

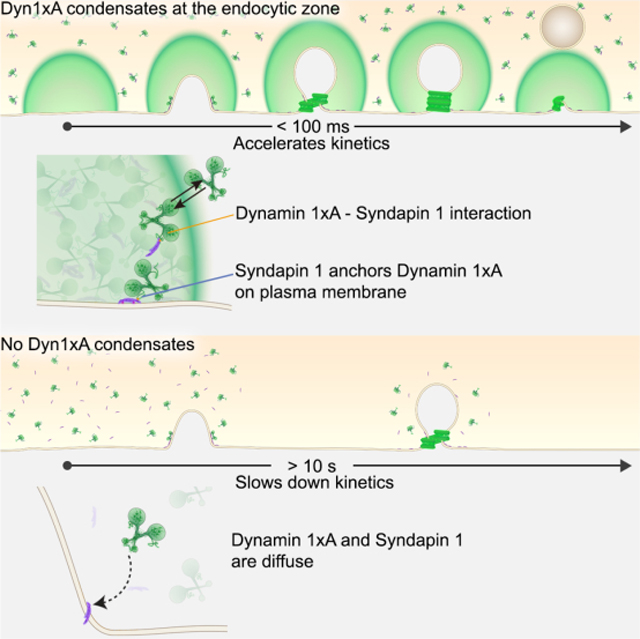
Summary:
Dynamin mediates fission of vesicles from the plasma membrane during endocytosis. Typically, dynamin is recruited from the cytosol to endocytic sites, requiring seconds to tens of seconds. However, ultrafast endocytosis in neurons internalizes vesicles as quickly as 50 ms during synaptic vesicle recycling. Here we demonstrate that Dynamin 1 is pre-recruited to endocytic sites for ultrafast endocytosis. Specifically, Dynamin 1xA, a splice variant of Dynamin 1, interacts with Syndapin 1 to form molecular condensates on the plasma membrane. Single-particle tracking of Dynamin 1xA molecules confirms the liquid-like property of condensates in vivo. When Dynamin 1xA is mutated to disrupt its interaction with Syndapin 1, the condensates do not form, and consequently, ultrafast endocytosis slows down by 100-fold. Mechanistically, Syndapin 1 acts as an adaptor by binding the plasma membrane and stores Dynamin 1xA at endocytic sites. This cache bypasses the recruitment step and accelerates endocytosis at synapses.
Keywords: Ultrafast endocytosis, Dynamin, Dynamin splice variants, endocytosis, flash-and-freeze, Syndapin, synaptic vesicle recycling, phase separation, liquid condensates
eTOC Blurb
Imoto et al. demonstrate that a splice variant of Dynamin 1, Dyn1xA, mediates vesicle scission during ultrafast endocytosis. For such a rapid event, Dyn1xA molecules are concentrated at endocytic zones through molecular condensation with Syndapin 1. This cache of Dyn1xA accelerates the kinetics of endocytosis by 100-fold.
Graphical Abstract
Introduction:
Dynamin GTPase (Shpetner and Vallee, 1989) catalyzes membrane fission during endocytosis (Praefcke and McMahon, 2004). However, Dynamin is recruited from the cytosol, requiring ~10–30 s (Cocucci et al., 2014; Taylor et al., 2011a, 2012), and thus, endocytosis is inherently slow. Despite the slow kinetics, dynamin is thought to play a key role in synaptic vesicle recycling. During ultrafast endocytosis, synaptic vesicle exocytosis triggers the formation of endocytic pits next to the active zone where synaptic vesicles fuse, and these pits are internalized between 50 ms to 300 ms at mouse synapses. Ultrafast endocytosis likely depends on dynamin in C. elegans (Watanabe et al., 2013a) and in mouse hippocampal synapses (Watanabe et al., 2013b). However, given the slow recruitment of dynamin from the cytosol, it is not clear how dynamin can pinch off endocytic intermediates on this time scale.
Dynamin function is regulated by Syndapin 1 binding through phosphorylation of the proline-rich motif (PRM). The PRM is phosphorylated by multiple kinases including GSK3ß and CDK5 and dephosphorylated by Calcineurin. Under resting conditions, approximately 75% of Dynamin 1 (Dyn1) present at synaptic terminals is dephosphorylated (Graham et al., 2007; Liu et al., 1994), and this fraction is increased or decreased based on the activity level of neurons (Clayton et al., 2009). The dephosphorylated PRM of Dyn1 binds the Src-Homology 3 (SH3) domain of Bin, Amphiphysin, Rvs (BAR) proteins, Syndapin 1, Amphiphysin 1, and Endophilin A (Ferguson and De Camilli, 2012). The interaction between Dyn1 and Syndapin 1 or Endophilin A is reduced by phosphorylation within the PRM of dynamin (Anggono et al., 2006) (Xue et al., 2011). The endocytic defect in Dyn1,3 DKO can be rescued by the expression of wild-type Dyn1 or phospho-deficient Dyn1 (Armbruster et al., 2013). However, even when Dyn1 is dephosphorylated, the time constant of synaptic vesicle endocytosis is ~10 s (Armbruster et al., 2013), and thus, how dynamin mediates ultrafast endocytosis remains unclear.
Results:
Dynamin 1 is required for ultrafast endocytosis
To test how dynamin mediates ultrafast endocytosis, we first performed flash-and-freeze experiments (see STAR Methods) in neurons lacking the two brain-enriched isoforms of dynamin, Dyn1 and 3 (Figure 1). Dyn3 KO served as a control since Dyn3 KO itself does not show apparent functional or structural phenotypes (Ferguson et al., 2007; Raimondi et al., 2011), although it exacerbates the Dyn1 KO phenotypes (Raimondi et al., 2011). Like in wild-type neurons (Watanabe et al., 2013b, 2014), ferritin particles were first observed in large endocytic vesicles and synaptic endosomes at 1 s after stimulation and then in synaptic vesicles at 10 s in Dyn3 KO neurons (Figure 1A, B), suggesting that ultrafast endocytosis is intact and Dyn3 is likely dispensable for this process. However, ferritin-positive endosomes seemed to persist in these neurons, suggesting that Dyn3 may play a role in the resolution of synaptic endosomes (Figure 1A, B). By contrast, in Dyn1,3 DKO neurons, endocytic pits were stuck at the plasma membrane (Figure 1C, D) immediately next to the active zone, at the putative sites of ultrafast endocytosis (Figure S1A, D, E), and no ferritin particles were observed in endocytic vesicles and endosomes at either time point (Figure 1C, D, K, L), suggesting that endocytic structures cannot be internalized in the DKO neurons. No electron density indicative of clathrin-coats was observed around these pits (Figure 1C, Figure S1B, C). Occasionally, clathrin-coated pits were observed distant from the active zone (Figure S1B, D, translucent arrowhead), but the number did not increase following stimulation (Figure S1C) (Imig et al., 2020; Watanabe et al., 2013b, 2014). Endocytic pits in the DKO neurons were deeply invaginated (Figure 1M; pit depth, 44.2 ± 6.0 nm) and became slightly more tubulated by 10 s (Figure 1M; pit depth, 65.0 ± 7.7 nm). The width of the pit base did not change between 1–10 s (Figure 1N; the neck width; 1 s, 71.7 ± 3.2 nm; 10 s, 65.0 ± 3.7 nm). These results indicate that Dyn1 is likely necessary for constricting the neck of endocytic pits during ultrafast endocytosis. Serial reconstructions showed no connections from these pits to other structures such as clathrin-coated vesicles or endosomes (Figure S1D), suggesting that these are not bulk membrane invaginations, such as those previously observed following high-frequency stimulation or high potassium application (Raimondi et al., 2011; Wu et al., 2014). These phenotypes were not rescued by the overexpression of Dyn2 (Figure S2). These data suggest that ultrafast endocytosis is likely mediated by Dyn1.
Figure 1. Dyn1xA splice variant, but not xB variant, mediates ultrafast endocytosis.
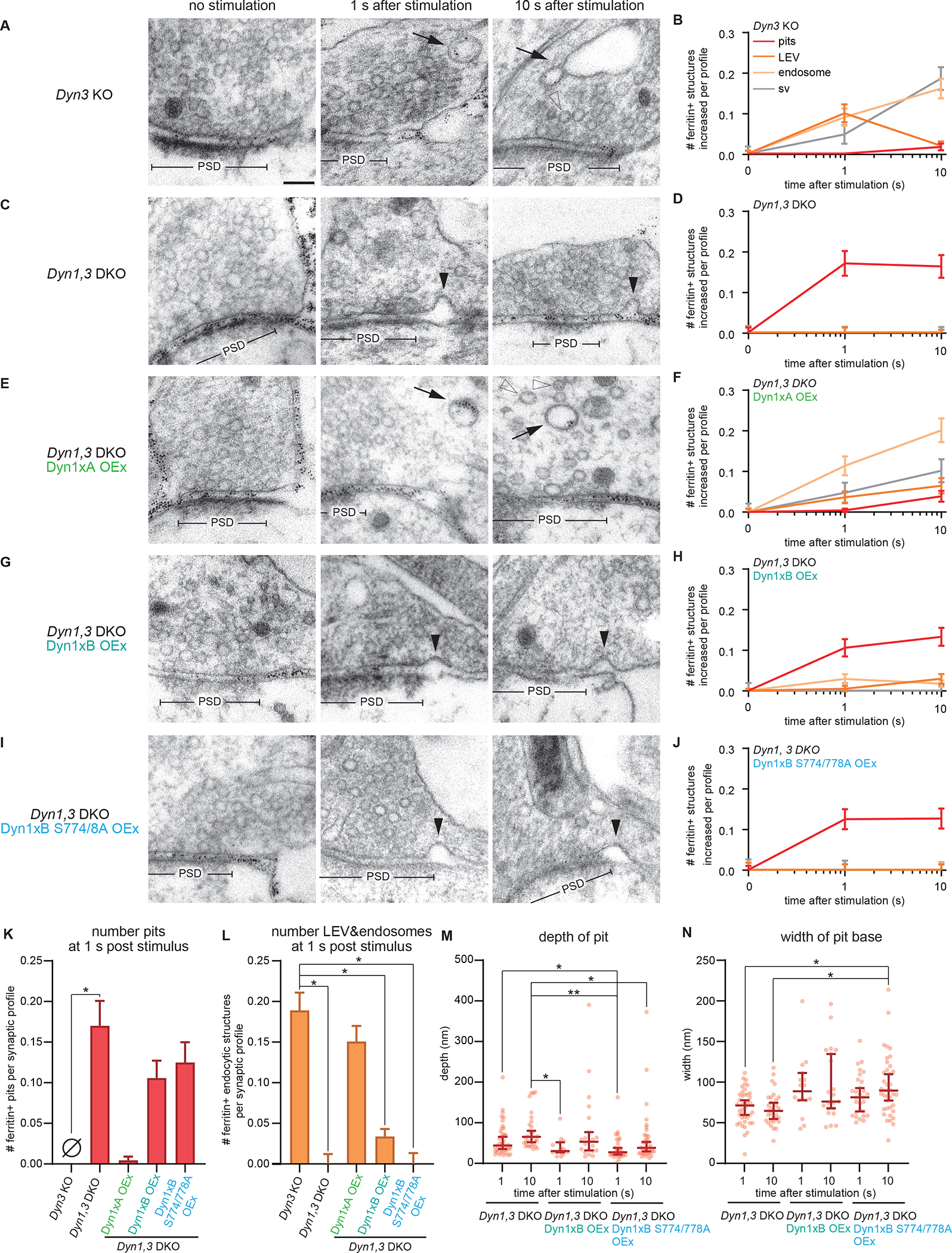
(A, C, E, G and I) Example micrographs showing endocytic pits and ferritin-containing endocytic structures at the indicated time points in Dyn3 KO (A), Dyn1, 3 DKO (C), Dyn1, 3 DKO, Dyn1xA overexpression (OEx) (E), Dyn1, 3 DKO, Dyn1xB OEx (G), Dyn1, 3 DKO, Dyn1xB S774/778A OEx (I). Black arrowheads, endocytic pits; black arrows, large endocytic vesicles (LEVs) or endosomes; white arrowheads, synaptic vesicles. Scale bar: 100 nm. PSD, post-synaptic density.
(B, D, F, H, and J) Plots showing the increase in the number of each endocytic structure per synaptic profile after a single stimulus in neurons with indicated genotypes.
(K) Number of endocytic pits at 1s after stimulation. The numbers are re-plotted from the 1 s time point in (B, D, F, H, and J).
(L) Number of LEVs and endosomes at 1s after stimulation. The numbers of LEVs and endosomes are summed from the data in (B, D, F, H, and J) and averaged.
(M and N) Plots showing the width (M) and depth (N) of endocytic pits at the 1s time point.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers for each time point.
The splice variant Dyn1xA, but not xB, mediates ultrafast endocytosis
DNM1 is alternatively spliced at the C-terminus after the proline-rich motif (Figure S3A, B). Two of these isoforms, xA and xB, are predominantly expressed in neurons (Chan et al., 2010). They share the same phospho-regulated interaction domain (S774 and S778), which binds Syndapin 1 when dephosphorylated (Anggono et al., 2006), but differ at the very end of the C-terminus after residue S845: xA has a highly disordered 20 amino acid extension that provides two additional SH3 binding motifs (Figure S3B, PSRP and PPRP), whereas xB is shorter but contains a calcineurin-binding motif (PxIxIT, Figure S3B). Previous studies suggest that Dyn1xB is specifically involved in bulk membrane uptake after intense neuronal activity (activity-dependent bulk endocytosis) (Cheung and Cousin, 2019; Xue et al., 2011). To confirm that Dyn1 is involved in ultrafast endocytosis, we performed the rescue experiments in Dyn1,3 DKO by expressing these two isoforms. Dyn1,3 DKO phenotypes were completely rescued by the overexpression of Dyn1xA in Dyn1,3 DKO neurons (Figure 1E, F, K, L). By contrast, the overexpression of Dyn1xB failed to rescue the defect in ultrafast endocytosis (Figure 1G, H, K, L). As in Dyn1,3 DKO, endocytic pits were arrested on the plasma membrane when Dyn1xB was expressed. Like the endocytic pits in the DKO neurons, these pits did not mature over time significantly, (Figure 1M, 1N), suggesting that Dyn1xB cannot actively participate in ultrafast endocytosis. Consequently, the number of ferritin-positive large endocytic vesicles and endosomes did not increase in these neurons (Figure 1K, L). These defects were not due to the phosphorylation status of the shared interaction domain near S774/778 since the expression of the phospho-deficient form (S774/778A) of Dyn1xB did not rescue these phenotypes (Figure 1I, J, M, N). Together these results suggest that the Dyn1xA splice variant is essential for ultrafast endocytosis, and the extended C-terminal domain likely plays a critical role.
Dyn1xA is associated with the plasma membrane
The ultrastructural data suggest that Dyn1xA mediates ultrafast endocytosis. However, the accumulation of Dyn1xA to endocytic pits may be too slow if it is recruited from the cytosol. To test how Dyn1xA mediates ultrafast endocytosis, we first checked its localization at synapses using confocal fluorescence microscopy. To avoid mislocalization of Dyn1-GFP from its endogenous location due to overexpression, primary hippocampal neurons were transfected with a small amount of the Dyn1-GFP plasmids for less than 20 hours (see STAR method). Cytosolic tdTomato was co-expressed to visualize neurites, and presynapses marked with the Synaptobrevin 2 (Syb2) antibody. Dyn1xA was localized to presynapses and formed distinct puncta in 51.1 ± 2.9 % of these synapses (Figure 2A, B). By contrast, Dyn1xB signals were diffuse throughout the axons (Figure 2A, C) and only occasionally formed puncta at synapses (Figure 2A, B; 22.4 % of presynaptic terminals). Moreover, Dyn1xB signals were dim within presynaptic terminals and at the remaining puncta (Figure 2C, D), indicating that Dyn1xB does not accumulate at presynaptic terminals or efficiently form puncta. Typically, there was only one Dyn1xA punctum per synaptic bouton, but in 10.9 ± 7.4 % of Dyn1xA puncta positive synapses, 2 or more puncta were observed (Figure 2E). These puncta most likely do not represent ongoing endocytic events at synapses since only 1% of synapses exhibit endocytic profiles in resting conditions (Watanabe et al., 2013b, 2014). These puncta were localized near the edge of Syb2 signals (Figure 2F), suggesting that Dyn1xA is localized at the periphery of a synaptic vesicle cluster. Given that ultrafast endocytosis takes place towards the edge of an active zone (Figure S1A) (Watanabe et al., 2013b), this localization pattern indicates that Dyn1xA is likely concentrated near endocytic sites.
Figure 2. Dyn1xA accumulates within presynapses.
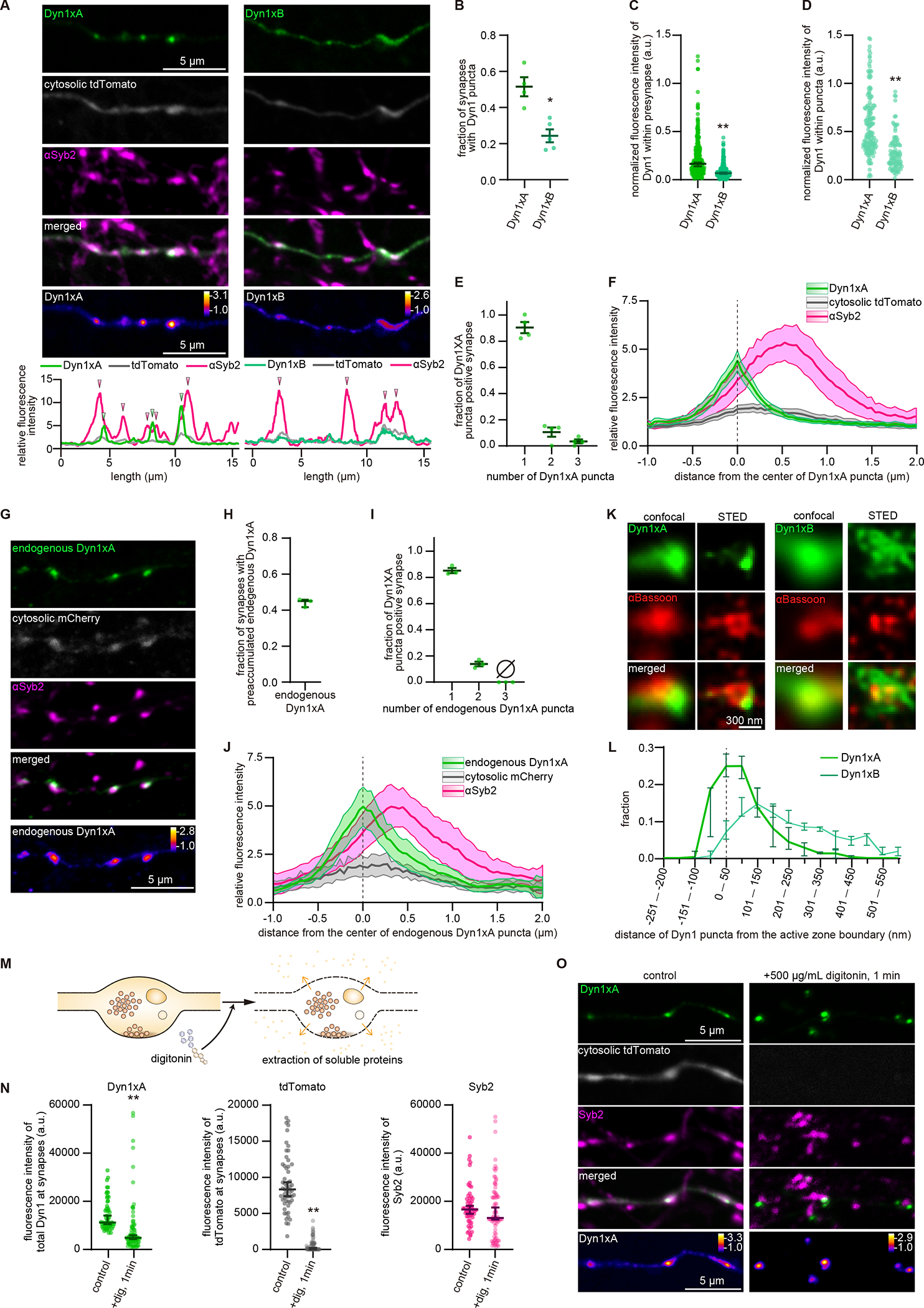
(A) Example confocal fluorescence micrographs showing the localization of GFP-tagged Dyn1xA or Dyn1xB, along with exogenously expressed cytosolic tdTomato and antibody staining of Synaptobrevin 2 (αSyb2). False-colored images of Dyn1xA and Dyn1xB (bottom panels) show the relative fluorescence intensity of these molecules. Line scan graphs represent relative fluorescence intensities of Dyn1xA, Dyn1xB, cytosolic tdTomato, and Syb2 and spatial organizations of these molecules. Green and magenta arrowheads indicating the peaks of Dyn1xA and Syb2 signals, respectively.
(B) The fraction of presynapses that contain Dyn1xA or Dyn1xB puncta. Dyn1xA puncta were defined as the puncta adjacent to or within Syb2 signals. Each dot indicates one neuron.
(C) The normalized fluorescence intensities of Dyn1xA or Dyn1xB within presynapse. Each dot represents one synapse.
(D) The normalized fluorescence intensities of Dyn1xA or Dyn1xB within puncta. Each dot represents one synapse.
(E) Relative frequency distributions of the number of puncta within presynaptic boutons among those that contain at least one punctum. Each dot represents one neuron.
(F) The distributions of cytosolic tdTomato and αSyb2 relative to the peak of Dyn1xA signals.
(G) Example confocal micrographs showing endogenous localization of Dyn1xA-GFP (endogenous Dyn1xA, stained with anti-GFP antibodies), cytosolic mCherry and αSyb2. False-colored images representing the relative fluorescence intensity of endogenous Dyn1xA.
(H) The fraction of presynapses that contain endogenous Dyn1xA puncta. The mean and SEM are shown. Each dot represents one neuron.
(I) Same as (E), but for endogenous Dyn1xA. The fraction is calculated from each neuron. The mean and SEM are shown. Each dot represents one neuron.
(J) Same as (F), but for endogenous Dyn1xA.
(K) Example STED micrographs showing the localization of Dyn1xA or Dyn1xB (stained with anti-GFP antibodies) and αBassoon (stained with anti-Bassoon antibody).
(L) The distributions of Dyn1xA or Dyn1xB against the active zone boundary, defined by αBassoon signals.
(M) Schematics showing the cytosolic extraction using digitonin.
(N) Fluorescence intensities of Dyn1xA, cytosolic tdTomato or αSyb2 within presynapses in control and digitonin treated neurons.
(O) Example confocal micrographs showing Dyn1xA, cytosolic tdTomato and αSyb2 in control and digitonin-treated neurons after fixation. False-colored images representing the relative fluorescence intensity of Dyn1xA.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
Since the overexpression of dynamin may result in mislocalization of proteins, we also localized Dyn1xA by endogenous GFP tagging CRISPR/Cas9 guided gene-editing (Willems et al., 2020) (Figure S3C). Sequencing confirmed that all GFP-positive neurons contain the correct Dyn1xA-GFP sequence in their genome (Figure S3D–F). In these neurons, Dyn1xA puncta were present in 43.5 ± 1.6 % of presynapses (Figure 2G, H). Of these, 85.1 ± 1.9 % of Dyn1xA positive synapses contained only one punctum (Figure 2I). Endogenous Dyn1xA puncta were also localized near the edge of Syb2 signals (Figure 2J). These results were nearly identical to the overexpressed exogenous Dyn1xA-GFP signals (Figure 2A, F), indicating that our expression scheme can probe the endogenous location of Dyn1xA and that Dyn1xA normally forms puncta within presynaptic terminals, presumably near endocytic sites.
To further test whether Dyn1xA localizes at the endocytic sites, we visualized Dyn1xA-GFP or Dyn1xB-GFP along with an active zone protein, Bassoon, using Stimulated Emission Depletion (STED) microscopy (Figure 2K, L). Dyn1xA signals were detected near the edge of Bassoon signals (Figure 2K). Using custom-written MATLAB codes, we defined the boundary of active zones based on Bassoon signals and quantified the distribution of Dyn1xA signals from the active zone edge (Figure S4A, B; see STAR Methods). Most Dyn1xA signals were found within 100 nm from the active zone edge (Figure 2L) where ultrafast endocytosis takes place (Figure S1A, E). In contrast, Dyn1xB signals were distributed broadly from the active zone edge (Figure 2L). These results suggest that Dyn1xA, but not xB isoform, accumulates around the putative endocytic zone.
To determine if Dyn1xA puncta are associated with the membrane at endocytic sites, we examined the exogenous Dyn1xA-GFP signals while applying digitonin in the external solution (see STAR methods). Digitonin is a non-ionic weak detergent that makes the plasma membrane porous and permits the diffusion of cytosolic materials from the cell, while retaining organelles inside the cell (Dubreuil et al., 2020; Gopal et al., 2017; Moore and Blobel, 1992) (Figure 2M). Thus, if Dyn1xA puncta were cytosolic, they would diffuse out of cells after the digitonin treatment. As controls, cytosolic tdTomato was co-expressed in these neurons, and synaptic vesicles marked with Syb2 antibodies. Cells were fixed immediately after the treatment. As expected, tdTomato was washed out completely, while synaptic vesicles marked by Syb2 remained within the terminals after the digitonin application for 1 min (Figure 2N, O). Although the total fluorescence intensity level decreased by ~40% (Figure 2N), Dyn1xA puncta remained near the Syb2 signals (Figure 2O), suggesting that some Dyn1xA are associated with membranes. Together, these results demonstrate the presence of pre-accumulated Dyn1xA molecules on the plasma membrane at the endocytic zone.
Dyn1xA puncta display liquid-like properties in neurons
Dyn1xA forms puncta on the plasma membrane in fixed neurons. To better observe the behavior of Dyn1xA signals, we performed live-cell imaging on neurons expressing Dyn1xA-GFP and Syb2-mCherry (Figure S4C). Immediately after the addition of digitonin, Dyn1xA signals outside the puncta disappeared (Figure S4C–D), indicating that there is a cytosolic pool of Dyn1xA present at presynaptic terminals. By contrast, the puncta remained intact over the course of the experiments (1 min), although the fluorescence intensity gradually decreased to ~61.9% (Figure S4D, E), suggesting that Dyn1xA molecules can slowly diffuse out of the puncta. Furthermore, these puncta occasionally underwent fusion and fission (Figure S4F). These data are indicative of a potential liquid droplet state of molecules (Hyman and Brangwynne, 2011; Hyman et al., 2014; Stone, 1994) and suggest that Dyn1xA may form molecular condensates. In fact, Dyn1xA formed liquid-like droplets with Syndapin 1 both in vitro and COS-7 cells (Figure 3 and Figure S5).
Figure 3. Dyn1xA exhibits liquid-like behaviors in vitro and in COS-7 cells.
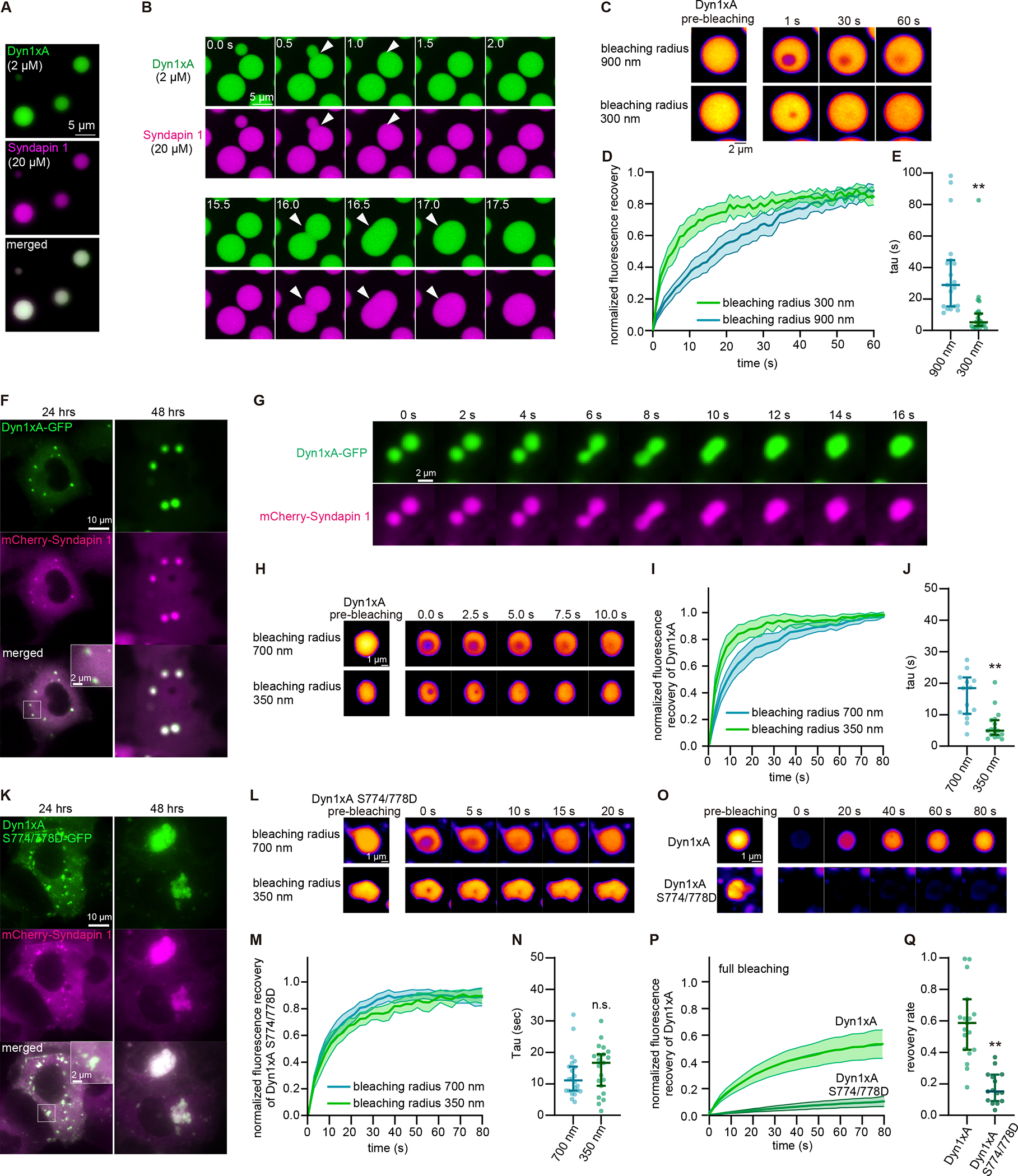
(A) Example images of purified Dyn1xA (labeled with Alexa488) and Syndapin1 (labeled with Alexa 549) forming droplets under the physiological salt concentration and with 5 % PEG.
(B) Example time-lapse images of Dyn1xA and Syndapin1 droplets undergoing fusion.
(C) Example time-lapse images of FRAP experiments in in vitro. Dyn1xA and Syndapin1 droplets were photobleached at 480 nm using the region-of-interest (ROI) radius of 900 nm or 300 nm.
(D) Normalized fluorescence recovery of Dyn1xA signals in the indicated ROI sizes. Fluorescence signals were normalized between immediately (0 s) and 60 s after the photobleaching.
(E) The recovery time constant of Dyn1xA signals following the photobleaching using the indicated ROI sizes. Each dot represents a Dyn1xA-Syndapin 1 droplet. The kinetics of fluorescence recovery after photobleaching is dependent on the radius of photobleaching (Figure 3C–E & S5K), suggesting that the molecular rearrangement is dominated by protein diffusion rather than binding and unbinding.
(F) Example images of COS-7 cells expressing Dyn1xA-GFP and mCherry-Syndapin1 at 24 hours and 48 hours after the transfection.
(G) Example time-lapse images showing fusion of Dyn1xA-GFP and mCherry-Syndapin1 droplets.
(H) Examples time-lapse images of FRAP experiments on Dyn1xA-GFP and mCherry-Syndapin1 droplets. Time indicates after the photobleaching. Dyn1xA signals were photobleached at 480 nm using the ROI radius of 700 nm or 350 nm ROI.
(I) Normalized fluorescence recovery of Dyn1xA signals in the indicated ROI sizes.
(J) The recovery time constant of Dyn1xA signals following the photobleaching using the indicated ROI sizes in (I).
(K) Example images of COS-7 cells expressing Dyn1xA S774/778D-GFP (which inhibits Syndapin1 binding) and mCherry-Syndapin1 at 24 hours and 48 hours after the transfection.
(L) Examples time-lapse images of FRAP experiments on Dyn1xA S774/778D-GFP and mCherry-Syndapin1 droplets. Dyn1xA signals were photobleached at 480 nm using the ROI radius of 700 nm or 350 nm.
(M) Normalized fluorescence recovery of Dyn1xA signals in the indicated ROI sizes.
(N) The recovery time constant of Dyn1xA S774/778D-GFP signals following the photobleaching using the indicated ROI sizes in (M). The recovery kinetics between two different ROI sizes are similar, indicating that the recovery is mediated by binding and unbinding of molecules rather than diffusion of proteins.
(O) Examples time-lapse images of FRAP experiments on Dyn1xA and mCherry-Syndapin1 droplets or Dyn1xA S774/778D-GFP and mCherry-Syndapin1 droplets with the photobleaching laser on the entire droplets. Dyn1xA or Dyn1xA S774/778D-GFP signals were photobleached with 480-nm laser.
(P) Normalized fluorescence recovery of Dyn1xA or Dyn1xA S774/778D-GFP signals.
(Q) The recovery rate of Dyn1xA or Dyn1xA S774/778D-GFP signals at 80 s after the photobleaching in (P), indicating that Dyn1xA S774/778D-GFP molecules form aggregates.
In (I, M, P, Q), fluorescence signals were normalized between immediately (0 s) and 80 s after the photobleaching. Times above FRAP time-lapse images indicate after the photobleaching.
All data are examined by n > 15 droplets from two independent protein preparations or cultures. **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
To test if Dyn1xA puncta at synapses exhibit the same behavior, we first performed FRAP experiments in neurons expressing Dyn1xA-GFP. When we photo-bleached the Dyn1xA signals at intersynaptic regions along the axon, the signals recovered quickly (Figure 4A, B). By contrast, when entire Dyn1xA puncta at synapses were photobleached, fluorescence recovery was significantly slower (Figure 4C, D). These data suggest that Dyn1xA can exchange between the puncta and the cytosol, similarly to the experiments in COS-7 cells. To further investigate the possibility of liquid-like phase separation, we applied aliphatic alcohols, 1,6-hexanediol, 2,5-hexanediol, and 1,4-butanediol, which disrupt weak hydrophobic interactions and dissolve liquid droplets to varying degrees (Gopal et al., 2017; Kroschwald et al., 2015; Patel et al., 2007; Kozak and Kaksonen, 2019; Wilfling et al., 2020). These treatments dispersed the puncta, and Dyn1xA became diffuse along the axon within 30–60 s (Figure 4E, F). In control neurons, the puncta were stable over 1 min (Figure 4E, F). These results are all consistent with the characteristic features of phase separation (Park et al., 2021).
Figure 4. Dyn1xA exhibits liquid-like behaviors in presynapses.
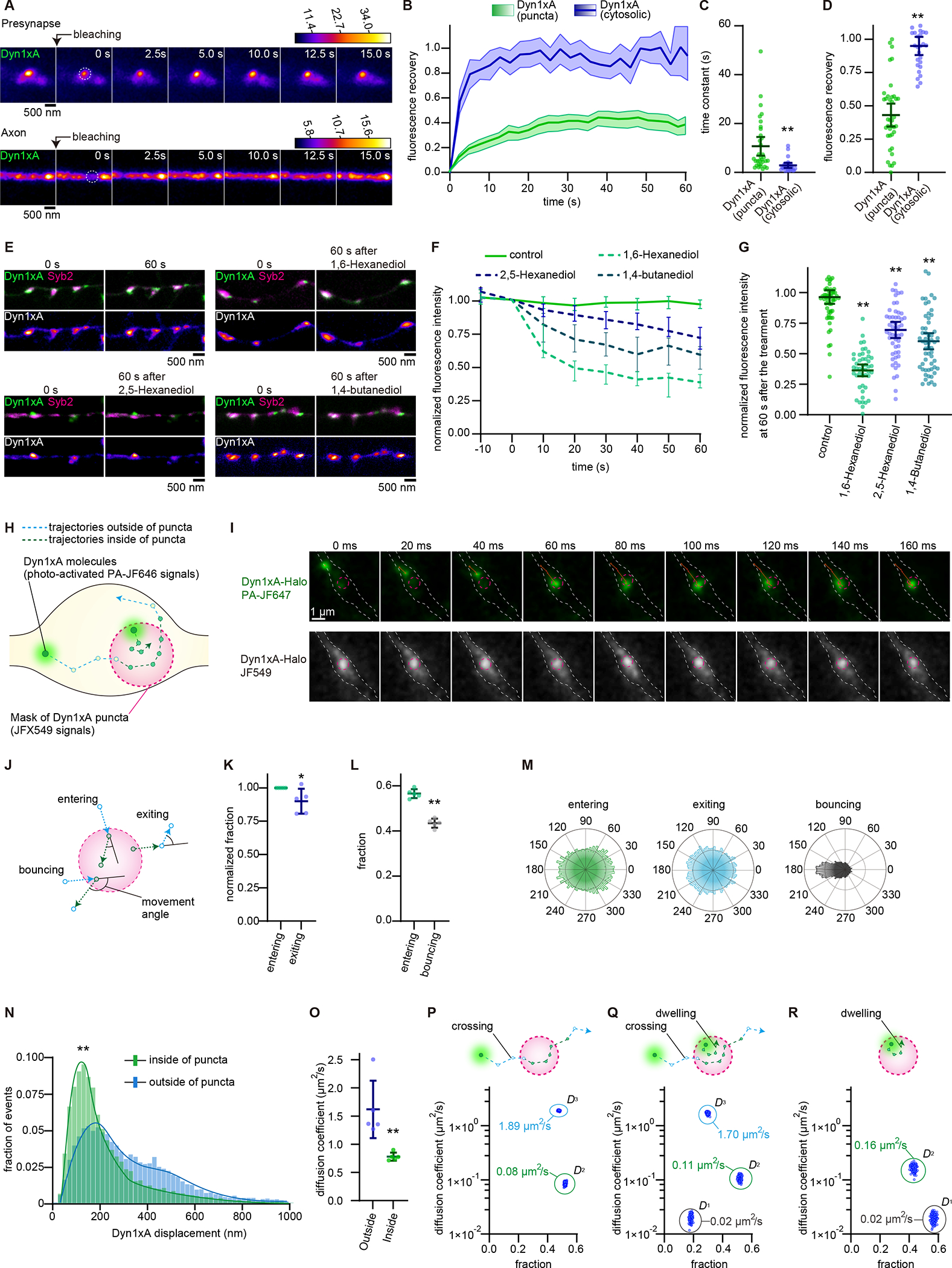
(A) Example time-lapse fluorescence images showing Dyn1xA puncta pre- and post-photobleaching. A dashed circle indicates the photobleached region.
(B) Fluorescence recovery of Dyn1xA signals when signals at the entire puncta or cytosolic Dyn1xA in axons were photobleached.
(C) The recovery half-time of Dyn1xA signals following the photobleaching of the entire puncta or cytosolic Dyn1xA in axons. The half-time is calculated at the recovery period of 60 s after photobleaching. Each dot represents a punctum.
(D) The fraction of fluorescence recovery 60 s after photobleaching the entire puncta and cytosolic Dyn1xA in axons. Each dot represents a punctum. n = Dyn1xA puncta, 71; and cytosolic Dyn1xA, 26; in (A-D). 5 different neurons were examined in each condition from 2 independent cultures.
(E) Example time-lapse fluorescence micrographs showing Dyn1xA signals over 1 min (control), before and 1 min after the addition of 4% 1,6-hexanediol, 4% 2,5-hexanediol or 4% 1,4-buntanediol.
(F) Averaged normalized fluorescence intensities of Dyn1xA puncta in control and aliphatic alcohol treatments. Fluorescence is normalized to the pre-treatment (0 s). Time indicates after the addition.
(G) The fraction of Dyn1xA puncta fluorescence intensities 60 s after the treatment with aliphatic alcohol. Each dot represents one synapse.
(H) Schematic showing hypothetical movements of Dyn1xA single molecules and their trajectories (See Materials and Methods).
(I) Example time-lapse images of a single molecule of Dyn1xA in a presynaptic bouton after photo-activation.
(J) Schematic showing hypothetical movements of Dyn1xA single molecules when they cross the boundary. Entering; two consecutive trajectories that move from outside to inside and then stay within the punctum. Bouncing; two consecutive trajectories that move from outside to inside and then back to outside. Exiting; two consecutive trajectories that move from inside to outside and then stay outside.
(K) Fraction of entering and bouncing events. Each dot represents one culture.
(L) Fraction of entering and exiting events normalized to entering events. Each dot represents one culture.
(M) Angular distribution histograms extracted from (J) and (K).
(N) Histogram showing displacement distance of trajectories inside (green) or outside the puncta (light blue).
(O) Diffusion coefficient calculated based on molecules crossing the boundary either from outside to inside or inside to outside. Each dot represents one culture.
(P) Schematic showing entire trajectories of a molecule entering and exiting the boundary (crossing). Dot plot shows two diffusional states of dyn1xA molecules (D2, D3) based on the SMAUG algorithm when crossing molecules are considered. n > 150 iterations.
(Q) Schematic showing entire trajectories of two molecules: one is crossing, and another is initiating and terminating within the boundary (dwelling). Dot plot shows three diffusional states of dyn1xA molecules (D1, D2, D3) based on the SMAUG algorithm when both crossing and dwelling molecules are considered. n > 150 iterations.
(R) Schematic showing entire trajectories of a dwelling molecule. Dot plot shows two diffusional states of dyn1xA molecules (D1, D2) based on the SMAUG algorithm when only dwelling molecules are considered. n > 150 iterations.
Times above FRAP time-lapse images indicate after the photobleaching.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
To further probe the liquid property of condensates, we tested whether the diffusion coefficient of molecules changes when they are inside the puncta. Molecules diffuse slower inside liquid-like condensates due to molecular crowding and intermolecular interactions (Ladouceur et al., 2020; McSwiggen et al., 2019; Miné-Hattab et al., 2021). To this end, we performed stroboscopic photoactivatable single particle tracking (spaSPT) to monitor the behavior of individual Dyn1xA molecules within and around the puncta (McSwiggen et al., 2019; Miné-Hattab et al., 2021). We labeled exogenously-expressed Dyn1xA::HaloTag with JFX549 and PA-JF646 at the 2:1 ratio (Grimm et al., 2016, 2021) (see STAR methods). With this scheme, the majority of Dyn1xA molecules are labelled with JFX549, visualizing the Dyn1xA puncta (Figure 4G, H). Stochastic photo-activation of PA-JF646 is then used to probe the behavior of individual molecules relative to the puncta (Figure 4H, I; McSwiggen et al., 2019b; Miné-Hattab et al., 2021). PA-JF646 Halo-ligand increases its photon absorption upon binding to HaloTag (Grimm et al., 2016) similar to fluorogenic Janelia Fluor dyes (Grimm et al., 2020). As a control for blinking from nonspecific binding of PA-JF646, we included Dyn1xA::GFP-expressing neurons in our experiments - almost no PA-JF646 signals were detected within neurites of these neurons (Dyn1xA::Halo-expressing neurons, 27699 molecules in 88 boutons, Dyn1xA::GFP-expressing neurons, 50 molecules in 49 boutons) (Figure S7A, B). Images are acquired at 50 Hz on a custom-built lattice-light sheet microscope (see STAR Methods). Custom-written MATLAB codes delineate the boundary of Dyn1xA-JFX549 puncta and generate trajectories (a molecule movement between two consecutive frames) of Dyn1xA-PA-JF646 molecules (Figure 4H, S7B–D; see STAR Methods). The photo-activated Dyn1xA-PA-JF646 molecules can enter and exit the puncta, suggesting that molecules are exchanged between the cytosol and puncta. As expected for the liquid condensates (McSwiggen et al., 2019), the molecules are trapped in the puncta upon entry while some escaping from the puncta (Figure 4J, K). Upon entry, they diffused in all directions (Figure 4J, M). Occasionally, molecules entered the puncta but bounced right back to where they came from (Figure 4J, L, M). Nonetheless, when they successfully entered and stayed within the puncta, their displacement distance (distance traveled between two consecutive camera frames) became shorter and remained so until they exited the puncta (Figure 4N), indicating an apparent change in the diffusion coefficient. In fact, the weighted diffusion coefficient was down after the entry into the puncta when we fitted the histogram of the displacement distance using maximum likelihood estimation (Figure 4O). These results are consistent with the liquid like behavior in previous studies (Ladouceur et al., 2020; Miné-Hattab et al., 2021).
Since diffusion of Dyn1xA molecules changes upon entry into the puncta, conventional calculation methods such as curve fitting on displacement histograms (Figure 4N, O) and mean squared displacement (not shown) are not appropriate for calculating the diffusion coefficients. To more accurately calculate the diffusion coefficients of molecules, we used the Single-Molecule Analysis by Unsupervised Gibbs sampling (SMAUG) algorithm, which provides the number of diffusion components, their average diffusion coefficient, and the fraction of single molecules in each component (Karslake et al., 2021). The trajectories that crossed the boundary at least once (crossing) (Figure S7C, S7D) had two diffusional states: D2 (intermediate, 0.0848 ± 0.0543 μm2/s) and D3 (1.885 ± 0.0416 μm2/s) (Figure 4P, S7E, S7F), suggesting that the molecules indeed slow down when they enter the puncta. The diffusion coefficient of the D3 component is similar to the reported value of free diffusion of soluble proteins at presynaptic boutons (~1 – 3 μm2/s) (Reshetniak et al., 2020) and greater than the values of plasma membrane interacting proteins (0.47–0.50 μm2/s) (Reshetniak et al., 2020) and membrane-bound dynamin (0.5–0.8 μm2/s) (Foley et al., 2021), suggesting that this component likely represents free diffusion. The intermediate component D2 is within the range of the diffusion within the liquid-like environment but not immobile molecules (Ladouceur et al., 2020; Miné-Hattab et al., 2021). Interestingly, when the trajectories that initiated and terminated inside the puncta (dwelling) (Figure S7C, S7D) were included in the analysis (crossing + dwelling), three diffusional states were present (Figure 4Q, S7E, S7F): D1 (slow, 0.0201 ± 0.0027 μm2/s,), D2 (intermediate, 0.1066 ± 0.0103 μm2/s) and D3 (free diffusion, 1.6960 ± 0.0758 μm2/s). This result suggests that some of the dwelling molecules (~18%) are likely bound to other proteins or lipids. In fact, the analysis with only dwelling molecules showed two diffusional states: D1 (slow, 0.0197 ± 0.0002 μm2/s) and D2 (intermediate, 0.1663 ± 0.0017 μm2/s) (Figure 4R, S7E, S7F), suggesting that the molecules are either bound or moving slowly within the puncta. These data indicate that the Dyn1xA puncta are liquid-like with a small fraction of Dyn1xA molecules possibly bound to proteins or membranes within the puncta.
Dyn1xA localization at endocytic zone requires dephosphorylation of the PRM
The phosphorylation state of Dyn1xA modulates its ability to interact with Syndapin 1. Thus, altering the phosphorylation level of its PRM may affect condensate formation. To test this possibility, we applied inhibitors of the kinase and phosphatase responsible for controlling Dyn1xA phosphorylation. Under resting conditions, approximately 75% of Dyn1 at synaptic terminals is dephosphorylated (Graham et al., 2007; Liu et al., 1994), and this fraction increases or decreases based on the activity level of neurons (Clayton et al., 2009). Downstream of neuronal activity is calcineurin-dependent dephosphorylation of Dyn1 S774 and S778, which are phosphorylated by GSK3ß (S774) and CDK5 (S778) (Anggono et al., 2006). We applied the calcineurin inhibitor FK506 (2 μM, 30 min) to block further dephosphorylation of Dyn1xA, and the GSK3b inhibitor CHIR990201 (10 μM, 30 min) to block the phosphorylation of S774. DMSO (0.05%) was applied as a control. Consistent with the results in untreated neurons (Figure S8A–C), Dyn1xA-GFP signals recovered to median 39.0% within 1 min of photobleaching in the DMSO-treated cells (Figure S8A–C). The FK506 treatment had no effect on recovery (Figure S8A–C), presumably because ~75% of Dyn1xA is already not phosphorylated in the resting condition (Anggono et al., 2006; Liu et al., 1994). By contrast, the level of fluorescence recovery was enhanced in CHIR990201-treated neurons (median 59.1 %) (Figure S8A–C). These data indicate that dephosphorylation increases the amount of cytosolic Dyn1xA that can be exchanged with those in puncta.
To further examine the requirement for dephosphorylation of Dyn1xA in phase separation, we localized wild-type, phosphomimetic (S774/778D), and phospho-deficient (S774/778A) forms of Dyn1xA-GFP in neurons. Cytosolic tdTomato was co-expressed, and synaptic vesicle clusters visualized with Syb2 antibodies. Wild-type Dyn1xA-GFP and phospho-deficient Dyn1xA-GFP appeared punctate along the axons and localized at the edge of the synaptic vesicle clusters (Figure 5A). More than a half of putative synapses (58.2% of Syb2-positive boutons) contained at least one punctum when a phospho-deficient form was expressed, similarly to when a wild-type form was expressed (65.1%) (Figure 5A, B). The total fluorescence levels of Dyn1xA in boutons were similar between neurons expressing wild-type and phospho-deficient Dyn1xA-GFP (Figure 5C), but fluorescence signals within puncta were brighter by 25.8% in neurons expressing the phospho-deficient form (Figure S8D, E), suggesting that phosphodeficient Dyn1xA accumulate in the puncta efficiently. By contrast, the phosphomimetic form appeared diffuse throughout the axons; only 26.9% of Syb2-positive boutons contained puncta (Figure 5A, B, p = 0.02). The total fluorescence level of Dyn1xA within boutons was reduced by 44.5% (Figure 5C). The remaining puncta in these boutons were dim: the fluorescence intensity was reduced by 42% (Figure S8D, E), and did not show recovery after the photobleaching (Figure S8F, G), suggesting that these are aggregates, consistent with experiments in vitro (Figure S5L) and in COS-7 cells (Figure 3O–Q). Sub-synaptically, phosphodeficient Dyn1xA accumulated within 100 nm from the active zone edge, similarly to Dyn1xA (Figure 5E, F). By contrast, the phosphomimetic form was distributed broadly within boutons (Figure 5E, F). Together, these results suggest that Dyn1xA forms liquid condensates at the endocytic zone in presynaptic boutons, and this formation is promoted by dephosphorylation.
Figure 5. Phase separation of Dyn1xA requires dephosphorylation of the proline-rich motif.
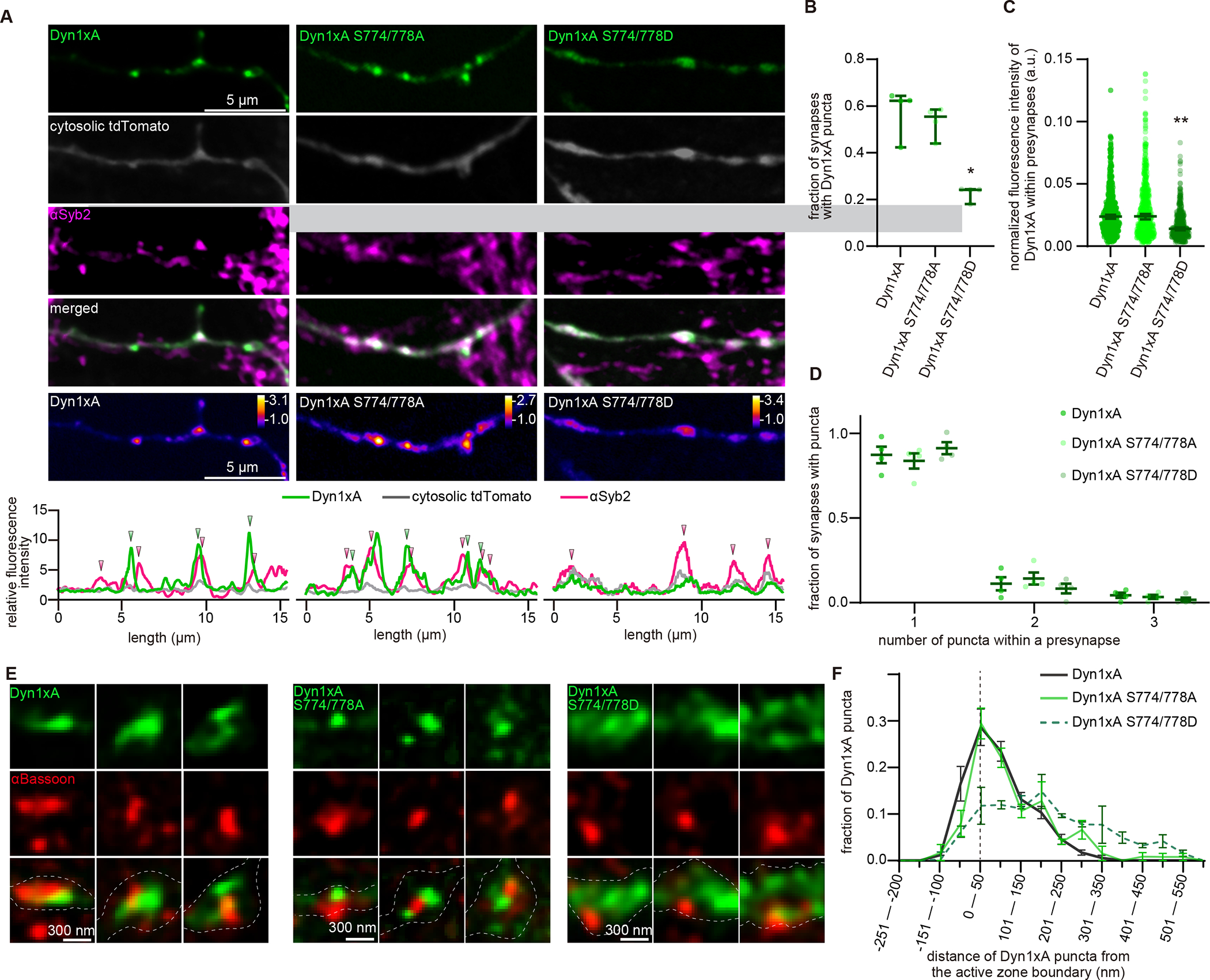
(A) Example confocal micrographs showing overexpression of GFP-tagged Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D, along with exogenously expressed cytosolic tdTomato and immuno-stained αSyb2 in fixed neurons. False-colored images (bottom panels) show the relative fluorescence intensity of Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D. Line scan graphs represent the relative locations of Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D to cytosolic tdTomato and αSyb2.
(B) The fraction of presynapses that contain Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D puncta. Each dot represents one neuron.
(C) The normalized fluorescence intensities of Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D within presynaptic terminals. Each dot represents one synapse.
(D) Relative frequency distributions of the number of puncta within presynaptic boutons among those that contain at least one punctum in neurons expressing Dyn1xA, Dyn1xA S774/778A or Dyn1xA S774/778D puncta within presynapse. The fraction is calculated from each neuron.
(E) Example STED micrographs showing the localization of Dyn1xA or Dyn1xB (stained with anti-GFP antibodies) and αBassoon (stained with anti-Bassoon antibody).
(F) The distribution of Dyn1xA or Dyn1xB relative to the active zone edge.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
Dephosphorylated Dyn1xA is required for the kinetics of ultrafast endocytosis
The phosphorylation status of Dyn1xA controls its localization around the endocytic zone and the condensate formation. This membrane localization would likely accelerate endocytosis by bypassing the slow, diffusion-limited recruitment of molecules to endocytic sites. Since the phospho-deficient form of Dyn1xA is localized to the endocytic zone similarly to wild-type Dyn1xA (Figure 5E, F), ultrafast endocytosis is expected to take place normally when this form is expressed. By contrast, the phosphomimetic form is largely cytosolic or aggregates (Figure 5E, F, S8F, S8G). Having to recruit these molecules from the cytosol would likely slow down ultrafast endocytosis. In fact, our theoretical calculations suggest that it will take ~257 ms at the fastest to nucleate a tetramer of dynamin on the neck of endocytic pits when Dyn1xA is diffusely localized within a synaptic bouton with the estimated concentration of 3.5 μM (see STAR methods for details). An additional 94–147 ms is necessary to generate a helix of dynamin polymer, and thus, at the fastest, 351 ms is required after the neck of endocytic pits becomes ~36 nm in diameter. Given that Dyn1xA is required at the early stage of the neck constriction when the neck diameter is still wide open (>60 nm) (Figure 1N), this process will be much slower if Dyn1xA is cytosolic. By contrast, with liquid condensates, Dyn1xA is concentrated to ~11.6 μM, which is close to the critical concentration of dynamin oligomerization (Roux et al., 2010). At this concentration, the time required for the nucleation process becomes negligible, and dynamin would polymerize spontaneously. Thus, only ~28–39 ms is needed to polymerize dynamin around the neck of endocytic pits (regardless of the neck diameter) – kinetics consistent with ultrafast endocytosis (Watanabe et al., 2013a). Therefore, we predicted that condensate formation is essential for ultrafast endocytosis.
To test these possibilities, we performed ‘flash-and-freeze’ experiments in Dyn1,3 DKO neurons, expressing wild-type, phospho-deficient (S774/778A), or phosphomimetic (S774/778D) forms of Dyn1xA (Figure 6). The results from the controls were as expected: Dyn3KO showed normal ultrafast endocytosis, while Dyn1,3 DKO exhibited stalled endocytic pits on the plasma membrane (Figure 6A–D, K, L). The endocytic defect of Dyn1,3 DKO was rescued by the phospho-deficient and wild-type forms. Ferritin particles were found in endocytic vesicles and endosomes by 1 s and additionally in synaptic vesicles by 10 s (Figure 6E–H, K, L). However, in neurons expressing the phosphomimetic form, endocytic pits were arrested at the plasma membrane up to 10 s after stimulation (Figure 6I–L). Consequently, the number of ferritin-positive vesicles and endosomes did not increase over time, suggesting that the phosphomimetic form cannot rescue the ultrafast endocytic defect. Interestingly, unlike endocytic pits in Dyn1,3 DKO, which exhibited a wide opening at their base (Figure 6M; pit width, 1s, median 61.67 nm; 10 s, median 58.33 nm), the base of endocytic pits in Dyn1xA S774/778D neurons was constricted over time and formed a neck (Figure 6I, M, N; pit width, 1 s: pit width, median 58.33 nm; 10 s, median 33.33 nm), suggesting that endocytosis may complete in the presence of Dyn1xA S774/778D but at a much slower rate. These results are consistent with previous studies using pHluorin assays (Armbruster et al., 2013), indicating that these phosphorylation sites control the kinetics of endocytosis. Given that the majority of Dyn1xA S774/778D is cytosolic and diffuse throughout axons (Figure 5A), ultrafast endocytosis is likely delayed due to the slow recruitment of these molecules to endocytic sites. Altogether, these data suggest that the pre-accumulated Dyn1xA accelerates endocytosis at synapses by bypassing the recruitment step of endocytosis.
Figure 6. Dephosphorylation of Dyn1xA is required for the kinetics of ultrafast endocytosis.
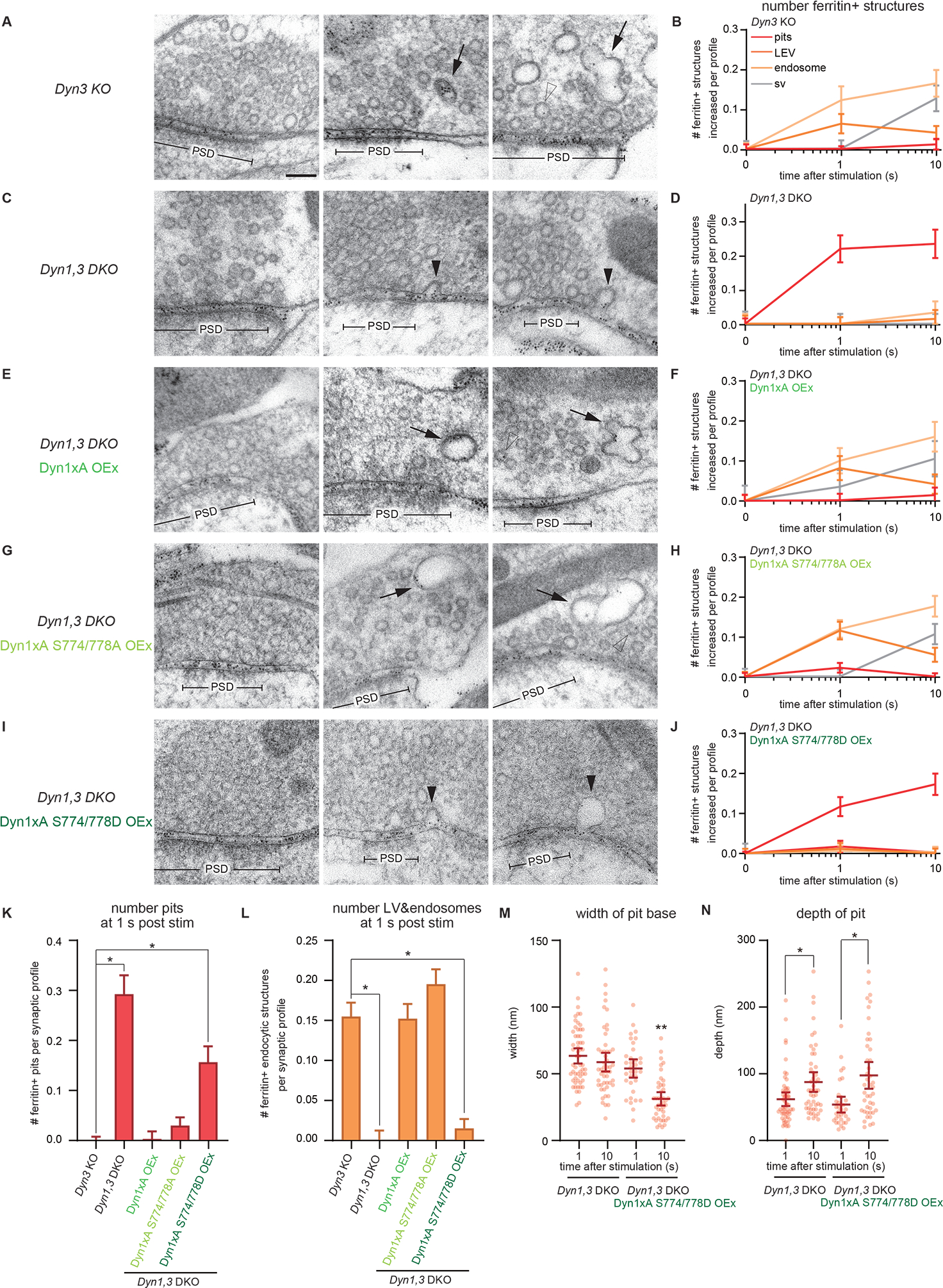
(A, C, E, G and I) Example micrographs showing endocytic pits and ferritin-containing endocytic structures at the indicated time points in Dyn3 KO (A), Dyn1,3 DKO (C), Dyn1,3 DKO, Dyn1xA overexpression (OEx) (E), Dyn1,3 DKO, Dyn1xA S774/778A OEx (G), Dyn1,3 DKO, Dyn1xA S774/778D OEx (I). Black arrowheads, endocytic invaginations; black arrows, LEVs or endosomes; white arrowheads, synaptic vesicles. Note that the endocytic defect can be rescued with Dyn1xA S774/778A but not with Dyn1xA S774/778D. Scale bar: 100 nm. PSD, post-synaptic density.
(B, D, F, H and J) Plots showing the increase in the number of each ferritin-positive endocytic structure per synaptic profile after a single stimulus in neurons with the indicated genotypes. (J). The mean and SEM are shown in each graph.
(K) Number of endocytic invaginations at 1s after the stimulations. The numbers are re-plotted as a bar graph from the 1 s time point in (B, D, F, H, and J).
(L) Number of LEVs and endosomes at 1s after stimulation. The numbers of LEVs and endosomes are summed from the data presented in (B, D, F, H, and J) and averaged.
(M and N) Plots showing the width (M) and depth (N) of endocytic pits at the 1s time point.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
Syndapin 1 acts as an adaptor between the plasma membrane and Dyn1xA
How does Dyn1xA accumulates on the membrane at the endocytic zone? Dyn1 binds to BAR protein Syndapin 1 which targets to plasma membrane (Dharmalingam et al., 2009; Qualmann et al., 1999). We tested whether Syndapin 1 has a role in the formation of Dyn1xA puncta in neurons (Figure 7). We generated shRNA against Syndapin 1 (Syndapin 1 knock-down or KD, hereafter)(Figure S9A, B). Scramble shRNA was used as a control (Figure S9A, B). In scramble shRNA controls, Dyn1xA-GFP formed puncta along the axons and localized next to Syb2 signals (Figure 7A, B; 53.2% of Syb2-positive boutons) – consistent with the results in the wild type (Figure 2). However, in Syndapin 1 KD, Dyn1xA-GFP signals were diffuse along axons and only occasionally formed puncta (Figure 7A–E; 20.3% of Syb2-positive boutons), suggesting that Syndapin 1 is necessary for the localization of Dyn1xA. The total fluorescence level of Dyn1xA in boutons was reduced (Figure 7C). The remaining puncta were dim (Figure 7D). The number of puncta per bouton did not change in Syndapin 1 KD neurons (Figure 7E). Sub-synaptically, Dyn1xA localized within 100 nm from the active zone edge in the scramble shRNA infected neurons (Figure 7F, G), as in wild type (Figure 2K, L). By contrast, in Syndapin 1 KD neurons, Dyn1xA was distributed broadly within presynapses and did not accumulate at the edge of active zones (Figure 7F, G). These results suggest that Syndapin 1 is likely an essential interaction partner of Dyn1xA for its accumulation.
Figure 7. Syndapin 1 is essential for the phase separation of Dyn1xA.
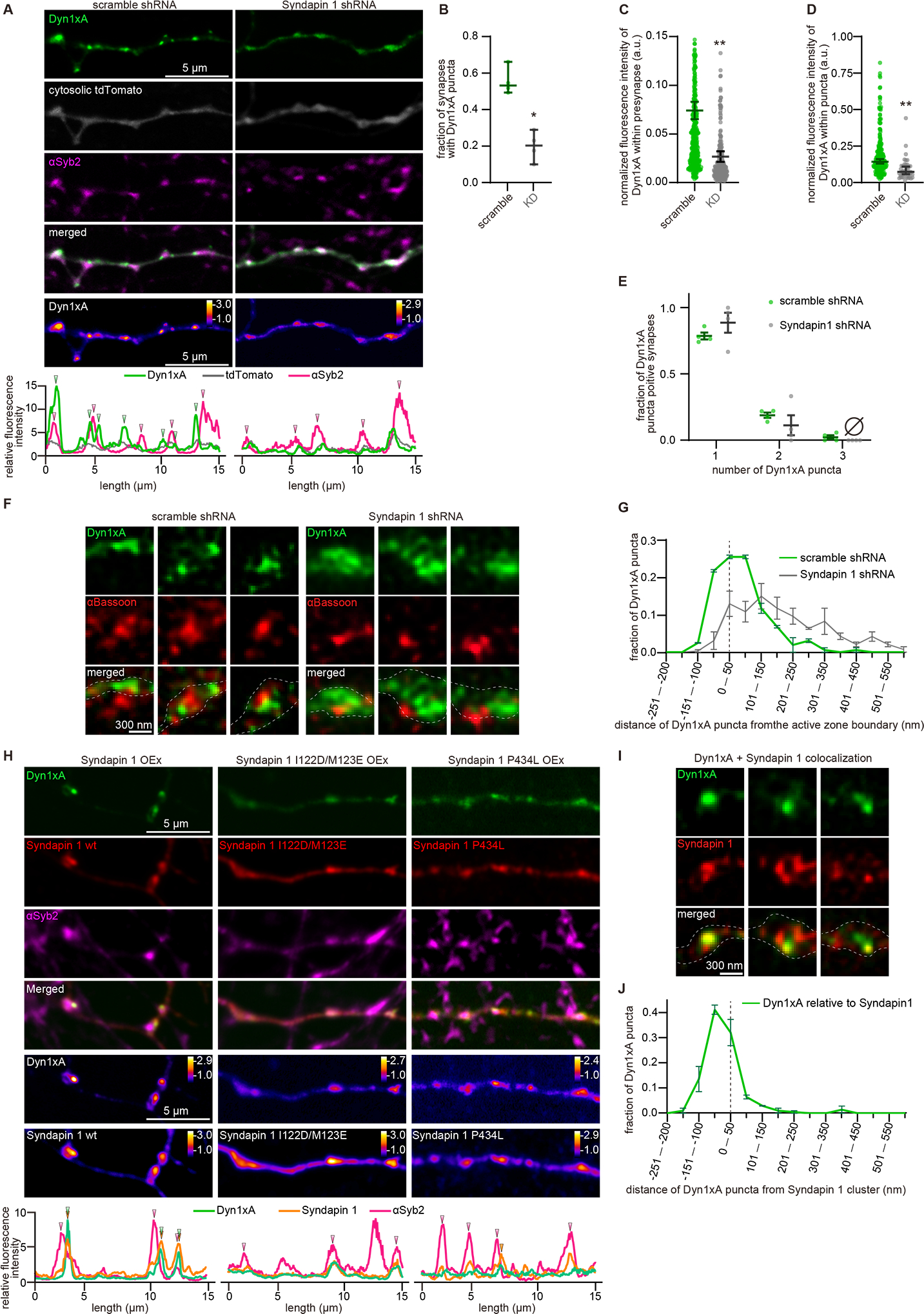
(A) Example confocal immunofluorescence micrographs showing overexpression of Dyn1xA, cytosolic tdTomato, and αSyb2 in neurons expressing scramble shRNA or Syndapin 1 shRNA. False-colored images (the bottom panels) show the relative fluorescence intensity of Dyn1xA. Line scan graphs represent the localization of Dyn1xA relative to cytosolic tdTomato and αSyb2.
(B) The fraction of presynapses that contain Dyn1xA in neurons expressing scramble shRNA or Syndapin 1 shRNA. Each dot represents one neuron.
(C) The normalized fluorescence intensities of Dyn1xA within the presynapse in neurons expressing scramble shRNA or Syndapin 1 shRNA. Each dot represents a punctum.
(D) The normalized fluorescence intensities of Dyn1xA within the puncta in neurons expressing scramble shRNA or Syndapin 1 shRNA. Each dot represents a punctum.
(E) Relative frequency distributions of the number of puncta within presynaptic boutons among those that contain at least one punctum in neurons expressing scramble shRNA or Syndapin 1 shRNA. The fraction is calculated from each neuron.
(F) Example STED micrographs showing the localization of Dyn1xA (stained with anti-GFP antibodies) and αBassoon (stained with anti-Bassoon antibody) in scramble RNA or Syndapin 1 shRNA infected neurons.
(G) The distributions of Dyn1xA relative to the active zone boundary in scramble RNA or Syndapin 1 shRNA infected neurons. The active zone boundary was defined by αBassoon signals.
(H) Example confocal immunofluorescence micrographs showing overexpression of Dyn1xA along with mCherry-tagged Syndapin 1 wild type (Syndapin 1 wt), Syndapin 1 I122D/123ME (Syndapin 1 ID/ME), or Syndapin 1 P434L. The endogenous Syndapin 1 is knocked-down by shRNA, and all mCherry-Syndapin 1 constructs were rendered shRNA-resistant. Syb2 is immuno-stained to visualize synaptic vesicle clusters (αSyb2). Line scan graphs show colocalization of Dyn1xA with αSyb2 and wild-type or mutants Syndapin.
(I) Example STED micrographs showing the localization of Dyn1xA (stained with anti-GFP antibody) and Syndapin1 (stained with anti-DsRed antibody).
(J) The distributions of Dyn1xA against the Syndapin 1 signals.
*p < 0.05, **p < 0.0001. See Data S1 for the n values, statistical test, and detailed numbers.
Since Syndapin 1 interacts with both membranes and Dynamin, it may act as the hub between the plasma membrane and Dyn1xA. To test this possibility, we investigated the localization of Syndapin 1 relative to Dyn1xA, while mutating the membrane binding domain (I122D/M123E) (Rao et al., 2010) or the Dyn1-binding SH3 domain (P434L) (Widagdo et al., 2016). To reduce the overexpression of Syndapin 1, the experiments were performed in Syndapin 1 KD neurons, and the rescue constructs were rendered shRNA-resistant (see STAR Method; Figure S9A). The wild-type Syndapin 1 formed puncta along the axons and colocalized with Dyn1xA near the Syb2 signals (Figure 7H, S9C–E; 45.0 % of Syb2 positive boutons). STED microscopy analysis also showed colocalization of Syndapin 1 with Dyn1xA (Figure 7I, J). By contrast, the Syndapin 1 BAR domain mutation (I122D/M123E) caused both Syndapin 1 and Dyn1xA to be diffuse along the axons (Figure 7H, S9D–I); colocalization was only observed in 12.4 % of Syb2-positive boutons (Figure S9D, E). The total fluorescence levels of Dyn1xA and Syndapin 1 in boutons as well as within puncta were reduced in these neurons (Figure S9F–I). Likewise, Dyn1xA and Syndapin 1 were diffuse along the axons when Syndapin 1-P434L was expressed (Figure 7H), but the phenotypes were less severe than those observed in the neurons expressing Syndapin 1-I122D/M123E (Figure 7H, S9D–I). The number of Dyn1xA or Syndapin 1 puncta per bouton in mutant neurons was similar to the number observed in wild type (Figure S9J, K). These data suggest that both membrane and protein interacting abilities of Syndapin 1 are necessary for the proper Dyn1xA localization. Thus, Syndapin 1 acts as an adaptor between the plasma membrane and Dyn1xA.
Syndapin 1 is necessary for the kinetics of ultrafast endocytosis
Syndapin 1 is essential for the Dyn1xA condensate formation on the plasma membrane. When Syndapin 1 is absent or its membrane or protein interacting domains are mutated, Dyn1xA becomes cytosolic. To determine whether the change in Dyn1xA localization due to these mutations has functional consequences, we performed flash-and-freeze experiments in Syndapin 1 KD neurons, expressing shRNA-resistant wild-type, I122D/M123E, or P434L Syndapin 1 (Figure 8). Ultrafast endocytosis and subsequent endosomal sorting were all normal in neurons expressing scrambled shRNA (Figure 8A, B). By contrast, Syndapin 1 KD neurons exhibited an accumulation of endocytic pits on the plasma membrane at 1 s (Figure 8C, D, K, L) – the full resolution of these intermediates required ~10 s (Figure 8C, D, K, L), likely because the base of pits was slowly constricted in Syndapin 1 KD (Figure 8M, N: pit width, 100 ms, mean 84.7 ± 5.5 nm; 1 s, mean 77.6 ± 3.7 nm; 10 s, mean 58.5 ± 8.8 nm). These defects were rescued by the overexpression of wild-type Syndapin 1, but not with Syndapin BAR domain mutant (I122D/M123E) or SH3 domain mutant (P434L) (Figure 8E–L). These results were similar to the endocytic defects observed in Dyn1,3 DKO neurons expressing the phosphomimetic form of Dyn1xA (Figure 6), which cannot interact with Syndapin 1 (Figure S5A). Together, these data suggest that the kinetics of ultrafast endocytosis is controlled by the interaction between Syndapin 1 and Dyn1xA and the liquid-like condensate formation at the endocytic zone before endocytosis.
Figure 8. Syndapin 1 is necessary for the kinetics of ultrafast endocytosis.
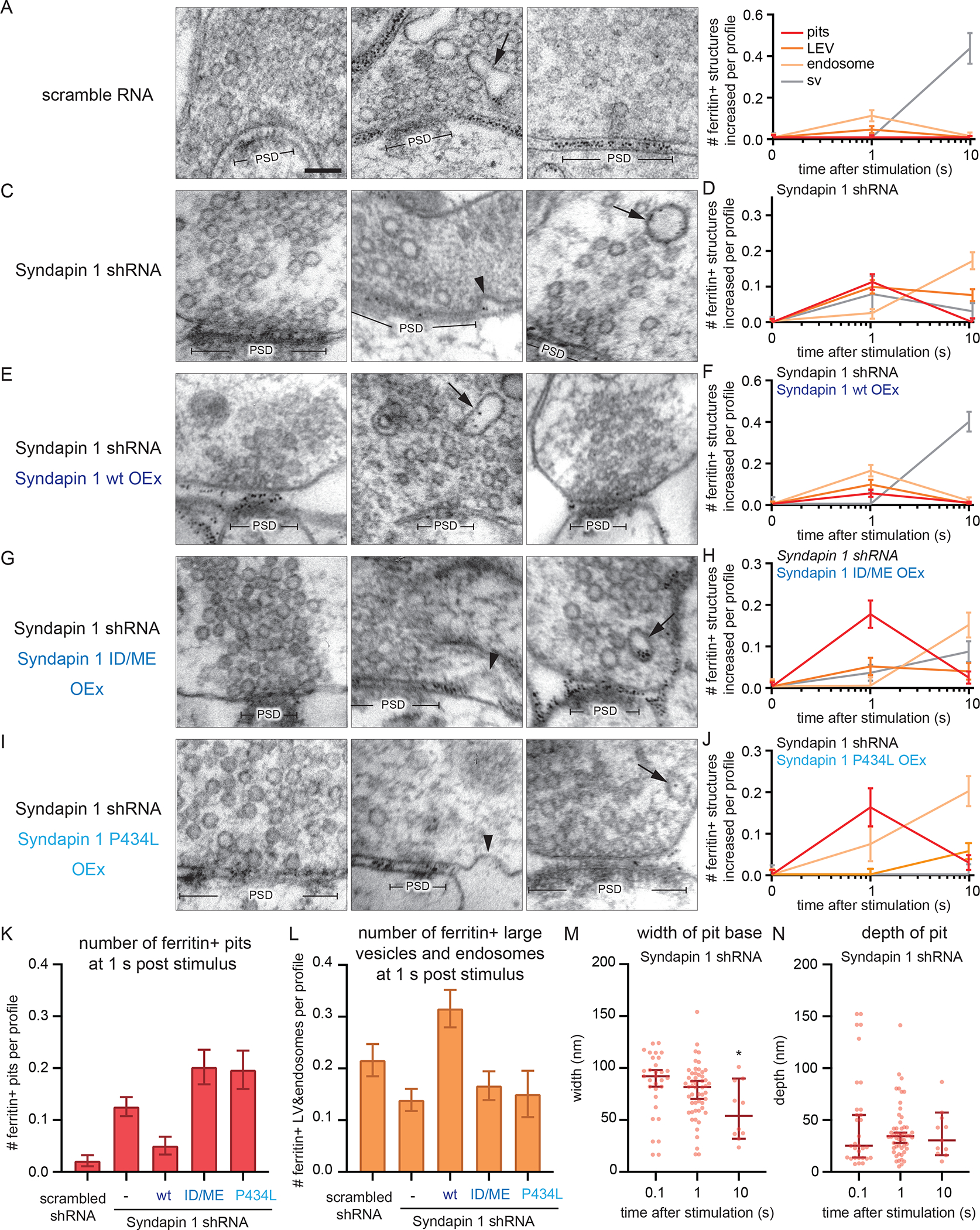
(A, C, E, G and I) Example transmission electron micrographs showing endocytic pits and ferritin-containing endocytic structures at the indicated time points in neurons expressing scramble RNA (A), Syndapin 1 shRNA (C), Syndapin 1 shRNA and wild-type Syndapin 1 overexpression (Syndapin 1 wt OEx) (E), Syndapin 1 shRNA and Syndapin 1 I122D/M123E (Syndapin 1 ID/ME OEx) (G), Syndapin 1 shRNA and Syndapin 1 P434L (Syndapin 1 P434L OEx) (I). In all cases, Syndapin 1 is rendered shRNA-resistant. Black arrowheads, endocytic invaginations; black arrows, ferritin-positive large endocytic vesicles (LEVs) or endosomes; white arrowheads, ferritin-positive synaptic vesicles. Scale bar: 100 nm. PSD, post-synaptic density.
(B, D, F, H and J) Plots showing the increase in the number of each endocytic structure per synaptic profile after a single stimulus in neurons with the indicated genotypes.
(K) Number of endocytic invaginations at 1s after stimulation. The numbers are re-plotted from the 1 s time point in (B, D, F, H, and J).
(L) Number of ferritin-positive LEVs and endosomes at 1s after stimulation. The mean and SEM are shown. The numbers of LEVs and endosomes are summed from the data in (B, D, F, H, and J) and averaged.
(M and N) Plots showing the width (M) and depth (N) of endocytic pits at the 1s time point. The median and 95% confidence interval are shown in each graph.
*p < 0.05. See Data S1 for the n values, statistical test, and detailed numbers.
Discussion
The mode of endocytosis at synapses
A large body of literature suggests that clathrin-mediated endocytosis is responsible for retrieving synaptic vesicle components from the plasma membrane. This conclusion relies on genetic or molecular perturbations that indicate a requirement for clathrin-associated proteins (Dittman and Ryan, 2009; Saheki and De Camilli, 2012). Our data suggest that proteins associated with clathrin-mediated endocytosis also function in clathrin-independent endocytosis. Moreover, these proteins must be clustered at sites of endocytosis to support ultrafast endocytosis. Specifically, we demonstrate that Syndapin I anchors Dynamin 1 in a molecular condensate prior to endocytosis (Figure S10 and Supplementary movie).
Other proteins involved in endocytosis are likely to be associated with this matrix. Syndapin also binds Synaptojanin 1 (Qualmann et al., 1999) and Synaptojanin and Dynamin 1 bind Endophilin A (Micheva et al., 1997; Verstreken et al., 2003)(Ringstad et al., 1999). Like Dynamin 1, Endophilin A and Synaptojanin 1 cinch the neck at the base of endocytic pit during ultrafast endocytosis (Watanabe et al., 2018) and are therefore likely to be localized within the Dyn1xA condensate. In the absence of Endophilin A, Synaptojanin 1 becomes diffusely distributed (Milosevic et al., 2011), and consequently, endocytosis slows down substantially when assayed with pHluorin and flash-and-freeze electron microscopy (Watanabe et al., 2018). This delay may be caused by the loss of pre-assembled protein complexes at endocytic sites and slow recruitment of these proteins to the site of endocytosis. Consistent with this idea, recent studies indicate that ultrafast endocytosis is fast after a single stimulus but slows with high-frequency stimulation, consistent with a depletion of the depot of endocytic proteins (Delvendahl et al., 2016; Imig et al., 2020; Soykan et al., 2017). When the activity level increases dramatically, another mode of clathrin-independent endocytosis, bulk endocytosis, kicks-in to clear out excess membranes using the cytosolic Dyn1xB isoform (Clayton and Cousin, 2009) (Xue et al., 2011). Thus, we propose that clathrin-independent endocytosis is the predominant mechanism for synaptic vesicle retrieval, and the speed is determined by pre-assembly of endocytic proteins at synapses.
The kinetic control of endocytosis
How does stockpiling Dyn1xA accelerate endocytosis? The simplest model is that liquid-like condensation maintains the critical concentration of Dyn1xA at endocytic sites for efficient oligomerization and fission. In vitro experiments suggest that oligomerization (Stowell et al., 1999), phospholipid binding (Powell et al., 2000) and rate of membrane fission (Pucadyil and Schmid, 2008) are all enhanced when dynamin concentration is increased. Such a concentration is typically not achieved during clathrin-mediated endocytosis – dynamin gradually accumulates near endocytic pits and is recruited to the neck by other proteins such as Endophilin and Amphiphysin (Aguet et al., 2013; Cocucci et al., 2014; Macia et al., 2006; Merrifield et al., 2002; Taylor et al., 2011b, 2012). Phase separation bypasses such a recruitment phase, which normally takes seconds to tens of seconds (Cocucci et al., 2014; Taylor et al., 2012), and perhaps constantly maintains Dyn1xA at the critical concentration near endocytic sites for efficient oligomerization, phospholipid binding, and membrane fission.
In addition to enhancing Dyn1 activity, pre-recruitment may allow Dyn1xA to participate in the early phase of endocytosis during cinching of the neck, not just in scission. Unlike Dyn2, which requires a narrow neck generated by multiple BAR domain proteins for it to function (Neumann and Schmid, 2013), Dyn1 can be recruited to shallow endocytic pits and promote the transition of shallow pits to deep invaginations (Liu et al., 2011). This process can bypass the requirement for BAR proteins in neck formation. Endocytosis occurs, albeit slowly, in the absence of the BAR domain protein Endophilin A, suggesting Dyn1 might be capable of acting without Endophilin (Watanabe et al., 2018). Thus, Dyn1 might assist in neck formation and accelerate neck cinching. Indeed, when Dyn1 is activated, the rate of clathrin-coated pit initiation and endocytic kinetics are both elevated (Srinivasan et al., 2018). Thus, pre-recruitment of Dyn1xA molecules allows them to participate in in neck formation with Synaptojanin and Endophilin A (Watanabe et al., 2018) during ultrafast endocytosis. Endocytic pits arrested on the plasma membrane in Dyn1,3 DKO neurons have a wide opening at their base – similar to the pits observed in Synaptojanin 1 KO and Endophilin A triple KO (Watanabe et al., 2018). By contrast, when the GTPase activity of dynamin, but not GTP-binding, is inhibited by the application of Dynasore (Macia et al., 2006) in wild-type neurons, the membrane gap at the base of endocytic pits is almost closed (Watanabe et al., 2013b). These data support in vitro experiments suggesting that the presence of dynamin is likely involved in neck formation (Liu et al., 2011). Thus, pre-recruitment of Dyn1xA can potentially accelerate ultrafast endocytosis by inducing rapid oligomerization and membrane remodeling by Dyn1, Endophilin and Synaptojanin.
For other discussion materials, please see Supplementary Information.
STAR Methods:
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shigeki Watanabe (shigeki.watanabe@jhmi.edu)
Materials availability
All plasmid generated in this study are available from the Lead Contact without restriction
Data and code availability
All original data have been deposited at Mendeley and publicly available as of the date of publication (https://data.mendeley.com/datasets/bkn78y232y/draft?preview=1).
All original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All experiments were performed according to the rules and regulations of the National Institute of Health, USA, the UK Animal (Scientific Procedures) Act 1986, under Project and Personal Licence authority (Home Office project licence – 7008878), and Berlin, Germany authorities. Animal protocols were approved by committee of animal care, use of the Johns Hopkins University, the University of Edinburgh Animal Welfare and Ethical Review Body, and welfare committee of the Charite, Berlin.
In mammals, there are three isoforms of dynamin; Dynamin 1, 2, 3, encoded by DNM1, 2, 3, genes in mouse, respectively (Cook et al., 1996). All dynamin isoforms contain an N-terminal GTPase domain, a Pleckstrin homology (PH) domain, and a C-terminal proline-rich motif (PRM) (Cao et al., 1998). Dynamin 2 (or Dyn2) is ubiquitously expressed and essential for the development of a nervous system (Ferguson et al., 2009), while Dynamin 1 and 3 (or Dyn1,3) are highly expressed in brain and primarily involved in synaptic vesicle recycling (Ferguson et al., 2007; Raimondi et al., 2011).
Primary cultures of mouse hippocampal neurons with the following genotypes were used in this study: C57/BL6-N; C57/BL6-J, DNM3 KO (Raimondi et al., 2011), and DNM1,3 DKO (Raimondi et al., 2011). The animals of the genotype DNM1+/+, DNM3−/− and DNM−/−, DNM3−/− were generated by crossing DNM1+/−, -3−/− animals. DNM1+/−, DNM3−/− animals were not included in the study to avoid any confound results due to the haploinsufficiency. The sex of pups was not assessed because it is difficult to distinguish it at E18 or P0.
Method details
Primary neuronal cultures
To prepare primary neuronal cultures, the following procedures (Itoh et al., 2019) were carried out. Newborn or embryonic day 18 (E18) mice of both genders were decapitated. The brain is dissected from these animals and placed on ice cold dissection medium (1 x HBSS, 1 mM sodium pyruvate, 10 mM HEPES, 30 mM glucose, and 1% penicillin-streptomycin). For the knockout experiments, DNM1+/−,3−/− mice were bred to obtain DNM1+/+,3−/− (Dyn3 KO) and DNM1−/−,3−/− (Dyn1,3 DKO). DNM1+/−,3−/− mice were not used in this study. Because Dyn1,3 DKO mice die perinatally (Raimondi et al., 2011), we cultured hippocampal neurons from animals of embryonic days 18 (E18) or postnatal day 0 (P0) immediately after birth.
For high pressure freezing, hippocampal neurons were cultured on a feeder layer of astrocytes. Astrocytes were harvested from cortices with treatment of trypsin (0.05%) for 20 min at 37 °C, followed by trituration and seeding on T-75 flasks containing DMEM supplemented with 10% FBS and 0.2% penicillin-streptomycin. After 2 weeks, astrocytes were plated onto 6-mm sapphire disks (Technotrade Inc) coated with poly-D-lysine (0.1 mg/ml), collagen (0.6 mg/ml) and 17 mM acetic acid at a density of 13 × 103 cells/cm2. After 1 week, astrocytes were incubated with 5-Fluoro-2′-deoxyuridine (81 μM) and uridine (204 μM) for at least 2 hours to stop the cell growth, and then medium was switched to Neurobasal-A (Gibco) supplemented with 2 mM GlutaMax, 2% B27 and 0.2% penicillin-streptomycin prior to addition of hippocampal neurons. Hippocampi were dissected under a binocular microscope and digested with papain (0.5 mg/ml) and DNase (0.01%) in the dissection medium for 25 min at 37 °C. After trituration, neurons were seeded onto astrocyte feeder layers at density of 20 × 103 cells/cm2. Cultures were incubated at 37 °C in humidified 5% CO2/95% air atmosphere. At DIV14–15, neurons were used for high pressure freezing experiments.
For fluorescence imaging, dissociated hippocampal neurons were seeded on 18-mm or 25-mm coverslips coated with poly-L-lysine (1 mg/ml) in 0.1 M Tris-HCl (pH8.5) at a density of 25–40 × 103 cells/cm2. Neurons were cultured in Neurobasal media (Gibco) supplemented with 2 mM GlutaMax, 2% B27, 5% horse serum and 1% penicillin-streptomycin at 37 °C in 5% CO2. Next day, medium was switched to Neurobasal with 2 mM GlutaMax and 2% B27 (NM0), and neurons maintained thereafter in this medium. For biochemical experiments, dissociated cortical neurons were seeded on poly-L-lysine coated plates with Neurobasal media supplemented with 2 mM GlutaMax, 2% B27, 5% horse serum and 1% penicillin-streptomycin, at a density of 1 × 105 cells/cm2. Next day, the medium was switched to Neurobasal medium with 2 mM GlutaMax and 2% B27, and neurons maintained in this medium thereafter. A half of the medium was refreshed every week.
Expression constructs
All the plasmids used in this study and primers to make these construct was listed in Table S1. DNA cloning was performed using transformation into competent DH5α cells. For the dynamin rescue constructs, wild-type Dyn1xA, Dyn1xB and Dyn2 from human sequences were amplified from plasmids (generously provided by Pietro De Camilli’s lab) and fused by gibson assembly (NEW ENGLAND BioLabs) after a self-cleaving P2A site within a pFUGW derived plasmid, which encodes nuclear RFP controlled by the human synapsin 1 promoter (f(syn)NLS-RFP-P2A), to form f(syn)NLS-RFP-P2A-Dyn1xA/Dyn1xB/Dyn2. Using the same approach, we generated Dyn1xA S774/778A, S774/778D, Dyn1xB S774/778A.
For Syndapin 1 shRNA, oligos containing the target sequence were annealed with T4 DNA ligase (NEW ENGLAND BioLabs) and phosphorylated with T4 polynucleotide kinase (NEW ENGLAND BioLabs). The annealed oligos were ligated downstream of a human U6 promoter using BamHI and PacI restriction sites within the modified FUGW vector (f(U6).hSyn-NLS-RFP-WPRE) that further contained a human synapsin-1 promoter controlled NLS-RFP reporter construct (f(U6)Synapsin 1-shRNA.hSyn-NLS-RFP-WPRE). For the Syndapin 1 rescue constructs, mouse Syndapin 1 sequence was amplified and ligated into NheI and AscI restriction sites of f(syn)NLS-RFP-P2A (f(syn)NLS-RFP-P2A- Syndapin 1). f(syn)NLS-RFP-P2A- Syndapin 1 construct was linearized using primers with shRNA Syndapin 1 resistant sequence and re-circularized using In-Fusion HD cloning kit (TAKARA) (f(syn)NLS-RFP-P2A-Syndapin 1 shRNA resistant). For the Syndapin 1 I122D/M123E rescue construct, f(syn)NLS-RFP-P2A-Syndapin 1 shRNA resistant construct was amplified using primers with I122D/M123E mutations. The resulting PCR solution was digested with DpnI to degrade template DNA followed by ethanol precipitation. DNA was then transformed to competent DH5α cells. For the Syndapin 1 P434L rescue construct, f(syn)NLS-RFP-P2A-Syndapin 1 shRNA resistant construct was amplified using primers with P434L mutations and re-circularized using In-Fusion HD cloning kit (TAKARA).
To generate mCherry-Syndapin 1 with the shRNA resistant sequence (mCherry-shRNA resistant Syndapin 1), mouse Syndapin 1 sequence was amplified and combined with linearized mCherry construct using In-Fusion HD cloning kit. Then, mCherry-Syndapin 1 construct was linearized using primers with shRNA syndapin resistant sequence and re-circularized using In-Fusion HD cloning kit (TAKARA). To make mCherry-shRNA resistant Syndapin 1 122D/M123E and mCherry-shRNA resistant Syndapin 1 P434L, mCherry-shRNA resistant Syndapin construct was linearized using the primers with corresponding mutations and were re-circularized using In-Fusion HD cloning kit.
Dyn1xA-GFP was purchased from addgene. To generate Dyn1xA S774/778A-GFP and Dyn1xA-S774/778D GFP, Dyn1xA-GFP construct was linearized using the primers with corresponding mutations and were re-circularized using In-Fusion HD cloning kit. To generate Dyn1xA-GFP_mCherry-Syb2, rat Syb2 (addgene) and mCherry (addgene) sequence were amplified and combined with linearized Dyn1xA construct using In-Fusion HD cloning kit. As a result, mCherry-Syb2 sequence are inserted in to downstream of Dyn1xA-GFP sequence. To generate cytosolic tdTomato, tdTomato sequence was amplified from tdTomato vector (Clontech) and was ligated at the XhoI and KpnI digestion sites of pCAGGS construct by DNA ligation kit Solution I (TAKARA).
For pull-down assays, wild-type rat Dyn1xA-PRM (C-terminus residues 746–864), S774/778E and S774/778A mutants were generated by amplifying the required region from Dyn1aa-GFP (rat) in pEGFP-N1. The amplified product was inserted into pGEX4T-1 vector (Amersham Biosciences) using the restriction enzymes EcoRI and NotI (underlined in sequence). Subsequent modifications to generate S774/778E and S774/778A mutants used the QuickChange site-directed mutagenesis kit (Stratagene) and were confirmed by DNA sequencing (Anggono et al., 2006). Wild-type rat Dynamin-1xb C-terminus (residues 746–851) was generated by amplifying the required region from Dyn1ab in a pCR3.1 expression vector (Cao et al 1998). Similar to Dyn1xA, the amplified product was inserted into a pGEX-4T-1 using the restriction enzymes EcoRI and NotI (Xue et al., 2011). To generate Dyn1xA-PRM S851/857A and Dyn1xA-PRM S851/857E, Dyn1xA-PRM was linearized using the primers with corresponding mutations and were re-circularized using T4-ligase (Promega, M1801). Dyn1xB-PRM S774/778A and Dyn1xB-PRM S774/778E were generated via the digestion of Dyn1xA-PRM S774/778A or Dyn1xB-PRM S774/778E with BamHI. This excised a sequence encoding the mutated residues (cut sites in multi-cloning site and between G806/S807). This fragment was ligated into wild-type Dyn1xB-PRM previously digested with BamHI. An identical protocol as followed for the Dyn1xA-PRM S774/778/851/857A and Dyn1xA-PRM S774/778/851/857E mutants, except the corresponding fragment was ligated into BamHI-digested Dyn1xA-PRM S851/857A or Dyn1xA-PRM S851/857E respectively. The correct insertion of the fragment was confirmed by Sanger sequencing, which was also the case for site-directed mutagenesis.
For GFP knock-in constructs, oligos containing the 20-bp target sequences were annealed at gradient temperature from 95 °C to 20 °C for 30 min. The annealed oligos were ligated into the BbsI sites of pORANGE vector (addgene) by DNA ligation kit Solution I (TAKARA). Donor sequence, consisting of GFP and two Cas9 target sites flanking GFP, was PCR amplified from pEGFP-N1 plasmid and cloned into the HindIII and XhoI site of pORANGE vector containing the target sequence. First, mCherry was PCR amplified from pmCherry-N1 plasmid (Clontech) with mC-F and mC-R primers and ligated into the BamHI and EcoRI site of pFUGW-spCas9 replacing Cas9, generating pFUGW-mCherry. Then, knock-in cassettes containing the U6 promoter, gRNA, and donor DNA was PCR amplified from pORANGE constructs and cloned into PacI site of pFUGW vector using In-Fusion HD cloning kit.
For the recombinant protein expression constructs, wile-type Syndapin 1 and Syndapin 1 P434L sequences were amplified from mCherry-shRNA resistant Syndapin 1 and mCherry-shRNA resistant Syndapin 1 P434L, respectively. In this design, the forward primer inserts a BamHI site and TEV cleavage sequence (ENLYFQ/SG) in the upstream of Syndapin 1, and the reverse primer inserts a NotI site after the stop codon of Syndapin 1. The PCR products and pET28a were digested by BamHI-HF and NotI-HF, and ligated by T4 ligase (NEB, #M0202). Wild-type Dyn1xA and Dyn1xA S774/778D sequences were amplified from Dyn1xA -GFP and Dyn1xA S774/778A-GFP, respectively. Then, combined with linearized pGex6T using In-Fusion HD cloning kit.
Lentivirus production and infection
For lentivirus production, HEK293T cells were maintained in DMEM supplemented with 10% FBS and 0.2% penicillin-streptomycin. One day after plating 6.5 × 106 cells in T75 flask (Corning), medium was replaced with Neurobasal-A media supplemented with 2 mM GlutaMax, 2% B27 and 0.2% penicillin-streptomycin. Cells were transfected using polyethylenimine with a pFUGW plasmid encoding the RFP co-infection marker and insert of interest, and two helper plasmids (pHR-CMV8.2 deltaR and pCMV-VSVG) at a 4:3:2 molar ratio. Three days after transfection, supernatant was collected and concentrated to 20-fold using Amicon Ultra-15 10K centrifuge filter (Millipore). ChetaTC and dynamin rescue viruses were added to each well of neurons at DIV 3 with 15 μl per well (12-well plates). Syndapin 1 shRNA virus was added to each well of neurons at DIV 3 with 5 μl per well (12-well plates, 75 K neurons / well). Syndapin 1 rescue viruses were added to each well of neurons at DIV 3 with 5 μl per well (12-well plates, 75K neurons / well) or 20 μl per well (6-well plates, 300 K neurons / well). In all cases, infection efficiency of over 95% was achieved. Neurons were fixed or used for high pressure freezing at DIV13–16.
Transient transfection of neurons
For transient protein expression, neurons were transfected at DIV13–14 using Lipofectamine 2000 (Invitrogen) in accordance with manufacture’s manual with minor modifications (Araki et al., 2015). Before transfections, a half of medium in each well was transferred to 15 mL tubes and mixed with the same volume of fresh NM0 and warmed to 37 °C in CO2 incubator. This solution later served as a conditioned medium. Briefly, 0.25–2 μg of plasmids was mixed well with 2 μl Lipofectamine in 100 μl Neurobasal media and incubated for 20 min. For Dyn1 expression, 0.5 μg of constructs were used to reduce the expression level. For tdTomato expression, 2.0 μg of constructs were used. For Syndapin 1 expression, 0.25 μg of constructs were used in the Syndapin 1 knock-down neurons. The plasmid mixture was added to each well with 1 ml of fresh Neurobasal media supplemented with 2 mM GlutaMax and 2% B27. After 4 hours, medium was replaced with the pre-warmed conditioned media. Neurons were incubated for less than 20 hours and fixed for imaging or subjected to live imaging. For GFP knock-in experiments, hippocampal neurons were transfected at DIV 3 with pORANGE construct (1.5 μg) and pmCherry-N1 (0.5 μg) as cell-fill using Lipofectamine 2000 and fixed at DIV15–18.
Sequencing of genomic sites of GFP integration
Genomic DNA was isolated from the neurons at DIV15 using DNeasy Blood & Tissue Kit (Qiagen). Genomic PCR was performed to amplify the 5’ and 3’ junctions of the integrated GFP by KOD Hot Start DNA Polymerase (Novagen) using the primers listed in Table S1. PCR products were separated by agarose gel electrophoresis and the fragments were purified using QIAquick Gel extraction kit (Qiagen). Purified DNA was cloned into pCR4Blunt TOPO vector using Zero Blunt TOPO PCR Cloning Kit (Invitrogen). The sequences of 18 clones were analyzed.
Immunofluorescence staining
For immunofluorescence, 125 k neurons were seeded on 18 mm poly-L-lysins (1 mg/mL, overnight) coated coverslips (Thickness 0.09–0.12 mm, Caroline Biological) in 12-well plate (Corning). Neurons were fixed at DIV14–15 with pre-warmed (37 °C) 4% paraformaldehyde and 4% sucrose in PBS for 20 min and wash three times with PBS to remove remaining PFA. Next, fixed neurons were permeabilized with 0.2% Triton X-100 in PBS for 8 min at room temperature followed by three times wash with PBS. After blocking with 1% BSA in PBS for 30 min, cells were incubated with primary antibodies diluted in 60 μl of 1% BSA/PBS overnight at 4 °C in a humidity chamber. Unbound antibodies were removed by washing with PBS three times followed by appropriate secondary antibodies diluted 1:400 in 60 μl of 1% BSA/PBS for 1 hour at room temperature in a humidity chamber. Excess antibodies were removed by washing with PBS three times. Coverslips were rinsed with MilliQ water three times to remove salts and were mounted in ProLong Gold Antifade Mountant (Invitrogen) and stored at 4 °C until imaging. For the STED imaging, Dyn1xA-GFP or Dyn1xB-GFP was stained with 1:500 dilution of anti-GFP antibodies, Rabbit polyclonal (MBL International) and endogenous Bassoon was stained with 1:500 dilution of anti-Bassoon antibody, mouse monoclonal (Synaptic Systems). 50 μM of anti-rabbit ATTO647 (Rockland) and anti-mouse Alexa594 (Invitrogen) was used for the secondary antibodies. Coverslips were rinsed with MilliQ water three times to remove salts and mounted in ProLong Diamond Antifade Mountant (Thermo Fisher) and stored at 4 °C until imaging.
Confocal microscopy imaging and analysis
For fluorescence imaging, all samples were imaged using a confocal microscope Zeiss LSM880 (Carl Zeiss). Fluorescence was acquired using a 63x objective lens (NA = 1.4) at 2048×2048 pixel resolution with the following settings: pixel Dwell 1.02 μs and pin hole size at 2 airy unit. For experiments comparing fluorescence intensity, these settings were left constant between samples. Snapshots were captured at the cell body first and then 3–7 different locations along the axon of the same neurons, typically covering 1–3 mm length from each neuron. All Dyn1-GFP transfected neurons in each coverglass were imaged. Axons were distinguished from dendritic processes based on their morphology (thin and lacking spines). If GFP signals were aberrantly saturated throughout the processes, they were dismissed to avoid overexpression artifact. Apparently dying neurons and glia cells were also excluded. We quantified all presynaptic varicosities along axons in each image. Presynaptic regions were confirmed with the Syb2-Alexa647 signals. The Syb2-Alexa647 signals were used to define the boundaries of regions-of-interest (ROIs) for quantifications, and all Dyn1-GFP and mCherry-Syndapin 1 signals within ROIs were measured as the total signals at each synapse. To define puncta within the boutons, we applied Gaussian smoothing (σ = 0.2 ) on images, performed threshold cut-off on the average fluorescence intensity of Dyn1xA and Syndapin 1 in the intersynaptic axonal regions. The circumference of each punctum adjacent to or within Syb2 signals were delineated and set as ROIs, and fluorescence intensity measured. The background signals were subtracted using the same size of ROI but 40–60 pixels away from the original ROI and outside of neural processes. For the quantification of the colocalization between Dyn1xA and Syndapin 1 puncta, ROIs of overlapped Dyn1xA and Syndapin 1 were counted. To avoid bias from the expression variability between neurons, fluorescence intensity was normalized to the average signal within the corresponding cell body of each axon. False colored images were made by applying LUT colors and ratio signal intensities are measured by calibrate bar function in Fiji.
Live imaging and analysis
For live-cell imaging, 250 k neurons were seeded on 25 mm coverslips (thickness 0.13–0.17 mm, Carolina Biological) in 6-well plate (Corning). Dyn1xA-GFP and mCherry-Syb2 were expressed on the same construct using pDyn1xA-GFP_P2A_mCherry-Syb2 construct by transient transfection. For the digitonin treatment, 10 mg/mL of digitonin (Sigma-Aldrich) was prepared in MilliQ water and diluted into final concentration 5 μg/mL or 500 μg/mL in NM0. For the 1,6-Hexanediol treatment, 4 % of 1,6-Hexanediol was directly dissolved in NM0. For imaging, cover slips were transferred to live cell round chamber (ALA science) filled with NM0. FK506 (Tocris) or CHIR99021 (Sigma-Aldrich) were dissolved in DMSO and added to the imaging chamber to achieve the final concentrations of 2 μM and 10 μM, respectively, 30 min before imaging. 0.05 % of DMSO were added to the chamber for control experiments. All live imaging experiments were carried out on a Zeiss LSM880 confocal microscope at 37 °C in humidified 5% CO2/95% air atmosphere, and live imaging data was acquired by raster scan. Neurons were not imaged more than 30 min to avoid phototoxic artifacts.
For photobleaching experiments, 33 frames were collected with a time interval of 2.5 s. After 3 frames, the maximum laser power was applied to bleach fluorescence. The 4th frame was started immediately after bleaching. 30 additional frames were collected after bleaching. A circular ROI was placed around each Dyn1xA punctum found adjacent to the mCherry-Syb2 signal, and Dyn1xA-GFP signals within the ROI was photobleached with a 488-nm laser. For quantifications, average fluorescence intensity was measured from each ROI and a random region outside of the cell, which was used for the background subtraction. A circular ROI of the same size was used to measure signals from an unbleached region to measure the degree of imaging-related photobleaching. The fluorescence intensity at the ROIs was normalized to the intensity before bleaching (3rd frame) and after bleaching (4th frame). To correct signal loss due to the bleaching during the imaging, the normalized fluorescence recovery over time, F(t), was then fitted to an exponential function:
where the corrected normalized fluorescence over time is Fcorr(t), time after the photo bleaching is t, and K is the rate constant acquired from the unbleached region and measured exponential curve fitting tool in Fiji.
The Fcorr(t), was then fitted to an exponential function using GraphPad Prism (v8);
where the recovery is A, half-recovery time is .
For the cytosolic extraction experiments with digitonin, a total of 17 frames were collected with a time interval of 5 s: 5 frames were collected prior to the application of digitonin (500 μg/mL in NM0) (Sigma-Aldrich), and additional 12 frames were collected thereafter. For the aliphatic alcohol treatment, a total of 12 frames were collected with a time interval of 10 s: 2 frames before the addition of 4 % 1,6,-hexanediol, 2,5,-hexanediol or 1,4,-butanediol (Sigma-Aldrich) in NM0 and 10 frames afterwards. For the purpose of imaging-related photobleaching corrections, the same number of frames were collected at additional regions distant from the imaged before the experiments. For quantifications, the average fluorescence intensities before and after the treatment were measured within the same ROIs, which were selected as described in the previous section: one ROI covering the entire Syb2 signals and another ROI along the circumference of the Dyn1xA-GFP puncta. For presentations, the brightness and the contrast in each image were adjusted in Fiji and cropped in Adobe photoshop 2021 (Adobe).
Stimulated emission depletion microscopy (STED) imaging
All STED images were obtained using a home-built two-color STED microscope (Han and Ha, 2015; Ma and Ha, 2019). Basically, a femtosecond laser beam with repetition rate of 80 MHz from a Ti:Sapphire laser head (Mai Tai HP, Spectra-Physics) is split into two parts: one part is used for producing the excitation beam, which is coupled into a photonic crystal fiber (Newport) for wide-spectrum light generation and is further filtered by a frequency-tunable acoustic optical tunable filter (AA Opto-Electronic) for multi-color excitation. The other part of the laser pulse is temporally stretched to ~300 ps (with two 15-cm-long glass rods and a 100-m long polarization-maintaining single-mode fiber, OZ optics), collimated and expanded, and wave-front modulated with a vortex phase plate (VPP-1, RPC photonics) for hollow STED spot generation to de-excite the fluorophores at the periphery of the excitation focus, thus improving the lateral resolution. The STED beam is set at 765 nm with power of 120 mW at back focal plane of the objective (NA=1.4 HCX PL APO 100×, Leica), and the excitation wavelengths are set as 594 nm and 650 nm for imaging Alexa-594 and Atto-647N labeled targets, respectively. The fluorescent photons are detected by two avalanche photodiodes (SPCM-AQR-14-FC, Perkin Elmer). The images are obtained by scanning a piezo-controlled stage (Max311D, Thorlabs) controlled with the Imspector data acquisition program.
STED image analysis
A custom MATLAB code package was used to analyze the endocytic protein distribution relative to the active zone marked by Bassoon in STED images. First, the STED images were blurred with a Gaussian filter with radius of 1.2 pixels to reduce the Poisson noise, and then deconvoluted twice using the built-in deconvblind function: the first point spread function (PSF) input is measured from the unspecific antibodies in the STED images, and the second PSF input is chosen as the returned PSF from the first run of blind deconvolution (Sapoznik et al., 2020). Each time 10 iterations are performed. All of presynaptic bouton in each deconvoluted images were selected within 30×30-pixel (0.81 μm2) ROIs based on the varicosity shape and bassoon signals. The active zone boundary was identified as the contour that represents half of the intensity of each local intensity peak in the Bassoon channel, and the Dyn1 clusters are picked as the local maxima. The distances between the Dyn1 cluster centers and active zone boundary are automatically calculated correspondingly. Dyn1 cluster over crossing the ROIs and the Bassoon signals outside of the transfected neurons were excluded from the analysis. The MATLAB scripts are available by request.
Lattice light sheet microscopy (LLSM) imaging
For LLMS imaging, 120 k neurons were seeded on 18 mm coverslips (thickness 0.13–0.17 mm, Carolina Biological) in 12-well plate (Corning). Dyn1xA-Halo was expressed transient transfection at DIV13. At DIV14, 100 nM JFX-549 (Grimm et al., 2021) and 50 nM JF-PA-647 (Grimm et al., 2016) (Gifts from Janelia Research Campus) were incubated at 37 °C for 30 min in 1 ml NM0 followed by 4 times wash (30 min each) in the 12-well plate. Before imaging, a coverslip was rinsed three times by pre-warmed imaging buffer [140 mM NaCl, 2.4 mM KCl, 10 mM HEPES, 10 mM Glucose (pH adjusted to 7.3 with NaOH, 300 mOsm, 1 mM CaCl2, 4 mM MgCl2, 3mM NBQX and 30 mM Bicuculline]. All these processes were performed under dark room to reduce pre-activated JF-PA647. For the LLSM imaging, a home-built LLSM setup was used (Chen et al., 2014). The detailed blueprints are provided by the Betzig group at the Howard Hughes Medical Institute Janelia Research Campus. Basically, four laser beams (405nm/250mW, RPMC; 488nm/300 mW, MPB Communications; 560nm/500 mW, MPB Communications and 642nm/500 mW, MPB Communications) were expanded and collimated independently to a size of 2.5 mm, and then were combined and sent into an acoustic optical tunable filter (AA Opto-Electronic) for rapid channel switching and power adjustment. To achieve single particle detection sensitivity, two pairs of cylindrical lenses (25mm /200 mm and 250mm/50 mm) were then used to convert the Gaussian beam into a thin stripe illumination projected onto a binary phase spatial light modulator (SLM, Forth Dimension, SXGA-3DM). The squared lattice patterns with an inner NA of 0.44 and outer NA of 0.55 was projected on the SLM, which is relayed by a 500-mm lens onto an annular mask conjugated to the back pupil plane of the excitation (Special Optics, 0.65 NA, 3.74 mm WD). Two galvo mirrors (Cambridge Technology, 6215H) were used for the excitation light sheet positioning along the axis of the detection objective (Nikon, CFI Apo LWD 25XW, 1.1 NA, 2 mm WD) and dithering along the orthogonal direction. The power of the 642-nm excitation beam measured at the back pupil plane of the excitation objective is 3.3 mW. The emitted fluorescence was collected by the detection objective (Nikon, CFI Apo LWD 25XW, 1.1 NA, 2 mm WD) orthogonally mounted and projected onto an sCMOS camera (Hamamatsu, Orca Flash 4.0 v3) by a 500-mm tube lens. A quad-band filter (Semrock) was placed before the camera. Before imaging, 642-nm excitation beam was illuminated to completely bleach pre-activated PA-JF646 dyes. Single-particle tracking was recorded by illuminating the sample by the 642-nm laser with 20-ms exposure time (50 Hz) interleaved with 1–2 ms 405-nm activation for 300 frames. 10 frames with 560-nm excitation were collected before and after recording the single particle dynamics for generating the Dyn1xA pre-accumulation mask channel.
Single particle tracking analysis
A custom MATLAB code package (available upon request) was used to analyze single particle dynamics of the Dyn1xA. Source data including all trajectory files are available from sharing server (https://data.mendeley.com/datasets/bkn78y232y/draft?preview=1). First, the individual particles were detected and linked into trajectories by running the Fiji plugin TrackMate (Tinevez et al., 2017) in batch to generate xml files recording the spatiotemporal coordinates of each particle in each trajectory. The xml files were then read by the customized MATLAB code. To generate the mask boundaries representing the pre-accumulation of Dyn1xA, the 560-nm channel images were averaged, and the boundaries were selected as the contours with 65% of the local intensity maxima. Only the trajectories that have overlap with the pre-accumulation mask were selected and further analyzed. This is to reduce the bias imposed by the differences in available spaces between the cytosol and puncta. In addition, when a trajectory crosses the boundary from outside, they are counted as the movement outside the punctum, while a trajectory crossing the boundary from inside are quantified as movement inside the punctum.
To calculate the average diffusion coefficients inside and outside the puncta, we placed a mask on each punctum at the 50% intensity of the peak fluorescence. Displacements (distance traveled between two consecutive frames) with the start and end point purely inside or outside the mask were collected with the Pythagorean Theorem based on the x-, y-coordinates of molecules displaced in two consecutive images.
Then, displacement is fitted with the following distribution with 3 diffusive states:
where r is displacement, D1, D2 and D3 are the diffusion coefficients with the fraction of f1, f2 and f3. Δt is the exposure time, and σ is the localization precision. The average diffusion coefficient was calculated as f1D1 + f2D2 + (1 − f1 − f2)D3
To extract the full information within single-particle trajectories collected in our experiments, which potentially contain multiple interconverting species with different diffusive state, we also applied the unsupervised Gibbs sampling (SMAUG) algorithm using nonparametric Bayesian statistics with default setting (Karslake et al., 2021).
Protein expression and GST-pulldown assay
Isolated nerve terminals, or synaptosomes, were prepared from rat brains of both sexes (Cousin and Robinson 2000). GST fusion proteins were expressed in bacteria at 19 °C for 16–18 hours in 500 ml cultures of supermedia (5g/L NaCl, 25g/L yeast extract (Alfa Aesar J60287), 15g/L Tryptone (Merk 1.11931), pH7.4) and coupled to glutathione-Sepharose beads (GE healthcare, 17–0756-01).
Nerve terminals were solubilized for 5 minutes at 4 °C in 25 mM Tris, pH 7.4, with 1% Triton X-100, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM PMSF and protease inhibitor cocktail (Sigma, P58849) and centrifuged at 20,442 g for 5 minutes at 4 °C. The subsequent supernatant was incubated with GST-fusion proteins for 2 hours at 4 °C under constant rotation. After washing in lysis buffer (including a 500 mM NaCl wash), beads were washed twice in cold 20 mM Tris (pH 7.4) and boiled in SDS sample buffer (67 mM SDS, 2 mM EGTA, 9.3% glycerol, 12% β-mercaptoethanol, 0.7 mg/mL Bromophenol blue, 67 mM Tris-HCl, pH 6.8). The released proteins were separated by SDS-PAGE and analyzed by Western blotting. Primary antibodies were incubated with the blots overnight at 4°C under constant rotation (anti-endophillin, sc10874, 1:3000; anti-syndapin/PACSIN1, ab137390, 1:5000; anti-endophilin A2, a gift from Pietro De Camilli (Milosevic et al., 2011). IRDye secondary antibodies (800CW anti-goat IgG, #926–32214 and 680RD anti-rabbit, #926–68073 both 1:10000) were applied in Odyssey blocking PBS buffer for 2 hours at room temperature (LI-COR Biosciences, Nebraska, USA). Blots were visualized using a LiCOR Odyssey fluorescent imaging system, with band densities quantified using Fiji (ImageJ) software. Both syndapin and endophilin bands were normalized to the GST fusion protein band revealed by Ponceau-S staining.
Protein expression and purification for in vitro phase separation assay
Expression and purification of dynamin proteins were performed as previously described with minor modifications (Imoto et al., 2018). Wild-type Dyn1xA and Dyn1xA S774/778D constructs were transformed into E. coli BL21-CodonPlus (DE3)-RIPL (Agilent Technologies, # 230280). Transformed BL21-CodonPlus strain cells were cultured at 37 °C for 12 hours in 200 mL of LB medium, scaled up to 2 L of LB medium, and further incubated at 37 °C for 2 hours and at 18 °C for 1 hour. IPTG was added to a final concentration of 0.1 mM, and cells were harvested after a further 12-h incubation at 18 °C by centrifugation at 5000×g for 15 min. Cell pellets were resuspended in 200 mL of HEPES buffer (HDB800) containing 800 mM NaCl, 20mM HEPES-NaOH, pH 7.5, 2 mM EGTA, 1mM MgCl2, 1 mM DTT, and a cOmplete protease inhibitor cocktail. Resuspended pellets were lysed by the microfluidizer (80 psi, 3 times). Soluble fraction was collected by centrifugation at 20,000× g, 4 °C, 30 min and rotated at 4 °C with 0.5 mL bed volume of glutathione Sepharose 4B beads (GE Healthcare) for 1 hour. The beads were collected by centrifugation at 1,500× g, 4 °C, 1 min and resuspended in 25 mL of HBD800. The beads were collected again by the centrifugation and resuspended in 25 mL of cOmplete-free HDB800. Then, beads resuspension was loaded onto a 10 mL Poly-Prep Chromatography column (BioRad) and washed with 25 mL of cOmplete-free HDB800 at 4 °C. After the washing, Sepharose beads were treated with 20 μL of PreScission Protease (GE Healthcare) in 5 mL of cOmplete-free HDB800 at 4 °C for 2 days and the cleaved-off proteins were eluted. Expression of wild-type and P434L Syndapin 1 was performed in E. coli BL21-CodonPlus (DE3)-RIPL (Agilent Technologies, # 230280). After overnight culture in LB media containing Kanamycin and Chloramphenicol (LB/Kan/Cam), 5 mL preculture was inoculated to 1 L LB/Kan/Cam and cultured at 37 °C for 4 hours. At OD600 = 0.450–0.550, 500 μM IPTG was added, and the proteins expressed at 23 °C for 26 hours.
For the Syndapin 1 purification, two-liter culture cell pellet was resuspended by 75 mL of buffer [50 mM HEPES-NaOH (pH 7.5 at room temperature), 200 mM NaCl, 1 mM DTT] supplemented with 5 mM MgCl2, 1 mM PMSF, and cOmplete protease inhibitor cocktail (Roche, #4693159001) (1 tablet/75 mL) and lysed by microfluidizer (Microfluidics, Model M-110Y, 80 psi, 3 times). Soluble fraction was collected by centrifugation at 10,000× g, 4 °C, 60 min and applied to pre-equilibrated Ni-NTA agarose (QIAGEN, #30230, 3 mL resin/2 L culture). The proteins were washed and eluted by the stepwise imidazole gradient in Buffer at 20, 30, 40, 50, 100, 200, and 500 mM Imidazole; 3 column volume each. Forty to five hundred imidazole fractions were collected and mixed with 1/50 weight of 6xHis-MBP-TEV-protease-S219V (purified in house) to remove N-terminal 6xHis tag of Syndapin 1 during 2 overnight dialysis against 25 mM HEPES-NaOH (pH 7.5 at room temperature), 200 mM NaCl, 1 mM DTT. His-tag-cleaved Synapin-1 was separated from un-cleaved sample and TEV protease by passing through Ni-NTA agarose resin (3 mL) and diluted into half with 25 mM HEPES-NaOH (pH 7.5) 1 mM DTT. The protein was further applied to pre-equilibrated HiTrapQ anion exchange column (Cytiva, #17115301, 1 mL) and eluted by linear gradient of NaCl from 100 mM to 500 mM in buffer containing 25 mM HEPES-NaOH (pH 7.5), 1 mM DTT. Higher purity fractions from 227.5 mM to 272.5 mM NaCl were then collected, concentrated by Amicon-Ultra (Millipore, #UP901024) (10 kDa cut off), snap frozen, and stored at −80 °C (final buffer condition: 25 mM HEPES-NaOH (pH 7.5), 250 mM NaCl, 1 mM DTT). Proteins were concentrated by Amicon-Ultra (Millipore, #UP901024) (10 kDa cut off), snap frozen, and stored at −80 °C. The purity of wild-type Syndapin 1, P434L, w-type Dyn1xA and Dyn1xA S774/778D was evaluated by SDS-PAGE.
Protein labelling
Proteins were labelled using Alexa Fluor™ 488 or Alexa Fluor™ 568 NHS Ester (Succinimidyl Ester) (Thermo Fisher) in HDB. Dye and proteins were reacted at 2;1 molar ratio. Reaction mixtures were rotated for 60 min at room temperature, then labelled protein was separated from unconjugated dye using Zeba™ Spin Desalting Columns, 7K MWCO, 0.5 mL (ThermoFischer). Reaction and the column washing buffers were as same as protein elution buffer of Dynamin 1xA or Syndapin 1.
In vitro phase separation assay
In most of the experiments, Dyn1xA or Dyn1xA-S774/778D was used at 2 μM, which was the maximum concentration without having protein aggregates. Syndapin1 or Syndapin1 P434L was used at 20 μM, reflecting physiological concentration within synapses (Wilhelm et al., 2014). Before experiments, the proteins were centrifuged at 15,000× rpm at 4 °C for 10 min to remove aggregates. For the droplet formation assay, Dyn1xA and Syndapin1 are mixed at room temperature in 50 μl of HDB200 followed by addition of 5% PEG (Sigma) to their final concentrations. After 5min incubation, reaction mixture was transferred to 8-well glass chamber (LAB-TEK). The fluorescence images were collected at room temperature using 100x objective lens (NA=1.45) and LED light source at 2048×2048 pixel resolution by Nikon ECLIPSE Ti2 equipped with ORCA-Fusion BT Digital CMOS camera (HAMAMATSU). NIS-Elements AR software were used for the image acquisition. For the live imaging of the droplet fusion, confocal microscope Zeiss LSM880 was used as described above. For the sedimentation assay, 5% PEG was added to protein mixtures with the total volume of 100 μl. After the incubation at room temperature for 5 min, the protein mixtures were centrifuged 15,000× rpm for 10 min. Pellets were dissolved in 100 μl of SDS sample buffer. Supernatant and pellet fractions were examined by 4–20% SDS-PAGE gels (Biorad) and gel code staining (Thermo Fisher). For the FRAP experiments and analysis, images were acquired at room temperature using confocal microscope Zeiss LSM880 as described above. Images were acquired at 2.5 s/frame, and photobleaching with the 488 nm laser was performed just after the third frame. Droplets of similar size were selected for the experiments. For the FRAP using different ROI size, ROIs radius were setup as small as possible compared to droplet radius, which was enough to ignore influx of fluorescent molecules from outside. The intensity range was normalized to maximum and minimum intensity values after post-bleaching frames. For the bleaching entire droplet, the ROI with same radius as the droplets was used. The intensity range was normalized to the 1st frame of post-bleaching images.
Phase separation assay in COS-7 cells
COS-7 cells were seeded at 80k on 18 mm cover slips (thickness 0.13–0.17 mm, Carolina Biological) coated with 100 μg/ml fibronectin (Sigma). On next day, the 0.5 μg each of plasmid DNA were transfected using Lipofectamine (Invitrogen) in accordance with manufacture’s manual. At ~24 or ~48 hours post transfection, coverslips were transferred to live cell round chamber (ALA science) filled with DMEM. FRAP experiments and analysis were performed at 37 °C using a confocal microscope Zeiss LSM880 as described above. Cells were not imaged more than 30 min to avoid phototoxic artifacts.
Evaluations of Syndapin 1 shRNA efficiency
To test the efficiency of Syndapin 1 KD and the amount of overexpression, cultured hippocampal neurons were harvested, lysed by addition of lysis buffer (50 mM Tris pH 8.0 and 1% SDS containing cOmplete Mini Protease Inhibitor (Roche)) and boiling at 95 °C for 5 min. Lysates were centrifuged at 15,000x g for 15 min at 4 °C, and the supernatants collected. Proteins were separated by SDS–PAGE and transferred onto Immobilon-FL membranes (Millipore). After blocking with 5% skim milk in PBS containing 0.05% Tween-20 (PBST) for 30 min, membranes were incubated with anti PACSIN 1 rabbit polyclonal antibody diluted at 1:1000 in 3% BSA/PBST overnight at 4 °C, followed by IRDye secondary antibodies (LiCor) diluted in 1:10,000 in 3% BSA/PBST for 45 min at room temperature. Visualization was performed using LiCor Odyssey Clx and quantification was done by Image Studio from LiCor.
Flash-and-freeze experiments
For flash-and-freeze experiments (Watanabe et al., 2013a, 2014). sapphire disks with cultured neurons (DIV14–15) were mounted in the freezing chamber of the high-pressure freezer (HPM100 or EM ICE, Leica), which was set at 37 °C. The physiological saline solution contained 140 mM NaCl, 2.4 mM KCl, 10 mM HEPES, 10 mM Glucose (pH adjusted to 7.3 with NaOH, 300 mOsm, 4 mM CaCl2, and 1 mM MgCl2. Additionally, NBQX (3 mM) and Bicuculline (30 mM) were added to suppress recurrent network activity following optogenetic stimulation of neurons. To minimize the exposure to room temperature, solutions were kept at 37 °C water bath prior to use. The table attached to the high-pressure freezer was heated to 37 °C while mounting specimens on the high-pressure freezer. The transparent polycarbonate sample cartridges were also warmed to 37 °C. Immediately after the sapphire disk was mounted on the sample holder, recording solution kept at 37 °C was applied to the specimen and the cartridge was inserted into the freezing chamber. The specimens were left in the chamber for 30 s to recover from the exposure to ambient light. We applied a single light pulse (10 ms) to the specimens (20 mW/mm2). This stimulation protocol was chosen based on the results from previous experiments showing approximately 90% of cells fire at least one action potential (Watanabe et al., 2013b). The non-stimulation controls for each experiment were always frozen on the same day from the same culture. We set the device such that the samples were frozen at 1 or 10 s after the initiation of the first stimulus. These time points are chosen because the majority of ultrafast endocytosis complete by 1s – under normal conditions ferritin particles are in endocytic vesicles or endosomes by this time point and subsequently in synaptic vesicles by ~10 s (Watanabe et al., 2013b, 2014). For ferritin-loading experiments, cationized ferritin (Sigma-Aldrich) was added in the saline solution at 0.25 mg/ml. The calcium concentration was reduced to 1mM to suppress spontaneous activity during the loading. The cells were incubated in the solution for 5 min at 37 °C. After ferritin incubation, the cells were immersed in the saline solution containing 4 mM Ca2+. For dynamin experiments, all samples were incubated with 1 μM sodium channel blocker tetrodotoxin (TTX) for overnight to block spontaneous network activity and reduce the number of pits arrested on the plasma membrane prior to flash-and-freeze experiments (Raimondi et al., 2011; Wu et al., 2014). On the next day, TTX was washed off thoroughly, and a fluid phase marker, cationized ferritin, was applied as described above.
Following high-pressure freezing, samples were transferred into vials containing 1% osmium tetroxide (EMS),1% glutaraldehyde (EMS), and 1% milliQ water, in anhydrous acetone (EMS). The freeze-substitution was performed in an automated freeze-substitution device (AFS2, Leica) with the following program: −90C for 5–7 h, 5C per hour to −20 °C, 12 hours at −20 °C, and 10 °C per hour to 20 °C. Following en bloc staining with 0.1% uranyl acetate and infiltration with plastic (30% for 3 hours, 70% for 4 hours, and 90% for overnight), the samples were embedded into 100% Epon-Araldite resin (Araldite 4.4 g, Epon 6.2 g, DDSA 12.2 g, and BDMA 0.8 ml) and cured for 48 hours in a 60 °C oven. Serial 40-nm sections were cut using a microtome (Leica) and collected onto pioloform-coated single-slot grids. Sections were stained with 2.5% uranyl acetate before imaging.
Electron microscopy
Ultrathin sections of samples were imaged at 80 kV on the Philips CM120 at 93,000x magnification or on the Hitachi 7600 at 150,000x. At these magnifications, one synapse essentially occupies the whole screen, and thus, with the bidirectional raster scanning of a section, it is difficult to pick certain synapses, reducing bias while collecting the data. In all cases, microscopists were additionally blinded to specimens and conditions of experiments. Both microscopes were equipped with an AMT XR80 camera, which is operated by AMT capture software v6. About 120 images per sample were acquired. If synapses do not contain a prominent postsynaptic density, they were excluded from the analysis – typically a few images from each sample fall into this category.
Analysis of electron micrographs
Electron micrographs were analyzed using SynpasEM. Briefly, images were pooled into a single folder from one set of experiments, randomized, and annotated blind. Using custom macros (Watanabe et al., 2020), the x- and y- coordinates of the following features were recorded in Fiji and exported as text files: plasma membrane, postsynaptic density, synaptic vesicles, large vesicles, endosomes, and pits. By visual inspection, large vesicles were defined as any vesicle with a diameter of 60–100 nm. Endosomes were distinguished by any circular structures larger than large vesicles or irregular membrane-bound structures that were not mitochondria or endoplamic reticulum. Late endosomes and multivesicular bodies were not annotated in this study. Pits were defined as smooth membrane invaginations within or next to the active zone, which were not mirrored by the postsynaptic membranes. After annotations, the text files were imported into Matlab (MathWorks). The number and locations of vesicles, pits, endosomes were calculated using custom scripts (Watanabe et al., 2020). The example micrographs shown were adjusted in brightness and contrast to different degrees, rotated and cropped in Adobe Photoshop (v21.2.1) or Illustrator (v24.2.3). Raw images and additional examples are provided in Figshare.com/projects/dynamin_phase_separation. The macros and Matlab scripts are available at https://github.com/shigekiwatanabe/SynapsEM.
Scientific Animation
The axon and dendrite were modeled using 3DEM images from previous studies as reference (Bromer et al., 2018; Harris et al., 2015). The bouton and its contents were modeled by referring to the 3D model of synaptic architecture (Wilhelm et al., 2014). The structure and composition of a synaptic vesicle were based on the molecular model generated by previous studies (Takamori et al., 2006; Tao et al., 2018). Structures of proteins in the animation were obtained from the following files from the Protein Data Bank - 2KOG for synaptobrevin (VAMP-2), 2R83 for synaptotagmin-1, 6BAT for glutamine transporter, 5VOX for V-ATPase, 1PK8 for synapsin I, 3RAB for RAB3A, 3C98 for syntaxin-1A, 6MDM for SNAP-25, 5GJV for voltage-gated calcium channel, 5W5C for complexin-1, 3C98 for munc18a, 5UE8 for munc13a, 4PE5 for NMDA receptor, 3KG2 for AMPA receptor, 2X3X for syndapin-1, 1ZWW and 3IQL for endophilin-A1, 1I9Z and 3LWT for synaptojanin-1, 6DLU, 6DLV and 5A3F for dynamin-1 and 3IYV for clathrin. The 3D structure file of glutamine was acquired from PubChem (CID 5961). Calcium and protons were represented as spheres. The axon, dendrite, bouton, the membranes, and the missing regions of proteins were modeled in the 3D graphics application, Autodesk Maya (https://www.autodesk.com/products/maya/). Surface meshes of protein structures were obtained from the molecular visualization application, UCSF Chimera (Pettersen et al., 2004), and imported into Autodesk Maya for animation. The overall ultrafast synaptic vesicle cycling process depicted in this animation was built on information from previous studies (Chanaday et al., 2019; Watanabe et al., 2013b). The dynamic movement of synaptic vesicles at the active zone was inferred from a previous study (Neher and Brose, 2018). The details of calcium triggered fusion mediated by SNAREs, including formation of a primed complex with synaptotagmin and complexin facilitated by Munc proteins, were derived from previous studies (Bai et al., 2016; Chang et al., 2018; Palfreyman and Jorgensen, 2017; Wang et al., 2019; Zhou et al., 2017). Research that was referred to for endocytic pit formation, membrane bending by syndapin and endophilin, synaptojanin hydrolysis and membrane fission by dynamin, are published in previous studies (Chang-Ileto et al., 2011; Chappie et al., 2011; Colom et al., 2017; Frost et al., 2008; Mahmood et al., 2019; Mim et al., 2012; Watanabe et al., 2018). Clathrin-mediated endosomal budding was referenced from previous studies (Morris et al., 2019; Paraan et al., 2020), and vATPase inactivity in the presence of a clathrin coat was based on findings from a previous study (Farsi et al., 2018). The animated scenes were rendered into a series of images and imported into the post production application, Adobe After Effects (https://www.adobe.com/products/aftereffects) for compositing, and including annotation and audio to create the final video.
Theoretical calculations of dynamin nucleation and polymerization kinetics
We estimated the concentration of Dyn1xA in the cytosol based on fluorescence from endogenous proteins (Figure 2G–J) and also the data from a previous study (Wilhelm et al., 2014). The concentration is about 3.5 μM at a presynaptic terminal. The average volume of a synaptic terminal is 0.37 μm3 (Wilhelm et al., 2014). Based on the area of Dyn1xA puncta (0.1586 μm2), we calculated the volume of the condensate to be 0.05 μm3. From these values, the concentration of Dyn1xA in the puncta was estimated to be 11.6 μM, which is close the critical concentration for dynamin oligomerization (12.6 μM). At this concentration, the time that takes to recruit dynamin to the membrane becomes negligible since there are a sufficient number of molecules near the membrane to drive rapid polymerization. Based on the previous experiment (Roux et al., 2010), the polymerization speed is expected to be ~ 361 nm/s with the concentration of 11.6 μM, assuming that dynamin polymerization if a first order reaction. Since dynamin pitch consists of 13 dimers of ~ 10–14 nm (Chen et al., 2004; Cocucci et al., 2014), it will likely take 28 – 39 ms to form one dynamin pitch around the collar of endocytic pits when the liquid condensate is present.
When the concentration of dynamin is low, the critical number (N*>1) of dynamin dimers must nucleate on the membrane before they can polymerize spontaneously. Hereby, the nucleation process is the rate-limiting step. According to the established theory (Lenz, 2009), the nucleation time can be determined analytically by relating the dynamin concentration (C), the critical number of dynamin dimer (N*), and membrane mechanics. Consider the scenario with sufficient dynamin dimers in solution that are in chemical equilibrium with the membrane-bound dynamin molecules. According to this theory, the analytic formula of the nucleation time can be derived in the two extreme limits, and the real nucleation time will reside in-between. In the limit #1, where the membrane is soft and the dynamin polymer is stiff, the nucleation time, . According to the above theory, in the limit #1, the nucleation time at 3.4 μM of dynamin will be ~ 257 ms if the critical number of dynamin dimer, N*, is 2 (i.e., dynamin tetrameter) (Ramachandran et al., 2007). In the limit #2, where the membrane is stiff and the dynamin polymer is soft, the nucleation time, Tnucleation is insensitive to the dynamin dimer concentration. The in vitro reconstitution experiments show that with 0.44 μM of dynamin in solution, the dynamin nucleation on a narrow membrane tubule (~ 10–35 nm in radius) takes ~ 2–5 seconds (Roux et al., 2010), which is the upper limit. Taken together, if the dynamin molecules do not form liquid droplets at a presynaptic terminal, the dynamin nucleation time alone is expected to range from ~ 257 ms to several seconds. An additional ~ 94–147 ms is necessary to polymerize dynamin around the collar once the nucleation initiates. Note that this estimated time is based on the optimal curvature-sensing condition that speeds up the membrane recruitment of dynamin. If the membrane curvature is less than optimal, then it will take even a longer time for dynamin to nucleate and polymerize on the membrane. Given that the ultrafast endocytosis completes within ~ 50–100 ms (Watanabe et al., 2013b), the nucleation of freely-diffusing dynamin molecules may not be sufficiently fast to drive the ultrafast endocytosis.
Quantification and statistical analysis
Detailed statistical information was summarized in Data S1. All EM data are pooled from multiple experiments after examined on a per-experiment basis (with all freezing on the same day); none of the pooled data show significant deviation from each replicate. Sample sizes were based on our prior flash-and-freeze experiments (~2–3 independent cultures, over 200 images), not power analysis. An alpha was set at 0.05 for statistical hypothesis testing. Normality was determined by D’Agostino-Pearson omnibus test. Comparisons between two groups were performed using a two-tailed Welch two-sample t-test if parametric or Wilcoxon rank-sum test and Mann-Whiteney Test if nonparametric. For groups, full pairwise comparisons were performed using one-way analysis of variance (ANOVA) followed by Holm-Šídák’s multiple comparisons test if parametric or Kruskal-Wallis test followed by Dunn’s multiple comparisons test if nonparametric. All statistical analyses were performed and all graphs created in Graphpad Prism (v8).
All fluorescence microscopy data were first examined on a per-experiment basis. For Figure 1B, 1D, 1F, 1H, 1J–N, 2C, 2D, 2F, 2N, 2L, 3D, 3E, 3I, 3J, 3M, 3N, 3P, 3Q, 4B, 4C, 4D, 4F, 4G, 4M, 4O–Q, 5C, 6C, 6D, 6G, 6J–M, 7C, 7D, 8B, 8D, 8F, 8H, 8J, 8K–N, S1A, S1C, S2B, S2D, S2F, S2H–2L, S4D–E, S5K, S6D, S6E, S6G, S6H, S6K, S6L, S6N, S6M, S7E, S7F, S8A–C, S8E, S8G, S9D, S9G–J, the data were pooled; none of the pooled data show significant deviation from each replicate. Sample sizes were ~2–3 independent cultures, at least 50 synapses from 4 different neurons in each condition. An alpha was set at 0.05 for statistical hypothesis testing. The skewness was determined by Pearson’s skewness test in GraphPad Prism (v8). Since data were all nonparametric, Mann–Whitney test or Kruskal–Wallis test were used. p-values in multiple comparison were adjusted with Bonferroni correction. Confidence levels were shown in each graph. All statistical analyses were performed, and all graphs created in Graphpad Prism (v8).
Supplementary Material
Data S1. List of statistics used in figures and supplemental figures.
Supplemental video 1. Molecular events occurring during exocytosis and ultrafast endocytosis of synaptic vesicles. Related to figure 1–8.
Supplemental table 1. List of primers used in this study. Related to START methods.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Synaptobrevin 2 antibody, mouse monoclonal | Synaptic Systems | Cat# 104 211 |
| anti-PACSIN1-1 antibody, rabbit polyclonal | Abcam | Cat# ab137390 |
| anti-PACSIN1-1 antibody, rabbit polyclonal | Synaptic Systems | Cat# 196 002 |
| anti-GFP antibody, rabbit polyclonal | MBL International | Cat# 598 |
| anti-GFP antibody, mouse polyclonal | Roche | Cat# 11814460001 |
| anti-Bassoon antibody, mouse monoclonal | Synaptic Systems | Cat# 141 011 |
| Rabbit IgG (H&L) Antibody ATTO 647N Conjugated Pre-Adsorbed | Rockland | Cat# 611-156-122S |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat# A11005 |
| Bacterial and virus strains | ||
| pLenti_f(syn)_hChR2_E123T-T159C-eYFP | (Watanabe et al., 2013) | N/A |
| pLenti_f(syn)-NLS-RFP-P2A-Dyn1xA | This paper | N/A |
| pLenti_f(syn)-NLS-RFP -P2A- Dyn1xA S774/778A | This paper | N/A |
| pLenti_f(syn)-NLS-RFP -P2A- Dyn1xA S774/778D | This paper | N/A |
| pLenti_f(syn)-NLS-RFP -P2A-Dyn1xB | This paper | N/A |
| pLenti_f(syn)-NLS-RFP -P2A-Dyn1xB S774/778D | N/A | |
| pLenti_f(syn)-NLS-RFP -P2A-Dyn2 | This paper | N/A |
| pLenti_f(U6) Scramble shRNA –(syn)-NLS-RFP-P2A-WPRE | (Watanabe et al., 2014) | |
| pLenti_f(U6) Syndapin shRNA –(syn)-NLS-RFP-P2A-WPRE | This paper | N/A |
| pLenti_f(syn)-NLS-GFP-P2A-Syndapin 1 | This paper | N/A |
| pLenti_f(syn)-NLS-GFP-P2A-Syndapin 1 shRNA resistant | This paper | N/A |
| pLenti_f(syn)-NLS-GFP-P2A-Syndapin 1 SH3 domain | This paper | N/A |
| pLenti_f(syn)-NLS-GFP-P2A-Syndapin 1 F-BAR domain | This paper | N/A |
| pLenti_f(syn)-NLS-GFP-P2A-Syndapin 1 P434L shRNA resistant | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trypsin-EDTA | Gibco | Cat# 25300-054 |
| DMEM | Gibco | Cat# 10569-044 |
| FBS | Gibco | Cat# 26140-079 |
| Penicillin-streptomycin | Gibco | Cat# 1540-122 |
| Poly-D-lysin hydrobromide | Sigma-Aldrich | Cat# P6407 |
| Poly-L-lysin hydrobromide | Sigma-Aldrich | Cat# P2636 |
| Rat tail collagen I | Gibco | Cat# A10483-01 |
| Acetic acid | Merck | Cat# 1.00063.1011 |
| 5-Fluoro-2′-deoxyuridine | Sigma-Aldrich | Cat# F0503 |
| Neurobasal A Medium | Gibco | Cat# 10888-022 |
| Neurobasal Medium | Gibco | Cat# 21103-049 |
| Glutamax supplement | Gibco | Cat# 35050-061 |
| B-27 plus supplement | Gibco | Cat# A35828-01 |
| Tris-HCL | Roth | Cat# 90903 |
| Cysteine | Sigma-Aldrich | Cat# C7352 |
| CaCl2 | Fluka | Cat# 21114 |
| tetrodotoxin | Tocris | Cat# 1078 |
| NaCl | Sigma-Aldrich | Cat# S7653 |
| polyethylenimine | Polysciences | Cat# 23966-2 |
| cOMPLETE protease inhibitor cocktail tablet | Roche | Cat# 11836153001 |
| HEPES | Merck | Cat# 1.10110.0250 |
| KCl | Sigma-Aldrich | Cat# P9333 |
| Glucose | Sigma-Aldrich | Cat# 49159 |
| MgCl2 | Fluka | Cat# 63020 |
| NBQX | Tocris | Cat# 1044 |
| Bicuculline | Tocris | Cat# 0130 |
| Cationized ferritin | Sigma-Aldrich | Cat# F7879 |
| Glutaraldehyde in anhydrous acetone | EMS | Cat# 16530 |
| Osmium tetroxide | EMS | Cat# RT19132 |
| Uranyl acetate | EMS | Cat# 22400 |
| Acetone | EMS | Cat# RT10016 |
| InFusion HD Cloning Kit | Thermal Scientific | Cat# 63649 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| 1,6-Hexanediol | Sigma-Aldrich | Cat# 240117 |
| 2,5-Hexanediol | Sigma-Aldrich | Cat# H11904 |
| 1,4-Butanediol | Sigma-Aldrich | Cat# 493732 |
| FK506 | Tocris | Cat# 3631 |
| CHIR99021 | Sigma-Aldrich | Cat# SML 1046 |
| ProLong Diamond Antifade Mountant | Thermo Fisher | Cat# P36971 |
| Gold Seal Microscope Slides, 3”×1”, 1mm thick | Thermo Fisher | Cat# 3010 |
| JFX549 | Luke Davis’s lab (Janelia Research Campus) (Grimm et al., 2016) | N/A |
| PA-JF646 | Luke Davis’s lab (Janelia Research Campus) (Grimm et al., 2016) | N/A |
| Experimental Models: Organisms/Strains | ||
| Dynamin-1, -3 DKO | De Camilli’s Lab (Raimondi et al., 2011) | N/A |
| Dynamin-3 KO | De Camilli’s Lab (Raimondi et al., 2011) | N/A |
| Recombinant DNA | ||
| f(syn)NLS-RFP-P2A-hsDyn1xA | This paper | N/A |
| f(syn)NLS-RFP-P2A-hsDyn1xA S774/778A | This paper | N/A |
| f(syn)NLS-RFP-P2A-hsDyn1xA S774/778D | This paper | N/A |
| f(syn)NLS-RFP-P2A-hsDyn1xB | This paper | N/A |
| f(syn)NLS-RFP-P2A-hsDyn1xB S774/778A | This paper | N/A |
| f(syn)NLS-RFP-P2A-hsDyn2 | This paper | N/A |
| hDyn1-S774A-S778A in pcDNA3 | (Armbruster et al., 2013) | N/A |
| hDyn1-S774D-S778D in pcDNA3 | (Armbruster et al., 2013) | N/A |
| HA-hDyn2 in pcDNA3 | (Ferguson et al., 2007) | N/A |
| phsDyn1xA-EGFP-N1 | (Kong et al., 2018) | Addgene #120313 |
| phsDyn1xA S774/778A- EGFP-N1 | This paper | N/A |
| phsDyn1xA S774/778D- EGFP-N1 | This paper | N/A |
| phsDyn1xA I481D/H687D/L688S- EGFP-N1 | This paper | N/A |
| phsDyn1xA-Halo | This paper | N/A |
| pmCherry-Syb2 | This paper | N/A |
| phsDyn1xA- EGFP-N1_P2A_mCherr-rtSyb2 | This paper | N/A |
| phsDyn1xB- EGFP-N1 | This paper | N/A |
| pDyn2-mCherry-N1 | (Taylor et al., 2011) | Addgene #27689 |
| pVAMP2(Syb2)-SNAP | (Martineau et al., 2017) | Addgene #105288 |
| pCAGGS-tdTomato | This paper | N/A |
| pCMV-tdTomato | Clontech | Cat# 632534 |
| pORANGE | (Willems et al., 2020) | Addgene #131471 |
| pORANGE-msDyn1xA-GFP | This paper | N/A |
| pFUGW-mCherry | This paper | N/A |
| pFUGW-mCherry_ORANGE_msDyn1xA-GFP | This paper | N/A |
| pEGFP-N1 | Clontech | Cat# 6085-1 |
| pmCherry-N1 | Clontech | Cat# 632523 |
| pFUGW-spCas9 | (Willems et al., 2020) | Addgene #131506 |
| f(U6)scramble shRNA.hSyn-NLS-RFP-WPRE | (Watanabe et al., 2014) | N/A |
| f(U6)Syndapin 1-shRNA.hSyn-NLS-RFP-WPRE | This paper | N/A |
| f(syn)NLS-RFP-P2A-Syndapin 1 | This paper | N/A |
| f(syn)NLS-RFP-P2A-Syndapin 1 shRNA resistant | This paper | N/A |
| f(syn)NLS-RFP-P2A-Syndapin 1 I122D/M123E shRNA resistant | This paper | N/A |
| f(syn)NLS-RFP-P2A-Syndapin 1 P434L shRNA resistant | This paper | N/A |
| pmCherry-N1_Syndapin 1 shRNA resistant | This paper | N/A |
| pmCherry-N1_Syndapin 1 I122D/M123E shRNA resistant | This paper | N/A |
| pmCherry-N1-Syndapin 1 P434L shRNA resistant | This paper | N/A |
| pGex6T-Dyn1xA/Dyn1xA-S774/778D | This paper | N/A |
| pGex6T-Syndapin1 | This paper | N/A |
| pGex6T-Syndapin1 P434L | This paper | N/A |
| pDyn1xA-PRM | (Anggono et al., 2006) | N/A |
| pDyn1xA-PRM S774/778A | (Anggono et al., 2006) | N/A |
| pDyn1xA-PRM S774/778E | (Anggono et al., 2006) | N/A |
| Software and Algorithms | ||
| SynapsEM | (Watanabe et al., 2020) | https://github.com/shigekiwatanabe/SynapsEM |
| ImaegJ-Fiji | (Schindelin et al., 2012) | https://imagej.net/Fiji |
| Adobe Illustrator 2021 | Adobe | https://www.adobe.com/ |
| Adobe Photoshop 2021 | Adobe | https://www.adobe.com/ |
| Prism | GraphPad (Prism 8) | https://www.graphpad.com |
| Excel | Microsoft | https://products.office.com/en-us/explore-office-for-home |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Microscopy | ||
| Hitachi 7600 TEM | Hitachi | N/A |
| LSM880 | Carl Zeiss | N/A |
| Nikon Eclipse Ti2 | Nikon | N/A |
| ORCA-Fusion BT Digital CMOS camera | Hamamatsu | N/A |
| Ti:Sapphire laser head | Mai Tai HP, Spectra-Physics | N/A |
| photonic crystal fiber | Newport | N/A |
| acoustic optical tunable filter | AA Opto-Electronic | N/A |
| 100-m long polarization-maintaining single-mode fiber | OZ optics | N/A |
| vortex phase plate | VPP-1, RPC photonics | N/A |
| avalanche photodiodes SPCM-AQR-14-FC | Perkin Elmer | N/A |
| piezo-controlled stage Max311D | Thorlabs | N/A |
| 405nm/250 mW laser | RPMC Lasers Inc. | N/A |
| 488nm/300 mW laser | MPB Communications | N/A |
| 560nm/500 mW laser | MPB Communications | N/A |
| 642nm/500 mW laser | MPB Communications | N/A |
| acoustic optical tunable filter | AA Opto-Electronic | N/A |
| galvo mirrors | Cambridge Technology | 6215H |
| CFI Apo LWD 25XW, 1.1 NA, 2 mm WD | Nikon | N/A |
| Orca Flash 4.0 v3 | Hamamatsu | N/A |
| Other | ||
| Sapphire Disks, 6-nm | Technotrade | Cat# 616-100 |
| AFS2 | Leica microsystem | N/A |
| EM ICE | Leica microsystem | N/A |
Highlights.
A splice variant of Dynamin 1, Dyn1xA, specifically mediate ultrafast endocytosis
Dyn1xA forms condensates with Syndapin 1 and pre-accumulates at endocytic zones
Syndapin 1 acts as a hub between Dyn1xA and the plasma membrane
Disruption of Dyn1xA condensates slows down the kinetics of endocytosis by 100-fold
Acknowledgements:
We thank Pietro De Camilli and Shawn Ferguson for sharing reagents and mice, Ira Milosevic for advice on antibodies, Geraldine Seydoux, Philip Robinson and Jie Xiao for discussion. We are also indebted to M. Delanoy, B. Smith and Hoku West-Foyle at the Johns Hopkins Microscopy Facility for technical assistance in electron and optical microscopy, Tyler Ogunmowo, Quan Gan and Sydney Brown for animal husbandry and the preparation of cells, Sebastian Markert and Yuta Nihongaki for the helpful discussion on in vitro protein assay, and Grant F. Kusick for editing of the manuscript, Kyu Young Han for the initial building of the STED microscope. S.W. and this work were supported by start-up funds from the Johns Hopkins University School of Medicine, Johns Hopkins Discovery funds, Johns Hopkins Catalyst award, the National Science Foundation (1727260), and the National Institutes of Health (1DP2 NS111133-01 and 1R01 NS105810-01A1) awarded to S.W, and German research council funded grants CRG958/A5, Exc257 and the Reinhard Koselleck project awarded to C.R.. S.W. is an Alfred P. Sloan fellow, a McKnight Foundation Scholar and a Klingenstein and Simons Foundation scholar. Y.I. was supported by JSPS. M.A.C. is supported by The Wellcome Trust (204954/Z/16/Z). E.M.J. is supported by the NIH grant NS034307, and is an Investigator of the Howard Hughes Medical Institute. T.I. and J.L. are supported by National Science Foundation (NSF 2148534).
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Aguet F, Antonescu CN, Mettlen M, Schmid SL, and Danuser G (2013). Advances in Analysis of Low Signal-to-Noise Images Link Dynamin and AP2 to the Functions of an Endocytic Checkpoint. Developmental Cell 26, 279–291. 10.1016/j.devcel.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Smillie KJ, Graham ME, Valova VA, Cousin MA, and Robinson PJ (2006). Syndapin I is the phosphorylation-regulated dynamin I partner in synaptic vesicle endocytosis. Nature Neuroscience 9, 752–760. 10.1038/nn1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, and Huganir RL (2015). Rapid Dispersion of SynGAP from Synaptic Spines Triggers AMPA Receptor Insertion and Spine Enlargement during LTP. Neuron 85, 173–189. 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster M, Messa M, Ferguson SM, De Camilli P, and Ryan TA (2013). Dynamin phosphorylation controls optimization of endocytosis for brief action potential bursts. ELife 2, e00845. 10.7554/eLife.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, and Chapman ER (2016). Different states of synaptotagmin regulate evoked versus spontaneous release. Nature Communications 7, 10971. 10.1038/ncomms10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromer C, Bartol TM, Bowden JB, Hubbard DD, Hanka DC, Gonzalez PV, Kuwajima M, Mendenhall JM, Parker PH, Abraham WC, et al. (2018). Long-term potentiation expands information content of hippocampal dentate gyrus synapses. PNAS 115, E2410–E2418. 10.1073/pnas.1716189115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Garcia F, and McNiven MA (1998). Differential Distribution of Dynamin Isoforms in Mammalian Cells. MBoC 9, 2595–2609. 10.1091/mbc.9.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L-S, Hansra G, Robinson PJ, and Graham ME (2010). Differential Phosphorylation of Dynamin I Isoforms in Subcellular Compartments Demonstrates the Hidden Complexity of Phosphoproteomes. J. Proteome Res. 9, 4028–4037. 10.1021/pr100223n. [DOI] [PubMed] [Google Scholar]
- Chanaday NL, Cousin MA, Milosevic I, Watanabe S, and Morgan JR (2019). The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 39, 8209–8216. 10.1523/JNEUROSCI.1158-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S, Trimbuch T, and Rosenmund C (2018). Synaptotagmin-1 drives synchronous Ca 2+ -triggered fusion by C 2 B-domain-mediated synaptic-vesicle-membrane attachment. Nature Neuroscience 21, 33–40. 10.1038/s41593-017-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Ileto B, Frere SG, Chan RB, Voronov SV, Roux A, and Di Paolo G (2011). Synaptojanin 1-Mediated PI(4,5)P2 Hydrolysis Is Modulated by Membrane Curvature and Facilitates Membrane Fission. Developmental Cell 20, 206–218. 10.1016/j.devcel.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappie JS, Mears JA, Fang S, Leonard M, Schmid SL, Milligan RA, Hinshaw JE, and Dyda F (2011). A Pseudoatomic Model of the Dynamin Polymer Identifies a Hydrolysis-Dependent Powerstroke. Cell 147, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B-C, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998. 10.1126/science.1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-J, Zhang P, Egelman EH, and Hinshaw JE (2004). The stalk region of dynamin drives the constriction of dynamin tubes. Nat Struct Mol Biol 11, 574–575. 10.1038/nsmb762. [DOI] [PubMed] [Google Scholar]
- Cheung G, and Cousin MA (2019). Synaptic vesicle generation from activity-dependent bulk endosomes requires a dephosphorylation-dependent dynamin-syndapin interaction. J Neurochem 151, 570–583. 10.1111/jnc.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, and Cousin MA (2009). The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. Journal of Neurochemistry 111, 901–914. 10.1111/j.1471-4159.2009.06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Anggono V, Smillie KJ, Chau N, Robinson PJ, and Cousin MA (2009). The Phospho-Dependent Dynamin–Syndapin Interaction Triggers Activity-Dependent Bulk Endocytosis of Synaptic Vesicles. J. Neurosci. 29, 7706–7717. 10.1523/JNEUROSCI.1976-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocucci E, Gaudin R, and Kirchhausen T (2014). Dynamin recruitment and membrane scission at the neck of a clathrin-coated pit. MBoC 25, 3595–3609. 10.1091/mbc.e14-07-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom A, Redondo-Morata L, Chiaruttini N, Roux A, and Scheuring S (2017). Dynamic remodeling of the dynamin helix during membrane constriction. PNAS 114, 5449–5454. 10.1073/pnas.1619578114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T, Mesa K, and Urrutia R (1996). Three Dynamin-Encoding Genes Are Differentially Expressed in Developing Rat Brain. Journal of Neurochemistry 67, 927–931. 10.1046/j.1471-4159.1996.67030927.x. [DOI] [PubMed] [Google Scholar]
- Delvendahl I, Vyleta NP, von Gersdorff H, and Hallermann S (2016). Fast, Temperature-Sensitive and Clathrin-Independent Endocytosis at Central Synapses. Neuron 90, 492–498. 10.1016/j.neuron.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmalingam E, Haeckel A, Pinyol R, Schwintzer L, Koch D, Kessels MM, and Qualmann B (2009). F-BAR Proteins of the Syndapin Family Shape the Plasma Membrane and Are Crucial for Neuromorphogenesis. J. Neurosci. 29, 13315–13327. 10.1523/JNEUROSCI.3973-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman J, and Ryan TA (2009). Molecular Circuitry of Endocytosis at Nerve Terminals. Annual Review of Cell and Developmental Biology 25, 133–160. 10.1146/annurev.cellbio.042308.113302. [DOI] [PubMed] [Google Scholar]
- Dubreuil MM, Morgens DW, Okumoto K, Honsho M, Contrepois K, Lee-McMullen B, Traber GM, Sood RS, Dixon SJ, Snyder MP, et al. (2020). Systematic Identification of Regulators of Oxidative Stress Reveals Non-canonical Roles for Peroxisomal Import and the Pentose Phosphate Pathway. Cell Reports 30, 1417–1433.e7. 10.1016/j.celrep.2020.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsi Z, Gowrisankaran S, Krunic M, Rammner B, Woehler A, Lafer EM, Mim C, Jahn R, and Milosevic I (2018). Clathrin coat controls synaptic vesicle acidification by blocking vacuolar ATPase activity. ELife 7, e32569. 10.7554/eLife.32569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, and De Camilli P (2012). Dynamin, a membrane-remodelling GTPase. Nature Reviews Molecular Cell Biology 13, 75–88. 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, Destaing O, Ko G, Takasaki J, Cremona O, et al. (2009). Coordinated Actions of Actin and BAR Proteins Upstream of Dynamin at Endocytic Clathrin-Coated Pits. Developmental Cell 17, 811–822. 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wölfel M, Collesi C, Giovedi S, Raimondi A, Gong L-W, Ariel P, Paradise S, et al. (2007). A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 316, 570–574. 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Frost A, Perera R, Roux A, Spasov K, Destaing O, Egelman EH, De Camilli P, and Unger VM (2008). Structural Basis of Membrane Invagination by F-BAR Domains. Cell 132, 807–817. 10.1016/j.cell.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal PP, Nirschl JJ, Klinman E, and Holzbaur ELF (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. PNAS 114, E2466–E2475. 10.1073/pnas.1614462114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, Anggono V, Bache N, Larsen MR, Craft GE, and Robinson PJ (2007). The in Vivo Phosphorylation Sites of Rat Brain Dynamin I. J. Biol. Chem. 282, 14695–14707. 10.1074/jbc.M609713200. [DOI] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott-Schwartz J, Liu Z, Lionnet T, et al. (2016). Bright photoactivatable fluorophores for single-molecule imaging. Nat Methods 13, 985–988. 10.1038/nmeth.4034. [DOI] [PubMed] [Google Scholar]
- Grimm JB, Tkachuk AN, Xie L, Choi H, Mohar B, Falco N, Schaefer K, Patel R, Zheng Q, Liu Z, et al. (2020). A general method to optimize and functionalize red-shifted rhodamine dyes. Nat Methods 17, 815–821. 10.1038/s41592-020-0909-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Xie L, Casler JC, Patel R, Tkachuk AN, Falco N, Choi H, Lippincott-Schwartz J, Brown TA, Glick BS, et al. (2021). A General Method to Improve Fluorophores Using Deuterated Auxochromes. JACS Au 1, 690–696. 10.1021/jacsau.1c00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han KY, and Ha T (2015). Dual-color three-dimensional STED microscopy with a single high-repetition-rate laser. Opt. Lett., OL 40, 2653–2656. 10.1364/OL.40.002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Spacek J, Bell ME, Parker PH, Lindsey LF, Baden AD, Vogelstein JT, and Burns R (2015). A resource from 3D electron microscopy of hippocampal neuropil for user training and tool development. Scientific Data 2, 150046. 10.1038/sdata.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman AA, and Brangwynne CP (2011). Beyond Stereospecificity: Liquids and Mesoscale Organization of Cytoplasm. Developmental Cell 21, 14–16. 10.1016/j.devcel.2011.06.013. [DOI] [PubMed] [Google Scholar]
- Hyman AA, Weber CA, and Jülicher F (2014). Liquid-Liquid Phase Separation in Biology. Annual Review of Cell and Developmental Biology 30, 39–58. 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Imig C, López-Murcia FJ, Maus L, García-Plaza IH, Mortensen LS, Schwark M, Schwarze V, Angibaud J, Nägerl UV, Taschenberger H, et al. (2020). Ultrastructural Imaging of Activity-Dependent Synaptic Membrane-Trafficking Events in Cultured Brain Slices. Neuron 108, 843–860.e8. 10.1016/j.neuron.2020.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto Y, Abe Y, Honsho M, Okumoto K, Ohnuma M, Kuroiwa H, Kuroiwa T, and Fujiki Y (2018). Onsite GTP fuelling via DYNAMO1 drives division of mitochondria and peroxisomes. Nature Communications 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Murata D, Kato T, Yamada T, Araki Y, Saito A, Adachi Y, Igarashi A, Li S, Pletnikov M, et al. (2019). Brain-specific Drp1 regulates postsynaptic endocytosis and dendrite formation independently of mitochondrial division. ELife 8, e44739. 10.7554/eLife.44739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karslake JD, Donarski ED, Shelby SA, Demey LM, DiRita VJ, Veatch SL, and Biteen JS (2021). SMAUG: Analyzing single-molecule tracks with nonparametric Bayesian statistics. Methods 193, 16–26. 10.1016/j.ymeth.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Sochacki KA, Wang H, Fang S, Canagarajah B, Kehr AD, Rice WJ, Strub M-P, Taraska JW, and Hinshaw JE (2018). Cryo-EM of the dynamin polymer assembled on lipid membrane. Nature 560, 258–262. 10.1038/s41586-018-0378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M, and Kaksonen M (2019). Phase separation of Ede1 promotes the initiation of endocytic events. BioRxiv 861203. 10.1101/861203. [DOI] [Google Scholar]
- Kroschwald S, Maharana S, Mateju D, Malinovska L, Nüske E, Poser I, Richter D, and Alberti S (2015). Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. ELife 4, e06807. 10.7554/eLife.06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladouceur A-M, Parmar BS, Biedzinski S, Wall J, Tope SG, Cohn D, Kim A, Soubry N, Reyes-Lamothe R, and Weber SC (2020). Clusters of bacterial RNA polymerase are biomolecular condensates that assemble through liquid–liquid phase separation. PNAS 117, 18540–18549. 10.1073/pnas.2005019117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz M (2009). Shaping tubes in cells. phdthesis. Université Pierre et Marie Curie - Paris VI. [Google Scholar]
- Liu JP, Powell KA, Südhof TC, and Robinson PJ (1994). Dynamin I is a Ca(2+)-sensitive phospholipid-binding protein with very high affinity for protein kinase C. J. Biol. Chem. 269, 21043–21050. [PubMed] [Google Scholar]
- Liu Y-W, Neumann S, Ramachandran R, Ferguson SM, Pucadyil TJ, and Schmid SL (2011). Differential curvature sensing and generating activities of dynamin isoforms provide opportunities for tissue-specific regulation. PNAS 108, E234–E242. 10.1073/pnas.1102710108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Ha T (2019). Fight against background noise in stimulated emission depletion nanoscopy. Phys. Biol. 16, 051002. 10.1088/1478-3975/ab255c. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, and Kirchhausen T (2006). Dynasore, a Cell-Permeable Inhibitor of Dynamin. Developmental Cell 10, 839–850. 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Mahmood MI, Noguchi H, and Okazaki K (2019). Curvature induction and sensing of the F-BAR protein Pacsin1 on lipid membranes via molecular dynamics simulations. Scientific Reports 9, 14557. 10.1038/s41598-019-51202-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Somasundaram A, Grimm JB, Gruber TD, Choquet D, Taraska JW, Lavis LD, and Perrais D (2017). Semisynthetic fluorescent pH sensors for imaging exocytosis and endocytosis. Nat Commun 8, 1412. 10.1038/s41467-017-01752-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen DT, Hansen AS, Teves SS, Marie-Nelly H, Hao Y, Heckert AB, Umemoto KK, Dugast-Darzacq C, Tjian R, and Darzacq X (2019). Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. ELife 8, e47098. 10.7554/eLife.47098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Feldman ME, Wan L, and Almers W (2002). Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nature Cell Biology 4, 691–698. 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Kay BK, and McPherson PS (1997). Synaptojanin forms two separate complexes in the nerve terminal. Interactions with endophilin and amphiphysin. J. Biol. Chem. 272, 27239–27245. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O’Toole E, Ferguson S, Cremona O, et al. (2011). Recruitment of Endophilin to Clathrin-Coated Pit Necks Is Required for Efficient Vesicle Uncoating after Fission. Neuron 72, 587–601. 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, and Unger VM (2012). Structural Basis of Membrane Bending by the N-BAR Protein Endophilin. Cell 149, 137–145. 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné-Hattab J, Heltberg M, Villemeur M, Guedj C, Mora T, Walczak AM, Dahan M, and Taddei A (2021). Single molecule microscopy reveals key physical features of repair foci in living cells. ELife 10, e60577. 10.7554/eLife.60577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MS, and Blobel G (1992). The two steps of nuclear import, targeting to the nuclear envelope and translocation through the nuclear pore, require different cytosolic factors. Cell 69, 939–950. 10.1016/0092-8674(92)90613-H. [DOI] [PubMed] [Google Scholar]
- Morris KL, Jones JR, Halebian M, Wu S, Baker M, Armache J-P, Avila Ibarra A, Sessions RB, Cameron AD, Cheng Y, et al. (2019). Cryo-EM of multiple cage architectures reveals a universal mode of clathrin self-assembly. Nature Structural & Molecular Biology 26, 890–898. 10.1038/s41594-019-0292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, and Brose N (2018). Dynamically Primed Synaptic Vesicle States: Key to Understand Synaptic Short-Term Plasticity. Neuron 100, 1283–1291. 10.1016/j.neuron.2018.11.024. [DOI] [PubMed] [Google Scholar]
- Neumann S, and Schmid SL (2013). Dual Role of BAR Domain-containing Proteins in Regulating Vesicle Release Catalyzed by the GTPase, Dynamin-2. J. Biol. Chem. 288, 25119–25128. 10.1074/jbc.M113.490474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfreyman MT, and Jorgensen EM (2017). Unc13 Aligns SNAREs and Superprimes Synaptic Vesicles. Neuron 95, 473–475. 10.1016/j.neuron.2017.07.017. [DOI] [PubMed] [Google Scholar]
- Paraan M, Mendez J, Sharum S, Kurtin D, He H, and Stagg SM (2020). The Structures of Natively Assembled Clathrin Coated Vesicles. BioRxiv 2020.01.28.923128. 10.1101/2020.01.28.923128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Wu Y, Lee S-E, Kim G, Jeong S, Milovanovic D, De Camilli P, and Chang S (2021). Cooperative function of synaptophysin and synapsin in the generation of synaptic vesicle-like clusters in non-neuronal cells. Nat Commun 12, 263. 10.1038/s41467-020-20462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Belmont BJ, Sante JM, and Rexach MF (2007). Natively Unfolded Nucleoporins Gate Protein Diffusion across the Nuclear Pore Complex. Cell 129, 83–96. 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Powell KA, Valova VA, Malladi CS, Jensen ON, Larsen MR, and Robinson PJ (2000). Phosphorylation of Dynamin I on Ser-795 by Protein Kinase C Blocks Its Association with Phospholipids. J. Biol. Chem. 275, 11610–11617. 10.1074/jbc.275.16.11610. [DOI] [PubMed] [Google Scholar]
- Praefcke GJK, and McMahon HT (2004). The dynamin superfamily: universal membrane tubulation and fission molecules? Nature Reviews Molecular Cell Biology 5, 133–147. [DOI] [PubMed] [Google Scholar]
- Pucadyil TJ, and Schmid SL (2008). Real-Time Visualization of Dynamin-Catalyzed Membrane Fission and Vesicle Release. Cell 135, 1263–1275. 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann B, Roos J, DiGregorio PJ, and Kelly RB (1999). Syndapin I, a Synaptic Dynamin-binding Protein that Associates with the Neural Wiskott-Aldrich Syndrome Protein. Mol Biol Cell 10, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondi A, Ferguson SM, Lou X, Armbruster M, Paradise S, Giovedi S, Messa M, Kono N, Takasaki J, Cappello V, et al. (2011). Overlapping Role of Dynamin Isoforms in Synaptic Vesicle Endocytosis. Neuron 70, 1100–1114. 10.1016/j.neuron.2011.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Surka M, Chappie JS, Fowler DM, Foss TR, Song BD, and Schmid SL (2007). The dynamin middle domain is critical for tetramerization and higher-order self-assembly. EMBO J 26, 559–566. 10.1038/sj.emboj.7601491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Ma Q, Vahedi-Faridi A, Sundborger A, Pechstein A, Puchkov D, Luo L, Shupliakov O, Saenger W, and Haucke V (2010). Molecular basis for SH3 domain regulation of F-BAR–mediated membrane deformation. PNAS 107, 8213–8218. 10.1073/pnas.1003478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Gad H, Löw P, Di Paolo G, Brodin L, Shupliakov O, and De Camilli P (1999). Endophilin/SH3p4 Is Required for the Transition from Early to Late Stages in Clathrin-Mediated Synaptic Vesicle Endocytosis. Neuron 24, 143–154. 10.1016/S0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Roux A, Koster G, Lenz M, Sorre B, Manneville J-B, Nassoy P, and Bassereau P (2010). Membrane curvature controls dynamin polymerization. PNAS 107, 4141–4146. 10.1073/pnas.0913734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, and De Camilli P (2012). Synaptic Vesicle Endocytosis. Cold Spring Harb Perspect Biol 4. 10.1101/cshperspect.a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapoznik E, Chang B-J, Huh J, Ju RJ, Azarova EV, Pohlkamp T, Welf ES, Broadbent D, Carisey AF, Stehbens SJ, et al. (2020). A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics. ELife 9, e57681. 10.7554/eLife.57681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpetner HS, and Vallee RB (1989). Identification of dynamin, a novel mechanochemical enzyme that mediates interactions between microtubules. Cell 59, 421–432. 10.1016/0092-8674(89)90027-5. [DOI] [PubMed] [Google Scholar]
- Soykan T, Kaempf N, Sakaba T, Vollweiter D, Goerdeler F, Puchkov D, Kononenko NL, and Haucke V (2017). Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 93, 854–866.e4. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Burckhardt CJ, Bhave M, Chen Z, Chen P-H, Wang X, Danuser G, and Schmid SL (2018). A noncanonical role for dynamin-1 in regulating early stages of clathrin-mediated endocytosis in non-neuronal cells. PLOS Biology 16, e2005377. 10.1371/journal.pbio.2005377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone HA (1994). Dynamics of Drop Deformation and Breakup in Viscous Fluids. Annual Review of Fluid Mechanics 26, 65–102. 10.1146/annurev.fl.26.010194.000433. [DOI] [Google Scholar]
- Stowell MHB, Marks B, Wigge P, and McMahon HT (1999). Nucleotide-dependent conformational changes in dynamin: evidence for a mechanochemical molecular spring. Nature Cell Biology 1, 27–32. 10.1038/8997. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. (2006). Molecular Anatomy of a Trafficking Organelle. Cell 127, 831–846. 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Tao C-L, Liu Y-T, Sun R, Zhang B, Qi L, Shivakoti S, Tian C-L, Zhang P, Lau P-M, Zhou ZH, et al. (2018). Differentiation and Characterization of Excitatory and Inhibitory Synapses by Cryo-electron Tomography and Correlative Microscopy. J. Neurosci. 38, 1493–510. 10.1523/JNEUROSCI.1548-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Perrais D, and Merrifield CJ (2011a). A High Precision Survey of the Molecular Dynamics of Mammalian Clathrin-Mediated Endocytosis. PLOS Biology 9, e1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Perrais D, and Merrifield CJ (2011b). A High Precision Survey of the Molecular Dynamics of Mammalian Clathrin-Mediated Endocytosis. PLOS Biology 9, e1000604. 10.1371/journal.pbio.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Lampe M, and Merrifield CJ (2012). A Feedback Loop between Dynamin and Actin Recruitment during Clathrin-Mediated Endocytosis. PLOS Biology 10, e1001302. 10.1371/journal.pbio.1001302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J-Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh T-W, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, and Bellen HJ (2003). Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40, 733–748. [DOI] [PubMed] [Google Scholar]
- Wang S, Li Y, Gong J, Ye S, Yang X, Zhang R, and Ma C (2019). Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nature Communications 10, 69. 10.1038/s41467-018-08028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Liu Q, Davis MW, Hollopeter G, Thomas N, Jorgensen NB, and Jorgensen EM (2013a). Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. Elife 2, e00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Rost BR, Camacho-Pérez M, Davis MW, Söhl-Kielczynski B, Rosenmund C, and Jorgensen EM (2013b). Ultrafast endocytosis at mouse hippocampal synapses. Nature 504, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Trimbuch T, Camacho-Pérez M, Rost BR, Brokowski B, Söhl-Kielczynski B, Felies A, Davis MW, Rosenmund C, and Jorgensen EM (2014). Clathrin regenerates synaptic vesicles from endosomes. Nature 515, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Mamer LE, Raychaudhuri S, Luvsanjav D, Eisen J, Trimbuch T, Söhl-Kielczynski B, Fenske P, Milosevic I, and Rosenmund C (2018). Synaptojanin and endophilin mediate neck formation during ultrafast endocytosis. Neuron 98, 1184–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Davis MW, Kusick GF, Iwasa J, and Jorgensen EM (2020). SynapsEM: Computer-Assisted Synapse Morphometry. Front. Synaptic Neurosci. 12. 10.3389/fnsyn.2020.584549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Fang H, Jang SE, and Anggono V (2016). PACSIN1 regulates the dynamics of AMPA receptor trafficking. Scientific Reports 6, 31070. 10.1038/srep31070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Lee C-W, Erdmann PS, Zheng Y, Sherpa D, Jentsch S, Pfander B, Schulman BA, and Baumeister W (2020). A Selective Autophagy Pathway for Phase-Separated Endocytic Protein Deposits. Molecular Cell 80, 764–778.e7. 10.1016/j.molcel.2020.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BG, Mandad S, Truckenbrodt S, Kröhnert K, Schäfer C, Rammner B, Koo SJ, Claßen GA, Krauss M, Haucke V, et al. (2014). Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 344, 1023–1028. 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- Willems J, Jong A.P.H. de, Scheefhals N, Mertens E, Catsburg LAE, Poorthuis RB, Winter F. de, Verhaagen J, Meye FJ, and MacGillavry HD (2020). ORANGE: A CRISPR/Cas9-based genome editing toolbox for epitope tagging of endogenous proteins in neurons. PLOS Biology 18, e3000665. 10.1371/journal.pbio.3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, O’Toole ET, Girard M, Ritter B, Messa M, Liu X, McPherson PS, Ferguson SM, and De Camilli P (2014). A dynamin 1-, dynamin 3- and clathrin-independent pathway of synaptic vesicle recycling mediated by bulk endocytosis. ELife 3, e01621. 10.7554/eLife.01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Graham ME, Novelle AE, Sue N, Gray N, McNiven MA, Smillie KJ, Cousin MA, and Robinson PJ (2011). Calcineurin Selectively Docks with the Dynamin Ixb Splice Variant to Regulate Activity-dependent Bulk Endocytosis. J. Biol. Chem. 286, 30295–30303. 10.1074/jbc.M111.273110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Südhof TC, and Brunger AT (2017). The primed SNARE–complexin–synaptotagmin complex for neuronal exocytosis. Nature 548, 420–425. 10.1038/nature23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. List of statistics used in figures and supplemental figures.
Supplemental video 1. Molecular events occurring during exocytosis and ultrafast endocytosis of synaptic vesicles. Related to figure 1–8.
Supplemental table 1. List of primers used in this study. Related to START methods.
Data Availability Statement
All original data have been deposited at Mendeley and publicly available as of the date of publication (https://data.mendeley.com/datasets/bkn78y232y/draft?preview=1).
All original code is available from the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request