

Abstract
A fundamental component of cellular radioresponse is the translational control of gene expression. As a critical regulator of translational control is the eIF4F cap binding complex, we investigated whether eIF4A, the RNA helicase component of eIF4F, can serve as a target for radiosensitization. Knockdown of eIF4A using siRNA reduced translational efficiency, as determined from polysome profiles, and enhanced tumor cell radiosensitivity as determined by clonogenic survival. The increased radiosensitivity was accompanied by a delayed dispersion of radiation-induced γH2AX foci, suggestive of an inhibition of DNA double strand break repair. Studies were then extended to (−)-SDS-1-021, a pharmacological inhibitor of eIF4A. Treatment of cells with the rocaglate (−)-SDS-1-021 resulted in a decrease in translational efficiency as well as protein synthesis. (−)-SDS-1-021 treatment also enhanced the radiosensitivity of tumor cell lines. This (−)-SDS-1-021-induced radiosensitization was accompanied by a delay in radiation-induced γH2AX foci dispersal, consistent with a causative role for the inhibition of double strand break repair. In contrast, while (−)-SDS-1-021 inhibited translation and protein synthesis in a normal fibroblast cell line, it had no effect on radiosensitivity of normal cells. Subcutaneous xenografts were then used to evaluate the in vivo response to (−)-SDS-1-021 and radiation. Treatment of mice bearing subcutaneous xenografts with (−)-SDS-1-021 decreased tumor translational efficiency as determined by polysome profiles. While (−)-SDS-1-021 treatment alone had no effect on tumor growth, it significantly enhanced the radiation-induced growth delay. These results suggest that eIF4A is a tumor selective target for radiosensitization.
Keywords: translation initiation, eIF4A, radiosensitization
Introduction
Radiation therapy is a mainstay of cancer treatment, with approximately two-thirds of patients with solid tumors receiving radiation at some point during their treatment (1). Towards improving the efficacy of radiotherapy, one approach is the development of molecularly targeted radiation sensitizers, which requires elucidation of the mechanisms governing cellular radioresponse. The best understood processes regulating radiosensitivity involve the post-translational modification of constitutively expressed proteins. However, new gene expression also contributes to radioresponse (2,3). More specifically, radiation selectively regulates mRNA translation, a process that functions independently from transcription (2). In contrast to the radiation-induced transcriptome, the radiation-induced translatome correlates with protein expression and consists of genes in common among cell lines from the same tissue of origin (4,5). The establishment of translational control as a component of cellular radioresponse suggests that the translational machinery may provide a source of targets for radiosensitization.
Translational control primarily occurs at the level of initiation. In cap-dependent translation, the chief mechanism of translation in eukaryotic cells, the eukaryotic translation initiation factor 4F (eIF4F) complex interacts with the 5’ 7-methyl guanosine mRNA cap to recruit ribosomes to transcripts. eIF4F consists of three components: eIF4E, a cap binding protein; eIF4A, an RNA helicase; and eIF4G, a scaffold protein (6). Using siRNA, eIF4E was shown to participate in the radiation-induced translational control of gene expression and to be a tumor-selective target for radiosensitization (7). The cap binding function of eIF4E is regulated by mechanistic target of rapamycin kinase (mTOR), which phosphorylates eIF4E binding proteins (4E-BPs). As a clinically relevant approach to reducing eIF4E availability for eIF4F complex formation, competitive mTOR inhibitors were shown to inhibit radiation-induced translational control and to enhance the radiosensitivity of tumor cells grown in vitro and in vivo (8–10).
An alternative approach to targeting eIF4F function is to inhibit the helicase activity of eIF4A, the enzymatic component of the eIF4F complex. eIF4A mediates the unwinding of 5’ untranslated region (UTR) secondary structure, which is a key determinant of an mRNA’s translational efficiency (11). Translation of mRNAs with long, structured 5’ UTRs, which often encode for survival, proliferation, and tumor promoting proteins (12), are not only preferentially dependent on eIF4E availability but also critically dependent on eIF4A activity (13,14). Recent data suggest that eIF4A regulates the translation of a subset of genes specifically involved in oncogenesis (15–17). Along these lines, overexpression of eIF4A1, the predominant eIF4A isoform (18), has been shown to accelerate cancer development in mouse models of leukemia (15), and eIF4A1 overexpression has been detected clinically in a number of different types of solid tumors (17,19–22). Given eIF4A’s role as a critical component of the eIF4F complex and the previous results implicating eIF4E as a determinant of tumor cell radiosensitivity, we investigated the role of eIF4A in cellular radioresponse. The data shown here indicate that loss of eIF4A activity selectively enhances tumor cell radiosensitivity through the inhibition of double strand break (DSB) repair. Moreover, inhibition of eIF4A with the rocaglate translation inhibitor (−)-SDS-1-021 enhanced the radiosensitivity of human tumor cells grown in vitro and as subcutaneous tumor xenografts.
Materials and Methods
Cell lines and treatments.
U251 (glioblastoma) cells were obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository (DCTD, National Cancer Institute). PSN1 (pancreatic adenocarcinoma), HeLa (cervical carcinoma), and MRC9 (normal lung fibroblasts) cells were obtained from American Type Culture Collection (ATCC). DCTD and ATCC employ short tandem repeat DNA fingerprinting, karyotyping, and cytochrome C oxidase I testing to authenticate cell lines. All cell lines were cultured for less than 2 months after thawing frozen stocks. U251 and HeLa were cultured in DMEM supplemented with 10% FBS (Invitrogen). PSN1 was cultured in RPMI-1640 supplemented with 10% FBS. MRC9 was cultured in MEM supplemented with 10% FBS, sodium pyruvate, L-glutamine, and nonessential amino acids (all from Invitrogen). All cell lines were maintained at 37 °C in an atmosphere of 5%CO2/95% air. (−)-SDS-1-021 (23) was dissolved in dimethyl sulfoxide (DMSO) prior to use (see Supplementary Methods). Cells were irradiated using a 320 kV X-ray source (Precision X-ray, Inc.) with a 2.0 mm aluminum filtration (300 kV peak, 10 mA) at a dose rate of 2Gy/minute. Control cultures were mock irradiated.
siRNA transfection.
U251 and HeLa cells at 60–80% confluency were transfected with either a pool of four non-targeting siRNAs or four targeted siRNAs (Dharmacon) with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. All experiments were performed at 72 hours post-transfection.
Immunoblot analysis.
Cells were washed with PBS, pelleted by centrifugation, and incubated in lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.5; 2 mM EGTA, 2 mM EDTA, 0.3% Tween 20, 0.2% Triton X-100, 25 mM NaF, 25 mM β-glycerophosphate, 0.5 mM NaVO4, 1X HALT protease inhibitor cocktail [Thermo Scientific], 1X phosphatase inhibitor cocktail 2 [Sigma Aldrich], and 1X phosphatase inhibitor cocktail 3 [Sigma Aldrich]) on ice for 10 minutes. The resulting lysates were cleared by centrifugation at 12,000 rpm for five minutes at 4 °C. The protein concentration of the cleared lysates was determined by BCA protein assay (Thermo Scientific). Equal amounts of protein were separated on 4–20% SDS-PAGE gels (Bio-Rad), transferred to nitrocellulose membranes, and probed with primary antibodies. Antibodies used: BLM (Cell Signaling), eIF4A1 (Abcam), GAPDH (Thermo Scientific), Rad51C (GeneTex), RPA1 (Cell Signaling), β-tubulin (Cell Signaling), and XRCC1 (Cell Signaling). Proteins were visualized by IRDye secondary antibodies (LI-COR) and imaged on an Odyssey CLx (LI-COR). Bands were quantified using Image Studio software (LI-COR).
Polysome profiling.
In vitro polysome profiling was performed as previously described (3). For in vivo polysome profiling, U251 subcutaneous xenograft tumors (~150 mm3) were removed and immediately snap-frozen in liquid nitrogen. Subsequently, ~50 mm3 tumor pieces were homogenized in in vivo polysome extraction buffer (300 mM NaCl, 15 mM Tris-HCl, pH 7.5; 15 mM MgCl2, 500 U/mL recombinant RNasin [Promega], 1 mg/mL heparin, and 0.1 mg/mL cycloheximide) with a Polytron PT 2100. The homogenates were supplemented with Triton X-100 and sodium deoxycholate to a final concentration of 1% for each, incubated on ice for 10 min followed by clearing by centrifugation. Using the resulting supernatants for both in vitro and in vivo samples, polysome profiles were obtained as previously described (3). Translational efficiency (TE) was calculated by dividing the area under the curve of the polysome portion of the profile by area under the curve of the polysome and non-polysome portions of the profile (24).
Clonogenic survival.
To evaluate radiosensitivity, cells were plated in 6-well plates at clonal density and irradiated 16 hours later. Ten to 14 days after irradiation, colonies were stained with 0.5% crystal violet in methanol, the number of colonies was determined, and the surviving fractions were calculated. Radiation survival curves were generated after normalizing to the cytotoxicity induced by (−)-SDS-1-021 or eIF4A1 knockdown.
γH2AX foci formation.
γH2AX foci were analyzed as previously described (3) using antibodies against phosphorylated H2AX (Millipore), followed by anti-mouse Alexa Fluor 488 (Invitrogen). Slides were imaged on a Zeiss Axio Imager 2 with a 63✕ oil immersion lens.
Protein synthesis.
The effects of (−)-SDS-1-021 on protein synthesis were determined using the Click-iT Plus OPP Alexa Fluor 488 Protein Synthesis Assay Kit (Thermo Scientific), according to the manufacturer’s protocol and as described (25).
RNA-seq.
Polysome-bound RNA was collected from three sucrose gradients per treatment group and isolated using TRIzol LS (Invitrogen), according to the manufacturer’s protocol. RNA was further purified with two rounds of lithium chloride precipitation and on-column DNase digestion using the QIAGEN RNeasy kit with RNase-free DNase. RNA-seq was performed on the purified RNA by the Center for Cancer Research Sequencing Facility in Frederick, Maryland. Briefly, 200 ng of RNA was used as input for mRNA capture with oligo-dT coated magnetic beads. The mRNA was fragmented, followed by random-primed cDNA synthesis. The resulting double-stranded cDNA was used as the input to a standard Illumina library prep with end-repair, adapter ligation and PCR amplification to generate a sequencing ready library. The final library was then quantitated by qPCR before cluster generation and sequencing on the Illumina HiSeq4000 sequencer. All RNAseq NGS data processing occurred using the RNAseq pipelines implemented in the CCBR Pipeliner framework (https://github.com/CCBR/Pipeliner). For RNAseq gene expression analysis, contaminating adapter sequences were removed using Trimmomatic v0.36 (26) and reads were mapped to the hg38 human reference genome using STAR v2.5.3 (27) run in 2-pass mode. RSEM (28) was then used for gene-level expression quantification, and data processing was performed in R statistical program environment. Data was normalized by trimmed mean of M-values (TMM) implemented in edgeR software (29). Differential gene expression was calculated in edgeR using the GLM approach and likelihood ratio tests. Sequence data has been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO accession number GSE200162 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200162). Genes with an adjusted p-value < 0.05 were submitted for Ingenuity Pathway Analysis (IPA) (QIAGEN). Genes were also ssssranked by (sign of the fold change)*-log10(p-value) and submitted to GSEA (30,31) for pre-ranked analysis with 1000 permutations against the hallmarks gene set database.
Homologous recombination (HR) repair.
The Homologous Recombination Assay Kit (Norgen Biotek Corp.) was used to measure HR repair. Cells plated in a 24-well plate were transfected with 0.5 μg each of the dl-1 and dl-2 plasmids using Lipofectamine 3000 (Invitrogen). Five hours post-transfection, cells were treated with either vehicle or 10 nM (−)-SDS-1-021; 24 hours later DNA was isolated. The HR product and the plasmid backbone were detected by qPCR (Luna Universal qPCR Master Mix, New England Biolabs). The amount of the HR product was normalized to the amount of the plasmid backbone and fold change calculated using the ΔΔCt method.
Tumor growth delay.
A single cell suspension of 2×106 U251 cells was implanted subcutaneously in the right hind leg of seven- to eight-week-old athymic female nude mice (NCr nu/nu; NCI Animal Production Program). On day 13 post-implantation, mice were randomized into the following four treatment groups based on tumor volume, with each group having an average volume of approximately 180 mm3: vehicle control (5.2% Tween 80/5.2% PEG400 in sterile water), drug treated (0.35 mg/kg (−)-SDS-1-021), radiation (1.5Gy/day), and combination of drug and radiation. Each treatment group contained eight mice. Radiation was delivered locally for five consecutive days to animals restrained in a custom designed lead jig using an X-Rad 320 X-irradiator (Precision X-Rays, Inc.) with a 2.0 mm aluminum filtration (320 kV, 12.5 mA, 3.39 Gy/min dose rate). (−)-SDS-1-021 was administered one hour prior to radiation on days one and four. Perpendicular tumor measurements were taken twice a week with a digital caliper, and tumor volumes were calculated using the formula (length x width2)/2. GraphPad Prism 8 was used to determine the % mice with tumors less than 1000 mm3 as a function time after treatment with log-rank tests for significance. Fractional tumor volume (FTV) was calculated as the mean experimental tumor volume/mean control tumor volume. The expected FTV was calculated as FTV of drug treatment × FTV of radiation treated. A synergistic effect is suggested when the ratio of expected FTV/observed FTV is >1 (32). All animal studies were conducted in accordance with the principles and procedures outlined in the NIH Guide for Care and Use of Animals and approved by the Institutional Animal Care and Use Committee (IACUC).
Data Availability
Data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE200162.
Results
To determine whether eIF4A1 plays a role in tumor cell radiosensitivity, U251 and HeLa cells were treated with an siRNA pool specific to eIF4A1 (sieIF4A1) or a non-targeting siRNA (siNT) pool; 72 hours after transfection cells were subjected to analyses. As shown in Fig. 1A, this siRNA treatment of U251 and HeLa cells resulted in 79% and 60% knockdown of eIF4A1, respectively, as compared to cells transfected with siNT. Polysome profiling in eIF4A1 knockdown cells indicated an increase in the 40S and 60S peaks with a concomitant decrease in the polysome peaks (Fig. 1B), consistent with inhibition of translation initiation. Knockdown of eIF4A1 in both U251 and HeLa cells resulted in a 30–40% decrease in translational efficiency (TE) (24).
Figure 1.
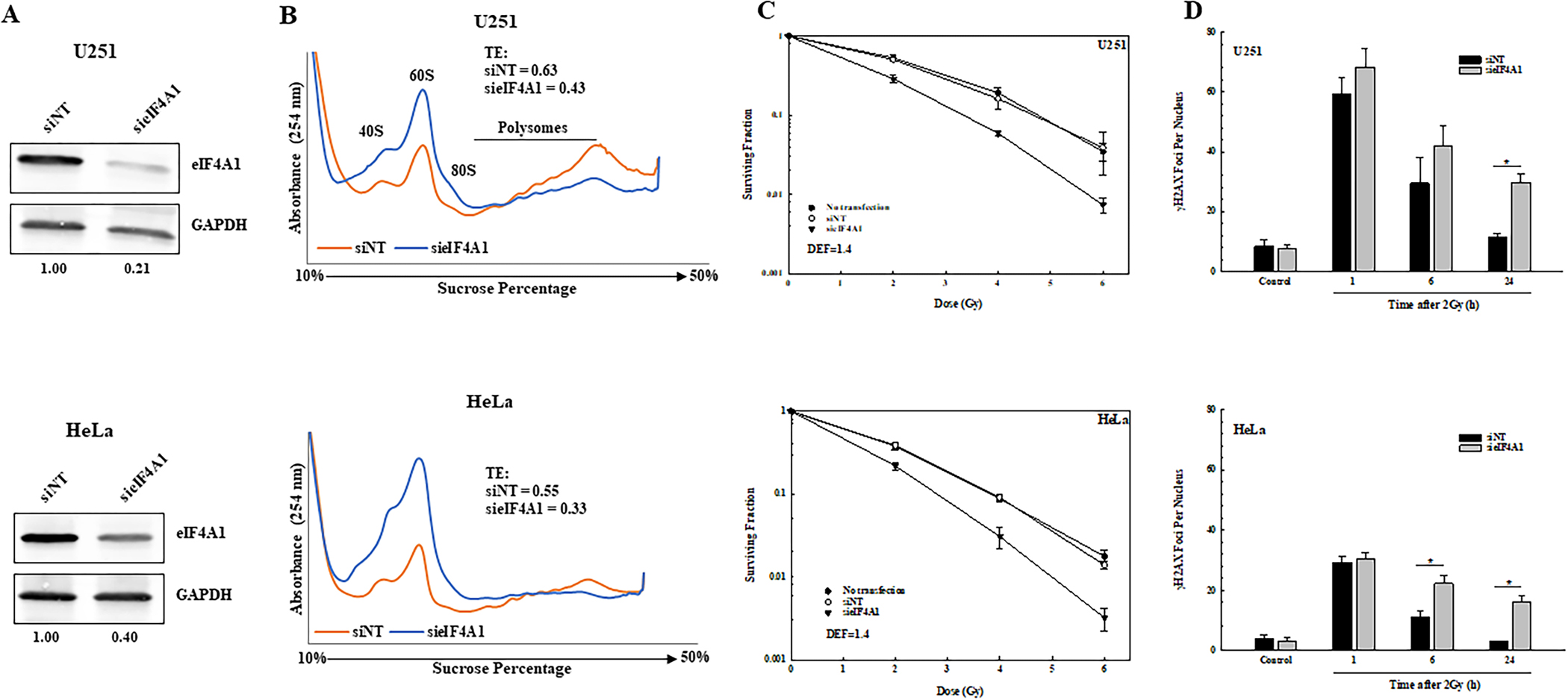
Effect of eIF4A1 knockdown on tumor cell translational efficiency, radiosensitivity, and radiation induced γH2AX foci. A. Tumor cell lines were transfected with non-targeting siRNA (siNT) or siRNA against eIF4A1 (sieIF4A1). 72 hours post-transfection, cell lysates were immunoblotted for eIF4A1 with GAPDH as a loading control. Normalized ratios of eIF4A1 to GAPDH are shown below the blots. B. Polysome profiling was performed on eIF4A1 knockdown U251 and HeLa cells 72 hours post-transfection. Translational efficiencies (TEs) calculated from the polysome profiles are shown. C. Clonogenic survival analysis of U251 and HeLa cells. 72 hours post-transfection, cells were plated at clonal density and colonies determined after 12–14 days. DEFs were calculated at a surviving fraction of 0.1. Values represent the mean ± SEM for 3 independent experiments. D. γH2AX foci analysis of U251 and HeLa cells. siNT and sieIF4A1 cells were plated 72 hours post-transfection, irradiated (2Gy) the next day, and collected at the indicated time points for foci analysis. γH2AX foci were counted in 50 cells per treatment group. Values represent the mean ± SEM for 3 independent experiments. * p<0.05 by Student’s t-test.
The effects of eIF4A1 knockdown on U251 and HeLa radiosensitivity are shown in Fig. 1C. For this study, cells were treated as described above, trypsinized, and irradiated 16 hours after seeding at clonal density. Transfection with siRNA to eIF4A1 resulted in an increase in the radiosensitivity of both tumor cell lines. The dose enhancement factors (DEF) at a surviving fraction of 0.1 for U251 and HeLa were both 1.4. Knockdown of eIF4A1 alone reduced the surviving fraction of U251 and HeLa cells to 0.37 ± 0.01 and 0.49 ± 0.13, respectively. These data suggest that eIF4A1 contributes to the survival of tumor cells after irradiation.
Because γH2AX foci correspond to radiation-induced DSBs, and their dispersal correlates with DSB repair, the effects of eIF4A1 knockdown on radiation-induced γH2AX foci were evaluated in U251 and HeLa cells (Fig. 1D). For both cell lines, no difference in foci levels was detected between control cells (non-targeting siRNA) and eIF4A1 knockdown cells at 1 hour after exposure to 2Gy, suggesting that eIF4A1 levels have no effect on the initial level of radiation-induced DSBs. However, at 24 hours after irradiation of U251 and HeLa cells, the number of γH2AX foci remaining in the eIF4A1 knockdown cells was significantly greater than in control cells. These data suggest that eIF4A1 knockdown results in an inhibition of radiation-induced DSB repair.
To extend this study to a therapeutic setting, the rocaglate hydroxamate (−)-SDS-1-021 was used to inhibit eIF4A activity (33,34). For these experiments, the pancreatic carcinoma cell line PSN1 was added, which results in a panel of three tumor cell lines corresponding to tumor types typically treated with radiotherapy. Initially, the effects of (−)-SDS-1-021 on overall translation were defined using polysome profiles. Cells were treated with 10 or 25 nM (−)-SDS-1-021 for 1 h and polysome profiles were generated (Fig. 2A). While exposure to each dose of (−)-SDS-1-021 reduced TE in all 3 cell lines, PSN1 exhibited the least translational suppression at 10 nM. Of note, treatment with (−)-SDS-1-021 for 1 h resulted in approximately the same reduction in translational efficiency as eIF4A1 knockdown in U251 and HeLa (Figs. 1B and 2A).
Figure 2.
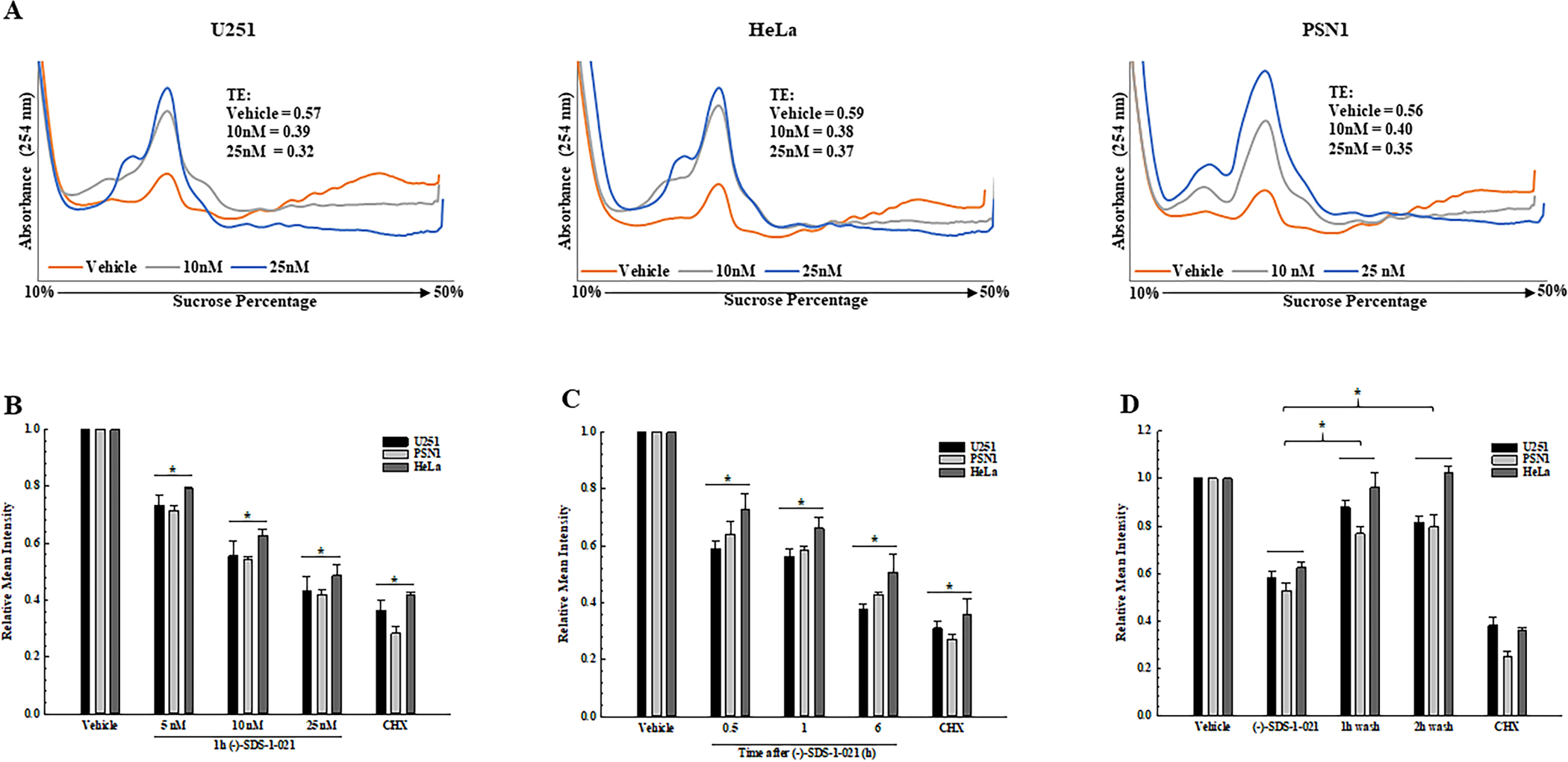
Translational efficiency and protein synthesis in tumor cells treated with (−)-SDS-1-021. A. Polysome profiles were generated from U251, HeLa, and PSN1 cell lines at 1 h after exposure to 10 or 25 nM (−)-SDS-1-021. TEs calculated from the polysome profiles are shown (single experiment). B. Protein synthesis measured as a function of (−)-SDS-1-021 dose (1 h). C. Protein synthesis (OPP incorporation) measured as a function of (−)-SDS-1-021 (10 nM) treatment time. D. Protein synthesis measured by OPP incorporation following (−)-SDS-1-021 wash out after treatment for 1 h with 10 nM. In B-D, values represent the mean of three independent experiments ± SEM. * p<0.05 by one-way ANOVA with Dunnett’s multiple comparison test; cycloheximide (CHX, 250 μM, 3 h) was used as a positive control for protein synthesis inhibition.
As a complement to polysome profiling, protein synthesis was analyzed using the OPP assay. (−)-SDS-1-021 exposure for 1 h resulted in a dose dependent decrease in protein synthesis in each cell line (Fig. 2B). Treatment with 10 nM (−)-SDS-1-021 induced a time dependent decrease in protein synthesis out to 6h (Fig. 2C). Thus, inhibition of protein synthesis by (−)-SDS-1-021 is both concentration and time dependent. As shown in Fig. 2D, recovery from the (−)-SDS-1-021-mediated decrease in protein synthesis was rapid, returning to control levels by 1 h after addition of drug-free media.
The effects of (−)-SDS-1-021 on tumor cell radiosensitivity are shown in Fig. 3A. Cells were plated at clonal density, and 16 hours later (−)-SDS-1-021 was added to culture medium immediately prior to irradiation and removed 24 hours post-irradiation. 10 nM (−)-SDS-1-021 enhanced U251 and HeLa radiosensitivity with a DEF of 1.2. Drug alone reduced the surviving fractions in U251 and HeLa to 0.55 ± 0.08, 0.75 ± 0.05, respectively. For PSN1 cells 25 nM enhanced radiation-induced cell killing with a DEF of 1.5. Treatment of PSN1 cells with (−)-SDS-1-021 alone reduced survival to 0.21 ± 0.04. These data indicate that (−)-SDS-1-021 enhances tumor cell radiosensitivity.
Figure 3.
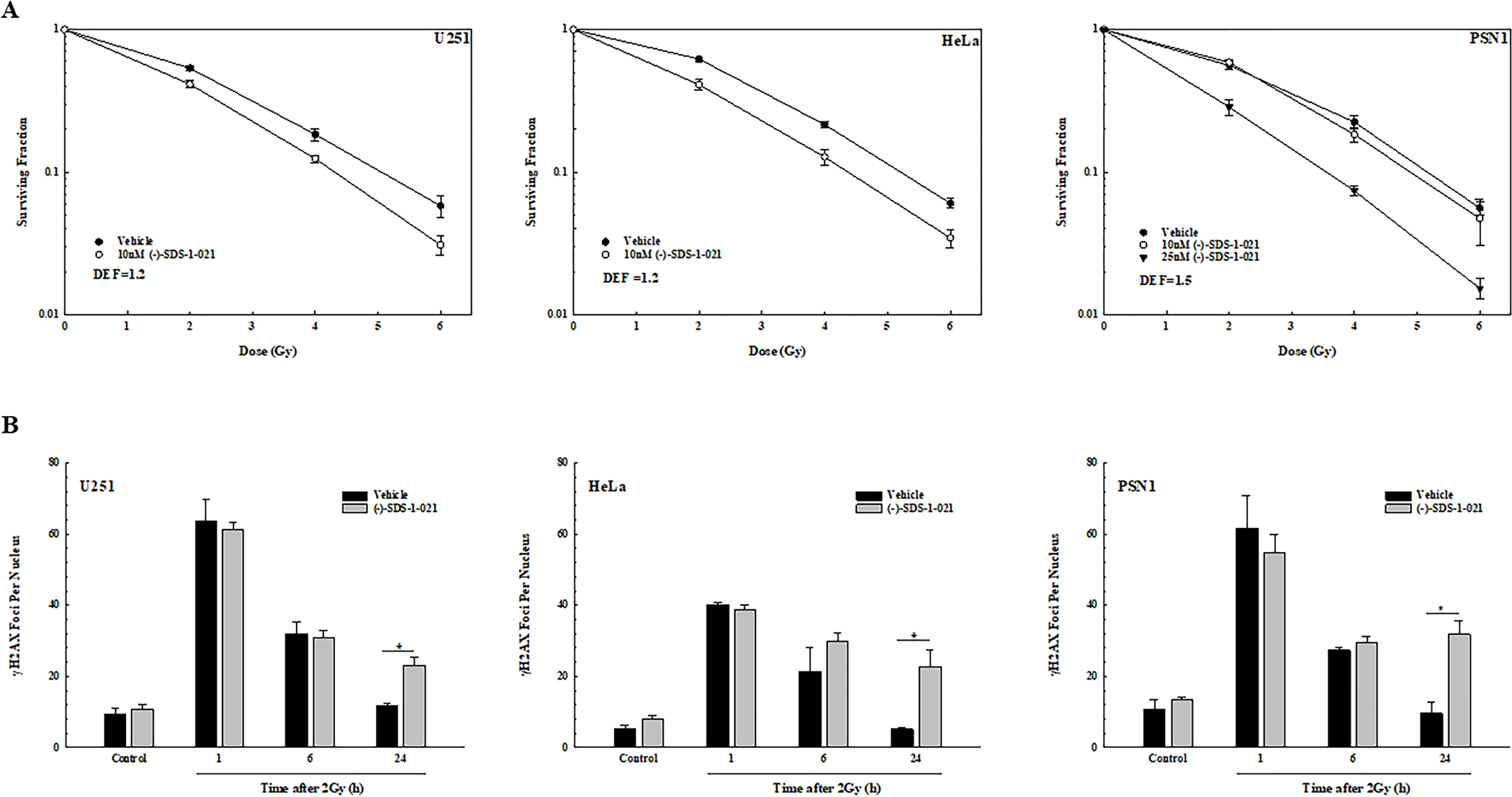
Effects of (−)-SDS-1-021 on tumor cell radiosensitivity and radiation-induced γH2AX foci. A. (−)-SDS-1-021 (10 or 25 nM) was added to U251, HeLa, and PSN1 cells immediately before irradiation. 24 h post-irradiation, drug containing media was removed, replaced with drug-free media and colonies determined after 10–14 days. DEFs were calculated at a surviving fraction of 0.1. Values represent the mean ± SEM for three to four independent experiments. B. γH2AX foci analysis of U251, HeLa, and PSN1 cells. (−)-SDS-1-021 (10 nM) was added to cultures immediately prior to irradiation (2Gy) and cells collected at the indicated time points for foci analysis. For unirradiated cells, vehicle or (−)-SDS-1-021 treatment time was 24 h. γH2AX foci were counted in 50 cells per treatment group. Values represent the mean ± SEM of three independent experiments. * p<0.05 by Student’s t-test.
To determine the effects of (−)-SDS-1-021 on radiation-induced DSBs, γH2AX foci were evaluated. The panel of cell lines was treated with 10 nM of (−)-SDS-1-021 immediately prior to irradiation (2Gy) with analyses at 1–24 hours later. In each cell line, (−)-SDS-1-021 alone had no effect on the number of γH2AX foci in unirradiated cells or on the initial level of radiation-induced γH2AX foci. However, the number of γH2AX foci remaining in each cell line at 24 hours after irradiation was significantly greater in the (−)-SDS-1-021 treated group as compared to irradiation alone (Fig. 3B). These data suggest that (−)-SDS-1-021 inhibits the repair of radiation-induced DSBs.
The effects of (−)-SDS-1-021 on the radiosensitivity of the normal lung fibroblast cell line MRC9 were then determined. As shown by polysome profiling (Fig. 4A), exposure of MRC9 cells to 10 or 25 nM (−)-SDS-1-021 resulted in reduction of TE to similar levels observed for the tumor cell lines. With respect to (−)-SDS-1-021-mediated decrease in protein synthesis, the dose dependence was similar between MRC9 and the tumor cells as was the recovery after addition of drug-free media (Fig. 4B). However, in contrast to the tumor cells, (−)-SDS-1-021 had no effect on MRC9 radiosensitivity as measured by clonogenic survival (Fig. 4C). Treatment with (−)-SDS-1-021 alone at 10 and 25 nM reduced the surviving fraction to 0.42 ± 0.02 and 0.30 ± 0.02, respectively.
Figure 4.
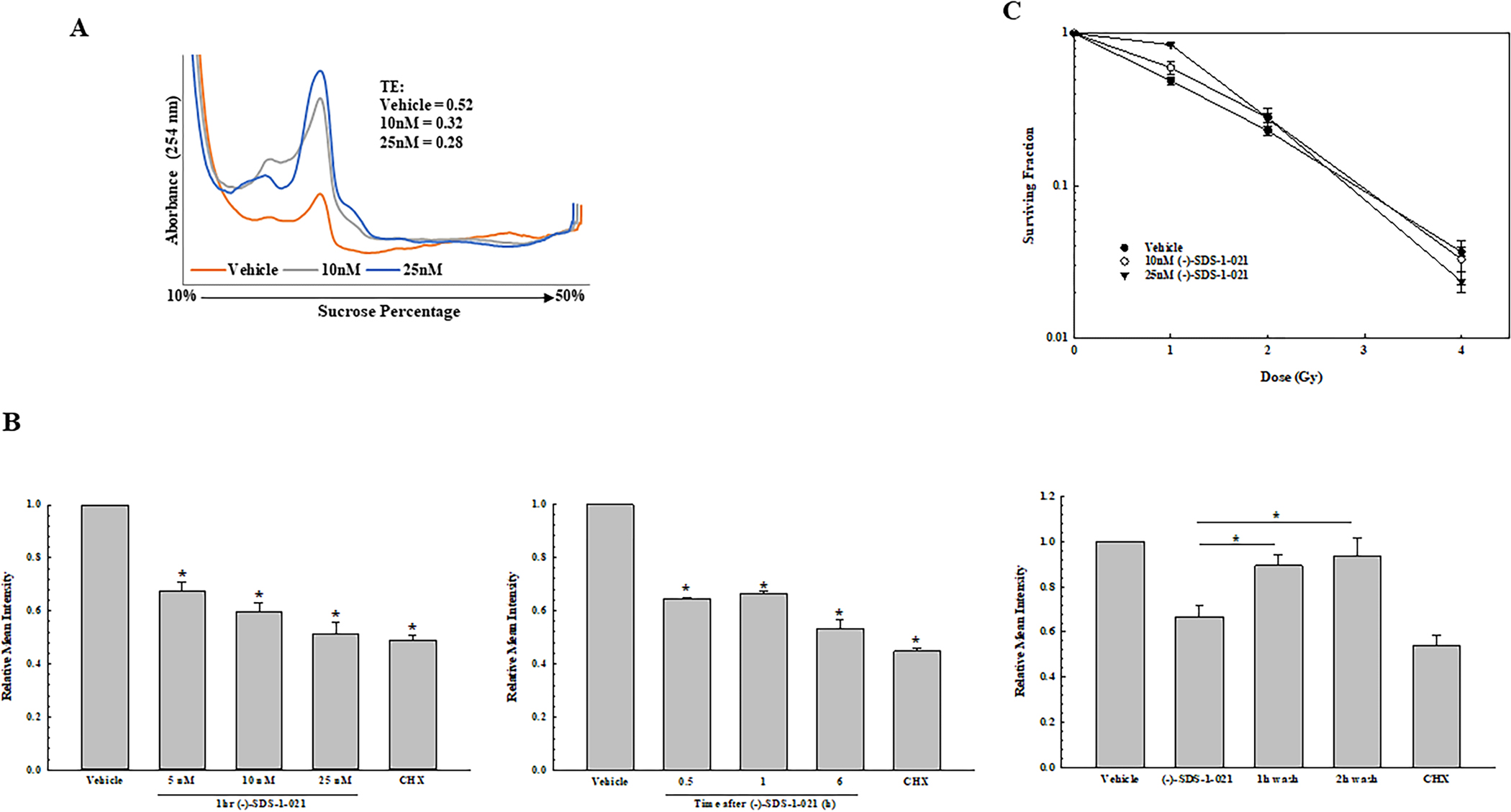
Effects of (−)-SDS-1-021 on translational efficiency, protein synthesis, and radiosensitivity in normal fibroblasts. A. Polysome profiles with (−)-SDS-1-021 were generated from MRC9 normal human fibroblasts. TEs calculated from the polysome profiles are shown. B. Protein synthesis measured by OPP incorporation as a function of (−)-SDS-1-021 dose (1 h) (left), treatment time (10 nM) (middle), and following (−)-SDS-1-021 wash out after treatment for 1 h with 10 nM (right). Cycloheximide (CHX, 250 μM, 3 h) was used as a positive control for protein synthesis inhibition. Values represent the mean ± SEM for three independent experiments. * p<0.05 by one-way ANOVA with Dunnett’s multiple comparison test. C. MRC9 radiosensitivity. (−)-SDS-1-021 was added to cells immediately before irradiation. 24 h post-irradiation, drug containing media was removed, replaced with drug-free media, and colonies were determined after 14 days. Values represent the mean ± SEM for three independent experiments. DEFs were calculated at a surviving fraction of 0.1.
To investigate the mechanisms mediating (−)-SDS-1-021-induced radiosensitization, polysome-bound mRNA was isolated by sucrose gradient fractionation and subjected to RNA-seq based gene expression analysis. This approach defines the effects of (−)-SDS-1-021 on the U251 translatome. Specifically, U251 cells were treated with 10nM (−)-SDS-1-021 and polysomes collected 6 hours later. As shown in Fig. 5A, (−)-SDS-1-021 significantly modified the U251 translatome, consistent with as previous reports of eIF4A inhibition (15–17). The functional significance of genes decreased and increased in polysome-binding after (−)-SDS-1-021 treatment were evaluated using GSEA and IPA. For genes whose polysome abundance decreased after (−)-SDS-1-021 treatment, which indicates decreased translation, the GSEA hallmark gene sets significantly enriched included DNA repair (Fig. 5B). The (−)-SDS-1-021 downregulated genes were also characterized using IPA. As shown in Fig. 5C, the top 20 significant molecular and cellular functions include DNA replication, recombination, and repair (Fig. 5C), which includes the significant subfunctions Repair of DNA, DNA recombination, DNA damage, DNA damage response of cells, and Recombination. The GSEA and IPA evaluations of genes whose translation was increased after (−)-SDS-1-021 treatment did not include DNA repair (GSEA) or DNA replication, recombination, and repair (IPA) (Supplementary Fig. S1). These results suggest that (−)-SDS-1-021 treatment inhibits the translation of genes participating in homologous recombination (HR) repair. Of the genes significantly decreased by (−)-SDS-1-021 treatment, 48 were annotated with the GO term “double-strand break repair by homologous recombination” (GO:0000724) (Supplementary Fig. S2A). Supporting the validity of the RNAseq analysis of polysome-bound mRNA, the levels of proteins corresponding to 4 representative genes within this GO network were decreased after (−)-SDS-1-021 treatment of U251 cells (Supplementary Fig. S2B). To evaluate the functional consequence of this decrease in gene translation, a plasmid-based assay (35) was used to test the effects of (−)-SDS-1-021 on HR repair (Fig. 5D). Treatment of U251 cells with (−)-SDS-1-021 resulted in a significant reduction in HR repair. These results suggest that (−)-SDS-1-021 decreases the translation of genes whose protein products participate in HR, which then contributes to the inhibition of DNA repair and increased radiosensitivity.
Figure 5.
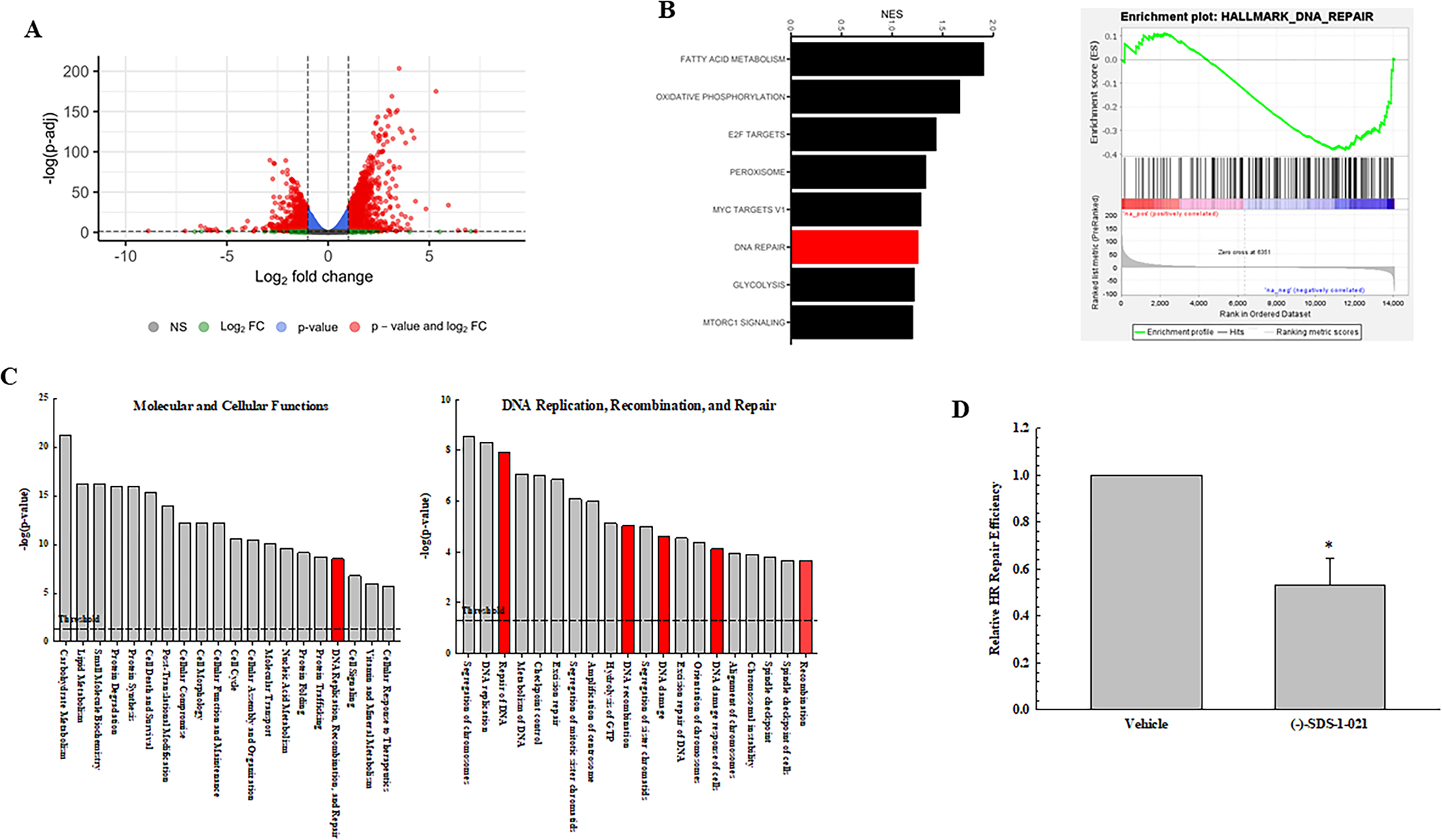
Effect of (−)-SDS-1-021 on the U251 translatome. Polysome-bound mRNA from U251 cells treated with vehicle or (−)-SDS-1-021 (10nM, 6 h) was collected and analyzed by RNA-seq (n=3 biological replicates). A. Volcano plot of transcripts affected by (−)-SDS-1-021. B. GSEA hallmarks enriched in (−)-SDS-1-021 downregulated genes (left) and the enrichment plot for DNA repair (right). C. IPA defined top 20 significant molecular and cellular functions in (−)-SDS-1-021 downregulated genes (adjusted p-value < 0.05) (left) with significant sub-functions within DNA replication, recombination, and repair (right). D. HR repair. U251 cells were transfected with HR assay plasmids and 5 h later treated with 10nM (−)-SDS-1-021; 24h later DNA was isolated. Values represent the mean ± SEM for three independent experiments.
To determine whether the tumor cell radiosensitization induced by (−)-SDS-1-021 observed in vitro could be extended to an in vivo model, we used a subcutaneous xenograft model initiated from U251 cells. To test the ability of (−)-SDS-1-021 to suppress tumor translation in vivo, polysome profiles were generated from U251 xenografts (Fig. 6A). When tumors were approximately 150mm3, mice were treated with either vehicle or a single dose of (−)-SDS-1-021; tumors were then harvested at times out to 24 h. A significant decrease in translational efficiency was detected at 1h post-treatment, which was maintained out to 24 h (Fig. 6A). Based on this information, a protocol was designed to test the antitumor effectiveness of the (−)-SDS-1-021/radiation combination. Mice bearing U251 xenografts in the right hind leg were randomized into four treatment groups: vehicle, (−)-SDS-1-021, radiation, and (−)-SDS-1-021 plus radiation. Radiation was delivered in five consecutive daily fractions of 1.5Gy. (−)-SDS-1-021 (0.35 mg/kg) or vehicle was injected 1 h before radiation on days 1 and 4. This drug dosing schedule was adapted from Manier, et al. (36), who reported that twice weekly dosing with (−)-SDS-1-021 for 4 weeks was effective in a mouse model of multiple myeloma with little to no toxicity. Growth curves for the individual tumors are shown in Fig. 6B, and the average tumor volumes at each timepoint are shown in Fig. 6C. The time for tumors to grow to 1000 mm3 after the initiation of treatment was determined for the individual mice in each group and used to calculate the absolute growth delay, which is defined as the time for treated tumors to reach 1000 mm3 minus the time for vehicle treated tumors to reach 1000 mm3. The absolute growth delay for (−)-SDS-1-021 alone and radiation alone were 2.6 ± 1.7 days (mean ± SEM) and 15.4 ± 3.1 days, respectively. The absolute growth delay for mice treated with the combination of (−)-SDS-1-021 and radiation was 28.5 ± 6.0, which is greater than the sum of the growth delays of the individual treatments. To determine a DEF, the growth delay induced by (−)-SDS-1-021 alone (2.6 days), was subtracted from the growth delay observed in mice treated with the (−)-SDS-1-021/radiation combination (28.5 days) and divided by the absolute growth delay of mice treated with radiation alone (15.4 days), which resulted in a DEF of 1.7. The (−)-SDS-1-021/radiation combination was also evaluated using fractional tumor volume (FTV) (32). At day 17, tumor growth was inhibited by 25% by SDS-1-021, 75% by radiation, and 90% by the (−)-SDS-1-021/radiation combination, resulting in a ratio of expected FTV (0.188)/observed FTV (0.10) of 2, which is consistent with a synergistic interaction (32).
Figure 6.
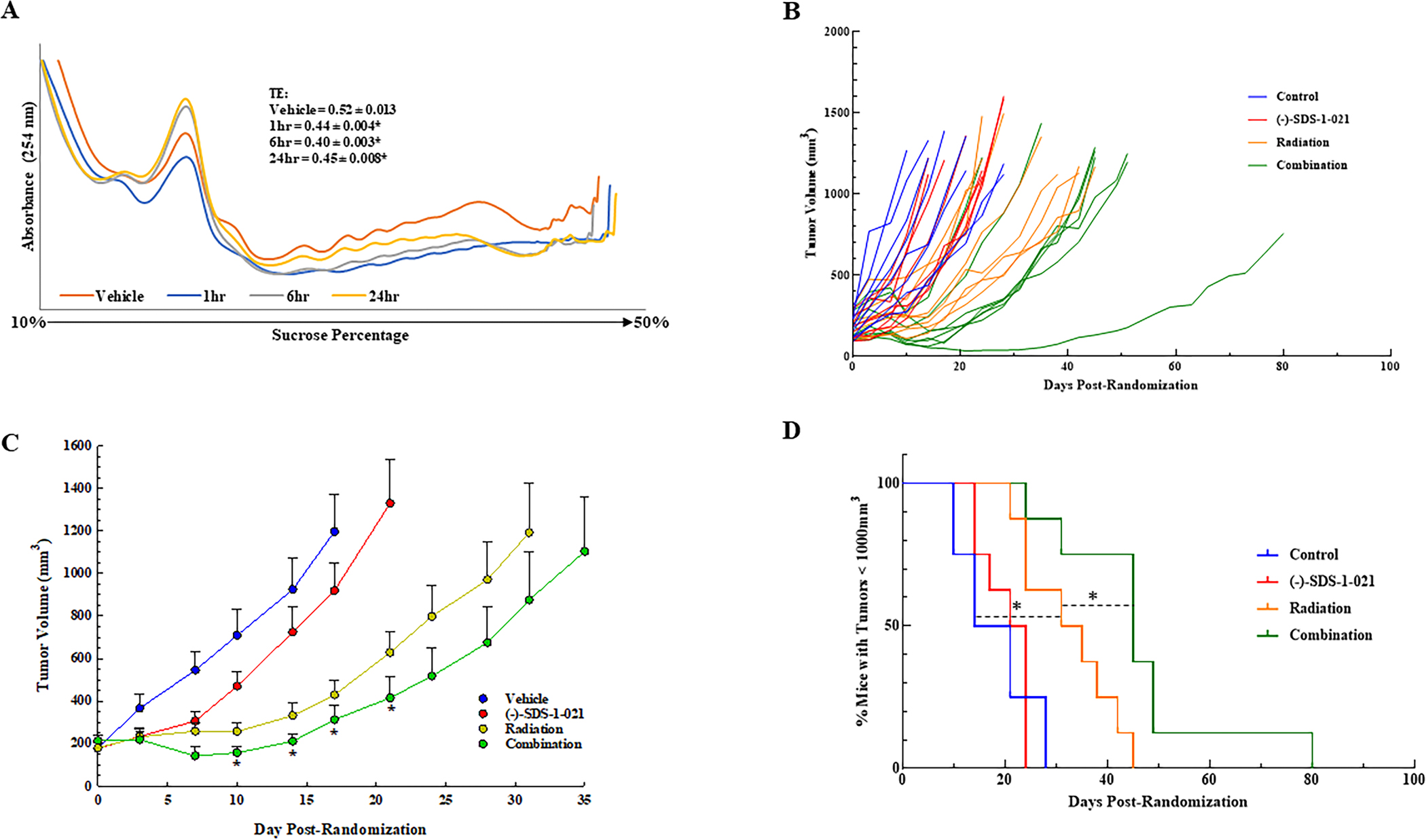
Effects of (−)-SDS-1-021 on tumor xenograft translational efficiency and radiation-induced tumor growth delay. A. Mice bearing subcutaneous xenografts were treated with vehicle or (−)-SDS-1-021 (0.35 mg/kg) by intraperitoneal injection. Tumors were collected at the indicated timepoints and polysome profiles generated. Representative profiles from each treatment group are shown. Translational efficiencies represent the mean ± SEM from three mice, * p<0.05 by one-way ANOVA with Dunnett’s multiple comparison test. B. Mice bearing U251 leg tumor xenografts were treated with 1.5Gy for five consecutive days and received (−)-SDS-1-021 (0.35 mg/kg) 1 h before tumor irradiation on days one and four. Tumors were measured twice weekly. C. Average growth curves of the individual mice shown in (B). Values shown represent the mean ± SEM from eight mice. * p<0.05 by Student’s t-test. D. Percent of mice with tumors below 1000 mm3 at each measurement (twice per week) after treatment initiation. * p<0.05 by log-rank (Mantel-Cox) test.
The tumor growth data were also evaluated according to the percent tumors less than 1000mm3 as determined on each day of measurement (twice per week). Log-rank analysis showed that (−)-SDS-1-021 treatment alone had no significant effect on tumor growth as compared to vehicle; radiation alone resulted in a significant decrease in growth rate. Tumor growth in mice receiving the combination protocol was significantly decreased as compared with control and, importantly, as compared with radiation alone (Fig. 6D). These data indicate that (−)-SDS-1-021, while having little effect alone on tumor growth, significantly enhanced radiation-induced tumor growth delay.
Discussion
Although a number of post-transcriptional processes mediate gene expression, (e.g., splicing, capping, and polyadenylation), the final event is translation. Translational control operates primarily at the initiation step via the assembly of eIF4E, eIF4G, and eIF4A into the eIF4F initiation complex. Previous studies showed that targeting eIF4E directly (7) or limiting its availability via mTORC1 inhibitors (8–10,37,38) enhances tumor cell radiosensitivity. Reducing eIF4G levels using shRNA or with small molecules that disrupt the eIF4G-eIF4E interaction has also been reported to increase radiosensitivity (39,40). Of the eIF4F components, eIF4A is unique in that it has enzymatic activity and thus more amenable to targeting with small molecule inhibitors. Knockdown of eIF4A1 levels using siRNA decreased translational efficiency in U251 and HeLa cells as measured from polysome profiles, indicative of the role of this helicase in mRNA translation. The same knockdown of eIF4A was then shown to enhance the radiosensitivity of these tumor cell lines as well as to inhibit the repair of radiation-induced DSBs. Consistent with these findings, shRNA mediated knockdown of eIF4A was also reported to inhibit DSB repair and enhance the radiosensitivity of HeLa cells, although no effect on translation was reported (41). Thus, based on the knockdown approach, eIF4A can serve as a determinant of cellular radiosensitivity. Of note, because eIFA1 is an essential gene (18), these studies were limited to the knockdown approach, rather than a complete knockout obtained with CRISPR/Cas9, and the potential for off target effects exists.
As an initial investigation of eIF4A as a clinically relevant target for tumor radiosensitization, (−)-SDS-1-021, a highly potent synthetic rocaglate (34), was investigated. Like other rocaglates, (−)-SDS-1-021 stabilizes the interaction between eIF4A and mRNA, which blocks ribosome scanning of the target mRNA. It can also trap eIF4F at the mRNA cap, which suppresses global translation by depleting available eIF4F (34). As shown, (−)-SDS-1-021 exposure of three tumor cell lines initiated from different solid tumor types resulted in rapid reductions in translational efficiency as well as protein synthesis, consistent with the inhibition of eIF4A. In addition, as was observed for siRNA targeting eIF4A1, (−)-SDS-1-021 treatment also induced an enhancement of tumor cell radiosensitivity. Regarding the mechanisms responsible, radiosensitization was induced when (−)-SDS-1-021 was added immediately prior to radiation, indicating that the effect was not the result of a redistribution of cells into a radiosensitive phase of the cell cycle. Based on γH2AX results, (−)-SDS-1-021 does not influence the initial level of radiation-induced DSBs but does inhibit their repair, which is similar to the radiosensitization induced by targeting of eIF4E (7–10,37). Thus, data suggest that the enhanced radiosensitivity induced by targeting translation initiation involves an inhibition of DSB repair. Moreover, (−)-SDS-1-021 decreased the translation of genes involved in DNA repair, specifically genes participating in HR repair, which is validated by the functional analysis showing that HR repair was reduced in (−)-SDS-1-021 treated cells (Fig. 5D). These results suggest that the radiosensitization induced by (−)-SDS-1-021, at least in part, is mediated by a decrease in the translation of HR repair related genes.
The clinical applicability of a radiosensitizing agent depends, at least in part, on the preferential sensitization of tumor cells over normal cells. Treatment of the normal human fibroblast cell line MRC9 with (−)-SDS-1-021 reduced translation efficiency and protein synthesis to similar degrees as in the tumor cell lines. Cell killing induced by (−)-SDS-1-021 treatment alone was also similar between the tumor cells and the normal fibroblasts. However, the eIF4A inhibitor only enhanced radiosensitivity of the tumor cell lines. In general, translational control is reprogramed in tumor cells to selectively enhance the translation of genes mediating cell proliferation, survival, and other processes critical to the neoplastic phenotype (42). This process may also be applicable to radioresponse in that radiation-induced translational control of gene expression differs between tumor and normal cells (4,43), suggesting that translation initiation may differentially influence the radiosensitivity of tumor and normal cells. Along these lines, studies to date have shown that inhibiting translation preferentially radiosensitizes tumor cells (7,8,10,25) suggesting that tumor cell radioresponse is more dependent on gene translation than that of normal cells.
A process critical to the preclinical evaluation of putative radiosensitizing agents is the extension of in vitro analyses to an in vivo tumor xenograft model. In the initial preclinical study of (−)-SDS-1-021 as a radiosensitizer described here, polysome profiles showed that a single dose reduced tumor translation efficiency for at least 24 h, consistent with the effective targeting of eIF4A under in vivo conditions. Combining two doses of (−)-SDS-1-021 with a 5-day fractionated irradiation protocol then resulted in a significant increase in the radiation-induced tumor growth delay, indicative of in vivo radiosensitization. Thus, these data suggest that delivery of this eIF4A inhibitor in combination with radiotherapy may improve tumor treatment response. Along these lines, while (−)-SDS-1-021 is in the pre-clinical stage of development, the eIF4A small molecule inhibitor eFT226 (44) has recently entered a phase I/II clinical trials (NCT04092673, NCT04632381).
Supplementary Material
Financial support:
Financial support provided by Intramural Program, National Cancer Institute (Z1ABC011373) to P.J. Tofilon. Work at Boston University is supported by NIH grants R35GM118173 and U01TR002625.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Liauw SL, Connell PP, Weichselbaum RR. New Paradigms and Future Challenges in Radiation Oncology: An Update of Biological Targets and Technology. Sci Transl Med 2013;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lü X, de la Pena L, Barker C, Camphausen K, Tofilon PJ. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res 2006;66:1052–61 [DOI] [PubMed] [Google Scholar]
- 3.Wahba A, Rath BH, Bisht K, Camphausen K, Tofilon PJ. Polysome Profiling Links Translational Control to the Radioresponse of Glioblastoma Stem-like Cells. Cancer Res 2016;76:3078–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumaraswamy S, Chinnaiyan P, Shankavaram UT, Lu X, Camphausen K, Tofilon PJ. Radiation-induced gene translation profiles reveal tumor type and cancer-specific components. Cancer Research 2008;68:3819–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szkanderova S, Port M, Stulik J, Hernychova L, Kasalova I, Van Beuningen D, et al. Comparison of the abundance of 10 radiation-induced proteins with their differential gene expression in L929 cells. Int J Radiat Biol 2003;79:623–33 [DOI] [PubMed] [Google Scholar]
- 6.Merrick WC, Pavitt GD. Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb Perspect Biol 2018;10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayman TJ, Williams ES, Jamal M, Shankavaram UT, Camphausen K, Tofilon PJ. Translation initiation factor eIF4E is a target for tumor cell radiosensitization. Cancer Res 2012;72:2362–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayman TJ, Kramp T, Kahn J, Jamal M, Camphausen K, Tofilon PJ. Competitive but Not Allosteric mTOR Kinase Inhibition Enhances Tumor Cell Radiosensitivity. Transl Oncol 2013;6:355–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K, et al. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol 2014;16:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayman TJ, Wahba A, Rath BH, Bae H, Kramp T, Shankavaram UT, et al. The ATP-competitive mTOR inhibitor INK128 enhances in vitro and in vivo radiosensitivity of pancreatic carcinoma cells. Clin Cancer Res 2014;20:110–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, et al. mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol 2011;12:235–45 [DOI] [PubMed] [Google Scholar]
- 12.Willis AE. Translational control of growth factor and proto-oncogene expression. Int J Biochem Cell Biol 1999;31:73–86 [DOI] [PubMed] [Google Scholar]
- 13.Pestova TV, Kolupaeva VG. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 2002;16:2906–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickering BM, Willis AE. The implications of structured 5’ untranslated regions on translation and disease. Semin Cell Dev Biol 2005;16:39–47 [DOI] [PubMed] [Google Scholar]
- 15.Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014;513:65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubio CA, Weisburd B, Holderfield M, Arias C, Fang E, DeRisi JL, et al. Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol 2014;15:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Modelska A, Turro E, Russell R, Beaton J, Sbarrato T, Spriggs K, et al. The malignant phenotype in breast cancer is driven by eIF4A1-mediated changes in the translational landscape. Cell Death Dis 2015;6:e1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galicia-Vazquez G, Cencic R, Robert F, Agenor AQ, Pelletier J. A cellular response linking eIF4AI activity to eIF4AII transcription. RNA 2012;18:1373–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Comtesse N, Keller A, Diesinger I, Bauer C, Kayser K, Huwer H, et al. Frequent overexpression of the genes FXR1, CLAPM1 and EIF4G located on amplicon 3q26–27 in squamous cell carcinoma of the lung. Int J Cancer 2007;120:2538–44 [DOI] [PubMed] [Google Scholar]
- 20.Liang S, Zhou Y, Chen Y, Ke G, Wen H, Wu X. Decreased expression of EIF4A1 after preoperative brachytherapy predicts better tumor-specific survival in cervical cancer. Int J Gynecol Cancer 2014;24:908–15 [DOI] [PubMed] [Google Scholar]
- 21.Gao C, Guo X, Xue A, Ruan Y, Wang H, Gao X. High intratumoral expression of eIF4A1 promotes epithelial-to-mesenchymal transition and predicts unfavorable prognosis in gastric cancer. Acta Biochim Biophys Sin (Shanghai) 2020 [DOI] [PubMed] [Google Scholar]
- 22.Oblinger JL, Burns SS, Huang J, Pan L, Ren Y, Shen R, et al. Overexpression of eIF4F components in meningiomas and suppression of meningioma cell growth by inhibiting translation initiation. Exp Neurol 2018;299:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone SD, Lajkiewicz NJ, Whitesell L, Hilmy A, Porco JA, Jr. Biomimetic kinetic resolution: highly enantio- and diastereoselective transfer hydrogenation of aglain ketones to access flavagline natural products. J Am Chem Soc 2015;137:525–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koritzinsky M, Seigneuric R, Magagnin MG, van den Beucken T, Lambin P, Wouters BG. The hypoxic proteome is influenced by gene-specific changes in mRNA translation. Radiother Oncol 2005;76:177–86 [DOI] [PubMed] [Google Scholar]
- 25.Wahba A, Rath BH, O’Neill JW, Camphausen K, Tofilon PJ. The XPO1 Inhibitor Selinexor Inhibits Translation and Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and In Vivo. Mol Cancer Ther 2018;17:1717–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oshlack A, Robinson MD, Young MD. From RNA-seq reads to differential expression results. Genome Biol 2010;11:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73 [DOI] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu DC, Chen Y, Dilley J, Li Y, Embry M, Zhang H, et al. Antitumor synergy of CV787, a prostate cancer-specific adenovirus, and paclitaxel and docetaxel. Cancer Res 2001;61:517–25 [PubMed] [Google Scholar]
- 33.Chu J, Cencic R, Wang W, Porco JA, Jr., Pelletier J. Translation Inhibition by Rocaglates Is Independent of eIF4E Phosphorylation Status. Mol Cancer Ther 2016;15:136–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu J, Zhang W, Cencic R, O’Connor PBF, Robert F, Devine WG, et al. Rocaglates Induce Gain-of-Function Alterations to eIF4A and eIF4F. Cell Rep 2020;30:2481–8 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bigot N, Mouche A, Preti M, Loisel S, Renoud ML, Le Guevel R, et al. Hypoxia Differentially Modulates the Genomic Stability of Clinical-Grade ADSCs and BM-MSCs in Long-Term Culture. Stem Cells 2015;33:3608–20 [DOI] [PubMed] [Google Scholar]
- 36.Manier S, Huynh D, Shen YJ, Zhou J, Yusufzai T, Salem KZ, et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci Transl Med 2017;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silvera D, Ernlund A, Arju R, Connolly E, Volta V, Wang J, et al. mTORC1 and −2 Coordinate Transcriptional and Translational Reprogramming in Resistance to DNA Damage and Replicative Stress in Breast Cancer Cells. Mol Cell Biol 2017;37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu CC, Huang HB, Hung SK, Liao HF, Lee CC, Lin HY, et al. AZD2014 Radiosensitizes Oral Squamous Cell Carcinoma by Inhibiting AKT/mTOR Axis and Inducing G1/G2/M Cell Cycle Arrest. PLoS One 2016;11:e0151942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Badura M, Braunstein S, Zavadil J, Schneider RJ. DNA damage and eIF4G1 in breast cancer cells reprogram translation for survival and DNA repair mRNAs. Proc Natl Acad Sci U S A 2012;109:18767–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang W, Li J, Wen Q, Luo J, Chu S, Chen L, et al. 4EGI-1 induces apoptosis and enhances radiotherapy sensitivity in nasopharyngeal carcinoma cells via DR5 induction on 4E-BP1 dephosphorylation. Oncotarget 2016;7:21728–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang S, Ju X, Zhou Y, Chen Y, Ke G, Wen H, et al. Downregulation of eukaryotic initiation factor 4A1 improves radiosensitivity by delaying DNA double strand break repair in cervical cancer. Oncol Lett 2017;14:6976–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robichaud N, Sonenberg N, Ruggero D, Schneider RJ. Translational Control in Cancer. Cold Spring Harb Perspect Biol 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wahba A, Lehman SL, Tofilon PJ. Radiation-induced translational control of gene expression. Translation (Austin) 2017;5:e1265703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson PA, Eam B, Young NP, Fish S, Chen J, Barrera M, et al. Targeting Oncogene mRNA Translation in B-Cell Malignancies with eFT226, a Potent and Selective Inhibitor of eIF4A. Mol Cancer Ther 2021;20:26–36 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE200162.