

Abstract
N‐Nitrosamine (NA) impurities are considered genotoxic and have gained attention due to the recall of several marketed drug products associated with higher‐than‐permitted limits of these impurities. Rifampicin is an index inducer of multiple cytochrome P450s (CYPs) including CYP2B6, 2C8, 2C9, 2C19, and 3A4/5 and an inhibitor of OATP1B transporters (single dose). Hence, rifampicin is used extensively in clinical studies to assess drug–drug interactions (DDIs). Despite NA impurities being reported in rifampicin and rifapentine above the acceptable limits, these critical anti‐infective drugs are available for therapeutic use considering their benefit–risk profile. Reports of NA impurities in rifampicin products have created uncertainty around using rifampicin in clinical DDI studies, especially in healthy volunteers. Hence, a systematic investigation through a literature search was performed to determine possible alternative index inducer(s) to rifampicin. The available strong CYP3A inducers were selected from the University of Washington DDI Database and their in vivo DDI potential assessed using the data from clinical DDI studies with sensitive CYP3A substrates. To propose potential alternative CYP3A inducers, factors including lack of genotoxic potential, adequate safety, feasibility of multiple dose administration to healthy volunteers, and robust in vivo evidence of induction of CYP3A were considered. Based on the qualifying criteria, carbamazepine, phenytoin, and lumacaftor were identified to be the most promising alternatives to rifampicin for conducting CYP3A induction DDI studies. Strengths and limitations of the proposed alternative CYP3A inducers, the magnitude of in vivo CYP3A induction, appropriate study designs for each alternative inducer, and future perspectives are presented in this paper.
INTRODUCTION
The pharmacokinetics (PKs) of a drug may be influenced by extrinsic and intrinsic factors that can lead to undesired or unexpected clinical outcomes. Extrinsic factors, such as smoking habits, diet, and drug–drug interactions (DDIs) are important sources of variability in drug exposures. 1 The DDIs can inadvertently lead to exaggeration of adverse drug effects or loss of efficacy. PK DDIs often result from inhibition and/or induction of drug metabolizing enzymes and/or transporter‐mediated disposition of a victim drug by a perpetrator drug. 2 , 3
CYP induction is an important mechanism for DDIs, and primarily occurs through the activation of xenobiotic‐sensing receptors, aryl hydrocarbon receptor (AHR), pregnane X receptor (PXR), and constitutive androstane receptor (CAR). 4 , 5 CYP induction increases metabolic clearance and thereby decreases the systemic exposure of a victim drug. Additionally, depending on the metabolizing enzymes involved, sometimes a perpetrator drug may induce its own metabolism (autoinduction). Induction is often nonselective and requires multiple doses to achieve the maximum effect. PXR regulates the expression of many CYPs (CYP3A, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP1A), non‐CYP (UGTs, SULTs, and GSTs), enzymes, and transporters (P‐gp and BCRP). 6 Many drugs and their metabolites regulate the transcription of enzymes and transporters through activation of PXR. 6
DDIs are one of the major causes for morbidity and mortality associated with the use of prescription drugs and have occasionally resulted even in the withdrawal of approved drugs. Quantitative assessments of the magnitude of DDIs for investigational agents both as potential victim and perpetrator are required to inform labeling language and clinical management by appropriate contraindications, dose adjustments, or staggered administration of drugs. This is generally accomplished by incorporating a well‐designed combination of in vitro, in vivo (clinical) studies, and quantitative model‐based assessment approaches at different stages of drug development. Regulatory agencies, such as the US Food and Drug Administration (FDA), European Medicines Agency (EMA), and Pharmaceuticals and Medical Devices Agency (PMDA) have issued guidelines related to the conduct of DDI studies including their timing and design, the interpretation of study results, and management of DDIs in patients. 7 , 8 , 9
For the assessment of the sensitivity of an investigational agent as a victim of CYP3A induction DDIs, a clinical study is generally performed with a strong index inducer, such as rifampicin as the primary choice. 10 Rifampicin, an antibiotic against gram‐positive bacteria, is a strong index inducer of CYP3A, and is most used in clinical DDI studies based on its established CYP3A induction and safety profiles. Rifampicin induces multiple drug metabolizing enzymes and certain transporters primarily via activation of PXR. In addition to hepatic CYP3A, rifampicin also induces intestinal CYP3A in a dose‐dependent manner. 11 The conduct of clinical DDI studies with rifampicin has been hampered recently by the presence of higher than acceptable limits of nitrosamine impurities. 12 In this review, we have surveyed the literature and presented a current view of fit‐for‐purpose rifampicin alternatives and associated design considerations for induction in DDI studies.
NITROSAMINE IMPURITIES IN DRUG PRODUCTS
N‐Nitrosoamines (NAs) are derivatives of secondary amines where a nitroso (‐NO) group is attached to an amino group and are found in food, water, tobacco, personal care, and consumer products. 13 NAs are classified as high potent mutagenic carcinogens as per the International Conference on Harmonization (ICH) M7(R1) guideline and categorized as group 2A (probably carcinogenic to humans) carcinogens by the International Agency for Research on Cancer (IARC). 14 , 15 Depending on their reactivity, NAs can form adducts with DNA directly or through bioactivation by cytochrome P450 enzymes via α‐hydroxylation to form highly reactive nitrosamide, alkyldiazohydroxide, or aldehyde intermediates (Figure 1). 16 , 17 These adducts, if not repaired or repaired incorrectly, can induce mutations, and ultimately cause cancer, especially if the adduct is in an oncogene or tumor suppressor gene. NA impurities gained attention recently due to the recall of many marketed products associated with higher than allowed limits of these impurities. The FDA first reported the presence of NA impurity, N‐nitrosodimethylamine (NDMA) from one of the manufacturers of valsartan in 2018. Since then, four other NA impurities, N‐nitrosodiethylamine (NDEA), N‐nitroso‐N‐methyl‐4‐aminobutanoic acid (NMBA), N‐nitrosoisopropylethylamine (NIPEA), and N‐nitrosomethylphenylamine (NMPA), were detected in many drug substances or drug products above the acceptable upper limits. This led to withdrawal or recall of drug products containing APIs of valsartan, irbesartan, losartan, metformin, ranitidine, and nizatidine. 18 The causes for the presence of these impurities are currently being investigated by regulators. Recently, the FDA issued a guidance on “Control of NA impurities in human drugs” and recommended acceptable intake limits for the NA impurities. 19
FIGURE 1.
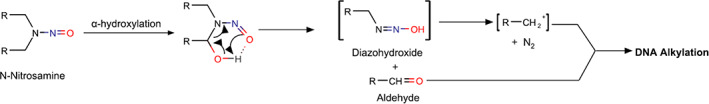
Example of metabolic activation and DNA alkylation by nitrosamine.
Two additional NA impurities, 1‐cyclopentyl‐4‐nitrosopiperazine (CPNP) and 1‐methyl‐4‐nitrosopiperazine (MNP), have been reported in rifapentine and rifampicin products, respectively, in 2020. 20 Both rifapentine and rifampicin are rifamycin derivatives used to treat bacterial infections, with a critical role in the pharmacotherapy of tuberculosis. 21 The acceptable intake limits are 0.16 parts per million (ppm) for MNP and 0.1 ppm for CPNP in rifampicin and rifapentine, respectively. 20 Despite the presence of NA impurities of rifampicin and rifapentine, the FDA permitted the marketing of these drugs with impurities below 5 ppm for MNP and 20 ppm for CPNP (above the acceptable intake limits), considering the benefit–risk profile of these life‐saving medications. 20
Rifampicin is a prototypical strong inducer of CYP3A and used extensively in clinical drug interaction studies to assess the sensitivity of CYP3A substrates to induction DDIs. The DDI studies with rifampicin are conducted primarily in healthy volunteers and occasionally in patients. 22 Recent issues with NA impurities in rifampicin products and restrictions around use of rifampicin in healthy volunteers and patients have challenged the design of DDI studies for investigational agents that are substrates of CYP3A to evaluate the magnitude of DDI with strong CYP3A inducers. Hence, there is a critical need for guidance for choosing the right alternative strong CYP3A inducers for conducting clinical DDI studies in drug development.
STRONG CYP3A INDUCERS
The University of Washington Drug–Drug Interaction Database (DIDB) was utilized to determine potential alternatives to rifampicin. A list of available strong CYP3A inducers were first prepared and their properties, including safety, enzymes responsible for metabolism, and CYPs that are induced, are summarized (Table 1). 23 To determine the maximum magnitude of CYP3A induction by strong inducers, a search was performed for clinical DDI studies conducted with strong CYP3A inducers and sensitive CYP3A substrates and results were compiled (Table 2). Published literature was then reviewed to determine the strengths and limitations of each strong CYP3A inducer and to propose alternatives to rifampicin. Finally, study designs for the identified alternative inducers were also proposed based on knowledge of their PK properties, typical considerations of the dynamics of CYP3A induction, and available literature data on the dose–response and time course of CYP3A induction by the identified alternative inducers. The properties of strong CYP3A inducers that are potential alternatives to rifampicin are summarized below.
TABLE 1.
Summary of safety and the enzymes involved in the metabolism and CYP induction of strong CYP3A inducers
| Therapeutic class | Metabolizing enzymes a | CYP induction | Mechanism of induction a | Safety | |||
|---|---|---|---|---|---|---|---|
| Strong | Moderate | Weak | |||||
| Apalutamide | Antiandrogen |
CYP2C8 CYP3A4 |
CYP2C19 CYP3A |
CYP2C9 | PXR | QT prolongation | |
| Avasimibe | Antilipemic | NA | CYP3A | CYP2C9 | PXR | ||
| Carbamazepine | Anticonvulsant |
CYP3A4 CYP2C8 UGT2B7 |
CYP2B6 CYP3A |
CYP2C8 CYP2C9 |
CYP1A2 CYP2C19 |
CAR/PXR | NTR |
| Enzalutamide | Antiandrogen |
CYP2C8 CYP3A |
CYP3A |
CYP2C9 CYP2C19 |
PXR/CAR | ||
| Ivosidenib b | Anticancer |
CYP3A4 CYP2B6 CYP2C8 |
CYP3A | CYP2C8 |
QT prolongation |
||
| Lumacaftor | Cystic Fibrosis Treatment | CYP3A4 (minor) | CYP3A | CYP2B6 c ; CYP2C8 c ; CYP2C9 c ; CYP2C19 c | PXR | ||
| Mitotane | Antineoplastic | NA | CYP3A | NTR | |||
| Phenytoin | Anticonvulsant |
CYP2C9 CYP2C19 |
CYP3A |
CYP1A CYP2C19 |
CAR/PXR | NTR | |
| Rifampicin d | Antibiotic |
AADAC UGT e |
CYP2C19 CYP3A |
CYP1A2 CYP2B6 CYP2C8 CYP2C9 |
PXR | ||
| Rifapentine | Antibiotic |
AADAC |
CYP3A |
CYP2C8 c CYP2C9 c |
PXR | ||
| St. John’s wort extract | Herbal Medication |
CYP3A4 f CYP2C8/9/19 |
CYP3A |
CYP2C9 CYP2C19 |
PXR | ||
Note: Gray boxes: None.
Abbreviations: NA, not available; NTR, narrow therapeutic range.
Bolded refers to primary pathway.
Physiologically‐based pharmacokinetic (PBPK) modeling was used to assess drug‐drug interaction potential.
Possible DDIs were assessed at clinically relevant concentration from in vitro data.
No definitive information on CYP3A involvement in metabolism of rifampicin.
Asaumi et al. assigned fm (0.76) through UGTs in PBPK modeling. 96
Enzyme responsible for hyperforin metabolism. Hyperforin, a constituent of St. John’s Wart, responsible for CYP3A induction.
TABLE 2.
Drug–drug interactions of strong CYP3A inducers (alternatives to rifampicin) and sensitive CYP3A substrates
| Dose/dosing regimen of strong CYP3A inducer | Sensitive CYP3A substrate | Population | CYP3A substrate dose | DDI | ||
|---|---|---|---|---|---|---|
| AUC ratio | Cmax ratio | |||||
| Apalutamide | 240 mg q.d., 28 days, oral | Midazolam (as part of cocktail) 45 | Patients | 2 mg, oral, SD (day 29) | 0.08 | 0.23 |
| Avasimibe | 50 mg or 750 mg, q.d., 7 days, oral | Midazolam 25 | HVs | 2 mg, oral, SD (day 7) |
50 mg: 0.27 750 mg: 0.07 |
50 mg: 0.43 750 mg: 0.18 |
| Carbamazepine | 200 mg q.d., (days 1–3) → 200 mg b.i.d. (days 4 until day 24), oral | Paritaprevir 30 | HVs | 150 mg, oral, SD (day 22) | 0.30 | 0.34 |
| 100 mg, b.i.d. (days 1–3) → 200 mg b.i.d. (day 4 to day 31), oral | Elvitegravir 31 | HVs | 150 mg (in combination with Cobistat a ), oral, q.d. (day 22 to day 31) | 0.31 | 0.55 | |
| 400 mg b.i.d., 24 days, oral (PBPK modeling) | Abemaciclib 36 | HVs | 200 mg, oral, SD (day 7) | 0.20 | NA | |
| 400 mg b.i.d., 7 days, oral (PBPK modeling) | Acalabrutinib 37 | HVs | 100 mg, oral, b.i.d. (7 days) | 0.39 | NA | |
| 200 mg q.d. (days 1 and 2) → 300 mg, b.i.d. (12 days), oral | Simvastatin 29 | HVs | 80 mg, oral, SD (day 15) | 0.25 | 0.32 | |
| 200 mg q.d. (day 1) → 200 mg b.i.d. (day 2 to day 4) → 200 mg t.i.d. (day 5 → 25), oral | Quetiapine 97 | Patients | 25 mg b.i.d., oral, MD | 0.13 | 0.20 | |
| 400 mg q.d., 16 days, oral | Ivabradine 35 | HVs | 10 mg, oral, SD (day 15) | 0.20 | 0.23 | |
| 100 mg b.i.d. → 300 mg b.i.d. (escalated to 300 mg b.i.d. over 7 days and continued until a total of 26 days), oral | Midazolam 33 | HVs | 2 mg, oral, SD (day 18, as part of cocktail) | 0.21 | 0.31 | |
| 1600 mg q.d., 14 days, oral (population‐based simulations) | Midazolam 40 | HVs | 8 mg, oral, SD | ~0.20 | – | |
|
400 mg q.d., multiple doses, oral (PBPK modeling) |
Ibrutinib 38 | HVs | 560 mg, oral, SD | 0.18 | 0.14 | |
|
200 mg b.i.d., oral, 14 days, oral (PBPK modeling) |
Zanubrutinib 39 | HVs | 160 mg, oral, b.i.d. (day 7 to day 14) | 0.42 | 0.39 | |
| Enzalutamide | 160 mg q.d., 50 days, oral | Midazolam (as part of cocktail) 44 | Patients | 2 mg, oral, SD (day 50) | 0.14 | 0.23 |
| Ivosidenib | 500 mg q.d., 15 days, oral | Midazolam 51 | Patients | 5 mg, oral, SD (day 15) | 0.18 | 0.27 |
| Lumacaftor | 200 mg q.d., 14 days, oral | Ivacaftor 53 | HVs | 150 mg, oral, b.i.d. (14 days) | 0.19 | 0.24 |
| Lumacaftor + ivacaftor | LUMA, 200 mg + IVA, 250 mg (q12h) 21 days, oral | Itraconazole a , 53 | HVs | 200 mg, oral, QD (day 15 to day 21; 7 days) | 0.1 | 0.097 |
| Mitotane | 0.5–3.5 g, t.i.d., oral, n = 4 (patient 4: 0.5 g q.d. or 0.5 g b.i.d. every other day) | Midazolam 56 | Patients | 7.5 mg, oral, SD | 0.055 | N/A |
| Phenytoin | 300 mg q.d., 11 days, oral |
Lopinavir/ Ritonavir 59 |
HVs | 400 mg/100 mg, oral, b.i.d. (11 days) |
0.67 (Lopinavir) |
0.76 (Lopinavir) |
| 4 mg/kg q.d., 21 days, oral | Atorvastatin 61 | HVs | 40 mg, oral, MD (21 days) | 0.46 | 0.76 | |
| 100 mg t.i.d., 10 days, oral | Quetiapine 64 | Patients | 250 mg t.i.d., oral, 10 days (co‐administration with phenytoin, 250 mg q.d. day 13 to day 22) | 0.20 | 0.34 | |
| 100 mg, b.i.d. (until reaching a Ctrough of 5.5 μg/ml), oral | Sirolimus 85 | One patient | 5 mg q.d., oral, (required escalation to 15 mg q.d.) | 0.61 | 0.87 | |
| 200–450 mg, q.d./(with stable phenytoin concentration: 9.2–25.9 μg/ml), oral | Nisoladipine 62 | Patients and HVs | 40 mg in patients and 20 mg in HVs, oral, SD | 0.11 | 0.18 | |
| 150 mg, b.i.d., 5 days, oral | Ivabradine 60 | HVs | 10 mg, oral, SD (day 5) | 0.31 | 0.35 | |
| 150–300 mg, q.d., for at least 2 months, oral | Midazolam 63 | Patients and HVs | 15 mg, oral, SD | 0.057 | 0.074 | |
| Rifapentine | 5, 10, 15, 20 mg/kg q.d., 14 days (days 2 to days 15), oral | Midazolam 66 | HVs |
15 mg, oral, SD (day 15) |
<0.1 | ≤0.2 |
| St John's Wort | 300 mg t.i.d., 15 days, oral (standardized to 0.3% hypericin) | Eplerenone 98 | HVs | 100 mg, oral, SD (day 15) | 0.68 | 0.81 |
| 300 mg t.i.d., 15 days, oral | Ivabradine 99 | HVs | 10 mg, oral, SD (day 15) | 0.39 | 0.49 | |
| 300 mg t.i.d., 14 days, oral | Indinavir 100 | HVs | 800 mg, oral, SD (day 15) | 0.43 | 0.72 | |
| 300 mg t.i.d., 14 days, oral | Midazolam 101 | HVs |
5 mg, oral 0.05 mg/kg, intravenous, SD |
0.79 (intravenous) 0.48 (oral) | 0.57 (oral) | |
| 300 mg t.i.d., 14 days, oral | Midazolam 102 | HVs | 5 mg, oral, SD | 0.72 | 0.88 | |
| 300 mg t.i.d., 8 weeks, oral | Midazolam 103 | HVs |
5 mg, oral 0.05 mg/kg, intravenous, SD |
0.94 (intravenous) 0.59 (oral) |
0.79 (oral) | |
| LI 160, 300 mg per coated tablet of an 80% (vol/vol) methanolic dry extract (41 mg/day, 14 days) b , oral | Midazolam 71 | HVs |
7.5 mg, oral, SD (day 14) |
0.20 | 0.35 | |
|
300 mg t.i.d., 14 days, oral |
Simvastatin 104 | HVs |
10 mg, oral, SD (day 14) |
0.38 (acid) 0.52 (lactone) |
0.48 (acid) 0.69 (lactone) |
|
| 300 mg q.d., 18 days, oral | Tacrolimus 105 | HVs |
0.1 mg/kg, SD (day 15) |
0.65 | 0.77 | |
Abbreviations: AUC, area under the curve; Cmax, maximum plasma concentration; DDI, drug‐drug interaction; HV, healthy volunteers; IVA, ivacaftor; LUMA, lumacaftor; PBPK, physiologically‐based pharmacokinetic; SD, single dose; MD, multiple doses; N/A, not applicable.
Itraconazole is a CYP3A substrate but not sensitive CYP3A substrate.
Apart from the enriched extract presented, various doses of Hypericum powder with different contents of hyperforin tested in this study and midazolam AUC and Cmax ranged from 0.52–0.79 and 0.61–1, respectively.
Avasimibe
Avasimibe is a potent inhibitor of acyl‐coenzyme A: cholesterol acyltransferase (ACAT), which was in development for the treatment of atherosclerosis and hyperlipidemia but was terminated at the phase II stage in 2003. It was shown that avasimibe has a similar inductive spectrum resembling rifampicin, as a potent inducer of CYP3A4, P‐glycoprotein (P‐gp), and CYP2C9. 24 , 25 Avasimibe known to exhibit CYP/P‐gp induction through potent activation human PXR. 25 Because avasimibe was discontinued from development, it is not available for use as an index perpetrator in DDI studies. Hence, avasimibe was not considered as a viable alternative to rifampicin.
Carbamazepine
Carbamazepine is an anticonvulsant and widely used to treat epileptic seizures, neuropathic pain, and schizophrenia. Carbamazepine is cleared almost entirely by metabolism (~72% is excreted in the urine, with only ~2% as unchanged drug). 26 As a low clearance drug, the elimination of carbamazepine is primarily governed by the intrinsic metabolic capacity of the liver. Metabolism via CYP3A4 was recognized as a main route of elimination with a minor role played by CYP2C8. 27 Even weak inhibitors of CYP3A4, such as isoniazid, have been known to increase carbamazepine exposures in a clinically relevant manner. 28 Carbamazepine is an inducer of multiple CYPs, P‐gp, and UDP‐glucuronosyltransferase (UGTs) and known to exhibit induction primarily by activating CAR and with minor contribution from PXR. The UW database classifies carbamazepine as a weak inducer of CYP1A2, CYP2C19, a moderate inducer of CYP2C8 and CYP2C9, and a strong inducer of CYP3A and CYP2B6. Clinically, carbamazepine reduces exposures of drugs that are substrates of CYP1A2, 2B6, 2C9, and 3A4 enzymes. Specifically, with sensitive substrates of CYP3A4, there are several reports that describe induction effects by carbamazepine (Table 2). For example, co‐administration of carbamazepine with simvastatin decreases exposures of simvastatin by ~75%. 29 Antivirals, such as paritaprevir, elvitegravir, and indinavir, have also been shown to have significantly reduced exposures when co‐administered with carbamazepine. 30 , 31 , 32 Carbamazepine has also been shown to significantly decrease the exposures (reduction in area under the curve [AUC] by ~79%) of oral midazolam, a sensitive probe substrate for CYP3A4 and significantly increase the clearance of lamotrigine, a substrate of UGT1A4 (major) and UGT2B7 (minor), upon co‐administration. 33 , 34 A study in healthy volunteers showed that carbamazepine decreased ~80% exposures of ivabradine, a sensitive substrate of CYP3A upon co‐administration for 400 mg q.d. of carbamazepine for 16 days. 35 Physiologically‐based pharmacokinetic (PBPK) modeling results supported the induction potential of carbamazepine with CYP3A substrates, such as abemaciclib, acalabrutinib, ibrutinib, zanabrutinib, and midazolam. 36 , 37 , 38 , 39 , 40 Carbamazepine also significantly reduced (>80%) AUC of other CYP3A (not sensitive) substrates, such as cobicistat, nefazodone, praziquantel, and ritonavir. 23 The exposure of P‐gp substrates ranged from no effect to 39% reduction upon co‐administration of carbamazepine. 41 In summary, there are sufficient clinical DDI data with CYP3A substrates for carbamazepine to be considered a potential alternative to rifampicin as an index inducer.
Enzalutamide and apalutamide
Enzalutamide and apalutamide are antiandrogens approved for the treatment of prostate cancers. 42 , 43 Both drugs are strong in vivo inducers of CYP3A4. 44 , 45
Enzalutamide is a moderate inducer of both CYP2C19 and CYP2C9. Enzalutamide and its primary active metabolite, N‐desmethyl enzalutamide, inhibit P‐gp and BCRP in vitro. A clinical cocktail DDI study investigated the CYP3A4, CYP2C19, and CYP2C9 induction potential of enzalutamide with daily dose of 160 mg enzalutamide for 50 days. 44 Here, the AUC of midazolam, (S)‐warfarin, and omeprazole decreased by 86%, 56%, and 71%, respectively, upon co‐administration of enzalutamide. The half‐life of enzalutamide is 5.8 days and steady‐state was achieved by 28 days. 46 A PBPK model also showed a strong CYP3A4 induction effect (predicted area under the concentration‐time curve ratio [AUCR]: 0.06–0.16) of enzalutamide and it takes up to 8 weeks to achieve baseline CYP3A4 activity after discontinuation of the drug. 47 Induction of CYP3A occurs via primarily by activation of the PXR receptor. Enzalutamide has been studied in healthy volunteers as a single dose, but not multiple doses. The product label has warnings of seizures, encephalopathy, and ischemic heart disease, that will have to be considered before any potential use in DDI studies.
Co‐administration (240 mg daily, 28 days) of apalutamide decreased AUC of midazolam (CYP3A), omeprazole (CYP2C19), and (S)‐warfarin AUC (CYP2C9) by 92%, 84%, and 46%, respectively. 45 Apalutamide is a strong inducer of CYP2C19 and a weak inducer of CYP2C9. Apalutamide is known to be an inducer of P‐gp and BCRP/OATP1B1, and noted to have the potential to induce UGTs. 43 The mechanism of induction by apalutamide is also mediated by activation of PXR. The mean effective half‐life of apalutamide is 3–4 days with steady‐state achieved after 4 weeks upon repeat daily dosing. Apalutamide exhibits a time‐dependent increase apparent clearance due to auto‐induction. 43 The QTc prolongation of apalutamide (although limited, with a mean change in QTc of 12.4 s at steady‐state) may limit its application as an index inducer in DDI studies. 48 Considering their safety profile and long half‐lives, both apalutamide and enzalutamide have limited potential to be alternative index inducers in place of rifampicin.
Ivosidenib
Ivosidenib (Tibsovo) is a selective inhibitor of mutant isocitrate dehydrogenase 1 (IDH1), approved in the United States for the treatment of adults with relapsed or refractory acute myeloid leukemia (AML) and newly diagnosed AML who are ≥ 75 years of age or are ineligible for intensive chemotherapy with a susceptible IDH1 mutation. 49 Ivosidenib is rapidly absorbed (median time to maximum concentration [T max] was typically 2–4 h) and concentrations slowly declined with a mean terminal half‐life (t ½) of ~72–138 h after single dosing and multiple doses in patients with hematologic malignancies. Ivosidenib exhibited an increase in apparent clearance after multiple dose administration which was attributed to a decrease in bioavailability due to auto‐induction of CYP3A4. 50 Ivosidenib is a substrate and an inducer of CYP3A4 and also induces CYP2B6, and CYP2C enzymes. PBPK modeling was used to predict magnitude of CYP2B6, CYP2C8, CYP2C9, and CYP3A4 induction of ivosidenib along with other DDI predictions in patients with cancer. 51 For CYP3A induction prediction, the in vivo induction parameters were optimized using 4β‐hydroxycholesterol (4‐OHC), an endogenous biomarker of CYP3A4 activity, data being obtained from patients. The model predicted the AUC and maximum plasma concentration (Cmax) ratios of midazolam (single dose) to be 0.18 and 0.27, respectively, after multiple doses (500 mg q.d., 15 days) suggesting ivosidenib to be a strong inducer of CYP3A. Ivosidenib was also predicted to decrease AUC of repaglinide (CYP2C8 and CYP3A4 substrate) and warfarin (CYP2C9 substrate) by 59% and 23%, respectively. Although a PBPK modeling approach was used to assess CYP3A induction potential, no clinical DDI study was conducted with a sensitive CYP3A index substrate. Mechanisms of pleiotropic effects of ivosidenib on CYPs and transporters are not fully understood at this time. In addition, multiple doses of ivosidenib were not studied in healthy participants. Taken together, ivosidenib has insufficient data to be a viable index inducer.
Lumacaftor
The fixed dose combination of lumacaftor and ivacaftor (LUM 400 mg/IVA 250 mg, b.i.d.) was approved for the treatment of cystic fibrosis in patients who have two copies of the F508del mutation in the CFTR gene. 52 Lumacaftor was not extensively metabolized and eliminated primarily as an unchanged parent via biliary/fecal excretion. Renal elimination of lumacaftor is minimal. In vitro studies suggested lumacaftor to be a strong inducer of CYP3A and it is also showed potential to induce CYP2B6, CYP2C8, CYP2C9, and CYP2C19 at clinically relevant concentrations. 51 Lumacaftor is an inhibitor of CYP2C8 and CYP2C9, however, net in vivo effect due to inhibition and induction of CYP2C8 and CYP2C9 is not known. A roughly proportional increase of steady‐state (multiple doses) exposure was observed after oral administration of lumacaftor from 50 to 1000 mg daily in healthy subjects with low apparent clearance (CL/F) and volume of distribution (V z/F), and long terminal half‐life (23–26 h). In healthy subjects, steady‐state concentrations were reached after 5–14 days of daily oral dose with an accumulation ratio of 1.9 to 2.2‐fold. In clinical DDI studies, lumacaftor exposure was not affected by co‐administration of CYP3A4 inhibitors (itraconazole and ciprofloxacin) or a strong inducer (rifampicin). The magnitude of CYP3A induction by lumacaftor was assessed in healthy participants with a sensitive CYP3A4 substrate and combination partner, ivacaftor. A DDI study conducted with lumacaftor (200 mg daily, 14 days) and ivacaftor (150 mg b.i.d., 14 days) showed significant decrease in ivacaftor and its metabolite exposure (AUC) by 81% and 72%, respectively. Lumacaftor showed consistent induction effect upon co‐administration with ivacaftor by reducing ivacaftor AUC by 75–78% with dosing regimens of either 400 mg q.d. (LUM)/150 mg b.i.d. (IVA) or 200 mg q.d. (LUM)/250 mg b.i.d. (IVA) for 14 days. A study conducted with co‐administration of itraconazole (200 mg daily) and lumacaftor (200 mg, twice a daily) plus ivacaftor (250 mg twice a daily) for 7 days, showed >80% decrease of AUC and Cmax of itraconazole further substantiating strong induction effect of lumacaftor/ivacaftor combination. 53 Lumacaftor concentrations were unaffected by multiple doses of ivacaftor or itraconazole. Lumacaftor showed dose dependent CYP3A induction which was demonstrated by a decrease in ivacaftor exposure with increased dose and frequency of lumacaftor (Figure 2).
FIGURE 2.
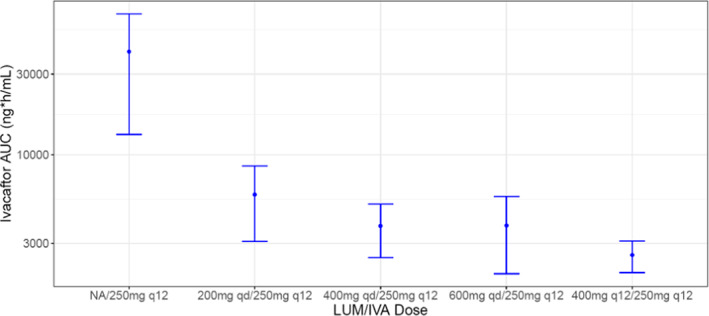
Decrease in AUC of ivacaftor due to dose dependent CYP3A induction by lumacaftor. AUC, area under the curve; IVA, ivacaftor; LUM, lumacaftor; NA, N‐Nitrosamine.
Lumacaftor is marketed in combination with ivacaftor (ORKAMBI) and is not available as a single agent to conduct clinical drug interaction studies. In terms of perpetrator drug interactions, ivacaftor is a weak inhibitor of CYP3A (increased midazolam AUC and Cmax by 1.54 and 1.38‐fold, respectively) and P‐gp (increased digoxin AUC and Cmax by 1.32‐ and 1.23‐fold, respectively). In addition, ivacaftor is an inhibitor of 2C9 in vitro. In summary, both lumacaftor and ivacaftor are inhibitors of P‐gp and 2C9 in vitro and the magnitude of drug interaction due to CYP3A inhibition by ivacaftor is minimal. The net DDI effect of combination (lumacaftor + ivacaftor) on CYP3A substrates was strong induction and hence fixed‐dose combination of lumacaftor and ivacaftor can be used in clinical DDI studies to determine the magnitude of drug interaction with CYP3A substrates. It is expected that ivacaftor would not introduce any bias in the final DDI outcome of combination compared to lumacaftor as a single agent. Based on the available information, lumacaftor is an activator of PXR in vitro and may cause induction of PXR‐dependent metabolic enzymes and transporters. 54 , 55 Although lumacaftor decreased exposure of ivacaftor and itraconazole in clinical DDI studies, the induction effect was not established on CYP3A clinical index substrate(s), such as midazolam. In addition, the relative contribution of gut and liver CYP3A4 induction to overall induction effect was also not established for lumacaftor. Taken together, lumacaftor can be a potential alternative to rifampicin; however, additional clinical DDI studies with standard CYP3A clinical index substrate(s) would be beneficial.
Mitotane
Mitotane (LYSODREN) is an adrenal cytotoxic agent approved for the treatment of inoperable, functional, or nonfunctional adrenal cortical carcinoma. The recommended initial dose is 2–3 g/day at 2‐week intervals with a recommendation to increase dose to achieve a plasma concentration of 14–20 mg/L or as tolerated. Serum drug concentrations >20 mg/L correlate with considerable adverse effects (AEs), especially neurologic toxicity. The half‐life of mitotane is very long, with a median of 53 days. The carcinogenicity and mutagenicity of mitotane are unknown.
A clinical study using single‐dose midazolam (7.5 mg) as the probe showed mitotane has a strong induction on CYP3A. 56 Both midazolam (AUC0–12h) and sunitinib (dose normalized AUC0–24h to 50 mg sunitinib) exposure were decreased, by 95% and 80%, respectively. Mitotane not only has a strong induction of CYP3A, but induction effect is long‐lasting due to its long elimination half‐life (18–159 days). Mitotane was not selected as an alternative to rifampicin due to its long half‐life and cytotoxicity.
Phenytoin
Phenytoin is indicated for the treatment of secondary generalized tonic–clonic and complex partial seizures and prevention and treatment of seizures occurring during or following neurosurgery. The recommended dose of phenytoin is 300 mg/day. Phenytoin is extensively bound to plasma proteins with an average half‐life of 14–22 h. Phenytoin is metabolized primarily by CYP2C9 with additional contribution from CYP2C19. Owing to saturation of metabolism at high serum concentrations, small increases in doses may substantially increase serum exposures with increase in half‐life. Most of the drug is excreted in the bile as inactive metabolites and then re‐absorbed from the intestinal tract and excreted in the urine. 57 The major metabolic pathways of phenytoin are hydroxylation and dihydrodiol formation accounting for 70–90% of the administered dose. Phenytoin induces several enzymes, including CYP1A2, CYP2C9, CYP2C19, CYP2B6, CYP3A4, and UGTs, and transporter, P‐gp. Phenytoin is a well‐characterized CAR activator and known to elicit induction effect on metabolic enzymes and P‐gp primarily through activation CAR with minor contribution from PXR. 58
Phenytoin has been evaluated in several studies as a CYP3A or P‐gp inducer and there are case reports on the interaction of phenytoin with sensitive CYP3A substrates. When multiple doses of phenytoin (300 mg q.d.) was used with lopinavir/ritonavir (400/100 mg b.i.d.) in healthy volunteers, it showed moderate induction with geometric mean ratios of 0.67 for AUC and 0.54 for Cmax. 59 Phenytoin (150 mg b.i.d.) showed moderate induction effect on ivabradine PKs (single dose of 10 mg) in healthy subjects with mean ratio of 0.31 for AUC and 0.35 for Cmax. 60 In another DDI study with phenytoin (4 mg/kg q.d.) and atorvastatin (40 mg/day), phenytoin showed moderate decrease in AUC for atorvastatin by 46% and its metabolites, 2‐OH‐atorvastain and 4‐OH‐atrovastain by 47% and 56%, respectively. 61 When comparing nisoldipine or midazolam PKs in patients with chronic administration of phenytoin (150–450 mg q.d.) versus controlled healthy subjects, the patients showed dramatically lower exposure with AUC ratio of 0.11 for nisoldipine and 0.06 for midazolam. 62 , 63 The interaction of phenytoin (100 mg t.i.d.) with quetiapine (25–250 mg t.i.d.) was reported in 17 patients with various disorders, AUC and Cmax of quetiapine were decreased by 81% and 72%, respectively. 64 Phenytoin also reduced itraconazole AUC and Cmax by >90%. Phenytoin (200 mg b.i.d.) was also reported to mildly induce P‐gp in a DDI study with digoxin in healthy volunteers, decreasing digoxin’s AUC by 22%. 65 Overall, phenytoin exhibits strong induction of CYP3A and has sufficient clinical DDI data to be considered as an alternative index inducer for clinical DDI studies.
Rifapentine
Rifapentine is a rifamycin antimycobacterial drug approved for tuberculosis (TB) treatment. For adults, 600 mg orally twice weekly is recommended during the intensive phase of TB treatment, followed by 600 mg once weekly during the continuation phase. Rifapentine is known to be an inducer of CYP3A4 and CYP2C8/9. Induction of enzyme activities occurred within 4 days after the first dose and returned to baseline levels 14 days after discontinuation. No significant auto‐induction effect was observed. A DDI study with midazolam and rifapentine (15 mg/kg q.d.) in healthy volunteers showed decrease in AUC and Cmax of midazolam by 93% and 84%, which indicates rifapentine is a strong inducer of CYP3A. 66 Similar to other rifamycin analogs, rifapentine also induces CYP3A through activation via PXR. 67 Presence of NA impurities is also reported in rifapentine and hence rifapentine cannot be an appropriate alternative to rifampicin.
St. John’s Wort
St. John’s Wort (SJW; Hypericum perforatum) is an herbal product and a dietary supplement that is used to treat mild to moderate depression. Although many diverse phytochemicals have been identified in SJW extracts, hypericin, pseudohypericin, and hyperforin are generally thought to be the pharmacologically active components. 68 Besides its antidepressant activity, profound drug interactions, mainly induction, have been reported with SJW and substrates of CYP3A4/5 and/or P‐gp. 68 Hyperforin, an acylphloroglucinol derivative, is thought to be a key component of the induction. Many in vitro investigations have revealed CYP3A induction by hyperforin is mediated by activation of PXR. 69 Clinical DDI studies conducted with SJW and oral administration of sensitive CYP3A or P‐gp substrates showed moderate induction with AUC ratio ranging from ~0.4 to 0.72 after 300 mg SJW, three times a day, ~14 to 18 days. 68 In a cocktail drug interaction study with SJW having a low hyperforin (≤0.2%), meaningful drug interactions were not observed with CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP3A4, and P‐gp substrates. 70 A study conducted with SJW preparations containing various amounts of hyperforin and midazolam showed a dose‐dependent induction effect on midazolam AUC with strong a induction effect being achieved at the highest hyperforin dose (41.25 mg/day). 71 The induction effect of SJW is more pronounced on intestinal CYP3A compared to hepatic CYP3A, as demonstrated by a significant difference in AUC of midazolam after oral and intravenous dose administration by PBPK modeling. 72 In herbal products, the concentration of active ingredients is expected to depend on geographical regions, growth location, time of harvest, and processing methodologies. Considering the variability in CYP3A induction and challenges in controlling consistent levels of hyperforin, the utility of SJW as an index inducer is limited.
ALTERNATIVE STRONG INDUCERS OF CYP3A
We initially identified 10 compounds that could serve as an alternative to rifampicin in clinical DDI studies as the index CYP3A inducer (Table 1). The data from clinical DDI studies with strong CYP3A inducers and sensitive CYP3A substrates were collected (Table 2). As clinical DDI studies in healthy subjects have clear advantages including easy recruitment, high compliance, and low PK variability, a few of these alternatives were eliminated based on their genotoxic and/or cytotoxic potential and safety concerns (mitotane, enzalutamide, apalutamide, and ivosidenib), due to unsuitability or questionable suitability for multiple dose administration to healthy subjects (mitotane, apalutamide, enzalutamide, and ivosidenib; Figure 3). However, for investigational drugs that cannot be dosed to healthy volunteers and are under evaluation in disease areas (e.g., oncology) where one of these drugs may offer potential therapeutic benefit, the selection of one of these agents may be justified based on broader benefit/risk considerations for the relevant patient population. A lack of adequate and robust clinical CYP3A induction data even with midazolam due to variations in hyperforin levels, St. John’s wort was eliminated as alternative to rifampicin (Figure 3). Avasimibe was terminated from development and rifapentine possesses NA impurities (similar to rifampicin); hence, these two drugs are not considered as viable alternatives. Therefore, carbamazepine, phenytoin, and lumacaftor were chosen as promising candidates for clinical DDI studies to assess CYP3A induction (Figure 3). A comparison of AUC decrease of sensitive CYP3A substrates upon co‐administration with the alternative inducers and rifampicin is presented in Figure 4. Each of these three drugs has strengths and limitations.
FIGURE 3.
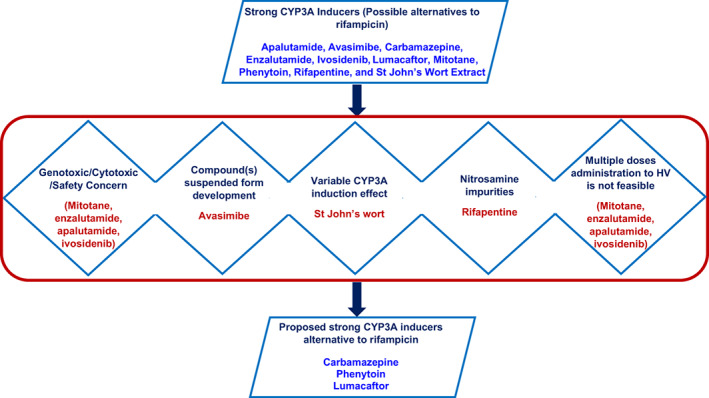
Strategy that was followed for recommendation of alternative CYP3A inducers. HV, healthy volunteers.
FIGURE 4.
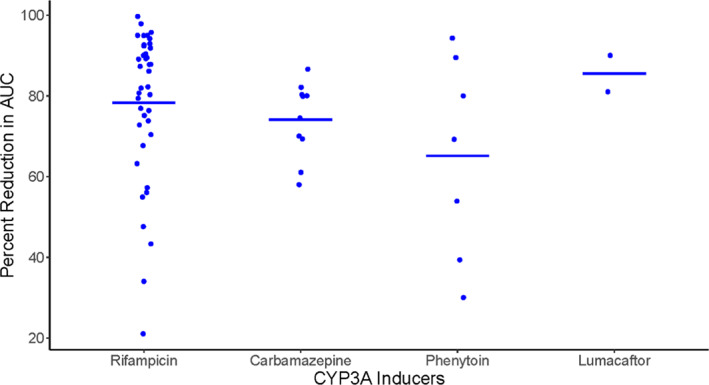
Comparison of percent decrease in AUC of sensitive CYP3A substrates (Y‐axis) upon coadministration of rifampicin and strong CYP3A inducers that are proposed as an alternative to rifampicin (X‐axis). The percent AUC reduction data for sensitive CYP3A substrates upon co‐administration of rifampicin (oral, 600 mg daily dose, 5–36 days) was obtained from the University of Washington DDI database. Doses and schedules for alternative strong CYP3A inducers are presented in Table 2. Lumacaftor data represents only two available (ivacaftor and itraconazole [not a sensitive substrate of CYP3A]) CYP3A induction studies. If more than one study was reported for the same substrate, the weighted mean across studies was calculated to represent one data point for each substrate. The horizontal line represents the mean of percent AUC reduction across all reported sensitive CYP3A substrates). AUC, area under the curve; DDI, drug‐drug interaction.
CYP3A induction can be achieved via activation of PXR and/or CAR, which binds with the promoter region of CYP3A genes. Rifampicin is known to be a strong PXR activator. Carbamazepine and phenytoin are well‐characterized CAR activators, whereas lumacaftor is an in vitro PXR activator. 55 , 58 PXR and CAR not only share sets of regulated genes, including CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, UGT1A1, P‐gp, and MRP2, but also regulate distinctive sets of genes, such as CYP4F12, UGT1A6, SULT2A1, and OATP1A2 by PXR, and CYP1A1, CYP1A2, UGT2B1, SULT2E1, and OATP1B3 by CAR. 73 For those dually regulated genes, CAR and PXR show different induction strengths and selectivity via binding to different motifs in the promoter region or binding the same motif with different affinity. PXR strongly binds with motifs in CYP2B6 and CYP3A4 promoters, whereas CAR strongly binds only with motifs in CYP2B6 promoter, and weakly binds with motifs in CYP3A4 promoter. With the same mechanism of activating PXR, lumacaftor is expected to have similar induction profile in vivo as rifampicin compared with carbamazepine/phenytoin. The differences caused by CAR or PXR activation should be considered in the design of CYP3A induction studies together with the metabolism profile of the victim drugs.
Besides the induction mechanism differences, carbamazepine was used most in clinical DDI studies after rifampicin for assessment of CYP3A induction, followed by phenytoin, with two reports of lumacaftor DDI studies with ivacaftor and itraconazole as CYP3A substrates. With the most abundant clinical data among the three alternatives (Table 2) and advances in PBPK modeling for DDI predictions, carbamazepine could be an immediate alternative to rifampicin. 74 However, carbamazepine is a narrow therapeutic index (NTI) drug and requires dose titration. With lesser clinical drug interaction studies, phenytoin ranks third in line as an index inducer to conduct CYP3A‐mediated drug interaction studies. Carbamazepine and phenytoin are extensively metabolized by CYP3A and CYP2C9, respectively, but lumacaftor is mainly eliminated as an unchanged drug. Lumacaftor shows great potential to replace rifampicin, however, more data needs to be generated in clinical DDI studies. Overall, based on our assessment, carbamazepine can be an immediate replacement to rifampicin followed by phenytoin and lumacaftor. The following section describes the strengths and limitations of alternative CYP3A inducers.
Carbamazepine
Strengths:
Sufficient clinical DDI experience with CYP3A substrates to guide dose and dosing schedule for future DDI studies.
Availability of PBPK models which can be utilized to predict DDIs of moderate and weak CYP3A inducers.
Correlation of carbamazepine CYP3A induction and endogenous biomarker, 4β‐hydroxycholesterol (4βOHC) levels have been established.
Limitations:
NTI and hence dose titration is required starting with 100 mg b.i.d. to 300 mg b.i.d. over 3–6 days.
Strong activator of CAR with weak hPXR activation and hence less potent inducer of CYP3A than rifampicin.
Carbamazepine is metabolized by CYP3A and is an auto‐inducer.
Quantitative phenotype of pleiotropic induction of non‐CYP3A enzymes/transporters is likely different from rifampicin due to a more predominant contribution of CAR versus PXR.
Phenytoin
Strengths:
Phenytoin ranks third in line for CYP3A induction studies after rifampicin and carbamazepine.
Sufficient experience is available with CYP3A substrates to guide dose and dosing schedule for future DDI studies. 75
Unlike carbamazepine, dose titration is not recommended for DDI studies with phenytoin. 75
Limitations:
NTI.
Strong activator of CAR with weak hPXR activation similar to carbamazepine and hence pleiotropic induction effect on non‐CYP3A enzymes/transporters could be different from rifampicin.
Lumacaftor
Strengths:
Safety was established in multiple clinical studies in healthy volunteers.
Activator of PXR in vitro and has potential to behave similar to rifampicin with respect to induction potential of CYPs and transporters.
Not extensively metabolized and primarily eliminated as an unchanged drug by biliary excretion and not a substrate of uptake transporters such as OATP1Bs, OATP2B1, or P‐gp. Hence limited scope of alteration of PK by victim drug.
Limitations:
Clinical data available with only one sensitive CYP3A substrate, ivacaftor and with itraconazole (CYP3A substrate). Hence, more data with probe CYP3A substrates such as midazolam are required to build confidence.
Additional data (in vitro and in vivo) are required for characterization of pleotropic induction effects of lumacaftor.
COMPARISON OF RIFAMPICIN AND ALTERNATIVE CYP3A INDUCERS ON THEIR UTILITY IN NON‐CYP3A MEDIATED CLINICAL DDI STUDIES
Rifampicin, carbamazepine, and phenytoin are all listed as strong inducers for CYP3A by the FDA. 76 In addition, rifampicin is also a strong inducer of CYP2C19, and a moderate inducer of CYP1A2, CYP2B6, CYP2C8, and CYP2C9. Carbamazepine is a strong inducer of CYP2B6 and a weak inducer of CYP2C9. Phenytoin is a moderate inducer of CYP1A2 and CYP2C19.
As for their induction mechanisms, rifampicin is a strong agonist of PXR, compared to carbamazepine and phenytoin being well‐characterized CAR activators. 73 Even though CYP2C8 was shown to be regulated by both PXR and CAR agonists, no reports could be identified regarding the induction of CYP2C8 by either carbamazepine or phenytoin, whereas rifampicin has been shown to induce CYP2C8. 77 As an agonist of PXR, lumacaftor induced CYP2B6, CYP2C8, CYP2C9, and CYP2C19 in vitro. However, comparative potencies and in vivo relevance of in vitro induction remains to be elucidated. Similar to CYP3A, induction of P‐gp occurs via perpetrators activating nuclear receptors PXR and/or CARs, followed by the nuclear receptors binding with the promoter region and thus upregulating the gene expression of P‐gp. PXR and CAR expression is tissue specific. Both PXR and CAR are expressed in the liver and CAR has little extrahepatic expression. PXR is also expressed in the intestine, but with limited expression in the kidneys. 73 As a result, PXR/CAR‐mediated P‐gp induction also shows tissue‐specific pattern by rifampicin, phenytoin, or carbamazepine. 41
Rifampicin is the strongest P‐gp inducer based on currently available clinical experience, and the induction is more evident in the enterocytes with greater impact. Rifampicin also increases total clearance of intravenously administered digoxin and talinolol, with no effects on renal clearance. Phenytoin caused a 22% decrease of AUC for digoxin, but no changes in renal clearance, which also indicates P‐gp induction is mainly in the liver and intestine, but not in the kidneys. Carbamazepine decreased fexofenadine AUC and Cmax by around 35%, but resulted in minimal changes in half‐life. This induction effects seems to be mainly driven via P‐gp and/or MRP2 induction in the gut. 44
Rifampicin (following single dose administration) has been used as an index inhibitor to assess the impact of inhibition on OATP1B substrates. Although there were inhibitors of OATP1B in vitro, no interaction with OATP1B1 substrates is expected at a clinically relevant concentration for carbamazepine (basic model calculations) and lumacaftor. 53 Rifampicin can also induce UGTs, of which UGT1A1 and UGT1A4 were most strongly induced. 11 , 78 With UGT induction being regulated by PXR and CAR, carbamazepine and phenytoin are also inducers of UGTs. In vitro studies showed carbamazepine could induce UGT1As and UGT2B7, and phenytoin could induce UGT1A1. 79 , 80 Clinical studies showed both carbamazepine and phenytoin increased clearance of lamotrigine (metabolized primarily UGT1A4 and UGT2B7) and acetaminophen via UGT induction, and the effects on lamotrigine were greater with phenytoin than with carbamazepine. 33 , 79 UGT induction of lumacaftor was not reported.
Although rifampicin is a pleiotropic inducer of many drug‐metabolizing enzymes and transporters, it remains uncertain whether multiple dose administration of rifampicin is associated with clinically meaningful induction of OATP transporters. The observed induction of the total apparent clearance of statins following rifampicin administration is likely multifactorial. Given the involvement of multiple molecular determinants of statin clearance (e.g., CYP3A3, P‐gp, MRP2, and BCRP) in addition to OATP transporters, the outcomes of interactions between rifampicin and the statins cannot be completely ascribed to OATP induction. 81 The comparative potential for inhibition and induction of OATP transporters by rifampicin versus the alternatives (e.g., phenytoin, carbamazepine, and lumacaftor) represents an area for future translational research.
PROPOSED CLINICAL STUDY DESIGN WITH ALTERNATIVE STRONG CYP3A INDUCERS
Both rifampicin and phenytoin have been recommended by the FDA and PMDA as strong index inducers of CYP3A for conducting clinical DDI studies. 9 , 76 Although rifampicin is being used extensively, the phenytoin usage in clinical DDI studies is limited.
The clinical DDI study design with rifampicin is well‐established, however, not much information is available on appropriate study design and recommendations with other strong CYP3A inducers. Hence, this section focuses on providing illustrative clinical study designs for the proposed alternate inducers based on available information. Various doses and dosing schedules were reported for both carbamazepine and phenytoin DDI studies (Table 2). In addition, dose titration is recommended to increase tolerability and decrease dropout rate in clinical DDI studies with carbamazepine. For carbamazepine DDI studies, 200–600 mg/day dose was used with sensitive CYP3A substrates along with 3–7 days of dose titration and 5–24 days of dose administration (Table 2). Xu et al. 40 utilized in vitro CYP3A induction data from hepatocytes and simulated various doses and duration of carbamazepine with population‐based dynamic modeling. The simulated results were successfully verified with the observed simvastatin and carbamazepine DDI results. Based on these results, authors concluded both 300 mg b.i.d. and 600 mg q.d. doses would achieve the near maximal CYP3A induction over 10 days treatment with carbamazepine.
Carbamazepine treatment is associated with dose‐ or concentration‐dependent AEs, such as somnolence, dizziness, gastrointestinal disturbance, and hematologic abnormalities. Other adverse events, including liver injury, and hypersensitivity reactions, such as maculopapular eruption (MPE), drug reaction with eosinophilia and systemic symptoms (DRESS), and Stevens–Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN), have a complex dose–response relationship. 82 The plasma concentration range of 4–12 μg/ml is the therapeutic range for carbamazepine and higher probability of AEs at the upper end of the therapeutic ranges were reported. 83 Hence, a cautious approach needs to be followed to escalate to the required dose for clinical DDI studies.
Patients who are positive for HLA‐B*1502, HLA‐A*3101, and HLA‐B*1511 are associated with SJS/TEN and HLA‐A*3101 allele also associated as DRESS and MPE upon treatment with carbamazepine. 84 The FDA label has warning signs for carbamazepine usage in patients who are positive for HLA‐B*1502 allele. Carbamazepine dosing guidelines based on HLA genotype have been published by the Dutch Pharmacogenetics Working Group (DPWG) of the Royal Dutch Association for the Advancement of Pharmacy (KNMP), the Clinical Pharmacogenetics Implementation Consortium (CPIC), and the Canadian Pharmacogenomics Network for Drug Safety (CPNDS). 84 The prevalence of positive HLA‐B*15:02 allele is 10–15% in Chinese populations and averages 2–4% in south Asian population. Hence, it is suggested to screen and exclude subjects with HLA‐B*1502 allele to minimize AEs associated with carbamazepine in drug interaction studies. 84
Based on the available clinical DDI studies with sensitive CYP3A substrates and population‐based modeling results, we propose a target carbamazepine dosage of 300 mg b.i.d. attained with a dose titration starting at 100 mg b.i.d. for 3 days followed by 200 mg b.i.d. for 3 days and continuous treatment with 300 mg b.i.d. for 14 days for conducting clinical DDI studies with carbamazepine as a strong CYP3A inducer. 33 , 39 The proposed study details are presented in Figure 5a. Briefly, an open label, fixed sequence, crossover study with two periods is proposed. Period 1 would evaluate PK of the investigational drug followed by a washout period and period 2 would assess DDI of carbamazepine with victim drug. The total time for period 1 and washout depends on the half‐life of the investigational compound. For period 2, carbamazepine should be started at 100 mg b.i.d. and can be titrated to 300 mg b.i.d. over a 7‐day period. To achieve maximum induction effect, 300 mg b.i.d. dose is suggested for 14 days prior to administration of the investigational drug, with carbamazepine dosing continued through the period of PK characterization of the victim drug.
FIGURE 5.
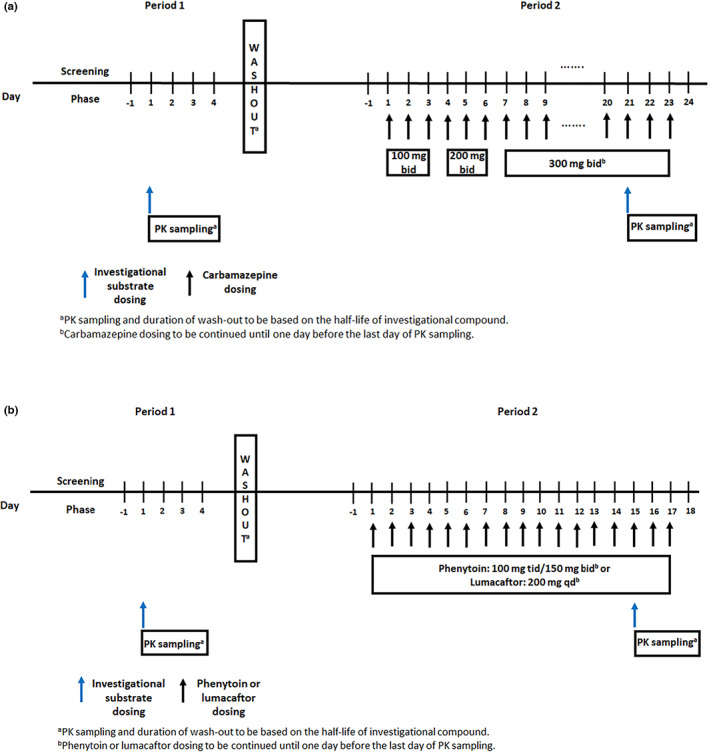
Proposed design for clinical DDI studies with alternative CYP3A inducers (a) carbamazepine; (b) phenytoin and lumacaftor with CYP3A substrates. PK, pharmacokinetic.
In the DDI studies of phenytoin with sensitive CYP3A substrates, various dosing regimens have been used, from q.d. to t.i.d., and the daily dose ranged from 200 to 450 mg. In several reports, phenytoin was targeted to reach a stable concentration, ranged from 5.5–25.9 μg/ml. 62 , 85 Of all the reports, the strongest CYP3A induction was reported in a DDI study with midazolam and nisoldipine, with ~90% lower AUC and ~80% to 90%, lower Cmax compared to the control group. However, both these studies were not controlled drug interaction studies. These studies compared midazolam/nisoldipine exposure in patients with epilepsy co‐administered phenytoin with the midazolam/nisoldipine exposure (without phenytoin) in healthy volunteers. In addition, patients with epilepsy were dosed with phenytoin for at least 2 months. 60 , 61 All of these factors confounded the interpretation of the induction strength of phenytoin on CYP3A. Similar to carbamazepine, phenytoin also has a narrow therapeutic window, between 10 and 20 μg/ml. Phenytoin treatment is associated with eosinophilia, DRESS syndrome, and SJS, and it is a more severe manifestation of TEN. Asian patients carrying HLAB*1502 and CYP2C9*3 genes are at high risk for phenytoin treatment. 86 The main AEs reported in the phenytoin treatment are due to central nervous system and cardiovascular toxicity. The symptoms of overdosing are nystagmus, ataxia, hyperactivity, and dysarthria, which should be closely monitored during the conduct of clinical DDI studies. 86
Although 300 mg q.d., 150 mg b.i.d., and 100 mg t.i.d. are known to produce similar CYP3A induction effects, considering the safety profile and therapeutic range of phenytoin, we propose 150 mg b.i.d. or 100 mg t.i.d. dose for 14 days in clinical DDI studies using phenytoin in healthy subjects (Figure 5b). Recently, PBPK models were developed based on 100 mg t.i.d. dose and a duration of 14 days was chosen to achieve maximum induction effect of phenytoin. 87 , 88 As described in the carbamazepine drug interaction study design, a similar open label, fixed sequence, crossover study with two periods is recommended for phenytoin DDI studies. Period 1 would evaluate PKs of the investigational drug followed by a washout period and period 2 would assess DDI effect of phenytoin on PK of the victim drug. The total time for period 1 and washout depends on the half‐life of an investigational compound.
Lumacaftor is a strong inducer of CYP3A and showed a dose‐dependent induction effect on ivacaftor, a sensitive CYP3A substrate. Ivacaftor AUC was decreased by 80–90% with co‐administration of 200–600 mg daily dose and >90% with 400 mg b.i.d. dosing. Two phase I studies were conducted to assess DDI interaction between lumacaftor and ivacaftor. 53 The first one was a randomized, double‐blind, placebo‐controlled study with a three‐treatment period (ivacaftor alone, lumacaftor alone, and their combination) in healthy subjects with a minimum washout period of 14 days between periods. This was a multiple dose DDI study for both agents for 14 days with 200 mg daily dose (q24 hours) of lumacaftor and 150 mg of ivacaftor twice daily (q12 hours). There was no effect of ivacaftor on lumacaftor PKs, however, AUC and Cmax of ivacaftor was decreased by 81% and 74%, respectively, upon co‐administration of both agents. Another phase I study was conducted to assess DDI between lumacaftor and ivacaftor with similar three‐period study design described earlier and two different doses of both lumacaftor (200 mg q.d. and 400 mg q.d.) and ivacaftor (250 mg b.i.d. and 150 mg b.i.d.). The reduction of AUC of ivacaftor ranged from 75–78% with lumacaftor and ivacaftor regimens, 400 mg lumacaftor q.d./150 mg ivacaftor q12h or 200 mg lumacaftor q.d./250 mg ivacaftor q12h for 14 days. A consistent reduction of AUC of ivacaftor was observed with treatment of 200 mg daily dose of lumacaftor for 14 days and hence this dose and dosing regimen can be used to assess DDI of other CYP3A substrates (Figure 5b). The approved dose of lumacaftor in combination with ivacaftor in adults is two tablets (each containing 200 mg lumacaftor /125 mg of ivacaftor) every 12 h. Hence, one tablet can be used for the proposed dose of 200 mg lumacaftor as a combination for DDI studies.
Lumacaftor as monotherapy or in combination with ivacaftor was generally well‐tolerated in healthy participants and patients with cystic fibrosis. The majority of AEs were mild or moderate in severity and resolved without discontinuing the treatment. 55 The most common drug‐related or possibly drug‐related AEs were diarrhea, upper abdominal pain, increased liver transaminases, vomiting, headache, and dyspnea. The proposed 200 mg dose is much lower than the approved daily dose of lumacaftor (800 mg) and is expected to be tolerated well in DDI studies. 55
FUTURE PERSPECTIVES
For investigational agents that are substrates of CYP3A and/or PXR‐inducible enzymes and transporters, rifampicin has traditionally been the most widely used index strong inducer in DDI studies to evaluate sensitivity as victim of induction DDIs. The dose–response for rifampicin induction of CYP3A, relative extent of induction of CYP3A versus other PXR‐inducible enzymes and transporters, and induction at both the hepatic and intestinal sites of the DDIs have all been extensively studied. 11 , 40 Progress in the field of enzyme and transporter induction, including the development of novel translational approaches using endogenous biomarkers and PBPK models, has largely relied on data from in vitro and clinical induction studies with rifampicin. With nitrosamine impurities prohibiting the use of currently available rifampicin formulations for induction DDI studies, the pharmaceutical industry at‐large is facing a challenge of having lost an important component of the clinical pharmacology toolkit to evaluate sensitivity of investigational agents to induction DDIs.
Rifampicin as a prototypic index strong inducer of PXR target genes is not without limitations. For example, selectivity is an acknowledged problem, with the most notable concerns stemming from its ability to potently inhibit OATP1B1‐mediated hepatic uptake transport. This can confound interpretation of induction DDI study results for dual substrates of inducible enzymes/transporters and OATP1B1, unless dose staggering is incorporated as part of the design to minimize effects on hepatic uptake. Despite these limitations, best practices for rifampicin DDI studies are well‐established based on decades of research that have characterized its potency and selectivity as a strong PXR‐mediated inducer.
Currently, the experience of the acceptance of the PBPK model predicted results in lieu of rifampicin DDI study by regulatory agencies is limited. Underprediction of DDIs with the rifampicin model along with lack of confidence in the predictability and reliable validation of the models were cited as reasons for not acceptability of modeling results. Nevertheless, it is important to note that the field of PBPK modeling of induction DDIs continues to advance, with notable recent progress made in the development and qualification of a model of rifampicin‐mediated induction, also incorporating P‐gp induction. 89 , 90 As the fidelity of PBPK models continues to grow, we trust that confidence in their application in lieu of clinical DDI studies should increase for suitable contexts of use.
Our research has identified carbamazepine, phenytoin, and lumacaftor as three potential alternatives to rifampicin. Clearly, the extent of characterization of these strong CYP3A inducers is more limited, and we do not fully understand their selectivity and “off‐target” effects beyond induction of CYP3A. Of note, the importance of CAR (vs. PXR alone) activation as a mechanism of induction by carbamazepine and phenytoin may have implications for the phenotype of pleiotropism in induction that would be observed compared to rifampicin. Looking ahead, we have an opportunity to characterize these alternate index inducers further leveraging all available tools. Comprehensive in vitro head‐to‐head concentration‐response studies, not only in human hepatocytes but also considering other emerging platforms, like liver spheroids, may help elucidate the quantitative and molecular pharmacology of induction. 91 Full profiling of these rifampicin alternatives with respect to their “off‐target” effects on a full panel of molecular determinants of absorption, distribution, metabolism, and excretion (ADME; e.g., drug‐metabolizing enzymes and transporters) will inform selectivity. Such comparative in vitro data with insights on mechanisms of induction and selectivity can be instrumental in selecting the right inducer for the right investigational agent based on knowledge of the molecular determinants of clearance of the investigational agent. Quantitative concentration‐response data can be further crucial to development and refinement of PBPK models for these inducers, which will be vital for model‐informed design of induction DDI studies as well as predictions of the effects of moderate inducers (e.g., efavirenz) based on data from strong inducer DDI studies conducted using one of these rifampicin alternatives. The roadmap for such PBPK model‐informed projections using a predict‐learn‐confirm‐apply paradigm is well‐established and precedented when the strong inducer DDI study was performed using rifampicin. 92 We will need to achieve a similar level of confidence in the future state when strong inducer DDI studies will be conducted with one of these alternatives instead of rifampicin. In addition to comprehensive in vitro profiling of pleiotropism in induction phenotype and concentration‐response for these inducers (vs. rifampicin), other recent advances in clinical pharmacology methods deserve to be exploited as well. For example, a well‐designed clinical pharmacology study evaluating the effects of these rifampicin alternatives can evaluate endogenous biomarkers of ADME protein activity (e.g., 4β‐hydroxycholesterol for CYP3A induction, CP‐I for OATP1B1 inhibition, etc.) to characterize the relative selectivity of these inducers. The endogenous biomarkers data could further inform PBPK modeling to evaluate or validate simulated results. Such an investigational clinical pharmacology study could additionally collect blood samples for liquid biopsy to enable exosome analyses using proteomics as well as activity measurements to characterize the pleiotropic induction effects and dose–response in vivo. 93 A cocktail DDI study may be embedded to quantify the strength of induction of non‐CYP3A pathways relative to CYP3A. The totality of results from such a suite of in vitro and clinical pharmacology evaluations together with literature data on existing clinical DDI studies with these rifampicin alternatives as inducers can provide excellent substrate for PBPK model development and refinement. This will support the fidelity of such modeling frameworks in serving relevant contexts of use (e.g., predictions to unstudied inducers that may vary in strength and/or pleiotropism in the induced phenotype). While we adapt to the current state of being unable to use rifampicin as an index inducer, we envision an important role for a multipronged quantitative translational research program (Figure 6) to increase confidence in use of data from induction DDI studies conducted with a suitable rifampicin alternative to inform labeling for our future medicines across contexts of clinical use. We have successfully done this when we faced a similar situation with inability to use ketoconazole as an index strong CYP3A inhibitor, with IQ consortium efforts leading to recommendations on optimal study designs with itraconazole as an alternative, informed by PBPK modeling and simulation. 94 We are in an analogous situation now with rifampicin. Additionally, given the limitations of some of the identified alternatives to rifampicin, we may need to use the results of DDI studies with these less established inducers to extrapolate and predict worst‐case scenarios for DDIs with rifampicin. This will require us to leverage PBPK platforms that will need to be qualified for cross‐inducer DDI predictions, considering not only differences between the inducers in potency/strength but also the specific pleiotropic phenotype of induction of molecular determinants of clearance beyond CYP3A4 that may be involved in disposition of the victim drug. Timely progress will require commitment to precompetitive collaboration, data sharing, and effective multidisciplinary partnerships with commitment to a totality of evidence mindset. 95
FIGURE 6.
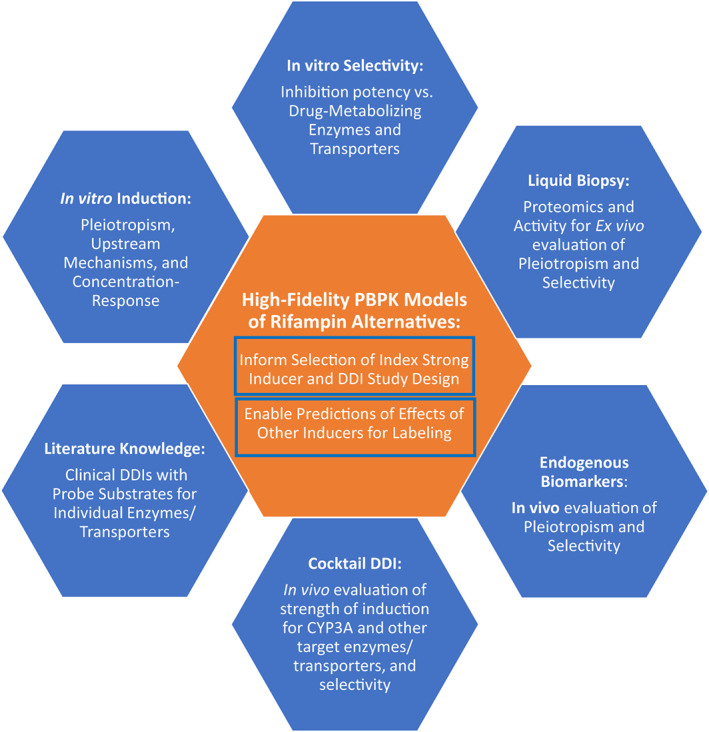
Multifaceted approach to enhance confidence on data generated from DDI studies conducted with alternative inducers of CYP3A. DDI, drug‐drug interaction; PBPK, physiologically‐based pharmacokinetic.
AUTHOR CONTRIBUTIONS
J.B., S.G., P.H., J.D., and K.V. reviewed the literature, and designed, wrote, and reviewed the manuscript.
CONFLICT OF INTEREST
J.B., P.H., J.D., and K.V. are employees of EMD Serono. S.G. is an employee of Merck KGAa and holds stock of the respective companies.
Bolleddula J, Gopalakrishnan S, Hu P, Dong J, Venkatakrishnan K. Alternatives to rifampicin: A review and perspectives on the choice of strong CYP3A inducers for clinical drug–drug interaction studies. Clin Transl Sci. 2022;15:2075‐2095. doi: 10.1111/cts.13357
Funding information
No funding was received for this work
REFERENCES
- 1. Reyner E, Lum B, Jing J, Kagedal M, Ware JA, Dickmann LJ. Intrinsic and extrinsic pharmacokinetic variability of small molecule targeted cancer therapy. Clin Transl Sci. 2020;13:410‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tornio A, Filppula AM, Niemi M, Backman JT. Clinical studies on drug‐drug interactions involving metabolism and transport: methodology, pitfalls, and interpretation. Clin Pharmacol Ther. 2019;105:1345‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hakkola J, Hukkanen J, Turpeinen M, Pelkonen O. Inhibition and induction of CYP enzymes in humans: an update. Arch Toxicol. 2020;94:3671‐3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mackowiak B, Wang H. Mechanisms of xenobiotic receptor activation: direct vs. indirect. Biochim Biophys Acta. 1859;2016:1130‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tolson AH, Wang H. Regulation of drug‐metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62:1238‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta. 2011;1812:956‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guidance for Industry (fda.gov) (Clinical Drug Interaction Studies – Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry). Accessed March 14, 2022. https://www.fda.gov/media/134581/download
- 8. Guideline on the investigation of drug interactions (europa.eu). Accessed March 14, 2022. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐investigation‐drug‐interactions‐revision‐1_en.pdf
- 9. Guideline on drug interaction for drug development and appropriate provision of information (000228122.pdf) pmda.go.jp. Accessed March 14, 2022. https://www.pmda.go.jp/files/000228122.pdf
- 10. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42:819‐850. [DOI] [PubMed] [Google Scholar]
- 11. Chen J, Raymond K. Roles of rifampicin in drug‐drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob. 2006;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Committee for medicinal products for human use (CHMP) minutes for the meeting on 19‐22 April 2021 (europa.eu). Accessed March 14, 2022. https://www.ema.europa.eu/en/documents/minutes/minutes‐chmp‐meeting‐19‐22‐april‐2021_en.pdf
- 13. Beard JC, Swager TM. An organic chemist's guide to N‐nitrosamines: their structure, reactivity, and role as contaminants. J Org Chem. 2021;86:2037‐2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. M7 (R1) Step 5 Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk (europa.eu). Accessed March 14, 2022. https://www.ema.europa.eu/en/documents/scientific‐guideline/ich‐guideline‐m7r1‐assessment‐control‐dna‐reactive‐mutagenic‐impurities‐pharmaceuticals‐limit_en.pdf
- 15. IARC . Accessed March 14, 2022. https://monographs.iarc.who.int/wp‐content/uploads/2018/06/mono94.pdf
- 16. Du H, Leng J, Wang P, Li L, Wang Y. Impact of tobacco‐specific nitrosamine‐derived DNA adducts on the efficiency and fidelity of DNA replication in human cells. J Biol Chem. 2018;293:11100‐11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nitrosamines EMEA‐H‐A5(3)‐1490 ‐ Assessment Report (europa.eu). Accessed March 14, 2022. https://www.ema.europa.eu/en/documents/referral/nitrosamines‐emea‐h‐a53‐1490‐assessment‐report_en.pdf
- 18. Information about Nitrosamine Impurities in Medications | FDA. https://www.fda.gov/drugs/drug‐safety‐and‐availability/information‐about‐nitrosamine‐impurities‐medications
- 19. Control of Nitrosamine Impurities in Human Drugs | FDA. Accessed March 14, 2022. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/control‐nitrosamine‐impurities‐human‐drugs Accessed March 14, 2022.
- 20. FDA Updates and Press Announcements on Nitrosamines in Rifampin and Rifapentine | FDA. Accessed March 14, 2022. https://www.fda.gov/drugs/drug‐safety‐and‐availability/fda‐updates‐and‐press‐announcements‐nitrosamines‐rifampin‐and‐rifapentine
- 21. Alfarisi O, Alghamdi WA, Al‐Shaer MH, Dooley KE, Peloquin CA. Rifampin vs. rifapentine: what is the preferred rifamycin for tuberculosis? Expert Rev Clin Pharmacol. 2017;10:1027‐1036. [DOI] [PubMed] [Google Scholar]
- 22. Ramsden D, Fung C, Hariparsad N, et al. Perspectives from the innovation and quality consortium induction working group on factors impacting clinical drug‐drug interactions resulting from induction: focus on cytochrome 3A substrates. Drug Metab Dispos. 2019;47:1206‐1221. [DOI] [PubMed] [Google Scholar]
- 23. UW Drug Interaction Solutions. Accessed March 14, 2022. https://www.druginteractionsolutions.org/
- 24. Sahi J, Stern RH, Milad MA, et al. Effects of avasimibe on cytochrome P450 2C9 expression in vitro and in vivo. Drug Metab Dispos. 2004;32:1370‐1376. [DOI] [PubMed] [Google Scholar]
- 25. Sahi J, Milad MA, Zheng X, et al. Avasimibe induces CYP3A4 and multiple drug resistance protein 1 gene expression through activation of the pregnane X receptor. J Pharmacol Exp Ther. 2003;306:1027‐1034. [DOI] [PubMed] [Google Scholar]
- 26. Faigle JW, Feldmann KF. Pharmacokinetic data of carbamazepine and its major metabolites in Man. Clinical pharmacology of anti‐epileptic drugs. Springer, Berlin, Heidelberg, 1975, 159–165. [Google Scholar]
- 27. Pelkonen O, Myllynen P, Taavitsainen P, et al. Carbamazepine: a 'blind' assessment of CYP‐associated metabolism and interactions in human liver‐derived in vitro systems. Xenobiotica. 2001;31:321‐343. [DOI] [PubMed] [Google Scholar]
- 28. Valsalan VC, Cooper GL. Carbamazepine intoxication caused by interaction with isoniazid. Br Med J (Clin Res ed). 1982;285:261‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ucar M, Neuvonen M, Luurila H, Dahlqvist R, Neuvonen PJ, Mjörndal T. Carbamazepine markedly reduces serum concentrations of simvastatin and simvastatin acid. Eur J Clin Pharmacol. 2004;59:879‐882. [DOI] [PubMed] [Google Scholar]
- 30. Menon RM, Badri PS, Wang T, et al. Drug‐drug interaction profile of the all‐oral anti‐hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J Hepatol. 2015;63:20‐29. [DOI] [PubMed] [Google Scholar]
- 31. 207561Orig1s000ClinPharmR.pdf (fda.gov) (Elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide clinical pharmacology and biopharmaceutics review). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207561Orig1s000ClinPharmR.pdf
- 32. Hugen PW, Burger DM, Brinkman K, et al. Carbamazepine‐indinavir interaction causes antiretroviral therapy failure. Ann Pharmacother. 2000;34:465‐470. [DOI] [PubMed] [Google Scholar]
- 33. Lutz JD, Kirby BJ, Wang L, et al. Cytochrome P450 3A induction predicts P‐glycoprotein induction; part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin Pharmacol Ther. 2018;104:1191‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson GD, Gidal BE, Messenheimer JA, Gilliam FG. Time course of lamotrigine de‐induction: impact of step‐wise withdrawal of carbamazepine or phenytoin. Epilepsy Res. 2002;49:211‐217. [DOI] [PubMed] [Google Scholar]
- 35. Vlase L, Neag M, Popa A, Muntean D, Bâldea I, Leucuta SE. Pharmacokinetic interaction between ivabradine and carbamazepine in healthy volunteers. J Clin Pharm Ther. 2011;36:225‐229. [DOI] [PubMed] [Google Scholar]
- 36. 208716Orig1s000MultidisciplineR.pdf (fda.gov) (multidisciplinary review of abemaciclib). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208716Orig1s000MultidisciplineR.pdf
- 37. Zhou D, Podoll T, Xu Y, et al. Evaluation of the drug‐drug interaction potential of acalabrutinib and its active metabolite, ACP‐5862, using a physiologically‐based pharmacokinetic modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8:489‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. de Zwart L, Snoeys J, De Jong J, Sukbuntherng J, Mannaert E, Monshouwer M. Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Ther. 2016;100:548‐557. [DOI] [PubMed] [Google Scholar]
- 39. Wang K, Yao X, Zhang M, et al. Comprehensive PBPK model to predict drug interaction potential of zanubrutinib as a victim or perpetrator. CPT Pharmacometrics Syst Pharmacol. 2021;10:441‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Y, Zhou Y, Hayashi M, Shou M, Skiles GL. Simulation of clinical drug‐drug interactions from hepatocyte CYP3A4 induction data and its potential utility in trial designs. Drug Metab Dispos. 2011;39:1139‐1148. [DOI] [PubMed] [Google Scholar]
- 41. Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P‐glycoprotein (P‐gp) inducers on exposure of P‐gp substrates: review of clinical drug‐drug interaction studies. Clin Pharmacokinet. 2020;59:699‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. XTANDI (enzalutamide) capsules Label (fda.gov). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203415lbl.pdf
- 43. label (fda.gov) (Apalutamide label). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210951s001lbl.pdf
- 44. Gibbons JA, de Vries M, Krauwinkel W, et al. Pharmacokinetic drug interaction studies with enzalutamide. Clin Pharmacokinet. 2015;54:1057‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Duran I, Carles J, Bulat I, et al. Pharmacokinetic drug‐drug interaction of apalutamide, part 1: clinical studies in healthy men and patients with castration‐resistant prostate cancer. Clin Pharmacokinet. 2020;59:1135‐1148. [DOI] [PubMed] [Google Scholar]
- 46. Gibbons JA, Ouatas J, Krauwinkel W, et al. Clinical pharmacokinetic studies of enzalutamide. Clin Pharmacokinet. 2015;54:1043‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Narayanan R, Hoffmann M, Kumar G, Surapaneni S. Application of a "fit for purpose" PBPK model to investigate the CYP3A4 induction potential of enzalutamide. Drug Metab Lett. 2016;10:172‐179. [DOI] [PubMed] [Google Scholar]
- 48. Belderbos BPSI, de Wit R, Chien C, et al. An open‐label, multicenter, phase Ib study investigating the effect of apalutamide on ventricular repolarization in men with castration‐resistant prostate cancer. Cancer Chemother Pharmacol. 2018;82:457‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. TIBSOVO (ivosidenib tablets (fda.gov)) . Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/211192_s008lbl.pdf
- 50. Jiang X, Wada R, Poland B, et al. Population pharmacokinetic and exposure‐response analyses of ivosidenib in patients with IDH1‐mutant advanced hematologic malignancies. Clin Transl Sci. 2021;14:942‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bolleddula J, Ke A, Yang H, Prakash C. PBPK modeling to predict drug‐drug interactions of ivosidenib as a perpetrator in cancer patients and qualification of the Simcyp platform for CYP3A4 induction. CPT Pharmacometrics Syst Pharmacol. 2021;10:577‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. label (fda.gov) (Lumacaftor label). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211358s000lbl.pdf
- 53. 0206038Orig1s000ClinPharmR.pdf (fda.gov) (Lumacaftor/ivacaftor: Clinical pharmacology and biopharmaceutical review). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/0206038Orig1s000ClinPharmR.pdf
- 54. https://didb.druginteractionsolutions.org/drug/monograph/12203/. Accessed March 14, 2022.
- 55. Orkambi, INN‐lumacaftor & ivacaftor (europa.eu). Accessed March 14, 2022. https://www.ema.europa.eu/en/documents/assessment‐report/orkambi‐epar‐public‐assessment‐report_en.pdf
- 56. van Erp NP, Guchelaar HJ, Ploeger BA, Romijn JA, Hartigh J, Gelderblom H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur J Endocrinol. 2011;164:621‐626. [DOI] [PubMed] [Google Scholar]
- 57. Dilantin (phenytoin sodium) (fda.gov). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/084349s060lbl.pdf
- 58. Brodie MJ, Mintzer S, Pack AM, Gidal BE, Vecht CJ, Schmidt D. Enzyme induction with antiepileptic drugs: cause for concern? Epilepsia. 2013;54:11‐27. [DOI] [PubMed] [Google Scholar]
- 59. Lim ML, Min SS, Eron JJ, et al. Coadministration of lopinavir/ritonavir and phenytoin results in two‐way drug interaction through cytochrome P‐450 induction. J Acquir Immune Defic Syndr. 2004;36:1034‐1040. [DOI] [PubMed] [Google Scholar]
- 60. Vlase L, Popa A, Neag M, Muntean D, Leucuta SE. Pharmacokinetic interaction between ivabradine and phenytoin in healthy subjects. Clin Drug Investig. 2012;32:533‐538. [DOI] [PubMed] [Google Scholar]
- 61. Bullman J, Nicholls A, Van Landingham K, et al. Effects of lamotrigine and phenytoin on the pharmacokinetics of atorvastatin in healthy volunteers. Epilepsia. 2011;52:1351‐1358. [DOI] [PubMed] [Google Scholar]
- 62. Michelucci R, Cipolla G, Passarelli D, et al. Reduced plasma nisoldipine concentrations in phenytoin‐treated patients with epilepsy. Epilepsia. 1996;37:1107‐1110. [DOI] [PubMed] [Google Scholar]
- 63. Backman JT, Olkkola KT, Ojala M, Laaksovirta H, Neuvonen PJ. Concentrations and effects of oral midazolam are greatly reduced in patients treated with carbamazepine or phenytoin. Epilepsia. 1996;37:253‐257. [DOI] [PubMed] [Google Scholar]
- 64. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady‐state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;2:89‐93. [DOI] [PubMed] [Google Scholar]
- 65. Rameis H. On the interaction between phenytoin and digoxin. Eur J Clin Pharmacol. 1985;29:49‐53. [DOI] [PubMed] [Google Scholar]
- 66. Dooley KE, Bliven‐Sizemore EE, Weiner M, et al. Safety and pharmacokinetics of escalating daily doses of the antituberculosis drug rifapentine in healthy volunteers. Clin Pharmacol Ther. 2012;91:881‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shehu AI, Li G, Xie W, Ma X. The pregnane X receptor in tuberculosis therapeutics. Expert Opin Drug Metab Toxicol. 2016;12:21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Borrelli F, Izzo AA. Herb‐drug interactions with St John's wort (Hypericum perforatum): an update on clinical observations. AAPS J. 2009;11:710‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gödtel‐Armbrust U, Metzger A, Kroll U, Kelber O, Wojnowski L. Variability in PXR‐mediated induction of CYP3A4 by commercial preparations and dry extracts of St. John's Wort. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:377‐382. [DOI] [PubMed] [Google Scholar]
- 70. Zahner C, Kruttschnitt E, Uricher J, et al. No clinically relevant interactions of St. John's wort extract ze 117 low in hyperforin with cytochrome P450 enzymes and P‐glycoprotein. Clin Pharmacol Ther. 2019;106:432‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mueller SC, Majcher‐Peszynska J, Uehleke B, et al. The extent of induction of CYP3A by St. John's wort varies among products and is linked to hyperforin dose. Eur J Clin Pharmacol. 2006;62:29‐36. [DOI] [PubMed] [Google Scholar]
- 72. Adiwidjaja J, Boddy AV, McLachlan AJ. Physiologically based pharmacokinetic modelling of hyperforin to predict drug interactions with St John's wort. Clin Pharmacokinet. 2019;58:911‐926. [DOI] [PubMed] [Google Scholar]
- 73. Wang YM, Ong SS, Chai SC, Chen T. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol. 2012;8:803‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fuhr LM, Marok FZ, Hanke N, Selzer D, Lehr T. Pharmacokinetics of the CYP3A4 and CYP2B6 inducer carbamazepine and its drug‐drug interaction potential: a physiologically based pharmacokinetic modeling approach. Pharmaceutics. 2021;13:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Haarst AV, Smith S, Garvin C, Benrimoh N, Paglialunga S. Rifampin drug‐drug‐interaction studies; reflections on the nitrosamine impurities issue. Clin Pharmacol Ther. 2022. (Online ahead of print). doi: 10.1002/cpt.2652 [DOI] [PubMed] [Google Scholar]
- 76. Drug Development and Drug Interactions | Table of Substrates, Inhibitors and Inducers | FDA. Accessed March 14, 2022. https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers
- 77. Ferguson SS, Chen Y, LeCluyse EL, Negishi M, Goldstein JA. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol Pharmacol. 2005;68:747‐757. [DOI] [PubMed] [Google Scholar]
- 78. Gufford BT, Robarge JD, Eadon MT, et al. Rifampin modulation of xeno‐ and endobiotic conjugating enzyme mRNA expression and associated microRNAs in human hepatocytes. Pharmacol Res Perspect. 2018;6:e00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Soars MG, Petullo DM, Eckstein JA, Kasper SC, Wrighton SA. An assessment of udp‐glucuronosyltransferase induction using primary human hepatocytes. Drug Metab Dispos. 2004;32:140‐148. [DOI] [PubMed] [Google Scholar]
- 80. Smith CM, Faucette SR, Wang H, LeCluyse EL. Modulation of UDP‐glucuronosyltransferase 1A1 in primary human hepatocytes by prototypical inducers. J Biochem Mol Toxicol. 2005;19:96‐108. [DOI] [PubMed] [Google Scholar]
- 81. Rodrigues AD, Lai Y, Shen H, Varma MVS, Rowland A, Oswald S. Induction of human intestinal and hepatic organic anion transporting polypeptides: where is the evidence for its relevance in drug‐drug interactions? Drug Metab Dispos. 2020;48:205‐216. [DOI] [PubMed] [Google Scholar]
- 82. Ferrell PB Jr, McLeod HL. Carbamazepine, HLA‐B*1502 and risk of Stevens‐Johnson syndrome and toxic epidermal necrolysis: US FDA recommendations. Pharmacogenomics. 2008;9:1543‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Greenberg RG, Melloni C, Wu H, et al. Therapeutic index estimation of antiepileptic drugs: a systematic literature review approach. Clin Neuropharmacol. 2016;39:232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dean L. Carbamazepine therapy and HLA genotype. Medical Genetics Summaries. National Center for Biotechnology Information (US). 2015. [PubMed] [Google Scholar]
- 85. Fridell JA, Jain AK, Patel K, et al. Phenytoin decreases the blood concentrations of sirolimus in a liver transplant recipient: a case report. Ther Drug Monit. 2003;25:117‐119. [DOI] [PubMed] [Google Scholar]
- 86. Iorga A, Horowitz BZ. Phenytoin toxicity (updated February 28, 2019). Treasure Island, FL: StatPearls Publishing. 2019. Accessed March 14, 2022. https://www.ncbi.nlm.nih.gov/books/NBK482444/ [Google Scholar]
- 87. Yamada M, Ishizuka T, Inoue SI, Rozehnal V, Fischer T, Sugiyama D. Drug‐drug interaction risk assessment of esaxerenone as a perpetrator by in vitro studies and static and physiologically based pharmacokinetic models. Drug Metab Dispos. 2020;48:769‐777. [DOI] [PubMed] [Google Scholar]
- 88. Djebli N, Buchheit V, Parrott N, et al. Physiologically‐based pharmacokinetic modelling of entrectinib parent and active metabolite to support regulatory decision‐making. Eur J Drug Metab Pharmacokinet. 2021;46:779‐791. [DOI] [PubMed] [Google Scholar]
- 89. Hariparsad N, Ramsden D, Taskar K, et al. Current practices, gap analysis, and proposed workflows for PBPK modeling of cytochrome P450 induction: an industry perspective. Clin Pharmacol Ther. 2021. (Online ahead of print). doi: 10.1002/cpt.2503 [DOI] [PubMed] [Google Scholar]
- 90. Pan X, Yamazaki S, Neuhoff S, Zhang M, Reddy VP. Unraveling pleiotropic effects of rifampicin by using physiologically based pharmacokinetic modeling: assessing the induction magnitude of P‐glycoprotein‐cytochrome P450 3A4 dual substrates. CPT Pharmacometrics Syst Pharmacol. 2021;10:1485‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hendriks DFG, Vorrink SU, Smutny T, et al. Clinically relevant cytochrome P450 3A4 induction mechanisms and drug screening in three‐dimensional spheroid cultures of primary human hepatocytes. Clin Pharmacol Ther. 2020;108:844‐855. [DOI] [PubMed] [Google Scholar]
- 92. Hanke N, Frechen S, Moj D, et al. PBPK models for CYP3A4 and P‐gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol. 2018;7:647‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rodrigues D, Rowland A. From endogenous compounds as biomarkers to plasma‐derived nanovesicles as liquid biopsy; has the golden age of translational pharmacokinetics‐absorption, distribution, metabolism, excretion‐drug‐drug interaction science finally arrived? Clin Pharmacol Ther. 2019;105:1407‐1420. [DOI] [PubMed] [Google Scholar]
- 94. Chen Y, Cabalu TD, Callegari E, et al. Recommendations for the design of clinical drug‐drug interaction studies with itraconazole using a mechanistic physiologically‐based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2019;8:685‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Venkatakrishnan K, Cook J. Driving access to medicines with a totality of evidence mindset: an opportunity for clinical pharmacology. Clin Pharmacol Ther. 2018;103:373‐375. [DOI] [PubMed] [Google Scholar]
- 96. Asaumi R, Toshimoto K, Tobe Y, et al. Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug‐drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacometrics Syst Pharmacol. 2018;7:186‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grimm SW, Richtand NM, Winter HR, Stams KR, Reele SB. Effects of cytochrome P450 3A modulators ketoconazole and carbamazepine on quetiapine pharmacokinetics. Br J Clin Pharmacol. 2006;61:58‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. 21‐437 Inspra Clinical Pharmacology Biopharmaceutics Review Part 6 (fda.gov). Accessed March 14, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21‐437_Inspra_BioPharmr_P6.pdf
- 99. Portolés A, Terleira A, Calvo A, Martínez I, Resplandy G. Effects of Hypericum perforatum on ivabradine pharmacokinetics in healthy volunteers: an open‐label, pharmacokinetic interaction clinical trial. J Clin Pharmacol. 2006;46:1188‐1194. [DOI] [PubMed] [Google Scholar]
- 100. Piscitelli SC, Burstein AH, Chaitt D, Alfaro RM, Falloon J. Indinavir concentrations and St John's wort. Lancet. 2000;355:547‐548. [DOI] [PubMed] [Google Scholar]
- 101. Wang Z, Gorski JC, Hamman MA, Huang SM, Lesko LJ, Hall SD. The effects of St John's wort (Hypericum perforatum) on human cytochrome P450 activity. Clin Pharmacol Ther. 2001;70:317‐326. [PubMed] [Google Scholar]
- 102. Imai H, Kotegawa T, Tsutsumi K, et al. The recovery time‐course of CYP3A after induction by St John's wort administration. Br J Clin Pharmacol. 2008;65:701‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hall SD, Wang Z, Huang SM, et al. The interaction between St John's wort and an oral contraceptive. Clin Pharmacol Ther. 2003;74:525‐535. [DOI] [PubMed] [Google Scholar]
- 104. Sugimoto K, Ohmori M, Tsuruoka S, et al. Different effects of St John's wort on the pharmacokinetics of simvastatin and pravastatin. Clin Pharmacol Ther. 2001;70:518‐524. [DOI] [PubMed] [Google Scholar]
- 105. Hebert MF, Park JM, Chen YL, Akhtar S, Larson AM. Effects of St. John's wort (Hypericum perforatum) on tacrolimus pharmacokinetics in healthy volunteers. J Clin Pharmacol. 2004;44:89‐94. [DOI] [PubMed] [Google Scholar]