

Abstract
Background:
Altered cerebral blood flow (CBF) has been linked to increased risk for Alzheimer’s disease (AD). However, whether altered CBF contributes to AD risk by accelerating cognitive decline remains unclear. It also remains unclear whether reductions in CBF accelerate neurodegeneration and development of small vessel cerebrovascular disease.
Objective:
To examine associations between CBF and trajectories of memory performance, regional brain atrophy, and global white matter hyperintensity (WMH) volume.
Method:
147 Alzheimer’s Disease Neuroimaging Initiative participants free of dementia underwent arterial spin labeling (ASL) magnetic resonance imaging (MRI) to measure CBF and serial neuropsychological and structural MRI examinations. Linear mixed effects models examined 5-year rate of change in memory and 4-year rate of change in regional brain atrophy and global WMH volumes as a function of baseline regional CBF. Entorhinal and hippocampal CBF were examined in separate models.
Results:
Adjusting for demographic characteristics, pulse pressure, apolipoprotein E ε4 positivity, cerebrospinal fluid p-tau/Aβ ratio, and neuronal metabolism (i.e., fluorodeoxyglucose standardized uptake value ratio), lower baseline entorhinal CBF predicted faster rates of decline in memory as well as faster entorhinal thinning and WMH progression. Hippocampal CBF did not predict cognitive or brain structure trajectories.
Conclusion:
Findings highlight the importance of early cerebrovascular dysfunction in AD risk and suggest that entorhinal CBF as measured by noninvasive ASL MRI is a useful biomarker predictive of future cognitive decline and of risk of both cerebrovascular and neuronal changes, even after adjusting for well-established AD risk factors and in a sample with relatively low vascular risk burden.
Keywords: Cognition, Entorhinal Cortex, White Matter Hyperintensities, Regional Blood Flow, Perfusion, Magnetic Resonance Imaging, Alzheimer’s Disease, Aging, Neuropsychology
INTRODUCTION
In recent years, Alzheimer’s disease (AD) research has increasingly focused on methods for the early detection of individuals at risk for future cognitive decline or progression to dementia, with a particular focus on the roles of amyloid and tau pathologies. In this context, cerebrovascular pathology has also garnered increased attention, as evidence has emerged that vascular changes may, in fact, be part of early AD pathogenesis or accelerate AD-related cognitive decline [1, 2]. One indicator of neurovascular function is cerebral blood flow (CBF), which is measured as the rate of delivery of arterial blood to the capillary beds. Arterial spin labeling (ASL) magnetic resonance imaging (MRI) is a non-invasive approach that directly measures CBF by magnetically labeling arterial water that is used as an endogenous tracer [3]. Vascular dysfunction is considered part of “hit one” in Zlokovic’s vascular two-hit hypothesis of AD pathogenesis, whereby hypoperfusion and blood brain barrier dysfunction precede and initiate further vascular damage involving leakage of neurotoxic molecules and accumulation of micro-ischemic injuries [4]. As a result of blood brain barrier breakdown and ischemia, there is subsequent accumulation of AD pathology such as amyloid (“hit two”), ultimately leading to cognitive impairment [1, 5]. Indeed, growing evidence suggests that CBF alterations may occur before irreversible parenchymal changes including brain atrophy and the development of white matter hyperintensities (WMH), an MRI marker of small vessel cerebrovascular disease [6, 7].
A complex pattern of associations between CBF and cognition has emerged such that both hypoperfusion and hyperperfusion can be associated with poorer outcomes across the clinical AD spectrum, depending on both the stage of AD (e.g., cognitively normal [CN] vs. preclinical/”at-risk” vs. mild cognitive impairment [MCI] vs. dementia) and brain region under investigation [8–13]. Several studies, however, have demonstrated cross-sectional relationships between both global and regional resting CBF (most often hypoperfusion) and memory [8, 14–18]. Relationships of CBF with cognition have also been shown to vary by amyloid status or other AD risk factors [10, 14, 18, 19] highlighting the complex nature of CBF effects within the context of aging and AD.
Relatively few longitudinal studies have examined the relationships between CBF and cognitive trajectories in older adults without dementia. In these few studies, the general finding has been that lower CBF is associated with faster declines in cognition, however, these studies have often performed whole-brain analyses rather than examined a priori regions of interest (ROIs) and have studied relatively short follow-up intervals (e.g., 1-2 years) [7, 20]. In the present study we examined entorhinal and hippocampal CBF and associations with longitudinal trajectories of memory performance across 5 years of follow up. Given that diminished CBF may affect cognition via neurodegeneration and/or vascular disease, we also examined associations between regional CBF and trajectories of cortical thickness, brain volume, and WMH volume.
MATERIALS AND METHODS
The ADNI Dataset
Data used in the preparation of this manuscript were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). ADNI was initiated in 2003 as a public-private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial MRI, positron emission tomography, other biological markers, and clinical and neuropsychological assessment can be combined to measure the latency of cognitive decline and the progression of MCI and early AD.
Participants
Enrollment criteria for ADNI have been previously described in detail [21]. Briefly, participants from ADNI were 55–90 years old and met the following criteria: (1) ≥6 years of education or work-history equivalent, (2) fluency in English or Spanish, (3) Geriatric Depression Scale (GDS) scores <6 (possible score range is 0-15) [22], (4) Hachinski Ischemic Scale (HIS) scores ≤4, (5) adequate vision and hearing to perform neuropsychological tests, (6) in generally good health and without significant head trauma or neurologic disease, (7) were stable on permitted medications, and (8) had a study partner. We included participants without dementia who had ASL MRI data (that passed quality control inspection) within 12 months of their baseline visit, and who also had available neuropsychological testing, structural MRI, and covariate data (n = 147). See Supplementary Figure 1. This study was approved by the Institutional Review Boards of all participating institutions. Informed written consent was obtained from all participants at each study site.
Neuropsychological Composite Scores
A composite scores measuring memory was developed and validated in previous studies within the ADNI cohort [23]. This composite score is based on the Rey Auditory Verbal Learning (RAVLT) learning and recall, word list learning and recognition from AD Assessment Scale – Cognitive Subscale (ADAS-Cog), immediate and delayed recall from Logical Memory of the Wechsler Memory Scale–Revised, and the 3-word recall item from the Mini-Mental State Examination (MMSE) [23] The composite score has a mean of 0 and a standard deviation of 1.
Arterial Spin Labeling and Structural MRI Data
Detailed information of ASL and structural MRI data acquisition and processing is available online (http://adni.loni.usc.edu/). All imaging data (e.g., ASL, structural MRI, Florbetapir (amyloid) PET, fluorodeoxyglucose [FDG] positron emission tomography [PET]) used in this study was previously processed and downloaded from www.loni.usc.edu. Briefly, MR imaging was performed on 3.0 Tesla MR scanners from a single vendor (MAGNETOM Trio, Verio, Skyra, and Siemens). Pulsed ASL scans were acquired using QUIPS II with thin-slice T11 periodic saturation sequence (“Q2TIPS”) with echo-planar imaging[24]: inversion time of arterial spins=700ms, total transit time of the spins=1900ms, tag thickness=100mm, tag to proximal slice gap=25.4mm, repetition time=3400ms, echo time=12ms, field of view=256mm, 64x64 matrix, 24 4-mm thick axial slices [52 tag + control image pairs], time lag between slices 22.5ms.
ASL data processing was largely automated and included motion correction, alignment of each ASL frame to the first frame using a rigid body transformation, and least squares fitting using SPM8. Perfusion-weighted images were computed as the difference of the mean-tagged and mean-untagged ASL images. Perfusion-weighted images were intensity scaled to account for signal decay during acquisition and to generate intensities in meaningful physiological units. After geometric distortion correction, ASL images were aligned to structural T1-weighted images. A partial volume correction that assumes that CBF in gray matter is 2.5 times greater than in white matter was performed. The partial volume corrected perfusion-weighted images were normalized by the reference image (i.e., an estimate of blood water magnetization) to convert the signal into physiological units (i.e., mL/100 g tissue/min). ADNI quality control procedures to determine a global pass/fail rating for each participant’s ASL data were based on visual inspection of signal uniformity, geometrical distortions, gray matter contrast, and presence of large, disruptive artifacts. A rating of “unusable” in any of these categories resulted in a global “fail” and those data were excluded from the present study.
A T1-weighted 3D MPRAGE structural MRI scan was acquired during the same session as the ASL scan with the following parameters: field of view=256mm, repetition time=230ms, echo time=2.98ms, flip angle=9, and resolution=1.1 x 1.1x 1.2 mm3. FreeSurfer was used to skull-strip, segment, and parcellate the structural scans. FreeSurfer-derived entorhinal cortical thickness and hippocampal volume were selected as an a priori dependent variables given their implication in early stages of AD (e.g., Braak stages I/II) [25, 26] FreeSurfer-derived hippocampal volume was normalized by total intracranial volume. FreeSurfer-derived ROIs were applied to extract mean CBF in the entorhinal cortex and hippocampus for each participant. Consistent with previous CBF work in ADNI, ROI CBF values were residualized by precentral gyrus CBF to adjust for individual variation in flow [11]. CBF of the precentral gyrus was selected to serve as a reference region as it is not thought to be impacted in early AD as well as its use as a reference region in past CBF work in ADNI [10, 11, 13]. Mean CBF corrected for partial volume effects was extracted for each ROI and the reference region for each hemisphere separately. However, to reduce the number of comparisons, averaged bilateral CBF estimates were used as the predictor variables in our models. Bilateral CBF estimates were calculated by averaging the mean CBF of each hemisphere. If participants were missing baseline ASL data but had ASL data within the first year of their baseline visit, the first occasion of ASL data was used in our analyses.
White Matter Hyperintensity (WMH) Volume
Detailed methods for WMH volumetric quantification have been previously described [27–30]. Briefly, WMH volume was calculated using a Bayesian approach to segmentation of the high resolution T1-weighted and fluid attenuated inversion recovery (FLAIR) scans. Non-brain tissues were removed from T1-weighted and FLAIR images, the FLAIR image was spatially aligned to the T1-weighted image, and MRI field artifacts were removed. Images were warped to a standard template space. The likelihood of WMH was estimated from FLAIR signal characteristics, prior probability maps of WMH occurrence calculated from previously supervised segmentations of independent FLAIR images from more than 700 individuals, and tissue class constraints. The segmented WMH masks were back-transformed to native space prior to volume calculation. Voxels labeled as WMH were summed and multiplied by voxel dimensions to obtain global WMH volumes and reported in units of cm3.
Covariates and clinical variables
Apolipoprotein E (APOE) ε4 carrier status was determined by the presence of at least one ε4 allele and was included as a covariate to adjust for the known effect of an ε4 allele on risk for AD, cognitive decline, and CBF. Cerebral metabolism was assessed using FDG-PET within a composite meta-ROI that is comprised of the standardized uptake value ratio (SUVR) of the left and right angular gyri, left and right middle/inferior temporal gyri, and bilateral posterior cingulate gyrus. These regions show metabolic changes in MCI and AD that are associated with cognition [31, 32]. The meta-ROI was intensity normalized by dividing by the mean value for a pons/cerebellar reference region [32]. FDG-PET of the meta-ROI was included as a covariate so that the CBF effects could be interpreted independent of brain metabolism. All participants underwent a lumbar puncture at baseline and AD CSF markers were processed using Elecsys® immunoassays. AD biomarker positivity was determined using the previously determined CSF p-tau/Aβ ratio cut-score of >0.0251pg/ml, which was optimized for the ADNI cohort [33]. Pulse pressure, a measure of arterial stiffening and vascular risk, was calculated as systolic blood pressure minus diastolic blood pressure [34].
To determine cognitive status (MCI versus normal cognition), Jak/Bondi actuarial neuropsychological MCI criteria were applied to all participants in this sample [35, 36]. Using this approach, participants were classified as MCI if they performed >1 SD below the age-/education-/sex-adjusted mean on (1) 2 neuropsychological measures within the same cognitive domain or (2) at least 1 measure across all 3 cognitive domains sampled. Six neuropsychological scores were considered in the MCI criteria and included two memory measures (RAVLT delayed free recall correct responses and RAVLT recognition [hits minus false positives]); two language measures (30-item Boston Naming Test [BNT] total correct and Animal Fluency total score), and two attention/executive functioning measures (Trail Making Test [TMT] Parts A and B times to completion). If neither criterion was met, participants were considered to have normal cognition.
Statistical Analyses
Baseline demographic and clinical characteristics were examined with descriptive statistics as well as t-tests for continuous variables and chi-square (X2) tests for categorical variables to compare p-tau/Aβ positive versus negative groups. For continuous variables, baseline group mean comparisons were inspected for homogeneity of variance assumptions using Levene’s test. When Levene’s test was significant (i.e., group variances were significantly different), the p-value associated with unequal variances was reported.
Multivariable linear mixed effects (LME) modeling was used to examine the longitudinal rate of change of memory performance; cortical thickness (for entorhinal cortex) or brain volume (for hippocampus); and global WMH volume as a function of baseline regional CBF. Cortical thickness and volume were examined in models including baseline CBF in the same region (e.g., the model with entorhinal thickness as the dependent variable included entorhinal CBF; the model with hippocampal volume as the dependent variable included hippocampal CBF). All models adjusted for age, sex, pulse pressure, APOE ε4 positivity, CSF p-tau/Aβ positivity, and FDG SUVR; education was also included as a covariate for the memory composite analyses. Random intercept and slope were included. Full information maximum likelihood estimation was used to allow for all available data to be included [37, 38]. All continuous independent variables and covariates in the model were standardized to have a mean of zero and standard deviation of one. Natural log transformation was used to improve distribution normality of WHM volumes.
Given reduction in available cognitive data at 72 months of follow-up that resulted in less than 20% of the sample having memory composite score data at that timepoint, our analyses for the cognitive composites focused on follow-up to 60 months. Given reduction in available MRI data at 60 months of follow-up (i.e., less than 5% of participants had entorhinal cortical thickness data available for download), analyses for MRI dependent variables focused on follow-up to 4 years.
A series of secondary analyses were performed. First, we performed analyses including continuous p-tau/Aβ rather than dichotomous p-tau/Aβ (positivity versus negativity) as a covariate. Second, we performed a series of analyses to determine whether the two-way CBF x time interaction was moderated by the following AD risk factors: cognitive status, p-tau/Aβ, APOE ε4 positivity, and female sex. For each of these AD risk factors, we re-ran the LME models described above but added the AD risk factor of interest (cognitive status, p-tau/Aβ, APOE ε4 positivity, or female sex), a two-way AD risk factor x time interaction, a two-way AD risk factor x CBF interaction, and three-way AD risk factor x CBF x time interaction.
All analyses were performed using Statistical Package for the Social Sciences (SPSS) version 25 (SPSS IBM, New York, USA). Graphs were made with the ggplot2 package in R (http://www.R-project.org/). An alpha=0.05 was set for statistical significance; all tests were two-tailed.
RESULTS
Participant characteristics
Participants’ baseline demographic and clinical data are presented in Table 1. One hundred and forty-seven older adults ranging in age from 55 to 85 (mean±SD=72.24±6.82) comprised the present study sample. There were 70 women (48%), the sample was approximately 94% white, and the average number of years of formal education was 16.67 (SD=2.58). Relative to p-tau/Aβ negative participants, the p-tau/Aβ positive group were significantly older at baseline; had a significantly higher proportion of men compared to women, APOE ε4 carriers compared to non-carriers, and individuals with MCI versus normal cognition; had lower entorhinal cortical thickness, hippocampal volume, and FDG-PET SUVR; had greater WMH volume; and exhibited poorer memory functioning at baseline. The mean memory z-scores was greater than 0 indicating that the group mean score was in the unimpaired range (all p-values < 0.05). There were no significant group differences in terms of education or pulse pressure (p-values > 0.05).
Table 1.
Baseline demographic and clinical data
| Whole Sample (n=147) |
p-tau/Aβ Negative (n=88) |
p-tau/Aβ Positive (n=59) |
t or X2 | d or Φ | p | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| Mean or % | SD or N | Min | Max | Mean or % | SD or N | Min | Max | Mean or % | SD or N | Min | Max | ||||
| Age, years | 71.24 | 6.82 | 55.00 | 85.39 | 69.55 | 6.45 | 55.00 | 84.70 | 73.76 | 6.60 | 58.50 | 85.30 | t=3.84 | d=0.64 | <0.001 |
| Education, years | 16.67 | 2.58 | 9.00 | 20.00 | 16.90 | 2.38 | 12.00 | 20.00 | 16.32 | 2.84 | 9.00 | 20.00 | t=1.33 | d=0.22 | 0.185 |
| Female, % (n) | 47.6% | N=70 | - | - | 54.5% | N=48 | - | - | 37.29 | N=22 | - | - | χ2=4.22 | Φ=0.17 | 0.040 |
| Race, % (n) | χ2=1.57 | Φ=0.10 | 0.666 | ||||||||||||
| Asian | 1.4% | N=2 | - | - | 2.3% | N=2 | - | - | 0.00% | N=0 | - | - | - | - | - |
| Black | 2.7% | N=4 | - | - | 2.3% | N=2 | - | - | 3.39% | N=2 | - | - | - | - | - |
| White | 93.9% | N=138 | - | - | 93.2% | N=82 | - | - | 94.92% | N=56 | - | - | - | - | - |
| More than one | 2.0% | N=3 | - | - | 2.3% | N=2 | - | - | 1.69% | N=1 | - | - | - | - | - |
| Ethnicity, % (n) | χ2=1.20 | Φ=0.03 | 0.729 | ||||||||||||
| Hispanic | 4.1% | N=6 | - | - | 4.5% | N=4 | - | - | 3.39% | N=2 | - | - | - | - | - |
| Non-Hispanic | 95.9% | N=141 | - | - | 95.5% | N=84 | - | - | 96.61% | N=57 | - | - | - | - | - |
| APOE ε4 carrier, % (n) | 39.5% | N=58 | - | - | 21.6% | N=19 | - | - | 66.10% | N=39 | - | - | χ2=29.29 | Φ=0.45 | <0.001 |
| MCI, % (n) | 31.3% | N=46 | - | - | 17.1% | N=15 | - | - | 52.54% | N=31 | - | - | χ2=20.70 | Φ=0.38 | <0.001 |
| Pulse pressure, mmHG | 59.11 | 16.01 | 17.00 | 109.00 | 58.93 | 14.10 | 36.00 | 94.00 | 59.37 | 18.63 | 17.00 | 109.00 | t=0.16 | d=0.03 | 0.871 |
| FDG-PET SUVR | 1.27 | 0.13 | 0.91 | 1.66 | 1.31 | 0.12 | 0.98 | 1.66 | 1.22 | 0.13 | 0.91 | 1.46 | t=2.24 | d=0.70 | <0.001 |
| Entorhinal cortical thickness, mm | 3.48 | 0.40 | 1.90 | 4.08 | 3.55 | 0.33 | 2.39 | 4.08 | 3.37 | 0.47 | 1.90 | 4.08 | t=2.64 | d=0.44 | 0.010 |
| Normalized Hippocampal volume | 47.81 | 7.56 | 27.16 | 63.92 | 50.30 | 7.14 | 27.16 | 63.92 | 44.09 | 6.65 | 29.31 | 59.48 | t=5.21 | d=0.90 | <0.001 |
| Log-transformed WMH volume | 0.68 | 0.37 | 0.04 | 1.87 | 0.55 | 0.30 | 0.04 | 1.38 | 0.86 | 0.38 | 0.04 | 1.87 | t=5.30 | d=0.88 | <0.001 |
| Memory | 0.68 | 0.80 | −1.53 | 3.14 | 1.02 | 0.68 | −0.28 | 3.14 | 0.16 | 0.69 | −1.53 | 1.80 | t=7.56 | d=1.26 | <0.001 |
Data reported are mean, standard deviation (SD), minimum value, and maximum value for continuous variables and percent and number of participants (N) for categorical variables.
Abbreviations: APOE = apolipoprotein E; MCI = mild cognitive impairment; mmHg = millimeters of mercury; FDG-PET = fluorodeoxyglucose positron emission tomography; SUVR = standardized uptake value ratio; mm = millimeter; WMH = white matter hyperintensity; p-tau = phosphorylated tau; Aβ = β-amyloid
Hippocampal volume was normalized by dividing by total intracranial volume and multiplying by 10,000
The memory composite score is based on the Rey Auditory Verbal Learning (RAVLT) learning and recall, word list learning and recognition from AD Assessment Schedule – Cognition (ADAS-Cog), immediate and delayed recall from Logical Memory of the Wechsler Memory Test–Revised, and the 3-word recall item from the Mini-Mental State Examination (MMSE) [23] The composite score has a mean of 0 and a standard deviation of 1.
Memory Trajectories
LME models, adjusting for baseline age, education, sex, pulse pressure, APOE ε4 genotype, p-tau/Aβ positivity, and FDG-PET, examined whether regional CBF predicted increased rate of memory decline over 60 months. There was a significant interaction between baseline entorhinal CBF and time, such that lower baseline entorhinal CBF predicted faster rates of decline in memory [t(144.78)=2.90, p=.004, r=0.23]. See Figure 1. Table 2 shows parameter estimates and test statistics. In contrast, there were no significant interaction between baseline hippocampal CBF and time on memory (p=.700). See Table 3.
Figure 1.
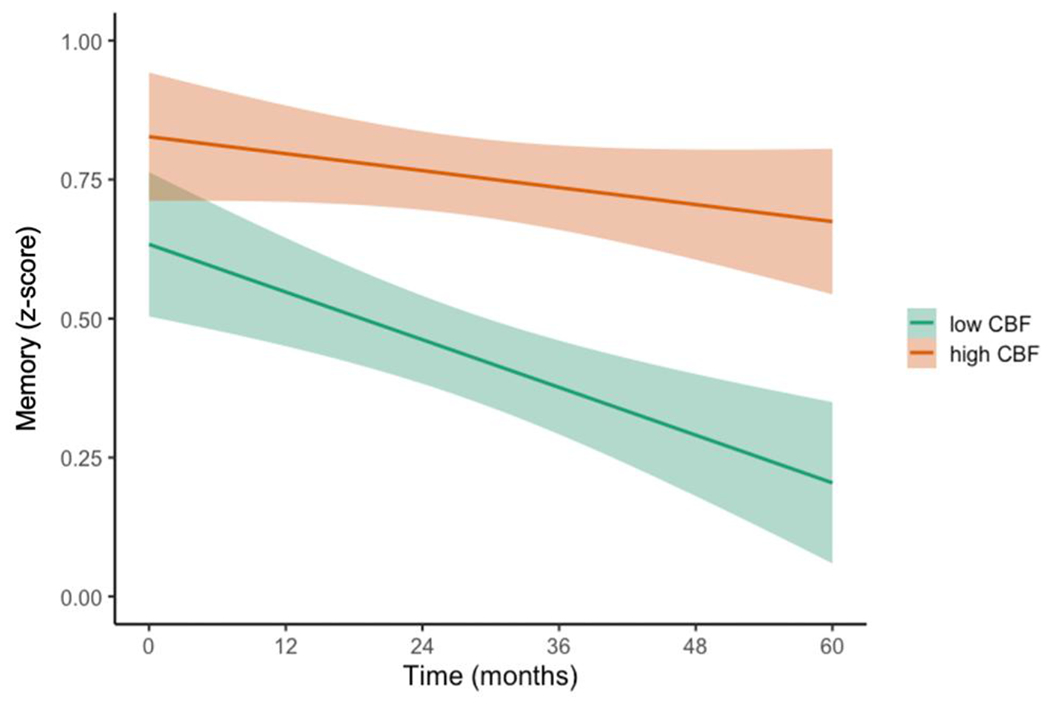
Trajectories of memory performance by baseline entorhinal CBF
Model-predicted values of performance in memory, adjusted for age, education, sex, APOE ε4 genotype, p-tau/Aβ positivity, and FDG-PET. For visual comparison, the graphs display results for low entorhinal cerebral blood flow (CBF) and high entorhinal CBF which were determined by a median split of the values in the analytic sample. Shaded area represents 95% confidence intervals.
Table 2.
Entorhinal CBF prediction of longitudinal performance in memory
| Memory | ||||
|---|---|---|---|---|
|
| ||||
| Estimate | SE | p | r | |
|
|
||||
| Intercept | 0.748 | 0.100 | <0.001 | 0.53 |
| Time | −0.103 | 0.021 | <0.001 | 0.38 |
| Age | −0.165 | 0.060 | 0.007 | 0.22 |
| Education | 0.047 | 0.054 | 0.394 | 0.07 |
| Sex | 0.310 | 0.112 | 0.006 | 0.22 |
| Pulse pressure | 0.107 | 0.057 | 0.062 | 0.15 |
| APOE ε4 | 0.018 | 0.130 | 0.889 | 0.01 |
| p-tau/Aβ | −0.753 | 0.136 | <0.001 | 0.42 |
| FDG-PET | 0.316 | 0.057 | <0.001 | 0.42 |
| Entorhinal CBF | 0.040 | 0.056 | 0.470 | 0.06 |
| Entorhinal CBF x Time | 0.064 | 0.022 | 0.004 | 0.23 |
APOE = apolipoprotein E; p-tau = phosphorylated tau; Aβ = β-amyloid; FDG-PET = fluorodeoxyglucose positron emission tomography; CBF = cerebral blood flow
Bold values are statistically significant (p<.05). Effect size (r-values) interpretation: small=0.10, medium=0.30, large=0.50.
Memory performance was measured using a composite score developed and validated within the ADNI sample [23]. The composite includes indices from the Rey Auditory Verbal Learning Test, Alzheimer’s Disease Assessment Scale – Cognitive Subscale, Logical Memory of the Wechsler Memory Scale–Revised, and the Mini-Mental State Examination.
Table 3.
Hippocampal CBF prediction of longitudinal performance in memory
| Memory | ||||
|---|---|---|---|---|
|
| ||||
| Estimate | SE | p | r | |
|
|
||||
| Intercept | 0.771 | 0.097 | <0.001 | 0.55 |
| Time | −0.098 | 0.022 | <0.001 | 0.37 |
| Age | −0.156 | 0.060 | 0.010 | 0.21 |
| Education | 0.054 | 0.055 | 0.326 | 0.08 |
| Sex | 0.311 | 0.111 | 0.006 | 0.23 |
| Pulse pressure | 0.100 | 0.057 | 0.083 | 0.14 |
| APOE ε4 | −0.014 | 0.132 | 0.916 | 0.01 |
| p-tau/Aβ | −0.765 | 0.135 | <0.001 | 0.43 |
| FDG-PET | 0.316 | 0.057 | <0.001 | 0.42 |
| Hippocampal CBF | −0.061 | 0.056 | 0.277 | 0.09 |
| Hippocampal CBF x Time | 0.009 | 0.024 | 0.700 | 0.03 |
APOE = apolipoprotein E; p-tau = phosphorylated tau; Aβ = β-amyloid; FDG-PET = fluorodeoxyglucose positron emission tomography; CBF = cerebral blood flow
Bold values are statistically significant (p<.05). Effect size (r-values) interpretation: small=0.10, medium=0.30, large=0.50.
Memory performance was measured using a composite score developed and validated within the ADNI sample [23]. The composite includes indices from the Rey Auditory Verbal Learning Test, Alzheimer’s Disease Assessment Scale – Cognitive Subscale, Logical Memory of the Wechsler Memory Scale–Revised, and the Mini-Mental State Examination.
Cortical Thickness, Volume, and WMH Volume Trajectories
Next, LME models, adjusting for baseline age, sex, pulse pressure, APOE ε4 genotype, p-tau/Aβ positivity, and FDG-PET, examined whether regional CBF predicted brain atrophy and global WMH accumulation over 48 months. There were significant interactions between entorhinal CBF and time such that lower baseline entorhinal CBF predicted faster rates of entorhinal thinning [t(130.80)=3.32, p=.001, r=0.28] and a faster increase in WMH volume [t(96.87)=−2.21, p=.029, r=0.22]. Figure 2 shows the trajectories of entorhinal thinning and WMH accumulation by baseline entorhinal CBF. Table 4 shows parameter estimates and test statistics. There were no significant interactions between baseline hippocampal CBF and time on normalized hippocampal volume or WMH volume (all p-values > 0.05). See Table 5.
Figure 2.
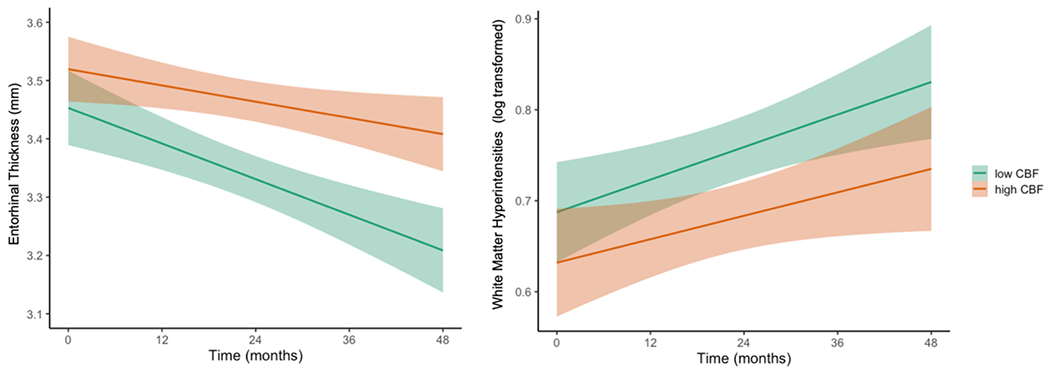
Trajectories of entorhinal cortical thinning and WMH progression by baseline entorhinal CBF
Model-predicted values of entorhinal cortical thickness and white matter hyperintensity volume, adjusted for age, sex, APOE ε4 genotype, p-tau/Aβ positivity, and FDG-PET. For visual comparison, the graphs display results for low entorhinal cerebral blood flow (CBF) and high entorhinal CBF which were determined by a median split of the values in the analytic sample. Shaded area represents 95% confidence intervals. mm = millimeters
Table 4.
Entorhinal CBF prediction of longitudinal entorhinal atrophy and white matter hyperintensity accumulation
| Entorhinal Thickness | WMH Volume | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Estimate | SE | p | r | Estimate | SE | p | r | |
|
|
||||||||
| Intercept | 3.524 | 0.051 | <0.001 | 0.99 | 0.626 | 0.046 | <0.001 | 0.75 |
| Time | −0.073 | 0.011 | <0.001 | 0.52 | 0.053 | 0.006 | <0.001 | 0.68 |
| Age | −0.097 | 0.031 | 0.003 | 0.25 | 0.118 | 0.028 | <0.001 | 0.33 |
| Sex | −0.164 | 0.058 | 0.005 | 0.23 | 0.067 | 0.052 | 0.194 | 0.11 |
| Pulse pressure | 0.053 | 0.030 | 0.078 | 0.15 | 0.040 | 0.026 | 0.130 | 0.13 |
| APOE ε4 | 0.026 | 0.068 | 0.706 | 0.03 | −0.080 | 0.060 | 0.187 | 0.11 |
| p-tau/Aβ | −0.182 | 0.071 | 0.011 | 0.21 | 0.258 | 0.064 | <0.001 | 0.32 |
| FDG-PET | 0.143 | 0.030 | <0.001 | 0.37 | −0.055 | 0.027 | 0.042 | 0.17 |
| Entorhinal CBF | 0.040 | 0.029 | 0.171 | 0.11 | −0.021 | 0.026 | 0.425 | 0.07 |
| Entorhinal CBF x Time | 0.039 | 0.012 | 0.001 | 0.28 | −0.013 | 0.006 | 0.029 | 0.22 |
APOE = apolipoprotein E; p-tau = phosphorylated tau; Aβ = β-amyloid; FDG-PET = fluorodeoxyglucose positron emission tomography; CBF = cerebral blood flow
Bold values are statistically significant (p<.05). Effect size (r-values) interpretation: small=0.10, medium=0.30, large=0.50.
Table 5.
Hippocampal CBF prediction of longitudinal hippocampal atrophy and white matter hyperintensity accumulation
| Hippocampal Volume | WMH Volume | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Estimate | SE | p | r | Estimate | SE | p | r | |
|
|
||||||||
| Intercept | 47.993 | 0.914 | <0.001 | 0.97 | 0.626 | 0.045 | <0.001 | 0.76 |
| Time | −1.256 | 0.125 | <0.001 | 0.70 | 0.051 | 0.006 | <0.001 | 0.66 |
| Age | −2.587 | 0.575 | <0.001 | 0.35 | 0.120 | 0.028 | <0.001 | 0.33 |
| Sex | −0.471 | 1.041 | 0.652 | 0.07 | 0.073 | 0.051 | 0.150 | 0.12 |
| Pulse pressure | 1.684 | 0.543 | 0.002 | 0.25 | 0.037 | 0.026 | 0.161 | 0.12 |
| APOE ε4 | −0.108 | 1.254 | 0.932 | 0.01 | −0.095 | 0.060 | 0.116 | 0.13 |
| p-tau/Aβ | −3.586 | 1.282 | 0.006 | 0.23 | 0.264 | 0.063 | <0.001 | 0.33 |
| FDG-PET | 2.966 | 0.545 | <0.001 | 0.41 | −0.056 | 0.026 | 0.034 | 0.16 |
| Hippocampal CBF | 0.591 | 0.523 | 0.260 | 0.09 | −0.039 | 0.025 | 0.126 | 0.12 |
| Hippocampal CBF x Time | 0.141 | 0.123 | 0.280 | 0.10 | 0.003 | 0.007 | 0.649 | 0.05 |
APOE = apolipoprotein E p-tau = phosphorylated tau; Aβ = β-amyloid; FDG-PET = fluorodeoxyglucose positron emission tomography; CBF = cerebral blood flow
Hippocampal volume was normalized by total intracranial volume. Bold values are statistically significant (p<.05). Effect size (r-values) interpretation: small=0.10, medium=0.30, large=0.50.
Attrition
Chi-square tests were performed to determine whether attrition rates differed by p-tau/Aβ positivity, APOE ε4 carrier status, and/or cognitive status (MCI versus CN). At 48 months follow-up, 90 participants had cognitive composite data, 73 had cortical thickness and volume data, and 68 had WMH data available. At 60 months follow-up, 42 participants had memory composite data. Attrition at 48 months did not differ for any of these variables (all p-values > 0.05). Similarly, attrition at 60 months did not differ for any of these variables (all p-values > 0.05).
Secondary Analyses
We performed secondary analyses where we re-ran the LME models described above but included continuous p-tau/Aβ as a covariate rather than the dichotomized variable based on established cut-offs for AD CSF marker positivity. Findings remained similar to those from the primary analyses. That is, when continuous (rather than dichotomous) p-tau/Aβ was included as a covariate, the entorhinal CBF x time interaction remained significant for the models with memory [t(147.52)=2.92, p=.004, r=.23], entorhinal cortical thickness [t(131.21)=3.30, p=.001, r=.28], and WMH volume [t(96.06)=−2.19, p=.031, r=.22] as dependent variables. Also similar to findings from the primary analyses, there were no significant hippocampal CBF x time interactions for the models with memory, hippocampal volume, and WMH volume as dependent variables (all p-values > 0.05).
In addition, we performed secondary analyses where we examined whether two-way CBF x time interactions were moderated by the following AD risk factors: cognitive status, p-tau/Aβ, APOE ε4 positivity, and female sex. Across models for the memory composite, entorhinal thickness/hippocampal volume, and WMH volume, there were no significant cognitive group x CBF x time interactions (all p-values > 0.05), indicating that the interaction between CBF and time was not significantly moderated by cognitive group (i.e., the CBF x time interaction did not differ between participants with normal cognition vs MCI).
When we examined whether the pattern of results may be influenced by CSF p-tau/Aβ, we observed a significant three-way p-tau/Aβ+ x entorhinal CBF x time interaction for memory performance [t(158.29)=2.52, p=.013, r=.20] suggesting that lower CBF disproportionally accelerated memory decline for those who were p-tau/Aβ+ relative to those who were p-tau/Aβ−. In contrast, there were no significant three-way p-tau/Aβ+ x entorhinal CBF x time interactions for models with entorhinal cortical thinning or WMH volume as the dependent variable and no significant three-way p-tau/Aβ+ x hippocampal CBF x time interactions across models with memory performance, hippocampal volume, and WMH volume as the dependent variable (all p-values > 0.05). When continuous p-tau/Aβ (rather than dichotomous p-tau/Aβ positivity versus negativity) was included in models, there were no significant three-way p-tau/Aβ x CBF x time interactions for any models (all p-values > 0.05).
When we examined whether two-way CBF x time interactions were moderated by APOE ε4 status, there was a significant three-way APOE ε4 x entorhinal CBF x time interaction for memory performance [t(153.41)=2.16, p=.032, r=.17] and a significant three-way APOE ε4 x hippocampal CBF x time interaction for WMH volume [t(103.47)=−2.03, p=.045, r=.20]. These findings suggest that lower entorhinal CBF disproportionally accelerated memory decline and lower hippocampal CBF disproportionally accelerated WMH accumulation for APOE ε4 carriers relative to noncarriers. There were no significant three-way APOE ε4 x entorhinal CBF x time interactions for entorhinal cortical thinning or WMH volume (all p-values > 0.05) or three-way APOE ε4 x hippocampal CBF x time interactions for memory performance or hippocampal volume (all p-values > 0.05).
Finally, sex moderated the two-way entorhinal CBF x time interaction for the model with entorhinal cortical thinning as the dependent variable such that lower CBF disproportionally accelerated entorhinal atrophy for women when compared with men [t(136.05)=2.12, p=.036, r=.19]. In contrast, sex did not moderate the time x CBF interactions for any of the other models examining effects of entorhinal or hippocampal CBF on memory, entorhinal thickness/hippocampal volume, or WMH volume (all p-values > 0.05)
DISCUSSION
Our study found that reduced entorhinal CBF predicts faster rates of memory decline, neurodegeneration (i.e., entorhinal thinning), and progression of small vessel cerebrovascular disease (i.e., WMH volume) in a well-characterized sample of older adults free of dementia. This pattern of findings was observed after adjusting for relevant AD risk factors and biomarkers with well-known and sizable effects on cognition, including CSF p-tau/Aβ, neuronal metabolism, and APOE ε4 genotype. Our findings add to the growing body of evidence demonstrating the utility of ASL MRI, and entorhinal CBF in particular, as an early biomarker predictive of cognitive and neurodegenerative changes in AD risk.
Relatively few longitudinal studies have examined the relationships between baseline CBF and cognitive trajectories in older adults free of dementia and existing findings have been mixed. For example, some studies indicate that lower baseline CBF is associated with cognitive decline [20, 39, 40] whereas other studies have observed associations only in particular subgroups who are at increased risk for decline such as APOE ε4 carriers [41]. In the present study, participants were followed for a longer period of time than these previously published studies; models took into account multiple AD risk factors; and the focus was on CBF of select regions including the entorhinal cortex, a region which few studies have included as an a priori ROI despite its role in early AD.
One recent study examined the complex relationship between baseline entorhinal cortex and hippocampal CBF and longitudinal memory performance in those at risk for AD [41], although there were no significant main effects of CBF on memory change. Baseline entorhinal CBF, however, was significantly associated with memory decline only among individuals who were APOE ε4 carriers and also had reduced entorhinal cortical thickness. Similar to the current results, this previous study found no associations between hippocampal CBF and memory change. In the present study, we examined entorhinal CBF and cognitive decline in a larger sample with longer follow up and more than two cognitive assessments allowing for statistical methods to examine trajectories. We found that baseline entorhinal CBF predicted faster memory decline across our sample. In the present study, results from secondary analyses showed that two-way CBF x time interactions were moderated by AD risk factors across a relatively small number of models. That is, three-way AD risk factor x CBF x time interaction effects were non-significant for the majority of models examined. In those cases where three-way interaction effects were significant, these interaction effects were relatively small in magnitude (i.e., all r’s ≤ .20) and did not show a consistent pattern across models. Nonetheless, lower entorhinal CBF disproportionally accelerated memory decline for those who were p-tau/Aβ+ (i.e., considered to be on the AD continuum) relative to those who were p-tau/Aβ- and also for APOE ε4 carriers compared to noncarriers. In addition, lower hippocampal CBF disproportionally accelerated WMH accumulation for APOE ε4 carriers relative to noncarriers. Finally, lower entorhinal CBF disproportionally accelerated entorhinal atrophy for women when compared with men. Future research may further elucidate potential moderating effects of AD risk factors on the associations between CBF and longitudinal outcomes.
In our primary analyses, we observed significant effects of entorhinal CBF but not hippocampal CBF on cognitive and brain structure trajectories. Given the established roles of the hippocampus in episodic memory, we expected to see effects of hippocampal CBF on memory trajectories. In the preclinical stages of AD, Braak and colleagues have demonstrated very early neurofibrillary tau tangle accumulations in the entorhinal cortex (e.g., Braak stage I) before spreading to other medial temporal lobe regions such as the hippocampus and neocortical regions [25] and it is possible that we are detecting subtle changes in cerebrovascular functioning in a region susceptible to pathology very early in the AD pathophysiological cascade. Notably, our findings in the present sample of older adults without dementia dovetail with those of previous studies that have shown that medial temporal lobe volumes were reduced in individuals with AD compared to cognitively normal older adults although there were no significant CBF difference in these regions for those with MCI relative to those with normal cognition [10]. In our own previous cross-sectional studies we have observed significant associations between hippocampal CBF and memory performance [14, 17], however, these studies focused on groups of participants with higher vascular risk burden relative to the current sample and were cross-sectional in design so did not focus on rate of cognitive decline as in the current study. Finally, it is worth noting that we have previously observed hyperperfusion in the hippocampus in older adults at elevated risk for AD relative to their lower risk peers [8, 12, 42] This pattern of increased CBF in those at-risk may reflect a neurovascular compensatory response and/or dynamic pathophysiologic processes and could contribute to the lack of observed associations in the hippocampus if both increased and decreased CBF in this particular region is associated with poorer cognitive outcomes depending on which stage of the AD pathophysiological process an individual may be in.
Entorhinal cortical thinning has repeatedly been shown to be a sensitive measure of structural change in prodromal AD [43]. Older adults who are positive for CSF Aβ and p-tau show entorhinal thinning [44, 45], and entorhinal cortical thinning precedes, and predicts, hippocampal atrophy [44, 46, 47]. A study that examined the accuracy of MRI and CSF biomarkers and neuropsychological tests for predicting progression to AD found that entorhinal thinning was one of best single predictors of MCI to AD conversion and surpassed models using a combination of biomarkers [48]. Previous research suggests that vascular defects precede neuronal changes [1], and our results suggest that reduced entorhinal CBF may be a useful predictor of future neurodegeneration even in a relatively healthy sample without dementia, and adjusting for important and potentially confounding variables.
Although WMH were once thought to represent benign changes in the underlying tissue occurring in “normal” aging, it is clear that WMH predict risk and progression of clinical symptoms in prodromal AD [49–53]. Indeed, growing evidence suggests that WMH may be a core feature of AD. In a study of individuals with autosomal dominant genetic mutations for AD, higher WMH volume was observed several years before estimated symptom onset indicating that WMH are an important feature of AD among younger to middle-aged individuals generally thought to be relatively free of non-AD pathologies [2]. The few studies have that have integrated ASL CBF and longitudinal WMH have shown that reduced CBF leads to WMH expansion [6, 54], although an ADNI study did not find a relationship between an ASL-derived measure of cerebral vascular resistance and WMH volume across 24 months of follow up [11]. Notably, the consequences of WMH are thought to be heterogeneous and have been linked to pathologic processes including demyelination, axonal loss due to ischemia or neuronal death, cerebral amyloid angiopathy, and microglia and endothelial activation [2, 55, 56]. A recent multimodal neuroimaging study integrating WMH, CBF, and diffusion tensor imaging indices of white matter microstructure, showed that WMH progression is often likely due to demyelinating injury secondary to hypoperfusion [6]. Our finding adds to the few existing studies linking reduced regional perfusion with the risk of developing WMH and extends this finding it to a large, well-characterized sample of older adults without dementia and low vascular risk burden.
Our findings that neurovascular changes in a region susceptible to early AD pathology predicts later cognitive decline, neurodegeneration, and WMH accumulation, is in line with vascular models such as the two-hit hypothesis [1], which posit that declines in capillary perfusion can trigger neurodegeneration, which can contribute to cognitive decline characteristic of AD. Regardless of the temporal precedence of vascular dysfunction in AD, although neurofibrillary tangles and Aβ plaques are considered the hallmark neuropathological changes in AD, it is now clear that “pure” AD is quite rare and dementia due to AD is often the result of multiple co-existing pathologies [57, 58]. Indeed, in a large neuropathological study that included 447 individuals clinically diagnosed with probable AD and examined degenerative (Lewy body, TAR DNA-binding protein 43 [TDP-43], hippocampal sclerosis) and vascular pathologies (microinfarcts, moderate to severe atherosclerosis, arteriolosclerosis and cerebral amyloid angiopathy) known to contribute to the dementia, over 95% of individuals with probable AD had mixed pathologies. Only 3% of these individuals had pure AD [59]. Furthermore, in another recent study, although AD was the most common (65%) pathology in an autopsy study of 1,079 individuals, it rarely occurred in isolation and, it is noteworthy that more than 230 different neuropathologic combinations were observed [58].
Although progress in AD biomarker research with amyloid and tau have been made and it is recognized that cerebrovascular disease may influence pathogenesis and clinical expression of AD, little attention has been paid to early biomarkers for vascular disease in AD and existing studies have generally focused on structural imaging markers (e.g., brain infarcts, WMH) rather than functional imaging markers such as CBF. Future research should supplement the existing biological definitions of AD [60, 61] by adding a cerebrovascular disease category. Notably, there are no widely accepted criteria for determining biomarker positivity for cerebrovascular disease and there is a need to develop such methodologies. Work has begun in this area with a metric proposed to identify participants with cerebrovascular imaging abnormalities based on a combination of infarcts and WMH burden in the context of absent or present abnormal amyloid [62]. These methods rely on signal changes that are associated with irreversible parenchymal damage and CBF, which is capable of detecting early cerebrovascular dysfunction prior to the manifestation of the frank lesions, may help inform the mechanisms that precede the development of irreversible parenchymal/structural damage.
Strengths of the current study include a large, well-characterized sample of older adults and the ability to characterize and adjust for metabolic markers and AD risk factors including FDG-PET SUVR, p-tau/Aβ ratio, and APOE ε4 carrier status that could impact CBF. It is noteworthy that our observed CBF effects adjusted for FDG-indexed neuronal metabolism, to which hypoperfusion is often attributed or linked [11, 63, 64]. Another strength includes partial volume correction of ASL data as a way to minimize effects of atrophy on CBF estimates, although this may not completely eliminate effects of atrophy on CBF. A limitation of this study is our inability to examine regional WMH volumes and future studies should examine regional WMH given the observed importance of posterior WMH in AD [2]. In addition, our study is limited in generalizability given that the ADNI sample is predominantly white, highly educated, and medically healthy. Selection criteria used by the ADNI exclude potential participants diagnosed with certain cardiovascular conditions, likely under-representing vascular risk burden relative to the broader population. It is possible that findings may look differently in populations with higher rates of cardiovascular diseases although it is notable that we observed associations with CBF despite ADNI participants being such a medically healthy sample (Hachinski scores at enrollment < 5). Future studies should examine the associations observed in the present study in a more representative community-based sample with greater racial diversity and vascular risk burden.
In closing, our findings add to an expanding literature suggesting that ASL MRI measures of CBF are useful markers of early neurovascular changes that may help clarify mechanisms that precede development of irreversible parenchymal damage and may also serve as a useful early biomarker of risk of cognitive decline. Further, ASL has advantages over PET imaging and lumbar puncture for CSF data collection given its noninvasive nature. Findings support the notion that even mild cerebrovascular dysfunction plays a role in progression of cognitive decline, neurodegeneration, and WMH even in a relatively healthy sample and after adjusting for AD risk factors. Future research focused on CBF may further improve our understanding of AD pathogenesis and develop interventions aimed at improving CBF may prevent cognitive decline, neurodegeneration, and WMH accumulation.
Supplementary Material
ACKNOWLEDGMENTS
Data used in this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). ADNI investigators contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this manuscript. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
The analyses in this manuscript were funded by the U.S. Department of Veterans Affairs Clinical Sciences Research and Development Service (Career Development Award-2 1IK2CX001865 to K.R.T. and 1IK2CX001415 to E.C.E.; and Merit Award 1I01CX001842 to K.J.B), NIH grants (R01 AG063782 to K.J.B. and R01 AG049810 and R01 AG054049 to M.W.B.), San Diego State University Advancing Diversity in Aging Research Program (R25AG043364), and the Alzheimer’s Association (AARF-17-528918 to K.R.T., AARG-18-566254 to K.J.B., AARG-17-500358 to E.C.E.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Footnotes
CONFLICT OF INTEREST/DISCLOSURE STATEMENT
Dr. Bondi receives royalties from Oxford University Press and serves as a consultant for Eisai, Novartis, and Roche Pharmaceutical. Other authors report no competing interests.
REFERENCES
- [1].Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12, 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC, Cairns NJ, Holtzman DM, Buckles V, Ghetti B, McDade E, Martins RN, Saykin AJ, Masters CL, Ringman JM, Ryan NS, Forster S, Laske C, Schofield PR, Sperling RA, Salloway S, Correia S, Jack C Jr., Weiner M, Bateman RJ, Morris JC, Mayeux R, Brickman AM (2016) White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol 79, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu TT, Brown GG (2007) Measurement of cerebral perfusion with arterial spin labeling: Part 1. Methods. J Int Neuropsychol Soc 13, 517–525. [DOI] [PubMed] [Google Scholar]
- [4].Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7, 11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Promjunyakul NO, Dodge HH, Lahna D, Boespflug EL, Kaye JA, Rooney WD, Silbert LC (2018) Baseline NAWM structural integrity and CBF predict periventricular WMH expansion over time. Neurology 90, e2119–e2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Staffaroni AM, Cobigo Y, Elahi FM, Casaletto KB, Walters SM, Wolf A, Lindbergh CA, Rosen HJ, Kramer JH (2019) A longitudinal characterization of perfusion in the aging brain and associations with cognition and neural structure. Hum Brain Mapp 40, 3522–3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bangen KJ, Restom K, Liu TT, Wierenga CE, Jak AJ, Salmon DP, Bondi MW (2012) Assessment of Alzheimer’s disease risk with functional magnetic resonance imaging: an arterial spin labeling study. J Alzheimers Dis 31 Suppl 3, S59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hays CC, Zlatar ZZ, Meloy MJ, Bondi MW, Gilbert PE, Liu TT, Helm JL, Wierenga CE (2019) APOE modifies the interaction of entorhinal cerebral blood flow and cortical thickness on memory function in cognitively normal older adults. Neuroimage 202, 116162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mattsson N, Tosun D, Insel PS, Simonson A, Jack CR Jr., Beckett LA, Donohue M, Jagust W, Schuff N, Weiner MW (2014) Association of brain amyloid-beta with cerebral perfusion and structure in Alzheimer’s disease and mild cognitive impairment. Brain 137, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yew B, Nation DA (2017) Cerebrovascular resistance: effects on cognitive decline, cortical atrophy, and progression to dementia. Brain 140, 1987–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wierenga CE, Dev SI, Shin DD, Clark LR, Bangen KJ, Jak AJ, Rissman RA, Liu TT, Salmon DP, Bondi MW (2012) Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J Cereb Blood Flow Metab 32, 1589–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thomas KR, Osuna JR, Weigand AJ, Edmonds EC, Clark AL, Holmqvist S, Cota IH, Wierenga CE, Bondi MW, Bangen KJ (2020) Regional hyperperfusion in older adults with objectively-defined subtle cognitive decline. J Cereb Blood Flow Metab, 271678x20935171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bangen KJ, Nation DA, Clark LR, Harmell AL, Wierenga CE, Dev SI, Delano-Wood L, Zlatar ZZ, Salmon DP, Liu TT, Bondi MW (2014) Interactive effects of vascular risk burden and advanced age on cerebral blood flow. Front Aging Neurosci 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, Birdsill AC, Palotti M, Wharton W, Hermann BP, LaRue A, Bendlin BB, Rowley HA, Asthana S, Sager MA, Johnson SC (2014) Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cereb Cortex 24, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Leeuwis AE, Benedictus MR, Kuijer JPA, Binnewijzend MAA, Hooghiemstra AM, Verfaillie SCJ, Koene T, Scheltens P, Barkhof F, Prins ND, van der Flier WM (2017) Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement 13, 531–540. [DOI] [PubMed] [Google Scholar]
- [17].Bangen KJ, Werhane ML, Weigand AJ, Edmonds EC, Delano-Wood L, Thomas KR, Nation DA, Evangelista ND, Clark AL, Liu TT, Bondi MW (2018) Reduced Regional Cerebral Blood Flow Relates to Poorer Cognition in Older Adults With Type 2 Diabetes. Frontiers in Aging Neuroscience 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bangen KJ, Clark AL, Edmonds EC, Evangelista ND, Werhane ML, Thomas KR, Locano LE, Tran M, Zlatar ZZ, Nation DA, Bondi MW, Delano-Wood L (2017) Cerebral Blood Flow and Amyloid-beta Interact to Affect Memory Performance in Cognitively Normal Older Adults. Front Aging Neurosci 9, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zlatar ZZ, Bischoff-Grethe A, Hays CC, Liu TT, Meloy MJ, Rissman RA, Bondi MW, Wierenga CE (2016) Higher Brain Perfusion May Not Support Memory Functions in Cognitively Normal Carriers of the ApoE ε4 Allele Compared to Non-Carriers. Front Aging Neurosci 8, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xekardaki A, Rodriguez C, Montandon ML, Toma S, Tombeur E, Herrmann FR, Zekry D, Lovblad KO, Barkhof F, Giannakopoulos P, Haller S (2015) Arterial spin labeling may contribute to the prediction of cognitive deterioration in healthy elderly individuals. Radiology 274, 490–499. [DOI] [PubMed] [Google Scholar]
- [21].Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR Jr., Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW (2010) Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sheikh JI, Yesavage JA (1986) Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version In Clinical Gerontology: a Guide to Assessment and Intervention The Haworth Press, New York, NY, pp. 165–173. [Google Scholar]
- [23].Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, Jones RN, Mukherjee S, Curtis SM, Harvey D, Weiner M, Mungas D (2012) Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 6, 502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Luh WM, Wong EC, Bandettini PA, Hyde JS (1999) QUIPSS II with thin-slice TI1 periodic saturation: a method for improving accuracy of quantitative perfusion imaging using pulsed arterial spin labeling. Magn Reson Med 41, 1246–1254. [DOI] [PubMed] [Google Scholar]
- [25].Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70, 960–969. [DOI] [PubMed] [Google Scholar]
- [26].Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- [27].DeCarli C, Miller BL, Swan GE, Reed T, Wolf PA, Garner J, Jack L, Carmelli D (1999) Predictors of brain morphology for the men of the NHLBI twin study. Stroke 30, 529–536. [DOI] [PubMed] [Google Scholar]
- [28].DeCarli C, Murphy DG, Teichberg D, Campbell G, Sobering GS (1996) Local histogram correction of MRI spatially dependent image pixel intensity nonuniformity. J Magn Reson Imaging 6, 519–528. [DOI] [PubMed] [Google Scholar]
- [29].Fletcher E, Carmichael O, Decarli C (2012) MRI non-uniformity correction through interleaved bias estimation and B-spline deformation with a template. Conf Proc IEEE Eng Med Biol Soc 2012, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Scott JA, Braskie MN, Tosun D, Thompson PM, Weiner M, DeCarli C, Carmichael OT (2015) Cerebral Amyloid and Hypertension are Independently Associated with White Matter Lesions in Elderly. Front Aging Neurosci 7, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Price JC, Reiman EM, Skovronsky D, Koeppe RA (2010) The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement 6, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ (2011) Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging 32, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hansson O, Seibyl J, Stomrud E, Zetterberg H, Trojanowski JQ, Bittner T, Lifke V, Corradini V, Eichenlaub U, Batrla R, Buck K, Zink K, Rabe C, Blennow K, Shaw LM (2018) CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement 14, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nation DA, Edmonds EC, Bangen KJ, Delano-Wood L, Scanlon BK, Han SD, Edland SD, Salmon DP, Galasko DR, Bondi MW (2015) Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol 72, 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jak AJ, Bondi MW, Delano-Wood L, Wierenga C, Corey-Bloom J, Salmon DP, Delis DC (2009) Quantification of five neuropsychological approaches to defining mild cognitive impairment. Am J Geriatr Psychiatry 17, 368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bondi MW, Edmonds EC, Jak AJ, Clark LR, Delano-Wood L, McDonald CR, Nation DA, Libon DJ, Au R, Galasko D, Salmon DP (2014) Neuropsychological criteria for mild cognitive impairment improves diagnostic precision, biomarker associations, and progression rates. J Alzheimers Dis 42, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Woodard JL (2017) A quarter century of advances in the statistical analysis of longitudinal neuropsychological data. Neuropsychology 31, 1020–1035. [DOI] [PubMed] [Google Scholar]
- [38].Singer J, Willett J (2003) Applied longitudinal data analysis: Modeling change and event occurrence, Oxford University Press, New York, NY. [Google Scholar]
- [39].Wolters FJ, Zonneveld HI, Hofman A, van der Lugt A, Koudstaal PJ, Vernooij MW, Ikram MA (2017) Cerebral Perfusion and the Risk of Dementia: A Population-Based Study. Circulation 136, 719–728. [DOI] [PubMed] [Google Scholar]
- [40].De Vis JB, Peng SL, Chen X, Li Y, Liu P, Sur S, Rodrigue KM, Park DC, Lu H (2018) Arterial-spin-labeling (ASL) perfusion MRI predicts cognitive function in elderly individuals: A 4-year longitudinal study. J Magn Reson Imaging 48, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hays CC, Zlatar ZZ, Meloy MJ, Bondi MW, Gilbert PE, Liu T, Helm JL, Wierenga CE (2020) Interaction of APOE, cerebral blood flow, and cortical thickness in the entorhinal cortex predicts memory decline. Brain Imaging Behav 14, 369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thomas KR, Bangen KJ, Weigand AJ, Edmonds EC, Wong CG, Cooper S, Delano-Wood L, Bondi MW (2020) Objective subtle cognitive difficulties predict future amyloid accumulation and neurodegeneration. Neurology 94, e397–e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Holland D, McEvoy LK, Dale AM (2012) Unbiased comparison of sample size estimates from longitudinal structural measures in ADNI. Hum Brain Mapp 33, 2586–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Desikan RS, McEvoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, Sperling RA, Dale AM (2012) Amyloid-β--associated clinical decline occurs only in the presence of elevated P-tau. Arch Neurol 69, 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Eskildsen SF, Coupé P, García-Lorenzo D, Fonov V, Pruessner JC, Collins DL (2013) Prediction of Alzheimer’s disease in subjects with mild cognitive impairment from the ADNI cohort using patterns of cortical thinning. Neuroimage 65, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Desikan RS, McEvoy LK, Thompson WK, Holland D, Roddey JC, Blennow K, Aisen PS, Brewer JB, Hyman BT, Dale AM (2011) Amyloid-β associated volume loss occurs only in the presence of phospho-tau. Ann Neurol 70, 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Desikan RS, Sabuncu MR, Schmansky NJ, Reuter M, Cabral HJ, Hess CP, Weiner MW, Biffi A, Anderson CD, Rosand J, Salat DH, Kemper TL, Dale AM, Sperling RA, Fischl B (2010) Selective disruption of the cerebral neocortex in Alzheimer’s disease. PLoS One 5, e12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ewers M, Walsh C, Trojanowski JQ, Shaw LM, Petersen RC, Jack CR Jr., Feldman HH, Bokde AL, Alexander GE, Scheltens P, Vellas B, Dubois B, Weiner M, Hampel H (2012) Prediction of conversion from mild cognitive impairment to Alzheimer’s disease dementia based upon biomarkers and neuropsychological test performance. Neurobiol Aging 33, 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bangen KJ, Preis SR, Delano-Wood L, Wolf PA, Libon DJ, Bondi MW, Au R, DeCarli C, Brickman AM (2018) Baseline White Matter Hyperintensities and Hippocampal Volume are Associated With Conversion From Normal Cognition to Mild Cognitive Impairment in the Framingham Offspring Study. Alzheimer Dis Assoc Disord 32, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brickman AM, Provenzano FA, Muraskin J, Manly JJ, Blum S, Apa Z, Stern Y, Brown TR, Luchsinger JA, Mayeux R (2012) Regional White Matter Hyperintensity Volume, Not Hippocampal Atrophy, Predicts Incident Alzheimer Disease in the Community. Arch Neurol, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brickman AM, Zahodne LB, Guzman VA, Narkhede A, Meier IB, Griffith EY, Provenzano FA, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Mayeux R (2015) Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer’s disease incidence. Neurobiol Aging 36, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tosto G, Zimmerman ME, Carmichael OT, Brickman AM (2014) Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol 71, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Silbert LC, Dodge HH, Perkins LG, Sherbakov L, Lahna D, Erten-Lyons D, Woltjer R, Shinto L, Kaye JA (2012) Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 79, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wong SM, Jansen JFA, Zhang CE, Hoff EI, Staals J, van Oostenbrugge RJ, Backes WH (2019) Blood-brain barrier impairment and hypoperfusion are linked in cerebral small vessel disease. Neurology 92, e1669–e1677. [DOI] [PubMed] [Google Scholar]
- [55].Wardlaw JM, Valdes Hernandez MC, Munoz-Maniega S (2015) What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4, 001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fernando MS, O’Brien JT, Perry RH, English P, Forster G, McMeekin W, Slade JY, Golkhar A, Matthews FE, Barber R, Kalaria RN, Ince PG (2004) Comparison of the pathology of cerebral white matter with post-mortem magnetic resonance imaging (MRI) in the elderly brain. Neuropathol Appl Neurobiol 30, 385–395. [DOI] [PubMed] [Google Scholar]
- [57].Schneider JA, Arvanitakis Z, Bang W, Bennett DA (2007) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- [58].Boyle PA, Yu L, Leurgans SE, Wilson RS, Brookmeyer R, Schneider JA, Bennett DA (2019) Attributable risk of Alzheimer’s dementia attributed to age-related neuropathologies. Ann Neurol 85, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kapasi A, DeCarli C, Schneider JA (2017) Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B (2016) A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R (2018) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vemuri P, Knopman DS (2016) The role of cerebrovascular disease when there is concomitant Alzheimer disease. Biochim Biophys Acta 1862, 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Musiek ES, Chen Y, Korczykowski M, Saboury B, Martinez PM, Reddin JS, Alavi A, Kimberg DY, Wolk DA, Julin P, Newberg AB, Arnold SE, Detre JA (2012) Direct comparison of fluorodeoxyglucose positron emission tomography and arterial spin labeling magnetic resonance imaging in Alzheimer’s disease. Alzheimers Dement 8, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Du AT, Jahng GH, Hayasaka S, Kramer JH, Rosen HJ, Gorno-Tempini ML, Rankin KP, Miller BL, Weiner MW, Schuff N (2006) Hypoperfusion in frontotemporal dementia and Alzheimer disease by arterial spin labeling MRI. Neurology 67, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.