

Keywords: barrier, fibrosis, hypoxia-inducible factor, inflammation, inflammatory bowel disease
Abstract
The healthy mammalian intestine is lined by a single layer of epithelial cells. These cells provide a selectively permeable barrier to luminal contents and normally do so in an efficient and effective manner. Barrier function in the healthy mucosa is provided via several mechanisms including epithelial junctional complexes, mucus production, as well as mucosal-derived antimicrobial proteins. As tissue metabolism is central to the maintenance of homeostasis in the mucosa, intestinal levels are uniquely low due to counter-current blood flow and the presence of the microbiota, resulting in the stabilization of the transcription factor hypoxia-inducible factor (HIF). Ongoing studies have revealed that HIF molds normal intestinal metabolism and is central to the coordination of barrier regulation during both homeostasis and active disease. During acute inflammation, HIF is central to controlling the rapid restitution of the epithelium consistent with normal wound healing responses. In contrast, HIF may also contribute to the fibrostenotic response associated with chronic, nonresolving inflammation. As such, HIF may function as a double-edged sword in the overall course of the inflammatory response. Here, we review recent literature on the contribution of HIF to mucosal barrier function, wound healing, and fibrosis.
INTRODUCTION
A healthy intestinal barrier function is an integral aspect of mucosal tissue homeostasis. Conversely, compromised barrier is a critical component of pathological inflammation seen in diseases such as inflammatory bowel disease (IBD) (1). Intestinal repair after inflammatory insult occurs via complex and incompletely understood mechanisms, orchestrated by epithelial cells, the stromal compartment, immune cells, and cytokines (2, 3). Tissue healing after an insult is generally thought to occur in three phases: “inflammatory,” “proliferative,” and “maturation” or “remodeling” (4, 5). When this process becomes uncontrolled or overactive, pathological tissue fibrosis can result. The hallmark of fibrosis is excessive production and deposition of extra cellular matrix (ECM) components that can lead to tissue dysfunction and distortion of tissue architecture (2, 4, 5). The importance of intestinal barrier and intestinal fibrosis converge in conditions that cause chronic inflammation such as inflammatory bowel disease (IBD) (6). IBD comprises a number of inflammatory intestinal diseases, chief among them being Crohn’s disease (CD) and ulcerative colitis (UC). Loss of intestinal barrier function and fibrosis are both critical aspects of the pathogenesis, progression, and natural history of both UC and CD (2, 7–9). Despite the availability of powerful biologic and small molecule anti-inflammatory therapies, there remains a significant number of patients who fail therapy (10–12). Furthermore, the ability of biologics to effectively alter the natural history of IBD has been called into question (10, 13). In addition to difficulties in achieving and maintaining remission with regards to inflammation, a majority of patients will eventually develop complications such as fibrostenosis and/or penetrating disease requiring surgery (2). It is therefore critical to understand the complex interplay between the intestinal barrier and the development of fibrosis, and both of these entities are heavily influenced by hypoxia.
Under normal homeostatic conditions, the intestinal mucosa faces immense challenges to maintain barrier function while performing critical functions of nutrient absorption and waste removal. One prominent challenge is maintaining mucosal barrier function in the face of luminal foreign material and organisms, luminal contents that would otherwise be in constant contact with lamina propria immune cells causing persistent low-grade inflammation (14, 15). This steady state can be perturbed by a number of factors, and dysregulated host responses to gut microbes is a major factor in the pathogenesis of inflammation as seen in IBD (16–18). A second challenge is the handling of tissue oxygenation. In the intestine, blood supply and, therefore, oxygenation originates from the celiac trunk and the superior and inferior mesenteric arteries (19). The blood supply then penetrates the intestinal wall on the serosal aspect via the mesentery to form a subserous and then submucous plexus that supplies the intestinal mucosa (20). Thus, the intestinal mucosa is nearly anoxic on the luminal aspect and is highly oxygenated on the vascularized subepithelial mucosa on the basal surface, creating a steep oxygen gradient (21). Further exacerbating the challenge of oxygenation is a dynamic intestinal perfusion profile that responds to a number of inputs such as fasting and meals (21). Lastly, the microbiota of the intestine plays an important role in the oxygen regulation (22), and thus local hypoxia is one mechanism by which dysbiosis may contribute to intestinal inflammation. Given these challenges, it is no surprise that regulatory pathways for oxygenation play critical roles in intestinal homeostasis, with mucosal hypoxia contributing to pathogenesis in IBD (21, 23).
Intestinal fibrosis is the result of long-term, unresolved inflammation. Fibrosis can impact patients with both CD and those with UC, and results in tissue dysfunction, stenosis, and the formation of fistulae (2, 24). In particular, intestinal fibrosis, a very common and highly morbid complication of CD (25). Despite the availability of an ever-growing armamentarium of powerful anti-inflammatory therapies for inflammatory bowel disease, the rate of progression and complications from intestinal fibrostenotic disease remains unacceptably high, and most patients will eventually experience clinically significant fibrosis (25). Intestinal fibrosis is an active area of both clinical and preclinical research (26–36), however, there are currently no medical therapies for intestinal fibrostenosis. This is in large part due to the fact that the underlying pathogenesis of intestinal fibrosis is complex, multifactorial, and poorly understood. Given what is known about hypoxia-inducible factor (HIF) signaling and fibrosis in other organs, there is strong rationale to suggest that HIF signaling plays a role in the pathogenesis of intestinal fibrosis as seen in IBD.
During homeostasis, the distal colon exists in a state of “physiologic hypoxia” in which the epithelium normally functions through increasing O2 consumption, as β-oxidation of short-chain fatty acids yields acetyl-CoA for oxidative respiration (37). In states of intestinal inflammation, such as in IBD, hypoxic-stress emerges due to vascular disruption, influx of oxygen consuming immune cells, and infiltration of microbes (22, 23, 38, 39). Several studies have demonstrated that intestinal hypoxia during inflammation plays a critical role in healing and inflammatory resolution (5, 40, 41). Intestinal hypoxia exerts an array of cellular and tissue effects primarily through activation of NF-κB and a family of transcription factors known as hypoxia-inducible factors (HIFs) (42). HIFs have been shown to be an important protective factor in intestinal inflammation (42–44). HIFs are heterodimeric and composed of a β subunit (HIF-1β) that is stable and constitutively expressed, and an α-subunit (HIF-1α, HIF-2α, and HIF3α) that is regulated by oxygen levels (23, 45). It is important to note that the different isotypes of the α subunit (HIF-1, 2, and 3) at times confer similar or synergistic effects, but can also have opposing actions (42, 46, 47). The relationship between the isoforms is complex. For example, HIF-1α and acute activation of HIF-2α have been convincingly shown to be anti-inflammatory in the intestine, while chronic HIF-2α overexpression is a pro-inflammatory mediator (42, 48). In this review, we refer broadly to HIF signaling where the exact relationships between the isoforms is unclear but will describe the role of separate isoforms where sufficient information is available.
Under normal-oxygenated conditions (normoxia), the HIFα subunits are hydroxylated by both factor-inhibiting HIF (FIH) and prolyl-hydroxylase domain-containing proteins (PHD 1, 2, and 3) (4, 23, 49, 50). FIH hydroxylation inhibits HIFα by disrupting the interaction of HIFα transcription coactivators, whereas PHDs create a substrate for the von Hippel–Lindau (VHL) E3 ubiquitin ligase that eventually leads to degradation (4, 23, 49, 50). HIF signaling has been shown to be protective in intestinal inflammation through a number of mechanisms including enhancing barrier function (40, 51, 52), promoting wound healing, and regulating innate immunity (53, 54). Upregulation of HIF via inhibition or loss of PHDs is recognized as being protective in numerous preclinical studies of intestinal inflammation (4). In addition to a critical role in intestinal inflammation, HIF signaling has been implicated in the development of fibrosis (4, 5).
The HIF pathway exerts proangiogenic effects through activation of a numerous genes, with classical examples being vascular endothelial growth factor (VEGF), angiopoietin 1 and 2 (ANGPT1 and ANGPT2), placental growth factor (PGF), and platelet-derived growth factor B (PDGFB) (55). Activation is executed directly via HIF binding to a hypoxia responsive element (HRE), or indirectly, and is cell and tissue specific (55). In addition to these classical HIF targets, in arterial endothelial cells, HIF has been shown to regulate numerous genes encoding collagens, cytokines, growth factors, oxidoreductases, receptor tyrosine kinases, G protein-coupled receptors, and other signaling proteins (55). These targets suggest a role for hypoxic signals through HIF in the regulation of fibrosis. Indeed, HIF has been implicated as a key modulator of fibrosis in several organs including the liver, lung, and kidney (4, 5, 56). As a form of dysregulated healing, much of the pathophysiology of fibrosis is thought to be conserved across multiple organ systems. Although direct evidence of HIF’s contribution to intestinal fibrosis is lacking, there are several molecular pathways that HIF has been shown to influence that may also be profibrotic in the intestine.
In this review, we will provide an overview of the contribution of HIF to intestinal barrier function, wound healing, and the potential contribution of HIF to the progression of intestinal fibrosis. It is worth noting the influence of hypoxia and HIF is cell-type specific with multiple cell type- and context-specific inputs shaping the HIF response (57). As the intestinal epithelium and stroma are highly heterogenous tissues, HIF function may likewise be heterogenous. Where possible, we will outline distinctions as they relate to cell type and tissue function within the mucosa.
INTESTINAL BARRIER FUNCTION
Intestinal epithelial cells (IECs) of the gastrointestinal tract make up the largest single-cell-layered dynamic physical and biochemical barrier in the human body covering ∼400 m2 of surface area specialized to promote intestinal homeostasis through separating the host and regulating immune system interactions with the external environment composed of both pathogenic and commensal microorganisms (58). HIF is a critical component to a number of mechanisms that the intestinal epithelium uses to maintain barrier function. These include controlled cell renewal, the maintenance of intercellular junctions, secretion of barrier proteins including trefoil factors and mucins, and support of appropriate immune responses (Fig. 1).
Figure 1.
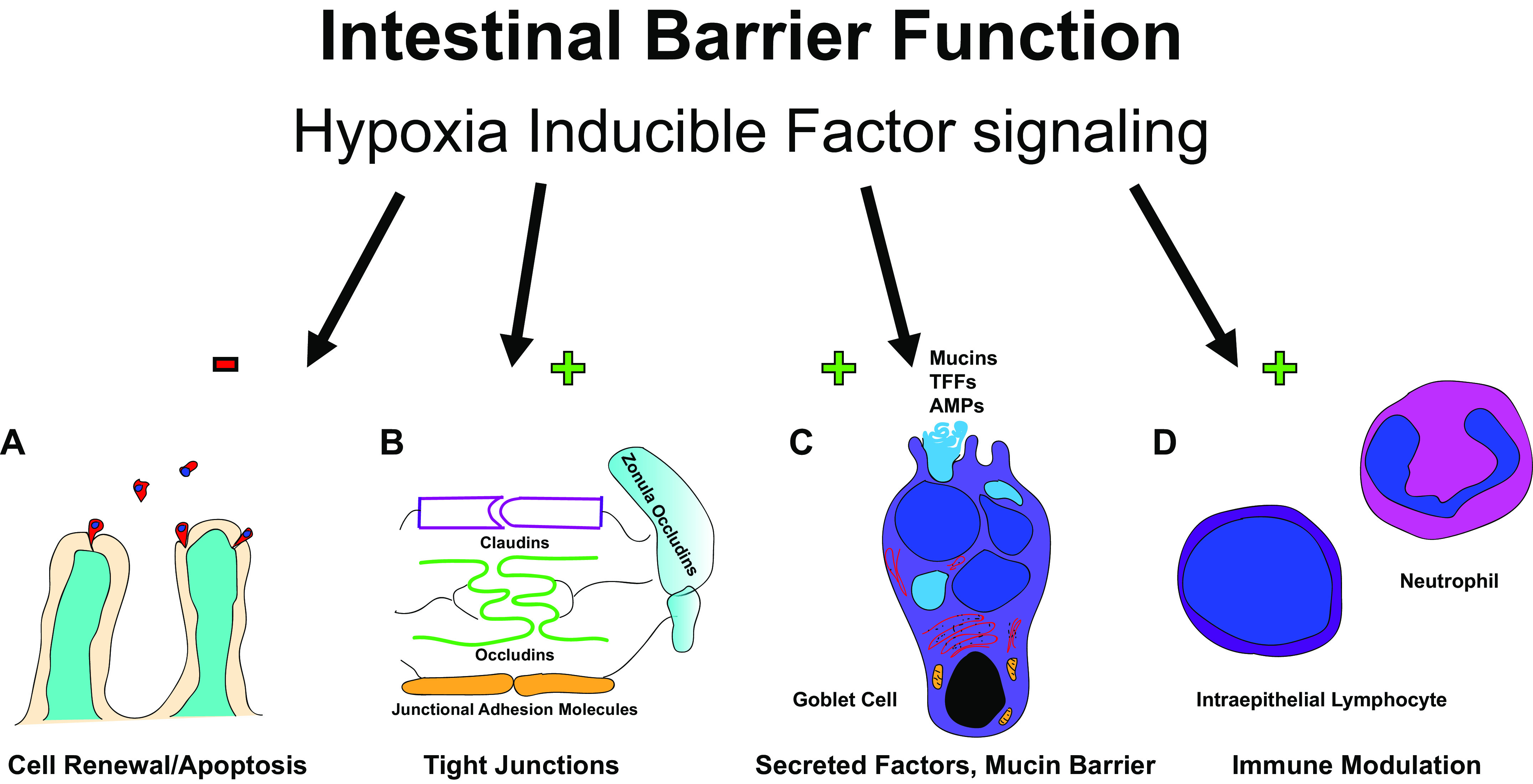
Hypoxia inducible factor (HIF) signaling effects the intestinal barrier function in a number of ways including via enhanced cellular renewal by prevention of apoptosis (A); tight junction function (B); secretion of mucous, trefoil factor proteins (TFF), and antimicrobial peptides (AMPs) (C); and modulation of intraepithelial lymphocyte and neutrophil function (D). A “+” indicates an upregulation effect, while a “–” indicates a downregulation effect.
Cell Renewal
Cell renewal is critical for the maintenance of the epithelial barrier considering the average lifespan of an IEC is 4–5 days (59). The cell renewal relies on a complex interplay between proliferation, differentiation, and apoptosis-dependent cell death. Fully differentiated IECs begin as stem cells located deep within the intestinal crypts and differentiate as they migrate towards the luminal surface. Stem cell self-renewal and migration from the crypt is a complex and highly regulated process (59, 60). As the stem cells proliferate and differentiate, mature IECs at the luminal surface undergo apoptosis to maintain tissue homeostasis. Maintenance of an effective epithelial barrier requires a balance between proliferation and apoptosis, even small alterations to this balance can result in barrier dysfunction and the potential development of disease (61, 62).
The importance of HIF signaling in the process of intestinal cellular renewal has been demonstrated in several studies. In murine models, genetic deletion of PHD1 is protective in dextran sodium sulfate (DSS) colitis through decreased apoptosis (62). The hydroxylase inhibitor dimethyloxalylglycine (DMOG) is nonspecific and likely inhibits multiple 2-oxoglutarate-dependent dioxygenases, but also robustly stabilizes HIF through inhibition of HIF prolyl hydroxylase and asparaginyl hydroxylase leading to induction of hypoxia-related genes (63). DMOG has been shown to attenuate intestinal inflammation via barrier protection from inhibiting TNFα-induced apoptosis (64). This was further explored to demonstrate a dependence on HIF-1α signaling via inhibition of Fas-associated death domain (FADD) protein. The FADD is a modulator of TNFα-mediated apoptosis, and the FADD promotor contains a HIF-1 binding site that controls repression of FADD (64). In this way, HIF signaling modulates intestinal barrier function in a protective manner.
Epithelial Tight Junctions
In addition to cell renewal, intercellular junctions are critical for epithelial barrier function. The intestinal barrier relies on tight junctions, adherens junctions, and desmosomes (65). The effect of HIF signaling on barrier is evident in in vivo models. Mice lacking intestinal epithelial HIF-1α are extremely sensitive to experimentally induced colitis using both 2,4,6-trinitrobenzene (TNBS)- and oxazalone-induced models (40). HIF-1α deficient mice have increased weight loss, decreased colon length, and increased intestinal permeability, indicating loss of barrier function. When HIF is overexpressed, mice are protected from TNBS-induced colitis and there is an overall decrease in intestinal permeability (40). The mechanisms of this protection are in large part related to the effects of hypoxia and HIF signaling on junctional proteins and functionality as has been shown using T84 epithelial cell monolayers exposed to hypoxia, PMN transmigration, and cytokine stimulation (66–68).
The sealing of the paracellular space is heavily dependent on tight junctions. Tight junctions are protein complexes formed by the interactions of multiple families of proteins including occludin, zonula occludens, and claudins (41, 69). Some aspects of this complex are membrane spanning, while others are peripheral and anchor the complex to the actin cytoskeleton (70). The functionality of tight junctions is responsive to hypoxia, and loss of HIF signaling results is loss of function and disruption of the epithelial barrier (68). The protective nature of HIF signaling on tight junctions appears to be multifactorial and includes both direct and indirect modulation of tight junction function. Claudin-1 (CLDN1) is a critical component of barrier function at tight junctions (71–73). CLDN1 is a transcriptional target of HIF and modulation of CLDN1 is a means by which HIF signaling directly effects tight junction functionality (74). Indirectly, HIF signaling promotes junctional barrier function through several processes including modulation of the autophagy pathway through ATG9 (75), nucleotide metabolism (51), overall increase in tissue energy usage, and integrity (76, 77). Thus, HIF signaling is critical in the function of tight junctions and integrity of the intestinal barrier.
Secretory Contributors to Barrier
The intestinal epithelium secretes several critical substances that contribute barrier function. Chief among them are a mucous layer and trefoil factor (TFF) peptides (68, 78, 79). There is evidence that HIF signaling is important in both of these secretory functions.
Mucins are a group of glycoproteins secreted by goblet cells that serve several functions in intestinal homeostasis. In addition to forming a physical barrier between luminal contents and the mucosa, mucins lubricate sequester antimicrobial peptides, form “decoy” sites for pathogenic bacteria to adhere, and neutralize free radicals (54, 80). Mucin 3 (MUC3) is a key cell surface mucin used by the gut. MUC3 is upregulated by hypoxia via HIF signaling in intestinal epithelial cells (52). MUC5AC is a gel-forming mucin that is an important aspect of the mucous layer in many organs (81, 82). Although not normally expressed in the intestine, MUC5AC has been shown to be upregulated in the cecum in response to intestinal nematode infection and is critical in the immune response to these enteric pathogens (83). HIF signaling also appears to have a regulatory function in the expression of mammalian MUC5AC, as the MUC5AC promotor contains a HIF-binding region (81). Conversely, MUC1 and MUC2 were not affected by hypoxia (52), and MUC1 expression has been shown to block activation and reduce stabilization of HIF-1α in the HCT116 colon cancer line (84). MUC2 is the primary gel-forming mucin in the intestine, while MUC1 is a transmembrane mucin expressed on the apical aspect of epithelial cells (85). Thus, the relationship between mucin expression and HIF signaling is a complicated one and more work is needed to elucidate the exact effects they have on one another.
Trefoil factor (TFF) peptides are a large family mucin-associated proteins that are important in the mucosal response to injury, inflammation, and host defense (78). These proteins modulate mucin structure and are resistant to proteolysis and low pH (86–88). TFF3 is the primary trefoil factor in the intestine and is sometimes referred to as intestinal trefoil factor (ITF) (78, 89). TFF3 has a demonstrable effect on the maintenance of intestinal homeostasis, as TFF3-null mice have been shown to have worse outcomes in dextran sodium sulfate models of intestinal inflammation and also fare worse when exposed to hypoxic conditions (79). Trefoil factor-3 is a known HIF-1α target gene, and is induced in hypoxic states (90, 91). This has been demonstrated in both CaCo and T84 intestinal epithelial cell lines, with induction of TFF3 by hypoxia and discovery of a HIF-1α binding site on the TFF3 promoter (92). Further, knockdown or over-expression of HIF-1α caused a subsequent decrease or increase in TFF3 protein levels, respectively. Thus, modulation of TFF expression is another mechanism by which HIF signaling contributes to barrier function in the intestinal mucosa.
Immune Function
HIF signaling has been shown to be important in both innate aspects of immune function and modulation of lymphocyte behavior at the intestinal epithelium. Antimicrobial peptides (AMPs) are a key component of the innate immune defense at the intestinal barrier. They are stably maintained in the mucous layer of the intestine and serve as a first-line defense against luminal organisms (93). Defensins, along with cathelicidins, represent the two most critical AMPs in mammals (93, 94). Human β defensin-1 (hBD1) is constitutively expressed in the intestine, and research has shown that its homeostatic expression is dependent on HIF-1α in multiple cell types (54, 95, 96). In neutrophils and macrophages, loss of HIF-1 resulted in dramatic reductions in ATP, impaired intracellular killing of pathogens, and impaired aggregation, motility, and invasion (97). This demonstrates that in myeloid populations, HIF-1 is critical for glycolysis and antimicrobial activity (97, 98). Other groups have shown that HIF-1α and HIF-2α have important roles in neutrophil functioning (98, 99). Macrophages contribute to innate immunity in the intestinal epithelium through their role in phagocytosis of pathogens and immune surveillance (100). Data from tissues other than the intestinal epithelium has shown that HIF signaling is critical in the activation of function of macrophages through several mechanisms, including activation through sphingosine 1-phosphate (S1P) signaling (101), metabolic modulation such as induction of glycolysis (102), and modulation of cytokine expression (103). This evidence supports an important role of HIF signaling in the innate immune aspect of the intestinal barrier.
In addition to innate immune components, HIF influences intraepithelial lymphocyte homeostasis and neutrophil contribution to inflammation. For example, conditional deletion of HIF-1α in IECs caused changes in the phenotype of IELs and increased susceptibility to inflammation in two murine models of intestinal inflammation (104). The phenotypic changes are due to changes in cytokine expression from IECs including increased IL-10 expression and decreased IL-7 and IL-15 expression, increased number of IELs in the small intestine and colon, and changes in the proportions of CD8αα+, CD8αβ+, TCRγδ+, and CD4+ IELs. Furthermore, some types of CD8+ cells with conditional deletion of HIF-1α had increased apoptosis, poor proliferation, reduced ability for gut homing, decreased IFN-γ and IL-2 expression, and/or increased keratinocyte growth factor (KGF) and regenerating islet-derived protein III γ (RegIIIγ) (104). In T cells, HIF-1 mediates cytokine regulation during inflammation, with loss of HIF-1 signaling in T cells resulting in worsening of chemically induced DSS colitis (105). The worsening colitis was demonstrable through increased weight loss, shortened and more swollen colon, and more severe histologic inflammation in the HIF-1α−/− mice treated with DSS compared with WT treated with DSS. Mechanistically, this is thought to be due to several changes in cytokine expression. These include increased Foxp3, IL-10, IL1β, IL-6, IL-17, IL-23, IFN-γ, IL-12a, and TNFα, but decreased RAR-related orphan receptor γ (RORγt). This is thought to result in an increase in Th17 cells and a decrease in Tregs, altering the balance in favor of a more proinflammatory milieu (105). In a separate study, HIF-1 was again shown to induce the transcriptional regulator of T-cells FoxP3, and HIF-1α-deficient regulatory T cells failed to effectively control intestinal inflammation (106). There is also clear evidence from nonintestinal studies that HIF signaling is critical in effector T-cell function in humoral immunity via actions on metabolism and modulation of cytokines (107, 108). The importance of HIF in lymphocytes is not limited to T cells, with emerging evidence that HIF signaling plays a key role in development, differentiation, and interleukin-10 production in B cells (95, 96). Thus, HIF signaling carries significance in a number of facets of the intestinal barrier’s immune characteristics and ability to deal with luminal microbes.
WOUND HEALING
Intestinal barrier function and appropriate wound healing responses require sufficient nucleotide generation to provide RNA for protein transcription and DNA for proliferation. Likewise, high levels of ATP are needed to maintain the balance of energy necessary for efficient cytoskeletal function to allow wound restitution (109). Since 1998, it has been known that adenine nucleotides (specifically ATP and ADP) significantly enhance IEC migration and promote structural and functional regeneration in vivo (110). Thus, nucleotides and their components are of tremendous importance in eukaryotic cells. Here we will review recent studies that implicate both host and microbial sources of metabolites in the promotion of mucosal wound healing responses (Fig. 2).
Figure 2.
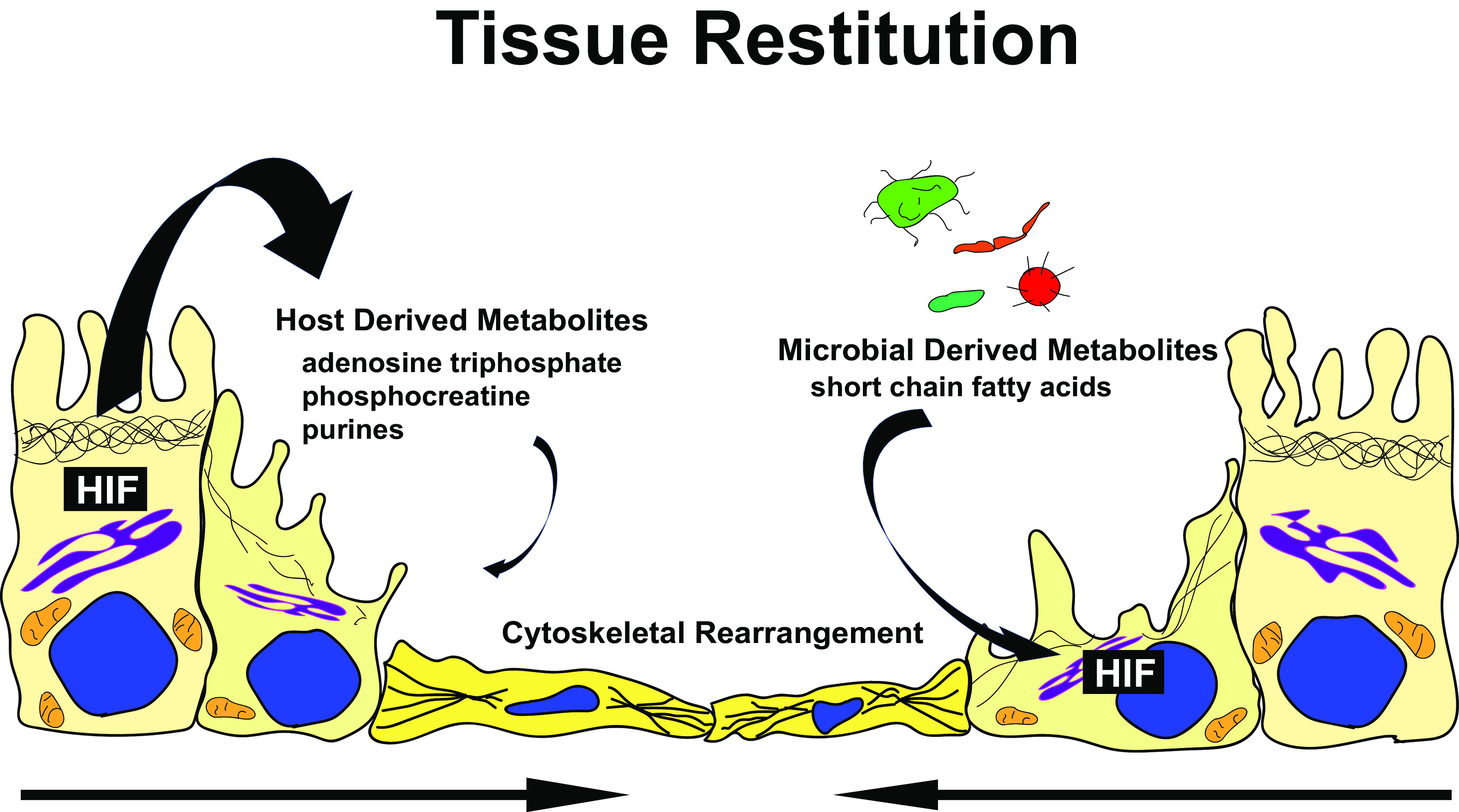
Hypoxia inducible factor (HIF) influences intestinal wound healing through modulating response to host and microbial-derived metabolites. Among other functions (see text), these metabolites significantly impact the epithelial cytoskeleton and promote cell migration and wound healing.
Host-Derived Metabolites
More than 40 years ago, it was proposed that defects in fatty acid oxidation by epithelial cells from ulcerative colitis patients implicated IBD is an “energy deficiency disease” (111). It is now appreciated that the provision of barrier function and wound healing places a particularly high energy demand on epithelial cells, requiring up to 25% of cellular ATP for homeostatic maintenance alone (112). Such metabolic demands have become an essential part of the overall conversation of mucosal barrier and particularly wound healing.
Original studies using models of ATP revealed an essential role for high-energy phosphates in barrier function (77). These studies inspired further analysis of hypoxia adaptation to such conditions where energy demands were increased. A genome-wide profiling of intestinal epithelial HIF-regulated gene promoters identified cytosolic creatine kinases (CK) as HIF-2 selective targets. Interestingly, CK localized within the adherens junction of confluent intestinal epithelia. Subsequent studies in the mucosa revealed that each of the CK isoforms (i.e., muscle, brain, and mitochondrial) were expressed in cultured intestinal epithelial cell lines, murine colonic epithelia, and human colonic epithelia (77). Creatine (Cr) and phosphocreatine (PCr) levels are regulated to near equilibrium, thereby buffering the high energy phosphate states of ADP and ATP and supporting the function numerous of cellular ATPases (113). Active inflammation represents a high-energy state accounted for by wound healing and restitution functions of epithelial cells following insult. Under such conditions, energy expenditure for cell migration and the re-establishment of epithelial cell–cell junction is tightly linked to the circumferential F-actin belt (70). At the expense of significant ATP, it has been shown that supplementation with Cr promotes wound healing in murine models of colitis. More recently, it was shown that creatine transport into intestinal epithelia is essential for both barrier function and effective wound healing responses (114). Indeed, extensions of the genome-wide profiling described above identified the major Cr transporter SLC6A8 as a HIF-regulated gene. Targeted knockdown and genetic knockout of SLC6A8 in cultured intestinal epithelia and murine colonoids, respectively, revealed a significant loss of barrier and wound healing responses. Likewise, the expression of the Cr components (CK and SLC6A8) has been shown to be substantially decreased in biopsies from patients with IBD (77, 114). Thus, it would appear that Cr–PCr ratios may serve as functional biomarkers of cellular energy demand that could be targetable in ways to promote tissue epithelial wound healing in the mucosa.
There is recent interest in understanding alternative sources of energy for purposes of wound healing. This has led to novel techniques such as using an HPLC-based profiling approach to monitor changes in high-energy phosphates and adenylate metabolites (112). This technique led to the discovery that hypoxanthine (Hpx) is a checkpoint metabolite in IEC function. These studies showed that Hpx promotes cellular energetics to support cytoskeletal function and significantly accelerated wound closure rates. In addition, it was shown that Hpx increases the epithelial cellular energetics to the extent that elevations in PCr and ATP result in overall increased total available energy. Finally, a metabolomic screen of healthy and colitic murine colon tissue revealed a >65% decrease in Hpx during active inflammation. Furthermore, the loss of Hpx strongly correlated with disease markers (e.g., weight loss, colon length) (112). Thus, purine salvage in the generation of energy resources appears to be an essential part of the normal wound healing response.
Microbial-Derived Metabolites
The mammalian gastrointestinal tract serves as a host to trillions of microbes, including fungi, viruses, and bacteria. This finely tuned host-microbe interaction depends upon complex metabolic interactions, and it is now appreciated that microbes are essential components of human health (115). One class of microbial metabolites of particular interest for barrier and wound healing are the short-chain fatty acids (SCFAs).
SCFAs are a requisite waste product to balance redox equivalent production in the anaerobic lumen of the gut. The SCFAs are classified as saturated aliphatic carboxylic aids that are between one to six carbons and consist of acetate (C2), propionate (C3), butyrate (C4), valerate (C5), and hexanoate (C6). Acetate, propionate, and butyrate are the most abundant and comprise >95% of SCFAs and exist in a molar colonic ratio of ∼60:20:20, with total SCFAs reaching up to 140 mM in the proximal colon and 70 mM in the distal colon (116). The majority of SCFAs are rapidly absorbed by the colonocytes with only 5%–10% secreted in feces. These SCFAs have a significant impact on host physiology as energy substrates, regulators of gene expression, and signaling molecules recognized by specific receptors (117–120). Butyrate, in particular, functions as the preferred energy source of the colonic epithelium, with oxidation of this SCFA accounting for over 70% of the cellular oxygen consumption in the distal colon (121). Colonocytes use butyrate over other SCFAs acetate and propionate, where it is oxidized to ketone bodies and CO2. Greater than 95% of produced butyrate is used by colonocytes for energy and functional analyses that have revealed that butyrate selectively promotes epithelial wound healing responses over other SCFAs. To define mechanisms of this response, an unbiased single-cell RNA sequencing approach has identified a cluster of actin-associated genes regulated by butyrate, and among these, showed the selective induction of synaptopodin (SYNPO) as a new intestinal tight junction protein with a central role in epithelial barrier restitution (122).
The relationship of intestinal butyrate and HIF lies at the intersection of metabolism and gene regulation. Although butyrate functions predominantly as a colonocyte fuel, it was shown to stabilize HIF at physiologic concentrations (123). Studies that limit β-oxidation and consequent oxygen consumption by butyrate, (e.g., methylenecyclopropylacetic acid) nonetheless revealed HIF stabilization and induction of HIF-target genes in colonic epithelial cells. Mechanisms of this response tracked to direct inhibition of HIF PHD protein by butyrate (124), thus identifying a unique evolutionary relationship where a microbial metabolite endogenously communicates transcriptional signaling as a means to promote mucosal barrier function.
INTESTINAL FIBROSIS
The pathogenesis of intestinal fibrosis is incompletely understood, but several known modulators are intricately linked to HIF signaling. Given the importance of oxygen on cellular activity and survival, as well as the role of hypoxia in inflammation that often precedes fibrosis, it is no surprise that hypoxia has been identified as a key component in the fibrotic microenvironment (5). Hypoxia exerts effects through NF-κB, TGFβ1, and HIF. The role of HIF signaling in this process includes induction of epithelial to mesenchymal transition, interactions with TGFβ1 signaling, effect on AXL signaling, and transglutaminase 2 (TG2) (Fig. 3).
Figure 3.
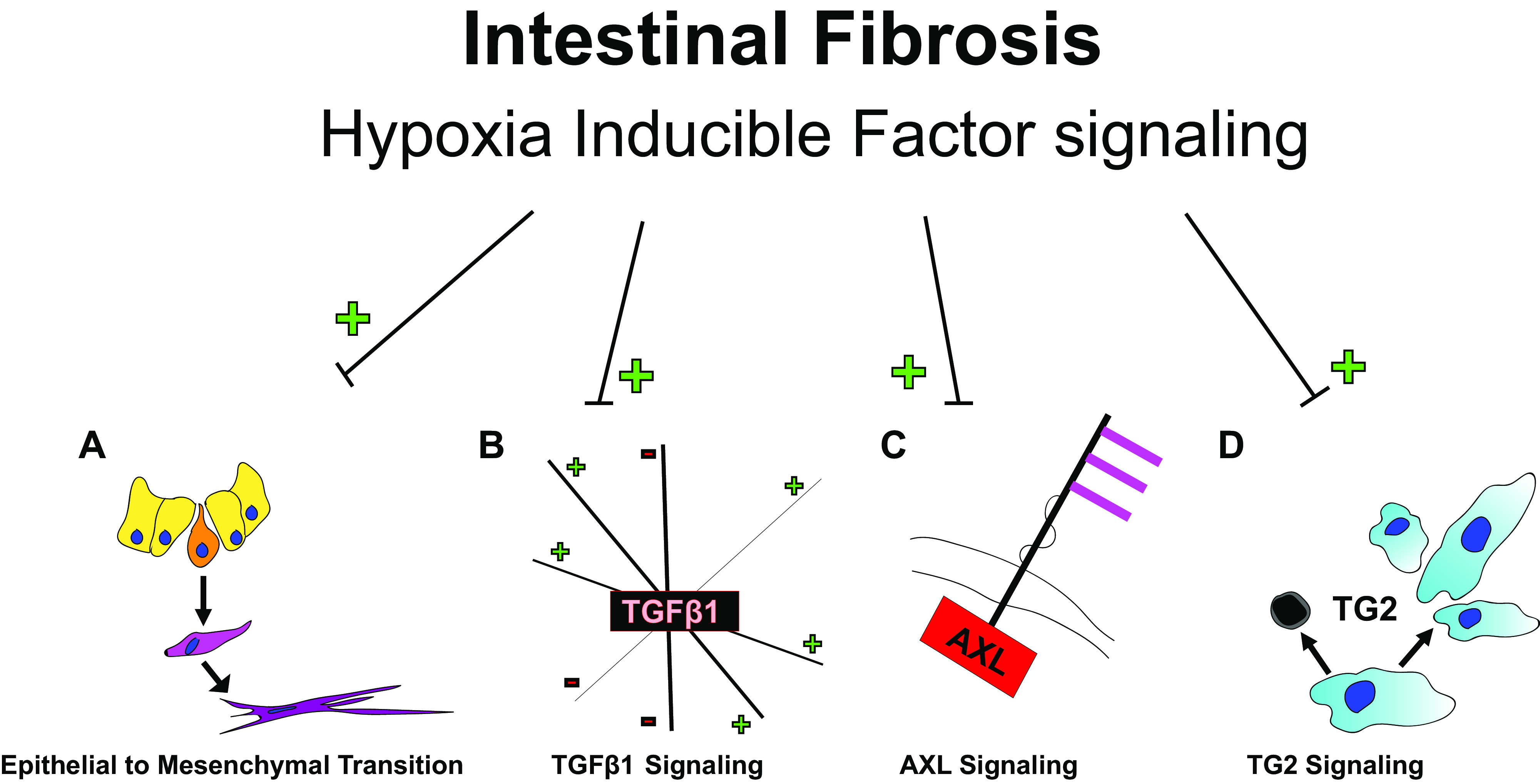
Hypoxia inducible factor (HIF) effects intestinal fibrosis in a number of ways including epithelial to mesenchymal transition (A); TGFβ1, which has numerous and complicated interactions that can be either upregulated or inhibitory (B); AXL (C); and transglutaminase 2 (TG2) (D). A “+” indicates an upregulation effect, while a “–” indicates a downregulation effect.
Epithelial to Mesenchymal Transition
Epithelial to mesenchymal transition (EMT) is the process by which a differentiated epithelial cell transforms into a mesenchymal phenotype (125). This transformation lends the cell increased capacity for migration, invasion, production of extracellular matrix, and resistance to apoptosis as well as loss of cell polarity (125, 126). This process is a critical component of embryogenesis as well as healing and regeneration (125, 127). EMT can be induced by a number of stimuli and is also thought to be a component of normal tissue restitution and healing (125, 126). EMT is also important in the pathogenesis of various cancers (128). In addition to its importance in normal physiology and the pathogenesis of numerous cancers, EMT is also a critical component in the pathogenesis of intestinal fibrosis, (126) as well as fibrosis of the liver (129), lung (130), and kidney (131, 132).
The transcription factors Snail (SNAI1), Slug (SNAI2), zinc finger E-box-binding homeobox 1/2 (ZEB1/2), survival of motor neuron protein-interacting protein 1 (SIP1), and Twist (TWIST1) are well described as inducers of EMT (125, 133–136). Snail expression is induced by hypoxia via an HRE within the promoter that is activated by HIF-1α and HIF-2α (137). In hepatocellular carcinoma (HCC), hypoxia-stabilized HIF-1α has been shown to induce Snail transcription and promote EMT (138). Lastly, TGFβ1 is a well-described inducer of EMT and is intimately intertwined with HIF signaling (139). In murine cells, coronary endothelial cells, ovarian cancer, and pancreatic cancer, snail has been shown to be regulated by and/or a direct target of HIF signaling (137, 140–142). In addition, slug and twist functioning have been linked to the actions of HIF signaling (143–145). It is, therefore, clear that HIF signaling plays a critical role in the regulation of EMT. Thus far, the role of HIF signaling in EMT in the intestine must be largely inferred, as direct studies in the intestine are lacking. Thus, HIF is an attractive target of future research into the role of hypoxia and HIF signaling in intestinal EMT.
TGFβ1
In addition to actions on EMT, TGFβ1 signaling is involved in a wide array of physiologic pathways and is a major regulator of fibrogenesis (146, 147). Although admittedly a gross over-simplification, TGFβ1 can be thought of as profibrotic, and is used experimentally to induce profibrotic states in the preclinical setting (148, 149). The relationship between hypoxia, HIF signaling, and TGFβ1 signaling has been studied extensively. In many studies, these pathways are found to be synergistic in their effects through several different mechanisms. Hypoxia has been shown to upregulate TGFβ1 expression, and the TGFβ1 promotor contains an HRE that interacts directly with HIF-1α (150). In addition, the TGFβ1 precursor is converted to mature TGFβ1 via the action of furin (151). The promoter of furin is also directly acted on by HIF-1 through a canonical hypoxia responsive element region (152). In return, TGFβ1 has been found to inhibit PHD2 through the suppressor of mothers against decapentaplegic (SMAD)-signaling pathway (153). In renal epithelial cells, TGFβ1/SMAD activity is upregulated by HIF-1α (154), and HIF-1α and SMAD3 signaling have been shown to be necessary for TGFβ1-mediated collagen deposition and fibrogenesis (155). Similarly, hypoxia-induced TGFβ1/SMAD activity via HIF signaling was shown to promote collagen deposition in dermal fibroblasts (156). In preclinical models of pulmonary fibrosis, HIF-1α has been shown to modulate TGFβ1 activity (157). The synergistic interaction of the TGFβ1 type 1 receptor (ALK5) and HIF signaling has been associated with a worse prognosis in clear cell renal cell carcinoma (158). HIF-1α has also been shown to alter the metabolic programming of non-small-cell lung cancer through interactions with TGFβ1/SMAD signaling. Through this cycle, TGFβ1 and HIF-1α signaling appear to potentiate each other.
The influence of these molecular interactions can be seen in several experimental models, and the synergistic effects of this interaction are prominent in the development and prognosis of several different malignancies. Conversely, hydroxylase inhibition has been shown to inhibit TGFβ1-related ERK activation of intestinal fibroblasts, (159) and in colorectal cancer patients, crosstalk between TGFβ1 and HIF-1α may actually improve patient survival (160). These seemingly opposing characteristics highlight the complexity of the relationship between HIF signaling and fibrosis.
Receptor Tyrosine Kinase AXL
AXL is a cell-surface tyrosine kinase named for the Greek word anexelkto, meaning uncontrolled. AXL is a member of the Tyro3-AXL-Mer (TAM) family and has a role in multiple cellular processes and functions such as migration, invasion, proliferation, survival, adhesion, and EMT. It has been shown to be a downstream effector of TGFβ1 signaling in both breast cancer and hepatocellular carcinoma. The primary ligand of AXL is thought to be growth arrest-specific 6 (GAS6). The GAS6/AXL pathway is known to contribute to human malignancy and metastasis (161), and several processes that are important in malignancy are also active in fibrosis such as cell survival, resistance to apoptosis, proliferation, and migration. Furthermore, AXL has been shown to be a direct HIF target in the renal cell carcinoma cell line RCC4, with a hypoxia-response element located in the AXL promotor (162). When evaluating fibrosis specifically, AXL signaling has been shown to contribute to hepatic fibrosis, and AXL inhibition resulted in inactivation of hepatic stellate cells (163). Translating to intestinal pathology, AXL is active in multiple models of intestinal fibrosis, and pharmacological inhibition of AXL is antifibrotic in vitro (148). More work is needed to elucidate the role of GAS6 and AXL and inhibition of GAS6/AXL in intestinal fibrosis, and with AXL inhibition already being actively studied for treatment in a number of malignancies, more information on this promising target is sure to come (164).
Transglutaminase 2
Transglutaminase 2 (TG2) is the most widely expressed member of the transglutaminase family of proteins with a wide array of functions including modulation of cell survival and death (165). TG2 can in fact be pro-cell death or pro-survival depending on the level of transamidating activity (166). Further, TG2 is known to be important in the pulmonary fibrosis (167) and inhibition of TG2 has been shown to ameliorate cardiac fibrosis (168). HIF-1β is a direct-binding partner of TG2, and interactions of TG2 and HIF can either induce or abrogate ischemic-related cell death depending on the context of the interaction (166, 169). This again represents a strong link between HIF signaling and fibrosis in organs other than the intestine and represents a promising avenue of investigation to better understand the mechanisms of intestinal fibrosis.
Conclusions
Epithelial cells that line the intestinal mucosa function in an austere and often harsh environment. The major function of the epithelium is to provide a selectively permeable barrier that allows for nutrient absorption and waste excretion while simultaneously preventing the recognition of luminal contents by the immune cells of the lamina propria and resultant inflammation. It is now appreciated that the steep gradient of O2 between the lumen and the serosa as well as shifts in energy requirements during mucosal injury have revealed important clues about tissue metabolism at homeostasis and in various disease states. Given its central role at the interface of metabolism and immunity, HIF has proven to be an important player at the interface of the mucosa. Notably, HIF may serve as a double-edged sword in this environment (Fig. 4). On the one hand, HIF transcriptional targets have proven to promote both barrier function in homeostasis and wound healing during acute inflammatory disease. On the other hand, existing studies in several tissues suggest that chronic HIF stabilization may support processes that promote fibrostenosis. This duality will be a major consideration as these agents are explored for treatment of IBD, especially for CD as a majority of CD patients do eventually develop penetrating and/or fibrostenotic disease (2). There is a possibility that any augmentation of HIF signaling in some IBD patients may hasten or worsen the development of fibrostenotic complications, although pre-clinical studies of colitis report a net antifibrotic effect (159). One potential approach is to clearly elucidate the role of timing and the interplay of HIF effects in acute versus chronic inflammation. Given the clear beneficial effects in early inflammation and restitution and role of chronicity in fibrosis, we hypothesize that there may be several opportunity windows for using agents that augment HIF signaling followed by withdrawal of these agents or even inhibition of HIF signaling. As clinical trials using both HIF-stabilizing agents (PHD inhibitors) (170) and direct HIF inhibitors (171) are currently ongoing, it would seem reasonable that intestinal diseases (e.g., IBD) could benefit from their further development and more research on the complex interplay of HIF signaling in barrier function, healing, and fibrosis of the intestine.
Figure 4.

Hypoxia inducible factor (HIF) signaling is a double-edged sword in the intestinal mucosa. HIF signaling has a positive effect on barrier and overall function during homeostasis and positive effects on healing and restitution during acute inflammation. Conversely, in the setting of chronic inflammation, HIF signaling is profibrotic and contributes to a dysregulated response to injury.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants DK104713, DK050189, DK122741, DK1200720, DK09549, VA Merit Award 1I01BX002182, and IK2BX005710.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.A.S. and S.P.C. conceived and designed research; C.A.S. and S.P.C. prepared figures; C.A.S., I.M.C., and S.P.C. drafted manuscript; C.A.S., I.M.C., C.T.T., and S.P.C. edited and revised manuscript; C.A.S., I.M.C., C.T.T., and S.P.C. approved final version of manuscript.
REFERENCES
- 1. Luissint AC, Parkos CA, Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology 151: 616–632, 2016. doi: 10.1053/j.gastro.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rieder F, Fiocchi C, Rogler G. Mechanisms, management, and treatment of fibrosis in patients with inflammatory bowel diseases. Gastroenterology 152: 340–350.e6, 2017. doi: 10.1053/j.gastro.2016.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xue X, Falcon DM. The role of immune cells and cytokines in intestinal wound healing. Int J Mol Sci 20: 6097, 2019. doi: 10.3390/ijms20236097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Strowitzki MJ, Ritter AS, Kimmer G, Schneider M. Hypoxia-adaptive pathways: a pharmacological target in fibrotic disease? Pharmacol Res 147: 104364, 2019. doi: 10.1016/j.phrs.2019.104364. [DOI] [PubMed] [Google Scholar]
- 5. Manresa MC, Godson C, Taylor CT. Hypoxia-sensitive pathways in inflammation-driven fibrosis. Am J Physiol Regul Integr Comp Physiol 307: R1369–R1380, 2014. doi: 10.1152/ajpregu.00349.2014. [DOI] [PubMed] [Google Scholar]
- 6. Sommer K, Wiendl M, Müller TM, Heidbreder K, Voskens C, Neurath MF, Zundler S. Intestinal mucosal wound healing and barrier integrity in IBD-crosstalk and trafficking of cellular players. Front Med (Lausanne) 8: 643973, 2021. doi: 10.3389/fmed.2021.643973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antoni L, Nuding S, Wehkamp J, Stange EF. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol 20: 1165–1179, 2014. doi: 10.3748/wjg.v20.i5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gordon IO, Agrawal N, Goldblum JR, Fiocchi C, Rieder F. Fibrosis in ulcerative colitis: mechanisms, features, and consequences of a neglected problem. Inflamm Bowel Dis 20: 2198–2206, 2014. doi: 10.1097/MIB.0000000000000080. [DOI] [PubMed] [Google Scholar]
- 9. Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol 4: 280, 2013. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Privitera G, Pugliese D, Lopetuso LR, Scaldaferri F, Neri M, Guidi L, Gasbarrini A, Armuzzi A. Novel trends with biologics in inflammatory bowel disease: sequential and combined approaches. Therap Adv Gastroenterol 14: 17562848211006669, 2021. doi: 10.1177/17562848211006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Volk N, Siegel CA. Defining failure of medical therapy for inflammatory bowel disease. Inflamm Bowel Dis 25: 74–77, 2019. doi: 10.1093/ibd/izy238. [DOI] [PubMed] [Google Scholar]
- 12. Roda G, Jharap B, Neeraj N, Colombel JF. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol 7: e135, 2016. doi: 10.1038/ctg.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Targownik LE, Tennakoon A, Leung S, Lix LM, Singh H, Bernstein CN. Temporal trends in initiation of therapy with tumor necrosis factor antagonists for patients with inflammatory bowel disease: a population-based analysis. Clin Gastroenterol Hepatol 15: 1061–1070.e1, 2017. doi: 10.1016/j.cgh.2017.01.035. [DOI] [PubMed] [Google Scholar]
- 14. Poonam P. The biology of oral tolerance and issues related to oral vaccine design. Curr Pharm Des 13: 2001–2007, 2007. doi: 10.2174/138161207781039814. [DOI] [PubMed] [Google Scholar]
- 15. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol 124: 3–20, 2009. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tamboli CP, Neut C, Desreumaux P, Colombel JF. Dysbiosis in inflammatory bowel disease. Gut 53: 1–4, 2004. doi: 10.1136/gut.53.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kaur N, Chen CC, Luther J, Kao JY. Intestinal dysbiosis in inflammatory bowel disease. Gut Microbes 2: 211–216, 2011. doi: 10.4161/gmic.2.4.17863. [DOI] [PubMed] [Google Scholar]
- 18. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol 11: 1–10, 2018. doi: 10.1007/s12328-017-0813-5. [DOI] [PubMed] [Google Scholar]
- 19. Geboes K, Geboes KP, Maleux G. Vascular anatomy of the gastrointestinal tract. Best Pract Res Clin Gastroenterol 15: 1–14, 2001. doi: 10.1053/bega.2000.0152. [DOI] [PubMed] [Google Scholar]
- 20. Kachlik D, Baca V, Stingl J. The spatial arrangement of the human large intestinal wall blood circulation. J Anat 216: 335–343, 2010. doi: 10.1111/j.1469-7580.2009.01199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) 85: 1295–1300, 2007. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 22. Singhal R, Shah YM. Oxygen battle in the gut: hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J Biol Chem 295: 10493–10505, 2020. doi: 10.1074/jbc.REV120.011188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Welden S, Selfridge AC, Hindryckx P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 14: 596–611, 2017. doi: 10.1038/nrgastro.2017.101. [DOI] [PubMed] [Google Scholar]
- 24. Rieder F, Fiocchi C. Intestinal fibrosis in IBD–a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol 6: 228–235, 2009. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 25. Li J, Mao R, Kurada S, Wang J, Lin S, Chandra J, Rieder F. Pathogenesis of fibrostenosing Crohn's disease. Transl Res 209: 39–54, 2019. doi: 10.1016/j.trsl.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 26. Bettenworth D, Bokemeyer A, Baker M, Mao R, Parker CE, Nguyen T, Ma C, Panes J, Rimola J, Fletcher JG, Jairath V, Feagan BG, Rieder F, Stenosis T, Anti-Fibrotic Research C; Stenosis Therapy and Anti-Fibrotic Research (STAR) Consortium. Assessment of Crohn’s disease-associated small bowel strictures and fibrosis on cross-sectional imaging: a systematic review. Gut 68: 1115–1126, 2019. doi: 10.1136/gutjnl-2018-318081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gordon IO, Bettenworth D, Bokemeyer A, Srivastava A, Rosty C, de Hertogh G, Robert ME, Valasek MA, Mao R, Li J, Harpaz N, Borralho P, Pai RK, Odze R, Feakins R, Parker CE, Guizzetti L, Nguyen T, Shackelton LM, Sandborn WJ, Jairath V, Baker M, Bruining D, Fletcher JG, Feagan BG, Pai RK, Rieder F; Stenosis Therapy and Anti-Fibrotic Research (STAR) Consortium. International consensus to standardise histopathological scoring for small bowel strictures in Crohn’s disease. Gut 71: 479–486, 2022. doi: 10.1136/gutjnl-2021-324374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steiner CA, Berinstein JA, Louissaint J, Higgins PDR, Spence JR, Shannon C, Lu C, Stidham RW, Fletcher JG, Bruining DH, Feagan BG, Jairath V, Baker ME, Bettenworth D, Rieder F, Stenosis T, Anti-Fibrotic Research C. Biomarkers for the prediction and diagnosis of fibrostenosing Crohn’s disease: a systematic review. Clin Gastroenterol Hepatol 20: 817–846, 2021. doi: 10.1016/j.cgh.2021.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Johnson LA, Luke A, Sauder K, Moons DS, Horowitz JC, Higgins PD. Intestinal fibrosis is reduced by early elimination of inflammation in a mouse model of IBD: impact of a “Top-Down” approach to intestinal fibrosis in mice. Inflamm Bowel Dis 18: 460–471, 2012.doi: 10.1002/ibd.21812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson LA, Rodansky ES, Moons DS, Larsen SD, Neubig RR, Higgins PDR. Optimisation of intestinal fibrosis and survival in the mouse S. typhimurium model for anti-fibrotic drug discovery and preclinical applications. J Crohns Colitis 11: 724–736, 2017. doi: 10.1093/ecco-jcc/jjw210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson LA, Rodansky ES, Sauder KL, Horowitz JC, Mih JD, Tschumperlin DJ, Higgins PD. Matrix stiffness corresponding to strictured bowel induces a fibrogenic response in human colonic fibroblasts. Inflamm Bowel Dis 19: 891–903, 2013. doi: 10.1097/MIB.0b013e3182813297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodansky ES, Johnson LA, Huang S, Spence JR, Higgins PD. Intestinal organoids: a model of intestinal fibrosis for evaluating anti-fibrotic drugs. Exp Mol Pathol 98: 346–351, 2015. doi: 10.1016/j.yexmp.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stidham RW, Xu J, Johnson LA, Kim K, Moons DS, McKenna BJ, Rubin JM, Higgins PD. Ultrasound elasticity imaging for detecting intestinal fibrosis and inflammation in rats and humans with Crohn’s disease. Gastroenterology 141: 819–826.e1, 2011. doi: 10.1053/j.gastro.2011.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Dejanovic D, Zangara MT, Chandra J, McDonald C, Rieder F. Mouse models of intestinal fibrosis. Methods Mol Biol 2299: 385–403, 2021. doi: 10.1007/978-1-0716-1382-5_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li XH, Feng ST, Cao QH, Coffey JC, Baker ME, Huang L, Fang ZN, Qiu Y, Lu BL, Chen ZH, Li Y, Bettenworth D, Iacucci M, Sun CH, Ghosh S, Rieder F, Chen MH, Li ZP, Mao R. Degree of creeping fat assessed by computed tomography enterography is associated with intestinal fibrotic stricture in patients with Crohn’s disease: a potentially novel mesenteric creeping fat index. J Crohns Colitis 15: 1161–1173, 2021. doi: 10.1093/ecco-jcc/jjab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vieujean S, Hu S, Bequet E, Salee C, Massot C, Bletard N, Pierre N, Quesada Calvo F, Baiwir D, Mazzucchelli G, De Pauw E, Coimbra Marques C, Delvenne P, Rieder F, Louis E, Meuwis MA. Potential role of epithelial endoplasmic reticulum stress and anterior gradient protein 2 homolog in Crohn’s disease fibrosis. J Crohns Colitis 15: 1737–1750, 2021. doi: 10.1093/ecco-jcc/jjab061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng L, Kelly CJ, Colgan SP. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol 309: C350–C360, 2015. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zeitouni NE, Chotikatum S, von Köckritz-Blickwede M, Naim HY. The impact of hypoxia on intestinal epithelial cell functions: consequences for invasion by bacterial pathogens. Mol Cell Pediatr 3: 14, 2016. doi: 10.1186/s40348-016-0041-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40: 66–77, 2014. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114: 1098–1106, 2004. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Manresa MC, Taylor CT. Hypoxia inducible factor (HIF) hydroxylases as regulators of intestinal epithelial barrier function. Cell Mol Gastroenterol Hepatol 3: 303–315, 2017. doi: 10.1016/j.jcmgh.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shah YM. The role of hypoxia in intestinal inflammation. Mol Cell Pediatr 3: 1, 2016. doi: 10.1186/s40348-016-0030-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134: 145–155, 2008. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134: 156–165, 2008. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 45. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92: 5510–5514, 1995. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest 117: 862–865, 2007. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Albadari N, Deng S, Li W. The transcriptional factors HIF-1 and HIF-2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov 14: 667–682, 2019. doi: 10.1080/17460441.2019.1613370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ramakrishnan SK, Shah YM. Role of Intestinal HIF-2α in Health and Disease. Annu Rev Physiol 78: 301–325, 2016. doi: 10.1146/annurev-physiol-021115-105202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399: 271–275, 1999. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 50. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG. Jr.. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292: 464–468, 2001. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 51. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110: 993–1002, 2002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem 99: 1616–1627, 2006. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 53. Keely S, Campbell EL, Baird AW, Hansbro PM, Shalwitz RA, Kotsakis A, McNamee EN, Eltzschig HK, Kominsky DJ, Colgan SP. Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal Immunol 7: 114–123, 2014. doi: 10.1038/mi.2013.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE, Bayless AJ, Saeedi BJ, Colgan SP. Fundamental role for HIF-1α in constitutive expression of human beta defensin-1. Mucosal Immunol 6: 1110–1118, 2013. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669, 2005. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 56. Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med 51: 1–13, 2019. doi: 10.1038/s12276-019-0235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol 18: 573–587, 2022. doi: 10.1038/s41581-022-00587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153, 2014. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 59. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71: 241–260, 2009. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 60. Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev 18: 1385–1390, 2004. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hall PA, Coates PJ, Ansari B, Hopwood D. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J Cell Sci 107: 3569–3577, 1994. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 62. Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, Fraisl P, Lasitschka F, Mollenhauer M, Saunders SP, Maxwell PH, Carmeliet P, Fallon PG, Schneider M, Taylor CT. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 139: 2093–2101, 2010. doi: 10.1053/j.gastro.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 63. Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM. Concordant regulation of gene expression by hypoxia and 2-oxoglutarate-dependent dioxygenase inhibition: the role of HIF-1α, HIF-2α, and other pathways. J Biol Chem 281: 15215–15226, 2006. doi: 10.1074/jbc.M511408200. [DOI] [PubMed] [Google Scholar]
- 64. Hindryckx P, De Vos M, Jacques P, Ferdinande L, Peeters H, Olievier K, Bogaert S, Brinkman B, Vandenabeele P, Elewaut D, Laukens D. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J Immunol 185: 6306–6316, 2010. doi: 10.4049/jimmunol.1002541. [DOI] [PubMed] [Google Scholar]
- 65. Giepmans BN, van Ijzendoorn SC. Epithelial cell-cell junctions and plasma membrane domains. Biochim Biophys Acta 1788: 820–831, 2009. doi: 10.1016/j.bbamem.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 66. Taylor CT, Dzus AL, Colgan SP. Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology 114: 657–668, 1998. doi: 10.1016/s0016-5085(98)70579-7. [DOI] [PubMed] [Google Scholar]
- 67. Friedman GB, Taylor CT, Parkos CA, Colgan SP. Epithelial permeability induced by neutrophil transmigration is potentiated by hypoxia: role of intracellular cAMP. J Cell Physiol 176: 76–84, 1998. doi:. [DOI] [PubMed] [Google Scholar]
- 68. Glover LE, Colgan SP. Epithelial barrier regulation by hypoxia-inducible factor. Ann Am Thorac Soc 14: S233–S236, 2017. doi: 10.1513/AnnalsATS.201608-610MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Günzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525–569, 2013. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol 177: 512–524, 2010. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Anderson JM. Molecular structure of tight junctions and their role in epithelial transport. News Physiol Sci 16: 126–130, 2001. doi: 10.1152/physiologyonline.2001.16.3.126. [DOI] [PubMed] [Google Scholar]
- 72. Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 141: 1539–1550, 1998. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 156: 1099–1111, 2002. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, Kendrick AA, Kelly CJ, Bayless AJ, Kominsky DJ, Campbell EL, Kuhn KA, Furuta GT, Colgan SP, Glover LE. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell 26: 2252–2262, 2015. doi: 10.1091/mbc.E14-07-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dowdell AS, Cartwright IM, Goldberg MS, Kostelecky R, Ross T, Welch N, Glover LE, Colgan SP. The HIF target ATG9A is essential for epithelial barrier function and tight junction biogenesis. Mol Biol Cell 31: 2249–2258, 2020. doi: 10.1091/mbc.E20-05-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Curtis VF, Cartwright IM, Lee JS, Wang RX, Kao DJ, Lanis JM, Burney KM, Welch N, Hall CHT, Goldberg MS, Campbell EL, Colgan SP. Neutrophils as sources of dinucleotide polyphosphates and metabolism by epithelial ENPP1 to influence barrier function via adenosine signaling. Mol Biol Cell 29: 2687–2699, 2018. doi: 10.1091/mbc.E18-06-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, Miller L, Kominsky DJ, Jedlicka P, Colgan SP. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis. Proc Natl Acad Sci USA 110: 19820–19825, 2013. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aihara E, Engevik KA, Montrose MH. Trefoil factor peptides and gastrointestinal function. Annu Rev Physiol 79: 357–380, 2017. doi: 10.1146/annurev-physiol-021115-105447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kelly CJ, Colgan SP. Targeting hypoxia to augment mucosal barrier function. J Epithel Biol Pharmacol 5: 67–76, 2012. doi: 10.2174/1875044301205010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol 1: 183–197, 2008. doi: 10.1038/mi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Young HW, Williams OW, Chandra D, Bellinghausen LK, Pérez G, Suárez A, Tuvim MJ, Roy MG, Alexander SN, Moghaddam SJ, Adachi R, Blackburn MR, Dickey BF, Evans CM. Central role of Muc5ac expression in mucous metaplasia and its regulation by conserved 5' elements. Am J Respir Cell Mol Biol 37: 273–290, 2007. doi: 10.1165/rcmb.2005-0460OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Carpenter J, Wang Y, Gupta R, Li Y, Haridass P, Subramani DB, Reidel B, Morton L, Ridley C, O'Neal WK, Buisine MP, Ehre C, Thornton DJ, Kesimer M. Assembly and organization of the N-terminal region of mucin MUC5AC: indications for structural and functional distinction from MUC5B. Proc Natl Acad Sci USA 118: e2104490118, 2021. doi: 10.1073/pnas.2104490118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hasnain SZ, Evans CM, Roy M, Gallagher AL, Kindrachuk KN, Barron L, Dickey BF, Wilson MS, Wynn TA, Grencis RK, Thornton DJ. Muc5ac: a critical component mediating the rejection of enteric nematodes. J Exp Med 208: 893–900, 2011. doi: 10.1084/jem.20102057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yin L, Kharbanda S, Kufe D. Mucin 1 oncoprotein blocks hypoxia-inducible factor 1α activation in a survival response to hypoxia. J Biol Chem 282: 257–266, 2007. doi: 10.1074/jbc.M610156200. [DOI] [PubMed] [Google Scholar]
- 85. Grondin JA, Kwon YH, Far PM, Haq S, Khan WI. Mucins in intestinal mucosal defense and inflammation: learning from clinical and experimental studies. Front Immunol 11: 2054, 2020. doi: 10.3389/fimmu.2020.02054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Thim L, Madsen F, Poulsen SS. Effect of trefoil factors on the viscoelastic properties of mucus gels. Eur J Clin Invest 32: 519–527, 2002. doi: 10.1046/j.1365-2362.2002.01014.x. [DOI] [PubMed] [Google Scholar]
- 87. Kjellev S. The trefoil factor family—small peptides with multiple functionalities. Cell Mol Life Sci 66: 1350–1369, 2009. doi: 10.1007/s00018-008-8646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kjellev S, Vestergaard EM, Nexø E, Thygesen P, Eghoj MS, Jeppesen PB, Thim L, Pedersen NB, Poulsen SS. Pharmacokinetics of trefoil peptides and their stability in gastrointestinal contents. Peptides 28: 1197–1206, 2007. doi: 10.1016/j.peptides.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 89. Suemori S, Lynch-Devaney K, Podolsky DK. Identification and characterization of rat intestinal trefoil factor: tissue- and cell-specific member of the trefoil protein family. Proc Natl Acad Sci USA 88: 11017–11021, 1991. doi: 10.1073/pnas.88.24.11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Guleng B, Han J, Yang JQ, Huang QW, Huang JK, Yang XN, Liu JJ, Ren JL. TFF3 mediated induction of VEGF via hypoxia in human gastric cancer SGC-7901 cells. Mol Biol Rep 39: 4127–4134, 2012. doi: 10.1007/s11033-011-1195-2. [DOI] [PubMed] [Google Scholar]
- 91. Hernandez C, Santamatilde E, McCreath KJ, Cervera AM, Diez I, Ortiz-Masiá D, Martinez N, Calatayud S, Esplugues JV, Barrachina MD. Induction of trefoil factor (TFF)1, TFF2 and TFF3 by hypoxia is mediated by hypoxia inducible factor-1: implications for gastric mucosal healing. Br J Pharmacol 156: 262–272, 2009. doi: 10.1111/j.1476-5381.2008.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193: 1027–1034, 2001. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Antoni L, Nuding S, Weller D, Gersemann M, Ott G, Wehkamp J, Stange EF. Human colonic mucus is a reservoir for antimicrobial peptides. J Crohns Colitis 7: e652–e664, 2013. doi: 10.1016/j.crohns.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 94. Jäger S, Stange EF, Wehkamp J. Antimicrobial peptides in gastrointestinal inflammation. Int J Inflam 2010: 910283, 2010. doi: 10.4061/2010/910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Meng X, Grötsch B, Luo Y, Knaup KX, Wiesener MS, Chen XX, Jantsch J, Fillatreau S, Schett G, Bozec A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun 9: 251, 2018. doi: 10.1038/s41467-017-02683-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McGettrick AF, O'Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab 32: 524–536, 2020. doi: 10.1016/j.cmet.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 97. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112: 645–657, 2003. [Erratum in Cell 113: 419, 2003]. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Colgan SP, Campbell EL. Oxygen metabolism and innate immune responses in the gut. J Appl Physiol (1985) 123: 1321–1327, 2017. doi: 10.1152/japplphysiol.00113.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, Williams L, Parmar S, Shaw G, McGrath EE, Formenti F, Van Eeden FJ, Kinnula VL, Pugh CW, Sabroe I, Dockrell DH, Chilvers ER, Robbins PA, Percy MJ, Simon MC, Johnson RS, Renshaw SA, Whyte MK, Walmsley SR. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood 123: 366–376, 2014. doi: 10.1182/blood-2013-05-500207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol 9: 2733, 2018. doi: 10.3389/fimmu.2018.02733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hutami IR, Izawa T, Khurel-Ochir T, Sakamaki T, Iwasa A, Tanaka E. Macrophage motility in wound healing is regulated by HIF-1α via S1P signaling. Int J Mol Sci 22: 8992, 2021. doi: 10.3390/ijms22168992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Wang T, Liu H, Lian G, Zhang SY, Wang X, Jiang C. HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm 2017: 9029327, 2017. doi: 10.1155/2017/9029327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Imtiyaz HZ, Williams EP, Hickey MM, Patel SA, Durham AC, Yuan LJ, Hammond R, Gimotty PA, Keith B, Simon MC. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest 120: 2699–2714, 2010. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sun L, Li T, Tang H, Yu K, Ma Y, Yu M, Qiu Y, Xu P, Xiao W, Yang H. Intestinal epithelial cells-derived hypoxia-inducible factor-1α is essential for the homeostasis of intestinal intraepithelial lymphocytes. Front Immunol 10: 806, 2019. doi: 10.3389/fimmu.2019.00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Higashiyama M, Hokari R, Hozumi H, Kurihara C, Ueda T, Watanabe C, Tomita K, Nakamura M, Komoto S, Okada Y, Kawaguchi A, Nagao S, Suematsu M, Goda N, Miura S. HIF-1 in T cells ameliorated dextran sodium sulfate-induced murine colitis. J Leukoc Biol 91: 901–909, 2012. doi: 10.1189/jlb.1011518. [DOI] [PubMed] [Google Scholar]
- 106. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA 109: E2784–E2793, 2012. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Phan AT, Goldrath AW. Hypoxia-inducible factors regulate T cell metabolism and function. Mol Immunol 68: 527–535, 2015. doi: 10.1016/j.molimm.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Cho SH, Raybuck AL, Blagih J, Kemboi E, Haase VH, Jones RG, Boothby MR. Hypoxia-inducible factors in CD4(+) T cells promote metabolism, switch cytokine secretion, and T cell help in humoral immunity. Proc Natl Acad Sci USA 116: 8975–8984, 2019. doi: 10.1073/pnas.1811702116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Lee JS, Wang RX, Goldberg MS, Clifford GP, Kao DJ, Colgan SP. Microbiota-sourced purines support wound healing and mucous barrier function. iScience 23: 101226, 2020. doi: 10.1016/j.isci.2020.101226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Dignass AU, Becker A, Spiegler S, Goebell H. Adenine nucleotides modulate epithelial wound healing in vitro. Eur J Clin Invest 28: 554–561, 1998. doi: 10.1046/j.1365-2362.1998.00330.x. [DOI] [PubMed] [Google Scholar]
- 111. Roediger WE. The colonic epithelium in ulcerative colitis: an energy-deficiency disease? Lancet 2: 712–715, 1980. doi: 10.1016/s0140-6736(80)91934-0. [DOI] [PubMed] [Google Scholar]
- 112. Lee JS, Wang RX, Alexeev EE, Lanis JM, Battista KD, Glover LE, Colgan SP. Hypoxanthine is a checkpoint stress metabolite in colonic epithelial energy modulation and barrier function. J Biol Chem 293: 6039–6051, 2018. doi: 10.1074/jbc.RA117.000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev 80: 1107–1213, 2000. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]
- 114. Hall CHT, Lee JS, Murphy EM, Gerich ME, Dran R, Glover LE, Abdulla ZI, Skelton MR, Colgan SP. Creatine transporter, reduced in colon tissues from patients with inflammatory bowel diseases, regulates energy balance in intestinal epithelial cells, epithelial integrity, and barrier function. Gastroenterology 159: 984–998, 2020. doi: 10.1053/j.gastro.2020.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Chun C, Zheng L, Colgan SP. Tissue metabolism and host-microbial interactions in the intestinal mucosa. Free Radic Biol Med 105: 86–92, 2017. doi: 10.1016/j.freeradbiomed.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 81: 1031–1064, 2001. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- 117. Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 19: 29–41, 2017. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- 118. Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut microbes 7: 189–200, 2016. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165: 1332–1345, 2016. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 120. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54: 2325–2340, 2013. doi: 10.1194/jlr.R036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 21: 793–798, 1980. doi: 10.1136/gut.21.9.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang RX, Lee JS, Campbell EL, Colgan SP. Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc Natl Acad Sci USA 117: 11648–11657, 2020. doi: 10.1073/pnas.1917597117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17: 662–671, 2015. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang RX, Henen MA, Lee JS, Vögeli B, Colgan SP. Microbiota-derived butyrate is an endogenous HIF prolyl hydroxylase inhibitor. Gut Microbes 13: 1938380, 2021. doi: 10.1080/19490976.2021.1938380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428, 2009. [Erratum in J Clin Invest 120: 1786, 2010]. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lovisa S, Genovese G, Danese S. Role of epithelial-to-mesenchymal transition in inflammatory bowel disease. J Crohns Colitis 13: 659–668, 2019. doi: 10.1093/ecco-jcc/jjy201. [DOI] [PubMed] [Google Scholar]
- 127. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 154: 8–20, 1995. doi: 10.1159/000147748. [DOI] [PubMed] [Google Scholar]
- 128. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer 18: 128–134, 2018. doi: 10.1038/nrc.2017.118. [DOI] [PubMed] [Google Scholar]
- 129. Zeisberg M, Yang C, Martino M, Duncan MB, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 282: 23337–23347, 2007. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 130. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA 103: 13180–13185, 2006. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110: 341–350, 2002. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 278: 21113–21123, 2003. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- 134. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-β3. Mol Biol Cell 19: 4875–4887, 2008. doi: 10.1091/mbc.e08-05-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939, 2004. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 136. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7: 1267–1278, 2001. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 137. Luo D, Wang J, Li J, Post M. Mouse snail is a target gene for HIF. Mol Cancer Res 9: 234–245, 2011. doi: 10.1158/1541-7786.MCR-10-0214. [DOI] [PubMed] [Google Scholar]
- 138. Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, Dong J, Qian C. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1α in hepatocellular carcinoma. BMC Cancer 13: 108, 2013. doi: 10.1186/1471-2407-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Tam SY, Wu VWC, Law HKW. Hypoxia-induced epithelial-mesenchymal transition in cancers: HIF-1α and beyond. Front Oncol 10: 486, 2020. doi: 10.3389/fonc.2020.00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Zhu GH, Huang C, Feng ZZ, Lv XH, Qiu ZJ. Hypoxia-induced snail expression through transcriptional regulation by HIF-1α in pancreatic cancer cells. Dig Dis Sci 58: 3503–3515, 2013. doi: 10.1007/s10620-013-2841-4. [DOI] [PubMed] [Google Scholar]
- 141. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem 290: 16653–16664, 2015. doi: 10.1074/jbc.M115.636944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang P, Liu Y, Feng Y, Gao S. SNAIL gene inhibited by hypoxia-inducible factor 1α (HIF-1α) in epithelial ovarian cancer. Int J Immunopathol Pharmacol 29: 364–375, 2016. doi: 10.1177/0394632016641423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wang N, Dong CR, Jiang R, Tang C, Yang L, Jiang QF, Chen GG, Liu ZM. Overexpression of HIF-1α, metallothionein and SLUG is associated with high TNM stage and lymph node metastasis in papillary thyroid carcinoma. Int J Clin Exp Pathol 7: 322–330, 2014. [PMC free article] [PubMed] [Google Scholar]
- 144. Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle 7: 2090–2096, 2008. doi: 10.4161/cc.7.14.6324. [DOI] [PubMed] [Google Scholar]
- 145. Gort EH, van Haaften G, Verlaan I, Groot AJ, Plasterk RH, Shvarts A, Suijkerbuijk KP, van Laar T, van der Wall E, Raman V, van Diest PJ, Tijsterman M, Vooijs M. The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2α. Oncogene 27: 1501–1510, 2008. doi: 10.1038/sj.onc.1210795. [DOI] [PubMed] [Google Scholar]
- 146. Yun SM, Kim SH, Kim EH. The molecular mechanism of transforming growth factor-beta signaling for intestinal fibrosis: a mini-review. Front Pharmacol 10: 162, 2019. doi: 10.3389/fphar.2019.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol 12: 325–338, 2016. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 148. Steiner CA, Rodansky ES, Johnson LA, Berinstein JA, Cushing KC, Huang S, Spence JR, Higgins PDR. AXL is a potential target for the treatment of intestinal fibrosis. Inflamm Bowel Dis 27: 303–316, 2021. doi: 10.1093/ibd/izaa169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Johnson LA, Rodansky ES, Tran A, Collins SG, Eaton KA, Malamet B, Steiner CA, Huang S, Spence JR, Higgins PDR. Effect of ABT-263 on intestinal fibrosis in human myofibroblasts, human intestinal organoids, and the mouse Salmonella typhimurium model. Inflamm Bowel Dis 28: 161–175, 2022. doi: 10.1093/ibd/izab166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hung SP, Yang MH, Tseng KF, Lee OK. Hypoxia-induced secretion of TGF-β1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant 22: 1869–1882, 2013. doi: 10.3727/096368912X657954. [DOI] [PubMed] [Google Scholar]
- 151. Blanchette F, Day R, Dong W, Laprise MH, Dubois CM. TGFβ1 regulates gene expression of its own converting enzyme furin. J Clin Invest 99: 1974–1983, 1997. doi: 10.1172/JCI119365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem 280: 6561–6569, 2005. doi: 10.1074/jbc.M413248200. [DOI] [PubMed] [Google Scholar]
- 153. McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem 281: 24171–24181, 2006. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- 154. Kushida N, Nomura S, Mimura I, Fujita T, Yamamoto S, Nangaku M, Aburatani H. Hypoxia-inducible factor-1α activates the transforming growth factor-beta/SMAD3 pathway in kidney tubular epithelial cells. Am J Nephrol 44: 276–285, 2016. doi: 10.1159/000449323. [DOI] [PubMed] [Google Scholar]
- 155. Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1α and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol 300: F898–F905, 2011. doi: 10.1152/ajprenal.00335.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Mingyuan X, Qianqian P, Shengquan X, Chenyi Y, Rui L, Yichen S, Jinghong X. Hypoxia-inducible factor-1α activates transforming growth factor-β1/Smad signaling and increases collagen deposition in dermal fibroblasts. Oncotarget 9: 3188–3197, 2018. doi: 10.18632/oncotarget.23225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, Tanaka T, Iso T, Suga T, Kurabayashi M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 300: L740–L752, 2011. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- 158. Mallikarjuna P, Raviprakash TS, Aripaka K, Ljungberg B, Landström M. Interactions between TGF-β type I receptor and hypoxia-inducible factor-α mediates a synergistic crosstalk leading to poor prognosis for patients with clear cell renal cell carcinoma. Cell Cycle 18: 2141–2156, 2019. doi: 10.1080/15384101.2019.1642069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Manresa MC, Tambuwala MM, Radhakrishnan P, Harnoss JM, Brown E, Cavadas MA, Keogh CE, Cheong A, Barrett KE, Cummins EP, Schneider M, Taylor CT. Hydroxylase inhibition regulates inflammation-induced intestinal fibrosis through the suppression of ERK-mediated TGF-β1 signaling. [corrected]. Am J Physiol Gastrointest Liver Physiol 311: G1076–G1090, 2016. [Erratum in Am J Physiol Gastrointest Liver Physiol 312: G405, 2017]. doi: 10.1152/ajpgi.00229.2016. [DOI] [PubMed] [Google Scholar]
- 160. Sulkowska M, Wincewicz A, Sulkowski S, Koda M, Kanczuga-Koda L. Relations of TGF-β1 with HIF-1α, GLUT-1 and longer survival of colorectal cancer patients. Pathology 41: 1–260, 2009. [Erratum in Pathology 41: 612, 2009]. doi: 10.1080/00313020802579318. [DOI] [PubMed] [Google Scholar]
- 161. Gay CM, Balaji K, Byers LA. Giving AXL the axe: targeting AXL in human malignancy. Br J Cancer 116: 415–423, 2017. doi: 10.1038/bjc.2016.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D, Axelson H, Miao YR, Krieg AJ, Giaccia AJ. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci USA 111: 13373–13378, 2014. doi: 10.1073/pnas.1404848111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Bárcena C, Stefanovic M, Tutusaus A, Joannas L, Menéndez A, García-Ruiz C, Sancho-Bru P, Marí M, Caballeria J, Rothlin CV, Fernandez-Checa JC, de Frutos PG, Morales A. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J Hepatol 63: 670–678, 2015. doi: 10.1016/j.jhep.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 18: 153, 2019. doi: 10.1186/s12943-019-1090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta 1823: 406–419, 2012. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Gundemir S, Colak G, Feola J, Blouin R, Johnson GV. Transglutaminase 2 facilitates or ameliorates HIF signaling and ischemic cell death depending on its conformation and localization. Biochim Biophys Acta 1833: 1–10, 2013. doi: 10.1016/j.bbamcr.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Olsen KC, Sapinoro RE, Kottmann RM, Kulkarni AA, Iismaa SE, Johnson GV, Thatcher TH, Phipps RP, Sime PJ. Transglutaminase 2 and its role in pulmonary fibrosis. Am J Respir Crit Care Med 184: 699–707, 2011. doi: 10.1164/rccm.201101-0013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Wang Z, Stuckey DJ, Murdoch CE, Camelliti P, Lip GYH, Griffin M. Cardiac fibrosis can be attenuated by blocking the activity of transglutaminase 2 using a selective small-molecule inhibitor. Cell Death Dis 9: 613, 2018. doi: 10.1038/s41419-018-0573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Filiano AJ, Bailey CD, Tucholski J, Gundemir S, Johnson GV. Transglutaminase 2 protects against ischemic insult, interacts with HIF1β, and attenuates HIF1 signaling. FASEB J 22: 2662–2675, 2008. doi: 10.1096/fj.07-097709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Requena-Ibáñez JA, Santos-Gallego CG, Rodriguez-Cordero A, Zafar MU, Badimon JJ. Prolyl hydroxylase inhibitors: a new opportunity in renal and myocardial protection. Cardiovasc Drugs Ther, 2021. doi: 10.1007/s10557-021-07257-0. [DOI] [PubMed] [Google Scholar]
- 171. DiGiacomo JW, Gilkes DM. Therapeutic strategies to block the hypoxic response. Adv Exp Med Biol 1136: 141–157, 2019. doi: 10.1007/978-3-030-12734-3_10. [DOI] [PubMed] [Google Scholar]