Figure 2B). We infected sgControl and sgUSP14 PK-15 cells with PRV-GFP and found that the rate of GFP-positive cells (Figure 2C a nd D) and the production of PRV progeny virus (Figure 2E) were significantly lower in sgUSP14 cells than in sgControl cells. USP14 deficiency decreased viral titer starting at 2 h post infection, which was similar to the observation from b-AP15 treatment (Figure 2F).
Figure 2.
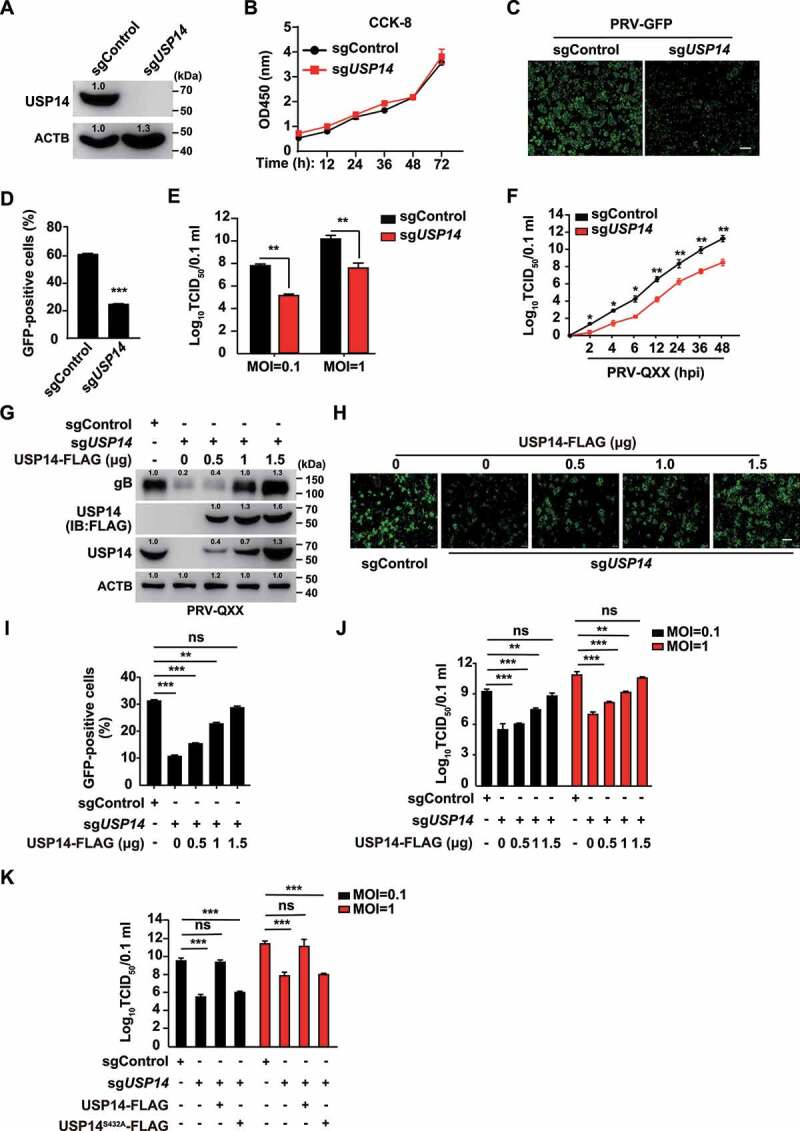
Knockout of USP14 inhibits PRV infection. (A) USP14 in sgControl and sgUSP14 PK-15 cells was assessed by immunoblot analysis. (B) sgControl and sgUSP14 PK-15 cells were cultured for 0–72 h. Cell proliferation was assessed using the CCK-8 cell counting assay. (C) sgControl and sgUSP14 PK-15 cells were infected with PRV-GFP (MOI = 0.01) for 36 h. The fluorescence of GFP was detected by fluorescent microscopy. Scale bar: 400 μm. (D) Quantification of the percentage of GFP-positive cells from C by flow cytometry. (E) sgControl and sgUSP14 PK-15 cells were infected with PRV-QXX (MOI = 0.1 and 1) for 24 h. Viral titers were assessed by the TCID50 assay. (F) sgControl and sgUSP14 PK-15 cells were infected with PRV-QXX (MOI = 0.01) for 0–48 h. One-step growth curves of PRV-QXX were assessed using a TCID50 assay of viral titers. hpi, hour post infection. (G) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding USP14-FLAG (0–1.5 μg) as indicated for 24 h, and then infected with PRV-QXX (MOI = 0.1) for 24 h. PRV gB, USP14-FLAG and USP14 were assessed by immunoblot analysis. (H) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding USP14-FLAG (0–1.5 μg) as indicated for 24 h, and then infected with PRV-GFP (MOI = 0.01) for 36 h. The fluorescence of GFP was detected by fluorescent microscopy. Scale bar: 400 μm. (I) Quantification of the percentage of GFP-positive cells from H by flow cytometry. (J) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding USP14-FLAG (0–1.5 μg) as indicated for 24 h, and then infected with PRV-QXX (MOI = 0.1 and 1) for 24 h. Viral titers were assessed by the TCID50 assay. (K) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding USP14-FLAG (1.5 μg) and USP14S432A-FLAG (1.5 μg) as indicated for 24 h, and then infected with PRV-QXX (MOI = 0.1 and 1) for 24 h. Viral titers were assessed by the TCID50 assay. Data were shown as mean ± SD based on three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test. ns, no significance.
To exclude off-target effects of sgRNA against USP14, we performed a rescue assay. Ectopic expression of USP14-FLAG in sgUSP14 PK-15 cells could restore PRV gB expression to a level comparable to that seen in sgControl cells (Figure 2G). Moreover, fluorescent microscopy demonstrated that GFP fluorescence was enhanced in a USP14-FLAG dose-dependent manner at 36 h post PRV-GFP infection in sgUSP14 PK-15 cells (Figure 2H a nd I). A viral titer assay suggested that ectopic expression of USP14-FLAG in sgUSP14 PK-15 cells could rescue the multiplication of PRV progeny virus (Figure 2J). However, the deubiquitinase activity mutant USP14S432A-FLAG [25] in sgUSP14 cells failed to bring viral titer to the level in sgControl cells (Figure 2K). These results demonstrated that USP14 played a bona fide role in PRV proliferation.
Inhibition of USP14 affects PRV replication
To characterize how b-AP15 inhibited PRV replication, we performed a time-of-drug-addition assay. We first performed a viral attachment assay. PK-15 cells were incubated with PRV and b-AP15 (0–1 μM) for 1 h at 4°C and shifted to 37°C for 24 h. Viral titer assay indicated that the production of PRV progeny virus was almost the same in control and b-AP15-treated cells (Figure 3A). We next carried out an entry assay. Cells bound with PRV were treated with b-AP15 (0–1 μM) for 1 h at 37°C and then shifted to maintenance medium for 24 h. The result demonstrated that b-AP15 did not influence viral entry as indicated by TCID50 assay (Figure 3B). In the replication assay, quantification of the PRV genome copy numbers by qRT-PCR indicated that b-AP15 treatment resulted in a decline in viral replication compared with DMSO treatment (Figure 3C). The viral titer assay also suggested that b-AP15 inhibited viral replication (Figure 3D). In addition, we assessed the expression of viral genes in response to b-AP15 treatment. The mRNA levels of PRV IE180 (immediate-early gene), EP0 (immediate-early gene), and UL9 (early gene) were decreased as early as 2 h post b-AP15 treatment (Figure 3E-G). Above all, our data suggested that inhibition of USP14 by b-AP15 disturbed PRV replication in vitro.
Figure 3.
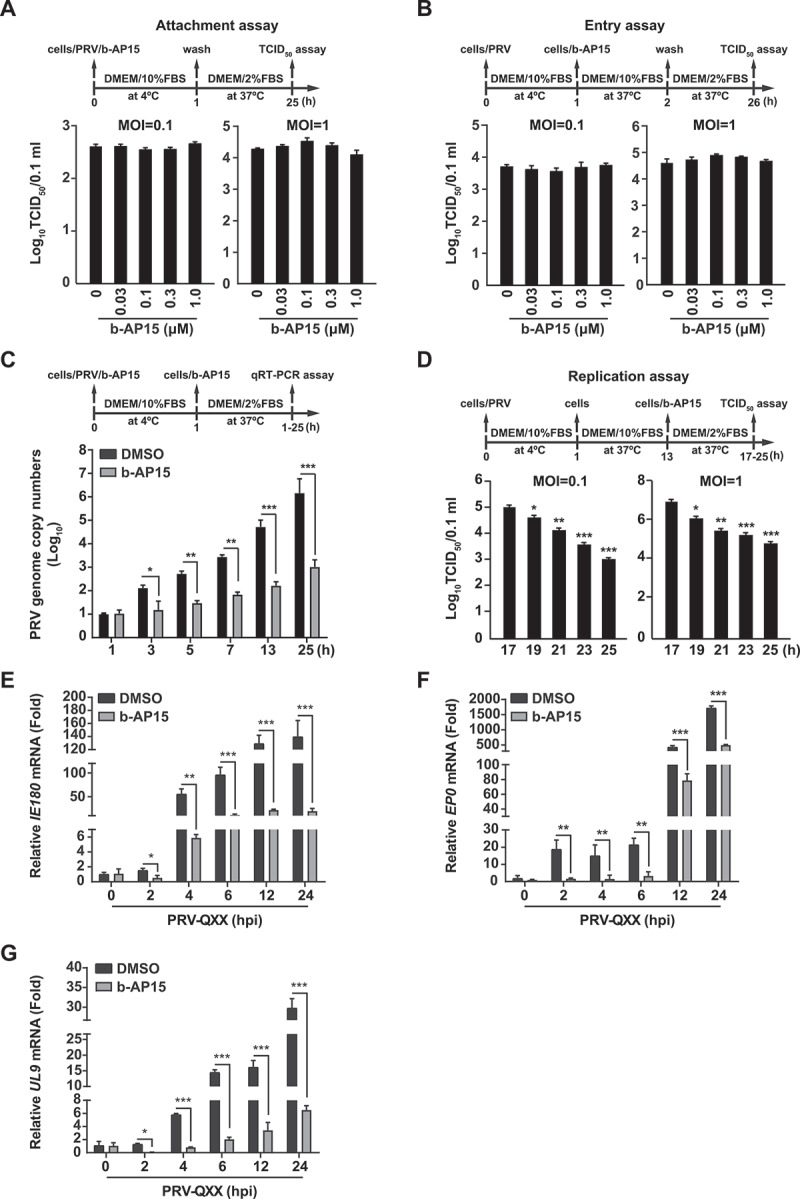
B-AP15 influences PRV replication. (A) PK-15 cells were incubated with PRV-QXX (MOI = 0.1 and 1) combined with b-AP15 (0–1 μM) for 1 h at 4°C. After washing with cold PBS 3 times, cells were cultured in DMEM with 2% FBS for 24 h at 37°C. An attachment assay was assessed using the TCID50 assay of viral titers. (B) PK-15 cells were incubated with PRV-QXX (MOI = 0.1 and 1) for 1 h at 4°C and then in DMEM with 10% FBS containing b-AP15 (0–1 μM) at 37°C. After 1 h to allow viral entry, cells were cultured in DMEM with 2% FBS for 24 h at 37°C. An entry assay was assessed using a TCID50 of viral titers. (C) PK-15 cells were incubated with PRV-QXX (MOI = 0.1) combined with b-AP15 (1 μM) for 1 h at 4°C. After washing with cold PBS 3 times, cells were cultured in DMEM with 2% FBS combined with b-AP15 (1 μM) for 0–24 h at 37°C. PRV genome copy numbers were assessed by qRT-PCR analysis. (D) PK-15 cells were incubated with PRV-QXX (MOI = 0.1 and 1) at 4°C for 1 h and then in DMEM with 10% FBS at 37°C for 12 h. Cells were then cultured in DMEM with 2% FBS containing b-AP15 (1 μM) for 4–12 h at 37°C. A replication assay was assessed using the TCID50 assay of viral titers. (E–G) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and simultaneously treated with DMSO or b-AP15 (1 μM) for 0–24 h. The mRNA levels of PRV IE180 (E), EP0 (F) and UL9 (G) were assessed by qRT-PCR analysis. hpi, hour post infection. Data were shown as mean ± SD based on three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test.
Inhibition of USP14 promotes the degradation of PRV VP16
We postulated that inhibition of USP14 might promote the degradation of PRV VP16, because VP16 regulates the transcription of the immediate early genes of alphaherpesviruses [26]. Our above results indicated that b-AP15 inhibited transcription of the PRV IE180 at 2 h post treatment (Figure 3E). In addition, we demonstrated that the mRNA levels of PRV IE180 and EP0 were downregulated as early as 10 min post b-AP15 treatment (Fig. S2A and B). We therefore assessed whether b-AP15 influenced VP16 expression by immunoblot analysis. b-AP15 treatment decreased the expressions of VP16 in PRV-infected cells and FLAG-VP16 (Figure 4A and B). Compared to the sgControl, the expression of VP16 in sgUSP14 cells was declined and was in line with b-AP15 treatment (Figure 4C and D).
Figure 4.
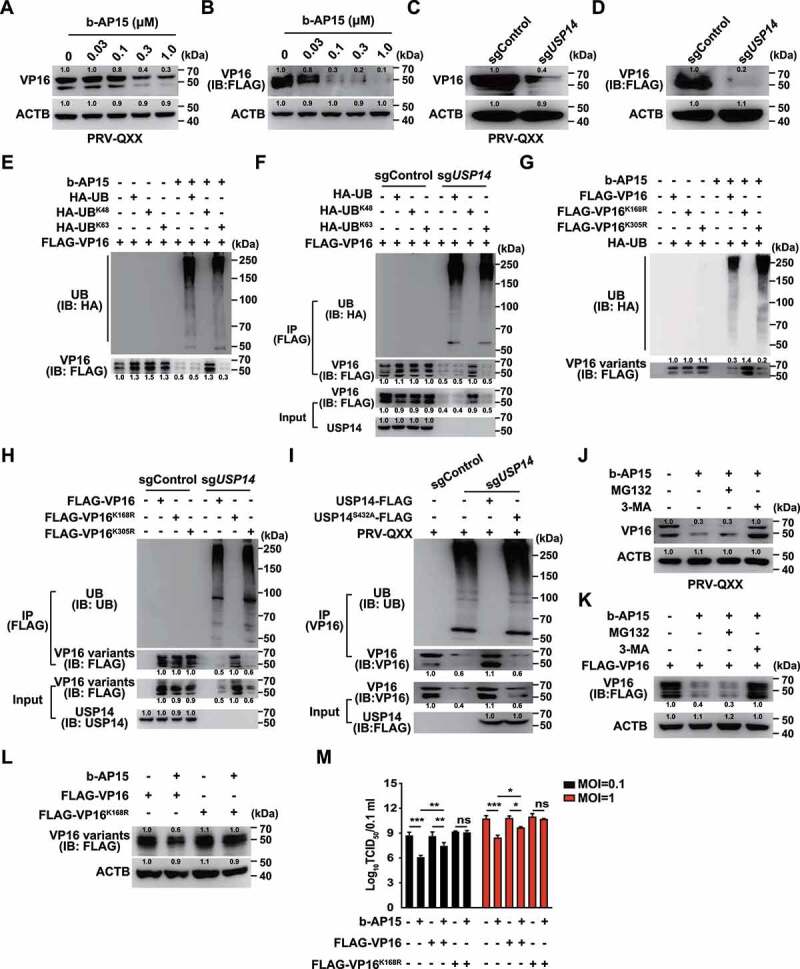
Inhibition of USP14 induces PRV VP16 ubiquitination and degradation that is not dependent on the proteasome. (A) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with b-AP15 (0–1 μM) for 24 h. PRV VP16 was assessed by immunoblot analysis. (B) PK-15 cells were transfected with plasmid encoding FLAG-VP16 and treated with b-AP15 (0–1 μM) for 24 h. FLAG-VP16 was assessed by immunoblot analysis. (C) sgControl and sgUSP14 PK-15 cells were infected with PRV-QXX (MOI = 0.1) for 24 h. PRV VP16 was assessed by immunoblot analysis. (D) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding FLAG-VP16 for 24 h. FLAG-VP16 was assessed by immunoblot analysis. (E) PK-15 cells were transfected with plasmids encoding FLAG-VP16, HA-UB, HA-UBK48 and HA-UBK63, and treated with b-AP15 (1 μM) as indicated for 24 h. Ubiquitination of FLAG-VP16 was assessed by the ubiquitination assay. (F) sgControl and sgUSP14 PK-15 cells were transfected with plasmids encoding FLAG-VP16, HA-UB, HA-UBK48 and HA-UBK63 as indicated for 24 h. Ubiquitination of FLAG-VP16 was assessed by the ubiquitination assay. (G) PK-15 cells were transfected with plasmids encoding FLAG-VP16, FLAG-VP16K168R, FLAG-VP16K305R, and HA-UB and treated with b-AP15 (1 μM) as indicated for 24 h. Ubiquitination of FLAG-VP16 variants was assessed by the ubiquitination assay. (H) sgControl and sgUSP14 PK-15 cells were transfected with plasmids encoding FLAG-VP16, FLAG-VP16K168R and FLAG-VP16K305R as indicated for 24 h. Ubiquitination of FLAG-VP16 variants was assessed by the ubiquitination assay. (I) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding USP14-FLAG and USP14-FLAGS432A as indicated for 24 h, and then infected with PRV-QXX (MOI = 0.1 and 1) for 24 h. Ubiquitination of VP16 was assessed by the ubiquitination assay. (J) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with b-AP15 (1 μM), MG132 (10 μM) and 3-MA (10 μM) as indicated for 24 h. PRV VP16 was assessed by immunoblot analysis. (K) PK-15 cells were transfected with plasmid encoding FLAG-VP16 and treated with b-AP15 (1 μM), MG132 (10 μM) and 3-MA (10 μM) as indicated for 24 h. FLAG-VP16 was assessed by immunoblot analysis. (L) PK-15 cells were transfected with plasmids encoding FLAG-VP16, FLAG-VP16K168R and treated with b-AP15 (1 μM) for 24 h. FLAG-VP16 and FLAG-VP16K168R were assessed by immunoblot analysis. (M) PK-15 cells were transfected with plasmids encoding FLAG-VP16 or FLAG-VP16K168R for 24 h. Then, cells were infected with PRV-QXX (MOI = 0.1 and 1) and treated with b-AP15 (1 μM) as indicated for 24 h. Viral titers were assessed by the TCID50 assay. Data were shown as mean ± SD based on three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test. ns, no significance.
Because USP14 belongs to a family of ubiquitin-specific proteases [27], we set out to determine whether inhibition of USP14 stimulated VP16 ubiquitination and proteasome-dependent degradation. We first determined the level of K48- or K63- linked ubiquitination of VP16. The plasmids encoding HA-UB, HA-UBK48, or HA-UBK63 were co-transfected with FLAG-VP16 into cells. The ubiquitination assay revealed that VP16 underwent K63-linked ubiquitination after challenge with b-AP15 and in sgUSP14 cells (Figure 4E and F). Furthermore, we identified which lysine residue in VP16 was modified by ubiquitination. PRV VP16 harbors two lysine residues (K168 and K305), which were mutated to arginine (VP16 K168R and VP16K305R) respectively. Inhibition of USP14 by b-AP15 or by USP14 knockout did not result in ubiquitination of VP16K168R, whereas VP16 and VP16K305R showed enhanced ubiquitination, suggesting that VP16 K168 was conjugated by ubiquitin chains (Figure 4G and H). VP16 was ubiquitinated in sgUSP14 cells and sgUSP14 cells expressing USP14S432A-FLAG, but not in sgControl cells and sgUSP14 cells expressing USP14-FLAG, which indicated that the deubiquitinase activity of USP14 was responsible for VP16 de-ubiquitination (Figure 4I).
We next treated cells with the proteasome inhibitor MG132 to determine if inhibition of USP14 induced proteasome-dependent degradation of VP16. Surprisingly, MG132 did not prevent b-AP15-induced degradation of either VP16 or FLAG-VP16 (Figure 4J and K). However, the autophagy inhibitor 3-MA suppressed VP16 degradation during b-AP15 treatment (Figure 4J and K). VP16K168R was resistant to b-AP15 treatment compared with WT VP16 (Figure 4L). Replenishment of WT VP16 could partially rescue and VP16K168R was able to fully restore the production of PRV progeny virus after b-AP15 treatment (Figure 4M). All of the results illustrated that inhibition of USP14 promoted VP16 degradation to prevent PRV replication.
USP14 binds to PRV VP16 at the early stage of viral infection
We hypothesized that USP14 interacted with PRV VP16 at the early stage of viral infection to stabilize VP16 for the transcription of viral immediate-early genes. To confirm this, we performed colocalization and co-IP assays of USP14 with VP16. We noted that punctate VP16 colocalized with USP14 from 5 to 20 min post viral entry (Figure 5A and Fig. S3C), which was further verified by co-IP assay (Figure 5B). The degradation of VP16 occurred as early as 5 min post viral entry in sgUSP14 PK-15 cells (Figure 5C).
Figure 5.
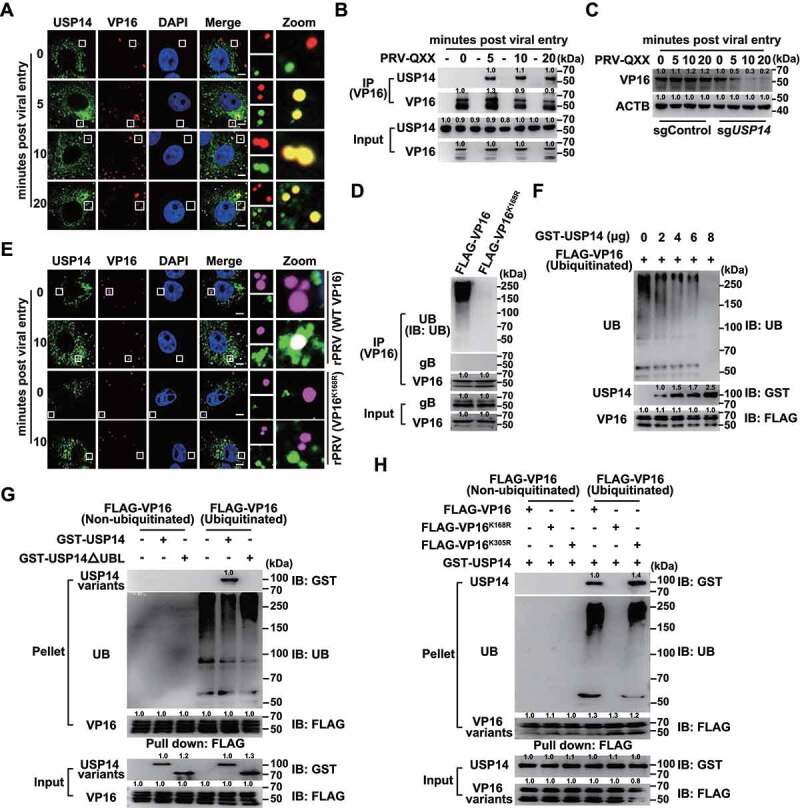
USP14 interacts with PRV VP16 at the early stage of viral infection. (A) PRV-QXX (MOI = 10) was incubated with PK-15 cells at 4°C for 1 h. After three times washing with ice-cold PBS, the cells were incubated at 37°C for 0–20 min. Colocalization of USP14 and VP16 was assessed by the immunofluorescence analysis. Scale bar: 10 μm. (B) Cells were treated as in A. The interaction of USP14 with VP16 was assessed by co-IP assay. (C) Cells were treated as in A. PRV VP16 was assessed by immunoblot analysis. (D) Ubiquitination of VP16 in rPRV ΔUL48 rescued by VP16 and VP16K168R was assessed by the ubiquitination assay. (E) rPRV (WT VP16, MOI = 10) and rPRV (VP16K168R, MOI = 10) were incubated with PK-15 cells at 4°C for 1 h. After three times washing with ice-cold PBS, the cells were incubated at 37°C for 10 min. The colocalization of USP14 and VP16 variants was assessed by the immunofluorescence analysis. Scale bar: 10 μm. (F) De-ubiquitination of FLAG-VP16 by GST-USP14 was assessed by the in vitro de-ubiquitination assay. (G) The interactions of non-ubiquitinated and ubiquitinated FLAG-VP16 with GST-USP14 and GST-USP14ΔUBL were assessed by an in vitro affinity-isolation assay. (H) The interactions of non-ubiquitinated and ubiquitinated FLAG-VP16, FLAG-VP16K168R, and FLAG-VP16K305R with GST-USP14 were assessed by an in vitro affinity-isolation assay.
VP16 is the late tegument protein of alphaherpesviruses and is encoded by the UL48 gene [28]. We generated rPRV ΔUL48 using CRISPR-Cas9 technology through homology-directed recombination to determine whether USP14 interacted with ubiquitin chains on VP16 (Fig. S3A). PRV lacking the UL48 gene fails to produce infectious virions [6]; hence, we produced rPRV ΔUL48 in cells expressing FLAG-VP16 (Fig. S3A). The infectivity of rPRV ΔUL48 rescued by FLAG-VP16, FLAG-VP16K168R, and FLAG-VP16K305R showed comparable viral titers, although they were lower than that of their parental strain (Fig. S3B). First, rPRV ΔUL48 rescued by WT VP16 and VP16K168R was purified, and ubiquitination of VP16 in virions was assessed. WT VP16, but not VP16K168R, was ubiquitinated (Figure 5D). We then infected cells with rPRV (WT VP16) and rPRV (VP16K168R) for 10 min and performed colocalization of USP14 with VP16. Fluorescent microscopy revealed that WT VP16 colocalized with USP14 and that VP16K168R failed to interact with USP14, suggesting that USP14 interacted with ubiquitin chains on VP16 (Figure 5E and Fig. S3D).
To clarify whether USP14 directly bound to VP16, we purified GST-USP14 and ubiquitinated FLAG-VP16 (Fig. S3E and F) to conduct in vitro de-ubiquitination assays. Ubiquitination of VP16 gradually decreased with the increase in the amount of USP14, thus suggesting that USP14 directly de-ubiquitinated VP16 (Figure 5F). We further performed in vitro affinity-isolation assays with recombinant USP14 and VP16. We showed that USP14 bound to ubiquitinated FLAG-VP16 with its UBL domain (Figure 5G). FLAG-VP16K168R, unlike FLAG-VP16 and FLAG-VP16K305R, failed to be ubiquitinated and could not associate with USP14 (Figure 5H). Taken together, all these results indicated that USP14 directly bound to VP16.
Inhibition of USP14 activates autophagy-dependent degradation of PRV VP16
Autophagy is another intracellular degradation system by which cytoplasmic materials are delivered to and degraded in the lysosome [15], so we examined whether autophagy was responsible for degradation of PRV VP16. We performed immunoblot analysis and observed that the expression levels of MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3 beta)-II (the lipidated form), ATG5, ATG12 and BECN1 were increased (Figure 6A). Ubiquitin-tagged substrates can be sequestered into an autophagosome by using the selective autophagy receptor SQSTM1 to enable their degradation in the lysosome [29]. When cells were treated with b-AP15, SQSTM1 expression was decreased, indicating that autophagy was activated (Figure 6A). Similar phenomena were also observed in sgUSP14 PK-15 cells (Figure 6B). Cytoplasmic LC3 puncta are characteristic of autophagosomal membrane formation and we noticed that b-AP15 and USP14 deficiency significantly induced LC3 puncta formation (Figure 6C-E). Furthermore, cells treated with b-AP15 formed more autophagosomes than DMSO-treated cells, as determined by transmission electron microscope analysis (Figure 6F and G). To measure autophagic flux, we first used the GFP-RFP-LC3 sensor [30]. We observed an increased RFP signal relative to the GFP signal in response to b-AP15 treatment, indicating that enhanced autophagic flux occurred after the inhibition of USP14 (Figure 6H and I). Furthermore, treatment of sgUSP14 cells with bafilomycin A1 (an inhibitor of late-stage fusion between autophagosomes and lysosomes [31]) showed greater accumulation of LC3-II than that without treatment (Figure 6J). These results indicated that inhibition of USP14 activated autophagy efflux.
Figure 6.
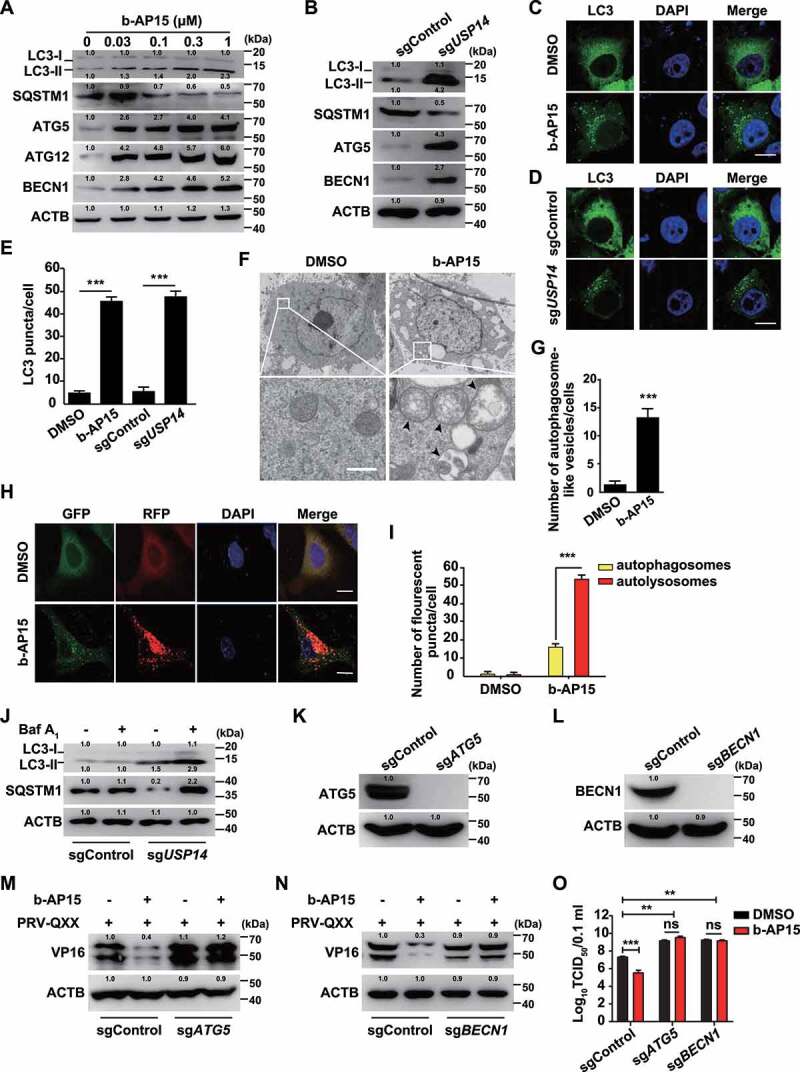
Inhibition of USP14 induces autophagy. (A) PK-15 cells were treated with b-AP15 (0–1 μM) for 24 h. LC3-I, LC3-II, SQSTM1, ATG5, ATG12 and BECN1 were assessed by immunoblot analysis. (B) LC3-I, LC3-II, SQSTM1, ATG5, and BECN1 were assessed by immunoblot analysis in sgControl and sgUSP14 PK-15 cells. (C) PK-15 cells were transfected with plasmid encoding GFP-LC3 and treated with DMSO or b-AP15 (1 μM) for 24 h. The fluorescence of GFP-LC3 was detected by fluorescent microscopy. Scale bar: 10 μm. (D) sgControl and sgUSP14 PK-15 cells were transfected with plasmid encoding GFP-LC3 for 24 h. The fluorescence of GFP-LC3 was detected by fluorescent microscopy. Scale bar: 10 μm. (E) Quantification of GFP-LC3 puncta per cell from C and D using ImageJ software. (F) PK-15 cells were treated with DMSO or b-AP15 (1 μM) for 24 h. The autophagosome-like vesicles were detected by transmission electron microscope. Scale bar: 500 nm. (G) Quantification of autophagosome-like vesicles from F using ImageJ software. (H) PK-15 cells were transfected with plasmid encoding GFP-RFP-LC3 and treated with DMSO or b-AP15 (1 μM) for 24 h. The fluorescence of GFP and RFP was detected by fluorescent microscopy. Scale bar: 10 μm. (I) Quantification of autophagosomes and autolysosomes from H using ImageJ software. (J) sgControl and sgUSP14 PK-15 cells were treated with bafilomycin A1 (10 μM) for 24 h. LC3-I, LC3-II and SQSTM1 were assessed by immunoblot analysis. (K) ATG5 in sgControl and sgATG5 PK-15 cells was assessed by immunoblot analysis. (L) BECN1 in sgControl and sgBECN1 PK-15 cells was assessed by immunoblot analysis. (M and N) sgControl, sgATG5 (M) and sgBECN1 (N) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with b-AP15 (1 μM) as indicated for 24 h. PRV VP16 was assessed by immunoblot analysis. (O) sgControl, sgATG5 and sgBECN1 PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with DMSO or b-AP15 (1 μM) as indicated for 24 h. Viral titers were assessed by the TCID50 assay. Data were shown as mean ± SD based on three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test. ns, no significance.
ATG5 and BECN1 are two autophagy-related proteins that are necessary for the formation of autophagosomes [32]. To determine whether autophagy was essential for b-AP15-induced degradation of PRV VP16, we ablated ATG5 and BECN1 using CRISPR-Cas9 (sgATG5 and sgBECN1, Figure 6K and L) to analyze whether inhibition of autophagy could abolish VP16 degradation. ATG5 and BECN1 deficiency inhibited VP16 degradation upon b-AP15 treatment (Figure 6M and N). We assessed the production of PRV progeny virus in sgControl, sgATG5 and sgBECN1 cells by the viral titer assay. As shown in (Figure 6O), ablation of either ATG5 or BECN1 enhanced viral titer and b-AP15 had no inhibitory effect on PRV replication in sgATG5 and sgBECN1 cells. These results suggested that inhibition of USP14 induced autophagy-mediated VP16 degradation.
Inhibition of USP14 activates ER stress response to trigger autophagy
ER stress is a potent trigger for autophagy [33], so we sought to determine whether inhibition of USP14 activated ER stress response to trigger autophagy. Three signal transducers located in the ER, EIF2AK3, ERN1 and ATF6, can be activated by ER stress [34]. Using immunoblot analysis, we observed that the expression of HSPA5/BiP and ATF6 was unchanged by b-AP15 treatment (Figure 7A). However, the phosphorylation of EIF2AK3 and its downstream effector EIF2A were all upregulated in response to b-AP15 treatment or USP14 deficiency (Figure 7A and B). Furthermore, we found that inhibition of USP14 enhanced XBP1 expression and impeded FOXO1 expression, consistent with the previous finding that XBP1 is regulated by ERN1 and negatively regulates FOXO1 expression [35] (Figure 7A and B). These data demonstrated that inhibition of USP14 induces ER stress and activates the EIF2AK3 and ERN1 signaling pathways. We further verified this finding by quantification of EIF2AK3 and ERN1 target genes with qRT-PCR analysis. EIF2A governs transcription of ATF4, PPP1R15A/GADD34 and DDIT3/CHOP [36], and they were all upregulated after b-AP15 treatment (Fig. S4A). ERN1 processes XBP1 mRNA [37], and qRT-PCR analysis indicated that XBP1(s):XBP1(t) was increased due to b-AP15 treatment (Fig. S4B). Transcription of DNAJB9/ERdj4 was enhanced and FOXO1 mRNA was decreased when USP14 was inhibited by b-AP15, suggesting that ERN1 signaling was activated (Fig. S4B and C).
Figure 7.
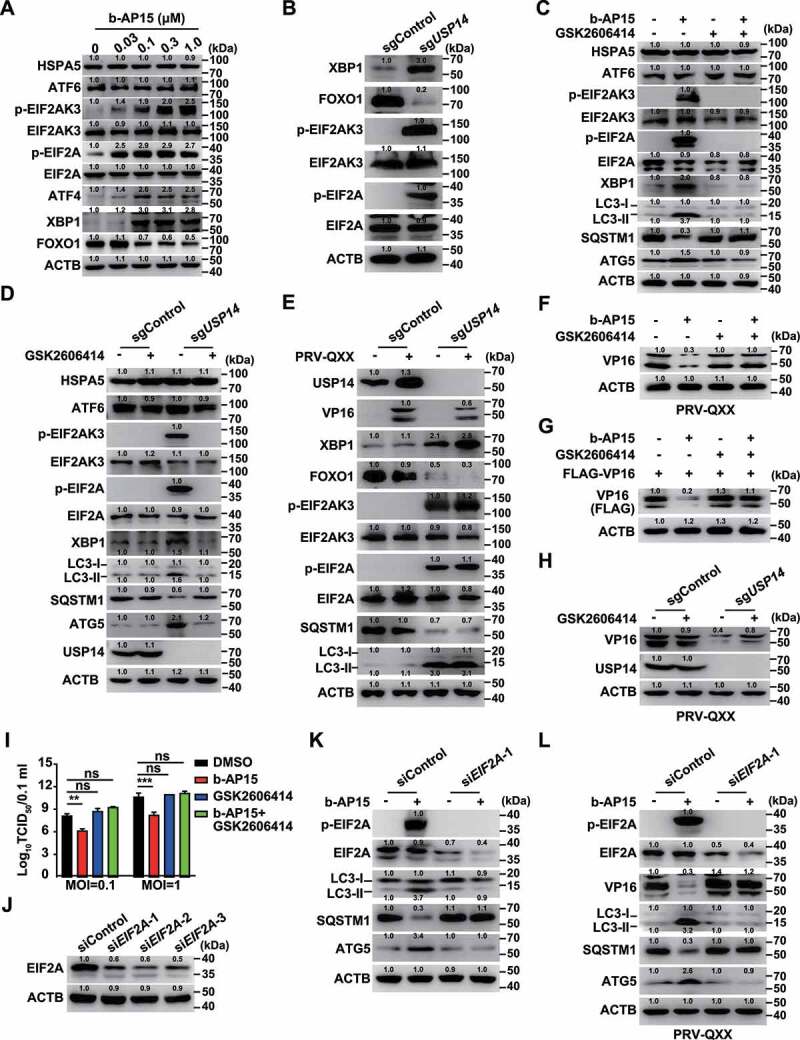
Inhibition of USP14 induces ER stress. (A) PK-15 cells were treated with b-AP15 (0–1 μM) for 24 h. HSPA5, ATF6, p-EIF2AK3, EIF2AK3, p-EIF2A, EIF2A, ATF4, XBP1 and FOXO1 levels were assessed by immunoblot analysis. (B) XBP1, FOXO1, p-EIF2AK3, EIF2AK3, p-EIF2A and EIF2A levels were assessed by immunoblot analysis in sgControl and sgUSP14 PK-15 cells. (C) PK-15 cells were treated with b-AP15 (1 μM) and GSK2606414 (10 μM) as indicated for 24 h. HSPA5, ATF6, p-EIF2AK, EIF2AK3, p-EIF2A, EIF2A, XBP1, LC3-I, LC3-II, SQSTM1 and ATG5 were assessed by immunoblot analysis. (D) sgControl and sgUSP14 PK-15 cells were treated with GSK2606414 (10 μM) as indicated for 24 h. HSPA5, ATF6, p-EIF2AK3, EIF2AK3, p-EIF2A, EIF2A, XBP1, LC3-I, LC3-II, SQSTM1 and ATG5 and USP14 were assessed by immunoblot analysis. (E) sgControl and sgUSP14 PK-15 cells were mock infected or infected with PRV-QXX (MOI = 0.1) for 24 h. USP14, VP16, XBP1, FOXO1, p-EIF2AK3, EIF2AK3, p-EIF2A, EIF2A, SQSTM1, LC3-I and LC3-II were assessed by immunoblot analysis. (F) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with b-AP15 (1 μM) and GSK2606414 (10 μM) as indicated for 24 h. PRV VP16 was assessed by immunoblot analysis. (G) PK-15 cells were transfected with plasmid encoding FLAG-VP16 and treated with b-AP15 (1 μM) and GSK2606414 (10 μM) as indicated for 24 h. FLAG-VP16 was assessed by immunoblot analysis. (H) sgControl and sgUSP14 PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with GSK2606414 (10 μM) for 24 h. PRV VP16 was assessed by immunoblot analysis. (I) PK-15 cells were infected with PRV-QXX (MOI = 0.1 and 1) and treated with DMSO, b-AP15 (1 μM), GSK2606414 (10 μM) and b-AP15 (1 μM) + GSK2606414 (10 μM) for 24 h. Viral titers were assessed by the TCID50 assay. (J) PK-15 cells were transfected with siControl, siEIF2A-1, siEIF2A-2 and siEIF2A-3 for 48 h. EIF2A was assessed by immunoblot analysis. (K) PK-15 cells were transfected with siControl or siEIF2A-1 and treated with b-AP15 (1 μM) as indicated for 48 h. p-EIF2A, EIF2A, LC3-I, LC3-II, SQSTM1 and ATG5 were assessed by immunoblot analysis. (L) PK-15 cells were transfected with siControl or siEIF2A-1 for 24 h. Then, cells were infected with PRV-QXX (MOI = 0.1) and treated with b-AP15 (1 μM) as indicated for 24 h. p-EIF2A, EIF2A, VP16, LC3-I, LC3-II, SQSTM1, and ATG5 were assessed by immunoblot analysis. Data were shown as mean ± SD based on three independent experiments. ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test. ns, no significance.
We next wanted to determine whether EIF2AK3 and ERN1 signaling pathways were essential for b-AP15-induced autophagy. GSK2606414 is a selective EIF2AK3 inhibitor [38]. During USP14 inhibition, GSK2606414 inhibited expression of the active XBP1s protein and phosphorylation of EIF2AK3 and EIF2A, but the expressions of HSPA5 and ATF6 were not affected (Figure 7C and D). Moreover, USP14 inhibition-induced autophagy was abolished by GSK2606414, as indicated by unaltered LC3-II, ATG5 and SQSTM1 expression (Figure 7C and D). PRV infection had no effect on the activation of ER stress and autophagy in sgControl cells (Figure 7E). Although inhibition of USP14 promoted VP16 degradation, EIF2AK3 inhibition by GSK2606414 abrogated this effect (Figure 7F–H). Production of infectious progeny virus in cells treated with DMSO, GSK2606414 and GSK2606414 + b-AP15 was not significantly different (Figure 7I). Next, we utilized siRNA to knockdown EIF2A to further confirm whether EIF2AK3 signaling was responsible for b-AP15-induced autophagy (Figure 7J). Interference of EIF2A impeded autophagy, because expression levels of LC3-II, ATG5, and SQSTM1 were unchanged (Figure 7K). Knockdown of EIF2A also blocked b-AP15-induced VP16 degradation and autophagy induction (Figure 7L). These data indicated that inhibition of USP14 activated ER stress to trigger autophagy.
Inhibition of USP14 stimulates the interaction of ubiquitinated VP16 with the selective autophagy receptor SQSTM1/p62
Because our data demonstrated that b-AP15 activated autophagy and induced VP16 ubiquitination and proteasome-independent degradation of VP16, we sought to examine whether VP16 was degraded by SQSTM1-mediated selective autophagy. SQSTM1 and ubiquitin colocalized with WT VP16, but not with VP16K168R, in response to b-AP15 treatment (Figure 8A and S5A). Co-IP assay indicated that SQSTM1 interacted with VP16 only in sgUSP14 cells (Figure 8B). We verified that SQSTM1-EGFP interacted with FLAG-VP16, and that inhibition of autophagy by 3-MA or by ablation of ATG5 abolished their interactions (Figure 8C and S5B). SQSTM1 interacts directly with ubiquitin on selected cargo through its C-terminal UBA domain [39], and we found that deletion of the SQSTM1 UBA domain prevented SQSTM1 from interacting with VP16 (Figure 8D). Furthermore, we purified SQSTM1 (Fig. S5C) and carried out in vitro affinity-isolation assays. SQSTM1 directly associated with ubiquitinated VP16 and VP16K305R, while VP16K168R that could not be modified by ubiquitination, failed to interact with SQSTM1 (Figure 8E). Meanwhile, the UBA domain of SQSTM1 was responsible for SQSTM1-VP16 association, which required VP16 to be ubiquitinated (Figure 8F). These results suggested that SQSTM1 bound to ubiquitin chains on VP16 through its UBA domain.
Figure 8.
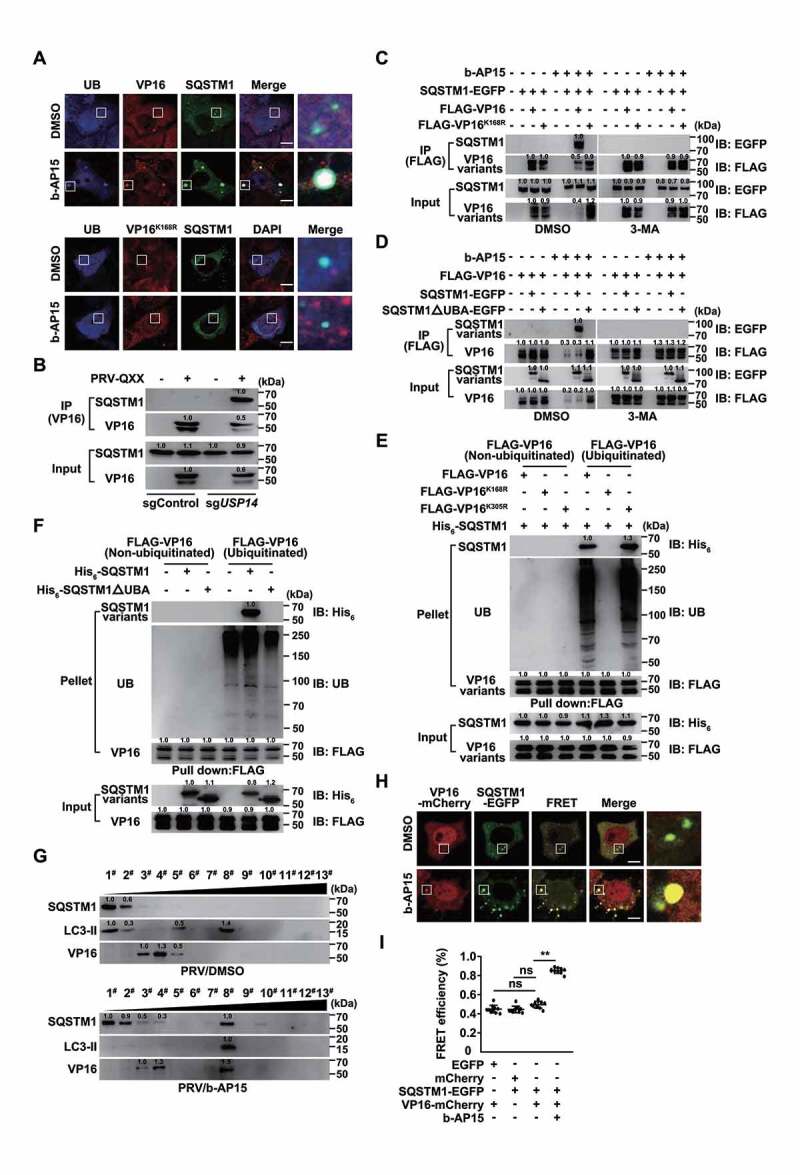
Inhibition of USP14 induces the interaction of SQSTM1/p62 with ubiquitinated VP16. (A) PK-15 cells were co-transfected with plasmids encoding VP16-mCherry and SQSTM1-EGFP, or VP16K168R-mCherry and SQSTM1-EGFP, and treated with DMSO or b-AP15 (1 μM) for 24 h. The colocalization of UB, SQSTM1 and VP16 variants was assessed by immunofluorescence of UB and fluorescent microscopy. Scale bar: 10 μm. (B) sgControl and sgUSP14 PK-15 cells were mock infected or infected with PRV-QXX (MOI = 0.1) for 24 h. The interaction of SQSTM1 with VP16 was assessed by co-IP assay. (C) PK-15 cells transfected with plasmids encoding SQSTM1-EGFP, FLAG-VP16 and FLAG-VP16K168R were treated with b-AP15 (1 μM) and 3-MA (10 μM) as indicated for 24 h. The interaction of SQSTM1-EGFP with the FLAG-VP16 variants was assessed by co-IP assay. (D) PK-15 cells transfected with plasmids encoding FLAG-VP16, SQSTM1-EGFP and SQSTM1ΔUBA-EGFP were treated with b-AP15 (1 μM) and 3-MA (10 μM) as indicated for 24 h. The interaction of FLAG-VP16 with SQSTM1-EGFP variants was assessed by a co-IP assay. (E) The interactions of His6-SQSTM1 with non-ubiquitinated and ubiquitinated FLAG-VP16, FLAG-VP16K168R and FLAG-VP16K305R were assessed by an in vitro affinity-isolation assay. (F) The interactions of non-ubiquitinated and ubiquitinated FLAG-VP16 with His6-SQSTM1 and His6-SQSTM1ΔUBA were assessed by an in vitro affinity-isolation assay. (G) PK-15 cells were infected with PRV-QXX (MOI = 0.1) and treated with DMSO or b-AP15 (1 μM) for 24 h. The cell lysates were subjected to iodixanol density gradient centrifugation. SQSTM1, LC3-II and PRV VP16 levels in each fraction were assessed by immunoblot analysis. (H) PK-15 cells were co-transfected with plasmids encoding VP16-mCherry and SQSTM1-EGFP and treated with DMSO or b-AP15 (1 μM) for 24 h. The interaction of SQSTM1 with VP16 was assessed by FRET analysis. Scale bar: 10 μm. (I) Quantification of FRET efficiency from H. Data were shown as mean ± SD based on three independent experiments. ** P < 0.01 determined by two-tailed Student’s t-test. ns, no significance.
Cell fraction analysis by iodixanol density gradient centrifugation detected that SQSTM1 and LC3-II were mainly located in the top fraction and VP16 was in fractions three–five, whereas b-AP15 treatment induced VP16 to shift to fraction eight, along with SQSTM1 and LC3-II (Figure 8G). Furthermore, significant FRET efficiency occurred between VP16-mCherry and SQSTM1-EGFP in cells treated with b-AP15 compared with that in cells treated with DMSO (Figure 8H and I). Together, these data demonstrated that inhibition of USP14 promoted degradation of VP16 through SQSTM1-mediated selective autophagy.
B-AP15 protects mice from PRV infection in vivo
On the basis of the above findings, we examined whether b-AP15 could be used as a potent antiviral against PRV in vivo. Mice were intraperitoneally injected with DMSO or b-AP15 every two days, and ER stress and autophagy in the lung were assessed by immunoblot and qRT-PCR analysis (Fig. S6A). Consistent with our in vitro data, b-AP15 could induce ER stress, which was indicated by phosphorylation of EIF2AK3 and EIF2A and upregulation of ATF4 and XBP1 (Fig. S6B). ERN1 and EIF2AK3 were activated in response to the b-AP15 challenge, as demonstrated by an increase in the processing of Xbp1 mRNA and transcription of Dnajb9, Atf4, Ppp1r15a, and Ddit3 (Fig. S6C). Challenge of mice with b-AP15 enhanced the expression of LC3-II, ATG5, and ATG12 and decreased SQSTM1 expression, suggesting that b-AP15 activated autophagy in vivo (Fig. S6D). These data suggested b-AP15 activated ER stress and autophagy in vivo.
We then determined the protective effect of b-AP15 on PRV infection in vivo (Figure 9A). We found that the mortality of mice injected with b-AP15 was significantly lower than that of mice injected with DMSO (Figure 9B). PRV VP16 was degraded in the lungs of b-AP15-treated mice (Figure 9C). Transcription of PRV gE mRNA from lungs was significantly lower in b-AP15-injected mice than that in PRV-infected mice (Figure 9D). Results from qRT-PCR analysis indicated that PRV genome copy numbers were decreased due to b-AP15 treatment, suggesting that b-AP15 inhibited PRV replication in vivo (Figure 9E). Lung injury caused by PRV infection was greatly attenuated upon b-AP15 treatment, because less infiltration of inflammatory cells was detected in the lungs in b-AP15-treated mice than DMSO-treated mice (Figure 9F a nd G). In addition, we tested the therapeutic effect of b-AP15 on PRV infection (Figure 9H). The survival rate of mice injected with b-AP15 was significantly higher than that of mice injected with DMSO (Figure 9I). These results demonstrated that USP14 inhibitors might be used as a potent antiviral against alphaherpesvirus.
Figure 9.
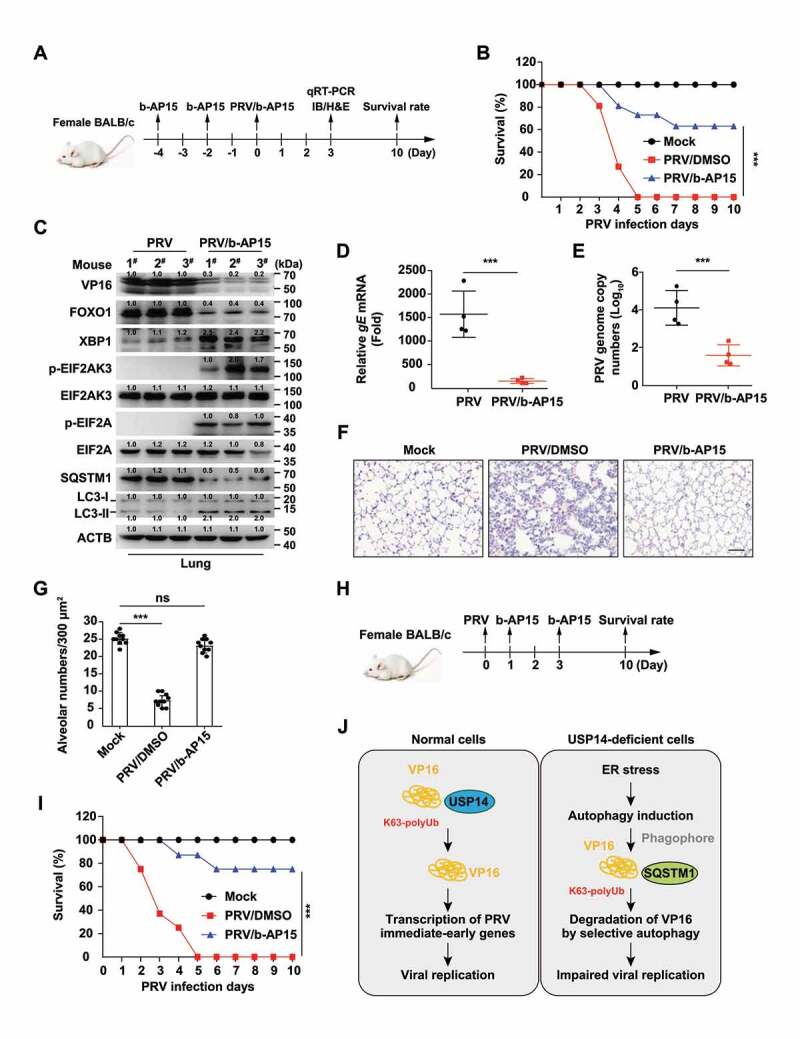
B-AP15 antagonizes PRV infection in vivo. Preventive strategy for PRV-QXX challenge and b-AP15 treatment in mice. (B) Mice were intraperitoneally injected with DMSO or b-AP15 (8 mg/kg) on day −4 and day −2. On day 0, mice were intraperitoneally injected with DMSO or b-AP15 (8 mg/kg) and intranasally infected with PRV-QXX (5 × 103 TCID50 per mouse). The survival rate was monitored daily for 10 days (n = 12 per group). (C) Mice were treated as in B. On day 3, VP16, FOXO1, XBP1, p-EIF2AK3, EIF2AK3, p-EIF2A, EIF2A, SQSTM1, LC3-I and LC3-II levels in the lung were assessed by immunoblot analysis (n = 3). (D and E) Mice were treated as in B. On day 3, PRV gE mRNA (D) and PRV genome copy numbers (E) in the lung were assessed by the qRT-PCR analysis (n = 4). (F) Sections of mouse lungs from D were stained by hematoxylin-eosin staining. Scale bar: 100 μm. (G) Quantification of alveolar numbers from F by ImageJ software. (H) Therapeutic strategy for PRV-QXX challenge and b-AP15 treatment in mice. (I) On day 0, mice were intranasally infected with PRV-QXX (5 × 103 TCID50 per mouse). Mice were intraperitoneally injected with DMSO or b-AP15 (8 mg/kg) on day 1 and day 3. The survival rate was monitored daily for 10 days. (n = 12 per group). (J) A schematic model showing inhibition of USP14 influenced alphaherpesvirus proliferation. Data were shown as mean ± SD based on three independent experiments. ** P < 0.01, *** P < 0.001 determined by two-tailed Student’s t-test.
Discussion
The development of new strategies to treat alphaherpesvirus infection is important for public health security. In this study, we revealed the mechanism of how inhibition of USP14 influenced alphaherpesvirus proliferation. USP14 directly bound to K63-linked polyubiquitin chains on PRV VP16 through its UBL domain at the early stage of viral infection. USP14 stabilized VP16 by de-ubiquitination of VP16 to facilitate the transcription of PRV immediate-early genes that were critical for viral proliferation. In contrast, USP14 deficiency induced ER stress-triggered autophagy induction. The selective autophagy receptor SQSTM1 interacted with ubiquitinated VP16 through its UBA domain to sequestrate VP16 into autolysosomes for subsequent degradation, which resulted in impaired viral replication (Figure 9J).
VP16 is essential for alphaherpesvirus replication. It not only regulates transcription of immediate early genes of alphaherpesviruses, but also participates in the assembly and maturation of nucleocapsids in the cytoplasm [6,40]. The assembly of a transcriptional regulatory complex, the VP16-induced complex, is critical for initiating the lytic program of alphaherpesviruses [26]. It has also been reported that VP16 can antagonize innate immune responses to facilitate viral replication. VP16 of duck enteritis virus directly binds to IRF7 (interferon regulatory factor 7) and antagonizes interferon-β-mediated antiviral innate immunity [41]. HSV-1 VP16 abrogates interferon-β production by inhibiting NFKB/NF-κB (nuclear factor kappa B) activation and blocking IRF3 (interferon regulatory factor 3) to recruit its co-activator CBP [42]. Moreover, alphaherpesvirus VHS is encoded by UL41 and it is able to degrade host cellular and viral mRNA [43]. However, VP16 negatively regulates VHS to inhibit its mRNA degradation activity and stimulate viral gene expression [44]. Intriguingly, our results indicated that inhibition of USP14 influenced PRV proliferation by degrading viral VP16 protein via ER stress-triggered selective autophagy. Under b-AP15 treatment, VP16K168R was not degraded and it could fully rescue PRV proliferation. Thus, our data suggested that VP16 was a promising therapeutic target to treat alphaherpesviruses infection.
Xenophagy is a selective form of autophagy that utilizes the cargo capture receptors, SQSTM1 for example, to bind both ubiquitin and LC3 to facilitate sequestration of an invasive microorganism to autolysosomes [45]. The HECT-type E3 ubiquitin ligase SMURF1 may be responsible for HSV-1 capsid ubiquitination, which is required for autophagic targeting [46]. We demonstrated that inhibition of USP14 enhanced VP16 ubiquitination and subsequent degradation by SQSTM1-mediated selective autophagy. Alphaherpesvirus, however, have evolved several mechanisms to modulate autophagy. The PRV US3 tegument protein may reduce the level of autophagy by activating the AKT-MTOR pathways [47]. Varicella zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of MTOR-mediated autophagic flux [48]. Inhibition of ULK1 and BECN1 by the HSV-1 AKT like Ser/Thr kinase limits autophagy to stimulate virus replication [49]. An HSV‑1 recombinant lacking the BECN1-binding domain of ICP34.5 is severely neuroattenuated and fails to inhibit xenophagy [50]. Therefore, augmentation of the autophagy-mediated degradation of essential proteins encoded by alphaherpesvirus could enable to develop a new class of antiviral therapies.
Previous studies have reported that alphaherpesviruses suppress ER stress to facilitate viral replication. The HSV-1 VHS protein suppresses the ERN1/XBP1 signaling pathway of the UPR through its RNase activity [51]. The HSV-1 gB protein associates with EIF2AK3 to maintain ER homeostasis in infected cells [52]. Moreover, the HSV-1 ICP0 protein is used as a sensor to disarm the UPR in the early stages of infection [53]. Both HSV and PRV infections can inhibit ER stress through dephosphorylation of EIF2A [54,55]. HSV-1 ICP34.5 binds to PPM1A (protein phosphatase 1A) to counter PKR-mediated phosphorylation of EIF2A to activate cellular transcription for viral replication and virulence [56]. Our study showed that inhibition of USP14 activated ER stress to trigger autophagy and VP16 degradation, which might explain why alphaherpesviruses suppressed ER stress to facilitate viral replication. Moreover, sustained, unresolved ER stress causes the UPR to switch from an adaptive response to a pro-apoptotic response [57]. Nonetheless, we showed that inhibition of USP14 had no inhibitory effect on cell proliferation, which suggested that USP14 inhibitors had limited side effects on cytotoxicity. It would be interesting to determine the role of ER stress induced by different stimuli in alphaherpesvirus replication.
Studies have shown that USP14 is involved in ER stress and autophagy. USP14 directly binds to ERN1 to inhibit ER-associated degradation [58]. Similar phenomena indicate that ER stress-mediated ERN1 activation is part of mutant huntingtin protein toxicity and that this is counteracted by USP14 expression [59,60]. It is reported that inhibition of USP14 significantly impairs cellular autophagic flux, especially at the autophagosome-lysosome fusion step [61]. Lee et al. also indicate that USP14 inhibition delays the fusion of autophagosomes with the lysosome [62]. However, other studies have demonstrated that USP14 regulates autophagy by negatively controlling K63 ubiquitination of BECN1 and inhibition of USP14 promotes autophagy in M1-like macrophages [63,64]. Our data showed that inhibition of USP14 enhanced autophagic flux. In addition, the USP14 inhibitor IU1 influences replication of several flaviviruses, possibly because of enhanced proteasome activity [65]. Our study indicated that the proteasome inhibitor MG132 could not suppress VP16 degradation under b-AP15 treatment. The roles of USP14 in ER stress and autophagy were complicated in distinct context. The detailed mechanisms by which USP14 induced ER stress and autophagy needed further investigation in the future.
Materials and Methods
Mice
Experiments involving animals were approved by the Committee on the Ethics of Animal Care and Use of National Research Center for Veterinary Medicine. The study was conducted in accordance with the Guide for the Care and Use of Animals in Research of the People’s Republic of China.
Female 6- to 8-weeks old BALB/c mice were purchased from the Center of Experimental Animal of Zhengzhou University (Zhengzhou, China) and maintained in a specific pathogen free animal facility according to the guide for the care and use of laboratory animals and the related ethical regulations at Henan Agricultural University.
Cells and viruses
PK-15 (American Type Culture Collection, ATCC, CCL-33), 3D4/21 (ATCC, CRL-2843), HEK293T (ATCC, CRL-11268), and Vero (ATCC, CL-81) were cultured in DMEM (Gibco, 10,566–016) supplemented with 10% FBS (Gibco, 10,099,141 C), 100 units/ml penicillin, and 100 mg/ml streptomycin sulfate (Sangon, B540732). All cells were grown in monolayers at 37°C in 5% CO2. PRV-QXX (virulent strain) and PRV-GFP (recombinant strain) was used as previously described [22].
Chemical reagents
SJB3-019A (HY-80012), SJB2-043 (HY-15757), ML-323 (HY-17543), PR-619 (HY-13814), USP7-USP47 inhibitor (HY-13487), USP7-IN-1 (HY-16709), P22077 (HY-13865), P005091 (HY-15667), DUBs-IN-1 (HY-50736), DUBs-IN-2 (HY-50737A), DUBs-IN-3 (HY-50737), b-AP15 (HY-13989), degrasyn (HY-13264), MG-132 (HY-13259), bafilomycin A1 (HY-100558) and 3-MA (HY-19312) were from MedChemExpress; and GSK2606414 (S7307) was from Selleck.
Antibodies
The antibodies anti-LC3 (12,741), anti-LC3-II (3868), anti-SQSTM1/p62 (5114), anti-ATG5 (12,994), anti-ATG12 (4180), anti-BECN1/beclin-1 (3495), anti-p-EIF2AK3/PERK (3179) and anti-p-EIF2A/eIF2α (3398) were from Cell Signaling Technology; anti-USP14 (14,517-1-AP), anti-HSPA5/Bip (11,587-1-AP), anti-ATF6 (24,169-1-AP), anti-EIF2AK3/PERK (24,390-1-AP), anti-EIF2A/eIF2α (11,170-1-AP), anti-ATF4 (10,835-1-AP), anti-XBP1 (25,997-1-AP), anti-FOXO1 (18,592-1-AP) and anti-EGFP (50,430-2-AP) were from Proteintech; anti-FLAG (F1804) and anti-ACTB/β-actin (A1978) were from Sigma-Aldrich; anti-His6 (A00186-100), anti-GST (A00865-100), and anti-HA (A01244-100) were from Genscript; anti-UB (sc-8017) was from Santa Cruz Biotechnology; Alexa Fluor 488-conjugated goat anti-rabbit IgG (A-11034), Alexa Fluor 568-conjugated goat anti-mouse IgG (A-11004), and Alexa Fluor 633-conjugated goat anti-mouse IgG (A-21052) were from Thermo Fisher Scientific. Mouse monoclonal antibody against PRV VP16 was a gift from Jing-Fei Wang (Harbin Veterinary Research Institute, Harbin, China). Anti-PRV gB was used as previously described [66].
Plasmids and transfection
The coding sequence of USP14 was amplified from the cDNA of PK-15 cells and cloned into p3 × FLAG-CMV-14 (Sigma-Aldrich, E4901). The coding sequence of PRV VP16 was amplified from the genomic DNA of PRV-QXX, and cloned into p3 × FLAG-CMV-10 (Sigma-Aldrich, E4401) and pmCherry-C1 (Clontech, 632,524) [67]. FLAG-VP16K168R, FLAG-VP16K305R, USP14S432A-FLAG and SQSTM1ΔUBA-EGFP were generated by site-directed mutagenesis, according to the manufacturer’s instructions (Stratagene, 200,519). SQSTM1-EGFP was a gift from Liang Ge (School of Life Sciences, Tsinghua University, China) [68]. HA-UB, HA-UBK48 and HA-UBK63 were gifts from Bo Zhong (College of Life Sciences, Wuhan University, China) [14,69]. GFP-LC3 and GFP-RFP-LC3 were used as previously described [67]. All plasmids were transfected with Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer’s instructions.
Cell viability and proliferation assays
PK-15 and 3D4/21 cells were seeded at 1 × 104 per well in 96-well plates and cell viability and proliferation levels were determined using a CCK-8 cell counting assay (DingGuo, GK3607).
Flow cytometry assay
PK-15 cells were infected with PRV-GFP (multiplicity of infection [MOI] = 0.01) and treated with compounds at indicated concentrations for 36 h. Cells were digested with trypsin-EDTA (Gibco, 25,200,072), collected by centrifugation, and re-suspended in PBS (Solarbio, P1010). The percentage of GFP-positive cells was measured by flow cytometry on a Beckman CytoFLEX instrument. All data were analyzed using the CytExpert software.
Immunoblot analysis
Cells were collected in lysis buffer (50 mM Tris-HCl [Solarbio, T8060], pH 8.0, 150 mM NaCl [Solarbio, S8210], 1% Triton X-100 [Solarbio, T8200], 1% sodium deoxycholate [Solarbio, ST9540], 0.1% SDS [Solarbio, S8010], 2 mM MgCl2 [Solarbio, M8161]) supplemented with protease and phosphatase inhibitor cocktail (MedChemExpress, HY-K0010 and HY-K0022). The protein concentrations of the lysates were quantified with a BCA Protein Assay Kit (DingGuo, BCA01). Protein samples were separated by SDS-PAGE (Genscript, M00660) and transferred to membranes (Millipore, ISEQ00010), which were incubated in 5% nonfat milk (Sangon, A600669) for 1 h at room temperature. The membrane was incubated with the primary antibody overnight at 4°C and then incubated with horseradish-peroxidase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, 709–035-149 or 715–035-150) for 1 h at room temperature. Immunoblot results were visualized with luminata crescendo western HRP substrate (Millipore, WBLUR0500) on a GE AI600 imaging system.
Immunofluorescence analysis
Cells grown on coverslips (Thermo Fisher Scientific, 12–545-80) were fixed with 4% paraformaldehyde (Sigma-Aldrich, 158,127) for 30 min, permeabilized in 0.1% Triton X-100 (Solarbio, T8200), and incubated with PBS containing 10% FBS (10%FBS-PBS) with the primary antibody for 1 h at room temperature. The cells were washed three times with PBS and then labeled with 10%FBS-PBS containing the appropriate fluorescent secondary antibody for 1 h. The cells were finally washed in PBS and mounted in ProLong Diamond with DAPI (Invitrogen, P36971). Images were captured on a Zeiss LSM 800 confocal microscope and processed in ImageJ software for quantitative image analysis.
Co-IP assay
Cells were transfected with the indicated plasmids for 24 h, harvested and lysed in 1 ml of lysis buffer (PBS, 1% NP-40 [Solarbio, N8032], 5 mM EDTA [Solarbio, E8030], 5 mM EGTA [Solarbio, E8050]) and clarified by centrifugation at 16,000 g for 10 min at 4°C. Next, 900 μl aliquots were incubated with 40 μl of a 1:1 slurry of sepharose conjugated with either IgG (GE Healthcare, 17–0969-01) or anti-FLAG mouse mAb (Sigma-Aldrich, A2220) for 4 h at 4°C. The beads were washed four times with lysis buffer and were eluted with SDS sample buffer (Solarbio, P1015) by boiling for 10 min before immunoblot analysis.
Ubiquitination assay
Cells were harvested and lysed in 1 ml of IP buffer (PBS, 1% NP-40 [Solarbio, N8032], 1% sodium deoxycholate [Solarbio, ST9540], 5 mM EDTA [Solarbio, E8030], 5 mM EGTA [Solarbio, E8050]) and clarified by centrifugation at 16,000 g for 10 min at 4°C. Next, 900 μl aliquots were incubated with 40 μl of a 1:1 slurry of sepharose conjugated with either anti-VP16 mouse mAb or anti-FLAG mouse mAb (Sigma-Aldrich, A2220) for 4 h at 4°C. The beads were washed four times with lysis buffer and eluted with SDS sample buffer by boiling for 10 min before immunoblot analysis.
In vitro affinity isolation assay
GST-tagged recombinant proteins (pGEX-4 T-3; GE Healthcare, 28–9545-52) and His6-tagged recombinant proteins (pET-21b; Novagen, 69,741–3) were expressed in E. coli BL21 and purified under non-denaturing conditions by using glutathione-Sepharose 4 Fast Flow (GE Healthcare, 17,513,202) or Ni-NTA-agarose (Qiagen, 30,210) according to the manufacturer’s instructions. Non-ubiquitinated FLAG-VP16, FLAG-VP16K168R and FLAG-VP16K305R were expressed in HEK293T and ubiquitinated FLAG-VP16, FLAG-VP16K168R and FLAG-VP16K305R were expressed in b-AP15 treated HEK293T cells. FLAG-VP16 variants were affinity purified in IP buffer using sepharose conjugated with anti-FLAG mouse mAb (Sigma-Aldrich, A2220). The ubiquitination of FLAG-VP16 variants was analyzed by immonoblot analysis of UB.
Aliquots of FLAG beads (Sigma-Aldrich, A2220) containing 1 nM of the recombinant FLAG-tagged proteins were mixed with 1 nM of each GST or His6 fusion protein in 500 μl of binding buffer (PBS, pH 7.2, 1% Triton X-100 [Solarbio, T8200]) and incubated for 45 min on a rotating platform at 4°C. The beads were collected by centrifugation at 1000 g for 3 min. Supernatants were removed and the beads were washed five times with binding buffer. The proteins bound to beads were eluted with SDS sample buffer by boiling for 10 min before immunoblot analysis.
In vitro de-ubiquitination assay
Ubiquitinated FLAG–VP16 was purified from b-AP15-treated HEK293T cells by immunoprecipitation. The immunoprecipitates were eluted using the FLAG peptide (0.5 mg/ml). GST-USP14 (0–8 μg) and FLAG–VP16 (1 μg) were incubated in a buffer containing 50 mM HEPES (Solarbio, H8090), pH 8.0, 0.01% Brij-35 (Sigma-Aldrich, P1254), 3 mM DTT (Solarbio, D8220), and 1 μM ATP (Solarbio, A8270) and incubated at 37°C for 2 h. Samples were prepared for immunoblot analysis as aforementioned.
Iodixanol density gradient centrifugation
Cells were homogenized in lysis buffer (5 mM MOPS [Solarbio, M8150], pH 7.5, 0.25 M sucrose [Solarbio, S8271], 5 mM EDTA [Solarbio, E8030], 5 mM EGTA [Solarbio, E8050]). The cells were broken to release intracellular compartments, organelles and proteins by 25 passages through a 25 G1 needle (Shanghai Kindly Medical Instruments, 60,016,674). After centrifugation at 1,500 g for 5 min, the post-nuclear supernatant was collected, and then mixed with 2 ml PBS and loaded in the bottom, and then 2 ml of 50%, 30%, 25%, 15%, 10% and 5% OptiPrep (Sigma-Aldrich, D1556) solutions were loaded. The separation of intracellular organelles was performed by ultracentrifugation for 5 h in an MLS-50 rotor at 94,000 g. The fractions were collected and analyzed for the presence of the indicated proteins by immunoblot analysis.
qRT-PCR
Total RNA was isolated with TRIzol Reagent (TaKaRa, 9108) and subjected to cDNA synthesis with a PrimeScript™ RT reagent Kit (TaKaRa, RR047A). qRT-PCR was performed in triplicate using SYBR Premix Ex Taq (TaKaRa, RR820A) according to the manufacturer’s instructions, and data were normalized to the level of ACTB/β-actin expression in each individual sample. Melting curve analysis indicated formation of a single product in all cases. The 2−ΔΔCt method was used to calculate relative expression changes. PRV genome copy numbers were quantified as previously described [22]. Primers used for qRT-PCR are as follows: PRV IE180-Fw: 5′- CATCGTGCTGGACACCATCGAG-3′; PRV IE180-Rv: 5′- ACGTAGACGTGGTAGTCCCCCA-3′; PRV EP0-Fw: 5′- GGGTGTGAACTATATCGACACGTC-3′; PRV EP0-Rv: 5′- TCAGAGTCAGAGTGTGCCTCG-3′; PRV UL9-Fw: 5′-CAAGTTCAAGCACCTGTTCGA-3′; PRV UL9-Rv: 5′-TGAGGCTGTCGTTGACGC-3′; PRV gH-Fw: 5′-CTCGCCATCGTCAGCAA-3-3′; PRV gH-Rv: 5′-GCTGCTCCTCCATGTCCTT-3′; PRV gE-Fw: 5′- GGCATCGCCAACTTCTTCC-3′; PRV gE-Rv: 5′- CCTCGTCCACGTCGTCCTC-3′; Porcine-ACTB-Fw: 5′-CTGAACCCCAAAGCCAACCGT-3′; Porcine-ACTB-Rv: 5′-TTCTCCTTGATGTCCCGCACG-3′; Porcine-ATF4-Fw: 5′-CCCTTTACGTTCTTGCAAACTC-3′; Porcine-ATF4-Rv: 5′-GCTTCCTATCTCCTTCCGAGA-3′; Porcine-PPP1R15A-Fw: 5′-AAGAGCCTGGAGAGAGGAGAG-3′; Porcine-PPP1R15A-Rv: 5′-GTCCCCAGGTTTCCAAAAGCA-3′; Porcine-DDIT3-Fw: 5′-CTCAGG AGGAAGAGGAGGAAG-3′; Porcine-DDIT3-Rv: 5′-GCTAGCTGTGCCACTTTCCTT-3′; Porcine-XBP1(s)-Fw: 5′-GAGTCCGCAGCAGGTG-3′; Porcine-XBP1(s)-Rv: 5′-CCGTCAGAATCCATGGGG-3′; Porcine-XBP1(t)-Fw: 5′-TCCGCAGCACTCAGACTACGT-3′; Porcine-XBP1(t)-Rv: 5′-ATGCCCAAGAGGATATCAGACTC-3′; Porcine-DNAJB9-Fw: 5′-CAGAGAGATTGCAGAAGCATATGA-3′; Porcine-DNAJB9-Rv: 5′-GCTTCTTGGATCGAGTGTTTT-3′; Porcine-FOXO1-Fw: 5′-TTCACCAGGCACCATCAT-3′; Porcine-FOXO1-Rv: 5′-GGAGGAGAGTCGGAAGTAA-3′; Mouse-Actb-Fw: 5′- CCCCATTGAACATGGCATTG-3′; Mouse-Actb-Rv: 5′- ACGACCAGAGGCATACAGG-3′; Mouse-Atf4-Fw: 5′- ACAAGACAGCAGCCACTA-3′; Mouse-Atf4-Rv: 5′- CTTACGGACCTCTTCTATCAG-3′; Mouse-Ppp1r15a-Fw: 5′- CTCCAACTCTCCTTCTTCAG-3′; Mouse-Ppp1r15a-Rv: 5′-GTCCCCAGGTTTCCAAAAGCA-3′; Mouse-Ddit3-Fw: 5′-CTCAGGAGGAAGAGGAGGAAG-3′; Mouse-Ddit3-Rv: 5′-GCTAGCTGTGCCACTTTCCTT-3′; Mouse-Xbp1(s)-Fw: 5′- GAGTCCGCAGCAGGTG-3′; Mouse-Xbp1(s)-Rv: 5′- GTGTCAGAGTCCATGGGA-3′; Mouse-Xbp1(t)-Fw: 5′- TCCGCAGCACTCAGACTATGT-3′; Mouse-Xbp1(t)-Rv: 5′- AGCTTGGCTGATGAGGTC-3′; Mouse-Dnajb9-Fw: 5′- CAGGATGGTTCTAGTAGACAA-3′; Mouse-Dnajb9-Rv: 5′- CTCTTCGTTGAGTGACAGT-3′.
RNA interference
Cells were seeded in 60-mm dishes at a density of 4 × 105 cells per dish and were transfected with indicated siRNAs (GenePharma, Shanghai, China) at a final concentration of 0.12 nM. Transfections were performed with Lipofectamine RNAiMAX Reagent (Invitrogen, 13,778,500) according to the manufacturer’s instructions in Opti-MEM reduced serum medium (Gibco, 31,985,062). The medium was replaced with DMEM containing 10% FBS at 8 h post-transfection. The knockdown efficacy was assessed by immunoblot analysis at 48 h post-transfection. The siRNA sequences were as follows: siControl: 5′-UUCUCCGAACGUGUCACGU-3′; siEIF2A-1: 5′-GGAAUACAACAACAUCGAAGG-3′; siEIF2A-2: 5′-GCAGAUAUUGAAGTGGCUUGU-3′; siEIF2A-3: 5′-CCCAAAGUGGUUACAGAUAC-3′.
FRET
Plasmids encoding SQSTM1-EGFP and VP16-mCherry were co-transfected into PK-15 cells for 24 h. Then, FRET between SQSTM1-EGFP (donor) and VP16-mCherry (acceptor) was measured by confocal microscopy (Zeiss LSM 800). FRET measurements were also made using a donor dequenching approach in which EGFP fluorescence was measured before and after photobleaching of VP16-mCherry. The FRET efficiency was calculated using ZEN black software (Zeiss).
Transmission electron microscope
PK-15 cells were treated with DMSO or b-AP15 (1 μM) for 24 h. Cells were then fixed with 1% glutaraldehyde (Sigma-Aldrich, G5882) for 30 min, postfixed with 1% osmium tetroxide (Sigma-Aldrich, 209,104) for 1 h, washed with PBS, dehydrated, and embedded in Epoxy embedding medium (Sigma-Aldrich, 45,345). Ultrathin sections were cut and post-stained with aqueous uranyl acetate (Electron Microscopy China, GZ02625) and lead citrate (Electron Microscopy China, GZ02618). Images were taken on an FEI Tecnai G2 Spirit Transmission Electron Microscope operated at 120 kV.
Generation of gene knockout cell lines using CRISPR-Cas9
sgRNAs targeting porcine USP14, ATG5 and BECN1 were synthesized and cloned into the lentiCRISPR v2 vector (Addgene, 52,961; deposited by Feng Zhang from Broad Institute of MIT and Harvard). On day 0, HEK293T cells were seeded in 10-cm dishes at 4 × 106 per dish. On day 1, the cells were transfected with 2 μg/dish lentiCRISPR v2 (Addgene, 52,961; deposited by Feng Zhang from Broad Institute of MIT and Harvard) with indicated sgRNAs, 1.5 μg/dish psPAX2 (Addgene, 12,260; deposited by Didier Trono from École Polytechnique Fédérale de Lausanne), and 0.5 μg/dish pMD2.G (Addgene, 12,259; deposited by Didier Trono from École Polytechnique Fédérale de Lausanne) using Lipofectamine 3000 (Invitrogen, L3000015). On day 3, the culture media containing the viruses were collected and were used for infection of PK-15 cells followed by selection in culture medium containing puromycin (4 μg/ml; Solarbio, P8230) for another 7 days. Single clonal knockout cells were obtained by serial dilution and verified by Sanger sequencing and immunoblot analysis. sgRNAs are as follows: sgUSP14: 5′-CACCGGGGGAAAGGAGAAATTTGA-3′; sgATG5: 5′-CACCGAGAAGACATTAGTGAGATATGG-3′; sgBECN1: 5′-CACCGACATCCAGCAGCACCATGCAGG-3′.
Generation of rPRV ∆UL48 using CRISPR-Cas9 through homology-directed recombination
sgRNA targeting PRV UL48 (5′-CACCGCTGCACCTGTACGTGGCCA-3′) was synthesized and cloned into the lentiCRISPR v2 vector (Addgene, 52,961; deposited by Feng Zhang from Broad Institute of MIT and Harvard). Lentivirus produced according to the aforementioned procedure was used to infect HEK293 cells. At 24 h post infection, cells were transfected with PRV-QXX genomic DNA, a donor plasmid containing the expression cassette of GFP [70], and FLAG-VP16. The recombinant virus was further purified by endpoint dilution in FLAG-VP16-transfected PK-15 cells.
TCID50 assay
On day 0, vero cells were seeded in a 96-well plate at 104 per well. On day 1, the cells were inoculated with serially diluted viruses (10−1–10−12 fold) for 1 h at 37°C. The excess virus inoculum was removed by washing with PBS. Then, 200 μl maintenance medium (DMEM with 2% FBS) was added to each well and the cells were cultured for 3–5 days. The cells demonstrating the expected cytopathic effect were observed daily and the TCID50 value was calculated using the Reed–Muench method.
Plaque assay
Vero cells were cultured to confluency in six-well plates and inoculated with serially diluted viruses (10−1–10−7 fold) for 1 h at 37°C. The excess viral inoculum was removed by washing with PBS. Next, 4 ml of DMEM-1%methylcellulose (Solarbio, M8070) was added to each well, and the cells were further cultured for 4–5 days. The cells were fixed with 4% paraformaldehyde (Sigma-Aldrich, 158,127) for 15 min and stained with 1% crystal violet (Sigma-Aldrich, C0775) for 30 min before the plaques were counted.
Histological analysis
The lung tissues dissected from mice were fixed in 4% paraformaldehyde (Sigma-Aldrich, 158,127) overnight, embedded in paraffin (Solarbio, YA0012), sectioned, and stained with hematoxylin (Sigma-Aldrich, MHS1) and eosin (Sigma-Aldrich, E4009) solution.
Statistical analysis
All data were analyzed in Prism 7 software (GraphPad Software) using a two-tailed Student’s t-test. P < 0.05 was considered statistically significant. For mouse survival studies, Kaplan-Meier survival curves were generated and analyzed for statistical significance.
Supplementary Material
Acknowledgments
We thank Jing-Fei Wang (Harbin Veterinary Research Institute, Harbin, China) for providing mouse monoclonal antibody against PRV VP16; Liang Ge (School of Life Sciences, Tsinghua University, China) for providing the SQSTM1-EGFP plasmid; and Bo Zhong (College of Life Sciences, Wuhan University, China) for providing the plasmids for HA-UB, HA-UBK48, and HA-UBK63.
Funding Statement
This work was supported by grants from National Natural Science Foundation of China (32072858), the Natural Science Foundation of Henan (202300410213), and Outstanding Talents of Henan Agricultural University (30600773).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Bharucha T, Houlihan CF, Breuer J, et al. Herpesvirus infections of the central nervous system. Semin Neurol. 2019;39(3):369–382. [DOI] [PubMed] [Google Scholar]
- [2].Wu Y, Cheng A, Wang M, et al. Comparative genomic analysis of duck enteritis virus strains. J Virol. 2012;86(24):13841–13842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Looker KJ, Magaret AS, May MT, et al. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One. 2015;10:e0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Koshizuka T, Suzutani T.. [Anti alpha-herpesvirus drugs]. Nihon Rinsho. 2012;70:558–563. [PubMed] [Google Scholar]
- [5].Muller T, Hahn EC, Tottewitz F, et al. Pseudorabies virus in wild swine: a global perspective. Arch Virol. 2011;156(10):1691–1705. [DOI] [PubMed] [Google Scholar]
- [6].Fuchs W, Granzow H, Klupp BG, et al. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J Virol. 2002;76(13):6729–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakamura N. Ubiquitin System. Int J Mol Sci. 2018;19(4):1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farshi P, Deshmukh RR, Nwankwo JO, et al. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat. 2015;25:1191–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhu HH, Zhao XB, Hu WW, et al. [Research progress on ubiquitin-specific protease in antiviral immunity]. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2015;44:578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yao J, Li C, Shi L, et al. Zebrafish ubiquitin-specific peptidase 5 (USP5) activates interferon resistance to the virus by increase the expression of RIG-I. Gene. 2020;751:144761. [DOI] [PubMed] [Google Scholar]
- [12].Sun H, Zhang Q, Jing YY, et al. USP13 negatively regulates antiviral responses by deubiquitinating STING. Nat Commun. 2017;8:15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kusakabe S, Suzuki T, and Sugiyama Y, et al. USP15 participates in hepatitis C virus propagation through regulation of viral RNA translation and lipid droplet formation. J Virol. 2019;93(6):e01708–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang Q, Tang Z, An R, et al. USP29 maintains the stability of cGAS and promotes cellular antiviral responses and autoimmunity. Cell Res. 2020;30(10):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- [16].Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13(11):681–696. [DOI] [PubMed] [Google Scholar]
- [17].Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. [DOI] [PubMed] [Google Scholar]
- [18].Margariti A, Li H, Chen T, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Adachi Y, Yamamoto K, Okada T, et al. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33(1):75–89. [DOI] [PubMed] [Google Scholar]
- [20].Rashid HO, Yadav RK, Kim HR, et al. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Davis ME, Gack MU. Ubiquitination in the antiviral immune response. Virology. 2015;479-480:52–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang J, Li GL, Ming SL, et al. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 2020;16:e1008429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].He W, Auclert LZ, Zhai X, et al. Interspecies transmission, genetic diversity, and evolutionary dynamics of pseudorabies virus. J Infect Dis. 2019;219(11):1705–1715. [DOI] [PubMed] [Google Scholar]
- [24].Tian Z, D’Arcy P, Wang X, et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood. 2014;123:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu D, Shan B, Lee BH, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife. 2015;4:e10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herr W. The herpes simplex virus VP16-induced complex: mechanisms of combinatorial transcriptional regulation. Cold Spring Harb Symp Quant Biol. 1998;63:599–607. [DOI] [PubMed] [Google Scholar]
- [27].Wang D, Ma H, and Zhao Y, et al. Ubiquitin-specific protease 14 is a new therapeutic target for the treatment of diseases. J Cell Physiol. 2020;236(5): 3396–3405 . [DOI] [PubMed] [Google Scholar]
- [28].Zhang Y, Chen A, Wang M, et al. [Research advances in VP16 of the herpes virus]. Bing Du Xue Bao. 2016;32(6):817–824. [PubMed] [Google Scholar]
- [29].Lamark T, Svenning S, Johansen T, et al. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–624. [DOI] [PubMed] [Google Scholar]
- [30].Kimura S, Noda T, Yoshimori T, et al. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [31].Yamamoto A, Tagawa Y, Yoshimori T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. [DOI] [PubMed] [Google Scholar]
- [32].Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. [DOI] [PubMed] [Google Scholar]
- [33].Lee WS, Yoo WH, Chae HJ, et al. ER stress and autophagy. Curr Mol Med. 2015;15(8):735–745. [DOI] [PubMed] [Google Scholar]
- [34].Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–181. [DOI] [PubMed] [Google Scholar]
- [35].Kishino A, Hayashi K, Hidai C, et al. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci Rep. 2017;7(1):4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen O, Manig F, Lehmann L, et al. Dual role of ER stress in response to metabolic co-targeting and radiosensitivity in head and neck cancer cells. Cell Mol Life Sci. 2020;78(6):3021–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Calfon M, Zeng H, Urano F, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415(6867):92–96. [DOI] [PubMed] [Google Scholar]
- [38].Axten JM, Medina JR, Feng Y, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012;55:7193–7207. [DOI] [PubMed] [Google Scholar]
- [39].Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- [40].Hall DB, Struhl K. The VP16 activation domain interacts with multiple transcriptional components as determined by protein-protein cross-linking in vivo. J Biol Chem. 2002;277:46043–46050. [DOI] [PubMed] [Google Scholar]
- [41].Li Y, Wang M, Cheng A, et al. Duck enteritis virus VP16 antagonizes IFN-beta-mediated antiviral innate immunity. J Immunol Res. 2020;2020:9630452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xing J, Ni L, Wang S, et al. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol. 2013;87:9788–9801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Smibert CA, Popova B, Xiao P, et al. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J Virol. 1994;68(4):2339–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Knez J, Bilan PT, Capone JP, et al. A single amino acid substitution in herpes simplex virus type 1 VP16 inhibits binding to the virion host shutoff protein and is incompatible with virus growth. J Virol. 2003;77(5):2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mostowy S, Sancho-Shimizu V, Hamon MA, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286(30):26987–26995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Orvedahl A, Sumpter R Jr., Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sun M, Hou L, Tang YD, et al. Pseudorabies virus infection inhibits autophagy in permissive cells in vitro. Sci Rep. 2017;7:39964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Graybill C, Morgan MJ, Levin MJ, et al. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology. 2018;522:220–227. [DOI] [PubMed] [Google Scholar]
- [49].Rubio RM, Mohr I. Inhibition of ULK1 and Beclin1 by an alpha-herpesvirus Akt-like Ser/Thr kinase limits autophagy to stimulate virus replication. Proc Natl Acad Sci U S A. 2019;116(52):26941–26950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1(1):23–35. [DOI] [PubMed] [Google Scholar]
- [51].Zhang P, Su C, and Jiang Z, et al. Herpes simplex virus 1 UL41 protein suppresses the IRE1/XBP1 signal pathway of the unfolded protein response via its RNase activity. J Virol. 2017;91(4):e02056–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Mulvey M, Arias C, Mohr I, et al. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol. 2007;81(7):3377–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Burnett HF, Audas TE, Liang G, et al. Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress Chaperones. 2012;17(4):473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cheng G, Feng Z, He B, et al. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J Virol. 2005;79:1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xu S, Chen D, Chen D, et al. Pseudorabies virus infection inhibits stress granules formation via dephosphorylating eIF2alpha. Vet Microbiol. 2020;247:108786. [DOI] [PubMed] [Google Scholar]
- [56].He B, Gross M, Roizman B, et al. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1997;94:843–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. [DOI] [PubMed] [Google Scholar]
- [58].Nagai A, Kadowaki H, Maruyama T, et al. USP14 inhibits ER-associated degradation via interaction with IRE1alpha. Biochem Biophys Res Commun. 2009;379:995–1000. [DOI] [PubMed] [Google Scholar]
- [59].Hyrskyluoto A, Bruelle C, Lundh SH, et al. Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: involvement of the proteasome and ER stress-activated kinase IRE1alpha. Hum Mol Genet. 2014;23(22):5928–5939. [DOI] [PubMed] [Google Scholar]
- [60].Qu J, Zou T, and Lin Z. The roles of the ubiquitin-proteasome system in the endoplasmic reticulum stress pathway. Int J Mol Sci. 2021;22(4):1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kim E, Park S, Lee JH, et al. Dual function of USP14 deubiquitinase in cellular proteasomal activity and autophagic flux. Cell Rep. 2018;24(3):732–743. [DOI] [PubMed] [Google Scholar]
- [62].Lee JH, Park S, Kim E, et al. Negative-feedback coordination between proteasomal activity and autophagic flux. Autophagy. 2019;15(4):726–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Xu F, Ma Y, Huang W, et al. Typically inhibiting USP14 promotes autophagy in M1-like macrophages and alleviates CLP-induced sepsis. Cell Death Dis. 2020;11(8):666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Xu D, Shan B, Sun H, et al. USP14 regulates autophagy by suppressing K63 ubiquitination of Beclin 1. Genes Dev. 2016;30(15):1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nag DK, Finley D. A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits Dengue virus replication. Virus Res. 2012;165(1):103–106. [DOI] [PubMed] [Google Scholar]
- [66].Wang J, Wang CF, Ming SL, et al. Porcine IFITM1 is a host restriction factor that inhibits pseudorabies virus infection. Int J Biol Macromol. 2020;151:1181–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang J, Liu JY, and Shao KY, et al. Porcine reproductive and respiratory syndrome virus activates lipophagy to facilitate viral replication through downregulation of NDRG1 expression. J Virol. 2019;93(17):e00526–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192(1):17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Cai Z, Zhang MX, Tang Z, et al. USP22 promotes IRF3 nuclear translocation and antiviral responses by deubiquitinating the importin protein KPNA2. J Exp Med. 2020;217(5):e20191174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Fu PF, Cheng X, Su BQ, et al. CRISPR/Cas9-based generation of a recombinant double-reporter pseudorabies virus and its characterization in vitro and in vivo. Vet Res. 2021;52:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.