

Abstract
Anti‐neutrophil cytoplasmic antibody‐associated vasculitides granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are rare, potentially organ‐ and life‐threatening autoimmune conditions affecting adult and pediatric patients. An open‐label phase II study was conducted to determine safe and effective dosing regimens of rituximab in pediatric patients with GPA/MPA. To determine the selection of an appropriate dose regimen in children for induction and maintenance, a population pharmacokinetic approach was used (nonlinear mixed‐effect modeling), combining pediatric data with data from adults with GPA/MPA. The time course of B‐cell depletion was assessed in both populations. The exposure‐effect relationship was assessed by logistic regression. Twenty‐five pediatric patients (80% female patients; age range, 6–17 years) were enrolled in the trial and received the induction regimen of intravenous rituximab 375 mg/m2 weekly for 4 weeks, which resulted in a similar exposure to that of adults. Based on pharmacokinetic modeling, a maintenance dosing regimen of 250 mg/m2 administered twice over 14 days followed by 250 mg/m2 every 6 months is expected to result in similar rituximab exposure as that of adults receiving the approved maintenance dose of 500 mg administered twice over 14 days followed by 500 mg every 6 months. The time course of B‐cell depletion was similar between the pediatric and adult populations, supporting the similarities in response in both populations and allowing extrapolation to patients less than 6 years old. Using a partial extrapolation approach helped identify safe and effective dosing regimens of rituximab in pediatric patients with GPA/MPA and lead to regulatory approval.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Rituximab was first approved for remission induction in adult patients with granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) and subsequently approved for remission maintenance. The approval of rituximab for pediatric patients was based on the PePRS trial, which used the same induction regimen as adults, whereas the follow‐up treatment and dosing regimen was left to the physician's discretion.
WHAT QUESTION DID THIS STUDY ADDRESS?
This open‐label phase II study was conducted to determine dosing regimens of rituximab in pediatric patients with GPA/MPA.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Modeling and simulations were used to confirm the dosing regimen used for induction in patients greater than 6 years old, extrapolate to younger patients aged ≥2 to < 6 years and suggest a dosing regimen for follow‐up maintenance treatment in pediatric patients aged ≥2 to <18 years.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The work presented here was instrumental in supporting the first approval of rituximab in pediatric patients with GPA/MPA and demonstrates the utility of model‐informed drug development to complement limited clinical trial data.
INTRODUCTION
Anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitides granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA) are rare, potentially organ‐ and life‐threatening autoimmune conditions that affect small blood vessels. 1 , 2 , 3 Left untreated, GPA and MPA can progress from a limited disease to multisystem involvement and/or renal complications that confer a high risk of end‐stage renal disease and death despite potent immunosuppressive therapy. 1 , 2 , 4 , 5
The pathophysiology and natural history of GPA and MPA and their immunosuppressant treatments are similar between the adult and childhood diseases. Pediatric patients with GPA or MPA often have clinical features, signs, and symptoms similar to those in adult patients, 6 although the relative frequencies of symptoms differ. Childhood‐onset GPA is associated with a higher prevalence of renal disease and nasal deformities at presentation and a higher overall incidence of subglottic stenosis over time than GPA in adults. 5 , 6 , 7 , 8 Pediatric patients are also more likely to have multiple organ involvement and disease‐related morbidity and mortality due to aggressive respiratory involvement or progressive renal failure. 5 , 7 , 8
Across age groups, the pathogenesis of GPA and MPA involves defects in innate and adaptive immunity, dysregulation of B‐cells and pathogenic production of ANCAs. 6 , 9 In support of a crucial role for B‐cells, the number of activated peripheral B‐cells correlates with disease activity in GPA and MPA. 10 B‐cells contribute to disease pathogenesis by acting as antigen‐presenting cells through the production of various cytokines or participation of their progeny in the production of autoantibodies. 10 , 11 The proportions of B‐cell subpopulations are similar in pediatric patients across age groups (18 months to 18 years). 12
Conventional treatment for severe disease is remission induction with cyclophosphamide in combination with glucocorticoids (GCs), followed by maintenance with azathioprine or methotrexate. 1 , 2 , 13 , 14 However, these treatments are associated with frequent relapses and substantial toxicity that can result in severe, permanent morbidity and fatal adverse events (AEs). 2 , 4 , 5 Hence, there is a major unmet need for pediatric patients with GPA or MPA.
Rituximab (Rituxan, South San Francisco, CA/MabThera, Basel, Switzerland) is an anti‐CD20 monoclonal antibody that induces rapid and sustained depletion of CD20+ B‐lymphocytes, thereby suppressing autoantibody production. 15 , 16 , 17 , 18 , 19 Rituximab was first approved for remission induction in adult patients with GPA or MPA and subsequently approved for remission maintenance. 15 , 16 The induction dose approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for adult patients with active GPA or MPA is, in combination with GCs, an intravenous (i.v.) infusion of rituximab 375 mg/m2 once weekly for 4 weeks, based on the results of the RAVE trial. 20 The follow‐up dose in adults who achieve disease control with induction treatment was based on the results of the MAINRITSAN trial, 21 which used, in combination with GCs, 2 i.v. infusions of rituximab 500 mg 2 weeks apart, followed by an i.v. infusion of rituximab 500 mg every 6 months per clinical evaluation. 15
The approval of rituximab for pediatric patients in the United States and European Union was based on the PePRS trial 22 of pediatric patients (≥2 years old) with GPA or MPA, 15 , 16 which used an induction dose of 375 mg/m2 body surface area (BSA) once weekly for 4 weeks; in the United States, a follow‐up dose of two 250 mg/m2 i.v. infusions separated by 2 weeks, followed by a 250 mg/m2 i.v. infusion every 6 months thereafter based on clinical evaluation, was also approved. When the PePRS trial began, the adult induction regimen was approved, but no follow‐up treatment data or approved maintenance regimen in adults was available. Therefore, the PePRS trial used the same induction regimen as in adults, whereas the follow‐up treatment and dosing regimen was left to the physician's discretion, based on factors like disease activity and response to induction treatment.
Drug development for pediatric patients can be challenging due to differences in drug response and metabolism between children and adults. Clinical trial design must also contend with ethical issues when conducting research in a vulnerable population. These challenges are compounded in orphan diseases like GPA and MPA, which have very low incidences in both adults and children. 5 Extrapolation from adult data to support findings of effectiveness, safety, and appropriate dosing regimen in pediatric patients can alleviate some of these issues; however, extrapolation is only justified when the following criteria are met: similar pharmacokinetic (PK) characteristics among patients (i.e., matching exposure), similar pathogenesis and disease progression, similar mechanism of action, similar efficacy, and similar exposure–response relationships. 23
To provide dosing recommendations for rituximab in pediatric patients with GPA or MPA, a population PK modeling approach was used to characterize the PK of rituximab in both adult and pediatric populations with GPA or MPA. Rituximab exposure following the PePRS induction regimen was simulated in pediatric patients (≥2 years old) with a population PK model and compared with predicted exposure in adults with GPA or MPA. PK simulations were used to extrapolate the dosing regimen for induction and maintenance therapy for patients aged greater than or equal to 2 to less than 6 years in accordance with the FDA and EMA guidelines. 24 , 25
METHODS
Patient population and study design
Data from the adult RAVE trial 20 and the pediatric PePRS trial 22 were combined. In RAVE, 197 ANCA‐positive adults with GPA or MPA were randomized to receive either rituximab (375 mg/m2 weekly for 4 weeks) or cyclophosphamide (2 mg/kg/day) with a primary end point of disease remission at month 6. Serum samples were collected in rituximab‐treated patients for measurement of rituximab concentrations, B‐cell counts, and anti‐drug antibodies (ADAs). For RAVE and PePRS, PK measurements for rituximab levels were evaluated prior to the first and third doses, then on days 29 (month 1), 60 (month 2), 120 (month 4), 180 (month 6), 270 (month 9), and 545 (month 18) from the first dose, and then every 6 months during the follow‐up period and at the end of the study. The PePRS trial 22 was an international, multicenter, open‐label, uncontrolled phase IIa study to assess safety, PK, pharmacodynamics (PD), and exploratory efficacy outcomes in pediatric patients with GPA or MPA treated with rituximab. Plasma/serum samples were collected for measurement of rituximab concentrations, B‐cell counts, and ADAs. Patients aged greater than or equal to 2 to less than or equal to 18 years with newly diagnosed or relapsing GPA or MPA received four weekly i.v. infusions of rituximab 375 mg/m2 BSA and a GC taper; after 6 months, patients could receive further rituximab doses and/or other immunosuppressants per investigator discretion. The study was conducted in accordance with the principles of the Declaration of Helsinki. Ethics approvals for this study were obtained from the respective institutional review boards/ethics committees.
Extrapolation strategy to support induction and maintenance labeling for pediatric patients (≥2 to 18 years old)
Population PK analysis
A two‐compartment population PK model with linear elimination was developed in NONMEM (version 7.3.0; ICON Development Solutions, Hanover, MD) 26 using data from 122 patients (97 adult patients [3 of whom were aged 16–17 years] from the RAVE trial, 20 and 25 pediatric patients from the PePRS trial 22 receiving greater than or equal to four weekly i.v. doses of rituximab 375 mg/m2). The pediatric sample size was considered sufficient to provide a reasonable estimate of variability for the mean PK parameters based on the observed interpatient variability from RAVE, ensured a 95% probability of observing at least one AE when the underlying incidence of that event is 11%, and allow for estimation of the percentage of patients in remission at 6 months within 20% of the end point. The PK model developed in adult and pediatric patients was qualified for simulation purposes.
Dosing recommendations for induction
Although GPA/MPA can occur in patients <6 years old, recruiting patients in this age group is challenging due to low prevalence, and none were enrolled in PePRS. Therefore, rituximab exposure following the PePRS induction regimen in pediatric patients (≥2 years old) was simulated with a population PK model and compared with predicted exposure in adults with GPA or MPA. In the absence of clinical data, PK simulations were used to provide dosing recommendations in patients ≥2 to <6 years old to ensure comparable exposure in this age group with the recruited patients >6 to <18 years old and adult patients from the RAVE trial.
In adults, the induction regimen was simulated using the individual empirical Bayes estimates (i.e., post hoc ETA estimates in NONMEM) from 94 adult patients from the RAVE trial and their respective BSA values and assuming no ADAs. For children, a similar sample size of 94 patients was simulated for each of the following BSA values: 0.5, 0.8, 1.2, and 1.8 m2, which correspond to the typical BSA of children aged 2, 6, 12, and 15 years, respectively. 27 The empirical Bayes estimates of those pediatric patients were simulated using the between‐patient variabilities of the population PK model. Residual variability was not used for simulations. For each patient from PePRS, the primary exposure variable considered for PK bridging was the cumulative area under the curve (AUC) over 6 months (i.e., over the induction period). The AUC represents the integrated overall rituximab exposure after the four rituximab infusions in the 6‐month remission induction phase and was used to assess similarity in exposure between children and adults. Minimum rituximab serum concentration (Cmin) at 6 months and maximum rituximab serum concentration (Cmax) at the end of the second infusion were also computed.
Dosing recommendations for follow‐up/maintenance treatment in pediatric patients (≥2 to 18 years old)
In the PePRS trial, after the 6‐month standardized rituximab induction regimen in all patients, various follow‐up regimens were used, according to physician and local clinical practice. In adults, a positive risk–benefit ratio of rituximab in the maintenance setting was demonstrated in the MAINRITSAN trial, 21 in which treatment with rituximab resulted in fewer relapses than azathioprine. Therefore, rituximab PK was simulated to ensure that the proposed maintenance dosing regimen for children aged 2 to less than 18 years would provide similar exposure as for adults who received the adult maintenance regimen in the MAINRITSAN trial. 21 Using the population PK model developed in adults and pediatric patients, different follow‐up doses (i.e., 2 i.v. infusions of rituximab 250, 275, and 300 mg/m2 administered 2 weeks apart) were simulated in pediatric patients to determine which dose would best match the adult exposure. 21
PK and PD sampling
In all clinical trials, rituximab concentrations in serum were determined using a validated enzyme‐linked immunosorbent method with a lower limit of quantitation of 500 ng/ml. Serum ADAs were measured using a validated enzyme‐linked immunosorbent method in which samples were first screened for ADAs; those that could be immunodepleted were considered ADA positive. Serum samples were provided to determine circulating CD19+ B‐cell populations, which were analyzed by fluorescence‐activated cell sorter analysis.
Safety, efficacy, PD, and exposure–response analysis
The cumulative AUC over 6 months was used to assess the relationships among rituximab exposure, efficacy, and safety, and compare those relationships between adult and pediatric patients. Efficacy was evaluated using the Pediatric Vasculitis Activity Score (PVAS) at/by month 6. 28 Logistic regression models assessed any correlation between the probability of being in PVAS remission by month 6 and rituximab exposure and any correlation between the probability of the occurrence of selected safety parameters of interest (i.e., serious AEs, AEs grade ≥3, infusion‐related reactions, serious infections, and prolonged low IgG levels [hypogammaglobulinemia] in the remission‐induction phase) and rituximab exposure in pediatric patients. The relationship between rituximab exposure and B‐cell depletion was also compared between adults and pediatric patients.
RESULTS
Patient population
PePRS enrolled 25 children from six countries; the median (range) age was 14 (range 6–17 years), and 80% were female patients (Table 1). 22 All patients completed the induction regimen, and 24 patients completed ≥18 months of follow‐up. A total of 204 rituximab concentrations from 25 patients were included in the analysis. Ninety‐seven patients who received rituximab in RAVE 20 were included in this population PK analysis; the median (range) age was 55 (16–92) years, and 54% were female patients (Table 1). A total of 487 concentrations from 97 patients were included in the data set. In RAVE, 97% of patients received greater than or equal to three study drug infusions, and among patients who received rituximab, 85% completed 6 months of treatment.
TABLE 1.
Summary of baseline characteristics in pediatric and adult patients with GPA/MPA who received rituximab in PePRS or RAVE
| Pediatric patients in PePRS 22 (N = 25) | Adults in RAVE 20 (N = 97 a ) | Total (N = 122) | |
|---|---|---|---|
| Age, median (range), years | 14 (6–17) | 55 (16–92) | 50 (6–92) |
| Sex, n (%) | |||
| Male | 5 (20) | 45 (46) | 50 (41) |
| Female | 20 (80) | 52 (54) | 72 (59) |
| Weight, median (range), kg | 50.9 (23.0–80.8) | 80.5 (47.3–128.0) | 73.9 (23.0–128.0) |
| BSA, median (range), m2 | 1.45 (0.9–1.9) | 1.9 (1.4–2.5) | 1.84 (0.9–2.5) |
| B‐cell count, median (range), 106 cells/L | 603 (197–2170) | 221 (10–2320) | 273 (10–2320) |
Abbreviations: BSA, body surface area; GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis.
Three of 97 patients were aged 16 and 17 years.
Matching PK exposure between adult and pediatric patients for induction
A linear two‐compartment model was used to describe rituximab PK during induction in both adult and pediatric patients. BSA and presence of ADAs were the only two covariates found to impact rituximab PK. Rituximab clearance (CL) and central volume of distribution were highly correlated with BSA. Because ADAs had no impact on the safety and efficacy of rituximab, and therefore no impact on the recommended dosing regimen, this covariate was not included in the simulations presented. 22 After the model was adjusted for BSA, no PK difference remained between pediatric and adult patients with GPA or MPA. The relationships among CL, AUC, and BSA in absence of ADAs have been described already (Figure S1). The final model parameter estimates are shown in Table S1. Visual predictive check plots (Figure S2) confirmed that the model was able to describe both adult and pediatric data. The model provided a good description of the observed data and was suitable for simulations; therefore, rituximab exposure following the induction regimen from PePRS was predicted in pediatric patients (≥2 to 15 years old) and compared with predicted exposure in adults with GPA or MPA (Figure 1). Based on these simulations, AUC is expected to be similar in patients aged greater than or equal to 2 to less than 6 years compared with those greater than or equal to 6 years. There was a trend towards lower Cmin in patients greater than or equal to 2 and less than or equal to 10 years of age compared with adults, whereas Cmax was similar in all age groups (Figure 1).
FIGURE 1.
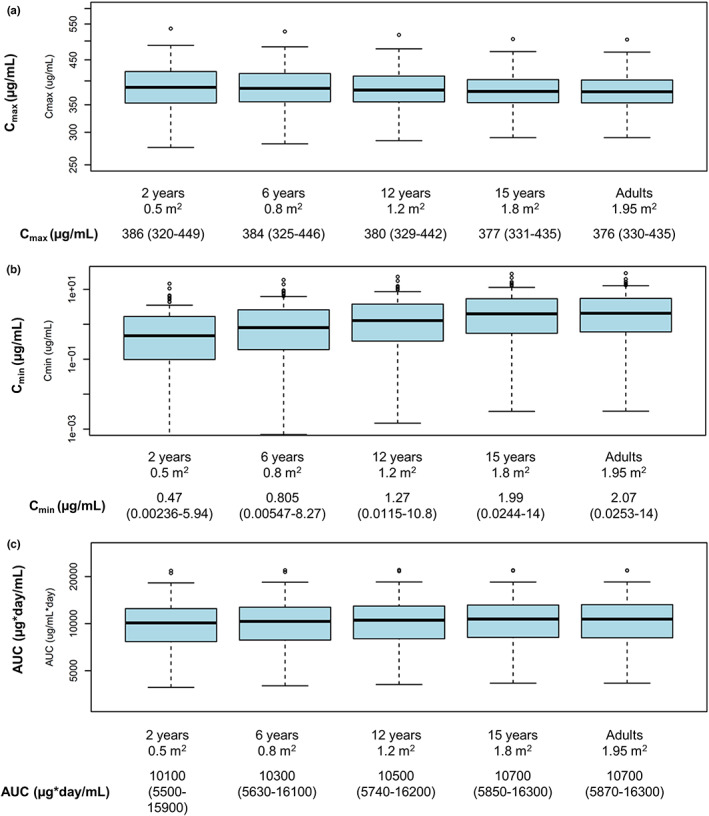
Predicted (a) Cmax, (b) Cmin, and (c) AUC in pediatric patients (≥2 to 15 years old) and adults with GPA and MPA in the induction setting. Results are presented as median (range). AUC, area under the curve; BSA, body surface area; Cmax, maximum serum concentration; Cmin, minimum serum concentration; GPA, granulomatosis with polyangiitis; MPA, microscopic polyangiitis.
Mechanism of action, exposure–response relationships, efficacy, and safety
In adults in the RAVE trial, rapid and prolonged peripheral CD19+ B‐cell depletion to below levels of quantification across the entire exposure range in all patients was observed after the first rituximab infusion. 20 In PePRS, peripheral B‐cells were depleted to below the lower laboratory limit of quantification of 20 cells/μl in all patients by week 1 after the first rituximab infusion, regardless of age. 22 B‐cell depletion was sustained until at least month 6, and in most cases, throughout the duration of follow‐up (≥12 months after remission induction and ≤4.5 years until the clinical close out; data not shown). At month 18, CD19+ B‐cell levels had increased but remained below baseline levels (mean, 160.10/μl; median, 58/μl). Younger patients (≥6 to <12 years old; n = 6) had baseline CD19+ B‐cell counts in the same range as patients aged greater than or equal to 12 to less than or equal to 17 years (n = 19), and their B‐cell counts were depleted below the lower laboratory limit of quantification after rituximab treatment. The magnitude of B‐cell depletion and the start time of repletion was comparable between adults and pediatric patients. Because no age‐related effect was observed in the study population, B‐cell depletion in patients greater than or equal to 2 to less than or equal to 6 years of age is expected to be similar to that in older patients, 12 supporting a similar PD effect across all ages. In the induction phase, the time course of B‐cell depletion was similar between pediatric and adult patients (Figure 2).
FIGURE 2.
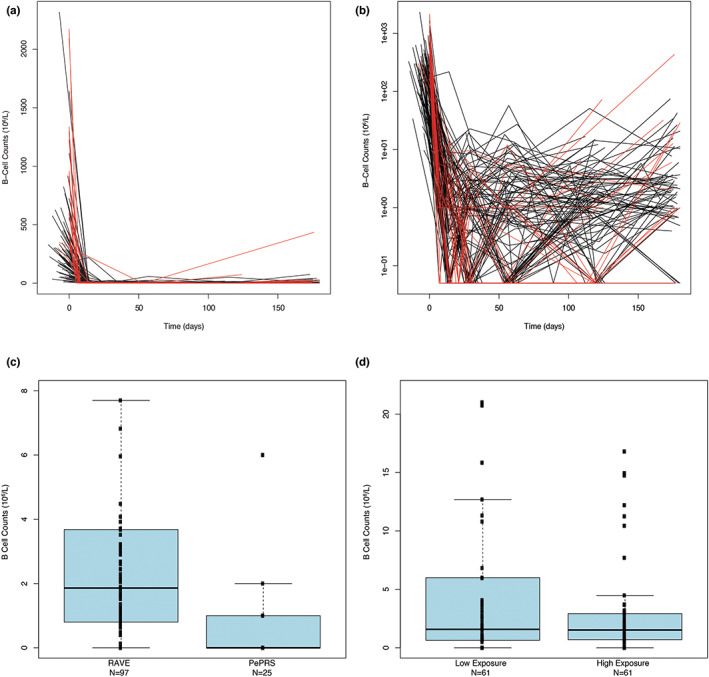
Observed CD19 B‐cell levels over time in adult and pediatric patients in the induction setting. (a) For the two studies superimposed. (b) For the two studies superimposed (zoomed). Red lines: B‐cell counts over time for pediatric patients; Black lines: B‐cell counts over time for adult patients. B‐cell counts equal to zero were assigned a value of 0.05 on the semi‐log scale plots. (c) Distributions of B‐cell count at 180 days by study (zoom through data). (d) Distributions of B‐cell count at baseline and at 180 days by exposure category for the two studies combined. The B‐cell counts are plotted versus an exposure category using a box and whisker plot. Median values of the B‐cells are designated by black lines in the center of the boxes. Boxes indicate IQR. Whiskers represent 1.5*IQR. Outliers are marked outside of the whiskers by circles. Exposure categories were defined by the values of AUC180. Low and high exposure categories included 61 patients each with AUC180 less or equal (greater) than the median of AUC180 values in patients of the combined set of RAVE and PePRS (equal to 10,201 μg*day/ml). One patient with a B‐cell count greater than 400 106/L was excluded for better figure visibility. AUC, area under the curve; IQR, interquartile range.
As in adults with GPA or MPA, no association was found between variability in exposure generated by four weekly infusions of 375 mg/m2 and clinical response at the end of induction in pediatric patients, suggesting that the induction regimen resulted in exposure at the plateau of the exposure–response relationship. Additionally, no association between rituximab exposure and efficacy was observed (Figure 3a).
FIGURE 3.
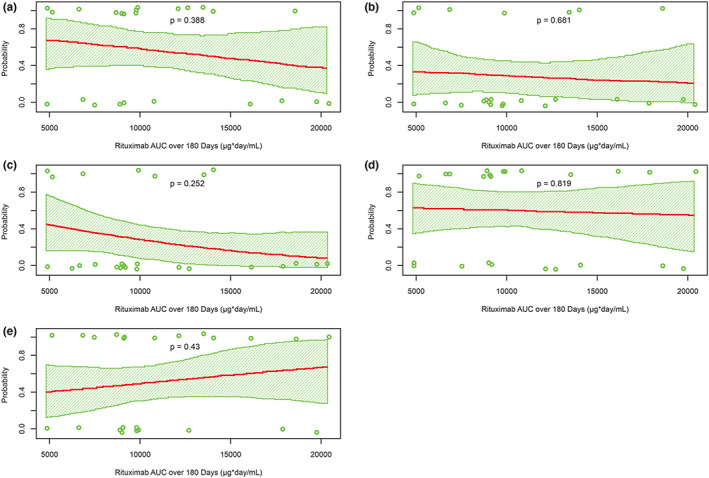
Probability of (a) remission by month 6, (b) AE greater than or equal to 3, (c) SAE, (d) infusion‐related reaction, and (e) hypogammaglobulinemia versus rituximab exposure over 6 months. AE, adverse event, AUC, area under the curve; SAE, serious adverse event.
No association between drug exposure and the selected safety parameters of interest was identified (Figure 3b–e). Overall, the safety profile of rituximab in pediatric patients was similar to the known safety profile of rituximab in adults with GPA/MPA, despite the prolonged durations of B‐cell depletion through month 18 and during the periods of repeat rituximab treatments.
Matching PK exposure between adult and pediatric patients for maintenance
Simulations showed that a follow‐up dose of rituximab 250 mg/m2 in pediatric patients was the most similar to the approved adult follow‐up dose of 500 mg. Comparison of the predicted PK parameters (AUC, Cmin, and Cmax) following two i.v. infusions administered 2 weeks apart of rituximab 250 mg/m2 in children with BSA of 0.5, 0.8, 1.2, and 1.8 m2 and rituximab 500 mg in adults is shown in Figure 4 and Figure S3. Predicted rituximab concentration–time profiles in pediatric patients following two i.v. infusions of rituximab 250 mg/m2 administered 2 weeks apart were compared with predicted PK profiles in adults following the rituximab regimen in the MAINRITSAN trial (rituximab 500 mg i.v. administered 2 weeks apart 21 ; Figure 1). The simulated PK time courses were summarized using median concentration–time profiles and 5th and 95th percentiles (90% prediction interval).
FIGURE 4.
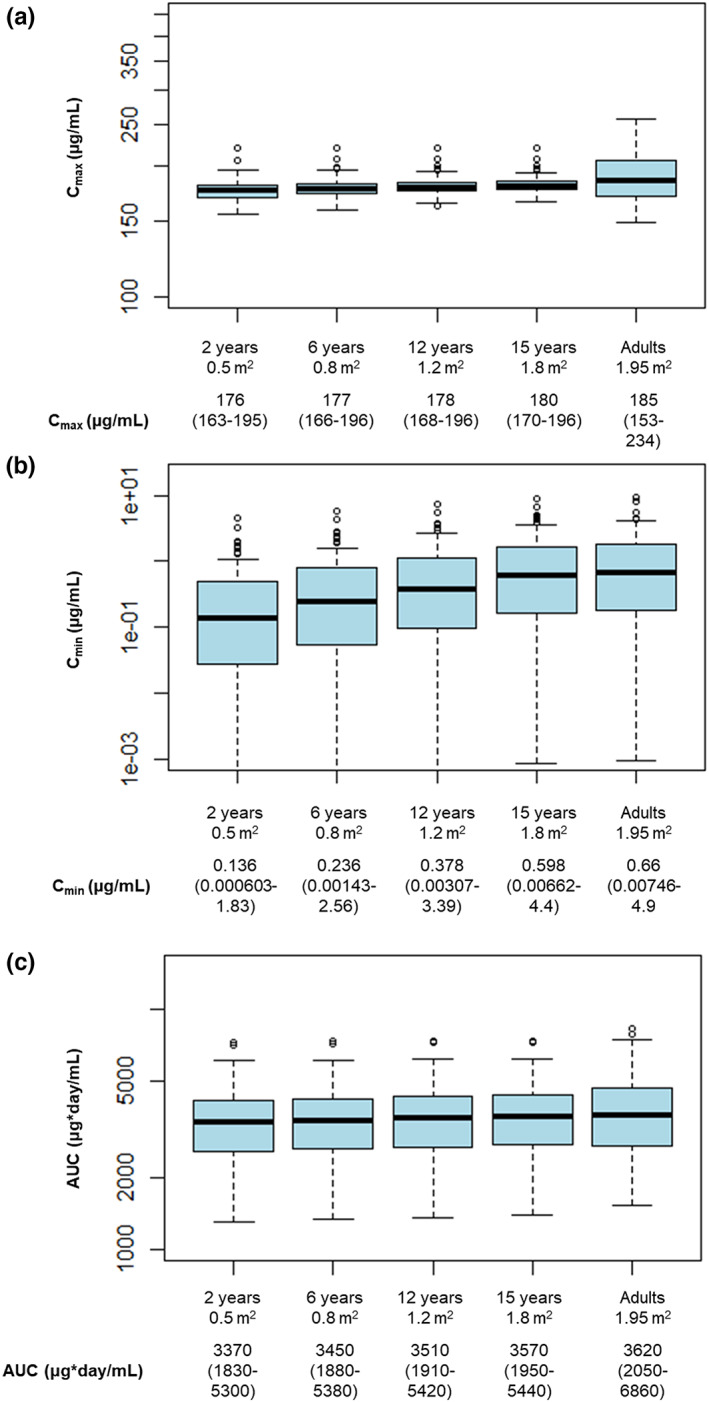
Comparison of predicted (a) Cmax, (b) Cmin, and (c) AUC following two i.v. infusions administered 2 weeks apart of 250 mg/m2 rituximab in pediatric patients with BSA of 0.5, 0.8, 1.2, and 1.8 m2 and 500 mg rituximab in adult patients (maintenance regimen). AUC, area under the curve; BSA, body surface area; Cmax, maximum serum concentration; Cmin, minimum serum concentration; GPA, granulomatosis with polyangiitis; IV, intravenous; MPA, microscopic polyangiitis. *Results are presented as median (range).
A BSA‐adjusted dosing regimen of rituximab 250 mg/m2 administered to children is expected to result in comparable exposure (AUC over 6 months) to adults treated with the approved follow‐up rituximab dosing regimen in GPA or MPA. The predicted Cmax in children is lower than in adults, whereas Cmin slightly increases with increasing BSA. For the maintenance phase, 375 mg/m2 infusions were given: eight patients had an infusion every 6–12 months (cycle range: 1–5), eight patients had weekly infusions for 4 weeks (range: 1–6), one patient had weekly infusions for 2 weeks, and eight patients had no further rituximab treatment; all of these regimens were well‐tolerated. The proportion of patients achieving PVAS remission during the maintenance phase increased in those who received repeated doses of rituximab (Table 2).
TABLE 2.
PVAS remission at key time points (after month 6 and to months 12 and 18) for patients in PePRS re‐treated or non–re‐treated with rituximab after the 6‐month remission induction phase, safety‐evaluable patients
| Rituximab (N = 25) | ||
|---|---|---|
| Re‐treated a (n = 14) | Non–re‐treated a (n = 11) | |
| Month 6 | ||
| n (%) | 8 (57.1) | 5 (45.5) |
| 95% CI | 28.9, 82.3 | 16.7, 76.6 |
| Month 12 | ||
| n (%) | 11 (78.6) | 7 (63.6) |
| 95% CI | 49.2, 95.3 | 30.8, 89.1 |
| Month 18 | ||
| n (%) | 10 (71.4) | 8 (72.7) |
| 95% CI | 41.9, 91.6 | 39.0, 94.0 |
Abbreviations: CI, confidence interval; PVAS, Pediatric Vasculitis Activity Score.
Re‐treated refers to patients who received rituximab between months 6 and 18. Non–re‐treated refers to patients who did not receive rituximab between months 6 and 18. Three of the 11 non–re‐treated patients received rituximab after month 18 and eight did not.
DISCUSSION
To provide dosing recommendations for induction and maintenance in pediatric patients with GPA or MPA, data were leveraged from adult and pediatric patients treated with rituximab. Modeling and simulations were used to confirm the dosing regimen used for induction in patients greater than 6 years old, extrapolate to younger patients aged greater than or equal to 2 to less than 6 years, and suggest a dosing regimen for follow‐up maintenance treatment in pediatric patients aged greater than or equal to 2 to less than 18 years. The rituximab PK, exposure‐safety, and exposure‐efficacy analyses were comparable between adult and pediatric patients with GPA or MPA who participated in RAVE and PePRS, respectively.
Rituximab's PK has been described previously in patients with non‐Hodgkin's lymphoma by a linear two‐compartment model with rituximab elimination composed of a time‐independent nonspecific catabolic antibody clearance, and a time‐dependent clearance associated with target‐mediated drug elimination, which decreases exponentially over time. 29 In the present analysis, rituximab dosing and sample collection did not allow capturing the nonlinear clearance, and the PK of rituximab was well‐described using a two‐compartment linear clearance only. Results of the population PK analysis showed a strong relationship (exponent = 0.952) between rituximab CL and BSA, with similar exposure (AUC over 6 months) in children and adolescents from PePRS compared with adults from RAVE across the entire range of BSA, following four weekly i.v. infusions of rituximab 375 mg/m2. In this analysis, the primary exposure variable considered for PK bridging was the AUC. Results from PePRS showed no association between variability in rituximab exposure and selected safety or efficacy end points, suggesting that the dosing regimen results in exposure at the plateau of the exposure–response relationship. 22
Whereas no patients aged less than 6 years were enrolled in PePRS, based on the model, exposure in pediatric patients aged less than 6 years is predicted to be similar to that of older pediatric patients. The results of the work presented here are supported by the following evidence:
IgG levels are low in children aged <1 year due to low synthesis (immature immune system at birth) and decline of maternal IgG levels after birth. 30 Endogenous IgG concentrations increase from childhood to adulthood; the increase primarily reflects an increase in production, rather than a decrease in IgG CL. 31 A prior study found that IgG concentrations decrease in infants within the first 3 months of age but increase to 85% of adult levels by ages 3–5 years. 32 The data suggest that IgG concentrations are slightly increased from age 2–5 years compared with older children and adults. Because the modest increase in IgG is due to an increase in IgG synthesis, the rituximab dose required in patients aged greater than or equal to 2 to less than 6 years is expected to be the same as in those aged greater than or equal to 6 years.
The neonatal Fc receptor (FcRn) protects IgG, both endogenous and therapeutic (i.e., monoclonal antibodies), from degradation. 33 As FcRn is present in fetuses, infants, children, and adults, rituximab's half‐life is expected to be as long in patients aged greater than or equal to 2 to less than 6 years as in older children. 34 , 35 A competition for recycling via FcRn exists between endogenous IgG and monoclonal antibodies 36 ; therefore, assuming constant FcRn expression and functionality, lower IgG levels in children (≥6 months to <3 years) could reduce rituximab CL and increase its exposure.
As children age, physiologic processes mature and can affect the disposition of monoclonal antibodies 37 , 38 ; however, these maturation processes are not expected to affect the PK of rituximab in patients greater than or equal to 6 years of age. Furthermore, rituximab PK data in patients receiving rituximab in the oncology setting demonstrated similar exposure in patients greater than 1 to less than 6 years old compared with older patients. 39
A PK extrapolation strategy was used to confirm rituximab dosing for induction and determine maintenance dosing for pediatric patients with GPA or MPA (accepted by the FDA) and for patients less than 6 years (accepted by the FDA and European Commission). This was the first regulatory approval of rituximab for any indication in pediatric patients. The approved induction dose for pediatric patients with GPA or MPA is, in combination with GCs, rituximab 375 mg/m2 once weekly for 4 weeks. 15 , 16 The follow‐up dosing approved by the FDA in pediatric patients with GPA or MPA who have achieved disease control with induction treatment is, in combination with GCs, two i.v. infusions of rituximab 250 mg/m2 separated by 2 weeks followed by an i.v. infusion of rituximab 250 mg/m2 every 6 months thereafter based on clinical evaluation. 15 Due to heterogeneity in repeat dosing and regimens used after induction of remission in PePRS, the determination of the optimal maintenance regimen was based on matching exposure between pediatric patients and adults receiving rituximab in MAINRITSAN. 21 , 22 The proposed dosing regimen uses a lower dose than the most intensive regimen (375 mg/m2/week for 4 weeks every 6 months for ≤6 cycles), which was well‐tolerated by patients in PePRS. 22 In PePRS, the proportion of patients achieving PVAS remission increased after repeated doses of rituximab (Table 2), supporting the use of additional rituximab therapy in the maintenance setting, for those who require it.
As B‐cell subpopulations are similar across pediatric age groups, 12 the PD of rituximab was expected to be similar in children less than 6 years old. No obvious differences in B‐cell depletion or the onset of repletion were found between adult patients in RAVE 20 and pediatric patients in PePRS 22 receiving rituximab for GPA or MPA. B‐cell depletion profiles in pediatric patients were as expected per the mechanism of action of rituximab and as observed for other autoimmune indications for which rituximab is approved in adults. 15 , 16 Return of peripheral B‐cells was not a prerequisite for repeat maintenance treatment with rituximab, and most patients received repeat rituximab treatment at investigators' discretion while maintaining peripheral B‐cell depletion.
No new or unexpected safety findings were observed during the overall study period, and no new safety concerns were observed in patients who were re‐treated with rituximab after the remission induction phase compared with the non–re‐treated patients. The safety profile over the duration of observation in the PePRS pediatric study, from baseline to 4.5 years, was expected and consistent with the known experience in adult patients with GPA or MPA and with the other approved autoimmune indications, 15 , 16 , 20 , 40 despite the prolonged durations of B‐cell depletion observed through month 18 and during the periods of repeat rituximab treatments. Given the lack of obvious differences around rituximab‐associated B‐cell depletion and mechanism of action in pediatric versus adult patients, extrapolation of safety data from adults to children in the maintenance setting is appropriate to support the safe use of rituximab for maintenance treatment in pediatric patients as observed in PePRS. The similarity of the rituximab safety profile that is expected between patients aged 2–5 years and those greater than or equal to 6 years further supports the use of rituximab in patients aged greater than or equal to 2 to less than 18 years.
This analysis has some limitations. Because GPA and MPA are orphan diseases, the trial sample size was necessarily small and this was an open‐label trial without a concurrent control arm; however, the results from this study provide key information for the pediatric population. In this study, the median age of 14 years (range: 6–17 years) was similar to published reports. 41 , 42 Given the extreme rarity of GPA and MPA in the younger pediatric (≥2 to <6 years old) population, no patients less than 6 years old were enrolled in this study. In addition, the PKs of mAbs in smaller children has not been fully elucidated. Differences in extracellular fluid could influence the volume of distribution, and variability in B‐cell counts could have an impact on CL. 38 , 43 However, these effects cannot be confirmed with existing PK models for pediatric populations, which show that compartmental volumes scale proportionally with body weight and fit the data well over a broad age range. 37 Moreover, these properties do not directly impact CL and are expected to have limited impact on AUC, and therefore on rituximab exposure during the induction phase. Although no phase III data in pediatric patients were available for the maintenance regimen, and no standard maintenance regimen was available at the time, the approved maintenance pediatric regimen is based on extrapolation from phase III data in adults with GPA/MPA. Considering that pediatric GPA/MPA is an orphan disease, this type of extrapolation enables treatment of patients with a dosing regimen, which is expected to have the same efficacy/safety profile as in adults.
CONCLUSION
A BSA‐adjusted induction regimen of four weekly i.v. infusions of rituximab 375 mg/m2, identical to the dosing regimen in adults, is recommended in children greater than 2 years old with GPA or MPA in the United States and Europe and is supported by similar PK characteristics and matching rituximab exposure between adult and pediatric patients (≥6 to <18 years). The PK extrapolation approach provided guidance for the recommended rituximab maintenance dosing regimen after induction therapy in pediatric patients aged greater than or equal to 2 years: a follow‐up dosing regimen consisting of two 250‐mg/m2 i.v. infusions separated by 2 weeks, followed by a 250‐mg/m2 i.v. infusion every 6 months, is approved in the United States. The full extrapolation strategy, applied from the established PK/PD, efficacy, and safety data in adults, avoided the need to conduct a dedicated randomized trial for evaluation of maintenance therapy in a pediatric population. This is especially important when considering pediatric development in rare conditions, such as GPA and MPA. The work presented here was instrumental in supporting the first approval of rituximab in pediatric patients with GPA/MPA and demonstrates the utility of model‐informed drug development to complement limited clinical trial data, especially in rare pediatric diseases.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript and performed the research. C.J., L.G., and J.G. designed the research. C.J., L.G., C.C., M.C., P.P., S.M., and J.G. analyzed the data.
CONFLICT OF INTEREST
C. Jamois, C. Chavanne, P. Pordeli, and S. Melega are employees of F. Hoffmann‐La Roche and C. Jamois owns stocks in F. Hoffmann‐La Roche. L. Gibiansky is an employee of QuantPharm, LLC. P. B. Lehane is an employee of Roche Products Ltd. M. Cheu is an employee of Genentech. J. Gaudreault is an employee of JJG Pharma Consulting, GmbH.
Supporting information
Supporting Information S1
ACKNOWLEDGMENTS
Support for third‐party writing assistance for this manuscript, furnished by Claire Stedden, PhD, Sarah Nordquist, PhD, and Ellen Mercado, PhD, of Health Interactions, Inc., was provided by F. Hoffmann‐La Roche.
Jamois C, Gibiansky L, Chavanne C, et al. Rituximab pediatric drug development: Pharmacokinetic and pharmacodynamic modeling to inform regulatory approval for rituximab treatment in patients with granulomatosis with polyangiitis or microscopic polyangiitis. Clin Transl Sci. 2022;15:2172‐2183. doi: 10.1111/cts.13351
Funding Information
This study was sponsored by F. Hoffmann‐La Roche.
REFERENCES
- 1. Yates M, Watts RA, Bajema IM, et al. EULAR/ERA‐EDTA recommendations for the management of ANCA‐associated vasculitis. Ann Rheum Dis. 2016;75:1583‐1594. [DOI] [PubMed] [Google Scholar]
- 2. de Graeff N, Groot N, Brogan P, et al. European consensus‐based recommendations for the diagnosis and treatment of rare paediatric vasculitides ‐ the SHARE initiative. Rheumatology (Oxford). 2019;58:656‐671. [DOI] [PubMed] [Google Scholar]
- 3. Finkielman JD, Lee AS, Hummel AM, et al. ANCA are detectable in nearly all patients with active severe Wegener's granulomatosis. Am J Med. 2007;120:643.e9‐14‐643.14. [DOI] [PubMed] [Google Scholar]
- 4. Yates M, Watts R. ANCA‐associated vasculitis. Clin Med (Lond). 2017;17:60‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jariwala MP, Laxer RM. Primary vasculitis in childhood: GPA and MPA in childhood. Front Pediatr. 2018;6:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Calatroni M, Oliva E, Gianfreda D, et al. ANCA‐associated vasculitis in childhood: recent advances. Ital J Pediatr. 2017;43:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Akikusa JD, Schneider R, Harvey EA, et al. Clinical features and outcome of pediatric Wegener's granulomatosis. Arthritis Rheum. 2007;57:837‐844. [DOI] [PubMed] [Google Scholar]
- 8. Cabral DA, Uribe AG, Benseler S, et al. Classification, presentation, and initial treatment of Wegener's granulomatosis in childhood. Arthritis Rheum. 2009;60:3413‐3424. [DOI] [PubMed] [Google Scholar]
- 9. Prendecki M, Pusey CD. Recent advances in understanding of the pathogenesis of ANCA‐associated vasculitis. F1000Res. 2018;7:1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B‐ and T‐cell activation in Wegener's granulomatosis. J Allergy Clin Immunol. 1999;103:885‐894. [DOI] [PubMed] [Google Scholar]
- 11. Martin F, Chan AC. Pathogenic roles of B cells in human autoimmunity; insights from the clinic. Immunity. 2004;20:517‐527. [DOI] [PubMed] [Google Scholar]
- 12. Duchamp M, Sterlin D, Diabate A, et al. B‐cell subpopulations in children: national reference values. Immun Inflamm Dis. 2014;2:131‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vanoni F, Bettinelli A, Keller F, Bianchetti MG, Simonetti GD. Vasculitides associated with IgG antineutrophil cytoplasmic autoantibodies in childhood. Pediatr Nephrol (Berlin, Germany). 2010;25:205‐212. [DOI] [PubMed] [Google Scholar]
- 14. Jones RB, Hiemstra TF, Ballarin J, et al. Mycophenolate mofetil versus cyclophosphamide for remission induction in ANCA‐associated vasculitis: a randomised, non‐inferiority trial. Ann Rheum Dis. 2019;78:399‐405. [DOI] [PubMed] [Google Scholar]
- 15. Rituxan (rituximab) injection, for intravenous use: US prescribing information. Genentech, Inc. (2018).
- 16. MabThera . Summary of Product Characteristics. Roche Registration, Limited; 2019. [Google Scholar]
- 17. Davies A, Berge C, Boehnke A, et al. Subcutaneous rituximab for the treatment of B‐cell hematologic malignancies: a review of the scientific rationale and clinical development. Adv Ther. 2017;34:2210‐2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weiner GJ. Rituximab: mechanism of action. Semin Hematol. 2010;47:115‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leidi M, Gotti E, Bologna L, et al. M2 macrophages phagocytose rituximab‐opsonized leukemic targets more efficiently than m1 cells in vitro. J Immunol. 2009;182:4415‐4422. [DOI] [PubMed] [Google Scholar]
- 20. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA‐associated vasculitis. N Engl J Med. 2010;363:221‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guillevin L, Pagnoux C, Karras A, et al. Rituximab versus azathioprine for maintenance in ANCA‐associated vasculitis. New Engl J Med. 2014;371:1771‐1780. [DOI] [PubMed] [Google Scholar]
- 22. Brogan P, Yeung RSM, Cleary G, et al. Phase IIa global study evaluating rituximab for the treatment of pediatric patients with granulomatosis with polyangiitis or microscopic polyangiitis. Arthritis Rheumatology. 2021;74:124‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watt, K. Application of Master Protocols to Pediatric Anesthetics and Analgesics. Accessed August 11, 2021. https://www.pharmacy.umaryland.edu/media/SOP/wwwpharmacyumarylandedu/centers/cersievents/pdf/Kevin%20Watt%20Pediatric%20Master%20Protocols.pdf.
- 24. U.S. Food and Drug Administration . Population pharmacokinetics guidance for industry 26 (2019).
- 25. Shepard T, Scott G, Cole S, Nordmark A, Bouzom F. Physiologically based models in regulatory submissions: output from the ABPI/MHRA forum on physiologically based modeling and simulation: pbpk models in regulatory submissions. CPT Pharmacometrics Syst Pharmacol. 2015;4:221‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beal, S. , Sheiner, L.B. , Boeckmann, A. & Bauer, R.J. NONMEM User's Guides (1989–2011) (Ellicott City, Maryland, USA, 2011).
- 27. CCLG ‐ Chemotherapy Standardisation Group 2008. Estimation of body‐surface area in infants and children. https://www.ouh.nhs.uk/oxparc/professionals/documents/Body‐surfaceareaCCLGChart1.pdf. Accessed March 30, 2021.
- 28. Dolezalova P, Price‐Kuehne FE, Özen S, et al. Disease activity assessment in childhood vasculitis: development and preliminary validation of the paediatric vasculitis activity score (PVAS). Ann Rheum Dis. 2013;72:1628‐1633. [DOI] [PubMed] [Google Scholar]
- 29. Li J, Zhi J, Wenger M, et al. Population pharmacokinetics of rituximab in patients with chronic lymphocytic leukemia. J Clin Pharmacol. 2012;52:1918‐1926. [DOI] [PubMed] [Google Scholar]
- 30. Siegrist CA, Aspinall R. B‐cell responses to vaccination at the extremes of age. Nat Rev Immunol. 2009;9:185‐194. [DOI] [PubMed] [Google Scholar]
- 31. Stoop JW, Zegers BJ, Sander PC, Ballieux RE. Serum immunoglobulin levels in healthy children and adults. Clin Exp Immunol. 1969;4:101‐112. [PMC free article] [PubMed] [Google Scholar]
- 32. Belldegrin A, Shoenfeld Y, Pick AI, Vana D. Age related distribution of serum immunoglobulin concentration in 1003 healthy children and adults. Biomedicine. 1980;33:8‐12. [PubMed] [Google Scholar]
- 33. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin Immunol. 2007;122:146‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Israel EJ, Taylor S, Wu Z, et al. Expression of the neonatal fc receptor, FcRn, on human intestinal epithelial cells. Immunology. 1997;92:69‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shah U, Dickinson BL, Blumberg RS, Simister NE, Lencer WI, Walker WA. Distribution of the IgG fc receptor, FcRn, in the human fetal intestine. Pediatr Res. 2003;53:295‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu L. Pharmacokinetics of monoclonal antibodies and fc‐fusion proteins. Protein Cell. 2018;9:15‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robbie GJ, Zhao L, Mondick J, Losonsky G, Roskos LK. Population pharmacokinetics of palivizumab, a humanized anti‐respiratory syncytial virus monoclonal antibody, in adults and children. Antimicrob Agents Chemother. 2012;56:4927‐4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malik P, Edginton A. Pediatric physiology in relation to the pharmacokinetics of monoclonal antibodies. Expert Opin Drug Metab Toxicol. 2018;14:585‐599. [DOI] [PubMed] [Google Scholar]
- 39. MabThera, European Medicine Agency Assessment Report . Assessment report: MabThera. European Medicines Agency 2020. https://www.ema.europa.eu/en/documents/variation‐report/mabthera‐h‐c‐165‐ii‐0168‐epar‐assessment‐report‐variation_en.pdf
- 40. Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA‐associated renal vasculitis. N Engl J Med. 2010;363:211‐220. [DOI] [PubMed] [Google Scholar]
- 41. Sacri AS, Chambaraud T, Ranchin B. et al. Clinical characteristics and outcomes of childhood‐onset ANCA‐associated vasculitis: a French nationwide study. Nephrol Dial Transplant. 2015;30(Suppl 1):i104‐i112. [DOI] [PubMed] [Google Scholar]
- 42. Iudici M, Pagnoux C, Quartier P, et al. Childhood‐ versus adult‐onset ANCA‐associated vasculitides: A nested, matched case‐control study from the French Vasculitis study group registry. Autoimmun Rev. 2018;17:108‐114. [DOI] [PubMed] [Google Scholar]
- 43. Edlund H, Melin J, Parra‐Guillen ZP, Kloft C. Pharmacokinetics and pharmacokinetic‐pharmacodynamic relationships of monoclonal antibodies in children. Clin Pharmacokinet. 2015;54:35‐80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1