

Abstract
Merck & Co. recently reported one of the first mRNA display-derived clinical candidates in a bioavailable inhibitor of proprotein convertase subtilisin/kexin type 9 (PCSK9). Herein, we discuss the chemical and pharmacological challenges surmounted in bringing this compound to trials and the current outlook for mRNA display-based therapeutic development.
Keywords: mRNA display, peptide therapeutics, macrocyclic peptides, noncanonical amino acids, proprotein convertase subtilisin/kexin type 9, hypercholesterolemia
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a well-characterized serine protease implicated in the progression of hypercholesterolemia and multiple cardiovascular diseases.1,2 Specifically, PCSK9 regulates levels of low-density lipoprotein receptors (LDLRs) on hepatocytes, which natively bind LDL cholesterol (LDLC) for uptake and breakdown by the liver.1 PCSK9-bound LDLRs, however, are removed via lysosomal degradation3 (Figure 1A); reduced LDLR levels correlate directly with reduced LDLC metabolism, contributing to hypercholesterolemia. Inhibition of PCSK9 allows LDLR recycling (Figure 1B) and thereby increases LDLC metabolism.4
Figure 1.
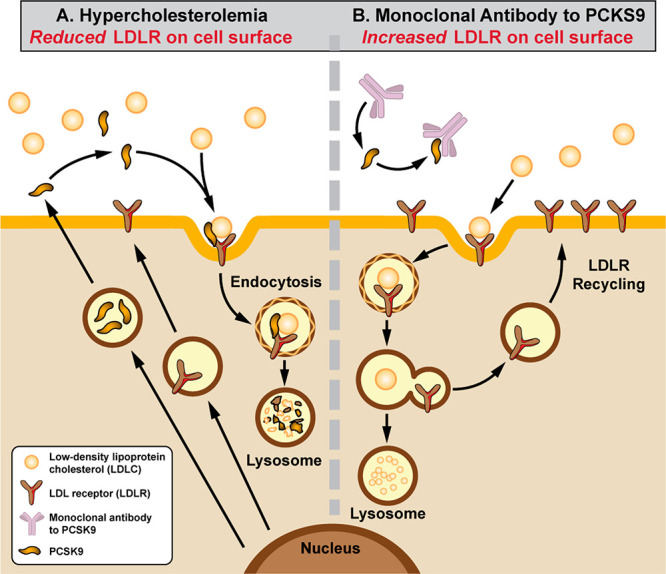
PCSK9 impacts LDLC metabolism by regulating LDLR levels. (A) PCSK9 binds LDLR on hepatocyte surfaces, triggering their internalization and degradation by the lysosome. (B) In the presence of a PCSK9 inhibitor (e.g., monoclonal antibody), the prevalence of LDLR increases, leading to improved LDLC metabolism. Adapted with permission from ref (4). Copyright 2022 Mayo Foundation for Medical Education and Research.
PCSK9 is well-suited for targeting by larger biomolecules because it is an extracellular protein, circulated in blood. To date, three biologics—two monoclonal antibodies (mAbs) and one siRNA—have been FDA-approved for PCSK9 inhibition. The two mAbs, alirocumab and evolocumab, are both effective in reducing LDLC levels in a variety of populations with hypercholesterolemia5 and in reducing cardiovascular events6−8 when combined with statin therapy. However, they are expensive (first $14 000/year,9 now $5850/year for alirocumab and $6056/year for evolocumab after pricing pressures) and require biweekly or monthly subcutaneous delivery, factors which have likely prevented the widespread adoption that was initially expected of these therapies. The more recently approved siRNA inclisiran produces sustained LDLC reduction 6 six months after one dose,10 thus requiring only two doses a year (three in the first year). However, its price still rivals that of the mAb treatments, at $3250 per dose, and it is still administered by injection. Several other biologics are in clinical development for PCSK9 inhibition, including a fusion protein containing an anti-PCSK9 binding domain11 and an orally available antisense oligonucleotide.12,13 Additionally, an anti-PCSK9 vaccine,14 CRISPR adenine base editors,15,16 and a variety of small molecules and peptidomimetics17−19 are in the pre-clinical pipeline. Currently, however, there is no orally bioavailable FDA-approved therapy for the treatment of hypercholesterolemia via PCSK9 inhibition.
Merck & Co. recently disclosed details on the development of an orally bioavailable peptide-based inhibitor of PCSK9 derived from an mRNA display selection.20,21 mRNA display enriches for high-affinity peptide ligands of protein targets from very large genetically encoded libraries. It is particularly useful in identifying inhibitors of protein–protein interactions,22,23 since peptides can extend over significantly larger surface areas than small molecules. Additionally, unprecedented library diversity is routinely achieved through the in vitro translation of natural and unnatural amino acids and through post-translational chemical modifications.24,25 Indeed, as an in vitro selection technique, mRNA display is uniquely equipped to access chemical diversity that far exceeds that possible with other cell-based display technologies, such as phage or yeast display.
Despite the demonstrated ability of mRNA display to quickly identify potent peptide ligands of protein targets, this PCSK9 inhibitor is one of the first clinical candidates to result from an mRNA display campaign. Herein we will summarize the steps that proved necessary to transform the initial mRNA display hit into an orally bioavailable clinical candidate, and we will briefly discuss the future potential and remaining challenges for mRNA display as a tool for development of peptide therapeutics.
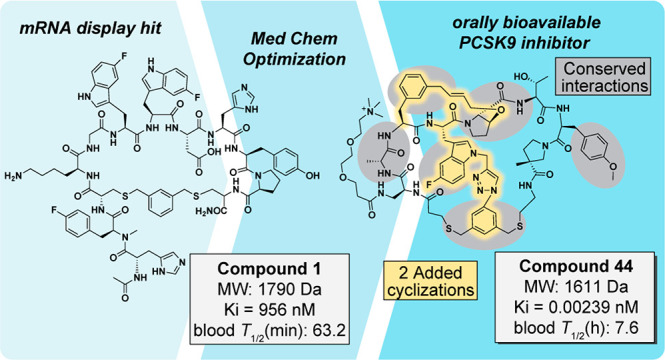
Merck’s initial mRNA display selection was done in collaboration with Ra Pharmaceuticals, one of the first mRNA display biotech companies, and screened 7–12mer peptide libraries flanked by cysteine residues that were chemically cyclized via dibromoxylene (DBX) alkylation. Of the many robust cyclization strategies in display technologies,25 DBX chemistry is one with well-documented success in both phage and mRNA display.26,27 Upon validation of a successful selection with initial hit compound 1 (Figure 2a, all numbering based on the original paper, ref (20)) in an LDLR FRET assay, a medicinal chemistry campaign was initiated to improve potency, stability, specificity, and bioavailability. Figure 2 summarizes the rationale behind the major modifications made at each residue to arrive at the optimized lead, compound 44, as well as key functional groups within this compound that make important contacts with PCSK9.
Figure 2.
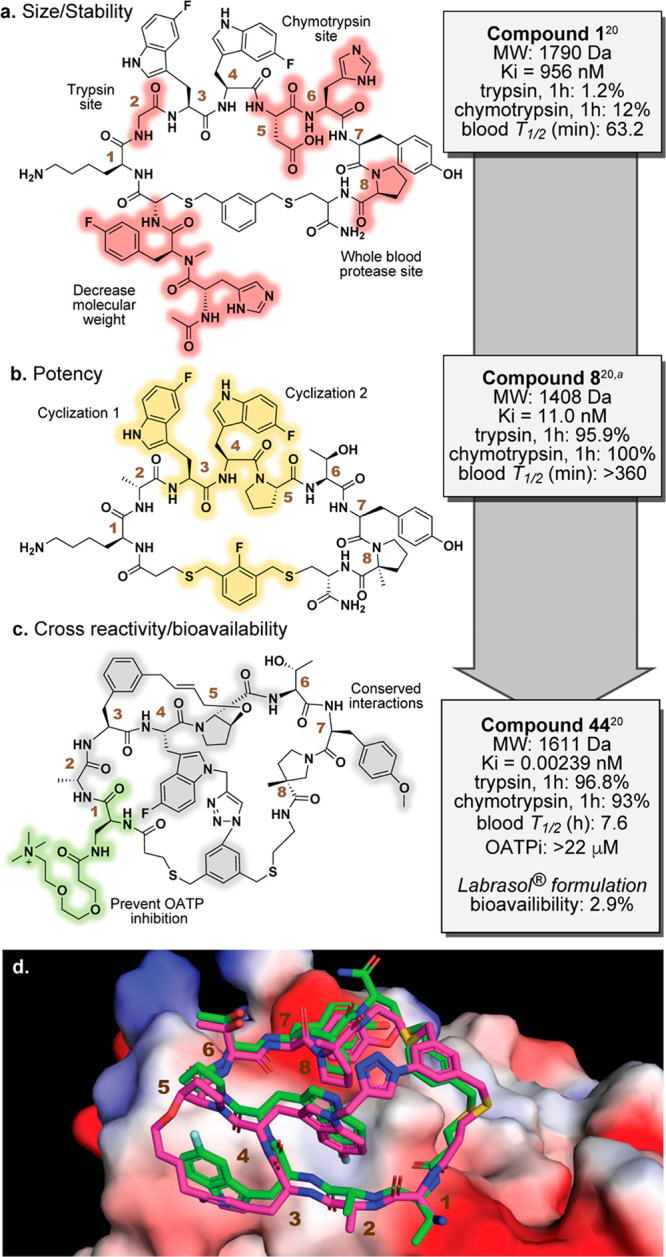
Development of macrocyclic PCSK9 inhibitor from mRNA display hit compound 1 to clinically relevant analog compound 44 via intermediate analog 8. (a–c) Medicinal chemistry efforts improved potency, proteolytic stability, and pharmacokinetic properties such as bioavailability, target specificity, and immunogenicity. Key alterations made to compound 1 to arrive at compound 44 are highlighted and summarized. (d) Overlay of structures of compounds 8 (green, PDB: 6XID) and 44 (magenta, PDB: 7S5H) bound to PCSK9, showing the close conformational conservation of binding poses between the mono- and tricyclic compounds. aStability data are for the reported homolog closest to compound 8.
One of the first changes involved reducing the size and complexity of the molecule by removing the N-terminal tail, which also improved the potency by almost 9-fold (Ki = 110 nM). Significant efforts were then made to improve the resistance against gut and whole blood proteases. Metabolite identification studies revealed a susceptible site between the Pro8 and Cys9 residues. Addition of a methyl group at the proline α position relieved this vulnerability while simultaneously bringing potency into the low nanomolar range (Ki = 4.2 nM). This drastic improvement may be rationalized by added rigidification from the new quaternary carbon, reducing the entropic cost of adopting the binding conformation. Subsequent removal of the C-terminal portion of Cys9 maintained potency while eliminating a hydrogen bond donor/acceptor pair. Another protease-susceptible site at Lys1 was alleviated by replacing Gly2 with a d-Ala residue.
With potency now in the low nanomolar range, crystal structures for many of the subsequent derivatives became much more feasible. The first structure revealed that the macrocycle adopted a “donut-like” conformation, with the fluorine of 5F-Trp at position 4 extending from the center of the donut and anchoring the peptide through several interactions with a shallow pocket on the relatively flat protein surface. Notably, subsequent analog work demonstrated the specificity of this interaction by the aryl fluoride, highlighting the importance of the unnatural amino acid choice in mRNA display selections. Several areas of the peptide backbone, including the amine and carbonyl groups of the position 2 residue, the amine of 5F-Trp3, the carbonyl of 5F-Trp4, and the backbone carbonyl Asp5, make important hydrogen bonds with PCSK9. Tyr6 forms hydrophobic contacts with another shallow binding pocket, while the DBX linker sits directly atop a hydrophobic Ile residue (Figure 2d). This initial crystal structure also revealed solvent-exposed sites that would serve as handles to improve potency, stability, and other pharmacokinetic (PK) properties.
Resistance to degradation by gut proteases, especially chymotrypsin, remained a concern due to the necessity of the 5F-Trp4 for potency. Replacing the residues immediately following 5F-Trp4 with Pro-Thr introduced a turn-inducing tertiary amine that helped substantially on this front. Still further improvements in chymotrypsin stability and significant leaps in potency were achieved through the rather bold addition of two new macrocyclic linkages. New structures revealed that the indole ring of 5F-Trp3 and the beta carbon of newly installed Pro5 might be appropriately positioned to allow cross-linking. Thus, 5F-Trp3 was replaced with m-allyl-Phe, and an o-allyl group was added to the beta carbon of Pro5; cyclization via olefin cross-metathesis to the trans-alkene resulted in a 10-fold increase in potency, breaking into the sub-nanomolar range (Ki = 0.39 nM). A crystal structure of this bicycle showed no new contacts with PCSK9, suggesting that potency gain was likely entropic. Structures also suggested that another linkage might be possible between the indole ring of 5F-Trp4 and the DBX linker. These two positions were joined through a click cyclization between the indole nitrogen and the meta-DBX aryl ring, installing a polar triazole. Gratifyingly, the now tricyclic compound demonstrated a >40-fold improvement in potency (Ki = 0.0094 nM). Structures again showed these improvements to be entropic, speaking to the power of conformational rigidity in peptide–protein interactions. Notably, click cyclization alone did not cause a drastic potency increase (Ki = 1.36 nM), suggesting a clear synergistic stabilization between the click and metathesis reactions.
Remaining analogs were aimed at improving PK parameters such as oral bioavailability, clearance, immunogenicity, and specificity. Two important off-target activities observed in earlier analogs were inhibition of a hepatic organic anion transporter protein (OATP), a known contributor to renal secretion and clearance, and mast cell degranulation, which has been implicated in pseudoallergic drug reactions.28−30 The authors noted that, in previous empirical evidence, they had observed (1) a direct correlation between lipophilicity and OATP inhibition and (2) lysine basicity contributing to off-target effects such as mast cell degranulation and histamine release. Lys1 proved key in efforts to circumvent both off-target activities while also improving bioavailability, as it was completely solvent exposed and could be readily manipulated without perturbing other interactions. Thus, a PEG linker equipped with a trimethylammonium group was installed at Lys1 to arrive at the late-stage analog compound 44 (Figure 2c). Pleasingly, this removed both OATP inhibition and degranulation concerns while maintaining the desired potency (Ki = 0.00239 nM). When dosed with the excipient Labrasol in rats, compound 44 showed 2.7% bioavailability, low clearance, and a reasonable half-life of 4.6 h. Similar lipoidal excipients have previously been used for oral delivery of the GLP-1 analog semaglutide in the treatment of type 2 diabetes,31 showing the prior utility of emulsion systems in achieving oral peptide therapeutics. Indeed, the excipient was critical in achieving desired pharmacological effects in this campaign, as compound 44 did not show any bioavailability when dosed intraduodenally with standard vehicles.
The overall goal of this campaign was to develop a lead molecule with antibody-like affinity that demonstrated PCSK9 and LDLC reduction comparable to the standard of care (>80% reduction of PCSK9 and >50% reduction of LDLC). Having achieved the target potency, compound 44 was dosed intravenously (0.2 mg/kg) into cynomolgus monkeys to effect >50% LDLC reduction at 48 h, low clearance, and a 7.6 h half-life. When 44 was orally dosed with Labrasol (1 mg/kg), >80% reduction of PCSK9 was observed between 30 min and 6 h, with 60% reduction observed at 24 h. Due to its exquisite potency, oral bioavailability was satisfactory at 2.9%.
At an American Heart Association meeting in late 2021, Merck & Co. reported promising data from a phase I clinical trial with their final candidate, MK-0616. When dosed in healthy men, MK-0616 reduced PCSK9 blood levels by >90%, with no serious side effects. When added to an existing statin therapy for 2 weeks in men and women with high cholesterol, MK-0616 decreased LDLC levels by ∼65%. With this encouraging data, phase II studies are currently underway in patients with renal impairment.
MK-0616 marks one of the first clinical candidates born from an mRNA display selection. The initial hit compound binds a large but relatively shallow surface of PCSK9, reinforcing the utility of peptides as therapeutics against proteins with superficial binding pockets. Notably, the selection campaign incorporated several unnatural amino acids into its libraries, including what proved to be an indispensable 5F-Trp residue. This exemplifies the advantage and even necessity of diversifying display libraries beyond canonical amino acids to increase the likelihood of successful selections.
Yet, the journey from the initial mRNA display hit to final clinical candidate certainly required extensive efforts. Like many small-molecule hit-to-lead optimization campaigns, substantial medicinal chemistry was required to arrive at a lead compound with desired potency, clearance, and selectivity. However, unlike small molecules, additional factors such as proteolytic stability, immunogenicity, and bioavailability were also tackled in a specific fashion during the development campaign. Additionally, and in further contrast to small molecules, potency improvements were not caused by new interactions between peptide analogs and PCSK9. Instead, the picomolar potency of compound 44 was largely achieved by reducing the entropic cost required to reach the correct peptide binding conformation, while intermolecular interactions remained mostly unchanged. Indeed, the addition of two stabilizing cyclizations alone synergistically increased the potency by almost 500-fold (from Ki = 4.2 nM to Ki = 0.0094 nM). Importantly, dosing 44 with Labrasol granted the minimal but crucial bioavailability required for oral delivery of this therapeutic. 44 showed no intraduodenal bioavailability with standard vehicles, a common challenge facing many peptide inhibitors, so its picomolar affinity was essential to achieve the desired pharmacological response with the nominal bioavailability it was awarded.
This development campaign details one of the first instances where an mRNA display hit was successfully developed into an orally bioavailable clinical candidate. Exhaustive medicinal chemistry, reliance on excipient strategies, and superb inhibitor potency were all imperative factors for success and thus may prove to be standard features in future oral peptide therapeutic development. To potentially minimize these considerable efforts, however, it will be fruitful to continue endeavors in creating chemically diverse mRNA display libraries, such as those with increased proteolytic resistance or lipophilicity. Enhanced bioavailability may also be achieved by creating smaller macrocycles with extensively reprogrammed genetic codes to maximize library diversity within a small number of residues. These types of advances will likely be crucial in making mRNA display a reliable tool for the quick identification of clinically relevant peptide ligands.
Acknowledgments
S.E.I. and A.A.B. were supported by a grant from NIGMS (NIH 1R35GM125005).
Glossary
Abbreviations
- PCSK9
proprotein convertase subtilisin/kexin type 9
- LDLR
low-density lipoprotein receptor
- LDLC
low-density lipoprotein cholesterol
- mAb
monoclonal antibody
- DBX
dibromoxylene
- FRET
fluorescence resonance energy transfer
- OATP
organic anion transporter protein
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
References
- Seidah N. G.; Awan Z.; Chrétien M.; Mbikay M. PCSK9: A Key Modulator of Cardiovascular Health. Circ. Res. 2014, 114 (6), 1022–1036. 10.1161/CIRCRESAHA.114.301621. [DOI] [PubMed] [Google Scholar]
- Norata G. D.; Tibolla G.; Catapano A. L. Targeting PCSK9 for Hypercholesterolemia. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 273–93. 10.1146/annurev-pharmtox-011613-140025. [DOI] [PubMed] [Google Scholar]
- Lagace T. A. PCSK9 and LDLR Degradation: Regulatory Mechanisms in Circulation and in Cells. Curr. Opin. Lipidol. 2014, 25 (5), 387–393. 10.1097/MOL.0000000000000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PCSK9 inhibition: A game changer in cholesterol management. Mayo Clinic, Nov 20, 2015.https://www.mayoclinic.org/medical-professionals/cardiovascular-diseases/news/pcsk9-inhibition-a-game-changer-in-cholesterol-management/mac-20430713
- McDonagh M.; Peterson K.; Holzhammer B.; Fazio S. A Systematic Review of PCSK9 Inhibitors Alirocumab and Evolocumab. J. Manag. Care Spec. Pharm. 2016, 22 (6), 641–653. 10.18553/jmcp.2016.22.6.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarek M.; White H. D.; Schwartz G. G.; Alings M.; Bhatt D. L.; Bittner V. A.; Chiang C. E.; Diaz R.; Edelberg J. M.; Goodman S. G.; Hanotin C.; Harrington R. A.; Jukema J. W.; Kimura T.; Kiss R. G.; Lecorps G.; Mahaffey K. W.; Moryusef A.; Pordy R.; Roe M. T.; Tricoci P.; Xavier D.; Zeiher A. M.; Steg P. G. Alirocumab Reduces Total Nonfatal Cardiovascular and Fatal Events: The ODYSSEY OUTCOMES Trial. J. Am. Coll. Cardiol. 2019, 73 (4), 387–396. 10.1016/j.jacc.2018.10.039. [DOI] [PubMed] [Google Scholar]
- Schwartz G. G.; Steg P. G.; Szarek M.; Bhatt D. L.; Bittner V. A.; Diaz R.; Edelberg J. M.; Goodman S. G.; Hanotin C.; Harrington R. A.; Jukema J. W.; Lecorps G.; Mahaffey K. W.; Moryusef A.; Pordy R.; Quintero K.; Roe M. T.; Sasiela W. J.; Tamby J.-F.; Tricoci P.; White H. D.; Zeiher A. M. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379 (22), 2097–2107. 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- Sabatine M. S.; Giugliano R. P.; Keech A. C.; Honarpour N.; Wiviott S. D.; Murphy S. A.; Kuder J. F.; Wang H.; Liu T.; Wasserman S. M.; Sever P. S.; Pedersen T. R. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376 (18), 1713–1722. 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- Nasir K. Just Price for PCSK9 Inhibitors: No Less, No More. J. Am. Heart Assoc. 2018, 7 (21), 1–3. 10.1161/JAHA.118.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K. K.; Landmesser U.; Leiter L. A.; Kallend D.; Dufour R.; Karakas M.; Hall T.; Troquay R. P. T.; Turner T.; Visseren F. L. J.; Wijngaard P.; Wright R. S.; Kastelein J. J. P. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376 (15), 1430–1440. 10.1056/NEJMoa1615758. [DOI] [PubMed] [Google Scholar]
- Stein E. A.; Turner T.; Butcher B.; Mangu P.; Kereiakes D.; Zhou R.; Fu R.; Mitchell T.; Mitchell T. Efficacy and Safety of a 52 Week Open Label Phase 2B Extension Trial With Monthly Dosing of an Anti-Pcsk9 Small Binding Protein. J. Am. Coll. Cardiol. 2020, 75 (11), 2077. 10.1016/S0735-1097(20)32704-2. [DOI] [Google Scholar]
- Gennemark P.; Walter K.; Clemmensen N.; Rekić D.; Nilsson C. A. M.; Knöchel J.; Hölttä M.; Wernevik L.; Rosengren B.; Kakol-Palm D.; Wang Y.; Yu R. Z.; Geary R. S.; Riney S. J.; Monia B. P.; Isaksson R.; Jansson-Löfmark R.; Rocha C. S. J.; Lindén D.; Hurt-Camejo E.; Crooke R.; Tillman L.; Rydén-Bergsten T.; Carlsson B.; Andersson U.; Elebring M.; Tivesten A.; Davies N. An Oral Antisense Oligonucleotide for PCSK9 Inhibition. Sci. Transl. Med. 2021, 13 (593), 1–13. 10.1126/scitranslmed.abe9117. [DOI] [PubMed] [Google Scholar]
- Rifai M. A.; Ballantyne C. M. PCSK9-Targeted Therapies: Present and Future Approaches. Nat. Rev. Cardiol. 2021, 18 (12), 805–806. 10.1038/s41569-021-00634-0. [DOI] [PubMed] [Google Scholar]
- Momtazi-Borojeni A. A.; Jaafari M. R.; Banach M.; Gorabi A. M.; Sahraei H.; Sahebkar A. Pre-Clinical Evaluation of the Nanoliposomal Antipcsk9 Vaccine in Healthy Non-Human Primates. Vaccines 2021, 9 (7), 749. 10.3390/vaccines9070749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothgangl T.; Dennis M. K.; Lin P. J. C.; Oka R.; Witzigmann D.; Villiger L.; Qi W.; Hruzova M.; Kissling L.; Lenggenhager D.; Borrelli C.; Egli S.; Frey N.; Bakker N.; Walker J. A.; Kadina A. P.; Victorov D. V.; Pacesa M.; Kreutzer S.; Kontarakis Z.; Moor A.; Jinek M.; Weissman D.; Stoffel M.; van Boxtel R.; Holden K.; Pardi N.; Thöny B.; Häberle J.; Tam Y. K.; Semple S. C.; Schwank G. In Vivo Adenine Base Editing of PCSK9 in Macaques Reduces LDL Cholesterol Levels. Nat. Biotechnol. 2021, 39 (8), 949–957. 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K.; Chadwick A. C.; Mizoguchi T.; Garcia S. P.; DeNizio J. E.; Reiss C. W.; Wang K.; Iyer S.; Dutta C.; Clendaniel V.; Amaonye M.; Beach A.; Berth K.; Biswas S.; Braun M. C.; Chen H. M.; Colace T. V.; Ganey J. D.; Gangopadhyay S. A.; Garrity R.; Kasiewicz L. N.; Lavoie J.; Madsen J. A.; Matsumoto Y.; Mazzola A. M.; Nasrullah Y. S.; Nneji J.; Ren H.; Sanjeev A.; Shay M.; Stahley M. R.; Fan S. H. Y.; Tam Y. K.; Gaudelli N. M.; Ciaramella G.; Stolz L. E.; Malyala P.; Cheng C. J.; Rajeev K. G.; Rohde E.; Bellinger A. M.; Kathiresan S. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593 (7859), 429–434. 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- Taechalertpaisarn J.; Zhao B.; Liang X.; Burgess K. Small Molecule Inhibitors of the PCSK9·LDLR Interaction. J. Am. Chem. Soc. 2018, 140 (9), 3242–3249. 10.1021/jacs.7b09360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Luo S.; Zhu Z.; Xu J. Small Molecules as Inhibitors of PCSK9: Current Status and Future Challenges. Eur. J. Med. Chem. 2019, 162, 212–233. 10.1016/j.ejmech.2018.11.011. [DOI] [PubMed] [Google Scholar]
- Ahamad S.; Mathew S.; Khan W. A.; Mohanan K.. Development of Small-Molecule PCSK9 Inhibitors for the Treatment of Hypercholesterolemia. Drug Discovery Today 2022, 27 (). 1332. 10.1016/j.drudis.2022.01.014. [DOI] [PubMed] [Google Scholar]
- Alleyne C.; Amin R. P.; Bhatt B.; Bianchi E.; Blain J. C.; Boyer N.; Branca D.; Embrey M. W.; Ha S. N.; Jette K.; Johns D. G.; Kerekes A. D.; Koeplinger K. A.; Laplaca D.; Li N.; Murphy B.; Orth P.; Ricardo A.; Salowe S.; Seyb K.; Shahripour A.; Stringer J. R.; Sun Y.; Tracy R.; Wu C.; Xiong Y.; Youm H.; Zokian H. J.; Tucker T. J. Series of Novel and Highly Potent Cyclic Peptide PCSK9 Inhibitors Derived from an MRNA Display Screen and Optimized via Structure-Based Design. J. Med. Chem. 2020, 63 (22), 13796–13824. 10.1021/acs.jmedchem.0c01084. [DOI] [PubMed] [Google Scholar]
- Tucker T. J.; Embrey M. W.; Alleyne C.; Amin R. P.; Bass A.; Bhatt B.; Bianchi E.; Branca D.; Bueters T.; Buist N.; Ha S. N.; Hafey M.; He H.; Higgins J.; Johns D. G.; Kerekes A. D.; Koeplinger K. A.; Kuethe J. T.; Li N.; Murphy B.; Orth P.; Salowe S.; Shahripour A.; Tracy R.; Wang W.; Wu C.; Xiong Y.; Zokian H. J.; Wood H. B.; Walji A. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem. 2021, 64 (22), 16770–16800. 10.1021/acs.jmedchem.1c01599. [DOI] [PubMed] [Google Scholar]
- Yu H.; Dranchak P.; Li Z.; Macarthur R.; Munson M. S.; Mehzabeen N.; Baird N. J.; Battalie K. P.; Ross D.; Lovell S.; Carlow C. K. S.; Suga H.; Inglese J. Macrocycle Peptides Delineate Locked-Open Inhibition Mechanism for Microorganism Phosphoglycerate Mutases. Nat. Commun. 2017, 8, 14932. 10.1038/ncomms14932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura A.; Münzel M.; Kojima T.; Yapp C.; Bhushan B.; Goto Y.; Tumber A.; Katoh T.; King O. N. F.; Passioura T.; Walport L. J.; Hatch S. B.; Madden S.; Müller S.; Brennan P. E.; Chowdhury R.; Hopkinson R. J.; Suga H.; Schofield C. J. Highly Selective Inhibition of Histone Demethylases by de Novo Macrocyclic Peptides. Nat. Commun. 2017, 8 (1), 14773. 10.1038/ncomms14773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado J. N.; Ngo P.; Anslyn E. V.; Ellington A. D. Chemical Insights into Flexizyme-Mediated TRNA Acylation. Cell Chem. Biol. 2022, 29 (7), 1071–1112. 10.1016/j.chembiol.2022.03.012. [DOI] [PubMed] [Google Scholar]
- Iskandar S. E.; Haberman V. A.; Bowers A. A. Expanding the Chemical Diversity of Genetically Encoded Libraries. ACS Comb. Sci. 2020, 22, 712. 10.1021/acscombsci.0c00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen Schlippe Y. V.; Hartman M. C. T.; Josephson K.; Szostak J. W. In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012, 134 (25), 10469–10477. 10.1021/ja301017y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinis C.; Rutherford T.; Freund S.; Winter G. Phage-Encoded Combinatorial Chemical Libraries Based on Bicyclic Peptides. Nat. Chem. Biol. 2009, 5 (7), 502–507. 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- Mathialagan S.; Piotrowski M. A.; Tess D. A.; Feng B.; Litchfield J.; Varma M. V. Quantitative Prediction of Human Renal Clearance and Drug-Drug Interactions of Organic Anion Transporter Substrates Using In Vitro Transport Data : A Relative Activity Factor Approach S. Drug Metab. Dispos. 2017, 45, 409–417. 10.1124/dmd.116.074294. [DOI] [PubMed] [Google Scholar]
- Subramanian H.; Gupta K.; Ali H. Roles of Mas-Related G Protein-Coupled Receptor X2 on Mast Cell-Mediated Host Defense, Pseudoallergic Drug Reactions, and Chronic Inflammatory Diseases. J. Allergy Clin. Immunol. 2016, 138 (3), 700–710. 10.1016/j.jaci.2016.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil B. D.; Pundir P.; Meeker S.; Han L.; Undem B. J.; Kulka M.; Dong X. Identification of a Mast-Cell-Specific Receptor Crucial for Pseudo-Allergic Drug Reactions. Nature 2015, 519 (7542), 237–241. 10.1038/nature14022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T. D.; Whitehead K. A.; Mitragotri S. Materials for Oral Delivery of Proteins and Peptides. Nat. Rev. Mater. 2020, 5 (2), 127–148. 10.1038/s41578-019-0156-6. [DOI] [Google Scholar]