

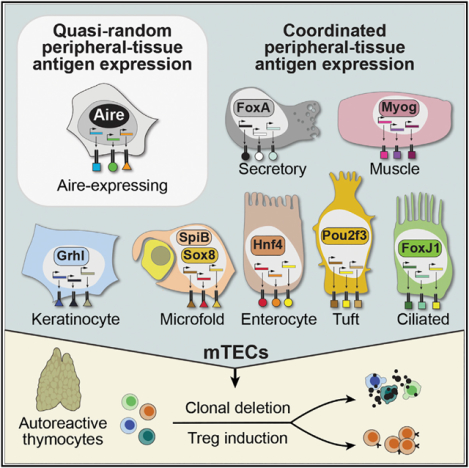
SUMMARY
Medullary thymic epithelial cells (mTECs) ectopically express thousands of peripheral-tissue antigens (PTAs), which drive deletion or phenotypic diversion of self-reactive immature T cells during thymic differentiation. Failure of PTA expression causes multiorgan autoimmunity. By assaying chromatin accessibility in individual mTECs, we uncovered signatures of lineage-defining transcription factors (TFs) for skin, lung, liver, and intestinal cells—including Grhl, FoxA, FoxJ1, Hnf4, Sox8, and SpiB—in distinct mTEC subtypes. Transcriptomic and histologic analyses showed that these subtypes, which we collectively term mimetic cells, expressed PTAs in a biologically logical fashion, mirroring extra-thymic cell types while maintaining mTEC identity. Lineage-defining TFs bound to mimetic-cell open chromatin regions and were required for mimetic cell accumulation, whereas the tolerogenic factor Aire was partially and variably required. Expression of a model antigen in mimetic cells sufficed to induce cognate T cell tolerance. Thus, mTECs co-opt lineage-defining TFs to drive mimetic cell accumulation, PTA expression, and self-tolerance.
Graphical Abstract
In brief
Medullary thymic epithelial cells repurpose the lineage-defining transcription factors of diverse extra-thymic cell types to create cellular mimics of the peripheral self within the thymus, allowing for self-tolerization of maturing T cells.
INTRODUCTION
The thymic epithelium plays an indispensable role in multiple phases of T cell maturation (reviewed in Abramson and Anderson, 2017). Following T cell receptor (TCR) formation, immature T cells are first positively selected for self-major histocompatibility complex (MHC) recognition by cortical thymic epithelial cells (cTECs), then negatively selected for self-antigen reactivity by medullary thymic epithelial cells (mTECs) and other antigen-presenting cells (APCs). mTECs are uniquely important effectors of negative selection because they ectopically express thousands of peripheral-tissue antigens (PTAs) in a mosaic fashion (Derbinski et al., 2001), allowing immature T cells to broadly sample self-antigens prior to their release into the periphery, promoting deletion of autoreactive T cell clones or their conversion into regulatory T cells (Tregs), and thereby preventing autoimmunity.
Given the importance of PTA expression in self-tolerance, its controlling mechanisms have invited considerable interest. Nearly two decades ago, the transcription factor (TF) Aire was shown to be an important driver of PTA expression: null mutations in Aire impair the expression of a large repertoire of ectopic transcripts in mTECs, and autoimmunity against Aire-dependent antigens subsequently develops in mice and humans (Aaltonen et al., 1997; Nagamine et al., 1997; Anderson et al., 2002). Common polymorphisms in Aire also predispose to autoimmune diseases, including Addison’s disease, rheumatoid arthritis, and type 1 diabetes (Oftedal et al., 2015; Eriksson et al., 2021; Terao et al., 2011). However, Aire lacks DNA sequence-specificity and does not appear to function as a conventional TF (Koh et al., 2008). Instead, several studies have suggested that Aire probabilistically induces PTAs in individual mTECs in a quasi-random fashion (Derbinski et al., 2008; Villaseñor et al., 2008; Meredith et al., 2015; Brennecke et al., 2015), without regard to PTAs’ tissue of origin or biological significance, by repurposing general transcriptional mechanisms like pause release and chromatin looping (Abramson et al., 2010; Giraud et al., 2012; Sansom et al., 2014; Chuprin et al., 2015; Yoshida et al., 2015; Bansal et al., 2017, 2021). Although consistent with the available data, this model raises perplexing questions, such as how a single protein can reproducibly control the expression of thousands of biologically disparate genes and why the expression of most PTAs is diminished, but not extinguished, in the absence of Aire.
Although recent mechanistic studies of PTA expression have focused on Aire, an old body of literature dating to the mid-1800s reported a variety of histologically distinct epithelial cell types in the thymic medulla, visible by light microscopy. The most prominent of these were cornified, skin-like Hassall’s corpuscles, but other cell types, including multiciliated cells, striated myoid cells, and electron-dense neurosecretory cells, were also observed (Hassall, 1846; Remak, 1855; Watney, 1882; Mayer, 1888). Indeed, one proposed model for PTA expression predating the discovery of Aire was that the thymus acts as a “mosaic of epithelial self,” serving up archetypal epithelial cells against which maturing T cells can be tolerized (Farr and Rudensky, 1998). However, a holistic understanding of these histologically defined cells—their ontogeny, detailed phenotype, relationship to Aire, roles in PTA expression and tolerance, and other functions—remains lacking.
Here, we used the single-cell assay for transposase-accessible chromatin with sequencing (scATAC-seq) to investigate mechanisms of PTA expression in individual mTECs. Although Aire had a strong and specific effect on mTEC chromatin, we were surprised to also find a variety of highly distinct mTEC subtypes characterized by lineage-defining TFs for diverse peripheral cell types. These mTECs, which we collectively term mimetic cells, expressed PTAs in a biologically logical fashion, required lineage-defining TFs for their accumulation, and sufficed to induce antigen-specific T cell tolerance.
RESULTS
Individual mTECs occupy diverse chromatin states
To understand how changes in chromatin accessibility might influence PTA expression in individual mTECs, we performed scATAC-seq in duplicate on purified MHC class II-high mTECs (mTEChi) from Aire+/+ and Aire −/− mice (Figure 1A). The scATAC-seq libraries were of high quality, showing nucleosomal periodicity in fragment length, signal enrichment in promoters, and concordance in aggregate with bulk ATAC-seq of mTECs (Bansal et al., 2017; Figures S1A–S1D). After filtering, we retained 11,582 mTECs for analysis, of which 5,690 were derived from Aire+/+ and 5,892 from Aire −/− mice, with a mean of 10,680 unique fragments per cell.
Figure 1. Individual mTECs occupy diverse chromatin states, some strongly influenced by Aire.
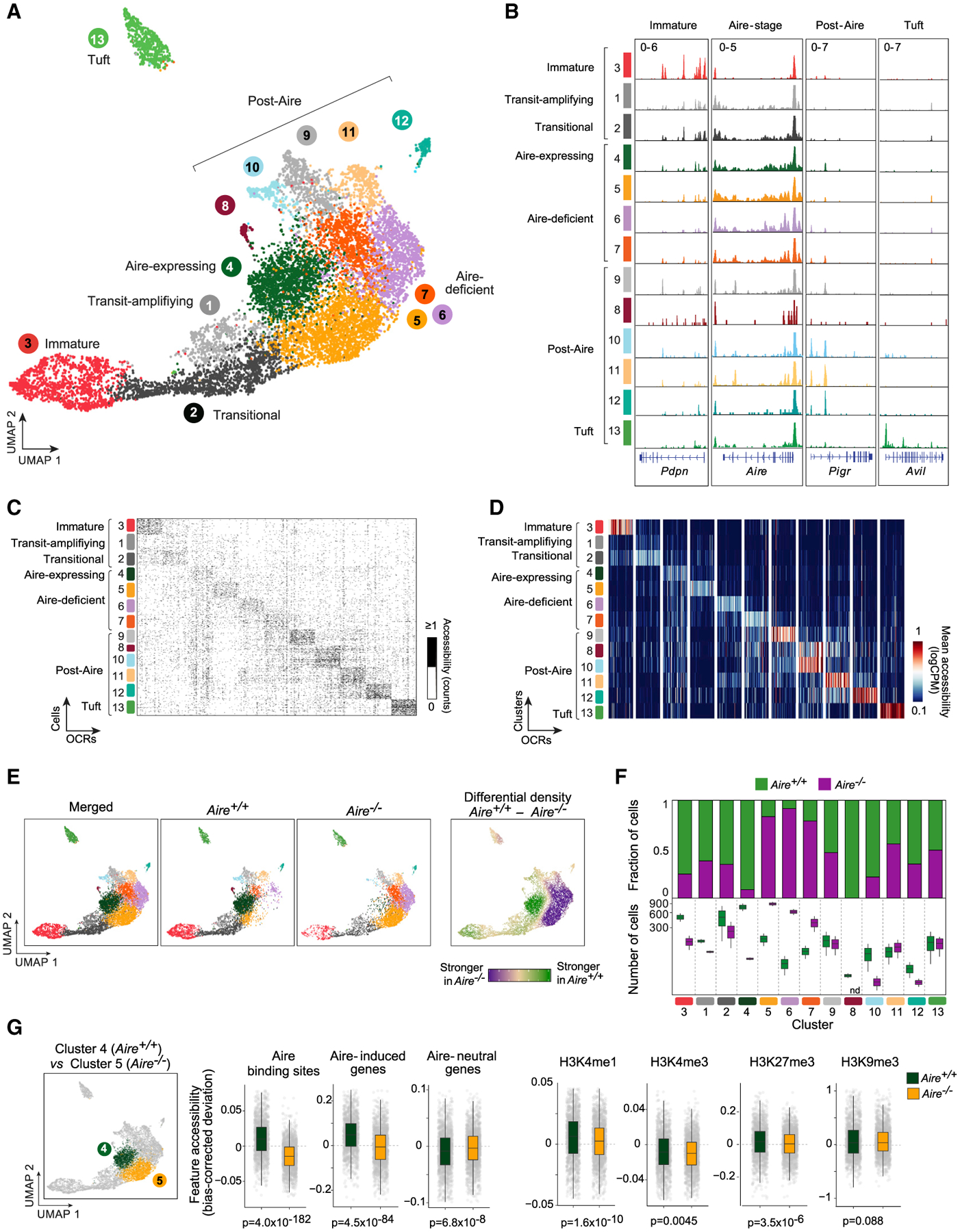
(A) Merged UMAP of scATAC-seq of mTEChi from Aire+/+ (n = 2) and Aire–/–(n = 2) mice. For all UMAPs, each dot is a single cell.
(B) Chromatin-accessibility tracks for mTEC clusters at the indicated loci. Signal is in counts per million (CPM).
(C and D) Single-cell (C) and cluster-level (D) chromatin accessibility at unique OCRs identified for each mTEC cluster. For each cluster, the top 50 unique OCRs and (for C) up to 100 randomly sampled cells are shown.
(E) Comparison of the distributions of mTECs from Aire+/+ and Aire−/− mice in UMAP space.
(F) Mean fraction (top) and number (bottom) of mTECs in each cluster from Aire+/+ (green) and Aire−/− (purple) mice. nd, not detected.
(G) Comparisons of chromatin accessibility at the indicated genomic features between Aire-expressing mTECs from Aire+/+ mice and Aire-deficient mTECs (cluster 5) from Aire−/− mice. Each gray dot is a single cell. p values were calculated by the Wilcoxon rank-sum test with Bonferroni correction.
See also Figure S1.
Dimensionality reduction and clustering yielded 13 distinct clusters, which could be broadly annotated as known mTEC subtypes based on chromatin accessibility at marker genes (Figures 1A, 1B, and S1E). Clustering was not driven by batch or other technical factors (Figures S1F–S1I). The main body of cells (clusters 4–7) had strong accessibility at the Aire gene body and at loci encoding antigen-presenting molecules like H2-Ab1. We inferred that these cells were Aire-stage mTECs, which constitute roughly 60% of mTEChi in adult mice (Gray et al., 2007) and labeled these clusters “Aire-expressing” or “Aire-deficient,” depending on their enrichment in Aire+/+ or Aire −/− mice (explored in detail below). A smaller cluster of cells (cluster 3) with enhanced accessibility at Pdpn and Krt5 corresponded to an mTEC subtype variously referred to as immature, junctional, or intertypical (Onder et al., 2015; Baran-Gale et al., 2020). Connecting the immature and Aire-stage subtypes were two clusters that we surmised to be transit-amplifying TEC and their immediate progeny (clusters 1 and 2), which have been shown by lineage tracing to give rise to both immature and Aire-stage mTECs (Wells et al., 2020). Finally, six clusters (clusters 8–13) were initially identified as post-Aire mTECs, given their accessibility at post-Aire markers like Pigr and Spink5, and their proximity to Aire-stage mTECs. One of these subtypes (cluster 13) was specifically enriched in tuft-cell markers like Avil and Chat; we annotated these cells as the recently described tuft mTECs, which have a post-Aire provenance (Panneck et al., 2014; Bornstein et al., 2018; Miller et al., 2018).
To more globally assess chromatin accessibility differences between mTEC subtypes, we used the irreproducible discovery rate (IDR) framework (Qunhua et al., 2011) to define sets of open chromatin regions (OCRs) unique to each scATAC-seq cluster. We defined unique OCRs for all subtypes except clusters 1 and 8, which had few OCRs after IDR analysis. Each subtype showed strong and specific chromatin accessibility at its cluster-specific OCRs, as assayed in single cells (Figure 1C) and at the cluster level (Figure 1D).
Aire promotes chromatin accessibility at its binding sites and target genes
We next explored what effect Aire has on chromatin accessibility in individual mTECs. Previous studies using bulk ATAC-seq have reached diverging conclusions on this question: one study argued that Aire enhances chromatin accessibility at Aire binding sites and mTEC enhancers, whereas a second report contended that Aire’s effect on chromatin accessibility is largely repressive and targeted to tissue-specific genes (Bansal et al., 2017; Koh et al., 2018). Dividing our scATAC-seq data by genotype, we observed major differences in the cell distribution in uniform manifold approximation and projection (UMAP) space, primarily localized to the Aire-stage mTEC chromatin states (Figure 1E). In Aire+/+ mice, nearly all Aire-stage mTECs were localized to a single Aire-expressing cluster (cluster 4), whereas in Aire −/− mice, Aire-stage mTECs segregated into three Aire-deficient clusters (clusters 5–7). Quantification of fractional and numeric differences in each cluster between the two genotypes confirmed these visual observations (Figure 1F). It also demonstrated that immature, transit-amplifying, and post-Aire mTECs tended to be less affected by the presence or absence of Aire, although a few post-Aire clusters were markedly reduced or absent in Aire-deficient mice. Thus, Aire had a strong effect on chromatin accessibility, and logically, this effect was observed primarily in mTECs that expressed Aire.
To better understand the nature of chromatin accessibility differences between Aire-expressing and Aire-deficient mTECs, we quantified chromatin accessibility changes at important genomic features previously mapped or defined in mTECs, such as Aire-induced and Aire-neutral genes, Aire binding sites, and a variety of active or repressive histone marks (Figure 1G; Bansal et al., 2017; Meredith et al., 2015; Handel et al., 2018; Sansom et al., 2014). We limited our comparisons to the first Aire-deficient cluster (cluster 5), reasoning that these cells lay closest to their putative precursors, transit-amplifying mTECs, and were therefore least likely to have experienced secondary effects related to Aire’s absence. Although some modest changes in chromatin accessibility were evident at histone marks, by far the strongest changes occurred at Aire binding sites and Aire-induced genes, where Aire significantly enhanced chromatin accessibility. No such changes were evident at expression-matched Aire-neutral genes. Thus, by resolving the mTEC compartment at the single-cell level, we found that Aire specifically promoted chromatin accessibility at its binding sites and its target genes, without strongly affecting genes neutral to its presence or active or repressive chromatin more generally.
We also investigated the additional Aire-deficient clusters that accumulated in the absence of Aire (clusters 6 and 7). TF-motif-enrichment analyses revealed that accessible chromatin in these cells was enriched for interferon regulatory factor (IRF) motifs (Figure S1J). Reanalysis of RNA sequencing (RNA-seq) data from Aire+/+ versus Aire −/− mTECs (Bansal et al., 2021) identified Irf8 as highly induced in Aire-deficient mTECs, which we confirmed by flow cytometry (Figures S1K and S1L). IRF8 is essential for the differentiation of several subsets of APCs (Tamura et al., 2005), and we wondered if it might similarly potentiate the antigen-presenting capabilities of mTECs. Indeed, mTECs with high MHCII and CD80 were increased in Aire-deficient mice (Figure S1M). Thus, absent Aire, mTECs accumulated in an IRF8hi state with strong antigen-presentation character.
Small subsets of mTECs harbor the lineage-defining TFs and chromatin landscapes of peripheral cell types
We next turned to a second, somewhat unexpected aspect of our scATAC-seq data, the striking diversity in chromatin states among post-Aire mTECs. TF-motif-enrichment analysis at the cluster level revealed that in addition to enhancement for known TFs in mTEC subtypes (i.e., Pou2f3 in tuft mTECs), each of the post-Aire clusters showed enrichment for specific TF families (Figures 2A and S2A), including forkhead box A (FoxA; clusters 8 and 10), grainyhead-like (Grhl; cluster 9), hepatocyte nuclear factor 4 (Hnf4; clusters 11 and 12), and SRY-related HMG-box (Sox; cluster 12). These TF families contain a number of lineage-defining TFs from extra-thymic tissues: for instance, Hnf4α/γ are essential for liver and gut epithelia (Parviz et al., 2003; Chen et al., 2019); Grhl1/2/3 control skin development (Ting et al., 2005; Cangkrama et al., 2016); and FoxA1/2/3 drive differentiation of various neuroendocrine and secretory cell types (Golson and Kaestner, 2016). Single-cell TF-motif-enrichment analysis confirmed that these motifs were enriched in subsets of post-Aire mTECs and that many of these motifs were among the most variable in mTECs (Figures 2B and S2B). Single-cell analysis also revealed additional features of interest: for instance, forkhead box J (FoxJ) motifs, which are similar to FoxA motifs but play a distinct and essential role in ciliogenesis, were also enriched in clusters 8 and 10, and Ets sub-family Spi (PU.1, SpiB, and SpiC) motifs were enriched in cluster 12. This cluster 12—enriched for Hnf4, Spi, and Sox family motifs—was thus reminiscent of gut microfold (M) cells, professional antigen-trancytosing cells that differentiate in Peyer’s patches from Hnf4-expressing intestinal epithelial cells in a SpiB- and Sox8-dependent fashion (Kanaya et al., 2012; Kimura et al., 2019).
Figure 2. Small subsets of mTECs harbor the lineage-defining TFs and chromatin landscapes of peripheral cell types.
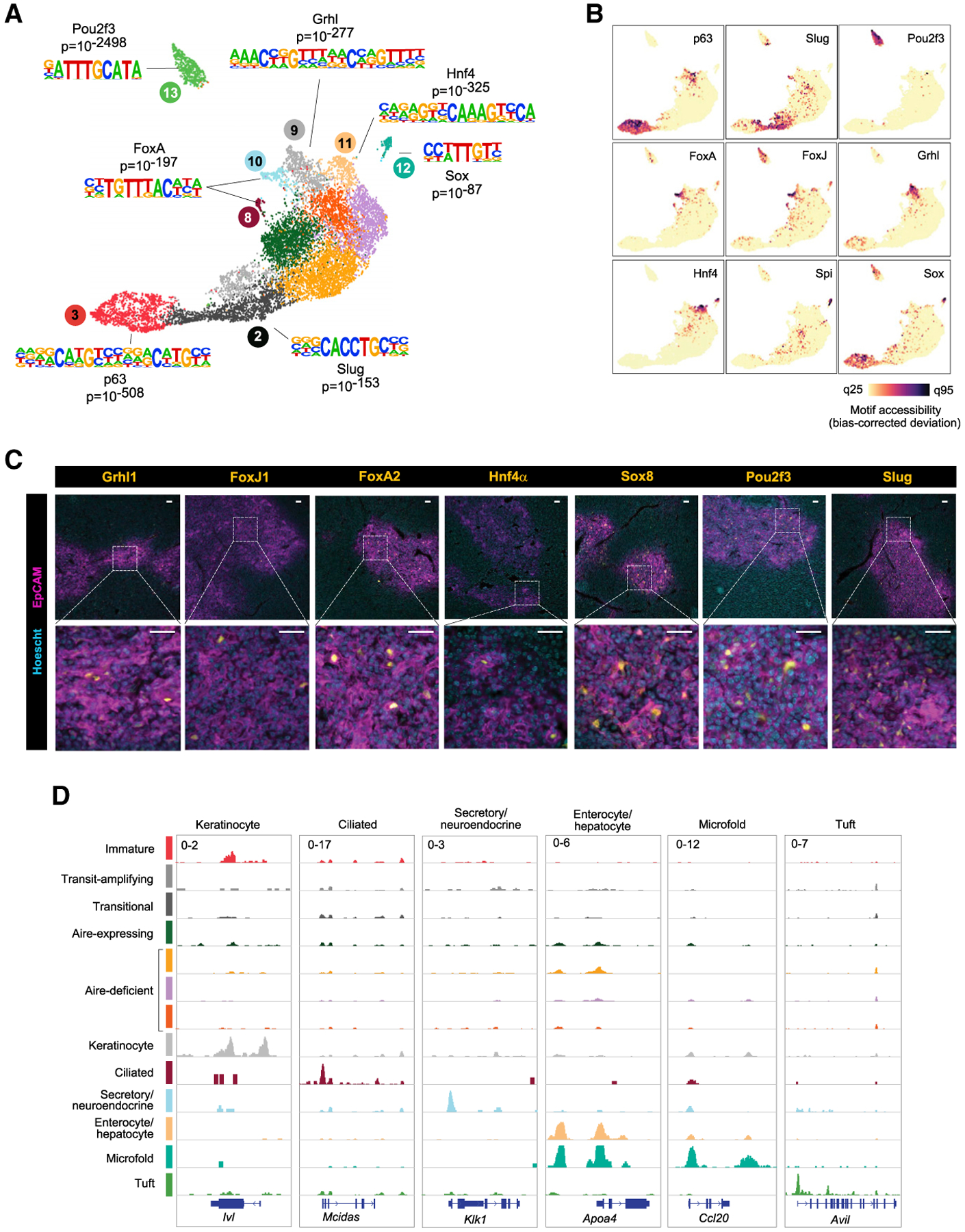
(A) Cluster-level TF-motif-enrichment analysis of mTEC scATAC-seq data.
(B) Single-cell TF-motif-enrichment analysis of scATAC-seq data. q25, 25th quantile; q95, 95th.
(C) Immunofluorescence of the indicated markers in thymic sections, showing wide (top) and zoomed (bottom) views. Scale bars, 50 μm.
(D) Chromatin-accessibility tracks for mTEC clusters at the indicated loci. Signal is in CPM.
See also Figure S2.
According to footprinting analysis, each post-Aire cluster showed enriched protection of its corresponding TF motif in its respective OCRs (Figure S2C). RNA-seq of mTECs confirmed that TFs with motif enrichment were themselves expressed (Figure S2D). Immunofluorescence of thymic sections confirmed nuclear expression of select members of each TF family, largely confined to the EpCAM+ thymic medulla (Figures 2C and S2E). (Note: although many TF families have multiple members, we performed in-depth characterization of individual members for which high-quality antibodies were commercially available. However, we suspect that, as in the periphery, families with multiple members likely exhibit some degree of functional redundancy. Some motifs are shared across TF families as well; here, we focused on families with the strongest motif enrichments and orthogonal support from RNA-seq and immunofluorescence data.)
What is the significance of expressing the lineage-defining TFs of peripheral cell types in mTECs? Remarkably, each post-Aire cluster also displayed an accessible-chromatin landscape resembling that of the peripheral cell type corresponding to each lineage-defining TF (Figures 2D and S2F). To wit: the Grhl-defined cluster 9, but not other clusters, showed enriched accessibility at keratinocyte-specific genes like Ivl and Cnfn, encoding key components of the cornified cell envelope in the skin; the FoxJ-defined cluster 8 had enriched accessibility at ciliated-cell-specific genes like Mcidas and Spag8, encoding essential regulators of ciliogenesis; the FoxA-defined cluster 10 exhibited enriched accessibility at secretory-cell-specific genes like Klk1 and Krt7, encoding characteristic markers of glandular epithelium; the Hnf4-defined cluster 11 had enriched accessibility at enterocyte- and hepatocyte-specific genes like Apoa4 and Aldob, encoding lipid- and sugar-processing proteins produced by the gut and liver; and the Sox8- and SpiB-defined cluster 12 showed enriched accessibility at M cell-specific genes like Ccl20 and Tnfaip2, encoding a Peyer’s patch chemokine and a classic M cell marker, respectively. The Pou2f3-defined cluster 13 exhibited enriched accessibility at tuft-cell-specific genes like Il25 and Avil, consistent with previous results (Bornstein et al., 2018). Thus, not only did post-Aire clusters show specific enrichment for lineage-defining TFs, but they also bore chromatin-accessibility landscapes that mirrored the peripheral cell types whose differentiation is driven by these TFs.
In sum, multiple mTEC subtypes initially annotated as post-Aire mTECs showed specific enrichment of lineage-defining TF motifs in their accessible chromatin, including those of Grhl, FoxA, FoxJ1, Hnf4, Sox8, and SpiB. These TFs were expressed in mTECs and were specifically associated with chromatin-accessibility patterns corresponding to the peripheral cell types defined by each TF. We named each cluster after its peripheral counterpart (keratinocyte, ciliated, secretory/neuroendocrine, enterocyte/hepatocyte, microfold, and tuft mTECs) and collectively termed these peripheral-cell-mimicking mTECs “mimetic cells.”
Mimetic cells comprise a diverse mTEC compartment with biologically logical PTA expression
We wondered how to reconcile the discovery of multiple mimetic cell types with previous studies concluding that PTA expression is “probabilistic,” “stochastic,” or “biologically indeterminate” (Villaseñor et al., 2008; Derbinski et al., 2008; Meredith et al., 2015; Brennecke et al., 2015; Dhalla et al., 2020). One potential explanation was that these studies had focused on Aire-expressing mTEChi but that the mimetic cells uncovered in our scATAC-seq data occurred in the post-Aire compartment, which occupies both MHCII-high and MHCII-intermediate/low (mTEClo) states (Metzger et al., 2013). The mimetic-cell chromatin states in our mTEChi scATAC-seq data might have presaged transcriptional states evident primarily in subsequent mTEClo stages.
We thus sought to identify markers that would allow us to enrich for mimetic cells in the mTEClo compartment, which had been thought to consist primarily of immature mTECs. By reanalyzing Aire-lineage-tracing RNA-seq data (Miller et al., 2018), we identified two proteins, podoplanin (Pdpn) and integrin β4 (CD104), that were downregulated in post-Aire mTECs; by flow cytometry, the mTEClo population separated into Pdpn+CD104+ (pre-Aire) and Pdpn−CD104− (post-Aire) compartments (Figures 3A, S3A, and S3B). The post-Aire compartment expanded from less than 20% of the mTEClo compartment in perinates to roughly 50% in adults (Figures 3A and S3C). Bulk RNA-seq followed by correlation and principal-component analysis (PCA) demonstrated that each mTEC subtype was cohesive and distinct from the other subtypes (Figures 3B and S3D). Fold-change/fold-change (FC/FC) comparison with Aire lineage tracing demonstrated that the Pdpn/CD104 gating scheme captured much of the same variation (Figure S3E; r = 0.76, p < 0.0001). Compared with pre-Aire mTEClo, post-Aire mTEClo more strongly expressed loci encoding lineage-defining TFs and Aire-induced, but not Aire-neutral, genes (Figure S3F; Table S1). Thus, we concluded that mTEClo could be divided into pre- and post-Aire compartments based on Pdpn and CD104 expression and that the post-Aire compartment was enriched for PTAs and mimetic-cell TFs.
Figure 3. Mimetic cells comprise a diverse mTEC compartment with biologically logical PTA expression.
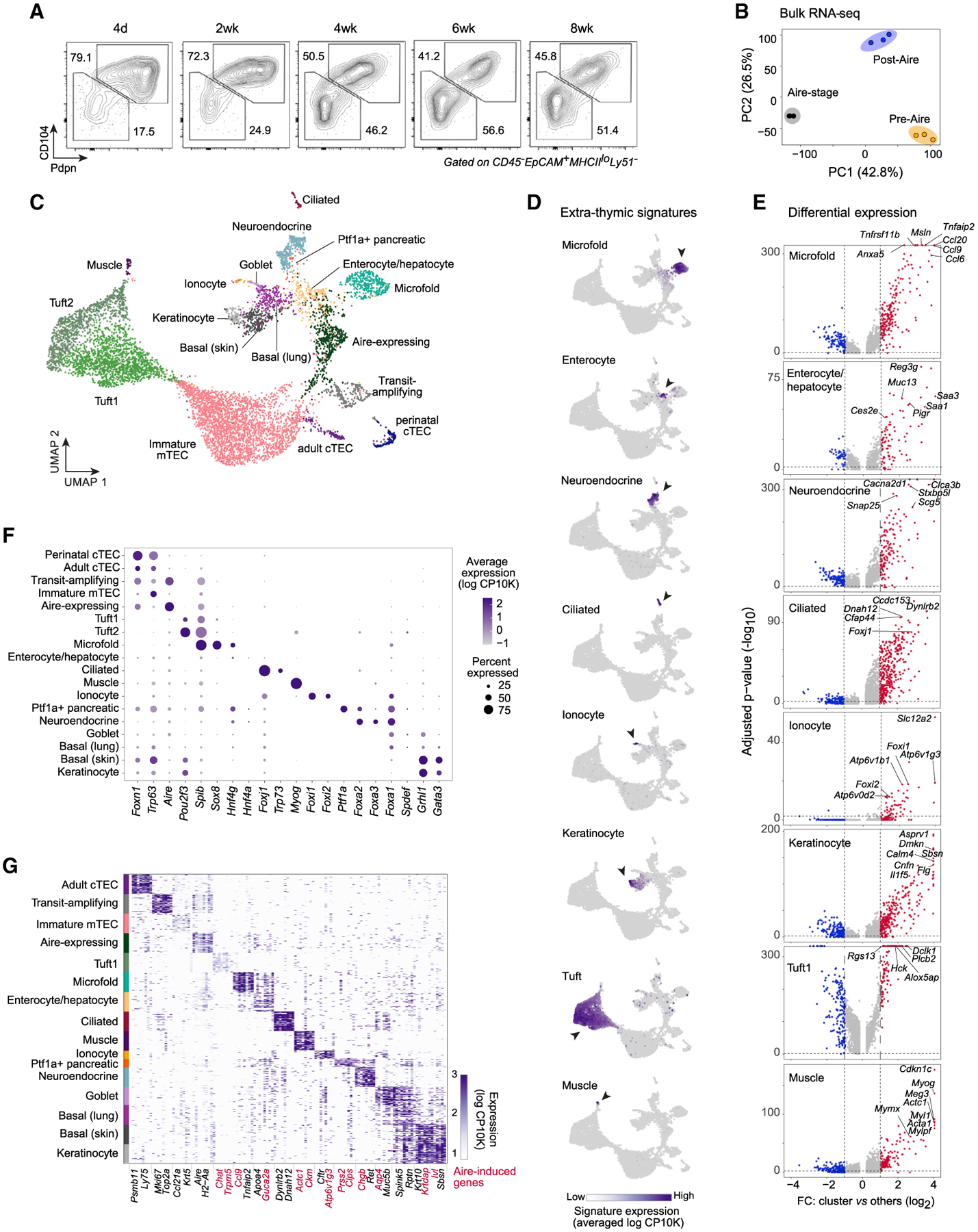
(A) Representative flow plots showing Pdpn and CD104 expression in mTEClo across mouse development.
(B) PCA of bulk RNA-seq of pre-Aire mTEClo, Aire-stage mTEChi, and post-Aire mTEClo (n = 3 for each). Each dot is one biological replicate.
(C) Merged UMAP of scRNA-seq of Pdpn–CD104– mTEClo from perinatal (n = 3) and adult (n = 3) mice.
(D) UMAPs of gene-signature expression from the indicated extra-thymic cell types in mTECs, assayed by scRNA-seq. Log CP10K, natural log1p of counts per 10,000 counts.
(E) Volcano plots of scRNA-seq of the indicated mTEC subtypes versus all other cells. Per-gene p values were BH-corrected. FC, fold change.
(F) Cluster-level expression of transcripts encoding various TFs, assayed by scRNA-seq.
(G) Heatmap of expression of transcripts encoding marker genes for each mTEC subtype, assayed by scRNA-seq. For each subtype, up to 50 randomly sampled cells are shown. Rows are cells, columns are genes, and two genes per subtype are labeled. Aire-induced genes are highlighted in red.
See also Figures S3 and S4 and Tables S1 and S2.
To address whether the mimetic cells revealed in our scATAC-seq data had transcriptional correlates in the mTEClo compartment, we performed single-cell RNA-seq (scRNA-seq) in triplicate on Pdpn−CD104 mTEClo from perinatal and adult mice (Figure 3C). After quality control, we retained 8,236 mTECs for downstream analysis, 1,083 from perinates and 7,153 from adult mice, with a mean of 6,716 unique fragments per cell. Dimensionality reduction and clustering revealed a remarkable diversity of mTECs contained within the post-Aire mTEClo compartment (Figure 3C), including correlates of all the mimetic cells observed in our scATAC-seq data as well as additional subtypes of mimetic cells not detected by scATAC-seq (skin/lung basal, ionocyte, goblet, Ptf1a+ pancreatic, and skeletal muscle mTECs). By overlaying published gene signatures from extra-thymic cell types (Joost et al., 2016; Haber et al., 2017; Montoro et al., 2018; Cao et al., 2020) onto our scRNA-seq data, we could observe clear compartmentalization of signature expression in subsets of mimetic cells (Figure 3D). Differential expression analysis showed that the most differential genes for each mimetic cell subtype corresponded to key marker genes for peripheral cell types, such as Tnfaip2 and Tnfrsf11b in microfold mTECs, Reg3g and Saa1/Saa3 in enterocyte/hepatocyte mTECs, Snap25 and Stxbp5l in neuroendocrine mTECs, Dynlrb2 and Dnah12 in ciliated mTECs, Slc12a2 and Atp6v1b1 in ionocyte mTECs, Cnfn and Flg in keratinocyte mTECs, and Myl1 and Actc1 in muscle mTECs (Figure 3E; Table S2). Each mimetic cell subtype readily expressed a lineage-defining TF or set of TFs corresponding to its peripheral counterpart (Figure 3F). Focused analysis of PTA expression demonstrated that many PTAs expressed in mimetic cells were Aire-induced genes (Figure 3G). Thus, PTA expression in mimetic cells was modular, mirrored peripheral cell types, followed a lineage-defining-TF logic, and included numerous Aire-induced transcripts.
Because we profiled mTECs from perinates and adults, we were able to examine the effect of age on mTEC composition. Compared with those of adults, perinatal Pdpn−CD104−mTEClo were relatively enriched for Aire-stage mTECs and relatively depleted of immature and tuft mTECs (Figures S4A and S4B). Perinates also showed a more modest enrichment for some mimetic cell subtypes, especially muscle, enterocyte/hepatocyte, and ciliated mTECs (Figure S4B). Considering the importance of the perinatal period in central tolerance induction (Miller, 1961; Guerau-de-Arellano et al., 2009; Yang et al., 2015), these biases may have functional implications for the tolerization of T cells during early life.
We also investigated whether there was a precursor-product relationship between mimetic cells. RNA velocity analysis, which suggests differentiation trajectories based on mRNA splicing, showed that analogous to how gut M cells differentiate from intestinal epithelial precursors, enterocyte/hepatocyte mTECs seemed to give rise to microfold mTECs, with enterocyte/hepatocyte markers like Aldob, Saa3, and Pigr preceding microfold markers like Gp2, Ccl9, and Ccl20 (Figure S4C). Similarly, the splicing dynamics of basal and keratinocyte mTECs indicated that expression of basal markers, including Krt5, Itgb1, and H2-Aa, preceded that of keratinized markers, including Ivl, Flg, and Il1f5 (Figure S4D).
To assess whether these various mimetic cells were bona fide post-Aire mTECs, we constructed gene signatures for each mimetic cell subtype and overlaid them onto our previously generated FC/FC plots of Aire-lineage-tracing and Pdpn/CD104 RNA-seq data (Figure S4E). Consistent with published results (Miller et al., 2018), the keratinocyte and tuft mTEC gene signatures were enriched in post-Aire mTECs, whereas the immature mTEC signature was depleted. Nearly every one of the mimetic cell subtypes we identified was similarly enriched in post-Aire mTECs, excepting only muscle mTECs. Orthogonal analysis of another published Aire-lineage-tracing experiment, this one using scRNA-seq (Wells et al., 2020), yielded a similar result: small numbers of mTECs with high expression of mimetic-cell gene signatures were detectable, and a substantial fraction of nearly all mimetic cell subtypes had previously expressed Aire (Figure S4F). Thus, most mimetic cell subtypes appeared to be downstream of Aire expression, although they may not strictly depend on Aire (for instance, tuft mTECs do not; Miller et al., 2018).
Finally, are mimetic cells also present in the human thymus? Reanalysis of scRNA-seq data from human fetal thymic epithelium (Park et al., 2020) revealed focused expression of mimetic-cell gene-signature orthologues in distinct subsets of human TECs (Figure S4G). Thus, the human thymus also hosts a rich constellation of mimetic cells, consistent with some more limited previous observations (Park et al., 2020; Bautista et al., 2021).
Lineage-defining TFs bind to mimetic-cell OCRs
To understand the mechanistic basis of the mimetic cell phenomenon, we sought to evaluate the role of lineage-defining TFs in mimetic cells. If they are necessary to produce and/or maintain mimetic cells, these TFs ought to bind mimetic-cell chromatin, and their absence should impair mimetic cell accumulation.
To examine the first hypothesis, we mapped the chromatin binding of several lineage-defining TFs—Pou2f3 (tuft cells), Hnf4α (enterocytes/hepatocytes), and Grhl1 (keratinocytes)—in primary mTECs using cleavage under targets and tagmentation (CUT&Tag) (Kaya-Okur et al., 2019; Figure 4). We also mapped H3K27 acetylation (H3K27ac) as a proxy for active enhancers and promoters, and IgG binding as a negative control. We contextualized our results using published Aire binding data (Bansal et al., 2017). The CUT&Tag data were of good quality, showing enrichment of the mouse genome, depletion of the E. coli spike-in genome, reproducible fragment-length patterns, and enriched binding to promoters (Figures S5A–S5C; Table S3).
Figure 4. Lineage-defining TFs bind to mimetic-cell OCRs.
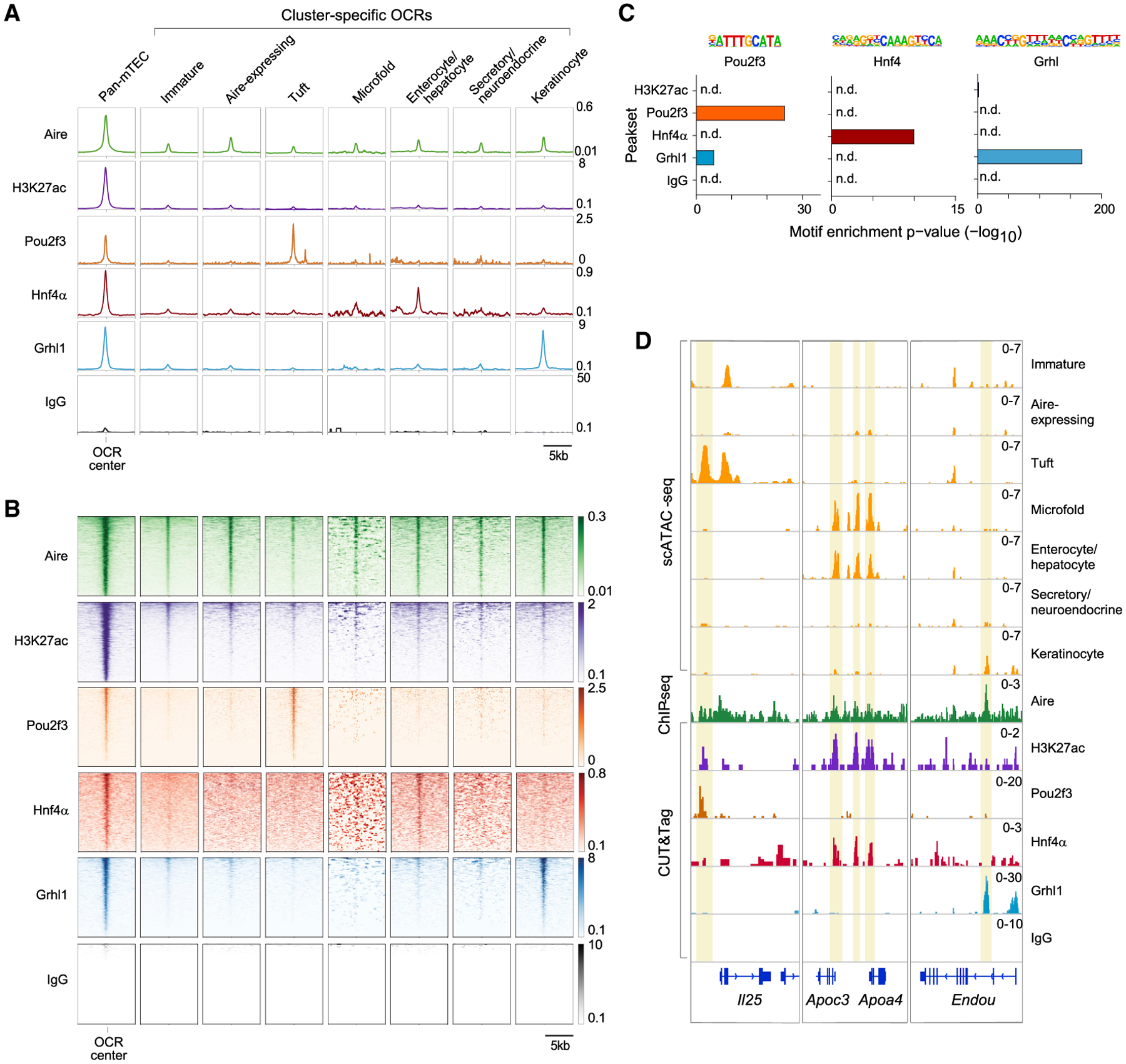
(A and B) Profile plots (A) and heatmaps (B) of binding of the indicated factors to the indicated OCRs previously defined by scATAC-seq, assayed by CUT&Tag (H3K27ac, Pou2f3, Hnf4α, Grhl1, and IgG) or ChIP-seq (Aire). For profile plots, mean signal is shown. For heatmaps, each row is one OCR.
(C) TF-motif enrichment in the peaksets of the indicated factors.
(D) Genome tracks showing chromatin accessibility in mTEC subtypes (yellow) and binding of the indicated factors at the indicated loci. In (A), (B), and (D), signal is in CPM and was merged from n = 2 (Aire, H3K27ac, and IgG), n = 3 (Grhl1), n = 4 (Hnf4α), or n = 8 (Pou2f3) independent replicates.
We examined binding of Aire, H3K27ac, Pou2f3, Hnf4α, and Grhl1 at OCRs accessible in all mTECs (pan-mTEC OCRs) and at subtype-specific OCRs previously defined by scATAC-seq (Figures 4A, 4B, and S5D; see also Figures 1C and 1D). Aire and H3K27ac bound all OCRs, most strongly at pan-mTEC OCRs but also at subtype-specific OCRs. In contrast to Aire and H3K27ac, however, the binding of lineage-defining TFs was restricted to pan-mTEC OCRs and their respective mimetic-cell OCRs: Pou2f3 bound tuft-specific OCRs, Hnf4α bound enterocyte/hepatocyte-specific OCRs, and Grhl1 bound keratinocyte-specific OCRs, with little overlap between the three. Few OCRs appreciably bound IgG. Pou2f3, Hnf4α, and Grhl1 peaks were each substantially enriched for their respective motifs (Figure 4C), demonstrating that we were detecting specific binding of each factor. Lineage-defining TF binding was evident at many mimetic-cell-specific loci (Figures 4D and S5E): Pou2f3 bound tuft-specific OCRs at Il25 and Alox5ap; Hnf4α bound enterocyte/hepatocyte- and microfold-specific OCRs at Apoa4/Apoc3 and Muc13; and Grhl1 bound keratinocyte-specific OCRs at Endou and Sbsn. Thus, although Aire bound to most mTEC OCRs, lineage-defining TFs bound only to pan-mTEC OCRs and their respective mimetic-cell OCRs, but not to other mimetic-cell OCRs, indicating that lineage-defining TFs were binding chromatin within their respective mimetic cells.
We also used the chromatin binding data to probe the molecular relationship between Aire and lineage-defining TFs. By stratifying TF signals at mimetic-cell OCRs by Aire co-binding, two distinct modes of TF behavior were apparent: for Pou2f3 and Grhl1, the amount of TF bound to chromatin was enhanced by Aire co-binding, whereas for Hnf4α, TF binding was similar irrespective of Aire co-binding (Figure S5F). For all three factors, substantial binding to mimetic-cell OCRs was observed even when Aire was not co-bound. Thus, Aire appeared to enhance the binding of some lineage-defining TFs but in no case was required for basal TF binding.
Mimetic cell accumulation requires lineage-defining TFs
We then addressed whether lineage-defining TFs were necessary for mimetic cell accumulation. We focused on microfold mTECs for several reasons: (1) little is known about this subtype, (2) we were able to establish a reliable marker scheme to isolate microfold mTECs by flow cytometry, and most importantly, (3) SpiB and Sox8, the lineage-defining TFs of M cells, are neither lethal nor redundant when deleted in the germ line.
We began by more deeply characterizing microfold mTECs. According to scRNA-seq, these cells expressed high levels of transcripts encoding classic M cell markers (Figure 5A). To localize microfold mTECs in the thymus, we performed whole-mount immunofluorescence, which revealed GP2+ cells scattered diffusely throughout the medullary regions (Figure 5B). Immunofluorescence of thymic sections showed GP2+ cells confined to medullary islets, often co-staining with or located near Villin+ epithelial cells (Figure 5C). The GP2+ cells often adopted dendritic morphologies and encircled nearby lymphocytes in a manner reminiscent of the “lymphocyte pocket” of gut M cells (Figure 5C, inset) (Wolf and Bye, 1984).
Figure 5. Mimetic cell accumulation requires lineage-defining TFs.
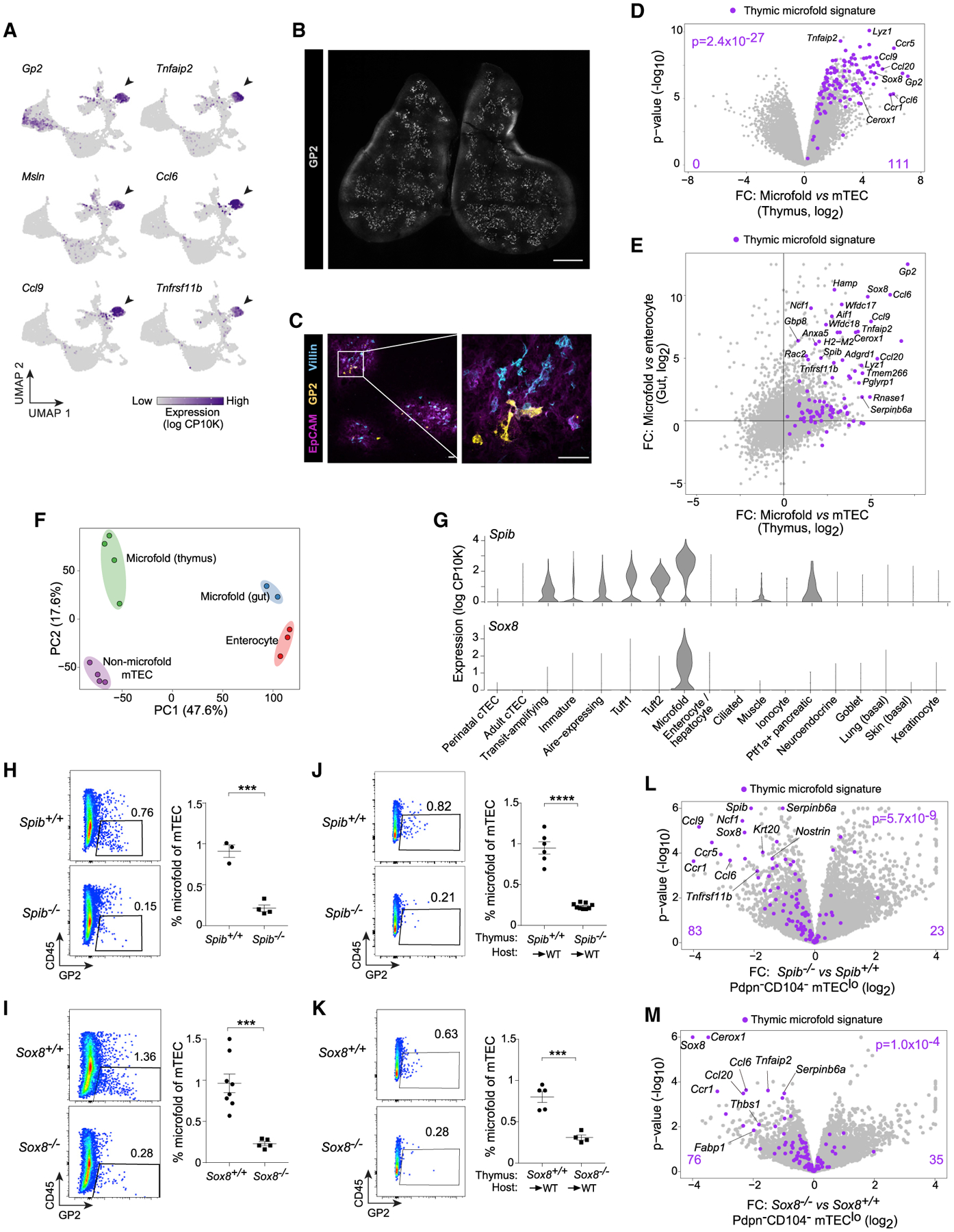
(A) UMAPs showing expression of microfold-associated transcripts in mTECs, assayed by scRNA-seq.
(B) Representative 2D slice of thymus whole-mount immunofluorescence of GP2. Scale bar, 1 mm.
(C) Immunofluorescence of EpCAM, GP2, and Villin in thymic sections. Scale bars, 30 μm.
(D) Volcano plot of bulk RNA-seq of purified GP2+CD45neg mTEC (n = 4) versus GP2–CD45neg mTEC (n = 4).
(E) FC/FC plot comparing thymic microfold mTECs and gut M cells versus their respective epithelial counterparts.
(F) PCA of bulk RNA-seq of non-microfold mTEC (n = 4), microfold mTEC (n = 4), gut M cells (n = 2), and enterocytes (n = 3). Each dot is one biological replicate.
(G) Violin plots showing the distribution of Spib and Sox8 expression in mTEC subtypes, assayed by scRNA-seq.
(H–K) Representative flow plots (left) and summarized data (right) of microfold mTEC abundance in (H) Spib+/+ (n = 3) versus Spib–/–(n = 4) thymi; (I) Sox8+/+ (n = 8) versus Sox8–/–(n = 5) thymi; (J) Spib+/+ (n = 6) versus Spib–/–(n = 9) thymi grafted into WT hosts; and (K) Sox8+/+ (n = 5) versus Sox8–/– (n = 4) thymi grafted into WT hosts. For (H)–(K), data were pooled from two independent experiments, each dot is one mouse, bars show mean ± SEM, and p values were calculated by unpaired, two-sided Student’s t tests.
(L and M) Volcano plots of bulk RNA-seq of Pdpn–CD104– mTEClo from (L) Spib–/– versus Spib+/+ mice and (M) Sox8–/– versus Sox8+/+ mice (n = 3 for each). For (D), (L), and (M), signature p values were calculated by chi-square tests.
See also Figures S6 and S7 and Table S4.
Next, we devised a gating scheme to isolate microfold mTECs by flow cytometry. Because a subpopulation of tuft mTECs also expressed Gp2 (Figure 5A), we sought a second marker to permit purer flow cytometric discrimination of microfold mTECs. Interestingly, Gp2-expressing tuft mTECs also expressed Ptprc, encoding CD45 (Figure S6A). By flow cytometry, we could see distinct GP2+CD45low and GP2+CD45neg mTEC populations (Figure S6A); we excluded the former as tuft mTECs and gated the latter as microfold mTECs. Bulk RNA-seq confirmed this cell population was enriched for every microfold mTEC marker derived from scRNA-seq (Figure 5D; Table S4). To determine how similar microfold mTECs were to gut M cells, we purified gut M cells for bulk RNA-seq (Figures S6B and S6C). FC/FC analysis demonstrated that both thymic and gut microfold cells shared a substantial upregulation of the microfold signature compared with their non-microfold epithelial neighbors (Figure 5E). Whole-transcriptome integration by PCA showed that among non-microfold mTECs, microfold mTECs, gut M cells, and enterocytes, the greatest component of the variance (47.6%) was organotypic, separating the thymus from the gut, but that the microfold program contributed a substantial minor fraction (17.6%) as well (Figure 5F). Thus, microfold mTECs could be purified as GP2+CD45neg mTECs and upregulated a transcriptional signature resembling that of gut M cells while still maintaining their mTEC identity.
Do microfold mTECs, thus defined, depend on the lineage-defining TFs of peripheral M cells, SpiB and Sox8? To address this question, we analyzed mice lacking Spib or Sox8, cross-checking the results between strains to minimize the possibility of confounding effects from germline deletion. scRNA-seq confirmed that microfold mTECs highly expressed the genes encoding these two TFs (Figure 5G). Analysis of microfold mTEC accumulation in SpiB- and Sox8-deficient mice showed a significant loss of microfold mTECs in both strains (Figures 5H and 5I). To test whether this requirement for SpiB and Sox8 was TEC-intrinsic, we performed thymic grafting experiments, wherein we transplanted thymi from newborn Spib−/−, Sox8−/−, or control mice into wild-type (WT) hosts. All grafts were efficiently reconstituted by host thymocytes, and microfold mTECs accumulated from barely detectable in newborn thymi to normal levels in grafted control thymi (Figures 5J, 5K, and S6D–S6F). Grafted Spib−/−and Sox8−/−thymi again had major defects in microfold mTEC accumulation (Figures 5J and 5K). Non-TEC thymic stromal cells expressed negligible levels of Spib or Sox8 (Figure S6G), suggesting this was a TEC-intrinsic effect. We also performed bulk RNA-seq of Pdpn−CD104−mTEClo from Spib−/−, Sox8−/−, and control mice to assess the role of these TFs in PTA expression. Signature analysis demonstrated significant loss of microfold-associated transcripts in both Spib−/− and Sox8−/−mTECs relative to their controls (Figures 5L, 5M, and S6H). This downregulation may reflect cellular loss of microfold mTECs from the mTEC pool, failure to directly induce microfold-associated transcripts, or some combination of the two. Altogether, we concluded that SpiB and Sox8 were necessary for microfold mTEC accumulation and for microfold-associated PTA expression in the thymic epithelium. Given that tuft mTEC accumulation also depends on the tuft-cell lineage-defining TF, Pou2f3 (Bornstein et al., 2018; Miller et al., 2018), we suggest that the requirement for lineage-defining TFs in mimetic cell accumulation is likely a general phenomenon.
Several other points related to microfold mTEC biology were addressed. First, we asked whether microfold mTEC accumulation might also require Aire. Indeed, there was a diminished frequency of microfold mTECs in Aire-deficient mice, albeit not to the same extent as in Spib−/−or Sox8−/−mice (Figure S6I). Bulk RNA-seq of microfold mTECs from Aire−/−versus Aire+/+ thymi showed that loss of Aire did not significantly perturb expression of the microfold signature in microfold mTECs, suggesting that Aire promotes microfold PTA expression indirectly, by enhancing microfold mTEC accumulation, rather than by direct transactivation (Figure S6J).
We also hypothesized that microfold mTECs might have other roles in thymic biology beyond serving as a source of microfold antigens for maturing T cells. SpiB- and Sox8-deficiency had relatively minor impacts on thymocyte subsets but did increase the frequency and number of thymic B cells roughly 2-fold relative to controls (Figures S7A–S7F). It remains to be seen whether these effects stem directly from loss of microfold mTECs or are secondary impacts of SpiB- and Sox8-deficiency.
For a more global picture of the impacts of lineage-defining TFs and Aire on mimetic cells, we performed scRNA-seq on Pdpn−CD104−mTEClo from WT, Spib−/−, Sox8−/−, and Aire−/−mice, hashtagging across genotypes to avoid batch effects (Stoeckius et al., 2018). After quality control, we retained 6,375 cells for analysis, distributed roughly evenly across replicates, with a mean of 4,053 unique fragments per cell (Figure 6A). We could readily re-identify most mimetic cell subtypes discovered in the previous scRNA-seq experiment.
Figure 6. Mimetic cells depend strictly on lineage-defining TFs and variably on Aire.
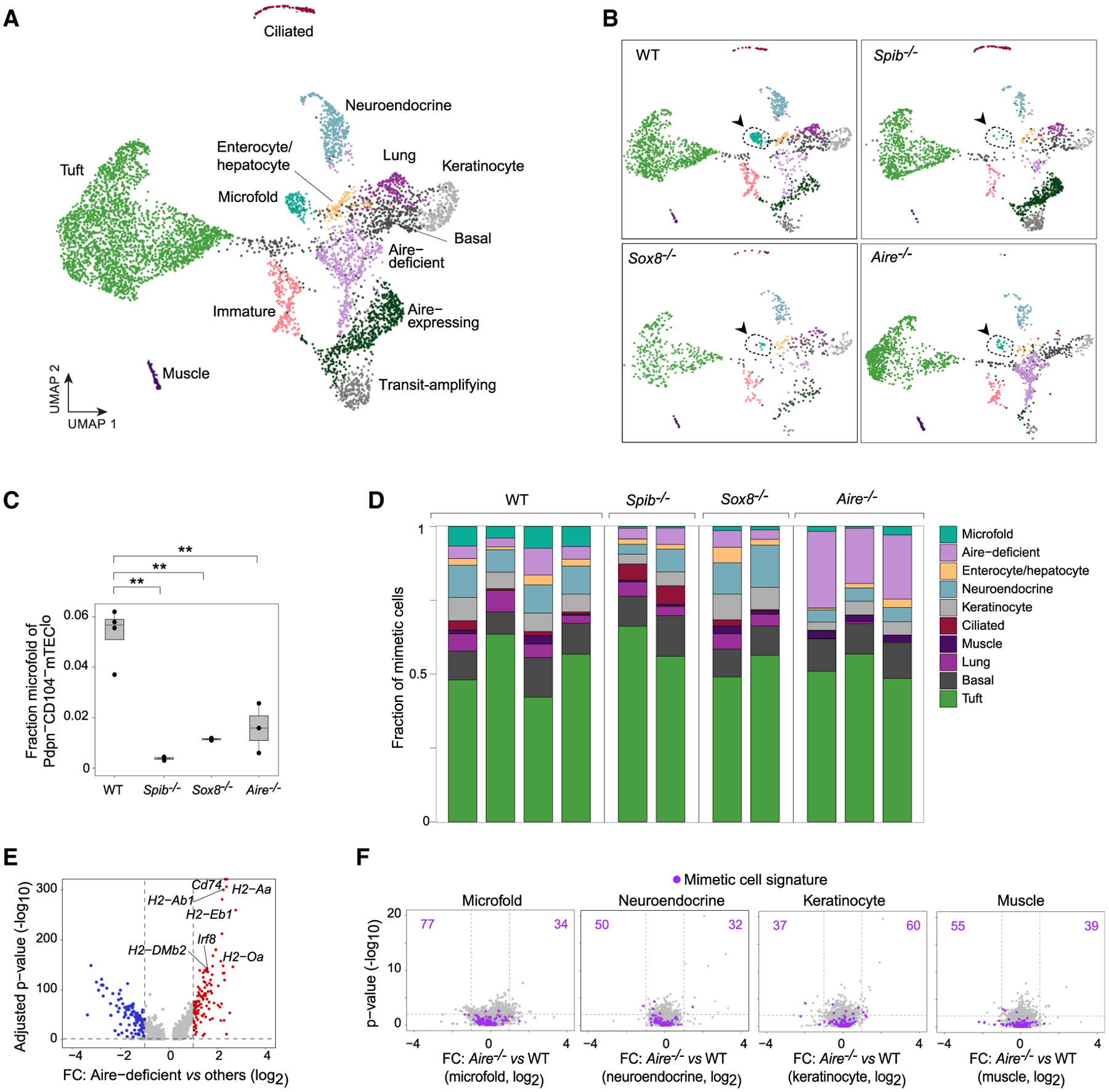
(A) Merged UMAP of scRNA-seq of Pdpn–CD104– mTEClo from WT (n = 4), Spib–/– (n = 2), Sox8–/– (n = 2), and Aire–/– (n = 3) mice.
(B) Data from (A), split by genotype. Arrows and dashed lines indicate the microfold mTEC cluster.
(C) Fraction of microfold mTECs among Pdpn–CD104– mTEClo, assayed by scRNA-seq and split by genotype. Each dot is one mouse, and p values are from two-sided, unpaired Student’s t tests.
(D) Fraction of each mimetic cell subtype among all mimetic cells in each scRNA-seq replicate from the indicated genotypes.
(E) Volcano plot of scRNA-seq of Aire-deficient mTECs versus all other cells. Per-gene p values were BH-corrected.
(F) Volcano plots of scRNA-seq of the indicated mimetic cell subtypes from Aire–/– versus WT mice. For each subtype, the corresponding gene signature is highlighted in purple.
Clear differences in the cell distribution in UMAP space were evident across the genotypes, with near-total ablation of microfold mTECs in Spib−/−and Sox8−/−mice and wholesale shifts in cell distributions in Aire−/−mice (Figure 6B). Focused quantification of microfold mTECs confirmed that SpiB- and Sox8-deficiency significantly diminished the microfold mTEC compartment (Figure 6C). Aire-deficiency also reduced microfold mTECs, albeit more variably, again consistent with our earlier results. The other mimetic cell subtypes were not substantially diminished in either SpiB- or Sox8-deficient mice (Figure 6D), supporting the idea that specific lineage-defining TFs promote the generation of specific mimetic cell subtypes.
In contrast to SpiB- and Sox8-deficient mice, mimetic cells from Aire-deficient mice did not show focused loss of a single subtype but rather accumulation in an Aire-deficient state that grew to nearly 25% of Pdpn−CD104− mTEClo (Figures 6B and 6D). This state resembled the IRF8hi antigen-presenting state uncovered by scATAC-seq (Figure 6E). Aire-deficient mTECs crowded out the accumulation of mimetic cells, with marked, although variable, loss of mimetic cell subtypes, most pronounced for ciliated, lung (including goblet and ionocyte), microfold, and neuroendocrine mTECs. Interestingly, however, when we directly compared the transcriptomes of mimetic cell subtypes from Aire−/− versus WT mice, few transcriptional differences were observed (Figure 6F). Thus, Aire indirectly promoted the accumulation of mimetic cells but was not required for direct transactivation of mimetic-cell PTAs, consistent with our more focused analysis of microfold mTECs.
In summary, microfold mTECs were characterized as prototypical mimetic cells that adopted many characteristics of their extra-thymic counterparts while maintaining their mTEC identity. Lineage-defining TFs were required for mimetic cell accumulation, whereas Aire was partially and variably required.
Expression of a model antigen in mimetic cells suffices to induce T cell tolerance
Finally, we asked whether mimetic cells were physiologically important sources of antigen for negative selection of autoreactive T cells and/or positive selection of Tregs, given the broad implication of mTECs in these processes. We adapted a previously developed system wherein the size of an antigen-specific CD4+ T cell repertoire can be estimated as a function of thymic selection in a fully polyclonal repertoire by expressing a model antigen (here, yellow fluorescent protein [YFP]) in diverse thymic cell compartments, immunizing mice with said antigen, and quantifying cognate T cells using peptide:MHC tetramers (Moon et al., 2007; Malhotra et al., 2016). We drove YFP expression in ciliated, muscle, or total mTECs by crossing mice expressing Cre recombinase under the Foxj1, Ckm, or Foxn1 promoters with Rosa26-LSL-eYFP reporter mice (hereafter referred to as Foxj1YFP, CkmYFP, and Foxn1YFP). Foxn1YFP mice served as positive controls for tolerization, whereas WT mice lacking YFP acted as negative controls.
On average, ciliated mTECs represented 0.15%, muscle mTECs 0.06%, and Foxn1+ mTECs 64.6% of total mTECs (Figure 7A). We verified labeling of the desired mimetic cells by bulk RNA-seq of YFP+ versus YFP− mTECs from Foxj1YFP and CkmYFP mice (Figures 7B and 7C; Table S4). PCA of ciliated and muscle mTECs versus their peripheral equivalents again showed that the major component of variance was organotypic, separating thymic from peripheral epithelia, and a significant minor component consisted of the lineage-specific programs adopted by mimetic cells (Figures 7D and 7E). Immunofluorescence of Foxj1YFP and CkmYFP thymi revealed distinct YFP+ structures within the thymic medulla for each line: in Foxj1YFP mice, YFP expression localized to clusters of polarized, acetylated-tubulin-positive, ciliated cells surrounding cysts (Figure 7F), consistent with old reports of ciliated cysts in the thymus (Remak, 1855), whereas in CkmYFP mice, YFP expression marked individual, scattered, desmin-positive cells (Figure 7G), reminiscent of old descriptions of thymic “myoidzellen” (Mayer, 1888; Van de Velde and Friedman, 1966). Thus, as for microfold mTECs, ciliated and muscle mTECs retained their mTEC identity while layering on cell-type-specific signatures and adopting histological characteristics of their peripheral counterparts.
Figure 7. Expression of a model antigen in mimetic cells suffices to induce T cell tolerance.
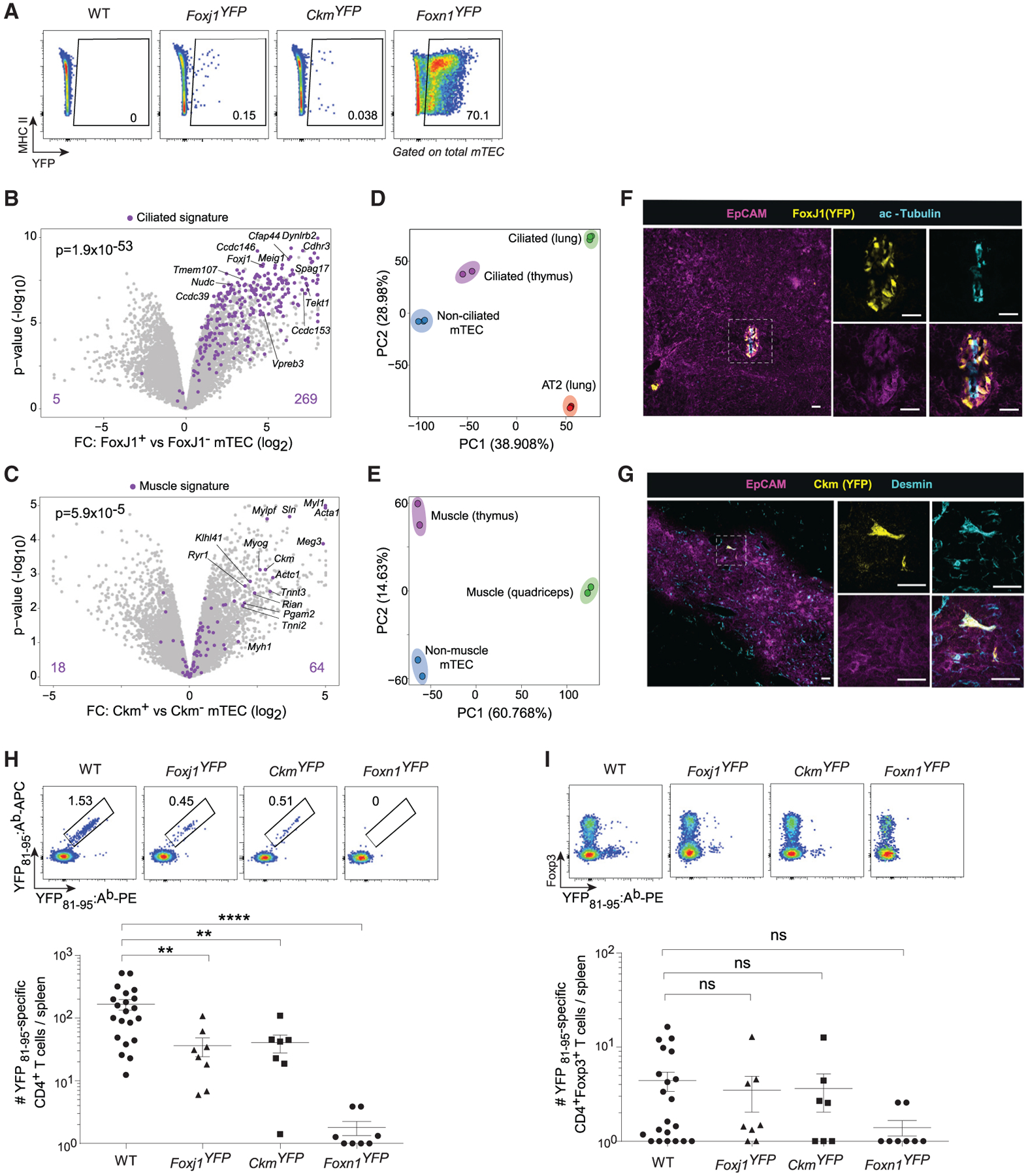
(A) Representative flow plots showing YFP expression in mTECs from the indicated strains.
(B and C) Volcano plots of bulk RNA-seq of purified YFP+ versus YFP– mTECs from (B) Foxj1YFP and (C) CkmYFP mice. Signature p values were calculated by chi-square tests.
(D and E) PCA of bulk RNA-seq of (D) non-ciliated mTEC (n = 3), ciliated mTEC (n = 2), ciliated airway cells (n = 3), and alveolar type 2 (AT2) lung epithelial cells (n = 3), and (E) non-muscle mTEC (n = 2), muscle mTEC (n = 2), and quadriceps skeletal muscle (n = 2). Each dot is one biological replicate.
(F and G) Immunofluorescence of thymic sections from (F) Foxj1YFP and (G) CkmYFP mice, stained for the indicated markers. Scale bars, 30 μm.
(H and I) Representative flow plots (top) and summarized data (bottom) of the number of YFP81–95‒specific CD4+ T cells (H) and CD4+Foxp3+ Tregs (I) elicited in the indicated strains. For summarized data in (H) and (I), each dot is one mouse, data are pooled from 6 independent experiments, bars show mean ± SEM, and p values were calculated by one-way ANOVA with Dunnett’s test on log-transformed cell numbers. Mice with zero antigen-specific T cells were assigned a value of 1 to allow for log-transformation.
See also Table S4.
With the specificity of YFP expression in these lines established, we tested the ability of mimetic cells to mediate T cell tolerance by quantitating YFP-specific peripheral CD4+ T cells in WT, Foxj1YFP, CkmYFP, and Foxn1YFP mice using double-tetramer staining of splenic T cells reactive against a YFP peptide (YFP81–95) (Figure 7H; please note log scale). WT mice, to which YFP is a foreign antigen, possessed a sizable pool of YFP81–95‒ specific CD4+ T cells. Both Foxj1YFP and CkmYFP mice displayed significant reductions in the number of YFP81–95‒specific CD4+ T cells (4-fold fewer in both strains). As expected, Foxn1YFP mice showed near-total deletion of peptide-specific CD4+ T cells (100-fold reduction). Interestingly, and consistent with prior work (Van Santen et al., 2004; Malhotra et al., 2016), normal YFP81–95‒specific Treg numbers were maintained in all but Foxn1YFP mice under these assay conditions (Figure 7I), suggesting that positive selection of Tregs may also play a role in enforcing tolerance to mimetic-cell antigens.
In summary, we genetically labeled two subtypes of mimetic cells, ciliated and muscle mTECs, and found that they had similar general properties to microfold mTECs. Expression of a model antigen in mimetic cells was sufficient to induce T cell tolerance to said antigen.
DISCUSSION
Our study aimed to address the mechanism of PTA expression by mTECs, initially focusing on Aire. However, we unexpectedly discovered an impressive constellation of mTEC subtypes marked by the lineage-defining TFs, chromatin-accessibility landscapes and transcriptional programs of peripheral cell types. These mTEC subtypes, which we term mimetic cells, appeared largely downstream of Aire expression, required lineage-defining TFs for their accumulation and mediated tolerance to a model antigen.
Given the prevailing model of probabilistic, quasi-random expression of PTAs in the thymic epithelium (Derbinski et al., 2008; Villaseñor et al., 2008; Meredith et al., 2015; Brennecke et al., 2015; Anderson and Su, 2016; Abramson and Anderson, 2017; Kadouri et al., 2020; Dhalla et al., 2020; Kaiser et al., 2022), we were surprised to discover numerous mimetic cell subtypes in which PTA expression was organized according to a coherent biological logic. Why had this phenomenon escaped previous attention? In fact, some of the earliest work on mechanisms of PTA expression proposed that certain histologically distinct thymic epithelial cells provided self-antigen for the selection of maturing T cells (Farr and Rudensky, 1998). However, with the discovery of Aire, the focus of the field shifted primarily to Aire-mediated PTA induction, and the early histological work largely fell by the wayside. Only recently, with the development of single-cell genomic technologies, have groups begun to detect some molecular equivalents of these early histological cell types (Miller et al., 2018; Bornstein et al., 2018; Dhalla et al., 2020; Park et al., 2020; Baran-Gale et al., 2020; Bautista et al., 2021). In our study, scATAC-seq was crucial in shifting our mindset about how PTAs might be induced: because we observed discrete mTEC chromatin states corresponding to peripheral cell types, we were able to deduce a mechanism for PTA expression driven by lineage-defining TFs, which then served as an illuminating framework for organizing the diverse mimetic cells we revealed through scRNA-seq.
However, it is incumbent on us to reconcile these results on lineage-defining TFs with the last two decades of work on Aire. scATAC-seq showed that Aire and lineage-defining TFs operated in distinct mTEC subtypes, raising the possibility that PTA expression mediated by Aire and by lineage-defining TFs might be separate processes. However, scRNA-seq revealed that many Aire-induced PTAs were predominantly expressed in mimetic cells and that Aire-deficiency impaired accumulation of several mimetic cell subtypes. One possibility, then, is that Aire can both directly induce PTAs in Aire-stage mTECs by established mechanisms (i.e., Pol II pause release, enhancer-promoter looping) and indirectly promote PTA expression in mimetic cells by enhancing their accumulation. Alternatively, Aire’s actions may be guided even in Aire-stage mTECs by TFs whose fingerprints may not be detectable by the methods employed here. We favor the first hypothesis, which might explain why previous studies of PTA co-expression patterns in mTECs had found significantly smaller co-expression groups and a lack of biological coherence (Pinto et al., 2013; Brennecke et al., 2015; Meredith et al., 2015; Dhalla et al., 2020).
Beyond PTA expression, understanding the specific functions of each of these mimetic cell subtypes will be an exciting area for future inquiry. Certainly, one possibility is that they have no further function: mimetic cells may exist solely to provide antigens for T cell selection. However, given the proposed role of tuft mTECs in controlling innate lymphoid and natural killer T cell subsets (Bornstein et al., 2018; Miller et al., 2018; Lucas et al., 2020) and the potential impact of microfold mTECs on thymic B cells described here, we suspect that at least some mimetic cell subtypes may assume additional functions beyond the provision of PTAs. For instance, tuft mTECs express Il10 and ll25, microfold mTECs express Ccl6, Ccl9, and Ccl20, and other mimetic cells similarly express cytokines, chemokines, and other molecules that may influence the quality of T cell selection, either by direct effects on T cells or by impacts on surrounding APCs.
More generally, the ectopic activity of lineage-defining TFs in the thymic epithelium, outside of their native tissue contexts, is a remarkable phenomenon fertile for future investigation. For example, Aire and many of the lineage-defining TFs highlighted here are also expressed in the early embryo; comparative analyses of embryonic stem cells and mTECs may yield insight into mechanisms controlling or restricting lineage choice. Furthermore, the thymic activity of lineage-defining TFs may serve as an unappreciated mechanism of autoimmune disease risk, as impaired PTA induction by lineage-defining TFs could result in impaired tolerance to specific sets of antigens. Consistent with this hypothesis, genome-wide association studies have implicated several of the lineage-defining TFs identified here as risk loci in autoimmune disease, including HNF4α in inflamma-tory bowel disease (UK IBD Genetics Consortium et al., 2009) and SPIB in primary biliary cirrhosis (Liu et al., 2010). Finally, there may be extra-thymic biology to be learned from thymic mimetic cells, especially for rare subtypes like ionocytes, tuft cells, and microfold cells, for which technical challenges have until recently precluded detailed study. Additional characterizations of mimetic cells, like we provided here for microfold mTECs, are likely to shed light on the biology of the thymus and peripheral tissues alike.
Limitations of the study
Here, we showed that germline deletion of two TFs, SpiB and Sox8, impaired microfold mTEC accumulation, as does Pou2f3 for tuft mTECs per prior reports (Bornstein et al., 2018; Miller et al., 2018). These effects appeared to be TEC-intrinsic, as similar results were obtained with thymic grafts. However, conditional deletion of these TFs in mTECs will be useful to formally rule out systemic or indirect effects of SpiB, Sox8, and Pou2f3 deletion on mimetic cells. Because many of the other TFs highlighted here are lethal and/or redundant when deleted in the germ line, complex transgenic approaches will be required to comprehensively test the necessity of lineage-defining TFs in mimetic cell accumulation. Finally, conditional short-term deletion may prove useful to dissect the precise mechanism by which lineage-defining TFs control mimetic cell accumulation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Diane Mathis (dm@hms.harvard.edu
Materials availability
This study did not generate new unique reagents.
Data and code availability
scATAC-seq, scRNA-seq, bulk RNA-seq and CUT&Tag data have been deposited at the Gene Expression Omnibus (GEO: GSE194253). Aire-lineage-tracing scRNA-seq (GEO: GSE137699), human fetal thymus scRNA-seq (E-MTAB-8581), Aire−/− bulk RNA-seq (GEO: GSE180935), Aire ChIP-seq (GEO: GSE92597) and histone mark ChIP-seq (GEO: GSE114713, GEO: GSE53109), all from mTECs, were reanalyzed from previous work. This study did not generate original code; scripts used for major analyses are available via Github (github.com/dmichelson). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were maintained in accordance with Harvard Medical School’s Animal Care and Use Committee guidelines (IACUC protocol #IS00001257). Strains used were wildtype C57BL/6J (B6; JAX), Aire−/− (our colony), B6/CD45.1 (JAX), Foxn1-cre (JAX), Foxj1-creERT2-eGFP (JAX), Ckm-cre (JAX), Rosa26-LSL-eYFP (JAX), Sox8−/− (O’Bryan et al., 2008), and Spib−/− (Sasaki et al., 2012). Strains were maintained on a B6 background and were crossed as appropriate to generate the desired genotypes. Mice were generally used for experiments between 4–6 weeks of age. Littermates were used for comparisons of WT and knockout mice unless otherwise noted, and both male and female mice were used for flow cytometry and immunofluorescence experiments, after confirming no difference between sexes. Female mice were used for sequencing experiments, except for ciliated-cell RNA-seq, for which male mice were used because of breeding considerations. Age-matched male mice were used in the YFP immunization experiments to permit comparison across multiple strains and to control for sex-specific variation in immunization responses. All mice were housed under specific-pathogen-free conditions.
METHOD DETAILS
Isolation, analysis, and sorting of mTECs
Mice were sacrificed and thymi removed. Individual thymi were finely chopped using scissors, lymphocyte-rich supernatant was removed, and thymic fragments were digested sequentially in 0.5mg/mL collagenase (Sigma) and 0.1mg/mL DNase (Sigma) in Dulbecco’s Modified Eagle Media (DMEM; Gibco) plus 2% fetal calf serum (FCS; Gibco) for 15 minutes, then 0.5mg/mL collagenase/dispase (Roche) and 0.1mg/mL DNase in DMEM/FCS for 15 minutes, as previously described (Bansal et al., 2017). Ethylenediaminetetraacetic acid (EDTA) was added to 10mM. Cells were then spun down and stained with primary antibodies (anti-A/E, -Ly51, -CD45, -EpCAM; all Biolegend) in phosphate-buffered saline (PBS) plus 2% FCS (flow buffer). CD45+ cells were magnetically depleted using anti-PE or -CD45 beads and MACS LS columns (all Miltenyi). 4′,6-diamidino-2-phenylindole (DAPI) was added to exclude dead cells. mTECs were cytofluorometrically analyzed and/or sorted for scATAC-seq, scRNA-seq, CUT&Tag or bulk RNA-seq. Cell analysis was performed using FACSymphony and LSRII instruments (BD). Cell sorting was performed using FACSAria (BD) or MoFlo Astrios EQ (Beckman Coulter) instruments. In some cases, such as anti-IRF8 (Miltenyi) staining, mTECs were labeled with a fixable viability dye (Invitrogen) following surface staining, fixed for 1 hour at 4°C with fixation/permeabilization buffer (eBioscience), and stained intracellularly for 1 hour at 40°C in permeabilization buffer (eBioscience) prior to analysis. Flow cytometry data was analyzed with FlowJo.
scATAC-seq library preparation
Cells were processed following the 10X Genomics scATAC-seq protocol using 10X buffers. Briefly, ~50,000 sorted mTECs were spun down and examined for cell number and viability. Nuclei were isolated in 50ul ice-cold lysis buffer (10mM Tris-HCl [pH 7.4], 10mM NaCl, 3mM MgCl2, 0.1% Tween-20, 0.1% NP-40, 0.01% digitonin, 1% bovine serum albumin) for 5 minutes (first replicate) or 1 minute (second replicate). Nuclei were washed and resuspended in nuclei buffer to target 5,000 recovered nuclei per replicate. Nuclei were transposed, loaded into gel beads-in-emulsion, and incubated for linear amplification. Barcoded single nucleus fragments were purified and amplified by polymerase chain reaction (PCR). All four replicates were pooled for paired-end sequencing on an Illumina NovaSeq at the Harvard Bauer Core Facility.
scATAC-seq preprocessing, visualization, and clustering
Sequenced libraries were demultiplexed, assigned to individual cells, and aligned to the mm10 genome using Cell Ranger ATAC. Each sample was preprocessed to remove low-quality and duplicate reads, then converted into a snap file, a hdf5 file structure for storing and manipulating scATAC-seq data, using the package SnapTools. Most downstream scATAC-seq analyses were performed using the package SnapATAC. Cells were further filtered to keep only cells with at least 103.5 unique fragments and at least 20% of their reads in promoters. Reads were initially binned into 5kb genomic windows for data visualization and clustering, excluding ENCODE blacklisted regions and bins with extremely high or low coverage. We also removed cells that ended up with coverage of fewer than 1,000 bins following filtering. Dimensionality reduction was performed in two steps to account for the sparsity of scATAC-seq data and the computationally large feature set of genomic bins, following a standard approach (Fang et al., 2021). Briefly, the cell-by-bin matrix was first binarized and transformed into a cell-by-cell similarity matrix, with each entry representing the pairwise Jaccard index of bin accessibility between two cells. The similarity matrix was normalized for sequencing depth, then subjected to a second dimensionality reduction step using diffusion maps. The top 20 eigenvectors, which represented most of the variance, were kept for downstream analysis. 2D UMAPs were generated for visualization. Louvain clustering was performed on the k-nearest neighbor graph (k=40). Three small outlying clusters with strong accessibility at the canonical dendritic, T, and B cell marker genes Itgax, Cd3e, and Cd19 were presumed to be contaminating cells and were removed. The ciliated cluster was initially called as part of the secretory/neuroendocrine cluster, but after its distinct nature became clear, we subclustered the ciliated mTECs by subsetting the combined cluster from the larger dataset, performing principal component analysis (PCA) on the cell-by-bin matrix, and separating ciliated mTECs from secretory/neuroendocrine mTECs using k-means clustering (k=2). Differential density UMAPs were generated by estimating the 2D kernel density using the ‘kde2d’ function in the R package MASS for Aire+/+ and Aire−/− samples separately, then subtracting the densities to calculate differential density.
scATAC-seq peak calling
To ensure detection of peaks specific to smaller clusters, peaks were called independently for each cluster, then merged to generate a master peakset. Briefly, reads corresponding to the cells belonging to each cluster were extracted from snap files, then used as inputs for peak calling with macs2 using the parameters: –nomodel –shift 100 –ext 200 –qval 0.05 -B –SPMR. Depth-normalized pileups from macs2 were used for genome-browser visualization in IGV. For downstream analyses requiring cell-by-peak matrix inputs (i.e., single-cell motif analysis), we merged cluster-specific peaks into a combined peakset and computed a cell-by-peak matrix from the aligned reads. To define high-confidence cluster-specific OCRs, we called peaks for each cluster for each replicate, used the irreproducible discovery rate (IDR) framework (Qunhua et al., 2011) to identify peaks with good concordance between replicates (global IDR<0.05), and filtered for peaks unique to each cluster using bedtools. This method yielded a reasonably sized set of peaks for all clusters except the transit-amplifying cluster (due to its heterogeneity and/or intrinsic biology) and the ciliated cluster (due to its small size). These high-confidence peaks were used for chromatin accessibility heatmap and CUT&Tag binding analyses. We also defined pan-mTEC OCRs by retaining IDR-replicated peaks found in all clusters, excluding those overlapping non-specific IgG CUT&Tag peaks.
scATAC-seq motif and genomic-feature analysis
We analyzed transcription factor motifs in scATAC-seq data in three ways. First, we looked for motif enrichment on a pseudobulk per-cluster basis. Second, we looked for single-cell motif enrichment. Third, we performed TF footprinting in cluster-specific peaks. For the first approach, we used cluster-specific peaks as input to the HOMER routine ‘findMotifsGenome’, with the parameters: motif length 10, scan size 300, automatic background, and only known motifs. For the second approach, we used chromVAR, which calculates the relative enrichment or depletion of peaks containing a particular motif within each single cell as compared with a GC-matched set of background peaks. We used the combined JASPAR core motifs for Mus musculus and Homo sapiens as reference motifs, with preference for mouse motifs when available. Peaks found in at least 10 cells were used as input for motif analysis. Single-cell motif deviations were used for UMAP visualization. To examine differential accessibility of genomic features (e.g., histone marks, Aire peaks, Aire-induced genes, Aire-neutral genes) in single cells, a similar approach was employed, using peaks overlapping said genomic features as the input to chromVAR. For the third approach, we used the program HINT-ATAC to find and plot TF footprints in the scATAC-seq data (Li et al., 2019). Briefly, HINT-ATAC normalizes the ATAC-seq signal for sequencing depth and Tn5 insertion bias, uses a hidden Markov model to detect TF footprints in a defined peakset, and aggregates signal for each TF. To normalize to a common background and facilitate multi-way comparison, we further measured TF footprints in a random sample of 1,000 mTECs and subtracted this signal from each cluster’s aggregate signal. In general, we refer to motifs at the family level due to motif redundancy among individual family members.
Immunofluorescence of thymic sections
Mice were sacrificed and dissected for their thymi, which were fixed with 4% paraformaldehyde for 1 hour and progressively dehydrated in 5%, 15%, and 30% sucrose in PBS. Thymi were embedded in blocks, flash frozen, sectioned at 8μm and stored at −80°C until use. For staining, thymic sections were rinsed and permeabilized in PBS plus 0.05% Tween-20 (PBS-T), blocked for 30 minutes in PBS-T plus 5% donkey serum, and incubated with primary antibodies for either 1 hour at room temperature or overnight at 4°C. The primary antibodies used were anti-EpCAM (Biolegend), -FoxA2 (Cell Signaling), -Hnf4α (Abcam), -FoxJ1 (Novus), -GP2 (MBL), -Grhl1 (Novus), -Sox8 (Proteintech), -GFP (Abcam, Aves Labs), -Slug/Snai2 (Cell Signaling), -Pou2f3 (Sigma), -Villin (Abcam), -Desmin (Cell Signaling), and -acetylated-Tubulin (Sigma). Thymic sections were washed in PBS-T and incubated for 1 hour at room temperature in the presence of FITC-, Cy3-, Alexa Fluor 647-, or Cy5-labeled secondary antibodies against rat, mouse, rabbit, or chicken IgG as appropriate, all produced in donkey (Jackson ImmunoResearch). Thymic sections were washed, counterstained with Hoescht 33342 (Sigma), mounted in ProLong Diamond mountant (Invitrogen), and imaged by widefield microscopy using a Nikon Ti inverted microscope, Andor Zyla 4.2 Plus sCMOS camera, Plan Apo 20X air objective, and Nikon Elements acquisition software; or by spinning-disk confocal microscopy across multiple Z-planes using a Nikon Ti inverted microscope, W1 Yokogawa spinning disk with 50μm pinholes and Plan Apo 20X air, 60X oil and 100X oil objectives. Images were analyzed in ImageJ. Confocal images with multiple Z-planes were processed as maximum intensity projections. All images shown are representative of at least two independent experiments.
Bulk RNA-seq library preparation
Cytofluorometrically purified cell populations were directly sorted into 5ul TCL buffer (Qiagen) supplemented with 1% 2-mercaptoethanol (Sigma) for cell lysis. 250–1000 cells were typically used for each replicate, double-sorting for purity when cell numbers permitted. Samples were subjected to Smart-seq2 RNA-seq library preparation and sequencing by the Broad Genomics Platform, following the standard ImmGen ultra-low-input RNA-seq protocol (immgen.org).
Bulk RNA-seq preprocessing and analysis
Reads were aligned to the mm10 genome by STAR and counts quantified using featureCounts (Subread). Samples with fewer than 8,000 genes with more than ten reads, high contamination by hematopoietic-cell-specific transcripts, median transcript integrity number for housekeeping genes below 45, or poor intra-replicate correlation were excluded from downstream analyses. For PCA, calculation of inter-sample Pearson correlations, and analyses of the expression of individual genes, we used DESeq2-normalized expression values (median of ratios method, the default ImmGen approach), after removing lowly expressed genes. Transcriptome-wide differential expression analyses were performed using edgeR, excluding lowly expressed genes (‘filterByExpr’), normalizing libraries by the trimmed mean of M-values method (‘calcNormFactors’), and testing for differential expression using the quasi-likelihood F-test method (‘glmQLFTest’). For some volcano and FC/FC plots, off-scale points were collapsed to the nearest axis. For published datasets, count matrices were downloaded directly and used for differential expression analyses.
scRNA-seq library preparation
For analysis of adult and perinatal Pdpn–CD104– mTEClo, we dissected thymi from 3 adult mice (6 weeks old) and 3 perinatal mice (5 days old), hashed each sample using TotalSeq-A anti-biotin hashtags (Biolegend) against biotinylated antibodies targeting CD29 and H2-Kb, isolated mTECs, and submitted purified cells to the Broad Institute Genomics Platform, which performed encapsulation and RNA and hash library preparation following 10X Genomics protocols. For analysis of WT, Spib−/−, Sox8−/−, and Aire−/− Pdpn–CD104– mTEClo, we followed the same procedure but hashed with only with H2-Kb, using commercial TotalSeq-A anti-mouse hashtags (Biolegend).
scRNA-seq preprocessing, visualization and clustering
Sequenced reads were demultiplexed, aligned, assigned to cells, and output as transcript-by-cell matrices using Cell Ranger. Hash-by-cell matrices were computed using CITE-seq-count. Mimetic cell scRNA-seq analysis was largely performed using the Seurat package. For the adult vs perinate experiment, hash counts were normalized by the centered log ratio method, and single cells were assigned hash identities by high expression of a single hash. Cells lacking hashes, cells with multiple hashes, and cells with high mitochondrial reads were removed. Gene-expression matrices were log-normalized as the natural log1p of counts per 10,000 counts (log CP10K), the top 2000 variable genes were selected by the variance-stabilizing transform method, data were scaled and centered, and PCA was performed on variable genes. The top 40 PCs were retained for shared-nearest-neighbor graph construction (k=20) and 2D UMAP visualization on the basis of jackstraw and elbow plots. Several small clusters expressing canonical T cell, B cell, myeloid, fibroblast, or endothelial markers were assumed to be contaminants and removed—and the remaining data re-normalized—prior to further analysis. Cell clustering was first performed using the Louvain method (resolution=1.8). To highlight some substructure within the data (i.e., Ptf1a+ pancreatic mTEC), we subset mimetic cells and subclustered as above, then performed supervised integration of subclusters into the larger dataset.
scRNA-seq analysis
Module scores for peripheral-cell-type signatures were calculated using the Seurat function ‘AddModuleScore’, which computes enrichment of module expression against a set of expression-matched control genes. Cluster-based differential expression was performed using the Seurat function ‘FindMarkers’ with the likelihood-ratio test for single-cell gene expression method. Mimetic-cell gene signatures were derived from differentially expressed genes by filtering for genes unique to each cluster with fold-change > 2, minimum percent expression ≥ 10%, and adjusted p-value < 0.01. For some volcano plots, off-scale points were collapsed to the nearest axis. The differential density UMAP was computed using the same procedure as for scATAC-seq to compare adult vs perinatal mTEC densities. For RNA velocity analyses, briefly, fastq files were reprocessed using kallisto-bustools to map spliced and un-spliced transcripts, then renormalized using the package scVelo. Only cells included in the prior analyses were kept, and cluster assignments were made by direct carryover from Seurat. Cells of interest were further subsetted, RNA velocities were calculated using the dynamical mode, and the resultant predicted cell trajectories were plotted on a 2D diffusion map. For analysis of thymic stromal Spib and Sox8 expression, data were reprocessed as above, but the thymic fibroblast and endothelial clusters were retained for analysis. For scRNA-seq of WT, Spib−/−, Sox8−/−, and Aire−/− mTECs, we largely followed the same procedure for preprocessing and assigned cluster identities based on correspondence to the adult vs neonate experiment.
Reanalysis of published scRNA-seq datasets
We reanalyzed two published scRNA-seq datasets, one from Aire-lineage-traced mTECs to assay mimetic cell enrichment downstream of Aire expression (Wells et al., 2020) and one from human fetal thymus to search for mimetic cells in humans (Park et al., 2020). For Aire-lineage-traced mTECs, we downloaded count matrices and preprocessed as above, retaining 20 PCs for low-dimensional analysis. Mimetic cells were identified empirically by high expression of the mimetic-cell gene signatures derived from our scRNA-seq data, and co-expression with the Aire-lineage-tracing marker zsGreen was assessed for each mimetic cell subtype. For human fetal thymus, we downloaded the processed scRNA-seq data corresponding to thymic epithelium and overlaid orthologue-converted mimetic-cell gene signatures from our scRNA-seq data onto the human dataset. Cell clusters with elevated signature-specific expression were assessed to correspond to mimetic cell subtypes.
CUT&Tag library preparation
CUT&Tag libraries were prepared as described elsewhere (Kaya-Okur et al., 2019), with some modifications. Briefly, 50,000–100,000 mTECs were sorted cytofluorometrically, washed in wash buffer (20mM HEPES pH 7.5, 150mM NaCl, 0.5mM spermidine [Sigma]), bound to concavalin A beads (Bangs Laboratories), permeabilized in wash buffer plus 0.05% digitonin (Sigma), and incubated overnight with 1:50 primary antibody (anti-H3K27ac [Abcam], -Hnf4α [Abcam], -Grhl1 [Novus], -Pou2f3 [Sigma], or -IgG [Rockland]) at 4°C in wash buffer plus 0.05% digitonin, 2mM EDTA and 0.1% bovine serum albumin (BSA). The next day, H3K27ac, Grhl1, IgG, and some Pou2f3 samples were lightly fixed with 0.1% paraformaldehyde at room temperature for 2 minutes before proceeding. (We and others have observed improved CUT&Tag signal-to-noise ratio for some TFs with light fixation.) Samples were incubated with secondary antibody (guinea pig anti-rabbit IgG [Rockland] for H3K27ac, Grhl1, Pou2f3, and IgG; rabbit anti-mouse IgG [Abcam] for Hnf4α) at 1:100 for 1 hour at room temperature in wash buffer plus 0.05% digitonin. Samples were washed and incubated with pA-Tn5 (Addgene #124601, purified in-house) at 1:200 for 1 hour at room temperature in 300mM NaCl wash buffer plus 0.01% digitonin. Samples were washed twice then tagmented for 1 hour at 37°C in 300mM NaCl wash buffer plus 0.01% digitonin and 10mM MgCl2. Tagmentation was halted with EDTA, sodium dodecyl sulfate and proteinase K for 1 hour at 55°C. Tagmented DNA was phenol-chloroform extracted and amplified by PCR with NEBNext 2X Master Mix (NEB) using indexed primers (Buenrostro et al., 2015) and the following program: 72°C for 2min, 98°C for 30s, 16 cycles of 98°C for 10s and 63°C for 10s, 72°C for 1min, hold. Amplified libraries were quantified by Qubit (Thermo) and Tapestation (Agilent), pooled, and paired-end sequenced on an Illumina MiSeq, NextSeq, or NovaSeq at the Harvard Biopolymers Core Facility.
CUT&Tag preprocessing and analysis
Fastq files were trimmed for adapters and low-quality reads by Trimmomatic and mapped to the mm10 genome by bowtie2 using the parameters: –local –very-sensitive -no-mixed –no-discordant -I 10 -X 700. Reads were simultaneously mapped to the E. coli genome to quantify spike-in reads carried over from the pA-Tn5. Bam files were generated and multimapping reads removed by SAMtools, and duplicate reads were removed by picard. Pileups were generated as bigwig files using deeptools with counts per million (CPM) normalization and visualized in IGV. Note that CPM values, while useful for internal signal comparison within each sample, depend on total read number and should not be used to compare signal between samples, as samples with very few reads (i.e., IgG) will have inflated CPM values. Profile plots and heatmaps at cluster-specific OCRs were generated using the deeptools functions ‘compute-Matrix’ and ‘plotHeatmap’. To call peaks in CUT&Tag data, we used SEACR, a peak caller specifically designed for sparse CUT&RUN/CUT&Tag data, following a standard procedure (Meers et al., 2019). We retained only the top 1% of peaks ranked by signal over global background called under “stringent” mode on the merged data for each factor. We found that merging replicates prior to peak calling was essential for sensitive and accurate detection of peaks, as peak calling on individual replicates resulted in a superlinear decline in the fraction of consensus peaks detected, likely due to the sparsity of TF CUT&Tag data (see Table S3). Enrichment of different genomic elements in peaksets was calculated in HOMER using the function ‘annotatePeaks’. Cluster-specific OCRs were stratified as overlapping or not overlapping different peaksets using the bedtools function ‘intersect’. Motif-enrichment analysis was performed in HOMER using the same procedure as for scATAC-seq.
Whole-mount immunofluorescence of thymi
Whole mount was performed following the iDISCO method (Renier et al., 2014), with some modifications. Mice were sacrificed, and thymi were removed and fixed with 4% paraformaldehyde for 1 hour. Thymi were sequentially dehydrated in methanol, bleached for 1 hour in 6% hydrogen peroxide at 4°C, and rehydrated in PBS. Thymi were then permeabilized and blocked in PBS-GT (PBS, 0.2% gelatin, 0.5% Triton X-100) supplemented with 1:100 Fc block and 1% donkey serum for 2 days at 37°C with shaking. Samples were incubated with rat anti-GP2 (1:1000) in PBS-GT for 3 days at 37°C with shaking, extensively washed, and incubated with Cy5 donkey anti-rat (1:1000) in PBS-GT for 3 days at 37°C with shaking. Samples were again extensively washed, then dehydrated with methanol, delipidated in dichloromethane, cleared in 1:2 benzyl alcohol:benzyl benzoate, and stored at 4°C until imaging. Full-thickness imaging of thymi was performed using the spinning-disk confocal microscope previously described and a Plan Apo 10X air objective. Post-processing, including 2D visualization and 3D rendering, was performed in arivis Vision4D.
Comparison of gut and thymic epithelial cells
Enterocytes and microfold cells were isolated from duodenal and ileal Peyer’s patches (PPs). Mice were sacrificed, small intestines removed and flushed, and PPs isolated. PPs were washed several times in PBS and several times in PBS plus 20mM EDTA, then were incubated for 75 minutes on ice in PBS/EDTA. PPs were then shaken extremely vigorously and this first epithelial fraction discarded. Next, we performed sequential 5-minute incubations in PBS/EDTA followed by vigorous shaking and retention of liberated epithelial cells, up to 5–6 times. These cells were pooled, trypsinized for 1 minute at 37°C in 0.05% trypsin (Gibco), then stained with anti-CD45, -CD31, -EpCAM (all Biolegend), -GP2 (MBL), and NKM16-2-4 (Miltenyi) on ice in flow buffer for 30 minutes. Microfold cells were sorted as live CD45– CD31– EpCAM+ GP2+ NKM16-2-4+; enterocytes as live CD45− CD31− EpCAM+ GP2− NKM16-2-4−. Microfold mTECs were prepared as detailed above for mTECs and sorted as live CD45lo EpCAM+ Ly51− GP2+ CD45neg.
Thymic grafts
Thymic grafts were performed following established protocols (Anderson et al., 2002; Miller et al., 2018). Briefly, CD45.2+ donor thymi of the appropriate genotypes were isolated from newborn (postnatal day 0) mice. Adult, sex-matched, CD45.1+ congenic recipient mice were anesthetized with ketamine (7 mg/kg) and xylazine (1.4 mg/kg), and the left kidney was exposed by laparotomy. Individual donor thymic lobes were carefully placed under the capsule of the recipient kidney, and the incision was closed. Recipient mice received buprenorphine (75 μg/kg) twice daily for two days following surgery. Grafts were allowed to reconstitute for 4 weeks, then graft reconstitution efficiency and microfold mTEC accumulation were assessed by flow cytometry.
Analysis of thymic hematopoietic compartments
Thymi were isolated and mashed over 70μm filters. A small fraction of the thymus was stained on ice for 30 minutes for T cells (anti-CD45, -CD8α, -TCRβ, -CD25, -CD73 [all Biolegend], and -CD4 [eBioscience]) or B cells and dendritic cells (anti-CD45, -CD19, -B220, -A/E, -CD8α, -CD44 [all Biolegend], and -CD11c [eBioscience]). Cells were stained with fixable viability dye (Invitrogen), fixed for 1 hour at 4°C using fixation/permeabilization buffer (eBioscience), and T cells were further stained for 1 hour at room temperature with anti-Foxp3 in permeabilization buffer (both eBioscience). Cells were analyzed by flow cytometry, normalizing cell counts using counting beads (Invitrogen).
Comparison of lung and thymic epithelial cells
Lung epithelial cells were isolated following a published protocol (Slyper et al., 2020), with some modifications. To acutely label ciliated cells, adult Foxj1-creERT2-eGFP R26-LSL-eYFP mice were injected intraperitoneally (ip) with 75mg/kg tamoxifen in peanut oil (both Sigma) every two days for six days, then sacrificed on the seventh day. Mice were perfused through the right ventricle with 15mL ice cold PBS; then both lungs, including the tracheal tree, were removed from the thorax. Lungs were extensively chopped, then incubated for 20 minutes at 37°C with agitation in DMEM plus 2% FCS, 0.5mg/mL Liberase TM (Roche), 0.5mg/mL elastase (Worthington Biochem), and 0.5mg/mL DNase. Digested fragments were passed through a 70μm filter and incubated for 1 minute in ACK lysis buffer (Lonza) to lyse red blood cells. Lungs were then stained on ice for 30 minutes in flow buffer with anti-CD45, -CD31, -EpCAM, and -A/E (all Biolegend). Ciliated cells were sorted as live CD45– CD31– EpCAM+ A/E− Foxj1-YFP+; alveolar type 2 cells were sorted for comparison as live CD45− CD31− EpCAM+ A/E+, which were all YFP−. Ciliated mTECs were prepared as detailed above for mTECs and sorted as live CD45− EpCAM+ Ly51− Foxj1-YFP+, pooling 2–3 mice per replicate to obtain adequate cell numbers for RNA-seq.
Comparison of skeletal muscle and thymic epithelial cells
Muscle RNA was prepared by isolating quadriceps from healthy mice, flash freezing the tissue in liquid nitrogen, and homogenizing the frozen tissue in TRIzol (Invitrogen). RNA was phenol-chloroform extracted and quantified on a Nanodrop (ThermoFisher), then diluted into TCL lysis buffer plus 1% 2-mercaptoethanol for bulk RNA-seq. Thymic muscle mTECs were prepared as detailed above for mTECs and sorted as live CD45− EpCAM+ Ly51− Ckm-YFP+, pooling 5 mice per replicate to obtain adequate cell numbers for RNA-seq.
Quantification of YFP-specific T cells
CD4+ T cells reactive against the peptide YFP81–95 (HDFFKSAMPEGYVQE; Genscript; note that this sequence is perfectly conserved between GFP and YFP) on the Ab background were quantified in WT, Foxj1-creERT2-eGFP R26-LSL-eYFP, Ckm-cre R26-LSL-eYFP, and Foxn1-cre R26-LSL-eYFP mice using tetramer reagents, adapting methods described previously (Malhotra et al., 2016; Moon et al., 2009; Dolton et al., 2015). Briefly, mice were ip-immunized with 50μg YFP81–95 emulsified 1:1 in PBS/complete Freund’s adjuvant (Sigma). Seven days after immunization, mice were sacrificed and their spleens taken for analysis. Whole spleens were doubly stained with 10nM APC- and PE-labeled YFP81–95:Ab tetramers (NIH Tetramer Core) for 1 hour at room temperature in DMEM plus 2% FCS and 50nM dasatinib (LC Laboratories). Tetramer-labeled cells were then incubated with anti-PE microbeads and anti-APC antibody (Biolegend) for 15 minutes on ice, then magnetically enriched using MACS LS columns. The tetramer-enriched cell fraction was stained for 30 minutes on ice in flow buffer with surface antibodies (anti-CD45, -B220, -CD11b, -CD3ε, -CD44 [all Biolegend], and -CD4 [eBioscience]), fixed for 1 hour at 4°C in fixation/permeabilization buffer, and stained for 1 hour at room temperature with anti-Foxp3 in permeabilization buffer. The number of YFP81–95‒ -specific CD4+ T cells (gated as live CD45+ B220− CD11b− CD3ε+ CD4+ Tetramer-PE+ Tetramer-APC+) per spleen was quantified by flow cytometry, normalizing cell counts using counting beads. Most Foxj1YFP mice were ip-injected with 75mg/kg tamoxifen (Sigma) once weekly from ages 2–5 weeks to induce YFP expression prior to immunization. We confirmed that CD4− T cells did not bind the tetramers, tamoxifen did not impact the anti-YFP81–95 response, and R26-LSL-eYFP (no Cre) mice did not differ in their responses from WT mice.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cluster-level TF-motif-enrichment p-values were calculated in HOMER by binomial test, p-values for scRNA-seq differential expression were calculated in Seurat using the likelihood ratio test for single-cell gene expression, p-values for bulk RNA-seq differential expression were calculated in edgeR using the quasi-likelihood F-test, p-values for enrichment of gene signatures in bulk RNA-seq data were calculated by one-way chi-square test, and p-values for flow cytometry data were calculated by unpaired, two-sided Student’s t-test, unless otherwise noted in the figure legends. p-values were adjusted for multiple comparisons by the Benjamini-Hochberg (BH) or Bonferroni methods where indicated in the figure legends. Boxplots show median and interquartile range (IQR) as boxes and minimum and maximum values (up to ±1.5*IQR from hinge) as whiskers. Sample sizes and other statistical tests are noted in the figure legends. p=*, <0.05; **, <0.01; ***, <0.001; ****, <0.0001. Statistical analyses were performed using R or GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-A/E (clone M5/114.15.2), Pacific Blue, APC, BV605 | Biolegend | Cat# 107620 |
| Cat# 107614 | ||
| Cat# 107639 | ||
| Rat monoclonal anti-Ly51 (clone 6C3), PE, Alexa Fluor 647 | Biolegend | Cat# 108308 |
| Cat# 108312 | ||
| Rat monoclonal anti-CD45 (clone 30-F11), Pacific Blue, Alexa Fluor 647, BV605, PE-Cy7 | Biolegend | Cat# 103126 |
| Cat# 103124 | ||
| Cat# 103155 | ||
| Cat# 103114 | ||
| Rat monoclonal anti-EpCAM (clone G8.8), biotinylated, PE, APC, APC-Cy7 | Biolegend | Cat# 118204 |
| Cat# 118206 | ||
| Cat# 118214 | ||
| Cat# 118218 | ||
| Human monoclonal anti-IRF8, PE | Miltenyi | Cat# 130-122-971 |
| Rabbit monoclonal anti-FoxA2 (clone D56D6) | Cell Signaling | Cat# 8186T |
| Mouse monoclonal anti-Hnf4a (clone K9218; CUT&Tag) | Abcam | Cat# ab41898 |
| Rabbit monoclonal anti-Hnf4a (clone EPR16885; IF) | Abcam | Cat# ab181604 |
| Rabbit polyclonal anti-FoxJ1 | Novus | Cat# NBP1-87928 |
| Rat monoclonal anti-GP2 (clone 2F11-C3), Alexa Fluor 488 | MBL | Cat# D278-A48 |
| Rabbit polyclonal anti-Grhl1 | Novus | Cat# NBP1-81321 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat# ab4729 |
| Rabbit polyclonal anti-Sox8 | Proteintech | Cat# 20627-1-AP |
| Rabbit polyclonal anti-GFP | Abcam | Cat# ab290 |
| Chicken polyclonal anti-GFP | Aves Labs | Cat# GFP-1010 |
| Rabbit monoclonal anti-Slug (clone C19G7) | Cell Signaling | Cat# 9585 |
| Rabbit polyclonal anti-Pou2f3 | Sigma-Aldrich | Cat# HPA019562 |
| Rabbit monoclonal anti-Villin (clone SP145) | Abcam | Cat# ab130751 |
| Rabbit monoclonal anti-Desmin (clone D93F5) | Cell Signaling | Cat# 5332 |
| Mouse monoclonal anti-Acetylated-Tubulin (clone 6–11B-1) | Sigma-Aldrich | Cat# T7451 |
| Donkey polyclonal anti-rat IgG, FITC, Cy3, Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 712-095-153 |
| Cat# 712-166-153 | ||
| Cat# 712-606-153 | ||
| Donkey polyclonal anti-mouse IgG, Cy5 | Jackson ImmunoResearch | Cat #715-175-150 |
| Donkey polyclonal anti-rabbit IgG, FITC, Cy3, Alexa Fluor 647 | Jackson ImmunoResearch | Cat# 711-096-152 |
| Cat# 711-165-152 | ||
| Cat# 711-606-152 | ||
| Donkey polyclonal anti-chicken IgY, FITC | Jackson ImmunoResearch | Cat# 703-095-155 |
| Armenian hamster monoclonal anti-CD29 (clone HMB1-1), biotinylated | Biolegend | Cat# 102203 |
| Rat monoclonal anti-H-2 (clone AF6-88.5), biotinylated | Biolegend | Cat# 116504 |
| Guinea pig polyclonal anti-rabbit IgG | Rockland | Cat# 611-201-122 |
| Rabbit polyclonal anti-mouse IgG | Abcam | Cat# ab46540 |
| Rat monoclonal anti-microfold (clone NKM16-2-4), PE | Miltenyi | Cat#130-102-150 |
| Rat monoclonal anti-CD31 (clone 390), Alexa Fluor 647 | Biolegend | Cat# 102416 |
| Mouse monoclonal anti-CD45.1 (clone A20), PE-Cy7 | Biolegend | Cat# 3110730 |
| Mouse monoclonal anti-CD45.2 (clone 104), APC-Cy7 | Biolegend | Cat# 109824 |
| Rat monoclonal anti-CD3e (clone KT3.1.1), FITC | Biolegend | Cat# 155604 |
| Rat monoclonal anti-CD4 (clone RM4–5), PE-eFluor 610 | eBioscience | Cat# 61-0042-82 |
| Rat monoclonal anti-CD8a (clone 53–6.7), FITC | Biolegend | Cat# 100706 |
| Rat monoclonal anti-TCRb (clone H57-597), PE-Cy7 | Biolegend | Cat# 109222 |
| Rat monoclonal anti-CD25 (clone 3C7), APC-Cy7 | Biolegend | Cat# 101918 |
| Rat monoclonal anti-CD73 (clone TY/11.8), PE | Biolegend | Cat# 127206 |
| Rat monoclonal anti-Foxp3 (clone FJK-16s), APC, PE-Cy7 | eBioscience | Cat# 17-5773-82 |
| Cat# 25-5773-82 | ||
| Armenian hamster monoclonal anti-CD11c (clone N418), PE-eFluor 610 | eBioscience | Cat# 61-0114-82 |
| Rat monoclonal anti-CD11b (clone M1/70), PerCP-Cy5.5 | Biolegend | Cat# 101228 |
| Rat monoclonal anti-CD19 (clone 6D5), PE | Biolegend | Cat# 152408 |
| Rat monoclonal anti-CD45R/B220 (clone RA3-6B2), FITC, PerCP-Cy5.5 | Biolegend | Cat# 103206 |
| Cat# 103236 | ||
| Rat monoclonal anti-CD44 (clone IM7), APC-Cy7 | Biolegend | Cat# 103028 |
| Mouse monoclonal anti-APC (clone APC003) | Biolegend | Cat# 408004 |
| TotalSeq-A anti-biotin hashtags | Biolegend | N/A |
| TotalSeq-A anti-mouse hashtags | Biolegend | Cat# 155801 |
| Cat# 155803 | ||
| Cat# 155805 | ||
| Cat# 155807 | ||
| Cat# 155809 | ||
| Cat# 155811 | ||
| Cat# 155813 | ||
| Cat# 155815 | ||
| Cat# 155817 | ||
| Cat# 155819 | ||
| Cat# 155821 | ||
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase, from Clostridium histolyticum | Sigma-Aldrich | Cat# C6885 |
| Collagenase/Dispase | Roche | Cat# 10269638001 |
| Deoxyribonuclease I, from bovine pancreas | Sigma-Aldrich | Cat# D4527 |
| LIVE/DEAD Fixable Yellow | Invitrogen | Cat# L34959 |
| Hoescht 33342 | Sigma-Aldrich | Cat# B2261 |
| ProLong Diamond Antifade Mountant | Invitrogen | Cat# P36965 |
| Digitonin | Sigma-Aldrich | Cat# D141 |
| Spermidine | Sigma-Aldrich | Cat# S2501 |
| Concavalin A beads | Bangs Labs | Cat# BP531 |
| TCL RNA lysis buffer | Qiagen | Cat# 1031576 |
| Liberase TM | Roche | Cat# 5401119001 |
| Elastase, from porcine pancreas | Worthington Biochem | Cat# LS002292 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| TRIzol | Invitrogen | Cat# 15596026 |
| Complete Freund’s adjuvant | Sigma-Aldrich | Cat# F5881 |
| YFP81–95 peptide (HDFFKSAMPEGYVQE) | Genscript | N/A |
| Dasatinib | LC Laboratories | Cat# D-3307 |
| PE- and APC-labeled YFP81–95:Ab tetramers | NIH Tetramer Core | N/A |
| Critical commercial assays | ||
| Anti-CD45 microbeads | Miltenyi | Cat# 130-052-301 |
| Anti-PE microbeads | Miltenyi | Cat# 130-048-801 |
| MACS LS columns | Miltenyi | Cat# 130-042-401 |
| Intracellular fixation/permeabilization buffer | eBioscience | Cat# 88-8824-00 |
| Permeabilization buffer (10X) | eBioscience | Cat# 00-8333-56 |
| Chromium Single Cell ATAC | 10X Genomics | Cat# 1000176 |
| Chromium Single Cell Gene Expression 3’ v3 | 10X Genomics | Cat# 1000269 |
| NEBNext 2X Master Mix | NEB | Cat# M0541 |
| 123count eBeads | Invitrogen | Cat# 01-1234-42 |
| Deposited data | ||
| Raw and analyzed scATAC-seq | This paper | GEO: GSE194233 |
| Raw and analyzed scRNA-seq | This paper | GEO: GSE194252 |
| Raw and analyzed bulk RNA-seq | This paper | GEO: GSE194232 |
| Raw and analyzed CUT&Tag | This paper | GEO: GSE194231 |
| Histone mark ChIP-seq | (Handel et al., 2018; Sansom et al., 2014) | GEO: GSE114713, GEO: GSE53109 |
| Human fetal scRNA-seq | (Park et al., 2020) | E-MTAB-8581 |
| Aire−/− RNA-seq | (Bansal et al., 2021) | GEO: GSE180935 |
| Aire ChIP-seq | (Bansal et al., 2017) | GEO: GSE92597 |
| Aire lineage-tracing scRNA-seq | (Wells et al., 2020) | GEO: GSE137699 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX #000664 |
| Mouse: B6.Aire | Our colony | (Anderson et al., 2002) |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | JAX #002014 |
| Mouse: B6(Cg)-Foxn1tm3(cre)Nrm/J | The Jackson Laboratory | JAX #018448 |
| Mouse: Foxj1tm1.1(cre/ERT2/GFP)Htg/J (backcrossed to B6) | The Jackson Laboratory | JAX #027012 |
| Mouse: B6.FVB(129S4)-Tg(Ckmm-cre)5Khn/J | The Jackson Laboratory | JAX #000664 |
| Mouse: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | The Jackson Laboratory | JAX #006148 |
| Mouse: B6.Sox8 | Dr. Koji Hase | (O’Bryan et al., 2008) |
| Mouse: B6.Spib | Dr. Tsuneyasu Kaisho | (Sasaki et al., 2012) |
| Recombinant DNA | ||
| 3Xflag-pA-Tn5-Fl | Dr. Steve Henikoff | Addgene #124601 |
| Software and algorithms | ||
| Cell Ranger ATAC v1.1.0 | 10X Genomics | N/A |
| SnapTools v1.4.1 | (Fang et al., 2021) | https://github.com/r3fang/SnapTools |
| SnapATAC v1.0.0 | (Fang et al., 2021) | https://github.com/r3fang/SnapATAC |
| macs2 v2.1.1.2 | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| IGV v2.4.14 | (Robinson et al., 2011) | https://software.broadinstitute.org/software/igv/ |
| bedtools v2.26.0 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io |
| HOMER v4.9 | (Heinz et al., 2010) | http://homer.ucsd.edu |
| chromVAR v1.4.1 | (Schep et al., 2017) | https://github.com/GreenleafLab/chromVAR |
| HINT-ATAC v0.13.1 | (Li et al., 2019) | https://www.regulatory-genomics.org |
| STAR v2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Subread v2.0.0 | (Liao et al., 2013) | http://subread.sourceforge.net/ |
| R v4.1.0 | R Core Team | https://www.r-project.org/ |
| DESeq2 v1.28.1 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR v3.34.0 | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Cell Ranger v6.1.0 | 10X Genomics | N/A |
| CITE-seq-count v1.4.3 | doi:10.5281/zenodo.2590196 | https://hoohm.github.io/CITE-seq-Count/ |
| Seurat v4.0.2 | (Hao et al., 2021) | https://satijalab.org/seurat/ |
| kallisto-bustools v0.24.4 | (Melsted et al., 2021) | https://www.kallistobus.tools/ |
| scVelo v0.2.2 | (Bergen et al., 2020) | https://scvelo.readthedocs.io/ |
| Trimmomatic v0.36 | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| bowtie2 v2.2.9 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/ |
| SAMtools v1.3.1 | (Li et al., 2009) | http://www.htslib.org/ |
| picard v2.8.0 | N/A | http://broadinstitute.github.io/picard |
| deeptools v3.0.2 | (Ramírez et al., 2016) | https://deeptools.readthedocs.io |
| SEACR v1.3 | (Meers et al., 2019) | https://github.com/FredHutch/SEACR |
| Nikon Elements v5.02 | Nikon | N/A |
| Vision 4D v3.4 | Arivis | N/A |
| Flowjo v10.7.1 | BD Biosciences | N/A |
| Prism v7.0 | GraphPad | N/A |
| ImageJ v1.52 | (Schindelin et al., 2012) | https://imagej.net/software/fiji/ |
Highlights.
The thymic stroma includes a constellation of peripheral cell mimics
Lineage-defining transcription factors drive mimetic cell accumulation
Aire is partially and variably required for mimetic cell accumulation
Expression of self-antigen in specific mimetic cell types induces T cell tolerance
ACKNOWLEDGMENTS
We gratefully acknowledge: N. Ramirez and the Harvard Bauer Core for scATAC-seq; A. Baysov, J. Lee, I. Magill, and the Broad Genomics Platform for RNA-seq; the HMS Biopolymers Sequencing Facility; the HMS Immunology Flow Core; the HMS MiCRoN Core; the NIH Tetramer Core; L. Du and the HMS Transgenic Mouse Core; K. Hattori and A. Ortiz-Lopez for experimental help; L. Yang, B. Vijaykumar, and N. Patel for computational help; C. Laplace for graphics; and O. Yaghi, G. Wang, G. Burgin, K. Langston, A. Mann, and K. Bansal for discussions. This work was supported by NIH grants R01AI088204 and R01DK060027 (to D.M.) and T32GM007753 (for D.A.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.05.018
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aaltonen J, Bjorses P, Perheentupa J, Horelli-Kuitunen N, Palotie A, Peltonen L, Lee YS, Francis F, Henning S, Thiel C, et al. (1997). An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet 17, 399–403. [DOI] [PubMed] [Google Scholar]
- Abramson J, and Anderson G (2017). Thymic epithelial cells. Annu. Rev. Immunol 35, 85–118. [DOI] [PubMed] [Google Scholar]
- Abramson J, Giraud M, Benoist C, and Mathis D (2010). Aire’s partners in the molecular control of immunological tolerance. Cell 140, 123–135. [DOI] [PubMed] [Google Scholar]
- Anderson MS, and Su MA (2016). AIRE expands: new roles in immune tolerance and beyond. Nat. Rev. Immunol 16, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, et al. (2002). Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401. [DOI] [PubMed] [Google Scholar]
- Bansal K, Michelson DA, Ramirez RN, Viny AD, Levine RL, Benoist C, and Mathis D (2021). Aire regulates chromatin looping by evicting CTCF from domain boundaries and favoring accumulation of cohesin on superenhancers. Proc. Natl. Acad. Sci. USA 118. e2110991118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal K, Yoshida H, Benoist C, and Mathis D (2017). The transcriptional regulator Aire binds to and activates super-enhancers. Nat. Immunol 18, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran-Gale J, Morgan MD, Maio S, Dhalla F, Calvo-Asensio I, Deadman ME, Handel AE, Maynard A, Chen S, Green F, et al. (2020). Ageing compromises mouse thymus function and remodels epithelial cell differentiation. Elife 9, e56221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista JL, Cramer NT, Miller CN, Chavez J, Berrios DI, Byrnes LE, Germino J, Ntranos V, Sneddon JB, Burt TD, et al. (2021). Single-cell transcriptional profiling of human thymic stroma uncovers novel cellular heterogeneity in the thymic medulla. Nat. Commun 12, 1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein C, Nevo S, Giladi A, Kadouri N, Pouzolles M, Gerbe F, David E, Machado A, Chuprin A, Tóth B, et al. (2018). Single-cell mapping of the thymic stroma identifies IL-25-producing tuft epithelial cells. Nature 559, 622–626. [DOI] [PubMed] [Google Scholar]
- Brennecke P, Reyes A, Pinto S, Rattay K, Nguyen M, Küchler R, Huber W, Kyewski B, and Steinmetz LM (2015). Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat. Immunol 16, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cangkrama M, Darido C, Georgy SR, Partridge D, Auden A, Srivastava S, Wilanowski T, and Jane SM (2016). Two ancient gene families are critical for maintenance of the mammalian skin barrier in postnatal life. J. Invest. Dermatol 136, 1438–1448. [DOI] [PubMed] [Google Scholar]
- Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, et al. (2020). A human cell atlas of fetal gene expression. Science 370, eaba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, Perekatt AO, and Verzi MP (2019). A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat. Genet 51, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, Sela A, Katz S, Grossman M, Guyon C, et al. (2015). The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat. Immunol 16, 737–745. [DOI] [PubMed] [Google Scholar]
- Derbinski J, Pinto S, Rösch S, Hexel K, and Kyewski B (2008). Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc. Natl. Acad. Sci. USA 105, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbinski J, Schulte A, Kyewski B, and Klein L (2001). Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat. Immunol 2, 1032–1039. [DOI] [PubMed] [Google Scholar]
- Dhalla F, Baran-Gale J, Maio S, Chappell L, Holländer GA, and Ponting CP (2020). Biologically indeterminate yet ordered promiscuous gene expression in single medullary thymic epithelial cells. EMBO J. 39, e101828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolton G, Tungatt K, Lloyd A, Bianchi V, Theaker SM, Trimby A, Holland CJ, Donia M, Godkin AJ, Cole DK, et al. (2015). More tricks with tetramers: a practical guide to staining T cells with peptide-MHC multi-mers. Immunology 146, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson D, Røyrvik EC, Aranda-Guillén M, Berger AH, Landegren N, Artaza H, Hallgren Å, Grytaas MA, Ström S, Bratland E, et al. (2021). GWAS for autoimmune Addison’s disease identifies multiple risk loci and highlights AIRE in disease susceptibility. Nat. Commun 12, 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with snapATAC. Nat. Commun 12, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr AG, and Rudensky A (1998). Medullary thymic epithelium: a mosiac of epithelial “self”? J. Exp. Med 188, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud M, Yoshida H, Abramson J, Rahl PB, Young RA, Mathis D, and Benoist C (2012). Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc. Natl. Acad. Sci. USA 109, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golson ML, and Kaestner KH (2016). Fox transcription factors: from development to disease. Development 143, 4558–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D, Abramson J, Benoist C, and Mathis D (2007). Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J. Exp. Med 204, 2521–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerau-de-Arellano M, Martinic M, Benoist C, and Mathis D (2009). Neonatal tolerance revisited: a perinatal window for Aire control of autoimmunity. J. Exp. Med 206, 1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Shikama-Dorn N, Zhanybekova S, Maio S, Graedel AN, Zuklys S, Ponting CP, and Holländer GA (2018). Comprehensively profiling the chromatin architecture of tissue restricted antigen expression in thymic epithelial cells over development. Front. Immunol 9, 2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM III, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassall AH (1846). The Microscopic Anatomy of the Human Body: in Health and Disease (S. Highley; ). [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, and Kasper M (2016). Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 3, 221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadouri N, Nevo S, Goldfarb Y, and Abramson J (2020). Thymic epithelial cell heterogeneity: TEC by TEC. Nat. Rev. Immunol 20, 239–253. [DOI] [PubMed] [Google Scholar]
- Kaiser C, Bradu A, Gamble N, Caldwell JA, and Koh AS (2022). AIRE in context: leveraging chromatin plasticity to trigger ectopic gene expression. Immunol. Rev 305, 59–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya T, Hase K, Takahashi D, Fukuda S, Hoshino K, Sasaki I, Hemmi H, Knoop KA, Kumar N, Sato M, et al. (2012). The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat. Immunol 13, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Kobayashi N, Nakamura Y, Kanaya T, Takahashi D, Fujiki R, Mutoh M, Obata Y, Iwanaga T, Nakagawa T, et al. (2019). Sox8 is essential for M cell maturation to accelerate IgA response at the early stage after weaning in mice. J. Exp. Med 216, 831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh AS, Kuo AJ, Park SY, Cheung P, Abramson J, Bua D, Carney D, Shoelson SE, Gozani O, Kingston RE, et al. (2008). Aire employs a his-tone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc. Natl. Acad. Sci. USA 105, 15878–15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh AS, Miller EL, Buenrostro JD, Moskowitz DM, Wang J, Greenleaf WJ, Chang HY, and Crabtree GR (2018). Rapid chromatin repression by Aire provides precise control of immune tolerance. Nat. Immunol 19, 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, Podda M, Xu C, Xie G, Macciardi F, et al. (2010). Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet 42, 658–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas B, White AJ, Cosway EJ, Parnell SM, James KD, Jones ND, Ohigashi I, Takahama Y, Jenkinson WE, and Anderson G (2020). Diversity in medullary thymic epithelial cells controls the activity and availability of iNKT cells. Nat. Commun 11, 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Linehan JL, Dileepan T, Lee YJ, Purtha WE, Lu JV, Nelson RW, Fife BT, Orr HT, Anderson MS, et al. (2016). Tolerance is established in polyclonal CD4+ T cells by distinct mechanisms, according to self-peptide expression patterns. Nat. Immunol 17, 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S (1888). Zur Lehre von der Schilddrüse und Thymus bei den Amphibien. Anat. Anz 3, 97–103. [Google Scholar]
- Meers MP, Tenenbaum D, and Henikoff S (2019). Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melsted P, Booeshaghi AS, Liu L, Gao F, Lu L, Min KHJ, da Veiga Beltrame E, Hjörleifsson KE, Gehring J, and Pachter L (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol 39, 813–818. [DOI] [PubMed] [Google Scholar]
- Meredith M, Zemmour D, Mathis D, and Benoist C (2015). Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat. Immunol 16, 942–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger TC, Khan IS, Gardner JM, Mouchess ML, Johannes KP, Krawisz AK, Skrzypczynska KM, and Anderson MS (2013). Lineage tracing and cell ablation identify a post-Aire-expressing thymic epithelial cell population. Cell Rep. 5, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CN, Proekt I, von Moltke J, Wells KL, Rajpurkar AR, Wang H, Rattay K, Khan IS, Metzger TC, Pollack JL, et al. (2018). Thymic tuft cells promote an IL-4-enriched medulla and shape thymocyte development. Nature 559, 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JFAP (1961). Immunological function of the thymus. Lancet 2, 748–749. [DOI] [PubMed] [Google Scholar]
- Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, et al. (2018). A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Hataye J, Pagán AJ, Pepper M, McLachlan JB, Zell T, and Jenkins MK (2009). Tracking epitope-specific T cells. Nat. Protoc 4, 565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK (2007). Naive CD4+ T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, et al. (1997). Positional cloning of the APECED gene. Nat. Genet 17, 393–398. [DOI] [PubMed] [Google Scholar]
- O’Bryan MK, Takada S, Kennedy CL, Scott G, Harada S, Ray MK, Dai Q, Wilhelm D, de Kretser DM, Eddy EM, et al. (2008). Sox8 is a critical regulator of adult Sertoli cell function and male fertility. Dev. Biol 316, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oftedal BE, Hellesen A, Erichsen MM, Bratland E, Vardi A, Perheentupa J, Kemp EH, Fiskerstrand T, Viken MK, Weetman AP, et al. (2015). Dominant mutations in the autoimmune regulator AIRE are associated with common organ-specific autoimmune diseases. Immunity 42, 1185–1196. [DOI] [PubMed] [Google Scholar]
- Onder L, Nindl V, Scandella E, Chai Q, Cheng HW, Caviezel-Firner S, Novkovic M, Bomze D, Maier R, Mair F, et al. (2015). Alternative NF-κB signaling regulates mTEC differentiation from podoplanin-expressing precursors in the cortico-medullary junction. Eur. J. Immunol 45, 2218–2231. [DOI] [PubMed] [Google Scholar]
- Panneck AR, Rafiq A, Schütz B, Soultanova A, Deckmann K, Chubanov V, Gudermann T, Weihe E, Krasteva-Christ G, Grau V, et al. (2014). Cholinergic epithelial cell with chemosensory traits in murine thymic medulla. Cell Tissue Res. 358, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Botting RA, Domínguez Conde CC, Popescu DM, Lavaert M, Kunz DJ, Goh I, Stephenson E, Ragazzini R, Tuck E, et al. (2020). A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parviz F, Matullo C, Garrison WD, Savatski L, Adamson JW, Ning G, Kaestner KH, Rossi JM, Zaret KS, and Duncan SA (2003). Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet 34, 292–296. [DOI] [PubMed] [Google Scholar]
- Pinto S, Michel C, Schmidt-Glenewinkel H, Harder N, Rohr K, Wild S, Brors B, and Kyewski B (2013). Overlapping gene coexpression patterns in human medullary thymic epithelial cells generate self-antigen diversity. Proc. Natl. Acad. Sci. USA 110, E3497–E3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qunhua L, James BB, Haiyan H, and Peter JB (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779. [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remak R (1855). Untersuchungen über die Entwickelung der Wirbelthiere (G. Reimer; ). [Google Scholar]
- Renier N, Wu Z, Simon DJ, Yang J, Ariel P, and Tessier-Lavigne M (2014). iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, and Holländer GA (2014). Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24, 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki I, Hoshino K, Sugiyama T, Yamazaki C, Yano T, Iizuka A, Hemmi H, Tanaka T, Saito M, Sugiyama M, et al. (2012). Spi-B is critical for plasmacytoid dendritic cell function and development. Blood 120, 4733–4743. [DOI] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, Smillie C, Smith-Rosario G, Wu J, Dionne D, et al. (2020). A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM III, Smibert P, and Satija R (2018). Cell hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 19, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, and Ozato K (2005). IFN regulatory factor-4 and −8 govern dendritic cell subset development and their functional diversity. J. Immunol 174, 2573–2581. [DOI] [PubMed] [Google Scholar]
- Terao C, Yamada R, Ohmura K, Takahashi M, Kawaguchi T, Kochi Y, Human Disease Genomics Working Group; RA Clinical and Genetic Study Consortium, Okada Y, Nakamura Y, et al. (2011). The human AIRE gene at chromosome 21q22 is a genetic determinant for the predisposition to rheumatoid arthritis in Japanese population. Hum. Mol. Genet 20, 2680–2685. [DOI] [PubMed] [Google Scholar]
- Ting SB, Caddy J, Hislop N, Wilanowski T, Auden A, Zhao LL, Ellis S, Kaur P, Uchida Y, Holleran WM, et al. (2005). A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science 308, 411–413. [DOI] [PubMed] [Google Scholar]
- UK IBD Genetics Consortium, Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, et al. (2009). Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet 41, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Velde RL, and Friedman NB (1966). The thymic “myoidzellen” and myasthenia gravis. JAMA 198, 287–288. [DOI] [PubMed] [Google Scholar]
- Van Santen HM, Benoist C, and Mathis D (2004). Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J. Exp. Med 200, 1221–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villaseñor J, Besse W, Benoist C, and Mathis D (2008). Ectopic expression of peripheral-tissue antigens in the thymic epithelium: probabilistic, monoallelic, misinitiated. Proc. Natl. Acad. Sci. USA 105, 15854–15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watney H (1882). The minute anatomy of the thymus. London. Philos. Trans. R. Soc. Lond 173, 1063. [Google Scholar]
- Wells KL, Miller CN, Gschwind AR, Wei W, Phipps JD, Anderson MS, and Steinmetz LM (2020). Combined transient ablation and single-cell RNA-sequencing reveals the development of medullary thymic epithelial cells. Elife 9, e60188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JL, and Bye WA (1984). The membranous epithelial (M) cell and the mucosal immune system. Annu. Rev. Med 35, 95–112. [DOI] [PubMed] [Google Scholar]
- Yang S, Fujikado N, Kolodin D, Benoist C, and Mathis D (2015). Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Bansal K, Schaefer U, Chapman T, Rioja I, Proekt I, Anderson MS, Prinjha RK, Tarakhovsky A, Benoist C, et al. (2015). Brd4 bridges the transcriptional regulators, Aire and P-TEFb, to promote elongation of peripheral-tissue antigen transcripts in thymic stromal cells. Proc. Natl. Acad. Sci. USA 112, E4448–E4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scATAC-seq, scRNA-seq, bulk RNA-seq and CUT&Tag data have been deposited at the Gene Expression Omnibus (GEO: GSE194253). Aire-lineage-tracing scRNA-seq (GEO: GSE137699), human fetal thymus scRNA-seq (E-MTAB-8581), Aire−/− bulk RNA-seq (GEO: GSE180935), Aire ChIP-seq (GEO: GSE92597) and histone mark ChIP-seq (GEO: GSE114713, GEO: GSE53109), all from mTECs, were reanalyzed from previous work. This study did not generate original code; scripts used for major analyses are available via Github (github.com/dmichelson). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.