

The title compound exists with one protonated imidazolium ring, one neutral imidazole ring, and a benzenesulfonate anion in the asymmetric unit. The imidazole rings are held together through hydrogen bonding via a protonated nitrogen on the ring.
Keywords: crystal structure, tosylate, imidazolium, ionic crystal
Abstract
In the title co-crystal, C5H9N2
+·C6H5O3S−·C5H8N2, the two 1,2-dimethylimidazole rings exist as partially protonated moieties in the asymmetric unit as a two-part disordered unit wherein the acidic hydrogen atom is bound to each ring. The two imidazolium cations share a strong hydrogen bond via the acidic hydrogen atom, which is disordered between two positions, being bonded to the first versus second imidazole ring in a 0.33 (2) to 0.67 (2) ratio. A benzene sulfonate anion is present for charge balance and interacts with the aromatic H atoms on both imidazole rings as well as with the methyl groups on the rings.

Structure description
The title compound (Fig. 1 ▸) crystallizes with two 1,2-dimethylimidazolium cations in the asymmetric unit. The two imidazole rings are each partially protonated, wherein the acidic hydrogen atom is bound between the two N atoms of the aromatic ring in a 0.33 (2) to 0.67 (2) ratio. Hydrogen bonding appears to the dominant intermolecular interaction with each molecule or ion exhibiting interactions (Fig. 2 ▸). For instance, the shortest hydrogen bonds are N—H⋯N links between the imidazolium rings with H⋯N = 1.83 (8) and 1.90 (8) Å. This bonding arises from the disordered hydrogen atom, which appears to be shared between the two rings. Further, cation–anion C—H⋯O interactions occur between the aromatic H atoms and the sulfonate O atoms. Finally, there are anion–anion interactions wherein O atoms of the sulfonate group interact with hydrogens on the benzene rings. A summary of the distances for the hydrogen bonds is found in Table 1 ▸.
Figure 1.

The title compound shown with 50% probability ellipsoids. Only the major component is shown.
Figure 2.

Packing diagram of the title compound viewed down the (010) plane showing a layered network of ion pairs held together through hydrogen interactions. Both parts of the disorder are shown.
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N⋯N3 | 0.83 | 1.90 | 2.6970 (11) | 163 |
| N3—H3N⋯N1 | 0.89 | 1.81 | 2.6970 (11) | 170 |
| C1—H1⋯O1 | 0.95 | 2.43 | 3.3741 (13) | 170 |
| C4—H4B⋯O3i | 0.98 | 2.33 | 3.2956 (12) | 170 |
| C6—H6⋯O2 | 0.95 | 2.60 | 3.4288 (14) | 146 |
| C7—H7⋯O2ii | 0.95 | 2.41 | 3.3254 (12) | 162 |
| C9—H9A⋯O3iii | 0.98 | 2.53 | 3.4846 (14) | 164 |
| C9—H9B⋯O3ii | 0.98 | 2.46 | 3.4381 (14) | 173 |
| C13—H13⋯O1i | 0.95 | 2.67 | 3.4738 (14) | 143 |
| C14—H14⋯O1iv | 0.95 | 2.48 | 3.3920 (13) | 162 |
Symmetry codes: (i)
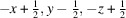 ; (ii)
; (ii)
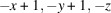 ; (iii)
; (iii)
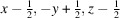 ; (iv)
; (iv)
 .
.
For a related structure with a chloride anion, see Kelley et al. (2013 ▸).
Synthesis and crystallization
The reaction was conducted in a Biotage Initiator+ microwave reactor. To a microwave vial was added a stir bar as well as 7.5 mmol (721 mg) of 1,2 dimethylimidazole and 7.5 mmol (901 μL) of benzenesulfonyl fluoride. The vial was sealed and placed into the microwave reactor. The reaction was performed at 105°C for 5 minutes and 38 s with very high microwave absorption, stirring at 600 rpm. Once finished and cooled to room temperature, the solution was transferred to an oven-dried amber scintillation vial and sealed with parafilm. After one week, crystals suitable for diffraction were found growing in the vial.
A proposed mechanism leading to the formation of the crystallized product reported herein is shown in Fig. 3 ▸.
Figure 3.

Proposed mechanism leading to the formation of the crystallized product reported herein.
1H NMR (400 MHz, chloroform-D) δ 8.01–7.99 (m, 1H), 7.89–7.87 (m, 1H), 7.77 (dd, J = 8.1, 6.7 Hz, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.35 (t, J = 2.6 Hz, 1H), 7.25 (s, 1H), 6.98–6.83 (m, 2H), 3.61 (d, J = 9.1 Hz, 3H), 2.46 (d, J = 14.0 Hz, 3H)
13C NMR (101 MHz, chloroform-D) δ 206.7, 144.8, 135.7, 130.0, 129.8, 128.5, 128.3, 126.0, 121.2, 121.0, 77.4, 77.1, 76.8, 76.6, 33.5, 12.0, −1.6.
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 2 ▸.
Table 2. Experimental details.
| Crystal data | |
| Chemical formula | C5H9N2 +·C6H5O3S−·C5H8N2 |
| M r | 350.43 |
| Crystal system, space group | Monoclinic, P21/n |
| Temperature (K) | 150 |
| a, b, c (Å) | 10.8820 (6), 8.4029 (4), 18.9678 (11) |
| β (°) | 95.440 (2) |
| V (Å3) | 1726.61 (16) |
| Z | 4 |
| Radiation type | Mo Kα |
| μ (mm−1) | 0.21 |
| Crystal size (mm) | 0.55 × 0.42 × 0.33 |
| Data collection | |
| Diffractometer | Bruker AXS D8 Quest CMOS diffractometer |
| Absorption correction | Multi-scan (SADABS; Krause et al., 2015 ▸) |
| T min, T max | 0.716, 0.747 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 74762, 6612, 5787 |
| R int | 0.032 |
| (sin θ/λ)max (Å−1) | 0.771 |
| Refinement | |
| R[F 2 > 2σ(F 2)], wR(F 2), S | 0.035, 0.095, 1.05 |
| No. of reflections | 6612 |
| No. of parameters | 225 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement |
| Δρmax, Δρmin (e Å−3) | 0.40, −0.44 |
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2414314620006896/bv4031sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314620006896/bv4031Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314620006896/bv4031Isup3.cml
CCDC reference: 2005097
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
This material is based upon work supported by the National Science Foundation through the Major Research Instrumentation Program under grant No. CHE 1625543 (funding for the single-crystal X-ray diffractometer). Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for support of this research. The authors gratefully acknowledge the Communities in Transition Initiative for the generous support.
full crystallographic data
Crystal data
| C5H9N2+·C6H5O3S−·C5H8N2 | F(000) = 744 |
| Mr = 350.43 | Dx = 1.348 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| a = 10.8820 (6) Å | Cell parameters from 9689 reflections |
| b = 8.4029 (4) Å | θ = 2.7–33.2° |
| c = 18.9678 (11) Å | µ = 0.21 mm−1 |
| β = 95.440 (2)° | T = 150 K |
| V = 1726.61 (16) Å3 | Block, colourless |
| Z = 4 | 0.55 × 0.42 × 0.33 mm |
Data collection
| Bruker AXS D8 Quest CMOS diffractometer | 6612 independent reflections |
| Radiation source: fine focus sealed tube X-ray source | 5787 reflections with I > 2σ(I) |
| Triumph curved graphite crystal monochromator | Rint = 0.032 |
| Detector resolution: 10.4167 pixels mm-1 | θmax = 33.2°, θmin = 2.3° |
| ω and phi scans | h = −16→16 |
| Absorption correction: multi-scan (SADABS; Krause et al., 2015) | k = −12→12 |
| Tmin = 0.716, Tmax = 0.747 | l = −29→28 |
| 74762 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.035 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.095 | w = 1/[σ2(Fo2) + (0.0417P)2 + 0.6618P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.05 | (Δ/σ)max = 0.001 |
| 6612 reflections | Δρmax = 0.40 e Å−3 |
| 225 parameters | Δρmin = −0.44 e Å−3 |
| 0 restraints | Extinction correction: SHELXL-2018/3 (Sheldrick 2015), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0085 (10) |
Special details
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. H atoms attached to carbon atoms were positioned geometrically and constrained to ride on their parent atoms. C—H bond distances were constrained to 0.95 Å for aromatic and alkene C—H moieties, and to 0.98 Å for CH3 moieties, respectively. The N—H proton hydrogen bonding between atoms N1 and N3 was found to be disordered and was refined as split between two positions. The H atoms were assigned as bonded to a planar (sp2 hybridized) N atom, respectively with fixed bond angles and torsion angles, but the N—H bond distances were allowed to refine to account for asymmetry induced by charge and hydrogen bonding (AFIX 44 command). N—H distances refined to 0.83 (5) for N1—H1 and to 0.89 (2) for N3—H3, occupancies refined to 0.33 (2) for H1 and 0.67 (2) for H3. Methyl CH3 were allowed to rotate but not to tip to best fit the experimental electron density. Uiso(H) values were set to a multiple of Ueq(C/N) with 1.5 for CH3 and 1.2 for C—H and NH+, respectively. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| C1 | 0.07995 (8) | 0.11974 (12) | 0.12505 (5) | 0.02370 (17) | |
| H1 | 0.162507 | 0.154570 | 0.136425 | 0.028* | |
| C2 | 0.01650 (8) | 0.02111 (12) | 0.16550 (5) | 0.02268 (16) | |
| H2 | 0.045537 | −0.025178 | 0.209661 | 0.027* | |
| C3 | −0.10216 (8) | 0.08832 (10) | 0.06876 (4) | 0.01832 (14) | |
| C4 | −0.20228 (9) | −0.08561 (12) | 0.15402 (5) | 0.02410 (17) | |
| H4A | −0.264761 | −0.010298 | 0.167446 | 0.036* | |
| H4B | −0.172925 | −0.150361 | 0.195187 | 0.036* | |
| H4C | −0.238614 | −0.154975 | 0.116071 | 0.036* | |
| C5 | −0.21283 (8) | 0.09673 (13) | 0.01670 (5) | 0.02515 (18) | |
| H5A | −0.194381 | 0.162346 | −0.023687 | 0.038* | |
| H5B | −0.281506 | 0.144135 | 0.039231 | 0.038* | |
| H5C | −0.235631 | −0.010751 | 0.000206 | 0.038* | |
| C6 | 0.23459 (9) | 0.40162 (12) | −0.00734 (5) | 0.02566 (18) | |
| H6 | 0.285770 | 0.372447 | 0.034004 | 0.031* | |
| C7 | 0.26716 (8) | 0.49589 (12) | −0.06067 (6) | 0.02502 (18) | |
| H7 | 0.345007 | 0.545291 | −0.063695 | 0.030* | |
| C8 | 0.07312 (8) | 0.42021 (10) | −0.08640 (4) | 0.01863 (14) | |
| C9 | 0.15832 (10) | 0.59191 (13) | −0.17707 (5) | 0.02847 (19) | |
| H9A | 0.142896 | 0.516294 | −0.216246 | 0.043* | |
| H9B | 0.236534 | 0.647282 | −0.181302 | 0.043* | |
| H9C | 0.090972 | 0.669653 | −0.178780 | 0.043* | |
| C10 | −0.05084 (9) | 0.40053 (13) | −0.12482 (5) | 0.02596 (18) | |
| H10A | −0.090249 | 0.504931 | −0.131824 | 0.039* | |
| H10B | −0.101453 | 0.332570 | −0.097120 | 0.039* | |
| H10C | −0.042876 | 0.351054 | −0.170954 | 0.039* | |
| C11 | 0.43346 (7) | 0.00325 (9) | 0.14618 (4) | 0.01514 (13) | |
| C12 | 0.36079 (8) | −0.06738 (10) | 0.19405 (4) | 0.01852 (14) | |
| H12 | 0.326061 | −0.004339 | 0.228696 | 0.022* | |
| C13 | 0.33919 (9) | −0.23035 (11) | 0.19101 (5) | 0.02475 (18) | |
| H13 | 0.289972 | −0.278763 | 0.223810 | 0.030* | |
| C14 | 0.38933 (10) | −0.32262 (12) | 0.14020 (6) | 0.0293 (2) | |
| H14 | 0.375570 | −0.434269 | 0.138722 | 0.035* | |
| C15 | 0.45951 (9) | −0.25157 (13) | 0.09161 (6) | 0.02817 (19) | |
| H15 | 0.492304 | −0.314441 | 0.056220 | 0.034* | |
| C16 | 0.48217 (8) | −0.08834 (11) | 0.09445 (5) | 0.02151 (16) | |
| H16 | 0.530578 | −0.039928 | 0.061247 | 0.026* | |
| N1 | 0.00522 (7) | 0.16129 (10) | 0.06471 (4) | 0.02100 (14) | |
| H1N | 0.0238 (11) | 0.220 (3) | 0.0325 (19) | 0.025* | 0.33 (2) |
| N2 | −0.09869 (7) | 0.00209 (9) | 0.12923 (4) | 0.01850 (13) | |
| N3 | 0.11314 (7) | 0.35581 (10) | −0.02425 (4) | 0.02125 (14) | |
| H3N | 0.0690 (11) | 0.2936 (16) | 0.0021 (7) | 0.026* | 0.67 (2) |
| N4 | 0.16511 (7) | 0.50600 (9) | −0.10967 (4) | 0.02018 (14) | |
| O1 | 0.35734 (8) | 0.28443 (9) | 0.17511 (6) | 0.0439 (2) | |
| O2 | 0.49875 (9) | 0.26229 (11) | 0.08516 (4) | 0.0383 (2) | |
| O3 | 0.57130 (9) | 0.21999 (10) | 0.20780 (5) | 0.0400 (2) | |
| S1 | 0.46799 (2) | 0.20953 (2) | 0.15392 (2) | 0.01791 (6) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0187 (4) | 0.0277 (4) | 0.0242 (4) | −0.0014 (3) | −0.0009 (3) | 0.0018 (3) |
| C2 | 0.0207 (4) | 0.0263 (4) | 0.0202 (4) | 0.0008 (3) | −0.0024 (3) | 0.0030 (3) |
| C3 | 0.0179 (3) | 0.0208 (4) | 0.0163 (3) | 0.0012 (3) | 0.0019 (3) | 0.0019 (3) |
| C4 | 0.0218 (4) | 0.0268 (4) | 0.0244 (4) | −0.0017 (3) | 0.0057 (3) | 0.0059 (3) |
| C5 | 0.0206 (4) | 0.0326 (5) | 0.0214 (4) | −0.0007 (3) | −0.0025 (3) | 0.0057 (3) |
| C6 | 0.0217 (4) | 0.0257 (4) | 0.0285 (4) | −0.0007 (3) | −0.0032 (3) | −0.0017 (3) |
| C7 | 0.0176 (4) | 0.0236 (4) | 0.0338 (5) | −0.0029 (3) | 0.0020 (3) | −0.0033 (3) |
| C8 | 0.0182 (3) | 0.0197 (3) | 0.0183 (3) | −0.0019 (3) | 0.0032 (3) | −0.0005 (3) |
| C9 | 0.0367 (5) | 0.0263 (4) | 0.0240 (4) | −0.0061 (4) | 0.0108 (4) | 0.0020 (3) |
| C10 | 0.0209 (4) | 0.0326 (5) | 0.0237 (4) | −0.0051 (3) | −0.0017 (3) | 0.0037 (3) |
| C11 | 0.0138 (3) | 0.0151 (3) | 0.0160 (3) | −0.0013 (2) | −0.0009 (2) | 0.0004 (2) |
| C12 | 0.0195 (3) | 0.0185 (3) | 0.0174 (3) | −0.0033 (3) | 0.0012 (3) | 0.0012 (3) |
| C13 | 0.0264 (4) | 0.0195 (4) | 0.0271 (4) | −0.0066 (3) | −0.0035 (3) | 0.0064 (3) |
| C14 | 0.0310 (5) | 0.0154 (4) | 0.0388 (5) | 0.0007 (3) | −0.0104 (4) | −0.0017 (3) |
| C15 | 0.0254 (4) | 0.0259 (4) | 0.0320 (5) | 0.0068 (3) | −0.0036 (3) | −0.0108 (4) |
| C16 | 0.0171 (3) | 0.0271 (4) | 0.0203 (4) | 0.0017 (3) | 0.0017 (3) | −0.0031 (3) |
| N1 | 0.0185 (3) | 0.0238 (3) | 0.0208 (3) | −0.0010 (3) | 0.0027 (2) | 0.0026 (3) |
| N2 | 0.0179 (3) | 0.0203 (3) | 0.0174 (3) | 0.0002 (2) | 0.0017 (2) | 0.0026 (2) |
| N3 | 0.0206 (3) | 0.0230 (3) | 0.0199 (3) | −0.0013 (3) | 0.0008 (2) | 0.0010 (3) |
| N4 | 0.0199 (3) | 0.0193 (3) | 0.0219 (3) | −0.0027 (2) | 0.0051 (2) | −0.0011 (3) |
| O1 | 0.0321 (4) | 0.0167 (3) | 0.0868 (7) | −0.0008 (3) | 0.0267 (5) | −0.0041 (4) |
| O2 | 0.0559 (5) | 0.0311 (4) | 0.0288 (4) | −0.0159 (4) | 0.0089 (4) | 0.0088 (3) |
| O3 | 0.0445 (5) | 0.0265 (4) | 0.0438 (5) | −0.0125 (3) | −0.0235 (4) | 0.0020 (3) |
| S1 | 0.01652 (9) | 0.01532 (9) | 0.02171 (10) | −0.00380 (6) | 0.00082 (7) | 0.00194 (6) |
Geometric parameters (Å, º)
| C1—C2 | 1.3612 (13) | C9—H9A | 0.9800 |
| C1—N1 | 1.3845 (12) | C9—H9B | 0.9800 |
| C1—H1 | 0.9500 | C9—H9C | 0.9800 |
| C2—N2 | 1.3809 (11) | C10—H10A | 0.9800 |
| C2—H2 | 0.9500 | C10—H10B | 0.9800 |
| C3—N1 | 1.3283 (11) | C10—H10C | 0.9800 |
| C3—N2 | 1.3541 (11) | C11—C16 | 1.3910 (12) |
| C3—C5 | 1.4848 (12) | C11—C12 | 1.3921 (11) |
| C4—N2 | 1.4613 (11) | C11—S1 | 1.7767 (8) |
| C4—H4A | 0.9800 | C12—C13 | 1.3897 (12) |
| C4—H4B | 0.9800 | C12—H12 | 0.9500 |
| C4—H4C | 0.9800 | C13—C14 | 1.3883 (15) |
| C5—H5A | 0.9800 | C13—H13 | 0.9500 |
| C5—H5B | 0.9800 | C14—C15 | 1.3866 (16) |
| C5—H5C | 0.9800 | C14—H14 | 0.9500 |
| C6—C7 | 1.3580 (14) | C15—C16 | 1.3937 (14) |
| C6—N3 | 1.3847 (12) | C15—H15 | 0.9500 |
| C6—H6 | 0.9500 | C16—H16 | 0.9500 |
| C7—N4 | 1.3815 (12) | N1—H1N | 0.83 (5) |
| C7—H7 | 0.9500 | N3—H3N | 0.89 (2) |
| C8—N3 | 1.3322 (11) | O1—S1 | 1.4489 (8) |
| C8—N4 | 1.3418 (11) | O2—S1 | 1.4465 (8) |
| C8—C10 | 1.4805 (12) | O3—S1 | 1.4484 (8) |
| C9—N4 | 1.4639 (12) | ||
| C2—C1—N1 | 109.27 (8) | C8—C10—H10C | 109.5 |
| C2—C1—H1 | 125.4 | H10A—C10—H10C | 109.5 |
| N1—C1—H1 | 125.4 | H10B—C10—H10C | 109.5 |
| C1—C2—N2 | 105.94 (8) | C16—C11—C12 | 120.15 (8) |
| C1—C2—H2 | 127.0 | C16—C11—S1 | 120.42 (6) |
| N2—C2—H2 | 127.0 | C12—C11—S1 | 119.39 (6) |
| N1—C3—N2 | 110.00 (7) | C13—C12—C11 | 119.83 (8) |
| N1—C3—C5 | 127.01 (8) | C13—C12—H12 | 120.1 |
| N2—C3—C5 | 122.99 (8) | C11—C12—H12 | 120.1 |
| N2—C4—H4A | 109.5 | C14—C13—C12 | 120.20 (9) |
| N2—C4—H4B | 109.5 | C14—C13—H13 | 119.9 |
| H4A—C4—H4B | 109.5 | C12—C13—H13 | 119.9 |
| N2—C4—H4C | 109.5 | C15—C14—C13 | 119.89 (9) |
| H4A—C4—H4C | 109.5 | C15—C14—H14 | 120.1 |
| H4B—C4—H4C | 109.5 | C13—C14—H14 | 120.1 |
| C3—C5—H5A | 109.5 | C14—C15—C16 | 120.31 (9) |
| C3—C5—H5B | 109.5 | C14—C15—H15 | 119.8 |
| H5A—C5—H5B | 109.5 | C16—C15—H15 | 119.8 |
| C3—C5—H5C | 109.5 | C11—C16—C15 | 119.60 (9) |
| H5A—C5—H5C | 109.5 | C11—C16—H16 | 120.2 |
| H5B—C5—H5C | 109.5 | C15—C16—H16 | 120.2 |
| C7—C6—N3 | 107.48 (8) | C3—N1—C1 | 106.66 (8) |
| C7—C6—H6 | 126.3 | C3—N1—H1N | 126.7 |
| N3—C6—H6 | 126.3 | C1—N1—H1N | 126.7 |
| C6—C7—N4 | 106.69 (8) | C3—N2—C2 | 108.13 (7) |
| C6—C7—H7 | 126.7 | C3—N2—C4 | 125.59 (7) |
| N4—C7—H7 | 126.7 | C2—N2—C4 | 126.12 (7) |
| N3—C8—N4 | 108.53 (8) | C8—N3—C6 | 108.47 (8) |
| N3—C8—C10 | 126.61 (8) | C8—N3—H3N | 125.8 |
| N4—C8—C10 | 124.85 (8) | C6—N3—H3N | 125.8 |
| N4—C9—H9A | 109.5 | C8—N4—C7 | 108.83 (8) |
| N4—C9—H9B | 109.5 | C8—N4—C9 | 125.08 (8) |
| H9A—C9—H9B | 109.5 | C7—N4—C9 | 126.06 (8) |
| N4—C9—H9C | 109.5 | O2—S1—O3 | 112.77 (6) |
| H9A—C9—H9C | 109.5 | O2—S1—O1 | 112.73 (6) |
| H9B—C9—H9C | 109.5 | O3—S1—O1 | 112.82 (7) |
| C8—C10—H10A | 109.5 | O2—S1—C11 | 106.84 (4) |
| C8—C10—H10B | 109.5 | O3—S1—C11 | 105.15 (4) |
| H10A—C10—H10B | 109.5 | O1—S1—C11 | 105.78 (4) |
| N1—C1—C2—N2 | 0.06 (11) | C1—C2—N2—C3 | −0.06 (10) |
| N3—C6—C7—N4 | −0.20 (11) | C1—C2—N2—C4 | −175.75 (9) |
| C16—C11—C12—C13 | 1.40 (12) | N4—C8—N3—C6 | −0.12 (10) |
| S1—C11—C12—C13 | −176.27 (7) | C10—C8—N3—C6 | 179.13 (9) |
| C11—C12—C13—C14 | −0.33 (13) | C7—C6—N3—C8 | 0.20 (11) |
| C12—C13—C14—C15 | −1.04 (14) | N3—C8—N4—C7 | 0.00 (10) |
| C13—C14—C15—C16 | 1.35 (15) | C10—C8—N4—C7 | −179.28 (9) |
| C12—C11—C16—C15 | −1.09 (12) | N3—C8—N4—C9 | 178.37 (8) |
| S1—C11—C16—C15 | 176.55 (7) | C10—C8—N4—C9 | −0.91 (14) |
| C14—C15—C16—C11 | −0.28 (14) | C6—C7—N4—C8 | 0.13 (10) |
| N2—C3—N1—C1 | 0.00 (10) | C6—C7—N4—C9 | −178.22 (9) |
| C5—C3—N1—C1 | 179.55 (9) | C16—C11—S1—O2 | 24.77 (8) |
| C2—C1—N1—C3 | −0.04 (11) | C12—C11—S1—O2 | −157.57 (7) |
| N1—C3—N2—C2 | 0.03 (10) | C16—C11—S1—O3 | −95.30 (8) |
| C5—C3—N2—C2 | −179.54 (9) | C12—C11—S1—O3 | 82.36 (8) |
| N1—C3—N2—C4 | 175.76 (8) | C16—C11—S1—O1 | 145.10 (8) |
| C5—C3—N2—C4 | −3.81 (14) | C12—C11—S1—O1 | −37.24 (9) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···N3 | 0.83 | 1.90 | 2.6970 (11) | 163 |
| N3—H3N···N1 | 0.89 | 1.81 | 2.6970 (11) | 170 |
| C1—H1···O1 | 0.95 | 2.43 | 3.3741 (13) | 170 |
| C4—H4B···O3i | 0.98 | 2.33 | 3.2956 (12) | 170 |
| C6—H6···O2 | 0.95 | 2.60 | 3.4288 (14) | 146 |
| C7—H7···O2ii | 0.95 | 2.41 | 3.3254 (12) | 162 |
| C9—H9A···O3iii | 0.98 | 2.53 | 3.4846 (14) | 164 |
| C9—H9B···O3ii | 0.98 | 2.46 | 3.4381 (14) | 173 |
| C13—H13···O1i | 0.95 | 2.67 | 3.4738 (14) | 143 |
| C14—H14···O1iv | 0.95 | 2.48 | 3.3920 (13) | 162 |
Symmetry codes: (i) −x+1/2, y−1/2, −z+1/2; (ii) −x+1, −y+1, −z; (iii) x−1/2, −y+1/2, z−1/2; (iv) x, y−1, z.
Funding Statement
Funding for this research was provided by: National Science Foundation (grant No. CHE 11625543); American Chemical Society Petroleum Research Fund (grant No. PRF 58975-UR4); Ave Maria University Department of Chemistry and Physics .
References
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst. 37, 335–338.
- Bruker (2018). APEX3 and SAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Hübschle, C. B., Sheldrick, G. M. & Dittrich, B. (2011). J. Appl. Cryst. 44, 1281–1284. [DOI] [PMC free article] [PubMed]
- Kelley, S. P., Narita, A., Holbrey, J. D., Green, K. D., Reichert, W. M. & Rogers, R. D. (2013). Cryst. Growth Des. 13, 965–975.
- Krause, L., Herbst-Irmer, R., Sheldrick, G. M. & Stalke, D. (2015). J. Appl. Cryst. 48, 3–10. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Sheldrick, G. M. (2015). Acta Cryst. C71, 3–8.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S2414314620006896/bv4031sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2414314620006896/bv4031Isup2.hkl
Supporting information file. DOI: 10.1107/S2414314620006896/bv4031Isup3.cml
CCDC reference: 2005097
Additional supporting information: crystallographic information; 3D view; checkCIF report