

SUMMARY
In Huntington’s disease (HD), the uninterrupted CAG repeat length, but not the polyglutamine length, predicts disease onset. However, the underlying pathobiology remains unclear. Here, we developed bacterial artificial chromosome (BAC) transgenic mice expressing human mutant huntingtin (mHTT) with uninterrupted, and somatically unstable, CAG repeats that exhibit progressive disease-related phenotypes. Unlike prior mHTT transgenic models with stable, CAA-interrupted, polyglutamine-encoding repeats, BAC-CAG mice show robust striatum-selective nuclear inclusions and transcriptional dysregulation resembling those in murine huntingtin knockin models and HD patients. Importantly, the striatal transcriptionopathy in HD models is significantly correlated with their uninterrupted CAG repeat length but not polyglutamine length. Finally, among the pathogenic entities originating from mHTT genomic transgenes and only present or enriched in the uninterrupted CAG repeat model, somatic CAG repeat instability and nuclear mHTT aggregation are best correlated with early-onset striatum-selective molecular pathogenesis and locomotor and sleep deficits, while repeat RNA-associated pathologies and repeat-associated non-AUG (RAN) translation may play less selective or late pathogenic roles, respectively.
Graphical abstract
In brief
Using a novel human genomic BAC transgenic mouse model of HD with long uninterrupted CAG repeats, Gu et al. provided molecular, pathological, and behavioral data to demonstrate critical pathogenic roles of uninterrupted CAG repeats in mutant HTT, beyond its encoded polyglutamine protein, in eliciting striatum-selective pathogenesis in vivo.
INTRODUCTION
Huntington’s disease (HD) is one of the most common autosomal dominant neurodegenerative disorders characterized by progressive movement disorder (i.e., chorea and dystonia), cognitive impairment, and psychiatric symptoms (Ross et al., 2014). Neuropathologically, HD is characterized by selective degeneration of the striatal medium spiny neurons (MSNs) and, to a lesser extent, the deep layer cortical neurons and associated astrocytosis and microgliosis (Vonsattel and DiFiglia, 1998). HD is caused by a CAG trinucleotide repeat expansion encoding a polyglutamine (polyQ) stretch near the N terminus of huntingtin (HTT) (The Huntington’s Disease Collaborative Research Group, 1993). Importantly, the age of HD motor disease onset is inversely correlated with the CAG repeat length, a clinical feature shared with other neurodegenerative disorders characterized by CAG repeat and polyQ expansion (Orr and Zoghbi, 2007).
Genetic mouse models of HD and other polyQ disorders have been instrumental in showing how gain-of-function toxicities from mutant polyQ proteins elicit behavioral, neuropathological, and molecular deficits in vivo (e.g., Rousseaux et al., 2018; Gray et al., 2008; Langfelder et al., 2016). However, emerging evidence suggests the potential for mutant CAG repeats to contribute to disease pathogenesis beyond encoding polyQ peptides (Lieberman et al., 2019; Malik et al., 2021). This may include distinct toxic molecular entities originating from the expanded CAG repeat locus, including somatic DNA repeat instability (Jones et al., 2017), RNA gain-of-function toxicities (Nalavade et al., 2013; Chung et al., 2011), and toxic protein products due to repeat-associated non-ATG translation (RAN; Bañez-Coronel et al., 2015). The precise pathogenic roles of these toxic mutant huntingtin (mHTT)-locus-derived toxic entities in the age-dependent, striatum-selective pathogenesis remain unclear.
Recent genome-wide association studies (GWASs) for modifiers of HD age of onset provide compelling evidence that uninterrupted CAG repeat length, instead of polyQ repeat length, is more closely associated with motor disease onset in HD (GeM-HD, 2019; Wright et al., 2019). By examining the presence, absence, or duplication of the terminal CAA-CAG sequences (both encoding glutamine residues) in HTT, it was found that HD onset is best predicted by uninterrupted CAG repeat length and less well predicted by the polyQ repeat length (GeM-HD, 2019). Moreover, the GWAS studies (GeM-HD, 2015, 2019) also identified significant modifier loci containing DNA repair genes, particularly those involved in mismatch repair (MMR), which were shown to modulate the somatic repeat instability in HD (Ciosi et al., 2019) and the striatal repeat instability and mHTT aggregation in the knockin (KI) model of HD (Wheeler and Dion, 2021). Together, these studies support the hypothesis that both the DNA repair enzymes and uninterrupted HTT CAG repeat may converge on somatic CAG repeat instability in mHTT in the vulnerable striatal neurons in HD (GeM-HD, 2019; Jones et al., 2017; Hong et al., 2021). Despite the genetic evidence thus far, it remains unclear how the uninterrupted CAG repeat in mHTT may elicit the progressive and selective pathogenesis in HD.
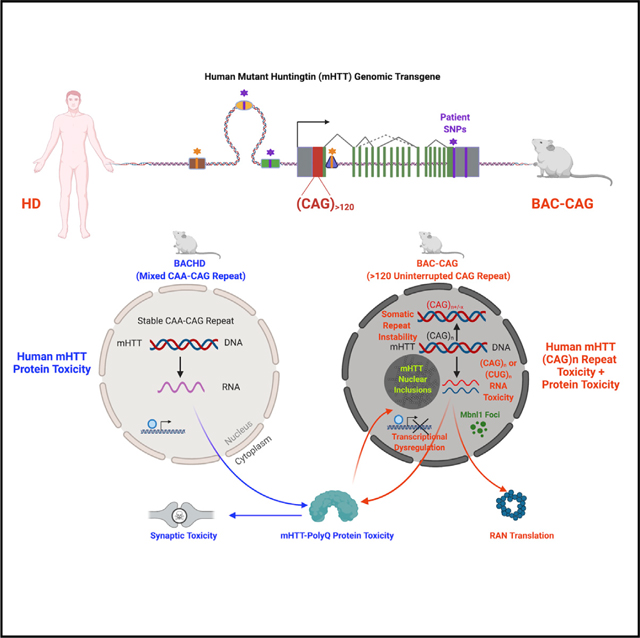
Multiple genetic mouse models expressing full-length mutant Huntingtin have been generated to investigate HD pathogenesis and test candidate therapeutics (Ehrnhoefer et al., 2011; Yang and Gray, 2011). The KI mouse models have an allelic series of expanded CAG repeat in the endogenous murine Huntingtin (Htt) locus either in the context of human HTT exon1 (Ehrnhoefer et al., 2011; Langfelder et al., 2016) or murine mutant Huntingtin (mHtt) exon 1 (Kumar et al., 2016). The KI models with human mHTT exon 1 and high CAG repeat lengths (e.g., Q111, Q140, and Q175) show striatum-selective nuclear mHTT aggregation and nuclear inclusions (NIs), CAG-length-dependent transcriptionopathy, somatic CAG repeat instability, and some behavioral impairment (Ehrnhoefer et al., 2011; Yang and Gray, 2011). Other widely used full-length mHTT mouse models are the human HTT genomic DNA transgenic mouse models, including BACHD and YAC128, which display progressive behavioral deficits, selective cortical and striatal atrophies (Gray et al., 2008; Slow et al., 2003; Menalled et al., 2009). These models have the intact human HTT genomic locus as transgenes, and the BACHD model also has several HD-patient-associated single-nucleotide polymorphisms (SNPs) (Carroll et al., 2011; Southwell et al., 2013). Thus, the BACHD and YAC128 models play pivotal roles in preclinical testing of therapeutics, specifically targeting human HTT sequences (Kordasiewicz et al., 2012; Lu and Yang, 2012; Southwell et al, 2018), including those selectively targeting the patient-associated allele in BACHD (Monteys et al., 2017; Southwell et al, 2018). Both BACHD and YAC128 contain CAA-interrupted CAG repeats, with BACHD having a polyQ stretch encoded by a mixed CAA-CAG repeat sequence (Gray et al., 2008) and YAC128 having 9 CAA interruptions among CAG repeats (its longest uninterrupted repeat being 81; Pouladi et al., 2012). Therefore, both BACHD and YAC128 mice have stable HTT repeat DNA sequences in somatic tissues and germline (Gray et al., 2008; Farshim and Bates, 2018). Moreover, both BACHD and YAC128 mice show significant weight gain due to human HTT overexpression (Pouladi et al., 2010) and lack robust striatal and cortical NIs and transcriptional dysregulation (Gray et al., 2008; Bayram-Weston et al, 2015). Currently, it is unclear whether the phenotypic differences between the murine KI models and human genomic transgenic models of HD are due to the many species-specific sequence differences in the huntingtin genomic DNA, RNA, or protein or due to the multiple CAA interruptions in the BAC and YAC models.
This study describes the generation and characterization of a new human HTT genomic BAC transgenic mouse model of HD expressing full-length human mHTT with about 120 uninterrupted CAG repeats (BAC-CAG). This model demonstrates that long uninterrupted CAG repeats in human mHTT transgene can elicit multiple HD-like features that eluded previous human genomic transgenic mouse models, including minimal body weight gain, somatic CAG repeat instability that is significantly correlated with behavioral deficits, striatum-selective and progressive NIs and transcriptional dysregulation, and striatal astrocytosis and microgliosis. Moreover, by analyzing the spatiotemporal emergence of mHTT repeat instability, repeat-containing sense and antisense transcripts, nuclear foci formation of Mbnl1, which binds to CG-rich repeat expansions, and RAN translation products, our study provides new insights into the relationships between these mHTT repeat-associated toxic entities in striatum-selective and progressive neuropathogenesis in vivo. The latter includes possible early contributing roles of CAG-and CUG-repeat-associated RNA toxicities leading to striatal and cortical Mbnl1 foci formation and the late-onset striatal and cortical accumulation of aggregated mHTT RAN products, which are unlikely to play an early pathogenic role in this model but may contribute to disease progression.
RESULTS
A BAC transgenic mouse model expressing human mHTT with a long uninterrupted CAG repeat
To create a human genomic transgenic mouse model of HD with a long uninterrupted CAG repeat in HTT, we engineered a human mHTT-exon 1 construct with about 120 pure CAG repeats followed by one CAA, 8 CAGs, and another CAA-CAG, encoding about 131 glutamine repeats (Figure 1A; Gray et al., 2008; Gu et al., 2009, Gu et al., 2015; Yang et al., 1997). Besides the long uninterrupted CAG repeat, another key difference in the new mHTT BAC construct from the BACHD is the absence of LoxP sites and some silent coding variants for the N17 domain of mHTT. Thus, the new BAC construct contains only the human HTT genomic DNA and is free of other noncognate genetic footprints in or near mHTT-exon 1. Through pronuclear injections of FvB/NJ inbred mouse embryos, we obtained multiple transgenic founders that gave rise to germline transmission of the transgenes. We identified two lines expressing the full-length human mHTT protein without any artificial mHTT fragments, and mHTT repeat sizing and sequencing revealed a single uninterrupted CAG repeat size above 120 and with a total polyQ repeat length of about 131. Quantitative genomic DNA PCR assay suggests that these two lines have two copies of the BAC transgene inserted into their genomes (data not shown). We focused our study on one of the two lines, named BAC-CAG, based on its relatively high level of full-length mHTT expression and good breeding characteristics.
Figure 1. BAC-CAG mouse model hosts a full-length human mutant huntingtin BAC with over 120 uninterrupted CAG repeats in exon 1.

(A) Schematic representation of the human huntingtin BAC with its exon 1 replaced with mHTT-exon 1 containing over 120 CAG repeat and encoding over 130 glutamine repeat.
(B) Representative western blot assay of brainstriatal tissue lysates from BAC-CAG mice at 2 months of age, probed with anti-polyQ 1C2 antibody. Striatal tissue lysates from WT and BACHD set as negative and positive controls, respectively. α-tubulin was set as the loading control.
(C) BAC-CAG mice display mild body weight gainin males only at 12 months of age (2 months of age: 25 WT males and 15 BAC-CAG males, 26 WT females and 20 BAC-CAG females; 6 months of age: 16 WT males and 15 BAC-CAG males, 24 WT females and 26 BAC-CAG females; 12 months of age: 12 WT males and 24 BAC-CAG males, 23 WT females; 29 BAC-CAG females).
(D and E) Significantly reduced performance in rotarod testing in 6- and 12-month BAC-CAG mice (black bar) compared with WT (white bar) mice. Results are shown as mean ± SEM; *p < 0.05, **p < 0.005. Two-way ANOVA test indicates no genotypes and times interactions (6 months F(48,66) = 0.2808, p > 0.9999; 12 months F(52,78) = 0.4349, p = 0.9991). Unpaired t test for 6 months, day 1 (p = 0.3255), day 2 (p = 0.0068), day 3 (p = 0.0063), BAC-CAG (n = 22), WT (n = 25); for 12 months, day 1 (p = 0.0388), day 2 (p = 0.0044), day 3 (p = 0.001), BAC-CAG (n = 27), WT (n = 26).
(F) Diminished grip strength in 12-month BAC-CAG mice (black bar) compared with WT (white bar) mice. WT (n = 11), BAC-CAG (n = 13).
(G) Compromised rhythms in cage activity in 12month BAC-CAG mice. Running averages (1-h bin) of immobility-defined sleep in wild-type (WT, open circles) and BAC-CAG (filled circles) mice are plotted. ZT, zeitgeber time. 24-h profiles of locomotor activity were analyzed using a two-way ANOVA with genotype and time as factors, followed by Holm-Sidak’s multiple comparisons test, *p < 0.05. Data points are shown as mean ± SEM, WT (n = 9), BAC-CAG (n = 11). The white/black bar on the top of the graph indicates the 12:12-h LD cycle.
(H) Altered timing of sleep behavior in BAC-CAG mice. 24-h profiles of sleep behavior were analyzed using a two-way ANOVA with genotype and time asfactors, followed by Holm-Sidak’s multiple comparisons test, *p < 0.05. Data points are shown as mean ± SEM, WT (n = 10), BAC-CAG (n = 13). The white/ black bar on the top of the graph indicates the 12:12-h LD cycle. See also Figures S1 and S2.
Western blot analysis with an anti-polyQ antibody (1C2) revealed that BAC-CAG mice express intact mHTT in the striatum at 2 months of age, which showed minimal soluble mHTT fragments at this age (Figure 1B). Despite BAC-CAG mice expressing polyQ protein with a longer glutamine repeat than BACHD mice (130 versus 97), quantitation of western blots revealed that the 1C2 signals in BAC-CAG mice is about half the protein level that of BACHD mice (Figure S1A). To more accurately quantify the relative protein level of BAC-CAG mice versus BACHD, we used MAB2166, which recognizes an N-terminal epitope that is conserved between human and mouse (Cong et al., 2005) but shows stronger binding to wild-type (WT) murine Htt than to mHTT (Macdonald et al., 2014; Franich et al., 2018). Since endogenous murine Htt, as detected by MAB2166, does not appear to differ from that of WT mice (Macdonald et al., 2014), we reasoned that we can accurately compare the human mHTT levels in BAC-CAG and BACHD mice by normalizing the mHTT levels to endogenous murine Htt. In this assay, BAC-CAG mice express about 26% mHTT compared with BACHD in the striatum (Figures S1B and S1C). Since BACHD mice express about 2-fold of the endogenous HTT level (Gray et al., 2008), we estimated that BAC-CAG mice express approximately 50% of the endogenous HTT level. At the transcript level, the total human HTT mRNA in the striatum of BAC-CAG mice is less than 50% of that in BACHD mice (Figure S2A; based on RNAseq reads; STAR Methods). Moreover, the transcript level of total human HTT in BAC-CAG mice is relatively constant between 2 and 6 months and has modest increase between 6 and 12 months (Figure S2B). Together, we conclude that BAC-CAG mice express much lower levels of human mHTT RNA and protein than BACHD mice.
Unlike HD patients and KI mice, earlier human mHTT transgenic models (BACHD and YAC128) show 20%–30% body weight gain due to human HTT overexpression (Gray et al., 2008; Pouladi et al, 2010). Interestingly, BAC-CAG female mice did not show any body weight gain at 2, 6, or 12 months of age compared with WT females, and BAC-CAG male mice show only a modest, but significant, weight gain at 12 months of age (about 5%; p < 0.05) (Figure 1C). The lack of body weight gain could be due to the relatively low level of human mHTT overexpression in BAC-CAG mice, but one cannot rule out the possible effects of the uninterrupted CAG repeats. Thus, unlike prior human mHTT transgenic mice models (Menalled et al., 2009), the behavioral phenotypes of the BAC-CAG model could not be confounded by body weight changes.
Motor and sleep deficits in BAC-CAG mice
Motor deficits are major symptoms of HD patients (Ross et al., 2014), and impaired motor coordination (i.e., accelerating rotarod test) and locomotor activities have been shown in previous mouse models, such as BACHD (Gray et al, 2008; Menalled et al., 2009). The BAC-CAG mice showed normal accelerating rotarod performance at 2 months of age (Figure S2C). At 6 and 12 months of age, BAC-CAG mice showed significant impairment in this test compared with WT controls (Figures 1D and 1E). Moreover, we found that 12 months BAC-CAG mice showed highly significant decrease in weight-adjusted grip strength, a test of muscle strength (Figure 1F). Similar grip strength deficits were found in mHTT fragment models (i.e., R6/2) and homozygous Q150 and Q175 KI mice but not in BACHD, YAC128, or heterozygous KI models (Woodman et al, 2007; Menalled et al., 2009).
One of the early and common impairments in HD patients is sleep disturbance (Morton, 2013), and disrupted circadian locomotor activities have been found in R6/2 and BACHD models but were not apparent in heterozygous Q140 or Q175 KI mice (Morton et al., 2005; Kudo et al., 2011; Loh et al., 2013). We first evaluated the diurnal rhythms of locomotor activities of 12-month-old BAC-CAG mice and WT littermate controls over a 10-day period (Figure 1G). Both WT and mutant mice displayed diurnal rhythms of activities. BAC-CAG mice exhibited reduced total locomotor activity (t test, t = 2.367, p = 0.03), which was mostly driven by reduced nighttime activity (t test, t = 2.679, p = 0.01) without daytime changes (Mann-Whitney rank-sum test, U =33.0, p = 0.224). The time of activity onset was delayed in the BAC-CAG mice (ZT 12.4 ± 0.3) compared with that in the WT controls (ZT 11.7 ± 0.4), but this delay was not significant (rank-sum test, U = 34.0, p = 0.109). The BAC-CAG mice exhibited significantly (t test, t = −2.531, p = 0.02) more activity bouts (8.7 ± 0.5) than WT controls did (6.7 ± 0.6), but the average bout duration did not vary between the genotypes. Thus, the BAC-CAG mice exhibit significant deficits in their activity rhythms with less activity during the night and more fragmented activity patterns compared with WT littermates.
Next, we sought to determine the impact of the BAC-CAG mutation on the temporal pattern of sleep behavior in regular light-dark (LD) conditions. Both WT and mutant mice exhibited robust daily rhythms in sleep behavior (Figure 1H). The BAC-CAG mice exhibited significantly reduced total sleep (Mann-Whitney rank-sum test, U = 32.000, p = 0.044) that was driven by reduced daytime sleep (rank-sum test, U = 30.000, p = 0.032) without changes at night (t test, t = 1.5, p = 0.138). Analysis using 1-h bins (two-way ANOVA) found a significant effect of genotype (F(1,551) = 47.510, p < 0.001) and time (F(23,551) = 15.410; p < 0.001) on sleep behavior (Figure 1H). The percentage of daytime sleep was significantly lower (t test, t = 2.7; p = 0.0123) in the mutants (56 ± 1.2%) than in WT (61% ± 0.7%) mice. BAC-CAG mice also exhibited significantly (t test, t = 3.767, p = 0.001) more sleep bouts (9.4 ± 0.6) than WT controls (5.9 ± 0.7), but the average bout duration did not vary between the genotypes. Thus, the BAC-CAG mice exhibited an abnormal sleep distribution, characterized by less sleep during the day and a more fragmented pattern of sleep than WT littermates. Importantly, this HD-like sleep disruption phenotype is similar to that found in BACHD mice and is absent in mHtt heterozygous KI mice (Kudo et al., 2011; Loh et al., 2013).
Striatal MSNs and glial cell pathology in BAC-CAG mice
An important question is whether BAC-CAG mice demonstrate HD-like neuropathology, such as cortical and striatal atrophy, MSN synapse loss, astrocytosis, and microgliosis (Vonsattel and DiFiglia, 1998). We first evaluated forebrain and cerebellar weights as readouts of forebrain-selective atrophy, as seen in aged BACHD and YAC128 mice (Gray et al., 2008; Pouladi et al., 2012). We did not detect any significant forebrain or cerebellar weight loss in BAC-CAG mice at 12 or 22 months of age compared with age-matched WT controls, suggesting lack of overt brain atrophy in this model (Figures S2D and S2E).
A consistent neuropathological finding in HD postmortem brains and some HD mouse models is synapse loss in the striatal MSNs (Veldman and Yang, 2018). Prior studies have shown loss of MSN postsynaptic marker proteins, such as Actn2 in BACHD and Q140 mice (Wang et al., 2014; Langfelder et al., 2016; Wood et al., 2019). We found a significant reduction of immunostained Actn2 levels in the striata of BAC-CAG compared with WT mice at 12 months of age (Figures 2A and 2B, p < 0.01, Student’s t test). To more rigorously quantify the MSN dendritic spines in these mice, we crossed the genetic sparse-labeling MORF3 mice (Veldman et al., 2020) and Camk2a-CreER onto the BAC-CAG or WT background. The MORF3/Camk2a-CreER mice confer sparse and bright labeling of the striatal MSNs with a membrane-bound tandem smFP-V5 reporter, enabling the imaging of MSN spines (Veldman et al., 2020). We quantified the spine densities of MORF3-labeled MSNs in the dorsolateral striatum of BAC-CAG and WT mice at 12 months of age and found a significant reduction in the spine densities in BAC-CAG mice (Figures 2C and 2D). Together, the two independent methods demonstrate a significant loss of striatal MSN synapses in BAC-CAG mice at 12 months of age compared with WT controls.
Figure 2. Neurodegenerative pathology changes in 12-month-old BAC-CAG mice.

(A and B) Significant reduction of Actn2(+) intensity in striatum of 12-month BAC-CAG mice. Results are shown as mean ± SEM from 5 brains for each group; *p < 0.05. Unpaired t test. Scale bars, 10 mm.
(C and D) Significant spine loss in striatal medium spiny neurons in 12-month-old BAC-CAG mice. Representative micrographs of striatum. Results are shown as mean ± SEM from 15 neurons from 3 mice for each group; **p < 0.01. Scale bars, 10 mm.
(E–I) Representative images of GFAP immunohistochemical staining of striatum (E) and cortex/corpus callosum (F) in WT and BAC-CAG brains. Quantification of the intensities of GFAP staining for striatum (G), cortex (H), and corpus callosum (I) is shown. Results are shown as mean ± SEM from 3 brains for each group. **p < 0.005. Unpaired t test. Scale bars, 20 μm in (E) and 100 μm in (F). See also Figure S3.
We next examined the BAC-CAG mice for the presence of reactive astrogliosis or microgliosis, which are neuropathological features of HD (Vonsattel and DiFiglia, 1998; Sapp et al., 2001). The 12-month BAC-CAG striata and corpus callosum exhibited significant increase in reactive astrogliosis, as indicated by elevated levels of glial fibrillary acidic protein (GFAP) expression and hyper trophic morphology of the reactive astrocytes (Figures 2E–2I). Such an increase in astrogliosis was not observed in the cortex (Figures 2F and 2H). We evaluated microgliosis using immunostaining for general microglial marker Iba1 and activated microglia marker galectin-3 (Gal3, also known as Mac-2 or Lgals3). These genes are elevated in the brains of patients with Alzheimer’s disease and HD and of mouse models (Rahimian et al., 2021; Lee et al., 2018; Siew et al., 2019; Gu et al., 2015). As shown in Figure S3A, the Iba1 staining revealed that microglia in BAC-CAG striata at 12 months showed more heterogenous and activated morphology compared with those in the WT controls. Moreover, significantly higher number of Gal3+-activated microglia were observed in BAC-CAG striata at 12 months and 18–22 months compared with age-matched WT controls (Figures S3B and S3C; p < 0.001, Student’st test).Thesereactive gliosis phenotypes were not found in prior mHTT genomic transgenic or KI mouse models (Ehrnhoefer et al., 2011; Yang and Gray, 2011).
Progressive striatum-selective nuclear mHTT inclusion and aggregation pathology in BAC-CAG mice
NIs containing mHTT are a hallmark of HD (DiFiglia et al., 1997) and prominent striatum-selective pathological features of multiple mHtt KI mice (Li et al., 2000; Menalled et al., 2003; Carty et al., 2015). However, NIs or aggregated nuclear mHTT are rare in BACHD striata (Gray et al., 2008), and YAC128 mice show predominantly diffuse striatal and cortical nuclear mHTT staining (Pouladi et al., 2012; Van Raamsdonk et al., 2005) and a few NIs at 15 months or older age (Bayram-Weston et al., 2015). We stained BAC-CAG mice at multiple ages using an aggregate-sensitive human HTT antibody, S830 (Sathasivam et al., 2001; Figures 3A–3D). Although we did not detect any S830+mHTT aggregates in 6-month BAC-CAGstriata (FigureS4), we found diffuse nuclear mHTT accumulation in about 36% of striatal NeuN+ neurons (178/500) in 12-month BAC-CAG mice (Figure 3B), with no such staining in 6- to 18-month WT mice (Figure 3A; data not shown). Importantly, at 18 months, about 98% of the striatal NeuN+ neurons in BAC-CAG showed NIs (491/500) (Figure 3C). In the cortex, we did not see any diffuse nuclear mHTT staining at 6 or 12 months of age (Figure S4; data not shown). At 18 months, we observed mHTT NIs and diffuse nuclear staining in only about 5%–10% of deep layer cortical pyramidal neurons (Figure 3D) but not in upper cortical layer neurons (Figure S4). The deep-cortical-layer distribution of NIs in BAC-CAG mice is reminiscent of that found in the patients (DiFiglia et al., 1997) and is unlike the predominantly upper cortical layer distribution of mHTT nuclear aggregation in the Q140 and Q175 KI mice (Carty et al., 2015; Menalled et al., 2003).
Figure 3. Progressive S830(+) mHTT nuclear accumulation and aggregation in striatum and deeper cortical layers of BAC-CAG mice.
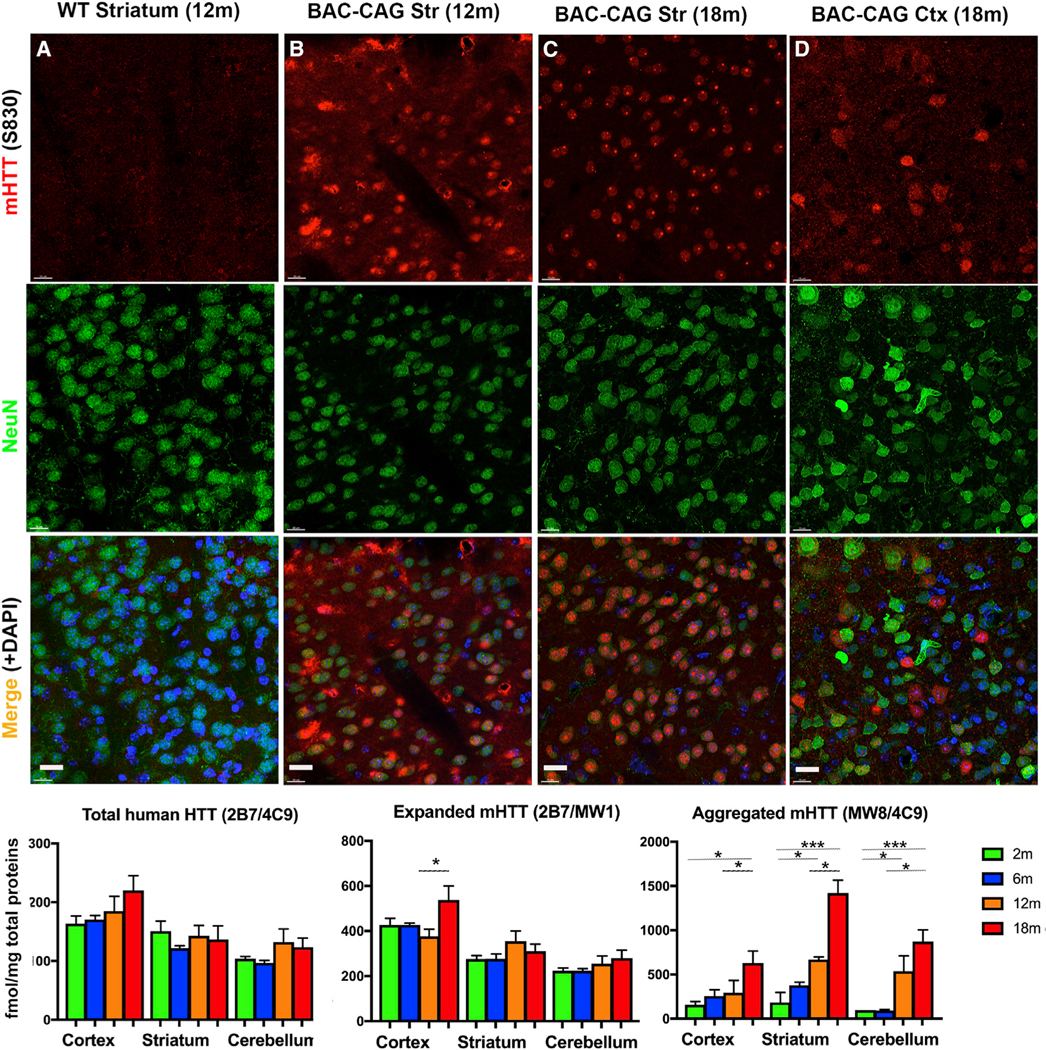
(A) No detection of S830 signal in WT control mice.
(B) Nuclear accumulation and aggregation of mHTT in striatum of 12-month-old mice.
(C) mHTT Nuclear aggregates and inclusions in striatum of 18-month-old mice.
(D) Nuclear and cytosolic mHTT accumulation and aggregates in deeper cortical layers of 18-month-old mice.
(E–G) MSD assay for total, expanded, and aggregated mHTT forms in 2- to 18-month-old cortex, striatum, and cerebellum tissues. Results are shown as mean ± SEM, n = 4 for each age groups. *p < 0.05, ***p < 0.001. Scale bars, 20 mm. See also Figure S4
To quantitatively evaluate the soluble and aggregated mHTT in BAC-CAG mice, we used the Meso Scale Discovery (MSD) assay, which can accurately report different HTT species, including soluble human HTT monomer, soluble expanded mHTT, and aggregated mHTT, based on distinct capture and detection antibody pairs (Macdonald et al., 2014; Reindl et al., 2019) (Figures 3E–3G). We did not detect significant changes in the levels of total human HTT or expanded mHTT in the cortex, striatum, or cerebellum of BAC-CAG mice at four ages (2, 6, 12, and 18 months), with the exception of a significant increase in cortical expanded mHTT between 12 and 18 months (Figures 3F and 3G). Since these two assays measure the same soluble mHTT species, we concluded that there was no consistent increase in the soluble mHTT species in the three brain regions between 2 and 18 months. When we measured aggregated mHTT with MSD assay, we found a significant increase in the BAC-CAG striatum at 12 months compared with that at 2 months, and this further increased at 18 months compared with that at 6 and 12 months (Figure 3G). In the cortex, aggregated mHTT was significantly increased only at 18 months compared with that at 2 months. Lastly, in the cerebellum, the aggregated mHTT was increased at 12 months compared with that at 2 months and increased at 18 months compared with that at 12 and 6 months. Overall, the MSD assays reveal that the earliest and most robust mHTT aggregation occurs in the striatum of BAC-CAG mice, with other HD-relevant brain regions (e.g., cortex) also showing progressive mHTT aggregation, albeit at a slower pace.
Age-dependent and striatum-selective transcriptional dysregulation in BAC-CAG mice
A salient molecular pathogenic feature of HD patients and allelic series mHtt KI mice is age- and Htt-CAG-length-dependent, striatum-selective transcriptional dysregulation (Langfelder et al., 2016; Kuhn et al., 2007). Studies thus far have not been able to discern whether transcriptional dysregulation in HD is due to only polyQ protein toxicities or whether the uninterrupted CAG repeats, which encode the polyQ protein, also play a role. To examine this question, we first performed RNA-seq studies of BACHD mice at 12 months and found only 50 significantly differentially expressed (DE) genes (false discovery rate or FDR < 0.1) in the striatum compared with WT littermates, 31 downregulated and 19 upregulated genes (Figure S5; Table S1). Moreover, the BACHD cortex at 12 months showed only 13 significant DE genes (Figure S5; Table S1). Enrichment analysis using striatal DE genes with less stringent statistical threshold (p < 0.01) showed that the DE genes between BACHD and WT are enriched with those found in mHtt KI mice and two of the mHtt CAG length-dependent coexpression modules, M2 and M7 (Langfelder et al., 2016). The downregulated DE genes are enriched with general neuronal marker genes, and upregulated DE genes are enriched with oligodendrocyte marker genes (Figure S5). We also reanalyzed the published striatal transcriptomic data for 12-month YAC128 mice (Bayram-Weston et al., 2015; Table S1; see STAR Methods for reanalysis of the YAC128 transcriptomic data) and found 81 significant DE genes (FDR < 0.1) compared with WT controls, 63 downregulated and 18 upregulated genes. Overall, the number of DE genes in the BACHD and YAC128 striata at 12 months is far fewer than the significant DE genes found in the striatum of the allelic series mHtt KI mice with comparable polyQ lengths at 6 and 10 months of ages (Langfelder et al., 2016).
We next performed RNA-seq analysis of the striatum and cortex of BAC-CAG mice at 2, 6, and 12 months of age (N = 6 per genotype, sex balanced). Interestingly, we observed an age-dependent transcriptionopathy in the striatum of BAC-CAG mice, with only 7 and 36 significant DE genes in 2- and 6-month striatum (FDR < 0.1), respectively but 820 DE genes (538 downregulated and 282 upregulated genes) at 12 months of age (FDR < 0.1; Figure 4A; Table S2). The transcriptomic deficits in this model are highly striatum selective, as we only detected 4, 3, and 14 DE genes (FDR < 0.1) at 2, 6, and 12 months, respectively, in the cortices of BAC-CAG compared with WT mice (Figure 4A; Table S2). Remarkably, we found a highly significant positive transcriptome-wide correlation of Z statistics of DE genes (r = 0.62, p < 1E™200) between BAC-CAG versus WT at 12 months and Q140 versus WT at 6 months (Figure 4B). This result demonstrates a high concordance of both upregulated and downregulated genes in the BAC-CAG striatum compared with those found in the Q140 KI mice. The BAC-CAG and Q140 KI models share 402 significantly downregulated genes and 188 significantly upregulated genes in the striatum (Table S2). The top shared DE genes in BAC-CAG and Q140 have different kinetics with regard to age dependency; in Q140 mice, the main changes emerge at 6 months of age, whereas in BAC-CAG mice, the changes emerge at 12 months of age (Figure 4C). It is notable that even at 6 months of age, several DE genes are already significantly downregulated (i.e., Actn2, Hcn2, Pcp4, Phex, Rgs4, Sema7a, St8sia2, Shank3, Lrrk2, Cdk19, Odf4, and Myh7b) or upregulated (i.e., Adnp, Col6a2, Htr2c, Ksr1, Tnnt2, Ly6h, Pcdh20, Phlpp2, and Gpr101) in both BAC-CAG (FDR < 0.1) and allelic series KI Q140 and Q175 mice (Table S2 and S3; Langfelder et al., 2016). These genes may represent an early and high-confidence set of dysregulated gene expression signature in the BAC-CAG striatum at 6 months of age.
Figure 4. RNA-seq analysis of striatum and cortex data from 2-, 6-, and 12-month-old BAC-CAG mice.

(A) Number of significantly DE genes at FDR < 0.1 in striatum and cortex at 2, 6, and 12 months. Blue (red) represent downregulated/upregulated genes.
(B) Scatterplot of DE Z statistics for Q140 versus Q20 from (Langfelder et al., 2016; y axis) against BAC-CAG versus WT (x axis). Each point represents a gene. Gray lines show approximate locations of FDR < 0.1 thresholds. Insets in corners give the number of genes and hypergeometric enrichment p value for each of the regions with genes DE at FDR < 0.1 in both datasets.
(C and D) Heatmaps of DE Z statistics of top downregulated genes in 12-month BAC-CAG versus WT (C), top upregulated genes in 12-month BAC-CAG versus WT (D).
(E and F) Selected terms (reference gene sets) with high enrichment significance for downregulated genes (E) and upregulated genes (F). Bar length represents –log10 of the Bonferroni-corrected enrichment p value. Red line marks the threshold PBonferroni = 0.05. See also Figures S5–S8.
Enrichment analysis showed that both the downregulated and upregulated striatal DE genes (FDR < 0.1) in 12-month BAC-CAG mice compared with those in WT controls are strongly enriched with those in the allelic series KI mice (FDR = 4.46E-241 for downregulated genes in the striatum of Q175; FDR = 9.31E−100 for upregulated in the striatum of Q140) (Langfelder et al., 2016; Figures 4E and 4F; Table S3). They are also enriched with several top mHtt CAG length-dependent coexpression modules, including the downregulated modules (M2, FDR = 2.56E−197; M11, FDR = 1.78E−13) and upregulated modules (M20, FDR = 2.68E−35; M39, FDR = 8.42E−13) (Langfelder et al., 2016; Figures 4E and 4F; Table S3). Interestingly, similar to the allelic series KI mice, BAC-CAG mice also showed a selective downregulation of MSN identity genes, including D2-MSN marker genes (FDR = 7.61E−21) and striatum-specific Allen Brain Atlas (ABA) marker genes (FDR = 5.90E−26) but not ABA general neuronal marker genes (Figures 4E, 4F, and S6A; Table S3). Moreover, similar to the allelic series mHtt KI mice, BAC-CAG also showed a significant upregulation of clustered protocadherin genes compared with WT controls (FDR = 3.99E−07; Figures 4E, 4F, and S6B). Moreover, we performed Ingenuity pathway analysis (IPA, Qiagen) of striatal DE genes (p < 0.01) in the 12-month BAC-CAG versus WT mice and found significantly enriched terms, including G-protein-coupled receptor signaling, dopamine-DARPP32 feedback in cAMP signaling, CREB signaling in neurons, and glutamate receptor signaling and IPA upstream regulators significantly enriched for levodopa (i.e., dopamine signaling), HTT, and TCF7L2 (Table S4). The IPA enrichment terms are highly analogous to those found in the allelic series KI mice (Langfelder et al., 2016). Finally, comparison of shared significantly DE transcription factors, chromatin and RNA-binding proteins, revealed 20 genes that are significantly downregulated (e.g., Zfp365, Rarb, Dbp, Kdm3a, Zswim6, Id4, Ciart, Celf1, Celf2, Tcf7, Taf4, Npas2, and Dlx6) or upregulated (e.g., Zfp488, Isl1, Myt1, tet1, Olig1, and Sox11) in the striatum of both BAC-CAG and allelic series KI mice (Figures S6C and S6D), which constitute an independently verified set of candidate transcription or chromatin regulators that may play more direct roles in striatal transcriptionopathy in HD.
To assess whether the striatal DE genes in BAC-CAG mice are similar to those found in the caudate and putamen of HD postmortem brains, we performed an enrichment analysis of striatal DE genes in BAC-CAG, BACHD, and Q140 KI mice using two independent patient-derived datasets (Hodges et al., 2006; Durrenberger et al., 2015). Importantly, we found that downregulated DE genes in the striatum of BAC-CAG at 12 months (but not at 2 or 6 months) and in Q140 KI mice at 6 and 10 months (but not at 2 months) significantly overlap with downregulated genes in the HD patient datasets (Figure S7). However, upregulated genes in both BAC-CAG and Q140 mice do not significantly overlap with those found in the HD patients (Figure S7), which may reflect the lack of robust HD-like neuroinflammation in these mouse models (Langfelder et al., 2016). Finally, BACHD striatal DE genes do not significantly overlap with those found in the HD patient caudate and putamen. To further validate a subset of the DE genes found in BAC-CAG mice at 12 months, we performed quantitative reverse-transcriptase PCR analysis and confirmed 9 significant DE genes between BAC-CAG and WT mice (Figure S8).
In summary, compared with BACHD and YAC128, the BAC-CAG is the first full-length human genomic, transgenic mouse model with robust striatum-selective transcriptionopathy that closely resembles the mHtt CAG length-dependent transcriptionopathy defined by the allelic series KI mice (Langfelder et al., 2016) and partially overlap with those found in HD patient brains.
Uninterrupted CAG repeat length, but not glutamine repeat length, in mHTT predicts striatal transcriptionopathy in HD mouse models
To compare all the available full-length murine and human mutant huntingtin models in terms of their transcriptional dysregulation, we correlated transcriptome-wide DE gene Z statistics in mutant mice versus controls for the allelic series KI mice (Langfelder et al., 2016) at 6 months and human genomic transgenic models at 12 months (Figures 5A and S5). The ages were chosen based on the onset of significant transcriptional dysregulation in these models. Interestingly, hierarchical clustering based on the DE gene correlations revealed that only BAC-CAG striatal DE gene clustered within the allelic series KI mice between Q111 and Q92, while YAC128 and BACHD clustered outside of Q80 (Figure 5A). The close clustering of the BAC-CAG DE genes with the allelic series KI mice was only observed in the striatal transcriptomes, as the cortical BAC-CAG transcriptome-wide DE genes Z statistics clustered outside the allelic series KI mice (Figure S9A).
Figure 5. Comparison of striatal transcriptionopathy between BAC-CAG and other full-length HD mouse models.

(A) Heatmap representation of genome-wide correlations of DE z statistics across 8 HD mouse models. Correlations larger than 0.2 are also shown as numbers in the heatmap. The clustering tree on the left represents average-linkage hierarchical clustering based on the correlations. (B and C) Scatterplots of uninterrupted CAG length (B) and Q-length (C) versus mean concordance for each HD model (the mean concordance is the column-wise mean of the nondiagonal elements in the heatmap (A). Each point represents a full-length HD mouse model. See also Figure S9.
We next asked whether the transcriptomic concordance among the full-length huntingtin genomic transgenic or knockin models could be related to either the uninterrupted CAG repeat length or the glutamine repeat (Q) length. Although the uninterrupted CAG length and Q-length match well in the allelic series KI mice, they are quite divergent in the human genomic models (i.e., the longest continuous CAG repeat length of 2 within a Q-length of 97Q in BACHD, the longest continuous CAG repeat length of 81 in 125Q in YAC128, and the longest continuous CAG repeat length of about 123 and 133Q in BAC-CAG; Gray et al., 2008; Pouladi et al., 2012; Figure 1A). To answer this question, we defined an average concordance for each model as the mean of the correlations of DE Z statistics of the model with all other models. Interestingly, there is a robust positive correlation between the average concordance and uninterrupted mutant huntingtin CAG repeat length in all these models (Cor = 0.91, p = 0.00017; Figure 5B), while the correlation with glutamine repeat length is much weaker and is not statistically significant (Figure 5C). Moreover, the correlation between uninterrupted mutant huntingtin CAG repeat length and average DE concordance was not observed in the cortical transcriptomic datasets from these HD mouse models (Figures S9B and S9C).
BAC-CAG mice exhibit mHTT somatic repeat instability that is correlated with behavioral impairment
To examine whether BAC-CAG mice exhibit CAG repeat instability in somatic tissues, we calculated repeat instability index scores, which represent the size and distribution of PCR products that are amplified from the human mHTT transgene and encompassing the expanded CAG repeat region of mHTT alleles (Lee et al., 2010; STAR Methods). Unlike BACHD, which has a single peak due to its mixed CAA-CAG repeats (Gray et al., 2008), BAC-CAG mice show a broad distribution of peaks in GeneMapper traces, representing mHTT CAG repeat instability in somatic tissues, at 2 and 12 months (Figures 6A and 6B). Importantly, between 2 and 12 months of ages, the striatum and liver, but not cortex, cerebellum, heart, testis, or tail, showed significant increases in repeat instability indices (Figure 6B). Such tissue-specific increases in repeat instability indices have been shown in other HD mouse models, including Q111 KI (Lee et al., 2011), R6/2 (Mangiarini et al, 1997), and Q140 mice (data not shown).
Figure 6. Somatic CAG instability in BAC-CAG mice and repeat instability correlation with behavior phenotypes.

(A) Representative GeneMapper traces of PCR amplified mHTT CAG repeats from genomic DNAs isolated from somatic tissues of 2- and 12-month BAC-CAG mice.
(B) Somatic CAG instability indices from different tissues. Only aging striatum and liver tissues show significant increase of indices from 2 to 12 months (see thenumber of mice used in STAR Methods).
(C) Heatmap representation showing association of somatic CAG instability with locomotor and sleep behaviors. Color in the heatmap represents association z statistics; nominal p values are shown for cells where p < 0.05.
(D) Scatterplot of night sleep time versus somatic CAG instability in striatum.
(E) Scatterplot of nighttime locomotor activity versus cortical CAG instability.
Prior studies in these models have not established a link between mHtt CAG repeat instability indices and disease pathophysiology. To explore this association, we took advantage of the behavioral phenotyping data of 12-month-old BAC-CAG mice (n = 13) used for the 10-day continuous monitoring of sleep and locomotor activities (Figures 1G and 1H) and sized the repeat instability in the striatum, cortex, and cerebellum of these mice. The repeat instability index of the cerebellum was not significantly associated with day/night locomotor activities or sleep patterns (Figure 6C). However, we found a significant negative correlation of the cortical CAG repeat instability index with locomotor activity during day and night, as well as with total locomotor activity (Figures 6C and 6E). In addition, we found that the striatal instability index was significantly negatively correlated with sleep during the night (Figures 6C and 6D). These findings establish novel connections between both striatal and cortical mHTT CAG repeat instability and behavioral impairment in the BAC-CAG mice.
Evaluating human HTT RNA nuclear retention and Mbnl1 nuclear foci formation in BAC-CAG mice
In addition to the canonical polyQ mHTT protein, the CAG repeat expansion in the mHTT locus is also known to elicit additional putative toxic species (Malik et al., 2021), which include (1) sense CAG repeat containing RNA or antisense CUG-containing transcripts that can be processed into toxic small RNAs (Murmann et al., 2018) or accumulate in the nuclei and sequester RNA-binding proteins, such as muscleblind 1 (Mbnl1) (Sun et al., 2015; Chung et al., 2011), (2) aberrant cryptic polyadenylation in intron 1 resulting in an mHTT-exon1 toxic protein (Neueder et al., 2017; Sathasivam et al., 2013), and (3) repeat-associated non-AUG (RAN) translation from either CUG or CAG expansion transcripts to generate RAN proteins expressed in non-polyQ reading frames (Bañ ez-Coronel et al., 2015). RAN proteins have been shown to be toxic independent of the RNA gain-of-function effects and to accumulate as aggregates in HD patients’ brains (Bañez-Coronel et al., 2015). Further work is needed to understand the spatiotemporal accumulation patterns of RAN proteins during disease and their contribution to the progressive and selective pathogenesis in HD. Here, we evaluated the repeat-associated mHTT sense/antisense RNA-related pathologies and RAN protein products in BAC-CAG mouse brains. Where possible, we compared phenotypes with those of BACHD mouse brains. These comparisons may uncover non-polyQ mechanisms that contribute to the age-dependent and striatum-selective disease processes in BAC-CAG mice.
We first assessed mHTT sense and antisense transcripts and their associated nuclear pathologies. Both the sense and antisense transcripts surrounding the mHTT-exon 1 genomic region can be assessed by mapping the BAC-CAG and BACHD RNAseq reads onto the human genome (see STAR Methods). For the sense transcripts, we focused on those containing mHTT intron 1, which is an estimate of the aberrant mHTT-exon 1 containing transcripts as a result of CAG repeat expansion (Sathasivam et al., 2013; Neueder et al., 2017) but could also represent mHTT pre-mRNA with an unspliced intron 1. In both striatum and cortex at 12 months of age, we found mHTT intron 1 reads in both BAC-CAG and BACHD mice, with BACHD showing greater than 23 higher levels of the normalized mHTT intron 1 reads (Figures S10A and S10C). This result suggests that the levels of mHTT sense transcripts, including aberrant mHTT-exon 1 product, are Figure S2 likely dependent on the BAC transgene dosage (Figures S2, S10A, and S10C). Interestingly, the transcript reads that mapped to the human HTT antisense transcript (HTT-AS) are about 3-fold higher (p < 0.01) in both the striatum and cortex of BAC-CAG compared with BACHD mice (Figures S10B and S10D). We next mapped human HTTintron 1 and HTT-AS transcripts over time in BAC-CAG mice and found that these transcripts were relatively stable and did not appear to change at 2, 6, and 12 months (Figure S11). Thus, BAC-CAG mice show both mHTT intron 1 transcripts, which is consistent with the presence of the aberrant mHTT-exon 1 product found in HD patients and other HD mouse models (Sathasivam et al., 2013; Neueder et al., 2017), and elevated levels of HTT-AS transcripts compared with BACHD mice, suggesting that this difference could contribute to the differential phenotypes between these models.
In addition to the transcription of repeat-containing mHTT sense and antisense transcripts, recent studies have also shown that mHTT mRNA is preferentially retained in the neuronal nuclei as foci (Didiot et al., 2018). Moreover, such RNA foci formed by CAG (or CUG) repeat-containing transcripts are known to be neurotoxic by sequestering RNA-binding proteins, such as Mbnl1 (Sun et al., 2015; Li et al., 2018). We first used RNA-scope to selectively detect the nuclear foci formation of murine Htt and human HTT, a method shown to be more specificthan the conventional repeat-containing FISH probes (Didiot et al., 2018). The murine Htt foci are detected at comparable levels in the striatal and cortical nuclei of WT, BAC-CAG, and BACHD mice at 12 months of age (Figures S12A, S12B, and S12D). However, the human HTT nuclear foci are only detected in BAC-CAG and BACHD mice, and quantitation of HTT nuclear foci signals revealed that BACHD nuclei show about 43 higher nuclear HTT signals compared with BAC-CAG mice (p < 0.001; Figures S12A–S12C).
We next examined Mbnl1 nuclear foci levels, which has been shown to be a CAG or CUG repeat RNA-mediated pathogenic mechanism in multiple CAG/CTG repeat expansion disorders (Kanadia et al., 2006; Li et al., 2008; Malik et al., 2021), including HD (Sun et al., 2015; de Mezer et al., 2011). We performed immunostaining of striatal and cortical sections of BAC-CAG, BACHD, and WT mice for Mbnl1 and NeuN (a neuronal marker protein) at 12 months of age. Interestingly, in NeuN+ cells in the striatum (Figure 7A) and cortex (Figure S13), we detected significantly more nuclear Mbnl1+ foci in BAC-CAG neurons than in WT or BACHD neurons. Moreover, BACHD mice do not appear to show a significant difference in neuronal nuclear Mbnl1+ foci compared with WT controls. Finally, we confirmed another HD model with long CAG repeats, the Q140 KI mice, also has striatal and cortical Mbnl1 foci at 12 months of age (Figure S14).
Figure 7. BAC-CAG mice exhibit brain-wide brain cells nuclear accumulation of Mbnl1 and late-onset of RAN translation.
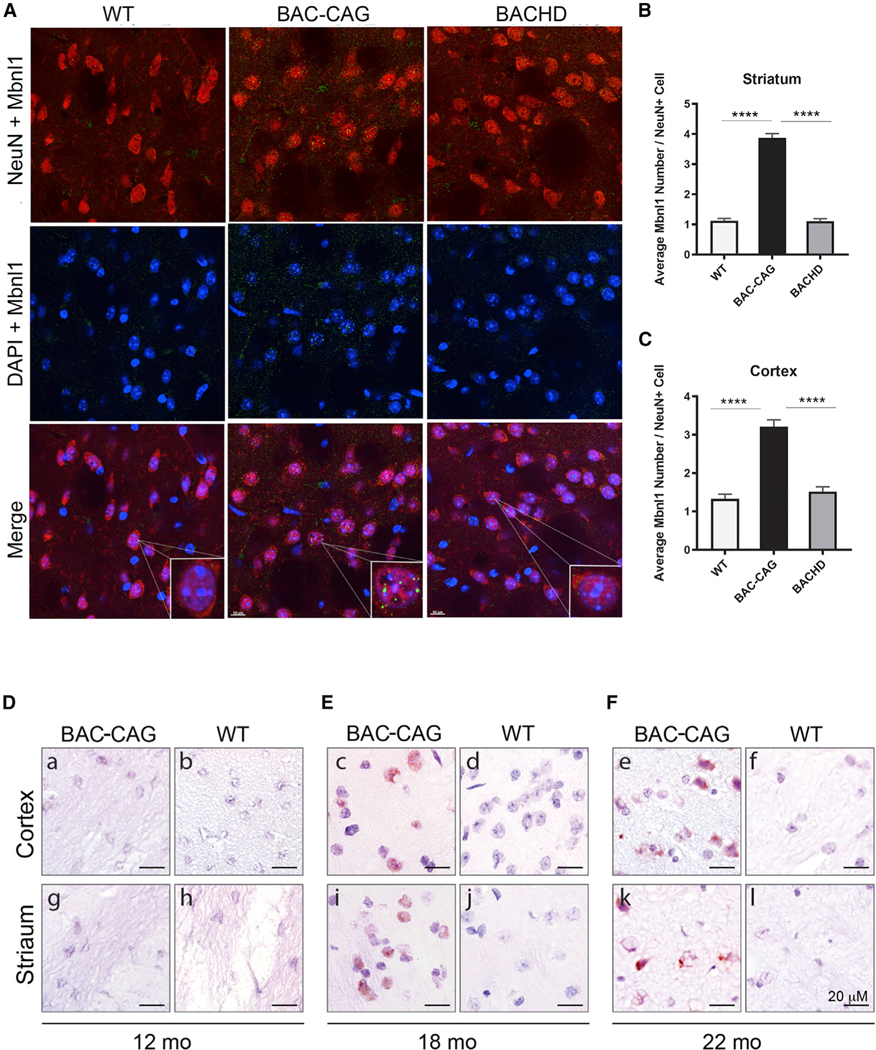
(A and B) BAC-CAG mice display significantly more dot-like Mbnl1 nuclear accumulation in striatal cells than BACHD and WT mice do. Results are shown as mean ± SEM, n = 5 for each age groups. ****p < 0.0001. Scale bars: 10 μm.
(C–E) RAN translation (in reading frame of polySer) in cortex and striatum only detected in BAC-CAG mice after 18 months of age. Scale bars, 20 μm. See also Figures S10–S17.
In summary, BAC-CAG mice show bi-directional transcripts surrounding mHTT-exon 1, with the mHTT antisense transcript, but not sense transcript, appearing to be elevated in an uninterrupted repeat-associated manner (i.e., higher in BAC-CAG than in BACHD). Moreover, BAC-CAG but not BACHD mice show significantly higher levels of neuronal nuclear Mbnl1 foci in striatal and cortical neurons, consistent with a role of repeat-associated RNA toxicities in this mouse model.
Detection of RAN proteins in BAC-CAG mouse brains
RAN translation has emerged as a shared pathogenic mechanism in many repeat expansion disorders, including HD (Nguyen et al., 2019). In HD postmortem brains, four different RAN proteins have been found in brain regions with neurodegenerative changes. HD-RAN proteins are expressed from CAG but not CAA repeats and are toxic to transfected neuronal and glial cells (Bañez-Coronel et al., 2015). Because RAN polySerine proteins accumulate in brain regions showing markers of neuropathology in HD patients , we next tested if polySer RAN proteins accumulate in the brains of BAC-CAG mice, using a polyclonal antibody against the C-terminal region of polySer (Bañez-Coronel et al., 2015). We performed immunohistochemistry on brain sections of BAC-CAG mice at 12, 18, and 22 months of age to evaluate if polySer RAN proteins accumulate with disease progression in this model (Figures 7D–7F and S15). While we did not observe any RAN polySer-specific signal in the striatum or cortex of BAC-CAG mice at 12 months, we consistently observed frequent polySer RAN-positive cells in both brain regions in BAC-CAG mice at 18 and 22 months of age (Figures S15 and S16). No similar staining was found in WT controls (Figures 7D–7F, S15, and S16). PolySer protein staining was found in BAC-CAG neurons and glial cells, accumulating as mostly diffuse nuclear staining at 18 months of age in both cortex and striatum (Figures S15 and S16). At 22 months of age, robust polySer nuclear and perinuclear aggregates were detected in the striatum, with less frequent aggregates found in the cortex. Since silent codon changes in the mHTT-exon 1 region of BACHD alter the sequence encoding the epitope of anti-HTT polySer antibody, we did not expect any RAN polySer protein products in 12month or older BACHD mouse brains. Consistent with this prediction, we did not detect polySer staining in BACHD striatum or cortex at 12 months of age (n = 3; Figure S17).
Together, our results show that RAN polySer accumulates and aggregates in the striatum and cortex of BAC-CAG mice in an age-dependent manner. The age at which RAN polySer accumulation occurs is later than 12 months, an age at which multiple HD-related pathogenic phenotypes are already evident, including striatal nuclear polyQ mHTT aggregation, striatum-specific transcriptionopathy, and accumulation of striatal and cortical neuronal Mbnl1 foci. Thus, our finding suggests that accumulation of HTT polySer RAN products does not appear to contribute to the early stages of pathogenesis in the striatum and cortex of this HD mouse model. However, our study is consistent with the possibility that polySer RAN proteins could contribute to the progression of the striatal neuropathogenesis as we observed a progression of mHTT nuclear polyQ aggregates evolved from diffuse staining in a subset of MSNs at 12 months to NIs in most MSNs at 18 months (Figure 3).
DISCUSSION
In this study, we developed a novel human genomic transgenic mouse model of HD (BAC-CAG) with about 120 uninterrupted CAG repeats encoding an expanded polyQ stretch. We report that BAC-CAG mice exhibit multiple phenotypes similar to prior full-length human mHTT models, including progressive rotarod deficits, hypolocomotion, sleep disturbance, and striatal synapse loss (Yang and Gray, 2011; Veldman and Yang, 2018; Kudo et al., 2011). However, this new model exhibits additional HD-like phenotypes that absent or weak in the prior human genomic transgenic mouse models with numerous CAA interruptions in the DNA sequences encoding the mHTT polyQ stretch (i.e., BACHD and YAC128). Such phenotypes include minimal body weight gain, somatic CAG repeat instability, striatal astrogliosis and microgliosis, striatum-selective NIs, and robust age-dependent and striatum-selective transcriptional dysregulation. These latter two phenotypes closely resemble those found in the allelic series mHtt KI mice. Importantly, taking all the full-length mHtt KI models and human mHTT genomic transgenic models together, the striatal but not cortical transcriptomic DE genes are best predicted by the uninterrupted CAG repeat length and not by the uninterrupted glutamine repeat length in mutant HTT/Htt. Additionally, the BAC-CAG model represents the first human genomic transgenic mouse model of HD with somatically unstable mHTT CAG repeats and the first HD animal model that shows significant correlations between somatic CAG repeat instability indices in the striatum or cortex and behavioral impairments. By analyzing the spatiotemporal emergence of mHTT CAG repeat-associated candidate toxic species, we identified CAG repeat instability, sense and antisense mHTT RNA around exon 1 of the mHTT gene, Mbnl1 nuclear accumulation, and mHTT nuclear aggregation and NIs as candidate contributors to the onset of striatum-selective pathogenesis in BAC-CAG mice. Moreover, the polySer RAN proteins are detectable at a later age (18–22 months) and hence unlikely to be involved in the early stage of the disease but could contribute to disease progression in this HD model. Together, our study demonstrates novel pathogenic roles of uninterrupted CAG repeat, beyond its encoded polyQ protein, in mHTT-induced striatum-selective pathogenesis in vivo.
GWAS study of modifiers of HD age of onset first identified the significance of uninterrupted CAG length, rather than the overall Q-length, in mHTT, in determining the disease onset in patients (GeM-HD, 2019). However, the study in patients did not uncover any mechanisms or neuropathological correlates to explain why uninterrupted CAG repeat length has an important role in HD. Prior studies in the allelic series KI mouse models revealed the impact of CAG repeat expansion on multiple pathological processes, such as somatic CAG repeat instability (Wheeler and Dion, 2021) and striatum-selective transcriptional dysregulation (Langfelder et al., 2016). However, these studies could not differentiate the roles of CAG repeat length and Q-length, as each KI model has identical number of CAG versus glutamine repeats. Taking advantage of the broad divergence between the uninterrupted CAG length and overall Q-length in the three full-length human mHTT transgenic models (Figures 8A and 8B), our study is the first to provide strong evidence on the neuropathological and molecular pathogenic impact of uninterrupted CAG repeats in inducing multiple disease processes in the striatum, the most vulnerable brain region in HD. This conclusion is based on close phenotypic similarities of BAC-CAG with allelic series KI mice, which also have pure CAG repeats encoding polyQ stretch, in the phenotypes that are divergent between BACHD/YAC128 and the KI mice (Figure S18). Such phenotypes include the striatum-selective nuclear aggregation and NIs (Gray et al., 2008; Pouladi et al., 2012; Bayram-Weston et al., 2012, 2016) and somatic CAG repeat instability with the age-dependent increase in the striatum and liver (Wheeler and Dion, 2021). Moreover, analyses of the transcriptomic data from BAC-CAG, BACHD, YAC128, and allelic series KI mice (Langfelder et al., 2016) allow us to draw a strong conclusion on the pivotal role of uninterrupted mutant huntingtin CAG repeat length, beyond the encoded polyQ repeat, in the pathogenesis of striatum-selective transcriptional dysregulation. These advances greatly extend our understanding of the role of uninterrupted CAG repeats in HD pathogenesis beyond our prior knowledge based on the human genetic study (GeM-HD, 2019).
Figure 8. Phenotypic comparison of BAC-CAG model with prior human genomic transgenic mouse models (e.g. BACHD) and mHtt knockin mouse models provide new insights into the role of uninterrupted CAG repeat in striatum-selective pathogenesis in HD models.

(A) A schematic showing the current full-lengthmutant huntingtin-expressing mouse models ordered by their uninterrupted CAG repeat length. For models with CAA-interrupted and stable repeats (i.e., BACHD and YAC128), key disease-like phenotypes likely elicited by mHTT protein include corticostriatal synaptic toxicity. For the models with long uninterrupted CAG repeats, including allelic series KI mice and BAC-CAG, the models showed additional pathogenic phenotypes mostly in the nuclei. Such phenotypes include CAG repeat instability, bi-directional transcription of sense and antisense CAG or CUG repeat-containing RNAs, nuclear Mbnl1 foci, striatum-selective transcriptionopathy, nuclear mHTT aggregates, and inclusions. Additional pathologies found in the latter group of models, including RAN translation products and corticostriatal synaptic toxicities.
(B) Comparison of BAC-CAG and BACHD molecular and pathological phenotype indemnities that are selective to BAC-CAG model with a long uninterrupted CAG repeat and those that are more selective to the striatum, the brain region that is most vulnerable to neurodegeneration in HD. See also Figure S18.
Our study also indicates that certain disease phenotypes are likely dependent on its polyQ protein and may be independent of the uninterrupted CAG repeat length. Prior human mHTT genomic transgene models of HD (Figure 7B), particularly the BACHD mice, show robust phenotypic deficits that are dependent on mHTT expression in the cortical and striatal neurons (Wang et al., 2014). These include progressive corticostriatal synaptic marker loss and striatal MSN electrophysiological deficits, similar to those found in the mHtt KI mice (Indersmitten et al., 2015; Plotkin et al., 2014; Wang et al., 2014; Veldman and Yang, 2018; Smith-Dijak et al., 2019). The synaptic marker loss of BACHD mice is recapitulated in BAC-CAG mice (Figures 2A–2D). These findings suggest that the corticostriatal synaptic deficits in HD mice can be induced with both CAA-interrupted and uninterrupted CAG repeats in full-length mHtt/mHTT mouse models and are, therefore, likely due to the polyQ protein toxicities (Figure 8A).
Our study describes the spatiotemporal emergence of distinct pathogenic entities, including multiple non-polyQ species, that originate from the mHTT locus and correlates the presence of such species with disease phenotypes (Figure 8B). Historically, the HD field has been focused on the study of pathogenesis elicited by the mHTT protein, such as proteolysis to generate toxic mHTT fragments or posttranslational modifications that significantly alter disease progression (Ross and Tabrizi, 2011). However, such a protein-centric view of HD pathogenesis was revised by the GWAS finding that the CAG repeat length play a distinct pathogenic role beyond the polyQ protein in HD (GeM-HD, 2019; Wheeler and Dion, 2021). Our study defines a critical disease-modifying role of uninterrupted mHTT CAG repeat in eliciting striatum-selective nuclear pathogenesis and transcriptionopathy. These findings raise important questions on what the CAG-repeat-associated, non-polyQ species derived from the mHTT locus are and what their roles are in the disease. To begin addressing this question, we performed the first spatiotemporal analyses of multiple non-polyQ mHTT-transgene-locus-derived candidate toxic species in the context of disease pathogenesis in vivo (Figure 8B). Among such disease mechanisms, prior studies primarily focused on the somatic repeat instability (Wheeler and Dion, 2021), and limited studies have been carried out to explore the in vivo roles of CAG/CUG repeat-containing RNAs and RAN translation (Bañez-Coronel et al., 2015; Neueder and Bates, 2018). Our analysis revealed two mHTT-related pathogenic entities, the CAG repeat instability and nuclear mHTT aggregation and inclusion formation, show high striatum specificity and age dependency that coincide with the emergence of other key disease-related phenotypes (i.e., transcriptionopathy, reactive gliosis, locomotor, and sleep deficits). Thus, our study suggests a possible causal relationship between the somatic mHTT CAG repeat instability and/or nuclear mHTT aggregation in eliciting behavioral deficits and striatum-selective molecular pathogenesis in HD.
At the RNA level, the HTT locus is known to confer bi-directional transcription to generate CAG- or CUG-repeat-containing RNAs (Chung et al., 2011). These RNA species are postulated to be toxic through multiple mechanisms, including phase transition (Jain and Vale, 2017) and formation of nuclear RNA foci (de Mezer et al., 2011; Didiot et al., 2018), and sequestration of RNA-binding proteins (Sun et al., 2015). Our study shows that both sense and antisense mHTT RNA transcripts, at the expanded repeat region, are present in BAC-CAG mice, with levels relatively steady across ages and comparable in cortex and striatum (Figures 8B and S11). However, interestingly, the HTT antisense transcript levels and Mbnl1 nuclear foci are more abundant in BAC-CAG than in BACHD mice (Figures 7A, S12, and S14). Thus, despite the lack of brain region or temporal specificity in relationship to the disease, the presence of mHTT CAG- and CUG-repeat-containing RNAs can selectively induce RNA toxicity (Mbnl1 foci) in the BAC-CAG striatum and cortex, which could contribute to the disease pathogenesis in this HD model. Another RNA-related mechanism as a result of mHTT CAG repeat expansion is the generation of aberrantly polyadenylated mHTT-exon 1 transcript that encodes a toxic mHTT-exon 1 protein (Sathasivam et al., 2013; Neueder et al., 2017). Our analysis based on RNA-seq reads showed such transcripts exist in both BAC-CAG and BACHD mice, and their levels in BACHD are significantly higher than those in BAC-CAG (Figures S10 and S12). Notwithstanding, the polyQ repeat length in the BACHD is shorter than that in BAC-CAG, and BACHD also appears to have fewer nuclear mHtt aggregation and transcriptionopathy than Q92 KI mice (Gray et al., 2008; Langfelder et al., 2016; Wheeler et al., 2000), a model expressing mHTT at the endogenous level. Therefore, BACHD model reveals that the aberrant mHTT-exon 1 transcript alone, without expanded and uninterrupted CAG repeats, appears insufficient to elicit striatum-selective nuclear aggregation and transcriptionopathy. Since we do observe a correlation between nuclear mHTT aggregation/NI formation and transcriptionopathy in BAC-CAG and most of the KI mice (Wheeler et al., 2002; Langfelder et al., 2016), we favor the hypothesis that mHTT N-terminal fragments (including the fragment generated by the aberrant mHTT-exon 1 transcript) may play a role in the nuclear pathogenic process.
Finally, our study revealed that polySer RAN protein products, which were originally found in the patients’ brains (Bañez-Coronel et al., 2015), are also found in BAC-CAG striata and cortices at 18–22 months of age but not at 12 months of age (Figures 7D–7F, S15, and S16). Such RAN products were not found in BACHD mice at 12 months or older because the silent coding region alters the epitope for the polySer RAN antibody (Figure S17, data not shown). Since BAC-CAG mice did show disease progressivity between 12 and 18 months (e.g., striatal NI formation and aggregation in MSD assays; Figures 3B, 3C, and 3G), our finding is consistent with the interpretation that toxic RAN products may not contribute to the onset of striatal disease phenotypes and behavioral impairment, but they may play a role in the disease progression.
Our study represents an important advance in resolving the phenotypic discrepancies between the two major types of HD mouse models—mHtt KI models and full-length human mHTT genomic transgenic models (Figure S18; Yang and Gray, 2011). The former models, with an allelic series of human mHTT-exon 1 with increased pure CAG repeats replacing the murine endogenous Htt exon 1, exhibit phenotypes that are absent or very weakly expressed in prior full-length human mHTT genomic transgenic mice (i.e., BACHD and YAC128), most notably, the CAG repeat instability, progressive accumulation of striatal nuclear mHTT aggregates and NIs, and striatum-selective robust transcriptionopathy (Yang and Gray, 2011; Langfelder et al., 2016; Slow et al., 2003; Gray et al., 2008). The lack of consistency between the full-length human mHTT and murine mHtt KI models has led to the speculation that the numerous species-specific sequence differences between human HTT and murine Htt could account for the overall phenotypic divergence of the two types of HD mouse models (Ehrnhoefer et al., 2009). Despite the overall 91% identity and 95% similarity between human and murine huntingtin proteins, there are still 270 amino acid differences and 151 nonconservative changes. Such protein sequence differences may alter certain HD pathogenic processes, such as calpain cleavage of full-length mutant huntingtin to create toxic N-terminal fragments (Ehrnhoefer et al., 2009). Additionally, differences in human and murine huntingtin genomic DNA or RNA sequences may alter gene regulation, polyadenylation, alternative splicing (Franich et al., 2019), and RNA metabolism, which may also alter disease phenotypes in these models (Ehrnhoefer et al., 2009). Our study created BAC-CAG mice to unequivocally demonstrate that the major phenotypic divergence between the prior human genomic transgenic mouse models and KI models of HD are due to the numerous CAA interruptions in the polyQ coding regions of BACHD and YAC128 and are not due to the inherent species-specific differences in the human and murine huntingtin genes or due to the types of genetic models (i.e. transgenic vs knockin models).
Additionally, our study demonstrates that the BAC-CAG model offers certain advantages in terms of modeling disease-related phenotypes and its applications in therapeutic studies. First, compared with the KI mice, the BAC-CAG model has behavioral deficits, such as sleep disruption that is absent in the Q140 or Q175 KI mice (Loh et al., 2013; Kudo et al., 2011). Moreover, similar to BACHD, BAC-CAG not only has the human genomic mHTT transgene but also patient-associated SNPs, which can be used for testing the vast majority of human HTT-lowering therapeutics (Kordasiewicz et al., 2012; Tabrizi et al., 2019), including patient allele-specific silencing (Caroll et al., 2011; Skotte et al., 2014; Southwell et al., 2018; Monteys et al., 2017; Zeitler et al., 2019) or human-specific alternative splicing modulation (Bhattacharyya et al., 2021; Tabrizi et al., 2019), which cannot be tested in KI models with predominantly murine Htt sequences. Compared with BACHD and YAC128, BAC-CAG shows superior construct validity and face validity (Figure S18; Yang and Gray, 2011). First and foremost, BAC-CAG is the only human genomic transgenic mouse model with long uninterrupted CAG repeat and demonstrated somatic repeat instability, which are correlated with behavioral impairment. Additionally, BAC-CAG mice exhibit multiple HD-like phenotypes that are absent or weak in BACHD and YAC128 models, including minimal body weight gain, robust striatal astrogliosis and microgliosis, striatum-specific mHTT aggregation and NIs, neuronal nuclear Mbnl1 retention, and an accumulation of HD RAN proteins. Finally, the BAC-CAG model has unique advantages in preclinical therapeutic testing applications over all the prior HD mouse models. The BAC-CAG mouse is the first and only HD mouse model that has both full-length human mHTT genomic transgene sequences (including some patient-associated SNPs) and shows mHTT CAG repeat instability. Therefore, BAC-CAG is uniquely positioned to test the interactions of therapeutics that lower human HTT (or patient-associated mHTT allele) and those that target mHTT CAG repeat instability (e.g., targeting the GWAS-associated DNA repair genes). Thus, the BAC-CAG mouse model may enable the advancement of polypharmaco-logical therapies against multiple high-value patient-derived therapeutic targets.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and request for reagents should be directed the lead contact Dr. X. William Yang (xwyang@mednet.ucla.edu).
Materials availability
The BAC-CAG mouse model is being deposited at the Jackson Laboratory for distribution to the scientific community. JAX has approved the acceptance of the BAC-CAG strain (JAX Stock # 037050) for inclusion in The Jackson Laboratory Mouse Repository.
Data and code availability
The RNA-seq data reported in this paper is has been deposited at GEO (GEO access number GSE153827).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental animals
All the experiments were approved by the Committee for Animal Care of the Division of Laboratory Animal Management at University of California at Los Angeles (UCLA) and carried out at UCLA. BACHD, allele series of Hdh KI mice were obtained from the Jackson laboratory. BAC-CAG mouse model and MORF3 mice were generated in our lab. All the transgenic and KI mice were bred and maintained in our animal facility. Mice were bred and maintained with male BAC-CAG crossing FvB/NJ females. All mice were housed in standard mouse cages under conventional laboratory conditions, with constant temperature and humidity, 7 AM to 7 PM light/dark cycle and food and water ad libitum. We used both male and female mice with uninterrupted CAG repeats over 120 for the experiments. All mice were group housed (2– 5 mice per cage). All experiments were done using age-matched littermates.
METHOD DETAILS
Transgene constructs and generation of BAC-CAG mice
mHTT exon1 containing expanded CAG repeats was PCR amplified from DNA template of pBC-mHTT-Ex1-Q140 plasmid (gift from Dr. G. Bates, UCL) and cloned into shuttle vector pLD53-SCAB. Multiple clones were picked up and CAG repeat sizes were sequenced at Laragen (Los Angeles, CA). We selected the clone with the longest CAG repeat size [(CAG)130-CAA-(CAG)10-CAACAG] and cultured in LB medium in 30 °C incubator. Purified plasmid DNAs were used to modify human huntingtin BAC (RP11–866L6) clone as reported previously (Gray et al., 2008; Gu et al., 2009; Yang et al., 1997). The modified BAC DNAs were confirmed by sequencing before pronuclear injections into FvB/NJ FvB/NJ inbred mouse embryos, at the Cedars Sinai Transgenic Core, to generate BAC-CAG mice. Ten founders were identified by PCR amplification and sequencing, and two germline-transmitted lines with a single CAG repeats size were identified and propagated. One such line (i.e. BAC-CAG) with relatively high-level of human mHTT transgene expression by western blot were used in this study.
Through pronuclear injections of FvB/NJ inbred mouse embryos, we obtained multiple transgenic founders that gave rise to germline transmission of the transgenes.
Tissue collection and genomic DNA preparation
Mice were deeply anesthetized with Nembutal (50 mg/kg IP). For western blot and MSD assays, mouse brains were quickly dissected without perfusion in PBS solution. The cortex, striatum and cerebellum were separated under a dissecting microscope, snap frozen in dry ice and stored at −80°C until further processing. For immunohistochemistry staining and other usage, anesthetized mice were transcardially perfused first with 0.1M PBS, followed by 4% paraformaldehyde (PFA) in 0.1M PBS, pH 7.4. The brains were dissected and post-fixed with 4% PFA for overnight at 4°C. Subsequently, the brains were transferred to tubes containing 30% sucrose in cold 0.1M PBS. After they sank to the bottom of the tubes, the brains were removed, briefly washed with distilled water and dried with soft tissue paper and snap frozen in dry ice. The brains were transferred and stored in the chamber of the Leica Cryostat for one hour; coronal and sagittal brain sections were cut at 40 μm and stored at −20°C in cryopreservation solution (30% ethylene glycol, 30% glycerol in 0.02M PB) for further processing.
Phenol-chloroform purified mouse genomic DNA samples were used for transgene copy number estimation and somatic CAG instability assessment in this study. Briefly, about 100 – 500 μg mouse tissues (tail, liver, heart and various brain region tissues) were dissected and placed in 1.5 ml centrifuge tubes, digested overnight in 0.5 ml digestion buffer with proteinase K solution (final concentration 2 mg/ml) in a 55 °C water bath. Equal amount of phenol-chloroform solution (Sigma) was added to the mixture (1:1) and the solutions were mixed vigorously for 5 minutes. After centrifugation at max speed for 20 mins at room temperature on a bench top centrifuge, supernatants were transferred to new tubes. One tenth (by volume) 3M sodium acetate solution and 2.5 times (by volume) 100% ethanol were added to the supernatants in the tubes and mixed. Precipitated DNAs were washed extensively with 70% ethanol. Relatively pure genomic DNA pellets at bottom of the tubes look like transparent. If the DNA pellets look like white and not transparent, the pellets were re-dissolved in autoclaved distilled water and the phenol-chloroform purification procedure was repeated. Genomic DNA samples were dissolved in DNase free water and DNA concentrations were measured with Nanodrop (Nanodrop ND-8000, Thermo Scientific). Purified genomic DNAs were used for transgene copy number quantification and somatic CAG instability measurement.
Quantitative PCR to estimate transgene copy number
Genomic DNA was purified from tail samples (about 1cm length) of 3 young BAC-CAG and BACHD mice using phenol-chloroform extraction. Primers for detection of human-specific HTT (5’- GCAACGTGCGTGTCTCTG-3’ and 5’- TGTTCCCAAAGCCTGCTC-3’) and primers for detection of GAPDH of both human and mouse origins (5’- ACCACAGTCCATGCCATCAC-3’ and 5’- TCCAC CACCCTGTTGCTGTA-3’) were designed and tested before applying to real time quantitative PCR, to estimate integrated BAC transgene copy numbers (Gray et al., 2008). Briefly, tail genomic DNA samples from three BAC-CAG and three BACHD mice, as well as 1 human genomic DNA sample (Clontech) were set up in 3 different concentrations (1:1, 1:100 and 1:1000) and DNA samples and primer pairs were mixed with SYBR™ Green PCR Master Mix (Applied Biosystems #4309155), as recommended by the manufacturer. Triplicate PCR reactions for each sample and triplicate PCR reactions without genomic DNA samples as negative controls were used. Transgene copy numbers were estimated using the CΔtΔt method with human genomic samples (having 2 copies) as the standard.
Quantitation of mHTT expression by western blot and MSD assays
To compare transgene expression in different founder lines, three mice from each founder line, BACHD positive control and wildtype negative control were used for mHTT quantification by western blot assay, probed with 1C2, and α-tubulin as reported previously (Gray et al., 2008; Gu et al., 2009). Briefly, brains from 6-week old mice, of each line, were dissected, 40 μg protein from lysates of cortex and striatum of each brain was mixed with NuPAGE LDS loading buffer (Invitrogen), and heated for 10 min at 70°C, then resolved on 3–8% Tris-Acetate NuPAGE gel (Invitrogen). After protein transfer to PVDF membrane, blots were probed with the respective antibodies. Densitometric values from scanned Western blot films were obtained using ImageQuantTL software (GE Healthcare). To test age-dependent mHTT expression, striatal proteins from 2 male mice at each age (2m, 12m and 18m) were blotted and assayed as above.
Quantification of mHTT protein levels in different brain tissues of 2, 6, 12 and 18 month old mice were performed at Evotec (Hamburg, Germany) using a customized Mesoscale Discovery (MSD) electrochemiluminescence assays (Reindl et al., 2019). Briefly, 50 μg tissues of cortex, striatum and cerebellum were dissected from BAC-CAG or WT littermates (N=4 per age per genotype), then lysed and quantified for protein concentration. Homogenates were loaded onto MSD plate, and then coated with the following antibodies: 2B7/4C9 antibody pair for total mHTT proteins; 2B7/MW1 antibody pair for expanded mHTT proteins; and MW8/4C9 antibody pair for aggregated mHTT. Recombinant HTT-Q46_GST N-terminal fragments were used to make the standard curve and calculate the mHTT levels.
iDISCO+ staining and spine quantification
The 12 months old BAC-CAG/CamK2a-CreERT2/WT/MORF3 and CamK2a-CreERT2/WT/MORF3 mice were induced by one dose of Tamoxifen (20mg/kg body weight, i.p.). Mice were perfused, sectioned, and stained as previously described (Veldman et al., 2020). The striatum of each brain sections was imaged by the Andor DragonFly spinning disk confocal (Belfast, UK) with 60x silicon lens. The images were stitched by Imaris stitcher (Oxford Instruments). The spines on the terminal 30 μm of the dendrites, which has clear dendrite terminals in the top 50 μm sections, were reconstructed and analyzed using Aivia (Bellevue,US). 5~6 medium spiny neurons were analyzed per mouse. For each neuron, the spine density of the terminal 30 μm in 3~5 dendrites were analyzed. Unpaired t-test were used to compare the average spine density per neuron in BAC-CAG and WT mice.
RNAscope
The RNAscope was performed as the manufacture’s protocol described, using the RNAscope® Multiplex Fluorescent Kit v2 (ACDBio cat#323100). Briefly, the %4 PFA fixed mouse brain were dehydrated and sectioned to 20¼m sections using the cryostat. The brain sections from the BAC-CAG, WT, and BAC-HD mice were mounted to the same slide. The sections were treated by the proteinase III for 30min at 40°C. After wash, the sections were hybridized with the HTT specific (ACDBio cat# 420231-C2) and Htt specific (ACDBio cat# 473001) probes for 2hrs at 40°C. After Amplification steps, the HTT was developed with the Opal 570 (Perkinelmer cat#FP1488001KT) and Htt was developed with Opal 520 (Perkinelmer cat#FP1487001KT). After developing, the sections were stained with DAPI and mounted with Prolong Diamond (ThermoFisher cat#P36970). The dorsal lateral striatum of each sections was imaged under 60x silicon objectives, using Andor DragonFly spinning disk confocal (Belfast, UK). 10 intact nuclei from each section were segmented and the mRNA foci were identified and quantified using Imaris 9.3 (Oxford Instruments). Unpaired t-test was applied to compare HTT average pixel. One-way ANOVA was applied to compare the Htt average pixel.
RAN protein detection by immunohistochemical staining
Three mice from each group of BAC-CAG, BACHD and their wildtype littermates at ages of 12m, 18m and 22m, respectively, were perfused; brains were dissected, treated with 30% sucrose. After paraffin-embedding, the brains were sectioned at 6 micrometer and mounted on Superfrost Plus microscope slides (#12-550-125, Fishersci). Brain sections were processed for RAN protein detection using rabbit anti-human C-terminal HD polySerine polyclonal antibody as described (Bañez-Coronel et al., 2015). Briefly, 6 micrometer sections were deparaffinized in xylene (15 min, twice) and rehydrated through an alcohol gradient (100%, 95% and 80%), rinsed in distilled water, and subjected to a triple antigen retrieval process: 1) Proteinase K treatment (1ug/mL proteinase K in 1mM CaCl2, 50mM Tris buffer, pH=7.6) for 40 minutes at 37°C. 2) Pressure cooked in 10mM EDTA (pH=6.5) for 15minutes (when the pressure knob goes up). 3) Formic acid treatment for 7 min. Slides were then washed with distilled water for 10 minutes and incubated in 3% H2O2 in methanol for 10 minutes to block the endogenous peroxidase activity. Non-specific Ig binding was blocked by incubating with Background Snipper (BS966M, Biocare Medical) for 17 minutes, then incubated with polySer polyclonal antibody (1:20,000) in 10% Background Snipper in distilled water overnight at 4 °C.
Slides were washed in PBS three times and incubated with rabbit Linking Reagent (#93030, BioLegend) for 30 minutes at room temperature. Biotinylated goat anti-rabbit secondary antibody was applied for 30 minutes and detected using ABC reagent (PK-6100, Vector laboratories, Inc.). Peroxidase activity was developed with NovaRed Substrate Kit (Vector Laboratories, Inc.) for 5 minutes. Slides were washed in water (10minutes) and later counterstained with hematoxylin. After a last wash in water, slides were dehydrated and glass coverslip were mounted using Cytoseal 60 (Electron microscopy sciences).
Motor assessments using accelerated rotarod
Accelerating rotarod testing was performed at 2, 6 and 12 months of age based on our published protocol (Wang et al., 2014). Briefly, mice are placed on the rubber covered axle of the Rotarod apparatus (Ugo Basile). When mice move at the pace set by the rotation rate of the axle they will remain on the axle. When mice no longer move at the selected pace they drop a short distance to the platform below. The time it takes for a mouse to drop to the platform is called the latency to fall and is a widely accepted measure of motor coordination. Naive mice were trained at 2 months of age. During the training period, mice were placed on the slowly rotating axle, at a constant speed of 4 rpm, for 120 seconds. They were placed back on the axle if they fell off during this time. Each mouse had three trials per day for two constitutive days for training. One week after training, mice were tested by accelerating rotarod. Briefly, mice were placed on the axle. The axle gradually accelerates from 4 rpm to 40 rpm over 5 minutes. The length of time the mouse stays on the axle is recorded. The test ends when the mouse drops to the platform after 20s or stays on the axle for 600s. Each mouse completes three trials per day for 3 consecutive days. Fifteen-minute breaks were given between each trial. Mice were returned to their home cage during breaks. The apparatus was cleaned with 70% alcohol and allowed to dry completely between trials.
Locomotion and sleeping activity
Monitoring of locomotor activity: Twelve-month-old mice were singly housed in cages containing IR motion sensors (Mini Mitter, Bend, OR), and locomotor activity recorded as previously described (Kudo et al., 2011, Loh et al., 2013). Mice were entrained to a 12:12 hr light:dark (LD) cycle for a minimum of 2 weeks prior to collection of 10–14 days of data under LD conditions, followed by 10–14 days in constant darkness (DD) to obtain free-running activity. Locomotor activity data was recorded using Mini Mitter data loggers in 3 min bins, and 10 days of data under each condition averaged for analysis. Free-running period (tau, τ) was determined using the χ2 periodogram and the power of the rhythm was determined by multiplying the amplitude, Qp, by 100/n, where n = number of datapoints examined using the El Temps program (A. Diez-Noguera, Barcelona, Spain). Activity amount was determined by averaging 10 days of wheel revolutions (rev/hr). Activity duration (alpha, α) was determined by the duration of activity over the threshold of the mean using an average waveform, of 10 days of activity. Nocturnality was determined from the average percentage of activity conducted during the dark. Precision was determined by calculating the daily variation in onset from best-fit regression line drawn through 10 days of activity in LD using Clocklab (Actimetrics, Wilmette, IL). Fragmentation was defined by the number of bouts/ day with a maxgap of 21 min.
Immobility-defined sleep measurements: Mice were individually housed in automated feeding cages and their behavior recorded using continuous video recording and automated mouse tracking as previously described (Loh et al., 2013). Side-on views of the cage were acquired using CCTV cameras (Gadspot, GS-335C, City of Industry, CA), and the ANY-maze software (Stoelting Co., Wood Dale, IL) was used to track the animals. Immobility-defined sleep in 1 min bins were averaged, and day (ZT 0–12) and night (ZT 12–24) sleep compared. Sleep and wake onset, and fragmentation of behavioral sleep were determined using Clocklab.
Grip strength for testing neuromuscular function
The grip strength ergometer (Santa Cruz Biotechnology, Santa Cruz, CA) is set up on a flat surface with a mouse grid firmly secured in place. The maximal strength exerted by a mouse to remain gripped on the grid is measured. Mice were acclimatized to the testing room and tested during their active phase (night). Mice underwent three to five trials with an inter-trial interval of at least two minutes. The sensor was reset and the grid cleaned with 70% ethanol between mice and allowed to dry. For each trial, the mouse was removed from its home cage by gripping the base of the tail between the thumb and the forefinger. The mouse was lowered slowly over the grid, and only its forepaws were allowed to grip the grid. The experimenter then steadily pulled the mouse back by its tail, ensuring that the torso remained horizontal, until the mouse was no longer able to grip the grid. The mouse was then returned to its home cage. The maximal grip strength value of the mouse was recorded. Sometimes mice may not be adequately motivated to grip the apparatus. This was minimized by holding the mice by the tail for a brief time before bringing within gripping distance. The maximum value obtained per animal was reported as maximal strength normalized to body weight.
Immunohistochemical staining and imaging
Immunostaining was carried out using published methods (Gu et al., 2009). S830 sheep anti-HTT antibody (from Dr. G. Bates; 1:1,000 dilution) antibody was used for mHTT aggregate detection; GFAP antibody (Sigma, 1:20,000 dilution) was used for detection of reactive astrocytes; NeuN antibody (Millipore, 1:1,000) was used to identify neurons; ubiquitin antibody (Dako, 1:1,000 dilution) was used for detection of ubiquitin; ACTN2 antibody (Millipore, 1:500) was used to quantify alpha-actinin 2 expression in striatum. For double immunofluorescent staining (e.g. S830/NeuN) we added the S830 antibody and incubated at 4°C for one day, then added NeuN or other antibodies and incubated at 4°C overnight. Images were acquired using a Dragonfly High-Speed Confocal Microscope 200 and the Fusion 2.0 software package (Andor, Oxford Instrument). Laser and detector settings were maintained at constant for the acquisition of each immunostaining. For all analyses, at least three images were taken per brain region and slide using 360 oil objective lens, at 2,048 × 2,048 pixel resolution, with z-step size of 1 μm at 10 μm thickness.
Quantification of Astrogliosis
Five randomly selected coronal brain sections, containing both cortex and striatum, from each mouse at 12 months of age were selected. Three mice from each group for BAC-CAG and WT littermates were stained with polyclonal rabbit anti-mouse GFAP primary antibody and goat anti- rabbit secondary antibody, AlexaFluor 488, and counter stained with DAPI. Sections were mounted with anti-fading reagent (ProLong Gold Antifade Mountant, Invitrogen). Images of dorsal striatum and deeper cortical layers with corpus callosum of each brain section were acquired at 40x magnificent using a Dragonfly High-Speed Confocal Microscope 200 and the Fusion 2.0 software package (Andor, Oxford Instrument). Laser and detector settings were maintained constant for the acquisition of each immunostaining. The intensity of GFAP staining of each image was estimated using the Analyze Particles plugin of ImageJ software (NIH). Values from each section were averaged to generate a mean GFAP intensity for each animal.
Quantification of nuclear Mbnl1 in NeuN+ cells
Four mice each from WT, BAC-CAG and BACHD groups were selected for the quantification. Three adjacent coronal brain sections (45 um thick) about 200 um postal bregma point were selected and double stained with Mbnl1 (1:1000. ab45899, Abcam) and NeuN antibodies, then count stained with DAPI. Nine images of dorsal striatum and motor sensory cortex of each brain sections were captured at 60x magnificent using the Dragonfly Confocal microscope. All images were taken with the same conditions for Laser and detector settings. We trimmed the top and bottom of the images and took the 5 μm thick trimmed confocal images for Mbnl1 quantification using the Analyze Particles plugin of the Image J Software (NIH). We set the “Size” at “260-Infinity” and “Circularity” to “0.00–1.00” to count only those relatively big Mbnl1 dots inside the nucleus of NeuN+ cells. All NeuN+ cells in focus of each image were counted. About 500 – 600 cells in total in each group were counted for each group.
Analysis of CAG repeat instability
About 100 μg tissues of cortex, striatum, cerebellum, heart, liver and gonad, dissected from 2m and 12mold BAC-CAG mice, were used for HTT CAG repeat instability study. The number of mice per genotype and ages are the following: striatum (6 at 2m, 26 at 12m), cortex (6 at 2m, 28 at 12m), cerebellum (6 at 2m, 20 at 12m), liver (4 at 2m, 4 at 12m), heart (6 at 2m, 13 at 12m), testis (5 at 2m, 16 at 12m), and tail (10 at 2m, 8 at 12m). The dissected tissue was digested with tissue digestion buffer containing proteinase K overnight in a 55°C water bath. Genomic DNA was isolated using phenol: chloroform extraction and precipitated with sodium acetate /ethanol, washed extensively with 70% ethanol. The mHTT CAG repeat was amplified using a human-specific PCR assay. The forward primer was fluorescently labeled with 6-FAM (Applied Biosystems) and products were resolved using the ABI 3730xl DNA analyzer (Applied Biosystems) with GeneScan 500 LIZ as internal size standard (Applied Biosystems). GeneMapper v5 (Applied Biosystems) was used to generate CAG repeat size distribution traces. Expansion and contraction indices were quantified from the GeneMapper CAG repeat distributions as reported (Lee et al., 2010). Briefly, the highest peak in each trace was used to determine a relative threshold of 10% and peaks falling below this threshold were excluded from analysis. Peak heights normalized to the sum of all peak heights were multiplied by the change in CAG length of each peak, relative to the highest peak, and summed to generate an instability index representing the mean repeat length change.
Quantification for HTT intron and exon readings from RNAseq datasets
Readings were mapped to the human genome GRCh38 using STAR spliced read aligner (Dobin et al., 2013)with default parameters. The counts of read-fragments for introns and exons aligned to gene HTT region, were derived using the featureCounts program ($http://bioinf.wehi.edu.au/featureCounts/) with genome assembly GRCh38 (hg38) as reference, at exon and intron levels, respectively. Only uniquely mapped reads were used for subsequent analyses.
RNA extraction and sequencing
For BACHD cohort, 4 BACHD and 4 WT littermate mice (sex balanced) at 12 months were used for RNAseq. For BAC-CAG cohort, 8 BAC-CAG and 8 WT littermates at ages 2, 6 and 12 months (sex balanced) were used for RNAseq. Animals were deeply anesthetized with pentobarbital prior to transcardial perfusion with 0.1 mM phosphate buffered saline (PBS), pH7.4, made with DEPC-treated water. Cortex and striatum from each mouse were dissected in ice cold PBS, and flash-frozen on dry ice. Brain issues were stored at −80°C until tissues for all the mice were ready for RNA preparation. Tissues were lysed in Trizol (Invitrogen; Carlsbad, CA) before total RNA was extracted using Qiagen (Valencia, CA) RNeasy kit with QIAshredder columns, per manufacturer recommendations, including on-column DNase digestion. Total RNAs were stored at −80°C until use, avoiding freeze-thaw cycles. Library preparation and RNAseq were performed by the UCLA Neuroscience Genomics Core (UNGC). RNA quality was measured using the Agilent Bioanalyzer system according to the manufacturer’s recommendations. cDNA libraries were generated using Illumina TruSeq RNA Library Prep Kit v2 and sequenced using an Illumina HiSeq 4000 sequencer with strand-specific, paired-end, 69-mer sequencing protocols to a minimum read depth of 37.5 million reads per sample. Clipped reads were aligned to mouse genome mm10 using the STAR aligner using default settings. Read counts for individual genes were obtained using HTSeq. RNAseq data is in the process of being deposited to the Gene Expression Omnibus (GEO) repository (www.ncbi.nlm.nih.gov/geo).
Preprocessing of mRNA expression data
Expression data from BACHD and BAC-CAG at 2, 6 and 12 months were each analyzed as a separate set. We retained only genes with at least 1 count per million aligned reads in at least ¼ of the total number of samples. The rationale is to only include genes that are likely to be expressed in at least half of the samples of each genotype group. We used a modified version of the sample network methodology originally described in (Oldham et al., 2012). Specifically, to quantify inter-sample connectivity, we first transformed the raw counts using variance stabilization (R function variance Stabilizing Transformation) and then used Euclidean inter-sample distance based on the scaled profiles of the 8000 genes with highest mean expression. The intersample connectivities k were transformed to Z scores using robust standardization
where index a labels samples, MAD is the median absolute deviation, a robust analog of standard deviation, and the constant 1.4826 ensures asymptotic consistency (approximate equality of MAD and standard deviation for large, normally distributed samples). Finally, samples with Za < −6 were flagged as outliers. This procedure resulted in the removal of 1 sample in 6 month striatum and 2 samples in 12 month striatum; no samples were removed in 2 month striatum, BACHD striatum or any of the cortex data sets.
Differential expression analysis
To make DE analysis more robust against potential outlier measurements (counts) that may remain even after outlier sample removal, we defined individual observation weights designed to downweigh potential outliers. The weights are constructed separately for each gene. First, Tukey bi-square-like weights λ (Wilcox, 2013) are calculated for each (variance-stabilized) observation xa (index a labels samples) as
where
The median is calculated separately for each gene across all samples. MMAD stands for modified MAD, calculated as follows. For each gene, we first set MMAD = MAD. The following conditions are then checked separately for each gene: (1) 10th percentile of the weights λ is at least 0.1 (that is, the proportion of observations with weights <0.1 is less than 10%) (Langfelder and Horvath, 2012) and (2) for each individual genotype, 40th percentile of the weights λ is at least 0.9 (that is, at least 40% of the observation have a high coefficient of at least 0.9). If both conditions are met, MMAD = MAD. If either condition is not met, MMAD equals the lowest value for which both conditions are met. The rationale is to exclude outliers but ensure that the number of outliers is not too large either overall or in each genotype group. This approach has previously been used in (Lee et al., 2018). DE analysis was carried out using DESeq2 (Love et al., 2014) version 1.22.2 with default arguments except for disabling outlier replacement (since we use weights to downweigh potential outliers) and independent filtering (since we have pre-filtered genes based on expression levels).
Enrichment analysis
Enrichment analysis were performed using our published methods (Langfelder et al., 2016; Lee et al., 2018). Briefly, enrichment calculations for DE genes with FDR<0.1 or P<0.01 were carried out using our in-house R package anRichment
(https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/GeneAnnotation/) that implements standard Fisher exact test and a multiple-testing correction across all query and refence gene sets. Ingenuity Pathway Analysis (IPA, Qiagen) were performed using genes DE at P<0.01.
QUANTIFICATION AND STATISTICAL ANALYSIS
Multiple group analyses were performed by one-way or two-way ANOVA followed by Tukey’s post hoc test for normally distributed datasets. Unpaired t test with Welch’s correction was performed for normally distributed datasets. statistical analysis was performed using the GraphPad Prism 7.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Sheep polyclonal anti-mHTT (S830) | Sathasivam K. et al., 2001 | N/A |
| Mouse anti-polyglutamine-expansion diseases marker antibody (clone 5TF1–1C2) | Millipore | Cat# MAB1574; RRID: AB_94263 |
| Mouse monoclonal anti-NeuN (clone A60) | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Polyclonal Rabbit Anti-Glial Fibrillary Acidic Protein (GFAP) antibody |
Agilent | Cat# N1506; RRID:AB_10013482 |
| Anti-Alpha-actinin-2 / ACTN2 antibody | Abcam | Cat# ab68167; RRID: AB_11157538 |
| Rabbit polyclonal anti-Ubiquitin antibody | Sigma-Aldrich | Cat# U5379, RRID:AB_477667 |
| Mouse anti-Huntingtin protein antibody (clone mEM48) |
Millipore | Cat#MAB5374;RRIDAB_2307353 |
| V5 Tag Monoclonal Antibody | Invitrogen | Cat # R960–25 |
| Ib1 antibody | Wako | Cat#019–19741 |
| Lgals3 antibody | Santa Cruz Biotechnology | Cat# sc-19283 RRID:AB_2136897 |
| Mbnl1 antibody | Abcam | Cat# ab45899 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Proteinase K | Sigma-Aldrich | Cat# P6556 |
| Triton X-100 | Sigma Aldrich | Cat# X100 |
| Protease inhibitor cocktail tablets | Roche | Cat# 11873580001 |
| ProLong Gold Antifade Mountant with DAPI | Invitrogen | Cat# P36935 |
| Vector SG Peroxidase Substrate Kit | Vector Lab. | Cat# SK4700 |
| Critical Commercial Assays | ||
| QIAshredder | QIAGEN | Cat# 79656 |
| RNAeasy Plus Micro Kit | QIAGEN | Cat# 74034 |
| TruSeq Stranded mRNA LT Prep Kit | Illumina | Cat# RS-122–2101 |
| Agilent RNA 6000 Pico Chip | Agilent Technologies | Cat# 5067–1513 |
| SYBR™ Green PCR Master Mix | Thermo Fisher Sci. | Cat# 4309155 |
| Deposited Data | ||
| Raw data files for BAC-CAG and BACHD RNA-seq |
This paper | In the process of data deposition at GEO GSE153827 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BAC-CAG | This paper | JAX: 037050 |
| Mouse: BACHD | The Jackson Laboratory | JAX: 008197 |
| Mouse: MORF3 | The Jackson Laboratory | JAX: 035403 |
| Mouse: CaMK2a-CreERT2 | The Jackson laboratory | JAX: 012362 |
| Recombinant DNA | ||
| RP11–866L6 BAC clone | BACPAC Resource |
https://bacpacresources.org/cloneInfoSearch.php |
| pLD53. SC-AB Shuttle Vector | Gong S. et al., 2002 | US Patent No. 6,821,759 (Rockefeller University) |
| pBC-mHTTEx1–140Q | Dr. Bates G. Uni. Col. London | Upon request |
| pLD53. mHTTEx1–135Q | This paper | Upon request |
| BAC-CAG BAC clone | This paper | Upon request |
| WGCNA an R package for weighted correlation network analysis |
University of California at Los Angeles |
http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/ ; RRID:SCR_003302 |
| ImageJ | NIH |
https://imagej.nih.gov/ij/index.html; RRID:SCR_003070 |
| STAR | N/A |
https://github.com/alexdobin/STAR; RRID:SCR_015899 |
| HTSeq | N/A | https://htseq.readthedocs.io/en/release_0.10.0/ ; RRID:SCR_005514 |
| GraphPad Prism 7 | GraphPad Software |
https://www.graphpad.com RRID:SCR_002798 |
| Adobe Photoshop CC19 | Adobe |
https://www.adobe.com; RRID:SCR_014199 |
| GeneMapper v5 | Applied Biosystem | Cat# 4370784 |
| Imaris 9.7 | Oxford Instrument |
http://www.bitplane.com/imaris/imaris RRID:SCR_007370 |
Highlights.
A novel human mHTT BAC transgenic model of HD with long uninterrupted CAG repeats
BAC-CAG shows striatum-selective pathogenesis that evaded previous mHTT models
Uninterrupted mHTT CAG repeat length is key to striatal transcriptionopathy in HD
Repeat instability and nuclear mHTT best correlate with striatal disease onset
ACKNOWLEDGMENTS
The generation of the BAC-CAG model was supported by generous donations to X.W.Y. lab from HD patient families, and HD research in the X.W.Y. lab was also supported by NIH/NINDS R01 grants (NS084298 and NS113612), CHDI Foundation, Inc., and the Hereditary Disease Foundation. M.G. is supported by NIH/NINDS RO1 (NS089750) and M.B.C. and L.P.W.R. by NIH/NINDS (R01NS117910). We acknowledge Y. Kamata and M. Katsumata at Cedar Sinai Medical Center for performing the transgenic microinjections; J. DeYoung and UCLA Neuroscience Genomics Core for its RNA-seq service; R.M. Pinto and V. Wheeler at Massachusetts General Hospital/Harvard Medical School for advice on the analysis of repeat instability index; and K. Kuhlbrodt, M. Charpenay, and J.-P. Herault at Evotec for performing the MSD assays. We are indebted to J. Rosinski, J. Aaronson, and T. Vogt at CHDI and members of the Yang lab for feedback on the study and the manuscript.
Footnotes
DECLARATION OF INTERESTS
X.W.Y. is a scientific advisory board (SAB) member for Triplet Therapeutics and Ophidion Bio and is a former SAB member and stockholder for Mitokinin.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2022.01.006.
REFERENCES
- Bañez-Coronel M, Ayhan F, Tarabochia AD, Zu T, Perez BA, Tusi SK, Pletnikova O, Borchelt DR, Ross CA, Margolis RL, et al. (2015). RAN translation in Huntington disease. Neuron 88, 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram-Weston Z, Jones L, Dunnett SB, and Brooks SP (2012). Light and electron microscopic characterization of the evolution of cellular pathology in HdhQ92 Huntington’s disease knock-in mice. Brain Res. Bull. 88, 171–181. [DOI] [PubMed] [Google Scholar]
- Bayram-Weston Z, Jones L, Dunnett SB, and Brooks SP (2016). Comparison of mHTT antibodies in Huntington’s disease mouse models reveal specific binding profiles and steady-state ubiquitin levels with disease development. PLoS One 11, e0155834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram-Weston Z, Stone TC, Giles P, Elliston L, Janghra N, Higgs GV, Holmans PA, Dunnett SB, Brooks SP, and Jones L (2015). Similar striatal gene expression profiles in the striatum of the YAC128 and HdhQ150 mouse models of Huntington’s disease are not reflected in mutant Huntingtin inclusion prevalence. BMC Genomics 16, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Trotta CR, Narasimhan J, Wiedinger KJ, Li W, Effenberger KA, Woll MG, Jani MB, Risher N, Yeh S, et al. (2021). Small molecule splicing modifiers with systemic HTT-lowering activity.Nature communications 12, 7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JB, Warby SC, Southwell AL, Doty CN, Greenlee S, Skotte N, Hung G, Bennett CF, Freier SM, and Hayden MR (2011). Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin.Mol. Ther. 19, 2178–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carty N, Berson N, Tillack K, Thiede C, Scholz D, Kottig K, Sedaghat Y, Gabrysiak C, Yohrling G, von der Kammer H, et al. (2015). Characterization of HTT inclusion size, location, and timing in the zQ175 mouse model of Huntington’s disease: an in vivo high-content imaging study. PLoS One 10, e0123527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. [DOI] [PubMed] [Google Scholar]
- Chung DW, Rudnicki DD, Yu L, and Margolis RL (2011). A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 20, 3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, Durr A, Leavitt BR, Roos R TRACK-HD team, et al. (2019). A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine 48, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong SY, Pepers BA, Roos RA, Van Ommen GJ, and Dorsman JC (2005). Epitope mapping of monoclonal antibody 4C8 recognizing the protein huntingtin. Hybridoma (Larchmt) 24, 231–235. [DOI] [PubMed] [Google Scholar]
- de Mezer M, Wojciechowska M, Napierala M, Sobczak K, and Krzyzosiak WJ (2011). Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res 39, 3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didiot MC, Ferguson CM, Ly S, Coles AH, Smith AO, Bicknell AA, Hall LM, Sapp E, Echeverria D, Pai AA, et al. (2018). Nuclear localization of Huntingtin mRNA is specific to cells of neuronal origin. Cell Rep 24, 2553– 2560.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, and Aronin N (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B, Schmitt A, Gebicke-Haerter PJ, Falkai P, Gru€nblatt, E., et al. (2015). Common mechanisms in neurodegeneration and neuroinflammation: a BrainNet Europe gene expression microarray study. J. Neural Transm. (Vienna) 122, 1055–1068. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Butland SL, Pouladi MA, and Hayden MR (2009). Mouse models of Huntington disease: variations on a theme. Dis. Model. Mech. 2, 123–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Sutton L, and Hayden MR (2011). Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 17, 475–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshim PP, and Bates GP (2018). Mouse models of Huntington’s disease. Methods Mol. Biol. 1780, 97–120. [DOI] [PubMed] [Google Scholar]
- Franich NR, Basso M, André EA, Ochaba J, Kumar A, Thein S, Fote G, Kachemov M, Lau AL, Yeung SY, et al. (2018). Striatal mutant huntingtin protein levels decline with age in homozygous Huntington’s disease knockin mouse models. J. Huntingtons Dis. 7, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franich NR, Hickey MA, Zhu C, Osborne GF, Ali N, Chu T, Bove NH, Lemesre V, Lerner RP, Zeitlin SO, et al. (2019). Phenotype onset in Huntington’s disease knock-in mice is correlated with the incomplete splicing of the mutant huntingtin gene. J. Neurosci. Res. 97, 1590–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium (2015). Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 162, 516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium (2019). CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell 178, 887–900.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, Andre VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, et al. (2008). Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Cantle JP, Greiner ER, Lee CYD, Barth AM, Gao F, Park CS, Zhang Z, Sandoval-Miller S, Zhang RL, et al. (2015). N17 modifies mutant huntingtin nuclear pathogenesis and severity of disease in HD BAC transgenic mice. Neuron 85, 726–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, and Yang XW (2009). Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. (2006). Regional and cellular gene expression changes in human Huntington’s disease brain.Hum. Mol. Genet. 15, 965–977. [DOI] [PubMed] [Google Scholar]
- Hong EP, Chao MJ, Massey T, McAllister B, Lobanov S, Jones L, Holmans P, Kwak S, Orth M, Ciosi M, et al. (2021). Association analysis of chromosome X to identify genetic modifiers of Huntington’s disease. J. Huntingtons Dis. 10, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indersmitten T, Tran CH, Cepeda C, and Levine MS (2015). Altered excitatory and inhibitory inputs to striatal medium-sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington’s disease. J. Neurophysiol. 113, 2953–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, and Vale RD (2017). RNA phase transitions in repeat expansion disorders. Nature 546, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L, Houlden H, and Tabrizi SJ (2017). DNA repair in the trinucleotide repeat disorders. Lancet Neurol 16, 88–96. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, and Swanson MS (2006). Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 103, 11748–11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, et al. (2012). Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, and Colwell CS (2011). Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp. Neurol. 228, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha JH, et al. (2007). Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol.Genet. 16, 1845–1861. [DOI] [PubMed] [Google Scholar]
- Kumar A, Zhang J, Tallaksen-Greene S, Crowley MR, Crossman DK, Morton AJ, Van Groen T, Kadish I, Albin RL, Lesort M, and Detloff PJ (2016). Allelic series of Huntington’s disease knock-in mice reveals expression discorrelates. Hum. Mol. Genet. 25, 1619–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2012). Fast R Functions for Robust Correlations and Hierarchical Clustering. Journal of statistical software 46 (11), i11. [PMC free article] [PubMed] [Google Scholar]
- Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X, Wang N, Zhao Y, Park CS, Cooper Y, et al. (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer’s disease models. Neuron 97, 1032–1048.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Pinto RM, Gillis T, St Claire JC, and Wheeler VC (2011). Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver.PLoS One 6, e23647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-M, Zhang J, Su AI, Walker JR, Wiltshire T, Kang K, Dragileva E, Gillis T, Lopez ET, Boily M-J, et al. (2010). A novel approach to investigate tissue-specific trinucleotide repeat instability. BMC Syst. Biol. 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, and Li XJ (2000). Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25, 385–389. [DOI] [PubMed] [Google Scholar]
- Li J, Nakamori M, Matsumoto J, Murata A, Dohno C, Kiliszek A, Taylor K, Sobczak K, and Nakatani K (2018). A dimeric 2,9-diamino-1,10-phenanthroline derivative improves alternative splicing in myotonic dystrophy type 1 cell and mouse models. Chemistry 24, 18115–18122. [DOI] [PubMed] [Google Scholar]
- Li LB, Yu Z, Teng X, and Bonini NM (2008). RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 453, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Shakkottai VG, and Albin RL (2019). Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol. 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh DH, Kudo T, Truong D, Wu Y, and Colwell CS (2013). The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS One 8, e69993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XH, and Yang XW (2012). Huntingtin holiday: progress toward an antisense therapy for Huntington’s disease. Neuron 74, 964–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald D, Tessari MA, Boogaard I, Smith M, Pulli K, Szynol A, Albertus F, Lamers MB, Dijkstra S, Kordt D, et al. (2014). Quantification assays for total and polyglutamine-expanded huntingtin proteins. PLoS One 9, e96854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik I, Kelley CP, Wang ET, and Todd PK (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, and Bates GP (1997). Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat. Genet. 15, 197–200. [DOI] [PubMed] [Google Scholar]
- Menalled L, El-Khodor BF, Patry M, Suá rez-Fariñ as M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, et al. (2009). Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 35, 319–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, and Chesselet MF (2003). Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. The Journal of comparative neurology 465, 11–26. [DOI] [PubMed] [Google Scholar]
- Monteys AM, Ebanks SA, Keiser MS, and Davidson BL (2017). CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 25, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ (2013). Circadian and sleep disorder in Huntington’s disease.Exp. Neurol. 243, 34–44. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, and Maywood ES (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci. 25, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murmann AE, Gao QQ, Putzbach WE, Patel M, Bartom ET, Law CY, Bridgeman B, Chen S, McMahon KM, Thaxton CS, and Peter ME (2018). Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Rep 19, e45336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalavade R, Griesche N, Ryan DP, Hildebrand S, and Krauss S (2013). Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4, e752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neueder A, and Bates GP (2018). RNA related pathology in Huntington’s disease. Adv. Exp. Med. Biol. 1049, 85–101. [DOI] [PubMed] [Google Scholar]
- Neueder A, Landles C, Ghosh R, Howland D, Myers RH, Faull RLM, Tabrizi SJ, and Bates GP (2017). The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep.7, 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Cleary JD, and Ranum LPW (2019). Repeat-associated non-ATG translation: molecular mechanisms and contribution to neurological disease. Annu. Rev. Neurosci. 42, 227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Langfelder P, and Horvath S (2012). Network methods for describing sample relationships in genomic datasets: application to Huntington’s disease. BMC Syst. Biol. 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, and Zoghbi HY (2007). Trinucleotide repeat disorders. Annu. Rev. Neurosci. 30, 575–621. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, et al. (2014). Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 83, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouladi MA, Stanek LM, Xie Y, Franciosi S, Southwell AL, Deng Y, Butland S, Zhang W, Cheng SH, Shihabuddin LS, and Hayden MR (2012). Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum. Mol. Genet. 21, 2219–2232. [DOI] [PubMed] [Google Scholar]
- Pouladi MA, Xie Y, Skotte NH, Ehrnhoefer DE, Graham RK, Kim JE, Bissada N, Yang XW, Paganetti P, Friedlander RM, et al. (2010). Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum. Mol. Genet. 19, 1528–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimian R, Bé land LC, Sato S, and Kriz J. (2021). Microglia-derived galectin-3 in neuroinflammation; a bittersweet ligand? Med. Res. Rev. 41, 2582–2589. [DOI] [PubMed] [Google Scholar]
- Reindl W, Baldo B, Schulz JJ, Janack I, Lindner I, Kleinschmidt M, Sedaghat Y, Thiede C, Tillack K, et al. (2019). Meso scale discoverybased assays for the detection of aggregated huntingtin. PLoS One 14, e0213521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 10, 204–216. [DOI] [PubMed] [Google Scholar]
- Ross CA, and Tabrizi SJ (2011). Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10, 83–98. [DOI] [PubMed] [Google Scholar]
- Rousseaux MWC, Tschumperlin T, Lu HC, Lackey EP, Bondar VV, Wan YW, Tan Q, Adamski CJ, Friedrich J, Twaroski K, et al. (2018). ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron 97, 1235–1243.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, and DiFiglia M (2001). Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropathol. Exp. Neurol. 60, 161–172. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RL, Roos RA, Howland D, et al. (2013). Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A. 110, 2366–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathasivam K, Woodman B, Mahal A, Bertaux F, Wanker EE, Shima DT, and Bates GP (2001). Centrosome disorganization in fibroblast cultures derived from R6/2 Huntington’s disease (HD) transgenic mice and HD patients. Hum. Mol. Genet. 10, 2425–2435. [DOI] [PubMed] [Google Scholar]
- Siew JJ, Chen H-M, Chen H-Y, Chen H-L, Chen C-M, Soong B-W, Wu Y-R, Chang C-P, Chan Y-C, Lin C-H, et al. (2019). Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat. Commun. 10, 3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotte NH, Southwell AL, Østergaard ME, Carroll JB, Warby SC, Doty CN, Petoukhov E, Vaid K, Kordasiewicz H, Watt AT, et al. (2014). Allele-specific suppression of mutant huntingtin using antisense oligo PLoS One 9, e107434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang Y-Z, et al. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567. [DOI] [PubMed] [Google Scholar]
- Smith-Dijak AI, Sepers MD, and Raymond LA (2019). Alterations in synaptic function and plasticity in Huntington disease. J. Neurochem. 150, 346–365. [DOI] [PubMed] [Google Scholar]
- Southwell AL, Kordasiewicz HB, Langbehn D, Skotte NH, Parsons MP, Villanueva EB, Caron NS, Østergaard ME, Anderson LM, Xie Y, et al. (2018). Huntingtin suppression restores cognitive function in a mouse model of Huntington’s disease. Sci. Transl. Med. 10, eaar3959. [DOI] [PubMed] [Google Scholar]
- Southwell AL, Warby SC, Carroll JB, Doty CN, Skotte NH, Zhang W, Villanueva EB, Kovalik V, Xie Y, Pouladi MA, et al. (2013). A fully humanized transgenic mouse model of Huntington disease. Hum. Mol. Genet. 22, 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Li PP, Zhu S, Cohen R, Marque LO, Ross CA, Pulst SM, Chan HY, Margolis RL, and Rudnicki DD (2015). Nuclear retention of full-length HTT RNA is mediated by splicing factors MBNL1 and U2AF65. Sci. Rep. 5, 12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, Blair NF, Craufurd D, Priller J, Rickards H, et al. (2019). Targeting Huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 380, 2307–2316. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, and Hayden MR (2005). Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 14, 3823–3835. [DOI] [PubMed] [Google Scholar]
- Veldman MB, Park CS, Eyermann CM, Zhang JY, Zuniga-Sanchez E, Hirano AA, Daigle TL, Foster NN, Zhu M, Langfelder P, et al. (2020). Brainwide genetic sparse cell labeling to illuminate the morphology of neurons and glia with Cre-dependent MORF mice. Neuron 108, 111–127.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman MB, and Yang XW (2018). Molecular insights into cortico-striatal miscommunications in Huntington’s disease. Curr. Opin. Neurobiol. 48, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, and DiFiglia M (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369. [DOI] [PubMed] [Google Scholar]
- Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, Gu X, Shirasaki D, Cepeda C, Li Y, et al. (2014). Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat. Med. 20, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, and Dion V (2021). Modifiers of CAG/CTG repeat instability: insights from mammalian models. J. Huntingtons. Dis. 10, 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, et al. (2000). Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 9, 503–513. [DOI] [PubMed] [Google Scholar]
- Wilcox RR (2013). Introduction to Robust Estimation and Hypothesis Testing, 3rd (Amsterdam; Boston: Academic Press; ). [Google Scholar]
- Wood TE, Barry J, Yang Z, Cepeda C, Levine MS, and Gray M (2019). Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 28, 487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman B, Butler R, Landles C, Lupton MK, Tse J, Hockly E, Moffitt H, Sathasivam K, and Bates GP (2007). The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 72, 83–97. [DOI] [PubMed] [Google Scholar]
- Wright GEB, Collins JA, Kay C, McDonald C, Dolzhenko E, Xia Q, Becanovic K, Drö gemö ller BI, Semaka A, Nguyen CM, et al. (2019). Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am. J. Hum. Genet. 104, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XW, and Gray M (2011). In Mouse model of for validating preclinical candidates for Huntington’s disease, Lo Dand Hughes RE, eds. (CRC Press Press; ), pp. 165–196. [PubMed] [Google Scholar]
- Yang XW, Model P, and Heintz N (1997). Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nature biotechnology 15, 859–865. [DOI] [PubMed] [Google Scholar]
- Zeitler B, Froelich S, Marlen K, Shivak DA, Yu Q, Li D, Pearl JR, Miller JC, Zhang L, Paschon DE, et al. (2019). Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 25, 1131–1142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data reported in this paper is has been deposited at GEO (GEO access number GSE153827).