

ABSTRACT
The cytoplasmic accumulation and aggregates of TARDBP/TDP-43 (TAR DNA binding protein) are a pathological hallmark in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. We previously reported that the primate specific cleavage of TARDBP accounts for its cytoplasmic mislocalization in the primate brains, prompting us to further investigate how the cytoplasmic TARDBP mediates neuropathology. Here we reported that cytoplasmic mutant TARDBP reduced SQSTM1 expression selectively in the monkey brain, when compared with the mouse brain, by inducing SQSTM1 mRNA instability via its binding to the unique 3ʹUTR sequence (GU/UG)n of the primate SQSTM1 transcript. Overexpression of SQSTM1 could diminish the cytoplasmic C-terminal TARDBP accumulation in the monkey brain by augmenting macroautophagy/autophagy activity. Our findings provide additional clues for the pathogenesis of cytoplasmic TARDBP and a potential therapy for mutant TARDBP-mediated neuropathology.
KEYWORDS: Aggregate, non-human primate, SQSTM1, RNA instability, TARDBP
Introduction
TARDBP/TDP-43 (TAR DNA binding protein) forms aggregates in the brain of patients with amyotrophic lateral sclerosis (ALS), frontotemporal lobar degeneration (FTLD) [1,2], and other neurological disorders [3–6]. Two conserved RNA recognition motifs RRM1 and RRM2 in TARDBP render its nucleic acid-binding activity [7,8] to regulate different functions of gene transcription or RNA processing [7,9–11]. Through binding single-stranded RNA in the low nanomolar range with high specificity for UG-rich conserved sequences [12,13], TARDBP is recruited to splice sites to inhibit exon recognition [7,14] or mRNA instability [13,15]. TARDBP also interacts with the (UG)n repeat elements, including VEGF (vascular endothelial growth factor), GRN (granulin precursor), NEFL (neurofilament light chain), and ADD2 (adducin 2), in the 3ʹ-untranslated region (3ʹ-UTR) to mediate the degradation of mRNA, even its own mRNA, to suppress gene expression [12,16–20]. Thus, the pathological accumulation of TARDBP can cause abnormal gene expression.
It is known that a large proportion of FTD-ALS-causing genes are related, either directly or indirectly, to pathways that maintain protein homeostasis via macroautophagy/autophagy and involve proteins such as UBQLN2 (ubiquilin 2), OPTN (optineurin), VCP (valosin containing protein) and SQSTM1 (sequestosome 1) [21–23]. Of these proteins, SQSTM1 is an N-recognin of the N-end rule pathway to modulate autophagosome biogenesis [24]. Mutations on the SQSTM1 gene were firstly identified in Paget disease of bone [25], and more recently identified in both ALS and FTLD patients [21,26–28]. Evidence of the involvement of SQSTM1 in ALS and FTLD also includes the presence of SQSTM1-positive inclusions in patient brains, which leads to the inhibition of proteasome and autophagy as well as the accumulation of toxic proteins [29–31]. However, the role of ALS and FTLD-associated SQSTM1 variants in disease onset and progression remain unclear.
Current understanding of SQSTM1 associated-pathogenesis was largely based on cultured cellular or small animal models. Mice with an sqstm1 knockout exhibit increased accumulation of hyperphosphorylated MAPT/tau and subsequent neurodegeneration [32,33], while the absence of SQSTM1 leads to increased degeneration of motor neurons [34], enhanced SNCA/α-synuclein pathology [35], and exacerbated motor phenotypes and neuropathological outcomes in mice [36]. Although mouse models provided valuable information for important pathological events and mechanistic insights, the species differences prevent small animals from recapitulating some important pathologic changes seen in patient brains. For example, in contrast to ALS patient brains that display obvious cytoplasmic TARDBP inclusions [1,10], most transgenic rodent models of ALS show the predominant nuclear localization of mutant TARDBP [37–42] or some minimal level of cytoplasmic TARDBP [41,43,44]. However, in transgenic mutant TARDBP pig model [45] and the monkey brain expressing mutant TARDBP [46,47], TARDBP is localized in the cytoplasm to form aggregates with pathological features, including highly ubiquitinated, phosphorylated, and cleaved TARDBP forms, which are similar to those seen in the patient brains [3–6]. In addition, human specific TARDBP-missplicing was not found in mice [48–50].
The effects of the cytoplasmic TARDBP in the primate brain remain to be fully investigated, though toxic gain-of-function effects were observed in cell cultures [37]. Because mutant TARDBP is accumulated in the cytoplasm in the brains of large animals and because the non-human primates are closer to humans, we investigated the effects of cytoplasmic mutant TARDBP in the monkey brain. Our studies revealed that cytoplasmic mutant TARDBP impaired autophagy function by affecting SQSTM1 expression and that overexpressing SQSTM1 could reduce the accumulation of cytoplasmic TARDBP aggregates by promoting autophagy function. Our findings provide additional clues for the pathogenesis and potential treatment of TARDBP- associated neurological diseases.
Results
Mutant TARDBP reduces SQSTM1 gene expression in the monkey brain
We previously reported that expressing mutant TARDBPM337V in substantial nigra or motor cortex of rhesus monkeys led to the distribution of cytoplasmic and cleaved TARDBP in the primate brain [46]. The emerging issue is how cytoplasmic TARDBP is accumulated and elicits neuropathology. Addressing these issues needs to use the mutant TARDBP form that can accumulate in the cytoplasm of cells. We therefore injected the cerebral cortex of monkey with AAV-TARDBPM337V, which was tagged with FLAG, to examine its distribution and association with autophagy marker proteins (SQSTM1, BECN1 and LC3) (Figure 1A). We used AAV-GFP injection as a control and also included the injection of the mouse cerebral cortex for comparison. The exogenous mutant TARDBP was distributed in neuronal and glial cells (Fig. S1A) and showed cytoplasmic distribution in the injected monkey brain (Figure 1B and S1A). Moreover, SQSTM1, but not LC3, was reduced and colocalized with the cytoplasmic mutant TARDBP in the monkey brain (Figure 1B and S1B), which was consistent with SQSTM1-positive inclusions in patients with TARDBP mutations [29–31]. However, in the mouse brain, mutant TARDBP remained in the nuclei and did not recruit SQSTM1 (Figure 1C, quantification in 1D; Fig. S1C). Western blotting revealed that the truncated TARDBP fragments TARDBP-35 (35 kDa) and TARDBP-25 (25 kDa) were more abundant in the injected monkeys than in the injected mouse brain (Figure 1E), which was consistent with our earlier study [46,47]. When the same samples were probed with antibody to SQSTM1, a dramatic reduction of SQSTM1, but not BECN1, was seen in the AAV-TARDBP-injected monkey brain as compared with the AAV-GFP control-injected monkey brain and AAV-TARDBP-injected mouse brain (Figure 1E, quantified in 1 F). Analysis of mRNA expression via qRT-PCR showed that the SQSTM1 transcript level was selectively decreased in the AAV-TARDBP-injected monkey brain as compared with LC3B and BECN1 transcripts (Figure 1G), suggesting that mutant TARDBP affects SQSTM1 expression in the monkey brain via transcriptional regulation.
Figure 1.
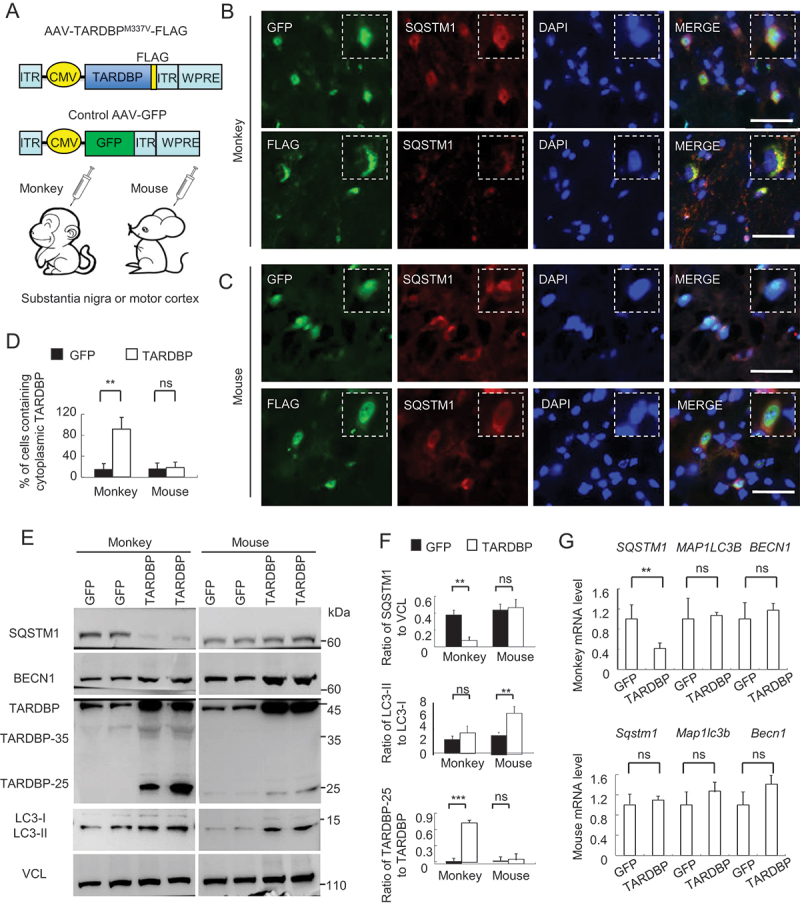
Cytoplasmic mutant TARDBP reduced SQSTM1 expression in the monkey brain. (A) A schematic diagram for the injection of adeno-associated viruses, which express mutant TARDBPM337V or control GFP under the CMV promoter, into the substantia nigra or motor cortex of monkeys and mouse brain. (B) Immunofluorescent staining of the monkey substantia nigra injected with AAV- GFP control (upper panel) or -TARDBPM337V (lower panel), using antibodies to GFP or FLAG (green) and SQSTM1 (red). The nuclei were stained with DAPI (blue). Representative images were obtained from three 8- to 12-years-old monkeys. The monkey brain showed significant cytoplasmic colocalization of mutant TARDBP as compared with that expressing GFP. Scale bars: 40 μm. (C) Representative immunofluorescent images of the substantia nigra in 6- to 9-months-old mice injected with AAV-TARDBPM337V or -GFP control, using antibodies to TARDBP or GFP (green) and SQSTM1 (red). The nuclei were stained with DAPI (blue). Scale bars: 40 μm. (D) Quantitative analysis of the relative numbers of cells containing cytoplasmic TARDBP over total DAPI-staining injected with AAV-TARDBPM337V or -GFP control. Twenty random fields (40X) in each section were examined from 3 samples in each group. Data are mean ±SEM. (**P < 0.01, ns: not significant). (E) Western blotting analysis of the soluble fractions in AAV-TARDBP or -GFP injected monkey and mouse tissues. Probing with antibodies to the autophagy related proteins BECN1, SQSTM1 and LC3 revealed that mutant TARDBP decreased endogenous SQSTM1 expression in the monkey brain as compared with the mouse brain. (F) The ratios of SQSTM1 to VCL, LC3-II to LC3-I, and truncated TARDBP (25-kDa) to full-length TARDBP (43-kDa) on Western blots in (E) are presented. The data were obtained from independent Western blotting analyses of tissues from AAV-GFP- or AAV-TARDBP-injected monkeys and mice (n = 3 animals per group). **P < 0.01; ns: not significant. (G) Quantitative PCR of expression of the autophagy related genes SQSTM1, LC3 and BECN1 in AAV-TARDBP monkey and mouse substantia nigra. AAV-GFP injection served as a control. Note that SQSTM1-encoding mRNA level was selectively reduced by mutant TARDBP in the monkey brain, but not in the mouse brain. The data (mean ± SE) were obtained from three independent experiments (n = 3 animals per group, *P < 0.05; **P < 0.01; ns: not significant).
TARDBP binds to 3ʹUTR of the primate SQSTM1 transcript
Since TARDBP interacts with the UG repeats of 3ʹ-untranslated region (3ʹ-UTR) to affect gene expression [12,16,18,20], we examined (GU/UG)n motifs of 3ʹ-UTR in the human, monkey and mouse SQSTM1 genes (Figure 2A). We found that the putative RNA binding sequences “GUGUGUGUGUGU” or “GUGAGGGUGUGU” were different in a species dependent manner. The GU/UG repeat numbers were 10 and 8 in human and macaque, respectively, but 4 in mouse (Figure 2A). Because of the preferential accumulation of mutant TARDBP in the cytoplasm, the (UG)n repeats having more than 6 units would likely to interact with mutant TARDBP [51–53]. To confirm this, we transfected FLAG-tagged TARDBP-ΔNLS, which deletes the nuclear localization signal (NLS), truncated TARDBP-25 or TARDBP-35, and full-length TARDBP into human SH-SY5Y cells (Figure 2B). The NLS-deleted and truncated TARDBP (TARDBP-25 or TARDBP-35) that also lacks NLS were distributed in the cytoplasm, and the smallest fragment (TARDBP-25) was accumulated as cytoplasmic inclusions (Fig. S2A). Through RNA immunoprecipitation (RIP) assay with the FLAG antibody, we captured the transfected and FLAG-tagged mutant TARDBP in the human SH-SY5Y or mouse Neuro-2a cells (Fig. S2B). The RNAs associated with the precipitated TARDBP were identified by RT-PCR and DNA sequencing, revealing that specific RNA bound mutant TARDBP in SH-SY5Y, but not mouse Neuro-2a, cells and contained (GU/UG) repeats in the human SQSTM1 3ʹUTR segment (Figure 2C and S2C). Expressing different TARDBP fragments without NLS but tagged with GFP in transfected HEK293 cells (Figure 2D) and GFP immunoprecipitation of transfected TARDBP showed that these cytoplasmic TARDBP could bind the 3ʹUTR repeats in the human SQSTM1 gene (Figure 2E). The RNA immunoprecipitation sequencing (RIP-Seq) indicated that 3ʹUTR segment (28.91%) was the second enriched component, followed by 5ʹ UTR segment (14.55%), while the coding sequences (CDS) are the most abundant binding component (46.55%) (Figure 2F). Using the UCSC genome browser (https://genome.ucsc.edu) to analyze the transcript reads of TARDBP associated RNAs, we verified that the 3ʹUTR in the SQSTM1 gene on chromosome 5 was indeed precipitated by TARDBP (Figure 2G).
Figure 2.
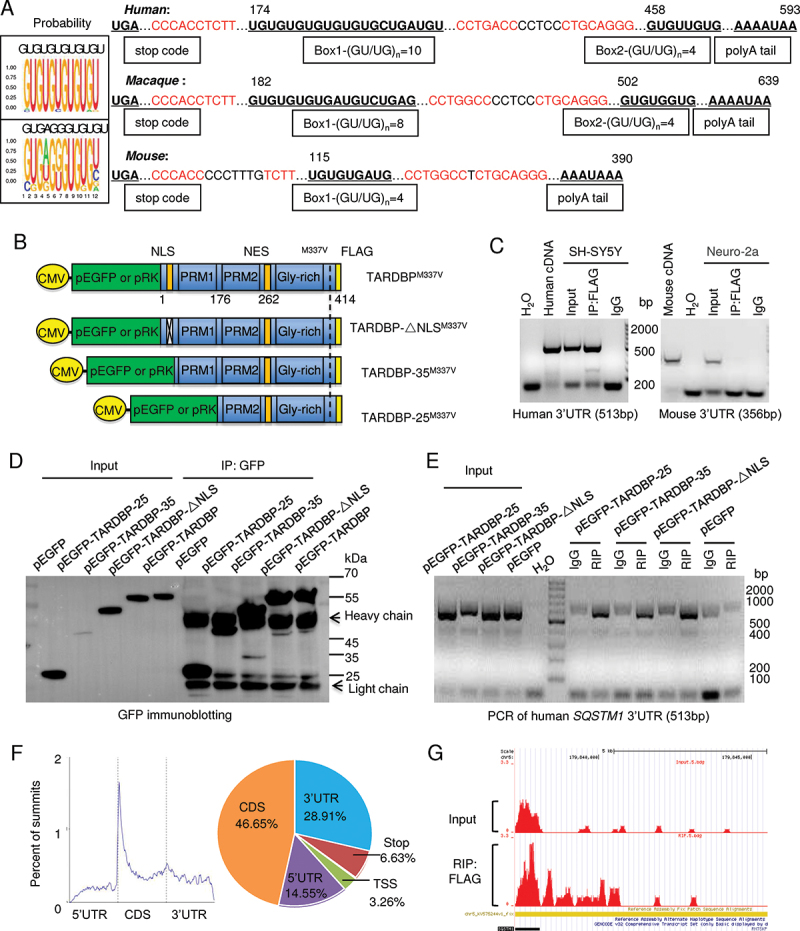
Preferential binding of TARDBP to the primate SQSTM1 transcript 3ʹUTR to suppress SQSTM1 expression. (A) The putative RNA binding conserved sequences “GUGUGUGUGUGU” or “GUGAGGGUGUGU” of TARDBP were determined in the box. In the 3ʹUTR region of the human, macaque and mouse SQSTM1 gene, each (GU/UG)n motif and polyA tail at the downstream of message RNA stop code were predicated. The numbers of GU/UG repeats were indicated in the boxes under the sequences. The consensus motifs with the species were indicated in red. (B) The plasmid DNA structures for expressing full-length TARDBPM337V, truncated C-terminal-TARDBP-(90-414) as TARDBP-35M337V, C-terminal-TARDBP-(184-414) as TARDBP-25M337V; or the nuclear localization signal (NLS) deletion TARDBP-ΔNLSM337V, under the same CMV promoter in pEGFP or pRK vector. (C) Ribonucleoprotein complex (RNA) immunoprecipitation (RIP) assay was performed using TARDBPM337V-FLAG transfected human SH-SY5Y and mouse Neuro-2a cells. The transfected TARDBP was immunoprecipitated by FLAG antibody and the associated mRNAs were then detected by RT-PCR. The human and mouse SQSTM1 3ʹ-UTR cDNAs were used for comparison, and their expression was seen in transfected cells and input. IgG and H2O served as control for the immunoprecipitation and RT-PCR. Compared with mouse Neuro-2a cells, human SH-SY5Y cells show that transfected TARDBP interacted with the human SQSTM1 3ʹ-UTR. (D) Western blotting analysis of the immunoprecipitated pEGFP, pEGFP-TARDBP, pEGFP-TARDBP-35, pEGFP-TARDBP-25 and pEGFP-TARDBP-ΔNLS in HEK293 cells, showed that transfected C-terminal TARDBP fragments could be precipitated by the GFP antibody. (E) The TARDBP-associated RNAs precipitated by anti-GFP (RIP) in SH-SY5Y cells were analyzed by RT-PCR with specific primers for identifying human SQSTM1 3ʹUTR (513 bp) segment. The input or IgG groups served as control. The H2O group was the negative control for the RT-PCR process. The results revealed that all the truncated cytoplasmic TARDBP, but not the GFP control, could interact with human SQSTM1 mRNA 3ʹUTR. (F) TARDBPM337V-associated RNAs were analyzed by deep sequencing (RIP-Seq) to identify the putative pre-mRNA targets. The overall distribution (left panel) and the statistic annotation (right panel) of TARDBPM337V-associated RNAs in RIP-Seq data show the enriched RNA components consisting of the CDS segment (46.65%) and the 3ʹ-UTR segment (28.91%). (G) The UCSC genome browser was used to analyze the general transcript reads of TARDBP associated RNAs. The 3ʹ-UTR segment on the human SQSTM1 gene, localized on the chromosome 5: (179,806,353 … 179,838,078), was successfully captured and amplified by deep sequencing.
Mutant TARDBP promotes the instability of human SQSTM1 mRNA
We then investigated whether the preferential binding of TARDBP to 3ʹUTR of human SQSTM1 transcript affects SQSTM1 mRNA stability. Ranking the most associated genes consisting of 3ʹ-UTR segments from the RIP-Seq data showed the peak annotation of TARDBP-associated RNAs distribution among all chromosomes (Fig. S3A). The mRNA expression in human SH-SY5Y cells transfected with cytoplasmic TARDBP-25 or pRK control vector was also detected by RNA-Seq. Through combining analyses of the RIP-Seq and RNA-Seq, the heatmap was obtained and indicated that cytoplasmic TARDBP-25 decreased the mRNA expression of most associated genes with 3ʹUTR segments (Fig. S3B).
To further investigate whether the human SQSTM1 gene was down-regulated by mutant TARDBP, we generated psiCHECK reporters that express 3ʹUTR segments of the human or mouse SQSTM1 gene in HEK293 cells. Luciferase report assay showed that only human SQSTM1 3ʹUTR-mediated renilla value was dramatically down-regulated by mutant TARDBP when compared with the mouse Sqstm1 3ʹUTR (Figure 3A). To confirm the role of (GU/UG)n repeats in this down-regulation, we deleted (GU/UG)n-boxes in human SQSTM1 3ʹUTR segment for luciferase report assay. After deleting the first “(GU/UG)n = 10” box, the cytoplasmic TARDBP-25 was no longer to change the renilla activity when compared with the intact human SQSTM1 3ʹUTR or mouse Sqstm1 3ʹUTR containing a shorter “(GU/UG)n = 4” (Figure 3B and S3C). When the 4 “(GU/UG) repeats of mouse Sqstm1-3ʹUTR were extended to 10 repeats, the extended mouse Sqstm1-3ʹUTR was also decreased by cytoplasmic TARDBP-25 (Figure 3C and S3C).
Figure 3.
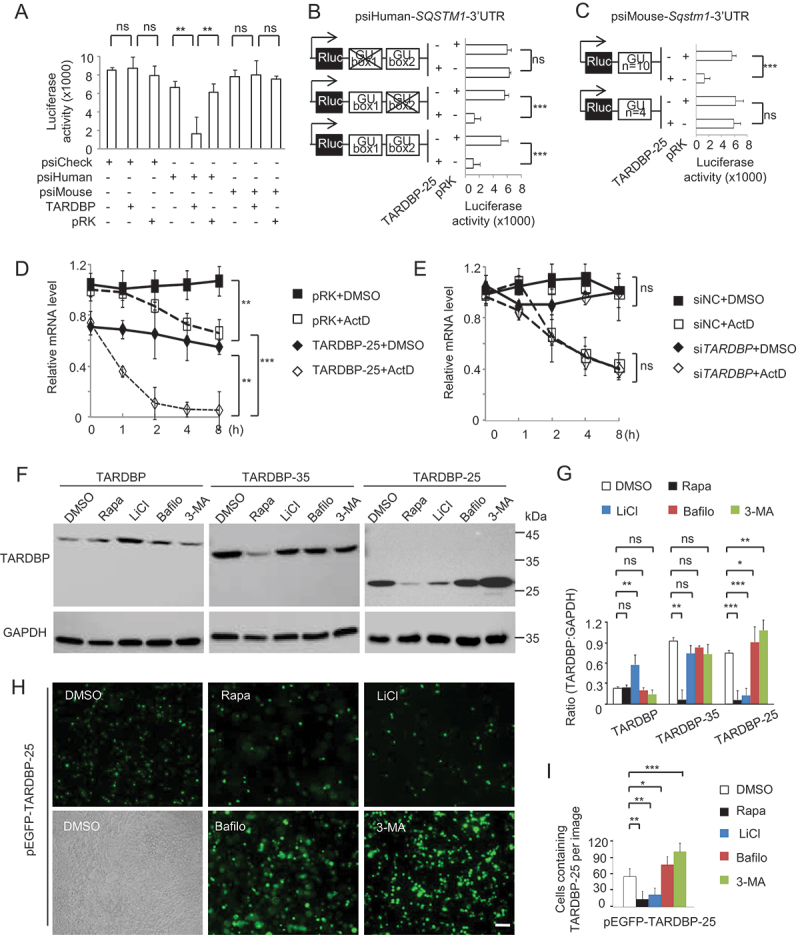
TARDBP-25 promoted human SQSTM1 mRNA instability and was eliminated by autophagic process. (A) The human and mouse SQSTM1 3ʹUTR segments were transfected into HEK293 cells for detection of Renilla luciferase activity. The values of luciferase report assay obtained from three independent experiments indicate that only the human SQSTM1 3ʹUTR was significantly down-regulated by mutant TARDBP, compared with the empty vector control. Data are mean ±SEM (**P < 0.01. ns: not significant). (B) The (GU/UG)n-box deletion vectors of human SQSTM1 3ʹUTR segment were generated by Site-Directed Mutagenesis Kit, then co-transfected with pRK-TARDBP-25 or empty pRK vector in HEK293 cell. The result showed that after deleting the first “(GU/UG)n = 10”, the cytoplasmic TARDBP-25 did not decrease the activity of the human SQSTM1 3ʹ-UTR when compared with the truncation of “(GU/UG)n = 4” and wild type human SQSTM1 3ʹ-UTR treatment. The values of luciferase activity were obtained from three independent experiments. Data are mean ±SEM (***P < 0.001. ns: not significant). (C) The (GU/UG)n-box (4 repeats) in the mouse Sqstm1 3ʹUTR psiCheck vector was replaced by (GU/UG)n = 10 repeats) and then co-transfected with pRK-TARDBP-25M337V into HEK293 cells. The Renilla activity of the extended mouse Sqstm1 3ʹUTR containing (GU/UG)n = 10, but not the wild type “(GU/UG)n = 4, was inhibited by cytoplasmic TARDBP-25. The values of luciferase activity were obtained from three independent experiments. Data are mean ±SE (***P < 0.001. ns: not significant). (D) The actinomycin D (ActD) chase experiment to determine endogenous mRNA’s half-lives in vitro. The human SH-SY5Y cells were transfected with pRK-TARDBP-25M337V or pRK control for 24 h and then treated for 8 h with 10 µg/ml ActD for blocking transcription. Total RNA was extracted and quantified by qPCR with specific primers for human SQSTM1. The RNA levels at different times of transfection by pPK-TARDBP-25 or pRK control were obtained by comparing that at 0 h. TARDBP-25 decreased the SQSTM1 mRNA level via its influence on mRNA stability. (E) Human SH-SY5Y cells were transfected with TARDBP siRNA or its scramble siRNA control for 48 h. Ten µg/ml ActD was added up to 8 h. The chase experiment showed that the human SQSTM1 mRNA level or its instability was not influenced by knockdown of endogenous wild type TARDBP. (F) Western blotting analysis of transfected pRK-TARDBP-25, pRK-TARDBP-35 and pRK-TARDBP in HEK293 cells after treatments with autophagic activators (rapamicin [Rapa] and LiCl) or inhibitors (bafilomycin A1 [Bafilo] and 3-MA). The blots were probed with C-terminal TARDBP antibody, revealing that TARDBP-25, but not TARDBP-35 and full-length TARDBP, was markedly reduced by autophagic activators and accumulated by the inhibitors. (G) Quantitative representation of the band intensity ratios of TARDBP-25, TARDBP-35, or TARDBP to GAPDH in (F). The data (mean ±SE) were obtained from three independent experiments. (*P < 0.05; **P < 0.01; ***P < 0.001, ns: not significant). (H) Representative staining of transfected pEGFP-TARDBP-25M337V in HEK293 cells treated with autophagic activators (rapamycin [Rapa] and LiCl) or inhibitors (bafilomycin A1 [Bafilo] and 3-MA). The immunofluorescent (upper) and phase (lower) images of the control (DMSO-treated) cells are presented. Scale bars: 20 μm. (I) Quantitative analysis of the numbers of cells in (C) containing pEGFP-TARDBP-25 per image (40X). Twenty random images in each section were examined from 3 experiments. The data are mean ±SE (*P < 0.05; **P < 0.01; ***P < 0.001, ns: not significant).
The binding of TARDBP to human SQSTM1 3ʹUTR could mediate the degradation of SQSTM1 transcripts. Thus, we used the actinomycin D (ActD) chase assay to determine mRNA’s half-lives in vitro. In SH-SY5Y cells, transfection of mutant TARDBP-25 accelerated the degradation of endogenous human SQSTM1 mRNA (Figure 3D), However, suppressing endogenous TARDBP by siRNA did not alter SQSTM1 transcript stability (Figure 3E), supporting the idea that cytoplasmic mutant TARDBP binds the repeats to mediate toxic gain of function.
As an important receptor in autophagy process, SQSTM1 can shuttle ubiquitinated cargo for lysosomal degradation [54–56]. It is possible that suppression of SQSTM1 expression by mutant TARDBP could lead to the impairment of autophagy function and the alteration of cytoplasmic TARDBP accumulation. To test this possibility, we firstly added the autophagy activators or inhibitors to HEK293 cells that were transfected with mutant TARDBP-25. Western blotting with a C-terminal TARDBP antibody (Figure 3F, quantified in 3 G) and staining of the pEGFP-TARDBP-25M337V transfection (Figure 3H, quantified in 3I) revealed that the accumulated TARDBP-25, when compared with full-length TARDBP, was markedly reduced by the autophagic activator rapamycin (Rapa) or LiCl and increased by the autophagic inhibitor bafilomycin A1 (Bafilo) or 3-MA. Taken together, SQSTM1-involved autophagy may be important for the selective clearance of pathologic TARDBP-25 in the cytoplasm.
SQSTM1 promotes the clearance of cytoplasmic TARDBP inclusions in vivo
The above findings led us to investigate whether overexpression of SQSTM1 enhances the clearance of cytoplasmic TARDBP in vivo. We generated the AAV9 vector expressing human SQSTM1, which is tagged with MYC at its C-terminus and expressed under the control by the CMV promoter (Figure 4A). We first performed experiments to overexpress human SQSTM1 or used SQSTM1 siRNA to inhibit its expression in cultured HEK293 cells. Suppression of SQSTM1 by its siRNA promoted the accumulation of TARDBP-25 and overexpression of SQSTM1 increased TARDBP-25 accumulation in human HEK293 cells (Figure 4B, quantified in 4C; Fig. S4). However, no significant or coincident alteration of TARDBP-35 and intact TARDBP was observed after altering SQSTM1 expression when compared with the vector alone or control scrambled siRNA (Figure 4B, quantified in 4C).
Figure 4.
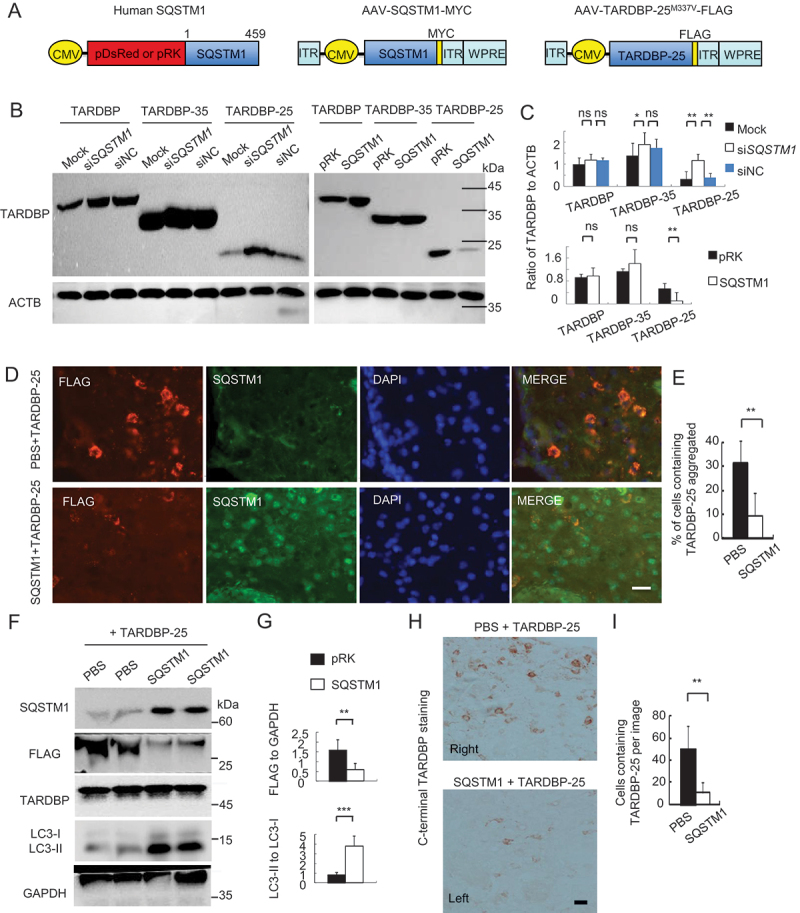
SQSTM1 overexpression inhibits TARDBP-25 accumulation in vitro and in mouse brain. (A) Schematic diagrams of SQSTM1-expressing plasmid and AAV vector. The truncated AAV-TARDBP-25M337V tagged with FLAG was also expressed using AAV vector. (B) Western blotting analysis of different TARDBP fragments co-transfected with the human SQSTM1 siRNA (siSQSTM1) or control siRNA (siNC) in human SH-SY5Y cells. The knockdown of SQSTM1 increased TARDBP-25 whereas SQSTM1 overexpression decreased TARDBP-25 with no effect on TARDBP-35 or TARDBP. (C) Quantitative analysis of the band intensity ratios of TARDBP-25, TARDBP-35 and TARDBP to ACTB in (B). The data (mean ± SE) were obtained from three independent experiments (**P < 0.01; ns: not significant). (D) Immunofluorescent staining with anti-FLAG (Red) to detect TARDBP-25-FLAG and anti-SQSTM1 (green) indicated that the cytoplasmic TARDBP-25 was diminished by SQSTM1 overexpression. The nuclei were stained with DAPI (blue). Scale bar: 40 μm. (E) Quantitative analysis of the relative numbers of cells containing TARDBP-25 positive aggregates over total DAPI-nuclei per image in the injected brain region. Twenty random fields (40X) in each section were examined. n = 3 samples in each group. Data are mean ±SE. (**P < 0.01). (F) Western blotting analysis of the mouse brains injected with AAV TARDBP-25-FLAG at 6–9 months of age. The exogenous TARDBP-25-FLAG, but not endogenous TARDBP, was reduced by AAV-SQSTM1’s overexpression, as compared with the TARDBP-25-FLAG expression alone (PBS co-injection). (G) Quantitative representation of the band intensity ratios of the exogenous TARDBP-25-FLAG:GAPDH and LC3-II:LC3-I in (F). The data (mean ±SE) were obtained from three independent experiments. (**P < 0.01; ***P < 0.001, ns: not significant). (H) Immunohistochemical staining with antibody to C-terminal TARDBP showed that TARDBP-25-FLAG was also reduced by SQSTM1 overexpression. Scale bar: 20 μm. (I) Quantitative analysis of the numbers of cytoplasmic TARDBP-25-positive cells in (H). Twenty random fields (20X) in each section were examined from 3 samples in each group. Data are mean ±SEM. (**P < 0.01).
hen stereotaxically co-injected AAV-TARDBP-25M337V and AAV-SQSTM1 into the left side cortex of mouse at 6–9 months of age. The right side was the AAV-TARDBP-25 injection alone. Expression of the cytoplasmic TARDBP-25 was evidenced by obvious inclusions in the cytoplasm 3 weeks after the injection, which were reduced by co-expression of SQSTM1 (Figure 4D, quantified in 4E). Western blotting verified FLAG-tagged TARDBP-25 was dramatically reduced by overexpression of SQSTM1 (Figure 4F, quantified in 4G). Immunohistochemical staining also showed that SQSTM1 overexpression significantly reduced the accumulation of TARDBP-25 (Figure 4H, quantified in 4I).
We found that SQSTM1 overexpression did not influence the soluble GFP expression, though SQSTM1 overexpression could increase LC3 level (Fig. S5A-C). Next, AAV-SQSTM1 was co-injected with AAV-TARDBPM337V into the right (R) substantia nigra and motor cortex of 8–12 years old monkeys. For the control, the left (L) side in the same animal was co-injected with the same titers of AAV-GFP and AAV-TARDBPM337V (Figure 5A). Six weeks later, MRI analysis revealed the damage in the substantia nigra or motor cortex by TARDBPM337V, and this damage was diminished by SQSTM1 overexpression to some extent (Figure 5B). Western blotting of freshly isolated monkey brain tissues showed that SQSTM1 overexpression indeed promoted TARDBP-25 clearance, though there was no obvious effect on TARDBP-35 or full-length TARDBP. The protective effects of SQSTM1 were apparently associated with activation of autophagy, which was indicated by the increased ratio of LC3II to LC3I (Figure 5C, quantified in 5D). Immunofluorescent (Figure 5E, quantified in 5F) and immunohistochemical (Figure 5G, quantified in 5H) staining also showed that over-expressing SQSTM1 augmented the clearance of cytoplasmic TARDBP in the monkey substantial nigra.
Figure 5.
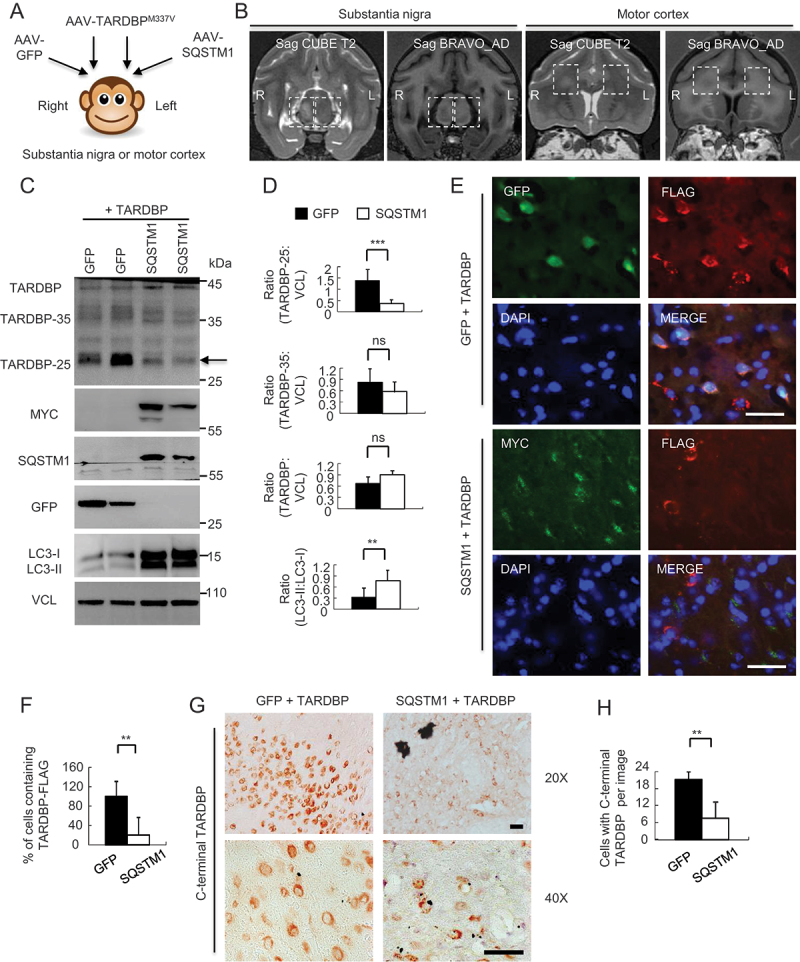
Overexpressing SQSTM1 promoted the clearance of TARDBPM337V in the monkey brain. (A) A schematic diagram for the stereotaxic co-injection of AAV-TARDBPM337V with AAV-SQSTM1 into the left substantia nigra and motor cortex of 10-years-old monkeys. The right side was the co-injection of TARDBPM337V with GFP control. (B) Magnetic resonance imaging (MRI) analysis revealing the BRAVO T1 and CUBE T2-weighted coronal images of the injected monkeys at the age of 10 years. The boxed areas indicate the injected regions. The right side of substantia nigra with AAV-TARDBP and AAV-GFP co-injection show damages (increased density in Sag CUBE images or reduced density in Sag BRAVO images) as compared with the left side that was co-injected with AAV-SQSTM1. (C) Western blotting with anti-C-terminal TARDBP showing that SQSTM1 overexpression promoted TARDBP-25 (arrow) clearance. Transgenic SQSTM1-MYC was detected by antibodies to MYC-Tag and SQSTM1. Note that SQSTM1 expression also increased LC3 expression. (D) Quantitative analysis of the band intensity ratios of TARDBP-25, TARDBP-35, TARDBP and LC3 to VCL in (C). The data are presented as mean ±SEM and obtained from 3 independent experiments. (*P < 0.05; **P < 0.01; ***P < 0.001, ns: not significant). (E) Immunofluorescent staining with antibodies to MYC-Tag for transgenic SQSTM1-MYC and FLAG for TARDBP-FLAG showing that SQSTM1-MYC (green) co-injection decreased the cytoplasmic accumulation of mutant TARDBP (red), in the monkey substantia nigra as compared with AAV-GFP control. Scale bar: 40 μm. (F) Quantitative analysis of the relative numbers of cells containing the cytoplasmic TARDBP-FLAG-positive cells over total DAPI-nuclei per image in the monkey substantia nigra injected with AAV-SQSTM1 or AAV-GFP control (E). Twenty random fields (40X) in each section were examined from 3 samples in each group. Data are mean ±SEM. (**P < 0.01). (G) Immunohistochemical staining with antibody to C-terminal TARDBP showing that overexpressing SQSTM1 reduced mutant TARDBP in the cytoplasm of monkey substantia nigra. Scale bar: 40 μm. (H) Quantitative analysis of the numbers of cytoplasmic TARDBP-positive cells per image (40X) in (G). Twenty random images in each section were examined from 3 samples in each group. Data are mean ±SEM. (**P < 0.01).
Taken together, our findings indicated that mutant TARDBP in the cytoplasm affects autophagic function by reducing SQSTM1 mRNA expression. This effect is species-dependent because cytoplasmic truncated TARDBP-25 was able to bind 3ʹ-UTR of the primate SQSTM1 transcript, but not the mouse SQSTM1 transcript, to inhibit SQSTM1 transcription. Since SQSTM1 mediated autophagy is responsible for the clearance of misfolded proteins, our findings provide mechanistic insight into the pathogenesis of cytoplasmic mutant TARDBP and a potential therapeutic target for alleviating TARDBP associated neuropathology, especially in the primate brains (Figure 6).
Figure 6.
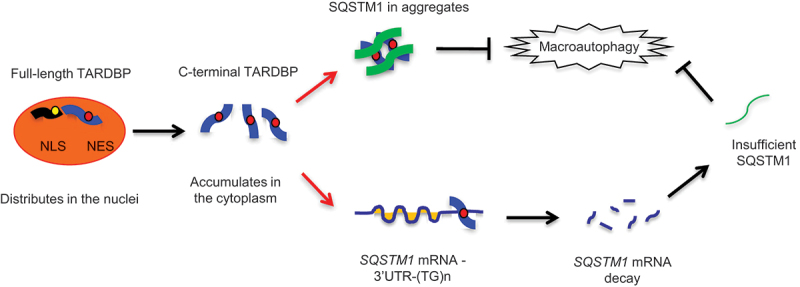
A proposed model for the role of SQSTM1 in clearance of cytoplasmic mutant TARDBP in the primate brain. The primate specific cleavage of TARDBP accounts for its cytoplasmic mislocalization in the primate brain. The cytoplasmic mutant TARDBP impaired the autophagic function by binding the 3ʹ-UTR of the SQSTM1 transcript to suppress SQSTM1 expression, leading to the autophagy dysfunction that can reduce TARDBP clearance in the cytoplasm. Overexpressing SQSTM1 can enhance the clearance of cytoplasmic mutant TARDBP and may be beneficial for treating TARDBP-associated neuropathology.
Discussion
TARDBP is a major component of cytoplasmic aggregates in the brains and spinal cords of nearly all patient with (~97%) ALS and (~45%) FTLD [1,2]. In addition, 57% of Alzheimer’s disease cases and some dementia patients with Lewy bodies also show TARDBP proteinopathies in their brains [4–6]. However, only <5% of ALS patients carry mutations in TARDBP, thus pathological conditions other than TARDBP mutations are the frequent factors responsible for the cytoplasmic accumulation of TARDBP [3,10,57,58]. It is known that the increased level of TARDBP in the nucleus and cytoplasm contributes to the neuropathological development [59–61]. The cytoplasmic accumulation of TARDBP appears to be independent of its mutations but is regulated by species-related factors. This is because transgenic rodent models overexpressing either normal or mutant TARDBP show the predominantly nuclear accumulation of TARDBP [37–39,41,62], whereas expression of TARDBP in the brain and spinal cords of monkeys by injecting AAV vector led to the cytoplasmic distribution of TARDBP [46,63]. Also, a transgenic TARDBP pig model expressed mutant TARDBP in the cytoplasm [45]. These findings led us to use the non-human primates to investigate the effects of cytoplasmic mutant TARDBP.
Autophagic cargo SQSTM1 plays a critical role in protein degradation, which is highly relevant to the accumulation of misfolded proteins in neurodegenerative diseases [26]. SQSTM1 was found in inclusions along with polyubiquitinated proteins in neuronal cells of familial ALS (FALS) and sporadic ALS (SALS) patient samples [29,31] and also in TARDBP aggregates colocalized with markers of autophagy [64,65]. Although it is not clear whether SQSTM1 initiates aggregate formation, SQSTM1 does appear to bind toxic misfolded proteins, a process that is enhanced by substrate ubiquitination [66,67]. We found that in the monkey brain, mutant TARDBP was distributed in the cytoplasm with significant colocalization of SQSTM1, consistent with the previous finding that TARDBP is associated with SQSTM1-positive inclusions in the patient brain [29–31]. This colocalization was not seen in the mouse brain because the majority of mutant TARDBP remained in the nucleus in the mouse brain. Our earlier studies identified the primate-specific CASP4 as an important enzyme to cleave mutant TARDBP, leading to a small TARDBP fragment such as TARDBP-25 that is able to accumulate in the cytoplasm [46]. Thus, cytoplasmic mutant TARDBP is likely to mediate species-dependent effects to affect neuronal function.
In support of the above hypothesis, we found that cytoplasmic mutant TARDBP can reduce SQSTM1 expression in the monkey brain, but not in the mouse brain, via transcriptional regulation. This selective effect also appears to depend on the unique sequences of 3ʹ-UTR of the primate SQSTM1 transcript. The 3ʹ-UTR-mediated mRNA instability was essential for suppressing gene expression by TARDBP through the (UG)n repeats [12,16,18,20]. Due to the greater number of repeats in the 3ʹ-UTR segments of the primate SQSTM1 transcript than the rodent SQSTM1 transcript, cytoplasmic mutant TARDBP is able to bind the 3ʹ-UTR of the primate SQSTM1 transcript to suppress its expression. Loss of SQSTM1 has been found to associate with neurodegeneration in many disease models. For example, sqstm1 knockout mice exhibited accumulation of hyperphosphorylated tau and subsequent neurodegeneration [32,33]. In SOD1H46R mice, absence of SQSTM1 leads to an increase in insoluble SOD1 and ubiquitinated proteins with obvious motor neuron degeneration [34]. Since SQSTM1 was partially reduced rather than the complete absent in our monkey model, it would be interesting to know whether SQSTM1 deficiency has any influence on the cytoplasmic accumulation of mutant TARDBP. The rationale of this investigation was also strengthened by the recent findings that induction of autophagy can mitigate TARDBP aggregates in the mouse brains [68,69], which raised an important issue regarding the effects of SQSTM1-associated autophagy on the cytoplasmic TARDBP in the primate brain. The function of SQSTM1 is complex. Although overexpression of SQSTM1 was found to be toxic in some mouse models [70,71], beneficial effects of overexpressed SQSTM1 in improving behaviors and protection of cell damage in the rodent and Drosophila brains have been reported [36,72,73]. It is clear that the effects of SQSTM1 are dependent on its level and the time course of its action. In addition, SQSTM1 in different cell types and species is likely to have differential functions. Our study demonstrated for the first time that overexpression of SQSTM1 can reduce mutant TARDBP accumulation in the non-human primate brain, consistent with the previous findings that SQSTM1 interacts with cytoplasmic misfolded proteins in the human brains to remove misfolded proteins [65,67,74].
By comparing TARDBPM337V monkey and mouse models, we identified two lines of evidence supporting the idea that dysfunctional SQSTM1 mediates TARDBP pathogenesis. First, in the monkey brains, cytoplasmic mutant TARDBP induced SQSTM1 mRNA instability because of its preferential binding to the conserved sequence (UG)n of the 3ʹ-UTR of the primate SQSTM1 transcript. Second, SQSTM1 overexpression could diminish mutant TARDBP’s effect to facilitate the removal of cytoplasmic TARDBP-25. Our findings thus provide additional clues for the cytoplasmic TARDBP-mediated gain-of-toxicity function and pointed out a potential therapeutic strategy to prevent or reduce TARDBP-associated neuropathology.
Materials and methods
Plasmids and virus
The cDNAs encoding full-length human mutant TARDBPM337V [42,46], truncated C-terminal TARDBPM337V-(77-414) (TARDBP-35) or TARDBPM337V-(174-414) (TARDBP-25) were subcloned into pAAV-MCS, pEGFP or pRK vector (Addgene, 46,954, 165,830 and 10,883; deposited by Steve Jackson, Koen Venken and BD PharMingen). SQSTM1 vector, encoding the human SQSTM1 cDNA, was constructed into the pDsRed (Addgene, 54,502; deposited by Michael Davidson) or pRK vector. The human SQSTM1 cDNA was also subcloned into pAAV-MCS backbone at BamH1 and Xba1 restriction sites to generate AAV-SQSTM1. The control DsRed and pAAV-GFP vectors consisted of the same promoter. Titers of AAV-TARDBPM337V, -TARDBP-35M337V, -TARDBP-25M337V, -SQSTM1 or -GFP viruses were confirmed by determining the number of packaged vector genomes using quantitative PCR.
Antibodies and reagents
Primary antibodies used for immunoblot, immunofluorescence, and immunoprecipitation assays were as follows: rabbit anti-TARDBP (Cell Signaling Technology, 3448) for a synthetic peptide corresponding to residues surrounding Gly400 of human TARDBP, mouse anti-GFP (GeneTex, GTX113617), mouse anti-VCL/vinculin (Abcam, ab18058), mouse anti-GAPDH (GeneTex,GTX100118), rabbit anti-FLAG-Tag (Cell Signaling Technology, 14,793), mouse anti-SQSTM1 (Abcam, ab56416), rabbit anti-LC3 (Sigma, L8918), rabbit anti-BECN1 (Cell Signaling Technology, 3738) and rabbit anti-MYC-Tag (Cell Signaling Technology, 2272). Secondary antibodies were HRP-labeled donkey anti-mouse, donkey anti-rabbit (Jackson ImmunoResearch, 715,035,150, 711,035,152), donkey anti-mouse Alexa Fluor, or donkey anti-rabbit Alexa Fluor (Jackson ImmunoResearch, 715,546,150, 109,585,003).
Animals
Cynomolgus monkeys were raised at Guangdong Landau Biotechnology Co. Ltd., which is an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility. Water, temperature, humidity in the home cage in which monkeys were kept were closely monitored. Monkey health was daily examined by veterinarians. Mice were kept at the animal facility at Jinan University. All animal-related protocols were approved in advance by the Animal Care and Use Committee of Guangdong Landau Biotechnology Co. Ltd and Jinan University. This study occurred in strict compliance with the “Guide for the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Science (est. 2006)” and “The use of non-human primates in research of the Institute of Laboratory Animal Science (est. 2006)” to ensure the safety of personnel and animal welfare.
Stereotaxic injection
AAV viral injection into monkey brain was performed as reported previously [46]. We injected AAV-GFP, -TARDBP or -SQSTM1 into the substantia nigra of wild-type monkeys at the age of 8–12 years (n = 6 each group). Each monkey was anesthetized by intraperitoneal injection of 0.3–0.5 mg of atropine, followed by 10–12 mg of ketamine, and 15–20 mg of pelltobarbitalumnatricum per kg body weight. The monkeys were stabilized on a stereotaxic instrument (RWD Life Science, 68,901). The precise position of substantia nigra or motor cortex for stereotaxic injection was determined by MRI before injection. Ten μl viruses were injected at different locations in the right brain with the depth of needle insertion, which was calculated from the pre-operatively taken MRI. For injection of the mouse brain, the C57BL/6 mice at 8 months of age (n = 12 each group, 6 males and 6 females per group) were anesthetized by i.p. injection of 2.5% Avertin, and their heads were placed in a Kopf stereotaxic frame (Model 1900) equipped with a digital manipulator, a UMP3-1 Ultra pump, and a 10 μl Hamilton microsyringe. A 33 G needle was inserted through a 1 mm drill hole on the scalp. Injections occurred at the following stereotaxic coordinates: 3.1 mm posterior to bregma, 1.0 mm lateral to the midline, 4.7 mm ventral to the dura, with bregma set at zero. The microinjections were carried out at the rate of 0.2 μl/min. The microsyringe was left in place for an additional 10 min before and after each injection. The viruses (0.5 μl) of AAV-C-terminal TARDBP-25M337V or -SQSTM1 were stereotaxically injected into the right motor cortex of mice.
Cell culture and treatment
Mouse neural crest-derived N2a cell line and human neuroblastoma SH-SY5Y cell line were purchased from ATCC (CCL-131, CRL-2266) and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium (Thermo Fisher Scientific, 11,320,033), containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific, 15,140,163) and 0.25 μg/ml amphotericin B (Thermo Fisher Scientific, 15,290,026). The cells were maintained at 37°C in 5% CO2 incubators. Medium was changed every 2 days. For transient transfection, cells were plated at 70% confluence and transfected with plasmid DNA by using Lipofectamine 3000 (Invitrogen, L3000001) for 24–48 h. For chemical treatment, rapamycin (Selleck Chemicals, S1039), LiCl (Sigma, 7,447,418), bafilomycin A1 (Selleck Chemicals, S1413) or 3-MA (Selleck Chemicals, S2767) were used for autophagy activation or inhibition; actinomycin D (Sigma, 50,760) was used for mRNA stability assays.
siRNA knockdown
The cells were transiently transfected with human SQSTM1 target siRNA (Genepham Co. sequence CAUCUUCCGCAUCUACAUUtt or GAGGAAUUGACAAUGGCCAUGUCCU), human TARDBP target siRNA (GGUGGUGCAUAAUGGAUAUtt or GCUUUGGCUCAAGCAUGGAUUtt), or control siRNA (scrambled sequence) by using RNAi Max transfection reagent (Thermo Fisher Scientific, 13,778,150) according to the manufacturer’s protocol. At 72 h following transfection, cells were harvested and lysed in ice-cold 0.5% Triton X-100 (Sigma, 9,036,195) in 1x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) solution lysis buffer on ice. Following this, the lysates were spun at 16,873 × g for 15 min, and protein concentrations in the supernatants were determined by using BCA (bicinchoninic acid) assay (Thermo Fisher Scientific, 23,225).
Western blotting and qRT-PCR
The cultured cell or brain tissues were homogenized in the soluble fraction buffer (10 mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% SDS and 1% Triton X-100) with protease inhibitors (Sigma, P8340). The lysates were diluted in 1x SDS sample buffer and sonicated for 10 s after incubation at 100°C for 5 min. The total lysates (20 μg) were loaded in Tris-glycine gel and blotted to a nitrocellulose membrane. The Western blotting was developed by using the ECL (electro chemiluminescence) Prime Chemiluminescence kit (GE Healthcare Amersham, 28,980,926). For qRT-PCR, reverse transcription reactions were performed with 1.5 µg of RNA that was isolated using the Superscript III First-Strand Synthesis System (Invitrogen, 18,080,051). One ul of cDNA was combined with 10 µl SYBR Select Master Mix (Applied Biosystems, 4,472,908) and 1 µl of each primer in a 20 µl reaction. The reaction was performed in a real time thermal cycler (Eppendorf, Realplex Mastercycler). The sequences of primers for both monkey and mouse SQSTM1 cDNA were: Forward: CAGCCAAGCAGCTGCT and Reverse: CCGACTCCATCTGTTCCTC. The GAPDH served as an internal control. Relative expression levels were calculated using 2^-ΔΔCT, with AAV-GFP injection set at 1.
Ribonucleoprotein complex (RNP) Isolation and Microarray
Isolation of endogenous RNP complexes for RIP-chip analysis was conducted using EZ-Magna Nuclear RIP (MBL Life Science, RN1001). Briefly, the treated cells were harvested and resuspended in RNP buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, pH 7.4, 0.5% Nonidet P-40 [Solarbio Science and Technology, N8030], 10 uM dithiothreitol [DTT]) plus 400 units of RNase inhibitor (Promega, 2313B) and a protease inhibitor mixture (Sigma, P8465). Lysates were incubated for 2 h at room temperature in NT2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, 20 mM EDTA, 0.05% Nonidet P-40). The lysates were then incubated with protein G-Sepharose beads (Invitrogen, 101,241) pre-coated overnight with anti-FLAG (Cell Signaling Technology, 14,793) and anti-GFP antibodies (GeneTex, GTX113617) for exogenous TARDBP or anti-IgG (Millipore, PP64) for the negative control, to immuneprecipitate associated RNPs. After washes with ice-cold NT2 buffer, mRNA was phenol-chloroform-extracted from the immunoprecipitated RNPs after digestion with proteinase K (Invitrogen, 25,530,015) for 30 min. In parallel, proteins recovered from the immunoprecipitated RNPs were analyzed by Western blotting. The IP and input RNAs were sent to Novogene (Beijing, China) for library construction and sequencing on Illumina HiSeq 2500 or used for PCR analysis. For comparison and visualization of splicing efficiency, the intron, 3ʹUTR or 5ʹUTR retention rate were determined by calculating the ratio of fragments per kilobase million (FPKM) to the gene’s overall FPKM. The UCSC genome browser was used to analyze the general transcript reads of the associated RNAs.
For the microarray assay, the cells transfected with pRK-TARDBP-25 or pRK control were used to generate mRNA profiling. Total RNA was isolated using the RNeasy Lipid Tissue Mini Kit (Qiagen, 74,804) and sent to Novogene for library construction and sequencing on Illumina Hiseq X-Ten sequencer. For each sample, 35–50 M clean RNA-seq reads were obtained and mapped. To filter out the most captured genes, only genes with a minimum expression level of 1 count per million were included according to the “PeakScore” numbers of the previous RIP-seq analysis. The 3ʹUTR retention rate was filtered for an adjusted P value < 0.05 in the pRK-TARDBP-25 or pRK samples. After completing the above filtering, data were subsequently subjected to Ingenuity Pathway Analysis (Ingenuity Systems), and probe sets with at least 1.5-fold change were considered significant. The peak annotation of TOP-50 genes from TARDBP precipitated RNAs were distributed among all the chromosomes. The extracted heatmap indicated the changes of the most enriched TOP-50 genes.
Luciferase report assay
For luciferase experiments, the human or mouse SQSTM1 3ʹUTR was subcloned from SH-SY5Y or Neuro-2a cell line into psiCHECK (Promega, C8021) vector separately using double digest enzymes Xhol and NotI. For the human SQSTM1 3ʹUTR, the primers were: hSQSTM1-Forward: CCGCTCGAGTGACCACTTTTGCCCACCT, and hSQSTM1-Reverse: ATTTGCGGCCGCTTATTTTAAAAGCAGC; For the mouse Sqstm1 3ʹUTR, the primers were: mSqstm1-Forward: CCGCTCGAGTGATAGTGCTGTGGCCAA and mSqstm1-Reverse: ATTTGCGGCCGCTTTTATTACAGGTTACTT. HEK293 cells were plated in duplicate for each experiment and were transfected with 500 ng of each firefly luciferase SQSTM1-3ʹUTR construct (psiCHECK-human-3ʹUTR and psiCHECK-mouse-3ʹUTR). Measurement of the luciferase activity was performed using the Dual Luciferase Reporter Assay System (Promega, E1910). The firefly luciferase activity of SQSTM1 3ʹUTR constructs was normalized against the renilla luciferase output of the same psiCHECK vector.
Immunofluorescence and immunohistochemistry
Mice were anaesthetized with 5% chloral hydrate and perfused with 0.9% NaCl, followed by 4% paraformaldehyde (PFA). The brains were removed and post-fixed in 4% PFA overnight at 4°C. The brains were transferred to 30% sucrose for 48 h, then cut to 20- or 40-μm sections with the cryostat at −20°C. Sections were blocked in 4% donkey serum with 0.2% Triton X-100 and 3% BSA (bovine serum albumin, Roche Applied Science, BSAV-RO) in 1X PBS for 1 h. For immunofluorescent staining, 20-μm sections were incubated with primary antibodies in the same buffer at 4°C overnight. After washing with 1X PBS, the sections were incubated with fluorescent secondary antibodies. Fluorescent images were acquired with a Zeiss microscope (Carl Zeiss Imaging, Axiovert 200 MOT) and either a 40X lens (LD-Achroplan 40X/0.6) with a digital camera (Hamamatsu, Orca-100) and Openlab software (Improvision). For immunohistochemistry with DAB staining, 40-μm sections were incubated with antibody for at least 48 h at 4°C after pre-blocking. A biotin/avidin immuno-assay (Vector Laboratories, PK-4001) and DAB kit (Thermo Scientific, 34,002) was used for visualizing the staining. Images were acquired with a Zeiss microscope (Carl Zeiss Imaging, Imager A.2) and either a 40X or 63X lens (Plan-Apochromat 40X/0.95 or 63X/1.4) with a digital camera (Carl Zeiss Imaging, AxioCam HRc) and AxioVision software.
Statistical analyses
Statistical significance was assessed using the 2-tailed Student’s t-test whenever 2 groups were compared. When analyzing multiple groups, we used one-way ANOVA to determine statistical significance. Data were presented as mean ± SEM. Calculations were performed with GraphPad Prism software (GraphPad Inc.). A P-value less than 0.05 was considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by Key Field Research and Development Program of Guangdong province (2018B030337001), Guangzhou Key Research Program on Brain Science (202007030008), The National Key Research and Development Program of China Stem Cell and Translational Research [2017YFA0105102, 2017YFA0105201], The National Natural Science Foundation of China [81830032, 31872779, 82071421, 32070534], Department of Science and Technology of Guangdong Province [2021ZT09Y007; 2020B121201006].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
All the key data supporting the findings of this study are presented within the article and the Supplemental Information. The associated RIP and RNA sequencing reported in this paper can be found at https://doi.org/10.6084/m9.figshare.16836643.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. [DOI] [PubMed] [Google Scholar]
- [2].Chen-Plotkin AS, Lee VM, Trojanowski JQ, et al. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cohen TJ, Lee VM, Trojanowski JQ, et al. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hasegawa M, Nonaka T, Masuda-Suzukake M, et al. Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther. 2017;172:22–33. [DOI] [PubMed] [Google Scholar]
- [5].Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127(6):811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McAleese KE, Walker L, Erskine D, et al. TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 2016;27(4):472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Buratti E, Brindisi A, Giombi M, et al. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail - An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. [DOI] [PubMed] [Google Scholar]
- [8].Prakash A, Kumar V, Banerjee A, et al. Structural heterogeneity in RNA recognition motif 2 (RRM2) of TAR DNA-binding protein 43 (TDP-43): clue to amyotrophic lateral sclerosis. J Biomol Struct Dyn. 2020;39(1):357–367. [DOI] [PubMed] [Google Scholar]
- [9].Freibaum BD, Chitta RK, High AA, et al. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9(2):1104–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lagier-Tourenne C, Cleveland DW.. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14(4):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ayala YM, De Conti L, Avendano-Vazquez SE, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30(2):277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14(4):452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. Journal of Biological Chemistry. 2001;276(39):36337–36343. [DOI] [PubMed] [Google Scholar]
- [15].Tank EM, Figueroa-Romero C, Hinder LM, et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun. 2018;9(1):2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Colombrita C, Onesto E, Megiorni F, et al. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem. 2012;287(19):15635–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Costessi L, Porro F, Iaconcig A, et al. TDP-43 regulates beta-adducin (Add2) transcript stability. RNA Biol. 2014;11(10):1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Strong MJ, Volkening K, Hammond R, et al. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35(2):320–327. [DOI] [PubMed] [Google Scholar]
- [19].Volkening K, Leystra-Lantz C, Yang W, et al. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009;1305:168–182. [DOI] [PubMed] [Google Scholar]
- [20].Wang IF, Wu LS, Chang HY, et al. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105(3):797–806. [DOI] [PubMed] [Google Scholar]
- [21].Blauwendraat C, Wilke C, Simon-Sanchez J, et al. The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med. 2018;20(2):240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Deng Z, Lim J, Wang Q, et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy. 2020;16(5):917–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27(7):491–504. [DOI] [PubMed] [Google Scholar]
- [24].Cha-Molstad H, Yu JE, Feng Z, et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun. 2017;8(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Laurin N, Brown JP, Morissette J, et al. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet. 2002;70(6):1582–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446. [DOI] [PubMed] [Google Scholar]
- [27].Rubino E, Rainero I, Chio A, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79(15):1556–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].van der Zee J, Van Langenhove T, Kovacs GG, et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014;128(3):397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arai T, Nonaka T, Hasegawa M, et al. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett. 2003;342(1–2):41–44. [DOI] [PubMed] [Google Scholar]
- [30].Kawakami I, Arai T, Hasegawa M, et al. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019;138(5):751–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mann DM, Rollinson S, Robinson A, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Babu JR, Geetha T, Wooten MW, et al. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94(1):192–203. [DOI] [PubMed] [Google Scholar]
- [33].Ramesh Babu J, Lamar Seibenhener M, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;06(1):107–120. [DOI] [PubMed] [Google Scholar]
- [34].Hadano S, Mitsui S, Pan L, et al. Functional links between SQSTM1 and ALS2 in the pathogenesis of ALS: cumulative impact on the protection against mutant SOD1-mediated motor dysfunction in mice. Hum Mol Genet. 2016;25(15):3321–3340. [DOI] [PubMed] [Google Scholar]
- [35].Tanji K, Odagiri S, Miki Y, et al. p62 deficiency enhances alpha-synuclein pathology in mice. Brain Pathol. 2015;25(5):552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Doi H, Adachi H, Katsuno M, et al. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013;33(18):7710–7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gendron TF, Rademakers R, Petrucelli L, et al. TARDBP mutation analysis in TDP-43 proteinopathies and deciphering the toxicity of mutant TDP-43. J Alzheimers Dis. 2013;33(Suppl 1):S35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang C, Tong J, Bi F, et al. Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J Clin Invest. 2012;122(1):107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shan X, Chiang PM, Price DL, et al. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci U S A. 2010;107(37):16325–16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Watanabe S, Oiwa K, Murata Y, et al. ALS-linked TDP-43(M337V) knock-in mice exhibit splicing deregulation without neurodegeneration. Mol Brain. 2020;13(1). DOI: 10.1186/s13041-020-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wegorzewska I, Bell S, Cairns NJ, et al. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2009;106(44):18809–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yan S, Wang CE, Wei W, et al. TDP-43 causes differential pathology in neuronal versus glial cells in the mouse brain. Hum Mol Genet. 2014;23(10):2678–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mitchell JC, Constable R, So E, et al. Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol Commun. 2015;3(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wils H, Kleinberger G, Janssens J, et al. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci U S A. 2010;107(8):3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang G, Yang H, Yan S, et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol Neurodegener. 2015;10(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yin P, Guo XY, Yang WL, et al. Caspase-4 mediates cytoplasmic accumulation of TDP-43 in the primate brains. Acta Neuropathol. 2019;137(6):919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yin P, Bai DZ, Zhu LH, et al. Cytoplasmic TDP-43 impairs the activity of the ubiquitin-proteasome system. Exp Neurol. 2021;345:113833. [DOI] [PubMed] [Google Scholar]
- [48].Melamed Z, Lopez-Erauskin J, Baughn MW, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Klim JR, Williams LA, Limone F, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ling JP, Pletnikova O, Troncoso JC, et al. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Chang CK, Wu TH, Wu CY, et al. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem Biophys Res Commun. 2012;425(2):219–224. [DOI] [PubMed] [Google Scholar]
- [52].Mompean M, Romano V, Pantoja-Uceda D, et al. The TDP-43 N-terminal domain structure at high resolution. FEBS J. 2016;283(7):1242–1260. [DOI] [PubMed] [Google Scholar]
- [53].Qin H, Lim LZ, Wei Y, et al. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc Natl Acad Sci U S A. 2014;111(52):18619–18624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163. [DOI] [PubMed] [Google Scholar]
- [55].Ma S, Attarwala IY, Xie XQ, et al. SQSTM1/p62: a potential target for neurodegenerative disease. ACS Chem Neurosci. 2019;10(5):2094–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shin WH, Park JH, Chung KC, et al. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020;53(1):56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kabashi E, Valdmanis PN, Dion P, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. [DOI] [PubMed] [Google Scholar]
- [58].Renton AE, Chio A, Traynor BJ, et al. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Koyama A, Sugai A, Kato T, et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016;44(12):5820–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].White MA, Kim E, Duffy A, et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat Neurosci. 2018;21(4):552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gitcho MA, Bigio EH, Mishra M, et al. TARDBP 3ʹ-UTR variant in autopsy-confirmed frontotemporal lobar degeneration with TDP-43 proteinopathy. Acta Neuropathol. 2009;118(5):633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Swarup V, Phaneuf D, Dupre N, et al. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J Exp Med. 2011;208(12):2429–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Uchida A, Sasaguri H, Kimura N, et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain. 2012;135(Pt 3):833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Brady OA, Meng P, Zheng Y, et al. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116(2):248–259. [DOI] [PubMed] [Google Scholar]
- [65].Tanji K, Zhang HX, Mori F, et al. p62/sequestosome 1 binds to TDP-43 in brains with frontotemporal lobar degeneration with TDP-43 inclusions. J Neurosci Res. 2012;90(10):2034–2042. [DOI] [PubMed] [Google Scholar]
- [66].Gal J, Strom AL, Kilty R, et al. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282(15):11068–11077. [DOI] [PubMed] [Google Scholar]
- [67].Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160(1):255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kumar S, Phaneuf D, Cordeau P, et al. Induction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD. Mol Neurodegener. 2021;16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ormeno F, Hormazabal J, Moreno J, et al. Chaperone Mediated Autophagy Degrades TDP-43 Protein and Is Affected by TDP-43 Aggregation. Front Mol Neurosci. 2020;13:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jackson LK, Lin W-L, Miriyala S, et al. p62 pathology model in the rat substantia nigra with filamentous inclusions and progressive neurodegeneration. PLoS One. 2017;12(1):e0169291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mitsui S, Otomo A, Nozaki M, et al. Systemic over expression of SQSTM1/p62 accelerates disease onset in a SOD1H46R-expressing ALS mouse model. Mol Brain. 2018;11(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Seibenhener ML, Zhao T, Yf D, et al. Behavioral effects of SQSTM1/p62 overexpression in mice: support for a mitochondrial role in depression and anxiety. Behav Brain Res. 2013;248:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Aparicio R, Rana A, Walker DW, et al. Upregulation of the autophagy adaptor p62/SQSTM1 prolongs health and lifespan in middle-aged Drosophila. Cell Rep. 2019;28(4):1029–1040.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Du YF, Wooten MC, Gearing M, et al. Age-associated oxidative damage to the p62 promoter: implications for Alzheimer disease. Free Radic Biol Med. 2009;46(4):492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the key data supporting the findings of this study are presented within the article and the Supplemental Information. The associated RIP and RNA sequencing reported in this paper can be found at https://doi.org/10.6084/m9.figshare.16836643.